User login
Beta-blockers for hypertension: Are they going out of style?
In recent years the role of beta-blockers as a primary tool to treat hypertension has come under question. These drugs have shown disappointing results when used as antihypertensive therapy in patients without heart disease, ie, when used as primary prevention. At the same time, beta-blockers clearly reduce the risk of future cardiovascular events in patients who already have heart disease, eg, who already have had a myocardial infarction or who have congestive heart failure.
Several meta-analyses and a few clinical trials have shown that beta-blockers may have no advantage over other antihypertensive drugs, and in fact may not reduce the risk of stroke as effectively as other classes of blood pressure medications.
Why should this be? Is it that the patients in the antihypertensive trials were mostly older, and that beta-blockers do not work as well in older patients as in younger ones? Or does it have to do with the fact that atenolol (Tenormin) was the drug most often used in the trials? Would newer, different beta-blockers be better?
Hypertension experts currently disagree on how to interpret the available data, and this has led to conflict and confusion among clinicians as to the role of beta-blockers in managing hypertension. Current evidence suggests that older beta-blockers may not be the preferred first-line antihypertensive drugs for hypertensive patients who have no compelling indications for them (eg, heart failure, myocardial infarction, diabetes, high risk of coronary heart disease). However, newer beta-blockers with vasodilatory properties should be considered in cases of uncontrolled or resistant hypertension, especially in younger patients.
Further, while controversy and debate continue over the benefits and adverse effects of one class of antihypertensive drugs vs another, it is indisputable that controlling arterial blood pressure to the recommended goal offers major protection against cardiovascular and renal events in patients with hypertension.1,2
MECHANISM OF ACTION OF BETA-BLOCKERS
Beta-blockers effectively reduce blood pressure in both systolic-diastolic hypertension and isolated systolic hypertension.3–5 Exactly how is not known, but it has been proposed that they may do so by:
Reducing the heart rate and cardiac output. When catecholamines activate beta-1 receptors in the heart, the heart rate and myocardial contractility increase. By blocking beta-1 receptors, beta-blockers reduce the heart rate and myocardial contractility, thus lowering cardiac output and arterial blood pressure.6
Inhibiting renin release. Activation of the renin-angiotensin system is another major pathway that can lead to elevated arterial blood pressure. Renin release is mediated through the sympathetic nervous system via beta-1 receptors on the juxtaglomerular cells of the kidney. Beta-blockers can therefore lower blood pressure by inhibiting renin release.7
Inhibiting central nervous sympathetic outflow, thereby inducing presynaptic blockade, which in turn reduces the release of catecholamines.
Reducing venous return and plasma volume.
Generating nitric oxide, thus reducing peripheral vascular resistance (some agents).8
Reducing vasomotor tone.
Reducing vascular tone.
Improving vascular compliance.
Resetting baroreceptor levels.
Attenuating the pressor response to catecholamines with exercise and stress.
HETEROGENEITY OF BETA-BLOCKERS
Selectivity
Beta-blockers are not all the same. They can be classified into three categories.
Nonselective beta-blockers block both beta-1 and beta-2 adrenergic receptors. It is generally accepted that beta-blockers exert their primary antihypertensive effect by blocking beta-1 adrenergic receptors.6 Of interest, nonselective beta-blockers inhibit beta-2 receptors on arteries and thus cause an unopposed alpha-adrenergic effect, leading to increased peripheral vascular resistance.9 Examples of this category:
- Nadolol (Corgard)
- Pindolol (Visken)
- Propranolol (Inderal)
- Timolol (Blocadren).
Selective beta-blockers specifically block beta-1 receptors alone, although they are known to be nonselective at higher doses. Examples:
- Atenolol (Tenormin)
- Betaxolol (Kerlone)
- Bisoprolol (Zebeta)
- Esmolol (Brevibloc)
- Metoprolol (Lopressor, Toprol).
Beta-blockers with peripheral vasodilatatory effects act either via antagonism of the alpha-1 receptor, as with labetolol (Normodyne) and carvedilol (Coreg),10 or via enhanced release of nitric oxide, as with nebivolol (Bystolic).8
Lipid and water solubility
The lipid solubility and water solubility of each beta-blocker determine its bioavailability and side-effect profile.
Lipid solubility determines the degree to which a beta-blocker penetrates the blood-brain barrier and thereby leads to central nervous system side effects such as lethargy, nightmares, confusion, and depression. Propranolol is highly lipid-soluble; metoprolol and labetalol are moderately so.
Water-soluble beta-blockers such as atenolol have less tissue permeation, have a longer half-life, and cause fewer central nervous system effects and symptoms.11
Routes of elimination
Beta-blockers also differ in their route of elimination.
Atenolol and nadolol are eliminated by the kidney and require dose adjustment in patients with impaired renal function.12,13
On the other hand, propranolol, metoprolol, labetalol, carvedilol, and nebivolol are excreted primarily via hepatic metabolism.13
BETA-BLOCKERS IN THE MANAGEMENT OF HYPERTENSION
Beta-blockers were initially used to treat arrhythmias, but by the early 1970s they were also widely accepted for managing hypertension. 14 Their initial acceptance as one of the first-line classes of drugs for hypertension was based on their better side-effect profile compared with other antihypertensive drugs available at that time.
In the 1980s and 1990s, beta-blockers were listed as preferred first-line antihypertensive drugs along with diuretics in national hypertension guidelines.15 Subsequent updates of the guidelines favored diuretics as initial therapy and relegated all other classes of antihypertensive medications to be alternatives to diuretics.16 Although beta-blockers remain alternative first-line drugs in the latest guidelines (published in 2003; see reference 66), they are the preferred antihypertensive agents for patients with cardiac disease.
The current recommendations reflect the findings from hypertension trials in which patients with myocardial infarction and congestive heart failure had better cardiovascular outcomes if they received these drugs,17–19 including a lower risk of death.20,21 It was widely assumed that beta-blockers would also prevent first episodes of cardiovascular events.
However, to date, there is no evidence that beta-blockers are effective as primary prevention. Several large randomized controlled trials showed no benefit with beta-blockers compared with other antihypertensive drugs—in fact, there were more cardiovascular events with beta-blockers (see below).
Beta-blockers are well tolerated in clinical practice, although they can have side effects that include fatigue, depression, impaired exercise tolerance, sexual dysfunction, and asthma attacks.
Wiysonge et al22 analyzed how many patients withdrew from randomized trials of antihypertensive treatment because of drug-related adverse events. There was no significant difference in the incidence of fatigue, depressive symptoms, or sexual dysfunction with beta-blockers compared with placebo, and trial participants on a beta-blocker were not statistically significantly more likely to discontinue treatment than those receiving a placebo in three trials with 22,729 participants (relative risk [RR] 2.34, 95% confidence interval [CI] 0.84–6.52).
THE CONTROVERSY: WHAT THE TRIALS SHOWED
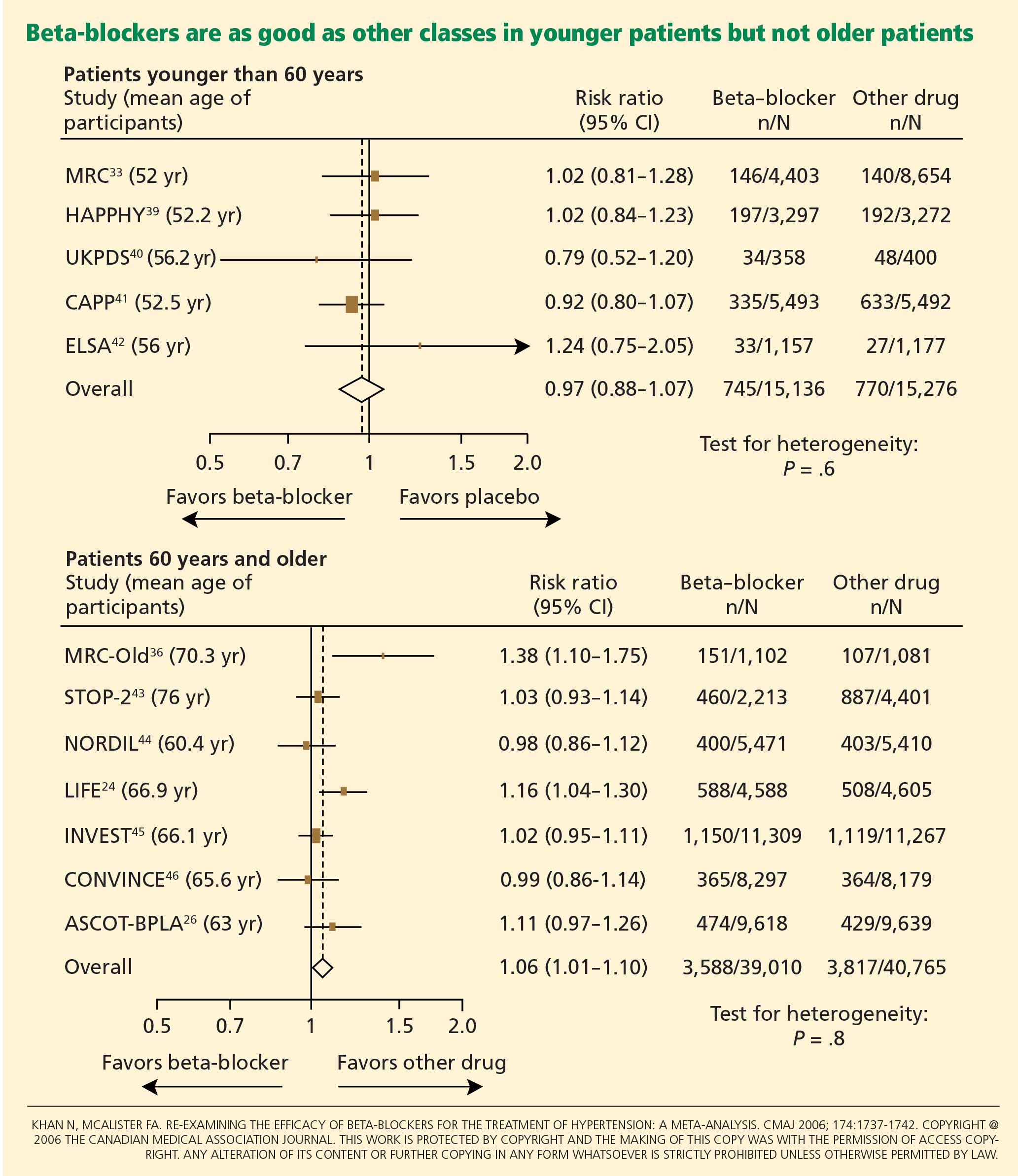
Messerli et al23 performed a meta-analysis published in 1998 that suggested that beta-blockers may not be as effective as diuretics in preventing cardiovascular events when used as first-line antihypertensive therapy in elderly patients. In 10 randomized controlled trials in 16,164 patients who were treated with either a diuretic or a beta-blocker (atenolol), blood pressure was normalized in two-thirds of diuretic-treated patients but only one-third of patients treated with atenolol as monotherapy. Diuretic therapy was superior with regard to all end points, and beta-blockers were found to be ineffective except in reducing cerebrovascular events.
The LIFE study (Losartan Intervention for Endpoint Reduction in Hypertension)24 compared the angiotensin-receptor blocker losartan (Cozaar) and atenolol in 9,193 patients with hypertension and left ventricular hypertrophy. At 4 years of follow-up, the rate of primary cardiovascular events (death, myocardial infarction, or stroke) was lower in the losartan group than in the atenolol group. The difference was mainly due to a 25% lower incidence of stroke, which was statistically significant. The rates of myocardial infarction and death from cardiovascular causes were not significantly different between the two treatment groups. The systolic blood pressure was 1 mm Hg lower in the losartan group than in the atenolol group, which was statistically significant.
Carlberg et al25 performed another important meta-analysis that questioned whether atenolol reduces rates of cardiovascular morbidity and death in hypertensive patients. The results were surprising: eight randomized controlled trials including more than 6,000 patients and comparing atenolol with placebo or no treatment showed no differences between the treatment groups with regard to the outcomes of all-cause mortality (RR 1.01, 95% CI 0.89–1.15), cardiovascular mortality (RR 0.99, 95% CI 0.83–1.18), or myocardial infarction (RR 0.99, 95% CI 0.83–1.19).
In addition, when atenolol was compared with other antihypertensives in five other randomized controlled trials that included more than 14,000 patients, those treated with atenolol had a higher risk of stroke (RR 1.30, 95% CI 1.12–1.50) and death (RR 1.13, 95% CI 1.02–1.25).
The ASCOT-BPLA trial (Anglo-Scandinavian Cardiac Outcomes Trial—Blood Pressure Lowering Arm)26 had similar results. This trial compared the combination of atenolol plus the diuretic bendroflumethiazide against the combination of the calcium channel blocker amlodipine (Norvasc) plus the angiotensin-converting enzyme (ACE) inhibitor perindopril (Aceon). Although no significant difference was seen in the primary outcome of nonfatal myocardial infarction or fatal coronary heart disease (unadjusted hazard ratio [HR] with amlodipine-perindopril 0.90, 95% CI 0.79–1.02, P = .1052), the amlodipine-plus-perindopril group had significantly fewer strokes (327 vs 422, HR 0.77, 95% CI 0.66–0.89, P = .0003), fewer total cardiovascular events (1,362 vs 1,602, HR 0.84, 95% CI 0.78–0.90, P = .0001), and fewer deaths from any cause (738 vs 820; HR 0.89, 95% CI 0.81–0.99, P = .025).
Lindholm et al27 performed a meta-analysis that included studies of selective beta-blockers (including atenolol) and nonselective beta-blockers, with a follow-up time of more than 2 years. Compared with placebo or no treatment, beta-blockers reduced the risk of stroke by 19% but had no effect on myocardial infarction or all-cause mortality. Compared with other antihypertensive drugs, beta-blockers were less than optimum, and the relative risk of stroke was 16% higher. Atenolol was the beta-blocker used in most of the randomized clinical trials included in this meta-analysis.
The Cochrane group22 found beta-blockers to be inferior to all other antihypertensive drugs with respect to the ability to lower the risk of stroke.
WHY WERE THE RESULTS SO DISAPPOINTING?
Problems with atenolol
Most of the trials in the meta-analyses discussed above used atenolol and other beta-blockers that had no vasodilatory properties.
Further, in most of the trials atenolol was used in a once-daily dosage, whereas ideally it needs to be taken more frequently, based on its pharmacokinetic and pharmacodynamic properties (a half-life of 6–9 hours).3 Neutel et al28 confirmed that atenolol, when taken once daily, leaves the patient unprotected in the last 6 hours of a 24-hour period, as demonstrated by 24-hour ambulatory blood pressure monitoring. It is possible that this short duration of action of atenolol may have contributed to the results observed in clinical trials that used atenolol to treat hypertension.
Differences between older and younger patients
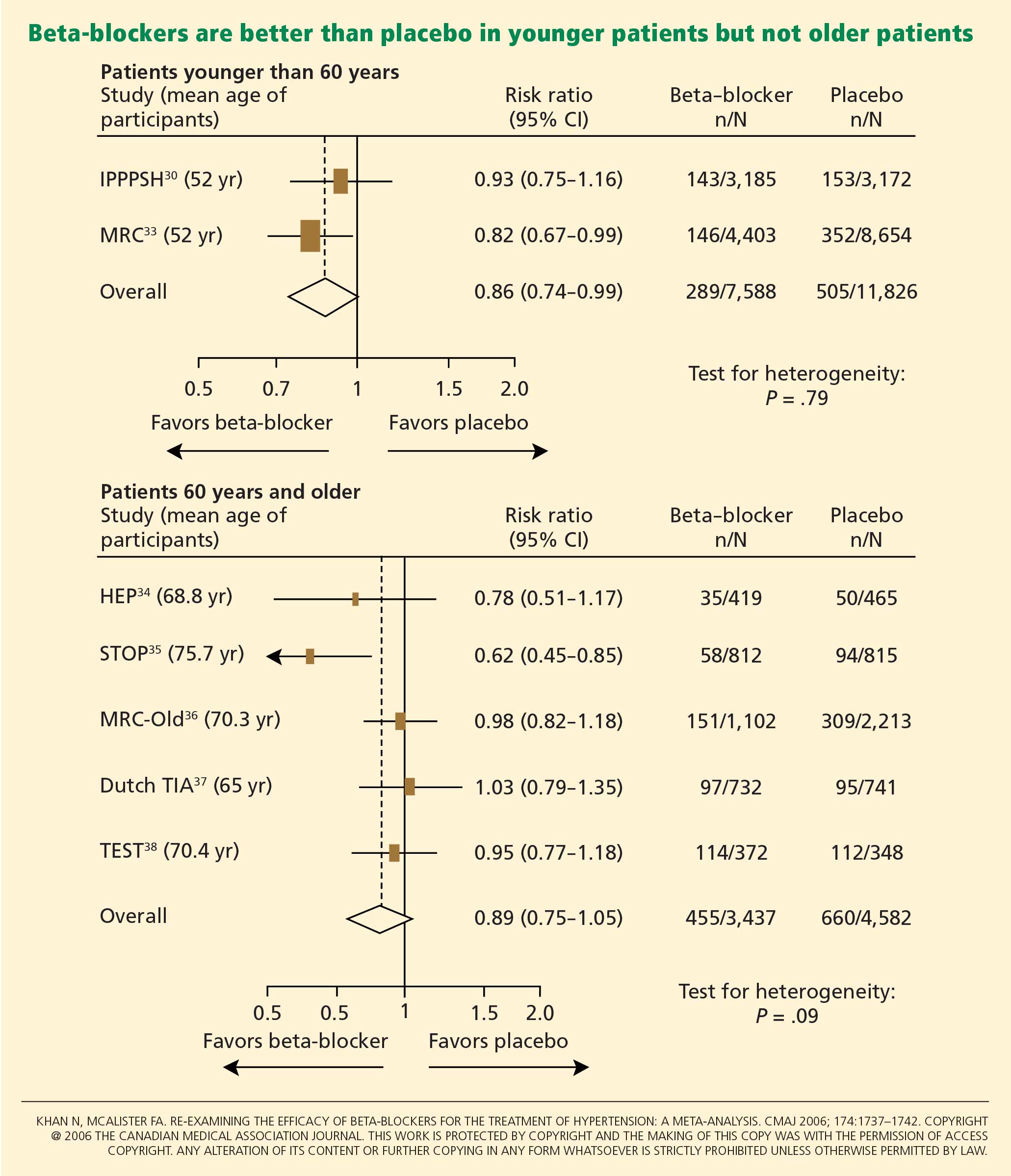
Another possible reason for the disappointing results is that the trials included many elderly patients, in whom beta-blockers may not be as effective. The pathophysiology of hypertension in younger people is different from that in older patients.29 Hemodynamic characteristics of younger hypertensive patients include a high cardiac output and hyperdynamic circulation with a low pulse pressure, while older patients have lower arterial compliance with an elevated vascular resistance.
The notion of choosing antihypertensive medications on the basis of age and age-related pathophysiology is supported by several clinical studies. Randomized controlled trials appear to show that beta-blockers are effective in younger hypertensive patients.30
Conversely, the CAFE (Conduit Artery Function Evaluation) trial,31 a substudy of the main ASCOT trial,26 indicated that betablocker-based therapy was less effective in reducing central aortic pressure than were regimens based on an ACE inhibitor or a calcium channel blocker.
The CAFE researchers recruited 2,073 patients from five ASCOT centers and used radial artery applanation tonometry and pulse-wave analysis to derive central aortic pressures and hemodynamic indices during study visits up to a period of 4 years. Although the two treatment groups achieved similar brachial systolic blood pressures, the central aortic systolic pressure was 4.3 mm Hg lower in the amlodipine group (95% CI 3.3–5.4; P < .0001), and the central aortic pulse pressure was 3.0 mm Hg lower (95% CI 2.1–3.9; P < .0001).
Pulse-wave dyssynchrony
Bangalore et al47 offer an interesting hypothesis to explain the probable adverse effect of beta-blockers. Their theory concerns the effect of these drugs on the arterial pulse wave.
Normally, with each contraction of the left ventricle during systole, an arterial pulse wave is generated and propagated forward to the peripheral arteries. This wave is then reflected back to the heart from the branching points of peripheral arteries. The final form of the pressure wave at the aortic root is a synchronized summation of the forward-traveling wave and the backward-reflected wave.
In healthy people with normal arteries, the reflected wave merges with the forward-traveling wave in diastole and augments coronary blood flow. In patients whose arteries are stiff due to aging or vascular comorbidities, the reflected wave returns faster and merges with the incident wave in systole, resulting in higher left ventricular afterload and less coronary perfusion.48
Bangalore et al47 propose that artificially reducing the heart rate with beta-blockers may further dyssynchronize the pulse wave, adversely affecting coronary perfusion and leading to an increased risk of cardiovascular events and death.
Metabolic side effects
Older beta-blockers, and especially atenolol, have well-known metabolic adverse effects, particularly impairment of glycemic control. This adverse effect appears to occur only with beta-blockers that do not possess vasodilatory properties and thus increase peripheral vascular resistance, which results in lower glucose availability and reduced uptake by skeletal muscles.49
Bangalore et al50 evaluated the effect of beta-blockers in a meta-analysis of 12 studies in 94,492 patients followed up for more than 1 year. Beta-blocker therapy resulted in a 22% higher risk of new-onset diabetes mellitus (RR 1.22, 95% CI 1.12–1.33) than with other nondiuretic antihypertensive agents.
Of note, however, the meta-analysis did not show a significantly higher risk of the onset of diabetes with propranolol or metoprolol than with other nondiuretic antihypertensives when studies of these beta-blockers were separated from atenolol-based studies.
Further, the United Kingdom Prospective Diabetes Study40 found that cardiovascular outcomes in patients with good blood pressure control were similar when atenolol-based therapy was compared with therapy with the ACE inhibitor captopril (Capoten).
A meta-analysis conducted by Balamuthusamy et al51 in 2009 found no higher risk of stroke in patients with hypertension and diabetes mellitus who received beta-blockers than in those who received other antihypertensive medications. However, beta-blockers were associated with a higher risk of death from cardiovascular causes (RR 1.39, 95% CI 1.07–1.804; P < .01) compared with reninangiotensin blockade.
NEWER BETA-BLOCKERS MAY BE BETTER
In the United States, more than 40 million prescriptions for atenolol are written every year, making it by far the most commonly used beta-blocker for the treatment of hypertension. 52 It is clear, however, that atenolol is not an ideal representative of this class of antihypertensive medications.
Preliminary data from studies of newer beta-blockers that possess beneficial vasodilatory properties are encouraging. Animal studies and preliminary human studies find that these new-generation beta-blockers cause fewer adverse metabolic effects and improve endothelial function, measures of arterial stiffness, and cardiovascular outcomes.
Carvedilol
Carvedilol is a nonselective beta-blocker with vasodilatory effects that are thought to be due to its ability to concurrently block alpha-1 receptors in addition to beta receptors. 53 In experiments in vitro and in trials in patients with diabetes and hypertension, carvedilol increased endothelial vasodilation and reduced inflammation and platelet aggregation. These effects may be achieved though antioxidant actions, thereby preserving nitric oxide bioactivity.54,55
In the Glycemic Effects in Diabetes Mellitus: Carvedilol-Metoprolol Comparison in Hypertensives (GEMINI) trial,56 carvedilol was associated with better maintenance of glycemic control in diabetic hypertensive patients than was metoprolol. Insulin sensitivity improved with carvedilol but not with metoprolol, and fewer patients on carvedilol progressed to microalbuminuria.
Nebivolol
Nebivolol is a novel selective beta-blocker with a much higher affinity for beta-1 adrenergic receptors than for beta-2 adrenergic receptors. Among all the beta-blockers in clinical use today, nebivolol has the highest selectivity for beta-1 receptors.8
Nebivolol causes vasodilation through activation of the l-arginine/nitric oxide pathway.57–59 Blockade of synthesis of nitric oxide leads to local arterial stiffness. Endothelial dysfunction is characterized by decreased bioavailability of nitric oxide and has been shown to be a strong predictor of cardiovascular outcomes. By generating nitric oxide, nebivolol reduces peripheral vascular resistance, overcoming a significant side effect of earlier beta-blockers that lowered blood pressure but ultimately increased peripheral vascular tone and resistance.8
In an experiment in a bovine model,60 nebivolol significantly reduced the pulse-wave velocity (a measure of arterial stiffness), while atenolol had no effect. Moreover, evidence for the role of the l-arginine/nitric oxide pathway in the vasodilatory effect of nebivolol was demonstrated by co-infusion of NG-monomethyl-L-arginine, a specific endothelial nitric oxide synthetase inhibitor that attenuated the reduction of pulse-wave velocity by nebivolol.
In studies in hypertensive patients, nebivolol was associated with a better metabolic profile than atenolol, with none of the adverse effects on insulin sensitivity that atenolol had.61 In the Study of Effects of Nebivolol Interventions on Outcomes and Rehospitalization in Seniors With Heart Failure (SENIORS) trial, significantly fewer patients receiving nebivolol died or were admitted to the hospital for cardiovascular reasons compared with those receiving placebo.62
Although these findings are encouraging, we do not yet know if these effects will translate into a significant reduction in cardiovascular outcomes in clinical trials. Large, prospective hypertension outcome trials, particularly to evaluate primary prevention of cardiovascular outcomes, are needed for an evidence-based approach to using the newer beta-blockers as preferred first-line therapy for hypertension.
WHAT RECENT GUIDELINES SAY ABOUT BETA-BLOCKERS
The British National Institute for Health and Clinical Excellence and the British Hypertension Society, in their 2004 guidelines, recommended beta-blockers as one of several first-line antihypertensive medications in young, nonblack patients.63 On the other hand, they advised clinicians to be aware of the reported increase in onset of diabetes mellitus in patients treated with these medications. After the LIFE24 and ASCOT26 study results were published, these guidelines were amended to exclude beta-blockers as preferred routine initial therapy for hypertension.64
More recently, the 2007 European Society of Hypertension and European Society of Cardiology reconsidered the role of beta-blockers, recommending them as an option in both initial and subsequent antihypertensive treatment strategies.65
The current guidelines from the National Heart, Lung, and Blood Institute,66 which were published in 2003, were highly influenced by the results of the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT),2 and favor diuretics as the first-line therapy. However, they indicate that beta-blockers are a suitable alternative, particularly when a compelling cardiac indication is present.53 We hope that the next update, expected late in 2009, will re-address this issue in the light of more recent data.
- Staessen JA, Wang JG, Thijs L. Cardiovascular protection and blood pressure reduction: a meta-analysis. Lancet 2001; 358:1305–1315.
- ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002; 288:2981–2997.
- Neutel JM, Smith DH, Ram CV, et al. Application of ambulatory blood pressure monitoring in differentiating between antihypertensive agents. Am J Med 1993; 94:181–187.
- Materson BJ, Reda DJ, Cushman WC, et al. Single-drug therapy for hypertension in men. A comparison of six antihypertensive agents with placebo. The Department of Veterans Affairs Cooperative Study Group on Antihypertensive Agents. N Engl J Med 1993; 328:914–921.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- Frishman W, Silverman R. Clinical pharmacology of the new beta-adrenergic blocking drugs. Part 2. Physiologic and metabolic effects. Am Heart J 1979; 97:797–807.
- Garrett BN, Kaplan NM. Plasma renin activity suppression: duration after withdrawal from beta-adrenergic blockade. Arch Intern Med 1980; 140:1316–1318.
- Pedersen ME, Cockcroft JR. The latest generation of beta-blockers: new pharmacologic properties. Curr Hypertens Rep 2006; 8:279–286.
- Man in’t Veld AJ, Van den Meiracker AH, Schalekamp MA. Do beta-blockers really increase peripheral vascular resistance? Review of the literature and new observations under basal conditions. Am J Hypertens 1988; 1:91–96.
- Pearce CJ, Wallin JD. Labetalol and other agents that block both alpha- and beta-adrenergic receptors. Cleve Clin J Med 1994; 61:59–69.
- Dimsdale JE, Newton RP, Joist T. Neuropsychological side effects of beta-blockers. Arch Intern Med 1989; 149:514–525.
- Agarwal R. Supervised atenolol therapy in the management of hemodialysis hypertension. Kidney Int 1999; 55:1528–1535.
- Sica DA, Black HR. Pharmacologic considerations in the positioning of beta-blockers in antihypertensive therapy. Curr Hypertens Rep 2008; 10:330–335.
- Prichard BN, Gillam GP. Use of propranolol (Inderal) in treatment of hypertension. Br Med J 1964; 19; 2:725–727.
- The fifth report of the Joint National Committee on Detection, Evaluation, and Treatment of High Blood Pressure (JNC V). Arch Intern Med 1993; 153:154–183.
- Moser M. Evolution of the treatment of hypertension from the 1940s to JNC V. Am J Hypertens 1997; 10:2S–8S.
- Houghton T, Freemantle N, Cleland JG. Are beta-blockers effective in patients who develop heart failure soon after myocardial infarction? A meta-regression analysis of randomised trials. Eur J Heart Fail 2000; 2:333–340.
- The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet 1999; 353:9–13.
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999; 353:2001–2007.
- Yusuf S, Peto R, Lewis J, Collins R, Sleight P. Beta blockade during and after myocardial infarction: an overview of the randomized trials. Prog Cardiovasc Dis 1985; 27:335–371.
- Brophy JM, Joseph L, Rouleau JL. Beta-blockers in congestive heart failure. A Bayesian meta-analysis. Ann Intern Med 2001; 134:550–560.
- Wiysonge CS, Bradley H, Mayosi BM, et al. Beta-blockers for hypertension. Cochrane Database Syst Rev 2007;CD002003.
- Messerli FH, Grossman E, Goldbourt U. Are beta-blockers efficacious as first-line therapy for hypertension in the elderly? A systematic review. JAMA 1998; 279:1903–1907.
- Dahlöf B, Devereux RB, Kjeldsen SE, et al; for the LIFE study group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint Reduction in Hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002; 359:995–1003.
- Carlberg B, Samuelsson O, Lindholm LH. Atenolol in hypertension: is it a wise choice? Lancet 2004; 364:1684–1689.
- Dahlöf B, Sever PS, Poulter NR, et al; ASCOT Investigators. Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA): a multicentre randomised controlled trial. Lancet 2005; 366:895–906.
- Lindholm LH, Carlberg B, Samuelsson O. Should beta-blockers remain first choice in the treatment of primary hypertension? A meta-analysis. Lancet 2005; 366:1545–1553.
- Neutel JM, Schnaper H, Cheung DG, Graettinger WF, Weber MA. Antihypertensive effects of beta-blockers administered once daily: 24-hour measurements. Am Heart J 1990; 120:166–171.
- Franklin SS, Gustin W, Wong ND, et al. Hemodynamic patterns of age-related changes in blood pressure. The Framingham Heart Study. Circulation 1997; 96:308–315.
- The IPPPSH Collaborative Group. Cardiovascular risk and risk factors in a randomized trial of treatment based on the beta-blocker oxprenolol: the International Prospective Primary Prevention Study in Hypertension (IPPPSH). J Hypertens 1985; 3:379–392.
- Williams B, Lacy PS, Thom SM, et al; CAFE Investigators. Differential impact of blood pressure-lowering drugs on central aortic pressure and clinical outcomes: principal results of the Conduit Artery Function Evaluation (CAFE) study. Circulation 2006; 113:1213–1225.
- Khan N, McAlister FA. Re-examining the efficacy of beta-blockers for the treatment of hypertension: a meta-analysis. CMAJ 2006; 174:1737–1742.
- Medical Research Council Working Party. MRC trial of treatment of mild hypertension: principal results. BMJ 1985; 291:97–104.
- Coope J, Warrender TS. Randomised trial of treatment of hypertension in elderly patients in primary care. BMJ 1986; 293:1145–1151.
- Dahlöf B, Lindholm LH, Hansson L, et al. Morbidity and mortality in the Swedish Trial in Old Patients with Hypertension (STOP-Hypertension). Lancet 1991; 338:1281–1285.
- MRC Working Party. Medical Research Council trial of treatment of hypertension in older adults: principal results. BMJ 1992; 304:405–412.
- The Dutch TIA Study Group. Trial of secondary prevention with atenolol after transient ischemic attack or nondisabling ischemic stroke. Stroke 1993; 24:543–548.
- Eriksson S, Olofsson B-O, Wester P-O; for the TEST Study Group. Atenolol in secondary prevention after stroke. Cerebrovasc Dis 1995; 5:21–25.
- Wilhelmsen L, Berglund G, Elmfeldt D, et al. Beta-blockers versus diuretics in hypertensive men: main results from the HAPPHY trial. J Hypertens 1987; 5:561–572.
- UK Prospective Diabetes Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. BMJ 1998; 317:713–720.
- Hansson L, Lindholm LH, Niskanen L, et al. Effect of angiotensin-converting enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPP) randomised trial. Lancet 1999; 353:611–616.
- Lanchetti A, Bond MG, Henning M, et al. Calcium antagonist lacidipine slow down progression of asymptomatic carotid atherosclerosis. Principal results of the European Lacidipine Study on Atherosclerosis (ELSA), a randomized, double-blind, long-term trial. Circulation 2002; 106:2422–2427.
- Hansson L, Lindholm LH, Ekbom T, et al. Randomised trial of old and new antihypertensive drugs in elderly patients: cardiovascular mortality and morbidity in the Swedish Trial in Old Patients with Hypertension-2 study. Lancet 1999; 354:1751–1756.
- Hansson L, Hedner T, Lund-Johansen P, et al. Randomised trial of effects of calcium antagonists compared with diuretics and ß blockers on cardiovascular morbidity and mortality in hypertension: the Nordic Diltiazem (NORDIL) study. Lancet 2000; 356:359–365.
- Pepine CJ, Handsberg EM, Cooper-DeHoff RM, et al. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290:2805–2816.
- Black HR, Elliott WJ, Grandits G, et al. Principal results of the Controlled Onset Verapamil Investigation of Cardiovascular End Points (CONVINCE) Trial. JAMA 2003; 289:2073–2082.
- Bangalore S, Sawhney S, Messerli FH. Relation of beta-blocker-induced heart rate lowering and cardioprotection in hypertension. J Am Coll Cardiol 2008; 52:1482–1489.
- Boutouyrie P, Vermersch S, Laurent S, Briet M. Cardiovascular risk assessment through target organ damage: role of carotid to femoral pulse wave velocity. Clin Exp Pharmacol Physiol 2008; 35:530–533.
- Kveiborg B, Christiansen B, Major-Petersen A, Torp-Pedersen C. Metabolic effects of beta-adrenoceptor antagonists with special emphasis on carvedilol. Am J Cardiovasc Drugs 2006; 6:209–217.
- Bangalore S, Parkar S, Grossman E, Messerli FH. A meta-analysis of 94,492 patients with hypertension treated with beta-blockers to determine the risk of new-onset diabetes mellitus. Am J Cardiol 2007; 100:1254–1262.
- Balamuthusamy S, Molnar J, Adigopula S, Arora R. Comparative analysis of beta-blockers with other antihypertensive agents on cardiovascular outcomes in hypertensive patients with diabetes mellitus: a systematic review and meta-analysis. Am J Ther 2009; 16:133–142.
- Berenson A. Big drug makers see sales decline with their image. New York Times 2005 Nov 14.
- Bristow MR. Beta-adrenergic receptor blockade in chronic heart failure. Circulation 2000; 101:558–569.
- Giugliano D, Marfella R, Acampora R, Giunta R, Coppola L, D’Onofrio F. Effects of perindopril and carvedilol on endothelium-dependent vascular functions in patients with diabetes and hypertension. Diabetes Care 1998; 21:631–636.
- Lopez BL, Christopher TA, Yue TL, Ruffolo R, Feuerstein GZ, Ma XL. Carvedilol, a new beta-adrenoreceptor blocker antihypertensive drug, protects against free-radical-induced endothelial dysfunction. Pharmacology 1995; 51:165–173.
- Bakris GL, Fonseca V, Katholi RE, et al; GEMINI Investigators. Metabolic effects of carvedilol vs metoprolol in patients with type 2 diabetes mellitus and hypertension: a randomized controlled trial. JAMA 2004; 292:2227–2236.
- Georgescu A, Pluteanu F, Flonta ML, Badila E, Dorobantu M, Popov D. The cellular mechanisms involved in the vasodilator effect of nebivolol on the renal artery. Eur J Pharmacol 2005; 508:159–166.
- Kalinowski L, Dobrucki LW, Szczepanska-Konkel M, et al. Third-generation beta-blockers stimulate nitric oxide release from endothelial cells through ATP efflux: a novel mechanism for antihypertensive action. Circulation 2003; 107:2747–2752.
- Cockcroft JR, Chowienczyk PJ, Brett SE, et al. Nebivolol vasodilates human forearm vasculature: evidence for an L-arginine/NO-dependent mechanism. J Pharmacol Exp Ther 1995; 274:1067–1071.
- McEniery CM, Schmitt M, Qasem A, et al. Nebivolol increases arterial distensibility in vivo. Hypertension 2004; 44:305–310.
- Poirier L, Cleroux J, Nadeau A, Lacourciere Y. Effects of nebivolol and atenolol on insulin sensitivity and haemodynamics in hypertensive patients. J Hypertens 2001; 19:1429–1435.
- Flather MD, Shibata MC, Coats AJ, et al; SENIORS Investigators. Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (SENIORS). Eur Heart J 2005; 26:215–225.
- Williams B, Poulter NR, Brown MJ, et al; BHS guidelines working party, for the British Hypertension Society. British Hypertension Society guidelines for hypertension management 2004 (BHS-IV): summary. BMJ 2004; 328:634–640.
- Sever P. New hypertension guidelines from the National Institute for Health and Clinical Excellence and the British Hypertension Society. J Renin Angiotensin Aldosterone Syst 2006; 7:61–63.
- Mancia G, De Backer G, Dominiczak A, et al; Management of Arterial Hypertension of the European Society of Hypertension. 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens 2007; 25:1105–1187.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289:2560–2572.
In recent years the role of beta-blockers as a primary tool to treat hypertension has come under question. These drugs have shown disappointing results when used as antihypertensive therapy in patients without heart disease, ie, when used as primary prevention. At the same time, beta-blockers clearly reduce the risk of future cardiovascular events in patients who already have heart disease, eg, who already have had a myocardial infarction or who have congestive heart failure.
Several meta-analyses and a few clinical trials have shown that beta-blockers may have no advantage over other antihypertensive drugs, and in fact may not reduce the risk of stroke as effectively as other classes of blood pressure medications.
Why should this be? Is it that the patients in the antihypertensive trials were mostly older, and that beta-blockers do not work as well in older patients as in younger ones? Or does it have to do with the fact that atenolol (Tenormin) was the drug most often used in the trials? Would newer, different beta-blockers be better?
Hypertension experts currently disagree on how to interpret the available data, and this has led to conflict and confusion among clinicians as to the role of beta-blockers in managing hypertension. Current evidence suggests that older beta-blockers may not be the preferred first-line antihypertensive drugs for hypertensive patients who have no compelling indications for them (eg, heart failure, myocardial infarction, diabetes, high risk of coronary heart disease). However, newer beta-blockers with vasodilatory properties should be considered in cases of uncontrolled or resistant hypertension, especially in younger patients.
Further, while controversy and debate continue over the benefits and adverse effects of one class of antihypertensive drugs vs another, it is indisputable that controlling arterial blood pressure to the recommended goal offers major protection against cardiovascular and renal events in patients with hypertension.1,2
MECHANISM OF ACTION OF BETA-BLOCKERS
Beta-blockers effectively reduce blood pressure in both systolic-diastolic hypertension and isolated systolic hypertension.3–5 Exactly how is not known, but it has been proposed that they may do so by:
Reducing the heart rate and cardiac output. When catecholamines activate beta-1 receptors in the heart, the heart rate and myocardial contractility increase. By blocking beta-1 receptors, beta-blockers reduce the heart rate and myocardial contractility, thus lowering cardiac output and arterial blood pressure.6
Inhibiting renin release. Activation of the renin-angiotensin system is another major pathway that can lead to elevated arterial blood pressure. Renin release is mediated through the sympathetic nervous system via beta-1 receptors on the juxtaglomerular cells of the kidney. Beta-blockers can therefore lower blood pressure by inhibiting renin release.7
Inhibiting central nervous sympathetic outflow, thereby inducing presynaptic blockade, which in turn reduces the release of catecholamines.
Reducing venous return and plasma volume.
Generating nitric oxide, thus reducing peripheral vascular resistance (some agents).8
Reducing vasomotor tone.
Reducing vascular tone.
Improving vascular compliance.
Resetting baroreceptor levels.
Attenuating the pressor response to catecholamines with exercise and stress.
HETEROGENEITY OF BETA-BLOCKERS
Selectivity
Beta-blockers are not all the same. They can be classified into three categories.
Nonselective beta-blockers block both beta-1 and beta-2 adrenergic receptors. It is generally accepted that beta-blockers exert their primary antihypertensive effect by blocking beta-1 adrenergic receptors.6 Of interest, nonselective beta-blockers inhibit beta-2 receptors on arteries and thus cause an unopposed alpha-adrenergic effect, leading to increased peripheral vascular resistance.9 Examples of this category:
- Nadolol (Corgard)
- Pindolol (Visken)
- Propranolol (Inderal)
- Timolol (Blocadren).
Selective beta-blockers specifically block beta-1 receptors alone, although they are known to be nonselective at higher doses. Examples:
- Atenolol (Tenormin)
- Betaxolol (Kerlone)
- Bisoprolol (Zebeta)
- Esmolol (Brevibloc)
- Metoprolol (Lopressor, Toprol).
Beta-blockers with peripheral vasodilatatory effects act either via antagonism of the alpha-1 receptor, as with labetolol (Normodyne) and carvedilol (Coreg),10 or via enhanced release of nitric oxide, as with nebivolol (Bystolic).8
Lipid and water solubility
The lipid solubility and water solubility of each beta-blocker determine its bioavailability and side-effect profile.
Lipid solubility determines the degree to which a beta-blocker penetrates the blood-brain barrier and thereby leads to central nervous system side effects such as lethargy, nightmares, confusion, and depression. Propranolol is highly lipid-soluble; metoprolol and labetalol are moderately so.
Water-soluble beta-blockers such as atenolol have less tissue permeation, have a longer half-life, and cause fewer central nervous system effects and symptoms.11
Routes of elimination
Beta-blockers also differ in their route of elimination.
Atenolol and nadolol are eliminated by the kidney and require dose adjustment in patients with impaired renal function.12,13
On the other hand, propranolol, metoprolol, labetalol, carvedilol, and nebivolol are excreted primarily via hepatic metabolism.13
BETA-BLOCKERS IN THE MANAGEMENT OF HYPERTENSION
Beta-blockers were initially used to treat arrhythmias, but by the early 1970s they were also widely accepted for managing hypertension. 14 Their initial acceptance as one of the first-line classes of drugs for hypertension was based on their better side-effect profile compared with other antihypertensive drugs available at that time.
In the 1980s and 1990s, beta-blockers were listed as preferred first-line antihypertensive drugs along with diuretics in national hypertension guidelines.15 Subsequent updates of the guidelines favored diuretics as initial therapy and relegated all other classes of antihypertensive medications to be alternatives to diuretics.16 Although beta-blockers remain alternative first-line drugs in the latest guidelines (published in 2003; see reference 66), they are the preferred antihypertensive agents for patients with cardiac disease.
The current recommendations reflect the findings from hypertension trials in which patients with myocardial infarction and congestive heart failure had better cardiovascular outcomes if they received these drugs,17–19 including a lower risk of death.20,21 It was widely assumed that beta-blockers would also prevent first episodes of cardiovascular events.
However, to date, there is no evidence that beta-blockers are effective as primary prevention. Several large randomized controlled trials showed no benefit with beta-blockers compared with other antihypertensive drugs—in fact, there were more cardiovascular events with beta-blockers (see below).
Beta-blockers are well tolerated in clinical practice, although they can have side effects that include fatigue, depression, impaired exercise tolerance, sexual dysfunction, and asthma attacks.
Wiysonge et al22 analyzed how many patients withdrew from randomized trials of antihypertensive treatment because of drug-related adverse events. There was no significant difference in the incidence of fatigue, depressive symptoms, or sexual dysfunction with beta-blockers compared with placebo, and trial participants on a beta-blocker were not statistically significantly more likely to discontinue treatment than those receiving a placebo in three trials with 22,729 participants (relative risk [RR] 2.34, 95% confidence interval [CI] 0.84–6.52).
THE CONTROVERSY: WHAT THE TRIALS SHOWED
Messerli et al23 performed a meta-analysis published in 1998 that suggested that beta-blockers may not be as effective as diuretics in preventing cardiovascular events when used as first-line antihypertensive therapy in elderly patients. In 10 randomized controlled trials in 16,164 patients who were treated with either a diuretic or a beta-blocker (atenolol), blood pressure was normalized in two-thirds of diuretic-treated patients but only one-third of patients treated with atenolol as monotherapy. Diuretic therapy was superior with regard to all end points, and beta-blockers were found to be ineffective except in reducing cerebrovascular events.
The LIFE study (Losartan Intervention for Endpoint Reduction in Hypertension)24 compared the angiotensin-receptor blocker losartan (Cozaar) and atenolol in 9,193 patients with hypertension and left ventricular hypertrophy. At 4 years of follow-up, the rate of primary cardiovascular events (death, myocardial infarction, or stroke) was lower in the losartan group than in the atenolol group. The difference was mainly due to a 25% lower incidence of stroke, which was statistically significant. The rates of myocardial infarction and death from cardiovascular causes were not significantly different between the two treatment groups. The systolic blood pressure was 1 mm Hg lower in the losartan group than in the atenolol group, which was statistically significant.
Carlberg et al25 performed another important meta-analysis that questioned whether atenolol reduces rates of cardiovascular morbidity and death in hypertensive patients. The results were surprising: eight randomized controlled trials including more than 6,000 patients and comparing atenolol with placebo or no treatment showed no differences between the treatment groups with regard to the outcomes of all-cause mortality (RR 1.01, 95% CI 0.89–1.15), cardiovascular mortality (RR 0.99, 95% CI 0.83–1.18), or myocardial infarction (RR 0.99, 95% CI 0.83–1.19).
In addition, when atenolol was compared with other antihypertensives in five other randomized controlled trials that included more than 14,000 patients, those treated with atenolol had a higher risk of stroke (RR 1.30, 95% CI 1.12–1.50) and death (RR 1.13, 95% CI 1.02–1.25).
The ASCOT-BPLA trial (Anglo-Scandinavian Cardiac Outcomes Trial—Blood Pressure Lowering Arm)26 had similar results. This trial compared the combination of atenolol plus the diuretic bendroflumethiazide against the combination of the calcium channel blocker amlodipine (Norvasc) plus the angiotensin-converting enzyme (ACE) inhibitor perindopril (Aceon). Although no significant difference was seen in the primary outcome of nonfatal myocardial infarction or fatal coronary heart disease (unadjusted hazard ratio [HR] with amlodipine-perindopril 0.90, 95% CI 0.79–1.02, P = .1052), the amlodipine-plus-perindopril group had significantly fewer strokes (327 vs 422, HR 0.77, 95% CI 0.66–0.89, P = .0003), fewer total cardiovascular events (1,362 vs 1,602, HR 0.84, 95% CI 0.78–0.90, P = .0001), and fewer deaths from any cause (738 vs 820; HR 0.89, 95% CI 0.81–0.99, P = .025).
Lindholm et al27 performed a meta-analysis that included studies of selective beta-blockers (including atenolol) and nonselective beta-blockers, with a follow-up time of more than 2 years. Compared with placebo or no treatment, beta-blockers reduced the risk of stroke by 19% but had no effect on myocardial infarction or all-cause mortality. Compared with other antihypertensive drugs, beta-blockers were less than optimum, and the relative risk of stroke was 16% higher. Atenolol was the beta-blocker used in most of the randomized clinical trials included in this meta-analysis.
The Cochrane group22 found beta-blockers to be inferior to all other antihypertensive drugs with respect to the ability to lower the risk of stroke.
WHY WERE THE RESULTS SO DISAPPOINTING?
Problems with atenolol
Most of the trials in the meta-analyses discussed above used atenolol and other beta-blockers that had no vasodilatory properties.
Further, in most of the trials atenolol was used in a once-daily dosage, whereas ideally it needs to be taken more frequently, based on its pharmacokinetic and pharmacodynamic properties (a half-life of 6–9 hours).3 Neutel et al28 confirmed that atenolol, when taken once daily, leaves the patient unprotected in the last 6 hours of a 24-hour period, as demonstrated by 24-hour ambulatory blood pressure monitoring. It is possible that this short duration of action of atenolol may have contributed to the results observed in clinical trials that used atenolol to treat hypertension.
Differences between older and younger patients
Another possible reason for the disappointing results is that the trials included many elderly patients, in whom beta-blockers may not be as effective. The pathophysiology of hypertension in younger people is different from that in older patients.29 Hemodynamic characteristics of younger hypertensive patients include a high cardiac output and hyperdynamic circulation with a low pulse pressure, while older patients have lower arterial compliance with an elevated vascular resistance.
The notion of choosing antihypertensive medications on the basis of age and age-related pathophysiology is supported by several clinical studies. Randomized controlled trials appear to show that beta-blockers are effective in younger hypertensive patients.30
Conversely, the CAFE (Conduit Artery Function Evaluation) trial,31 a substudy of the main ASCOT trial,26 indicated that betablocker-based therapy was less effective in reducing central aortic pressure than were regimens based on an ACE inhibitor or a calcium channel blocker.
The CAFE researchers recruited 2,073 patients from five ASCOT centers and used radial artery applanation tonometry and pulse-wave analysis to derive central aortic pressures and hemodynamic indices during study visits up to a period of 4 years. Although the two treatment groups achieved similar brachial systolic blood pressures, the central aortic systolic pressure was 4.3 mm Hg lower in the amlodipine group (95% CI 3.3–5.4; P < .0001), and the central aortic pulse pressure was 3.0 mm Hg lower (95% CI 2.1–3.9; P < .0001).
Pulse-wave dyssynchrony
Bangalore et al47 offer an interesting hypothesis to explain the probable adverse effect of beta-blockers. Their theory concerns the effect of these drugs on the arterial pulse wave.
Normally, with each contraction of the left ventricle during systole, an arterial pulse wave is generated and propagated forward to the peripheral arteries. This wave is then reflected back to the heart from the branching points of peripheral arteries. The final form of the pressure wave at the aortic root is a synchronized summation of the forward-traveling wave and the backward-reflected wave.
In healthy people with normal arteries, the reflected wave merges with the forward-traveling wave in diastole and augments coronary blood flow. In patients whose arteries are stiff due to aging or vascular comorbidities, the reflected wave returns faster and merges with the incident wave in systole, resulting in higher left ventricular afterload and less coronary perfusion.48
Bangalore et al47 propose that artificially reducing the heart rate with beta-blockers may further dyssynchronize the pulse wave, adversely affecting coronary perfusion and leading to an increased risk of cardiovascular events and death.
Metabolic side effects
Older beta-blockers, and especially atenolol, have well-known metabolic adverse effects, particularly impairment of glycemic control. This adverse effect appears to occur only with beta-blockers that do not possess vasodilatory properties and thus increase peripheral vascular resistance, which results in lower glucose availability and reduced uptake by skeletal muscles.49
Bangalore et al50 evaluated the effect of beta-blockers in a meta-analysis of 12 studies in 94,492 patients followed up for more than 1 year. Beta-blocker therapy resulted in a 22% higher risk of new-onset diabetes mellitus (RR 1.22, 95% CI 1.12–1.33) than with other nondiuretic antihypertensive agents.
Of note, however, the meta-analysis did not show a significantly higher risk of the onset of diabetes with propranolol or metoprolol than with other nondiuretic antihypertensives when studies of these beta-blockers were separated from atenolol-based studies.
Further, the United Kingdom Prospective Diabetes Study40 found that cardiovascular outcomes in patients with good blood pressure control were similar when atenolol-based therapy was compared with therapy with the ACE inhibitor captopril (Capoten).
A meta-analysis conducted by Balamuthusamy et al51 in 2009 found no higher risk of stroke in patients with hypertension and diabetes mellitus who received beta-blockers than in those who received other antihypertensive medications. However, beta-blockers were associated with a higher risk of death from cardiovascular causes (RR 1.39, 95% CI 1.07–1.804; P < .01) compared with reninangiotensin blockade.
NEWER BETA-BLOCKERS MAY BE BETTER
In the United States, more than 40 million prescriptions for atenolol are written every year, making it by far the most commonly used beta-blocker for the treatment of hypertension. 52 It is clear, however, that atenolol is not an ideal representative of this class of antihypertensive medications.
Preliminary data from studies of newer beta-blockers that possess beneficial vasodilatory properties are encouraging. Animal studies and preliminary human studies find that these new-generation beta-blockers cause fewer adverse metabolic effects and improve endothelial function, measures of arterial stiffness, and cardiovascular outcomes.
Carvedilol
Carvedilol is a nonselective beta-blocker with vasodilatory effects that are thought to be due to its ability to concurrently block alpha-1 receptors in addition to beta receptors. 53 In experiments in vitro and in trials in patients with diabetes and hypertension, carvedilol increased endothelial vasodilation and reduced inflammation and platelet aggregation. These effects may be achieved though antioxidant actions, thereby preserving nitric oxide bioactivity.54,55
In the Glycemic Effects in Diabetes Mellitus: Carvedilol-Metoprolol Comparison in Hypertensives (GEMINI) trial,56 carvedilol was associated with better maintenance of glycemic control in diabetic hypertensive patients than was metoprolol. Insulin sensitivity improved with carvedilol but not with metoprolol, and fewer patients on carvedilol progressed to microalbuminuria.
Nebivolol
Nebivolol is a novel selective beta-blocker with a much higher affinity for beta-1 adrenergic receptors than for beta-2 adrenergic receptors. Among all the beta-blockers in clinical use today, nebivolol has the highest selectivity for beta-1 receptors.8
Nebivolol causes vasodilation through activation of the l-arginine/nitric oxide pathway.57–59 Blockade of synthesis of nitric oxide leads to local arterial stiffness. Endothelial dysfunction is characterized by decreased bioavailability of nitric oxide and has been shown to be a strong predictor of cardiovascular outcomes. By generating nitric oxide, nebivolol reduces peripheral vascular resistance, overcoming a significant side effect of earlier beta-blockers that lowered blood pressure but ultimately increased peripheral vascular tone and resistance.8
In an experiment in a bovine model,60 nebivolol significantly reduced the pulse-wave velocity (a measure of arterial stiffness), while atenolol had no effect. Moreover, evidence for the role of the l-arginine/nitric oxide pathway in the vasodilatory effect of nebivolol was demonstrated by co-infusion of NG-monomethyl-L-arginine, a specific endothelial nitric oxide synthetase inhibitor that attenuated the reduction of pulse-wave velocity by nebivolol.
In studies in hypertensive patients, nebivolol was associated with a better metabolic profile than atenolol, with none of the adverse effects on insulin sensitivity that atenolol had.61 In the Study of Effects of Nebivolol Interventions on Outcomes and Rehospitalization in Seniors With Heart Failure (SENIORS) trial, significantly fewer patients receiving nebivolol died or were admitted to the hospital for cardiovascular reasons compared with those receiving placebo.62
Although these findings are encouraging, we do not yet know if these effects will translate into a significant reduction in cardiovascular outcomes in clinical trials. Large, prospective hypertension outcome trials, particularly to evaluate primary prevention of cardiovascular outcomes, are needed for an evidence-based approach to using the newer beta-blockers as preferred first-line therapy for hypertension.
WHAT RECENT GUIDELINES SAY ABOUT BETA-BLOCKERS
The British National Institute for Health and Clinical Excellence and the British Hypertension Society, in their 2004 guidelines, recommended beta-blockers as one of several first-line antihypertensive medications in young, nonblack patients.63 On the other hand, they advised clinicians to be aware of the reported increase in onset of diabetes mellitus in patients treated with these medications. After the LIFE24 and ASCOT26 study results were published, these guidelines were amended to exclude beta-blockers as preferred routine initial therapy for hypertension.64
More recently, the 2007 European Society of Hypertension and European Society of Cardiology reconsidered the role of beta-blockers, recommending them as an option in both initial and subsequent antihypertensive treatment strategies.65
The current guidelines from the National Heart, Lung, and Blood Institute,66 which were published in 2003, were highly influenced by the results of the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT),2 and favor diuretics as the first-line therapy. However, they indicate that beta-blockers are a suitable alternative, particularly when a compelling cardiac indication is present.53 We hope that the next update, expected late in 2009, will re-address this issue in the light of more recent data.
In recent years the role of beta-blockers as a primary tool to treat hypertension has come under question. These drugs have shown disappointing results when used as antihypertensive therapy in patients without heart disease, ie, when used as primary prevention. At the same time, beta-blockers clearly reduce the risk of future cardiovascular events in patients who already have heart disease, eg, who already have had a myocardial infarction or who have congestive heart failure.
Several meta-analyses and a few clinical trials have shown that beta-blockers may have no advantage over other antihypertensive drugs, and in fact may not reduce the risk of stroke as effectively as other classes of blood pressure medications.
Why should this be? Is it that the patients in the antihypertensive trials were mostly older, and that beta-blockers do not work as well in older patients as in younger ones? Or does it have to do with the fact that atenolol (Tenormin) was the drug most often used in the trials? Would newer, different beta-blockers be better?
Hypertension experts currently disagree on how to interpret the available data, and this has led to conflict and confusion among clinicians as to the role of beta-blockers in managing hypertension. Current evidence suggests that older beta-blockers may not be the preferred first-line antihypertensive drugs for hypertensive patients who have no compelling indications for them (eg, heart failure, myocardial infarction, diabetes, high risk of coronary heart disease). However, newer beta-blockers with vasodilatory properties should be considered in cases of uncontrolled or resistant hypertension, especially in younger patients.
Further, while controversy and debate continue over the benefits and adverse effects of one class of antihypertensive drugs vs another, it is indisputable that controlling arterial blood pressure to the recommended goal offers major protection against cardiovascular and renal events in patients with hypertension.1,2
MECHANISM OF ACTION OF BETA-BLOCKERS
Beta-blockers effectively reduce blood pressure in both systolic-diastolic hypertension and isolated systolic hypertension.3–5 Exactly how is not known, but it has been proposed that they may do so by:
Reducing the heart rate and cardiac output. When catecholamines activate beta-1 receptors in the heart, the heart rate and myocardial contractility increase. By blocking beta-1 receptors, beta-blockers reduce the heart rate and myocardial contractility, thus lowering cardiac output and arterial blood pressure.6
Inhibiting renin release. Activation of the renin-angiotensin system is another major pathway that can lead to elevated arterial blood pressure. Renin release is mediated through the sympathetic nervous system via beta-1 receptors on the juxtaglomerular cells of the kidney. Beta-blockers can therefore lower blood pressure by inhibiting renin release.7
Inhibiting central nervous sympathetic outflow, thereby inducing presynaptic blockade, which in turn reduces the release of catecholamines.
Reducing venous return and plasma volume.
Generating nitric oxide, thus reducing peripheral vascular resistance (some agents).8
Reducing vasomotor tone.
Reducing vascular tone.
Improving vascular compliance.
Resetting baroreceptor levels.
Attenuating the pressor response to catecholamines with exercise and stress.
HETEROGENEITY OF BETA-BLOCKERS
Selectivity
Beta-blockers are not all the same. They can be classified into three categories.
Nonselective beta-blockers block both beta-1 and beta-2 adrenergic receptors. It is generally accepted that beta-blockers exert their primary antihypertensive effect by blocking beta-1 adrenergic receptors.6 Of interest, nonselective beta-blockers inhibit beta-2 receptors on arteries and thus cause an unopposed alpha-adrenergic effect, leading to increased peripheral vascular resistance.9 Examples of this category:
- Nadolol (Corgard)
- Pindolol (Visken)
- Propranolol (Inderal)
- Timolol (Blocadren).
Selective beta-blockers specifically block beta-1 receptors alone, although they are known to be nonselective at higher doses. Examples:
- Atenolol (Tenormin)
- Betaxolol (Kerlone)
- Bisoprolol (Zebeta)
- Esmolol (Brevibloc)
- Metoprolol (Lopressor, Toprol).
Beta-blockers with peripheral vasodilatatory effects act either via antagonism of the alpha-1 receptor, as with labetolol (Normodyne) and carvedilol (Coreg),10 or via enhanced release of nitric oxide, as with nebivolol (Bystolic).8
Lipid and water solubility
The lipid solubility and water solubility of each beta-blocker determine its bioavailability and side-effect profile.
Lipid solubility determines the degree to which a beta-blocker penetrates the blood-brain barrier and thereby leads to central nervous system side effects such as lethargy, nightmares, confusion, and depression. Propranolol is highly lipid-soluble; metoprolol and labetalol are moderately so.
Water-soluble beta-blockers such as atenolol have less tissue permeation, have a longer half-life, and cause fewer central nervous system effects and symptoms.11
Routes of elimination
Beta-blockers also differ in their route of elimination.
Atenolol and nadolol are eliminated by the kidney and require dose adjustment in patients with impaired renal function.12,13
On the other hand, propranolol, metoprolol, labetalol, carvedilol, and nebivolol are excreted primarily via hepatic metabolism.13
BETA-BLOCKERS IN THE MANAGEMENT OF HYPERTENSION
Beta-blockers were initially used to treat arrhythmias, but by the early 1970s they were also widely accepted for managing hypertension. 14 Their initial acceptance as one of the first-line classes of drugs for hypertension was based on their better side-effect profile compared with other antihypertensive drugs available at that time.
In the 1980s and 1990s, beta-blockers were listed as preferred first-line antihypertensive drugs along with diuretics in national hypertension guidelines.15 Subsequent updates of the guidelines favored diuretics as initial therapy and relegated all other classes of antihypertensive medications to be alternatives to diuretics.16 Although beta-blockers remain alternative first-line drugs in the latest guidelines (published in 2003; see reference 66), they are the preferred antihypertensive agents for patients with cardiac disease.
The current recommendations reflect the findings from hypertension trials in which patients with myocardial infarction and congestive heart failure had better cardiovascular outcomes if they received these drugs,17–19 including a lower risk of death.20,21 It was widely assumed that beta-blockers would also prevent first episodes of cardiovascular events.
However, to date, there is no evidence that beta-blockers are effective as primary prevention. Several large randomized controlled trials showed no benefit with beta-blockers compared with other antihypertensive drugs—in fact, there were more cardiovascular events with beta-blockers (see below).
Beta-blockers are well tolerated in clinical practice, although they can have side effects that include fatigue, depression, impaired exercise tolerance, sexual dysfunction, and asthma attacks.
Wiysonge et al22 analyzed how many patients withdrew from randomized trials of antihypertensive treatment because of drug-related adverse events. There was no significant difference in the incidence of fatigue, depressive symptoms, or sexual dysfunction with beta-blockers compared with placebo, and trial participants on a beta-blocker were not statistically significantly more likely to discontinue treatment than those receiving a placebo in three trials with 22,729 participants (relative risk [RR] 2.34, 95% confidence interval [CI] 0.84–6.52).
THE CONTROVERSY: WHAT THE TRIALS SHOWED
Messerli et al23 performed a meta-analysis published in 1998 that suggested that beta-blockers may not be as effective as diuretics in preventing cardiovascular events when used as first-line antihypertensive therapy in elderly patients. In 10 randomized controlled trials in 16,164 patients who were treated with either a diuretic or a beta-blocker (atenolol), blood pressure was normalized in two-thirds of diuretic-treated patients but only one-third of patients treated with atenolol as monotherapy. Diuretic therapy was superior with regard to all end points, and beta-blockers were found to be ineffective except in reducing cerebrovascular events.
The LIFE study (Losartan Intervention for Endpoint Reduction in Hypertension)24 compared the angiotensin-receptor blocker losartan (Cozaar) and atenolol in 9,193 patients with hypertension and left ventricular hypertrophy. At 4 years of follow-up, the rate of primary cardiovascular events (death, myocardial infarction, or stroke) was lower in the losartan group than in the atenolol group. The difference was mainly due to a 25% lower incidence of stroke, which was statistically significant. The rates of myocardial infarction and death from cardiovascular causes were not significantly different between the two treatment groups. The systolic blood pressure was 1 mm Hg lower in the losartan group than in the atenolol group, which was statistically significant.
Carlberg et al25 performed another important meta-analysis that questioned whether atenolol reduces rates of cardiovascular morbidity and death in hypertensive patients. The results were surprising: eight randomized controlled trials including more than 6,000 patients and comparing atenolol with placebo or no treatment showed no differences between the treatment groups with regard to the outcomes of all-cause mortality (RR 1.01, 95% CI 0.89–1.15), cardiovascular mortality (RR 0.99, 95% CI 0.83–1.18), or myocardial infarction (RR 0.99, 95% CI 0.83–1.19).
In addition, when atenolol was compared with other antihypertensives in five other randomized controlled trials that included more than 14,000 patients, those treated with atenolol had a higher risk of stroke (RR 1.30, 95% CI 1.12–1.50) and death (RR 1.13, 95% CI 1.02–1.25).
The ASCOT-BPLA trial (Anglo-Scandinavian Cardiac Outcomes Trial—Blood Pressure Lowering Arm)26 had similar results. This trial compared the combination of atenolol plus the diuretic bendroflumethiazide against the combination of the calcium channel blocker amlodipine (Norvasc) plus the angiotensin-converting enzyme (ACE) inhibitor perindopril (Aceon). Although no significant difference was seen in the primary outcome of nonfatal myocardial infarction or fatal coronary heart disease (unadjusted hazard ratio [HR] with amlodipine-perindopril 0.90, 95% CI 0.79–1.02, P = .1052), the amlodipine-plus-perindopril group had significantly fewer strokes (327 vs 422, HR 0.77, 95% CI 0.66–0.89, P = .0003), fewer total cardiovascular events (1,362 vs 1,602, HR 0.84, 95% CI 0.78–0.90, P = .0001), and fewer deaths from any cause (738 vs 820; HR 0.89, 95% CI 0.81–0.99, P = .025).
Lindholm et al27 performed a meta-analysis that included studies of selective beta-blockers (including atenolol) and nonselective beta-blockers, with a follow-up time of more than 2 years. Compared with placebo or no treatment, beta-blockers reduced the risk of stroke by 19% but had no effect on myocardial infarction or all-cause mortality. Compared with other antihypertensive drugs, beta-blockers were less than optimum, and the relative risk of stroke was 16% higher. Atenolol was the beta-blocker used in most of the randomized clinical trials included in this meta-analysis.
The Cochrane group22 found beta-blockers to be inferior to all other antihypertensive drugs with respect to the ability to lower the risk of stroke.
WHY WERE THE RESULTS SO DISAPPOINTING?
Problems with atenolol
Most of the trials in the meta-analyses discussed above used atenolol and other beta-blockers that had no vasodilatory properties.
Further, in most of the trials atenolol was used in a once-daily dosage, whereas ideally it needs to be taken more frequently, based on its pharmacokinetic and pharmacodynamic properties (a half-life of 6–9 hours).3 Neutel et al28 confirmed that atenolol, when taken once daily, leaves the patient unprotected in the last 6 hours of a 24-hour period, as demonstrated by 24-hour ambulatory blood pressure monitoring. It is possible that this short duration of action of atenolol may have contributed to the results observed in clinical trials that used atenolol to treat hypertension.
Differences between older and younger patients
Another possible reason for the disappointing results is that the trials included many elderly patients, in whom beta-blockers may not be as effective. The pathophysiology of hypertension in younger people is different from that in older patients.29 Hemodynamic characteristics of younger hypertensive patients include a high cardiac output and hyperdynamic circulation with a low pulse pressure, while older patients have lower arterial compliance with an elevated vascular resistance.
The notion of choosing antihypertensive medications on the basis of age and age-related pathophysiology is supported by several clinical studies. Randomized controlled trials appear to show that beta-blockers are effective in younger hypertensive patients.30
Conversely, the CAFE (Conduit Artery Function Evaluation) trial,31 a substudy of the main ASCOT trial,26 indicated that betablocker-based therapy was less effective in reducing central aortic pressure than were regimens based on an ACE inhibitor or a calcium channel blocker.
The CAFE researchers recruited 2,073 patients from five ASCOT centers and used radial artery applanation tonometry and pulse-wave analysis to derive central aortic pressures and hemodynamic indices during study visits up to a period of 4 years. Although the two treatment groups achieved similar brachial systolic blood pressures, the central aortic systolic pressure was 4.3 mm Hg lower in the amlodipine group (95% CI 3.3–5.4; P < .0001), and the central aortic pulse pressure was 3.0 mm Hg lower (95% CI 2.1–3.9; P < .0001).
Pulse-wave dyssynchrony
Bangalore et al47 offer an interesting hypothesis to explain the probable adverse effect of beta-blockers. Their theory concerns the effect of these drugs on the arterial pulse wave.
Normally, with each contraction of the left ventricle during systole, an arterial pulse wave is generated and propagated forward to the peripheral arteries. This wave is then reflected back to the heart from the branching points of peripheral arteries. The final form of the pressure wave at the aortic root is a synchronized summation of the forward-traveling wave and the backward-reflected wave.
In healthy people with normal arteries, the reflected wave merges with the forward-traveling wave in diastole and augments coronary blood flow. In patients whose arteries are stiff due to aging or vascular comorbidities, the reflected wave returns faster and merges with the incident wave in systole, resulting in higher left ventricular afterload and less coronary perfusion.48
Bangalore et al47 propose that artificially reducing the heart rate with beta-blockers may further dyssynchronize the pulse wave, adversely affecting coronary perfusion and leading to an increased risk of cardiovascular events and death.
Metabolic side effects
Older beta-blockers, and especially atenolol, have well-known metabolic adverse effects, particularly impairment of glycemic control. This adverse effect appears to occur only with beta-blockers that do not possess vasodilatory properties and thus increase peripheral vascular resistance, which results in lower glucose availability and reduced uptake by skeletal muscles.49
Bangalore et al50 evaluated the effect of beta-blockers in a meta-analysis of 12 studies in 94,492 patients followed up for more than 1 year. Beta-blocker therapy resulted in a 22% higher risk of new-onset diabetes mellitus (RR 1.22, 95% CI 1.12–1.33) than with other nondiuretic antihypertensive agents.
Of note, however, the meta-analysis did not show a significantly higher risk of the onset of diabetes with propranolol or metoprolol than with other nondiuretic antihypertensives when studies of these beta-blockers were separated from atenolol-based studies.
Further, the United Kingdom Prospective Diabetes Study40 found that cardiovascular outcomes in patients with good blood pressure control were similar when atenolol-based therapy was compared with therapy with the ACE inhibitor captopril (Capoten).
A meta-analysis conducted by Balamuthusamy et al51 in 2009 found no higher risk of stroke in patients with hypertension and diabetes mellitus who received beta-blockers than in those who received other antihypertensive medications. However, beta-blockers were associated with a higher risk of death from cardiovascular causes (RR 1.39, 95% CI 1.07–1.804; P < .01) compared with reninangiotensin blockade.
NEWER BETA-BLOCKERS MAY BE BETTER
In the United States, more than 40 million prescriptions for atenolol are written every year, making it by far the most commonly used beta-blocker for the treatment of hypertension. 52 It is clear, however, that atenolol is not an ideal representative of this class of antihypertensive medications.
Preliminary data from studies of newer beta-blockers that possess beneficial vasodilatory properties are encouraging. Animal studies and preliminary human studies find that these new-generation beta-blockers cause fewer adverse metabolic effects and improve endothelial function, measures of arterial stiffness, and cardiovascular outcomes.
Carvedilol
Carvedilol is a nonselective beta-blocker with vasodilatory effects that are thought to be due to its ability to concurrently block alpha-1 receptors in addition to beta receptors. 53 In experiments in vitro and in trials in patients with diabetes and hypertension, carvedilol increased endothelial vasodilation and reduced inflammation and platelet aggregation. These effects may be achieved though antioxidant actions, thereby preserving nitric oxide bioactivity.54,55
In the Glycemic Effects in Diabetes Mellitus: Carvedilol-Metoprolol Comparison in Hypertensives (GEMINI) trial,56 carvedilol was associated with better maintenance of glycemic control in diabetic hypertensive patients than was metoprolol. Insulin sensitivity improved with carvedilol but not with metoprolol, and fewer patients on carvedilol progressed to microalbuminuria.
Nebivolol
Nebivolol is a novel selective beta-blocker with a much higher affinity for beta-1 adrenergic receptors than for beta-2 adrenergic receptors. Among all the beta-blockers in clinical use today, nebivolol has the highest selectivity for beta-1 receptors.8
Nebivolol causes vasodilation through activation of the l-arginine/nitric oxide pathway.57–59 Blockade of synthesis of nitric oxide leads to local arterial stiffness. Endothelial dysfunction is characterized by decreased bioavailability of nitric oxide and has been shown to be a strong predictor of cardiovascular outcomes. By generating nitric oxide, nebivolol reduces peripheral vascular resistance, overcoming a significant side effect of earlier beta-blockers that lowered blood pressure but ultimately increased peripheral vascular tone and resistance.8
In an experiment in a bovine model,60 nebivolol significantly reduced the pulse-wave velocity (a measure of arterial stiffness), while atenolol had no effect. Moreover, evidence for the role of the l-arginine/nitric oxide pathway in the vasodilatory effect of nebivolol was demonstrated by co-infusion of NG-monomethyl-L-arginine, a specific endothelial nitric oxide synthetase inhibitor that attenuated the reduction of pulse-wave velocity by nebivolol.
In studies in hypertensive patients, nebivolol was associated with a better metabolic profile than atenolol, with none of the adverse effects on insulin sensitivity that atenolol had.61 In the Study of Effects of Nebivolol Interventions on Outcomes and Rehospitalization in Seniors With Heart Failure (SENIORS) trial, significantly fewer patients receiving nebivolol died or were admitted to the hospital for cardiovascular reasons compared with those receiving placebo.62
Although these findings are encouraging, we do not yet know if these effects will translate into a significant reduction in cardiovascular outcomes in clinical trials. Large, prospective hypertension outcome trials, particularly to evaluate primary prevention of cardiovascular outcomes, are needed for an evidence-based approach to using the newer beta-blockers as preferred first-line therapy for hypertension.
WHAT RECENT GUIDELINES SAY ABOUT BETA-BLOCKERS
The British National Institute for Health and Clinical Excellence and the British Hypertension Society, in their 2004 guidelines, recommended beta-blockers as one of several first-line antihypertensive medications in young, nonblack patients.63 On the other hand, they advised clinicians to be aware of the reported increase in onset of diabetes mellitus in patients treated with these medications. After the LIFE24 and ASCOT26 study results were published, these guidelines were amended to exclude beta-blockers as preferred routine initial therapy for hypertension.64
More recently, the 2007 European Society of Hypertension and European Society of Cardiology reconsidered the role of beta-blockers, recommending them as an option in both initial and subsequent antihypertensive treatment strategies.65
The current guidelines from the National Heart, Lung, and Blood Institute,66 which were published in 2003, were highly influenced by the results of the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT),2 and favor diuretics as the first-line therapy. However, they indicate that beta-blockers are a suitable alternative, particularly when a compelling cardiac indication is present.53 We hope that the next update, expected late in 2009, will re-address this issue in the light of more recent data.
- Staessen JA, Wang JG, Thijs L. Cardiovascular protection and blood pressure reduction: a meta-analysis. Lancet 2001; 358:1305–1315.
- ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002; 288:2981–2997.
- Neutel JM, Smith DH, Ram CV, et al. Application of ambulatory blood pressure monitoring in differentiating between antihypertensive agents. Am J Med 1993; 94:181–187.
- Materson BJ, Reda DJ, Cushman WC, et al. Single-drug therapy for hypertension in men. A comparison of six antihypertensive agents with placebo. The Department of Veterans Affairs Cooperative Study Group on Antihypertensive Agents. N Engl J Med 1993; 328:914–921.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- Frishman W, Silverman R. Clinical pharmacology of the new beta-adrenergic blocking drugs. Part 2. Physiologic and metabolic effects. Am Heart J 1979; 97:797–807.
- Garrett BN, Kaplan NM. Plasma renin activity suppression: duration after withdrawal from beta-adrenergic blockade. Arch Intern Med 1980; 140:1316–1318.
- Pedersen ME, Cockcroft JR. The latest generation of beta-blockers: new pharmacologic properties. Curr Hypertens Rep 2006; 8:279–286.
- Man in’t Veld AJ, Van den Meiracker AH, Schalekamp MA. Do beta-blockers really increase peripheral vascular resistance? Review of the literature and new observations under basal conditions. Am J Hypertens 1988; 1:91–96.
- Pearce CJ, Wallin JD. Labetalol and other agents that block both alpha- and beta-adrenergic receptors. Cleve Clin J Med 1994; 61:59–69.
- Dimsdale JE, Newton RP, Joist T. Neuropsychological side effects of beta-blockers. Arch Intern Med 1989; 149:514–525.
- Agarwal R. Supervised atenolol therapy in the management of hemodialysis hypertension. Kidney Int 1999; 55:1528–1535.
- Sica DA, Black HR. Pharmacologic considerations in the positioning of beta-blockers in antihypertensive therapy. Curr Hypertens Rep 2008; 10:330–335.
- Prichard BN, Gillam GP. Use of propranolol (Inderal) in treatment of hypertension. Br Med J 1964; 19; 2:725–727.
- The fifth report of the Joint National Committee on Detection, Evaluation, and Treatment of High Blood Pressure (JNC V). Arch Intern Med 1993; 153:154–183.
- Moser M. Evolution of the treatment of hypertension from the 1940s to JNC V. Am J Hypertens 1997; 10:2S–8S.
- Houghton T, Freemantle N, Cleland JG. Are beta-blockers effective in patients who develop heart failure soon after myocardial infarction? A meta-regression analysis of randomised trials. Eur J Heart Fail 2000; 2:333–340.
- The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet 1999; 353:9–13.
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999; 353:2001–2007.
- Yusuf S, Peto R, Lewis J, Collins R, Sleight P. Beta blockade during and after myocardial infarction: an overview of the randomized trials. Prog Cardiovasc Dis 1985; 27:335–371.
- Brophy JM, Joseph L, Rouleau JL. Beta-blockers in congestive heart failure. A Bayesian meta-analysis. Ann Intern Med 2001; 134:550–560.
- Wiysonge CS, Bradley H, Mayosi BM, et al. Beta-blockers for hypertension. Cochrane Database Syst Rev 2007;CD002003.
- Messerli FH, Grossman E, Goldbourt U. Are beta-blockers efficacious as first-line therapy for hypertension in the elderly? A systematic review. JAMA 1998; 279:1903–1907.
- Dahlöf B, Devereux RB, Kjeldsen SE, et al; for the LIFE study group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint Reduction in Hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002; 359:995–1003.
- Carlberg B, Samuelsson O, Lindholm LH. Atenolol in hypertension: is it a wise choice? Lancet 2004; 364:1684–1689.
- Dahlöf B, Sever PS, Poulter NR, et al; ASCOT Investigators. Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA): a multicentre randomised controlled trial. Lancet 2005; 366:895–906.
- Lindholm LH, Carlberg B, Samuelsson O. Should beta-blockers remain first choice in the treatment of primary hypertension? A meta-analysis. Lancet 2005; 366:1545–1553.
- Neutel JM, Schnaper H, Cheung DG, Graettinger WF, Weber MA. Antihypertensive effects of beta-blockers administered once daily: 24-hour measurements. Am Heart J 1990; 120:166–171.
- Franklin SS, Gustin W, Wong ND, et al. Hemodynamic patterns of age-related changes in blood pressure. The Framingham Heart Study. Circulation 1997; 96:308–315.
- The IPPPSH Collaborative Group. Cardiovascular risk and risk factors in a randomized trial of treatment based on the beta-blocker oxprenolol: the International Prospective Primary Prevention Study in Hypertension (IPPPSH). J Hypertens 1985; 3:379–392.
- Williams B, Lacy PS, Thom SM, et al; CAFE Investigators. Differential impact of blood pressure-lowering drugs on central aortic pressure and clinical outcomes: principal results of the Conduit Artery Function Evaluation (CAFE) study. Circulation 2006; 113:1213–1225.
- Khan N, McAlister FA. Re-examining the efficacy of beta-blockers for the treatment of hypertension: a meta-analysis. CMAJ 2006; 174:1737–1742.
- Medical Research Council Working Party. MRC trial of treatment of mild hypertension: principal results. BMJ 1985; 291:97–104.
- Coope J, Warrender TS. Randomised trial of treatment of hypertension in elderly patients in primary care. BMJ 1986; 293:1145–1151.
- Dahlöf B, Lindholm LH, Hansson L, et al. Morbidity and mortality in the Swedish Trial in Old Patients with Hypertension (STOP-Hypertension). Lancet 1991; 338:1281–1285.
- MRC Working Party. Medical Research Council trial of treatment of hypertension in older adults: principal results. BMJ 1992; 304:405–412.
- The Dutch TIA Study Group. Trial of secondary prevention with atenolol after transient ischemic attack or nondisabling ischemic stroke. Stroke 1993; 24:543–548.
- Eriksson S, Olofsson B-O, Wester P-O; for the TEST Study Group. Atenolol in secondary prevention after stroke. Cerebrovasc Dis 1995; 5:21–25.
- Wilhelmsen L, Berglund G, Elmfeldt D, et al. Beta-blockers versus diuretics in hypertensive men: main results from the HAPPHY trial. J Hypertens 1987; 5:561–572.
- UK Prospective Diabetes Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. BMJ 1998; 317:713–720.
- Hansson L, Lindholm LH, Niskanen L, et al. Effect of angiotensin-converting enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPP) randomised trial. Lancet 1999; 353:611–616.
- Lanchetti A, Bond MG, Henning M, et al. Calcium antagonist lacidipine slow down progression of asymptomatic carotid atherosclerosis. Principal results of the European Lacidipine Study on Atherosclerosis (ELSA), a randomized, double-blind, long-term trial. Circulation 2002; 106:2422–2427.
- Hansson L, Lindholm LH, Ekbom T, et al. Randomised trial of old and new antihypertensive drugs in elderly patients: cardiovascular mortality and morbidity in the Swedish Trial in Old Patients with Hypertension-2 study. Lancet 1999; 354:1751–1756.
- Hansson L, Hedner T, Lund-Johansen P, et al. Randomised trial of effects of calcium antagonists compared with diuretics and ß blockers on cardiovascular morbidity and mortality in hypertension: the Nordic Diltiazem (NORDIL) study. Lancet 2000; 356:359–365.
- Pepine CJ, Handsberg EM, Cooper-DeHoff RM, et al. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290:2805–2816.
- Black HR, Elliott WJ, Grandits G, et al. Principal results of the Controlled Onset Verapamil Investigation of Cardiovascular End Points (CONVINCE) Trial. JAMA 2003; 289:2073–2082.
- Bangalore S, Sawhney S, Messerli FH. Relation of beta-blocker-induced heart rate lowering and cardioprotection in hypertension. J Am Coll Cardiol 2008; 52:1482–1489.
- Boutouyrie P, Vermersch S, Laurent S, Briet M. Cardiovascular risk assessment through target organ damage: role of carotid to femoral pulse wave velocity. Clin Exp Pharmacol Physiol 2008; 35:530–533.
- Kveiborg B, Christiansen B, Major-Petersen A, Torp-Pedersen C. Metabolic effects of beta-adrenoceptor antagonists with special emphasis on carvedilol. Am J Cardiovasc Drugs 2006; 6:209–217.
- Bangalore S, Parkar S, Grossman E, Messerli FH. A meta-analysis of 94,492 patients with hypertension treated with beta-blockers to determine the risk of new-onset diabetes mellitus. Am J Cardiol 2007; 100:1254–1262.
- Balamuthusamy S, Molnar J, Adigopula S, Arora R. Comparative analysis of beta-blockers with other antihypertensive agents on cardiovascular outcomes in hypertensive patients with diabetes mellitus: a systematic review and meta-analysis. Am J Ther 2009; 16:133–142.
- Berenson A. Big drug makers see sales decline with their image. New York Times 2005 Nov 14.
- Bristow MR. Beta-adrenergic receptor blockade in chronic heart failure. Circulation 2000; 101:558–569.
- Giugliano D, Marfella R, Acampora R, Giunta R, Coppola L, D’Onofrio F. Effects of perindopril and carvedilol on endothelium-dependent vascular functions in patients with diabetes and hypertension. Diabetes Care 1998; 21:631–636.
- Lopez BL, Christopher TA, Yue TL, Ruffolo R, Feuerstein GZ, Ma XL. Carvedilol, a new beta-adrenoreceptor blocker antihypertensive drug, protects against free-radical-induced endothelial dysfunction. Pharmacology 1995; 51:165–173.
- Bakris GL, Fonseca V, Katholi RE, et al; GEMINI Investigators. Metabolic effects of carvedilol vs metoprolol in patients with type 2 diabetes mellitus and hypertension: a randomized controlled trial. JAMA 2004; 292:2227–2236.
- Georgescu A, Pluteanu F, Flonta ML, Badila E, Dorobantu M, Popov D. The cellular mechanisms involved in the vasodilator effect of nebivolol on the renal artery. Eur J Pharmacol 2005; 508:159–166.
- Kalinowski L, Dobrucki LW, Szczepanska-Konkel M, et al. Third-generation beta-blockers stimulate nitric oxide release from endothelial cells through ATP efflux: a novel mechanism for antihypertensive action. Circulation 2003; 107:2747–2752.
- Cockcroft JR, Chowienczyk PJ, Brett SE, et al. Nebivolol vasodilates human forearm vasculature: evidence for an L-arginine/NO-dependent mechanism. J Pharmacol Exp Ther 1995; 274:1067–1071.
- McEniery CM, Schmitt M, Qasem A, et al. Nebivolol increases arterial distensibility in vivo. Hypertension 2004; 44:305–310.
- Poirier L, Cleroux J, Nadeau A, Lacourciere Y. Effects of nebivolol and atenolol on insulin sensitivity and haemodynamics in hypertensive patients. J Hypertens 2001; 19:1429–1435.
- Flather MD, Shibata MC, Coats AJ, et al; SENIORS Investigators. Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (SENIORS). Eur Heart J 2005; 26:215–225.
- Williams B, Poulter NR, Brown MJ, et al; BHS guidelines working party, for the British Hypertension Society. British Hypertension Society guidelines for hypertension management 2004 (BHS-IV): summary. BMJ 2004; 328:634–640.
- Sever P. New hypertension guidelines from the National Institute for Health and Clinical Excellence and the British Hypertension Society. J Renin Angiotensin Aldosterone Syst 2006; 7:61–63.
- Mancia G, De Backer G, Dominiczak A, et al; Management of Arterial Hypertension of the European Society of Hypertension. 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens 2007; 25:1105–1187.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289:2560–2572.
- Staessen JA, Wang JG, Thijs L. Cardiovascular protection and blood pressure reduction: a meta-analysis. Lancet 2001; 358:1305–1315.
- ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002; 288:2981–2997.
- Neutel JM, Smith DH, Ram CV, et al. Application of ambulatory blood pressure monitoring in differentiating between antihypertensive agents. Am J Med 1993; 94:181–187.
- Materson BJ, Reda DJ, Cushman WC, et al. Single-drug therapy for hypertension in men. A comparison of six antihypertensive agents with placebo. The Department of Veterans Affairs Cooperative Study Group on Antihypertensive Agents. N Engl J Med 1993; 328:914–921.
- Psaty BM, Smith NL, Siscovick DS, et al. Health outcomes associated with antihypertensive therapies used as first-line agents. A systematic review and meta-analysis. JAMA 1997; 277:739–745.
- Frishman W, Silverman R. Clinical pharmacology of the new beta-adrenergic blocking drugs. Part 2. Physiologic and metabolic effects. Am Heart J 1979; 97:797–807.
- Garrett BN, Kaplan NM. Plasma renin activity suppression: duration after withdrawal from beta-adrenergic blockade. Arch Intern Med 1980; 140:1316–1318.
- Pedersen ME, Cockcroft JR. The latest generation of beta-blockers: new pharmacologic properties. Curr Hypertens Rep 2006; 8:279–286.
- Man in’t Veld AJ, Van den Meiracker AH, Schalekamp MA. Do beta-blockers really increase peripheral vascular resistance? Review of the literature and new observations under basal conditions. Am J Hypertens 1988; 1:91–96.
- Pearce CJ, Wallin JD. Labetalol and other agents that block both alpha- and beta-adrenergic receptors. Cleve Clin J Med 1994; 61:59–69.
- Dimsdale JE, Newton RP, Joist T. Neuropsychological side effects of beta-blockers. Arch Intern Med 1989; 149:514–525.
- Agarwal R. Supervised atenolol therapy in the management of hemodialysis hypertension. Kidney Int 1999; 55:1528–1535.
- Sica DA, Black HR. Pharmacologic considerations in the positioning of beta-blockers in antihypertensive therapy. Curr Hypertens Rep 2008; 10:330–335.
- Prichard BN, Gillam GP. Use of propranolol (Inderal) in treatment of hypertension. Br Med J 1964; 19; 2:725–727.
- The fifth report of the Joint National Committee on Detection, Evaluation, and Treatment of High Blood Pressure (JNC V). Arch Intern Med 1993; 153:154–183.
- Moser M. Evolution of the treatment of hypertension from the 1940s to JNC V. Am J Hypertens 1997; 10:2S–8S.
- Houghton T, Freemantle N, Cleland JG. Are beta-blockers effective in patients who develop heart failure soon after myocardial infarction? A meta-regression analysis of randomised trials. Eur J Heart Fail 2000; 2:333–340.
- The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet 1999; 353:9–13.
- Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999; 353:2001–2007.
- Yusuf S, Peto R, Lewis J, Collins R, Sleight P. Beta blockade during and after myocardial infarction: an overview of the randomized trials. Prog Cardiovasc Dis 1985; 27:335–371.
- Brophy JM, Joseph L, Rouleau JL. Beta-blockers in congestive heart failure. A Bayesian meta-analysis. Ann Intern Med 2001; 134:550–560.
- Wiysonge CS, Bradley H, Mayosi BM, et al. Beta-blockers for hypertension. Cochrane Database Syst Rev 2007;CD002003.
- Messerli FH, Grossman E, Goldbourt U. Are beta-blockers efficacious as first-line therapy for hypertension in the elderly? A systematic review. JAMA 1998; 279:1903–1907.
- Dahlöf B, Devereux RB, Kjeldsen SE, et al; for the LIFE study group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint Reduction in Hypertension study (LIFE): a randomised trial against atenolol. Lancet 2002; 359:995–1003.
- Carlberg B, Samuelsson O, Lindholm LH. Atenolol in hypertension: is it a wise choice? Lancet 2004; 364:1684–1689.
- Dahlöf B, Sever PS, Poulter NR, et al; ASCOT Investigators. Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA): a multicentre randomised controlled trial. Lancet 2005; 366:895–906.
- Lindholm LH, Carlberg B, Samuelsson O. Should beta-blockers remain first choice in the treatment of primary hypertension? A meta-analysis. Lancet 2005; 366:1545–1553.
- Neutel JM, Schnaper H, Cheung DG, Graettinger WF, Weber MA. Antihypertensive effects of beta-blockers administered once daily: 24-hour measurements. Am Heart J 1990; 120:166–171.
- Franklin SS, Gustin W, Wong ND, et al. Hemodynamic patterns of age-related changes in blood pressure. The Framingham Heart Study. Circulation 1997; 96:308–315.
- The IPPPSH Collaborative Group. Cardiovascular risk and risk factors in a randomized trial of treatment based on the beta-blocker oxprenolol: the International Prospective Primary Prevention Study in Hypertension (IPPPSH). J Hypertens 1985; 3:379–392.
- Williams B, Lacy PS, Thom SM, et al; CAFE Investigators. Differential impact of blood pressure-lowering drugs on central aortic pressure and clinical outcomes: principal results of the Conduit Artery Function Evaluation (CAFE) study. Circulation 2006; 113:1213–1225.
- Khan N, McAlister FA. Re-examining the efficacy of beta-blockers for the treatment of hypertension: a meta-analysis. CMAJ 2006; 174:1737–1742.
- Medical Research Council Working Party. MRC trial of treatment of mild hypertension: principal results. BMJ 1985; 291:97–104.
- Coope J, Warrender TS. Randomised trial of treatment of hypertension in elderly patients in primary care. BMJ 1986; 293:1145–1151.
- Dahlöf B, Lindholm LH, Hansson L, et al. Morbidity and mortality in the Swedish Trial in Old Patients with Hypertension (STOP-Hypertension). Lancet 1991; 338:1281–1285.
- MRC Working Party. Medical Research Council trial of treatment of hypertension in older adults: principal results. BMJ 1992; 304:405–412.
- The Dutch TIA Study Group. Trial of secondary prevention with atenolol after transient ischemic attack or nondisabling ischemic stroke. Stroke 1993; 24:543–548.
- Eriksson S, Olofsson B-O, Wester P-O; for the TEST Study Group. Atenolol in secondary prevention after stroke. Cerebrovasc Dis 1995; 5:21–25.
- Wilhelmsen L, Berglund G, Elmfeldt D, et al. Beta-blockers versus diuretics in hypertensive men: main results from the HAPPHY trial. J Hypertens 1987; 5:561–572.
- UK Prospective Diabetes Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. BMJ 1998; 317:713–720.
- Hansson L, Lindholm LH, Niskanen L, et al. Effect of angiotensin-converting enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPP) randomised trial. Lancet 1999; 353:611–616.
- Lanchetti A, Bond MG, Henning M, et al. Calcium antagonist lacidipine slow down progression of asymptomatic carotid atherosclerosis. Principal results of the European Lacidipine Study on Atherosclerosis (ELSA), a randomized, double-blind, long-term trial. Circulation 2002; 106:2422–2427.
- Hansson L, Lindholm LH, Ekbom T, et al. Randomised trial of old and new antihypertensive drugs in elderly patients: cardiovascular mortality and morbidity in the Swedish Trial in Old Patients with Hypertension-2 study. Lancet 1999; 354:1751–1756.
- Hansson L, Hedner T, Lund-Johansen P, et al. Randomised trial of effects of calcium antagonists compared with diuretics and ß blockers on cardiovascular morbidity and mortality in hypertension: the Nordic Diltiazem (NORDIL) study. Lancet 2000; 356:359–365.
- Pepine CJ, Handsberg EM, Cooper-DeHoff RM, et al. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA 2003; 290:2805–2816.
- Black HR, Elliott WJ, Grandits G, et al. Principal results of the Controlled Onset Verapamil Investigation of Cardiovascular End Points (CONVINCE) Trial. JAMA 2003; 289:2073–2082.
- Bangalore S, Sawhney S, Messerli FH. Relation of beta-blocker-induced heart rate lowering and cardioprotection in hypertension. J Am Coll Cardiol 2008; 52:1482–1489.
- Boutouyrie P, Vermersch S, Laurent S, Briet M. Cardiovascular risk assessment through target organ damage: role of carotid to femoral pulse wave velocity. Clin Exp Pharmacol Physiol 2008; 35:530–533.
- Kveiborg B, Christiansen B, Major-Petersen A, Torp-Pedersen C. Metabolic effects of beta-adrenoceptor antagonists with special emphasis on carvedilol. Am J Cardiovasc Drugs 2006; 6:209–217.
- Bangalore S, Parkar S, Grossman E, Messerli FH. A meta-analysis of 94,492 patients with hypertension treated with beta-blockers to determine the risk of new-onset diabetes mellitus. Am J Cardiol 2007; 100:1254–1262.
- Balamuthusamy S, Molnar J, Adigopula S, Arora R. Comparative analysis of beta-blockers with other antihypertensive agents on cardiovascular outcomes in hypertensive patients with diabetes mellitus: a systematic review and meta-analysis. Am J Ther 2009; 16:133–142.
- Berenson A. Big drug makers see sales decline with their image. New York Times 2005 Nov 14.
- Bristow MR. Beta-adrenergic receptor blockade in chronic heart failure. Circulation 2000; 101:558–569.
- Giugliano D, Marfella R, Acampora R, Giunta R, Coppola L, D’Onofrio F. Effects of perindopril and carvedilol on endothelium-dependent vascular functions in patients with diabetes and hypertension. Diabetes Care 1998; 21:631–636.
- Lopez BL, Christopher TA, Yue TL, Ruffolo R, Feuerstein GZ, Ma XL. Carvedilol, a new beta-adrenoreceptor blocker antihypertensive drug, protects against free-radical-induced endothelial dysfunction. Pharmacology 1995; 51:165–173.
- Bakris GL, Fonseca V, Katholi RE, et al; GEMINI Investigators. Metabolic effects of carvedilol vs metoprolol in patients with type 2 diabetes mellitus and hypertension: a randomized controlled trial. JAMA 2004; 292:2227–2236.
- Georgescu A, Pluteanu F, Flonta ML, Badila E, Dorobantu M, Popov D. The cellular mechanisms involved in the vasodilator effect of nebivolol on the renal artery. Eur J Pharmacol 2005; 508:159–166.
- Kalinowski L, Dobrucki LW, Szczepanska-Konkel M, et al. Third-generation beta-blockers stimulate nitric oxide release from endothelial cells through ATP efflux: a novel mechanism for antihypertensive action. Circulation 2003; 107:2747–2752.
- Cockcroft JR, Chowienczyk PJ, Brett SE, et al. Nebivolol vasodilates human forearm vasculature: evidence for an L-arginine/NO-dependent mechanism. J Pharmacol Exp Ther 1995; 274:1067–1071.
- McEniery CM, Schmitt M, Qasem A, et al. Nebivolol increases arterial distensibility in vivo. Hypertension 2004; 44:305–310.
- Poirier L, Cleroux J, Nadeau A, Lacourciere Y. Effects of nebivolol and atenolol on insulin sensitivity and haemodynamics in hypertensive patients. J Hypertens 2001; 19:1429–1435.
- Flather MD, Shibata MC, Coats AJ, et al; SENIORS Investigators. Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (SENIORS). Eur Heart J 2005; 26:215–225.
- Williams B, Poulter NR, Brown MJ, et al; BHS guidelines working party, for the British Hypertension Society. British Hypertension Society guidelines for hypertension management 2004 (BHS-IV): summary. BMJ 2004; 328:634–640.
- Sever P. New hypertension guidelines from the National Institute for Health and Clinical Excellence and the British Hypertension Society. J Renin Angiotensin Aldosterone Syst 2006; 7:61–63.
- Mancia G, De Backer G, Dominiczak A, et al; Management of Arterial Hypertension of the European Society of Hypertension. 2007 Guidelines for the Management of Arterial Hypertension: The Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens 2007; 25:1105–1187.
- Chobanian AV, Bakris GL, Black HR, et al; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289:2560–2572.
KEY POINTS
- No evidence exists that beta-blockers prevent first episodes of cardiovascular events in patients with hypertension, and in some trials, outcomes were worse with beta-blockers than with antihypertensive drugs of other classes.
- Younger hypertensive patients have hemodynamic characteristics that would seem to be amenable to beta-blocker therapy. However, most clinical trials of beta blockers did not stratify patients by age.
- Most trials of the antihypertensive effects of beta-blockers used atenolol (Tenormin), which is not an ideal representative of this class of drugs.
- Newer beta-blockers with vasodilatory properties may overcome the adverse effect of increased peripheral vascular resistance that occurs with older agents such as atenolol.
Does measuring natriuretic peptides have a role in patients with chronic kidney disease?
Yes, measuring the levels of certain natriuretic peptides can help diagnose decompensated heart failure and predict the risk of death and cardiac hospitalization in patients across a wide spectrum of renal function.
However, at this time, it is unclear whether routinely measuring natriuretic peptides will result in any change in the management of patients with chronic kidney disease. Additionally, using these peptides to monitor volume status in dialysis patients has not yet been deemed useful, although it may be complementary to echocardiography in evaluating cardiac risk in patients with end-stage renal disease.
A BRIEF REVIEW OF NATRIURETIC PEPTIDES
Natriuretic peptides include atrial natriuretic peptide, brain natriuretic peptide (BNP), C-type natriuretic peptide, and urodilantin.
BNP, which is homologous to atrial natriuretic peptide, is present in the brain and the heart. The circulating concentration of BNP is less than 20% of the atrial natriuretic peptide level in healthy people, but equals or exceeds that of atrial natriuretic peptide in patients with congestive heart failure.
BNP starts as a precursor protein. This is modified within the cell into a prohormone, proBNP, which is secreted from the left ventricle in response to myocardial wall stress. In the circulation, proBNP is cleaved into a biologically active C-terminal fragment—BNP—and a biologically inactive N-terminal fragment (NT-proBNP).1 NT-proBNP is primarily cleared by the kidney. BNP is cleared by receptor-mediated binding and removed by neutral endopeptidase, as well as by the kidney.
Both BNP and NT-proBNP have been investigated as diagnostic markers of suspected heart disease.
PEPTIDE LEVELS ARE HIGH IN CHRONIC KIDNEY DISEASE AND HEART FAILURE
An estimated 8.3 million people in the United States have stage 3, 4, or 5 chronic kidney disease,2 defined as an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2. Approximately 50% of patients with heart failure have chronic kidney disease, and almost 60% of patients with chronic kidney disease have some abnormality in ventricular function.
A few years ago, researchers began investigating the benefits and limitations of using natriuretic peptides to diagnose cardiac dysfunction (left ventricular structural and functional abnormalities) in patients with chronic kidney disease.
One important study3 was conducted in almost 3,000 patients from the Dallas Heart Study who were between the ages of 30 and 65 years—a relatively young, mostly healthy population. The authors found that natriuretic peptide levels did not vary as long as the estimated glomerular filtration rate was within the normal range. However, when the estimated glomerular filtration rate dropped below a threshold of 90 mL/min/1.73 m2, the concentrations of both NT-proBNP and BNP increased exponentially. NT-proBNP levels rose more than BNP levels, as NT-proBNP is primarily cleared by the kidney.
More recent studies found that the high levels of NT-proBNP in patients with chronic kidney disease do not simply reflect the reduced clearance of this peptide; they also reflect compromised ventricular function.2,4 This relationship was supported by studies of the fractional renal excretion of NT-proBNP and BNP in several populations with and without renal impairment.5 Interestingly, fractional excretion of both peptides remained equivalent across a wide spectrum of renal function. Seemingly, cardiac disease drove the increase in values rather than the degree of renal impairment.
HIGH PEPTIDE LEVELS PREDICT DEATH, HOSPITALIZATION
Both BNP and NT-proBNP are strong predictors of death and cardiac hospitalization in kidney patients.1,4,6
In patients with end-stage renal disease, the risk of cardiovascular disease and death is significantly higher than that in the general population, and BNP has been found to be a valuable prognostic indicator of cardiac disease.7
Multiple studies showed that high levels of natriuretic peptides are associated with a higher risk of death in patients with acute coronary syndrome, independent of traditional cardiovascular risk factors such as electrocardiographic changes and levels of other biomarkers. However, these data were derived from patients with mild renal impairment.2
Apple et al8 compared the prognostic value of NT-proBNP with that of cardiac troponin T in hemodialysis patients who had no symptoms and found that NT-proBNP was more strongly associated with left ventricular systolic dysfunction and subsequent cardiovascular death.
PEPTIDE LEVELS ARE HIGHER IN ANEMIA
A significant number of patients with congestive heart failure have renal insufficiency and low hemoglobin levels, which may increase natriuretic peptide levels. It is unclear why anemia is associated with elevated levels of natriuretic peptides, even in the absence of clinical heart failure and independent of other cardiovascular risk factors.9 Nevertheless, anemia should be taken into consideration and treated effectively when evaluating patients with renal impairment and possible congestive heart failure.
PEPTIDES COMPLEMENT CARDIAC ECHO IN END-STAGE RENAL DISEASE
Numerous studies have found a close association between BNP and NT-proBNP levels and left ventricular mass and systolic function in patients with end-stage renal disease.10,11 Data from the Cardiovascular Risk Extended Evaluation in Dialysis Patients study12 suggest that BNP measurement can be reliably applied in end-stage renal disease to rule out systolic dysfunction and to detect left ventricular hypertrophy, but it has a very low negative predictive value for left ventricular hypertrophy in this patient population: someone with a normal BNP level can still have left ventricular hypertrophy.
In addition, volume status is harder to assess with BNP alone than with echocardiography, and an elevated BNP value is not very specific.13
In essence, both BNP and NT-proBNP can be used to complement echocardiography in evaluating cardiac risk in patients with end-stage renal disease. With additional data, it may be possible in the future to use them as substitutes for echocardiography when managing ventricular abnormalities in patients with end-stage renal disease.
USING SPECIFIC CUT POINTS IN RENAL DISEASE
When evaluating a patient with acute dyspnea and either chronic kidney disease or end-stage renal disease who is receiving dialysis, both BNP and NT-proBNP are affected similarly and necessitate a higher level of interpretation to diagnose decompensated heart failure. Currently, researchers disagree about specific cut points for natriuretic peptides. However, deFilippi and colleagues4 suggested the following cut points for NT-proBNP for diagnosing heart failure in patients of different ages with or without renal impairment:
- Younger than 50 years—450 ng/L
- Age 50 to 75 years—900 ng/L
- Older than 75 years—1,800 ng/L.
A BNP cutoff point of 225 pg/mL can be used for patients with an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2, based on data from the Breathing Not Properly multinational study.14
There is no set cut-point for either BNP or NT-proBNP for predicting death and cardiac hospitalization in renal patients, but abnormally high levels should signal the need to optimize medical management and to monitor more closely.
- Austin WJ, Bhalla V, Hernandez-Arce I, et al. Correlation and prognostic utility of B-type natriuretic peptide and its amino-terminal fragment in patients with chronic kidney disease. Am J Clin Pathol 2006; 126:506–512.
- DeFilippi C, van Kimmenade RR, Pinto YM. Amino-terminal pro-B-type natriuretic peptide testing in renal disease. Am J Cardiol 2008; 101:82–88.
- Das SR, Abdullah SM, Leonard D, et al. Association between renal function and circulating levels of natriuretic peptides (from the Dallas Heart Study). Am J Cardiol 2008; 102:1394–1398.
- DeFilippi CR, Seliger SL, Maynard S, Christenson RH. Impact of renal disease on natriuretic peptide testing for diagnosing decompensated heart failure and predicting mortality. Clin Chem 2007; 53:1511–1519.
- Goetze JP, Jensen G, Møller S, Bendtsen F, Rehfeld JF, Henriksen JH. BNP and N-terminal proBNP are both extracted in the normal kidney. Eur J Clin Invest 2006; 36:8–15.
- Zoccali C. Biomarkers in chronic kidney disease: utility and issues towards better understanding. Curr Opin Nephrol Hypertens 2005; 14:532–537.
- Haapio M, Ronco C. BNP and a renal patient: emphasis on the unique characteristics of B-type natriuretic peptide in end-stage kidney disease. Contrib Nephrol 2008; 161:68–75.
- Apple FS, Murakami MM, Pearce LA, Herzog CA. Multibiomarker risk stratification of N-terminal pro-B-type natriuretic peptide, high-sensitivity C-reactive protein, and cardiac troponin T and I in end-stage renal disease for all-cause death. Clin Chem 2004: 50:2279–2285.
- Hogenhuis J, Voors AA, Jaarsma T, et al. Anemia and renal dysfunction are independently associated with BNP and NT-proBNP levels in patients with heart failure. Eur J Heart Fail 2007; 9:787–794.
- Madsen LH, Ladefoged S, Corell P, Schou M, Hildebrandt PR, Atar D. N-terminal pro brain natriuretic peptide predicts mortality in patients with end-stage renal disease on hemodialysis. Kidney Int 2007; 71:548–554.
- Wang AY, Lai KN. Use of cardiac biomarkers in end-stage renal disease. J Am Soc Nephrol 2008; 19:1643–1652.
- Mallamaci F, Zoccali C, Tripepi G, et al; on behalf of the CREED Investigators. Diagnostic potential of cardiac natriuretic peptides in dialysis patients. Kidney Int 2001; 59:1559–1566.
- Biasioli S, Zamperetti M, Borin D, Guidi G, De Fanti E, Schiavon R. Significance of plasma B-type natriuretic peptide in hemodialysis patients: blood sample timing and comorbidity burden. ASAIO J 2007; 53:587–591.
- McCullough PA, Duc P, Omland T, et al. B-type natriuretic peptide and renal function in the diagnosis of heart failure: an analysis from the Breathing Not Properly multinational study. Am J Kidney Dis 2003; 41:571–579.
Yes, measuring the levels of certain natriuretic peptides can help diagnose decompensated heart failure and predict the risk of death and cardiac hospitalization in patients across a wide spectrum of renal function.
However, at this time, it is unclear whether routinely measuring natriuretic peptides will result in any change in the management of patients with chronic kidney disease. Additionally, using these peptides to monitor volume status in dialysis patients has not yet been deemed useful, although it may be complementary to echocardiography in evaluating cardiac risk in patients with end-stage renal disease.
A BRIEF REVIEW OF NATRIURETIC PEPTIDES
Natriuretic peptides include atrial natriuretic peptide, brain natriuretic peptide (BNP), C-type natriuretic peptide, and urodilantin.
BNP, which is homologous to atrial natriuretic peptide, is present in the brain and the heart. The circulating concentration of BNP is less than 20% of the atrial natriuretic peptide level in healthy people, but equals or exceeds that of atrial natriuretic peptide in patients with congestive heart failure.
BNP starts as a precursor protein. This is modified within the cell into a prohormone, proBNP, which is secreted from the left ventricle in response to myocardial wall stress. In the circulation, proBNP is cleaved into a biologically active C-terminal fragment—BNP—and a biologically inactive N-terminal fragment (NT-proBNP).1 NT-proBNP is primarily cleared by the kidney. BNP is cleared by receptor-mediated binding and removed by neutral endopeptidase, as well as by the kidney.
Both BNP and NT-proBNP have been investigated as diagnostic markers of suspected heart disease.
PEPTIDE LEVELS ARE HIGH IN CHRONIC KIDNEY DISEASE AND HEART FAILURE
An estimated 8.3 million people in the United States have stage 3, 4, or 5 chronic kidney disease,2 defined as an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2. Approximately 50% of patients with heart failure have chronic kidney disease, and almost 60% of patients with chronic kidney disease have some abnormality in ventricular function.
A few years ago, researchers began investigating the benefits and limitations of using natriuretic peptides to diagnose cardiac dysfunction (left ventricular structural and functional abnormalities) in patients with chronic kidney disease.
One important study3 was conducted in almost 3,000 patients from the Dallas Heart Study who were between the ages of 30 and 65 years—a relatively young, mostly healthy population. The authors found that natriuretic peptide levels did not vary as long as the estimated glomerular filtration rate was within the normal range. However, when the estimated glomerular filtration rate dropped below a threshold of 90 mL/min/1.73 m2, the concentrations of both NT-proBNP and BNP increased exponentially. NT-proBNP levels rose more than BNP levels, as NT-proBNP is primarily cleared by the kidney.
More recent studies found that the high levels of NT-proBNP in patients with chronic kidney disease do not simply reflect the reduced clearance of this peptide; they also reflect compromised ventricular function.2,4 This relationship was supported by studies of the fractional renal excretion of NT-proBNP and BNP in several populations with and without renal impairment.5 Interestingly, fractional excretion of both peptides remained equivalent across a wide spectrum of renal function. Seemingly, cardiac disease drove the increase in values rather than the degree of renal impairment.
HIGH PEPTIDE LEVELS PREDICT DEATH, HOSPITALIZATION
Both BNP and NT-proBNP are strong predictors of death and cardiac hospitalization in kidney patients.1,4,6
In patients with end-stage renal disease, the risk of cardiovascular disease and death is significantly higher than that in the general population, and BNP has been found to be a valuable prognostic indicator of cardiac disease.7
Multiple studies showed that high levels of natriuretic peptides are associated with a higher risk of death in patients with acute coronary syndrome, independent of traditional cardiovascular risk factors such as electrocardiographic changes and levels of other biomarkers. However, these data were derived from patients with mild renal impairment.2
Apple et al8 compared the prognostic value of NT-proBNP with that of cardiac troponin T in hemodialysis patients who had no symptoms and found that NT-proBNP was more strongly associated with left ventricular systolic dysfunction and subsequent cardiovascular death.
PEPTIDE LEVELS ARE HIGHER IN ANEMIA
A significant number of patients with congestive heart failure have renal insufficiency and low hemoglobin levels, which may increase natriuretic peptide levels. It is unclear why anemia is associated with elevated levels of natriuretic peptides, even in the absence of clinical heart failure and independent of other cardiovascular risk factors.9 Nevertheless, anemia should be taken into consideration and treated effectively when evaluating patients with renal impairment and possible congestive heart failure.
PEPTIDES COMPLEMENT CARDIAC ECHO IN END-STAGE RENAL DISEASE
Numerous studies have found a close association between BNP and NT-proBNP levels and left ventricular mass and systolic function in patients with end-stage renal disease.10,11 Data from the Cardiovascular Risk Extended Evaluation in Dialysis Patients study12 suggest that BNP measurement can be reliably applied in end-stage renal disease to rule out systolic dysfunction and to detect left ventricular hypertrophy, but it has a very low negative predictive value for left ventricular hypertrophy in this patient population: someone with a normal BNP level can still have left ventricular hypertrophy.
In addition, volume status is harder to assess with BNP alone than with echocardiography, and an elevated BNP value is not very specific.13
In essence, both BNP and NT-proBNP can be used to complement echocardiography in evaluating cardiac risk in patients with end-stage renal disease. With additional data, it may be possible in the future to use them as substitutes for echocardiography when managing ventricular abnormalities in patients with end-stage renal disease.
USING SPECIFIC CUT POINTS IN RENAL DISEASE
When evaluating a patient with acute dyspnea and either chronic kidney disease or end-stage renal disease who is receiving dialysis, both BNP and NT-proBNP are affected similarly and necessitate a higher level of interpretation to diagnose decompensated heart failure. Currently, researchers disagree about specific cut points for natriuretic peptides. However, deFilippi and colleagues4 suggested the following cut points for NT-proBNP for diagnosing heart failure in patients of different ages with or without renal impairment:
- Younger than 50 years—450 ng/L
- Age 50 to 75 years—900 ng/L
- Older than 75 years—1,800 ng/L.
A BNP cutoff point of 225 pg/mL can be used for patients with an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2, based on data from the Breathing Not Properly multinational study.14
There is no set cut-point for either BNP or NT-proBNP for predicting death and cardiac hospitalization in renal patients, but abnormally high levels should signal the need to optimize medical management and to monitor more closely.
Yes, measuring the levels of certain natriuretic peptides can help diagnose decompensated heart failure and predict the risk of death and cardiac hospitalization in patients across a wide spectrum of renal function.
However, at this time, it is unclear whether routinely measuring natriuretic peptides will result in any change in the management of patients with chronic kidney disease. Additionally, using these peptides to monitor volume status in dialysis patients has not yet been deemed useful, although it may be complementary to echocardiography in evaluating cardiac risk in patients with end-stage renal disease.
A BRIEF REVIEW OF NATRIURETIC PEPTIDES
Natriuretic peptides include atrial natriuretic peptide, brain natriuretic peptide (BNP), C-type natriuretic peptide, and urodilantin.
BNP, which is homologous to atrial natriuretic peptide, is present in the brain and the heart. The circulating concentration of BNP is less than 20% of the atrial natriuretic peptide level in healthy people, but equals or exceeds that of atrial natriuretic peptide in patients with congestive heart failure.
BNP starts as a precursor protein. This is modified within the cell into a prohormone, proBNP, which is secreted from the left ventricle in response to myocardial wall stress. In the circulation, proBNP is cleaved into a biologically active C-terminal fragment—BNP—and a biologically inactive N-terminal fragment (NT-proBNP).1 NT-proBNP is primarily cleared by the kidney. BNP is cleared by receptor-mediated binding and removed by neutral endopeptidase, as well as by the kidney.
Both BNP and NT-proBNP have been investigated as diagnostic markers of suspected heart disease.
PEPTIDE LEVELS ARE HIGH IN CHRONIC KIDNEY DISEASE AND HEART FAILURE
An estimated 8.3 million people in the United States have stage 3, 4, or 5 chronic kidney disease,2 defined as an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2. Approximately 50% of patients with heart failure have chronic kidney disease, and almost 60% of patients with chronic kidney disease have some abnormality in ventricular function.
A few years ago, researchers began investigating the benefits and limitations of using natriuretic peptides to diagnose cardiac dysfunction (left ventricular structural and functional abnormalities) in patients with chronic kidney disease.
One important study3 was conducted in almost 3,000 patients from the Dallas Heart Study who were between the ages of 30 and 65 years—a relatively young, mostly healthy population. The authors found that natriuretic peptide levels did not vary as long as the estimated glomerular filtration rate was within the normal range. However, when the estimated glomerular filtration rate dropped below a threshold of 90 mL/min/1.73 m2, the concentrations of both NT-proBNP and BNP increased exponentially. NT-proBNP levels rose more than BNP levels, as NT-proBNP is primarily cleared by the kidney.
More recent studies found that the high levels of NT-proBNP in patients with chronic kidney disease do not simply reflect the reduced clearance of this peptide; they also reflect compromised ventricular function.2,4 This relationship was supported by studies of the fractional renal excretion of NT-proBNP and BNP in several populations with and without renal impairment.5 Interestingly, fractional excretion of both peptides remained equivalent across a wide spectrum of renal function. Seemingly, cardiac disease drove the increase in values rather than the degree of renal impairment.
HIGH PEPTIDE LEVELS PREDICT DEATH, HOSPITALIZATION
Both BNP and NT-proBNP are strong predictors of death and cardiac hospitalization in kidney patients.1,4,6
In patients with end-stage renal disease, the risk of cardiovascular disease and death is significantly higher than that in the general population, and BNP has been found to be a valuable prognostic indicator of cardiac disease.7
Multiple studies showed that high levels of natriuretic peptides are associated with a higher risk of death in patients with acute coronary syndrome, independent of traditional cardiovascular risk factors such as electrocardiographic changes and levels of other biomarkers. However, these data were derived from patients with mild renal impairment.2
Apple et al8 compared the prognostic value of NT-proBNP with that of cardiac troponin T in hemodialysis patients who had no symptoms and found that NT-proBNP was more strongly associated with left ventricular systolic dysfunction and subsequent cardiovascular death.
PEPTIDE LEVELS ARE HIGHER IN ANEMIA
A significant number of patients with congestive heart failure have renal insufficiency and low hemoglobin levels, which may increase natriuretic peptide levels. It is unclear why anemia is associated with elevated levels of natriuretic peptides, even in the absence of clinical heart failure and independent of other cardiovascular risk factors.9 Nevertheless, anemia should be taken into consideration and treated effectively when evaluating patients with renal impairment and possible congestive heart failure.
PEPTIDES COMPLEMENT CARDIAC ECHO IN END-STAGE RENAL DISEASE
Numerous studies have found a close association between BNP and NT-proBNP levels and left ventricular mass and systolic function in patients with end-stage renal disease.10,11 Data from the Cardiovascular Risk Extended Evaluation in Dialysis Patients study12 suggest that BNP measurement can be reliably applied in end-stage renal disease to rule out systolic dysfunction and to detect left ventricular hypertrophy, but it has a very low negative predictive value for left ventricular hypertrophy in this patient population: someone with a normal BNP level can still have left ventricular hypertrophy.
In addition, volume status is harder to assess with BNP alone than with echocardiography, and an elevated BNP value is not very specific.13
In essence, both BNP and NT-proBNP can be used to complement echocardiography in evaluating cardiac risk in patients with end-stage renal disease. With additional data, it may be possible in the future to use them as substitutes for echocardiography when managing ventricular abnormalities in patients with end-stage renal disease.
USING SPECIFIC CUT POINTS IN RENAL DISEASE
When evaluating a patient with acute dyspnea and either chronic kidney disease or end-stage renal disease who is receiving dialysis, both BNP and NT-proBNP are affected similarly and necessitate a higher level of interpretation to diagnose decompensated heart failure. Currently, researchers disagree about specific cut points for natriuretic peptides. However, deFilippi and colleagues4 suggested the following cut points for NT-proBNP for diagnosing heart failure in patients of different ages with or without renal impairment:
- Younger than 50 years—450 ng/L
- Age 50 to 75 years—900 ng/L
- Older than 75 years—1,800 ng/L.
A BNP cutoff point of 225 pg/mL can be used for patients with an estimated glomerular filtration rate of less than 60 mL/min/1.73 m2, based on data from the Breathing Not Properly multinational study.14
There is no set cut-point for either BNP or NT-proBNP for predicting death and cardiac hospitalization in renal patients, but abnormally high levels should signal the need to optimize medical management and to monitor more closely.
- Austin WJ, Bhalla V, Hernandez-Arce I, et al. Correlation and prognostic utility of B-type natriuretic peptide and its amino-terminal fragment in patients with chronic kidney disease. Am J Clin Pathol 2006; 126:506–512.
- DeFilippi C, van Kimmenade RR, Pinto YM. Amino-terminal pro-B-type natriuretic peptide testing in renal disease. Am J Cardiol 2008; 101:82–88.
- Das SR, Abdullah SM, Leonard D, et al. Association between renal function and circulating levels of natriuretic peptides (from the Dallas Heart Study). Am J Cardiol 2008; 102:1394–1398.
- DeFilippi CR, Seliger SL, Maynard S, Christenson RH. Impact of renal disease on natriuretic peptide testing for diagnosing decompensated heart failure and predicting mortality. Clin Chem 2007; 53:1511–1519.
- Goetze JP, Jensen G, Møller S, Bendtsen F, Rehfeld JF, Henriksen JH. BNP and N-terminal proBNP are both extracted in the normal kidney. Eur J Clin Invest 2006; 36:8–15.
- Zoccali C. Biomarkers in chronic kidney disease: utility and issues towards better understanding. Curr Opin Nephrol Hypertens 2005; 14:532–537.
- Haapio M, Ronco C. BNP and a renal patient: emphasis on the unique characteristics of B-type natriuretic peptide in end-stage kidney disease. Contrib Nephrol 2008; 161:68–75.
- Apple FS, Murakami MM, Pearce LA, Herzog CA. Multibiomarker risk stratification of N-terminal pro-B-type natriuretic peptide, high-sensitivity C-reactive protein, and cardiac troponin T and I in end-stage renal disease for all-cause death. Clin Chem 2004: 50:2279–2285.
- Hogenhuis J, Voors AA, Jaarsma T, et al. Anemia and renal dysfunction are independently associated with BNP and NT-proBNP levels in patients with heart failure. Eur J Heart Fail 2007; 9:787–794.
- Madsen LH, Ladefoged S, Corell P, Schou M, Hildebrandt PR, Atar D. N-terminal pro brain natriuretic peptide predicts mortality in patients with end-stage renal disease on hemodialysis. Kidney Int 2007; 71:548–554.
- Wang AY, Lai KN. Use of cardiac biomarkers in end-stage renal disease. J Am Soc Nephrol 2008; 19:1643–1652.
- Mallamaci F, Zoccali C, Tripepi G, et al; on behalf of the CREED Investigators. Diagnostic potential of cardiac natriuretic peptides in dialysis patients. Kidney Int 2001; 59:1559–1566.
- Biasioli S, Zamperetti M, Borin D, Guidi G, De Fanti E, Schiavon R. Significance of plasma B-type natriuretic peptide in hemodialysis patients: blood sample timing and comorbidity burden. ASAIO J 2007; 53:587–591.
- McCullough PA, Duc P, Omland T, et al. B-type natriuretic peptide and renal function in the diagnosis of heart failure: an analysis from the Breathing Not Properly multinational study. Am J Kidney Dis 2003; 41:571–579.
- Austin WJ, Bhalla V, Hernandez-Arce I, et al. Correlation and prognostic utility of B-type natriuretic peptide and its amino-terminal fragment in patients with chronic kidney disease. Am J Clin Pathol 2006; 126:506–512.
- DeFilippi C, van Kimmenade RR, Pinto YM. Amino-terminal pro-B-type natriuretic peptide testing in renal disease. Am J Cardiol 2008; 101:82–88.
- Das SR, Abdullah SM, Leonard D, et al. Association between renal function and circulating levels of natriuretic peptides (from the Dallas Heart Study). Am J Cardiol 2008; 102:1394–1398.
- DeFilippi CR, Seliger SL, Maynard S, Christenson RH. Impact of renal disease on natriuretic peptide testing for diagnosing decompensated heart failure and predicting mortality. Clin Chem 2007; 53:1511–1519.
- Goetze JP, Jensen G, Møller S, Bendtsen F, Rehfeld JF, Henriksen JH. BNP and N-terminal proBNP are both extracted in the normal kidney. Eur J Clin Invest 2006; 36:8–15.
- Zoccali C. Biomarkers in chronic kidney disease: utility and issues towards better understanding. Curr Opin Nephrol Hypertens 2005; 14:532–537.
- Haapio M, Ronco C. BNP and a renal patient: emphasis on the unique characteristics of B-type natriuretic peptide in end-stage kidney disease. Contrib Nephrol 2008; 161:68–75.
- Apple FS, Murakami MM, Pearce LA, Herzog CA. Multibiomarker risk stratification of N-terminal pro-B-type natriuretic peptide, high-sensitivity C-reactive protein, and cardiac troponin T and I in end-stage renal disease for all-cause death. Clin Chem 2004: 50:2279–2285.
- Hogenhuis J, Voors AA, Jaarsma T, et al. Anemia and renal dysfunction are independently associated with BNP and NT-proBNP levels in patients with heart failure. Eur J Heart Fail 2007; 9:787–794.
- Madsen LH, Ladefoged S, Corell P, Schou M, Hildebrandt PR, Atar D. N-terminal pro brain natriuretic peptide predicts mortality in patients with end-stage renal disease on hemodialysis. Kidney Int 2007; 71:548–554.
- Wang AY, Lai KN. Use of cardiac biomarkers in end-stage renal disease. J Am Soc Nephrol 2008; 19:1643–1652.
- Mallamaci F, Zoccali C, Tripepi G, et al; on behalf of the CREED Investigators. Diagnostic potential of cardiac natriuretic peptides in dialysis patients. Kidney Int 2001; 59:1559–1566.
- Biasioli S, Zamperetti M, Borin D, Guidi G, De Fanti E, Schiavon R. Significance of plasma B-type natriuretic peptide in hemodialysis patients: blood sample timing and comorbidity burden. ASAIO J 2007; 53:587–591.
- McCullough PA, Duc P, Omland T, et al. B-type natriuretic peptide and renal function in the diagnosis of heart failure: an analysis from the Breathing Not Properly multinational study. Am J Kidney Dis 2003; 41:571–579.
Preventing renal disease progression: Can complete renin-angiotensin-aldosterone blockade work?
Perhaps the most daunting challenge for any primary care physician, nephrologist, or other internal medicine specialist is how to prevent the progression of chronic kidney disease.
A MAJOR HEALTH CARE CRISIS
Ten to 20 million people in the United States have chronic kidney disease, with diabetic nephropathy and arterial hypertension accounting for two-thirds of cases. In 2007, the US Renal Data System1 reported that, at the end of 2005, 341,319 patients were receiving dialysis and another 143,693 had received renal transplants.
The National Kidney Foundation’s Kidney Disease Outcomes Quality Initiatives2 has raised the level of awareness of chronic kidney disease among physicians and the general public. We have become more adept at diagnosing chronic kidney disease, in particular by calculating the estimated glomerular filtration rate, and we are starting to learn how to sort out the patients designated as having chronic kidney disease by this calculation but without “true” kidney disease. Nevertheless, the medical profession is still struggling to determine the best way to prevent progression in chronic kidney disease, and no single innovative approach currently exists. Should the emphasis be on the blood pressure target, the level of proteinuria reduction, the classes of medications to be used, or on other factors such as lipid control, vitamin D repletion,3 or glycemic control?
WHY INHIBIT THE RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM?
Over the last 20 years, investigators have devoted much effort to controlling the adverse effects of the renin-angiotensin-aldosterone system on the renal vasculature and parenchyma. We now understand that this system is a complex cascade and that angiotensin II plays a key role.
Angiotensin II enhances the vascular tone of both the afferent and the efferent glomerular arterioles, helps regulate intraglomerular pressure and glomerular filtration, and stimulates the adrenal cortex to release aldosterone. In addition, it has several nonhemodynamic effects. In particular, it may alter the selective permeability of the glomerular capillary barrier by influencing podocyte morphology and by directing a reorganization of its actin cytostructure.
Podocytes are highly differentiated pericyte-like cells that are essential for normal kidney function, but they have limited regenerative ability. Angiotensin II stimulation can lead to podocyte injury via mechanical stress due to increased intraglomerular pressure or an increase in cytosolic calcium,4 formation of bridging between the parietal basement membrane and the glomerular basement membrane,5 and extension of the extracapillary disease process to the glomerular-proximal tubular junction.6 These alterations can result in progressive atrophy, cell death, subsequent fibrosis, and irreversible loss in functioning renal parenchyma.
EVIDENCE FOR AND AGAINST COMBINATION THERAPY
In theory, by completely inhibiting the renin-angiotensin-aldosterone system in some patients with proteinuric chronic kidney disease (as Dr. Sheldon Hirsch suggests in this issue of the Cleveland Clinic Journal of Medicine7), we might be better able to prevent progressive renal injury than with an incomplete blockade of this system.
The rationale for complete blockade stems from evidence that long-term treatment with an angiotensin-converting enzyme (ACE) inhibitor results in the accumulation of angiotensin I, the escape of angiotensin II generation by ACE-independent enzymes (chymases), and the inhibition of angiotensin-(1–7) formation that partially antagonizes the effects of angiotensin II. In addition, aldosterone may injure the kidney by its rapid nongenomic effect on the renal vasculature, resulting in increased renal vascular resistance, with afferent and efferent vasoconstriction. Therefore, treatment with either an ACE inhibitor or an angiotensin receptor blocker (ARB) by itself may delay but not prevent end-stage renal disease for most patients with proteinuric chronic kidney disease.8
Combining an ACE inhibitor and an ARB
Regimens in which an ACE inhibitor is combined with an ARB may achieve their therapeutic benefit of lowering proteinuria by modulating the compensatory events in kidney injury that stress “normal” nephrons, inhibiting the podocyte injury responsible for contiguous damage in the tubulointerstitial area, and limiting fibrosis and inflammation. However, few trials actually showed that combining an ACE inhibitor with an ARB leads to greater renal protection in the long term than with either agent alone, despite a greater chance of lowering the protein excretion rate.9,10
The COOPERATE study. The Combination Treatment of Angiotensin II Receptor Blocker and Angiotensin-Converting-Enzyme Inhibitor in Non-diabetic Renal Disease (COOPERATE) study11 evaluated the renoprotective effects of the combination of trandolapril (Mavik, an ACE inhibitor) and losartan (Cozaar, an ARB). Significantly fewer patients reached one of the end points (doubling of the serum creatinine concentration or end-stage renal disease) with the combined therapy than with either agent alone.
Kunz et al12 recently performed a meta-analysis, which indicated that the combination of an ACE inhibitor and an ARB reduces proteinuria to a greater extent than either drug alone. However, the total number of patients in each trial was less than 30 on average, the duration of therapy rarely exceeded 1 year, and the effect on changes in the glomerular filtration rate or the need for dialysis was not reported.
ONTARGET. In the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET),13 combination therapy had no clear benefit in the group at the highest renal risk (ie, with overt diabetic nephropathy), and it was associated with a trend toward worse results in the low-risk group. Most participants in ONTARGET did not have microalbuminuria or macroalbuminuria, and of interest, these patients without protein excretion were at increased risk for renal events, such as acute renal failure requiring dialysis.
Phillips et al14 recently reported on the safety profile of patients with symptomatic left ventricular dysfunction treated with the combination of an ACE inhibitor and an ARB. Even in these nonrenal patients there was a significantly higher risk of worsening renal dysfunction (relative risk 4.87, 95% confidence interval 2.39–9.94) and hyperkalemia (relative risk 4.87, 95% confidence interval 2.39–9.94) with combination therapy.
Adding an aldosterone blocker to an ACE inhibitor, ARB, or both
There is little evidence that aldosterone plays a role in the progression of chronic kidney disease. However, several studies found that combining an aldosterone blocker with an ACE inhibitor, ARB, or both had an additional impact on reducing proteinuria and modulating the rate of change in the glomerular filtration rate.15–17
When aldosterone antagonists were added to an ACE inhibitor, an ARB, or both combined, proteinuria was reduced, but there was little effect on preserving the glomerular filtration rate.17 However, most of the studies were small, with short observation periods. Hyperkalemia is a risk when using aldosterone antagonists in combination with ACE inhibitors and ARBs, especially in patients with glomerular filtration rates less than 30 mL/minute.18
Adding a renin inhibitor to an ACE inhibitor or an ARB
Few studies have examined combination therapy with either an ACE inhibitor or ARB plus a renin inhibitor, the newest class of agents that block this system.
Parving et al19 recently reported the results of combining aliskiren (Tekturna, a renin inhibitor) with losartan in 599 patients with type 2 diabetes and nephropathy. At 6 months, the renin inhibitor showed a renoprotective effect that was independent of its blood-pressure-lowering effect in those who were receiving maximal recommended doses of the ARB.
OTHER FACTORS ALSO INFLUENCE PROGRESSION
Even though there is broad agreement that an approach that neutralizes the effects of the renin-angiotensin-aldosterone system on the kidney would lower blood pressure and protein excretion rates, whether it would change the natural history of chronic kidney disease and prevent progression is less clear. In reality, a number of factors other than the renin-angiotensin-aldosterone system are responsible for the progression of chronic kidney disease. These other factors may help explain why control of this system does not totally prevent deterioration of chronic kidney disease, although the rate may be slowed.
MORE QUESTIONS THAN ANSWERS
A number of provocative questions arise from Dr. Hirsch’s discussion of complete renin-angiotensin-aldosterone system blockade to prevent disease progression:
- Will decreasing proteinuria to a specific target (< 500 mg/day) prevent progression?
- How low should the blood pressure target be set to modulate progression, and should it be the same in all age groups?
- Should complete blockade be applied all at once or in a stepwise fashion depending on the glomerular filtration rate, the level of proteinuria, or both?
- Which patients would benefit most from complete blockade?
- Is direct renin inhibition a critical component of complete blockade?
- What model of chronic disease management is required to avoid unexpected complications if this treatment approach is embraced?
Currently, therefore, there are more questions than answers. This strategy is an intriguing, opinion-based option, but for now it should only be applied to patients with proteinuria and evidence of early progression despite standard therapy who can be closely monitored, and it is not for the faint of heart. In view of the risks of hyperkalemia, hypotension, and perhaps even worsening renal function, more data from carefully designed trials are needed before the general medical community widely applies a complete blockade of the renin-angiotensin-aldosterone pathway to prevent progressive chronic kidney disease.
- United States Renal Data System. Annual data report. www.usrds.org/adr.htm. Accessed 9/5/2008.
- National Kidney Foundation. NKF K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. www.kidney.org/Professionals/Kdoqi/guidelines_ckd/toc.htm. Accessed 9/5/2008.
- Remuzzi A. Vitamin D, insulin resistance, and renal disease. Kidney Int. 2007; 71:96–98.
- Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003; 83:253–307.
- Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int 1998; 54:687–697.
- Endlich N, Endlich K. Stretch, tension and adhesion—adaptive mechanisms of the actin cytoskeleton in podocytes. Eur J Cell Biol. 2006; 85:229–234.
- Hirsch S. An update on proteinuric chronic kidney disease: the dual-goal approach. Cleve Clin J Med. 2008; 75:705–713.
- Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993; 329:1456–1462.
- Wolf G, Ritz E. Combination therapy with ACE inhibitors and angiotensin II receptor blockers to halt progression of chronic renal disease: pathophysiology and indications. Kidney Int. 2005; 67:799–812.
- Campbell R, Sangalli F, Perticucci E, et al. Effects of combined ACE inhibitor and angiotensin II antagonist treatment in human chronic nephropathies. Kidney Int. 2003; 63:1094–1103.
- Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet. 2003; 361:117–124.
- Kunz R, Friedrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med. 2008; 148:30–48.
- Mann JF, Schmieder RE, McQueen M, et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet. 2008; 372:547–553.
- Phillips CO, Kashani A, Ko DK, Francis G, Krumholz HM. Adverse effects of combination angiotensin II receptor blockers plus angiotensin-converting enzyme inhibitors for left ventricular dysfunction: a quantitative review of data from randomized clinical trials. Arch Intern Med. 2007; 167:1930–1936.
- Epstein M. Adding spironolactone to conventional antihypertensives reduces albuminuria in patients with diabetic nephropathy. Nat Clin Pract Nephrol. 2006; 2:310–311.
- Rossing K, Schjoedt KJ, Smidt UM, Boomsma F, Parving HH. Beneficial effects of adding spironolactone to recommended antihypertensive treatment in diabetic nephropathy: a randomized, double-masked, cross-over study. Diabetes Care. 2005; 28:2106–2112.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006; 70:2116–2123.
- Bomback AS, Kshirsagar AV, Amamoo MA, Klemmer PJ. Change in proteinuria after adding aldosterone blockers to ACE inhibitors or angiotensin receptor blockers in CKD: a systematic review. Am J Kidney Dis. 2008; 51:199–211.
- Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med. 2008; 358:2433–2446.
Perhaps the most daunting challenge for any primary care physician, nephrologist, or other internal medicine specialist is how to prevent the progression of chronic kidney disease.
A MAJOR HEALTH CARE CRISIS
Ten to 20 million people in the United States have chronic kidney disease, with diabetic nephropathy and arterial hypertension accounting for two-thirds of cases. In 2007, the US Renal Data System1 reported that, at the end of 2005, 341,319 patients were receiving dialysis and another 143,693 had received renal transplants.
The National Kidney Foundation’s Kidney Disease Outcomes Quality Initiatives2 has raised the level of awareness of chronic kidney disease among physicians and the general public. We have become more adept at diagnosing chronic kidney disease, in particular by calculating the estimated glomerular filtration rate, and we are starting to learn how to sort out the patients designated as having chronic kidney disease by this calculation but without “true” kidney disease. Nevertheless, the medical profession is still struggling to determine the best way to prevent progression in chronic kidney disease, and no single innovative approach currently exists. Should the emphasis be on the blood pressure target, the level of proteinuria reduction, the classes of medications to be used, or on other factors such as lipid control, vitamin D repletion,3 or glycemic control?
WHY INHIBIT THE RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM?
Over the last 20 years, investigators have devoted much effort to controlling the adverse effects of the renin-angiotensin-aldosterone system on the renal vasculature and parenchyma. We now understand that this system is a complex cascade and that angiotensin II plays a key role.
Angiotensin II enhances the vascular tone of both the afferent and the efferent glomerular arterioles, helps regulate intraglomerular pressure and glomerular filtration, and stimulates the adrenal cortex to release aldosterone. In addition, it has several nonhemodynamic effects. In particular, it may alter the selective permeability of the glomerular capillary barrier by influencing podocyte morphology and by directing a reorganization of its actin cytostructure.
Podocytes are highly differentiated pericyte-like cells that are essential for normal kidney function, but they have limited regenerative ability. Angiotensin II stimulation can lead to podocyte injury via mechanical stress due to increased intraglomerular pressure or an increase in cytosolic calcium,4 formation of bridging between the parietal basement membrane and the glomerular basement membrane,5 and extension of the extracapillary disease process to the glomerular-proximal tubular junction.6 These alterations can result in progressive atrophy, cell death, subsequent fibrosis, and irreversible loss in functioning renal parenchyma.
EVIDENCE FOR AND AGAINST COMBINATION THERAPY
In theory, by completely inhibiting the renin-angiotensin-aldosterone system in some patients with proteinuric chronic kidney disease (as Dr. Sheldon Hirsch suggests in this issue of the Cleveland Clinic Journal of Medicine7), we might be better able to prevent progressive renal injury than with an incomplete blockade of this system.
The rationale for complete blockade stems from evidence that long-term treatment with an angiotensin-converting enzyme (ACE) inhibitor results in the accumulation of angiotensin I, the escape of angiotensin II generation by ACE-independent enzymes (chymases), and the inhibition of angiotensin-(1–7) formation that partially antagonizes the effects of angiotensin II. In addition, aldosterone may injure the kidney by its rapid nongenomic effect on the renal vasculature, resulting in increased renal vascular resistance, with afferent and efferent vasoconstriction. Therefore, treatment with either an ACE inhibitor or an angiotensin receptor blocker (ARB) by itself may delay but not prevent end-stage renal disease for most patients with proteinuric chronic kidney disease.8
Combining an ACE inhibitor and an ARB
Regimens in which an ACE inhibitor is combined with an ARB may achieve their therapeutic benefit of lowering proteinuria by modulating the compensatory events in kidney injury that stress “normal” nephrons, inhibiting the podocyte injury responsible for contiguous damage in the tubulointerstitial area, and limiting fibrosis and inflammation. However, few trials actually showed that combining an ACE inhibitor with an ARB leads to greater renal protection in the long term than with either agent alone, despite a greater chance of lowering the protein excretion rate.9,10
The COOPERATE study. The Combination Treatment of Angiotensin II Receptor Blocker and Angiotensin-Converting-Enzyme Inhibitor in Non-diabetic Renal Disease (COOPERATE) study11 evaluated the renoprotective effects of the combination of trandolapril (Mavik, an ACE inhibitor) and losartan (Cozaar, an ARB). Significantly fewer patients reached one of the end points (doubling of the serum creatinine concentration or end-stage renal disease) with the combined therapy than with either agent alone.
Kunz et al12 recently performed a meta-analysis, which indicated that the combination of an ACE inhibitor and an ARB reduces proteinuria to a greater extent than either drug alone. However, the total number of patients in each trial was less than 30 on average, the duration of therapy rarely exceeded 1 year, and the effect on changes in the glomerular filtration rate or the need for dialysis was not reported.
ONTARGET. In the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET),13 combination therapy had no clear benefit in the group at the highest renal risk (ie, with overt diabetic nephropathy), and it was associated with a trend toward worse results in the low-risk group. Most participants in ONTARGET did not have microalbuminuria or macroalbuminuria, and of interest, these patients without protein excretion were at increased risk for renal events, such as acute renal failure requiring dialysis.
Phillips et al14 recently reported on the safety profile of patients with symptomatic left ventricular dysfunction treated with the combination of an ACE inhibitor and an ARB. Even in these nonrenal patients there was a significantly higher risk of worsening renal dysfunction (relative risk 4.87, 95% confidence interval 2.39–9.94) and hyperkalemia (relative risk 4.87, 95% confidence interval 2.39–9.94) with combination therapy.
Adding an aldosterone blocker to an ACE inhibitor, ARB, or both
There is little evidence that aldosterone plays a role in the progression of chronic kidney disease. However, several studies found that combining an aldosterone blocker with an ACE inhibitor, ARB, or both had an additional impact on reducing proteinuria and modulating the rate of change in the glomerular filtration rate.15–17
When aldosterone antagonists were added to an ACE inhibitor, an ARB, or both combined, proteinuria was reduced, but there was little effect on preserving the glomerular filtration rate.17 However, most of the studies were small, with short observation periods. Hyperkalemia is a risk when using aldosterone antagonists in combination with ACE inhibitors and ARBs, especially in patients with glomerular filtration rates less than 30 mL/minute.18
Adding a renin inhibitor to an ACE inhibitor or an ARB
Few studies have examined combination therapy with either an ACE inhibitor or ARB plus a renin inhibitor, the newest class of agents that block this system.
Parving et al19 recently reported the results of combining aliskiren (Tekturna, a renin inhibitor) with losartan in 599 patients with type 2 diabetes and nephropathy. At 6 months, the renin inhibitor showed a renoprotective effect that was independent of its blood-pressure-lowering effect in those who were receiving maximal recommended doses of the ARB.
OTHER FACTORS ALSO INFLUENCE PROGRESSION
Even though there is broad agreement that an approach that neutralizes the effects of the renin-angiotensin-aldosterone system on the kidney would lower blood pressure and protein excretion rates, whether it would change the natural history of chronic kidney disease and prevent progression is less clear. In reality, a number of factors other than the renin-angiotensin-aldosterone system are responsible for the progression of chronic kidney disease. These other factors may help explain why control of this system does not totally prevent deterioration of chronic kidney disease, although the rate may be slowed.
MORE QUESTIONS THAN ANSWERS
A number of provocative questions arise from Dr. Hirsch’s discussion of complete renin-angiotensin-aldosterone system blockade to prevent disease progression:
- Will decreasing proteinuria to a specific target (< 500 mg/day) prevent progression?
- How low should the blood pressure target be set to modulate progression, and should it be the same in all age groups?
- Should complete blockade be applied all at once or in a stepwise fashion depending on the glomerular filtration rate, the level of proteinuria, or both?
- Which patients would benefit most from complete blockade?
- Is direct renin inhibition a critical component of complete blockade?
- What model of chronic disease management is required to avoid unexpected complications if this treatment approach is embraced?
Currently, therefore, there are more questions than answers. This strategy is an intriguing, opinion-based option, but for now it should only be applied to patients with proteinuria and evidence of early progression despite standard therapy who can be closely monitored, and it is not for the faint of heart. In view of the risks of hyperkalemia, hypotension, and perhaps even worsening renal function, more data from carefully designed trials are needed before the general medical community widely applies a complete blockade of the renin-angiotensin-aldosterone pathway to prevent progressive chronic kidney disease.
Perhaps the most daunting challenge for any primary care physician, nephrologist, or other internal medicine specialist is how to prevent the progression of chronic kidney disease.
A MAJOR HEALTH CARE CRISIS
Ten to 20 million people in the United States have chronic kidney disease, with diabetic nephropathy and arterial hypertension accounting for two-thirds of cases. In 2007, the US Renal Data System1 reported that, at the end of 2005, 341,319 patients were receiving dialysis and another 143,693 had received renal transplants.
The National Kidney Foundation’s Kidney Disease Outcomes Quality Initiatives2 has raised the level of awareness of chronic kidney disease among physicians and the general public. We have become more adept at diagnosing chronic kidney disease, in particular by calculating the estimated glomerular filtration rate, and we are starting to learn how to sort out the patients designated as having chronic kidney disease by this calculation but without “true” kidney disease. Nevertheless, the medical profession is still struggling to determine the best way to prevent progression in chronic kidney disease, and no single innovative approach currently exists. Should the emphasis be on the blood pressure target, the level of proteinuria reduction, the classes of medications to be used, or on other factors such as lipid control, vitamin D repletion,3 or glycemic control?
WHY INHIBIT THE RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM?
Over the last 20 years, investigators have devoted much effort to controlling the adverse effects of the renin-angiotensin-aldosterone system on the renal vasculature and parenchyma. We now understand that this system is a complex cascade and that angiotensin II plays a key role.
Angiotensin II enhances the vascular tone of both the afferent and the efferent glomerular arterioles, helps regulate intraglomerular pressure and glomerular filtration, and stimulates the adrenal cortex to release aldosterone. In addition, it has several nonhemodynamic effects. In particular, it may alter the selective permeability of the glomerular capillary barrier by influencing podocyte morphology and by directing a reorganization of its actin cytostructure.
Podocytes are highly differentiated pericyte-like cells that are essential for normal kidney function, but they have limited regenerative ability. Angiotensin II stimulation can lead to podocyte injury via mechanical stress due to increased intraglomerular pressure or an increase in cytosolic calcium,4 formation of bridging between the parietal basement membrane and the glomerular basement membrane,5 and extension of the extracapillary disease process to the glomerular-proximal tubular junction.6 These alterations can result in progressive atrophy, cell death, subsequent fibrosis, and irreversible loss in functioning renal parenchyma.
EVIDENCE FOR AND AGAINST COMBINATION THERAPY
In theory, by completely inhibiting the renin-angiotensin-aldosterone system in some patients with proteinuric chronic kidney disease (as Dr. Sheldon Hirsch suggests in this issue of the Cleveland Clinic Journal of Medicine7), we might be better able to prevent progressive renal injury than with an incomplete blockade of this system.
The rationale for complete blockade stems from evidence that long-term treatment with an angiotensin-converting enzyme (ACE) inhibitor results in the accumulation of angiotensin I, the escape of angiotensin II generation by ACE-independent enzymes (chymases), and the inhibition of angiotensin-(1–7) formation that partially antagonizes the effects of angiotensin II. In addition, aldosterone may injure the kidney by its rapid nongenomic effect on the renal vasculature, resulting in increased renal vascular resistance, with afferent and efferent vasoconstriction. Therefore, treatment with either an ACE inhibitor or an angiotensin receptor blocker (ARB) by itself may delay but not prevent end-stage renal disease for most patients with proteinuric chronic kidney disease.8
Combining an ACE inhibitor and an ARB
Regimens in which an ACE inhibitor is combined with an ARB may achieve their therapeutic benefit of lowering proteinuria by modulating the compensatory events in kidney injury that stress “normal” nephrons, inhibiting the podocyte injury responsible for contiguous damage in the tubulointerstitial area, and limiting fibrosis and inflammation. However, few trials actually showed that combining an ACE inhibitor with an ARB leads to greater renal protection in the long term than with either agent alone, despite a greater chance of lowering the protein excretion rate.9,10
The COOPERATE study. The Combination Treatment of Angiotensin II Receptor Blocker and Angiotensin-Converting-Enzyme Inhibitor in Non-diabetic Renal Disease (COOPERATE) study11 evaluated the renoprotective effects of the combination of trandolapril (Mavik, an ACE inhibitor) and losartan (Cozaar, an ARB). Significantly fewer patients reached one of the end points (doubling of the serum creatinine concentration or end-stage renal disease) with the combined therapy than with either agent alone.
Kunz et al12 recently performed a meta-analysis, which indicated that the combination of an ACE inhibitor and an ARB reduces proteinuria to a greater extent than either drug alone. However, the total number of patients in each trial was less than 30 on average, the duration of therapy rarely exceeded 1 year, and the effect on changes in the glomerular filtration rate or the need for dialysis was not reported.
ONTARGET. In the Ongoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial (ONTARGET),13 combination therapy had no clear benefit in the group at the highest renal risk (ie, with overt diabetic nephropathy), and it was associated with a trend toward worse results in the low-risk group. Most participants in ONTARGET did not have microalbuminuria or macroalbuminuria, and of interest, these patients without protein excretion were at increased risk for renal events, such as acute renal failure requiring dialysis.
Phillips et al14 recently reported on the safety profile of patients with symptomatic left ventricular dysfunction treated with the combination of an ACE inhibitor and an ARB. Even in these nonrenal patients there was a significantly higher risk of worsening renal dysfunction (relative risk 4.87, 95% confidence interval 2.39–9.94) and hyperkalemia (relative risk 4.87, 95% confidence interval 2.39–9.94) with combination therapy.
Adding an aldosterone blocker to an ACE inhibitor, ARB, or both
There is little evidence that aldosterone plays a role in the progression of chronic kidney disease. However, several studies found that combining an aldosterone blocker with an ACE inhibitor, ARB, or both had an additional impact on reducing proteinuria and modulating the rate of change in the glomerular filtration rate.15–17
When aldosterone antagonists were added to an ACE inhibitor, an ARB, or both combined, proteinuria was reduced, but there was little effect on preserving the glomerular filtration rate.17 However, most of the studies were small, with short observation periods. Hyperkalemia is a risk when using aldosterone antagonists in combination with ACE inhibitors and ARBs, especially in patients with glomerular filtration rates less than 30 mL/minute.18
Adding a renin inhibitor to an ACE inhibitor or an ARB
Few studies have examined combination therapy with either an ACE inhibitor or ARB plus a renin inhibitor, the newest class of agents that block this system.
Parving et al19 recently reported the results of combining aliskiren (Tekturna, a renin inhibitor) with losartan in 599 patients with type 2 diabetes and nephropathy. At 6 months, the renin inhibitor showed a renoprotective effect that was independent of its blood-pressure-lowering effect in those who were receiving maximal recommended doses of the ARB.
OTHER FACTORS ALSO INFLUENCE PROGRESSION
Even though there is broad agreement that an approach that neutralizes the effects of the renin-angiotensin-aldosterone system on the kidney would lower blood pressure and protein excretion rates, whether it would change the natural history of chronic kidney disease and prevent progression is less clear. In reality, a number of factors other than the renin-angiotensin-aldosterone system are responsible for the progression of chronic kidney disease. These other factors may help explain why control of this system does not totally prevent deterioration of chronic kidney disease, although the rate may be slowed.
MORE QUESTIONS THAN ANSWERS
A number of provocative questions arise from Dr. Hirsch’s discussion of complete renin-angiotensin-aldosterone system blockade to prevent disease progression:
- Will decreasing proteinuria to a specific target (< 500 mg/day) prevent progression?
- How low should the blood pressure target be set to modulate progression, and should it be the same in all age groups?
- Should complete blockade be applied all at once or in a stepwise fashion depending on the glomerular filtration rate, the level of proteinuria, or both?
- Which patients would benefit most from complete blockade?
- Is direct renin inhibition a critical component of complete blockade?
- What model of chronic disease management is required to avoid unexpected complications if this treatment approach is embraced?
Currently, therefore, there are more questions than answers. This strategy is an intriguing, opinion-based option, but for now it should only be applied to patients with proteinuria and evidence of early progression despite standard therapy who can be closely monitored, and it is not for the faint of heart. In view of the risks of hyperkalemia, hypotension, and perhaps even worsening renal function, more data from carefully designed trials are needed before the general medical community widely applies a complete blockade of the renin-angiotensin-aldosterone pathway to prevent progressive chronic kidney disease.
- United States Renal Data System. Annual data report. www.usrds.org/adr.htm. Accessed 9/5/2008.
- National Kidney Foundation. NKF K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. www.kidney.org/Professionals/Kdoqi/guidelines_ckd/toc.htm. Accessed 9/5/2008.
- Remuzzi A. Vitamin D, insulin resistance, and renal disease. Kidney Int. 2007; 71:96–98.
- Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003; 83:253–307.
- Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int 1998; 54:687–697.
- Endlich N, Endlich K. Stretch, tension and adhesion—adaptive mechanisms of the actin cytoskeleton in podocytes. Eur J Cell Biol. 2006; 85:229–234.
- Hirsch S. An update on proteinuric chronic kidney disease: the dual-goal approach. Cleve Clin J Med. 2008; 75:705–713.
- Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993; 329:1456–1462.
- Wolf G, Ritz E. Combination therapy with ACE inhibitors and angiotensin II receptor blockers to halt progression of chronic renal disease: pathophysiology and indications. Kidney Int. 2005; 67:799–812.
- Campbell R, Sangalli F, Perticucci E, et al. Effects of combined ACE inhibitor and angiotensin II antagonist treatment in human chronic nephropathies. Kidney Int. 2003; 63:1094–1103.
- Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet. 2003; 361:117–124.
- Kunz R, Friedrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med. 2008; 148:30–48.
- Mann JF, Schmieder RE, McQueen M, et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet. 2008; 372:547–553.
- Phillips CO, Kashani A, Ko DK, Francis G, Krumholz HM. Adverse effects of combination angiotensin II receptor blockers plus angiotensin-converting enzyme inhibitors for left ventricular dysfunction: a quantitative review of data from randomized clinical trials. Arch Intern Med. 2007; 167:1930–1936.
- Epstein M. Adding spironolactone to conventional antihypertensives reduces albuminuria in patients with diabetic nephropathy. Nat Clin Pract Nephrol. 2006; 2:310–311.
- Rossing K, Schjoedt KJ, Smidt UM, Boomsma F, Parving HH. Beneficial effects of adding spironolactone to recommended antihypertensive treatment in diabetic nephropathy: a randomized, double-masked, cross-over study. Diabetes Care. 2005; 28:2106–2112.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006; 70:2116–2123.
- Bomback AS, Kshirsagar AV, Amamoo MA, Klemmer PJ. Change in proteinuria after adding aldosterone blockers to ACE inhibitors or angiotensin receptor blockers in CKD: a systematic review. Am J Kidney Dis. 2008; 51:199–211.
- Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med. 2008; 358:2433–2446.
- United States Renal Data System. Annual data report. www.usrds.org/adr.htm. Accessed 9/5/2008.
- National Kidney Foundation. NKF K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. www.kidney.org/Professionals/Kdoqi/guidelines_ckd/toc.htm. Accessed 9/5/2008.
- Remuzzi A. Vitamin D, insulin resistance, and renal disease. Kidney Int. 2007; 71:96–98.
- Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev. 2003; 83:253–307.
- Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int 1998; 54:687–697.
- Endlich N, Endlich K. Stretch, tension and adhesion—adaptive mechanisms of the actin cytoskeleton in podocytes. Eur J Cell Biol. 2006; 85:229–234.
- Hirsch S. An update on proteinuric chronic kidney disease: the dual-goal approach. Cleve Clin J Med. 2008; 75:705–713.
- Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993; 329:1456–1462.
- Wolf G, Ritz E. Combination therapy with ACE inhibitors and angiotensin II receptor blockers to halt progression of chronic renal disease: pathophysiology and indications. Kidney Int. 2005; 67:799–812.
- Campbell R, Sangalli F, Perticucci E, et al. Effects of combined ACE inhibitor and angiotensin II antagonist treatment in human chronic nephropathies. Kidney Int. 2003; 63:1094–1103.
- Nakao N, Yoshimura A, Morita H, Takada M, Kayano T, Ideura T. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet. 2003; 361:117–124.
- Kunz R, Friedrich C, Wolbers M, Mann JF. Meta-analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med. 2008; 148:30–48.
- Mann JF, Schmieder RE, McQueen M, et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet. 2008; 372:547–553.
- Phillips CO, Kashani A, Ko DK, Francis G, Krumholz HM. Adverse effects of combination angiotensin II receptor blockers plus angiotensin-converting enzyme inhibitors for left ventricular dysfunction: a quantitative review of data from randomized clinical trials. Arch Intern Med. 2007; 167:1930–1936.
- Epstein M. Adding spironolactone to conventional antihypertensives reduces albuminuria in patients with diabetic nephropathy. Nat Clin Pract Nephrol. 2006; 2:310–311.
- Rossing K, Schjoedt KJ, Smidt UM, Boomsma F, Parving HH. Beneficial effects of adding spironolactone to recommended antihypertensive treatment in diabetic nephropathy: a randomized, double-masked, cross-over study. Diabetes Care. 2005; 28:2106–2112.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int. 2006; 70:2116–2123.
- Bomback AS, Kshirsagar AV, Amamoo MA, Klemmer PJ. Change in proteinuria after adding aldosterone blockers to ACE inhibitors or angiotensin receptor blockers in CKD: a systematic review. Am J Kidney Dis. 2008; 51:199–211.
- Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med. 2008; 358:2433–2446.