User login
The medical community has struggled with two important questions for the past 10 years: When it comes to the low-density lipoprotein cholesterol (LDL-C) level, how low should one go and at what cost? And are there other markers of risk that can identify a higher-risk subpopulation in relatively healthy people? The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin) provided partial answers for these questions by finding that a highly potent statin lowered the risk of cardiovascular events in patients with “normal” LDL-C but elevated levels of high-sensitivity C-reactive protein (hs-CRP).1
In this article, we will critically evaluate the methods, results, and conclusions of the JUPITER trial. Additionally, we will discuss its limitations and areas of uncertainty.
BEFORE JUPITER
The LDL-C-lowering drugs called statins have revolutionized cardiovascular medicine.2 They are beneficial in both the primary prevention setting and in acute coronary syndromes, stable angina, and unstable angina and can halt the progression of coronary artery disease—in some cases even resulting in modest regression of plaque.3–6
Many experts have credited the reduction in LDL-C as being the sole factor responsible for the decrease in major adverse events seen with statin therapy.7 However, statins have other, non-lipid-lowering properties, including anti-inflammatory and antioxidant effects, that may also contribute to their benefits.8–15
One of the anti-inflammatory actions of statins is evidenced by lower levels of the acute-phase reactant CRP.10,11,15,16 Measuring systemic CRP levels with a highly sensitive assay (yielding the so-called high-sensitivity or hs-CRP level) provides significant clinical prognostic value across a spectrum of clinical situations, ranging from risk screening in apparently healthy people to stable and unstable angina.17–22 People with higher hs-CRP levels are, on average, at higher risk of adverse cardiovascular events. However, controversy remains as to whether hs-CRP plays a mechanistic role in plaque formation and acute complications. Indeed, recent genetic studies argue strongly that hs-CRP lies outside the mechanistic path of atherosclerosis.23 Nonetheless, an overwhelming amount of data indicates that hs-CRP serves as a marker of disease.17–21
Nissen et al10 showed that the rate of progression of atherosclerosis is lower when the levels of atherogenic lipoproteins and hs-CRP are both lowered with statin therapy. Simultaneously, Ridker et al11 showed that patients who have lower hs-CRP levels after statin therapy have better clinical outcomes than those with higher hs-CRP levels, regardless of their achieved level of LDL-C.
Collectively, these studies and others have led some to believe that, in people with relatively low LDL-C but persistently elevated hs-CRP, statin therapy may reduce the rate of events.15,24 The JUPITER trial was undertaken to test this hypothesis.
JUPITER STUDY DESIGN
JUPITER was designed to see whether highly potent statin therapy is beneficial in people with elevated hs-CRP who otherwise do not meet the criteria for lipid-lowering therapy. The study was conducted at 1,315 sites in 26 countries. It was sponsored by AstraZeneca, the maker of rosuvastatin (Crestor).
Inclusion and exclusion criteria
All participants had to be free of known cardiovascular disease, have an LDL-C level lower than 130 mg/dL, and have an hs-CRP level of 2.0 mg/L or greater. Patients were excluded if they were previous or current users of lipid-lowering drugs; had severe arthritis, lupus, or inflammatory bowel disease; or were taking immune-modulating drugs such as cyclosporine (Sandimmune, others), tacrolimus (Prograf), azathioprine (Azasan, Imuran), or long-term oral corticosteroids.
Rosuvastatin therapy
Participants were randomly assigned in a 1:1 ratio to receive rosuvastatin 20 mg daily or a matching placebo in a double-blind fashion.
End points
The primary end point was the composite of nonfatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina, an arterial revascularization procedure, or confirmed death from cardiovascular causes. Secondary end points were the individual components of the primary end point.
Statistical analysis
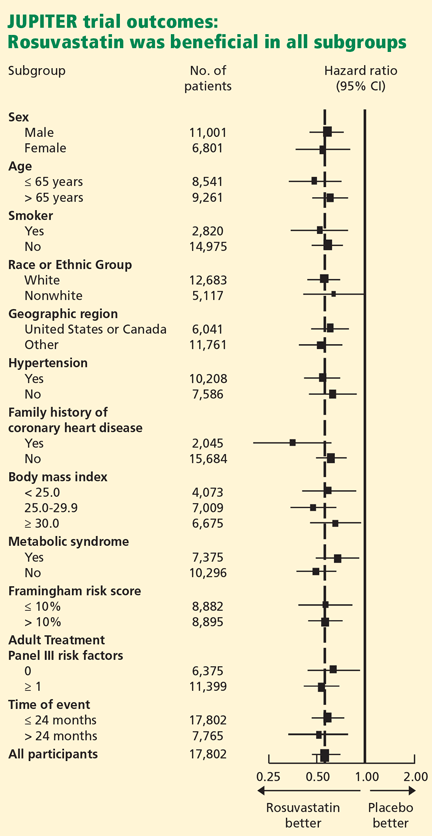
The study was powered to detect a 25% reduction in the primary end point among those treated with rosuvastatin. The trial was designed to run until 520 end point events had occurred. However, on March 29, 2008, after the first prespecified interim analysis, the Data and Safety Monitoring Board stopped the trial due to a significant reduction in the primary end point in the rosuvastatin group. As in most randomized clinical trials, all analyses were done on an intention-to-treat basis. Prespecified subgroup analyses were also performed.
STUDY RESULTS
Patient recruitment and eligibility
Between February 4, 2003, and December 15, 2006, a total of 89,890 people were screened. Of these, 17,802 met the inclusion and exclusion criteria and were included in the study. Of the 72,088 people who were excluded, 25,993 (36.1%) had an hs-CRP level below 2 mg/L and 37,611 (52.2%) had an LDL-C level of 130 mg/dL or higher.
A not-so-healthy population
The aim of the investigators was to include relatively healthy people. The median age was 66 years, about 16% of participants were current smokers, about 11% had a family history of heart disease, and about 41% met the criteria for metabolic syndrome, all conditions that are associated with elevated hs-CRP.25 Of note, the median hs-CRP level was 4.2 mg/L, a level indicating higher global risk according to the American College of Cardiology/American Heart Association consensus statement.26
Reduction in lipid levels and hs-CRP
By 12 months, in the rosuvastatin group, the median LDL-C level had fallen by 50% (from 108 to 55 mg/dL), and the median hs-CRP level had fallen by 37% (from 4.2 to 2.2 mg/L). Additionally, the triglyceride level had fallen by 17%. The high-density lipoprotein cholesterol levels did not change significantly.
Impact on end points
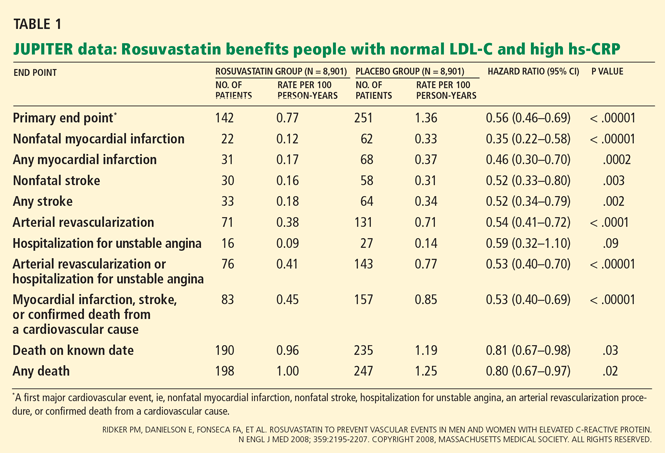
Adverse events
WHAT DOES THIS MEAN?
Is lower LDL-C better?
The JUPITER trial is the latest of several statin trials that have shown significant reductions in major adverse cardiovascular events when LDL-C was lowered below what has been recommended by the current guidelines.27,28
In 2002, the Heart Protection Study29 showed a significant reduction in major adverse cardiovascular events in patients at high risk of coronary artery disease if they received simvastatin (Zocor), even if they had LDL-C levels lower than 100 mg/dL at baseline. Similarly, the Pravastatin or Atorvastatin Evaluation and Infection-Thrombolysis in Myocardial Infarction 22 (PROVE-IT TIMI 22) trial30 showed a 16% relative risk reduction in a composite end point in patients presenting with acute coronary syndrome if they received intensive statin therapy.
These two studies led to an update by the National Cholesterol Education Program (Adult Treatment Panel III), suggesting an optimal LDL-C goal of less than 70 mg/dL in those with coronary artery disease or its risk equivalent (ie, diabetes mellitus, peripheral vascular disease). Furthermore, in support of the “lower is better” theory, a number of studies that used intravascular ultrasonography have shown regression of coronary plaque with aggressive LDL-C lowering. Notably, in a Study to Evaluate the Effect of Rosuvastatin on Intravascular Ultrasound-Derived Coronary Atheroma Burden (the ASTEROID trial),5 rosuvastatin 40 mg daily caused significant plaque regression while lowering LDL-C to 61 mg/dL over a 24-month period.
A number of high-dose statin trials have shown that lowering LDL-C to less than 70 mg/dL significantly reduces major adverse cardiovascular events.31–39 The JUPITER trial was unique in that it extended these findings to people without known coronary disease (ie, primary prevention) or elevated cholesterol but with elevated levels of a marker of inflammation—hs-CRP. In view of the JUPITER results and of studies using intravascular ultrasonography in the primary prevention setting, it seems clear that lowering LDL-C to levels less than 70 mg/dL also reduces both atherosclerotic plaque progression and the rate of first major adverse cardiovascular events in primary prevention in patients at higher global risk.
Did the study prove that reducing hs-CRP lowers risk?
Measuring hs-CRP levels has been extensively studied in apparently healthy populations, stable angina, unstable angina, and other cardiovascular settings.18,21,40–43 It has been shown to have significant prognostic implications in a number of primary and secondary trials.44 Additionally, those with elevated LDL-C and hs-CRP levels benefit the most from statin therapy.16,45,46 Animal studies have also provided some evidence that CRP may play a role in atherogenesis.47,48 However, recent clinical and genetic studies have raised doubt about the direct causal relationship between CRP and coronary artery disease,23,49,50 and epidemiologic studies have questioned its usefulness as a marker of risk.51,52
The JUPITER study adds little to clear up the controversy about whether hs-CRP is a mechanistic participant in atherosclerotic disease. However, it also shows that this issue is somewhat irrelevant, in that selection of patients for high-potency statin therapy solely on the basis of high hs-CRP without other indications for lipid-lowering therapy clearly reduces risk and improves survival.
JUPITER did not examine whether people with higher hs-CRP levels benefited more from statin therapy than those with lower levels. The hypothesis-generating data for JUPITER came from an analysis of changes in hs-CRP and LDL-C in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS).16 Thus, JUPITER did not include people with both low LDL-C and low hs-CRP because, in the AFCAPS/TexCAPS analysis, those with low LDL-C and low hs-CRP had extremely low event rates and no clinical efficacy of statin therapy, despite good LDL-C reduction. In marked contrast, those with low LDL-C but elevated hs-CRP had high event rates and large relative risk reductions— hence the need for JUPITER to prospectively test this hypothesis. Nevertheless, the initial results of JUPITER as presented do not yet make it clear that there is a dose-response relationship between higher levels of hs-CRP and a greater reduction in events, even in a cohort with elevated hs-CRP at baseline. This analysis will no doubt be forthcoming in another manuscript from Ridker and colleagues. Specifically, it will be of interest to examine whether those with the highest hs-CRP levels benefited the most from rosuvastatin on both an absolute and relative scale, and whether those with the greatest hs-CRP reduction also benefited more. With the present data available from JUPITER, a reasonable interpretation is that an elevated hs-CRP simply widens the inclusion criterion for those for whom high-potency statin therapy improves clinical outcomes.53
Better markers are needed
Even with a nonspecific marker such as hs-CRP, patients at higher global risk and with LDL-C below the recommended levels could be identified and treated aggressively. This benefit, however, required that approximately 100 people be treated with rosuvastatin for 2 years to prevent one event. Additionally, only 20% of all patients screened were eligible for the trial. Therefore, one could argue that its generalizability is limited.
Markers of risk that are more specific and sensitive are needed to identify people at higher global risk who would otherwise be considered to be at low risk with the current risk assessment tools. A number of such inflammatory and oxidative markers are under development.54–60
Absolute vs relative risk reduction and the public health burden
The 44% reduction in the number of primary end point events in the rosuvastatin group was considerable in relative terms. However, in absolute terms, 95 people had to be treated for up to 2 years in order to prevent one event.53 In making recommendations, the United States Department of Health and Human Services has to consider the clinical benefit of a test or a drug in light of its cost. With health care costs increasing, many agencies are refusing to pay for therapies on the basis of cost or small absolute benefit.
While we do not have the answer as to whether treating 95 people for 2 years to see one benefit is cost-effective, one thing is clear: the field of medicine is in desperate need of a better way to identify individuals who may benefit from a test or therapy.61 Additionally, we think it is important to note that the “numbers-needed-to-treat” (95 at 2 years and 25 at 5 years) derived from JUPITER are actually smaller than the values observed in the AFCAPS/TexCAPS and the West of Scotland Coronary Prevention Study.62,63 This suggests that statin therapy is at least as cost-effective in those with elevated hs-CRP as in those with elevated LDL-C. Even our most robust therapies are effective in only a minority of patients treated.61
Should ‘healthy’ people be tested for hs-CRP?
In 2003, we wrote in this journal21 that measuring hs-CRP may add to the current risk-prediction models by identifying people at increased risk who would otherwise not be considered as such by current risk models. The US Centers for Disease Control and Prevention and the American Heart Association have also stated that measuring hs-CRP in those at intermediate risk may be reasonable.26
The JUPITER investigators intended to study a relatively healthy population, but, as we mentioned, a close look at the cohort’s baseline characteristics indicates a substantial proportion met the criteria for metabolic syndrome. Therefore, one could challenge whether we really need hs-CRP in such a population to identify who will benefit from statin therapy.
We agree with the recommendation from the Centers for Disease Control and Prevention and the American Heart Association that measuring hs-CRP in people at intermediate risk is a reasonable option.26 We also believe that hs-CRP should be tested as a secondary risk factor, in combination with blood pressure, lipids, diabetes, smoking, serum creatinine, and fasting blood glucose. Factors such as obesity, sedentary lifestyle, family history of heart disease, and emotional and physical stress should also be considered.
Safety of high-dose statin therapy
High-dose statin therapy has been well tolerated in clinical trials, but rates of discontinuation have been higher (7%–10%) than with moderate-dose therapy (4%–5%).64 Fortunately, the rates of serious adverse events have in general been low. For example, with simvastatin 80 mg, the rates of myopathy and rhabdomyolysis were quite low.31
Rates of elevations in serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) with high-dose statin therapy have been reported to be below 1.3%. Studies have shown that reducing LDL-C to below 100 mg/dL is associated with a higher incidence of ALT and AST elevations. However, these elevations have usually been benign and often return to normal when the drug is reduced in dose or withdrawn.
In previous studies of rosuvastatin,65 the incidence of myopathy and liver function abnormalities was less than 0.1%. Rates of proteinuria were similarly low, and in many patients renal function actually improved on rosuvastatin.66,67 Furthermore, rosuvastatin may have different pharmacokinetic properties than atorvastatin (Lipitor) and simvastatin, which may result in a lower incidence of musculoskeletal toxicity.68,69
In general, the incidence of cancer has been similar in those treated with high-dose statins and those treated with placebo. The Treating to New Targets trial70 suggested that the incidence of cancer was higher with atorvastatin 80 mg daily than with 20 mg daily. However, a meta-analysis of 14 trials of moderate-dose statin therapy did not show any evidence of increased cancer rates with these agents.70 Indeed, in JUPITER, there was a reduction in cancer-related mortality rates, which could have been due to chance.
The JUPITER trial also showed an increase in the physician-reported incidence of diabetes mellitus with rosuvastatin. This is an important finding, and it may be a class effect because modest increases have similarly been reported with other statins in other major trials, eg, with pravastatin (Pravachol) in PROSPER, simvastatin in the Heart Protection Study, and atorvastatin in PROVE-IT. However, even in those with diabetes or impaired fasting glucose, the reduction in the rate of major adverse events is significant. For example, in JUPITER, almost all of the cases of “incident diabetes” were in those with impaired fasting glucose at baseline, and this group had nearly a 50% reduction in rates of myocardial infarction, stroke, and cardiovascular death. Therefore, on balance, the modest risk of earlier diagnosis of diabetes with statin therapy seems substantially offset by the marked reduction in rates of major adverse cardiovascular events in people with diabetes and impaired fasting glucose on statin therapy.
TAKE-HOME POINTS
The JUPITER trial, like previous high-dose statin trials, calls into question whether current LDL-C guidelines are appropriate for people at higher global risk with otherwise “normal” LDL-C levels.27,28 This trial heralds a new era in preventive therapy because it extends beyond LDL-C as an indication for statin therapy within the primary prevention setting. Statins have revolutionized the therapy of cardiovascular disease, and they continue to show benefit even in the “healthy.”
Clearly, hs-CRP serves as a nonlipid marker to identify those who may benefit from statin therapy. Nonetheless, more specific and sensitive markers (or panels) of cardiovascular risk are necessary. In the future, we will need markers that not only identify people at higher global risk, but that also tell us who would benefit from certain medical or surgical therapies. Elevated hs-CRP in a patient who otherwise would not be a candidate for statin therapy should trigger a reassessment of the risks vs benefits of statin therapy—JUPITER teaches us that statin therapy will benefit these patients.
Aggressive lifestyle modification that encompasses a balanced diet, routine exercise, and smoking cessation should be applied in both primary and secondary prevention. Additionally, risk factors such as elevated blood pressure and hyperlipidemia should be aggressively treated with appropriate medications.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Topol EJ. Intensive statin therapy—a sea change in cardiovascular prevention. N Engl J Med 2004; 350:1562–1564.
- Cannon CP, Murphy SA, Braunwald E. Intensive lipid lowering with atorvastatin in coronary disease. N Engl J Med 2005; 353:93–96.
- Cohen DJ, Carrozza JP, Baim DS. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. N Engl J Med 1999; 341:1853–1854.
- Nissen SE, Nicholls SJ, Sipahi I, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556–1565.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA 2004; 291:1071–1080.
- Robinson JG, Smith B, Maheshwari N, Schrott H. Pleiotropic effects of statins: benefit beyond cholesterol reduction? A meta-regression analysis. J Am Coll Cardiol 2005; 46:1855–1862.
- Aikawa M, Rabkin E, Sugiyama S, et al. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation 2001; 103:276–283.
- Liao JK. Effects of statins on 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition beyond low-density lipoprotein cholesterol. Am J Cardiol 2005; 96:24F–33F.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N Engl J Med 2005; 352:29–38.
- Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med 2005; 352:20–28.
- Shishehbor MH, Aviles RJ, Brennan ML, et al. Association of nitrotyrosine levels with cardiovascular disease and modulation by statin therapy. JAMA 2003; 289:1675–1680.
- Shishehbor MH, Brennan ML, Aviles RJ, et al. Statins promote potent systemic antioxidant effects through specific inflammatory pathways. Circulation 2003; 108:426–431.
- Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol 2001; 21:1712–1719.
- Shishehbor MH, Patel T, Bhatt DL. Using statins to treat inflammation in acute coronary syndromes: Are we there yet? Cleve Clin J Med 2006; 73:760–766.
- Ridker PM, Rifai N, Clearfield M, et al. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med 2001; 344:1959–1965.
- Ridker PM, Buring JE, Shih J, Matias M, Hennekens CH. Prospective study of C-reactive protein and the risk of future cardiovascular events among apparently healthy women. Circulation 1998; 98:731–733.
- Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 2000; 342:836–843.
- Ridker PM, Stampfer MJ, Rifai N. Novel risk factors for systemic atherosclerosis: a comparison of C-reactive protein, fibrinogen, homocysteine, lipoprotein(a), and standard cholesterol screening as predictors of peripheral arterial disease. JAMA 2001; 285:2481–2485.
- Rifai N, Ridker PM. High-sensitivity C-reactive protein: a novel and promising marker of coronary heart disease. Clin Chem 2001; 47:403–411.
- Shishehbor MH, Bhatt DL, Topol EJ. Using C-reactive protein to assess cardiovascular disease risk. Cleve Clin J Med 2003; 70:634–640.
- Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med 1997; 336:973–979.
- Zacho J, Tybjaerg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med 2008; 359:1897–1908.
- Ridker PM. Are statins anti-inflammatory? Issues in the design and conduct of the pravastatin inflammation C-reactive protein evaluation. Curr Cardiol Rep 2000; 2:269–273.
- Ridker PM, Buring JE, Cook NR, Rifai N. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy American women. Circulation 2003; 107:391–397.
- Pearson TA, Mensah GA, Alexander RW, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003; 107:499–511.
- Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001; 285:2486–2497.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- Collins R, Peto R, Armitage J. The MRC/BHF Heart Protection Study: preliminary results. Int J Clin Pract 2002; 56:53–56.
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- de Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA 2004; 292:1307–1316.
- LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Liem AH, van Boven AJ, Veeger NJ, et al. Effect of fluvastatin on ischaemia following acute myocardial infarction: a randomized trial. Eur Heart J 2002; 23:1931–1937.
- Pedersen TR, Faergeman O, Kastelein JJ, et al. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437–2445.
- Pitt B, Waters D, Brown WV, et al. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. Atorvastatin versus Revascularization Treatment Investigators. N Engl J Med 1999; 341:70–76.
- Ray KK, Cannon CP, McCabe CH, et al. Early and late benefits of highdose atorvastatin in patients with acute coronary syndromes: results from the PROVE IT-TIMI 22 trial. J Am Coll Cardiol 2005; 46:1405–1410.
- Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
- Serruys PW, de Feyter P, Macaya C, et al. Fluvastatin for prevention of cardiac events following successful first percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 287:3215–3222.
- Patel TN, Shishehbor MH, Bhatt DL. A review of high-dose statin therapy: targeting cholesterol and inflammation in atherosclerosis. Eur Heart J 2007; 28:664–672.
- Ridker PM. Novel risk factors and markers for coronary disease. Adv Intern Med 2000; 45:391–418.
- Ridker PM. High-sensitivity C-reactive protein: potential adjunct for global risk assessment in the primary prevention of cardiovascular disease. Circulation 2001; 103:1813–1818.
- Ridker PM, Glynn RJ, Hennekens CH. C-reactive protein adds to the predictive value of total and HDL cholesterol in determining risk of first myocardial infarction. Circulation 1998; 97:2007–2011.
- Cushman M, Arnold AM, Psaty BM, et al. C-reactive protein and the 10-year incidence of coronary heart disease in older men and women: the cardiovascular health study. Circulation 2005; 112:25–31.
- Ridker PM. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol 2007; 49:2129–2138.
- Albert MA, Danielson E, Rifai N, Ridker PM. Effect of statin therapy on C-reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): a randomized trial and cohort study. JAMA 2001; 286:64–70.
- Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E. Long-term effects of pravastatin on plasma concentration of C-reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation 1999; 100:230–235.
- Bisoendial RJ, Kastelein JJ, Levels JH, et al. Activation of inflammation and coagulation after infusion of C-reactive protein in humans. Circ Res 2005; 96:714–716.
- Schwedler SB, Amann K, Wernicke K, et al. Native C-reactive protein increases whereas modified C-reactive protein reduces atherosclerosis in apolipoprotein E-knockout mice. Circulation 2005; 112:1016–1023.
- Pfister R, Hellmich M. Multiple biomarkers and cardiovascular risk. N Engl J Med 2008; 359:760.
- Schunkert H, Samani NJ. Elevated C-reactive protein in atherosclerosis— chicken or egg? N Engl J Med 2008; 359:1953–1955.
- Danesh J, Wheeler JG, Hirschfield GM, et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med 2004; 350:1387–1397.
- Kushner I, Sehgal AR. Is high-sensitivity C-reactive protein an effective screening test for cardiovascular risk? Arch Intern Med 2002; 162:867–869.
- Hlatky MA. Expanding the orbit of primary prevention—moving beyond JUPITER. N Engl J Med 2008; 359:2280–2282.
- Shishehbor MH, Hazen SL. Inflammatory and oxidative markers in atherosclerosis: relationship to outcome. Curr Atheroscler Rep 2004; 6:243–250.
- Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol 2005; 25:1102–1111.
- Bhattacharyya T, Nicholls SJ, Topol EJ, et al. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA 2008; 299:1265–1276.
- Choi SH, Chae A, Miller E, et al. Relationship between biomarkers of oxidized low-density lipoprotein, statin therapy, quantitative coronary angiography, and atheroma: volume observations from the REVERSAL (Reversal of Atherosclerosis with Aggressive Lipid Lowering) study. J Am Coll Cardiol 2008; 52:24–32.
- Hakonarson H, Thorvaldsson S, Helgadottir A, et al. Effects of a 5-lipoxygenase-activating protein inhibitor on biomarkers associated with risk of myocardial infarction: a randomized trial. JAMA 2005; 293:2245–2256.
- Ky B, Burke A, Tsimikas S, et al. The influence of pravastatin and atorvastatin on markers of oxidative stress in hypercholesterolemic humans. J Am Coll Cardiol 2008; 51:1653–1662.
- Levy AP, Levy JE, Kalet-Litman S, et al. Haptoglobin genotype is a determinant of iron, lipid peroxidation, and macrophage accumulation in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol 2007; 27:134–140.
- Mukherjee D, Topol EJ. Pharmacogenomics in cardiovascular diseases. Curr Probl Cardiol 2003; 28:317–347
- West of Scotland Coronary Prevention Study: identification of highrisk groups and comparison with other cardiovascular intervention trials. Lancet 1996; 348:1339–1342.
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998; 279:1615–1622.
- Davidson MH, Robinson JG. Safety of aggressive lipid management. J Am Coll Cardiol 2007; 49:1753–1762.
- Davidson MH. Rosuvastatin safety: lessons from the FDA review and post-approval surveillance. Expert Opin Drug Saf 2004; 3:547–557.
- Kasiske BL, Wanner C, O’Neill WC. An assessment of statin safety by nephrologists. Am J Cardiol 2006; 97:82C–85C.
- McTaggart F, Buckett L, Davidson R, et al. Preclinical and clinical pharmacology of rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Am J Cardiol 2001; 87:28B–32B.
- Jacobson TA. Statin safety: lessons from new drug applications for marketed statins. Am J Cardiol 2006; 97:44C–51C.
- Jacobson TA. Comparative pharmacokinetic interaction profiles of pravastatin, simvastatin, and atorvastatin when coadministered with cytochrome P450 inhibitors. Am J Cardiol 2004; 94:1140–1146.
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
The medical community has struggled with two important questions for the past 10 years: When it comes to the low-density lipoprotein cholesterol (LDL-C) level, how low should one go and at what cost? And are there other markers of risk that can identify a higher-risk subpopulation in relatively healthy people? The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin) provided partial answers for these questions by finding that a highly potent statin lowered the risk of cardiovascular events in patients with “normal” LDL-C but elevated levels of high-sensitivity C-reactive protein (hs-CRP).1
In this article, we will critically evaluate the methods, results, and conclusions of the JUPITER trial. Additionally, we will discuss its limitations and areas of uncertainty.
BEFORE JUPITER
The LDL-C-lowering drugs called statins have revolutionized cardiovascular medicine.2 They are beneficial in both the primary prevention setting and in acute coronary syndromes, stable angina, and unstable angina and can halt the progression of coronary artery disease—in some cases even resulting in modest regression of plaque.3–6
Many experts have credited the reduction in LDL-C as being the sole factor responsible for the decrease in major adverse events seen with statin therapy.7 However, statins have other, non-lipid-lowering properties, including anti-inflammatory and antioxidant effects, that may also contribute to their benefits.8–15
One of the anti-inflammatory actions of statins is evidenced by lower levels of the acute-phase reactant CRP.10,11,15,16 Measuring systemic CRP levels with a highly sensitive assay (yielding the so-called high-sensitivity or hs-CRP level) provides significant clinical prognostic value across a spectrum of clinical situations, ranging from risk screening in apparently healthy people to stable and unstable angina.17–22 People with higher hs-CRP levels are, on average, at higher risk of adverse cardiovascular events. However, controversy remains as to whether hs-CRP plays a mechanistic role in plaque formation and acute complications. Indeed, recent genetic studies argue strongly that hs-CRP lies outside the mechanistic path of atherosclerosis.23 Nonetheless, an overwhelming amount of data indicates that hs-CRP serves as a marker of disease.17–21
Nissen et al10 showed that the rate of progression of atherosclerosis is lower when the levels of atherogenic lipoproteins and hs-CRP are both lowered with statin therapy. Simultaneously, Ridker et al11 showed that patients who have lower hs-CRP levels after statin therapy have better clinical outcomes than those with higher hs-CRP levels, regardless of their achieved level of LDL-C.
Collectively, these studies and others have led some to believe that, in people with relatively low LDL-C but persistently elevated hs-CRP, statin therapy may reduce the rate of events.15,24 The JUPITER trial was undertaken to test this hypothesis.
JUPITER STUDY DESIGN
JUPITER was designed to see whether highly potent statin therapy is beneficial in people with elevated hs-CRP who otherwise do not meet the criteria for lipid-lowering therapy. The study was conducted at 1,315 sites in 26 countries. It was sponsored by AstraZeneca, the maker of rosuvastatin (Crestor).
Inclusion and exclusion criteria
All participants had to be free of known cardiovascular disease, have an LDL-C level lower than 130 mg/dL, and have an hs-CRP level of 2.0 mg/L or greater. Patients were excluded if they were previous or current users of lipid-lowering drugs; had severe arthritis, lupus, or inflammatory bowel disease; or were taking immune-modulating drugs such as cyclosporine (Sandimmune, others), tacrolimus (Prograf), azathioprine (Azasan, Imuran), or long-term oral corticosteroids.
Rosuvastatin therapy
Participants were randomly assigned in a 1:1 ratio to receive rosuvastatin 20 mg daily or a matching placebo in a double-blind fashion.
End points
The primary end point was the composite of nonfatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina, an arterial revascularization procedure, or confirmed death from cardiovascular causes. Secondary end points were the individual components of the primary end point.
Statistical analysis
The study was powered to detect a 25% reduction in the primary end point among those treated with rosuvastatin. The trial was designed to run until 520 end point events had occurred. However, on March 29, 2008, after the first prespecified interim analysis, the Data and Safety Monitoring Board stopped the trial due to a significant reduction in the primary end point in the rosuvastatin group. As in most randomized clinical trials, all analyses were done on an intention-to-treat basis. Prespecified subgroup analyses were also performed.
STUDY RESULTS
Patient recruitment and eligibility
Between February 4, 2003, and December 15, 2006, a total of 89,890 people were screened. Of these, 17,802 met the inclusion and exclusion criteria and were included in the study. Of the 72,088 people who were excluded, 25,993 (36.1%) had an hs-CRP level below 2 mg/L and 37,611 (52.2%) had an LDL-C level of 130 mg/dL or higher.
A not-so-healthy population
The aim of the investigators was to include relatively healthy people. The median age was 66 years, about 16% of participants were current smokers, about 11% had a family history of heart disease, and about 41% met the criteria for metabolic syndrome, all conditions that are associated with elevated hs-CRP.25 Of note, the median hs-CRP level was 4.2 mg/L, a level indicating higher global risk according to the American College of Cardiology/American Heart Association consensus statement.26
Reduction in lipid levels and hs-CRP
By 12 months, in the rosuvastatin group, the median LDL-C level had fallen by 50% (from 108 to 55 mg/dL), and the median hs-CRP level had fallen by 37% (from 4.2 to 2.2 mg/L). Additionally, the triglyceride level had fallen by 17%. The high-density lipoprotein cholesterol levels did not change significantly.
Impact on end points
Adverse events
WHAT DOES THIS MEAN?
Is lower LDL-C better?
The JUPITER trial is the latest of several statin trials that have shown significant reductions in major adverse cardiovascular events when LDL-C was lowered below what has been recommended by the current guidelines.27,28
In 2002, the Heart Protection Study29 showed a significant reduction in major adverse cardiovascular events in patients at high risk of coronary artery disease if they received simvastatin (Zocor), even if they had LDL-C levels lower than 100 mg/dL at baseline. Similarly, the Pravastatin or Atorvastatin Evaluation and Infection-Thrombolysis in Myocardial Infarction 22 (PROVE-IT TIMI 22) trial30 showed a 16% relative risk reduction in a composite end point in patients presenting with acute coronary syndrome if they received intensive statin therapy.
These two studies led to an update by the National Cholesterol Education Program (Adult Treatment Panel III), suggesting an optimal LDL-C goal of less than 70 mg/dL in those with coronary artery disease or its risk equivalent (ie, diabetes mellitus, peripheral vascular disease). Furthermore, in support of the “lower is better” theory, a number of studies that used intravascular ultrasonography have shown regression of coronary plaque with aggressive LDL-C lowering. Notably, in a Study to Evaluate the Effect of Rosuvastatin on Intravascular Ultrasound-Derived Coronary Atheroma Burden (the ASTEROID trial),5 rosuvastatin 40 mg daily caused significant plaque regression while lowering LDL-C to 61 mg/dL over a 24-month period.
A number of high-dose statin trials have shown that lowering LDL-C to less than 70 mg/dL significantly reduces major adverse cardiovascular events.31–39 The JUPITER trial was unique in that it extended these findings to people without known coronary disease (ie, primary prevention) or elevated cholesterol but with elevated levels of a marker of inflammation—hs-CRP. In view of the JUPITER results and of studies using intravascular ultrasonography in the primary prevention setting, it seems clear that lowering LDL-C to levels less than 70 mg/dL also reduces both atherosclerotic plaque progression and the rate of first major adverse cardiovascular events in primary prevention in patients at higher global risk.
Did the study prove that reducing hs-CRP lowers risk?
Measuring hs-CRP levels has been extensively studied in apparently healthy populations, stable angina, unstable angina, and other cardiovascular settings.18,21,40–43 It has been shown to have significant prognostic implications in a number of primary and secondary trials.44 Additionally, those with elevated LDL-C and hs-CRP levels benefit the most from statin therapy.16,45,46 Animal studies have also provided some evidence that CRP may play a role in atherogenesis.47,48 However, recent clinical and genetic studies have raised doubt about the direct causal relationship between CRP and coronary artery disease,23,49,50 and epidemiologic studies have questioned its usefulness as a marker of risk.51,52
The JUPITER study adds little to clear up the controversy about whether hs-CRP is a mechanistic participant in atherosclerotic disease. However, it also shows that this issue is somewhat irrelevant, in that selection of patients for high-potency statin therapy solely on the basis of high hs-CRP without other indications for lipid-lowering therapy clearly reduces risk and improves survival.
JUPITER did not examine whether people with higher hs-CRP levels benefited more from statin therapy than those with lower levels. The hypothesis-generating data for JUPITER came from an analysis of changes in hs-CRP and LDL-C in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS).16 Thus, JUPITER did not include people with both low LDL-C and low hs-CRP because, in the AFCAPS/TexCAPS analysis, those with low LDL-C and low hs-CRP had extremely low event rates and no clinical efficacy of statin therapy, despite good LDL-C reduction. In marked contrast, those with low LDL-C but elevated hs-CRP had high event rates and large relative risk reductions— hence the need for JUPITER to prospectively test this hypothesis. Nevertheless, the initial results of JUPITER as presented do not yet make it clear that there is a dose-response relationship between higher levels of hs-CRP and a greater reduction in events, even in a cohort with elevated hs-CRP at baseline. This analysis will no doubt be forthcoming in another manuscript from Ridker and colleagues. Specifically, it will be of interest to examine whether those with the highest hs-CRP levels benefited the most from rosuvastatin on both an absolute and relative scale, and whether those with the greatest hs-CRP reduction also benefited more. With the present data available from JUPITER, a reasonable interpretation is that an elevated hs-CRP simply widens the inclusion criterion for those for whom high-potency statin therapy improves clinical outcomes.53
Better markers are needed
Even with a nonspecific marker such as hs-CRP, patients at higher global risk and with LDL-C below the recommended levels could be identified and treated aggressively. This benefit, however, required that approximately 100 people be treated with rosuvastatin for 2 years to prevent one event. Additionally, only 20% of all patients screened were eligible for the trial. Therefore, one could argue that its generalizability is limited.
Markers of risk that are more specific and sensitive are needed to identify people at higher global risk who would otherwise be considered to be at low risk with the current risk assessment tools. A number of such inflammatory and oxidative markers are under development.54–60
Absolute vs relative risk reduction and the public health burden
The 44% reduction in the number of primary end point events in the rosuvastatin group was considerable in relative terms. However, in absolute terms, 95 people had to be treated for up to 2 years in order to prevent one event.53 In making recommendations, the United States Department of Health and Human Services has to consider the clinical benefit of a test or a drug in light of its cost. With health care costs increasing, many agencies are refusing to pay for therapies on the basis of cost or small absolute benefit.
While we do not have the answer as to whether treating 95 people for 2 years to see one benefit is cost-effective, one thing is clear: the field of medicine is in desperate need of a better way to identify individuals who may benefit from a test or therapy.61 Additionally, we think it is important to note that the “numbers-needed-to-treat” (95 at 2 years and 25 at 5 years) derived from JUPITER are actually smaller than the values observed in the AFCAPS/TexCAPS and the West of Scotland Coronary Prevention Study.62,63 This suggests that statin therapy is at least as cost-effective in those with elevated hs-CRP as in those with elevated LDL-C. Even our most robust therapies are effective in only a minority of patients treated.61
Should ‘healthy’ people be tested for hs-CRP?
In 2003, we wrote in this journal21 that measuring hs-CRP may add to the current risk-prediction models by identifying people at increased risk who would otherwise not be considered as such by current risk models. The US Centers for Disease Control and Prevention and the American Heart Association have also stated that measuring hs-CRP in those at intermediate risk may be reasonable.26
The JUPITER investigators intended to study a relatively healthy population, but, as we mentioned, a close look at the cohort’s baseline characteristics indicates a substantial proportion met the criteria for metabolic syndrome. Therefore, one could challenge whether we really need hs-CRP in such a population to identify who will benefit from statin therapy.
We agree with the recommendation from the Centers for Disease Control and Prevention and the American Heart Association that measuring hs-CRP in people at intermediate risk is a reasonable option.26 We also believe that hs-CRP should be tested as a secondary risk factor, in combination with blood pressure, lipids, diabetes, smoking, serum creatinine, and fasting blood glucose. Factors such as obesity, sedentary lifestyle, family history of heart disease, and emotional and physical stress should also be considered.
Safety of high-dose statin therapy
High-dose statin therapy has been well tolerated in clinical trials, but rates of discontinuation have been higher (7%–10%) than with moderate-dose therapy (4%–5%).64 Fortunately, the rates of serious adverse events have in general been low. For example, with simvastatin 80 mg, the rates of myopathy and rhabdomyolysis were quite low.31
Rates of elevations in serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) with high-dose statin therapy have been reported to be below 1.3%. Studies have shown that reducing LDL-C to below 100 mg/dL is associated with a higher incidence of ALT and AST elevations. However, these elevations have usually been benign and often return to normal when the drug is reduced in dose or withdrawn.
In previous studies of rosuvastatin,65 the incidence of myopathy and liver function abnormalities was less than 0.1%. Rates of proteinuria were similarly low, and in many patients renal function actually improved on rosuvastatin.66,67 Furthermore, rosuvastatin may have different pharmacokinetic properties than atorvastatin (Lipitor) and simvastatin, which may result in a lower incidence of musculoskeletal toxicity.68,69
In general, the incidence of cancer has been similar in those treated with high-dose statins and those treated with placebo. The Treating to New Targets trial70 suggested that the incidence of cancer was higher with atorvastatin 80 mg daily than with 20 mg daily. However, a meta-analysis of 14 trials of moderate-dose statin therapy did not show any evidence of increased cancer rates with these agents.70 Indeed, in JUPITER, there was a reduction in cancer-related mortality rates, which could have been due to chance.
The JUPITER trial also showed an increase in the physician-reported incidence of diabetes mellitus with rosuvastatin. This is an important finding, and it may be a class effect because modest increases have similarly been reported with other statins in other major trials, eg, with pravastatin (Pravachol) in PROSPER, simvastatin in the Heart Protection Study, and atorvastatin in PROVE-IT. However, even in those with diabetes or impaired fasting glucose, the reduction in the rate of major adverse events is significant. For example, in JUPITER, almost all of the cases of “incident diabetes” were in those with impaired fasting glucose at baseline, and this group had nearly a 50% reduction in rates of myocardial infarction, stroke, and cardiovascular death. Therefore, on balance, the modest risk of earlier diagnosis of diabetes with statin therapy seems substantially offset by the marked reduction in rates of major adverse cardiovascular events in people with diabetes and impaired fasting glucose on statin therapy.
TAKE-HOME POINTS
The JUPITER trial, like previous high-dose statin trials, calls into question whether current LDL-C guidelines are appropriate for people at higher global risk with otherwise “normal” LDL-C levels.27,28 This trial heralds a new era in preventive therapy because it extends beyond LDL-C as an indication for statin therapy within the primary prevention setting. Statins have revolutionized the therapy of cardiovascular disease, and they continue to show benefit even in the “healthy.”
Clearly, hs-CRP serves as a nonlipid marker to identify those who may benefit from statin therapy. Nonetheless, more specific and sensitive markers (or panels) of cardiovascular risk are necessary. In the future, we will need markers that not only identify people at higher global risk, but that also tell us who would benefit from certain medical or surgical therapies. Elevated hs-CRP in a patient who otherwise would not be a candidate for statin therapy should trigger a reassessment of the risks vs benefits of statin therapy—JUPITER teaches us that statin therapy will benefit these patients.
Aggressive lifestyle modification that encompasses a balanced diet, routine exercise, and smoking cessation should be applied in both primary and secondary prevention. Additionally, risk factors such as elevated blood pressure and hyperlipidemia should be aggressively treated with appropriate medications.
The medical community has struggled with two important questions for the past 10 years: When it comes to the low-density lipoprotein cholesterol (LDL-C) level, how low should one go and at what cost? And are there other markers of risk that can identify a higher-risk subpopulation in relatively healthy people? The JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin) provided partial answers for these questions by finding that a highly potent statin lowered the risk of cardiovascular events in patients with “normal” LDL-C but elevated levels of high-sensitivity C-reactive protein (hs-CRP).1
In this article, we will critically evaluate the methods, results, and conclusions of the JUPITER trial. Additionally, we will discuss its limitations and areas of uncertainty.
BEFORE JUPITER
The LDL-C-lowering drugs called statins have revolutionized cardiovascular medicine.2 They are beneficial in both the primary prevention setting and in acute coronary syndromes, stable angina, and unstable angina and can halt the progression of coronary artery disease—in some cases even resulting in modest regression of plaque.3–6
Many experts have credited the reduction in LDL-C as being the sole factor responsible for the decrease in major adverse events seen with statin therapy.7 However, statins have other, non-lipid-lowering properties, including anti-inflammatory and antioxidant effects, that may also contribute to their benefits.8–15
One of the anti-inflammatory actions of statins is evidenced by lower levels of the acute-phase reactant CRP.10,11,15,16 Measuring systemic CRP levels with a highly sensitive assay (yielding the so-called high-sensitivity or hs-CRP level) provides significant clinical prognostic value across a spectrum of clinical situations, ranging from risk screening in apparently healthy people to stable and unstable angina.17–22 People with higher hs-CRP levels are, on average, at higher risk of adverse cardiovascular events. However, controversy remains as to whether hs-CRP plays a mechanistic role in plaque formation and acute complications. Indeed, recent genetic studies argue strongly that hs-CRP lies outside the mechanistic path of atherosclerosis.23 Nonetheless, an overwhelming amount of data indicates that hs-CRP serves as a marker of disease.17–21
Nissen et al10 showed that the rate of progression of atherosclerosis is lower when the levels of atherogenic lipoproteins and hs-CRP are both lowered with statin therapy. Simultaneously, Ridker et al11 showed that patients who have lower hs-CRP levels after statin therapy have better clinical outcomes than those with higher hs-CRP levels, regardless of their achieved level of LDL-C.
Collectively, these studies and others have led some to believe that, in people with relatively low LDL-C but persistently elevated hs-CRP, statin therapy may reduce the rate of events.15,24 The JUPITER trial was undertaken to test this hypothesis.
JUPITER STUDY DESIGN
JUPITER was designed to see whether highly potent statin therapy is beneficial in people with elevated hs-CRP who otherwise do not meet the criteria for lipid-lowering therapy. The study was conducted at 1,315 sites in 26 countries. It was sponsored by AstraZeneca, the maker of rosuvastatin (Crestor).
Inclusion and exclusion criteria
All participants had to be free of known cardiovascular disease, have an LDL-C level lower than 130 mg/dL, and have an hs-CRP level of 2.0 mg/L or greater. Patients were excluded if they were previous or current users of lipid-lowering drugs; had severe arthritis, lupus, or inflammatory bowel disease; or were taking immune-modulating drugs such as cyclosporine (Sandimmune, others), tacrolimus (Prograf), azathioprine (Azasan, Imuran), or long-term oral corticosteroids.
Rosuvastatin therapy
Participants were randomly assigned in a 1:1 ratio to receive rosuvastatin 20 mg daily or a matching placebo in a double-blind fashion.
End points
The primary end point was the composite of nonfatal myocardial infarction, nonfatal stroke, hospitalization for unstable angina, an arterial revascularization procedure, or confirmed death from cardiovascular causes. Secondary end points were the individual components of the primary end point.
Statistical analysis
The study was powered to detect a 25% reduction in the primary end point among those treated with rosuvastatin. The trial was designed to run until 520 end point events had occurred. However, on March 29, 2008, after the first prespecified interim analysis, the Data and Safety Monitoring Board stopped the trial due to a significant reduction in the primary end point in the rosuvastatin group. As in most randomized clinical trials, all analyses were done on an intention-to-treat basis. Prespecified subgroup analyses were also performed.
STUDY RESULTS
Patient recruitment and eligibility
Between February 4, 2003, and December 15, 2006, a total of 89,890 people were screened. Of these, 17,802 met the inclusion and exclusion criteria and were included in the study. Of the 72,088 people who were excluded, 25,993 (36.1%) had an hs-CRP level below 2 mg/L and 37,611 (52.2%) had an LDL-C level of 130 mg/dL or higher.
A not-so-healthy population
The aim of the investigators was to include relatively healthy people. The median age was 66 years, about 16% of participants were current smokers, about 11% had a family history of heart disease, and about 41% met the criteria for metabolic syndrome, all conditions that are associated with elevated hs-CRP.25 Of note, the median hs-CRP level was 4.2 mg/L, a level indicating higher global risk according to the American College of Cardiology/American Heart Association consensus statement.26
Reduction in lipid levels and hs-CRP
By 12 months, in the rosuvastatin group, the median LDL-C level had fallen by 50% (from 108 to 55 mg/dL), and the median hs-CRP level had fallen by 37% (from 4.2 to 2.2 mg/L). Additionally, the triglyceride level had fallen by 17%. The high-density lipoprotein cholesterol levels did not change significantly.
Impact on end points
Adverse events
WHAT DOES THIS MEAN?
Is lower LDL-C better?
The JUPITER trial is the latest of several statin trials that have shown significant reductions in major adverse cardiovascular events when LDL-C was lowered below what has been recommended by the current guidelines.27,28
In 2002, the Heart Protection Study29 showed a significant reduction in major adverse cardiovascular events in patients at high risk of coronary artery disease if they received simvastatin (Zocor), even if they had LDL-C levels lower than 100 mg/dL at baseline. Similarly, the Pravastatin or Atorvastatin Evaluation and Infection-Thrombolysis in Myocardial Infarction 22 (PROVE-IT TIMI 22) trial30 showed a 16% relative risk reduction in a composite end point in patients presenting with acute coronary syndrome if they received intensive statin therapy.
These two studies led to an update by the National Cholesterol Education Program (Adult Treatment Panel III), suggesting an optimal LDL-C goal of less than 70 mg/dL in those with coronary artery disease or its risk equivalent (ie, diabetes mellitus, peripheral vascular disease). Furthermore, in support of the “lower is better” theory, a number of studies that used intravascular ultrasonography have shown regression of coronary plaque with aggressive LDL-C lowering. Notably, in a Study to Evaluate the Effect of Rosuvastatin on Intravascular Ultrasound-Derived Coronary Atheroma Burden (the ASTEROID trial),5 rosuvastatin 40 mg daily caused significant plaque regression while lowering LDL-C to 61 mg/dL over a 24-month period.
A number of high-dose statin trials have shown that lowering LDL-C to less than 70 mg/dL significantly reduces major adverse cardiovascular events.31–39 The JUPITER trial was unique in that it extended these findings to people without known coronary disease (ie, primary prevention) or elevated cholesterol but with elevated levels of a marker of inflammation—hs-CRP. In view of the JUPITER results and of studies using intravascular ultrasonography in the primary prevention setting, it seems clear that lowering LDL-C to levels less than 70 mg/dL also reduces both atherosclerotic plaque progression and the rate of first major adverse cardiovascular events in primary prevention in patients at higher global risk.
Did the study prove that reducing hs-CRP lowers risk?
Measuring hs-CRP levels has been extensively studied in apparently healthy populations, stable angina, unstable angina, and other cardiovascular settings.18,21,40–43 It has been shown to have significant prognostic implications in a number of primary and secondary trials.44 Additionally, those with elevated LDL-C and hs-CRP levels benefit the most from statin therapy.16,45,46 Animal studies have also provided some evidence that CRP may play a role in atherogenesis.47,48 However, recent clinical and genetic studies have raised doubt about the direct causal relationship between CRP and coronary artery disease,23,49,50 and epidemiologic studies have questioned its usefulness as a marker of risk.51,52
The JUPITER study adds little to clear up the controversy about whether hs-CRP is a mechanistic participant in atherosclerotic disease. However, it also shows that this issue is somewhat irrelevant, in that selection of patients for high-potency statin therapy solely on the basis of high hs-CRP without other indications for lipid-lowering therapy clearly reduces risk and improves survival.
JUPITER did not examine whether people with higher hs-CRP levels benefited more from statin therapy than those with lower levels. The hypothesis-generating data for JUPITER came from an analysis of changes in hs-CRP and LDL-C in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS).16 Thus, JUPITER did not include people with both low LDL-C and low hs-CRP because, in the AFCAPS/TexCAPS analysis, those with low LDL-C and low hs-CRP had extremely low event rates and no clinical efficacy of statin therapy, despite good LDL-C reduction. In marked contrast, those with low LDL-C but elevated hs-CRP had high event rates and large relative risk reductions— hence the need for JUPITER to prospectively test this hypothesis. Nevertheless, the initial results of JUPITER as presented do not yet make it clear that there is a dose-response relationship between higher levels of hs-CRP and a greater reduction in events, even in a cohort with elevated hs-CRP at baseline. This analysis will no doubt be forthcoming in another manuscript from Ridker and colleagues. Specifically, it will be of interest to examine whether those with the highest hs-CRP levels benefited the most from rosuvastatin on both an absolute and relative scale, and whether those with the greatest hs-CRP reduction also benefited more. With the present data available from JUPITER, a reasonable interpretation is that an elevated hs-CRP simply widens the inclusion criterion for those for whom high-potency statin therapy improves clinical outcomes.53
Better markers are needed
Even with a nonspecific marker such as hs-CRP, patients at higher global risk and with LDL-C below the recommended levels could be identified and treated aggressively. This benefit, however, required that approximately 100 people be treated with rosuvastatin for 2 years to prevent one event. Additionally, only 20% of all patients screened were eligible for the trial. Therefore, one could argue that its generalizability is limited.
Markers of risk that are more specific and sensitive are needed to identify people at higher global risk who would otherwise be considered to be at low risk with the current risk assessment tools. A number of such inflammatory and oxidative markers are under development.54–60
Absolute vs relative risk reduction and the public health burden
The 44% reduction in the number of primary end point events in the rosuvastatin group was considerable in relative terms. However, in absolute terms, 95 people had to be treated for up to 2 years in order to prevent one event.53 In making recommendations, the United States Department of Health and Human Services has to consider the clinical benefit of a test or a drug in light of its cost. With health care costs increasing, many agencies are refusing to pay for therapies on the basis of cost or small absolute benefit.
While we do not have the answer as to whether treating 95 people for 2 years to see one benefit is cost-effective, one thing is clear: the field of medicine is in desperate need of a better way to identify individuals who may benefit from a test or therapy.61 Additionally, we think it is important to note that the “numbers-needed-to-treat” (95 at 2 years and 25 at 5 years) derived from JUPITER are actually smaller than the values observed in the AFCAPS/TexCAPS and the West of Scotland Coronary Prevention Study.62,63 This suggests that statin therapy is at least as cost-effective in those with elevated hs-CRP as in those with elevated LDL-C. Even our most robust therapies are effective in only a minority of patients treated.61
Should ‘healthy’ people be tested for hs-CRP?
In 2003, we wrote in this journal21 that measuring hs-CRP may add to the current risk-prediction models by identifying people at increased risk who would otherwise not be considered as such by current risk models. The US Centers for Disease Control and Prevention and the American Heart Association have also stated that measuring hs-CRP in those at intermediate risk may be reasonable.26
The JUPITER investigators intended to study a relatively healthy population, but, as we mentioned, a close look at the cohort’s baseline characteristics indicates a substantial proportion met the criteria for metabolic syndrome. Therefore, one could challenge whether we really need hs-CRP in such a population to identify who will benefit from statin therapy.
We agree with the recommendation from the Centers for Disease Control and Prevention and the American Heart Association that measuring hs-CRP in people at intermediate risk is a reasonable option.26 We also believe that hs-CRP should be tested as a secondary risk factor, in combination with blood pressure, lipids, diabetes, smoking, serum creatinine, and fasting blood glucose. Factors such as obesity, sedentary lifestyle, family history of heart disease, and emotional and physical stress should also be considered.
Safety of high-dose statin therapy
High-dose statin therapy has been well tolerated in clinical trials, but rates of discontinuation have been higher (7%–10%) than with moderate-dose therapy (4%–5%).64 Fortunately, the rates of serious adverse events have in general been low. For example, with simvastatin 80 mg, the rates of myopathy and rhabdomyolysis were quite low.31
Rates of elevations in serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) with high-dose statin therapy have been reported to be below 1.3%. Studies have shown that reducing LDL-C to below 100 mg/dL is associated with a higher incidence of ALT and AST elevations. However, these elevations have usually been benign and often return to normal when the drug is reduced in dose or withdrawn.
In previous studies of rosuvastatin,65 the incidence of myopathy and liver function abnormalities was less than 0.1%. Rates of proteinuria were similarly low, and in many patients renal function actually improved on rosuvastatin.66,67 Furthermore, rosuvastatin may have different pharmacokinetic properties than atorvastatin (Lipitor) and simvastatin, which may result in a lower incidence of musculoskeletal toxicity.68,69
In general, the incidence of cancer has been similar in those treated with high-dose statins and those treated with placebo. The Treating to New Targets trial70 suggested that the incidence of cancer was higher with atorvastatin 80 mg daily than with 20 mg daily. However, a meta-analysis of 14 trials of moderate-dose statin therapy did not show any evidence of increased cancer rates with these agents.70 Indeed, in JUPITER, there was a reduction in cancer-related mortality rates, which could have been due to chance.
The JUPITER trial also showed an increase in the physician-reported incidence of diabetes mellitus with rosuvastatin. This is an important finding, and it may be a class effect because modest increases have similarly been reported with other statins in other major trials, eg, with pravastatin (Pravachol) in PROSPER, simvastatin in the Heart Protection Study, and atorvastatin in PROVE-IT. However, even in those with diabetes or impaired fasting glucose, the reduction in the rate of major adverse events is significant. For example, in JUPITER, almost all of the cases of “incident diabetes” were in those with impaired fasting glucose at baseline, and this group had nearly a 50% reduction in rates of myocardial infarction, stroke, and cardiovascular death. Therefore, on balance, the modest risk of earlier diagnosis of diabetes with statin therapy seems substantially offset by the marked reduction in rates of major adverse cardiovascular events in people with diabetes and impaired fasting glucose on statin therapy.
TAKE-HOME POINTS
The JUPITER trial, like previous high-dose statin trials, calls into question whether current LDL-C guidelines are appropriate for people at higher global risk with otherwise “normal” LDL-C levels.27,28 This trial heralds a new era in preventive therapy because it extends beyond LDL-C as an indication for statin therapy within the primary prevention setting. Statins have revolutionized the therapy of cardiovascular disease, and they continue to show benefit even in the “healthy.”
Clearly, hs-CRP serves as a nonlipid marker to identify those who may benefit from statin therapy. Nonetheless, more specific and sensitive markers (or panels) of cardiovascular risk are necessary. In the future, we will need markers that not only identify people at higher global risk, but that also tell us who would benefit from certain medical or surgical therapies. Elevated hs-CRP in a patient who otherwise would not be a candidate for statin therapy should trigger a reassessment of the risks vs benefits of statin therapy—JUPITER teaches us that statin therapy will benefit these patients.
Aggressive lifestyle modification that encompasses a balanced diet, routine exercise, and smoking cessation should be applied in both primary and secondary prevention. Additionally, risk factors such as elevated blood pressure and hyperlipidemia should be aggressively treated with appropriate medications.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Topol EJ. Intensive statin therapy—a sea change in cardiovascular prevention. N Engl J Med 2004; 350:1562–1564.
- Cannon CP, Murphy SA, Braunwald E. Intensive lipid lowering with atorvastatin in coronary disease. N Engl J Med 2005; 353:93–96.
- Cohen DJ, Carrozza JP, Baim DS. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. N Engl J Med 1999; 341:1853–1854.
- Nissen SE, Nicholls SJ, Sipahi I, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556–1565.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA 2004; 291:1071–1080.
- Robinson JG, Smith B, Maheshwari N, Schrott H. Pleiotropic effects of statins: benefit beyond cholesterol reduction? A meta-regression analysis. J Am Coll Cardiol 2005; 46:1855–1862.
- Aikawa M, Rabkin E, Sugiyama S, et al. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation 2001; 103:276–283.
- Liao JK. Effects of statins on 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition beyond low-density lipoprotein cholesterol. Am J Cardiol 2005; 96:24F–33F.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N Engl J Med 2005; 352:29–38.
- Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med 2005; 352:20–28.
- Shishehbor MH, Aviles RJ, Brennan ML, et al. Association of nitrotyrosine levels with cardiovascular disease and modulation by statin therapy. JAMA 2003; 289:1675–1680.
- Shishehbor MH, Brennan ML, Aviles RJ, et al. Statins promote potent systemic antioxidant effects through specific inflammatory pathways. Circulation 2003; 108:426–431.
- Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol 2001; 21:1712–1719.
- Shishehbor MH, Patel T, Bhatt DL. Using statins to treat inflammation in acute coronary syndromes: Are we there yet? Cleve Clin J Med 2006; 73:760–766.
- Ridker PM, Rifai N, Clearfield M, et al. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med 2001; 344:1959–1965.
- Ridker PM, Buring JE, Shih J, Matias M, Hennekens CH. Prospective study of C-reactive protein and the risk of future cardiovascular events among apparently healthy women. Circulation 1998; 98:731–733.
- Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 2000; 342:836–843.
- Ridker PM, Stampfer MJ, Rifai N. Novel risk factors for systemic atherosclerosis: a comparison of C-reactive protein, fibrinogen, homocysteine, lipoprotein(a), and standard cholesterol screening as predictors of peripheral arterial disease. JAMA 2001; 285:2481–2485.
- Rifai N, Ridker PM. High-sensitivity C-reactive protein: a novel and promising marker of coronary heart disease. Clin Chem 2001; 47:403–411.
- Shishehbor MH, Bhatt DL, Topol EJ. Using C-reactive protein to assess cardiovascular disease risk. Cleve Clin J Med 2003; 70:634–640.
- Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med 1997; 336:973–979.
- Zacho J, Tybjaerg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med 2008; 359:1897–1908.
- Ridker PM. Are statins anti-inflammatory? Issues in the design and conduct of the pravastatin inflammation C-reactive protein evaluation. Curr Cardiol Rep 2000; 2:269–273.
- Ridker PM, Buring JE, Cook NR, Rifai N. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy American women. Circulation 2003; 107:391–397.
- Pearson TA, Mensah GA, Alexander RW, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003; 107:499–511.
- Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001; 285:2486–2497.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- Collins R, Peto R, Armitage J. The MRC/BHF Heart Protection Study: preliminary results. Int J Clin Pract 2002; 56:53–56.
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- de Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA 2004; 292:1307–1316.
- LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Liem AH, van Boven AJ, Veeger NJ, et al. Effect of fluvastatin on ischaemia following acute myocardial infarction: a randomized trial. Eur Heart J 2002; 23:1931–1937.
- Pedersen TR, Faergeman O, Kastelein JJ, et al. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437–2445.
- Pitt B, Waters D, Brown WV, et al. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. Atorvastatin versus Revascularization Treatment Investigators. N Engl J Med 1999; 341:70–76.
- Ray KK, Cannon CP, McCabe CH, et al. Early and late benefits of highdose atorvastatin in patients with acute coronary syndromes: results from the PROVE IT-TIMI 22 trial. J Am Coll Cardiol 2005; 46:1405–1410.
- Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
- Serruys PW, de Feyter P, Macaya C, et al. Fluvastatin for prevention of cardiac events following successful first percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 287:3215–3222.
- Patel TN, Shishehbor MH, Bhatt DL. A review of high-dose statin therapy: targeting cholesterol and inflammation in atherosclerosis. Eur Heart J 2007; 28:664–672.
- Ridker PM. Novel risk factors and markers for coronary disease. Adv Intern Med 2000; 45:391–418.
- Ridker PM. High-sensitivity C-reactive protein: potential adjunct for global risk assessment in the primary prevention of cardiovascular disease. Circulation 2001; 103:1813–1818.
- Ridker PM, Glynn RJ, Hennekens CH. C-reactive protein adds to the predictive value of total and HDL cholesterol in determining risk of first myocardial infarction. Circulation 1998; 97:2007–2011.
- Cushman M, Arnold AM, Psaty BM, et al. C-reactive protein and the 10-year incidence of coronary heart disease in older men and women: the cardiovascular health study. Circulation 2005; 112:25–31.
- Ridker PM. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol 2007; 49:2129–2138.
- Albert MA, Danielson E, Rifai N, Ridker PM. Effect of statin therapy on C-reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): a randomized trial and cohort study. JAMA 2001; 286:64–70.
- Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E. Long-term effects of pravastatin on plasma concentration of C-reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation 1999; 100:230–235.
- Bisoendial RJ, Kastelein JJ, Levels JH, et al. Activation of inflammation and coagulation after infusion of C-reactive protein in humans. Circ Res 2005; 96:714–716.
- Schwedler SB, Amann K, Wernicke K, et al. Native C-reactive protein increases whereas modified C-reactive protein reduces atherosclerosis in apolipoprotein E-knockout mice. Circulation 2005; 112:1016–1023.
- Pfister R, Hellmich M. Multiple biomarkers and cardiovascular risk. N Engl J Med 2008; 359:760.
- Schunkert H, Samani NJ. Elevated C-reactive protein in atherosclerosis— chicken or egg? N Engl J Med 2008; 359:1953–1955.
- Danesh J, Wheeler JG, Hirschfield GM, et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med 2004; 350:1387–1397.
- Kushner I, Sehgal AR. Is high-sensitivity C-reactive protein an effective screening test for cardiovascular risk? Arch Intern Med 2002; 162:867–869.
- Hlatky MA. Expanding the orbit of primary prevention—moving beyond JUPITER. N Engl J Med 2008; 359:2280–2282.
- Shishehbor MH, Hazen SL. Inflammatory and oxidative markers in atherosclerosis: relationship to outcome. Curr Atheroscler Rep 2004; 6:243–250.
- Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol 2005; 25:1102–1111.
- Bhattacharyya T, Nicholls SJ, Topol EJ, et al. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA 2008; 299:1265–1276.
- Choi SH, Chae A, Miller E, et al. Relationship between biomarkers of oxidized low-density lipoprotein, statin therapy, quantitative coronary angiography, and atheroma: volume observations from the REVERSAL (Reversal of Atherosclerosis with Aggressive Lipid Lowering) study. J Am Coll Cardiol 2008; 52:24–32.
- Hakonarson H, Thorvaldsson S, Helgadottir A, et al. Effects of a 5-lipoxygenase-activating protein inhibitor on biomarkers associated with risk of myocardial infarction: a randomized trial. JAMA 2005; 293:2245–2256.
- Ky B, Burke A, Tsimikas S, et al. The influence of pravastatin and atorvastatin on markers of oxidative stress in hypercholesterolemic humans. J Am Coll Cardiol 2008; 51:1653–1662.
- Levy AP, Levy JE, Kalet-Litman S, et al. Haptoglobin genotype is a determinant of iron, lipid peroxidation, and macrophage accumulation in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol 2007; 27:134–140.
- Mukherjee D, Topol EJ. Pharmacogenomics in cardiovascular diseases. Curr Probl Cardiol 2003; 28:317–347
- West of Scotland Coronary Prevention Study: identification of highrisk groups and comparison with other cardiovascular intervention trials. Lancet 1996; 348:1339–1342.
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998; 279:1615–1622.
- Davidson MH, Robinson JG. Safety of aggressive lipid management. J Am Coll Cardiol 2007; 49:1753–1762.
- Davidson MH. Rosuvastatin safety: lessons from the FDA review and post-approval surveillance. Expert Opin Drug Saf 2004; 3:547–557.
- Kasiske BL, Wanner C, O’Neill WC. An assessment of statin safety by nephrologists. Am J Cardiol 2006; 97:82C–85C.
- McTaggart F, Buckett L, Davidson R, et al. Preclinical and clinical pharmacology of rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Am J Cardiol 2001; 87:28B–32B.
- Jacobson TA. Statin safety: lessons from new drug applications for marketed statins. Am J Cardiol 2006; 97:44C–51C.
- Jacobson TA. Comparative pharmacokinetic interaction profiles of pravastatin, simvastatin, and atorvastatin when coadministered with cytochrome P450 inhibitors. Am J Cardiol 2004; 94:1140–1146.
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008; 359:2195–2207.
- Topol EJ. Intensive statin therapy—a sea change in cardiovascular prevention. N Engl J Med 2004; 350:1562–1564.
- Cannon CP, Murphy SA, Braunwald E. Intensive lipid lowering with atorvastatin in coronary disease. N Engl J Med 2005; 353:93–96.
- Cohen DJ, Carrozza JP, Baim DS. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. N Engl J Med 1999; 341:1853–1854.
- Nissen SE, Nicholls SJ, Sipahi I, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA 2006; 295:1556–1565.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosis: a randomized controlled trial. JAMA 2004; 291:1071–1080.
- Robinson JG, Smith B, Maheshwari N, Schrott H. Pleiotropic effects of statins: benefit beyond cholesterol reduction? A meta-regression analysis. J Am Coll Cardiol 2005; 46:1855–1862.
- Aikawa M, Rabkin E, Sugiyama S, et al. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation 2001; 103:276–283.
- Liao JK. Effects of statins on 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition beyond low-density lipoprotein cholesterol. Am J Cardiol 2005; 96:24F–33F.
- Nissen SE, Tuzcu EM, Schoenhagen P, et al. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N Engl J Med 2005; 352:29–38.
- Ridker PM, Cannon CP, Morrow D, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med 2005; 352:20–28.
- Shishehbor MH, Aviles RJ, Brennan ML, et al. Association of nitrotyrosine levels with cardiovascular disease and modulation by statin therapy. JAMA 2003; 289:1675–1680.
- Shishehbor MH, Brennan ML, Aviles RJ, et al. Statins promote potent systemic antioxidant effects through specific inflammatory pathways. Circulation 2003; 108:426–431.
- Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol 2001; 21:1712–1719.
- Shishehbor MH, Patel T, Bhatt DL. Using statins to treat inflammation in acute coronary syndromes: Are we there yet? Cleve Clin J Med 2006; 73:760–766.
- Ridker PM, Rifai N, Clearfield M, et al. Measurement of C-reactive protein for the targeting of statin therapy in the primary prevention of acute coronary events. N Engl J Med 2001; 344:1959–1965.
- Ridker PM, Buring JE, Shih J, Matias M, Hennekens CH. Prospective study of C-reactive protein and the risk of future cardiovascular events among apparently healthy women. Circulation 1998; 98:731–733.
- Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med 2000; 342:836–843.
- Ridker PM, Stampfer MJ, Rifai N. Novel risk factors for systemic atherosclerosis: a comparison of C-reactive protein, fibrinogen, homocysteine, lipoprotein(a), and standard cholesterol screening as predictors of peripheral arterial disease. JAMA 2001; 285:2481–2485.
- Rifai N, Ridker PM. High-sensitivity C-reactive protein: a novel and promising marker of coronary heart disease. Clin Chem 2001; 47:403–411.
- Shishehbor MH, Bhatt DL, Topol EJ. Using C-reactive protein to assess cardiovascular disease risk. Cleve Clin J Med 2003; 70:634–640.
- Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med 1997; 336:973–979.
- Zacho J, Tybjaerg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med 2008; 359:1897–1908.
- Ridker PM. Are statins anti-inflammatory? Issues in the design and conduct of the pravastatin inflammation C-reactive protein evaluation. Curr Cardiol Rep 2000; 2:269–273.
- Ridker PM, Buring JE, Cook NR, Rifai N. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy American women. Circulation 2003; 107:391–397.
- Pearson TA, Mensah GA, Alexander RW, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 2003; 107:499–511.
- Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001; 285:2486–2497.
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004; 110:227–239.
- Collins R, Peto R, Armitage J. The MRC/BHF Heart Protection Study: preliminary results. Int J Clin Pract 2002; 56:53–56.
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 2004; 350:1495–1504.
- de Lemos JA, Blazing MA, Wiviott SD, et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA 2004; 292:1307–1316.
- LaRosa JC, Grundy SM, Waters DD, et al. Intensive lipid lowering with atorvastatin in patients with stable coronary disease. N Engl J Med 2005; 352:1425–1435.
- Liem AH, van Boven AJ, Veeger NJ, et al. Effect of fluvastatin on ischaemia following acute myocardial infarction: a randomized trial. Eur Heart J 2002; 23:1931–1937.
- Pedersen TR, Faergeman O, Kastelein JJ, et al. High-dose atorvastatin vs usual-dose simvastatin for secondary prevention after myocardial infarction: the IDEAL study: a randomized controlled trial. JAMA 2005; 294:2437–2445.
- Pitt B, Waters D, Brown WV, et al. Aggressive lipid-lowering therapy compared with angioplasty in stable coronary artery disease. Atorvastatin versus Revascularization Treatment Investigators. N Engl J Med 1999; 341:70–76.
- Ray KK, Cannon CP, McCabe CH, et al. Early and late benefits of highdose atorvastatin in patients with acute coronary syndromes: results from the PROVE IT-TIMI 22 trial. J Am Coll Cardiol 2005; 46:1405–1410.
- Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA 2001; 285:1711–1718.
- Serruys PW, de Feyter P, Macaya C, et al. Fluvastatin for prevention of cardiac events following successful first percutaneous coronary intervention: a randomized controlled trial. JAMA 2002; 287:3215–3222.
- Patel TN, Shishehbor MH, Bhatt DL. A review of high-dose statin therapy: targeting cholesterol and inflammation in atherosclerosis. Eur Heart J 2007; 28:664–672.
- Ridker PM. Novel risk factors and markers for coronary disease. Adv Intern Med 2000; 45:391–418.
- Ridker PM. High-sensitivity C-reactive protein: potential adjunct for global risk assessment in the primary prevention of cardiovascular disease. Circulation 2001; 103:1813–1818.
- Ridker PM, Glynn RJ, Hennekens CH. C-reactive protein adds to the predictive value of total and HDL cholesterol in determining risk of first myocardial infarction. Circulation 1998; 97:2007–2011.
- Cushman M, Arnold AM, Psaty BM, et al. C-reactive protein and the 10-year incidence of coronary heart disease in older men and women: the cardiovascular health study. Circulation 2005; 112:25–31.
- Ridker PM. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol 2007; 49:2129–2138.
- Albert MA, Danielson E, Rifai N, Ridker PM. Effect of statin therapy on C-reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): a randomized trial and cohort study. JAMA 2001; 286:64–70.
- Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E. Long-term effects of pravastatin on plasma concentration of C-reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation 1999; 100:230–235.
- Bisoendial RJ, Kastelein JJ, Levels JH, et al. Activation of inflammation and coagulation after infusion of C-reactive protein in humans. Circ Res 2005; 96:714–716.
- Schwedler SB, Amann K, Wernicke K, et al. Native C-reactive protein increases whereas modified C-reactive protein reduces atherosclerosis in apolipoprotein E-knockout mice. Circulation 2005; 112:1016–1023.
- Pfister R, Hellmich M. Multiple biomarkers and cardiovascular risk. N Engl J Med 2008; 359:760.
- Schunkert H, Samani NJ. Elevated C-reactive protein in atherosclerosis— chicken or egg? N Engl J Med 2008; 359:1953–1955.
- Danesh J, Wheeler JG, Hirschfield GM, et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med 2004; 350:1387–1397.
- Kushner I, Sehgal AR. Is high-sensitivity C-reactive protein an effective screening test for cardiovascular risk? Arch Intern Med 2002; 162:867–869.
- Hlatky MA. Expanding the orbit of primary prevention—moving beyond JUPITER. N Engl J Med 2008; 359:2280–2282.
- Shishehbor MH, Hazen SL. Inflammatory and oxidative markers in atherosclerosis: relationship to outcome. Curr Atheroscler Rep 2004; 6:243–250.
- Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol 2005; 25:1102–1111.
- Bhattacharyya T, Nicholls SJ, Topol EJ, et al. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress and cardiovascular risk. JAMA 2008; 299:1265–1276.
- Choi SH, Chae A, Miller E, et al. Relationship between biomarkers of oxidized low-density lipoprotein, statin therapy, quantitative coronary angiography, and atheroma: volume observations from the REVERSAL (Reversal of Atherosclerosis with Aggressive Lipid Lowering) study. J Am Coll Cardiol 2008; 52:24–32.
- Hakonarson H, Thorvaldsson S, Helgadottir A, et al. Effects of a 5-lipoxygenase-activating protein inhibitor on biomarkers associated with risk of myocardial infarction: a randomized trial. JAMA 2005; 293:2245–2256.
- Ky B, Burke A, Tsimikas S, et al. The influence of pravastatin and atorvastatin on markers of oxidative stress in hypercholesterolemic humans. J Am Coll Cardiol 2008; 51:1653–1662.
- Levy AP, Levy JE, Kalet-Litman S, et al. Haptoglobin genotype is a determinant of iron, lipid peroxidation, and macrophage accumulation in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol 2007; 27:134–140.
- Mukherjee D, Topol EJ. Pharmacogenomics in cardiovascular diseases. Curr Probl Cardiol 2003; 28:317–347
- West of Scotland Coronary Prevention Study: identification of highrisk groups and comparison with other cardiovascular intervention trials. Lancet 1996; 348:1339–1342.
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA 1998; 279:1615–1622.
- Davidson MH, Robinson JG. Safety of aggressive lipid management. J Am Coll Cardiol 2007; 49:1753–1762.
- Davidson MH. Rosuvastatin safety: lessons from the FDA review and post-approval surveillance. Expert Opin Drug Saf 2004; 3:547–557.
- Kasiske BL, Wanner C, O’Neill WC. An assessment of statin safety by nephrologists. Am J Cardiol 2006; 97:82C–85C.
- McTaggart F, Buckett L, Davidson R, et al. Preclinical and clinical pharmacology of rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Am J Cardiol 2001; 87:28B–32B.
- Jacobson TA. Statin safety: lessons from new drug applications for marketed statins. Am J Cardiol 2006; 97:44C–51C.
- Jacobson TA. Comparative pharmacokinetic interaction profiles of pravastatin, simvastatin, and atorvastatin when coadministered with cytochrome P450 inhibitors. Am J Cardiol 2004; 94:1140–1146.
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
KEY POINTS
- LDL-C is the current gold standard diagnostic marker of risk, and elevated values should be aggressively treated in both primary and secondary prevention.
- The optional LDL-C goal of 70 mg/dL for patients at high risk may need to be extended to others at higher global risk, such as those with elevated hs-CRP.
- Although elevated hs-CRP may identify some people with low LDL-C who are nevertheless at higher global risk, more sensitive and specific markers of risk are needed.