User login
This patient's clinical presentation and laboratory findings are consistent with a diagnosis of pleomorphic mantle cell lymphoma (MCL).
MCL is a rare, clinically and biologically heterogeneous B-cell non-Hodgkin lymphoma. It accounts for approximately 5%-7% of all lymphomas. In North America and Europe, its incidence is akin to that of noncutaneous, peripheral T-cell lymphomas. The typical age at diagnosis is between 60 and 70 years. Approximately 70% of all cases are seen in men.
Little is known about risk factors for the development of MCL. Factors that have been associated with the development of other lymphomas (eg, familial risk, immunosuppression, other immune disorders, chemical and occupational exposures, and infectious agents) have not been convincingly identified as predisposing factors for MCL, with the possible exception of family history.
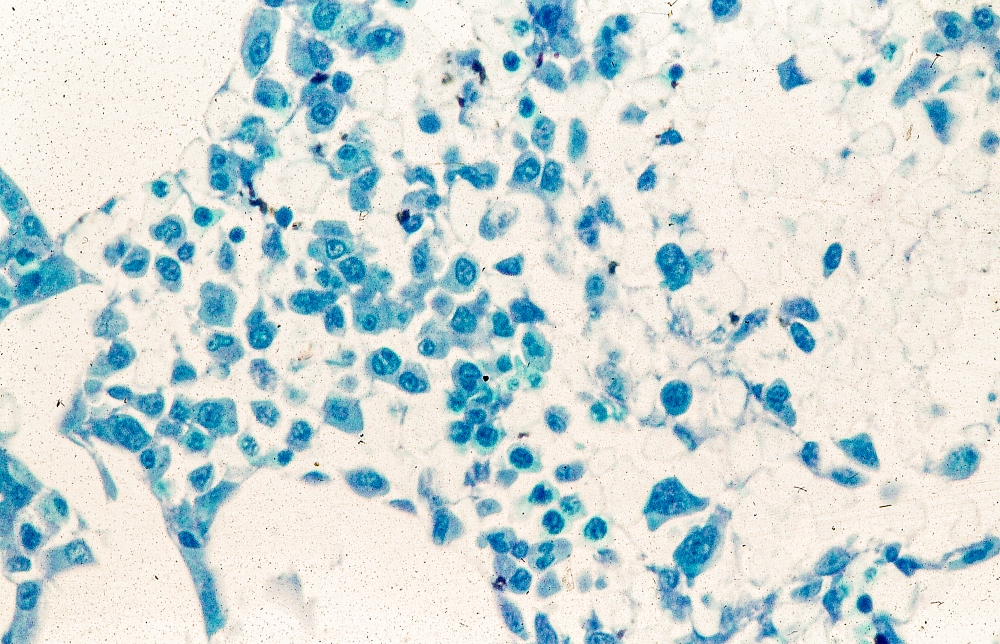
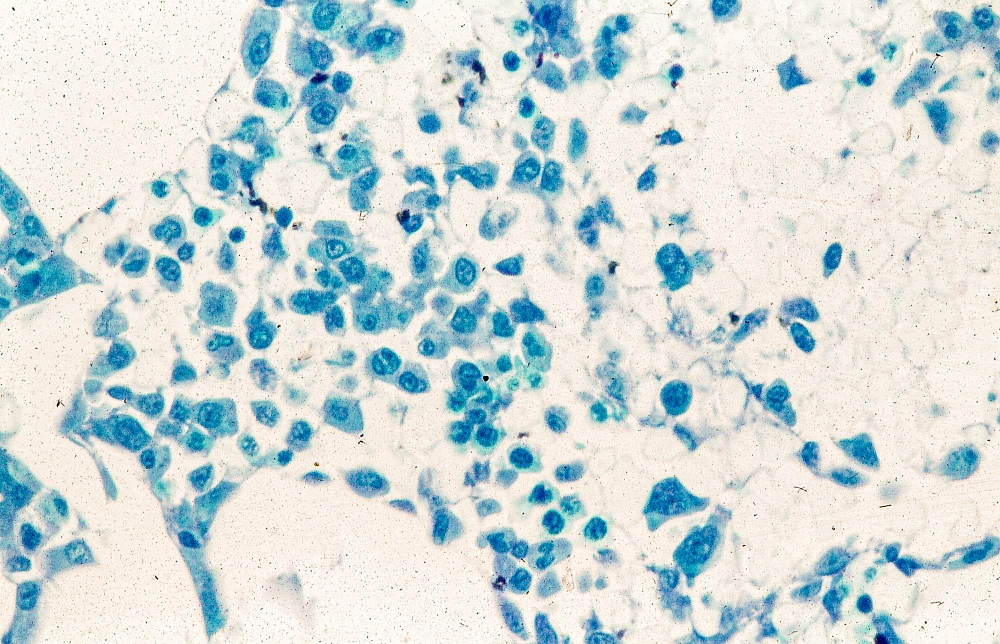
MCL is usually associated with reciprocal chromosomal translocation between chromosomes 11 and 14, t(11;14)(q13:q32), resulting in overexpression of cyclin D1, which plays a key role in tumor cell proliferation through cell-cycle dysregulation, chromosomal instability, and epigenetic regulation. Tumor cells (monoclonal B cells) express surface immunoglobulin, immunoglobulin M, or immunoglobulin D. Cells are usually CD5+ and pan B-cell antigen positive (eg, CD19, CD20, CD22) with no expression of CD10 and CD23. Histologic features include small-to-medium lymphocytes with scant cytoplasm, clumped chromatin, inconspicuous nucleoli, and prominent nuclear clefts. Cytologic subtypes include classic MCL, the blastoid variant (large cells, dispersed chromatin, and a high mitotic rate), and the pleomorphic variant (cells of varying size, although many are large, with pale cytoplasm, oval irregular nuclei, and prominent nucleoli). Blastoid and pleomorphic MCL typically have a more aggressive natural history and are associated with inferior clinical outcomes.
According to 2023 guidelines from the National Comprehensive Cancer Network (NCCN), an accurate pathologic diagnosis of the subtype is the most important initial step in the management of B-cell lymphomas, including pleomorphic MCL. The basic pathologic exam is the same for all subtypes, although additional testing may be needed in certain cases. An incisional or excisional lymph node biopsy is recommended. Fine-needle aspiration biopsy alone is typically not sufficient for the initial diagnosis of lymphoma; however, its diagnostic accuracy is significantly improved when it is used in combination with immunohistochemistry and flow cytometry. Immunohistochemistry is essential to differentiate MCL subtypes.
Essential workup procedures include a complete physical exam, with particular attention to node-bearing areas, including the Waldeyer ring, as well as the size of the liver and spleen, and assessment of performance status and B symptoms (fever, night sweats, unintentional weight loss). Laboratory studies should include complete blood count with differential, measurement of serum lactate dehydrogenase, hepatitis B virus testing, and a comprehensive metabolic panel. Required imaging studies include PET/CT (or chest/abdominal/pelvic CT with oral and intravenous contrast if PET/CT is not available) and multigated acquisition scanning or echocardiography when anthracyclines and anthracenedione-containing regimens are indicated.
A watch-and-wait approach may be appropriate for some patients with indolent MCL; however, patients with aggressive MCL, such as pleomorphic histology, require chemoimmunotherapy at diagnosis. For patients who relapse or achieve an incomplete response to first-line therapy, the NCCN guidelines recommend second-line treatment with a Bruton tyrosine kinase (BTK) inhibitor–containing regimen. Available BTK inhibitors include acalabrutinib, ibrutinib ± rituximab, zanubrutinib, and pirtobrutinib. Chemoimmunotherapy with lenalidomide + rituximab is another second-line option and may be particularly helpful for patients in whom a BTK inhibitor is contraindicated. Anti-CD19 CAR T-cell therapy is a recommended option for the third line and beyond.
Timothy J. Voorhees, MD, MSCR, Assistant Professor of Internal Medicine - Clinical, Division of Hematology, The Ohio State University James Comprehensive Cancer Center, Columbus, OH.
Timothy J. Voorhees, MD, MSCR, has disclosed the following relevant financial relationships:
Received research grant from: AstraZeneca; Morphosys; Incyte; Recordati.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's clinical presentation and laboratory findings are consistent with a diagnosis of pleomorphic mantle cell lymphoma (MCL).
MCL is a rare, clinically and biologically heterogeneous B-cell non-Hodgkin lymphoma. It accounts for approximately 5%-7% of all lymphomas. In North America and Europe, its incidence is akin to that of noncutaneous, peripheral T-cell lymphomas. The typical age at diagnosis is between 60 and 70 years. Approximately 70% of all cases are seen in men.
Little is known about risk factors for the development of MCL. Factors that have been associated with the development of other lymphomas (eg, familial risk, immunosuppression, other immune disorders, chemical and occupational exposures, and infectious agents) have not been convincingly identified as predisposing factors for MCL, with the possible exception of family history.
MCL is usually associated with reciprocal chromosomal translocation between chromosomes 11 and 14, t(11;14)(q13:q32), resulting in overexpression of cyclin D1, which plays a key role in tumor cell proliferation through cell-cycle dysregulation, chromosomal instability, and epigenetic regulation. Tumor cells (monoclonal B cells) express surface immunoglobulin, immunoglobulin M, or immunoglobulin D. Cells are usually CD5+ and pan B-cell antigen positive (eg, CD19, CD20, CD22) with no expression of CD10 and CD23. Histologic features include small-to-medium lymphocytes with scant cytoplasm, clumped chromatin, inconspicuous nucleoli, and prominent nuclear clefts. Cytologic subtypes include classic MCL, the blastoid variant (large cells, dispersed chromatin, and a high mitotic rate), and the pleomorphic variant (cells of varying size, although many are large, with pale cytoplasm, oval irregular nuclei, and prominent nucleoli). Blastoid and pleomorphic MCL typically have a more aggressive natural history and are associated with inferior clinical outcomes.
According to 2023 guidelines from the National Comprehensive Cancer Network (NCCN), an accurate pathologic diagnosis of the subtype is the most important initial step in the management of B-cell lymphomas, including pleomorphic MCL. The basic pathologic exam is the same for all subtypes, although additional testing may be needed in certain cases. An incisional or excisional lymph node biopsy is recommended. Fine-needle aspiration biopsy alone is typically not sufficient for the initial diagnosis of lymphoma; however, its diagnostic accuracy is significantly improved when it is used in combination with immunohistochemistry and flow cytometry. Immunohistochemistry is essential to differentiate MCL subtypes.
Essential workup procedures include a complete physical exam, with particular attention to node-bearing areas, including the Waldeyer ring, as well as the size of the liver and spleen, and assessment of performance status and B symptoms (fever, night sweats, unintentional weight loss). Laboratory studies should include complete blood count with differential, measurement of serum lactate dehydrogenase, hepatitis B virus testing, and a comprehensive metabolic panel. Required imaging studies include PET/CT (or chest/abdominal/pelvic CT with oral and intravenous contrast if PET/CT is not available) and multigated acquisition scanning or echocardiography when anthracyclines and anthracenedione-containing regimens are indicated.
A watch-and-wait approach may be appropriate for some patients with indolent MCL; however, patients with aggressive MCL, such as pleomorphic histology, require chemoimmunotherapy at diagnosis. For patients who relapse or achieve an incomplete response to first-line therapy, the NCCN guidelines recommend second-line treatment with a Bruton tyrosine kinase (BTK) inhibitor–containing regimen. Available BTK inhibitors include acalabrutinib, ibrutinib ± rituximab, zanubrutinib, and pirtobrutinib. Chemoimmunotherapy with lenalidomide + rituximab is another second-line option and may be particularly helpful for patients in whom a BTK inhibitor is contraindicated. Anti-CD19 CAR T-cell therapy is a recommended option for the third line and beyond.
Timothy J. Voorhees, MD, MSCR, Assistant Professor of Internal Medicine - Clinical, Division of Hematology, The Ohio State University James Comprehensive Cancer Center, Columbus, OH.
Timothy J. Voorhees, MD, MSCR, has disclosed the following relevant financial relationships:
Received research grant from: AstraZeneca; Morphosys; Incyte; Recordati.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's clinical presentation and laboratory findings are consistent with a diagnosis of pleomorphic mantle cell lymphoma (MCL).
MCL is a rare, clinically and biologically heterogeneous B-cell non-Hodgkin lymphoma. It accounts for approximately 5%-7% of all lymphomas. In North America and Europe, its incidence is akin to that of noncutaneous, peripheral T-cell lymphomas. The typical age at diagnosis is between 60 and 70 years. Approximately 70% of all cases are seen in men.
Little is known about risk factors for the development of MCL. Factors that have been associated with the development of other lymphomas (eg, familial risk, immunosuppression, other immune disorders, chemical and occupational exposures, and infectious agents) have not been convincingly identified as predisposing factors for MCL, with the possible exception of family history.
MCL is usually associated with reciprocal chromosomal translocation between chromosomes 11 and 14, t(11;14)(q13:q32), resulting in overexpression of cyclin D1, which plays a key role in tumor cell proliferation through cell-cycle dysregulation, chromosomal instability, and epigenetic regulation. Tumor cells (monoclonal B cells) express surface immunoglobulin, immunoglobulin M, or immunoglobulin D. Cells are usually CD5+ and pan B-cell antigen positive (eg, CD19, CD20, CD22) with no expression of CD10 and CD23. Histologic features include small-to-medium lymphocytes with scant cytoplasm, clumped chromatin, inconspicuous nucleoli, and prominent nuclear clefts. Cytologic subtypes include classic MCL, the blastoid variant (large cells, dispersed chromatin, and a high mitotic rate), and the pleomorphic variant (cells of varying size, although many are large, with pale cytoplasm, oval irregular nuclei, and prominent nucleoli). Blastoid and pleomorphic MCL typically have a more aggressive natural history and are associated with inferior clinical outcomes.
According to 2023 guidelines from the National Comprehensive Cancer Network (NCCN), an accurate pathologic diagnosis of the subtype is the most important initial step in the management of B-cell lymphomas, including pleomorphic MCL. The basic pathologic exam is the same for all subtypes, although additional testing may be needed in certain cases. An incisional or excisional lymph node biopsy is recommended. Fine-needle aspiration biopsy alone is typically not sufficient for the initial diagnosis of lymphoma; however, its diagnostic accuracy is significantly improved when it is used in combination with immunohistochemistry and flow cytometry. Immunohistochemistry is essential to differentiate MCL subtypes.
Essential workup procedures include a complete physical exam, with particular attention to node-bearing areas, including the Waldeyer ring, as well as the size of the liver and spleen, and assessment of performance status and B symptoms (fever, night sweats, unintentional weight loss). Laboratory studies should include complete blood count with differential, measurement of serum lactate dehydrogenase, hepatitis B virus testing, and a comprehensive metabolic panel. Required imaging studies include PET/CT (or chest/abdominal/pelvic CT with oral and intravenous contrast if PET/CT is not available) and multigated acquisition scanning or echocardiography when anthracyclines and anthracenedione-containing regimens are indicated.
A watch-and-wait approach may be appropriate for some patients with indolent MCL; however, patients with aggressive MCL, such as pleomorphic histology, require chemoimmunotherapy at diagnosis. For patients who relapse or achieve an incomplete response to first-line therapy, the NCCN guidelines recommend second-line treatment with a Bruton tyrosine kinase (BTK) inhibitor–containing regimen. Available BTK inhibitors include acalabrutinib, ibrutinib ± rituximab, zanubrutinib, and pirtobrutinib. Chemoimmunotherapy with lenalidomide + rituximab is another second-line option and may be particularly helpful for patients in whom a BTK inhibitor is contraindicated. Anti-CD19 CAR T-cell therapy is a recommended option for the third line and beyond.
Timothy J. Voorhees, MD, MSCR, Assistant Professor of Internal Medicine - Clinical, Division of Hematology, The Ohio State University James Comprehensive Cancer Center, Columbus, OH.
Timothy J. Voorhees, MD, MSCR, has disclosed the following relevant financial relationships:
Received research grant from: AstraZeneca; Morphosys; Incyte; Recordati.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
A 62-year-old man with no significant past medical history presents with a 3-month history of fever, night sweats, upper abdominal pain and bloating, and unintentional weight loss. He does not currently take any medications. His height and weight are 6 ft 2 in and 171 lb (BMI 22).
Physical examination reveals generalized lymphadenopathy and splenomegaly. Subsequently, an excisional lymph node biopsy is performed. Histologic examination of the specimen reveals sheets of mostly large cells of varying sizes, with nuclear overlap and extensive necrosis. Cytology findings include large lymphocytes with pale cytoplasm, clumped chromatin, oval irregular nuclei, and prominent nucleoli. Pertinent findings from immunohistochemical staining include the presence of t(11:14), Ki67 > 30%, CD5 and CD20 positivity, and CD10 and CD23 negativity. Centroblasts are absent.