User login
From the Department of Pediatrics, Boston University School of Medicine, Boston Medical Center, Boston, MA.
This article is the second in our Hemoglobinopathy Learning Collaborative series. See the related editorial by Oyeku et al in the February 2014 issue of JCOM. (—Ed.)
Abstract
- Objective: To describe the development and use of an electronic health record (EHR)–based sickle cell disease (SCD) registry for children with SCD to enhance case management and quality improvement (QI) efforts at an urban, academic, safety net institution.
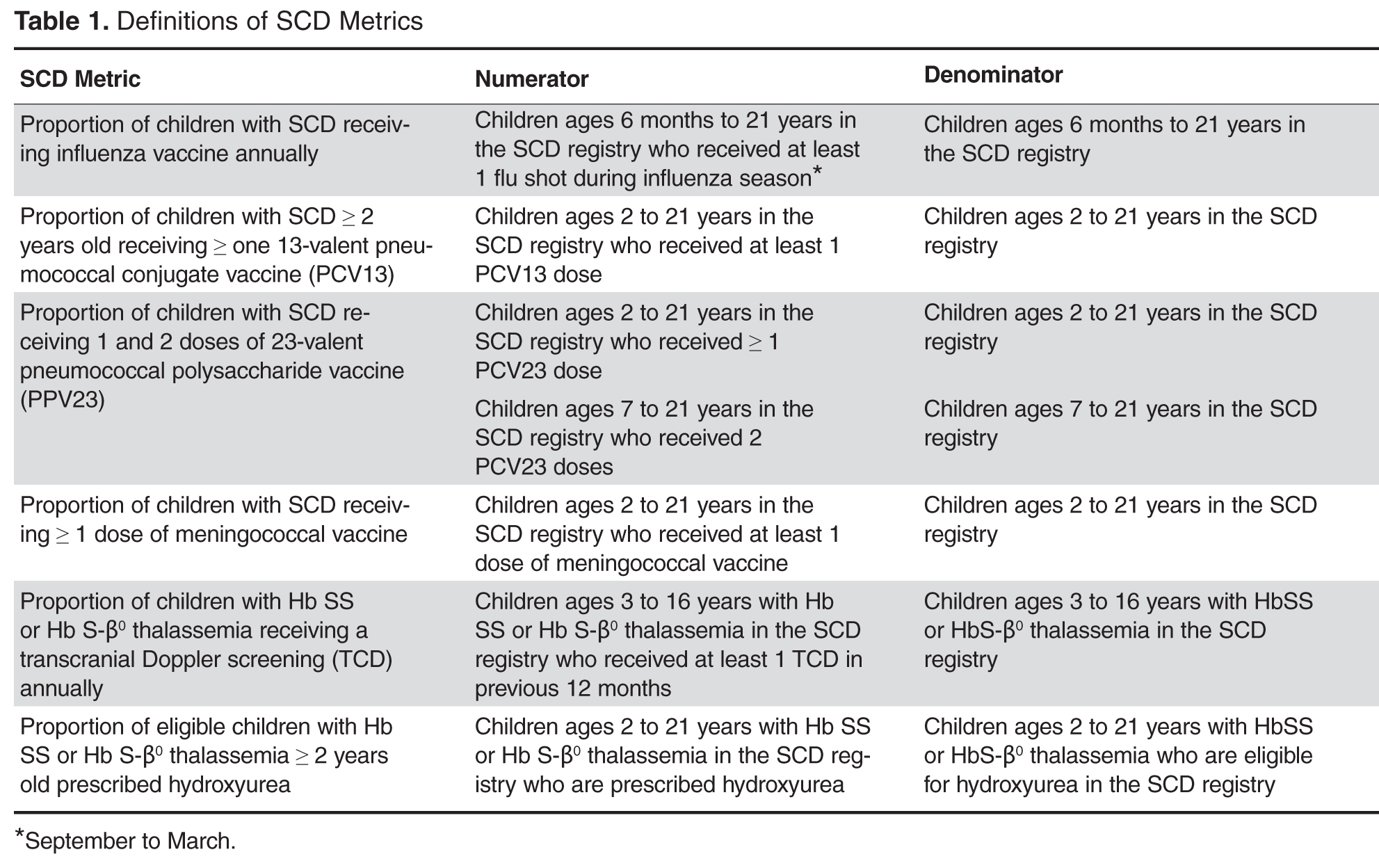
- Methods: Using national guidelines and the literature, we created quality metrics for pediatric SCD that focused on vaccination delivery and use of transcranial Doppler screening and hydroxyurea. We revised EHR forms for SCD care and created an EHR-based SCD registry that permitted monthly and annual reporting on quality metrics.
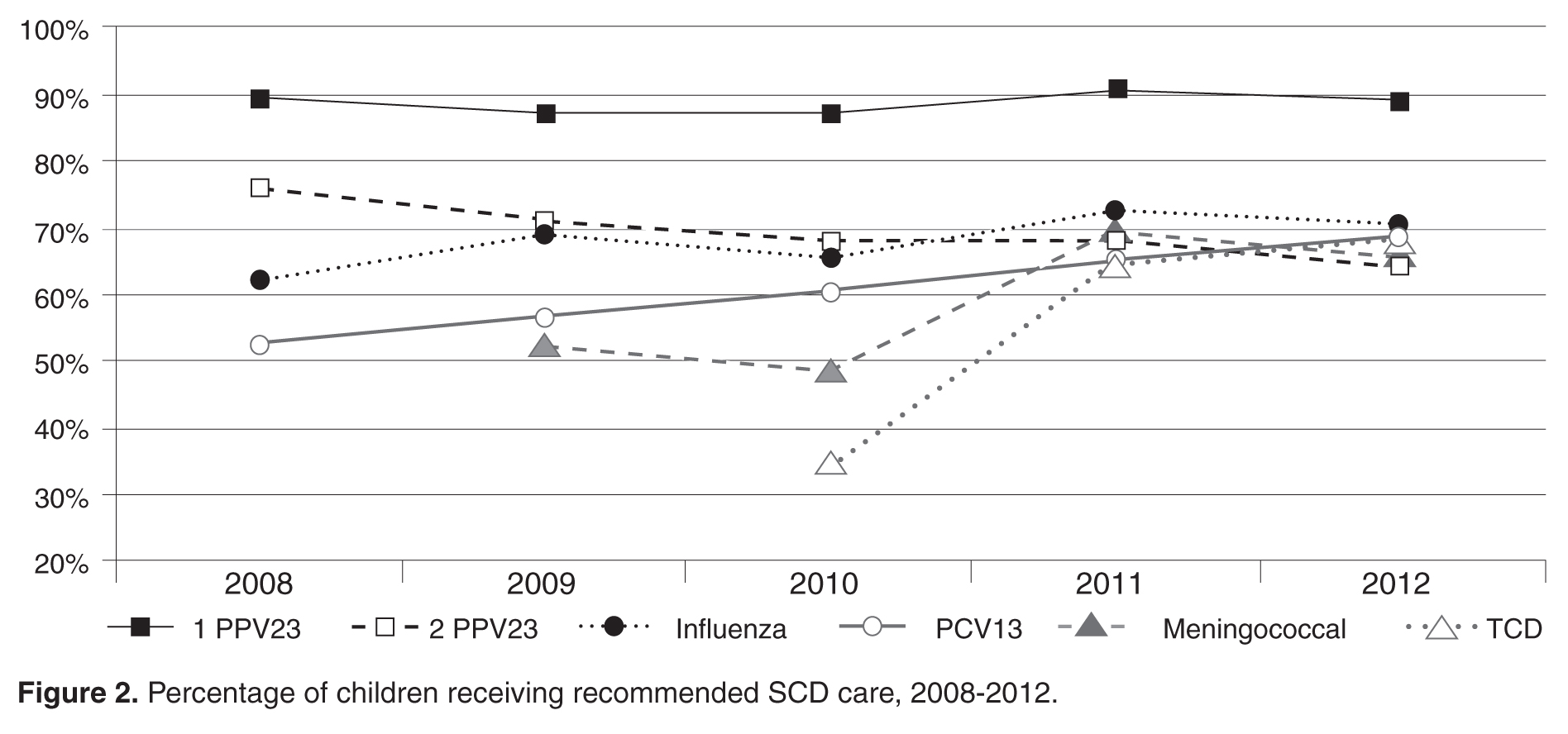
- Results: From 2008 to 2012, the percentage of children with SCD vaccinated for influenza increased from 52% to 65%, and for meningococcus from 53% to 70%. After licensure of PCV13 in 2010, the percentage of children vaccinated rose to 69% in 2012. Results for PPV23 were mixed: 87% to 91% received ≥1 dose, but the rate for receiving the second dose declined from 76% to 64%. Percentage of children screened annually with transcranial Doppler consistently ranged from 62% to 73% during the 5 years. QI initiatives in 2012–2013 led to increased influenza vaccination, from 65% to 83%, and increased hydroxyurea use, from 52% to 73%.
- Conclusion: In this study, a practical, replicable and feasible approach for improving the quality of SCD care combined the collaboration of a multidisciplinary team, an EHR-based disease registry, and QI initiatives. Additional work is needed to define and measure all elements of high-quality care for children with SCD and link process measures to clinical outcomes.
Sickle cell disease (SCD) is the most commonly inherited disorder in the United States, affecting approximately 100,000 individuals and 1 in 400 African American births [1,2]. The use of preventive strategies, such as immunizations [3], transcranial Doppler screening and transfusion protocols [4,5], and hydroxyurea therapy [6,7] has contributed to decreased morbidity and mortality among children with SCD [8,9]. However, a substantial gap exists between the care that children with SCD should receive and the care they actually receive [10–12]. An essential component of any effort that seeks to improve care is the ability to measure care processes and outcomes in a way that can drive quality improvement (QI) initiatives. Registries serve a vital role in quality improvement activities for many pediatric conditions, including inflammatory bowel disease [13] and cystic fibrosis [14]. However, there are no national or nationally representative registries currently available for children with SCD [15]. There is a pressing need for better information systems and tools that can be used in mainstream clinical settings to measure clinical performance with respect to quality indicators [16] if the goals of high quality care and better quality of life are to be achieved for children with SCD.
Electronic health records (EHRs) have been successfully used to improve the quality of care and enhance performance measurement in select institutions [17,18], and adoption of EHRs is growing. The 2009 American Recovery and Reinvestment Act allocated $20.8 billion in incentives to assist providers to adopt and “meaningfully use” EHRs [19,20]. As of 2011, 39% of office-based providers have implemented at least a basic EHR [21], up from 17% in 2008 [22]. The effective use of EHRs depends on collaboration between technical and medical experts so that functionality is achieved and clinical quality is appropriately measured. In addition, few EHRs contain specialized content for the care of persons with SCD.
While independent registries have been shown to be effective in improving care [13,14,23], they involve extra time and effort for data entry, can be difficult and expensive to maintain, and may not be feasible for many systems that care for SCD patients. In this paper, we describe the development and use of our EHR-based SCD registry for children with SCD, including our efforts to engage key technical and clinical experts to develop an EHR that is tailored to the outpatient workflow and data collection of quality measures and implement a fully functional system that collects data on quality measures to support case management and continuous QI.
Methods
This study was conducted at Boston Medical Center, New England’s largest safety net hospital, which cares for 190 children with SCD ages 0 to 21 years. The outpatient EHR (Centricity, GE) has been in use since 2000 and is used for all aspects of outpatient care, including ordering of immunizations and tests, electronic prescription writing, and referrals to specialty care.
Outcome Measures
Vaccines: The Centers for Disease Control and Prevention (CDC) recommends vaccinating children with SCD [26] against influenza annually, given their susceptibility to the influenza virus [24,27]. The CDC also recommends the 23-valent pneumococcal polysaccharide vaccine (PPV23 2-dose series) and 13-valent pneumococcal conjugate vaccine (PCV13, per childhood routine vaccine schedule for young children and 1 catch-up dose for children previously vaccinated with PCV7), and meningococcal vaccine (2-dose series), given patients’ functional asplenic status [25,28].
Transcranial Doppler screening can identify children with hemoglobin (Hb) SS and Hb S-β0 thalassemia at higher risk of stroke, which may be prevented through hypertransfusion programs [4]. Screening is recommended annually for these children ages 2 to 16 years [25].
Hydroxyurea use among children with Hb SS and Hb S-β0 thalassemia is an established practice [29,30]. We consider hydroxyurea therapy for all children 2 years and older with Hb SS and Hb S-β0 thalassemia, given the recently published safety data from the Baby-HUG trial [7] and the benefits of hydroxyurea among children and adults with SCD [6,31–35].
EHR-based Registry
Our EHR-based SCD registry includes 3 key components: (1) forms to support detailed documentation at the point-of-care (ie, clinic visit); (2) a registry management form to allow the QI team to identify patients to be included or excluded from the registry; and (3) a central data warehouse to support quality measurement and improvement.
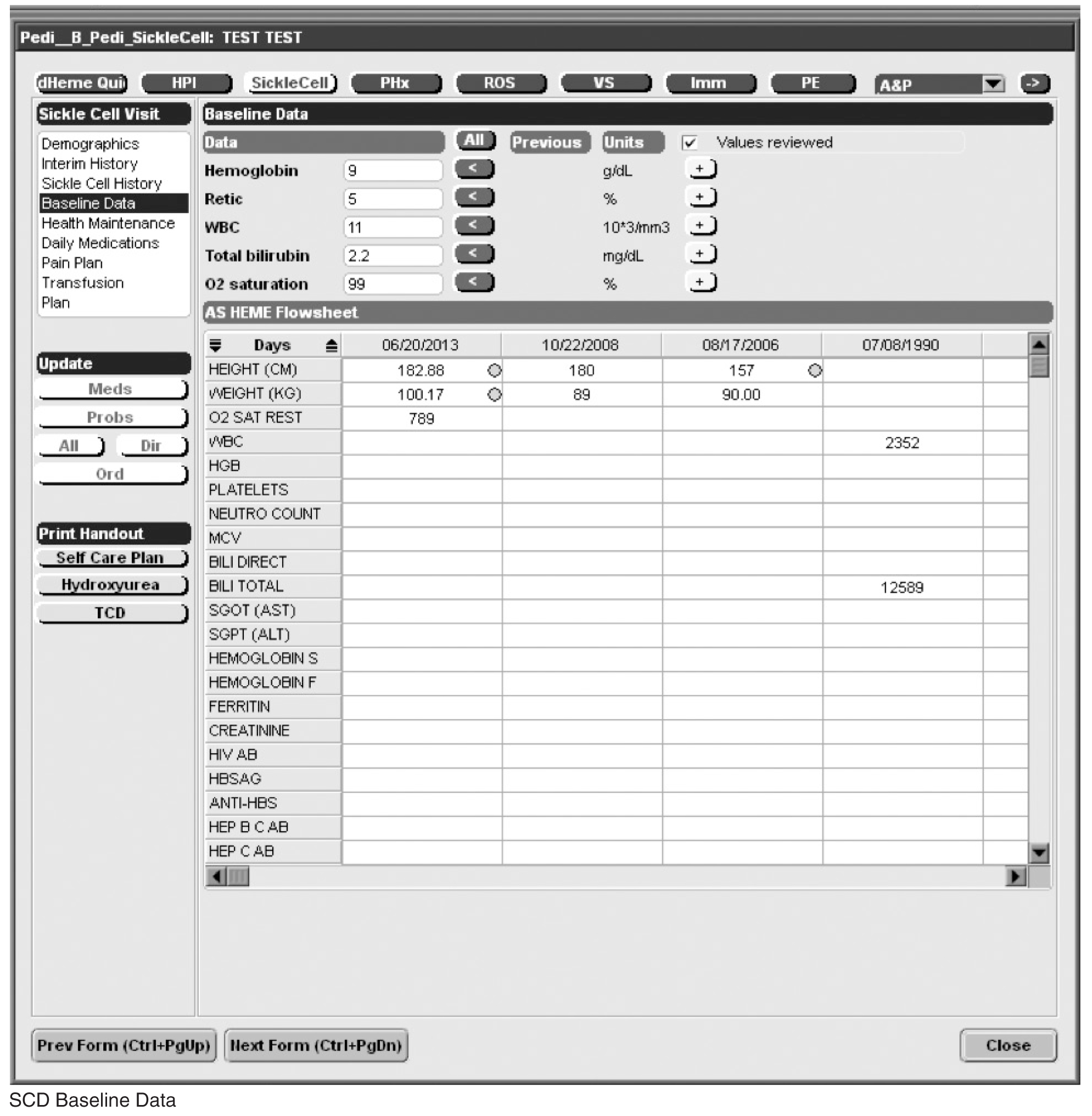
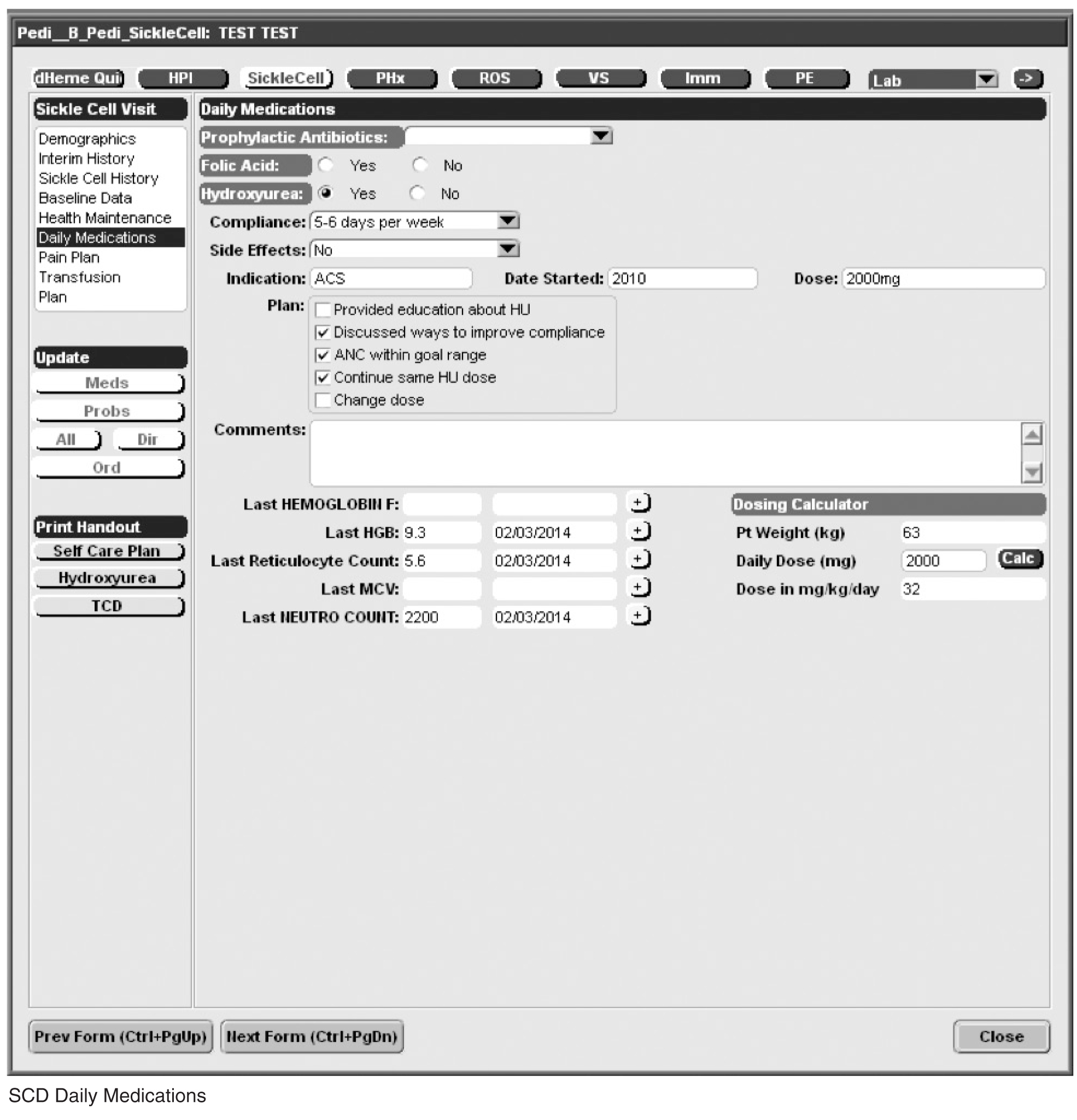
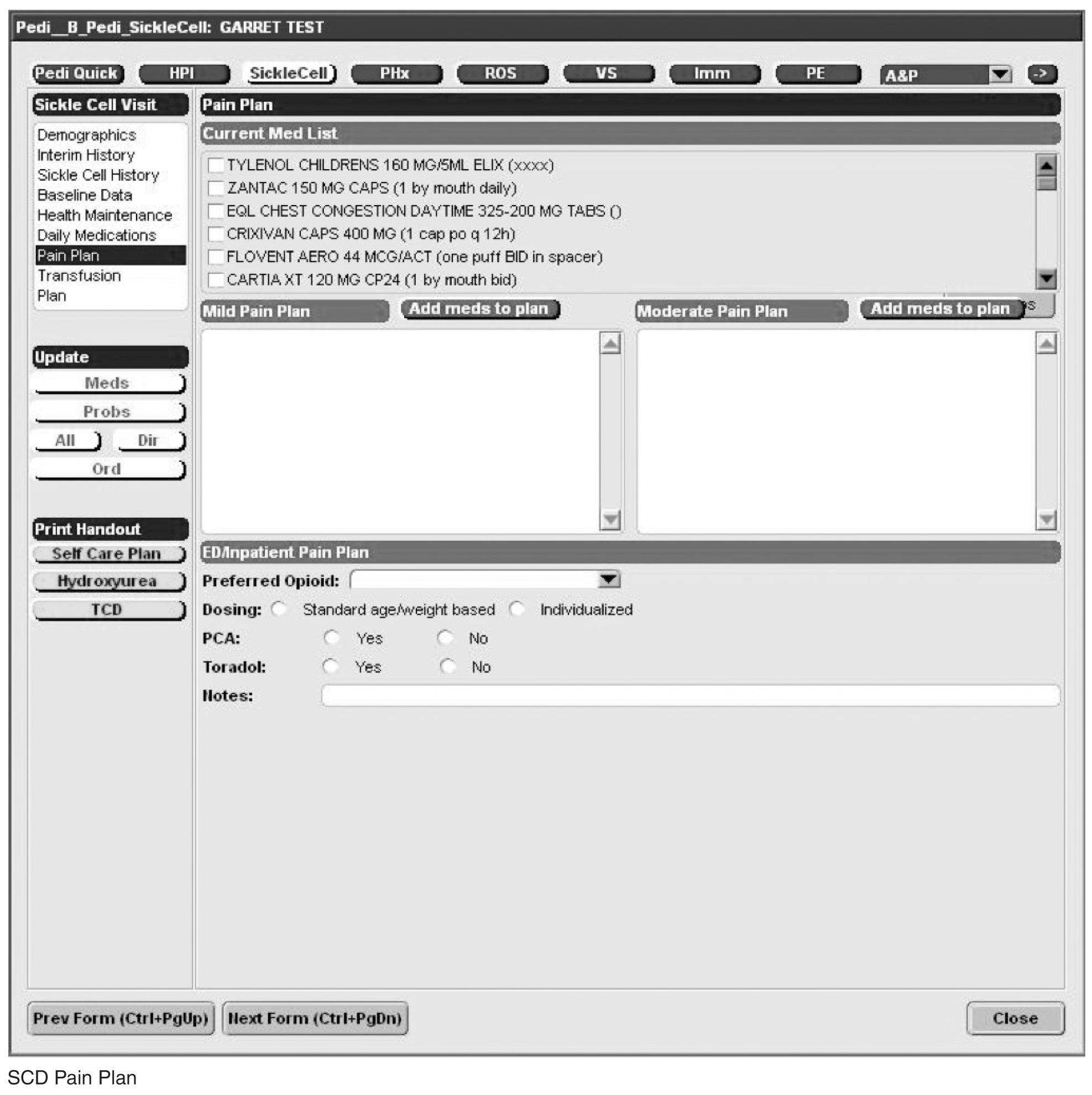
Documentation in the EHR is performed using a set of customized templates or “forms.” These forms allow documentation of care provision in a structured way. The discrete data elements are stored within the data warehousing system that supports the EHR. The SCD forms used in this project were a revised version of existing forms used by our pediatric hematologists for the past 6 years. The primary goal was to improve efficiency in a patient encounter and enhance data collection efforts. In particular, several changes were made to enhance data collection for quality measures included in the SCD registry. First, we collected genotype in a standardized way to better define subpopulations of SCD patients, as some of the care provided is dictated by genotype. We also expanded data capture for transcranial Doppler screening to include date of last screening to prompt scheduling. For hydroxyurea, the forms now capture if hydroxyurea has been prescribed, and if not, why (eg, declined, not indicated); adherence, current dose, and routine labs for monitoring are also listed to aid in clinical decision-making. Finally, the forms were revised to prominently display the subset of immunizations important to SCD (described above) to assess if the patient is current.
Within the new forms, we collected all data elements important to providing care to children with SCD. Several new items existed in other parts of the EHR and were automatically pulled into the forms, including laboratory results, medications and immunizations. Other new data elements required manual entry by providers based on EHR review, as they had previously not been documented, documented on an ad hoc basis, or found as free text within notes (eg, number of ED visits and hospitalizations in the past year). Initial completion of these forms took approximately 10 to 15 minutes per patient, as many of these data elements were not individually captured prior to this work; documentation for subsequent comprehensive visits required an additional 5 to 10 minutes per chart. Currently, the 3 pediatric hematologists regularly use the SCD forms for routine visits.
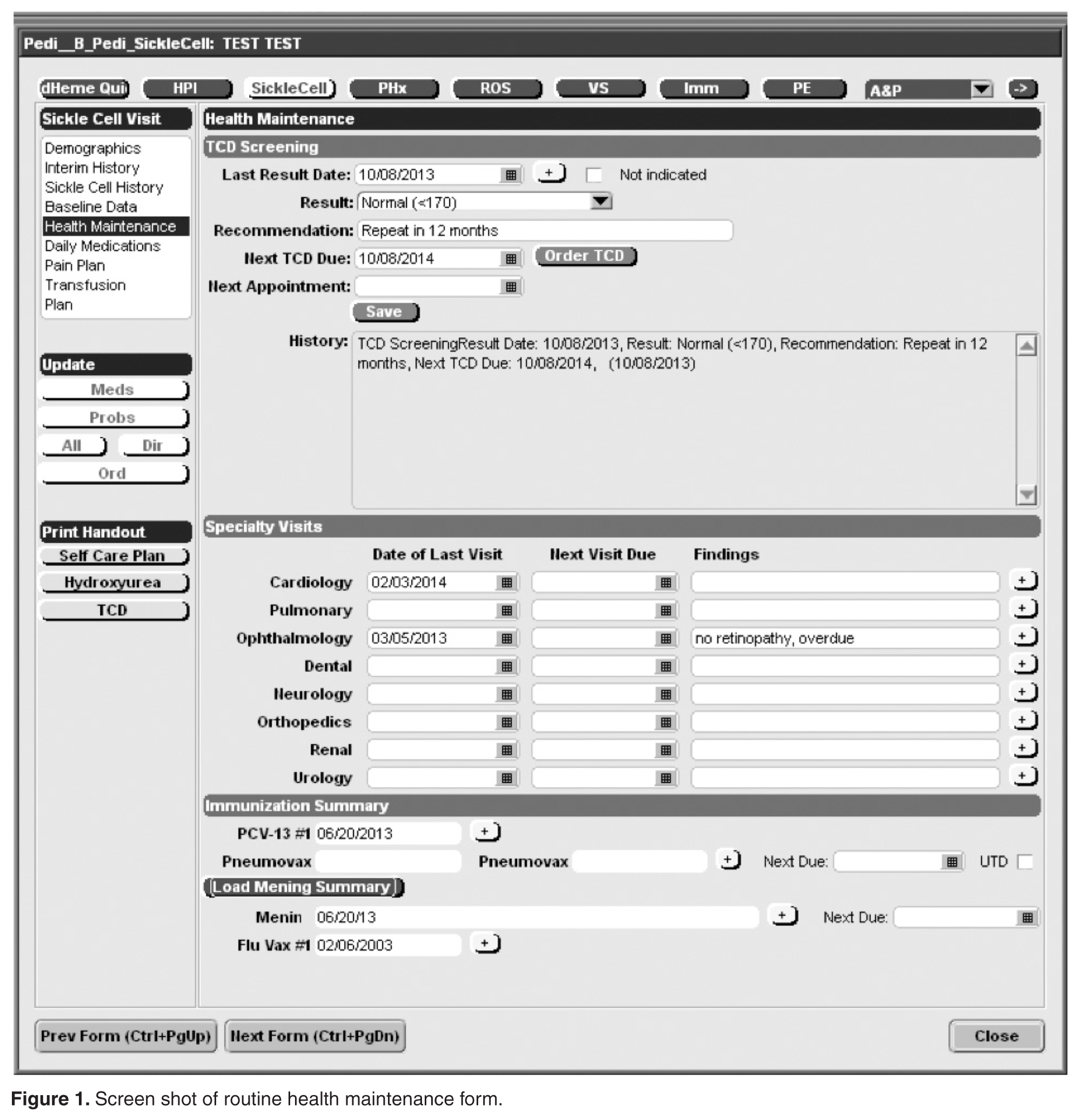
The registry management form was also created by the EHR design team. Although this form is separate from the SCD forms, it was readily accessible to the clinical team to quickly check whether patients should be included or excluded from the SCD registry. In this way, inactive patients could be removed and new patients could be included. This form was completed for all active pediatric patients with SCD as of February 2013 using data from a separately maintained clinical database. For patients who were new to the pediatric hematology practice between July 2012 and February 2013 (eg, infants born during this period, patients transferring care), we manually determined a registry start date in order to calculate accurate denominators for each measure. New patients were entered into the SCD registry by members of the care team on an ad hoc basis, and biannual searches of problem lists were planned to ensure the pediatric SCD registry was complete using the SCD-related ICD-9 codes 282.6, 282.41 and 282.4 to encompass all sickle hemoglobinopathies, including sickle cell thalassemia.
For this project, we were fortunate to have a well-established clinical data warehouse into which the medical center’s EHR data is copied nightly. In addition, the medical center already had multiple chronic disease registries and a framework for evaluating and sharing QI data. We were able to add SCD to this existing infrastructure, which was helpful since a secure and HIPAA-compliant location to post these patient-level reports had been previously identified.
We paid for 40 hours of technical staff time using grant funds to create reports using data collected in the EHR for patients who were actively in the SCD registry per the registry management form. Using these data, summary reports for our key SCD metrics were generated on both an annual and monthly basis. We tested and refined our key SCD metrics over a 4-month period to ensure that we had defined the numerators and denominators for each care process accurately. For example, children become eligible for influenza vaccine at 6 months of age, therefore, the eligible denominator would exclude infants < 6 months of age (Table 1). In addition, lists of patient names and phone numbers were automatically generated to identify those in need of care elements, facilitating both case management and continuous improvement for these measures, replacing the need for all external clinical databases.
Data Analysis
For children included in the SCD registry, we calculated the proportion who were appropriately vaccinated and received transcranial Doppler screening each year for the 5-year period 2008–2012. For the period July 2012–June 2013, we calculated the proportion of children with SCD in the registry who received influenza vaccine and children with Hb SS and Hb S-β0 thalassemia who were prescribed hydroxyurea.
This study was approved by the Boston University Medical Campus institutional review board.
Results
For influenza vaccination for the 2012–2013 season, only 49% of children were vaccinated as of November. This proportion increased after outreach 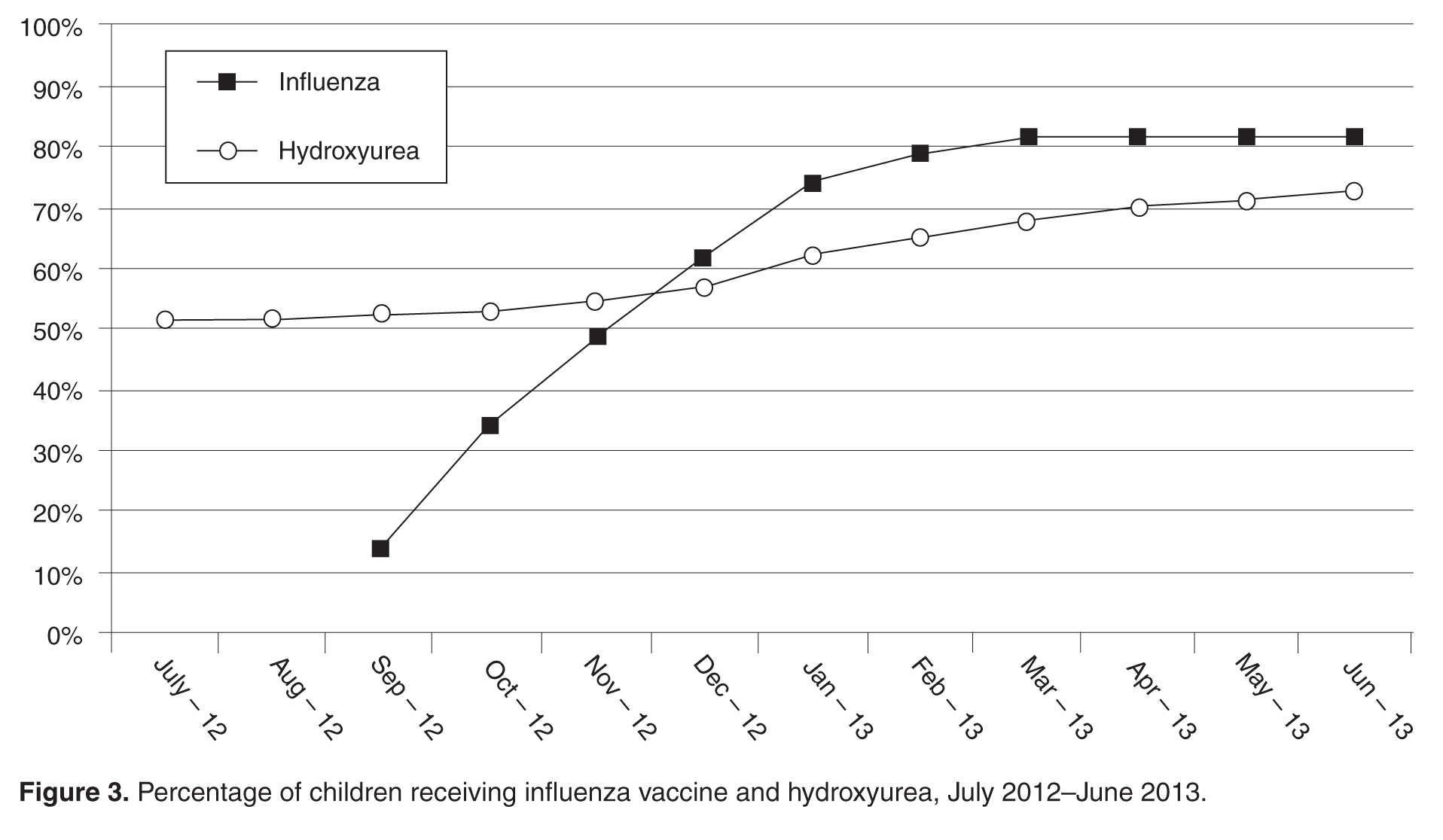
From July 2012 to June 2013, our rates of hydroxyurea use increased from 52% to 73% among eligible patients.
Discussion
In this paper we report on a practical approach for improving the quality of care for persons with SCD that combines the collaboration of a multidisciplinary team, the use of the EHR to create a disease registry, and QI initiatives. We identified where high-quality care is provided and where further attention is needed, and enhanced our case management capabilities with the generation of patient lists identifying those who are in need of care elements. We also used our registry to track care provision, achieving rates of influenza vaccination of 82% and hydroxyurea use to 73% as of June 2013. From these results, we have shown that our EHR can be used for registry management activities and provide real-time clinical data on the care that is provided, and can lead to improved performance on process measures important in the care for children with SCD.
After adjusting to the revised workflow required by the new SCD forms, the pediatric hematology team found them to be useful in tracking important clinical measures. They reported that the most important change was that all routine elements of SCD care, such as dates of last visits to pediatric subspecialists and receipt of recommended routine SCD care, were embedded into their note. This eliminated the need to search previous documents to find dates of the last cardiology visit or influenza immunizations and increased the likelihood that gaps in care would be addressed by the provider during the course of a clinic visit, thereby streamlining clinic workflow.
Healthy People 2020 recommend vaccination rates of 80% and 90% for influenza and PCV13 vaccines, respectively, in the general pediatric population [36]. We have met this goal for the influenza vaccine, but have room to improve for other recommended vaccines for children with SCD. Ultimately, our goal is to provide these vaccines to 100% of children with SCD at our institution. One barrier to achieving high vaccination rates is the lack of provider knowledge on the creation of catch-up vaccine schedules. A study of primary care providers showed that they frequently omitted vaccines when creating catch-up schedules, including the pneumococcal conjugate vaccine for healthy children [37]. Another hurdle is coordination of care between primary and specialty care, as these vaccines could be given in either setting. A recently published study found that only 20% of children with SCD had care coordination between primary and specialty care [38]. Promoting shared responsibility and information on the administration of vaccinations for children with SCD between primary and subspecialty care, and the development of state-wide immunization registries, may help alleviate these challenges.
In this study, our rates of hydroxyurea use among children with Hb SS and Hb S-β0 thalassemia are higher than in other reported studies [12]. We promote hydroxyurea use in this population of children based on the recently published safety data in infants and young children with Hb SS and Hb S-β0 thalassemia [7,32,39] and the significant benefits seen in adults, including improved survival [6,34,35,40]. Future efforts will include tracking outcomes, including the rates of acute chest syndrome and pain episodes, among children who are and are not taking hydroxyurea.
In this study, we found approximately 70% of eligible children were screened with transcranial Doppler each year from 2008–2012, which is higher than the 45% annual screening rate reported in the literature [10]. One reason our transcranial Doppler screening rates may be higher is that a technician is available to perform these tests on certain days that coincide with the pediatric hematology clinic, allowing patients and families to get this test and have a clinic visit on the same day. However, choosing a 12-month period for receipt of transcranial Doppler screening may be too conservative for centers who do not have such ready access to screening; reporting receipt of transcranial Doppler screening within a 15-month time period may be more appropriate and achievable.
Our study has several limitations. First, it was conducted in a single center with well-established electronic data systems, which are not available in many centers. Our hope is that this model can be replicated by others who seek to use EHR to improve the care of persons with SCD. Second, this work was performed in Massachusetts, a state with near-universal health care insurance coverage. As the Affordable Care Act is implemented nationally [41], other states may see improved performance on quality metrics as more people obtain health insurance. Third, although the EHR was designed to improve data capture for clinical care and quality initiatives, advanced clinical decision support systems were not incorporated due to the limitations of the EHR. The use of prompts for needed clinical care may further enhance performance on these measures. Fourth, this study is limited to children with SCD, who are traditionally monitored more closely than their adult counterparts. Efforts are currently underway to replicate these efforts with adults with SCD at our institution. Finally, the quality metrics in this study are process measures in the delivery of high quality SCD care. Future efforts will focus on linking outcomes to these measures, such as hydroxyurea use to reduce the frequency of acute chest syndrome and painful episodes.
Effective use of health information technology has proven challenging [42,43]. Although there are data that suggest that information technology has improved quality of care by increasing adherence to guidelines, enhancing disease surveillance, and decreasing medication errors, most of the high-quality literature to date comes from 4 research institutions [18]. We found that health IT can be effectively harnessed when end-users are engaged in the process of EHR design, there is a strong commitment to improve workflow and support documentation needs of end-users, the design of the EHR supports data collection for quality measures, and most importantly, there is close collaboration among those with overlapping technical, clinical, and health services research expertise.
There have been many calls for the creation of rare disease registries, as 6% to 8% of the population will develop one in their lifetime [44]. In 2010, the NIH’s Office of Rare Diseases Research funded 30 organizations with and without patient registries, and charged them with the creation of a common data collection template for rare diseases to be used internationally [45]. Common data collection elements for SCD, such as those used in our program, could be used in EHRs across US centers in an effort to improve the quality of care for these children. Although this work may be challenging for centers using large enterprise EHR systems, given the costs associated with modifications, once developed the content can often be shared easily with others using the same system. This would provide the opportunity to compare uniform data across institutions and facilitate learning nationally on ways to improve care. In addition, these efforts may serve as the beginnings of a national registry for pediatric SCD.
In conclusion, contemporary SCD care can lead to improved survival and quality of life, but only if the right care is delivered at the right time. In this study, we present our initial findings from the implementation of a population-based information system for children with SCD. Future efforts are needed to define and measure all elements of high quality care, and link improvements in the delivery of high quality care to outcomes for children and adults with SCD longitudinally.
Appendix. Additional Sickle Cell Disease Forms
Acknowledgments: We would like to thank David Botts for his tireless efforts in creating the sickle cell forms within our EHR. We would also like to thank Barry Zuckerman for his support of this project.
Corresponding author: Patricia Kavanagh, MD, Boston University School of Medicine/Boston Medical Center, 88 E Newton St, Vose Hall 3rd Fl, Boston, MA 02118.
Funding/support: This work was supported by the Health Resources and Services Administration Sickle Cell Disease and Newborn Screening Program, grant #U38MC22215. The authors have also actively participated in the Hemoglobinopathy Learning Collaborative, a quality improvement forum coordinated by HRSA and the National Initiative for Children’s Healthcare Quality.
Financial disclosures: None.
1. Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Preventive Med 2010;38(4 Suppl):S512–S521.
2. Steinberg MH. Management of sickle cell disease. N Engl J Med 1999;340:1021–30.
3. Adamkiewicz TV, Silk BJ, Howgate J, et al. Effectiveness of the 7-valent pneumococcal conjugate vaccine in children with sickle cell disease in the first decade of life. Pediatrics 2008;121:562–9.
4. Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial doppler ultrasonography. N Engl J Med 1998;339:5–1.
5. Adams RJ, Brambilla D, Optimizing Primary Stroke Prevention in Sickle Cell Anemia Trial I. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease.[see comment]. N Engl J Med 2005;353:2769–78.
6. Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. N Engl J Med 1995;332:1317–22.
7. Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: A multicentre, randomised, controlled trial (baby hug). Lancet 2011;377:1663–72.
8. Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood 2010;115:3447–52.
9. Hamideh D, Alvarez O. Sickle cell disease related mortality in the united states (1999–2009). Pediatr Blood Cancer 2013;60:1482–6.
10. Raphael JL, Shetty PB, Liu H, et al. A critical assessment of transcranial doppler screening rates in a large pediatric sickle cell center: Opportunities to improve healthcare quality. Pediatr Blood Cancer 2008;51:647–51.
11. Sox CM, Cooper WO, Koepsell TD, et al. Provision of pneumococcal prophylaxis for publicly insured children with sickle cell disease. JAMA 2003;290:1057–61.
12. Oyeku SO, Driscoll MC, Cohen HW, et al. Parental and other factors associated with hydroxyurea use for pediatric sickle cell disease. Pediatr Blood Cancer 2013;60:653–58.
13. Crandall WV, Margolis PA, Kappelman MD, et al. Improved outcomes in a quality improvement collaborative for pediatric inflammatory bowel disease. Pediatrics 2012;129:e1030–e1041.
14. Schechter MS, Margolis P. Improving subspecialty healthcare: Lessons from cystic fibrosis. J Pediatr 2005;147:295–301.
15. Smith LA, Oyeku SO, Homer C, Zuckerman B. Sickle cell disease: A question of equity and quality. Pediatrics 2006;117:1763–70.
16. Wang CJ, Kavanagh PL, Little AA, et al. Quality-of-care indicators for children with sickle cell disease. Pediatrics 2011;128:484–93.
17. Jha AK, Perlin JB, Kizer KW, Dudley RA. Effect of the transformation of the veterans affairs health care system on the quality of care. N Engl J Med 2003;348:2218–27.
18. Chaudhry B, Wang J, Wu S, et al. Systematic review: Impact of health information technology on quality, efficiency, and costs of medical care. Ann Intern Med 2006;144:742–52.
19. American recovery and reinvestment act of 2009. Obey D, Frank B, Gordon B, et al., trans. 111th Congress of the United States.
20. Blumenthal D, Tavenner M. The “meaningful use” regulation for electronic health records. N Engl J Med 2010;363:501–4.
21. Electronic health record adoption by office-based providers. Office of National Coordinator for Health Information Technology. U.S. Department of Health and Human Services. Accessed 15 Jul 2013.
22. DesRoches CM, Campbell EG, Rao SR, et al. Electronic health records in ambulatory care — a national survey of physicians. N Engl J Med 2008;359:50–60.
23. Tricco AC, Ivers NM, Grimshaw JM, et al. Effectiveness of quality improvement strategies on the management of diabetes: A systematic review and meta-analysis. Lancet 379:2252–61.
24. Bundy DG, Strouse JJ, Casella JF, Miller MR. Burden of influenza-related hospitalizations among children with sickle cell disease. Pediatrics 2010;125:234–43.
25. National Heart Lung and Blood Institute. The management of sickle cell disease. NIH Pub No. 02-2117. Bethesda, MD: National Institutes of Health; 2002.
26. Centers for Disease Control and Prevention. Immunization schedules. Accessed 5 Jan 2013 at www.cdc.gov/vaccines/schedules/index.html.
27. Strouse JJ, Reller ME, Bundy DG, et al. Severe pandemic h1n1 and seasonal influenza in children and young adults with sickle cell disease. Blood 2010;116:3431–4.
28. Pilishvili T, Zell ER, Farley MM, et al. Risk factors for invasive pneumococcal disease in children in the era of conjugate vaccine use. Pediatrics 2010;126:e9–17.
29. Heeney MM, Ware RE. Hydroxyurea for children with sickle cell disease. Pediatr Clin North Am 008;55:483–501.
30. Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood 2010;115:5300–11.
31. Ferster A, Vermylen C, Cornu G, et al. Hydroxyurea for treatment of severe sickle cell anemia: a pediatric clinical trial. Blood 1996;88:1960–4.
32. Strouse JJ, Lanzkron S, Beach MC, et al. Hydroxyurea for sickle cell disease: a systematic review for efficacy and toxicity in children. Pediatrics 2008;122:1332–42.
33. Hankins JS, Ware RE, Rogers ZR, et al. Long-term hydroxyurea therapy for infants with sickle cell anemia: the husoft extension study. Blood 2005;106:2269–75.
34. Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: a 17.5-year follow-up. Am J Hematol 2010;85:403–8.
35. Voskaridou E, Christoulas D, Bilalis A, et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (lashs). Blood 2010;115:2354–63.
36. Healthy people 2020. Immunization and infectious diseases. Accessed 3 Jun 2013 at www.healthypeople.gov/2020/topicsobjectives2020/objectiveslist.aspx?topicid=23.
37. Cohen NJ, Lauderdale DS, Shete PB, et al. Physician knowledge of catch-up regimens and contraindications for childhood immunizations. Pediatrics 2003;111:925–32.
38. Raphael JL, Rattler TL, Kowalkowski MA, et al. The medical home experience among children with sickle cell disease. Pediatr Blood Cancer 2013;60:275–80.
39. Strouse JJ, Heeney MM. Hydroxyurea for the treatment of sickle cell disease: efficacy, barriers, toxicity, and management in children. Pediatr Blood Cancer 2012;59:365–71.
40. Steinberg MH, Barton F, Castro O, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA 2003;289:1645–51.
41. Patient protection and affordable care act, US Pub. L. No. 111-148, §2702, 124 stat. 119, 318-319. 2010.
42. Harrison M, Koppel R, Bar-Lev S. Unintended consequences of information technologies in health care: an interactive sociotechnical analysis. J Am Med Inform Assoc 2007;14:542–9.
43. Haux R. Health information systems – past, present, future. Int J Med Informatics 2006;75:268–81.
44. Schieppati A, Henter J-I, Daina E, Aperia A. Why rare diseases are an important medical and social issue. Lancet 2008;371:2039–41.
45. Office of Rare Diseases Research National Institutes of Health. Rare diseases and related terms. Accessed 28 Jun 2013 at www.rarediseases.info.nih.gov/rarediseaselist.aspx.
From the Department of Pediatrics, Boston University School of Medicine, Boston Medical Center, Boston, MA.
This article is the second in our Hemoglobinopathy Learning Collaborative series. See the related editorial by Oyeku et al in the February 2014 issue of JCOM. (—Ed.)
Abstract
- Objective: To describe the development and use of an electronic health record (EHR)–based sickle cell disease (SCD) registry for children with SCD to enhance case management and quality improvement (QI) efforts at an urban, academic, safety net institution.
- Methods: Using national guidelines and the literature, we created quality metrics for pediatric SCD that focused on vaccination delivery and use of transcranial Doppler screening and hydroxyurea. We revised EHR forms for SCD care and created an EHR-based SCD registry that permitted monthly and annual reporting on quality metrics.
- Results: From 2008 to 2012, the percentage of children with SCD vaccinated for influenza increased from 52% to 65%, and for meningococcus from 53% to 70%. After licensure of PCV13 in 2010, the percentage of children vaccinated rose to 69% in 2012. Results for PPV23 were mixed: 87% to 91% received ≥1 dose, but the rate for receiving the second dose declined from 76% to 64%. Percentage of children screened annually with transcranial Doppler consistently ranged from 62% to 73% during the 5 years. QI initiatives in 2012–2013 led to increased influenza vaccination, from 65% to 83%, and increased hydroxyurea use, from 52% to 73%.
- Conclusion: In this study, a practical, replicable and feasible approach for improving the quality of SCD care combined the collaboration of a multidisciplinary team, an EHR-based disease registry, and QI initiatives. Additional work is needed to define and measure all elements of high-quality care for children with SCD and link process measures to clinical outcomes.
Sickle cell disease (SCD) is the most commonly inherited disorder in the United States, affecting approximately 100,000 individuals and 1 in 400 African American births [1,2]. The use of preventive strategies, such as immunizations [3], transcranial Doppler screening and transfusion protocols [4,5], and hydroxyurea therapy [6,7] has contributed to decreased morbidity and mortality among children with SCD [8,9]. However, a substantial gap exists between the care that children with SCD should receive and the care they actually receive [10–12]. An essential component of any effort that seeks to improve care is the ability to measure care processes and outcomes in a way that can drive quality improvement (QI) initiatives. Registries serve a vital role in quality improvement activities for many pediatric conditions, including inflammatory bowel disease [13] and cystic fibrosis [14]. However, there are no national or nationally representative registries currently available for children with SCD [15]. There is a pressing need for better information systems and tools that can be used in mainstream clinical settings to measure clinical performance with respect to quality indicators [16] if the goals of high quality care and better quality of life are to be achieved for children with SCD.
Electronic health records (EHRs) have been successfully used to improve the quality of care and enhance performance measurement in select institutions [17,18], and adoption of EHRs is growing. The 2009 American Recovery and Reinvestment Act allocated $20.8 billion in incentives to assist providers to adopt and “meaningfully use” EHRs [19,20]. As of 2011, 39% of office-based providers have implemented at least a basic EHR [21], up from 17% in 2008 [22]. The effective use of EHRs depends on collaboration between technical and medical experts so that functionality is achieved and clinical quality is appropriately measured. In addition, few EHRs contain specialized content for the care of persons with SCD.
While independent registries have been shown to be effective in improving care [13,14,23], they involve extra time and effort for data entry, can be difficult and expensive to maintain, and may not be feasible for many systems that care for SCD patients. In this paper, we describe the development and use of our EHR-based SCD registry for children with SCD, including our efforts to engage key technical and clinical experts to develop an EHR that is tailored to the outpatient workflow and data collection of quality measures and implement a fully functional system that collects data on quality measures to support case management and continuous QI.
Methods
This study was conducted at Boston Medical Center, New England’s largest safety net hospital, which cares for 190 children with SCD ages 0 to 21 years. The outpatient EHR (Centricity, GE) has been in use since 2000 and is used for all aspects of outpatient care, including ordering of immunizations and tests, electronic prescription writing, and referrals to specialty care.
Outcome Measures
Vaccines: The Centers for Disease Control and Prevention (CDC) recommends vaccinating children with SCD [26] against influenza annually, given their susceptibility to the influenza virus [24,27]. The CDC also recommends the 23-valent pneumococcal polysaccharide vaccine (PPV23 2-dose series) and 13-valent pneumococcal conjugate vaccine (PCV13, per childhood routine vaccine schedule for young children and 1 catch-up dose for children previously vaccinated with PCV7), and meningococcal vaccine (2-dose series), given patients’ functional asplenic status [25,28].
Transcranial Doppler screening can identify children with hemoglobin (Hb) SS and Hb S-β0 thalassemia at higher risk of stroke, which may be prevented through hypertransfusion programs [4]. Screening is recommended annually for these children ages 2 to 16 years [25].
Hydroxyurea use among children with Hb SS and Hb S-β0 thalassemia is an established practice [29,30]. We consider hydroxyurea therapy for all children 2 years and older with Hb SS and Hb S-β0 thalassemia, given the recently published safety data from the Baby-HUG trial [7] and the benefits of hydroxyurea among children and adults with SCD [6,31–35].
EHR-based Registry
Our EHR-based SCD registry includes 3 key components: (1) forms to support detailed documentation at the point-of-care (ie, clinic visit); (2) a registry management form to allow the QI team to identify patients to be included or excluded from the registry; and (3) a central data warehouse to support quality measurement and improvement.
Documentation in the EHR is performed using a set of customized templates or “forms.” These forms allow documentation of care provision in a structured way. The discrete data elements are stored within the data warehousing system that supports the EHR. The SCD forms used in this project were a revised version of existing forms used by our pediatric hematologists for the past 6 years. The primary goal was to improve efficiency in a patient encounter and enhance data collection efforts. In particular, several changes were made to enhance data collection for quality measures included in the SCD registry. First, we collected genotype in a standardized way to better define subpopulations of SCD patients, as some of the care provided is dictated by genotype. We also expanded data capture for transcranial Doppler screening to include date of last screening to prompt scheduling. For hydroxyurea, the forms now capture if hydroxyurea has been prescribed, and if not, why (eg, declined, not indicated); adherence, current dose, and routine labs for monitoring are also listed to aid in clinical decision-making. Finally, the forms were revised to prominently display the subset of immunizations important to SCD (described above) to assess if the patient is current.
Within the new forms, we collected all data elements important to providing care to children with SCD. Several new items existed in other parts of the EHR and were automatically pulled into the forms, including laboratory results, medications and immunizations. Other new data elements required manual entry by providers based on EHR review, as they had previously not been documented, documented on an ad hoc basis, or found as free text within notes (eg, number of ED visits and hospitalizations in the past year). Initial completion of these forms took approximately 10 to 15 minutes per patient, as many of these data elements were not individually captured prior to this work; documentation for subsequent comprehensive visits required an additional 5 to 10 minutes per chart. Currently, the 3 pediatric hematologists regularly use the SCD forms for routine visits.
The registry management form was also created by the EHR design team. Although this form is separate from the SCD forms, it was readily accessible to the clinical team to quickly check whether patients should be included or excluded from the SCD registry. In this way, inactive patients could be removed and new patients could be included. This form was completed for all active pediatric patients with SCD as of February 2013 using data from a separately maintained clinical database. For patients who were new to the pediatric hematology practice between July 2012 and February 2013 (eg, infants born during this period, patients transferring care), we manually determined a registry start date in order to calculate accurate denominators for each measure. New patients were entered into the SCD registry by members of the care team on an ad hoc basis, and biannual searches of problem lists were planned to ensure the pediatric SCD registry was complete using the SCD-related ICD-9 codes 282.6, 282.41 and 282.4 to encompass all sickle hemoglobinopathies, including sickle cell thalassemia.
For this project, we were fortunate to have a well-established clinical data warehouse into which the medical center’s EHR data is copied nightly. In addition, the medical center already had multiple chronic disease registries and a framework for evaluating and sharing QI data. We were able to add SCD to this existing infrastructure, which was helpful since a secure and HIPAA-compliant location to post these patient-level reports had been previously identified.
We paid for 40 hours of technical staff time using grant funds to create reports using data collected in the EHR for patients who were actively in the SCD registry per the registry management form. Using these data, summary reports for our key SCD metrics were generated on both an annual and monthly basis. We tested and refined our key SCD metrics over a 4-month period to ensure that we had defined the numerators and denominators for each care process accurately. For example, children become eligible for influenza vaccine at 6 months of age, therefore, the eligible denominator would exclude infants < 6 months of age (Table 1). In addition, lists of patient names and phone numbers were automatically generated to identify those in need of care elements, facilitating both case management and continuous improvement for these measures, replacing the need for all external clinical databases.
Data Analysis
For children included in the SCD registry, we calculated the proportion who were appropriately vaccinated and received transcranial Doppler screening each year for the 5-year period 2008–2012. For the period July 2012–June 2013, we calculated the proportion of children with SCD in the registry who received influenza vaccine and children with Hb SS and Hb S-β0 thalassemia who were prescribed hydroxyurea.
This study was approved by the Boston University Medical Campus institutional review board.
Results
For influenza vaccination for the 2012–2013 season, only 49% of children were vaccinated as of November. This proportion increased after outreach
From July 2012 to June 2013, our rates of hydroxyurea use increased from 52% to 73% among eligible patients.
Discussion
In this paper we report on a practical approach for improving the quality of care for persons with SCD that combines the collaboration of a multidisciplinary team, the use of the EHR to create a disease registry, and QI initiatives. We identified where high-quality care is provided and where further attention is needed, and enhanced our case management capabilities with the generation of patient lists identifying those who are in need of care elements. We also used our registry to track care provision, achieving rates of influenza vaccination of 82% and hydroxyurea use to 73% as of June 2013. From these results, we have shown that our EHR can be used for registry management activities and provide real-time clinical data on the care that is provided, and can lead to improved performance on process measures important in the care for children with SCD.
After adjusting to the revised workflow required by the new SCD forms, the pediatric hematology team found them to be useful in tracking important clinical measures. They reported that the most important change was that all routine elements of SCD care, such as dates of last visits to pediatric subspecialists and receipt of recommended routine SCD care, were embedded into their note. This eliminated the need to search previous documents to find dates of the last cardiology visit or influenza immunizations and increased the likelihood that gaps in care would be addressed by the provider during the course of a clinic visit, thereby streamlining clinic workflow.
Healthy People 2020 recommend vaccination rates of 80% and 90% for influenza and PCV13 vaccines, respectively, in the general pediatric population [36]. We have met this goal for the influenza vaccine, but have room to improve for other recommended vaccines for children with SCD. Ultimately, our goal is to provide these vaccines to 100% of children with SCD at our institution. One barrier to achieving high vaccination rates is the lack of provider knowledge on the creation of catch-up vaccine schedules. A study of primary care providers showed that they frequently omitted vaccines when creating catch-up schedules, including the pneumococcal conjugate vaccine for healthy children [37]. Another hurdle is coordination of care between primary and specialty care, as these vaccines could be given in either setting. A recently published study found that only 20% of children with SCD had care coordination between primary and specialty care [38]. Promoting shared responsibility and information on the administration of vaccinations for children with SCD between primary and subspecialty care, and the development of state-wide immunization registries, may help alleviate these challenges.
In this study, our rates of hydroxyurea use among children with Hb SS and Hb S-β0 thalassemia are higher than in other reported studies [12]. We promote hydroxyurea use in this population of children based on the recently published safety data in infants and young children with Hb SS and Hb S-β0 thalassemia [7,32,39] and the significant benefits seen in adults, including improved survival [6,34,35,40]. Future efforts will include tracking outcomes, including the rates of acute chest syndrome and pain episodes, among children who are and are not taking hydroxyurea.
In this study, we found approximately 70% of eligible children were screened with transcranial Doppler each year from 2008–2012, which is higher than the 45% annual screening rate reported in the literature [10]. One reason our transcranial Doppler screening rates may be higher is that a technician is available to perform these tests on certain days that coincide with the pediatric hematology clinic, allowing patients and families to get this test and have a clinic visit on the same day. However, choosing a 12-month period for receipt of transcranial Doppler screening may be too conservative for centers who do not have such ready access to screening; reporting receipt of transcranial Doppler screening within a 15-month time period may be more appropriate and achievable.
Our study has several limitations. First, it was conducted in a single center with well-established electronic data systems, which are not available in many centers. Our hope is that this model can be replicated by others who seek to use EHR to improve the care of persons with SCD. Second, this work was performed in Massachusetts, a state with near-universal health care insurance coverage. As the Affordable Care Act is implemented nationally [41], other states may see improved performance on quality metrics as more people obtain health insurance. Third, although the EHR was designed to improve data capture for clinical care and quality initiatives, advanced clinical decision support systems were not incorporated due to the limitations of the EHR. The use of prompts for needed clinical care may further enhance performance on these measures. Fourth, this study is limited to children with SCD, who are traditionally monitored more closely than their adult counterparts. Efforts are currently underway to replicate these efforts with adults with SCD at our institution. Finally, the quality metrics in this study are process measures in the delivery of high quality SCD care. Future efforts will focus on linking outcomes to these measures, such as hydroxyurea use to reduce the frequency of acute chest syndrome and painful episodes.
Effective use of health information technology has proven challenging [42,43]. Although there are data that suggest that information technology has improved quality of care by increasing adherence to guidelines, enhancing disease surveillance, and decreasing medication errors, most of the high-quality literature to date comes from 4 research institutions [18]. We found that health IT can be effectively harnessed when end-users are engaged in the process of EHR design, there is a strong commitment to improve workflow and support documentation needs of end-users, the design of the EHR supports data collection for quality measures, and most importantly, there is close collaboration among those with overlapping technical, clinical, and health services research expertise.
There have been many calls for the creation of rare disease registries, as 6% to 8% of the population will develop one in their lifetime [44]. In 2010, the NIH’s Office of Rare Diseases Research funded 30 organizations with and without patient registries, and charged them with the creation of a common data collection template for rare diseases to be used internationally [45]. Common data collection elements for SCD, such as those used in our program, could be used in EHRs across US centers in an effort to improve the quality of care for these children. Although this work may be challenging for centers using large enterprise EHR systems, given the costs associated with modifications, once developed the content can often be shared easily with others using the same system. This would provide the opportunity to compare uniform data across institutions and facilitate learning nationally on ways to improve care. In addition, these efforts may serve as the beginnings of a national registry for pediatric SCD.
In conclusion, contemporary SCD care can lead to improved survival and quality of life, but only if the right care is delivered at the right time. In this study, we present our initial findings from the implementation of a population-based information system for children with SCD. Future efforts are needed to define and measure all elements of high quality care, and link improvements in the delivery of high quality care to outcomes for children and adults with SCD longitudinally.
Appendix. Additional Sickle Cell Disease Forms
Acknowledgments: We would like to thank David Botts for his tireless efforts in creating the sickle cell forms within our EHR. We would also like to thank Barry Zuckerman for his support of this project.
Corresponding author: Patricia Kavanagh, MD, Boston University School of Medicine/Boston Medical Center, 88 E Newton St, Vose Hall 3rd Fl, Boston, MA 02118.
Funding/support: This work was supported by the Health Resources and Services Administration Sickle Cell Disease and Newborn Screening Program, grant #U38MC22215. The authors have also actively participated in the Hemoglobinopathy Learning Collaborative, a quality improvement forum coordinated by HRSA and the National Initiative for Children’s Healthcare Quality.
Financial disclosures: None.
From the Department of Pediatrics, Boston University School of Medicine, Boston Medical Center, Boston, MA.
This article is the second in our Hemoglobinopathy Learning Collaborative series. See the related editorial by Oyeku et al in the February 2014 issue of JCOM. (—Ed.)
Abstract
- Objective: To describe the development and use of an electronic health record (EHR)–based sickle cell disease (SCD) registry for children with SCD to enhance case management and quality improvement (QI) efforts at an urban, academic, safety net institution.
- Methods: Using national guidelines and the literature, we created quality metrics for pediatric SCD that focused on vaccination delivery and use of transcranial Doppler screening and hydroxyurea. We revised EHR forms for SCD care and created an EHR-based SCD registry that permitted monthly and annual reporting on quality metrics.
- Results: From 2008 to 2012, the percentage of children with SCD vaccinated for influenza increased from 52% to 65%, and for meningococcus from 53% to 70%. After licensure of PCV13 in 2010, the percentage of children vaccinated rose to 69% in 2012. Results for PPV23 were mixed: 87% to 91% received ≥1 dose, but the rate for receiving the second dose declined from 76% to 64%. Percentage of children screened annually with transcranial Doppler consistently ranged from 62% to 73% during the 5 years. QI initiatives in 2012–2013 led to increased influenza vaccination, from 65% to 83%, and increased hydroxyurea use, from 52% to 73%.
- Conclusion: In this study, a practical, replicable and feasible approach for improving the quality of SCD care combined the collaboration of a multidisciplinary team, an EHR-based disease registry, and QI initiatives. Additional work is needed to define and measure all elements of high-quality care for children with SCD and link process measures to clinical outcomes.
Sickle cell disease (SCD) is the most commonly inherited disorder in the United States, affecting approximately 100,000 individuals and 1 in 400 African American births [1,2]. The use of preventive strategies, such as immunizations [3], transcranial Doppler screening and transfusion protocols [4,5], and hydroxyurea therapy [6,7] has contributed to decreased morbidity and mortality among children with SCD [8,9]. However, a substantial gap exists between the care that children with SCD should receive and the care they actually receive [10–12]. An essential component of any effort that seeks to improve care is the ability to measure care processes and outcomes in a way that can drive quality improvement (QI) initiatives. Registries serve a vital role in quality improvement activities for many pediatric conditions, including inflammatory bowel disease [13] and cystic fibrosis [14]. However, there are no national or nationally representative registries currently available for children with SCD [15]. There is a pressing need for better information systems and tools that can be used in mainstream clinical settings to measure clinical performance with respect to quality indicators [16] if the goals of high quality care and better quality of life are to be achieved for children with SCD.
Electronic health records (EHRs) have been successfully used to improve the quality of care and enhance performance measurement in select institutions [17,18], and adoption of EHRs is growing. The 2009 American Recovery and Reinvestment Act allocated $20.8 billion in incentives to assist providers to adopt and “meaningfully use” EHRs [19,20]. As of 2011, 39% of office-based providers have implemented at least a basic EHR [21], up from 17% in 2008 [22]. The effective use of EHRs depends on collaboration between technical and medical experts so that functionality is achieved and clinical quality is appropriately measured. In addition, few EHRs contain specialized content for the care of persons with SCD.
While independent registries have been shown to be effective in improving care [13,14,23], they involve extra time and effort for data entry, can be difficult and expensive to maintain, and may not be feasible for many systems that care for SCD patients. In this paper, we describe the development and use of our EHR-based SCD registry for children with SCD, including our efforts to engage key technical and clinical experts to develop an EHR that is tailored to the outpatient workflow and data collection of quality measures and implement a fully functional system that collects data on quality measures to support case management and continuous QI.
Methods
This study was conducted at Boston Medical Center, New England’s largest safety net hospital, which cares for 190 children with SCD ages 0 to 21 years. The outpatient EHR (Centricity, GE) has been in use since 2000 and is used for all aspects of outpatient care, including ordering of immunizations and tests, electronic prescription writing, and referrals to specialty care.
Outcome Measures
Vaccines: The Centers for Disease Control and Prevention (CDC) recommends vaccinating children with SCD [26] against influenza annually, given their susceptibility to the influenza virus [24,27]. The CDC also recommends the 23-valent pneumococcal polysaccharide vaccine (PPV23 2-dose series) and 13-valent pneumococcal conjugate vaccine (PCV13, per childhood routine vaccine schedule for young children and 1 catch-up dose for children previously vaccinated with PCV7), and meningococcal vaccine (2-dose series), given patients’ functional asplenic status [25,28].
Transcranial Doppler screening can identify children with hemoglobin (Hb) SS and Hb S-β0 thalassemia at higher risk of stroke, which may be prevented through hypertransfusion programs [4]. Screening is recommended annually for these children ages 2 to 16 years [25].
Hydroxyurea use among children with Hb SS and Hb S-β0 thalassemia is an established practice [29,30]. We consider hydroxyurea therapy for all children 2 years and older with Hb SS and Hb S-β0 thalassemia, given the recently published safety data from the Baby-HUG trial [7] and the benefits of hydroxyurea among children and adults with SCD [6,31–35].
EHR-based Registry
Our EHR-based SCD registry includes 3 key components: (1) forms to support detailed documentation at the point-of-care (ie, clinic visit); (2) a registry management form to allow the QI team to identify patients to be included or excluded from the registry; and (3) a central data warehouse to support quality measurement and improvement.
Documentation in the EHR is performed using a set of customized templates or “forms.” These forms allow documentation of care provision in a structured way. The discrete data elements are stored within the data warehousing system that supports the EHR. The SCD forms used in this project were a revised version of existing forms used by our pediatric hematologists for the past 6 years. The primary goal was to improve efficiency in a patient encounter and enhance data collection efforts. In particular, several changes were made to enhance data collection for quality measures included in the SCD registry. First, we collected genotype in a standardized way to better define subpopulations of SCD patients, as some of the care provided is dictated by genotype. We also expanded data capture for transcranial Doppler screening to include date of last screening to prompt scheduling. For hydroxyurea, the forms now capture if hydroxyurea has been prescribed, and if not, why (eg, declined, not indicated); adherence, current dose, and routine labs for monitoring are also listed to aid in clinical decision-making. Finally, the forms were revised to prominently display the subset of immunizations important to SCD (described above) to assess if the patient is current.
Within the new forms, we collected all data elements important to providing care to children with SCD. Several new items existed in other parts of the EHR and were automatically pulled into the forms, including laboratory results, medications and immunizations. Other new data elements required manual entry by providers based on EHR review, as they had previously not been documented, documented on an ad hoc basis, or found as free text within notes (eg, number of ED visits and hospitalizations in the past year). Initial completion of these forms took approximately 10 to 15 minutes per patient, as many of these data elements were not individually captured prior to this work; documentation for subsequent comprehensive visits required an additional 5 to 10 minutes per chart. Currently, the 3 pediatric hematologists regularly use the SCD forms for routine visits.
The registry management form was also created by the EHR design team. Although this form is separate from the SCD forms, it was readily accessible to the clinical team to quickly check whether patients should be included or excluded from the SCD registry. In this way, inactive patients could be removed and new patients could be included. This form was completed for all active pediatric patients with SCD as of February 2013 using data from a separately maintained clinical database. For patients who were new to the pediatric hematology practice between July 2012 and February 2013 (eg, infants born during this period, patients transferring care), we manually determined a registry start date in order to calculate accurate denominators for each measure. New patients were entered into the SCD registry by members of the care team on an ad hoc basis, and biannual searches of problem lists were planned to ensure the pediatric SCD registry was complete using the SCD-related ICD-9 codes 282.6, 282.41 and 282.4 to encompass all sickle hemoglobinopathies, including sickle cell thalassemia.
For this project, we were fortunate to have a well-established clinical data warehouse into which the medical center’s EHR data is copied nightly. In addition, the medical center already had multiple chronic disease registries and a framework for evaluating and sharing QI data. We were able to add SCD to this existing infrastructure, which was helpful since a secure and HIPAA-compliant location to post these patient-level reports had been previously identified.
We paid for 40 hours of technical staff time using grant funds to create reports using data collected in the EHR for patients who were actively in the SCD registry per the registry management form. Using these data, summary reports for our key SCD metrics were generated on both an annual and monthly basis. We tested and refined our key SCD metrics over a 4-month period to ensure that we had defined the numerators and denominators for each care process accurately. For example, children become eligible for influenza vaccine at 6 months of age, therefore, the eligible denominator would exclude infants < 6 months of age (Table 1). In addition, lists of patient names and phone numbers were automatically generated to identify those in need of care elements, facilitating both case management and continuous improvement for these measures, replacing the need for all external clinical databases.
Data Analysis
For children included in the SCD registry, we calculated the proportion who were appropriately vaccinated and received transcranial Doppler screening each year for the 5-year period 2008–2012. For the period July 2012–June 2013, we calculated the proportion of children with SCD in the registry who received influenza vaccine and children with Hb SS and Hb S-β0 thalassemia who were prescribed hydroxyurea.
This study was approved by the Boston University Medical Campus institutional review board.
Results
For influenza vaccination for the 2012–2013 season, only 49% of children were vaccinated as of November. This proportion increased after outreach
From July 2012 to June 2013, our rates of hydroxyurea use increased from 52% to 73% among eligible patients.
Discussion
In this paper we report on a practical approach for improving the quality of care for persons with SCD that combines the collaboration of a multidisciplinary team, the use of the EHR to create a disease registry, and QI initiatives. We identified where high-quality care is provided and where further attention is needed, and enhanced our case management capabilities with the generation of patient lists identifying those who are in need of care elements. We also used our registry to track care provision, achieving rates of influenza vaccination of 82% and hydroxyurea use to 73% as of June 2013. From these results, we have shown that our EHR can be used for registry management activities and provide real-time clinical data on the care that is provided, and can lead to improved performance on process measures important in the care for children with SCD.
After adjusting to the revised workflow required by the new SCD forms, the pediatric hematology team found them to be useful in tracking important clinical measures. They reported that the most important change was that all routine elements of SCD care, such as dates of last visits to pediatric subspecialists and receipt of recommended routine SCD care, were embedded into their note. This eliminated the need to search previous documents to find dates of the last cardiology visit or influenza immunizations and increased the likelihood that gaps in care would be addressed by the provider during the course of a clinic visit, thereby streamlining clinic workflow.
Healthy People 2020 recommend vaccination rates of 80% and 90% for influenza and PCV13 vaccines, respectively, in the general pediatric population [36]. We have met this goal for the influenza vaccine, but have room to improve for other recommended vaccines for children with SCD. Ultimately, our goal is to provide these vaccines to 100% of children with SCD at our institution. One barrier to achieving high vaccination rates is the lack of provider knowledge on the creation of catch-up vaccine schedules. A study of primary care providers showed that they frequently omitted vaccines when creating catch-up schedules, including the pneumococcal conjugate vaccine for healthy children [37]. Another hurdle is coordination of care between primary and specialty care, as these vaccines could be given in either setting. A recently published study found that only 20% of children with SCD had care coordination between primary and specialty care [38]. Promoting shared responsibility and information on the administration of vaccinations for children with SCD between primary and subspecialty care, and the development of state-wide immunization registries, may help alleviate these challenges.
In this study, our rates of hydroxyurea use among children with Hb SS and Hb S-β0 thalassemia are higher than in other reported studies [12]. We promote hydroxyurea use in this population of children based on the recently published safety data in infants and young children with Hb SS and Hb S-β0 thalassemia [7,32,39] and the significant benefits seen in adults, including improved survival [6,34,35,40]. Future efforts will include tracking outcomes, including the rates of acute chest syndrome and pain episodes, among children who are and are not taking hydroxyurea.
In this study, we found approximately 70% of eligible children were screened with transcranial Doppler each year from 2008–2012, which is higher than the 45% annual screening rate reported in the literature [10]. One reason our transcranial Doppler screening rates may be higher is that a technician is available to perform these tests on certain days that coincide with the pediatric hematology clinic, allowing patients and families to get this test and have a clinic visit on the same day. However, choosing a 12-month period for receipt of transcranial Doppler screening may be too conservative for centers who do not have such ready access to screening; reporting receipt of transcranial Doppler screening within a 15-month time period may be more appropriate and achievable.
Our study has several limitations. First, it was conducted in a single center with well-established electronic data systems, which are not available in many centers. Our hope is that this model can be replicated by others who seek to use EHR to improve the care of persons with SCD. Second, this work was performed in Massachusetts, a state with near-universal health care insurance coverage. As the Affordable Care Act is implemented nationally [41], other states may see improved performance on quality metrics as more people obtain health insurance. Third, although the EHR was designed to improve data capture for clinical care and quality initiatives, advanced clinical decision support systems were not incorporated due to the limitations of the EHR. The use of prompts for needed clinical care may further enhance performance on these measures. Fourth, this study is limited to children with SCD, who are traditionally monitored more closely than their adult counterparts. Efforts are currently underway to replicate these efforts with adults with SCD at our institution. Finally, the quality metrics in this study are process measures in the delivery of high quality SCD care. Future efforts will focus on linking outcomes to these measures, such as hydroxyurea use to reduce the frequency of acute chest syndrome and painful episodes.
Effective use of health information technology has proven challenging [42,43]. Although there are data that suggest that information technology has improved quality of care by increasing adherence to guidelines, enhancing disease surveillance, and decreasing medication errors, most of the high-quality literature to date comes from 4 research institutions [18]. We found that health IT can be effectively harnessed when end-users are engaged in the process of EHR design, there is a strong commitment to improve workflow and support documentation needs of end-users, the design of the EHR supports data collection for quality measures, and most importantly, there is close collaboration among those with overlapping technical, clinical, and health services research expertise.
There have been many calls for the creation of rare disease registries, as 6% to 8% of the population will develop one in their lifetime [44]. In 2010, the NIH’s Office of Rare Diseases Research funded 30 organizations with and without patient registries, and charged them with the creation of a common data collection template for rare diseases to be used internationally [45]. Common data collection elements for SCD, such as those used in our program, could be used in EHRs across US centers in an effort to improve the quality of care for these children. Although this work may be challenging for centers using large enterprise EHR systems, given the costs associated with modifications, once developed the content can often be shared easily with others using the same system. This would provide the opportunity to compare uniform data across institutions and facilitate learning nationally on ways to improve care. In addition, these efforts may serve as the beginnings of a national registry for pediatric SCD.
In conclusion, contemporary SCD care can lead to improved survival and quality of life, but only if the right care is delivered at the right time. In this study, we present our initial findings from the implementation of a population-based information system for children with SCD. Future efforts are needed to define and measure all elements of high quality care, and link improvements in the delivery of high quality care to outcomes for children and adults with SCD longitudinally.
Appendix. Additional Sickle Cell Disease Forms
Acknowledgments: We would like to thank David Botts for his tireless efforts in creating the sickle cell forms within our EHR. We would also like to thank Barry Zuckerman for his support of this project.
Corresponding author: Patricia Kavanagh, MD, Boston University School of Medicine/Boston Medical Center, 88 E Newton St, Vose Hall 3rd Fl, Boston, MA 02118.
Funding/support: This work was supported by the Health Resources and Services Administration Sickle Cell Disease and Newborn Screening Program, grant #U38MC22215. The authors have also actively participated in the Hemoglobinopathy Learning Collaborative, a quality improvement forum coordinated by HRSA and the National Initiative for Children’s Healthcare Quality.
Financial disclosures: None.
1. Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Preventive Med 2010;38(4 Suppl):S512–S521.
2. Steinberg MH. Management of sickle cell disease. N Engl J Med 1999;340:1021–30.
3. Adamkiewicz TV, Silk BJ, Howgate J, et al. Effectiveness of the 7-valent pneumococcal conjugate vaccine in children with sickle cell disease in the first decade of life. Pediatrics 2008;121:562–9.
4. Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial doppler ultrasonography. N Engl J Med 1998;339:5–1.
5. Adams RJ, Brambilla D, Optimizing Primary Stroke Prevention in Sickle Cell Anemia Trial I. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease.[see comment]. N Engl J Med 2005;353:2769–78.
6. Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. N Engl J Med 1995;332:1317–22.
7. Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: A multicentre, randomised, controlled trial (baby hug). Lancet 2011;377:1663–72.
8. Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood 2010;115:3447–52.
9. Hamideh D, Alvarez O. Sickle cell disease related mortality in the united states (1999–2009). Pediatr Blood Cancer 2013;60:1482–6.
10. Raphael JL, Shetty PB, Liu H, et al. A critical assessment of transcranial doppler screening rates in a large pediatric sickle cell center: Opportunities to improve healthcare quality. Pediatr Blood Cancer 2008;51:647–51.
11. Sox CM, Cooper WO, Koepsell TD, et al. Provision of pneumococcal prophylaxis for publicly insured children with sickle cell disease. JAMA 2003;290:1057–61.
12. Oyeku SO, Driscoll MC, Cohen HW, et al. Parental and other factors associated with hydroxyurea use for pediatric sickle cell disease. Pediatr Blood Cancer 2013;60:653–58.
13. Crandall WV, Margolis PA, Kappelman MD, et al. Improved outcomes in a quality improvement collaborative for pediatric inflammatory bowel disease. Pediatrics 2012;129:e1030–e1041.
14. Schechter MS, Margolis P. Improving subspecialty healthcare: Lessons from cystic fibrosis. J Pediatr 2005;147:295–301.
15. Smith LA, Oyeku SO, Homer C, Zuckerman B. Sickle cell disease: A question of equity and quality. Pediatrics 2006;117:1763–70.
16. Wang CJ, Kavanagh PL, Little AA, et al. Quality-of-care indicators for children with sickle cell disease. Pediatrics 2011;128:484–93.
17. Jha AK, Perlin JB, Kizer KW, Dudley RA. Effect of the transformation of the veterans affairs health care system on the quality of care. N Engl J Med 2003;348:2218–27.
18. Chaudhry B, Wang J, Wu S, et al. Systematic review: Impact of health information technology on quality, efficiency, and costs of medical care. Ann Intern Med 2006;144:742–52.
19. American recovery and reinvestment act of 2009. Obey D, Frank B, Gordon B, et al., trans. 111th Congress of the United States.
20. Blumenthal D, Tavenner M. The “meaningful use” regulation for electronic health records. N Engl J Med 2010;363:501–4.
21. Electronic health record adoption by office-based providers. Office of National Coordinator for Health Information Technology. U.S. Department of Health and Human Services. Accessed 15 Jul 2013.
22. DesRoches CM, Campbell EG, Rao SR, et al. Electronic health records in ambulatory care — a national survey of physicians. N Engl J Med 2008;359:50–60.
23. Tricco AC, Ivers NM, Grimshaw JM, et al. Effectiveness of quality improvement strategies on the management of diabetes: A systematic review and meta-analysis. Lancet 379:2252–61.
24. Bundy DG, Strouse JJ, Casella JF, Miller MR. Burden of influenza-related hospitalizations among children with sickle cell disease. Pediatrics 2010;125:234–43.
25. National Heart Lung and Blood Institute. The management of sickle cell disease. NIH Pub No. 02-2117. Bethesda, MD: National Institutes of Health; 2002.
26. Centers for Disease Control and Prevention. Immunization schedules. Accessed 5 Jan 2013 at www.cdc.gov/vaccines/schedules/index.html.
27. Strouse JJ, Reller ME, Bundy DG, et al. Severe pandemic h1n1 and seasonal influenza in children and young adults with sickle cell disease. Blood 2010;116:3431–4.
28. Pilishvili T, Zell ER, Farley MM, et al. Risk factors for invasive pneumococcal disease in children in the era of conjugate vaccine use. Pediatrics 2010;126:e9–17.
29. Heeney MM, Ware RE. Hydroxyurea for children with sickle cell disease. Pediatr Clin North Am 008;55:483–501.
30. Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood 2010;115:5300–11.
31. Ferster A, Vermylen C, Cornu G, et al. Hydroxyurea for treatment of severe sickle cell anemia: a pediatric clinical trial. Blood 1996;88:1960–4.
32. Strouse JJ, Lanzkron S, Beach MC, et al. Hydroxyurea for sickle cell disease: a systematic review for efficacy and toxicity in children. Pediatrics 2008;122:1332–42.
33. Hankins JS, Ware RE, Rogers ZR, et al. Long-term hydroxyurea therapy for infants with sickle cell anemia: the husoft extension study. Blood 2005;106:2269–75.
34. Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: a 17.5-year follow-up. Am J Hematol 2010;85:403–8.
35. Voskaridou E, Christoulas D, Bilalis A, et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (lashs). Blood 2010;115:2354–63.
36. Healthy people 2020. Immunization and infectious diseases. Accessed 3 Jun 2013 at www.healthypeople.gov/2020/topicsobjectives2020/objectiveslist.aspx?topicid=23.
37. Cohen NJ, Lauderdale DS, Shete PB, et al. Physician knowledge of catch-up regimens and contraindications for childhood immunizations. Pediatrics 2003;111:925–32.
38. Raphael JL, Rattler TL, Kowalkowski MA, et al. The medical home experience among children with sickle cell disease. Pediatr Blood Cancer 2013;60:275–80.
39. Strouse JJ, Heeney MM. Hydroxyurea for the treatment of sickle cell disease: efficacy, barriers, toxicity, and management in children. Pediatr Blood Cancer 2012;59:365–71.
40. Steinberg MH, Barton F, Castro O, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA 2003;289:1645–51.
41. Patient protection and affordable care act, US Pub. L. No. 111-148, §2702, 124 stat. 119, 318-319. 2010.
42. Harrison M, Koppel R, Bar-Lev S. Unintended consequences of information technologies in health care: an interactive sociotechnical analysis. J Am Med Inform Assoc 2007;14:542–9.
43. Haux R. Health information systems – past, present, future. Int J Med Informatics 2006;75:268–81.
44. Schieppati A, Henter J-I, Daina E, Aperia A. Why rare diseases are an important medical and social issue. Lancet 2008;371:2039–41.
45. Office of Rare Diseases Research National Institutes of Health. Rare diseases and related terms. Accessed 28 Jun 2013 at www.rarediseases.info.nih.gov/rarediseaselist.aspx.
1. Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Preventive Med 2010;38(4 Suppl):S512–S521.
2. Steinberg MH. Management of sickle cell disease. N Engl J Med 1999;340:1021–30.
3. Adamkiewicz TV, Silk BJ, Howgate J, et al. Effectiveness of the 7-valent pneumococcal conjugate vaccine in children with sickle cell disease in the first decade of life. Pediatrics 2008;121:562–9.
4. Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial doppler ultrasonography. N Engl J Med 1998;339:5–1.
5. Adams RJ, Brambilla D, Optimizing Primary Stroke Prevention in Sickle Cell Anemia Trial I. Discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease.[see comment]. N Engl J Med 2005;353:2769–78.
6. Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. N Engl J Med 1995;332:1317–22.
7. Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: A multicentre, randomised, controlled trial (baby hug). Lancet 2011;377:1663–72.
8. Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood 2010;115:3447–52.
9. Hamideh D, Alvarez O. Sickle cell disease related mortality in the united states (1999–2009). Pediatr Blood Cancer 2013;60:1482–6.
10. Raphael JL, Shetty PB, Liu H, et al. A critical assessment of transcranial doppler screening rates in a large pediatric sickle cell center: Opportunities to improve healthcare quality. Pediatr Blood Cancer 2008;51:647–51.
11. Sox CM, Cooper WO, Koepsell TD, et al. Provision of pneumococcal prophylaxis for publicly insured children with sickle cell disease. JAMA 2003;290:1057–61.
12. Oyeku SO, Driscoll MC, Cohen HW, et al. Parental and other factors associated with hydroxyurea use for pediatric sickle cell disease. Pediatr Blood Cancer 2013;60:653–58.
13. Crandall WV, Margolis PA, Kappelman MD, et al. Improved outcomes in a quality improvement collaborative for pediatric inflammatory bowel disease. Pediatrics 2012;129:e1030–e1041.
14. Schechter MS, Margolis P. Improving subspecialty healthcare: Lessons from cystic fibrosis. J Pediatr 2005;147:295–301.
15. Smith LA, Oyeku SO, Homer C, Zuckerman B. Sickle cell disease: A question of equity and quality. Pediatrics 2006;117:1763–70.
16. Wang CJ, Kavanagh PL, Little AA, et al. Quality-of-care indicators for children with sickle cell disease. Pediatrics 2011;128:484–93.
17. Jha AK, Perlin JB, Kizer KW, Dudley RA. Effect of the transformation of the veterans affairs health care system on the quality of care. N Engl J Med 2003;348:2218–27.
18. Chaudhry B, Wang J, Wu S, et al. Systematic review: Impact of health information technology on quality, efficiency, and costs of medical care. Ann Intern Med 2006;144:742–52.
19. American recovery and reinvestment act of 2009. Obey D, Frank B, Gordon B, et al., trans. 111th Congress of the United States.
20. Blumenthal D, Tavenner M. The “meaningful use” regulation for electronic health records. N Engl J Med 2010;363:501–4.
21. Electronic health record adoption by office-based providers. Office of National Coordinator for Health Information Technology. U.S. Department of Health and Human Services. Accessed 15 Jul 2013.
22. DesRoches CM, Campbell EG, Rao SR, et al. Electronic health records in ambulatory care — a national survey of physicians. N Engl J Med 2008;359:50–60.
23. Tricco AC, Ivers NM, Grimshaw JM, et al. Effectiveness of quality improvement strategies on the management of diabetes: A systematic review and meta-analysis. Lancet 379:2252–61.
24. Bundy DG, Strouse JJ, Casella JF, Miller MR. Burden of influenza-related hospitalizations among children with sickle cell disease. Pediatrics 2010;125:234–43.
25. National Heart Lung and Blood Institute. The management of sickle cell disease. NIH Pub No. 02-2117. Bethesda, MD: National Institutes of Health; 2002.
26. Centers for Disease Control and Prevention. Immunization schedules. Accessed 5 Jan 2013 at www.cdc.gov/vaccines/schedules/index.html.
27. Strouse JJ, Reller ME, Bundy DG, et al. Severe pandemic h1n1 and seasonal influenza in children and young adults with sickle cell disease. Blood 2010;116:3431–4.
28. Pilishvili T, Zell ER, Farley MM, et al. Risk factors for invasive pneumococcal disease in children in the era of conjugate vaccine use. Pediatrics 2010;126:e9–17.
29. Heeney MM, Ware RE. Hydroxyurea for children with sickle cell disease. Pediatr Clin North Am 008;55:483–501.
30. Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood 2010;115:5300–11.
31. Ferster A, Vermylen C, Cornu G, et al. Hydroxyurea for treatment of severe sickle cell anemia: a pediatric clinical trial. Blood 1996;88:1960–4.
32. Strouse JJ, Lanzkron S, Beach MC, et al. Hydroxyurea for sickle cell disease: a systematic review for efficacy and toxicity in children. Pediatrics 2008;122:1332–42.
33. Hankins JS, Ware RE, Rogers ZR, et al. Long-term hydroxyurea therapy for infants with sickle cell anemia: the husoft extension study. Blood 2005;106:2269–75.
34. Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: a 17.5-year follow-up. Am J Hematol 2010;85:403–8.
35. Voskaridou E, Christoulas D, Bilalis A, et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (lashs). Blood 2010;115:2354–63.
36. Healthy people 2020. Immunization and infectious diseases. Accessed 3 Jun 2013 at www.healthypeople.gov/2020/topicsobjectives2020/objectiveslist.aspx?topicid=23.
37. Cohen NJ, Lauderdale DS, Shete PB, et al. Physician knowledge of catch-up regimens and contraindications for childhood immunizations. Pediatrics 2003;111:925–32.
38. Raphael JL, Rattler TL, Kowalkowski MA, et al. The medical home experience among children with sickle cell disease. Pediatr Blood Cancer 2013;60:275–80.
39. Strouse JJ, Heeney MM. Hydroxyurea for the treatment of sickle cell disease: efficacy, barriers, toxicity, and management in children. Pediatr Blood Cancer 2012;59:365–71.
40. Steinberg MH, Barton F, Castro O, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA 2003;289:1645–51.
41. Patient protection and affordable care act, US Pub. L. No. 111-148, §2702, 124 stat. 119, 318-319. 2010.
42. Harrison M, Koppel R, Bar-Lev S. Unintended consequences of information technologies in health care: an interactive sociotechnical analysis. J Am Med Inform Assoc 2007;14:542–9.
43. Haux R. Health information systems – past, present, future. Int J Med Informatics 2006;75:268–81.
44. Schieppati A, Henter J-I, Daina E, Aperia A. Why rare diseases are an important medical and social issue. Lancet 2008;371:2039–41.
45. Office of Rare Diseases Research National Institutes of Health. Rare diseases and related terms. Accessed 28 Jun 2013 at www.rarediseases.info.nih.gov/rarediseaselist.aspx.