User login
Frequent soaks ease pediatric atopic dermatitis
A regimen of twice-daily baths followed by occlusive moisturizer improved atopic dermatitis in children with moderate to severe disease more effectively than did a twice-weekly protocol, based on data from 42 children.
Guidelines for bathing frequency for children with atopic dermatitis are inconsistent and often confusing for parents, according to Ivan D. Cardona, MD, of Maine Medical Research Institute, Portland, and colleagues.
In a study published in the Journal of Allergy and Clinical Immunology: In Practice, the researchers randomized 42 children aged 6 months to 11 years with moderate to severe atopic dermatitis to a routine of twice-weekly “soak and seal” (SS) procedures consisting of soaking baths for 10 minutes or less, followed by an occlusive emollient, or to twice-daily SS with baths of 15-20 minutes followed by emollient. The groups were treated for 2 weeks, then switched protocols. The study included a total of four clinic visits over 5 weeks. All patients also received standard of care low-potency topical corticosteroids and moisturizer.
Overall, the frequent bathing (“wet method”) led to a decrease of 21.2 on the SCORing Atopic Dermatitis Index (SCORAD) compared with the less frequent bathing (“dry method”). Improvements in SCORAD (the primary outcome) correlated with a secondary outcome of improved scores on the parent-rated Atopic Dermatitis Quickscore.
The findings were limited by several factors including the small sample size, large rate of attrition prior to randomization among initially screened children, lack of data on environmental factors such as water temperature and quality, and the lack of a washout period between the treatment protocols, the researchers noted. They acknowledged that “twice-daily SS bathing in the real world can be time consuming, making adherence difficult for families.”
However, the results suggest that the frequent bathing protocol was safe and effective at improving symptoms of atopic dermatitis, and may reduce steroid use, they concluded.
The researchers had no financial conflicts to disclose.
SOURCE: Cardona ID et al. J Allergy Clin Immunol Pract. 2019 Nov 13. doi: 10.1016/j.jaip.2019.10.042.
A regimen of twice-daily baths followed by occlusive moisturizer improved atopic dermatitis in children with moderate to severe disease more effectively than did a twice-weekly protocol, based on data from 42 children.
Guidelines for bathing frequency for children with atopic dermatitis are inconsistent and often confusing for parents, according to Ivan D. Cardona, MD, of Maine Medical Research Institute, Portland, and colleagues.
In a study published in the Journal of Allergy and Clinical Immunology: In Practice, the researchers randomized 42 children aged 6 months to 11 years with moderate to severe atopic dermatitis to a routine of twice-weekly “soak and seal” (SS) procedures consisting of soaking baths for 10 minutes or less, followed by an occlusive emollient, or to twice-daily SS with baths of 15-20 minutes followed by emollient. The groups were treated for 2 weeks, then switched protocols. The study included a total of four clinic visits over 5 weeks. All patients also received standard of care low-potency topical corticosteroids and moisturizer.
Overall, the frequent bathing (“wet method”) led to a decrease of 21.2 on the SCORing Atopic Dermatitis Index (SCORAD) compared with the less frequent bathing (“dry method”). Improvements in SCORAD (the primary outcome) correlated with a secondary outcome of improved scores on the parent-rated Atopic Dermatitis Quickscore.
The findings were limited by several factors including the small sample size, large rate of attrition prior to randomization among initially screened children, lack of data on environmental factors such as water temperature and quality, and the lack of a washout period between the treatment protocols, the researchers noted. They acknowledged that “twice-daily SS bathing in the real world can be time consuming, making adherence difficult for families.”
However, the results suggest that the frequent bathing protocol was safe and effective at improving symptoms of atopic dermatitis, and may reduce steroid use, they concluded.
The researchers had no financial conflicts to disclose.
SOURCE: Cardona ID et al. J Allergy Clin Immunol Pract. 2019 Nov 13. doi: 10.1016/j.jaip.2019.10.042.
A regimen of twice-daily baths followed by occlusive moisturizer improved atopic dermatitis in children with moderate to severe disease more effectively than did a twice-weekly protocol, based on data from 42 children.
Guidelines for bathing frequency for children with atopic dermatitis are inconsistent and often confusing for parents, according to Ivan D. Cardona, MD, of Maine Medical Research Institute, Portland, and colleagues.
In a study published in the Journal of Allergy and Clinical Immunology: In Practice, the researchers randomized 42 children aged 6 months to 11 years with moderate to severe atopic dermatitis to a routine of twice-weekly “soak and seal” (SS) procedures consisting of soaking baths for 10 minutes or less, followed by an occlusive emollient, or to twice-daily SS with baths of 15-20 minutes followed by emollient. The groups were treated for 2 weeks, then switched protocols. The study included a total of four clinic visits over 5 weeks. All patients also received standard of care low-potency topical corticosteroids and moisturizer.
Overall, the frequent bathing (“wet method”) led to a decrease of 21.2 on the SCORing Atopic Dermatitis Index (SCORAD) compared with the less frequent bathing (“dry method”). Improvements in SCORAD (the primary outcome) correlated with a secondary outcome of improved scores on the parent-rated Atopic Dermatitis Quickscore.
The findings were limited by several factors including the small sample size, large rate of attrition prior to randomization among initially screened children, lack of data on environmental factors such as water temperature and quality, and the lack of a washout period between the treatment protocols, the researchers noted. They acknowledged that “twice-daily SS bathing in the real world can be time consuming, making adherence difficult for families.”
However, the results suggest that the frequent bathing protocol was safe and effective at improving symptoms of atopic dermatitis, and may reduce steroid use, they concluded.
The researchers had no financial conflicts to disclose.
SOURCE: Cardona ID et al. J Allergy Clin Immunol Pract. 2019 Nov 13. doi: 10.1016/j.jaip.2019.10.042.
FROM THE JOURNAL OF ALLERGY AND CLINICAL IMMUNOLOGY: IN PRACTICE
SZC passes extension test for hyperkalemia
Treatment with sodium zirconium cyclosilicate (SZC) led to lasting improvement of hyperkalemia, according to results from an 11-month open-label extension study of the HARMONIZE randomized clinical trial.
SZC selectively binds potassium ions in the colon, reducing absorption and promoting excretion. In the original study, 248 patients with mild hyperkalemia were randomized to SZC or placebo. Within 48 hours, the drug returned potassium to normal and maintained those levels out to 4 weeks.
In the extension study, 123 patients with measured potassium levels of 3.5-6.2 mmol/L, 48 of whom had previously been assigned to placebo, received a 5- to 10-g dose of SZC once per day for up to 337 days. Median daily dose was 10 g, with a dose range of 2.5-15 g (Am J Nephrol. 2019;50[6]:473-480).
Just under 65% of patients completed the 11 months of the open-label extension study, with 88.3% of those achieving the primary endpoint of mean serum potassium levels of 5.1 mmol/L or lower, according to Simon D. Roger, MD, a nephrologist based in Gosford, Australia, and colleagues.
Most patients (83) were taking renin–angiotensin–aldosterone system inhibitors at baseline of the extension study; 78.3% continued a stable dose throughout the open-label phase, 8.4% increased the dose, and 3.6% discontinued.
Two-thirds of patients reported adverse events, most commonly gastrointestinal disorders (18.7%). Constipation was the most frequent (5.7%), followed by nausea, vomiting, and diarrhea (3.3% each).
Adverse events that occurred in 5% or more of participants included hypertension (12.2%), urinary tract infection (8.9%), and peripheral edema (8.1%). Hypertension severity was either mild (46.7%) or moderate (53.3%), and only one case was believed to be associated with the study medication. Thirteen percent of participants reported a total of 17 SMQ edema events. Eleven of the 16 patients had baseline risk factors for edema, leading the authors to conclude that causality between SZC and edema could not be established.
Serious adverse events occurred in 19.5% of participants, and 4.9% of participants discontinued SZC as a result.
SZC is approved for the treatment of hyperkalemia in the United States and Europe. The study was funded by AstraZeneca.
SOURCE: Roger S et al. Am J Nephrol;2019:50(6):473-80.
Treatment with sodium zirconium cyclosilicate (SZC) led to lasting improvement of hyperkalemia, according to results from an 11-month open-label extension study of the HARMONIZE randomized clinical trial.
SZC selectively binds potassium ions in the colon, reducing absorption and promoting excretion. In the original study, 248 patients with mild hyperkalemia were randomized to SZC or placebo. Within 48 hours, the drug returned potassium to normal and maintained those levels out to 4 weeks.
In the extension study, 123 patients with measured potassium levels of 3.5-6.2 mmol/L, 48 of whom had previously been assigned to placebo, received a 5- to 10-g dose of SZC once per day for up to 337 days. Median daily dose was 10 g, with a dose range of 2.5-15 g (Am J Nephrol. 2019;50[6]:473-480).
Just under 65% of patients completed the 11 months of the open-label extension study, with 88.3% of those achieving the primary endpoint of mean serum potassium levels of 5.1 mmol/L or lower, according to Simon D. Roger, MD, a nephrologist based in Gosford, Australia, and colleagues.
Most patients (83) were taking renin–angiotensin–aldosterone system inhibitors at baseline of the extension study; 78.3% continued a stable dose throughout the open-label phase, 8.4% increased the dose, and 3.6% discontinued.
Two-thirds of patients reported adverse events, most commonly gastrointestinal disorders (18.7%). Constipation was the most frequent (5.7%), followed by nausea, vomiting, and diarrhea (3.3% each).
Adverse events that occurred in 5% or more of participants included hypertension (12.2%), urinary tract infection (8.9%), and peripheral edema (8.1%). Hypertension severity was either mild (46.7%) or moderate (53.3%), and only one case was believed to be associated with the study medication. Thirteen percent of participants reported a total of 17 SMQ edema events. Eleven of the 16 patients had baseline risk factors for edema, leading the authors to conclude that causality between SZC and edema could not be established.
Serious adverse events occurred in 19.5% of participants, and 4.9% of participants discontinued SZC as a result.
SZC is approved for the treatment of hyperkalemia in the United States and Europe. The study was funded by AstraZeneca.
SOURCE: Roger S et al. Am J Nephrol;2019:50(6):473-80.
Treatment with sodium zirconium cyclosilicate (SZC) led to lasting improvement of hyperkalemia, according to results from an 11-month open-label extension study of the HARMONIZE randomized clinical trial.
SZC selectively binds potassium ions in the colon, reducing absorption and promoting excretion. In the original study, 248 patients with mild hyperkalemia were randomized to SZC or placebo. Within 48 hours, the drug returned potassium to normal and maintained those levels out to 4 weeks.
In the extension study, 123 patients with measured potassium levels of 3.5-6.2 mmol/L, 48 of whom had previously been assigned to placebo, received a 5- to 10-g dose of SZC once per day for up to 337 days. Median daily dose was 10 g, with a dose range of 2.5-15 g (Am J Nephrol. 2019;50[6]:473-480).
Just under 65% of patients completed the 11 months of the open-label extension study, with 88.3% of those achieving the primary endpoint of mean serum potassium levels of 5.1 mmol/L or lower, according to Simon D. Roger, MD, a nephrologist based in Gosford, Australia, and colleagues.
Most patients (83) were taking renin–angiotensin–aldosterone system inhibitors at baseline of the extension study; 78.3% continued a stable dose throughout the open-label phase, 8.4% increased the dose, and 3.6% discontinued.
Two-thirds of patients reported adverse events, most commonly gastrointestinal disorders (18.7%). Constipation was the most frequent (5.7%), followed by nausea, vomiting, and diarrhea (3.3% each).
Adverse events that occurred in 5% or more of participants included hypertension (12.2%), urinary tract infection (8.9%), and peripheral edema (8.1%). Hypertension severity was either mild (46.7%) or moderate (53.3%), and only one case was believed to be associated with the study medication. Thirteen percent of participants reported a total of 17 SMQ edema events. Eleven of the 16 patients had baseline risk factors for edema, leading the authors to conclude that causality between SZC and edema could not be established.
Serious adverse events occurred in 19.5% of participants, and 4.9% of participants discontinued SZC as a result.
SZC is approved for the treatment of hyperkalemia in the United States and Europe. The study was funded by AstraZeneca.
SOURCE: Roger S et al. Am J Nephrol;2019:50(6):473-80.
FROM AMERICAN JOURNAL OF NEPHROLOGY
Supreme Court weighs ACA back pay case
U.S. Supreme Court Justices heard oral arguments Dec. 10 in a case that centers on whether the federal government owes insurers billions based on an Affordable Care Act provision intended to help health plans mitigate risk under the law.

Maine Community Health Options v. United States, which consolidates several lawsuits against the government, involves the ACA’s risk corridor program, which required the U.S. Department of Health & Human Services to collect funds from profitable insurers that offered qualified health plans under the exchanges and distribute the funds to insurers with excessive losses.
Collections from profitable insurers under the program fell short in 2014, 2015, and 2016, while losses steadily grew, resulting in the HHS paying about 12 cents on the dollar in payments to insurers. More than 150 insurers now allege they were shortchanged and they want the Supreme Court to force the government to reimburse them $12 billion.
The U.S. Department of Justice counters that the government is not required to pay the plans because of appropriations measures passed by Congress in 2014 and later years that limited the funding available to compensate insurers for their losses. The federal government is not obligated to pay the insurers back for the losses and the suits should be dismissed, the DOJ attorneys argue.
The federal government and insurers have each experienced wins and losses at the lower court level. Most recently, the U.S. Court of Appeals for the Federal Circuit decided in favor of the government, ruling that while the ACA required the government to compensate the insurers for their losses, the appropriations measures repealed or suspended that requirement.
Insurers, economists, and state government led by both parties have weighed in on the case via court briefs.
The case could have important ramifications that have an impact on more than just insurers, said Timothy Jost, a health law expert and retired professor from Washington and Lee University in Lexington, Va.
“The insurers are left holding the bag, but a number of insurers went insolvent [due to the losses], so in those cases, the states are left holding the bag,” Mr. Jost said in an interview.
The outcome could influence future partnerships between the federal government and private entities, according to Katie Keith, an attorney and health law analyst who writes for the Health Affairs Blog. She noted that the U.S. Chamber of Commerce, which historically has not supported the ACA, sided with insurers in the case. In a court brief, attorneys for the chamber wrote that businesses make substantial financial investments to participate in federal programs, and their willingness to do so is based on having assurance that the government will honor its statutory commitments. If left uncorrected, the circuit’s decision “will have far-reaching consequences for myriad areas in which U.S. businesses partner with the federal government to provide vital goods and services,” chamber attorneys wrote.
“This is about more than the ACA. It’s about the fundamentals of public/private partnerships,” Ms. Keith said in an interview. “If you’re contracting with the government, can the government just break those promises or break those obligations after the fact? Folks at the chamber are making it about bigger, broader issues about what it means when the government partners with private organizations.”
Mr. Jost said it’s hard to say which way the Supreme Court will rule, but the fact that the court is reviewing the case speaks volumes.
“Its kind of hard to believe they’re going to order the federal government to pay $12 billion,” he said. “On the other hand, they didn’t have to take the case.”
A Supreme Court decision is expected in early 2020.
U.S. Supreme Court Justices heard oral arguments Dec. 10 in a case that centers on whether the federal government owes insurers billions based on an Affordable Care Act provision intended to help health plans mitigate risk under the law.
Maine Community Health Options v. United States, which consolidates several lawsuits against the government, involves the ACA’s risk corridor program, which required the U.S. Department of Health & Human Services to collect funds from profitable insurers that offered qualified health plans under the exchanges and distribute the funds to insurers with excessive losses.
Collections from profitable insurers under the program fell short in 2014, 2015, and 2016, while losses steadily grew, resulting in the HHS paying about 12 cents on the dollar in payments to insurers. More than 150 insurers now allege they were shortchanged and they want the Supreme Court to force the government to reimburse them $12 billion.
The U.S. Department of Justice counters that the government is not required to pay the plans because of appropriations measures passed by Congress in 2014 and later years that limited the funding available to compensate insurers for their losses. The federal government is not obligated to pay the insurers back for the losses and the suits should be dismissed, the DOJ attorneys argue.
The federal government and insurers have each experienced wins and losses at the lower court level. Most recently, the U.S. Court of Appeals for the Federal Circuit decided in favor of the government, ruling that while the ACA required the government to compensate the insurers for their losses, the appropriations measures repealed or suspended that requirement.
Insurers, economists, and state government led by both parties have weighed in on the case via court briefs.
The case could have important ramifications that have an impact on more than just insurers, said Timothy Jost, a health law expert and retired professor from Washington and Lee University in Lexington, Va.
“The insurers are left holding the bag, but a number of insurers went insolvent [due to the losses], so in those cases, the states are left holding the bag,” Mr. Jost said in an interview.
The outcome could influence future partnerships between the federal government and private entities, according to Katie Keith, an attorney and health law analyst who writes for the Health Affairs Blog. She noted that the U.S. Chamber of Commerce, which historically has not supported the ACA, sided with insurers in the case. In a court brief, attorneys for the chamber wrote that businesses make substantial financial investments to participate in federal programs, and their willingness to do so is based on having assurance that the government will honor its statutory commitments. If left uncorrected, the circuit’s decision “will have far-reaching consequences for myriad areas in which U.S. businesses partner with the federal government to provide vital goods and services,” chamber attorneys wrote.
“This is about more than the ACA. It’s about the fundamentals of public/private partnerships,” Ms. Keith said in an interview. “If you’re contracting with the government, can the government just break those promises or break those obligations after the fact? Folks at the chamber are making it about bigger, broader issues about what it means when the government partners with private organizations.”
Mr. Jost said it’s hard to say which way the Supreme Court will rule, but the fact that the court is reviewing the case speaks volumes.
“Its kind of hard to believe they’re going to order the federal government to pay $12 billion,” he said. “On the other hand, they didn’t have to take the case.”
A Supreme Court decision is expected in early 2020.
U.S. Supreme Court Justices heard oral arguments Dec. 10 in a case that centers on whether the federal government owes insurers billions based on an Affordable Care Act provision intended to help health plans mitigate risk under the law.
Maine Community Health Options v. United States, which consolidates several lawsuits against the government, involves the ACA’s risk corridor program, which required the U.S. Department of Health & Human Services to collect funds from profitable insurers that offered qualified health plans under the exchanges and distribute the funds to insurers with excessive losses.
Collections from profitable insurers under the program fell short in 2014, 2015, and 2016, while losses steadily grew, resulting in the HHS paying about 12 cents on the dollar in payments to insurers. More than 150 insurers now allege they were shortchanged and they want the Supreme Court to force the government to reimburse them $12 billion.
The U.S. Department of Justice counters that the government is not required to pay the plans because of appropriations measures passed by Congress in 2014 and later years that limited the funding available to compensate insurers for their losses. The federal government is not obligated to pay the insurers back for the losses and the suits should be dismissed, the DOJ attorneys argue.
The federal government and insurers have each experienced wins and losses at the lower court level. Most recently, the U.S. Court of Appeals for the Federal Circuit decided in favor of the government, ruling that while the ACA required the government to compensate the insurers for their losses, the appropriations measures repealed or suspended that requirement.
Insurers, economists, and state government led by both parties have weighed in on the case via court briefs.
The case could have important ramifications that have an impact on more than just insurers, said Timothy Jost, a health law expert and retired professor from Washington and Lee University in Lexington, Va.
“The insurers are left holding the bag, but a number of insurers went insolvent [due to the losses], so in those cases, the states are left holding the bag,” Mr. Jost said in an interview.
The outcome could influence future partnerships between the federal government and private entities, according to Katie Keith, an attorney and health law analyst who writes for the Health Affairs Blog. She noted that the U.S. Chamber of Commerce, which historically has not supported the ACA, sided with insurers in the case. In a court brief, attorneys for the chamber wrote that businesses make substantial financial investments to participate in federal programs, and their willingness to do so is based on having assurance that the government will honor its statutory commitments. If left uncorrected, the circuit’s decision “will have far-reaching consequences for myriad areas in which U.S. businesses partner with the federal government to provide vital goods and services,” chamber attorneys wrote.
“This is about more than the ACA. It’s about the fundamentals of public/private partnerships,” Ms. Keith said in an interview. “If you’re contracting with the government, can the government just break those promises or break those obligations after the fact? Folks at the chamber are making it about bigger, broader issues about what it means when the government partners with private organizations.”
Mr. Jost said it’s hard to say which way the Supreme Court will rule, but the fact that the court is reviewing the case speaks volumes.
“Its kind of hard to believe they’re going to order the federal government to pay $12 billion,” he said. “On the other hand, they didn’t have to take the case.”
A Supreme Court decision is expected in early 2020.
FDA clears first fully disposable duodenoscope
The Food and Drug Administration on Dec. 13 cleared Boston Scientific’s single-use duodenoscope, the Exalt Model D, for endoscopic retrograde cholangiopancreatography.
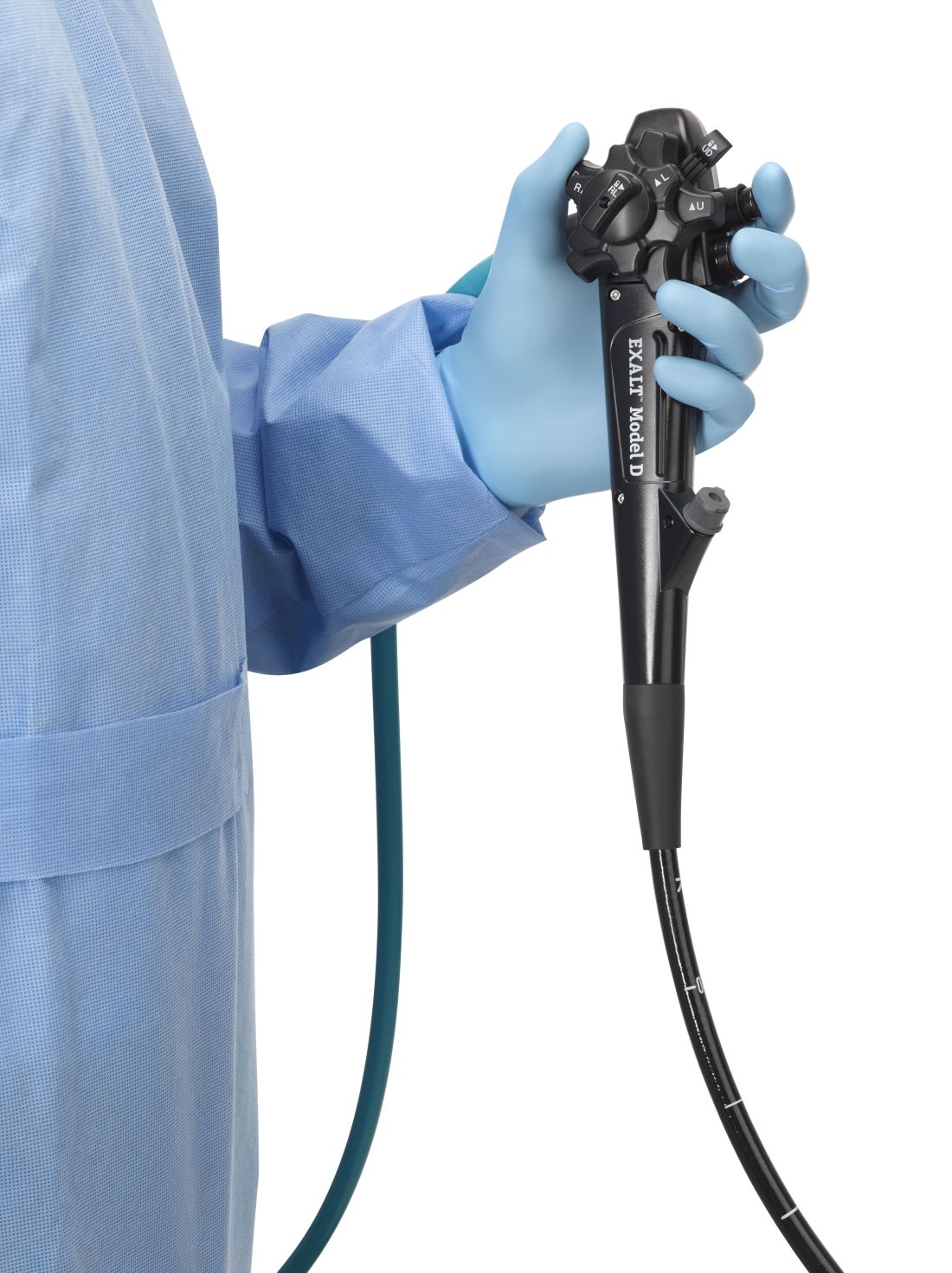
It’s the first disposable duodenoscope to hit the market in the wake of the agency’s August call for manufacturers and health care facilities to move to partially or fully disposable duodenoscopes. The goal is to eliminate the risk of spreading infections between patients from incomplete sterilization of traditional, multi-use scopes. The FDA also recently approved a Pentax duodenoscope with a disposable elevator, the most difficult part to clean.
The agency reported in April that 5.4% of samples from multi-use scopes test postive for Escherichia coli, Pseudomonas aeruginosa, or other “high-concern” organisms.
Boston Scientific spokesperson Kate Haranis said the Exalt Model D will be available in the first quarter of 2020, but the company is still working out how much it will charge.
Cost effectiveness will depend largely on the degree to which the price of the device is offset by the infections it prevents. It might prove particularly attractive to high-volume centers with higher than usual infection rates. It might also be of interest to smaller practices where the price of a multi-use scope doesn’t make sense for only a few procedures a year, said Gyanprakash Ketwaroo, MD, an interventional endoscopist and assistant professor of gastroenterology at Baylor University, Houston.
“The feel is a little different,” said Dr. Ketwaroo, who’s tried the new device, but “it’s pretty functional and probably okay to use in almost all endoscopic procedures that require ERCP.”
In a study funded by Boston Scientific, endoscopists reported a median overall satisfaction score of 9 out of 10 with the new scope (Clin Gastroenterol Hepatol. 2019 Nov 6. doi: 10.1016/j.cgh.2019.10.052).
As for using it at Baylor, Dr. Ketwaroo said, “we’re not sure yet; we are still evaluating it” and want to see if any problems emerge once it’s on the market. It’s also not clear if infection risks would be lower than with the disposable elevator model from Pentax, he added.
The Exalt Model D was granted breakthrough status by the FDA, and the agency worked closely with Boston Scientific to bring it to market.
“The availability of a fully disposable duodenoscope represents another major step forward for improving the safety of these devices, which are used in more than 500,000 procedures in the United States each year. The FDA continues to encourage innovative ways to improve the safety and effectiveness of these devices,” Jeff Shuren, MD, director of the FDA’s Center for Devices and Radiological Health, said in a statement.
Dr. Ketwaroo had no relevant financial disclosures.
The Food and Drug Administration on Dec. 13 cleared Boston Scientific’s single-use duodenoscope, the Exalt Model D, for endoscopic retrograde cholangiopancreatography.
It’s the first disposable duodenoscope to hit the market in the wake of the agency’s August call for manufacturers and health care facilities to move to partially or fully disposable duodenoscopes. The goal is to eliminate the risk of spreading infections between patients from incomplete sterilization of traditional, multi-use scopes. The FDA also recently approved a Pentax duodenoscope with a disposable elevator, the most difficult part to clean.
The agency reported in April that 5.4% of samples from multi-use scopes test postive for Escherichia coli, Pseudomonas aeruginosa, or other “high-concern” organisms.
Boston Scientific spokesperson Kate Haranis said the Exalt Model D will be available in the first quarter of 2020, but the company is still working out how much it will charge.
Cost effectiveness will depend largely on the degree to which the price of the device is offset by the infections it prevents. It might prove particularly attractive to high-volume centers with higher than usual infection rates. It might also be of interest to smaller practices where the price of a multi-use scope doesn’t make sense for only a few procedures a year, said Gyanprakash Ketwaroo, MD, an interventional endoscopist and assistant professor of gastroenterology at Baylor University, Houston.
“The feel is a little different,” said Dr. Ketwaroo, who’s tried the new device, but “it’s pretty functional and probably okay to use in almost all endoscopic procedures that require ERCP.”
In a study funded by Boston Scientific, endoscopists reported a median overall satisfaction score of 9 out of 10 with the new scope (Clin Gastroenterol Hepatol. 2019 Nov 6. doi: 10.1016/j.cgh.2019.10.052).
As for using it at Baylor, Dr. Ketwaroo said, “we’re not sure yet; we are still evaluating it” and want to see if any problems emerge once it’s on the market. It’s also not clear if infection risks would be lower than with the disposable elevator model from Pentax, he added.
The Exalt Model D was granted breakthrough status by the FDA, and the agency worked closely with Boston Scientific to bring it to market.
“The availability of a fully disposable duodenoscope represents another major step forward for improving the safety of these devices, which are used in more than 500,000 procedures in the United States each year. The FDA continues to encourage innovative ways to improve the safety and effectiveness of these devices,” Jeff Shuren, MD, director of the FDA’s Center for Devices and Radiological Health, said in a statement.
Dr. Ketwaroo had no relevant financial disclosures.
The Food and Drug Administration on Dec. 13 cleared Boston Scientific’s single-use duodenoscope, the Exalt Model D, for endoscopic retrograde cholangiopancreatography.
It’s the first disposable duodenoscope to hit the market in the wake of the agency’s August call for manufacturers and health care facilities to move to partially or fully disposable duodenoscopes. The goal is to eliminate the risk of spreading infections between patients from incomplete sterilization of traditional, multi-use scopes. The FDA also recently approved a Pentax duodenoscope with a disposable elevator, the most difficult part to clean.
The agency reported in April that 5.4% of samples from multi-use scopes test postive for Escherichia coli, Pseudomonas aeruginosa, or other “high-concern” organisms.
Boston Scientific spokesperson Kate Haranis said the Exalt Model D will be available in the first quarter of 2020, but the company is still working out how much it will charge.
Cost effectiveness will depend largely on the degree to which the price of the device is offset by the infections it prevents. It might prove particularly attractive to high-volume centers with higher than usual infection rates. It might also be of interest to smaller practices where the price of a multi-use scope doesn’t make sense for only a few procedures a year, said Gyanprakash Ketwaroo, MD, an interventional endoscopist and assistant professor of gastroenterology at Baylor University, Houston.
“The feel is a little different,” said Dr. Ketwaroo, who’s tried the new device, but “it’s pretty functional and probably okay to use in almost all endoscopic procedures that require ERCP.”
In a study funded by Boston Scientific, endoscopists reported a median overall satisfaction score of 9 out of 10 with the new scope (Clin Gastroenterol Hepatol. 2019 Nov 6. doi: 10.1016/j.cgh.2019.10.052).
As for using it at Baylor, Dr. Ketwaroo said, “we’re not sure yet; we are still evaluating it” and want to see if any problems emerge once it’s on the market. It’s also not clear if infection risks would be lower than with the disposable elevator model from Pentax, he added.
The Exalt Model D was granted breakthrough status by the FDA, and the agency worked closely with Boston Scientific to bring it to market.
“The availability of a fully disposable duodenoscope represents another major step forward for improving the safety of these devices, which are used in more than 500,000 procedures in the United States each year. The FDA continues to encourage innovative ways to improve the safety and effectiveness of these devices,” Jeff Shuren, MD, director of the FDA’s Center for Devices and Radiological Health, said in a statement.
Dr. Ketwaroo had no relevant financial disclosures.
FROM THE FDA
FDA clears first fully disposable duodenoscope
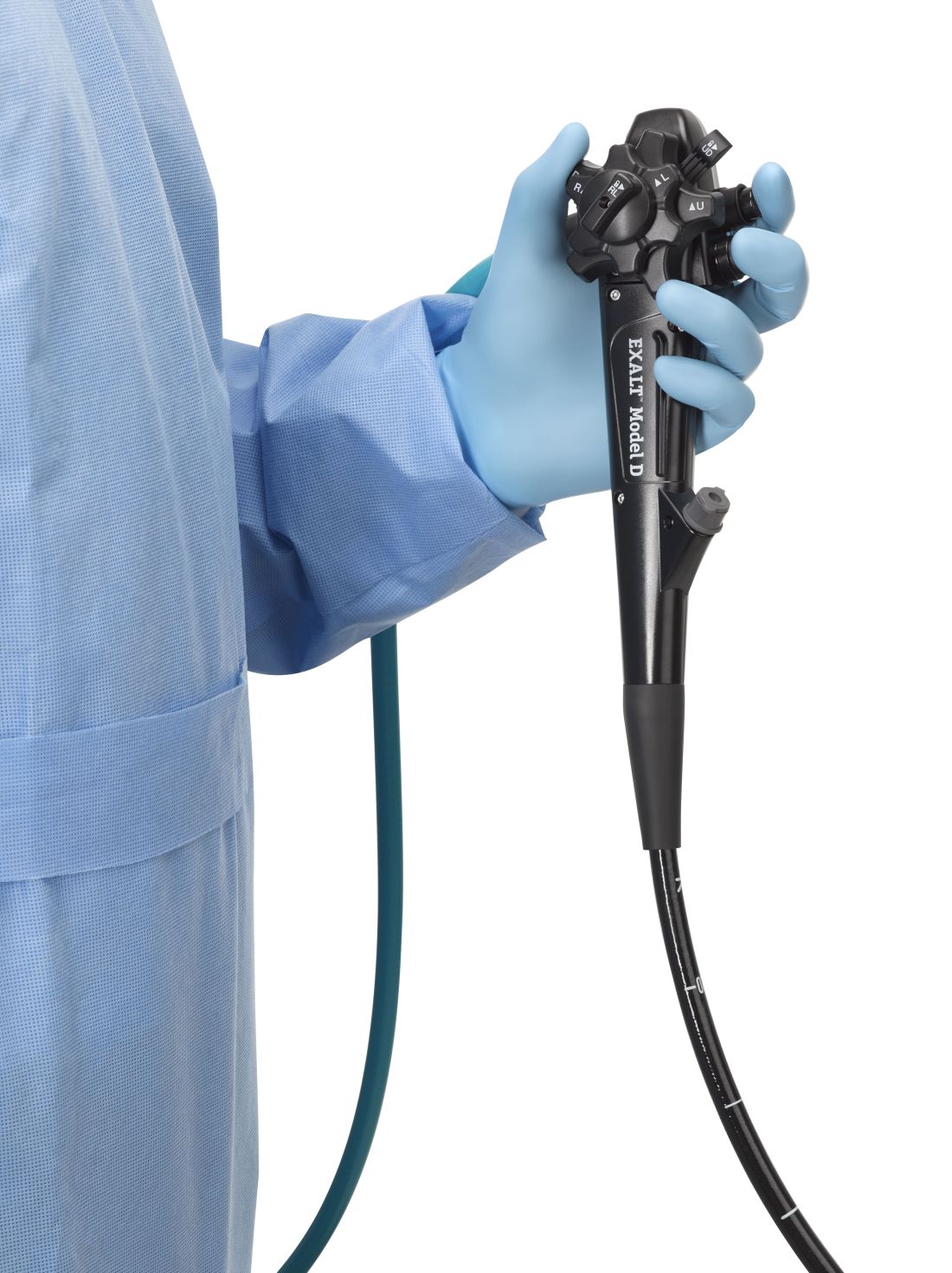
The Food and Drug Administration on Dec. 13 cleared Boston Scientific’s single-use duodenoscope, the Exalt Model D, for endoscopic retrograde cholangiopancreatography.
It’s the first disposable duodenoscope to hit the market in the wake of the agency’s August call for manufacturers and health care facilities to move to partially or fully disposable duodenoscopes. The goal is to eliminate the risk of spreading infections between patients from incomplete sterilization of traditional, multi-use scopes. The FDA also recently approved a Pentax duodenoscope with a disposable elevator, the most difficult part to clean.
The agency reported in April that 5.4% of samples from multi-use scopes test positive for Escherichia coli, Pseudomonas aeruginosa, or other “high-concern” organisms.
Boston Scientific spokesperson Kate Haranis said the Exalt Model D will be available in the first quarter of 2020, but the company is still working out how much it will charge.
Cost effectiveness will depend largely on the degree to which the price of the device is offset by the infections it prevents. It might prove particularly attractive to high-volume centers with higher than usual infection rates. It might also be of interest to smaller practices where the price of a multi-use scope doesn’t make sense for only a few procedures a year, said Gyanprakash Ketwaroo, MD, an interventional endoscopist and assistant professor of gastroenterology at Baylor University, Houston.
AGA is working with FDA to ensure physicians continue to have access to ERCP as new devices are introduced to the market and will continue to update members on the latest developments. The GI societies believe that device transitions can be incorporated over the life cycle of current instrumentation, to eliminate the potential for gaps in accessibility of care and to ensure that there is adequate efficacy and safety data to support the adoption of new technology. Review the GI societies’ guiding principles for continued scope evolution at https://www.gastro.org/news/gi-societies-advise-fda-on-duodenoscope-reprocessing.
Dr. Ketwaroo had no relevant financial disclosures.
The Food and Drug Administration on Dec. 13 cleared Boston Scientific’s single-use duodenoscope, the Exalt Model D, for endoscopic retrograde cholangiopancreatography.
It’s the first disposable duodenoscope to hit the market in the wake of the agency’s August call for manufacturers and health care facilities to move to partially or fully disposable duodenoscopes. The goal is to eliminate the risk of spreading infections between patients from incomplete sterilization of traditional, multi-use scopes. The FDA also recently approved a Pentax duodenoscope with a disposable elevator, the most difficult part to clean.
The agency reported in April that 5.4% of samples from multi-use scopes test positive for Escherichia coli, Pseudomonas aeruginosa, or other “high-concern” organisms.
Boston Scientific spokesperson Kate Haranis said the Exalt Model D will be available in the first quarter of 2020, but the company is still working out how much it will charge.
Cost effectiveness will depend largely on the degree to which the price of the device is offset by the infections it prevents. It might prove particularly attractive to high-volume centers with higher than usual infection rates. It might also be of interest to smaller practices where the price of a multi-use scope doesn’t make sense for only a few procedures a year, said Gyanprakash Ketwaroo, MD, an interventional endoscopist and assistant professor of gastroenterology at Baylor University, Houston.
AGA is working with FDA to ensure physicians continue to have access to ERCP as new devices are introduced to the market and will continue to update members on the latest developments. The GI societies believe that device transitions can be incorporated over the life cycle of current instrumentation, to eliminate the potential for gaps in accessibility of care and to ensure that there is adequate efficacy and safety data to support the adoption of new technology. Review the GI societies’ guiding principles for continued scope evolution at https://www.gastro.org/news/gi-societies-advise-fda-on-duodenoscope-reprocessing.
Dr. Ketwaroo had no relevant financial disclosures.
The Food and Drug Administration on Dec. 13 cleared Boston Scientific’s single-use duodenoscope, the Exalt Model D, for endoscopic retrograde cholangiopancreatography.
It’s the first disposable duodenoscope to hit the market in the wake of the agency’s August call for manufacturers and health care facilities to move to partially or fully disposable duodenoscopes. The goal is to eliminate the risk of spreading infections between patients from incomplete sterilization of traditional, multi-use scopes. The FDA also recently approved a Pentax duodenoscope with a disposable elevator, the most difficult part to clean.
The agency reported in April that 5.4% of samples from multi-use scopes test positive for Escherichia coli, Pseudomonas aeruginosa, or other “high-concern” organisms.
Boston Scientific spokesperson Kate Haranis said the Exalt Model D will be available in the first quarter of 2020, but the company is still working out how much it will charge.
Cost effectiveness will depend largely on the degree to which the price of the device is offset by the infections it prevents. It might prove particularly attractive to high-volume centers with higher than usual infection rates. It might also be of interest to smaller practices where the price of a multi-use scope doesn’t make sense for only a few procedures a year, said Gyanprakash Ketwaroo, MD, an interventional endoscopist and assistant professor of gastroenterology at Baylor University, Houston.
AGA is working with FDA to ensure physicians continue to have access to ERCP as new devices are introduced to the market and will continue to update members on the latest developments. The GI societies believe that device transitions can be incorporated over the life cycle of current instrumentation, to eliminate the potential for gaps in accessibility of care and to ensure that there is adequate efficacy and safety data to support the adoption of new technology. Review the GI societies’ guiding principles for continued scope evolution at https://www.gastro.org/news/gi-societies-advise-fda-on-duodenoscope-reprocessing.
Dr. Ketwaroo had no relevant financial disclosures.
Some MCL patients can safely stop venetoclax-ibrutinib, study suggests
ORLANDO – Updated trial results have revealed durable responses with venetoclax and ibrutinib in patients with mantle cell lymphoma (MCL), allowing some patients to stop treatment.

Five of 24 patients were able to stop treatment after achieving minimal residual disease (MRD)-negative complete responses (CRs). Four of these patients remain in CR at up to 18 months off treatment, although one patient ultimately progressed and died.
“Treatment cessation was feasible for patients in MRD-negative complete responses, raising the prospect of limited-duration, targeted-agent therapy in the management of relapsed/refractory mantle cell lymphoma,” said Sasanka M. Handunnetti, MBBS, of Peter MacCallum Cancer Centre in Melbourne. Dr. Handunnetti presented these results, from the AIM trial, at the annual meeting of the American Society of Hematology.
The phase 2 trial enrolled 24 patients. At baseline, patients had a median age of 68 years (range, 47-81 years), and 88% were men. One patient was treatment-naive, but the rest had relapsed/refractory MCL. These patients had received a median of two prior therapies (range, 1-6).
The patients received venetoclax at 400 mg daily and ibrutinib at 560 mg daily.
In the primary analysis, the CR rate was 62% at week 16 and 71% overall, according to positron-emission tomography/computed tomography. MRD negativity was achieved by 67% of patients according to flow cytometry and 38% according to allele-specific oligonucleotide polymerase chain reaction (N Engl J Med. 2018 Mar 29;378[13]:1211-23).
Response and survival
For the current analysis, the median follow up was 37.5 months (range, 1.4-45.3 months). The median duration of response has not been reached, the median progression-free survival is 29 months, and the median overall survival is 32 months.
Thirteen patients have died, 8 of them due to progressive disease. The remaining 11 patients are still alive, and 9 of them are still in CR. One patient is still in partial response, and one has not responded but remains on ibrutinib and venetoclax.
Dr. Handunnetti pointed out that 12 patients had TP53 aberrations, and 8 of them died, but 4 remain alive and in CR. All four patients with SMARCA4 aberrations died.
Treatment status
Five patients are still receiving treatment with ibrutinib and venetoclax, and one patient is receiving only venetoclax. One patient went off study treatment due to a diagnosis of myelodysplastic syndrome, but that patient’s MCL is still in CR.
Five patients were able to stop treatment after achieving MRD-negative CR and were placed under “stringent surveillance,” Dr. Handunnetti said.
One of the five patients who stopped treatment progressed at 7 months and died. The remaining four patients are still alive and in CR at 6 months, 13 months, 17 months, and 18 months off treatment.
Safety update
Within the first 56 weeks of treatment, 15 patients required dose adjustments. Twelve patients required an adjustment to ibrutinib, seven to venetoclax, and four to both drugs. After 56 weeks, there were no dose adjustments.
Two patients developed therapy-related myelodysplastic syndrome. One patient had previously received FCR (fludarabine, cyclophosphamide, and rituximab) and BR (bendamustine and rituximab). The other patient had received R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone).
This investigator-initiated trial was funded by Janssen and Abbvie. Dr. Handunnetti reported relationships with Abbvie and Gilead.
SOURCE: Handunnetti S et al. ASH 2019. Abstract 756.
ORLANDO – Updated trial results have revealed durable responses with venetoclax and ibrutinib in patients with mantle cell lymphoma (MCL), allowing some patients to stop treatment.
Five of 24 patients were able to stop treatment after achieving minimal residual disease (MRD)-negative complete responses (CRs). Four of these patients remain in CR at up to 18 months off treatment, although one patient ultimately progressed and died.
“Treatment cessation was feasible for patients in MRD-negative complete responses, raising the prospect of limited-duration, targeted-agent therapy in the management of relapsed/refractory mantle cell lymphoma,” said Sasanka M. Handunnetti, MBBS, of Peter MacCallum Cancer Centre in Melbourne. Dr. Handunnetti presented these results, from the AIM trial, at the annual meeting of the American Society of Hematology.
The phase 2 trial enrolled 24 patients. At baseline, patients had a median age of 68 years (range, 47-81 years), and 88% were men. One patient was treatment-naive, but the rest had relapsed/refractory MCL. These patients had received a median of two prior therapies (range, 1-6).
The patients received venetoclax at 400 mg daily and ibrutinib at 560 mg daily.
In the primary analysis, the CR rate was 62% at week 16 and 71% overall, according to positron-emission tomography/computed tomography. MRD negativity was achieved by 67% of patients according to flow cytometry and 38% according to allele-specific oligonucleotide polymerase chain reaction (N Engl J Med. 2018 Mar 29;378[13]:1211-23).
Response and survival
For the current analysis, the median follow up was 37.5 months (range, 1.4-45.3 months). The median duration of response has not been reached, the median progression-free survival is 29 months, and the median overall survival is 32 months.
Thirteen patients have died, 8 of them due to progressive disease. The remaining 11 patients are still alive, and 9 of them are still in CR. One patient is still in partial response, and one has not responded but remains on ibrutinib and venetoclax.
Dr. Handunnetti pointed out that 12 patients had TP53 aberrations, and 8 of them died, but 4 remain alive and in CR. All four patients with SMARCA4 aberrations died.
Treatment status
Five patients are still receiving treatment with ibrutinib and venetoclax, and one patient is receiving only venetoclax. One patient went off study treatment due to a diagnosis of myelodysplastic syndrome, but that patient’s MCL is still in CR.
Five patients were able to stop treatment after achieving MRD-negative CR and were placed under “stringent surveillance,” Dr. Handunnetti said.
One of the five patients who stopped treatment progressed at 7 months and died. The remaining four patients are still alive and in CR at 6 months, 13 months, 17 months, and 18 months off treatment.
Safety update
Within the first 56 weeks of treatment, 15 patients required dose adjustments. Twelve patients required an adjustment to ibrutinib, seven to venetoclax, and four to both drugs. After 56 weeks, there were no dose adjustments.
Two patients developed therapy-related myelodysplastic syndrome. One patient had previously received FCR (fludarabine, cyclophosphamide, and rituximab) and BR (bendamustine and rituximab). The other patient had received R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone).
This investigator-initiated trial was funded by Janssen and Abbvie. Dr. Handunnetti reported relationships with Abbvie and Gilead.
SOURCE: Handunnetti S et al. ASH 2019. Abstract 756.
ORLANDO – Updated trial results have revealed durable responses with venetoclax and ibrutinib in patients with mantle cell lymphoma (MCL), allowing some patients to stop treatment.
Five of 24 patients were able to stop treatment after achieving minimal residual disease (MRD)-negative complete responses (CRs). Four of these patients remain in CR at up to 18 months off treatment, although one patient ultimately progressed and died.
“Treatment cessation was feasible for patients in MRD-negative complete responses, raising the prospect of limited-duration, targeted-agent therapy in the management of relapsed/refractory mantle cell lymphoma,” said Sasanka M. Handunnetti, MBBS, of Peter MacCallum Cancer Centre in Melbourne. Dr. Handunnetti presented these results, from the AIM trial, at the annual meeting of the American Society of Hematology.
The phase 2 trial enrolled 24 patients. At baseline, patients had a median age of 68 years (range, 47-81 years), and 88% were men. One patient was treatment-naive, but the rest had relapsed/refractory MCL. These patients had received a median of two prior therapies (range, 1-6).
The patients received venetoclax at 400 mg daily and ibrutinib at 560 mg daily.
In the primary analysis, the CR rate was 62% at week 16 and 71% overall, according to positron-emission tomography/computed tomography. MRD negativity was achieved by 67% of patients according to flow cytometry and 38% according to allele-specific oligonucleotide polymerase chain reaction (N Engl J Med. 2018 Mar 29;378[13]:1211-23).
Response and survival
For the current analysis, the median follow up was 37.5 months (range, 1.4-45.3 months). The median duration of response has not been reached, the median progression-free survival is 29 months, and the median overall survival is 32 months.
Thirteen patients have died, 8 of them due to progressive disease. The remaining 11 patients are still alive, and 9 of them are still in CR. One patient is still in partial response, and one has not responded but remains on ibrutinib and venetoclax.
Dr. Handunnetti pointed out that 12 patients had TP53 aberrations, and 8 of them died, but 4 remain alive and in CR. All four patients with SMARCA4 aberrations died.
Treatment status
Five patients are still receiving treatment with ibrutinib and venetoclax, and one patient is receiving only venetoclax. One patient went off study treatment due to a diagnosis of myelodysplastic syndrome, but that patient’s MCL is still in CR.
Five patients were able to stop treatment after achieving MRD-negative CR and were placed under “stringent surveillance,” Dr. Handunnetti said.
One of the five patients who stopped treatment progressed at 7 months and died. The remaining four patients are still alive and in CR at 6 months, 13 months, 17 months, and 18 months off treatment.
Safety update
Within the first 56 weeks of treatment, 15 patients required dose adjustments. Twelve patients required an adjustment to ibrutinib, seven to venetoclax, and four to both drugs. After 56 weeks, there were no dose adjustments.
Two patients developed therapy-related myelodysplastic syndrome. One patient had previously received FCR (fludarabine, cyclophosphamide, and rituximab) and BR (bendamustine and rituximab). The other patient had received R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone).
This investigator-initiated trial was funded by Janssen and Abbvie. Dr. Handunnetti reported relationships with Abbvie and Gilead.
SOURCE: Handunnetti S et al. ASH 2019. Abstract 756.
REPORTING FROM ASH 2019
BCL11A-directed gene therapy advances in sickle cell disease
ORLANDO – A gene therapy approach that targets a major repressor of fetal hemoglobin appears to be acceptably safe and to mitigate the pathology of sickle cell disease among the five patients infused so far, an investigator reported at the annual meeting of the American Society of Hematology.

Knocking down BCL11A using a lentiviral vector-based approach resulted in effective induction of fetal hemoglobin and significant attenuation of the sickling phenotype, with no vector-related adverse events, investigator Erica B. Esrick, MD, of Children’s Hospital Boston, said during the meeting’s late-breaking abstracts session.
The single-center pilot and feasibility study, originally designed to include a total of seven patients, now has an expanded enrollment goal of 15 patients, and a multicenter phase 2/3 study is planned that will enroll a larger group of patients with sickle cell disease, according to Dr. Esrick.
BCL11A represents a promising target in sickle cell disease because of its regulation of the fetal-adult hemoglobin switch at the gamma-globin locus, investigators said in their late-breaking study abstract.
Dr. Esrick described BCH-BB694, a lentiviral vector encoding a BCL11A-targeting small hairpin RNA embedded in a microRNA scaffold (shmiR). “The advantage of this approach is that it harnesses the physiologic switch machinery, simultaneously increasing fetal hemoglobin and decreasing sickle hemoglobin, thus maintaining the alpha to beta globin ratio in the cell,” she said.
The results of the pilot study of the shmiR vector approach, although preliminary and in need of longer follow-up, contribute to a larger body of research showing that multiple gene therapy approaches hold promise in this disease, said Robert Brodsky, MD, professor of medicine and director of the division of hematology at Johns Hopkins School of Medicine, Baltimore.
“The exciting thing is that there are now multiple ways of going at this previously incurable disease,” Dr. Brodsky, who was not involved in the research, said during a press conference.
Development of the gene therapy described by Dr. Esrick involves mobilization of the patient’s peripheral stem cells using plerixafor, followed by selection of CD34+ cells that were transduced with the shmiR lentiviral vector, followed by infusion of gene modified cells into the patient after a busulfan conditioning regimen.
“In our treated patients, we’ve seen a consistent and substantial induction in fetal hemoglobin,” Dr. Esrick said, noting that the longest follow-up to date for the five treated patients is now 18 months.
The patients, who range in age from 12 to 26 years, are producing and maintaining very high numbers of F cells, or erythrocytes with measurable fetal hemoglobin, she said.
Total fetal hemoglobin has increased and remained stable at between 23% and 43% for the five patients, who are producing “stably high” average amounts of fetal hemoglobin per F cell, at 10 to 16 picograms of fetal hemoglobin per cell, while 37% to 62% of the F cells’ total hemoglobin is fetal hemoglobin, she added.
Following gene therapy, treated patients have had no instances of vaso-occlusive pain crises, respiratory events, or neurologic events. No patients have required transfusion, except one with severe underlying vascular disease for whom post–gene therapy transfusions were planned, she said.
Validated assays at the single-cell level are needed to better understand the effect of this gene therapy and eventually compare it to other therapeutic approaches in sickle cell disease, according to Dr. Esrick.
“We’re collaborating with several colleagues on exploratory assays to accomplish this,” she said, adding that the work is ongoing.
Dr. Esrick reported having no disclosures. Her coauthors reported disclosures related to Alerion Biosciences, Novartis, Orchard Therapeutics, Roche, AstraZeneca, and bluebird bio, among others.
SOURCE: Esrick EB et al. ASH 2019. Abstract LBA-5.
ORLANDO – A gene therapy approach that targets a major repressor of fetal hemoglobin appears to be acceptably safe and to mitigate the pathology of sickle cell disease among the five patients infused so far, an investigator reported at the annual meeting of the American Society of Hematology.
Knocking down BCL11A using a lentiviral vector-based approach resulted in effective induction of fetal hemoglobin and significant attenuation of the sickling phenotype, with no vector-related adverse events, investigator Erica B. Esrick, MD, of Children’s Hospital Boston, said during the meeting’s late-breaking abstracts session.
The single-center pilot and feasibility study, originally designed to include a total of seven patients, now has an expanded enrollment goal of 15 patients, and a multicenter phase 2/3 study is planned that will enroll a larger group of patients with sickle cell disease, according to Dr. Esrick.
BCL11A represents a promising target in sickle cell disease because of its regulation of the fetal-adult hemoglobin switch at the gamma-globin locus, investigators said in their late-breaking study abstract.
Dr. Esrick described BCH-BB694, a lentiviral vector encoding a BCL11A-targeting small hairpin RNA embedded in a microRNA scaffold (shmiR). “The advantage of this approach is that it harnesses the physiologic switch machinery, simultaneously increasing fetal hemoglobin and decreasing sickle hemoglobin, thus maintaining the alpha to beta globin ratio in the cell,” she said.
The results of the pilot study of the shmiR vector approach, although preliminary and in need of longer follow-up, contribute to a larger body of research showing that multiple gene therapy approaches hold promise in this disease, said Robert Brodsky, MD, professor of medicine and director of the division of hematology at Johns Hopkins School of Medicine, Baltimore.
“The exciting thing is that there are now multiple ways of going at this previously incurable disease,” Dr. Brodsky, who was not involved in the research, said during a press conference.
Development of the gene therapy described by Dr. Esrick involves mobilization of the patient’s peripheral stem cells using plerixafor, followed by selection of CD34+ cells that were transduced with the shmiR lentiviral vector, followed by infusion of gene modified cells into the patient after a busulfan conditioning regimen.
“In our treated patients, we’ve seen a consistent and substantial induction in fetal hemoglobin,” Dr. Esrick said, noting that the longest follow-up to date for the five treated patients is now 18 months.
The patients, who range in age from 12 to 26 years, are producing and maintaining very high numbers of F cells, or erythrocytes with measurable fetal hemoglobin, she said.
Total fetal hemoglobin has increased and remained stable at between 23% and 43% for the five patients, who are producing “stably high” average amounts of fetal hemoglobin per F cell, at 10 to 16 picograms of fetal hemoglobin per cell, while 37% to 62% of the F cells’ total hemoglobin is fetal hemoglobin, she added.
Following gene therapy, treated patients have had no instances of vaso-occlusive pain crises, respiratory events, or neurologic events. No patients have required transfusion, except one with severe underlying vascular disease for whom post–gene therapy transfusions were planned, she said.
Validated assays at the single-cell level are needed to better understand the effect of this gene therapy and eventually compare it to other therapeutic approaches in sickle cell disease, according to Dr. Esrick.
“We’re collaborating with several colleagues on exploratory assays to accomplish this,” she said, adding that the work is ongoing.
Dr. Esrick reported having no disclosures. Her coauthors reported disclosures related to Alerion Biosciences, Novartis, Orchard Therapeutics, Roche, AstraZeneca, and bluebird bio, among others.
SOURCE: Esrick EB et al. ASH 2019. Abstract LBA-5.
ORLANDO – A gene therapy approach that targets a major repressor of fetal hemoglobin appears to be acceptably safe and to mitigate the pathology of sickle cell disease among the five patients infused so far, an investigator reported at the annual meeting of the American Society of Hematology.
Knocking down BCL11A using a lentiviral vector-based approach resulted in effective induction of fetal hemoglobin and significant attenuation of the sickling phenotype, with no vector-related adverse events, investigator Erica B. Esrick, MD, of Children’s Hospital Boston, said during the meeting’s late-breaking abstracts session.
The single-center pilot and feasibility study, originally designed to include a total of seven patients, now has an expanded enrollment goal of 15 patients, and a multicenter phase 2/3 study is planned that will enroll a larger group of patients with sickle cell disease, according to Dr. Esrick.
BCL11A represents a promising target in sickle cell disease because of its regulation of the fetal-adult hemoglobin switch at the gamma-globin locus, investigators said in their late-breaking study abstract.
Dr. Esrick described BCH-BB694, a lentiviral vector encoding a BCL11A-targeting small hairpin RNA embedded in a microRNA scaffold (shmiR). “The advantage of this approach is that it harnesses the physiologic switch machinery, simultaneously increasing fetal hemoglobin and decreasing sickle hemoglobin, thus maintaining the alpha to beta globin ratio in the cell,” she said.
The results of the pilot study of the shmiR vector approach, although preliminary and in need of longer follow-up, contribute to a larger body of research showing that multiple gene therapy approaches hold promise in this disease, said Robert Brodsky, MD, professor of medicine and director of the division of hematology at Johns Hopkins School of Medicine, Baltimore.
“The exciting thing is that there are now multiple ways of going at this previously incurable disease,” Dr. Brodsky, who was not involved in the research, said during a press conference.
Development of the gene therapy described by Dr. Esrick involves mobilization of the patient’s peripheral stem cells using plerixafor, followed by selection of CD34+ cells that were transduced with the shmiR lentiviral vector, followed by infusion of gene modified cells into the patient after a busulfan conditioning regimen.
“In our treated patients, we’ve seen a consistent and substantial induction in fetal hemoglobin,” Dr. Esrick said, noting that the longest follow-up to date for the five treated patients is now 18 months.
The patients, who range in age from 12 to 26 years, are producing and maintaining very high numbers of F cells, or erythrocytes with measurable fetal hemoglobin, she said.
Total fetal hemoglobin has increased and remained stable at between 23% and 43% for the five patients, who are producing “stably high” average amounts of fetal hemoglobin per F cell, at 10 to 16 picograms of fetal hemoglobin per cell, while 37% to 62% of the F cells’ total hemoglobin is fetal hemoglobin, she added.
Following gene therapy, treated patients have had no instances of vaso-occlusive pain crises, respiratory events, or neurologic events. No patients have required transfusion, except one with severe underlying vascular disease for whom post–gene therapy transfusions were planned, she said.
Validated assays at the single-cell level are needed to better understand the effect of this gene therapy and eventually compare it to other therapeutic approaches in sickle cell disease, according to Dr. Esrick.
“We’re collaborating with several colleagues on exploratory assays to accomplish this,” she said, adding that the work is ongoing.
Dr. Esrick reported having no disclosures. Her coauthors reported disclosures related to Alerion Biosciences, Novartis, Orchard Therapeutics, Roche, AstraZeneca, and bluebird bio, among others.
SOURCE: Esrick EB et al. ASH 2019. Abstract LBA-5.
REPORTING FROM ASH 2019
KTE-X19 produces highest response rate in MCL subgroup
ORLANDO – KTE-X19, an anti-CD19 chimeric antigen receptor (CAR) T-cell therapy, demonstrated unprecedented efficacy in the ZUMA-2 trial, according to an investigator involved in the study.

KTE-X19 produced a 93% overall response rate in patients with relapsed/refractory mantle cell lymphoma (MCL). This is the highest reported response rate in patients who have failed treatment with a Bruton’s tyrosine kinase (BTK) inhibitor, said Michael L. Wang, MD, of the University of Texas MD Anderson Cancer Center in Houston.
Dr. Wang presented results from ZUMA-2 at the annual meeting of the American Society of Hematology.
“Patients with relapsed/refractory MCL have very poor outcomes,” Dr. Wang noted. “In patients who progress after BTK inhibition therapy, the overall response rate is only between 25% and 42%, and the overall survival is only between 6 and 10 months. Few patients proceed to allogeneic transplantation.”
The phase 2 ZUMA-2 trial was designed to test KTE-X19 in these patients. KTE-X19 is an anti-CD19 CAR T-cell therapy containing a CD3-zeta T-cell activation domain and a CD28 signaling domain. KTE-X19 is distinct from axicabtagene ciloleucel (KTE-C19) because the manufacturing process for KTE-X19 removes circulating tumor cells.
The trial enrolled 74 patients, and 68 of them received KTE-X19. Manufacturing failed for three patients, two patients died of progressive disease before they could receive KTE-X19, and one patient was found to be ineligible for treatment.
The 68 patients had a median age of 65 years (range, 38-79 years), and 84% were men. A majority of patients (85%) had stage IV disease and classical (59%) or blastoid (25%) morphology. Most patients (69%) had a Ki-67 proliferation index of 50% or greater, and most (56%) were intermediate- or high-risk according to the Mantle Cell Lymphoma International Prognostic Index (MIPI).
Patients had received a median of three prior therapies (range, one to five). All had been treated with a BTK inhibitor, with 85% receiving ibrutinib, 24% receiving acalabrutinib, and 9% receiving both. Most patients (68%) were refractory to BTK inhibition, and 32% relapsed on or after BTK inhibitor therapy.
In this study, patients could receive bridging therapy to keep their disease stable while KTE-X19 was being manufactured. There were 25 patients who received bridging therapy, which consisted of ibrutinib (n = 14), acalabrutinib (n = 5), dexamethasone (n = 12), and/or methylprednisolone (n = 2). Six patients received both BTK inhibitors and steroids.
All patients received conditioning with fludarabine and cyclophosphamide, followed by a single infusion of KTE-X19 at 2x106.
Efficacy
Sixty patients were evaluable for efficacy, and the median follow-up was 12.3 months (range, 7.0-32.3 months).
The overall response rate was 93%, with 67% of patients achieving a complete response and 27% achieving a partial response. Three percent of patients had stable disease, and 3% had progressive disease.
“The overall response rate was consistent across key subgroups, without any statistical difference,” Dr. Wang said. “This includes Ki-67, MIPI, and prior use of either steroids or bridging therapy.”
The median time to response was 1.0 month, and the median time to complete response was 3.0 months. Responses deepened over time, with 35% of patients converting from a partial response to a complete response, and 5% converting from stable disease to complete response.
The median duration of response has not been reached. At last follow-up, 57% of all patients and 78% of complete responders were still in response.
The median progression-free and overall survival have not been reached. At 12 months, the progression-free survival rate was 61%, and the overall survival rate was 83%.
Safety
All 68 patients were evaluable for safety. The most common adverse events were pyrexia (94%), neutropenia (87%), thrombocytopenia (74%), anemia (68%), and hypotension (51%).
Grade 3/4 adverse events included pyrexia (13%), neutropenia (85%), thrombocytopenia (51%), anemia (50%), hypotension (22%), hypoxia (21%), hypophosphatemia (22%), fatigue (1%), and headache (1%).
There were two grade 5 treatment-related adverse events – organizing pneumonia on day 37 and staphylococcal bacteremia on day 134.
Cytokine release syndrome (CRS) occurred in 91% of patients, with 15% experiencing grade 3 or higher CRS. Patients were treated with tocilizumab or corticosteroids, and all CRS events resolved.
Neurologic adverse events occurred in 63% of patients, with grade 3 or higher events occurring in 31%. Neurologic events were treated with tocilizumab or corticosteroids, and 86% of neurologic events resolved.
This trial was sponsored by Kite, a Gilead company. Dr. Wang reported financial relationships with Kite and other companies.
SOURCE: Wang M et al. ASH 2019. Abstract 754.
ORLANDO – KTE-X19, an anti-CD19 chimeric antigen receptor (CAR) T-cell therapy, demonstrated unprecedented efficacy in the ZUMA-2 trial, according to an investigator involved in the study.
KTE-X19 produced a 93% overall response rate in patients with relapsed/refractory mantle cell lymphoma (MCL). This is the highest reported response rate in patients who have failed treatment with a Bruton’s tyrosine kinase (BTK) inhibitor, said Michael L. Wang, MD, of the University of Texas MD Anderson Cancer Center in Houston.
Dr. Wang presented results from ZUMA-2 at the annual meeting of the American Society of Hematology.
“Patients with relapsed/refractory MCL have very poor outcomes,” Dr. Wang noted. “In patients who progress after BTK inhibition therapy, the overall response rate is only between 25% and 42%, and the overall survival is only between 6 and 10 months. Few patients proceed to allogeneic transplantation.”
The phase 2 ZUMA-2 trial was designed to test KTE-X19 in these patients. KTE-X19 is an anti-CD19 CAR T-cell therapy containing a CD3-zeta T-cell activation domain and a CD28 signaling domain. KTE-X19 is distinct from axicabtagene ciloleucel (KTE-C19) because the manufacturing process for KTE-X19 removes circulating tumor cells.
The trial enrolled 74 patients, and 68 of them received KTE-X19. Manufacturing failed for three patients, two patients died of progressive disease before they could receive KTE-X19, and one patient was found to be ineligible for treatment.
The 68 patients had a median age of 65 years (range, 38-79 years), and 84% were men. A majority of patients (85%) had stage IV disease and classical (59%) or blastoid (25%) morphology. Most patients (69%) had a Ki-67 proliferation index of 50% or greater, and most (56%) were intermediate- or high-risk according to the Mantle Cell Lymphoma International Prognostic Index (MIPI).
Patients had received a median of three prior therapies (range, one to five). All had been treated with a BTK inhibitor, with 85% receiving ibrutinib, 24% receiving acalabrutinib, and 9% receiving both. Most patients (68%) were refractory to BTK inhibition, and 32% relapsed on or after BTK inhibitor therapy.
In this study, patients could receive bridging therapy to keep their disease stable while KTE-X19 was being manufactured. There were 25 patients who received bridging therapy, which consisted of ibrutinib (n = 14), acalabrutinib (n = 5), dexamethasone (n = 12), and/or methylprednisolone (n = 2). Six patients received both BTK inhibitors and steroids.
All patients received conditioning with fludarabine and cyclophosphamide, followed by a single infusion of KTE-X19 at 2x106.
Efficacy
Sixty patients were evaluable for efficacy, and the median follow-up was 12.3 months (range, 7.0-32.3 months).
The overall response rate was 93%, with 67% of patients achieving a complete response and 27% achieving a partial response. Three percent of patients had stable disease, and 3% had progressive disease.
“The overall response rate was consistent across key subgroups, without any statistical difference,” Dr. Wang said. “This includes Ki-67, MIPI, and prior use of either steroids or bridging therapy.”
The median time to response was 1.0 month, and the median time to complete response was 3.0 months. Responses deepened over time, with 35% of patients converting from a partial response to a complete response, and 5% converting from stable disease to complete response.
The median duration of response has not been reached. At last follow-up, 57% of all patients and 78% of complete responders were still in response.
The median progression-free and overall survival have not been reached. At 12 months, the progression-free survival rate was 61%, and the overall survival rate was 83%.
Safety
All 68 patients were evaluable for safety. The most common adverse events were pyrexia (94%), neutropenia (87%), thrombocytopenia (74%), anemia (68%), and hypotension (51%).
Grade 3/4 adverse events included pyrexia (13%), neutropenia (85%), thrombocytopenia (51%), anemia (50%), hypotension (22%), hypoxia (21%), hypophosphatemia (22%), fatigue (1%), and headache (1%).
There were two grade 5 treatment-related adverse events – organizing pneumonia on day 37 and staphylococcal bacteremia on day 134.
Cytokine release syndrome (CRS) occurred in 91% of patients, with 15% experiencing grade 3 or higher CRS. Patients were treated with tocilizumab or corticosteroids, and all CRS events resolved.
Neurologic adverse events occurred in 63% of patients, with grade 3 or higher events occurring in 31%. Neurologic events were treated with tocilizumab or corticosteroids, and 86% of neurologic events resolved.
This trial was sponsored by Kite, a Gilead company. Dr. Wang reported financial relationships with Kite and other companies.
SOURCE: Wang M et al. ASH 2019. Abstract 754.
ORLANDO – KTE-X19, an anti-CD19 chimeric antigen receptor (CAR) T-cell therapy, demonstrated unprecedented efficacy in the ZUMA-2 trial, according to an investigator involved in the study.
KTE-X19 produced a 93% overall response rate in patients with relapsed/refractory mantle cell lymphoma (MCL). This is the highest reported response rate in patients who have failed treatment with a Bruton’s tyrosine kinase (BTK) inhibitor, said Michael L. Wang, MD, of the University of Texas MD Anderson Cancer Center in Houston.
Dr. Wang presented results from ZUMA-2 at the annual meeting of the American Society of Hematology.
“Patients with relapsed/refractory MCL have very poor outcomes,” Dr. Wang noted. “In patients who progress after BTK inhibition therapy, the overall response rate is only between 25% and 42%, and the overall survival is only between 6 and 10 months. Few patients proceed to allogeneic transplantation.”
The phase 2 ZUMA-2 trial was designed to test KTE-X19 in these patients. KTE-X19 is an anti-CD19 CAR T-cell therapy containing a CD3-zeta T-cell activation domain and a CD28 signaling domain. KTE-X19 is distinct from axicabtagene ciloleucel (KTE-C19) because the manufacturing process for KTE-X19 removes circulating tumor cells.
The trial enrolled 74 patients, and 68 of them received KTE-X19. Manufacturing failed for three patients, two patients died of progressive disease before they could receive KTE-X19, and one patient was found to be ineligible for treatment.
The 68 patients had a median age of 65 years (range, 38-79 years), and 84% were men. A majority of patients (85%) had stage IV disease and classical (59%) or blastoid (25%) morphology. Most patients (69%) had a Ki-67 proliferation index of 50% or greater, and most (56%) were intermediate- or high-risk according to the Mantle Cell Lymphoma International Prognostic Index (MIPI).
Patients had received a median of three prior therapies (range, one to five). All had been treated with a BTK inhibitor, with 85% receiving ibrutinib, 24% receiving acalabrutinib, and 9% receiving both. Most patients (68%) were refractory to BTK inhibition, and 32% relapsed on or after BTK inhibitor therapy.
In this study, patients could receive bridging therapy to keep their disease stable while KTE-X19 was being manufactured. There were 25 patients who received bridging therapy, which consisted of ibrutinib (n = 14), acalabrutinib (n = 5), dexamethasone (n = 12), and/or methylprednisolone (n = 2). Six patients received both BTK inhibitors and steroids.
All patients received conditioning with fludarabine and cyclophosphamide, followed by a single infusion of KTE-X19 at 2x106.
Efficacy
Sixty patients were evaluable for efficacy, and the median follow-up was 12.3 months (range, 7.0-32.3 months).
The overall response rate was 93%, with 67% of patients achieving a complete response and 27% achieving a partial response. Three percent of patients had stable disease, and 3% had progressive disease.
“The overall response rate was consistent across key subgroups, without any statistical difference,” Dr. Wang said. “This includes Ki-67, MIPI, and prior use of either steroids or bridging therapy.”
The median time to response was 1.0 month, and the median time to complete response was 3.0 months. Responses deepened over time, with 35% of patients converting from a partial response to a complete response, and 5% converting from stable disease to complete response.
The median duration of response has not been reached. At last follow-up, 57% of all patients and 78% of complete responders were still in response.
The median progression-free and overall survival have not been reached. At 12 months, the progression-free survival rate was 61%, and the overall survival rate was 83%.
Safety
All 68 patients were evaluable for safety. The most common adverse events were pyrexia (94%), neutropenia (87%), thrombocytopenia (74%), anemia (68%), and hypotension (51%).
Grade 3/4 adverse events included pyrexia (13%), neutropenia (85%), thrombocytopenia (51%), anemia (50%), hypotension (22%), hypoxia (21%), hypophosphatemia (22%), fatigue (1%), and headache (1%).
There were two grade 5 treatment-related adverse events – organizing pneumonia on day 37 and staphylococcal bacteremia on day 134.
Cytokine release syndrome (CRS) occurred in 91% of patients, with 15% experiencing grade 3 or higher CRS. Patients were treated with tocilizumab or corticosteroids, and all CRS events resolved.
Neurologic adverse events occurred in 63% of patients, with grade 3 or higher events occurring in 31%. Neurologic events were treated with tocilizumab or corticosteroids, and 86% of neurologic events resolved.
This trial was sponsored by Kite, a Gilead company. Dr. Wang reported financial relationships with Kite and other companies.
SOURCE: Wang M et al. ASH 2019. Abstract 754.
REPORTING FROM ASH 2019
Residual cancer burden linked to long-term survival
SAN ANTONIO – Residual cancer burden (RCB) is poised to alter American Joint Committee on Cancer (AJCC) staging standards in breast cancer, according to investigators.
A meta-analysis showed that RCB – which is calculated in the neoadjuvant setting through a combination of primary tumor bed cellularity, lymph node positivity, and size of largest metastasis – was significantly associated with long-term survival of more than 5,000 breast cancer patients, reported principal investigator W. Fraser Symmans, MD, of MD Anderson Cancer Center, Houston, who presented the findings at the San Antonio Breast Cancer Symposium.
Coinvestigator Laura J. Esserman, MD, of the University of California, San Francisco, who was in attendance at Dr. Symmans’ presentation, put the study in context.
“The reason we did this meta-analysis was to really change the joint commission – the AJCC staging – and we expect that will happen on the basis of these results,” Dr. Esserman said. “[RBC] is, moving forward, the standard of care.”
The investigators analyzed individual-level data from 5,160 patients treated at multiple institutions. For each patient, RCB, which is scored from 0 to III, from pathologic complete response (0) to high disease burden (III), was compared with event-free survival (EFS) and disease relapse-free survival (DRFS).
The results showed that both EFS and DRFS were strongly associated with RCB for the overall population and each subtype. For instance, for triple-negative breast cancer, patients with pathologic complete response had a 10-year EFS of 86%, a rate that decreased as RCB increased from RCB-I (75%) to RCB-II (61%), and finally, RCB-III (25%). This trend was similar for patients with HR-negative/HER2-positive or HR-positive/HER2-positive disease. Risk stratification for HR-positive/HER2-negative disease deviated slightly; pathologic complete response was associated with a 10-year EFS of 81%, compared with 86% for RCB-I.
Regarding this finding, Dr. Symmans noted that RCB is most prognostic when higher levels of residual disease are present. Still, reviewing long-term risk across subtypes, Dr. Fraser emphasized the reliability of RCB as a prognostic tool.
“The strong relationship [between RCB and] risk is very clear in each and every subtype of disease,” he said. “There are very tight confidence intervals around those risk estimates.”
Multivariate analysis provided further support for this conclusion, as RCB was independently prognostic for survival in each subtype of disease.
According to Dr. Esserman, the study findings should encourage implementation of RCB in daily practice.
“There has to be training and a commitment to using this in a consistent way so that we can all rely on the information,” Dr. Esserman said. “This is critical for our patients, and this is ... how we’re going to get people to the best outcome, and [it should be done] up front, when we still have the chance to cure people.”
Session moderator Virginia Kaklamani, MD, of Mays Cancer Center, San Antonio, agreed that clinicians should use RCB in practice; however, she also explained that RCB results for some patients are not yet actionable.

“I ask my pathologist to give me the RCB score,” Dr. Kaklamani said. “The question is, what do I do with the results? It’s a little limited. In triple-negative patients I may consider giving capecitabine in the adjuvant setting, or obviously enrolling them in clinical trials. In my HER2-positive patients, I will look at the results from the KATHERINE trial and the ExteNET trial to guide me. In my HR-positive patients, I’m still waiting for results from other trials to see how these results can be interpreted. What we’ve [found] is in many trials, achieving a pathologic complete response in HR-positive patients may not be as important, but as you can see here, it looks like RCB really is.”
The investigators disclosed relationships with Seattle Genetics, Almac, Syndax, and others.
SOURCE: Yau et al. SABCS. 2019 Dec 13. Abstract GS5-01.
SAN ANTONIO – Residual cancer burden (RCB) is poised to alter American Joint Committee on Cancer (AJCC) staging standards in breast cancer, according to investigators.
A meta-analysis showed that RCB – which is calculated in the neoadjuvant setting through a combination of primary tumor bed cellularity, lymph node positivity, and size of largest metastasis – was significantly associated with long-term survival of more than 5,000 breast cancer patients, reported principal investigator W. Fraser Symmans, MD, of MD Anderson Cancer Center, Houston, who presented the findings at the San Antonio Breast Cancer Symposium.
Coinvestigator Laura J. Esserman, MD, of the University of California, San Francisco, who was in attendance at Dr. Symmans’ presentation, put the study in context.
“The reason we did this meta-analysis was to really change the joint commission – the AJCC staging – and we expect that will happen on the basis of these results,” Dr. Esserman said. “[RBC] is, moving forward, the standard of care.”
The investigators analyzed individual-level data from 5,160 patients treated at multiple institutions. For each patient, RCB, which is scored from 0 to III, from pathologic complete response (0) to high disease burden (III), was compared with event-free survival (EFS) and disease relapse-free survival (DRFS).
The results showed that both EFS and DRFS were strongly associated with RCB for the overall population and each subtype. For instance, for triple-negative breast cancer, patients with pathologic complete response had a 10-year EFS of 86%, a rate that decreased as RCB increased from RCB-I (75%) to RCB-II (61%), and finally, RCB-III (25%). This trend was similar for patients with HR-negative/HER2-positive or HR-positive/HER2-positive disease. Risk stratification for HR-positive/HER2-negative disease deviated slightly; pathologic complete response was associated with a 10-year EFS of 81%, compared with 86% for RCB-I.
Regarding this finding, Dr. Symmans noted that RCB is most prognostic when higher levels of residual disease are present. Still, reviewing long-term risk across subtypes, Dr. Fraser emphasized the reliability of RCB as a prognostic tool.
“The strong relationship [between RCB and] risk is very clear in each and every subtype of disease,” he said. “There are very tight confidence intervals around those risk estimates.”
Multivariate analysis provided further support for this conclusion, as RCB was independently prognostic for survival in each subtype of disease.
According to Dr. Esserman, the study findings should encourage implementation of RCB in daily practice.
“There has to be training and a commitment to using this in a consistent way so that we can all rely on the information,” Dr. Esserman said. “This is critical for our patients, and this is ... how we’re going to get people to the best outcome, and [it should be done] up front, when we still have the chance to cure people.”
Session moderator Virginia Kaklamani, MD, of Mays Cancer Center, San Antonio, agreed that clinicians should use RCB in practice; however, she also explained that RCB results for some patients are not yet actionable.
“I ask my pathologist to give me the RCB score,” Dr. Kaklamani said. “The question is, what do I do with the results? It’s a little limited. In triple-negative patients I may consider giving capecitabine in the adjuvant setting, or obviously enrolling them in clinical trials. In my HER2-positive patients, I will look at the results from the KATHERINE trial and the ExteNET trial to guide me. In my HR-positive patients, I’m still waiting for results from other trials to see how these results can be interpreted. What we’ve [found] is in many trials, achieving a pathologic complete response in HR-positive patients may not be as important, but as you can see here, it looks like RCB really is.”
The investigators disclosed relationships with Seattle Genetics, Almac, Syndax, and others.
SOURCE: Yau et al. SABCS. 2019 Dec 13. Abstract GS5-01.
SAN ANTONIO – Residual cancer burden (RCB) is poised to alter American Joint Committee on Cancer (AJCC) staging standards in breast cancer, according to investigators.
A meta-analysis showed that RCB – which is calculated in the neoadjuvant setting through a combination of primary tumor bed cellularity, lymph node positivity, and size of largest metastasis – was significantly associated with long-term survival of more than 5,000 breast cancer patients, reported principal investigator W. Fraser Symmans, MD, of MD Anderson Cancer Center, Houston, who presented the findings at the San Antonio Breast Cancer Symposium.
Coinvestigator Laura J. Esserman, MD, of the University of California, San Francisco, who was in attendance at Dr. Symmans’ presentation, put the study in context.
“The reason we did this meta-analysis was to really change the joint commission – the AJCC staging – and we expect that will happen on the basis of these results,” Dr. Esserman said. “[RBC] is, moving forward, the standard of care.”
The investigators analyzed individual-level data from 5,160 patients treated at multiple institutions. For each patient, RCB, which is scored from 0 to III, from pathologic complete response (0) to high disease burden (III), was compared with event-free survival (EFS) and disease relapse-free survival (DRFS).
The results showed that both EFS and DRFS were strongly associated with RCB for the overall population and each subtype. For instance, for triple-negative breast cancer, patients with pathologic complete response had a 10-year EFS of 86%, a rate that decreased as RCB increased from RCB-I (75%) to RCB-II (61%), and finally, RCB-III (25%). This trend was similar for patients with HR-negative/HER2-positive or HR-positive/HER2-positive disease. Risk stratification for HR-positive/HER2-negative disease deviated slightly; pathologic complete response was associated with a 10-year EFS of 81%, compared with 86% for RCB-I.
Regarding this finding, Dr. Symmans noted that RCB is most prognostic when higher levels of residual disease are present. Still, reviewing long-term risk across subtypes, Dr. Fraser emphasized the reliability of RCB as a prognostic tool.
“The strong relationship [between RCB and] risk is very clear in each and every subtype of disease,” he said. “There are very tight confidence intervals around those risk estimates.”
Multivariate analysis provided further support for this conclusion, as RCB was independently prognostic for survival in each subtype of disease.
According to Dr. Esserman, the study findings should encourage implementation of RCB in daily practice.
“There has to be training and a commitment to using this in a consistent way so that we can all rely on the information,” Dr. Esserman said. “This is critical for our patients, and this is ... how we’re going to get people to the best outcome, and [it should be done] up front, when we still have the chance to cure people.”
Session moderator Virginia Kaklamani, MD, of Mays Cancer Center, San Antonio, agreed that clinicians should use RCB in practice; however, she also explained that RCB results for some patients are not yet actionable.
“I ask my pathologist to give me the RCB score,” Dr. Kaklamani said. “The question is, what do I do with the results? It’s a little limited. In triple-negative patients I may consider giving capecitabine in the adjuvant setting, or obviously enrolling them in clinical trials. In my HER2-positive patients, I will look at the results from the KATHERINE trial and the ExteNET trial to guide me. In my HR-positive patients, I’m still waiting for results from other trials to see how these results can be interpreted. What we’ve [found] is in many trials, achieving a pathologic complete response in HR-positive patients may not be as important, but as you can see here, it looks like RCB really is.”
The investigators disclosed relationships with Seattle Genetics, Almac, Syndax, and others.
SOURCE: Yau et al. SABCS. 2019 Dec 13. Abstract GS5-01.
REPORTING FROM SABCS 2019
House passes drug pricing bill, likely ending its journey
The House of Representatives passed a partisan drug pricing bill, a move that likely ends its legislative journey as Senate Majority Leader Mitch McConnell (R-Ky.) has already signaled he will not bring it to the Senate floor.

The Elijah E. Cummings Lower Drug Costs Now Act (H.R. 3) passed Dec. 12 on a near party-line vote of 230-192, with two Republicans crossing the aisle to join the Democrats in support of the bill, and no Democrats voting against it. Four members from each party did not record votes.
“The American people are fed up with paying 3, 4, or 10 times more than people in other countries for the exact same drug,” House Energy and Commerce Committee Chairman Frank Pallone (D-N.J) said in a statement following the passage. “I’m proud that the House took decisive action today to finally level the playing field and provide real relief to the American people.”
H.R. 3 would give the secretary of Health and Human Services the ability to negotiate with drug manufacturers on the price of pharmaceuticals in Medicare Part D (and available in the commercial markets) using an international pricing benchmark and would penalize manufacturers who do not negotiate or fail to lower prices to be more in line with generally lower costs internationally.
Drug prices would need to be within 120% of the average price in a reference group of six nations: Australia, Canada, France, Germany, Japan, and the United Kingdom.
Savings from the lower costs that result from negotiations would be reinvested into medical research.
Passage of H.R. 3 would “lower ... medication by 65%” per year for women with breast cancer, Rep. Haley Stevens (D-Mich.) said during the floor debate.
The Congressional Budget Office estimated that the drug price negotiation provision would lower spending on pharmaceuticals by $465 billion over the next 10 years, offset partially by an increase in spending by $358 billion associated with provisions to provide dental, vision, and hearing coverage.
The bill also includes mandatory rebates to the federal government when a drug’s price rises faster than the rate of inflation. It includes an annual cap on out-of-pocket spending on pharmaceuticals of $2,000 for Medicare Part D participants.
The bill was contentious from its introduction, which erased bipartisan work across the committees of jurisdiction, including a number of individual bills that passed at the committee level with bipartisan support.
The price negotiation scheme, using the international benchmark, was a key point of objection, which some argued was more akin to price-setting.
“Government price setting will kill innovation in clinical areas where it is most needed,” Rep. George Holding (R-N.C.) said during the floor debate. “The pricing scheme outlined in H.R. 3 would disincentivize research and development for drugs that are first in their class, such as the future cure for Alzheimer’s.”
The CBO estimates that if the bill were enacted, “8 fewer drugs would be introduced to the U.S. market over the 2020-2029 period, and about 30 fewer drugs over the subsequent decade.”
An amendment offered by House Energy and Commerce Committee Ranking Member Greg Walden (R-Ore.) attempted to replace the language of H.R. 3 with substitute language that collected all the individual drug pricing–related bills that had previously passed with bipartisan support at the committee level, but that was voted down by 223-201 vote with 12 members not voting.
Rep. Walden noted that policies within his substitute “unanimously passed the Energy and Commerce Committee earlier this year. They would have unanimously passed on this House floor had a poison pill not been put in up in the Rules Committee.”
Rep. Katie Porter (D-Calif.) countered that Rep. Walden’s amendment “doesn’t tackle the fundamental problem, which is reducing drug prices. This amendment fails to solve the main problem of actually lowering drug prices.”
The House of Representatives passed a partisan drug pricing bill, a move that likely ends its legislative journey as Senate Majority Leader Mitch McConnell (R-Ky.) has already signaled he will not bring it to the Senate floor.
The Elijah E. Cummings Lower Drug Costs Now Act (H.R. 3) passed Dec. 12 on a near party-line vote of 230-192, with two Republicans crossing the aisle to join the Democrats in support of the bill, and no Democrats voting against it. Four members from each party did not record votes.
“The American people are fed up with paying 3, 4, or 10 times more than people in other countries for the exact same drug,” House Energy and Commerce Committee Chairman Frank Pallone (D-N.J) said in a statement following the passage. “I’m proud that the House took decisive action today to finally level the playing field and provide real relief to the American people.”
H.R. 3 would give the secretary of Health and Human Services the ability to negotiate with drug manufacturers on the price of pharmaceuticals in Medicare Part D (and available in the commercial markets) using an international pricing benchmark and would penalize manufacturers who do not negotiate or fail to lower prices to be more in line with generally lower costs internationally.
Drug prices would need to be within 120% of the average price in a reference group of six nations: Australia, Canada, France, Germany, Japan, and the United Kingdom.
Savings from the lower costs that result from negotiations would be reinvested into medical research.
Passage of H.R. 3 would “lower ... medication by 65%” per year for women with breast cancer, Rep. Haley Stevens (D-Mich.) said during the floor debate.
The Congressional Budget Office estimated that the drug price negotiation provision would lower spending on pharmaceuticals by $465 billion over the next 10 years, offset partially by an increase in spending by $358 billion associated with provisions to provide dental, vision, and hearing coverage.
The bill also includes mandatory rebates to the federal government when a drug’s price rises faster than the rate of inflation. It includes an annual cap on out-of-pocket spending on pharmaceuticals of $2,000 for Medicare Part D participants.
The bill was contentious from its introduction, which erased bipartisan work across the committees of jurisdiction, including a number of individual bills that passed at the committee level with bipartisan support.
The price negotiation scheme, using the international benchmark, was a key point of objection, which some argued was more akin to price-setting.
“Government price setting will kill innovation in clinical areas where it is most needed,” Rep. George Holding (R-N.C.) said during the floor debate. “The pricing scheme outlined in H.R. 3 would disincentivize research and development for drugs that are first in their class, such as the future cure for Alzheimer’s.”
The CBO estimates that if the bill were enacted, “8 fewer drugs would be introduced to the U.S. market over the 2020-2029 period, and about 30 fewer drugs over the subsequent decade.”
An amendment offered by House Energy and Commerce Committee Ranking Member Greg Walden (R-Ore.) attempted to replace the language of H.R. 3 with substitute language that collected all the individual drug pricing–related bills that had previously passed with bipartisan support at the committee level, but that was voted down by 223-201 vote with 12 members not voting.
Rep. Walden noted that policies within his substitute “unanimously passed the Energy and Commerce Committee earlier this year. They would have unanimously passed on this House floor had a poison pill not been put in up in the Rules Committee.”
Rep. Katie Porter (D-Calif.) countered that Rep. Walden’s amendment “doesn’t tackle the fundamental problem, which is reducing drug prices. This amendment fails to solve the main problem of actually lowering drug prices.”
The House of Representatives passed a partisan drug pricing bill, a move that likely ends its legislative journey as Senate Majority Leader Mitch McConnell (R-Ky.) has already signaled he will not bring it to the Senate floor.
The Elijah E. Cummings Lower Drug Costs Now Act (H.R. 3) passed Dec. 12 on a near party-line vote of 230-192, with two Republicans crossing the aisle to join the Democrats in support of the bill, and no Democrats voting against it. Four members from each party did not record votes.
“The American people are fed up with paying 3, 4, or 10 times more than people in other countries for the exact same drug,” House Energy and Commerce Committee Chairman Frank Pallone (D-N.J) said in a statement following the passage. “I’m proud that the House took decisive action today to finally level the playing field and provide real relief to the American people.”
H.R. 3 would give the secretary of Health and Human Services the ability to negotiate with drug manufacturers on the price of pharmaceuticals in Medicare Part D (and available in the commercial markets) using an international pricing benchmark and would penalize manufacturers who do not negotiate or fail to lower prices to be more in line with generally lower costs internationally.
Drug prices would need to be within 120% of the average price in a reference group of six nations: Australia, Canada, France, Germany, Japan, and the United Kingdom.
Savings from the lower costs that result from negotiations would be reinvested into medical research.
Passage of H.R. 3 would “lower ... medication by 65%” per year for women with breast cancer, Rep. Haley Stevens (D-Mich.) said during the floor debate.
The Congressional Budget Office estimated that the drug price negotiation provision would lower spending on pharmaceuticals by $465 billion over the next 10 years, offset partially by an increase in spending by $358 billion associated with provisions to provide dental, vision, and hearing coverage.
The bill also includes mandatory rebates to the federal government when a drug’s price rises faster than the rate of inflation. It includes an annual cap on out-of-pocket spending on pharmaceuticals of $2,000 for Medicare Part D participants.
The bill was contentious from its introduction, which erased bipartisan work across the committees of jurisdiction, including a number of individual bills that passed at the committee level with bipartisan support.
The price negotiation scheme, using the international benchmark, was a key point of objection, which some argued was more akin to price-setting.
“Government price setting will kill innovation in clinical areas where it is most needed,” Rep. George Holding (R-N.C.) said during the floor debate. “The pricing scheme outlined in H.R. 3 would disincentivize research and development for drugs that are first in their class, such as the future cure for Alzheimer’s.”
The CBO estimates that if the bill were enacted, “8 fewer drugs would be introduced to the U.S. market over the 2020-2029 period, and about 30 fewer drugs over the subsequent decade.”
An amendment offered by House Energy and Commerce Committee Ranking Member Greg Walden (R-Ore.) attempted to replace the language of H.R. 3 with substitute language that collected all the individual drug pricing–related bills that had previously passed with bipartisan support at the committee level, but that was voted down by 223-201 vote with 12 members not voting.
Rep. Walden noted that policies within his substitute “unanimously passed the Energy and Commerce Committee earlier this year. They would have unanimously passed on this House floor had a poison pill not been put in up in the Rules Committee.”
Rep. Katie Porter (D-Calif.) countered that Rep. Walden’s amendment “doesn’t tackle the fundamental problem, which is reducing drug prices. This amendment fails to solve the main problem of actually lowering drug prices.”