User login
Moderate coffee intake associated with few seizures in drug-refractory patients with epilepsy
BALTIMORE – The effect of caffeine on seizures may be dose dependent, according to research presented at the annual meeting of the American Epilepsy Society. Moderate doses of caffeine may benefit patients with epilepsy, whereas high doses – four cups of coffee per day or more – may increase seizure susceptibility, said Julie Bourgeois-Vionnet, MD, of the department of functional neurology and epileptology at Hospices Civils de Lyon in France.

In rodent model studies, caffeine has been found in general to increase seizure susceptibility but with variable results according to dose and route of administration, but other studies of chronic low-dose exposure to caffeine have reported protective effects against seizures and sudden unexpected death in epilepsy (SUDEP; Epilepsy Behav. 2018 Mar;80:37-47). In patients, however, the relationship between caffeine consumption and seizure frequency has been less clear.
To examine the relationship between caffeine consumption and seizure frequency in patients with drug-resistant epilepsy, Dr. Bourgeois-Vionnet and colleagues analyzed data patients in the Safety of Antiepileptic Withdrawal in Long Term Video-EEG Monitoring (SAVE) study. This ongoing, multicenter, open-label trial is evaluating the management of antiepileptic drugs withdrawal during long-term monitoring in patients with drug-resistant focal epilepsy.
For the present analysis, the researchers examined data from 620 adults who were included in the SAVE study between 2016 and 2018 and had information available about coffee consumption and seizure frequency, including seizure frequency during the previous 3 months and number of focal seizure evolving to generalized tonic-clonic seizures (secondary generalized tonic-clonic seizures [sGTCS]) during the past year. Patients provided information about coffee consumption via a standardized questionnaire.
The investigators classified caffeine consumption as none, rare (less than 1 cup/week to up to 3 cups/week), moderate (between 4 cups/week and 3 cups/day) and high (more than 4 cups/day). The researchers evaluated risk of SUDEP using the revised SUDEP-7 inventory.
The patients had an average age of 36.2 years and an average duration of epilepsy of 18.1 years. In the 3 months preceding study inclusion, the median seizure frequency of any type was 4.33 per month. In all, 217 patients reported sGTCS in the past year.
Overall, 194 patients reported no coffee consumption, 149 reported rare coffee consumption, 177 moderate consumption, and 100 high consumption. The revised SUDEP-7 inventory was available for 607 patients, and the median score was 3.0.
Patients with moderate coffee consumption were more likely to not have any sGTCS (73.4%), compared with patients with no coffee consumption (64.4%), rare consumption (61.7%), and high consumption (56%). Likewise, patients with moderate coffee consumption were less likely to have more than three sGTCS per year (19.2%), compared with patients no coffee consumption (28.9%), rare consumption (24.8%), and high consumption (30%).
“There was no relation between caffeine consumption and seizure frequency of any type,” Dr. Bourgeois-Vionnet and colleagues reported. “However, we observed a bimodal association between frequency of sGTCS and coffee consumption. In contrast, no significant association was observed between score of the SUDEP-7 inventory and level of caffeine consumption.”
While these findings still need to be confirmed in prospective studies, they suggest possible guidance for patients, Dr. Bourgeois-Vionnet said. “They are allowed to drink coffee, but maybe avoid high doses,” she said.
The study was funded by the French Ministry of Health. The researchers had no disclosures.
SOURCE: Bourgeois-Vionnet J. AES 2019, Abstract 2.141.
BALTIMORE – The effect of caffeine on seizures may be dose dependent, according to research presented at the annual meeting of the American Epilepsy Society. Moderate doses of caffeine may benefit patients with epilepsy, whereas high doses – four cups of coffee per day or more – may increase seizure susceptibility, said Julie Bourgeois-Vionnet, MD, of the department of functional neurology and epileptology at Hospices Civils de Lyon in France.
In rodent model studies, caffeine has been found in general to increase seizure susceptibility but with variable results according to dose and route of administration, but other studies of chronic low-dose exposure to caffeine have reported protective effects against seizures and sudden unexpected death in epilepsy (SUDEP; Epilepsy Behav. 2018 Mar;80:37-47). In patients, however, the relationship between caffeine consumption and seizure frequency has been less clear.
To examine the relationship between caffeine consumption and seizure frequency in patients with drug-resistant epilepsy, Dr. Bourgeois-Vionnet and colleagues analyzed data patients in the Safety of Antiepileptic Withdrawal in Long Term Video-EEG Monitoring (SAVE) study. This ongoing, multicenter, open-label trial is evaluating the management of antiepileptic drugs withdrawal during long-term monitoring in patients with drug-resistant focal epilepsy.
For the present analysis, the researchers examined data from 620 adults who were included in the SAVE study between 2016 and 2018 and had information available about coffee consumption and seizure frequency, including seizure frequency during the previous 3 months and number of focal seizure evolving to generalized tonic-clonic seizures (secondary generalized tonic-clonic seizures [sGTCS]) during the past year. Patients provided information about coffee consumption via a standardized questionnaire.
The investigators classified caffeine consumption as none, rare (less than 1 cup/week to up to 3 cups/week), moderate (between 4 cups/week and 3 cups/day) and high (more than 4 cups/day). The researchers evaluated risk of SUDEP using the revised SUDEP-7 inventory.
The patients had an average age of 36.2 years and an average duration of epilepsy of 18.1 years. In the 3 months preceding study inclusion, the median seizure frequency of any type was 4.33 per month. In all, 217 patients reported sGTCS in the past year.
Overall, 194 patients reported no coffee consumption, 149 reported rare coffee consumption, 177 moderate consumption, and 100 high consumption. The revised SUDEP-7 inventory was available for 607 patients, and the median score was 3.0.
Patients with moderate coffee consumption were more likely to not have any sGTCS (73.4%), compared with patients with no coffee consumption (64.4%), rare consumption (61.7%), and high consumption (56%). Likewise, patients with moderate coffee consumption were less likely to have more than three sGTCS per year (19.2%), compared with patients no coffee consumption (28.9%), rare consumption (24.8%), and high consumption (30%).
“There was no relation between caffeine consumption and seizure frequency of any type,” Dr. Bourgeois-Vionnet and colleagues reported. “However, we observed a bimodal association between frequency of sGTCS and coffee consumption. In contrast, no significant association was observed between score of the SUDEP-7 inventory and level of caffeine consumption.”
While these findings still need to be confirmed in prospective studies, they suggest possible guidance for patients, Dr. Bourgeois-Vionnet said. “They are allowed to drink coffee, but maybe avoid high doses,” she said.
The study was funded by the French Ministry of Health. The researchers had no disclosures.
SOURCE: Bourgeois-Vionnet J. AES 2019, Abstract 2.141.
BALTIMORE – The effect of caffeine on seizures may be dose dependent, according to research presented at the annual meeting of the American Epilepsy Society. Moderate doses of caffeine may benefit patients with epilepsy, whereas high doses – four cups of coffee per day or more – may increase seizure susceptibility, said Julie Bourgeois-Vionnet, MD, of the department of functional neurology and epileptology at Hospices Civils de Lyon in France.
In rodent model studies, caffeine has been found in general to increase seizure susceptibility but with variable results according to dose and route of administration, but other studies of chronic low-dose exposure to caffeine have reported protective effects against seizures and sudden unexpected death in epilepsy (SUDEP; Epilepsy Behav. 2018 Mar;80:37-47). In patients, however, the relationship between caffeine consumption and seizure frequency has been less clear.
To examine the relationship between caffeine consumption and seizure frequency in patients with drug-resistant epilepsy, Dr. Bourgeois-Vionnet and colleagues analyzed data patients in the Safety of Antiepileptic Withdrawal in Long Term Video-EEG Monitoring (SAVE) study. This ongoing, multicenter, open-label trial is evaluating the management of antiepileptic drugs withdrawal during long-term monitoring in patients with drug-resistant focal epilepsy.
For the present analysis, the researchers examined data from 620 adults who were included in the SAVE study between 2016 and 2018 and had information available about coffee consumption and seizure frequency, including seizure frequency during the previous 3 months and number of focal seizure evolving to generalized tonic-clonic seizures (secondary generalized tonic-clonic seizures [sGTCS]) during the past year. Patients provided information about coffee consumption via a standardized questionnaire.
The investigators classified caffeine consumption as none, rare (less than 1 cup/week to up to 3 cups/week), moderate (between 4 cups/week and 3 cups/day) and high (more than 4 cups/day). The researchers evaluated risk of SUDEP using the revised SUDEP-7 inventory.
The patients had an average age of 36.2 years and an average duration of epilepsy of 18.1 years. In the 3 months preceding study inclusion, the median seizure frequency of any type was 4.33 per month. In all, 217 patients reported sGTCS in the past year.
Overall, 194 patients reported no coffee consumption, 149 reported rare coffee consumption, 177 moderate consumption, and 100 high consumption. The revised SUDEP-7 inventory was available for 607 patients, and the median score was 3.0.
Patients with moderate coffee consumption were more likely to not have any sGTCS (73.4%), compared with patients with no coffee consumption (64.4%), rare consumption (61.7%), and high consumption (56%). Likewise, patients with moderate coffee consumption were less likely to have more than three sGTCS per year (19.2%), compared with patients no coffee consumption (28.9%), rare consumption (24.8%), and high consumption (30%).
“There was no relation between caffeine consumption and seizure frequency of any type,” Dr. Bourgeois-Vionnet and colleagues reported. “However, we observed a bimodal association between frequency of sGTCS and coffee consumption. In contrast, no significant association was observed between score of the SUDEP-7 inventory and level of caffeine consumption.”
While these findings still need to be confirmed in prospective studies, they suggest possible guidance for patients, Dr. Bourgeois-Vionnet said. “They are allowed to drink coffee, but maybe avoid high doses,” she said.
The study was funded by the French Ministry of Health. The researchers had no disclosures.
SOURCE: Bourgeois-Vionnet J. AES 2019, Abstract 2.141.
REPORTING FROM AES 2019
Drug recall in U.S. causes extreme hardship for hypoparathyroid patients
The recall of injectable recombinant parathyroid hormone (PTH; Natpara) in the United States over safety concerns about the product is gravely affecting the lives of American patients with severe hypoparathyroidism who need replacement PTH therapy to adequately control symptoms.
Approximately 2,700 patients in the United States have been affected by the recall of Natpara, according to one of the manufacturers, Takeda. Those who were taking it have had to transition back to controlling their symptoms with just oral medications, including active vitamin D or calcitriol (Rocaltrol) plus calcium supplements, which has had a negative impact on the management of their disease in most cases, with some patients hospitalized because of severe hypocalcemia.
One patient related in an interview that even if the transition from injectable PTH back to calcium supplements and calcitriol goes smoothly, individuals can still feel pretty awful because the benefits of the Natpara are subtle – patients just feel better on it. And one report indicates at least 100 patients who had to stop Natpara because of the Food and Drug Administration recall have been hospitalized.
“It’s been a rough time for all patients previously on Natpara adjusting to oral medications after they have been on a replacement therapy,” endocrinologist Dolores Shoback, MD, University of California, San Francisco, said in an email.
Aliya Khan, MD, agrees: “This molecule ... is life changing ... [It] makes a huge difference in patients’ quality of life and in their ability to function normally.”
“The FDA in the United States made the recall decision, and we have to respect that,” added Dr. Khan, a professor of clinical medicine at McMaster University in Hamilton, Ontario.
“We need to address patients’ needs and [try to] reinstate it,” Dr. Khan added, who is the lead author of recent guidelines on the treatment of hypoparathyroidism and who has received research funding from Takeda and Ascendis Pharma, both of which make injectable PTH.
Dr. Kahn also has advice for U.S. endocrinologists who are having to deal with patients who were previously on Natpara and no longer have access to the drug.
She explains how they can transition patients back to other available therapies.
Oral treatments a 'Band-Aid'
Hypoparathyroidism is a rare endocrine disorder that affects approximately 60,000 people in the United States. The FDA approved Natpara as an orphan drug and as an adjunct to calcium and vitamin D to control hypocalcemia in patients with hypoparathyroidism in January 2015.
The FDA issued the recall for Natpara in September, out of concern that rubber particulate matter originating from the rubber septum of the drug’s cartridge might be contaminating the product.
But the European Medicines Agency, which approved the product under the brand name Natpar in 2017, has not reached the same decision as the FDA; patients in Europe who qualify for injectable PTH still have access to the drug.
As Dr. Khan explained in an interview, PTH is critical for the control of calcium and phosphate homeostasis.
When functioning normally, the parathyroid glands are able to regulate blood calcium levels very finely, sensing how much calcium is being resorbed through the kidney, how much is going in and out of bone, how active vitamin D levels are, and the amount the body is absorbing from the bowel and bone. “It’s all being very carefully fine-tuned continuously,” she said.
Patients with hypoparathyroidism suffer from very low levels of serum calcium, which can, in turn, lead to seizures and cardiac irregularities as well as bronchospasm and even respiratory failure. Magnesium levels can also be dysregulated by hypocalcemia.
“All these are serious results of very low calcium levels,” Dr. Khan emphasized.
These patients “are also not able to eliminate phosphate through the kidneys so their phosphate levels rise,” she added.
Standard treatment for hypoparathyroidism is active vitamin D or calcitriol plus oral calcium supplements.
However, for patients with severe hypoparathyroidism, for whom injectable PTH is indicated, standard treatment is like a “Band-Aid” because what they really need is replacement PTH to help them regulate calcium levels as finely as possible, Dr. Khan said.
High risk for severe hypocalcemia
If PTH is stopped abruptly, calcium levels will likely plummet, a phenomenon sometimes referred to as hungry bone syndrome, placing patients at risk for the consequences of severe hypocalcemia, Dr. Khan added.
(Hungry bone syndrome refers to the rapid, profound, and prolonged hypocalcemia associated with hypophosphatemia and hypomagnesemia, which is exacerbated by suppressed PTH).
One report suggests at least 100 patients who had to stop Natpara because of the FDA recall had to seek hospitalization to have calcium levels restored intravenously.
And even if the transition from injectable PTH back to calcium supplements and calcitriol goes smoothly, patients can still feel pretty awful.
“When you look at my blood levels on and off Natpara, they look very similar,” Danette Astolfi, a former Natpara user in the United States who recently transitioned from the injectable to standard of care, said in an interview.
“But I felt much, much better on Natpara, so this is an interesting ‘wild card’ that patients have with this drug – they just feel better on it. They don’t have the fatigue, they don’t have the brain fog, they don’t have the aches and pains even if their calcium levels are normal,” Ms. Astolfi said.
So patients who are candidates for injectable PTH therapy are by definition those who do not do very well on standard-of-care calcium and calcitriol.
“With the recall, patients are going back to this regimen they already did not have great success with,” Ms. Astolfi explained. “And that’s the tricky part because if you fall into this category, as I do, there is not much you can do about it.”
Ms. Astolfi, who is also on the board of directors of the Hypoparathyroidism Association in Pennsylvania, was lucky because she did not have to stop treatment with injectable PTH abruptly.
“I was fortunate. My physician did not want me to stop the drug right away, so we had time to develop a discontinuation plan,” she noted.
Transitioning safely to other therapies
Dr. Khan has advice for endocrinologists dealing with patients who were previously on Natpara and no longer have access to the drug.
First, “endocrinologists need to be aware that when you stop PTH treatment suddenly, patients’ need for calcium supplements and calcitriol may be as high as two times what they were on before they started PTH therapy,” she noted.
Physicians also need to follow patients closely by monitoring not only calcium and vitamin D levels but also phosphate and magnesium almost daily.
Dr. Khan said she does this because it’s difficult to predict how much calcium and calcitriol a patient will require and doses will likely have to be titrated up or down based on lab results.
It is noteworthy that a joint statement on the Natpara recall by the Endocrine Society and the American Society of Bone and Mineral Research (ASBMR) also emphasizes these points.
And Dr. Khan – as well as the Endocrine Society and ASBMR – also point out that patients may be transitioned from Natpara to teriparatide (Forteo), another recombinant form of PTH that is licensed for use in osteoporosis but has not been approved for hypoparathyroidism.
In Dr. Khan’s experience, patients often do quite well on teriparatide.
The big downside of prescribing teriparatide in the United States, however, is that the drug has to be used off label, and as such, insurance companies are unlikely to pick up the tab for what is an expensive treatment, she explained.
Furthermore, teriparatide has a very short shelf-life once injected so patients may need to use up to four needles a day to stabilize calcium levels.
If teriparatide is prescribed, subcutaneous injections given in the thigh are recommended for the treatment of hypoparathyroidism, according to the joint Endocrine Society/ASBMR statement.
Takeda vs. FDA
In the meantime, Takeda says it has been in regular contact with the FDA to try and work out how to bring Natpara back to U.S. patients who require it for symptom control.
Current outstanding issues between the company and FDA involve considerations related to dose accuracy, safety, and supply continuity, which the company hopes will be resolved in a timely manner.
Furthermore, shortly after the recall order, the company created a special use program that continues to support patients previously prescribed Natpara who would otherwise be at extreme risk of serious complications if forced to stop taking the drug.
“Originally intended for an extremely limited number of patients, the special use program has now enrolled approximately 300 patients,” Cheryl Schwartz, head, U.S. Hematology & Rare Business Unit, Takeda, said in a letter to members of the U.S. Hypoparathyroidism Association on Nov. 21.
The number is higher than expected and reflects the significant treatment needs of patients with serious disease, Ms. Schwartz noted.
“I want to reiterate that we do understand and sincerely regret the impact the Natpara recall is having on patients. We will provide another update as soon as we have more information to share. We continue to work around the clock to find ways to bring Natpara back to the broader hypoparathyroidism community,” she added.
“Patient safety always has been, and continues to be, the highest priority for Takeda,” she said.
Final efforts
Dr. Khan is currently involved in clinical trials evaluating Natpara in Canada (where it is not yet approved).
She was also involved in some of the pivotal studies that helped gain support of the product in the United States and European Union, including REPLACE, which showed that the injectable PTH molecule maintained serum calcium while reducing or eliminating requirements for calcium and active vitamin D supplementation.
“We now have 8 years of experience with this molecule, so we know that it’s safe and effective,” Dr. Khan emphasized.
Health Canada has not deemed Natpara problematic enough to stop the ongoing research program into injectable PTH therapies there, which is being headed up by Dr. Khan at McMaster University.
“The company has not yet applied for approval here, but when they do apply, I am sure they will have fixed this problem by then,” Dr. Khan said. “And I expect it will be fixed momentarily in the United States, where patients have been most affected.”
Asked if she was looking forward to getting Natpara back on track, Ms. Astolfi said “absolutely” several times.
“This recall has really had a large impact on our community,” she stressed. “And while it’s good that critically ill patients have access to the drug through the special-use program, we want to get everyone back to their best life and feeling great.”
A version of this story originally appeared on Medscape.com.
The recall of injectable recombinant parathyroid hormone (PTH; Natpara) in the United States over safety concerns about the product is gravely affecting the lives of American patients with severe hypoparathyroidism who need replacement PTH therapy to adequately control symptoms.
Approximately 2,700 patients in the United States have been affected by the recall of Natpara, according to one of the manufacturers, Takeda. Those who were taking it have had to transition back to controlling their symptoms with just oral medications, including active vitamin D or calcitriol (Rocaltrol) plus calcium supplements, which has had a negative impact on the management of their disease in most cases, with some patients hospitalized because of severe hypocalcemia.
One patient related in an interview that even if the transition from injectable PTH back to calcium supplements and calcitriol goes smoothly, individuals can still feel pretty awful because the benefits of the Natpara are subtle – patients just feel better on it. And one report indicates at least 100 patients who had to stop Natpara because of the Food and Drug Administration recall have been hospitalized.
“It’s been a rough time for all patients previously on Natpara adjusting to oral medications after they have been on a replacement therapy,” endocrinologist Dolores Shoback, MD, University of California, San Francisco, said in an email.
Aliya Khan, MD, agrees: “This molecule ... is life changing ... [It] makes a huge difference in patients’ quality of life and in their ability to function normally.”
“The FDA in the United States made the recall decision, and we have to respect that,” added Dr. Khan, a professor of clinical medicine at McMaster University in Hamilton, Ontario.
“We need to address patients’ needs and [try to] reinstate it,” Dr. Khan added, who is the lead author of recent guidelines on the treatment of hypoparathyroidism and who has received research funding from Takeda and Ascendis Pharma, both of which make injectable PTH.
Dr. Kahn also has advice for U.S. endocrinologists who are having to deal with patients who were previously on Natpara and no longer have access to the drug.
She explains how they can transition patients back to other available therapies.
Oral treatments a 'Band-Aid'
Hypoparathyroidism is a rare endocrine disorder that affects approximately 60,000 people in the United States. The FDA approved Natpara as an orphan drug and as an adjunct to calcium and vitamin D to control hypocalcemia in patients with hypoparathyroidism in January 2015.
The FDA issued the recall for Natpara in September, out of concern that rubber particulate matter originating from the rubber septum of the drug’s cartridge might be contaminating the product.
But the European Medicines Agency, which approved the product under the brand name Natpar in 2017, has not reached the same decision as the FDA; patients in Europe who qualify for injectable PTH still have access to the drug.
As Dr. Khan explained in an interview, PTH is critical for the control of calcium and phosphate homeostasis.
When functioning normally, the parathyroid glands are able to regulate blood calcium levels very finely, sensing how much calcium is being resorbed through the kidney, how much is going in and out of bone, how active vitamin D levels are, and the amount the body is absorbing from the bowel and bone. “It’s all being very carefully fine-tuned continuously,” she said.
Patients with hypoparathyroidism suffer from very low levels of serum calcium, which can, in turn, lead to seizures and cardiac irregularities as well as bronchospasm and even respiratory failure. Magnesium levels can also be dysregulated by hypocalcemia.
“All these are serious results of very low calcium levels,” Dr. Khan emphasized.
These patients “are also not able to eliminate phosphate through the kidneys so their phosphate levels rise,” she added.
Standard treatment for hypoparathyroidism is active vitamin D or calcitriol plus oral calcium supplements.
However, for patients with severe hypoparathyroidism, for whom injectable PTH is indicated, standard treatment is like a “Band-Aid” because what they really need is replacement PTH to help them regulate calcium levels as finely as possible, Dr. Khan said.
High risk for severe hypocalcemia
If PTH is stopped abruptly, calcium levels will likely plummet, a phenomenon sometimes referred to as hungry bone syndrome, placing patients at risk for the consequences of severe hypocalcemia, Dr. Khan added.
(Hungry bone syndrome refers to the rapid, profound, and prolonged hypocalcemia associated with hypophosphatemia and hypomagnesemia, which is exacerbated by suppressed PTH).
One report suggests at least 100 patients who had to stop Natpara because of the FDA recall had to seek hospitalization to have calcium levels restored intravenously.
And even if the transition from injectable PTH back to calcium supplements and calcitriol goes smoothly, patients can still feel pretty awful.
“When you look at my blood levels on and off Natpara, they look very similar,” Danette Astolfi, a former Natpara user in the United States who recently transitioned from the injectable to standard of care, said in an interview.
“But I felt much, much better on Natpara, so this is an interesting ‘wild card’ that patients have with this drug – they just feel better on it. They don’t have the fatigue, they don’t have the brain fog, they don’t have the aches and pains even if their calcium levels are normal,” Ms. Astolfi said.
So patients who are candidates for injectable PTH therapy are by definition those who do not do very well on standard-of-care calcium and calcitriol.
“With the recall, patients are going back to this regimen they already did not have great success with,” Ms. Astolfi explained. “And that’s the tricky part because if you fall into this category, as I do, there is not much you can do about it.”
Ms. Astolfi, who is also on the board of directors of the Hypoparathyroidism Association in Pennsylvania, was lucky because she did not have to stop treatment with injectable PTH abruptly.
“I was fortunate. My physician did not want me to stop the drug right away, so we had time to develop a discontinuation plan,” she noted.
Transitioning safely to other therapies
Dr. Khan has advice for endocrinologists dealing with patients who were previously on Natpara and no longer have access to the drug.
First, “endocrinologists need to be aware that when you stop PTH treatment suddenly, patients’ need for calcium supplements and calcitriol may be as high as two times what they were on before they started PTH therapy,” she noted.
Physicians also need to follow patients closely by monitoring not only calcium and vitamin D levels but also phosphate and magnesium almost daily.
Dr. Khan said she does this because it’s difficult to predict how much calcium and calcitriol a patient will require and doses will likely have to be titrated up or down based on lab results.
It is noteworthy that a joint statement on the Natpara recall by the Endocrine Society and the American Society of Bone and Mineral Research (ASBMR) also emphasizes these points.
And Dr. Khan – as well as the Endocrine Society and ASBMR – also point out that patients may be transitioned from Natpara to teriparatide (Forteo), another recombinant form of PTH that is licensed for use in osteoporosis but has not been approved for hypoparathyroidism.
In Dr. Khan’s experience, patients often do quite well on teriparatide.
The big downside of prescribing teriparatide in the United States, however, is that the drug has to be used off label, and as such, insurance companies are unlikely to pick up the tab for what is an expensive treatment, she explained.
Furthermore, teriparatide has a very short shelf-life once injected so patients may need to use up to four needles a day to stabilize calcium levels.
If teriparatide is prescribed, subcutaneous injections given in the thigh are recommended for the treatment of hypoparathyroidism, according to the joint Endocrine Society/ASBMR statement.
Takeda vs. FDA
In the meantime, Takeda says it has been in regular contact with the FDA to try and work out how to bring Natpara back to U.S. patients who require it for symptom control.
Current outstanding issues between the company and FDA involve considerations related to dose accuracy, safety, and supply continuity, which the company hopes will be resolved in a timely manner.
Furthermore, shortly after the recall order, the company created a special use program that continues to support patients previously prescribed Natpara who would otherwise be at extreme risk of serious complications if forced to stop taking the drug.
“Originally intended for an extremely limited number of patients, the special use program has now enrolled approximately 300 patients,” Cheryl Schwartz, head, U.S. Hematology & Rare Business Unit, Takeda, said in a letter to members of the U.S. Hypoparathyroidism Association on Nov. 21.
The number is higher than expected and reflects the significant treatment needs of patients with serious disease, Ms. Schwartz noted.
“I want to reiterate that we do understand and sincerely regret the impact the Natpara recall is having on patients. We will provide another update as soon as we have more information to share. We continue to work around the clock to find ways to bring Natpara back to the broader hypoparathyroidism community,” she added.
“Patient safety always has been, and continues to be, the highest priority for Takeda,” she said.
Final efforts
Dr. Khan is currently involved in clinical trials evaluating Natpara in Canada (where it is not yet approved).
She was also involved in some of the pivotal studies that helped gain support of the product in the United States and European Union, including REPLACE, which showed that the injectable PTH molecule maintained serum calcium while reducing or eliminating requirements for calcium and active vitamin D supplementation.
“We now have 8 years of experience with this molecule, so we know that it’s safe and effective,” Dr. Khan emphasized.
Health Canada has not deemed Natpara problematic enough to stop the ongoing research program into injectable PTH therapies there, which is being headed up by Dr. Khan at McMaster University.
“The company has not yet applied for approval here, but when they do apply, I am sure they will have fixed this problem by then,” Dr. Khan said. “And I expect it will be fixed momentarily in the United States, where patients have been most affected.”
Asked if she was looking forward to getting Natpara back on track, Ms. Astolfi said “absolutely” several times.
“This recall has really had a large impact on our community,” she stressed. “And while it’s good that critically ill patients have access to the drug through the special-use program, we want to get everyone back to their best life and feeling great.”
A version of this story originally appeared on Medscape.com.
The recall of injectable recombinant parathyroid hormone (PTH; Natpara) in the United States over safety concerns about the product is gravely affecting the lives of American patients with severe hypoparathyroidism who need replacement PTH therapy to adequately control symptoms.
Approximately 2,700 patients in the United States have been affected by the recall of Natpara, according to one of the manufacturers, Takeda. Those who were taking it have had to transition back to controlling their symptoms with just oral medications, including active vitamin D or calcitriol (Rocaltrol) plus calcium supplements, which has had a negative impact on the management of their disease in most cases, with some patients hospitalized because of severe hypocalcemia.
One patient related in an interview that even if the transition from injectable PTH back to calcium supplements and calcitriol goes smoothly, individuals can still feel pretty awful because the benefits of the Natpara are subtle – patients just feel better on it. And one report indicates at least 100 patients who had to stop Natpara because of the Food and Drug Administration recall have been hospitalized.
“It’s been a rough time for all patients previously on Natpara adjusting to oral medications after they have been on a replacement therapy,” endocrinologist Dolores Shoback, MD, University of California, San Francisco, said in an email.
Aliya Khan, MD, agrees: “This molecule ... is life changing ... [It] makes a huge difference in patients’ quality of life and in their ability to function normally.”
“The FDA in the United States made the recall decision, and we have to respect that,” added Dr. Khan, a professor of clinical medicine at McMaster University in Hamilton, Ontario.
“We need to address patients’ needs and [try to] reinstate it,” Dr. Khan added, who is the lead author of recent guidelines on the treatment of hypoparathyroidism and who has received research funding from Takeda and Ascendis Pharma, both of which make injectable PTH.
Dr. Kahn also has advice for U.S. endocrinologists who are having to deal with patients who were previously on Natpara and no longer have access to the drug.
She explains how they can transition patients back to other available therapies.
Oral treatments a 'Band-Aid'
Hypoparathyroidism is a rare endocrine disorder that affects approximately 60,000 people in the United States. The FDA approved Natpara as an orphan drug and as an adjunct to calcium and vitamin D to control hypocalcemia in patients with hypoparathyroidism in January 2015.
The FDA issued the recall for Natpara in September, out of concern that rubber particulate matter originating from the rubber septum of the drug’s cartridge might be contaminating the product.
But the European Medicines Agency, which approved the product under the brand name Natpar in 2017, has not reached the same decision as the FDA; patients in Europe who qualify for injectable PTH still have access to the drug.
As Dr. Khan explained in an interview, PTH is critical for the control of calcium and phosphate homeostasis.
When functioning normally, the parathyroid glands are able to regulate blood calcium levels very finely, sensing how much calcium is being resorbed through the kidney, how much is going in and out of bone, how active vitamin D levels are, and the amount the body is absorbing from the bowel and bone. “It’s all being very carefully fine-tuned continuously,” she said.
Patients with hypoparathyroidism suffer from very low levels of serum calcium, which can, in turn, lead to seizures and cardiac irregularities as well as bronchospasm and even respiratory failure. Magnesium levels can also be dysregulated by hypocalcemia.
“All these are serious results of very low calcium levels,” Dr. Khan emphasized.
These patients “are also not able to eliminate phosphate through the kidneys so their phosphate levels rise,” she added.
Standard treatment for hypoparathyroidism is active vitamin D or calcitriol plus oral calcium supplements.
However, for patients with severe hypoparathyroidism, for whom injectable PTH is indicated, standard treatment is like a “Band-Aid” because what they really need is replacement PTH to help them regulate calcium levels as finely as possible, Dr. Khan said.
High risk for severe hypocalcemia
If PTH is stopped abruptly, calcium levels will likely plummet, a phenomenon sometimes referred to as hungry bone syndrome, placing patients at risk for the consequences of severe hypocalcemia, Dr. Khan added.
(Hungry bone syndrome refers to the rapid, profound, and prolonged hypocalcemia associated with hypophosphatemia and hypomagnesemia, which is exacerbated by suppressed PTH).
One report suggests at least 100 patients who had to stop Natpara because of the FDA recall had to seek hospitalization to have calcium levels restored intravenously.
And even if the transition from injectable PTH back to calcium supplements and calcitriol goes smoothly, patients can still feel pretty awful.
“When you look at my blood levels on and off Natpara, they look very similar,” Danette Astolfi, a former Natpara user in the United States who recently transitioned from the injectable to standard of care, said in an interview.
“But I felt much, much better on Natpara, so this is an interesting ‘wild card’ that patients have with this drug – they just feel better on it. They don’t have the fatigue, they don’t have the brain fog, they don’t have the aches and pains even if their calcium levels are normal,” Ms. Astolfi said.
So patients who are candidates for injectable PTH therapy are by definition those who do not do very well on standard-of-care calcium and calcitriol.
“With the recall, patients are going back to this regimen they already did not have great success with,” Ms. Astolfi explained. “And that’s the tricky part because if you fall into this category, as I do, there is not much you can do about it.”
Ms. Astolfi, who is also on the board of directors of the Hypoparathyroidism Association in Pennsylvania, was lucky because she did not have to stop treatment with injectable PTH abruptly.
“I was fortunate. My physician did not want me to stop the drug right away, so we had time to develop a discontinuation plan,” she noted.
Transitioning safely to other therapies
Dr. Khan has advice for endocrinologists dealing with patients who were previously on Natpara and no longer have access to the drug.
First, “endocrinologists need to be aware that when you stop PTH treatment suddenly, patients’ need for calcium supplements and calcitriol may be as high as two times what they were on before they started PTH therapy,” she noted.
Physicians also need to follow patients closely by monitoring not only calcium and vitamin D levels but also phosphate and magnesium almost daily.
Dr. Khan said she does this because it’s difficult to predict how much calcium and calcitriol a patient will require and doses will likely have to be titrated up or down based on lab results.
It is noteworthy that a joint statement on the Natpara recall by the Endocrine Society and the American Society of Bone and Mineral Research (ASBMR) also emphasizes these points.
And Dr. Khan – as well as the Endocrine Society and ASBMR – also point out that patients may be transitioned from Natpara to teriparatide (Forteo), another recombinant form of PTH that is licensed for use in osteoporosis but has not been approved for hypoparathyroidism.
In Dr. Khan’s experience, patients often do quite well on teriparatide.
The big downside of prescribing teriparatide in the United States, however, is that the drug has to be used off label, and as such, insurance companies are unlikely to pick up the tab for what is an expensive treatment, she explained.
Furthermore, teriparatide has a very short shelf-life once injected so patients may need to use up to four needles a day to stabilize calcium levels.
If teriparatide is prescribed, subcutaneous injections given in the thigh are recommended for the treatment of hypoparathyroidism, according to the joint Endocrine Society/ASBMR statement.
Takeda vs. FDA
In the meantime, Takeda says it has been in regular contact with the FDA to try and work out how to bring Natpara back to U.S. patients who require it for symptom control.
Current outstanding issues between the company and FDA involve considerations related to dose accuracy, safety, and supply continuity, which the company hopes will be resolved in a timely manner.
Furthermore, shortly after the recall order, the company created a special use program that continues to support patients previously prescribed Natpara who would otherwise be at extreme risk of serious complications if forced to stop taking the drug.
“Originally intended for an extremely limited number of patients, the special use program has now enrolled approximately 300 patients,” Cheryl Schwartz, head, U.S. Hematology & Rare Business Unit, Takeda, said in a letter to members of the U.S. Hypoparathyroidism Association on Nov. 21.
The number is higher than expected and reflects the significant treatment needs of patients with serious disease, Ms. Schwartz noted.
“I want to reiterate that we do understand and sincerely regret the impact the Natpara recall is having on patients. We will provide another update as soon as we have more information to share. We continue to work around the clock to find ways to bring Natpara back to the broader hypoparathyroidism community,” she added.
“Patient safety always has been, and continues to be, the highest priority for Takeda,” she said.
Final efforts
Dr. Khan is currently involved in clinical trials evaluating Natpara in Canada (where it is not yet approved).
She was also involved in some of the pivotal studies that helped gain support of the product in the United States and European Union, including REPLACE, which showed that the injectable PTH molecule maintained serum calcium while reducing or eliminating requirements for calcium and active vitamin D supplementation.
“We now have 8 years of experience with this molecule, so we know that it’s safe and effective,” Dr. Khan emphasized.
Health Canada has not deemed Natpara problematic enough to stop the ongoing research program into injectable PTH therapies there, which is being headed up by Dr. Khan at McMaster University.
“The company has not yet applied for approval here, but when they do apply, I am sure they will have fixed this problem by then,” Dr. Khan said. “And I expect it will be fixed momentarily in the United States, where patients have been most affected.”
Asked if she was looking forward to getting Natpara back on track, Ms. Astolfi said “absolutely” several times.
“This recall has really had a large impact on our community,” she stressed. “And while it’s good that critically ill patients have access to the drug through the special-use program, we want to get everyone back to their best life and feeling great.”
A version of this story originally appeared on Medscape.com.
EVALI outbreak ongoing, but new cases decline
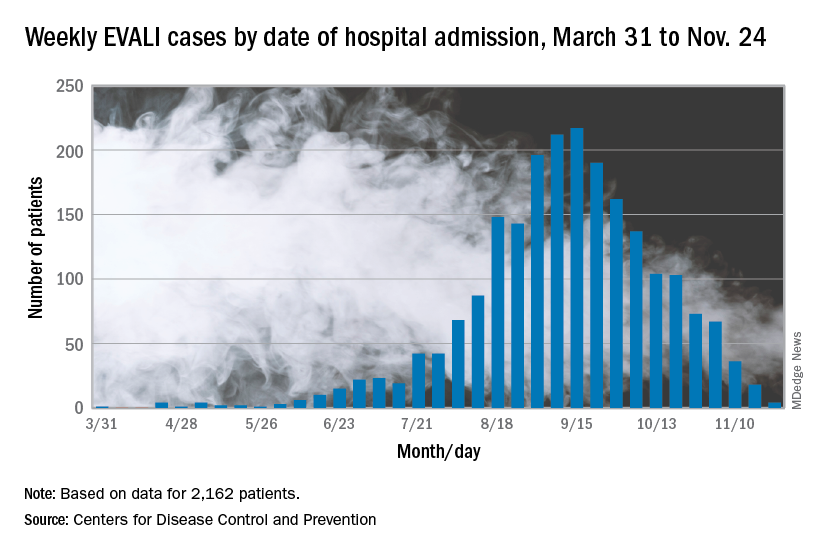
The vaping lung disease outbreak continues, but according to the Centers for Disease Control and Prevention, it may have peaked and the number of new hospitalized cases reported to the CDC may be decreasing.

In the Dec. 6, 2019, Morbidity and Mortality Weekly Report, the CDC has updated information about cases of e-cigarette, or vaping, product use–associated lung injury (EVALI): As of Dec. 3, there have been 2,291 cases reported from all 50 states, Washington, D.C., and two U.S. territories (Puerto Rico and U.S. Virgin Islands). A total of 48 deaths have been confirmed in 25 states and Washington, D.C., the CDC reported.
The largest number of weekly hospitalized cases occurred during the week of Sept. 15, 2019; since then, hospitalized cases have steadily declined. “Among all hospitalized EVALI patients reported to CDC weekly, the percentage of recent cases (patients hospitalized within the preceding 3 weeks) declined from 58% reported November 12 to 30% reported December 3,” the report stated.
About 80%of hospitalized EVALI patients reported using tetrahydrocannabinol (THC)–containing e-cigarette, or vaping, products. “Dank Vapes,” counterfeit THC-containing products of unknown origin, were the most commonly reported THC-containing branded products used. Dank Vapes were used by 56% of hospitalized EVALI patients nationwide, followed by TKO brand (15%), Smart Cart (13%), and Rove (12%).
Of EVALI patients for whom data were available, 67% were male, and the median age was 24 years (range, 13-77 years); 78% were aged under 35 years and 16% were under 18 years. About 75% of EVALI patients were non-Hispanic white and 16% were Hispanic. Among the 48 deaths, 54% of patients were male, and the median age was 52 years (range, 17-75 years).
CDC research on EVALI continues to be limited by the self-reported data, lack of data on substances used, missing data, loss to follow-up, and reporting lags, but the intensive investigation and data collection is ongoing.
The report concludes: “While the investigation continues, persons should consider refraining from the use of all e-cigarette, or vaping, products. Adults using e-cigarette, or vaping, products to quit smoking should not return to smoking cigarettes; they should weigh all risks and benefits and consider using [Food and Drug Administration]–approved cessation medications. Adults who continue to use e-cigarette, or vaping, products should carefully monitor themselves for symptoms and see a health care provider immediately if they develop symptoms similar to those reported in this outbreak. Irrespective of the ongoing investigation, e-cigarette, or vaping, products should never be used by youths, young adults or pregnant women.”
Information on the current investigation, reporting of cases, and other resources can be found on the CDC website.
SOURCE: Lozier MJ et al. MMWR Morb Mortal Wkly Rep. 2019 Dec 6. doi: 10.15585/mmwr.mm6849e1.
The vaping lung disease outbreak continues, but according to the Centers for Disease Control and Prevention, it may have peaked and the number of new hospitalized cases reported to the CDC may be decreasing.
In the Dec. 6, 2019, Morbidity and Mortality Weekly Report, the CDC has updated information about cases of e-cigarette, or vaping, product use–associated lung injury (EVALI): As of Dec. 3, there have been 2,291 cases reported from all 50 states, Washington, D.C., and two U.S. territories (Puerto Rico and U.S. Virgin Islands). A total of 48 deaths have been confirmed in 25 states and Washington, D.C., the CDC reported.
The largest number of weekly hospitalized cases occurred during the week of Sept. 15, 2019; since then, hospitalized cases have steadily declined. “Among all hospitalized EVALI patients reported to CDC weekly, the percentage of recent cases (patients hospitalized within the preceding 3 weeks) declined from 58% reported November 12 to 30% reported December 3,” the report stated.
About 80%of hospitalized EVALI patients reported using tetrahydrocannabinol (THC)–containing e-cigarette, or vaping, products. “Dank Vapes,” counterfeit THC-containing products of unknown origin, were the most commonly reported THC-containing branded products used. Dank Vapes were used by 56% of hospitalized EVALI patients nationwide, followed by TKO brand (15%), Smart Cart (13%), and Rove (12%).
Of EVALI patients for whom data were available, 67% were male, and the median age was 24 years (range, 13-77 years); 78% were aged under 35 years and 16% were under 18 years. About 75% of EVALI patients were non-Hispanic white and 16% were Hispanic. Among the 48 deaths, 54% of patients were male, and the median age was 52 years (range, 17-75 years).
CDC research on EVALI continues to be limited by the self-reported data, lack of data on substances used, missing data, loss to follow-up, and reporting lags, but the intensive investigation and data collection is ongoing.
The report concludes: “While the investigation continues, persons should consider refraining from the use of all e-cigarette, or vaping, products. Adults using e-cigarette, or vaping, products to quit smoking should not return to smoking cigarettes; they should weigh all risks and benefits and consider using [Food and Drug Administration]–approved cessation medications. Adults who continue to use e-cigarette, or vaping, products should carefully monitor themselves for symptoms and see a health care provider immediately if they develop symptoms similar to those reported in this outbreak. Irrespective of the ongoing investigation, e-cigarette, or vaping, products should never be used by youths, young adults or pregnant women.”
Information on the current investigation, reporting of cases, and other resources can be found on the CDC website.
SOURCE: Lozier MJ et al. MMWR Morb Mortal Wkly Rep. 2019 Dec 6. doi: 10.15585/mmwr.mm6849e1.
The vaping lung disease outbreak continues, but according to the Centers for Disease Control and Prevention, it may have peaked and the number of new hospitalized cases reported to the CDC may be decreasing.
In the Dec. 6, 2019, Morbidity and Mortality Weekly Report, the CDC has updated information about cases of e-cigarette, or vaping, product use–associated lung injury (EVALI): As of Dec. 3, there have been 2,291 cases reported from all 50 states, Washington, D.C., and two U.S. territories (Puerto Rico and U.S. Virgin Islands). A total of 48 deaths have been confirmed in 25 states and Washington, D.C., the CDC reported.
The largest number of weekly hospitalized cases occurred during the week of Sept. 15, 2019; since then, hospitalized cases have steadily declined. “Among all hospitalized EVALI patients reported to CDC weekly, the percentage of recent cases (patients hospitalized within the preceding 3 weeks) declined from 58% reported November 12 to 30% reported December 3,” the report stated.
About 80%of hospitalized EVALI patients reported using tetrahydrocannabinol (THC)–containing e-cigarette, or vaping, products. “Dank Vapes,” counterfeit THC-containing products of unknown origin, were the most commonly reported THC-containing branded products used. Dank Vapes were used by 56% of hospitalized EVALI patients nationwide, followed by TKO brand (15%), Smart Cart (13%), and Rove (12%).
Of EVALI patients for whom data were available, 67% were male, and the median age was 24 years (range, 13-77 years); 78% were aged under 35 years and 16% were under 18 years. About 75% of EVALI patients were non-Hispanic white and 16% were Hispanic. Among the 48 deaths, 54% of patients were male, and the median age was 52 years (range, 17-75 years).
CDC research on EVALI continues to be limited by the self-reported data, lack of data on substances used, missing data, loss to follow-up, and reporting lags, but the intensive investigation and data collection is ongoing.
The report concludes: “While the investigation continues, persons should consider refraining from the use of all e-cigarette, or vaping, products. Adults using e-cigarette, or vaping, products to quit smoking should not return to smoking cigarettes; they should weigh all risks and benefits and consider using [Food and Drug Administration]–approved cessation medications. Adults who continue to use e-cigarette, or vaping, products should carefully monitor themselves for symptoms and see a health care provider immediately if they develop symptoms similar to those reported in this outbreak. Irrespective of the ongoing investigation, e-cigarette, or vaping, products should never be used by youths, young adults or pregnant women.”
Information on the current investigation, reporting of cases, and other resources can be found on the CDC website.
SOURCE: Lozier MJ et al. MMWR Morb Mortal Wkly Rep. 2019 Dec 6. doi: 10.15585/mmwr.mm6849e1.
FROM THE MMWR
Oral BTK inhibitor shows continued promise for pemphigus
MADRID – A novel Dedee F. Murrell, MD, said at the annual congress of the European Academy of Dermatology and Venereology.

“In pemphigus, we have a considerable unmet medical need. We could do with a treatment that has rapid onset, is steroid sparing or avoiding, safe for chronic administration, avoids chronic B cell depletion – which is an issue with rituxumab – is efficacious in both newly diagnosed as well as in our commonly relapsing patients, and is convenient to administer,” observed Dr. Murrell, professor of dermatology at the University of New South Wales in Sydney.
In the phase 2 BELIEVE trial, the BTK inhibitor known for now only as PRN1008 appeared to check all the boxes. However, definitive evidence of the drug’s efficacy and safety must await the results of the ongoing, double-bind, placebo-controlled, pivotal phase 3 PEGASUS trial, which is enrolling a planned 120 patients with pemphigus vulgaris or foliaceus in 19 countries.
Pemphigus is driven by autoantibodies against desmogleins 1 and 3. Even in contemporary practice, this blistering disease has roughly a 5% mortality rate. Current management of the disease with high-dose corticosteroids at 1 mg/kg per day or more with or without rituximab (Rituxan) is challenging because of the associated pronounced toxicities. And even when rituximab is utilized, patients need to be on high-dose steroids for at least 3-6 months before a rituximab response is achieved, Dr. Murrell said.
PRN1008 has three mechanisms of action targeting the drivers of pemphigus and other immune-mediated diseases, she explained. The drug blocks inflammatory B cells, neutrophils, and macrophages; eliminates downstream signalling by antidesmoglein autoantibodies; and prevents production of new autoantibodies. The drug has a double lock-and-key mechanism which makes it highly specific for its target, so treated patients are much less likely to experience bruising, diarrhea, and other off-target effects than is the case with other tyrosine kinase inhibitors.
“Also, PRN1008 is reversible. It comes off its target receptor after about 12 hours, at which point serum levels become low. So if any side effects do develop, the patient can recover quickly, unlike with rituximab, which involves ongoing inhibition of B cells for a long period of time,” the dermatologist noted.
BELIEVE was a phase 2 dose-ranging study of 27 patients with pemphigus treated open-label with PRN1008 for 12 weeks. The primary endpoint was control of disease activity, meaning no new lesions while established lesions showed some evidence of healing on no more than 0.5 mg/kg per day of prednisone. This outcome was achieved in 27% of participants at 2 weeks, 54% at 4 weeks, and 73% at 12 weeks. Autoantibody levels dropped by a mean of 65% at 12 weeks, with a median 70% reduction in Pemphigus Disease Activity Index scores while patients were on an average of just 12 mg of prednisone per day.
Phase 2b of BELIEVE included a separate group of 15 patients on PRN1008 for 24 weeks. Nine achieved a Pemphigus Disease Activity Index score of 0 or 1. Six patients had a complete response, meaning an absence of both new and established lesions while on no or a very low dose of prednisone. Another five patients were unable to achieve a complete response, and the jury was still out on another four still on treatment.
The side effect profile was benign in comparison with that of current standard therapies, she said. It consisted of a handful of cases of mild, transient nausea, headache, or upper abdominal pain and a few Grade 1 infections. There have been no severe treatment-related adverse events in BELIEVE participants.
Patients enrolled in the ongoing phase 3 PEGASUS trial start with a short course of high-dose corticosteroids, followed by double-blind randomization to PRN1008 at 400 mg twice a day or placebo, with a corticosteroid taper. The primary endpoint is durable complete remission at week 37, defined as no lesions being present for at least the previous 8 weeks while on no more than 5 mg/day of prednisone.
Secondary endpoints include cumulative corticosteroid dose through 36 weeks and patient-reported quality of life measures assessed out to 61 weeks. The trial is scheduled for completion in the spring of 2022.
Dr. Murrell reported serving as a consultant to the study sponsor, Principia Biopharma, as well as numerous other pharmaceutical companies.
MADRID – A novel Dedee F. Murrell, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
“In pemphigus, we have a considerable unmet medical need. We could do with a treatment that has rapid onset, is steroid sparing or avoiding, safe for chronic administration, avoids chronic B cell depletion – which is an issue with rituxumab – is efficacious in both newly diagnosed as well as in our commonly relapsing patients, and is convenient to administer,” observed Dr. Murrell, professor of dermatology at the University of New South Wales in Sydney.
In the phase 2 BELIEVE trial, the BTK inhibitor known for now only as PRN1008 appeared to check all the boxes. However, definitive evidence of the drug’s efficacy and safety must await the results of the ongoing, double-bind, placebo-controlled, pivotal phase 3 PEGASUS trial, which is enrolling a planned 120 patients with pemphigus vulgaris or foliaceus in 19 countries.
Pemphigus is driven by autoantibodies against desmogleins 1 and 3. Even in contemporary practice, this blistering disease has roughly a 5% mortality rate. Current management of the disease with high-dose corticosteroids at 1 mg/kg per day or more with or without rituximab (Rituxan) is challenging because of the associated pronounced toxicities. And even when rituximab is utilized, patients need to be on high-dose steroids for at least 3-6 months before a rituximab response is achieved, Dr. Murrell said.
PRN1008 has three mechanisms of action targeting the drivers of pemphigus and other immune-mediated diseases, she explained. The drug blocks inflammatory B cells, neutrophils, and macrophages; eliminates downstream signalling by antidesmoglein autoantibodies; and prevents production of new autoantibodies. The drug has a double lock-and-key mechanism which makes it highly specific for its target, so treated patients are much less likely to experience bruising, diarrhea, and other off-target effects than is the case with other tyrosine kinase inhibitors.
“Also, PRN1008 is reversible. It comes off its target receptor after about 12 hours, at which point serum levels become low. So if any side effects do develop, the patient can recover quickly, unlike with rituximab, which involves ongoing inhibition of B cells for a long period of time,” the dermatologist noted.
BELIEVE was a phase 2 dose-ranging study of 27 patients with pemphigus treated open-label with PRN1008 for 12 weeks. The primary endpoint was control of disease activity, meaning no new lesions while established lesions showed some evidence of healing on no more than 0.5 mg/kg per day of prednisone. This outcome was achieved in 27% of participants at 2 weeks, 54% at 4 weeks, and 73% at 12 weeks. Autoantibody levels dropped by a mean of 65% at 12 weeks, with a median 70% reduction in Pemphigus Disease Activity Index scores while patients were on an average of just 12 mg of prednisone per day.
Phase 2b of BELIEVE included a separate group of 15 patients on PRN1008 for 24 weeks. Nine achieved a Pemphigus Disease Activity Index score of 0 or 1. Six patients had a complete response, meaning an absence of both new and established lesions while on no or a very low dose of prednisone. Another five patients were unable to achieve a complete response, and the jury was still out on another four still on treatment.
The side effect profile was benign in comparison with that of current standard therapies, she said. It consisted of a handful of cases of mild, transient nausea, headache, or upper abdominal pain and a few Grade 1 infections. There have been no severe treatment-related adverse events in BELIEVE participants.
Patients enrolled in the ongoing phase 3 PEGASUS trial start with a short course of high-dose corticosteroids, followed by double-blind randomization to PRN1008 at 400 mg twice a day or placebo, with a corticosteroid taper. The primary endpoint is durable complete remission at week 37, defined as no lesions being present for at least the previous 8 weeks while on no more than 5 mg/day of prednisone.
Secondary endpoints include cumulative corticosteroid dose through 36 weeks and patient-reported quality of life measures assessed out to 61 weeks. The trial is scheduled for completion in the spring of 2022.
Dr. Murrell reported serving as a consultant to the study sponsor, Principia Biopharma, as well as numerous other pharmaceutical companies.
MADRID – A novel Dedee F. Murrell, MD, said at the annual congress of the European Academy of Dermatology and Venereology.
“In pemphigus, we have a considerable unmet medical need. We could do with a treatment that has rapid onset, is steroid sparing or avoiding, safe for chronic administration, avoids chronic B cell depletion – which is an issue with rituxumab – is efficacious in both newly diagnosed as well as in our commonly relapsing patients, and is convenient to administer,” observed Dr. Murrell, professor of dermatology at the University of New South Wales in Sydney.
In the phase 2 BELIEVE trial, the BTK inhibitor known for now only as PRN1008 appeared to check all the boxes. However, definitive evidence of the drug’s efficacy and safety must await the results of the ongoing, double-bind, placebo-controlled, pivotal phase 3 PEGASUS trial, which is enrolling a planned 120 patients with pemphigus vulgaris or foliaceus in 19 countries.
Pemphigus is driven by autoantibodies against desmogleins 1 and 3. Even in contemporary practice, this blistering disease has roughly a 5% mortality rate. Current management of the disease with high-dose corticosteroids at 1 mg/kg per day or more with or without rituximab (Rituxan) is challenging because of the associated pronounced toxicities. And even when rituximab is utilized, patients need to be on high-dose steroids for at least 3-6 months before a rituximab response is achieved, Dr. Murrell said.
PRN1008 has three mechanisms of action targeting the drivers of pemphigus and other immune-mediated diseases, she explained. The drug blocks inflammatory B cells, neutrophils, and macrophages; eliminates downstream signalling by antidesmoglein autoantibodies; and prevents production of new autoantibodies. The drug has a double lock-and-key mechanism which makes it highly specific for its target, so treated patients are much less likely to experience bruising, diarrhea, and other off-target effects than is the case with other tyrosine kinase inhibitors.
“Also, PRN1008 is reversible. It comes off its target receptor after about 12 hours, at which point serum levels become low. So if any side effects do develop, the patient can recover quickly, unlike with rituximab, which involves ongoing inhibition of B cells for a long period of time,” the dermatologist noted.
BELIEVE was a phase 2 dose-ranging study of 27 patients with pemphigus treated open-label with PRN1008 for 12 weeks. The primary endpoint was control of disease activity, meaning no new lesions while established lesions showed some evidence of healing on no more than 0.5 mg/kg per day of prednisone. This outcome was achieved in 27% of participants at 2 weeks, 54% at 4 weeks, and 73% at 12 weeks. Autoantibody levels dropped by a mean of 65% at 12 weeks, with a median 70% reduction in Pemphigus Disease Activity Index scores while patients were on an average of just 12 mg of prednisone per day.
Phase 2b of BELIEVE included a separate group of 15 patients on PRN1008 for 24 weeks. Nine achieved a Pemphigus Disease Activity Index score of 0 or 1. Six patients had a complete response, meaning an absence of both new and established lesions while on no or a very low dose of prednisone. Another five patients were unable to achieve a complete response, and the jury was still out on another four still on treatment.
The side effect profile was benign in comparison with that of current standard therapies, she said. It consisted of a handful of cases of mild, transient nausea, headache, or upper abdominal pain and a few Grade 1 infections. There have been no severe treatment-related adverse events in BELIEVE participants.
Patients enrolled in the ongoing phase 3 PEGASUS trial start with a short course of high-dose corticosteroids, followed by double-blind randomization to PRN1008 at 400 mg twice a day or placebo, with a corticosteroid taper. The primary endpoint is durable complete remission at week 37, defined as no lesions being present for at least the previous 8 weeks while on no more than 5 mg/day of prednisone.
Secondary endpoints include cumulative corticosteroid dose through 36 weeks and patient-reported quality of life measures assessed out to 61 weeks. The trial is scheduled for completion in the spring of 2022.
Dr. Murrell reported serving as a consultant to the study sponsor, Principia Biopharma, as well as numerous other pharmaceutical companies.
REPORTING FROM THE EADV CONGRESS
Adjuvant denosumab falls short in early-stage breast cancer
Adjuvant denosumab did not improve bone metastasis–free survival and related outcomes in women with early-stage breast cancer, according to a phase 3 trial.
“We hypothesised that denosumab would modify the clinical course of early breast cancer, delaying the development of clinical bone metastases with or without disease recurrence at other sites,” reported Robert Coleman, MBBS, MD, of the University of Sheffield, England, and colleagues. Their report is in The Lancet Oncology.
The randomized, placebo-controlled, phase 3 D-CARE study included 4,509 women with early-stage, high-risk disease. The effects of adding denosumab to standard-of-care adjuvant or neoadjuvant chemotherapy was studied in 389 institutions around the globe. In the initial treatment phase, study patients received denosumab or placebo every 3-4 weeks in combination with adjuvant or neoadjuvant chemotherapy for approximately 6 months.
After completion of chemotherapy, the dosing interval was extended to every 12 weeks (range, 14 days) for a total of 5 years. The median age of women who received denosumab was 50 years (range, 44-59 years), 65% of whom were hormone receptor positive, HER2-negative. In the study, patients were stratified by various factors, including type of therapy, age, lymph node status, geographical region, and others. The primary outcome was a composite endpoint of bone metastasis–free survival.
At 5-year follow-up, the researchers found no significant difference in bone metastasis–free survival between the denosumab and placebo treatment arms (median survival not reached in either arm; P = .70). With respect to safety, the most frequently seen grade 3 or higher treatment-emergent adverse events were neutropenia (15% vs. 15%), febrile neutropenia (5% vs. 6%), and leukopenia (3% vs. 3%). Positively adjudicated osteonecrosis of the jaw occurred in 122 (5%) of 2,241 patients treated with denosumab versus 4 (less than 1%) of 2,218 patients treated with placebo, Dr. Coleman and colleagues wrote.
The researchers acknowledged that a key limitation of the study was the smaller than anticipated number of events for efficacy outcomes. As a result, the study protocol was modified, which could have limited the statistical power of the study. “The results of this study do not support a role for denosumab as an antitumour agent in this setting,” they concluded.
Amgen funded the study. The authors reported financial affiliations with AbbVie, Amgen, Astellas, Bristol-Myers Squibb, Celgene, Covance, Lilly, Medivation, Merck Serono, Merck Sharp and Dohme, Novartis, Pfizer, and several others.
SOURCE: Coleman R et al. Lancet Oncol. 2019 Dec 2. doi: 10.1016/S1470-2045(19)30687-4.
Adjuvant denosumab did not improve bone metastasis–free survival and related outcomes in women with early-stage breast cancer, according to a phase 3 trial.
“We hypothesised that denosumab would modify the clinical course of early breast cancer, delaying the development of clinical bone metastases with or without disease recurrence at other sites,” reported Robert Coleman, MBBS, MD, of the University of Sheffield, England, and colleagues. Their report is in The Lancet Oncology.
The randomized, placebo-controlled, phase 3 D-CARE study included 4,509 women with early-stage, high-risk disease. The effects of adding denosumab to standard-of-care adjuvant or neoadjuvant chemotherapy was studied in 389 institutions around the globe. In the initial treatment phase, study patients received denosumab or placebo every 3-4 weeks in combination with adjuvant or neoadjuvant chemotherapy for approximately 6 months.
After completion of chemotherapy, the dosing interval was extended to every 12 weeks (range, 14 days) for a total of 5 years. The median age of women who received denosumab was 50 years (range, 44-59 years), 65% of whom were hormone receptor positive, HER2-negative. In the study, patients were stratified by various factors, including type of therapy, age, lymph node status, geographical region, and others. The primary outcome was a composite endpoint of bone metastasis–free survival.
At 5-year follow-up, the researchers found no significant difference in bone metastasis–free survival between the denosumab and placebo treatment arms (median survival not reached in either arm; P = .70). With respect to safety, the most frequently seen grade 3 or higher treatment-emergent adverse events were neutropenia (15% vs. 15%), febrile neutropenia (5% vs. 6%), and leukopenia (3% vs. 3%). Positively adjudicated osteonecrosis of the jaw occurred in 122 (5%) of 2,241 patients treated with denosumab versus 4 (less than 1%) of 2,218 patients treated with placebo, Dr. Coleman and colleagues wrote.
The researchers acknowledged that a key limitation of the study was the smaller than anticipated number of events for efficacy outcomes. As a result, the study protocol was modified, which could have limited the statistical power of the study. “The results of this study do not support a role for denosumab as an antitumour agent in this setting,” they concluded.
Amgen funded the study. The authors reported financial affiliations with AbbVie, Amgen, Astellas, Bristol-Myers Squibb, Celgene, Covance, Lilly, Medivation, Merck Serono, Merck Sharp and Dohme, Novartis, Pfizer, and several others.
SOURCE: Coleman R et al. Lancet Oncol. 2019 Dec 2. doi: 10.1016/S1470-2045(19)30687-4.
Adjuvant denosumab did not improve bone metastasis–free survival and related outcomes in women with early-stage breast cancer, according to a phase 3 trial.
“We hypothesised that denosumab would modify the clinical course of early breast cancer, delaying the development of clinical bone metastases with or without disease recurrence at other sites,” reported Robert Coleman, MBBS, MD, of the University of Sheffield, England, and colleagues. Their report is in The Lancet Oncology.
The randomized, placebo-controlled, phase 3 D-CARE study included 4,509 women with early-stage, high-risk disease. The effects of adding denosumab to standard-of-care adjuvant or neoadjuvant chemotherapy was studied in 389 institutions around the globe. In the initial treatment phase, study patients received denosumab or placebo every 3-4 weeks in combination with adjuvant or neoadjuvant chemotherapy for approximately 6 months.
After completion of chemotherapy, the dosing interval was extended to every 12 weeks (range, 14 days) for a total of 5 years. The median age of women who received denosumab was 50 years (range, 44-59 years), 65% of whom were hormone receptor positive, HER2-negative. In the study, patients were stratified by various factors, including type of therapy, age, lymph node status, geographical region, and others. The primary outcome was a composite endpoint of bone metastasis–free survival.
At 5-year follow-up, the researchers found no significant difference in bone metastasis–free survival between the denosumab and placebo treatment arms (median survival not reached in either arm; P = .70). With respect to safety, the most frequently seen grade 3 or higher treatment-emergent adverse events were neutropenia (15% vs. 15%), febrile neutropenia (5% vs. 6%), and leukopenia (3% vs. 3%). Positively adjudicated osteonecrosis of the jaw occurred in 122 (5%) of 2,241 patients treated with denosumab versus 4 (less than 1%) of 2,218 patients treated with placebo, Dr. Coleman and colleagues wrote.
The researchers acknowledged that a key limitation of the study was the smaller than anticipated number of events for efficacy outcomes. As a result, the study protocol was modified, which could have limited the statistical power of the study. “The results of this study do not support a role for denosumab as an antitumour agent in this setting,” they concluded.
Amgen funded the study. The authors reported financial affiliations with AbbVie, Amgen, Astellas, Bristol-Myers Squibb, Celgene, Covance, Lilly, Medivation, Merck Serono, Merck Sharp and Dohme, Novartis, Pfizer, and several others.
SOURCE: Coleman R et al. Lancet Oncol. 2019 Dec 2. doi: 10.1016/S1470-2045(19)30687-4.
FROM LANCET ONCOLOGY
Lp(a) molar concentration flags CVD, diabetes risk
Lipoprotein(a) molar concentration, rather than apolipoprotein(a) size, appears to be the factor that drives lipoprotein(a)-based cardiovascular disease, according to research published in the Journal of the American College of Cardiology.
The causal association between lipoprotein(a), or Lp(a), and cardiovascular disease has been previously established, but exactly what attribute of Lp(a) is related to cardiovascular risk is not known, Daniel F. Gudbjartsson, PhD, of deCODE genetics and the University of Iceland in Reykjavik, and colleagues wrote in their study. The researchers set out to determine whether Lp(a) molar concentration or apolipoprotein(a), or apo(a), size affects cardiovascular risk. In addition, Dr. Gudbjartsson and colleagues examined the relationship between Lp(a) and type 2 diabetes. While low levels of Lp(a) have been linked to type 2 diabetes, the researchers sought to examine whether low Lp(a) molar concentration levels were also associated with type 2 diabetes risk.
“With Lp(a)-lowering drugs being developed, it is important to understand which attributes of Lp(a) best capture the cardiovascular risk and the consequences of Lp(a) lowering,” noted Dr. Gudbjartsson and colleagues.
Using Mendelian randomization, the researchers assessed Lp(a) molar concentration and kringle IV type 2 (KIV-2) repeat sequence variants to determine a causal relationship between both variants and disease risk. Lp(a) molar concentration serum samples were measured using particle-enhanced turbidimetric immunoassay, while KIV-2 repeats were genotyped with real-time polymerase chain reaction.
Overall, 143,087 participants from Iceland had their genetic information analyzed; of these, 17,715 participants had coronary artery disease, and 8,734 had type 2 diabetes. Lp(a) molecular concentration was analyzed in 12,137 participants and genetically imputed into 130,950 Icelanders, and KIV-2 repeats were estimated in 22,771 Icelanders and genetically imputed into 120,316 Icelanders.
Dr. Gudbjartsson and colleagues found there was a dose-dependent association between Lp(a) molar concentration and risk of coronary artery disease (CAD), peripheral artery disease, aortic valve stenosis, heart failure, and lifespan. In participants in whom Lp(a) molar concentration was at the 79th percentile (50 units of molarity [nM]), the odds ratio was 1.11, and for those in the 99th percentile (250 nM), there was an odds ratio of 2.01 when compared with participants with a median Lp(a) molar concentration of 14 nM. “Lp(a) molar concentration fully explained the Lp(a) association with CAD, and there was no residual association with apo(a) size,” the researchers said.
Participants who were not at increased risk for CAD included those with few KIV-2 repeats and participants with the splice variant G4925A. “This suggested that risk prediction based on Lp(a) should only depend on molar concentration and that treatment of Lp(a) should focus on lowering the molar concentration in subjects with high Lp(a) levels, regardless of the apo(a) size distribution,” Dr. Gudbjartsson and colleagues wrote.
Among participants with type 2 diabetes examined, the 10% of participants with Lp(a) molar concentrations of less than 3.5 nM were at the highest risk of developing type 2 diabetes.
In an accompanying editorial, Benoit J. Arsenault, PhD, of the Quebec Heart and Lung Institute, said that the findings of an association between Lp(a) concentration and atherosclerotic cardiovascular diseases (ASCVD) from Gudbjartsson et al. are important, particularly if they can be replicated in more diverse populations (doi: 10.1016/j.jacc.2019.06.083). “Investigating the association between Lp(a) levels, apo(a) isoform size, and ASCVD risk in different populations is important because the distribution of Lp(a) levels appears to be different across ethnic groups,” he said.
Despite the link between absolute Lp(a) concentrations and cardiovascular disease, cardiovascular outcome trials will need be conducted, Dr. Arsenault noted.
“In the post-statin and post-genomic era, finding much needed therapeutic targets for residual cardiovascular risk can be compared to a gold-digging expedition. Like a map to the location of the gold, GWAS [genome-wide association studies] and Mendelian randomization studies are consistently pointing us in the direction of Lp(a),” he said. “It is time to coordinate our efforts to dig where the map told us, to see once and for all if we will find the golden target of residual cardiovascular risk that we are hoping for and to give hope to high-risk patients with elevated Lp(a) levels.”
Dr. Gudbjartsson and 19 other authors reported being employees of deCODE genetics, owned by Amgen, which is developing Lp(a)-lowering drugs related to the study findings. The other authors reported no relevant conflict of interest. Dr. Arsenault reported being supported by the Fonds de recherche du Québec: Santé and the Canadian Institutes of Health Research; has received research funding from Pfizer, Merck, and Ionis; and was a former consultant for Pfizer and Novartis.
SOURCE: Gudbjartsson DF et al. J Am Coll Cardiol. 2019. doi: 10.1016/j.jacc.2019.10.019.
Lipoprotein(a) molar concentration, rather than apolipoprotein(a) size, appears to be the factor that drives lipoprotein(a)-based cardiovascular disease, according to research published in the Journal of the American College of Cardiology.
The causal association between lipoprotein(a), or Lp(a), and cardiovascular disease has been previously established, but exactly what attribute of Lp(a) is related to cardiovascular risk is not known, Daniel F. Gudbjartsson, PhD, of deCODE genetics and the University of Iceland in Reykjavik, and colleagues wrote in their study. The researchers set out to determine whether Lp(a) molar concentration or apolipoprotein(a), or apo(a), size affects cardiovascular risk. In addition, Dr. Gudbjartsson and colleagues examined the relationship between Lp(a) and type 2 diabetes. While low levels of Lp(a) have been linked to type 2 diabetes, the researchers sought to examine whether low Lp(a) molar concentration levels were also associated with type 2 diabetes risk.
“With Lp(a)-lowering drugs being developed, it is important to understand which attributes of Lp(a) best capture the cardiovascular risk and the consequences of Lp(a) lowering,” noted Dr. Gudbjartsson and colleagues.
Using Mendelian randomization, the researchers assessed Lp(a) molar concentration and kringle IV type 2 (KIV-2) repeat sequence variants to determine a causal relationship between both variants and disease risk. Lp(a) molar concentration serum samples were measured using particle-enhanced turbidimetric immunoassay, while KIV-2 repeats were genotyped with real-time polymerase chain reaction.
Overall, 143,087 participants from Iceland had their genetic information analyzed; of these, 17,715 participants had coronary artery disease, and 8,734 had type 2 diabetes. Lp(a) molecular concentration was analyzed in 12,137 participants and genetically imputed into 130,950 Icelanders, and KIV-2 repeats were estimated in 22,771 Icelanders and genetically imputed into 120,316 Icelanders.
Dr. Gudbjartsson and colleagues found there was a dose-dependent association between Lp(a) molar concentration and risk of coronary artery disease (CAD), peripheral artery disease, aortic valve stenosis, heart failure, and lifespan. In participants in whom Lp(a) molar concentration was at the 79th percentile (50 units of molarity [nM]), the odds ratio was 1.11, and for those in the 99th percentile (250 nM), there was an odds ratio of 2.01 when compared with participants with a median Lp(a) molar concentration of 14 nM. “Lp(a) molar concentration fully explained the Lp(a) association with CAD, and there was no residual association with apo(a) size,” the researchers said.
Participants who were not at increased risk for CAD included those with few KIV-2 repeats and participants with the splice variant G4925A. “This suggested that risk prediction based on Lp(a) should only depend on molar concentration and that treatment of Lp(a) should focus on lowering the molar concentration in subjects with high Lp(a) levels, regardless of the apo(a) size distribution,” Dr. Gudbjartsson and colleagues wrote.
Among participants with type 2 diabetes examined, the 10% of participants with Lp(a) molar concentrations of less than 3.5 nM were at the highest risk of developing type 2 diabetes.
In an accompanying editorial, Benoit J. Arsenault, PhD, of the Quebec Heart and Lung Institute, said that the findings of an association between Lp(a) concentration and atherosclerotic cardiovascular diseases (ASCVD) from Gudbjartsson et al. are important, particularly if they can be replicated in more diverse populations (doi: 10.1016/j.jacc.2019.06.083). “Investigating the association between Lp(a) levels, apo(a) isoform size, and ASCVD risk in different populations is important because the distribution of Lp(a) levels appears to be different across ethnic groups,” he said.
Despite the link between absolute Lp(a) concentrations and cardiovascular disease, cardiovascular outcome trials will need be conducted, Dr. Arsenault noted.
“In the post-statin and post-genomic era, finding much needed therapeutic targets for residual cardiovascular risk can be compared to a gold-digging expedition. Like a map to the location of the gold, GWAS [genome-wide association studies] and Mendelian randomization studies are consistently pointing us in the direction of Lp(a),” he said. “It is time to coordinate our efforts to dig where the map told us, to see once and for all if we will find the golden target of residual cardiovascular risk that we are hoping for and to give hope to high-risk patients with elevated Lp(a) levels.”
Dr. Gudbjartsson and 19 other authors reported being employees of deCODE genetics, owned by Amgen, which is developing Lp(a)-lowering drugs related to the study findings. The other authors reported no relevant conflict of interest. Dr. Arsenault reported being supported by the Fonds de recherche du Québec: Santé and the Canadian Institutes of Health Research; has received research funding from Pfizer, Merck, and Ionis; and was a former consultant for Pfizer and Novartis.
SOURCE: Gudbjartsson DF et al. J Am Coll Cardiol. 2019. doi: 10.1016/j.jacc.2019.10.019.
Lipoprotein(a) molar concentration, rather than apolipoprotein(a) size, appears to be the factor that drives lipoprotein(a)-based cardiovascular disease, according to research published in the Journal of the American College of Cardiology.
The causal association between lipoprotein(a), or Lp(a), and cardiovascular disease has been previously established, but exactly what attribute of Lp(a) is related to cardiovascular risk is not known, Daniel F. Gudbjartsson, PhD, of deCODE genetics and the University of Iceland in Reykjavik, and colleagues wrote in their study. The researchers set out to determine whether Lp(a) molar concentration or apolipoprotein(a), or apo(a), size affects cardiovascular risk. In addition, Dr. Gudbjartsson and colleagues examined the relationship between Lp(a) and type 2 diabetes. While low levels of Lp(a) have been linked to type 2 diabetes, the researchers sought to examine whether low Lp(a) molar concentration levels were also associated with type 2 diabetes risk.
“With Lp(a)-lowering drugs being developed, it is important to understand which attributes of Lp(a) best capture the cardiovascular risk and the consequences of Lp(a) lowering,” noted Dr. Gudbjartsson and colleagues.
Using Mendelian randomization, the researchers assessed Lp(a) molar concentration and kringle IV type 2 (KIV-2) repeat sequence variants to determine a causal relationship between both variants and disease risk. Lp(a) molar concentration serum samples were measured using particle-enhanced turbidimetric immunoassay, while KIV-2 repeats were genotyped with real-time polymerase chain reaction.
Overall, 143,087 participants from Iceland had their genetic information analyzed; of these, 17,715 participants had coronary artery disease, and 8,734 had type 2 diabetes. Lp(a) molecular concentration was analyzed in 12,137 participants and genetically imputed into 130,950 Icelanders, and KIV-2 repeats were estimated in 22,771 Icelanders and genetically imputed into 120,316 Icelanders.
Dr. Gudbjartsson and colleagues found there was a dose-dependent association between Lp(a) molar concentration and risk of coronary artery disease (CAD), peripheral artery disease, aortic valve stenosis, heart failure, and lifespan. In participants in whom Lp(a) molar concentration was at the 79th percentile (50 units of molarity [nM]), the odds ratio was 1.11, and for those in the 99th percentile (250 nM), there was an odds ratio of 2.01 when compared with participants with a median Lp(a) molar concentration of 14 nM. “Lp(a) molar concentration fully explained the Lp(a) association with CAD, and there was no residual association with apo(a) size,” the researchers said.
Participants who were not at increased risk for CAD included those with few KIV-2 repeats and participants with the splice variant G4925A. “This suggested that risk prediction based on Lp(a) should only depend on molar concentration and that treatment of Lp(a) should focus on lowering the molar concentration in subjects with high Lp(a) levels, regardless of the apo(a) size distribution,” Dr. Gudbjartsson and colleagues wrote.
Among participants with type 2 diabetes examined, the 10% of participants with Lp(a) molar concentrations of less than 3.5 nM were at the highest risk of developing type 2 diabetes.
In an accompanying editorial, Benoit J. Arsenault, PhD, of the Quebec Heart and Lung Institute, said that the findings of an association between Lp(a) concentration and atherosclerotic cardiovascular diseases (ASCVD) from Gudbjartsson et al. are important, particularly if they can be replicated in more diverse populations (doi: 10.1016/j.jacc.2019.06.083). “Investigating the association between Lp(a) levels, apo(a) isoform size, and ASCVD risk in different populations is important because the distribution of Lp(a) levels appears to be different across ethnic groups,” he said.
Despite the link between absolute Lp(a) concentrations and cardiovascular disease, cardiovascular outcome trials will need be conducted, Dr. Arsenault noted.
“In the post-statin and post-genomic era, finding much needed therapeutic targets for residual cardiovascular risk can be compared to a gold-digging expedition. Like a map to the location of the gold, GWAS [genome-wide association studies] and Mendelian randomization studies are consistently pointing us in the direction of Lp(a),” he said. “It is time to coordinate our efforts to dig where the map told us, to see once and for all if we will find the golden target of residual cardiovascular risk that we are hoping for and to give hope to high-risk patients with elevated Lp(a) levels.”
Dr. Gudbjartsson and 19 other authors reported being employees of deCODE genetics, owned by Amgen, which is developing Lp(a)-lowering drugs related to the study findings. The other authors reported no relevant conflict of interest. Dr. Arsenault reported being supported by the Fonds de recherche du Québec: Santé and the Canadian Institutes of Health Research; has received research funding from Pfizer, Merck, and Ionis; and was a former consultant for Pfizer and Novartis.
SOURCE: Gudbjartsson DF et al. J Am Coll Cardiol. 2019. doi: 10.1016/j.jacc.2019.10.019.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY
Key clinical point:
Major finding: There was a dose-dependent association between Lp(a) molar concentration and risk of coronary artery disease (CAD), peripheral artery disease, aortic valve stenosis, heart failure, and lifespan.
Study details: A case-control study of genetic information from 143,087 Icelandic participants.
Disclosures: Dr. Gudbjartsson and 19 other authors reported being employees of deCODE genetics, owned by Amgen, which is developing Lp(a)-lowering drugs related to the study findings. The other authors reported no relevant conflict of interest. Dr. Arsenault reported being supported by the Fonds de recherche du Québec: Santé and the Canadian Institutes of Health Research; has received research funding from Pfizer, Merck, and Ionis; and was a former consultant for Pfizer and Novartis.
Source: Gudbjartsson DF et al. J Am Coll Cardiol. 2019. doi: 10.1016/j.jacc.2019.10.019.
Women have fewer cardiovascular events after non–ST-segment elevation ACS
While women were found to be at lower risk of major cardiovascular events than men after non–ST-segment elevation acute coronary syndromes (NSTEACS), a new study has also shown that they remain undertreated with guideline-directed therapies.

“These findings underscore the fact that efforts to modify risk factors and implement appropriate treatment strategies in women and men may be a powerful means to further improve outcomes among patients with NSTEACS,” wrote Amy A. Sarma, MD, of Massachusetts General Hospital in Boston and coauthors. The study was published in the Journal of the American College of Cardiology.
To determine if outcomes differed between men and women after NSTEACS, the researchers analyzed 68,730 patients from 10 different clinical trials as part of the Thrombolysis In Myocardial Infarction (TIMI) Study Group. All trials enrolled patients with NSTEACS within 30 days of hospitalization. Across the 10 trials, there were a total of 19,827 women (29%).
Female patients had an average age of 67 years, compared with a mean age of 62 years for men. Women were also more likely to have had hypertension, diabetes, or prior heart failure, though less likely to have had a prior MI. In regard to treatment strategies for NSTEACS, women were less likely to receive aspirin, P2Y12 inhibitors, or statins. Women at high risk were also less likely to receive aspirin, P2Y12 inhibitors, or statins.
Before adjusted analysis, women and men had a similar risk of major adverse cardiovascular events (MACE) – defined as cardiovascular death, MI, or stroke – after NSTEACS (hazard ratio, 1.04; 95% confidence interval, 0.99-1.09; P = .16). However, women were found to be at increased risk of cardiovascular death (HR, 1.16; 95% CI, 1.02-1.32; P = .03), all-cause mortality (HR, 1.12; 95% CI, 1.01-1.24; P = .03), and stroke (HR, 1.19; 95% CI, 1.03-1.37; P = .02).
After adjusting for baseline risk predictors, women were found to have a 7% lower risk of MACE (adjusted HR, 0.93; 95% CI, 0.89-0.98; P = .005), along with a 15% lower risk of cardiovascular death (aHR, 0.85; 95% CI, 0.76-0.96; P = .008) and a 16% lower risk of all-cause death (aHR, 0.84; 95% CI, 0.78-0.90; P less than .0001). Additional adjustments for guideline-based therapies did not significantly alter the risk estimates.
In an accompanying editorial, Michael E. Farkouh, MD, and Wendy Tsang, MD, of the University of Toronto noted that this study from Sarma et al. underlines the “lack of progress made in addressing sex inequality in the care for women with ACS” (J Am Coll Cardiol. 2019 Dec 9. doi: 10.1016/j.jacc.2019.10.017).
Though Dr. Farkouh and Dr. Tsang were not surprised that the women in the study were older than the men, as women typically develop cardiovascular disease later, they noted that the women also presented with higher baseline risk. Could this be because of the “persistent perception that coronary disease is a male disease?” They cited the treatment-risk paradox and an emphasis on diagnosing obstructive coronary disease – typically seen in men – as potential contributors to underdiagnosis and undertreatment in at-risk women.
“That this care gap for women with cardiovascular diseases continues to persist, even in well-run contemporary landmark ACS trials, highlights how challenging it is to address,” they wrote.
Dr. Sarma and coauthors acknowledged their study’s potential limitations, including the 10 trials differing in study design, enrollment timing, treatments tested, and follow-up duration. They also noted that the cohort of patients were all at moderate to high risk, which may not make the findings generalizable to the broader NSTEACS patient population.
The study authors reported numerous potential conflicts of interest, including receiving research grants from – and serving as a consultant or on the advisory board of – various pharmaceutical and medical companies. Dr. Farkouh reported receiving research support from Amgen and Novo Nordisk, and Dr. Tsang is supported by a National New Investigator Award from the Heart and Stroke Foundation of Canada.
SOURCE: Sarma AA et al. J Am Coll Cardiol. 2019 Dec 9. doi: 10.1016/j.jacc.2019.09.065.
While women were found to be at lower risk of major cardiovascular events than men after non–ST-segment elevation acute coronary syndromes (NSTEACS), a new study has also shown that they remain undertreated with guideline-directed therapies.
“These findings underscore the fact that efforts to modify risk factors and implement appropriate treatment strategies in women and men may be a powerful means to further improve outcomes among patients with NSTEACS,” wrote Amy A. Sarma, MD, of Massachusetts General Hospital in Boston and coauthors. The study was published in the Journal of the American College of Cardiology.
To determine if outcomes differed between men and women after NSTEACS, the researchers analyzed 68,730 patients from 10 different clinical trials as part of the Thrombolysis In Myocardial Infarction (TIMI) Study Group. All trials enrolled patients with NSTEACS within 30 days of hospitalization. Across the 10 trials, there were a total of 19,827 women (29%).
Female patients had an average age of 67 years, compared with a mean age of 62 years for men. Women were also more likely to have had hypertension, diabetes, or prior heart failure, though less likely to have had a prior MI. In regard to treatment strategies for NSTEACS, women were less likely to receive aspirin, P2Y12 inhibitors, or statins. Women at high risk were also less likely to receive aspirin, P2Y12 inhibitors, or statins.
Before adjusted analysis, women and men had a similar risk of major adverse cardiovascular events (MACE) – defined as cardiovascular death, MI, or stroke – after NSTEACS (hazard ratio, 1.04; 95% confidence interval, 0.99-1.09; P = .16). However, women were found to be at increased risk of cardiovascular death (HR, 1.16; 95% CI, 1.02-1.32; P = .03), all-cause mortality (HR, 1.12; 95% CI, 1.01-1.24; P = .03), and stroke (HR, 1.19; 95% CI, 1.03-1.37; P = .02).
After adjusting for baseline risk predictors, women were found to have a 7% lower risk of MACE (adjusted HR, 0.93; 95% CI, 0.89-0.98; P = .005), along with a 15% lower risk of cardiovascular death (aHR, 0.85; 95% CI, 0.76-0.96; P = .008) and a 16% lower risk of all-cause death (aHR, 0.84; 95% CI, 0.78-0.90; P less than .0001). Additional adjustments for guideline-based therapies did not significantly alter the risk estimates.
In an accompanying editorial, Michael E. Farkouh, MD, and Wendy Tsang, MD, of the University of Toronto noted that this study from Sarma et al. underlines the “lack of progress made in addressing sex inequality in the care for women with ACS” (J Am Coll Cardiol. 2019 Dec 9. doi: 10.1016/j.jacc.2019.10.017).
Though Dr. Farkouh and Dr. Tsang were not surprised that the women in the study were older than the men, as women typically develop cardiovascular disease later, they noted that the women also presented with higher baseline risk. Could this be because of the “persistent perception that coronary disease is a male disease?” They cited the treatment-risk paradox and an emphasis on diagnosing obstructive coronary disease – typically seen in men – as potential contributors to underdiagnosis and undertreatment in at-risk women.
“That this care gap for women with cardiovascular diseases continues to persist, even in well-run contemporary landmark ACS trials, highlights how challenging it is to address,” they wrote.
Dr. Sarma and coauthors acknowledged their study’s potential limitations, including the 10 trials differing in study design, enrollment timing, treatments tested, and follow-up duration. They also noted that the cohort of patients were all at moderate to high risk, which may not make the findings generalizable to the broader NSTEACS patient population.
The study authors reported numerous potential conflicts of interest, including receiving research grants from – and serving as a consultant or on the advisory board of – various pharmaceutical and medical companies. Dr. Farkouh reported receiving research support from Amgen and Novo Nordisk, and Dr. Tsang is supported by a National New Investigator Award from the Heart and Stroke Foundation of Canada.
SOURCE: Sarma AA et al. J Am Coll Cardiol. 2019 Dec 9. doi: 10.1016/j.jacc.2019.09.065.
While women were found to be at lower risk of major cardiovascular events than men after non–ST-segment elevation acute coronary syndromes (NSTEACS), a new study has also shown that they remain undertreated with guideline-directed therapies.
“These findings underscore the fact that efforts to modify risk factors and implement appropriate treatment strategies in women and men may be a powerful means to further improve outcomes among patients with NSTEACS,” wrote Amy A. Sarma, MD, of Massachusetts General Hospital in Boston and coauthors. The study was published in the Journal of the American College of Cardiology.
To determine if outcomes differed between men and women after NSTEACS, the researchers analyzed 68,730 patients from 10 different clinical trials as part of the Thrombolysis In Myocardial Infarction (TIMI) Study Group. All trials enrolled patients with NSTEACS within 30 days of hospitalization. Across the 10 trials, there were a total of 19,827 women (29%).
Female patients had an average age of 67 years, compared with a mean age of 62 years for men. Women were also more likely to have had hypertension, diabetes, or prior heart failure, though less likely to have had a prior MI. In regard to treatment strategies for NSTEACS, women were less likely to receive aspirin, P2Y12 inhibitors, or statins. Women at high risk were also less likely to receive aspirin, P2Y12 inhibitors, or statins.
Before adjusted analysis, women and men had a similar risk of major adverse cardiovascular events (MACE) – defined as cardiovascular death, MI, or stroke – after NSTEACS (hazard ratio, 1.04; 95% confidence interval, 0.99-1.09; P = .16). However, women were found to be at increased risk of cardiovascular death (HR, 1.16; 95% CI, 1.02-1.32; P = .03), all-cause mortality (HR, 1.12; 95% CI, 1.01-1.24; P = .03), and stroke (HR, 1.19; 95% CI, 1.03-1.37; P = .02).
After adjusting for baseline risk predictors, women were found to have a 7% lower risk of MACE (adjusted HR, 0.93; 95% CI, 0.89-0.98; P = .005), along with a 15% lower risk of cardiovascular death (aHR, 0.85; 95% CI, 0.76-0.96; P = .008) and a 16% lower risk of all-cause death (aHR, 0.84; 95% CI, 0.78-0.90; P less than .0001). Additional adjustments for guideline-based therapies did not significantly alter the risk estimates.
In an accompanying editorial, Michael E. Farkouh, MD, and Wendy Tsang, MD, of the University of Toronto noted that this study from Sarma et al. underlines the “lack of progress made in addressing sex inequality in the care for women with ACS” (J Am Coll Cardiol. 2019 Dec 9. doi: 10.1016/j.jacc.2019.10.017).
Though Dr. Farkouh and Dr. Tsang were not surprised that the women in the study were older than the men, as women typically develop cardiovascular disease later, they noted that the women also presented with higher baseline risk. Could this be because of the “persistent perception that coronary disease is a male disease?” They cited the treatment-risk paradox and an emphasis on diagnosing obstructive coronary disease – typically seen in men – as potential contributors to underdiagnosis and undertreatment in at-risk women.
“That this care gap for women with cardiovascular diseases continues to persist, even in well-run contemporary landmark ACS trials, highlights how challenging it is to address,” they wrote.
Dr. Sarma and coauthors acknowledged their study’s potential limitations, including the 10 trials differing in study design, enrollment timing, treatments tested, and follow-up duration. They also noted that the cohort of patients were all at moderate to high risk, which may not make the findings generalizable to the broader NSTEACS patient population.
The study authors reported numerous potential conflicts of interest, including receiving research grants from – and serving as a consultant or on the advisory board of – various pharmaceutical and medical companies. Dr. Farkouh reported receiving research support from Amgen and Novo Nordisk, and Dr. Tsang is supported by a National New Investigator Award from the Heart and Stroke Foundation of Canada.
SOURCE: Sarma AA et al. J Am Coll Cardiol. 2019 Dec 9. doi: 10.1016/j.jacc.2019.09.065.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY
Key clinical point: Despite being undertreated with guideline-directed therapies, women have a lower risk of recurrent cardiovascular events after non–ST-segment elevation acute coronary syndromes.
Major finding: After adjustment for baseline risk predictors, women were found to have a 7% lower risk of major adverse cardiovascular events (adjusted hazard ratio, 0.93; 95% confidence interval, 0.89-0.98, P = 0.005).
Study details: A sex-specific analysis of cardiovascular outcomes in 68,730 patients with non–ST-segment elevation acute coronary syndromes across 10 clinical trials.
Disclosures: The authors reported numerous potential conflicts of interest, including receiving research grants from – and serving as a consultant or on the advisory board of – various pharmaceutical and medical companies.
Source: Sarma AA et al. J Am Coll Cardiol. 2019 Dec 9. doi: 10.1016/j.jacc.2019.09.065.
Genetic test stratified AFib patients with low CHA2DS2-VASc scores
PHILADELPHIA – A 32-gene screening test for stroke risk identified a subgroup of atrial fibrillation (AFib) patients with an elevated rate of ischemic strokes despite having a low stroke risk by conventional criteria by their CHA2DS2-VASc score in a post-hoc analysis of more than 11,000 patients enrolled in a recent drug trial.

Overall, AFib patients in the highest tertile for genetic risk based on a 32 gene-loci test had a 31% increase rate of ischemic stroke during a median 2.8 years of follow-up compared with patients in the tertile with the lowest risk based on the 32-loci screen, Nicholas A. Marston, MD, said at the American Heart Association scientific sessions. This suggested that the genetic test had roughly the same association with an increased stroke risk as several components of the CHA2DS2-VASc score, such as female sex, an age of 65-74 years old, or having heart failure as a comorbidity, each of which links with an increased risk for ischemic stroke of about 31%-38%, Dr. Marston noted.
The genetic test produced even sharper discrimination among the patients with the lowest stroke risk as measured by their CHA2DS2-VASc score (Circulation. 2012 Aug 14;126[7]: 860-5). Among the slightly more than 3,000 patients in the study with a CHA2DS2-VASc score of three or less, those in the subgroup with the highest risk by the 32-loci screen had a stroke rate during follow-up that was 76% higher than those in the low or intermediate tertile for their genetic score. Among the 796 patients with a CHA2DS2-VASc score of just 1 or 2, those who also fell into the highest level of risk on the 32-loci screen had a stroke rate 3.5-fold higher than those with a similar CHA2DS2-VASc score but in the low and intermediate tertiles by the 32-loci screen.
The additional risk prediction provided by the 32-loci test was statistically significant in the analysis of the 3,071 patients with a CHA2DS2-VASc score of 3 or less after adjustment for age, sex, ancestry, and the individual components of the CHA2DS2-VASc score, which includes factors such as hypertension, diabetes, and heart failure, said Dr. Marston, a cardiologist at Brigham and Women’s Hospital in Boston. The 3.5-fold elevation among patients with a high genetic-risk score in the cohort of 796 patients with a CHA2DS2-VASc score of 1 or 2 just missed statistical significance (P = .06), possibly because the number of patients in the analysis was relatively low. Future research should explore the predictive value of the genetic risk score in patients with a CHA2DS2-VASc score of 0 or 1, the “group where therapeutic decisions could be altered” depending on the genetic risk score, he explained.
Dr. Marston and his associates used data collected in the ENGAGE AF-TIMI 48 (Effective Anticoagulation with Factor Xa Next Generation in Atrial Fibrillation–Thrombolysis in Myocardial Infarction 48) trial, which was designed to assess the safety and efficacy of the direct-acting oral anticoagulant edoxaban in patients with AFib (New Engl J Med. 2013 Nov 28; 369[21]: 2091-2104). The 32-loci panel to measure a person’s genetic risk for stroke came from a 2018 report by a multinational team of researchers (Nature Genetics. 2018 Apr;50[4]: 524-37). The new analysis applied this 32-loci genetic test panel to 11.164 unrelated AFib patients with European ancestry from the ENGAGE AF-TIMIT 48 database. They divided this cohort into tertiles based on having a low, intermediate, or high stroke risk as assessed by the 32-loci genetic test. The analysis focused on patients enrolled in the trial who had European ancestry because the 32-loci screening test relied predominantly on data collected from people with this genetic background, Dr. Marston said.
SOURCE: Marston NA et al. AHA 2019, Abstract 336.
PHILADELPHIA – A 32-gene screening test for stroke risk identified a subgroup of atrial fibrillation (AFib) patients with an elevated rate of ischemic strokes despite having a low stroke risk by conventional criteria by their CHA2DS2-VASc score in a post-hoc analysis of more than 11,000 patients enrolled in a recent drug trial.
Overall, AFib patients in the highest tertile for genetic risk based on a 32 gene-loci test had a 31% increase rate of ischemic stroke during a median 2.8 years of follow-up compared with patients in the tertile with the lowest risk based on the 32-loci screen, Nicholas A. Marston, MD, said at the American Heart Association scientific sessions. This suggested that the genetic test had roughly the same association with an increased stroke risk as several components of the CHA2DS2-VASc score, such as female sex, an age of 65-74 years old, or having heart failure as a comorbidity, each of which links with an increased risk for ischemic stroke of about 31%-38%, Dr. Marston noted.
The genetic test produced even sharper discrimination among the patients with the lowest stroke risk as measured by their CHA2DS2-VASc score (Circulation. 2012 Aug 14;126[7]: 860-5). Among the slightly more than 3,000 patients in the study with a CHA2DS2-VASc score of three or less, those in the subgroup with the highest risk by the 32-loci screen had a stroke rate during follow-up that was 76% higher than those in the low or intermediate tertile for their genetic score. Among the 796 patients with a CHA2DS2-VASc score of just 1 or 2, those who also fell into the highest level of risk on the 32-loci screen had a stroke rate 3.5-fold higher than those with a similar CHA2DS2-VASc score but in the low and intermediate tertiles by the 32-loci screen.
The additional risk prediction provided by the 32-loci test was statistically significant in the analysis of the 3,071 patients with a CHA2DS2-VASc score of 3 or less after adjustment for age, sex, ancestry, and the individual components of the CHA2DS2-VASc score, which includes factors such as hypertension, diabetes, and heart failure, said Dr. Marston, a cardiologist at Brigham and Women’s Hospital in Boston. The 3.5-fold elevation among patients with a high genetic-risk score in the cohort of 796 patients with a CHA2DS2-VASc score of 1 or 2 just missed statistical significance (P = .06), possibly because the number of patients in the analysis was relatively low. Future research should explore the predictive value of the genetic risk score in patients with a CHA2DS2-VASc score of 0 or 1, the “group where therapeutic decisions could be altered” depending on the genetic risk score, he explained.
Dr. Marston and his associates used data collected in the ENGAGE AF-TIMI 48 (Effective Anticoagulation with Factor Xa Next Generation in Atrial Fibrillation–Thrombolysis in Myocardial Infarction 48) trial, which was designed to assess the safety and efficacy of the direct-acting oral anticoagulant edoxaban in patients with AFib (New Engl J Med. 2013 Nov 28; 369[21]: 2091-2104). The 32-loci panel to measure a person’s genetic risk for stroke came from a 2018 report by a multinational team of researchers (Nature Genetics. 2018 Apr;50[4]: 524-37). The new analysis applied this 32-loci genetic test panel to 11.164 unrelated AFib patients with European ancestry from the ENGAGE AF-TIMIT 48 database. They divided this cohort into tertiles based on having a low, intermediate, or high stroke risk as assessed by the 32-loci genetic test. The analysis focused on patients enrolled in the trial who had European ancestry because the 32-loci screening test relied predominantly on data collected from people with this genetic background, Dr. Marston said.
SOURCE: Marston NA et al. AHA 2019, Abstract 336.
PHILADELPHIA – A 32-gene screening test for stroke risk identified a subgroup of atrial fibrillation (AFib) patients with an elevated rate of ischemic strokes despite having a low stroke risk by conventional criteria by their CHA2DS2-VASc score in a post-hoc analysis of more than 11,000 patients enrolled in a recent drug trial.
Overall, AFib patients in the highest tertile for genetic risk based on a 32 gene-loci test had a 31% increase rate of ischemic stroke during a median 2.8 years of follow-up compared with patients in the tertile with the lowest risk based on the 32-loci screen, Nicholas A. Marston, MD, said at the American Heart Association scientific sessions. This suggested that the genetic test had roughly the same association with an increased stroke risk as several components of the CHA2DS2-VASc score, such as female sex, an age of 65-74 years old, or having heart failure as a comorbidity, each of which links with an increased risk for ischemic stroke of about 31%-38%, Dr. Marston noted.
The genetic test produced even sharper discrimination among the patients with the lowest stroke risk as measured by their CHA2DS2-VASc score (Circulation. 2012 Aug 14;126[7]: 860-5). Among the slightly more than 3,000 patients in the study with a CHA2DS2-VASc score of three or less, those in the subgroup with the highest risk by the 32-loci screen had a stroke rate during follow-up that was 76% higher than those in the low or intermediate tertile for their genetic score. Among the 796 patients with a CHA2DS2-VASc score of just 1 or 2, those who also fell into the highest level of risk on the 32-loci screen had a stroke rate 3.5-fold higher than those with a similar CHA2DS2-VASc score but in the low and intermediate tertiles by the 32-loci screen.
The additional risk prediction provided by the 32-loci test was statistically significant in the analysis of the 3,071 patients with a CHA2DS2-VASc score of 3 or less after adjustment for age, sex, ancestry, and the individual components of the CHA2DS2-VASc score, which includes factors such as hypertension, diabetes, and heart failure, said Dr. Marston, a cardiologist at Brigham and Women’s Hospital in Boston. The 3.5-fold elevation among patients with a high genetic-risk score in the cohort of 796 patients with a CHA2DS2-VASc score of 1 or 2 just missed statistical significance (P = .06), possibly because the number of patients in the analysis was relatively low. Future research should explore the predictive value of the genetic risk score in patients with a CHA2DS2-VASc score of 0 or 1, the “group where therapeutic decisions could be altered” depending on the genetic risk score, he explained.
Dr. Marston and his associates used data collected in the ENGAGE AF-TIMI 48 (Effective Anticoagulation with Factor Xa Next Generation in Atrial Fibrillation–Thrombolysis in Myocardial Infarction 48) trial, which was designed to assess the safety and efficacy of the direct-acting oral anticoagulant edoxaban in patients with AFib (New Engl J Med. 2013 Nov 28; 369[21]: 2091-2104). The 32-loci panel to measure a person’s genetic risk for stroke came from a 2018 report by a multinational team of researchers (Nature Genetics. 2018 Apr;50[4]: 524-37). The new analysis applied this 32-loci genetic test panel to 11.164 unrelated AFib patients with European ancestry from the ENGAGE AF-TIMIT 48 database. They divided this cohort into tertiles based on having a low, intermediate, or high stroke risk as assessed by the 32-loci genetic test. The analysis focused on patients enrolled in the trial who had European ancestry because the 32-loci screening test relied predominantly on data collected from people with this genetic background, Dr. Marston said.
SOURCE: Marston NA et al. AHA 2019, Abstract 336.
REPORTING FROM AHA 2019
E-cigarette use, interest in flavors remains high among youth
, according to new findings from the Centers for Disease Control and Prevention.

Just over half of high school students and about a quarter of middle school students have ever tried a tobacco product, and more than a third of students have ever tried an e-cigarette, according to results from the 2019 National Youth Tobacco Survey. These results were published in the Morbidity and Mortality Weekly Report on Dec. 6.
Adolescent cigarette smoking rates have continued their decline, hitting their lowest rate ever in 2019, but e-cigarette use, or “vaping,” has continued to increase. E-cigarette use surpassed that of all other tobacco products in 2014 and has remained the most common—as well as the least likely to be perceived as harmful, researchers reported.
“Although most current youth tobacco product users are not daily users, estimates of frequent e-cigarette use among high school students were comparable to those observed for cigarette and smokeless tobacco product users in 2019,” wrote Teresa W. Wang, PhD, of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, and associates at the CDC and Food and Drug Administration. “Youth use of tobacco products in any form is unsafe, regardless of whether the products are smoked, smokeless, or electronic.”
The high prevalence of e-cigarette use was no surprise to Karen Wilson, MD, chief of the division of general pediatrics at the Icahn School of Medicine at Mount Sinai and Mount Sinai Kravis Children’s Hospital, New York, and chair of the American Academy of Pediatrics’ Tobacco Consortium.
“It also fits with what we’re seeing anecdotally,” Dr. Wilson said in an interview. “We hear the statistic that 30% of high school students are using them, but high school students will say it’s much more than that.”
It’s therefore important for physicians to be proactive in talking to youth about these products. “They should absolutely be screening for vaping and know all about the different products,” including JUUL, Suorin, nicotine toothpicks, and candies and other products, Dr. Wilson said. “Pediatricians need to be asking their teenagers open-ended questions about what are kids using now.”
The American Academy of Pediatrics has resources available to help pediatricians and families of youth using e-cigarettes and vaping devices, she added.
Main findings
The researchers reported data from the annual, cross-sectional National Youth Tobacco Survey, administered to U.S. students in public and private schools in all 50 states and the District of Columbia. The results were divided into middle school (grades 6-8) and high school (grades 9-12) from 251 participating schools between February 2019 and May 2019.
The survey has been done using pencil and paper questionnaires since it began in 1999, but this year’s surveys were digital for the first time. Among the 19,018 questionnaires completed (student response rate 85.3%), 8,837 were middle school and 10,097 were high school. The weighted analysis of results represents 27 million students: 11.9 million in middle school and 15 million in high school.
More than half (53.3%) of high school students reported ever having tried a tobacco product, and 31.2% reported having used one in the past 30 days. In middle school, 24.3% of students reported ever using a tobacco product, and 12.5% have used one in the past month.
Tobacco products include cigarettes (traditional/combusted), electronic cigarettes, cigars, smokeless tobacco, hookahs, pipe tobacco, and bidis, which are small brown cigarettes wrapped in leaves. Among the electronic tobacco products mentioned in the survey were NJOY, Blu, Vuse, MarkTen, Logic, Vapin Plus, eGo and Halo.
The most common product for youth to try was e-cigarettes, which 35% of middle and high school students had ever tried. Just under a quarter of students (23%) had used a tobacco product in the past month, and e-cigarettes were again the most commonly used overall by that group, cited by 20% of recent users. Cigars (5.3%), cigarettes (4.3%), smokeless tobacco (3.5%), hookahs (2.6%) and pipes (under 1%) were used much less frequently.
Frequent use, defined as at least 20 of the previous 30 days, was most common among youth using smokeless tobacco (34.1% of current users) and e-cigarettes (30.4%) and least common among cigar smokers (16.8%). Among those currently using any tobacco product, 24.7% said they had cravings for a product within the past month, and 13.7% wanted to use it within a half hour of waking up.
More than half of those who currently used any tobacco products (57.8%) were seriously considering quitting, and a similar proportion (57.5%) had stopped using all tobacco products for at least 1 day in an attempt to quit.
“Many [adolescents] will tell you they will use it until they don’t have the availability of getting it,” Dr. Wilson said. “The problem is that they’re becoming so addicted to the high-nicotine products that they’re going farther and farther out of their way to try to get these products so that they can satisfy their addictions.”
Policies restricting access, such as increasing the age for sales to 21 and increasing taxes on products, can reduce tobacco use among youth, Dr. Wilson said.
“It will encourage teenagers to get help for their addiction by using FDA-approved devices or nicotine replacement therapy and behavioral interventions rather than relying on an unproven and potentially dangerous product,” she said.
Reasons for use, flavor, and harm perception
The most common flavored tobacco product used among youth was e-cigarettes, reported by 68.8% of current e-cigarette users, followed by smokeless tobacco (48%), cigarettes (46.7%, only menthol), cigars, pipe tobacco, and hookahs.
The top reasons youth cited for trying e-cigarettes were curiosity (55.3%), a friend or family member’s use (30.8%), and their availability in a wide range of flavors (22.4%). Almost as popular as flavor availability was e-cigarette users’ interest in doing “tricks” with the product (21.2%).
The cross-sectional questionnaire method of the study precluded the ability to draw conclusions about why students might perceive a particular tobacco product as more or less harmful. However, public health officials have expressed concern that flavors reduce the perceived harm that can come from the products. Dr. Wilson said the attraction to e-cigarette flavors is “huge.”
“If electronic cigarettes were only available in tobacco flavor, I do not believe that many teenagers at all would try them,” Dr. Wilson said. “They think because they’re sweet and flavored that they actually aren’t harmful. It makes the kids think these are safe products.”
More than one in four students (28.2%) perceived intermittent e-cigarette use as causing little to no harm, and only 16.4% similarly saw little or no harm from intermittent hookah use, compared with 11.5% for smokeless tobacco and 9.5% for cigarettes. Less than a third of respondents (32.3%) saw intermittent e-cigarette use as causing a lot of harm, compared with much higher percentages for cigarettes (54.9%) and smokeless tobacco (52.5%).
Part of the problem with harm perception is the narrative promoted by e-cigarette companies, Dr. Wilson said.
“From the very beginning, they started with a campaign that called this harmless water vapor, which it is absolutely not,” she said. “It’s an aerosol of toxic chemicals and nicotine, which is addictive. We know that nicotine that can impact scores of cognitive tests and impulsivity. We have no idea what these really high levels [of nicotine] will do.”
Further, potential long-term harm is still an open question, she pointed out.
“We also know that these are particulates and toxins that are being inhaled into the lungs,” Dr. Wilson said. “We know they have some impact on asthma, and we don’t know what the impact is for using for 10 or 20 years.”
Curiosity about e-cigarettes and about traditional cigarettes were prevalent in similar proportions among youth who had never tried a tobacco product: 39.1% of never-users were curious about e-cigarettes, and 37% about traditional cigarettes. In addition to curiosity, researchers assess susceptibility among those who have never tried a tobacco product and found nearly identical susceptibility to e-cigarettes (45%) and traditional cigarettes (45.9%).
The survey also asked students about their exposure to tobacco advertising or promotions from a wide range of sources: convenience stores, supermarkets, gas stations, the Internet, television, video streaming, cinemas, and newspapers or magazines. Among the students who reported going to these sources, 69.3% had seen e-cigarette marketing, and 81.7% had seen marketing for other tobacco products, including cigarettes.
SOURCE: Wang TW et al. MMWR Surveill Summ. 2019 Nov 6;68(12):1-22. doi: 10.15585/mmwr.ss6812a1.
, according to new findings from the Centers for Disease Control and Prevention.
Just over half of high school students and about a quarter of middle school students have ever tried a tobacco product, and more than a third of students have ever tried an e-cigarette, according to results from the 2019 National Youth Tobacco Survey. These results were published in the Morbidity and Mortality Weekly Report on Dec. 6.
Adolescent cigarette smoking rates have continued their decline, hitting their lowest rate ever in 2019, but e-cigarette use, or “vaping,” has continued to increase. E-cigarette use surpassed that of all other tobacco products in 2014 and has remained the most common—as well as the least likely to be perceived as harmful, researchers reported.
“Although most current youth tobacco product users are not daily users, estimates of frequent e-cigarette use among high school students were comparable to those observed for cigarette and smokeless tobacco product users in 2019,” wrote Teresa W. Wang, PhD, of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, and associates at the CDC and Food and Drug Administration. “Youth use of tobacco products in any form is unsafe, regardless of whether the products are smoked, smokeless, or electronic.”
The high prevalence of e-cigarette use was no surprise to Karen Wilson, MD, chief of the division of general pediatrics at the Icahn School of Medicine at Mount Sinai and Mount Sinai Kravis Children’s Hospital, New York, and chair of the American Academy of Pediatrics’ Tobacco Consortium.
“It also fits with what we’re seeing anecdotally,” Dr. Wilson said in an interview. “We hear the statistic that 30% of high school students are using them, but high school students will say it’s much more than that.”
It’s therefore important for physicians to be proactive in talking to youth about these products. “They should absolutely be screening for vaping and know all about the different products,” including JUUL, Suorin, nicotine toothpicks, and candies and other products, Dr. Wilson said. “Pediatricians need to be asking their teenagers open-ended questions about what are kids using now.”
The American Academy of Pediatrics has resources available to help pediatricians and families of youth using e-cigarettes and vaping devices, she added.
Main findings
The researchers reported data from the annual, cross-sectional National Youth Tobacco Survey, administered to U.S. students in public and private schools in all 50 states and the District of Columbia. The results were divided into middle school (grades 6-8) and high school (grades 9-12) from 251 participating schools between February 2019 and May 2019.
The survey has been done using pencil and paper questionnaires since it began in 1999, but this year’s surveys were digital for the first time. Among the 19,018 questionnaires completed (student response rate 85.3%), 8,837 were middle school and 10,097 were high school. The weighted analysis of results represents 27 million students: 11.9 million in middle school and 15 million in high school.
More than half (53.3%) of high school students reported ever having tried a tobacco product, and 31.2% reported having used one in the past 30 days. In middle school, 24.3% of students reported ever using a tobacco product, and 12.5% have used one in the past month.
Tobacco products include cigarettes (traditional/combusted), electronic cigarettes, cigars, smokeless tobacco, hookahs, pipe tobacco, and bidis, which are small brown cigarettes wrapped in leaves. Among the electronic tobacco products mentioned in the survey were NJOY, Blu, Vuse, MarkTen, Logic, Vapin Plus, eGo and Halo.
The most common product for youth to try was e-cigarettes, which 35% of middle and high school students had ever tried. Just under a quarter of students (23%) had used a tobacco product in the past month, and e-cigarettes were again the most commonly used overall by that group, cited by 20% of recent users. Cigars (5.3%), cigarettes (4.3%), smokeless tobacco (3.5%), hookahs (2.6%) and pipes (under 1%) were used much less frequently.
Frequent use, defined as at least 20 of the previous 30 days, was most common among youth using smokeless tobacco (34.1% of current users) and e-cigarettes (30.4%) and least common among cigar smokers (16.8%). Among those currently using any tobacco product, 24.7% said they had cravings for a product within the past month, and 13.7% wanted to use it within a half hour of waking up.
More than half of those who currently used any tobacco products (57.8%) were seriously considering quitting, and a similar proportion (57.5%) had stopped using all tobacco products for at least 1 day in an attempt to quit.
“Many [adolescents] will tell you they will use it until they don’t have the availability of getting it,” Dr. Wilson said. “The problem is that they’re becoming so addicted to the high-nicotine products that they’re going farther and farther out of their way to try to get these products so that they can satisfy their addictions.”
Policies restricting access, such as increasing the age for sales to 21 and increasing taxes on products, can reduce tobacco use among youth, Dr. Wilson said.
“It will encourage teenagers to get help for their addiction by using FDA-approved devices or nicotine replacement therapy and behavioral interventions rather than relying on an unproven and potentially dangerous product,” she said.
Reasons for use, flavor, and harm perception
The most common flavored tobacco product used among youth was e-cigarettes, reported by 68.8% of current e-cigarette users, followed by smokeless tobacco (48%), cigarettes (46.7%, only menthol), cigars, pipe tobacco, and hookahs.
The top reasons youth cited for trying e-cigarettes were curiosity (55.3%), a friend or family member’s use (30.8%), and their availability in a wide range of flavors (22.4%). Almost as popular as flavor availability was e-cigarette users’ interest in doing “tricks” with the product (21.2%).
The cross-sectional questionnaire method of the study precluded the ability to draw conclusions about why students might perceive a particular tobacco product as more or less harmful. However, public health officials have expressed concern that flavors reduce the perceived harm that can come from the products. Dr. Wilson said the attraction to e-cigarette flavors is “huge.”
“If electronic cigarettes were only available in tobacco flavor, I do not believe that many teenagers at all would try them,” Dr. Wilson said. “They think because they’re sweet and flavored that they actually aren’t harmful. It makes the kids think these are safe products.”
More than one in four students (28.2%) perceived intermittent e-cigarette use as causing little to no harm, and only 16.4% similarly saw little or no harm from intermittent hookah use, compared with 11.5% for smokeless tobacco and 9.5% for cigarettes. Less than a third of respondents (32.3%) saw intermittent e-cigarette use as causing a lot of harm, compared with much higher percentages for cigarettes (54.9%) and smokeless tobacco (52.5%).
Part of the problem with harm perception is the narrative promoted by e-cigarette companies, Dr. Wilson said.
“From the very beginning, they started with a campaign that called this harmless water vapor, which it is absolutely not,” she said. “It’s an aerosol of toxic chemicals and nicotine, which is addictive. We know that nicotine that can impact scores of cognitive tests and impulsivity. We have no idea what these really high levels [of nicotine] will do.”
Further, potential long-term harm is still an open question, she pointed out.
“We also know that these are particulates and toxins that are being inhaled into the lungs,” Dr. Wilson said. “We know they have some impact on asthma, and we don’t know what the impact is for using for 10 or 20 years.”
Curiosity about e-cigarettes and about traditional cigarettes were prevalent in similar proportions among youth who had never tried a tobacco product: 39.1% of never-users were curious about e-cigarettes, and 37% about traditional cigarettes. In addition to curiosity, researchers assess susceptibility among those who have never tried a tobacco product and found nearly identical susceptibility to e-cigarettes (45%) and traditional cigarettes (45.9%).
The survey also asked students about their exposure to tobacco advertising or promotions from a wide range of sources: convenience stores, supermarkets, gas stations, the Internet, television, video streaming, cinemas, and newspapers or magazines. Among the students who reported going to these sources, 69.3% had seen e-cigarette marketing, and 81.7% had seen marketing for other tobacco products, including cigarettes.
SOURCE: Wang TW et al. MMWR Surveill Summ. 2019 Nov 6;68(12):1-22. doi: 10.15585/mmwr.ss6812a1.
, according to new findings from the Centers for Disease Control and Prevention.
Just over half of high school students and about a quarter of middle school students have ever tried a tobacco product, and more than a third of students have ever tried an e-cigarette, according to results from the 2019 National Youth Tobacco Survey. These results were published in the Morbidity and Mortality Weekly Report on Dec. 6.
Adolescent cigarette smoking rates have continued their decline, hitting their lowest rate ever in 2019, but e-cigarette use, or “vaping,” has continued to increase. E-cigarette use surpassed that of all other tobacco products in 2014 and has remained the most common—as well as the least likely to be perceived as harmful, researchers reported.
“Although most current youth tobacco product users are not daily users, estimates of frequent e-cigarette use among high school students were comparable to those observed for cigarette and smokeless tobacco product users in 2019,” wrote Teresa W. Wang, PhD, of the CDC’s National Center for Chronic Disease Prevention and Health Promotion, and associates at the CDC and Food and Drug Administration. “Youth use of tobacco products in any form is unsafe, regardless of whether the products are smoked, smokeless, or electronic.”
The high prevalence of e-cigarette use was no surprise to Karen Wilson, MD, chief of the division of general pediatrics at the Icahn School of Medicine at Mount Sinai and Mount Sinai Kravis Children’s Hospital, New York, and chair of the American Academy of Pediatrics’ Tobacco Consortium.
“It also fits with what we’re seeing anecdotally,” Dr. Wilson said in an interview. “We hear the statistic that 30% of high school students are using them, but high school students will say it’s much more than that.”
It’s therefore important for physicians to be proactive in talking to youth about these products. “They should absolutely be screening for vaping and know all about the different products,” including JUUL, Suorin, nicotine toothpicks, and candies and other products, Dr. Wilson said. “Pediatricians need to be asking their teenagers open-ended questions about what are kids using now.”
The American Academy of Pediatrics has resources available to help pediatricians and families of youth using e-cigarettes and vaping devices, she added.
Main findings
The researchers reported data from the annual, cross-sectional National Youth Tobacco Survey, administered to U.S. students in public and private schools in all 50 states and the District of Columbia. The results were divided into middle school (grades 6-8) and high school (grades 9-12) from 251 participating schools between February 2019 and May 2019.
The survey has been done using pencil and paper questionnaires since it began in 1999, but this year’s surveys were digital for the first time. Among the 19,018 questionnaires completed (student response rate 85.3%), 8,837 were middle school and 10,097 were high school. The weighted analysis of results represents 27 million students: 11.9 million in middle school and 15 million in high school.
More than half (53.3%) of high school students reported ever having tried a tobacco product, and 31.2% reported having used one in the past 30 days. In middle school, 24.3% of students reported ever using a tobacco product, and 12.5% have used one in the past month.
Tobacco products include cigarettes (traditional/combusted), electronic cigarettes, cigars, smokeless tobacco, hookahs, pipe tobacco, and bidis, which are small brown cigarettes wrapped in leaves. Among the electronic tobacco products mentioned in the survey were NJOY, Blu, Vuse, MarkTen, Logic, Vapin Plus, eGo and Halo.
The most common product for youth to try was e-cigarettes, which 35% of middle and high school students had ever tried. Just under a quarter of students (23%) had used a tobacco product in the past month, and e-cigarettes were again the most commonly used overall by that group, cited by 20% of recent users. Cigars (5.3%), cigarettes (4.3%), smokeless tobacco (3.5%), hookahs (2.6%) and pipes (under 1%) were used much less frequently.
Frequent use, defined as at least 20 of the previous 30 days, was most common among youth using smokeless tobacco (34.1% of current users) and e-cigarettes (30.4%) and least common among cigar smokers (16.8%). Among those currently using any tobacco product, 24.7% said they had cravings for a product within the past month, and 13.7% wanted to use it within a half hour of waking up.
More than half of those who currently used any tobacco products (57.8%) were seriously considering quitting, and a similar proportion (57.5%) had stopped using all tobacco products for at least 1 day in an attempt to quit.
“Many [adolescents] will tell you they will use it until they don’t have the availability of getting it,” Dr. Wilson said. “The problem is that they’re becoming so addicted to the high-nicotine products that they’re going farther and farther out of their way to try to get these products so that they can satisfy their addictions.”
Policies restricting access, such as increasing the age for sales to 21 and increasing taxes on products, can reduce tobacco use among youth, Dr. Wilson said.
“It will encourage teenagers to get help for their addiction by using FDA-approved devices or nicotine replacement therapy and behavioral interventions rather than relying on an unproven and potentially dangerous product,” she said.
Reasons for use, flavor, and harm perception
The most common flavored tobacco product used among youth was e-cigarettes, reported by 68.8% of current e-cigarette users, followed by smokeless tobacco (48%), cigarettes (46.7%, only menthol), cigars, pipe tobacco, and hookahs.
The top reasons youth cited for trying e-cigarettes were curiosity (55.3%), a friend or family member’s use (30.8%), and their availability in a wide range of flavors (22.4%). Almost as popular as flavor availability was e-cigarette users’ interest in doing “tricks” with the product (21.2%).
The cross-sectional questionnaire method of the study precluded the ability to draw conclusions about why students might perceive a particular tobacco product as more or less harmful. However, public health officials have expressed concern that flavors reduce the perceived harm that can come from the products. Dr. Wilson said the attraction to e-cigarette flavors is “huge.”
“If electronic cigarettes were only available in tobacco flavor, I do not believe that many teenagers at all would try them,” Dr. Wilson said. “They think because they’re sweet and flavored that they actually aren’t harmful. It makes the kids think these are safe products.”
More than one in four students (28.2%) perceived intermittent e-cigarette use as causing little to no harm, and only 16.4% similarly saw little or no harm from intermittent hookah use, compared with 11.5% for smokeless tobacco and 9.5% for cigarettes. Less than a third of respondents (32.3%) saw intermittent e-cigarette use as causing a lot of harm, compared with much higher percentages for cigarettes (54.9%) and smokeless tobacco (52.5%).
Part of the problem with harm perception is the narrative promoted by e-cigarette companies, Dr. Wilson said.
“From the very beginning, they started with a campaign that called this harmless water vapor, which it is absolutely not,” she said. “It’s an aerosol of toxic chemicals and nicotine, which is addictive. We know that nicotine that can impact scores of cognitive tests and impulsivity. We have no idea what these really high levels [of nicotine] will do.”
Further, potential long-term harm is still an open question, she pointed out.
“We also know that these are particulates and toxins that are being inhaled into the lungs,” Dr. Wilson said. “We know they have some impact on asthma, and we don’t know what the impact is for using for 10 or 20 years.”
Curiosity about e-cigarettes and about traditional cigarettes were prevalent in similar proportions among youth who had never tried a tobacco product: 39.1% of never-users were curious about e-cigarettes, and 37% about traditional cigarettes. In addition to curiosity, researchers assess susceptibility among those who have never tried a tobacco product and found nearly identical susceptibility to e-cigarettes (45%) and traditional cigarettes (45.9%).
The survey also asked students about their exposure to tobacco advertising or promotions from a wide range of sources: convenience stores, supermarkets, gas stations, the Internet, television, video streaming, cinemas, and newspapers or magazines. Among the students who reported going to these sources, 69.3% had seen e-cigarette marketing, and 81.7% had seen marketing for other tobacco products, including cigarettes.
SOURCE: Wang TW et al. MMWR Surveill Summ. 2019 Nov 6;68(12):1-22. doi: 10.15585/mmwr.ss6812a1.
FROM THE MMWR

Perioperative antirheumatic drug use does not impact postsurgery infection rate in RA patients
ATLANTA – Patients with rheumatoid arthritis were more at risk of postoperative infection because of a high Charlson Comorbidity Index or longer surgery time than because of perioperative use of antirheumatic medications, according to a presentation at the annual meeting of the American College of Rheumatology.

Anna Shmagel, MD, of the University of Minnesota in Minneapolis and colleagues performed a retrospective cohort study of 154 patients with seropositive RA who were in the Fairview Health System between Jan. 2010 and Dec. 2017 and underwent either orthopedic or major organ surgery. The patients were classified based on their use of disease-modifying antirheumatic drugs (DMARDs) and biologics alone or in combination, with patients divided into “no DMARD or biologic,” “DMARD but no biologic” and “biologic with or without DMARD” groups.
The question of whether to discontinue antirheumatic medications before surgery is still controversial, with conflicting evidence across studies, Dr. Shmagel said in her presentation. A study by Giles and colleagues found 10 of 91 patients (11%) RA who underwent an orthopedic surgical procedure developed a postoperative infection, with patients receiving tumor necrosis factor (TNF) inhibitors more likely to develop an infection, compared with patients who were not receiving TNF inhibitors (Arthritis Care Res. 2006. doi: 10.1002/art.21841).
However, other studies have challenged that idea, and a 2018 study from Goodman and colleagues raised the issue of whether patients stopping biologics prior to surgery are at increased risk of flares. Of 120 RA patients in their study who underwent total hip or total knee arthroplasty, 75% of patients flared at 6 weeks after surgery. While patients who halted biologics before surgery were more likely to flare, stopping biologics did not predict flaring after surgery (J Rheumatol. 2018. doi: 10.3899/jrheum.170366).
“It’s not entirely clear whether these theories are related to what we do with antirheumatic medications, but we felt that it was pertinent to further study this question.” Dr. Shmagel said.
Dr. Shmagel and colleagues examined the 30-day infection rate of RA patients postoperatively, with 30-day readmission and 30-day mortality rates as secondary outcomes. Patient-associated factors such as age, gender, race, body mass index, smoking status, Charlson Comorbidity Index, income, and use of corticosteroids were analyzed as covariates in addition to factors involving surgery such as expected surgery time, perioperative antibiotic use, and whether the procedure was elective or emergency surgery.
A majority of the patients in the study across all groups were white women about 63 years old with a body mass index above 30 kg/m2 and almost all undergoing electing surgery compared with emergency surgery. While patients in each group were similar with regard to Charlson Comorbidity Index, expected length of surgery, and percentage of patients undergoing elective surgery, patients in the biologic with or without DMARD group had a significantly lower median income level compared with those in the other two groups (P = .01).
Overall, there were 244 surgeries in 154 patients, with 117 surgeries in the group not receiving biologics or DMARDs, 95 surgeries in the group receiving DMARDs but no biologics, and 32 surgeries in the biologics with or without DMARD group. In the DMARD but no biologics group, most patients were receiving methotrexate (45%) or hydroxychloroquine (44%), while the most common biologics in the biologics with or without DMARD group were infliximab (25%), tocilizumab (19%), abatacept (16%), etanercept (13%), rituximab (9%), and tofacitinib (9%).
There was an 11% overall rate of infection, with a similar rate of infection across all groups (P = .09). While there was a higher rate of surgical site infections among patients in the biologics with or without DMARD group (9%) and a higher percentage of urinary tract infections in the no DMARD and no biologics group (4%), the results were not statistically significant. When the rate of infections was examined by type of surgery, there were no significant differences between infections from musculoskeletal surgery (P = .7) and major organ surgery (P = .8).
The overall 30-day readmission rate was 12%, but there were no statistically significant differences between groups. Although there were five deaths in the study, four deaths were in the group not receiving DMARDs or biologics, and one death was in the biologic with or without DMARD group.
Higher Charlson Comorbidity Index did predict infection risk, with an odds ratio of 1.37 per 1-point increase in the index (95% confidence interval, 1.10-1.70). Length of surgery also increased the risk of infection, with an OR of 1.16 per 15-minute increase in surgery time (95% CI, 1.09-1.23).
Dr. Shmagel noted that the retrospective nature of the study and the midwestern cohort may mean the results are not generalizable to other populations and that larger randomized trials should be considered. “Certainly, a larger study with more events would be needed,” she said.
This study was funded by the University of Minnesota. Dr. Shmagel reported no relevant conflicts of interest.
SOURCE: Kerski M et al. Arthritis Rheumatol. 2019;71 (suppl 10), Abstract 1805.
ATLANTA – Patients with rheumatoid arthritis were more at risk of postoperative infection because of a high Charlson Comorbidity Index or longer surgery time than because of perioperative use of antirheumatic medications, according to a presentation at the annual meeting of the American College of Rheumatology.
Anna Shmagel, MD, of the University of Minnesota in Minneapolis and colleagues performed a retrospective cohort study of 154 patients with seropositive RA who were in the Fairview Health System between Jan. 2010 and Dec. 2017 and underwent either orthopedic or major organ surgery. The patients were classified based on their use of disease-modifying antirheumatic drugs (DMARDs) and biologics alone or in combination, with patients divided into “no DMARD or biologic,” “DMARD but no biologic” and “biologic with or without DMARD” groups.
The question of whether to discontinue antirheumatic medications before surgery is still controversial, with conflicting evidence across studies, Dr. Shmagel said in her presentation. A study by Giles and colleagues found 10 of 91 patients (11%) RA who underwent an orthopedic surgical procedure developed a postoperative infection, with patients receiving tumor necrosis factor (TNF) inhibitors more likely to develop an infection, compared with patients who were not receiving TNF inhibitors (Arthritis Care Res. 2006. doi: 10.1002/art.21841).
However, other studies have challenged that idea, and a 2018 study from Goodman and colleagues raised the issue of whether patients stopping biologics prior to surgery are at increased risk of flares. Of 120 RA patients in their study who underwent total hip or total knee arthroplasty, 75% of patients flared at 6 weeks after surgery. While patients who halted biologics before surgery were more likely to flare, stopping biologics did not predict flaring after surgery (J Rheumatol. 2018. doi: 10.3899/jrheum.170366).
“It’s not entirely clear whether these theories are related to what we do with antirheumatic medications, but we felt that it was pertinent to further study this question.” Dr. Shmagel said.
Dr. Shmagel and colleagues examined the 30-day infection rate of RA patients postoperatively, with 30-day readmission and 30-day mortality rates as secondary outcomes. Patient-associated factors such as age, gender, race, body mass index, smoking status, Charlson Comorbidity Index, income, and use of corticosteroids were analyzed as covariates in addition to factors involving surgery such as expected surgery time, perioperative antibiotic use, and whether the procedure was elective or emergency surgery.
A majority of the patients in the study across all groups were white women about 63 years old with a body mass index above 30 kg/m2 and almost all undergoing electing surgery compared with emergency surgery. While patients in each group were similar with regard to Charlson Comorbidity Index, expected length of surgery, and percentage of patients undergoing elective surgery, patients in the biologic with or without DMARD group had a significantly lower median income level compared with those in the other two groups (P = .01).
Overall, there were 244 surgeries in 154 patients, with 117 surgeries in the group not receiving biologics or DMARDs, 95 surgeries in the group receiving DMARDs but no biologics, and 32 surgeries in the biologics with or without DMARD group. In the DMARD but no biologics group, most patients were receiving methotrexate (45%) or hydroxychloroquine (44%), while the most common biologics in the biologics with or without DMARD group were infliximab (25%), tocilizumab (19%), abatacept (16%), etanercept (13%), rituximab (9%), and tofacitinib (9%).
There was an 11% overall rate of infection, with a similar rate of infection across all groups (P = .09). While there was a higher rate of surgical site infections among patients in the biologics with or without DMARD group (9%) and a higher percentage of urinary tract infections in the no DMARD and no biologics group (4%), the results were not statistically significant. When the rate of infections was examined by type of surgery, there were no significant differences between infections from musculoskeletal surgery (P = .7) and major organ surgery (P = .8).
The overall 30-day readmission rate was 12%, but there were no statistically significant differences between groups. Although there were five deaths in the study, four deaths were in the group not receiving DMARDs or biologics, and one death was in the biologic with or without DMARD group.
Higher Charlson Comorbidity Index did predict infection risk, with an odds ratio of 1.37 per 1-point increase in the index (95% confidence interval, 1.10-1.70). Length of surgery also increased the risk of infection, with an OR of 1.16 per 15-minute increase in surgery time (95% CI, 1.09-1.23).
Dr. Shmagel noted that the retrospective nature of the study and the midwestern cohort may mean the results are not generalizable to other populations and that larger randomized trials should be considered. “Certainly, a larger study with more events would be needed,” she said.
This study was funded by the University of Minnesota. Dr. Shmagel reported no relevant conflicts of interest.
SOURCE: Kerski M et al. Arthritis Rheumatol. 2019;71 (suppl 10), Abstract 1805.
ATLANTA – Patients with rheumatoid arthritis were more at risk of postoperative infection because of a high Charlson Comorbidity Index or longer surgery time than because of perioperative use of antirheumatic medications, according to a presentation at the annual meeting of the American College of Rheumatology.
Anna Shmagel, MD, of the University of Minnesota in Minneapolis and colleagues performed a retrospective cohort study of 154 patients with seropositive RA who were in the Fairview Health System between Jan. 2010 and Dec. 2017 and underwent either orthopedic or major organ surgery. The patients were classified based on their use of disease-modifying antirheumatic drugs (DMARDs) and biologics alone or in combination, with patients divided into “no DMARD or biologic,” “DMARD but no biologic” and “biologic with or without DMARD” groups.
The question of whether to discontinue antirheumatic medications before surgery is still controversial, with conflicting evidence across studies, Dr. Shmagel said in her presentation. A study by Giles and colleagues found 10 of 91 patients (11%) RA who underwent an orthopedic surgical procedure developed a postoperative infection, with patients receiving tumor necrosis factor (TNF) inhibitors more likely to develop an infection, compared with patients who were not receiving TNF inhibitors (Arthritis Care Res. 2006. doi: 10.1002/art.21841).
However, other studies have challenged that idea, and a 2018 study from Goodman and colleagues raised the issue of whether patients stopping biologics prior to surgery are at increased risk of flares. Of 120 RA patients in their study who underwent total hip or total knee arthroplasty, 75% of patients flared at 6 weeks after surgery. While patients who halted biologics before surgery were more likely to flare, stopping biologics did not predict flaring after surgery (J Rheumatol. 2018. doi: 10.3899/jrheum.170366).
“It’s not entirely clear whether these theories are related to what we do with antirheumatic medications, but we felt that it was pertinent to further study this question.” Dr. Shmagel said.
Dr. Shmagel and colleagues examined the 30-day infection rate of RA patients postoperatively, with 30-day readmission and 30-day mortality rates as secondary outcomes. Patient-associated factors such as age, gender, race, body mass index, smoking status, Charlson Comorbidity Index, income, and use of corticosteroids were analyzed as covariates in addition to factors involving surgery such as expected surgery time, perioperative antibiotic use, and whether the procedure was elective or emergency surgery.
A majority of the patients in the study across all groups were white women about 63 years old with a body mass index above 30 kg/m2 and almost all undergoing electing surgery compared with emergency surgery. While patients in each group were similar with regard to Charlson Comorbidity Index, expected length of surgery, and percentage of patients undergoing elective surgery, patients in the biologic with or without DMARD group had a significantly lower median income level compared with those in the other two groups (P = .01).
Overall, there were 244 surgeries in 154 patients, with 117 surgeries in the group not receiving biologics or DMARDs, 95 surgeries in the group receiving DMARDs but no biologics, and 32 surgeries in the biologics with or without DMARD group. In the DMARD but no biologics group, most patients were receiving methotrexate (45%) or hydroxychloroquine (44%), while the most common biologics in the biologics with or without DMARD group were infliximab (25%), tocilizumab (19%), abatacept (16%), etanercept (13%), rituximab (9%), and tofacitinib (9%).
There was an 11% overall rate of infection, with a similar rate of infection across all groups (P = .09). While there was a higher rate of surgical site infections among patients in the biologics with or without DMARD group (9%) and a higher percentage of urinary tract infections in the no DMARD and no biologics group (4%), the results were not statistically significant. When the rate of infections was examined by type of surgery, there were no significant differences between infections from musculoskeletal surgery (P = .7) and major organ surgery (P = .8).
The overall 30-day readmission rate was 12%, but there were no statistically significant differences between groups. Although there were five deaths in the study, four deaths were in the group not receiving DMARDs or biologics, and one death was in the biologic with or without DMARD group.
Higher Charlson Comorbidity Index did predict infection risk, with an odds ratio of 1.37 per 1-point increase in the index (95% confidence interval, 1.10-1.70). Length of surgery also increased the risk of infection, with an OR of 1.16 per 15-minute increase in surgery time (95% CI, 1.09-1.23).
Dr. Shmagel noted that the retrospective nature of the study and the midwestern cohort may mean the results are not generalizable to other populations and that larger randomized trials should be considered. “Certainly, a larger study with more events would be needed,” she said.
This study was funded by the University of Minnesota. Dr. Shmagel reported no relevant conflicts of interest.
SOURCE: Kerski M et al. Arthritis Rheumatol. 2019;71 (suppl 10), Abstract 1805.
REPORTING FROM ACR 2019