User login
Suicide deaths rising in children aged 10-19 years
according to the National Center for Health Statistics.
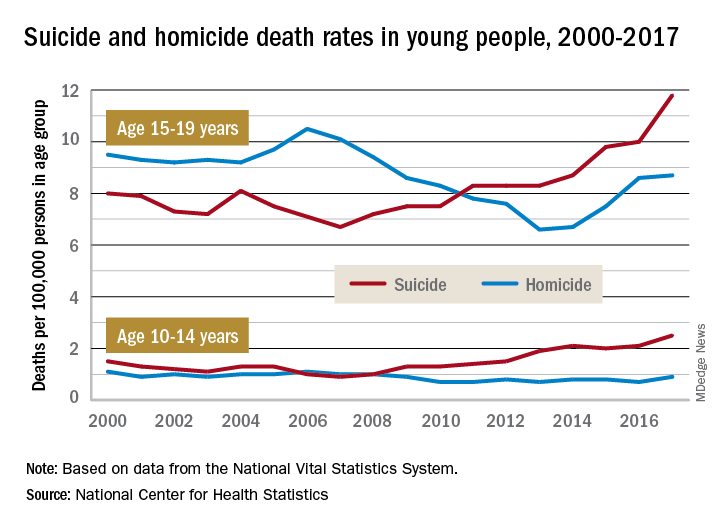
Death rates from suicide for children aged 10-14 years jumped by 178% from 2007 to 2017, while teenagers aged 15-19 years experienced a 76% increase over that period, with both changes reaching significance, the NCHS said in a recent data brief based on data from the National Vital Statistics System.
The actual rate for teens was higher to begin with, however, so in absolute terms the increase is larger for the older group. In 2007, deaths from suicide occurred at a rate of 6.7 per 100,000 persons for persons aged 15-19 years, and by 2017 that rate was up significantly to 11.8 per 100,000. Among children aged 10-14 years, the suicide-related death rate climbed from 0.9 per 100,000 in 2007 to 2.5 in 2014, the NCHS investigators reported.
The news was somewhat better on the other side of the violent death coin. Homicides are down by a significant 18% since 2000 among children aged 10-14 years, as the rate dropped from 1.1 per 100,000 in 2000 to 0.9 in 2017. The homicide rate since 2000 is down slightly for teens aged 15-19 years, but it has risen 32% in recent years, going from 6.6 deaths per 100,000 in 2013 to 8.7 in 2017, they said.
Suicide was the second-leading cause of death in both age groups in 2017, and homicide was third for those aged 15-19 and fifth among 10- to 14-year-olds, the investigators noted.
according to the National Center for Health Statistics.
Death rates from suicide for children aged 10-14 years jumped by 178% from 2007 to 2017, while teenagers aged 15-19 years experienced a 76% increase over that period, with both changes reaching significance, the NCHS said in a recent data brief based on data from the National Vital Statistics System.
The actual rate for teens was higher to begin with, however, so in absolute terms the increase is larger for the older group. In 2007, deaths from suicide occurred at a rate of 6.7 per 100,000 persons for persons aged 15-19 years, and by 2017 that rate was up significantly to 11.8 per 100,000. Among children aged 10-14 years, the suicide-related death rate climbed from 0.9 per 100,000 in 2007 to 2.5 in 2014, the NCHS investigators reported.
The news was somewhat better on the other side of the violent death coin. Homicides are down by a significant 18% since 2000 among children aged 10-14 years, as the rate dropped from 1.1 per 100,000 in 2000 to 0.9 in 2017. The homicide rate since 2000 is down slightly for teens aged 15-19 years, but it has risen 32% in recent years, going from 6.6 deaths per 100,000 in 2013 to 8.7 in 2017, they said.
Suicide was the second-leading cause of death in both age groups in 2017, and homicide was third for those aged 15-19 and fifth among 10- to 14-year-olds, the investigators noted.
according to the National Center for Health Statistics.
Death rates from suicide for children aged 10-14 years jumped by 178% from 2007 to 2017, while teenagers aged 15-19 years experienced a 76% increase over that period, with both changes reaching significance, the NCHS said in a recent data brief based on data from the National Vital Statistics System.
The actual rate for teens was higher to begin with, however, so in absolute terms the increase is larger for the older group. In 2007, deaths from suicide occurred at a rate of 6.7 per 100,000 persons for persons aged 15-19 years, and by 2017 that rate was up significantly to 11.8 per 100,000. Among children aged 10-14 years, the suicide-related death rate climbed from 0.9 per 100,000 in 2007 to 2.5 in 2014, the NCHS investigators reported.
The news was somewhat better on the other side of the violent death coin. Homicides are down by a significant 18% since 2000 among children aged 10-14 years, as the rate dropped from 1.1 per 100,000 in 2000 to 0.9 in 2017. The homicide rate since 2000 is down slightly for teens aged 15-19 years, but it has risen 32% in recent years, going from 6.6 deaths per 100,000 in 2013 to 8.7 in 2017, they said.
Suicide was the second-leading cause of death in both age groups in 2017, and homicide was third for those aged 15-19 and fifth among 10- to 14-year-olds, the investigators noted.
Trials examine T2T strategy in axial spondyloarthritis
Three international clinical trials in Europe are examining the effectiveness of treat-to-target (T2T) therapeutic regimens in patients with axial spondyloarthritis (axSpA), including two that will be the first randomized trial evidence to support or refute the T2T strategy for patients ranging from those with nonradiographic disease to patients with ankylosing spondylitis.
T2T has proved before to work in patients with rheumatoid arthritis and psoriatic arthritis with evidence from the TICORA (Tight Control of Rheumatoid Arthritis) and TICOPA (Tight Control in Psoriatic Arthritis) trials.
Two T2T trials in axSpA are still in the recruiting phase, and one has completed enrollment, with no results available yet.
Tight control in spondyloarthritis (TICOSPA)
TICOSPA is a 1-year, ongoing, multinational, cluster-randomized, prospective cohort study that has enrolled 163 patients with a diagnosis of active axial spondyloarthritis to evaluate the potential benefit of a T2T strategy in which the rheumatologist will agree to monitor very closely – at least every 4 weeks – and treat patients in accordance with a predefined strategy. The T2T strategy is compared with usual care as given by the treating rheumatologist. Prior to the trial, patients were on nonoptimal NSAID treatment.
“Tight control” in this study refers to the time from treatment initiation to adequate assessment of efficacy and safety, which for efficacy should be at 2-4 weeks for NSAIDs and 12-16 weeks for tumor necrosis factor inhibitors but can be a very short time frame for evaluating safety, “based on the occurrence of adverse events,” according to the study description at clinicaltrials.gov.
The primary endpoint is change on the Assessment of SpondyloArthritis international Society (ASAS) Health Index-Numerical Rating Score over the 1 year of follow-up.
There are 11 secondary endpoints, including:
- Percentage reaching major improvement in the Ankylosing Spondylitis Disease Activity Scale score (ASDAS).
- Percentage reaching 50% improvement of the initial Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score at 1 year.
- Change in the ASAS-NSAID score over 1 year.
- Change in the Work Productivity and Activity Impairment questionnaire.
The study is being conducted at 18 centers in Belgium, France, and the Netherlands and is sponsored by the Association de Recherche Clinique en Rhumatologie.
AScalate: Treat-to-target in axial spondyloarthritis
The Novartis-sponsored AScalate study seeks to enroll 300 patients with active disease despite NSAID therapy. The 36-week, randomized, parallel-group, open-label, multicenter trial will be conducted at seven sites in Germany.
The study will randomize patients to either of two arms: An active group will receive T2T therapy with secukinumab as a first-line biologic in escalating doses of 150-300 mg, determined by patient response until the T2T goal had been reached. Patients who don’t respond to secukinumab will be switched to an adalimumab biosimilar. The comparator group will receive standard-of-care therapy up to the maximum recommended dose at the discretion of the investigator.
The primary endpoint is the percentage of patients in each group who meet ASAS 40 response criteria by 24 weeks.
There are 11 secondary endpoints, including:
- Percentage achieving an ASAS40 response at 12 weeks.
- Percentage achieving ASAS20 and ASAS partial response at 12 and 24 weeks.
- Proportion of patients meeting the ASDAS definition of inactive disease, ASDAS clinically important and major improvement, and ASDAS low disease activity.
- Proportion of patients achieving 50% improvement of the initial BASDAI score.
Treat-to-target with secukinumab in axial spondyloarthritis (TRACE)
TRACE is a Novartis-sponsored phase 4 study examining reductions of inflammation seen on MRI of sacroiliac joints and spine at 16-24 weeks in patients who achieve ASDAS remission (score of less than 1.3) on 150 mg secukinumab by 16 weeks. The comparator group will be patients who are not in remission by week 16 and need a dose increase to 300 mg. The Danish trial seeks 88 participants with high disease activity and MRI signs of inflammation in the sacroiliac joints and/ or the spine.
After an initial four weekly doses of secukinumab 150 mg, patients will receive monthly secukinumab 150-mg doses out to week 16. Nonresponders at week 16 will escalate to 300 mg. If by 24 weeks these patients do not respond, they will be switched to a TNF inhibitor.
The primary outcome is the proportion of patients with a positive change in MRI-inflammation as measured by the sum of the Spondyloarthritis Research Consortium of Canada (SPARCC) sacroiliac joint and spine inflammation indices.
Three international clinical trials in Europe are examining the effectiveness of treat-to-target (T2T) therapeutic regimens in patients with axial spondyloarthritis (axSpA), including two that will be the first randomized trial evidence to support or refute the T2T strategy for patients ranging from those with nonradiographic disease to patients with ankylosing spondylitis.
T2T has proved before to work in patients with rheumatoid arthritis and psoriatic arthritis with evidence from the TICORA (Tight Control of Rheumatoid Arthritis) and TICOPA (Tight Control in Psoriatic Arthritis) trials.
Two T2T trials in axSpA are still in the recruiting phase, and one has completed enrollment, with no results available yet.
Tight control in spondyloarthritis (TICOSPA)
TICOSPA is a 1-year, ongoing, multinational, cluster-randomized, prospective cohort study that has enrolled 163 patients with a diagnosis of active axial spondyloarthritis to evaluate the potential benefit of a T2T strategy in which the rheumatologist will agree to monitor very closely – at least every 4 weeks – and treat patients in accordance with a predefined strategy. The T2T strategy is compared with usual care as given by the treating rheumatologist. Prior to the trial, patients were on nonoptimal NSAID treatment.
“Tight control” in this study refers to the time from treatment initiation to adequate assessment of efficacy and safety, which for efficacy should be at 2-4 weeks for NSAIDs and 12-16 weeks for tumor necrosis factor inhibitors but can be a very short time frame for evaluating safety, “based on the occurrence of adverse events,” according to the study description at clinicaltrials.gov.
The primary endpoint is change on the Assessment of SpondyloArthritis international Society (ASAS) Health Index-Numerical Rating Score over the 1 year of follow-up.
There are 11 secondary endpoints, including:
- Percentage reaching major improvement in the Ankylosing Spondylitis Disease Activity Scale score (ASDAS).
- Percentage reaching 50% improvement of the initial Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score at 1 year.
- Change in the ASAS-NSAID score over 1 year.
- Change in the Work Productivity and Activity Impairment questionnaire.
The study is being conducted at 18 centers in Belgium, France, and the Netherlands and is sponsored by the Association de Recherche Clinique en Rhumatologie.
AScalate: Treat-to-target in axial spondyloarthritis
The Novartis-sponsored AScalate study seeks to enroll 300 patients with active disease despite NSAID therapy. The 36-week, randomized, parallel-group, open-label, multicenter trial will be conducted at seven sites in Germany.
The study will randomize patients to either of two arms: An active group will receive T2T therapy with secukinumab as a first-line biologic in escalating doses of 150-300 mg, determined by patient response until the T2T goal had been reached. Patients who don’t respond to secukinumab will be switched to an adalimumab biosimilar. The comparator group will receive standard-of-care therapy up to the maximum recommended dose at the discretion of the investigator.
The primary endpoint is the percentage of patients in each group who meet ASAS 40 response criteria by 24 weeks.
There are 11 secondary endpoints, including:
- Percentage achieving an ASAS40 response at 12 weeks.
- Percentage achieving ASAS20 and ASAS partial response at 12 and 24 weeks.
- Proportion of patients meeting the ASDAS definition of inactive disease, ASDAS clinically important and major improvement, and ASDAS low disease activity.
- Proportion of patients achieving 50% improvement of the initial BASDAI score.
Treat-to-target with secukinumab in axial spondyloarthritis (TRACE)
TRACE is a Novartis-sponsored phase 4 study examining reductions of inflammation seen on MRI of sacroiliac joints and spine at 16-24 weeks in patients who achieve ASDAS remission (score of less than 1.3) on 150 mg secukinumab by 16 weeks. The comparator group will be patients who are not in remission by week 16 and need a dose increase to 300 mg. The Danish trial seeks 88 participants with high disease activity and MRI signs of inflammation in the sacroiliac joints and/ or the spine.
After an initial four weekly doses of secukinumab 150 mg, patients will receive monthly secukinumab 150-mg doses out to week 16. Nonresponders at week 16 will escalate to 300 mg. If by 24 weeks these patients do not respond, they will be switched to a TNF inhibitor.
The primary outcome is the proportion of patients with a positive change in MRI-inflammation as measured by the sum of the Spondyloarthritis Research Consortium of Canada (SPARCC) sacroiliac joint and spine inflammation indices.
Three international clinical trials in Europe are examining the effectiveness of treat-to-target (T2T) therapeutic regimens in patients with axial spondyloarthritis (axSpA), including two that will be the first randomized trial evidence to support or refute the T2T strategy for patients ranging from those with nonradiographic disease to patients with ankylosing spondylitis.
T2T has proved before to work in patients with rheumatoid arthritis and psoriatic arthritis with evidence from the TICORA (Tight Control of Rheumatoid Arthritis) and TICOPA (Tight Control in Psoriatic Arthritis) trials.
Two T2T trials in axSpA are still in the recruiting phase, and one has completed enrollment, with no results available yet.
Tight control in spondyloarthritis (TICOSPA)
TICOSPA is a 1-year, ongoing, multinational, cluster-randomized, prospective cohort study that has enrolled 163 patients with a diagnosis of active axial spondyloarthritis to evaluate the potential benefit of a T2T strategy in which the rheumatologist will agree to monitor very closely – at least every 4 weeks – and treat patients in accordance with a predefined strategy. The T2T strategy is compared with usual care as given by the treating rheumatologist. Prior to the trial, patients were on nonoptimal NSAID treatment.
“Tight control” in this study refers to the time from treatment initiation to adequate assessment of efficacy and safety, which for efficacy should be at 2-4 weeks for NSAIDs and 12-16 weeks for tumor necrosis factor inhibitors but can be a very short time frame for evaluating safety, “based on the occurrence of adverse events,” according to the study description at clinicaltrials.gov.
The primary endpoint is change on the Assessment of SpondyloArthritis international Society (ASAS) Health Index-Numerical Rating Score over the 1 year of follow-up.
There are 11 secondary endpoints, including:
- Percentage reaching major improvement in the Ankylosing Spondylitis Disease Activity Scale score (ASDAS).
- Percentage reaching 50% improvement of the initial Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score at 1 year.
- Change in the ASAS-NSAID score over 1 year.
- Change in the Work Productivity and Activity Impairment questionnaire.
The study is being conducted at 18 centers in Belgium, France, and the Netherlands and is sponsored by the Association de Recherche Clinique en Rhumatologie.
AScalate: Treat-to-target in axial spondyloarthritis
The Novartis-sponsored AScalate study seeks to enroll 300 patients with active disease despite NSAID therapy. The 36-week, randomized, parallel-group, open-label, multicenter trial will be conducted at seven sites in Germany.
The study will randomize patients to either of two arms: An active group will receive T2T therapy with secukinumab as a first-line biologic in escalating doses of 150-300 mg, determined by patient response until the T2T goal had been reached. Patients who don’t respond to secukinumab will be switched to an adalimumab biosimilar. The comparator group will receive standard-of-care therapy up to the maximum recommended dose at the discretion of the investigator.
The primary endpoint is the percentage of patients in each group who meet ASAS 40 response criteria by 24 weeks.
There are 11 secondary endpoints, including:
- Percentage achieving an ASAS40 response at 12 weeks.
- Percentage achieving ASAS20 and ASAS partial response at 12 and 24 weeks.
- Proportion of patients meeting the ASDAS definition of inactive disease, ASDAS clinically important and major improvement, and ASDAS low disease activity.
- Proportion of patients achieving 50% improvement of the initial BASDAI score.
Treat-to-target with secukinumab in axial spondyloarthritis (TRACE)
TRACE is a Novartis-sponsored phase 4 study examining reductions of inflammation seen on MRI of sacroiliac joints and spine at 16-24 weeks in patients who achieve ASDAS remission (score of less than 1.3) on 150 mg secukinumab by 16 weeks. The comparator group will be patients who are not in remission by week 16 and need a dose increase to 300 mg. The Danish trial seeks 88 participants with high disease activity and MRI signs of inflammation in the sacroiliac joints and/ or the spine.
After an initial four weekly doses of secukinumab 150 mg, patients will receive monthly secukinumab 150-mg doses out to week 16. Nonresponders at week 16 will escalate to 300 mg. If by 24 weeks these patients do not respond, they will be switched to a TNF inhibitor.
The primary outcome is the proportion of patients with a positive change in MRI-inflammation as measured by the sum of the Spondyloarthritis Research Consortium of Canada (SPARCC) sacroiliac joint and spine inflammation indices.
When patient autonomy gets in the way
“Why didn’t you see that patient?”

The hospitalist on the phone was angry. He’d wanted the patient seen by neurology and cleared for discharge. Apparently, I hadn’t complied.
Actually, that isn’t true. I was on call, so I had dutifully dragged myself in (with the help of some coffee), reviewed the chart, and gone in to see the fellow.
The patient, however, had other ideas. He said he was sick of doctors, didn’t like them, didn’t want to see me, and asked me to leave. So I did.
This threw off the hospitalist’s well-choreographed day of admissions and discharges. Without me seeing the patient, he had to either discharge him on his own decision or find another neurologist who would do it.
Sorry, but I’m not going to force this issue. If a patient doesn’t want to see me, it’s not worth fighting over. Believe me, I get paid to see patients, so I don’t have much incentive to just walk away.
But at the same time I have to respect patients’ decisions. While a neurology consult is pretty noninvasive, it’s still a part of medicine. If a patient doesn’t want to see me, I’m not going to force them to.
Granted, there are exceptions. Obviously, if the patient is fairly demented or otherwise not mentally competent to make such a decision, I’ll see them. In those cases, their deteriorating mental status is likely the reason for the consult.
But the fellow that day seemed alert and reasonable, and there was nothing in the chart about confusion. So I’m going to assume he knew what he was doing when he told me to go away.
The hospitalist didn’t see this as an issue, but I did. I’m sorry if it messes up the discharge planning, but that’s not my fault. It’s the patient’s decision.
While I may disagree at times with patients’ decisions, their autonomy is still central to medicine. I respect and believe in that, even if it makes things more difficult for those around them.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
“Why didn’t you see that patient?”
The hospitalist on the phone was angry. He’d wanted the patient seen by neurology and cleared for discharge. Apparently, I hadn’t complied.
Actually, that isn’t true. I was on call, so I had dutifully dragged myself in (with the help of some coffee), reviewed the chart, and gone in to see the fellow.
The patient, however, had other ideas. He said he was sick of doctors, didn’t like them, didn’t want to see me, and asked me to leave. So I did.
This threw off the hospitalist’s well-choreographed day of admissions and discharges. Without me seeing the patient, he had to either discharge him on his own decision or find another neurologist who would do it.
Sorry, but I’m not going to force this issue. If a patient doesn’t want to see me, it’s not worth fighting over. Believe me, I get paid to see patients, so I don’t have much incentive to just walk away.
But at the same time I have to respect patients’ decisions. While a neurology consult is pretty noninvasive, it’s still a part of medicine. If a patient doesn’t want to see me, I’m not going to force them to.
Granted, there are exceptions. Obviously, if the patient is fairly demented or otherwise not mentally competent to make such a decision, I’ll see them. In those cases, their deteriorating mental status is likely the reason for the consult.
But the fellow that day seemed alert and reasonable, and there was nothing in the chart about confusion. So I’m going to assume he knew what he was doing when he told me to go away.
The hospitalist didn’t see this as an issue, but I did. I’m sorry if it messes up the discharge planning, but that’s not my fault. It’s the patient’s decision.
While I may disagree at times with patients’ decisions, their autonomy is still central to medicine. I respect and believe in that, even if it makes things more difficult for those around them.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
“Why didn’t you see that patient?”
The hospitalist on the phone was angry. He’d wanted the patient seen by neurology and cleared for discharge. Apparently, I hadn’t complied.
Actually, that isn’t true. I was on call, so I had dutifully dragged myself in (with the help of some coffee), reviewed the chart, and gone in to see the fellow.
The patient, however, had other ideas. He said he was sick of doctors, didn’t like them, didn’t want to see me, and asked me to leave. So I did.
This threw off the hospitalist’s well-choreographed day of admissions and discharges. Without me seeing the patient, he had to either discharge him on his own decision or find another neurologist who would do it.
Sorry, but I’m not going to force this issue. If a patient doesn’t want to see me, it’s not worth fighting over. Believe me, I get paid to see patients, so I don’t have much incentive to just walk away.
But at the same time I have to respect patients’ decisions. While a neurology consult is pretty noninvasive, it’s still a part of medicine. If a patient doesn’t want to see me, I’m not going to force them to.
Granted, there are exceptions. Obviously, if the patient is fairly demented or otherwise not mentally competent to make such a decision, I’ll see them. In those cases, their deteriorating mental status is likely the reason for the consult.
But the fellow that day seemed alert and reasonable, and there was nothing in the chart about confusion. So I’m going to assume he knew what he was doing when he told me to go away.
The hospitalist didn’t see this as an issue, but I did. I’m sorry if it messes up the discharge planning, but that’s not my fault. It’s the patient’s decision.
While I may disagree at times with patients’ decisions, their autonomy is still central to medicine. I respect and believe in that, even if it makes things more difficult for those around them.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Supporting elimination of nonmedical vaccine exemptions
Let’s suppose your first patient of the morning is a 2-month-old you have never seen before. The family arrives 10 minutes late because they are still getting the dressing-undressing-diaper change-car seat–adjusting thing worked out. Father is a computer programmer. Mother lists her occupation as nutrition counselor. The child is gaining. Breastfeeding seems to come naturally to the dyad.
As the visit draws to a close, you take the matter-of-fact approach and say, “The nurse will be in shortly with the vaccines do you have any questions.” Well ... it turns out the parents don’t feel comfortable with vaccines. They claim to understand the science and feel that vaccines make sense for some families. But they feel that for themselves, with a healthy lifestyle and God’s benevolence their son will be protected without having to introduce a host of foreign substances into his body.
What word best describes your reaction? Anger? Frustration? Disappointment (in our education system)? Maybe you’re angry at yourself for failing to make it clear in your office pamphlet and social media feeds that to protect your other patients, you no longer accept families who refuse immunizations for the common childhood diseases.
The American Academy of Pediatrics says it feels your pain, and its Annual Leadership Forum made eliminating nonmedical vaccine exemption laws its top priority in 2019. As part of its effort to help, the AAP Board of Directors was asked to advocate for the creation of a toolkit of strategies for Academy chapters facing the challenge of nonmedical exemptions. As an initial step to this process, three physicians in the department of pediatrics at the Denver Health Medical Center have begun interviewing religious leaders in hopes of developing “clergy-specific vaccine educational materials and deriv[ing] best practices for engaging them as vaccination advocates.” The investigators describe their plan and initial findings in Pediatrics (2019 Oct. doi: 10.1542/peds.2019-0933). Although they acknowledged that their efforts may not provide a quick solution to the nonmedical exemption problem, they hope that including more stakeholders and engendering trust will help future discussions.
Fourteen pages deeper into that issue of Pediatrics is the runner-up submission of this year’s Section on Pediatric Trainees essay competition titled “What I Learned From the Antivaccine Movement” (2019 Oct. doi: 10.1542/peds.2019-2384). Alana C. Ju, MD, describes the 2-hour ordeal she endured to testify at the California State Capitol in support of a state Senate bill aimed at tightening the regulations for vaccine medical exemptions. Totally unprepared for the “level of vitriol” aimed at her and other supporters of the bill, she was “accused of violating her duty as” a pediatrician because she was failing to protect children. The supporters were called “greedy, ignorant, and negligent.”
To her credit, Dr. Ju was able to step back from this assault and began looking at the faces of her accusers and learned that, “they too, felt strongly about children’s health.” She realized that “focusing on perceived ignorance is counterproductive.” She now hopes that by focusing on the shared goal of what is best for children, “we can all be better advocates.”
Both of these articles have a warm sort of kumbaya feel about them. It never is a bad idea to learn more about those with whom we disagree. But before huddling up too close to the campfire, we must realize that there is good evidence that sharing the scientific data with vaccine-hesitant parents doesn’t convert them into vaccine acceptors. In fact, it may strengthen their resolve to resist (Nyhan et al. “Effective Messages in Vaccine Promotion: A Randomized Trial,” Pediatrics. 2014 Apr;133[4] e835-42).
We are unlikely to convert many anti-vaxxers by sitting down together. Our target audience needs to be legislators and the majority of people who do vaccinate their children. These are the voters who will support legislation to eliminate nonmedical vaccine exemptions. To characterize anti-vaxxers as despicable ignorants is untrue and serves no purpose. We all do care about the health of children. However,
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at pdnews@mdedge.com.
*This article has been updated 1/22/2020.
Let’s suppose your first patient of the morning is a 2-month-old you have never seen before. The family arrives 10 minutes late because they are still getting the dressing-undressing-diaper change-car seat–adjusting thing worked out. Father is a computer programmer. Mother lists her occupation as nutrition counselor. The child is gaining. Breastfeeding seems to come naturally to the dyad.
As the visit draws to a close, you take the matter-of-fact approach and say, “The nurse will be in shortly with the vaccines do you have any questions.” Well ... it turns out the parents don’t feel comfortable with vaccines. They claim to understand the science and feel that vaccines make sense for some families. But they feel that for themselves, with a healthy lifestyle and God’s benevolence their son will be protected without having to introduce a host of foreign substances into his body.
What word best describes your reaction? Anger? Frustration? Disappointment (in our education system)? Maybe you’re angry at yourself for failing to make it clear in your office pamphlet and social media feeds that to protect your other patients, you no longer accept families who refuse immunizations for the common childhood diseases.
The American Academy of Pediatrics says it feels your pain, and its Annual Leadership Forum made eliminating nonmedical vaccine exemption laws its top priority in 2019. As part of its effort to help, the AAP Board of Directors was asked to advocate for the creation of a toolkit of strategies for Academy chapters facing the challenge of nonmedical exemptions. As an initial step to this process, three physicians in the department of pediatrics at the Denver Health Medical Center have begun interviewing religious leaders in hopes of developing “clergy-specific vaccine educational materials and deriv[ing] best practices for engaging them as vaccination advocates.” The investigators describe their plan and initial findings in Pediatrics (2019 Oct. doi: 10.1542/peds.2019-0933). Although they acknowledged that their efforts may not provide a quick solution to the nonmedical exemption problem, they hope that including more stakeholders and engendering trust will help future discussions.
Fourteen pages deeper into that issue of Pediatrics is the runner-up submission of this year’s Section on Pediatric Trainees essay competition titled “What I Learned From the Antivaccine Movement” (2019 Oct. doi: 10.1542/peds.2019-2384). Alana C. Ju, MD, describes the 2-hour ordeal she endured to testify at the California State Capitol in support of a state Senate bill aimed at tightening the regulations for vaccine medical exemptions. Totally unprepared for the “level of vitriol” aimed at her and other supporters of the bill, she was “accused of violating her duty as” a pediatrician because she was failing to protect children. The supporters were called “greedy, ignorant, and negligent.”
To her credit, Dr. Ju was able to step back from this assault and began looking at the faces of her accusers and learned that, “they too, felt strongly about children’s health.” She realized that “focusing on perceived ignorance is counterproductive.” She now hopes that by focusing on the shared goal of what is best for children, “we can all be better advocates.”
Both of these articles have a warm sort of kumbaya feel about them. It never is a bad idea to learn more about those with whom we disagree. But before huddling up too close to the campfire, we must realize that there is good evidence that sharing the scientific data with vaccine-hesitant parents doesn’t convert them into vaccine acceptors. In fact, it may strengthen their resolve to resist (Nyhan et al. “Effective Messages in Vaccine Promotion: A Randomized Trial,” Pediatrics. 2014 Apr;133[4] e835-42).
We are unlikely to convert many anti-vaxxers by sitting down together. Our target audience needs to be legislators and the majority of people who do vaccinate their children. These are the voters who will support legislation to eliminate nonmedical vaccine exemptions. To characterize anti-vaxxers as despicable ignorants is untrue and serves no purpose. We all do care about the health of children. However,
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at pdnews@mdedge.com.
*This article has been updated 1/22/2020.
Let’s suppose your first patient of the morning is a 2-month-old you have never seen before. The family arrives 10 minutes late because they are still getting the dressing-undressing-diaper change-car seat–adjusting thing worked out. Father is a computer programmer. Mother lists her occupation as nutrition counselor. The child is gaining. Breastfeeding seems to come naturally to the dyad.
As the visit draws to a close, you take the matter-of-fact approach and say, “The nurse will be in shortly with the vaccines do you have any questions.” Well ... it turns out the parents don’t feel comfortable with vaccines. They claim to understand the science and feel that vaccines make sense for some families. But they feel that for themselves, with a healthy lifestyle and God’s benevolence their son will be protected without having to introduce a host of foreign substances into his body.
What word best describes your reaction? Anger? Frustration? Disappointment (in our education system)? Maybe you’re angry at yourself for failing to make it clear in your office pamphlet and social media feeds that to protect your other patients, you no longer accept families who refuse immunizations for the common childhood diseases.
The American Academy of Pediatrics says it feels your pain, and its Annual Leadership Forum made eliminating nonmedical vaccine exemption laws its top priority in 2019. As part of its effort to help, the AAP Board of Directors was asked to advocate for the creation of a toolkit of strategies for Academy chapters facing the challenge of nonmedical exemptions. As an initial step to this process, three physicians in the department of pediatrics at the Denver Health Medical Center have begun interviewing religious leaders in hopes of developing “clergy-specific vaccine educational materials and deriv[ing] best practices for engaging them as vaccination advocates.” The investigators describe their plan and initial findings in Pediatrics (2019 Oct. doi: 10.1542/peds.2019-0933). Although they acknowledged that their efforts may not provide a quick solution to the nonmedical exemption problem, they hope that including more stakeholders and engendering trust will help future discussions.
Fourteen pages deeper into that issue of Pediatrics is the runner-up submission of this year’s Section on Pediatric Trainees essay competition titled “What I Learned From the Antivaccine Movement” (2019 Oct. doi: 10.1542/peds.2019-2384). Alana C. Ju, MD, describes the 2-hour ordeal she endured to testify at the California State Capitol in support of a state Senate bill aimed at tightening the regulations for vaccine medical exemptions. Totally unprepared for the “level of vitriol” aimed at her and other supporters of the bill, she was “accused of violating her duty as” a pediatrician because she was failing to protect children. The supporters were called “greedy, ignorant, and negligent.”
To her credit, Dr. Ju was able to step back from this assault and began looking at the faces of her accusers and learned that, “they too, felt strongly about children’s health.” She realized that “focusing on perceived ignorance is counterproductive.” She now hopes that by focusing on the shared goal of what is best for children, “we can all be better advocates.”
Both of these articles have a warm sort of kumbaya feel about them. It never is a bad idea to learn more about those with whom we disagree. But before huddling up too close to the campfire, we must realize that there is good evidence that sharing the scientific data with vaccine-hesitant parents doesn’t convert them into vaccine acceptors. In fact, it may strengthen their resolve to resist (Nyhan et al. “Effective Messages in Vaccine Promotion: A Randomized Trial,” Pediatrics. 2014 Apr;133[4] e835-42).
We are unlikely to convert many anti-vaxxers by sitting down together. Our target audience needs to be legislators and the majority of people who do vaccinate their children. These are the voters who will support legislation to eliminate nonmedical vaccine exemptions. To characterize anti-vaxxers as despicable ignorants is untrue and serves no purpose. We all do care about the health of children. However,
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics, including “How to Say No to Your Toddler.” Email him at pdnews@mdedge.com.
*This article has been updated 1/22/2020.
Long catheters best standard-sized ones in trial of pediatric surgery patients
NEW ORLEANS – Children who require more than 48 hours of intravenous therapy should receive long peripheral catheters initially rather than standard peripheral intravenous catheters, results from a randomized, controlled trial suggest.

Long peripheral catheters (LPCs) are relatively new, 8- to 15-cm long peripheral vascular devices that have been investigated extensively in adults. “It has been shown that these devices are safe and reliable peripheral vascular access devices for use in this population,” lead study author Maurizio Pacilli, MD, said in an interview in advance of the annual meeting of the American Academy of Pediatrics.
“In addition, LPCs may represent an improvement to the quality of care in adults, as demonstrated by multiple randomized controlled trials, where they outperformed peripheral intravenous catheters (PIVCs). There remains, however, a lack of controlled studies to determine if this effect is replicable in children.”
In what he said is the first study of its kind, Dr. Pacilli, a senior research fellow at Monash Children’s Hospital and Monash University, Melbourne, and colleagues performed an open-label randomized trial of 72 pediatric surgical patients aged 1 year and older who required more than 48 hours of IV therapy. They assigned 36 to receive a PIVCs while the remaining 36 children received an 8-cm 22-gauge LPC.
The mean age of patients was 9 years and 71% were boys. Dr. Pacilli reported that the IV therapy duration was a mean of 5 days, and that gender, age, weight, emergency status, and IV therapy duration were similar between the two groups. However, the mean lifespan of catheters was 3 days in PIVC group, compared with 5 days in the LPC group, a difference that reached statistical significance (P = .003). Patients in the PIVC group received a median of two catheters, compared with one in the LPC group (P = .0002). The researchers found that patients in the PIVC group were less likely than were those in the LPC group to complete treatment with a single catheter (39% vs. 81%, relative risk [RR] 2.1, P = .0006), while the rate of catheter failure was higher for PIVCs than for LPCs (67% vs. 19%, RR 3.4, P = .0001; 187 vs. 43 failures per 1,000 catheter days). Infiltration was the most common complication, and occurred in 33% of patients in the PIVC group vs. 3% in LPC group (RR 12, P = .001).
“Our results showed for the first time, without doubt, that children requiring more than 48 hours of intravenous therapy benefit from receiving LPCs, compared with traditional PIVCs,” Dr. Pacilli said. “Of failed catheters, PIVCs are most likely to infiltrate, while LPCs are most likely to occlude. This indicates an additional benefit of LPCs as occlusion is a relatively benign complication. In addition, in all domains of satisfaction, parents favor LPCs. According to the parents, there is a significant improvement in ‘pain and discomfort’ and ‘overall satisfaction’ with the use of LPCs in children.”
He acknowledged certain limitations of the study, including the fact that children younger than 1 year of age were not included in the analysis. “New devices, suitable for very young children, need to be developed, and further studies are needed to confirm the findings from our trial in children younger than 1 year,” he said.
Dr. Pacilli reported having no financial disclosures.
NEW ORLEANS – Children who require more than 48 hours of intravenous therapy should receive long peripheral catheters initially rather than standard peripheral intravenous catheters, results from a randomized, controlled trial suggest.
Long peripheral catheters (LPCs) are relatively new, 8- to 15-cm long peripheral vascular devices that have been investigated extensively in adults. “It has been shown that these devices are safe and reliable peripheral vascular access devices for use in this population,” lead study author Maurizio Pacilli, MD, said in an interview in advance of the annual meeting of the American Academy of Pediatrics.
“In addition, LPCs may represent an improvement to the quality of care in adults, as demonstrated by multiple randomized controlled trials, where they outperformed peripheral intravenous catheters (PIVCs). There remains, however, a lack of controlled studies to determine if this effect is replicable in children.”
In what he said is the first study of its kind, Dr. Pacilli, a senior research fellow at Monash Children’s Hospital and Monash University, Melbourne, and colleagues performed an open-label randomized trial of 72 pediatric surgical patients aged 1 year and older who required more than 48 hours of IV therapy. They assigned 36 to receive a PIVCs while the remaining 36 children received an 8-cm 22-gauge LPC.
The mean age of patients was 9 years and 71% were boys. Dr. Pacilli reported that the IV therapy duration was a mean of 5 days, and that gender, age, weight, emergency status, and IV therapy duration were similar between the two groups. However, the mean lifespan of catheters was 3 days in PIVC group, compared with 5 days in the LPC group, a difference that reached statistical significance (P = .003). Patients in the PIVC group received a median of two catheters, compared with one in the LPC group (P = .0002). The researchers found that patients in the PIVC group were less likely than were those in the LPC group to complete treatment with a single catheter (39% vs. 81%, relative risk [RR] 2.1, P = .0006), while the rate of catheter failure was higher for PIVCs than for LPCs (67% vs. 19%, RR 3.4, P = .0001; 187 vs. 43 failures per 1,000 catheter days). Infiltration was the most common complication, and occurred in 33% of patients in the PIVC group vs. 3% in LPC group (RR 12, P = .001).
“Our results showed for the first time, without doubt, that children requiring more than 48 hours of intravenous therapy benefit from receiving LPCs, compared with traditional PIVCs,” Dr. Pacilli said. “Of failed catheters, PIVCs are most likely to infiltrate, while LPCs are most likely to occlude. This indicates an additional benefit of LPCs as occlusion is a relatively benign complication. In addition, in all domains of satisfaction, parents favor LPCs. According to the parents, there is a significant improvement in ‘pain and discomfort’ and ‘overall satisfaction’ with the use of LPCs in children.”
He acknowledged certain limitations of the study, including the fact that children younger than 1 year of age were not included in the analysis. “New devices, suitable for very young children, need to be developed, and further studies are needed to confirm the findings from our trial in children younger than 1 year,” he said.
Dr. Pacilli reported having no financial disclosures.
NEW ORLEANS – Children who require more than 48 hours of intravenous therapy should receive long peripheral catheters initially rather than standard peripheral intravenous catheters, results from a randomized, controlled trial suggest.
Long peripheral catheters (LPCs) are relatively new, 8- to 15-cm long peripheral vascular devices that have been investigated extensively in adults. “It has been shown that these devices are safe and reliable peripheral vascular access devices for use in this population,” lead study author Maurizio Pacilli, MD, said in an interview in advance of the annual meeting of the American Academy of Pediatrics.
“In addition, LPCs may represent an improvement to the quality of care in adults, as demonstrated by multiple randomized controlled trials, where they outperformed peripheral intravenous catheters (PIVCs). There remains, however, a lack of controlled studies to determine if this effect is replicable in children.”
In what he said is the first study of its kind, Dr. Pacilli, a senior research fellow at Monash Children’s Hospital and Monash University, Melbourne, and colleagues performed an open-label randomized trial of 72 pediatric surgical patients aged 1 year and older who required more than 48 hours of IV therapy. They assigned 36 to receive a PIVCs while the remaining 36 children received an 8-cm 22-gauge LPC.
The mean age of patients was 9 years and 71% were boys. Dr. Pacilli reported that the IV therapy duration was a mean of 5 days, and that gender, age, weight, emergency status, and IV therapy duration were similar between the two groups. However, the mean lifespan of catheters was 3 days in PIVC group, compared with 5 days in the LPC group, a difference that reached statistical significance (P = .003). Patients in the PIVC group received a median of two catheters, compared with one in the LPC group (P = .0002). The researchers found that patients in the PIVC group were less likely than were those in the LPC group to complete treatment with a single catheter (39% vs. 81%, relative risk [RR] 2.1, P = .0006), while the rate of catheter failure was higher for PIVCs than for LPCs (67% vs. 19%, RR 3.4, P = .0001; 187 vs. 43 failures per 1,000 catheter days). Infiltration was the most common complication, and occurred in 33% of patients in the PIVC group vs. 3% in LPC group (RR 12, P = .001).
“Our results showed for the first time, without doubt, that children requiring more than 48 hours of intravenous therapy benefit from receiving LPCs, compared with traditional PIVCs,” Dr. Pacilli said. “Of failed catheters, PIVCs are most likely to infiltrate, while LPCs are most likely to occlude. This indicates an additional benefit of LPCs as occlusion is a relatively benign complication. In addition, in all domains of satisfaction, parents favor LPCs. According to the parents, there is a significant improvement in ‘pain and discomfort’ and ‘overall satisfaction’ with the use of LPCs in children.”
He acknowledged certain limitations of the study, including the fact that children younger than 1 year of age were not included in the analysis. “New devices, suitable for very young children, need to be developed, and further studies are needed to confirm the findings from our trial in children younger than 1 year,” he said.
Dr. Pacilli reported having no financial disclosures.
REPORTING FROM AAP 2019
Key clinical point:
Major finding: The mean lifespan of catheters was 3 days in the peripheral intravenous catheters group, compared with 5 days in the long peripheral catheters group, a difference that reached statistical significance (P = .003).
Study details: An open-label, randomized trial of 72 pediatric surgical patients.
Disclosures: Dr. Pacilli reported having no financial disclosures.
Source: Pacilli M et al. AAP 2019, Section on Surgery program.
ACIP approves child and adolescent vaccination schedule for 2020
The Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices voted unanimously to approve the child and adolescent immunization schedule for 2020.
by busy providers,” Candice Robinson, MD, MPH, of the CDC’s National Center for Immunization and Respiratory Diseases, said at the CDC’s October meeting of ACIP. Updates reflect changes in language in the adult vaccination schedule, notably the change in the definition of “contraindication.” The updated wording in the Notes substitutes “not recommended or contraindicated” instead of the word “contraindicated” only.
Another notable change was the addition of information on adolescent vaccination of children who received the meningococcal ACWY vaccine before 10 years of age. For “children in whom boosters are not recommended due to an ongoing or increased risk of meningococcal disease” (such as a healthy child traveling to an endemic area), they should receive MenACWY according to the recommended adolescent schedule. But those children for whom boosters are recommended because of increased disease risk from conditions including complement deficiency, HIV, or asplenia should “follow the booster schedule for persons at increased risk.”
Other changes include restructuring of the notes for the live attenuated influenza vaccine (LAIV) in special situations. The schedule now uses a bulleted list to show that LAIV should not be used in the following circumstances:
- Having history of severe allergic reaction to a previous vaccine or vaccine component.
- Using aspirin or a salicylate-containing medication.
- Being aged 2-4 years with a history of asthma or wheezing.
- Having immunocompromised conditions.
- Having anatomic or functional asplenia.
- Having cochlear implants.
- Experiencing cerebrospinal fluid–oropharyngeal communication.
- Having immunocompromised close contacts or caregivers.
- Being pregnant.
- Having received flu antivirals within the previous 48 hours.
In addition, language on shared clinical decision-making was added to the notes on the meningococcal B vaccine for adolescents and young adults aged 18-23 years not at increased risk. Based on shared clinical decision making, the recommendation is a “two-dose series of Bexsero at least 1 month apart” or “two-dose series of Trumenba at least 6 months apart; if dose two is administered earlier than 6 months, administer a third dose at least 4 months after dose two.”
Several vaccines’ Notes sections, including hepatitis B and meningococcal disease, added links to detailed recommendations in the corresponding issues of the CDC’s Morbidity and Mortality Weekly Report, to allow clinicians easy access to additional information.
View the current Child & Adolescent Vaccination Schedule here.
The ACIP members had no financial conflicts to disclose.
The Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices voted unanimously to approve the child and adolescent immunization schedule for 2020.
by busy providers,” Candice Robinson, MD, MPH, of the CDC’s National Center for Immunization and Respiratory Diseases, said at the CDC’s October meeting of ACIP. Updates reflect changes in language in the adult vaccination schedule, notably the change in the definition of “contraindication.” The updated wording in the Notes substitutes “not recommended or contraindicated” instead of the word “contraindicated” only.
Another notable change was the addition of information on adolescent vaccination of children who received the meningococcal ACWY vaccine before 10 years of age. For “children in whom boosters are not recommended due to an ongoing or increased risk of meningococcal disease” (such as a healthy child traveling to an endemic area), they should receive MenACWY according to the recommended adolescent schedule. But those children for whom boosters are recommended because of increased disease risk from conditions including complement deficiency, HIV, or asplenia should “follow the booster schedule for persons at increased risk.”
Other changes include restructuring of the notes for the live attenuated influenza vaccine (LAIV) in special situations. The schedule now uses a bulleted list to show that LAIV should not be used in the following circumstances:
- Having history of severe allergic reaction to a previous vaccine or vaccine component.
- Using aspirin or a salicylate-containing medication.
- Being aged 2-4 years with a history of asthma or wheezing.
- Having immunocompromised conditions.
- Having anatomic or functional asplenia.
- Having cochlear implants.
- Experiencing cerebrospinal fluid–oropharyngeal communication.
- Having immunocompromised close contacts or caregivers.
- Being pregnant.
- Having received flu antivirals within the previous 48 hours.
In addition, language on shared clinical decision-making was added to the notes on the meningococcal B vaccine for adolescents and young adults aged 18-23 years not at increased risk. Based on shared clinical decision making, the recommendation is a “two-dose series of Bexsero at least 1 month apart” or “two-dose series of Trumenba at least 6 months apart; if dose two is administered earlier than 6 months, administer a third dose at least 4 months after dose two.”
Several vaccines’ Notes sections, including hepatitis B and meningococcal disease, added links to detailed recommendations in the corresponding issues of the CDC’s Morbidity and Mortality Weekly Report, to allow clinicians easy access to additional information.
View the current Child & Adolescent Vaccination Schedule here.
The ACIP members had no financial conflicts to disclose.
The Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices voted unanimously to approve the child and adolescent immunization schedule for 2020.
by busy providers,” Candice Robinson, MD, MPH, of the CDC’s National Center for Immunization and Respiratory Diseases, said at the CDC’s October meeting of ACIP. Updates reflect changes in language in the adult vaccination schedule, notably the change in the definition of “contraindication.” The updated wording in the Notes substitutes “not recommended or contraindicated” instead of the word “contraindicated” only.
Another notable change was the addition of information on adolescent vaccination of children who received the meningococcal ACWY vaccine before 10 years of age. For “children in whom boosters are not recommended due to an ongoing or increased risk of meningococcal disease” (such as a healthy child traveling to an endemic area), they should receive MenACWY according to the recommended adolescent schedule. But those children for whom boosters are recommended because of increased disease risk from conditions including complement deficiency, HIV, or asplenia should “follow the booster schedule for persons at increased risk.”
Other changes include restructuring of the notes for the live attenuated influenza vaccine (LAIV) in special situations. The schedule now uses a bulleted list to show that LAIV should not be used in the following circumstances:
- Having history of severe allergic reaction to a previous vaccine or vaccine component.
- Using aspirin or a salicylate-containing medication.
- Being aged 2-4 years with a history of asthma or wheezing.
- Having immunocompromised conditions.
- Having anatomic or functional asplenia.
- Having cochlear implants.
- Experiencing cerebrospinal fluid–oropharyngeal communication.
- Having immunocompromised close contacts or caregivers.
- Being pregnant.
- Having received flu antivirals within the previous 48 hours.
In addition, language on shared clinical decision-making was added to the notes on the meningococcal B vaccine for adolescents and young adults aged 18-23 years not at increased risk. Based on shared clinical decision making, the recommendation is a “two-dose series of Bexsero at least 1 month apart” or “two-dose series of Trumenba at least 6 months apart; if dose two is administered earlier than 6 months, administer a third dose at least 4 months after dose two.”
Several vaccines’ Notes sections, including hepatitis B and meningococcal disease, added links to detailed recommendations in the corresponding issues of the CDC’s Morbidity and Mortality Weekly Report, to allow clinicians easy access to additional information.
View the current Child & Adolescent Vaccination Schedule here.
The ACIP members had no financial conflicts to disclose.
FROM AN ACIP MEETING
PHM 19: PREP yourself for the PHM boards
Get ready for the first-ever ABP PHM exam
Presenters
Jared Austin, MD, FAAP
Ryan Bode, MD, FAAP
Jeremy Kern, MD, FAAP
Mary Ottolini, MD, MPH, MEd, FAAP
Stacy Pierson, MD, FAAP
Mary Rocha, MD, MPH, FAAP
Susan Walley, MD, CTTS, FAAP
Session summary
Professional development sessions at the Pediatric Hospital Medicine 2019 conference intended to further educate pediatric hospitalists and advance their careers. In November 2019, many pediatric hospitalists will be taking subspecialty PHM boards for the very first time. This PHM19 session had clear objectives: to describe the American Board of Pediatrics (ABP) PHM board content areas, to analyze common knowledge gaps in PREP PHM, and to examine different approaches to clinical management of PHM patients.

The session opened with a brief history of a vision of PHM and the story of its realization. In 2016, a group of eight stalwart writers, four new writers, and three editors created PREP 2018 and 2019 questions that were released in full prior to November 2019. The ABP will offer the board exam in 2019, 2021, and 2023
The exam content domains include the following:
- Medical conditions.
- Behavioral and mental health conditions.
- Newborn care.
- Children with medical complexity.
- Medical procedures.
- Patient and family centered care.
- Transitions of care.
- Quality improvement, patient safety and system based improvement.
- Evidence-based, high-value care.
- Advocacy and leadership.
- Ethics, legal issues, and human rights.
- Teaching and education.
- Core knowledge in scholarly activities.
Each question consists of a case vignette, question, response choices, critiques, PREP PEARLs, and references. There are also additional PREP Ponder Points that intend to prompt reflection on practice change.
For the remainder of the session presenters reviewed the PHM PREP questions that were most frequently answered incorrectly. Some of the topics included: asthma vs. anaphylaxis, venous thromboembolism prophylaxis in surgical patients, postoperative feeding regimens, transmission-based precautions, febrile neonates, Ebola, medical child abuse, absolute indications for intubation, toxic megacolon, palivizumab prophylaxis guidelines, key driver diagrams, and infantile hemangiomas.
Key takeaway
Pediatric hospitalists all over the United States will for the first time ever take PHM boards in November 2019. The exam content domains were demonstrated in detail, and several often incorrectly answered PREP questions were presented and discussed.
Dr. Giordano is assistant professor in pediatrics at Columbia University Medical Center, New York.
Get ready for the first-ever ABP PHM exam
Get ready for the first-ever ABP PHM exam
Presenters
Jared Austin, MD, FAAP
Ryan Bode, MD, FAAP
Jeremy Kern, MD, FAAP
Mary Ottolini, MD, MPH, MEd, FAAP
Stacy Pierson, MD, FAAP
Mary Rocha, MD, MPH, FAAP
Susan Walley, MD, CTTS, FAAP
Session summary
Professional development sessions at the Pediatric Hospital Medicine 2019 conference intended to further educate pediatric hospitalists and advance their careers. In November 2019, many pediatric hospitalists will be taking subspecialty PHM boards for the very first time. This PHM19 session had clear objectives: to describe the American Board of Pediatrics (ABP) PHM board content areas, to analyze common knowledge gaps in PREP PHM, and to examine different approaches to clinical management of PHM patients.
The session opened with a brief history of a vision of PHM and the story of its realization. In 2016, a group of eight stalwart writers, four new writers, and three editors created PREP 2018 and 2019 questions that were released in full prior to November 2019. The ABP will offer the board exam in 2019, 2021, and 2023
The exam content domains include the following:
- Medical conditions.
- Behavioral and mental health conditions.
- Newborn care.
- Children with medical complexity.
- Medical procedures.
- Patient and family centered care.
- Transitions of care.
- Quality improvement, patient safety and system based improvement.
- Evidence-based, high-value care.
- Advocacy and leadership.
- Ethics, legal issues, and human rights.
- Teaching and education.
- Core knowledge in scholarly activities.
Each question consists of a case vignette, question, response choices, critiques, PREP PEARLs, and references. There are also additional PREP Ponder Points that intend to prompt reflection on practice change.
For the remainder of the session presenters reviewed the PHM PREP questions that were most frequently answered incorrectly. Some of the topics included: asthma vs. anaphylaxis, venous thromboembolism prophylaxis in surgical patients, postoperative feeding regimens, transmission-based precautions, febrile neonates, Ebola, medical child abuse, absolute indications for intubation, toxic megacolon, palivizumab prophylaxis guidelines, key driver diagrams, and infantile hemangiomas.
Key takeaway
Pediatric hospitalists all over the United States will for the first time ever take PHM boards in November 2019. The exam content domains were demonstrated in detail, and several often incorrectly answered PREP questions were presented and discussed.
Dr. Giordano is assistant professor in pediatrics at Columbia University Medical Center, New York.
Presenters
Jared Austin, MD, FAAP
Ryan Bode, MD, FAAP
Jeremy Kern, MD, FAAP
Mary Ottolini, MD, MPH, MEd, FAAP
Stacy Pierson, MD, FAAP
Mary Rocha, MD, MPH, FAAP
Susan Walley, MD, CTTS, FAAP
Session summary
Professional development sessions at the Pediatric Hospital Medicine 2019 conference intended to further educate pediatric hospitalists and advance their careers. In November 2019, many pediatric hospitalists will be taking subspecialty PHM boards for the very first time. This PHM19 session had clear objectives: to describe the American Board of Pediatrics (ABP) PHM board content areas, to analyze common knowledge gaps in PREP PHM, and to examine different approaches to clinical management of PHM patients.
The session opened with a brief history of a vision of PHM and the story of its realization. In 2016, a group of eight stalwart writers, four new writers, and three editors created PREP 2018 and 2019 questions that were released in full prior to November 2019. The ABP will offer the board exam in 2019, 2021, and 2023
The exam content domains include the following:
- Medical conditions.
- Behavioral and mental health conditions.
- Newborn care.
- Children with medical complexity.
- Medical procedures.
- Patient and family centered care.
- Transitions of care.
- Quality improvement, patient safety and system based improvement.
- Evidence-based, high-value care.
- Advocacy and leadership.
- Ethics, legal issues, and human rights.
- Teaching and education.
- Core knowledge in scholarly activities.
Each question consists of a case vignette, question, response choices, critiques, PREP PEARLs, and references. There are also additional PREP Ponder Points that intend to prompt reflection on practice change.
For the remainder of the session presenters reviewed the PHM PREP questions that were most frequently answered incorrectly. Some of the topics included: asthma vs. anaphylaxis, venous thromboembolism prophylaxis in surgical patients, postoperative feeding regimens, transmission-based precautions, febrile neonates, Ebola, medical child abuse, absolute indications for intubation, toxic megacolon, palivizumab prophylaxis guidelines, key driver diagrams, and infantile hemangiomas.
Key takeaway
Pediatric hospitalists all over the United States will for the first time ever take PHM boards in November 2019. The exam content domains were demonstrated in detail, and several often incorrectly answered PREP questions were presented and discussed.
Dr. Giordano is assistant professor in pediatrics at Columbia University Medical Center, New York.
FDA approves onabotulinumtoxinA for pediatric lower limb spasticity
The Food and Drug Administration has approved onabotulinumtoxinA (Botox) for treatment of pediatric lower limb spasticity in patients aged 2-17 years, excluding those in whom it is associated with cerebral palsy, according to an announcement from Allergan.

The approval is based on a phase 3 study evaluating safety and efficacy in more than 300 patients with lower limb spasticity. Although patients with cerebral palsy were included in the study, they’re excluded from this indication. Orphan Drug Exclusivity prevents it from being indicated for lower limb spasticity in cerebral palsy because abobotulinumtoxinA (Dysport) already has marketing exclusivity for the indication. Botox also is indicated for children aged 2-17 years of age with upper limb spasticity, as well as nine other indications.
OnabotulinumtoxinA comes with warnings, including problems of swallowing, speaking, or breathing and even risk of spread of the toxin. It also may cause loss of strength or general muscle weakness, vision problems, or dizziness within hours or weeks of administration. Serious and sometimes immediate allergic reactions have been reported. Patients and health care professionals should discuss various concerns before treatment, including whether the patient has recently received antibiotics by injection, or has taken muscle relaxants, allergy or cold medicine, sleep medicine, and aspirinlike products or blood thinners. It’s important to note that the dose of onabotulinumtoxinA is not the same as that for other botulinum toxin products. The full prescribing information is available on the Allergan website.
The Food and Drug Administration has approved onabotulinumtoxinA (Botox) for treatment of pediatric lower limb spasticity in patients aged 2-17 years, excluding those in whom it is associated with cerebral palsy, according to an announcement from Allergan.
The approval is based on a phase 3 study evaluating safety and efficacy in more than 300 patients with lower limb spasticity. Although patients with cerebral palsy were included in the study, they’re excluded from this indication. Orphan Drug Exclusivity prevents it from being indicated for lower limb spasticity in cerebral palsy because abobotulinumtoxinA (Dysport) already has marketing exclusivity for the indication. Botox also is indicated for children aged 2-17 years of age with upper limb spasticity, as well as nine other indications.
OnabotulinumtoxinA comes with warnings, including problems of swallowing, speaking, or breathing and even risk of spread of the toxin. It also may cause loss of strength or general muscle weakness, vision problems, or dizziness within hours or weeks of administration. Serious and sometimes immediate allergic reactions have been reported. Patients and health care professionals should discuss various concerns before treatment, including whether the patient has recently received antibiotics by injection, or has taken muscle relaxants, allergy or cold medicine, sleep medicine, and aspirinlike products or blood thinners. It’s important to note that the dose of onabotulinumtoxinA is not the same as that for other botulinum toxin products. The full prescribing information is available on the Allergan website.
The Food and Drug Administration has approved onabotulinumtoxinA (Botox) for treatment of pediatric lower limb spasticity in patients aged 2-17 years, excluding those in whom it is associated with cerebral palsy, according to an announcement from Allergan.
The approval is based on a phase 3 study evaluating safety and efficacy in more than 300 patients with lower limb spasticity. Although patients with cerebral palsy were included in the study, they’re excluded from this indication. Orphan Drug Exclusivity prevents it from being indicated for lower limb spasticity in cerebral palsy because abobotulinumtoxinA (Dysport) already has marketing exclusivity for the indication. Botox also is indicated for children aged 2-17 years of age with upper limb spasticity, as well as nine other indications.
OnabotulinumtoxinA comes with warnings, including problems of swallowing, speaking, or breathing and even risk of spread of the toxin. It also may cause loss of strength or general muscle weakness, vision problems, or dizziness within hours or weeks of administration. Serious and sometimes immediate allergic reactions have been reported. Patients and health care professionals should discuss various concerns before treatment, including whether the patient has recently received antibiotics by injection, or has taken muscle relaxants, allergy or cold medicine, sleep medicine, and aspirinlike products or blood thinners. It’s important to note that the dose of onabotulinumtoxinA is not the same as that for other botulinum toxin products. The full prescribing information is available on the Allergan website.
FDA proposes new breast implant labeling with a boxed warning
Breast implants sold in the United States may soon require a boxed warning in their label, along with other label changes proposed by the Food and Drug Administration aimed at better informing prospective patients and clinicians of the potential risks from breast implants.

Other elements of the proposed labeling changes include creation of a patient-decision checklist, new recommendations for follow-up imaging to monitor for implant rupture, inclusion of detailed and understandable information about materials in the device, and provision of a device card to each patient with details on the specific implant they received.
These labeling changes all stemmed from a breast implant hearing held by the agency’s General and Plastic Surgery Devices Panel in March 2019, according to the draft guidance document officially released by the FDA on Oct. 24.
The proposed labeling changes were generally welcomed by patient advocates and by clinicians as a reasonable response to the concerns discussed at the March hearing. In an earlier move to address issues brought up at the hearing, the FDA in July arranged for a recall for certain Allergan models of textured breast implants because of their link with the development of breast implant–associated anaplastic large cell lymphoma (BIA-ALCL).
The boxed warning proposed by the FDA would highlight four specific facts that patients, physicians, and surgeons should know about breast implants: They are not considered lifetime devices, the chance of developing complications from implants increases over time, some complications require additional surgery, and placement of breast implants has been associated with development of BIA-ALCL and may also be associated with certain systemic symptoms.
The FDA also proposed four other notable labeling changes:
- Creation of a patient-decision checklist to better systematize the informed consent process and make sure that certain aspects of breast implant placement are clearly brought to patients’ attention. The FDA proposed that patients sign their checklist attesting to having read and understood the information and that patients receive a take-home copy for their future reference. Proposed elements of the checklist include situations to not use breast implants; considerations for successful implant recipients; the risks of breast implant surgery; the importance of appropriate physician education, training, and experience; the risk for developing BIA-ALCL or systemic symptoms; and discussion of options other than breast implants.
- A new scheme for systematically and serially using imaging to screen for implant rupture that designates for the first time that ultrasound is an acceptable alternative to MRI and relies on a schedule by which either method initially screens the implant 5-6 years post operatively and then every 2 years thereafter.
- Detailed and understandable information about each material component of the implant with further information on possible adverse health effects of these compounds.
- A device card that patients should receive after their surgery with the implant’s name, serial number, and other identifiers; the boxed warning information; and a web link for accessing more up-to-date information.
The patient group Breast Implant Victim Advocacy praised the draft guidance. “The March Advisory Committee meeting seems to have prompted a shift by the FDA, surgeons, and industry,” said Jamee Cook, cofounder of the group. “We are definitely seeing a change in patient engagement. The FDA has been cooperating with patients and listening to our concerns. We still have a long way to go in raising public awareness of breast implant issues, but progress over the past 1-2 years has been amazing.”
Diana Zuckerman, PhD, president of the National Center for Health Research in Washington, gave the draft guidance a mixed review. “The FDA’s draft includes the types of information that we had proposed to the FDA in recent months in our work with patient advocates and plastic surgeons,” she said. “However, it is not as informative as it should be in describing well-designed studies indicating a risk of systemic illnesses. Patients deserve to make better-informed decisions in the future than most women considering breast implants have been able to make” in the past.
Patricia McGuire, MD, a St. Louis plastic surgeon who specializes in breast surgery and has studied breast implant illness, declared the guidance to be “reasonable.”
“I think the changes address the concerns expressed by patients during the [March] hearing; I agree with everything the FDA proposed in the guidance document,” Dr. McGuire said. “The boxed warning is reasonable and needs to be part of the informed consent process. I also agree with the changes in screening implants postoperatively. Most patients do not get MRI examinations. High-resolution ultrasound is more convenient and cost effective.”
The boxed warning was rated as “reasonably strong” and “the most serious step the FDA can take short of taking a device off the market,” but in the case of breast implants, a wider recall of textured implants than what the FDA arranged last July would be even more appropriate, commented Sidney M. Wolfe, MD, founder and senior adviser to Public Citizen. He also faulted the agency for not taking quicker action in mandating inclusion of the proposed boxed warning.
Issuing the labeling changes as draft guidance “is a ministep forward,” but also a process that “guarantees delay” and “creeps along at a dangerously slow pace,” Dr. Wolfe said. “The FDA is delaying what should be inevitable. The agency could put the boxed warning in place right now if they had the guts to do it.”
Dr. McGuire has been a consultant to Allergan, Establishment Labs, and Hans Biomed. Ms. Cook, Dr. Zuckerman, and Dr. Wolfe reported having no commercial disclosures.
Breast implants sold in the United States may soon require a boxed warning in their label, along with other label changes proposed by the Food and Drug Administration aimed at better informing prospective patients and clinicians of the potential risks from breast implants.
Other elements of the proposed labeling changes include creation of a patient-decision checklist, new recommendations for follow-up imaging to monitor for implant rupture, inclusion of detailed and understandable information about materials in the device, and provision of a device card to each patient with details on the specific implant they received.
These labeling changes all stemmed from a breast implant hearing held by the agency’s General and Plastic Surgery Devices Panel in March 2019, according to the draft guidance document officially released by the FDA on Oct. 24.
The proposed labeling changes were generally welcomed by patient advocates and by clinicians as a reasonable response to the concerns discussed at the March hearing. In an earlier move to address issues brought up at the hearing, the FDA in July arranged for a recall for certain Allergan models of textured breast implants because of their link with the development of breast implant–associated anaplastic large cell lymphoma (BIA-ALCL).
The boxed warning proposed by the FDA would highlight four specific facts that patients, physicians, and surgeons should know about breast implants: They are not considered lifetime devices, the chance of developing complications from implants increases over time, some complications require additional surgery, and placement of breast implants has been associated with development of BIA-ALCL and may also be associated with certain systemic symptoms.
The FDA also proposed four other notable labeling changes:
- Creation of a patient-decision checklist to better systematize the informed consent process and make sure that certain aspects of breast implant placement are clearly brought to patients’ attention. The FDA proposed that patients sign their checklist attesting to having read and understood the information and that patients receive a take-home copy for their future reference. Proposed elements of the checklist include situations to not use breast implants; considerations for successful implant recipients; the risks of breast implant surgery; the importance of appropriate physician education, training, and experience; the risk for developing BIA-ALCL or systemic symptoms; and discussion of options other than breast implants.
- A new scheme for systematically and serially using imaging to screen for implant rupture that designates for the first time that ultrasound is an acceptable alternative to MRI and relies on a schedule by which either method initially screens the implant 5-6 years post operatively and then every 2 years thereafter.
- Detailed and understandable information about each material component of the implant with further information on possible adverse health effects of these compounds.
- A device card that patients should receive after their surgery with the implant’s name, serial number, and other identifiers; the boxed warning information; and a web link for accessing more up-to-date information.
The patient group Breast Implant Victim Advocacy praised the draft guidance. “The March Advisory Committee meeting seems to have prompted a shift by the FDA, surgeons, and industry,” said Jamee Cook, cofounder of the group. “We are definitely seeing a change in patient engagement. The FDA has been cooperating with patients and listening to our concerns. We still have a long way to go in raising public awareness of breast implant issues, but progress over the past 1-2 years has been amazing.”
Diana Zuckerman, PhD, president of the National Center for Health Research in Washington, gave the draft guidance a mixed review. “The FDA’s draft includes the types of information that we had proposed to the FDA in recent months in our work with patient advocates and plastic surgeons,” she said. “However, it is not as informative as it should be in describing well-designed studies indicating a risk of systemic illnesses. Patients deserve to make better-informed decisions in the future than most women considering breast implants have been able to make” in the past.
Patricia McGuire, MD, a St. Louis plastic surgeon who specializes in breast surgery and has studied breast implant illness, declared the guidance to be “reasonable.”
“I think the changes address the concerns expressed by patients during the [March] hearing; I agree with everything the FDA proposed in the guidance document,” Dr. McGuire said. “The boxed warning is reasonable and needs to be part of the informed consent process. I also agree with the changes in screening implants postoperatively. Most patients do not get MRI examinations. High-resolution ultrasound is more convenient and cost effective.”
The boxed warning was rated as “reasonably strong” and “the most serious step the FDA can take short of taking a device off the market,” but in the case of breast implants, a wider recall of textured implants than what the FDA arranged last July would be even more appropriate, commented Sidney M. Wolfe, MD, founder and senior adviser to Public Citizen. He also faulted the agency for not taking quicker action in mandating inclusion of the proposed boxed warning.
Issuing the labeling changes as draft guidance “is a ministep forward,” but also a process that “guarantees delay” and “creeps along at a dangerously slow pace,” Dr. Wolfe said. “The FDA is delaying what should be inevitable. The agency could put the boxed warning in place right now if they had the guts to do it.”
Dr. McGuire has been a consultant to Allergan, Establishment Labs, and Hans Biomed. Ms. Cook, Dr. Zuckerman, and Dr. Wolfe reported having no commercial disclosures.
Breast implants sold in the United States may soon require a boxed warning in their label, along with other label changes proposed by the Food and Drug Administration aimed at better informing prospective patients and clinicians of the potential risks from breast implants.
Other elements of the proposed labeling changes include creation of a patient-decision checklist, new recommendations for follow-up imaging to monitor for implant rupture, inclusion of detailed and understandable information about materials in the device, and provision of a device card to each patient with details on the specific implant they received.
These labeling changes all stemmed from a breast implant hearing held by the agency’s General and Plastic Surgery Devices Panel in March 2019, according to the draft guidance document officially released by the FDA on Oct. 24.
The proposed labeling changes were generally welcomed by patient advocates and by clinicians as a reasonable response to the concerns discussed at the March hearing. In an earlier move to address issues brought up at the hearing, the FDA in July arranged for a recall for certain Allergan models of textured breast implants because of their link with the development of breast implant–associated anaplastic large cell lymphoma (BIA-ALCL).
The boxed warning proposed by the FDA would highlight four specific facts that patients, physicians, and surgeons should know about breast implants: They are not considered lifetime devices, the chance of developing complications from implants increases over time, some complications require additional surgery, and placement of breast implants has been associated with development of BIA-ALCL and may also be associated with certain systemic symptoms.
The FDA also proposed four other notable labeling changes:
- Creation of a patient-decision checklist to better systematize the informed consent process and make sure that certain aspects of breast implant placement are clearly brought to patients’ attention. The FDA proposed that patients sign their checklist attesting to having read and understood the information and that patients receive a take-home copy for their future reference. Proposed elements of the checklist include situations to not use breast implants; considerations for successful implant recipients; the risks of breast implant surgery; the importance of appropriate physician education, training, and experience; the risk for developing BIA-ALCL or systemic symptoms; and discussion of options other than breast implants.
- A new scheme for systematically and serially using imaging to screen for implant rupture that designates for the first time that ultrasound is an acceptable alternative to MRI and relies on a schedule by which either method initially screens the implant 5-6 years post operatively and then every 2 years thereafter.
- Detailed and understandable information about each material component of the implant with further information on possible adverse health effects of these compounds.
- A device card that patients should receive after their surgery with the implant’s name, serial number, and other identifiers; the boxed warning information; and a web link for accessing more up-to-date information.
The patient group Breast Implant Victim Advocacy praised the draft guidance. “The March Advisory Committee meeting seems to have prompted a shift by the FDA, surgeons, and industry,” said Jamee Cook, cofounder of the group. “We are definitely seeing a change in patient engagement. The FDA has been cooperating with patients and listening to our concerns. We still have a long way to go in raising public awareness of breast implant issues, but progress over the past 1-2 years has been amazing.”
Diana Zuckerman, PhD, president of the National Center for Health Research in Washington, gave the draft guidance a mixed review. “The FDA’s draft includes the types of information that we had proposed to the FDA in recent months in our work with patient advocates and plastic surgeons,” she said. “However, it is not as informative as it should be in describing well-designed studies indicating a risk of systemic illnesses. Patients deserve to make better-informed decisions in the future than most women considering breast implants have been able to make” in the past.
Patricia McGuire, MD, a St. Louis plastic surgeon who specializes in breast surgery and has studied breast implant illness, declared the guidance to be “reasonable.”
“I think the changes address the concerns expressed by patients during the [March] hearing; I agree with everything the FDA proposed in the guidance document,” Dr. McGuire said. “The boxed warning is reasonable and needs to be part of the informed consent process. I also agree with the changes in screening implants postoperatively. Most patients do not get MRI examinations. High-resolution ultrasound is more convenient and cost effective.”
The boxed warning was rated as “reasonably strong” and “the most serious step the FDA can take short of taking a device off the market,” but in the case of breast implants, a wider recall of textured implants than what the FDA arranged last July would be even more appropriate, commented Sidney M. Wolfe, MD, founder and senior adviser to Public Citizen. He also faulted the agency for not taking quicker action in mandating inclusion of the proposed boxed warning.
Issuing the labeling changes as draft guidance “is a ministep forward,” but also a process that “guarantees delay” and “creeps along at a dangerously slow pace,” Dr. Wolfe said. “The FDA is delaying what should be inevitable. The agency could put the boxed warning in place right now if they had the guts to do it.”
Dr. McGuire has been a consultant to Allergan, Establishment Labs, and Hans Biomed. Ms. Cook, Dr. Zuckerman, and Dr. Wolfe reported having no commercial disclosures.
Acoustic pulse boosts laser tattoo removal
In tattoo removal, the impact of laser treatments in single office visits is limited because of the laser’s effects on the skin. Now, a new study suggests that a

“As a result,” the authors of the study wrote, “a lower total number of office visits will likely be required for complete tattoo removal leading to improved convenience and efficiency as well as increased satisfaction for both patients and clinicians.” The study, led by cosmetic surgeon Michael S. Kaminer, MD, of SkinCare Physicians in Chestnut Hill, Mass., appeared in Lasers in Surgery and Medicine.
In the study, he and his coauthors pointed out that tattoos are most frequently removed with short-pulse high-fluence lasers, such as a 1064-nm Nd:YAG Q‐switched (QS) laser. However, “the QS laser has a limited ability to affect the tattoo ink pigment particles in each treatment session due to shielding of the pigment particles caused by both the agglomeration of the pigment particles and laser‐induced epidermal and dermal vacuoles known as ‘whitening.’ ” Therefore, “use of the QS laser often requires 10 or more single‐pass office sessions to achieve acceptable fading results.”
The study evaluated whether the use of a rapid acoustic pulse (RAP) device could reduce the whitening effect by clearing vacuoles and make it possible to increase the number of potential laser passes. In the single-center, prospective trial, they treated 32 black-ink tattoos in 21 patients, dividing the tattoos into zones and treating them differently: One zone received at least three consecutive laser passes alternating with a minute of treatment with the RAP device, one received single-pass laser treatment, and one received no treatment.
Reviewers assessed the tattoos for fading at 12 weeks. Average percent fading was higher in the laser/RAP group, compared with the laser-only group (44% and 25%, respectively, P less than .01). The percentages of tattoos with more than 50% fading (38% vs. 9%, P less than .01) and more than 75% fading (22% vs. 3%, P less than .05) were also higher in the laser/RAP group, compared with the laser-only group.
“Further clinical studies will be performed to investigate the broader applicability of the RAP device as an accessory device to reduce the number of laser tattoo removal sessions for other tattoo ink colors in a broader range of skin types,” the researchers commented, noting that the study included patients with Fitzpatrick skin types I-III.
The study was funded by Soliton, which provided the equipment. One of the six authors is an employee of the company The other authors reported no relevant disclosures.
SOURCE: Kaminer MS et al. Lasers Surg Med. 2019 Sep 19. doi: 10.1002/lsm.23163.
In tattoo removal, the impact of laser treatments in single office visits is limited because of the laser’s effects on the skin. Now, a new study suggests that a
“As a result,” the authors of the study wrote, “a lower total number of office visits will likely be required for complete tattoo removal leading to improved convenience and efficiency as well as increased satisfaction for both patients and clinicians.” The study, led by cosmetic surgeon Michael S. Kaminer, MD, of SkinCare Physicians in Chestnut Hill, Mass., appeared in Lasers in Surgery and Medicine.
In the study, he and his coauthors pointed out that tattoos are most frequently removed with short-pulse high-fluence lasers, such as a 1064-nm Nd:YAG Q‐switched (QS) laser. However, “the QS laser has a limited ability to affect the tattoo ink pigment particles in each treatment session due to shielding of the pigment particles caused by both the agglomeration of the pigment particles and laser‐induced epidermal and dermal vacuoles known as ‘whitening.’ ” Therefore, “use of the QS laser often requires 10 or more single‐pass office sessions to achieve acceptable fading results.”
The study evaluated whether the use of a rapid acoustic pulse (RAP) device could reduce the whitening effect by clearing vacuoles and make it possible to increase the number of potential laser passes. In the single-center, prospective trial, they treated 32 black-ink tattoos in 21 patients, dividing the tattoos into zones and treating them differently: One zone received at least three consecutive laser passes alternating with a minute of treatment with the RAP device, one received single-pass laser treatment, and one received no treatment.
Reviewers assessed the tattoos for fading at 12 weeks. Average percent fading was higher in the laser/RAP group, compared with the laser-only group (44% and 25%, respectively, P less than .01). The percentages of tattoos with more than 50% fading (38% vs. 9%, P less than .01) and more than 75% fading (22% vs. 3%, P less than .05) were also higher in the laser/RAP group, compared with the laser-only group.
“Further clinical studies will be performed to investigate the broader applicability of the RAP device as an accessory device to reduce the number of laser tattoo removal sessions for other tattoo ink colors in a broader range of skin types,” the researchers commented, noting that the study included patients with Fitzpatrick skin types I-III.
The study was funded by Soliton, which provided the equipment. One of the six authors is an employee of the company The other authors reported no relevant disclosures.
SOURCE: Kaminer MS et al. Lasers Surg Med. 2019 Sep 19. doi: 10.1002/lsm.23163.
In tattoo removal, the impact of laser treatments in single office visits is limited because of the laser’s effects on the skin. Now, a new study suggests that a
“As a result,” the authors of the study wrote, “a lower total number of office visits will likely be required for complete tattoo removal leading to improved convenience and efficiency as well as increased satisfaction for both patients and clinicians.” The study, led by cosmetic surgeon Michael S. Kaminer, MD, of SkinCare Physicians in Chestnut Hill, Mass., appeared in Lasers in Surgery and Medicine.
In the study, he and his coauthors pointed out that tattoos are most frequently removed with short-pulse high-fluence lasers, such as a 1064-nm Nd:YAG Q‐switched (QS) laser. However, “the QS laser has a limited ability to affect the tattoo ink pigment particles in each treatment session due to shielding of the pigment particles caused by both the agglomeration of the pigment particles and laser‐induced epidermal and dermal vacuoles known as ‘whitening.’ ” Therefore, “use of the QS laser often requires 10 or more single‐pass office sessions to achieve acceptable fading results.”
The study evaluated whether the use of a rapid acoustic pulse (RAP) device could reduce the whitening effect by clearing vacuoles and make it possible to increase the number of potential laser passes. In the single-center, prospective trial, they treated 32 black-ink tattoos in 21 patients, dividing the tattoos into zones and treating them differently: One zone received at least three consecutive laser passes alternating with a minute of treatment with the RAP device, one received single-pass laser treatment, and one received no treatment.
Reviewers assessed the tattoos for fading at 12 weeks. Average percent fading was higher in the laser/RAP group, compared with the laser-only group (44% and 25%, respectively, P less than .01). The percentages of tattoos with more than 50% fading (38% vs. 9%, P less than .01) and more than 75% fading (22% vs. 3%, P less than .05) were also higher in the laser/RAP group, compared with the laser-only group.
“Further clinical studies will be performed to investigate the broader applicability of the RAP device as an accessory device to reduce the number of laser tattoo removal sessions for other tattoo ink colors in a broader range of skin types,” the researchers commented, noting that the study included patients with Fitzpatrick skin types I-III.
The study was funded by Soliton, which provided the equipment. One of the six authors is an employee of the company The other authors reported no relevant disclosures.
SOURCE: Kaminer MS et al. Lasers Surg Med. 2019 Sep 19. doi: 10.1002/lsm.23163.
FROM LASERS IN SURGERY AND MEDICINE