User login
When ultrasonography reveals a fetal abdominal wall defect
CASE Fetal anomalies detected on ultrasonography
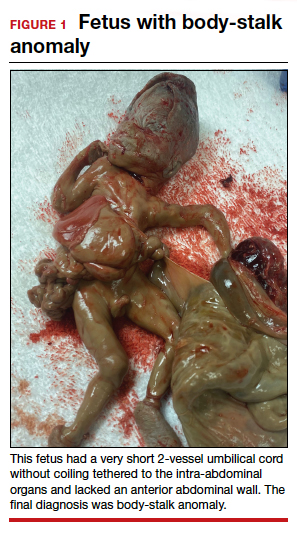
A 34-year-old woman (G2P1) at 19 weeks’ gestation presented for fetal anatomy ultrasonography evaluation. Ultrasonography demonstrated fetal demise with fetal size less than dates, oligohydramnios, and what appeared to be a full-thickness herniation of the thoracic and abdominal contents. Due to the positioning of the fetus and the oligohydramnios, the fetus appeared to have ectopia cordis and herniated liver and bowel; the bladder was not visualized. The patient was counseled regarding the findings and the suspected diagnosis of pentalogy of Cantrell. After counseling, the patient expressed desire to bury the fetus intact according to her religious custom. She underwent a successful uterine evacuation with misoprostol administration and delivered a nonviable fetus that had a closed thoracic cage without ectopia cordis. Key findings were a very short 2-vessel umbilical cord without coiling that was tethered to the intra-abdominal organs, “pulling” the internal organs out of the abdomen, and lack of an anterior abdominal wall (FIGURE 1). Given these findings, a final diagnosis of body-stalk anomaly was made.
Fetal abdominal wall defects (AWDs) encompass a wide array of congenital defects, although they all involve herniation of 1 or more intra-abdominal content through a ventral abdominal defect.1 Overall, the estimated incidence of AWDs is approximately 6 per 10,000 births.1 Gastroschisis and omphalocele are the most common of these defect types.2
The majority of AWDs can be diagnosed during the first trimester of pregnancy via ultrasonography; however, during the first trimester the physiologic midgut herniation resolves by 12 weeks of gestation. It is therefore important to repeat imaging at a later gestational age to confirm the suspicion. Furthermore, the differential diagnosis should include the relatively benign condition of umbilical hernia.
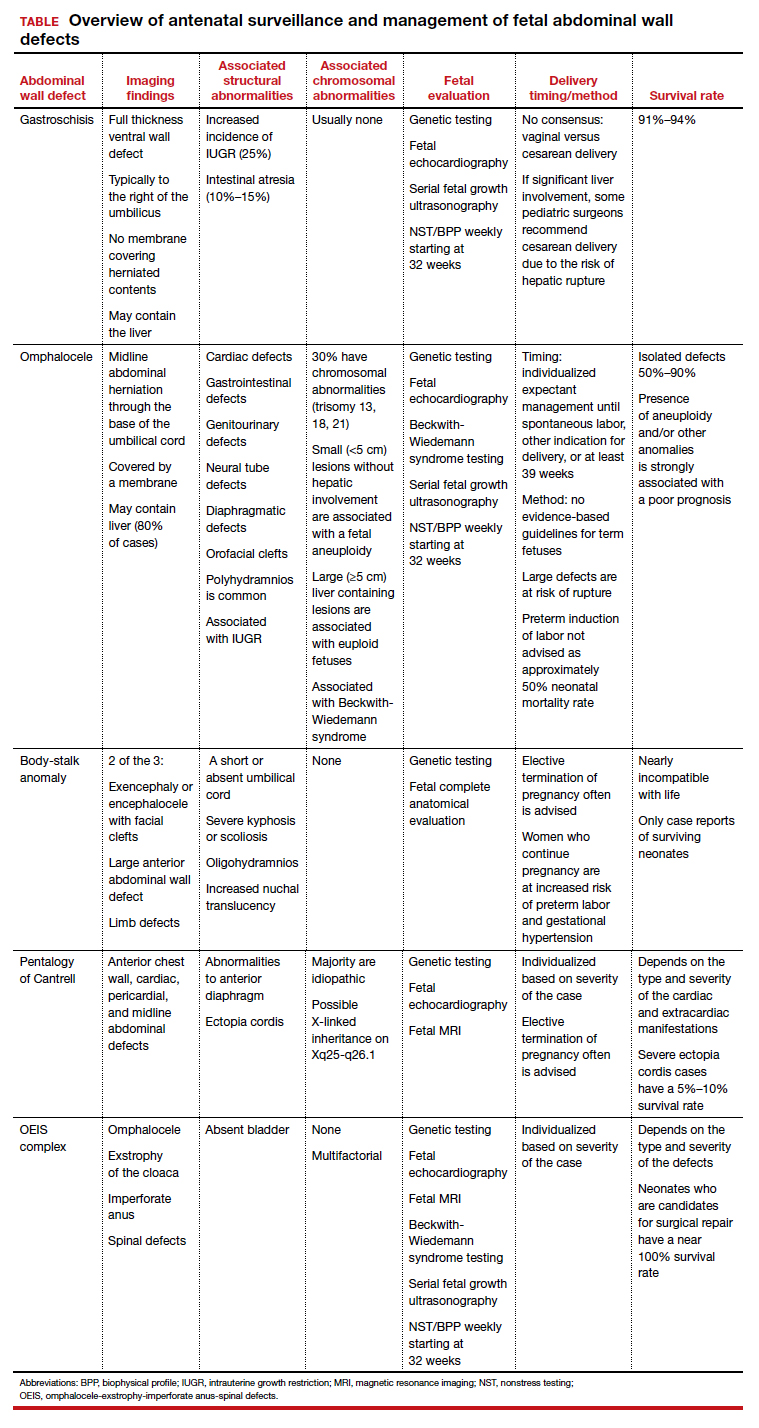
While many AWDs share similarities, they differ significantly in prognosis and management. Early detection is therefore crucial for fetal surveillance, prenatal testing, perinatal planning, and patient counseling (TABLE). In this article, we outline antenatal surveillance and management of AWDs based on recommendations from the American College of Obstetricians and Gynecologists and the Society for Maternal-Fetal Medicine as well as on our experience and practice.
Gastroschisis is an increasingly prevalent AWD
Gastroschisis is a full-thickness, ventral wall defect that results in bowel evisceration; it typically occurs to the right of the umbilical cord insertion.3 It is one of the most common AWDs and its prevalence has increased in the past few decades, from 2 to 3 cases per 10,000 live births in 1995 to as high as 6 cases per 10,000 live births in 2011.2,4,5
The cause of gastroschisis remains unclear. The main theory is that there is an ischemic disruption of the closure of the abdominal wall at or near the omphalomesenteric artery or the right umbilical vein.6,7 In addition, investigators have reported an increased incidence of gastroschisis in mothers exposed to cigarette smoking and certain medications, such as pseudoephedrine, salicylates, ibuprofen, and acetaminophen.8,9
Continue to: Making the diagnosis...
Making the diagnosis
Prenatal diagnosis using ultrasonography is possible at around 10 weeks of gestation. As previously mentioned, however, physiologic herniation of the midgut must be excluded by performing follow-up imaging at a later gestational age. In our practice, we typically do this at around 16 weeks of gestation.
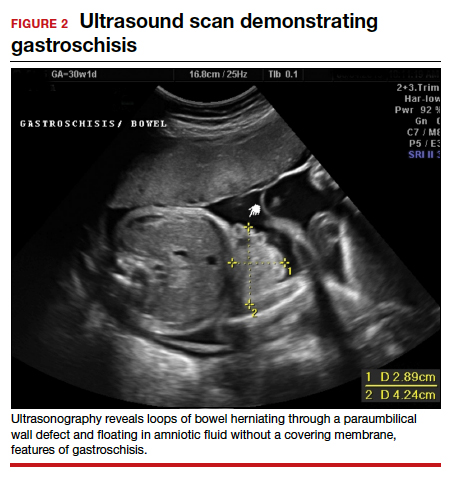
Ultrasonographic features of gastroschisis include loops of bowel herniating through a small paraumbilical wall defect (usually 2–3 cm) floating in amniotic fluid without a covering membrane4 (FIGURE 2). Direct exposure to amniotic fluid causes small bowel inflammation and fibrin deposition, leading to a thickened, echogenic appearance. Polyhydramnios and intra-abdominal bowel dilation have been associated with the presence of intestinal atresia.10
Management
There is no expert consensus regarding optimal prenatal management of gastroschisis.11-17 Prenatal care, patient counseling, and delivery planning should be individualized based on the defect and should be determined in a multidisciplinary discussion with specialists in maternal-fetal medicine, neonatology, and pediatric surgery, as necessary. In our practice, if the gastroschisis is isolated and uncomplicated, our generalist obstetricians manage the patient with maternal-fetal medicine consultation, increased fetal surveillance as described below, and delivery at our tertiary care institution.
Our standard practice is to use the initial ultrasonography imaging to evaluate the size and contents of the defect, measure the nuchal translucency, and evaluate for additional abnormalities. Serial ultrasonography monitoring of the fetus is required to assess the size and quality of the herniated intestine, amount of amniotic fluid, and fetal growth.10
As gastroschisis is a full-thickness defect of the anterior abdominal wall, the abdominal contents are exposed to amniotic fluid. This exposure causes progressive intestinal damage, which can be identified on ultrasonography as bowel thickening and dilation.12-14 Currently, intestinal thickening and dilation is not considered an indication for delivery as it is assumed that the intestinal damage has already occurred. It is debatable whether delivery around 37 weeks compared with delayed delivery beyond 37 weeks improves outcomes and decreases the stillbirth rate.11,13 Studies show that neonates delivered prior to 37 weeks have worse outcomes compared with those delivered after 37 weeks.14,15
Fetal surveillance. As standard practice, we evaluate the fetus at around 16 weeks and then again at around 20 weeks. In the absence of fetal growth restriction, which is associated with 25% of cases,16,17 our standard practice includes performing serial growth ultrasonography every 3 to 4 weeks starting at 28 weeks and biophysical profiles and nonstress testing weekly starting at 32 weeks. Fetal echocardiography can be offered. However, unlike with omphalocele, which has a high incidence of associated cardiac structural anomalies, gastroschisis has a low incidence of congenital cardiac anomalies, estimated to be between 2.5% and 4%.18,19
Delivery considerations. Little agreement exists regarding when and how to deliver pregnancies complicated by fetal gastroschisis. While some advocate for induction of labor at 36 to 38 weeks, most infants with gastroschisis can be delivered safely at term via either vaginal or cesarean delivery.14,15
Delivery timing should consider the clinical picture and incorporate performance on antenatal testing, fetal growth, the size and contents of the gastroschisis, and consultation with maternal-fetal medicine. Fetuses with gastroschisis often have non-reassuring antenatal testing. This can necessitate early delivery, although cesarean delivery should be reserved for obstetric indications, with the caveat that if there is large liver involvement, some pediatric surgeons recommend cesarean delivery due to the risk of hepatic rupture.
Neonate management. The survival rate of gastroschisis is reported to be as high as 91% to 94%.2 Morbidity is related to intestinal complications, such as strictures, adhesions, and volvulus.
In the case of simple gastroschisis, when the bowel is in good condition, the treatment method of choice is primary reduction.20 If performed in the operating room, an immediate sutured closure of the defect can be done. The benefits of primary repair include decreased length of stay, fewer intensive care bed days, and less time to achieve full feeds.20,21 Primary reduction has a reported success rate of 50% to 83%.22 A reduction with a delayed spontaneous closure also can be performed at bedside in the neonatal intensive care unit.22
For complex gastroschisis, characterized by bowel complications such as inflammation, perforation, ischemia, atresia, necrosis, or volvulus, primary closure may not be possible and reduction may need to be achieved through silo application.22-25 Additionally, further bowel surgery, such as stoma formation and bowel resection, may be required.25
Continue to: Omphalocele often is associated with abnormal karyotype...
Omphalocele often is associated with abnormal karyotype
Also known as exomphalos, omphalocele is a relatively common defect, with an estimated prevalence of 2 to 3 cases per 10,000 live births.2 In this condition, there is a midline defect in which intra-abdominal contents herniate through the base of the umbilical cord. Omphaloceles are covered by amniotic membranes, making them distinguishable from gastroschisis, which has no covering, and congenital umbilical hernias, which are covered by intact skin and subcutaneous tissue.26-33
Additionally, in omphalocele the umbilical cord insertion site varies, whereas in gastroschisis the umbilical cord insertion is usually to the right of midline. An omphalocele is often categorized based on whether or not it contains the liver (extracorporeal liver) or only the bowel (intracorporeal liver).
Genetic studies
Approximately 67% to 88% of all pregnancies with omphalocele have an abnormal karyotype and/or associated malformations, including Beckwith-Wiedemann syndrome.31 Of the aneuploidies, trisomy 18 is the one most commonly associated with omphalocele, accounting for approximately 62% to 75%, while trisomy 13 accounts for approximately 11% to 24%.32,33 The presence of other anomalies is strongly associated with poor prognosis, and increased defect size is an independent predictor of neonatal morbidity and mortality, as neonates with large omphaloceles with extracorporeal livers can develop respiratory insufficiency and require more complex surgical repairs. It is interesting, however, that the absence of an extracorporeal liver is associated with a higher risk of aneuploidy than are cases with an intracorporeal liver.33
We offer chorionic villus sampling or amniocentesis to all patients with omphalocele. If the patient undergoes invasive diagnostic testing, the sample then undergoes karyotyping, chromosomal microarray, and testing for Beckwith-Wiedemann syndrome. If the patient declines diagnostic sampling, we perform a cell-free DNA screening to rule out aneuploidy.
Continue to: Making the diagnosis...
Making the diagnosis
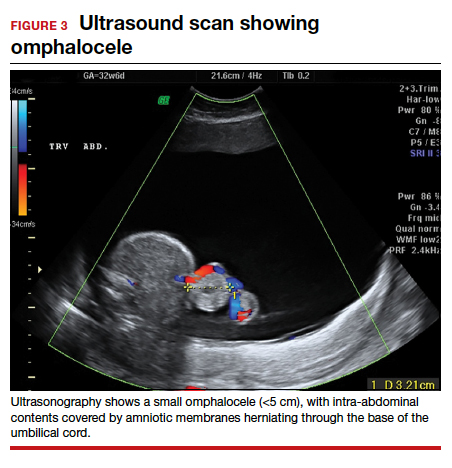
Omphaloceles can be diagnosed via prenatal ultrasonography as early as 11 to 14 weeks’ gestation.26 They are classified based on size, location, and contents of the sac.26,27 A small omphalocele is defined as a defect less than 5 cm with a sac that may contain a few loops of intestines (FIGURE 3).27 A giant omphalocele is a defect with more than 75% of the liver contained in the sac.29
Location can be epigastric, umbilical, or hypogastric, and both small and giant omphaloceles may have ruptured membranes that will result in exposure of the contained viscera.27 Omphaloceles are associated with such structural anomalies as cardiac, gastrointestinal, genitourinary, diaphragmatic, and neural tube defects. We do not routinely perform magnetic resonance imaging (MRI) for evaluation of omphaloceles, but MRI may be used to help predict postnatal outcomes in the case of giant omphaloceles.26
Management
Our standard practice is to use the initial ultrasonography imaging to evaluate the size and contents of defect, measure the nuchal translucency, and evaluate for additional abnormalities. As in cases of gastroschisis, serial ultrasonography monitoring of the fetus is required to assess the size and quality of the herniated intestine, amount of amniotic fluid, and fetal growth. We typically evaluate the fetus at around 16 weeks and then again at around 20 weeks. In the absence of fetal growth restriction, we recommend serial growth ultrasonography every 3 to 4 weeks starting at 28 weeks and biophysical profiles and nonstress testing weekly starting at 32 weeks. Additionally, we routinely obtain a fetal echocardiogram to rule out cardiac structural abnormalities.
Delivery considerations. Fetuses that do not undergo spontaneous abortion or medical termination of pregnancy often are born at term.26 We recommend expectant management until spontaneous labor, another indication for delivery arises, or at least 39 weeks’ estimated gestational age. There are no evidence-based guidelines for the optimal mode of delivery in fetuses with omphalocele, although we recommend cesarean delivery for fetuses with large defects to avoid postnatal sac rupture and liver damage. Preterm induction of labor is not indicated as infants born preterm have about a 50% mortality rate.26,27
Children born with isolated omphalocele typically have a good prognosis, with an estimated survival rate of 50% to 90%.32,33 However, compared to gastroschisis, omphaloceles are often associated with other anomalies.32,33
Management of omphaloceles depends on the size of the defect. In our institution, our generalist obstetricians manage the standard prenatal care with the addition of increased fetal surveillance and testing, interdisciplinary patient counseling with maternal-fetal medicine, pediatric surgeons, and neonatologists for delivery planning, and delivery is performed at our tertiary care center.
Neonate management. Small omphaloceles are amenable to primary early fascial closure.26-30 However, attempted primary closure of giant omphaloceles carries significant risks, including abdominal compartment syndrome and postoperative herniation.29,30 Instead, several options exist for staged surgical closure, in which there are multiple operations prior to final fascial closure, as well as nonoperative delayed closure for management of giant omphaloceles.29,30
Conservative management of giant omphaloceles has certain benefits, such as earlier first feeds, decreased risk of abdominal compartment syndrome, and lower risk of infection.30 Ruptured omphaloceles can be repaired through primary repair, employment of a synthetic or biologic mesh fascial bridge, or silo placement with delayed closure.28
Body-stalk anomaly: Multiple defects and poor prognosis
Also known as limb body wall complex, body-stalk anomaly is a rare malformation that has a reported prevalence of approximately 0.12 cases per 10,000 births (both live and stillbirths).34 Body-stalk anomaly is characterized by multiple defects, including severe kyphosis or scoliosis, a short or absent umbilical cord, and a large anterior abdominal wall defect.34-36 This malformation is almost entirely incompatible with life, resulting in abortion or stillbirth.35 Survival is extremely rare and limited to case reports.
While the exact etiology of body-stalk anomaly is unknown, 3 possible causes have been hypothesized: early amnion rupture, vascular compromise, and embryonic dysgenesis.37-40
Continue to: Making the diagnosis...
Making the diagnosis
Body-stalk anomaly typically can be diagnosed by 10 to 14 weeks’ gestation via ultrasonography.34-41 We currently follow the diagnostic criteria proposed by Van Allen and colleagues, which requires 2 of the following 3 anomalies34:
- exencephaly/encephalocele with facial clefts
- thoraco- and/or abdominoschisis (midline defect)
- limb defect.
Additional ultrasonographic findings can include the identification of evisceration of the abdominal contents, a short umbilical cord, and increased nuchal thickness.36,42 During the second and third trimesters, oligohydramnios may be seen.2
Management
Body-stalk anomaly is considered a fatal condition without specific therapeutic interventions. Maternal risks include an increased risk of preterm labor and gestational hypertension.35 Research on body-stalk anomaly has not shown any correlation with patients’ age, fetal sex, or abnormal karyotype, and the reported risk of recurrence for this anomaly is very low.42,43 Early diagnosis therefore is essential to provide families with information and counseling. Given the poor fetal prognosis, increased maternal risk, and low recurrence rates, mothers can be advised toward elective termination of pregnancy.
Should a patient desire expectant management, care can be provided by generalist obstetricians or care can be transferred to maternal-fetal medicine, with the addition of increased fetal surveillance and testing, interdisciplinary patient counseling with maternal-fetal medicine, pediatric surgeons, and neonatologists for delivery planning; delivery should be performed at a tertiary care center.
Pentalogy of Cantrell: Very rare, with variable prognosis
Pentalogy of Cantrell is characterized by a collection of defects in the midline abdominal wall, lower sternum, anterior diaphragm, diaphragmatic pericardium, and some manifestation of intra-cardiac defect.44 It is thought to arise early in gestation due to abnormal differentiation, migration, and fusion of the embryonic mesoderm.44 The condition is rare, with an incidence of about 1 in 5.5 million live births.45
Making the diagnosis
The diagnosis of pentalogy of Cantrell can be made via prenatal ultrasonography as early as the first trimester, although it is diagnosed more commonly in the second trimester.46 Three-dimensional ultrasonography and fetal MRI have been used to confirm the diagnosis.47
Management
Typically, corrective operations are performed during the neonatal period, and cases of successful staged and one-stage operations have been reported.48 Surgical treatment is determined based on the complexity of the condition and the presence of coexistent heart defects.49,50 However, very few patients survive surgical repair; mortality rates are estimated at around 50% to 60%, with high postsurgical morbidity risks for those who do survive.45
Prognosis varies depending on the type and severity of the associated malformations and intracardiac anomalies.46 Patients with partial ectopia cordis and incomplete presentation may have more favorable outcomes, but for patients with severe ectopia cordis, the survival rate is only 5% to 10%.47
Depending on the severity of the defects, mothers can be advised toward elective termination of pregnancy. In our institution, prenatal care usually is transferred to the maternal-fetal medicine service, and delivery is planned at our tertiary care institution.
OEIS complex comprises abdominal, pelvic, and spinal defects
Omphalocele-exstrophy-imperforate anus-spinal defects (OEIS) complex is a congenital malformation syndrome characterized by the combination of midline abdominal and pelvic defects (including omphalocele, exstrophy of the cloaca, and imperforate anus) and spinal defects.51 The condition’s etiology is unknown but is thought to be multifactorial.51-53 It is a rare condition, with an incidence of around 1 in 200,000 to 400,000 pregnancies.52
Making the diagnosis
Prenatal diagnosis of OEIS complex can be made as early as the first trimester via ultrasonographic identification of an infraumbilical abdominal wall defect with protruding mass, absent bladder, and spinal defects.52 When OEIS complex is suspected, fetal MRI can play a critical role in the diagnosis.
Management
As OEIS complex is rare, there are no evidence-based guidelines for optimal mode and timing of delivery. Cases are individualized based on their specific pathology, and we recommend cesarean delivery for fetuses with large defects to avoid postnatal sac rupture and liver damage.
The prognosis for infants with OEIS complex depends on the spectrum and severity of the structural defects.52,53 The many surgeries involved in the repair of OEIS have potential complications, such as urogenital and gastrointestinal dysfunction.52,53 Advances in medical and surgical treatment have resulted in improved survival and quality of life, and survival rates for OEIS complex are now close to 100%.53 While many OEIS patients live with a permanent colostomy, improvements in management mean that more patients are now candidates for gastrointestinal pull-through procedures, which allow for natural bowel control and a higher degree of bowel cleanliness.53
Prenatal care, patient counseling, and delivery planning should be individualized based on the defects present and determined in a multidisciplinary discussion with maternal-fetal medicine, neonatology, and pediatric surgery as necessary. In our institution, prenatal care usually is transferred to the maternal-fetal medicine service, and delivery is planned at our tertiary care institution.
Multidisciplinary team strategy is essential
Based on our experience, when faced with an anterior AWD in utero, prenatal imaging, genetic testing, increased fetal surveillance, and a multidisciplinary team approach improves outcomes. We must emphasize that careful patient counseling is paramount in our practice. ●
Acknowledgement: The authors would like to thank Ashley Tran, BS, for her assistance in the literature review and drafting of this article.
- Patients with fetuses with anterior wall defects should be referred to a maternal-fetal medicine specialist for co-management and advanced fetal imaging.
- The American College of Obstetricians and Gynecologists recommends microarray for all major fetal structural abnormalities, with the qualifier that karyotype can be offered if a specific aneuploidy is suspected based on the abnormality or prior genetic screening tests.
- If confirmatory testing is performed (amniocentesis or chorionic villus sampling), the sample should undergo karyotyping, chromosomal microarray, and if indicated, testing for Beckwith-Wiedemann syndrome. If the patient declines confirmatory sampling, performing cell-free DNA screening to rule out aneuploidy is recommended.
- Fetal echocardiography is recommended.
- Fetal magnetic resonance imaging should be considered in complex cases.
- Management should be individualized based on the type and severity of defect(s).
- Delivery timing and method should be individualized based on the defect(s) and determined in a multidisciplinary discussion with maternal-fetal medicine, neonatology, pediatric surgery, and pediatric cardiology, as necessary.
- The most common fetal abdominal wall defect is omphalocele, followed by gastroschisis.
- Maternal serum α-fetoprotein is usually elevated in all of the disorders.
- Victoria T, Andronikou S, Bowen D, et al. Fetal anterior abdominal wall defects: prenatal imaging by magnetic resonance imaging. Pediatr Radiol. 2018;48:499-512.
- Pakdaman R, Woodward PJ, Kennedy A. Complex abdominal wall defects: appearances at prenatal imaging. Radiographics. 2015;35:636-649.
- Oakes MC, Porto M, Chung JH. Advances in prenatal and perinatal diagnosis and management of gastroschisis. Semin Pediatr Surg. 2018;27:289-299.
- Mastroiacovo P, Lisi A, Castilla EE. The incidence of gastroschisis: research urgently needs resources. BMJ. 2006;332:423-424.
- Boyd PA, Haeusler M, Barisic I. EUROCAT report 9: surveillance of congenital anomalies in Europe 1980-2008. Birth Defects Res A Clin Mol Teratol. 2011;91(suppl 1):S1.
- Gamba P, Midrio P. Abdominal wall defects: prenatal diagnosis, newborn management, and long-term outcomes. Semin Pediatr Surg. 2014;23:283-290.
- Beaudoin S. Insights into the etiology and embryology of gastroschisis. Semin Pediatr Surg. 2018;27:283-288.
- Yazdy MM, Mitchell AA, Werler MM. Maternal genitourinary infections and the risk of gastroschisis. Am J Epidemiol. 2014;180:518-525.
- Werler MM, Sheehan JE, Mitchell AA. Maternal medication use and risks of gastroschisis and small intestinal atresia. Am J Epidemiol. 2002;155:26-31.
- D’Antonio F, Virgone C, Rizzo G, et al. Prenatal risk factors and outcomes in gastroschisis: a meta-analysis. Pediatrics. 2015;136:e159-e169.
- Baud D, Lausman A, Alfaraj MA, et al. Expectant management compared with elective delivery at 37 weeks for gastroschisis. Obstet Gynecol. 2013;121:990-998.
- Goetzinger KR, Tuuli MG, Longman RE, et al. Sonographic predictors of postnatal bowel atresia in fetal gastroschisis. Ultrasound Obstet Gynecol. 2014;43:420-425.
- Overton TG, Pierce MR, Gao H, et al. Antenatal management and outcomes of gastroschisis in the UK. Prenat Diagn. 2012;32:1256-1262.
- Ergün O, Barksdale E, Ergün FS, et al. The timing of delivery of infants with gastroschisis influences outcome. J Pediatr Surg. 2005;40:424-428.
- Overcash RT, DeUgarte DA, Stephenson ML, et al; University of California Fetal Consortium. Factors associated with gastroschisis outcomes. Obstet Gynecol. 2014;124:551-557.
- Wissanji H, Puligandla PS. Risk stratification and outcome determinants in gastroschisis. Semin Pediatr Surg. 2018;27: 300-303.
- Raynor BD, Richards D. Growth retardation in fetuses with gastroschisis. J Ultrasound Med. 1997;16:13-16.
- Mastroiacovo P, Lisi A, Castilla EE, et al. Gastroschisis and associated defects: an international study. Am J Med Genet A. 2007;143A:660-671.
- Kunz LH, Gilbert WM, Towner DR. Increased incidence of cardiac anomalies in pregnancies complicated by gastroschisis. Am J Obstet Gynecol. 2005;193(3 pt 2): 1248-1252.
- Lakshminarayanan B, Lakhoo K. Abdominal wall defects. Early Hum Dev. 2014;90:917-920.
- Prefumo F, Izzi C. Fetal abdominal wall defects. Best Pract Res Clin Obstet Gynaecol. 2014;28:391-402.
- Petrosyan M, Sandler AD. Closure methods in gastroschisis. Semin Pediatr Surg. 2018;27:304-308.
- Skarsgard ED. Management of gastroschisis. Curr Opin Pediatr. 2016;28:363-369.
- Bergholz R, Boettcher M, Reinshagen K, et al. Complex gastroschisis is a different entity to simple gastroschisis affecting morbidity and mortality—a systematic review and meta-analysis. J Pediatr Surg. 2014;49:1527-1532.
- Emil S. Surgical strategies in complex gastroschisis. Semin Pediatr Surg. 2018;27:309-315.
- Verla MA, Style CC, Olutoye OO. Prenatal diagnosis and management of omphalocele. Semin Pediatr Surg. 2019;28:84-88.
- Gonzalez KW, Chandler NM. Ruptured omphalocele: diagnosis and management. Semin Pediatr Surg. 2019;28:101-105.
- Sugandhi N, Saha M, Bhatnagar V, et al. Repair of ruptured omphalocele sac in the neonatal period and beyond. J Indian Assoc Pediatr Surg. 2020;25:46-48.
- Bauman B, Stephens D, Gershone H, et al. Management of giant omphaloceles: a systematic review of methods of staged surgical vs nonoperative delayed closure. J Pediatr Surg. 2016;51:1725-1730.
- Kogut KA, Fiore NF. Nonoperative management of giant omphalocele leading to early fascial closure. J Pediatr Surg. 2018;53:2404-2408.
- Conner P, Vejde JH, Burgos CM. Accuracy and impact of prenatal diagnosis in infants with omphalocele. Pediatr Surg Int. 2018;34:629-633.
- Iacovella C, Contro E, Ghi T, et al. The effect of the contents of exomphalos and nuchal translucency at 11-14 weeks on the likelihood of associated chromosomal abnormality. Prenat Diagn. 2012;32:1066-1070.
- Getachew MM, Goldstein RB, Edge V, et al. Correlation between omphalocele contents and karyotypic abnormalities: sonographic study in 37 cases. AJR Am J Roentgenol. 1992;158:133-136.
- Singh A, Singh J, Gupta K. Body stalk anomaly: antenatal sonographic diagnosis of this rare entity with review of literature. J Ultrason. 2017;17:133-135.
- Lazaroni TL, Cruzeiro PC, Piçarro C, et al. Body stalk anomaly: Three months of survival. Case report and literature review. J Pediatr Surg Case Rep. 2016;14:22-25.
- Gajzer DC, Hirzel AC, Saigal G, et al. Possible genetic origin of limb-body wall complex. Fetal Pediatr Pathol. 2015;34: 257–270.
- Maruyama H, Inagaki T, Nakata Y, et al. Minimally conjoined omphalopagus twins with a body stalk anomaly. AJP Rep. 2015;5:e124-e128.
- Bhat A, Ilyas M, Dev G. Prenatal sonographic diagnosis of limb-body wall complex: case series of a rare congenital anomaly. Radiol Case Rep. 2016;11:116-120.
- Quijano FE, Rey MM, Echeverry M, et al. Body stalk anomaly in a 9-week pregnancy. Case Rep Obstet Gynecol. 2014;2014:357285.
- Kocherla K, Kumari V, Kocherla PR. Prenatal diagnosis of body stalk complex: a rare entity and review of literature. Indian J Radiol Imaging. 2015;25:67-70.
- Panaitescu AM, Ushakov F, Kalaskar A, et al. Ultrasound features and management of body stalk anomaly. Fetal Diagn Ther. 2016;40:285-290.
- Routhu M, Thakkallapelli S, Mohan P, et al. Role of ultrasound in body stalk anomaly and amniotic band syndrome. Int J Reprod Med. 2016;2016:3974139.
- Costa ML, Couto E, Furlan E, et al. Body stalk anomaly: adverse maternal outcomes in a series of 21 cases. Prenat Diagn. 2012;32:264-267.
- Hubbard R, Hayes S, Gillis H, et al. Management challenges in an infant with pentalogy of Cantrell, giant anterior encephalocele, and craniofacial anomalies: a case report. A A Pract. 2018;11:238-240.
- Jnah AJ, Newberry DM, England A. Pentalogy of Cantrell: case report with review of the literature. Adv Neonatal Care. 2015;15:261-268.
- Williams AP, Marayati R, Beierle EA. Pentalogy of Cantrell. Semin Pediatr Surg. 2019;28:106-110.
- Restrepo MS, Cerqua A, Turek JW. Pentalogy of Cantrell with ectopia cordis totalis, total anomalous pulmonary venous connection, and tetralogy of Fallot: a case report and review of the literature. Congenit Heart Dis. 2014;9:E129–E134.
- Zhang X, Xing Q, Sun J, et al. Surgical treatment and outcomes of pentalogy of Cantrell in eight patients. J Pediatr Surg. 2014;49:1335-1340.
- Harring G, Weil J, Thiel C, et al. Management of pentalogy of Cantrell with complete ectopia cordis and double outlet right ventricle. Congenit Anom (Kyoto). 2015;55:121- 123.
- Mallula KK, Sosnowski C, Awad S. Spectrum of Cantrell’s pentalogy: case series from a single tertiary care center and review of the literature. Pediatr Cardiol. 2013;34:1703- 1710.
- Allam ES, Shetty VS, Farmakis SG. Fetal and neonatal presentation of OEIS complex. J Pediatr Surg. 2015;50:2155-2158.
- Neel N, Tarabay MS. Omphalocele, exstrophy of cloaca, imperforate anus, and spinal defect complex, multiple major reconstructive surgeries needed. Urol Ann. 2018;10:118-121.
- Sawaya D, Gearhart JP. Gastrointestinal reconstruction and outcomes for patients with the OEIS complex. Semin Pediatr Surg. 2011;20:123-125.
Dr. Juusela is an Obstetrician-Gynecologist, Department of Obstetrics and Gynecology, Newark Beth Israel Medical Center, Newark, New Jersey.
Dr. Gimovsky is a Perinatologist, Maternal-Fetal Medicine Department. Newark Beth Israel Medical Center, Newark, New Jersey.
The authors report no financial relationships relevant to this article.
Dr. Juusela is an Obstetrician-Gynecologist, Department of Obstetrics and Gynecology, Newark Beth Israel Medical Center, Newark, New Jersey.
Dr. Gimovsky is a Perinatologist, Maternal-Fetal Medicine Department. Newark Beth Israel Medical Center, Newark, New Jersey.
The authors report no financial relationships relevant to this article.
Dr. Juusela is an Obstetrician-Gynecologist, Department of Obstetrics and Gynecology, Newark Beth Israel Medical Center, Newark, New Jersey.
Dr. Gimovsky is a Perinatologist, Maternal-Fetal Medicine Department. Newark Beth Israel Medical Center, Newark, New Jersey.
The authors report no financial relationships relevant to this article.
CASE Fetal anomalies detected on ultrasonography
A 34-year-old woman (G2P1) at 19 weeks’ gestation presented for fetal anatomy ultrasonography evaluation. Ultrasonography demonstrated fetal demise with fetal size less than dates, oligohydramnios, and what appeared to be a full-thickness herniation of the thoracic and abdominal contents. Due to the positioning of the fetus and the oligohydramnios, the fetus appeared to have ectopia cordis and herniated liver and bowel; the bladder was not visualized. The patient was counseled regarding the findings and the suspected diagnosis of pentalogy of Cantrell. After counseling, the patient expressed desire to bury the fetus intact according to her religious custom. She underwent a successful uterine evacuation with misoprostol administration and delivered a nonviable fetus that had a closed thoracic cage without ectopia cordis. Key findings were a very short 2-vessel umbilical cord without coiling that was tethered to the intra-abdominal organs, “pulling” the internal organs out of the abdomen, and lack of an anterior abdominal wall (FIGURE 1). Given these findings, a final diagnosis of body-stalk anomaly was made.
Fetal abdominal wall defects (AWDs) encompass a wide array of congenital defects, although they all involve herniation of 1 or more intra-abdominal content through a ventral abdominal defect.1 Overall, the estimated incidence of AWDs is approximately 6 per 10,000 births.1 Gastroschisis and omphalocele are the most common of these defect types.2
The majority of AWDs can be diagnosed during the first trimester of pregnancy via ultrasonography; however, during the first trimester the physiologic midgut herniation resolves by 12 weeks of gestation. It is therefore important to repeat imaging at a later gestational age to confirm the suspicion. Furthermore, the differential diagnosis should include the relatively benign condition of umbilical hernia.
While many AWDs share similarities, they differ significantly in prognosis and management. Early detection is therefore crucial for fetal surveillance, prenatal testing, perinatal planning, and patient counseling (TABLE). In this article, we outline antenatal surveillance and management of AWDs based on recommendations from the American College of Obstetricians and Gynecologists and the Society for Maternal-Fetal Medicine as well as on our experience and practice.
Gastroschisis is an increasingly prevalent AWD
Gastroschisis is a full-thickness, ventral wall defect that results in bowel evisceration; it typically occurs to the right of the umbilical cord insertion.3 It is one of the most common AWDs and its prevalence has increased in the past few decades, from 2 to 3 cases per 10,000 live births in 1995 to as high as 6 cases per 10,000 live births in 2011.2,4,5
The cause of gastroschisis remains unclear. The main theory is that there is an ischemic disruption of the closure of the abdominal wall at or near the omphalomesenteric artery or the right umbilical vein.6,7 In addition, investigators have reported an increased incidence of gastroschisis in mothers exposed to cigarette smoking and certain medications, such as pseudoephedrine, salicylates, ibuprofen, and acetaminophen.8,9
Continue to: Making the diagnosis...
Making the diagnosis
Prenatal diagnosis using ultrasonography is possible at around 10 weeks of gestation. As previously mentioned, however, physiologic herniation of the midgut must be excluded by performing follow-up imaging at a later gestational age. In our practice, we typically do this at around 16 weeks of gestation.
Ultrasonographic features of gastroschisis include loops of bowel herniating through a small paraumbilical wall defect (usually 2–3 cm) floating in amniotic fluid without a covering membrane4 (FIGURE 2). Direct exposure to amniotic fluid causes small bowel inflammation and fibrin deposition, leading to a thickened, echogenic appearance. Polyhydramnios and intra-abdominal bowel dilation have been associated with the presence of intestinal atresia.10
Management
There is no expert consensus regarding optimal prenatal management of gastroschisis.11-17 Prenatal care, patient counseling, and delivery planning should be individualized based on the defect and should be determined in a multidisciplinary discussion with specialists in maternal-fetal medicine, neonatology, and pediatric surgery, as necessary. In our practice, if the gastroschisis is isolated and uncomplicated, our generalist obstetricians manage the patient with maternal-fetal medicine consultation, increased fetal surveillance as described below, and delivery at our tertiary care institution.
Our standard practice is to use the initial ultrasonography imaging to evaluate the size and contents of the defect, measure the nuchal translucency, and evaluate for additional abnormalities. Serial ultrasonography monitoring of the fetus is required to assess the size and quality of the herniated intestine, amount of amniotic fluid, and fetal growth.10
As gastroschisis is a full-thickness defect of the anterior abdominal wall, the abdominal contents are exposed to amniotic fluid. This exposure causes progressive intestinal damage, which can be identified on ultrasonography as bowel thickening and dilation.12-14 Currently, intestinal thickening and dilation is not considered an indication for delivery as it is assumed that the intestinal damage has already occurred. It is debatable whether delivery around 37 weeks compared with delayed delivery beyond 37 weeks improves outcomes and decreases the stillbirth rate.11,13 Studies show that neonates delivered prior to 37 weeks have worse outcomes compared with those delivered after 37 weeks.14,15
Fetal surveillance. As standard practice, we evaluate the fetus at around 16 weeks and then again at around 20 weeks. In the absence of fetal growth restriction, which is associated with 25% of cases,16,17 our standard practice includes performing serial growth ultrasonography every 3 to 4 weeks starting at 28 weeks and biophysical profiles and nonstress testing weekly starting at 32 weeks. Fetal echocardiography can be offered. However, unlike with omphalocele, which has a high incidence of associated cardiac structural anomalies, gastroschisis has a low incidence of congenital cardiac anomalies, estimated to be between 2.5% and 4%.18,19
Delivery considerations. Little agreement exists regarding when and how to deliver pregnancies complicated by fetal gastroschisis. While some advocate for induction of labor at 36 to 38 weeks, most infants with gastroschisis can be delivered safely at term via either vaginal or cesarean delivery.14,15
Delivery timing should consider the clinical picture and incorporate performance on antenatal testing, fetal growth, the size and contents of the gastroschisis, and consultation with maternal-fetal medicine. Fetuses with gastroschisis often have non-reassuring antenatal testing. This can necessitate early delivery, although cesarean delivery should be reserved for obstetric indications, with the caveat that if there is large liver involvement, some pediatric surgeons recommend cesarean delivery due to the risk of hepatic rupture.
Neonate management. The survival rate of gastroschisis is reported to be as high as 91% to 94%.2 Morbidity is related to intestinal complications, such as strictures, adhesions, and volvulus.
In the case of simple gastroschisis, when the bowel is in good condition, the treatment method of choice is primary reduction.20 If performed in the operating room, an immediate sutured closure of the defect can be done. The benefits of primary repair include decreased length of stay, fewer intensive care bed days, and less time to achieve full feeds.20,21 Primary reduction has a reported success rate of 50% to 83%.22 A reduction with a delayed spontaneous closure also can be performed at bedside in the neonatal intensive care unit.22
For complex gastroschisis, characterized by bowel complications such as inflammation, perforation, ischemia, atresia, necrosis, or volvulus, primary closure may not be possible and reduction may need to be achieved through silo application.22-25 Additionally, further bowel surgery, such as stoma formation and bowel resection, may be required.25
Continue to: Omphalocele often is associated with abnormal karyotype...
Omphalocele often is associated with abnormal karyotype
Also known as exomphalos, omphalocele is a relatively common defect, with an estimated prevalence of 2 to 3 cases per 10,000 live births.2 In this condition, there is a midline defect in which intra-abdominal contents herniate through the base of the umbilical cord. Omphaloceles are covered by amniotic membranes, making them distinguishable from gastroschisis, which has no covering, and congenital umbilical hernias, which are covered by intact skin and subcutaneous tissue.26-33
Additionally, in omphalocele the umbilical cord insertion site varies, whereas in gastroschisis the umbilical cord insertion is usually to the right of midline. An omphalocele is often categorized based on whether or not it contains the liver (extracorporeal liver) or only the bowel (intracorporeal liver).
Genetic studies
Approximately 67% to 88% of all pregnancies with omphalocele have an abnormal karyotype and/or associated malformations, including Beckwith-Wiedemann syndrome.31 Of the aneuploidies, trisomy 18 is the one most commonly associated with omphalocele, accounting for approximately 62% to 75%, while trisomy 13 accounts for approximately 11% to 24%.32,33 The presence of other anomalies is strongly associated with poor prognosis, and increased defect size is an independent predictor of neonatal morbidity and mortality, as neonates with large omphaloceles with extracorporeal livers can develop respiratory insufficiency and require more complex surgical repairs. It is interesting, however, that the absence of an extracorporeal liver is associated with a higher risk of aneuploidy than are cases with an intracorporeal liver.33
We offer chorionic villus sampling or amniocentesis to all patients with omphalocele. If the patient undergoes invasive diagnostic testing, the sample then undergoes karyotyping, chromosomal microarray, and testing for Beckwith-Wiedemann syndrome. If the patient declines diagnostic sampling, we perform a cell-free DNA screening to rule out aneuploidy.
Continue to: Making the diagnosis...
Making the diagnosis
Omphaloceles can be diagnosed via prenatal ultrasonography as early as 11 to 14 weeks’ gestation.26 They are classified based on size, location, and contents of the sac.26,27 A small omphalocele is defined as a defect less than 5 cm with a sac that may contain a few loops of intestines (FIGURE 3).27 A giant omphalocele is a defect with more than 75% of the liver contained in the sac.29
Location can be epigastric, umbilical, or hypogastric, and both small and giant omphaloceles may have ruptured membranes that will result in exposure of the contained viscera.27 Omphaloceles are associated with such structural anomalies as cardiac, gastrointestinal, genitourinary, diaphragmatic, and neural tube defects. We do not routinely perform magnetic resonance imaging (MRI) for evaluation of omphaloceles, but MRI may be used to help predict postnatal outcomes in the case of giant omphaloceles.26
Management
Our standard practice is to use the initial ultrasonography imaging to evaluate the size and contents of defect, measure the nuchal translucency, and evaluate for additional abnormalities. As in cases of gastroschisis, serial ultrasonography monitoring of the fetus is required to assess the size and quality of the herniated intestine, amount of amniotic fluid, and fetal growth. We typically evaluate the fetus at around 16 weeks and then again at around 20 weeks. In the absence of fetal growth restriction, we recommend serial growth ultrasonography every 3 to 4 weeks starting at 28 weeks and biophysical profiles and nonstress testing weekly starting at 32 weeks. Additionally, we routinely obtain a fetal echocardiogram to rule out cardiac structural abnormalities.
Delivery considerations. Fetuses that do not undergo spontaneous abortion or medical termination of pregnancy often are born at term.26 We recommend expectant management until spontaneous labor, another indication for delivery arises, or at least 39 weeks’ estimated gestational age. There are no evidence-based guidelines for the optimal mode of delivery in fetuses with omphalocele, although we recommend cesarean delivery for fetuses with large defects to avoid postnatal sac rupture and liver damage. Preterm induction of labor is not indicated as infants born preterm have about a 50% mortality rate.26,27
Children born with isolated omphalocele typically have a good prognosis, with an estimated survival rate of 50% to 90%.32,33 However, compared to gastroschisis, omphaloceles are often associated with other anomalies.32,33
Management of omphaloceles depends on the size of the defect. In our institution, our generalist obstetricians manage the standard prenatal care with the addition of increased fetal surveillance and testing, interdisciplinary patient counseling with maternal-fetal medicine, pediatric surgeons, and neonatologists for delivery planning, and delivery is performed at our tertiary care center.
Neonate management. Small omphaloceles are amenable to primary early fascial closure.26-30 However, attempted primary closure of giant omphaloceles carries significant risks, including abdominal compartment syndrome and postoperative herniation.29,30 Instead, several options exist for staged surgical closure, in which there are multiple operations prior to final fascial closure, as well as nonoperative delayed closure for management of giant omphaloceles.29,30
Conservative management of giant omphaloceles has certain benefits, such as earlier first feeds, decreased risk of abdominal compartment syndrome, and lower risk of infection.30 Ruptured omphaloceles can be repaired through primary repair, employment of a synthetic or biologic mesh fascial bridge, or silo placement with delayed closure.28
Body-stalk anomaly: Multiple defects and poor prognosis
Also known as limb body wall complex, body-stalk anomaly is a rare malformation that has a reported prevalence of approximately 0.12 cases per 10,000 births (both live and stillbirths).34 Body-stalk anomaly is characterized by multiple defects, including severe kyphosis or scoliosis, a short or absent umbilical cord, and a large anterior abdominal wall defect.34-36 This malformation is almost entirely incompatible with life, resulting in abortion or stillbirth.35 Survival is extremely rare and limited to case reports.
While the exact etiology of body-stalk anomaly is unknown, 3 possible causes have been hypothesized: early amnion rupture, vascular compromise, and embryonic dysgenesis.37-40
Continue to: Making the diagnosis...
Making the diagnosis
Body-stalk anomaly typically can be diagnosed by 10 to 14 weeks’ gestation via ultrasonography.34-41 We currently follow the diagnostic criteria proposed by Van Allen and colleagues, which requires 2 of the following 3 anomalies34:
- exencephaly/encephalocele with facial clefts
- thoraco- and/or abdominoschisis (midline defect)
- limb defect.
Additional ultrasonographic findings can include the identification of evisceration of the abdominal contents, a short umbilical cord, and increased nuchal thickness.36,42 During the second and third trimesters, oligohydramnios may be seen.2
Management
Body-stalk anomaly is considered a fatal condition without specific therapeutic interventions. Maternal risks include an increased risk of preterm labor and gestational hypertension.35 Research on body-stalk anomaly has not shown any correlation with patients’ age, fetal sex, or abnormal karyotype, and the reported risk of recurrence for this anomaly is very low.42,43 Early diagnosis therefore is essential to provide families with information and counseling. Given the poor fetal prognosis, increased maternal risk, and low recurrence rates, mothers can be advised toward elective termination of pregnancy.
Should a patient desire expectant management, care can be provided by generalist obstetricians or care can be transferred to maternal-fetal medicine, with the addition of increased fetal surveillance and testing, interdisciplinary patient counseling with maternal-fetal medicine, pediatric surgeons, and neonatologists for delivery planning; delivery should be performed at a tertiary care center.
Pentalogy of Cantrell: Very rare, with variable prognosis
Pentalogy of Cantrell is characterized by a collection of defects in the midline abdominal wall, lower sternum, anterior diaphragm, diaphragmatic pericardium, and some manifestation of intra-cardiac defect.44 It is thought to arise early in gestation due to abnormal differentiation, migration, and fusion of the embryonic mesoderm.44 The condition is rare, with an incidence of about 1 in 5.5 million live births.45
Making the diagnosis
The diagnosis of pentalogy of Cantrell can be made via prenatal ultrasonography as early as the first trimester, although it is diagnosed more commonly in the second trimester.46 Three-dimensional ultrasonography and fetal MRI have been used to confirm the diagnosis.47
Management
Typically, corrective operations are performed during the neonatal period, and cases of successful staged and one-stage operations have been reported.48 Surgical treatment is determined based on the complexity of the condition and the presence of coexistent heart defects.49,50 However, very few patients survive surgical repair; mortality rates are estimated at around 50% to 60%, with high postsurgical morbidity risks for those who do survive.45
Prognosis varies depending on the type and severity of the associated malformations and intracardiac anomalies.46 Patients with partial ectopia cordis and incomplete presentation may have more favorable outcomes, but for patients with severe ectopia cordis, the survival rate is only 5% to 10%.47
Depending on the severity of the defects, mothers can be advised toward elective termination of pregnancy. In our institution, prenatal care usually is transferred to the maternal-fetal medicine service, and delivery is planned at our tertiary care institution.
OEIS complex comprises abdominal, pelvic, and spinal defects
Omphalocele-exstrophy-imperforate anus-spinal defects (OEIS) complex is a congenital malformation syndrome characterized by the combination of midline abdominal and pelvic defects (including omphalocele, exstrophy of the cloaca, and imperforate anus) and spinal defects.51 The condition’s etiology is unknown but is thought to be multifactorial.51-53 It is a rare condition, with an incidence of around 1 in 200,000 to 400,000 pregnancies.52
Making the diagnosis
Prenatal diagnosis of OEIS complex can be made as early as the first trimester via ultrasonographic identification of an infraumbilical abdominal wall defect with protruding mass, absent bladder, and spinal defects.52 When OEIS complex is suspected, fetal MRI can play a critical role in the diagnosis.
Management
As OEIS complex is rare, there are no evidence-based guidelines for optimal mode and timing of delivery. Cases are individualized based on their specific pathology, and we recommend cesarean delivery for fetuses with large defects to avoid postnatal sac rupture and liver damage.
The prognosis for infants with OEIS complex depends on the spectrum and severity of the structural defects.52,53 The many surgeries involved in the repair of OEIS have potential complications, such as urogenital and gastrointestinal dysfunction.52,53 Advances in medical and surgical treatment have resulted in improved survival and quality of life, and survival rates for OEIS complex are now close to 100%.53 While many OEIS patients live with a permanent colostomy, improvements in management mean that more patients are now candidates for gastrointestinal pull-through procedures, which allow for natural bowel control and a higher degree of bowel cleanliness.53
Prenatal care, patient counseling, and delivery planning should be individualized based on the defects present and determined in a multidisciplinary discussion with maternal-fetal medicine, neonatology, and pediatric surgery as necessary. In our institution, prenatal care usually is transferred to the maternal-fetal medicine service, and delivery is planned at our tertiary care institution.
Multidisciplinary team strategy is essential
Based on our experience, when faced with an anterior AWD in utero, prenatal imaging, genetic testing, increased fetal surveillance, and a multidisciplinary team approach improves outcomes. We must emphasize that careful patient counseling is paramount in our practice. ●
Acknowledgement: The authors would like to thank Ashley Tran, BS, for her assistance in the literature review and drafting of this article.
- Patients with fetuses with anterior wall defects should be referred to a maternal-fetal medicine specialist for co-management and advanced fetal imaging.
- The American College of Obstetricians and Gynecologists recommends microarray for all major fetal structural abnormalities, with the qualifier that karyotype can be offered if a specific aneuploidy is suspected based on the abnormality or prior genetic screening tests.
- If confirmatory testing is performed (amniocentesis or chorionic villus sampling), the sample should undergo karyotyping, chromosomal microarray, and if indicated, testing for Beckwith-Wiedemann syndrome. If the patient declines confirmatory sampling, performing cell-free DNA screening to rule out aneuploidy is recommended.
- Fetal echocardiography is recommended.
- Fetal magnetic resonance imaging should be considered in complex cases.
- Management should be individualized based on the type and severity of defect(s).
- Delivery timing and method should be individualized based on the defect(s) and determined in a multidisciplinary discussion with maternal-fetal medicine, neonatology, pediatric surgery, and pediatric cardiology, as necessary.
- The most common fetal abdominal wall defect is omphalocele, followed by gastroschisis.
- Maternal serum α-fetoprotein is usually elevated in all of the disorders.
CASE Fetal anomalies detected on ultrasonography
A 34-year-old woman (G2P1) at 19 weeks’ gestation presented for fetal anatomy ultrasonography evaluation. Ultrasonography demonstrated fetal demise with fetal size less than dates, oligohydramnios, and what appeared to be a full-thickness herniation of the thoracic and abdominal contents. Due to the positioning of the fetus and the oligohydramnios, the fetus appeared to have ectopia cordis and herniated liver and bowel; the bladder was not visualized. The patient was counseled regarding the findings and the suspected diagnosis of pentalogy of Cantrell. After counseling, the patient expressed desire to bury the fetus intact according to her religious custom. She underwent a successful uterine evacuation with misoprostol administration and delivered a nonviable fetus that had a closed thoracic cage without ectopia cordis. Key findings were a very short 2-vessel umbilical cord without coiling that was tethered to the intra-abdominal organs, “pulling” the internal organs out of the abdomen, and lack of an anterior abdominal wall (FIGURE 1). Given these findings, a final diagnosis of body-stalk anomaly was made.
Fetal abdominal wall defects (AWDs) encompass a wide array of congenital defects, although they all involve herniation of 1 or more intra-abdominal content through a ventral abdominal defect.1 Overall, the estimated incidence of AWDs is approximately 6 per 10,000 births.1 Gastroschisis and omphalocele are the most common of these defect types.2
The majority of AWDs can be diagnosed during the first trimester of pregnancy via ultrasonography; however, during the first trimester the physiologic midgut herniation resolves by 12 weeks of gestation. It is therefore important to repeat imaging at a later gestational age to confirm the suspicion. Furthermore, the differential diagnosis should include the relatively benign condition of umbilical hernia.
While many AWDs share similarities, they differ significantly in prognosis and management. Early detection is therefore crucial for fetal surveillance, prenatal testing, perinatal planning, and patient counseling (TABLE). In this article, we outline antenatal surveillance and management of AWDs based on recommendations from the American College of Obstetricians and Gynecologists and the Society for Maternal-Fetal Medicine as well as on our experience and practice.
Gastroschisis is an increasingly prevalent AWD
Gastroschisis is a full-thickness, ventral wall defect that results in bowel evisceration; it typically occurs to the right of the umbilical cord insertion.3 It is one of the most common AWDs and its prevalence has increased in the past few decades, from 2 to 3 cases per 10,000 live births in 1995 to as high as 6 cases per 10,000 live births in 2011.2,4,5
The cause of gastroschisis remains unclear. The main theory is that there is an ischemic disruption of the closure of the abdominal wall at or near the omphalomesenteric artery or the right umbilical vein.6,7 In addition, investigators have reported an increased incidence of gastroschisis in mothers exposed to cigarette smoking and certain medications, such as pseudoephedrine, salicylates, ibuprofen, and acetaminophen.8,9
Continue to: Making the diagnosis...
Making the diagnosis
Prenatal diagnosis using ultrasonography is possible at around 10 weeks of gestation. As previously mentioned, however, physiologic herniation of the midgut must be excluded by performing follow-up imaging at a later gestational age. In our practice, we typically do this at around 16 weeks of gestation.
Ultrasonographic features of gastroschisis include loops of bowel herniating through a small paraumbilical wall defect (usually 2–3 cm) floating in amniotic fluid without a covering membrane4 (FIGURE 2). Direct exposure to amniotic fluid causes small bowel inflammation and fibrin deposition, leading to a thickened, echogenic appearance. Polyhydramnios and intra-abdominal bowel dilation have been associated with the presence of intestinal atresia.10
Management
There is no expert consensus regarding optimal prenatal management of gastroschisis.11-17 Prenatal care, patient counseling, and delivery planning should be individualized based on the defect and should be determined in a multidisciplinary discussion with specialists in maternal-fetal medicine, neonatology, and pediatric surgery, as necessary. In our practice, if the gastroschisis is isolated and uncomplicated, our generalist obstetricians manage the patient with maternal-fetal medicine consultation, increased fetal surveillance as described below, and delivery at our tertiary care institution.
Our standard practice is to use the initial ultrasonography imaging to evaluate the size and contents of the defect, measure the nuchal translucency, and evaluate for additional abnormalities. Serial ultrasonography monitoring of the fetus is required to assess the size and quality of the herniated intestine, amount of amniotic fluid, and fetal growth.10
As gastroschisis is a full-thickness defect of the anterior abdominal wall, the abdominal contents are exposed to amniotic fluid. This exposure causes progressive intestinal damage, which can be identified on ultrasonography as bowel thickening and dilation.12-14 Currently, intestinal thickening and dilation is not considered an indication for delivery as it is assumed that the intestinal damage has already occurred. It is debatable whether delivery around 37 weeks compared with delayed delivery beyond 37 weeks improves outcomes and decreases the stillbirth rate.11,13 Studies show that neonates delivered prior to 37 weeks have worse outcomes compared with those delivered after 37 weeks.14,15
Fetal surveillance. As standard practice, we evaluate the fetus at around 16 weeks and then again at around 20 weeks. In the absence of fetal growth restriction, which is associated with 25% of cases,16,17 our standard practice includes performing serial growth ultrasonography every 3 to 4 weeks starting at 28 weeks and biophysical profiles and nonstress testing weekly starting at 32 weeks. Fetal echocardiography can be offered. However, unlike with omphalocele, which has a high incidence of associated cardiac structural anomalies, gastroschisis has a low incidence of congenital cardiac anomalies, estimated to be between 2.5% and 4%.18,19
Delivery considerations. Little agreement exists regarding when and how to deliver pregnancies complicated by fetal gastroschisis. While some advocate for induction of labor at 36 to 38 weeks, most infants with gastroschisis can be delivered safely at term via either vaginal or cesarean delivery.14,15
Delivery timing should consider the clinical picture and incorporate performance on antenatal testing, fetal growth, the size and contents of the gastroschisis, and consultation with maternal-fetal medicine. Fetuses with gastroschisis often have non-reassuring antenatal testing. This can necessitate early delivery, although cesarean delivery should be reserved for obstetric indications, with the caveat that if there is large liver involvement, some pediatric surgeons recommend cesarean delivery due to the risk of hepatic rupture.
Neonate management. The survival rate of gastroschisis is reported to be as high as 91% to 94%.2 Morbidity is related to intestinal complications, such as strictures, adhesions, and volvulus.
In the case of simple gastroschisis, when the bowel is in good condition, the treatment method of choice is primary reduction.20 If performed in the operating room, an immediate sutured closure of the defect can be done. The benefits of primary repair include decreased length of stay, fewer intensive care bed days, and less time to achieve full feeds.20,21 Primary reduction has a reported success rate of 50% to 83%.22 A reduction with a delayed spontaneous closure also can be performed at bedside in the neonatal intensive care unit.22
For complex gastroschisis, characterized by bowel complications such as inflammation, perforation, ischemia, atresia, necrosis, or volvulus, primary closure may not be possible and reduction may need to be achieved through silo application.22-25 Additionally, further bowel surgery, such as stoma formation and bowel resection, may be required.25
Continue to: Omphalocele often is associated with abnormal karyotype...
Omphalocele often is associated with abnormal karyotype
Also known as exomphalos, omphalocele is a relatively common defect, with an estimated prevalence of 2 to 3 cases per 10,000 live births.2 In this condition, there is a midline defect in which intra-abdominal contents herniate through the base of the umbilical cord. Omphaloceles are covered by amniotic membranes, making them distinguishable from gastroschisis, which has no covering, and congenital umbilical hernias, which are covered by intact skin and subcutaneous tissue.26-33
Additionally, in omphalocele the umbilical cord insertion site varies, whereas in gastroschisis the umbilical cord insertion is usually to the right of midline. An omphalocele is often categorized based on whether or not it contains the liver (extracorporeal liver) or only the bowel (intracorporeal liver).
Genetic studies
Approximately 67% to 88% of all pregnancies with omphalocele have an abnormal karyotype and/or associated malformations, including Beckwith-Wiedemann syndrome.31 Of the aneuploidies, trisomy 18 is the one most commonly associated with omphalocele, accounting for approximately 62% to 75%, while trisomy 13 accounts for approximately 11% to 24%.32,33 The presence of other anomalies is strongly associated with poor prognosis, and increased defect size is an independent predictor of neonatal morbidity and mortality, as neonates with large omphaloceles with extracorporeal livers can develop respiratory insufficiency and require more complex surgical repairs. It is interesting, however, that the absence of an extracorporeal liver is associated with a higher risk of aneuploidy than are cases with an intracorporeal liver.33
We offer chorionic villus sampling or amniocentesis to all patients with omphalocele. If the patient undergoes invasive diagnostic testing, the sample then undergoes karyotyping, chromosomal microarray, and testing for Beckwith-Wiedemann syndrome. If the patient declines diagnostic sampling, we perform a cell-free DNA screening to rule out aneuploidy.
Continue to: Making the diagnosis...
Making the diagnosis
Omphaloceles can be diagnosed via prenatal ultrasonography as early as 11 to 14 weeks’ gestation.26 They are classified based on size, location, and contents of the sac.26,27 A small omphalocele is defined as a defect less than 5 cm with a sac that may contain a few loops of intestines (FIGURE 3).27 A giant omphalocele is a defect with more than 75% of the liver contained in the sac.29
Location can be epigastric, umbilical, or hypogastric, and both small and giant omphaloceles may have ruptured membranes that will result in exposure of the contained viscera.27 Omphaloceles are associated with such structural anomalies as cardiac, gastrointestinal, genitourinary, diaphragmatic, and neural tube defects. We do not routinely perform magnetic resonance imaging (MRI) for evaluation of omphaloceles, but MRI may be used to help predict postnatal outcomes in the case of giant omphaloceles.26
Management
Our standard practice is to use the initial ultrasonography imaging to evaluate the size and contents of defect, measure the nuchal translucency, and evaluate for additional abnormalities. As in cases of gastroschisis, serial ultrasonography monitoring of the fetus is required to assess the size and quality of the herniated intestine, amount of amniotic fluid, and fetal growth. We typically evaluate the fetus at around 16 weeks and then again at around 20 weeks. In the absence of fetal growth restriction, we recommend serial growth ultrasonography every 3 to 4 weeks starting at 28 weeks and biophysical profiles and nonstress testing weekly starting at 32 weeks. Additionally, we routinely obtain a fetal echocardiogram to rule out cardiac structural abnormalities.
Delivery considerations. Fetuses that do not undergo spontaneous abortion or medical termination of pregnancy often are born at term.26 We recommend expectant management until spontaneous labor, another indication for delivery arises, or at least 39 weeks’ estimated gestational age. There are no evidence-based guidelines for the optimal mode of delivery in fetuses with omphalocele, although we recommend cesarean delivery for fetuses with large defects to avoid postnatal sac rupture and liver damage. Preterm induction of labor is not indicated as infants born preterm have about a 50% mortality rate.26,27
Children born with isolated omphalocele typically have a good prognosis, with an estimated survival rate of 50% to 90%.32,33 However, compared to gastroschisis, omphaloceles are often associated with other anomalies.32,33
Management of omphaloceles depends on the size of the defect. In our institution, our generalist obstetricians manage the standard prenatal care with the addition of increased fetal surveillance and testing, interdisciplinary patient counseling with maternal-fetal medicine, pediatric surgeons, and neonatologists for delivery planning, and delivery is performed at our tertiary care center.
Neonate management. Small omphaloceles are amenable to primary early fascial closure.26-30 However, attempted primary closure of giant omphaloceles carries significant risks, including abdominal compartment syndrome and postoperative herniation.29,30 Instead, several options exist for staged surgical closure, in which there are multiple operations prior to final fascial closure, as well as nonoperative delayed closure for management of giant omphaloceles.29,30
Conservative management of giant omphaloceles has certain benefits, such as earlier first feeds, decreased risk of abdominal compartment syndrome, and lower risk of infection.30 Ruptured omphaloceles can be repaired through primary repair, employment of a synthetic or biologic mesh fascial bridge, or silo placement with delayed closure.28
Body-stalk anomaly: Multiple defects and poor prognosis
Also known as limb body wall complex, body-stalk anomaly is a rare malformation that has a reported prevalence of approximately 0.12 cases per 10,000 births (both live and stillbirths).34 Body-stalk anomaly is characterized by multiple defects, including severe kyphosis or scoliosis, a short or absent umbilical cord, and a large anterior abdominal wall defect.34-36 This malformation is almost entirely incompatible with life, resulting in abortion or stillbirth.35 Survival is extremely rare and limited to case reports.
While the exact etiology of body-stalk anomaly is unknown, 3 possible causes have been hypothesized: early amnion rupture, vascular compromise, and embryonic dysgenesis.37-40
Continue to: Making the diagnosis...
Making the diagnosis
Body-stalk anomaly typically can be diagnosed by 10 to 14 weeks’ gestation via ultrasonography.34-41 We currently follow the diagnostic criteria proposed by Van Allen and colleagues, which requires 2 of the following 3 anomalies34:
- exencephaly/encephalocele with facial clefts
- thoraco- and/or abdominoschisis (midline defect)
- limb defect.
Additional ultrasonographic findings can include the identification of evisceration of the abdominal contents, a short umbilical cord, and increased nuchal thickness.36,42 During the second and third trimesters, oligohydramnios may be seen.2
Management
Body-stalk anomaly is considered a fatal condition without specific therapeutic interventions. Maternal risks include an increased risk of preterm labor and gestational hypertension.35 Research on body-stalk anomaly has not shown any correlation with patients’ age, fetal sex, or abnormal karyotype, and the reported risk of recurrence for this anomaly is very low.42,43 Early diagnosis therefore is essential to provide families with information and counseling. Given the poor fetal prognosis, increased maternal risk, and low recurrence rates, mothers can be advised toward elective termination of pregnancy.
Should a patient desire expectant management, care can be provided by generalist obstetricians or care can be transferred to maternal-fetal medicine, with the addition of increased fetal surveillance and testing, interdisciplinary patient counseling with maternal-fetal medicine, pediatric surgeons, and neonatologists for delivery planning; delivery should be performed at a tertiary care center.
Pentalogy of Cantrell: Very rare, with variable prognosis
Pentalogy of Cantrell is characterized by a collection of defects in the midline abdominal wall, lower sternum, anterior diaphragm, diaphragmatic pericardium, and some manifestation of intra-cardiac defect.44 It is thought to arise early in gestation due to abnormal differentiation, migration, and fusion of the embryonic mesoderm.44 The condition is rare, with an incidence of about 1 in 5.5 million live births.45
Making the diagnosis
The diagnosis of pentalogy of Cantrell can be made via prenatal ultrasonography as early as the first trimester, although it is diagnosed more commonly in the second trimester.46 Three-dimensional ultrasonography and fetal MRI have been used to confirm the diagnosis.47
Management
Typically, corrective operations are performed during the neonatal period, and cases of successful staged and one-stage operations have been reported.48 Surgical treatment is determined based on the complexity of the condition and the presence of coexistent heart defects.49,50 However, very few patients survive surgical repair; mortality rates are estimated at around 50% to 60%, with high postsurgical morbidity risks for those who do survive.45
Prognosis varies depending on the type and severity of the associated malformations and intracardiac anomalies.46 Patients with partial ectopia cordis and incomplete presentation may have more favorable outcomes, but for patients with severe ectopia cordis, the survival rate is only 5% to 10%.47
Depending on the severity of the defects, mothers can be advised toward elective termination of pregnancy. In our institution, prenatal care usually is transferred to the maternal-fetal medicine service, and delivery is planned at our tertiary care institution.
OEIS complex comprises abdominal, pelvic, and spinal defects
Omphalocele-exstrophy-imperforate anus-spinal defects (OEIS) complex is a congenital malformation syndrome characterized by the combination of midline abdominal and pelvic defects (including omphalocele, exstrophy of the cloaca, and imperforate anus) and spinal defects.51 The condition’s etiology is unknown but is thought to be multifactorial.51-53 It is a rare condition, with an incidence of around 1 in 200,000 to 400,000 pregnancies.52
Making the diagnosis
Prenatal diagnosis of OEIS complex can be made as early as the first trimester via ultrasonographic identification of an infraumbilical abdominal wall defect with protruding mass, absent bladder, and spinal defects.52 When OEIS complex is suspected, fetal MRI can play a critical role in the diagnosis.
Management
As OEIS complex is rare, there are no evidence-based guidelines for optimal mode and timing of delivery. Cases are individualized based on their specific pathology, and we recommend cesarean delivery for fetuses with large defects to avoid postnatal sac rupture and liver damage.
The prognosis for infants with OEIS complex depends on the spectrum and severity of the structural defects.52,53 The many surgeries involved in the repair of OEIS have potential complications, such as urogenital and gastrointestinal dysfunction.52,53 Advances in medical and surgical treatment have resulted in improved survival and quality of life, and survival rates for OEIS complex are now close to 100%.53 While many OEIS patients live with a permanent colostomy, improvements in management mean that more patients are now candidates for gastrointestinal pull-through procedures, which allow for natural bowel control and a higher degree of bowel cleanliness.53
Prenatal care, patient counseling, and delivery planning should be individualized based on the defects present and determined in a multidisciplinary discussion with maternal-fetal medicine, neonatology, and pediatric surgery as necessary. In our institution, prenatal care usually is transferred to the maternal-fetal medicine service, and delivery is planned at our tertiary care institution.
Multidisciplinary team strategy is essential
Based on our experience, when faced with an anterior AWD in utero, prenatal imaging, genetic testing, increased fetal surveillance, and a multidisciplinary team approach improves outcomes. We must emphasize that careful patient counseling is paramount in our practice. ●
Acknowledgement: The authors would like to thank Ashley Tran, BS, for her assistance in the literature review and drafting of this article.
- Patients with fetuses with anterior wall defects should be referred to a maternal-fetal medicine specialist for co-management and advanced fetal imaging.
- The American College of Obstetricians and Gynecologists recommends microarray for all major fetal structural abnormalities, with the qualifier that karyotype can be offered if a specific aneuploidy is suspected based on the abnormality or prior genetic screening tests.
- If confirmatory testing is performed (amniocentesis or chorionic villus sampling), the sample should undergo karyotyping, chromosomal microarray, and if indicated, testing for Beckwith-Wiedemann syndrome. If the patient declines confirmatory sampling, performing cell-free DNA screening to rule out aneuploidy is recommended.
- Fetal echocardiography is recommended.
- Fetal magnetic resonance imaging should be considered in complex cases.
- Management should be individualized based on the type and severity of defect(s).
- Delivery timing and method should be individualized based on the defect(s) and determined in a multidisciplinary discussion with maternal-fetal medicine, neonatology, pediatric surgery, and pediatric cardiology, as necessary.
- The most common fetal abdominal wall defect is omphalocele, followed by gastroschisis.
- Maternal serum α-fetoprotein is usually elevated in all of the disorders.
- Victoria T, Andronikou S, Bowen D, et al. Fetal anterior abdominal wall defects: prenatal imaging by magnetic resonance imaging. Pediatr Radiol. 2018;48:499-512.
- Pakdaman R, Woodward PJ, Kennedy A. Complex abdominal wall defects: appearances at prenatal imaging. Radiographics. 2015;35:636-649.
- Oakes MC, Porto M, Chung JH. Advances in prenatal and perinatal diagnosis and management of gastroschisis. Semin Pediatr Surg. 2018;27:289-299.
- Mastroiacovo P, Lisi A, Castilla EE. The incidence of gastroschisis: research urgently needs resources. BMJ. 2006;332:423-424.
- Boyd PA, Haeusler M, Barisic I. EUROCAT report 9: surveillance of congenital anomalies in Europe 1980-2008. Birth Defects Res A Clin Mol Teratol. 2011;91(suppl 1):S1.
- Gamba P, Midrio P. Abdominal wall defects: prenatal diagnosis, newborn management, and long-term outcomes. Semin Pediatr Surg. 2014;23:283-290.
- Beaudoin S. Insights into the etiology and embryology of gastroschisis. Semin Pediatr Surg. 2018;27:283-288.
- Yazdy MM, Mitchell AA, Werler MM. Maternal genitourinary infections and the risk of gastroschisis. Am J Epidemiol. 2014;180:518-525.
- Werler MM, Sheehan JE, Mitchell AA. Maternal medication use and risks of gastroschisis and small intestinal atresia. Am J Epidemiol. 2002;155:26-31.
- D’Antonio F, Virgone C, Rizzo G, et al. Prenatal risk factors and outcomes in gastroschisis: a meta-analysis. Pediatrics. 2015;136:e159-e169.
- Baud D, Lausman A, Alfaraj MA, et al. Expectant management compared with elective delivery at 37 weeks for gastroschisis. Obstet Gynecol. 2013;121:990-998.
- Goetzinger KR, Tuuli MG, Longman RE, et al. Sonographic predictors of postnatal bowel atresia in fetal gastroschisis. Ultrasound Obstet Gynecol. 2014;43:420-425.
- Overton TG, Pierce MR, Gao H, et al. Antenatal management and outcomes of gastroschisis in the UK. Prenat Diagn. 2012;32:1256-1262.
- Ergün O, Barksdale E, Ergün FS, et al. The timing of delivery of infants with gastroschisis influences outcome. J Pediatr Surg. 2005;40:424-428.
- Overcash RT, DeUgarte DA, Stephenson ML, et al; University of California Fetal Consortium. Factors associated with gastroschisis outcomes. Obstet Gynecol. 2014;124:551-557.
- Wissanji H, Puligandla PS. Risk stratification and outcome determinants in gastroschisis. Semin Pediatr Surg. 2018;27: 300-303.
- Raynor BD, Richards D. Growth retardation in fetuses with gastroschisis. J Ultrasound Med. 1997;16:13-16.
- Mastroiacovo P, Lisi A, Castilla EE, et al. Gastroschisis and associated defects: an international study. Am J Med Genet A. 2007;143A:660-671.
- Kunz LH, Gilbert WM, Towner DR. Increased incidence of cardiac anomalies in pregnancies complicated by gastroschisis. Am J Obstet Gynecol. 2005;193(3 pt 2): 1248-1252.
- Lakshminarayanan B, Lakhoo K. Abdominal wall defects. Early Hum Dev. 2014;90:917-920.
- Prefumo F, Izzi C. Fetal abdominal wall defects. Best Pract Res Clin Obstet Gynaecol. 2014;28:391-402.
- Petrosyan M, Sandler AD. Closure methods in gastroschisis. Semin Pediatr Surg. 2018;27:304-308.
- Skarsgard ED. Management of gastroschisis. Curr Opin Pediatr. 2016;28:363-369.
- Bergholz R, Boettcher M, Reinshagen K, et al. Complex gastroschisis is a different entity to simple gastroschisis affecting morbidity and mortality—a systematic review and meta-analysis. J Pediatr Surg. 2014;49:1527-1532.
- Emil S. Surgical strategies in complex gastroschisis. Semin Pediatr Surg. 2018;27:309-315.
- Verla MA, Style CC, Olutoye OO. Prenatal diagnosis and management of omphalocele. Semin Pediatr Surg. 2019;28:84-88.
- Gonzalez KW, Chandler NM. Ruptured omphalocele: diagnosis and management. Semin Pediatr Surg. 2019;28:101-105.
- Sugandhi N, Saha M, Bhatnagar V, et al. Repair of ruptured omphalocele sac in the neonatal period and beyond. J Indian Assoc Pediatr Surg. 2020;25:46-48.
- Bauman B, Stephens D, Gershone H, et al. Management of giant omphaloceles: a systematic review of methods of staged surgical vs nonoperative delayed closure. J Pediatr Surg. 2016;51:1725-1730.
- Kogut KA, Fiore NF. Nonoperative management of giant omphalocele leading to early fascial closure. J Pediatr Surg. 2018;53:2404-2408.
- Conner P, Vejde JH, Burgos CM. Accuracy and impact of prenatal diagnosis in infants with omphalocele. Pediatr Surg Int. 2018;34:629-633.
- Iacovella C, Contro E, Ghi T, et al. The effect of the contents of exomphalos and nuchal translucency at 11-14 weeks on the likelihood of associated chromosomal abnormality. Prenat Diagn. 2012;32:1066-1070.
- Getachew MM, Goldstein RB, Edge V, et al. Correlation between omphalocele contents and karyotypic abnormalities: sonographic study in 37 cases. AJR Am J Roentgenol. 1992;158:133-136.
- Singh A, Singh J, Gupta K. Body stalk anomaly: antenatal sonographic diagnosis of this rare entity with review of literature. J Ultrason. 2017;17:133-135.
- Lazaroni TL, Cruzeiro PC, Piçarro C, et al. Body stalk anomaly: Three months of survival. Case report and literature review. J Pediatr Surg Case Rep. 2016;14:22-25.
- Gajzer DC, Hirzel AC, Saigal G, et al. Possible genetic origin of limb-body wall complex. Fetal Pediatr Pathol. 2015;34: 257–270.
- Maruyama H, Inagaki T, Nakata Y, et al. Minimally conjoined omphalopagus twins with a body stalk anomaly. AJP Rep. 2015;5:e124-e128.
- Bhat A, Ilyas M, Dev G. Prenatal sonographic diagnosis of limb-body wall complex: case series of a rare congenital anomaly. Radiol Case Rep. 2016;11:116-120.
- Quijano FE, Rey MM, Echeverry M, et al. Body stalk anomaly in a 9-week pregnancy. Case Rep Obstet Gynecol. 2014;2014:357285.
- Kocherla K, Kumari V, Kocherla PR. Prenatal diagnosis of body stalk complex: a rare entity and review of literature. Indian J Radiol Imaging. 2015;25:67-70.
- Panaitescu AM, Ushakov F, Kalaskar A, et al. Ultrasound features and management of body stalk anomaly. Fetal Diagn Ther. 2016;40:285-290.
- Routhu M, Thakkallapelli S, Mohan P, et al. Role of ultrasound in body stalk anomaly and amniotic band syndrome. Int J Reprod Med. 2016;2016:3974139.
- Costa ML, Couto E, Furlan E, et al. Body stalk anomaly: adverse maternal outcomes in a series of 21 cases. Prenat Diagn. 2012;32:264-267.
- Hubbard R, Hayes S, Gillis H, et al. Management challenges in an infant with pentalogy of Cantrell, giant anterior encephalocele, and craniofacial anomalies: a case report. A A Pract. 2018;11:238-240.
- Jnah AJ, Newberry DM, England A. Pentalogy of Cantrell: case report with review of the literature. Adv Neonatal Care. 2015;15:261-268.
- Williams AP, Marayati R, Beierle EA. Pentalogy of Cantrell. Semin Pediatr Surg. 2019;28:106-110.
- Restrepo MS, Cerqua A, Turek JW. Pentalogy of Cantrell with ectopia cordis totalis, total anomalous pulmonary venous connection, and tetralogy of Fallot: a case report and review of the literature. Congenit Heart Dis. 2014;9:E129–E134.
- Zhang X, Xing Q, Sun J, et al. Surgical treatment and outcomes of pentalogy of Cantrell in eight patients. J Pediatr Surg. 2014;49:1335-1340.
- Harring G, Weil J, Thiel C, et al. Management of pentalogy of Cantrell with complete ectopia cordis and double outlet right ventricle. Congenit Anom (Kyoto). 2015;55:121- 123.
- Mallula KK, Sosnowski C, Awad S. Spectrum of Cantrell’s pentalogy: case series from a single tertiary care center and review of the literature. Pediatr Cardiol. 2013;34:1703- 1710.
- Allam ES, Shetty VS, Farmakis SG. Fetal and neonatal presentation of OEIS complex. J Pediatr Surg. 2015;50:2155-2158.
- Neel N, Tarabay MS. Omphalocele, exstrophy of cloaca, imperforate anus, and spinal defect complex, multiple major reconstructive surgeries needed. Urol Ann. 2018;10:118-121.
- Sawaya D, Gearhart JP. Gastrointestinal reconstruction and outcomes for patients with the OEIS complex. Semin Pediatr Surg. 2011;20:123-125.
- Victoria T, Andronikou S, Bowen D, et al. Fetal anterior abdominal wall defects: prenatal imaging by magnetic resonance imaging. Pediatr Radiol. 2018;48:499-512.
- Pakdaman R, Woodward PJ, Kennedy A. Complex abdominal wall defects: appearances at prenatal imaging. Radiographics. 2015;35:636-649.
- Oakes MC, Porto M, Chung JH. Advances in prenatal and perinatal diagnosis and management of gastroschisis. Semin Pediatr Surg. 2018;27:289-299.
- Mastroiacovo P, Lisi A, Castilla EE. The incidence of gastroschisis: research urgently needs resources. BMJ. 2006;332:423-424.
- Boyd PA, Haeusler M, Barisic I. EUROCAT report 9: surveillance of congenital anomalies in Europe 1980-2008. Birth Defects Res A Clin Mol Teratol. 2011;91(suppl 1):S1.
- Gamba P, Midrio P. Abdominal wall defects: prenatal diagnosis, newborn management, and long-term outcomes. Semin Pediatr Surg. 2014;23:283-290.
- Beaudoin S. Insights into the etiology and embryology of gastroschisis. Semin Pediatr Surg. 2018;27:283-288.
- Yazdy MM, Mitchell AA, Werler MM. Maternal genitourinary infections and the risk of gastroschisis. Am J Epidemiol. 2014;180:518-525.
- Werler MM, Sheehan JE, Mitchell AA. Maternal medication use and risks of gastroschisis and small intestinal atresia. Am J Epidemiol. 2002;155:26-31.
- D’Antonio F, Virgone C, Rizzo G, et al. Prenatal risk factors and outcomes in gastroschisis: a meta-analysis. Pediatrics. 2015;136:e159-e169.
- Baud D, Lausman A, Alfaraj MA, et al. Expectant management compared with elective delivery at 37 weeks for gastroschisis. Obstet Gynecol. 2013;121:990-998.
- Goetzinger KR, Tuuli MG, Longman RE, et al. Sonographic predictors of postnatal bowel atresia in fetal gastroschisis. Ultrasound Obstet Gynecol. 2014;43:420-425.
- Overton TG, Pierce MR, Gao H, et al. Antenatal management and outcomes of gastroschisis in the UK. Prenat Diagn. 2012;32:1256-1262.
- Ergün O, Barksdale E, Ergün FS, et al. The timing of delivery of infants with gastroschisis influences outcome. J Pediatr Surg. 2005;40:424-428.
- Overcash RT, DeUgarte DA, Stephenson ML, et al; University of California Fetal Consortium. Factors associated with gastroschisis outcomes. Obstet Gynecol. 2014;124:551-557.
- Wissanji H, Puligandla PS. Risk stratification and outcome determinants in gastroschisis. Semin Pediatr Surg. 2018;27: 300-303.
- Raynor BD, Richards D. Growth retardation in fetuses with gastroschisis. J Ultrasound Med. 1997;16:13-16.
- Mastroiacovo P, Lisi A, Castilla EE, et al. Gastroschisis and associated defects: an international study. Am J Med Genet A. 2007;143A:660-671.
- Kunz LH, Gilbert WM, Towner DR. Increased incidence of cardiac anomalies in pregnancies complicated by gastroschisis. Am J Obstet Gynecol. 2005;193(3 pt 2): 1248-1252.
- Lakshminarayanan B, Lakhoo K. Abdominal wall defects. Early Hum Dev. 2014;90:917-920.
- Prefumo F, Izzi C. Fetal abdominal wall defects. Best Pract Res Clin Obstet Gynaecol. 2014;28:391-402.
- Petrosyan M, Sandler AD. Closure methods in gastroschisis. Semin Pediatr Surg. 2018;27:304-308.
- Skarsgard ED. Management of gastroschisis. Curr Opin Pediatr. 2016;28:363-369.
- Bergholz R, Boettcher M, Reinshagen K, et al. Complex gastroschisis is a different entity to simple gastroschisis affecting morbidity and mortality—a systematic review and meta-analysis. J Pediatr Surg. 2014;49:1527-1532.
- Emil S. Surgical strategies in complex gastroschisis. Semin Pediatr Surg. 2018;27:309-315.
- Verla MA, Style CC, Olutoye OO. Prenatal diagnosis and management of omphalocele. Semin Pediatr Surg. 2019;28:84-88.
- Gonzalez KW, Chandler NM. Ruptured omphalocele: diagnosis and management. Semin Pediatr Surg. 2019;28:101-105.
- Sugandhi N, Saha M, Bhatnagar V, et al. Repair of ruptured omphalocele sac in the neonatal period and beyond. J Indian Assoc Pediatr Surg. 2020;25:46-48.
- Bauman B, Stephens D, Gershone H, et al. Management of giant omphaloceles: a systematic review of methods of staged surgical vs nonoperative delayed closure. J Pediatr Surg. 2016;51:1725-1730.
- Kogut KA, Fiore NF. Nonoperative management of giant omphalocele leading to early fascial closure. J Pediatr Surg. 2018;53:2404-2408.
- Conner P, Vejde JH, Burgos CM. Accuracy and impact of prenatal diagnosis in infants with omphalocele. Pediatr Surg Int. 2018;34:629-633.
- Iacovella C, Contro E, Ghi T, et al. The effect of the contents of exomphalos and nuchal translucency at 11-14 weeks on the likelihood of associated chromosomal abnormality. Prenat Diagn. 2012;32:1066-1070.
- Getachew MM, Goldstein RB, Edge V, et al. Correlation between omphalocele contents and karyotypic abnormalities: sonographic study in 37 cases. AJR Am J Roentgenol. 1992;158:133-136.
- Singh A, Singh J, Gupta K. Body stalk anomaly: antenatal sonographic diagnosis of this rare entity with review of literature. J Ultrason. 2017;17:133-135.
- Lazaroni TL, Cruzeiro PC, Piçarro C, et al. Body stalk anomaly: Three months of survival. Case report and literature review. J Pediatr Surg Case Rep. 2016;14:22-25.
- Gajzer DC, Hirzel AC, Saigal G, et al. Possible genetic origin of limb-body wall complex. Fetal Pediatr Pathol. 2015;34: 257–270.
- Maruyama H, Inagaki T, Nakata Y, et al. Minimally conjoined omphalopagus twins with a body stalk anomaly. AJP Rep. 2015;5:e124-e128.
- Bhat A, Ilyas M, Dev G. Prenatal sonographic diagnosis of limb-body wall complex: case series of a rare congenital anomaly. Radiol Case Rep. 2016;11:116-120.
- Quijano FE, Rey MM, Echeverry M, et al. Body stalk anomaly in a 9-week pregnancy. Case Rep Obstet Gynecol. 2014;2014:357285.
- Kocherla K, Kumari V, Kocherla PR. Prenatal diagnosis of body stalk complex: a rare entity and review of literature. Indian J Radiol Imaging. 2015;25:67-70.
- Panaitescu AM, Ushakov F, Kalaskar A, et al. Ultrasound features and management of body stalk anomaly. Fetal Diagn Ther. 2016;40:285-290.
- Routhu M, Thakkallapelli S, Mohan P, et al. Role of ultrasound in body stalk anomaly and amniotic band syndrome. Int J Reprod Med. 2016;2016:3974139.
- Costa ML, Couto E, Furlan E, et al. Body stalk anomaly: adverse maternal outcomes in a series of 21 cases. Prenat Diagn. 2012;32:264-267.
- Hubbard R, Hayes S, Gillis H, et al. Management challenges in an infant with pentalogy of Cantrell, giant anterior encephalocele, and craniofacial anomalies: a case report. A A Pract. 2018;11:238-240.
- Jnah AJ, Newberry DM, England A. Pentalogy of Cantrell: case report with review of the literature. Adv Neonatal Care. 2015;15:261-268.
- Williams AP, Marayati R, Beierle EA. Pentalogy of Cantrell. Semin Pediatr Surg. 2019;28:106-110.
- Restrepo MS, Cerqua A, Turek JW. Pentalogy of Cantrell with ectopia cordis totalis, total anomalous pulmonary venous connection, and tetralogy of Fallot: a case report and review of the literature. Congenit Heart Dis. 2014;9:E129–E134.
- Zhang X, Xing Q, Sun J, et al. Surgical treatment and outcomes of pentalogy of Cantrell in eight patients. J Pediatr Surg. 2014;49:1335-1340.
- Harring G, Weil J, Thiel C, et al. Management of pentalogy of Cantrell with complete ectopia cordis and double outlet right ventricle. Congenit Anom (Kyoto). 2015;55:121- 123.
- Mallula KK, Sosnowski C, Awad S. Spectrum of Cantrell’s pentalogy: case series from a single tertiary care center and review of the literature. Pediatr Cardiol. 2013;34:1703- 1710.
- Allam ES, Shetty VS, Farmakis SG. Fetal and neonatal presentation of OEIS complex. J Pediatr Surg. 2015;50:2155-2158.
- Neel N, Tarabay MS. Omphalocele, exstrophy of cloaca, imperforate anus, and spinal defect complex, multiple major reconstructive surgeries needed. Urol Ann. 2018;10:118-121.
- Sawaya D, Gearhart JP. Gastrointestinal reconstruction and outcomes for patients with the OEIS complex. Semin Pediatr Surg. 2011;20:123-125.