User login
In reply: Wilson disease
In Reply: We thank Dr. Mirrakhimov and colleagues for bringing important questions to our attention.
In terms of the differential diagnosis of cholestatic liver injury, we agree that pathologic processes such choledocholithiasis, cholangitis, primary biliary cirrhosis, and primary sclerosing cholangitis should be generally considered. However, in the case we described, the patient had no abdominal pain or fever, which makes choledocholithiasis or cholangitis very unlikely. Primary biliary cirrhosis and primary sclerosing cholangitis can cause chronic liver disease but should not be considered in the differential diagnosis of acute liver injury (acute hepatitis), such as in the case we described.
We agree that the hemolytic anemia typically seen in patients with Wilson disease is Coombs-negative, and that Coombs testing and a peripheral smear should be performed. Both were negative in our patient.
We also agree with Dr. Mirrakhimov and colleagues that Kayser-Fleischer rings are not necessarily specific for Wilson disease and can be seen in patients with other forms of cholestatic liver disease such as primary biliary cirrhosis. However, Kayser-Fleischer rings are pathognomonic for acute liver failure from Wilson disease. In other words, when Kayser-Fleischer rings are seen in a patient with acute liver failure, the diagnosis is Wilson disease until proven otherwise.
We discussed on page 112 of our article other treatments such as plasmapheresis as adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant. However, liver transplant is still the only definitive and potentially curative treatment.
In Reply: We thank Dr. Mirrakhimov and colleagues for bringing important questions to our attention.
In terms of the differential diagnosis of cholestatic liver injury, we agree that pathologic processes such choledocholithiasis, cholangitis, primary biliary cirrhosis, and primary sclerosing cholangitis should be generally considered. However, in the case we described, the patient had no abdominal pain or fever, which makes choledocholithiasis or cholangitis very unlikely. Primary biliary cirrhosis and primary sclerosing cholangitis can cause chronic liver disease but should not be considered in the differential diagnosis of acute liver injury (acute hepatitis), such as in the case we described.
We agree that the hemolytic anemia typically seen in patients with Wilson disease is Coombs-negative, and that Coombs testing and a peripheral smear should be performed. Both were negative in our patient.
We also agree with Dr. Mirrakhimov and colleagues that Kayser-Fleischer rings are not necessarily specific for Wilson disease and can be seen in patients with other forms of cholestatic liver disease such as primary biliary cirrhosis. However, Kayser-Fleischer rings are pathognomonic for acute liver failure from Wilson disease. In other words, when Kayser-Fleischer rings are seen in a patient with acute liver failure, the diagnosis is Wilson disease until proven otherwise.
We discussed on page 112 of our article other treatments such as plasmapheresis as adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant. However, liver transplant is still the only definitive and potentially curative treatment.
In Reply: We thank Dr. Mirrakhimov and colleagues for bringing important questions to our attention.
In terms of the differential diagnosis of cholestatic liver injury, we agree that pathologic processes such choledocholithiasis, cholangitis, primary biliary cirrhosis, and primary sclerosing cholangitis should be generally considered. However, in the case we described, the patient had no abdominal pain or fever, which makes choledocholithiasis or cholangitis very unlikely. Primary biliary cirrhosis and primary sclerosing cholangitis can cause chronic liver disease but should not be considered in the differential diagnosis of acute liver injury (acute hepatitis), such as in the case we described.
We agree that the hemolytic anemia typically seen in patients with Wilson disease is Coombs-negative, and that Coombs testing and a peripheral smear should be performed. Both were negative in our patient.
We also agree with Dr. Mirrakhimov and colleagues that Kayser-Fleischer rings are not necessarily specific for Wilson disease and can be seen in patients with other forms of cholestatic liver disease such as primary biliary cirrhosis. However, Kayser-Fleischer rings are pathognomonic for acute liver failure from Wilson disease. In other words, when Kayser-Fleischer rings are seen in a patient with acute liver failure, the diagnosis is Wilson disease until proven otherwise.
We discussed on page 112 of our article other treatments such as plasmapheresis as adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant. However, liver transplant is still the only definitive and potentially curative treatment.
A tale of two sisters with liver disease
A 25-year-old woman presents to the emergency department with a 7-day history of fatigue and nausea. On presentation she denies having abdominal pain, headache, fever, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss. She recalls changes in the colors of her eyes and darkening urine over the last few days. Her medical history before this is unremarkable. She takes no prescription, over-the-counter, or herbal medications. She works as a librarian and has no occupational toxic exposures. She is single and has one sister with no prior medical history. She denies recent travel, sick contacts, smoking, recreational drug use, or pets at home.
On physical examination, her vital signs are temperature 37.3°C (99.1°F), heart rate 90 beats per minute, blood pressure 125/80 mm Hg, respiration rate 14 per minute, and oxygen saturation 97% on room air. She has icteric sclera and her skin is jaundiced. Cardiac examination is normal. Lungs are clear to auscultation and percussion bilaterally. Her abdomen is soft with no visceromegaly, masses, or tenderness. Extremities are normal with no edema. She is alert and oriented, but she has mild asterixis of the outstretched hands. The neurologic examination is otherwise unremarkable.
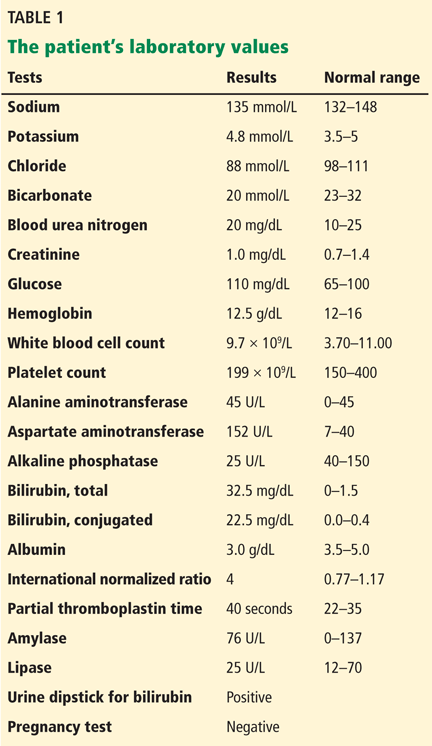
The patient’s basic laboratory values are listed in Table 1. Shortly after admission, she develops changes in her mental status, remaining alert but becoming agitated and oriented to person only. In view of her symptoms and laboratory findings, acute liver failure is suspected.
ACUTE LIVER FAILURE
1. The diagnostic criteria for acute liver failure include all of the following except which one?
- Acute elevation of liver biochemical tests
- Presence of preexisting liver disease
- Coagulopathy, defined by an international normalized ratio (INR) of 1.5 or greater
- Encephalopathy
- Duration of symptoms less than 26 weeks
Acute liver failure is defined by acute onset of worsening liver tests, coagulopathy (INR ≥ 1.5), and encephalopathy in patients with no preexisting liver disease and with symptom duration of less than 26 weeks.1 With a few exceptions, a history of preexisting liver disease negates the diagnosis of acute liver failure. Our patient meets the diagnostic criteria for acute liver failure.
Immediate management
Once acute liver failure is identified or suspected, the next step is to transfer the patient to the intensive care unit for close monitoring of mental status. Serial neurologic evaluations permit early detection of cerebral edema, which is considered the most common cause of death in patients with acute liver failure. Additionally, close monitoring of electrolytes and plasma glucose is necessary since these patients are susceptible to electrolyte disturbances and hypoglycemia.
Patients with acute liver failure are at increased risk of infections and should be routinely screened by obtaining urine and blood cultures.
Gastrointestinal bleeding is not uncommon in patients with acute liver failure and is usually due to gastric stress ulceration. Prophylaxis with a histamine 2 receptor antagonist or proton pump inhibitor should be considered in order to prevent gastrointestinal bleeding.
Treatment with N-acetylcysteine is beneficial, not only in patients with acute liver failure due to acetaminophen overdose, but also in those with acute liver failure from other causes.
CASE CONTINUES:
TRANSFER TO THE INTENSIVE CARE UNIT
The patient, now diagnosed with acute liver failure, is transferred to the intensive care unit. Arterial blood gas measurement shows:
- pH 7.38 (reference range 7.35–7.45)
- Pco2 40 mm Hg (36–46)
- Po2 97 mm Hg (85–95)
- Hco3 22 mmol/L (22–26).
A chest radiograph is obtained and is clear. Computed tomography (CT) of the brain reveals no edema. Transcranial Doppler ultrasonography does not show any intracranial fluid collections.
Blood and urine cultures are negative. Her hemoglobin level remains stable, and she does not develop signs of bleeding. She is started on a proton pump inhibitor for stress ulcer prophylaxis and is empirically given intravenous N-acetylcysteine until the cause of acute liver failure can be determined.
CAUSES OF ACUTE LIVER FAILURE
2. Which of the following can cause acute liver failure?
- Acetaminophen overdose
- Viral hepatitis
- Autoimmune hepatitis
- Wilson disease
- Alcoholic hepatitis
Drug-induced liver injury is the most common cause of acute liver failure in the United States,2,3 and of all drugs, acetaminophen overdose is the number-one cause. In acetaminophen-induced liver injury, serum aminotransferase levels are usually elevated to more than 1,000 U/L, while serum bilirubin remains normal in the early stages. Antimicrobial agents, antiepileptic drugs, and herbal supplements have also been implicated in acute liver failure. Our patient has denied taking herbal supplements or medications, including over-the-counter ones.
Acute viral hepatitis can explain the patient’s condition. It is a common cause of acute liver failure in the United States.2 Hepatitis A and E are more common in developing countries. Other viruses such as cytomegalovirus, Epstein-Barr virus, herpes simplex virus type 1 and 2, and varicella zoster virus can also cause acute liver failure. Serum aminotransferase levels may exceed 1,000 U/L in patients with viral hepatitis.
Autoimmune hepatitis is a rare cause of acute liver failure, but it should be considered in the differential diagnosis, particularly in middle-aged women with autoimmune disorders such as hypothyroidism. Autoimmune hepatitis can cause marked elevation in aminotransferase levels (> 1,000 U/L).
Wilson disease is an autosomal-recessive disease in which there is excessive accumulation of copper in the liver and other organs because of an inherited defect in the biliary excretion of copper. Wilson disease can cause acute liver failure and should be excluded in any patient, particularly if under age 40 with acute onset of unexplained hepatic, neurologic, or psychiatric disease.
Alcoholic hepatitis usually occurs in patients with a long-standing history of heavy alcohol use. As a result, most patients with alcoholic hepatitis have manifestations of chronic liver disease due to alcohol use. Therefore, by definition, it is not a cause of acute liver failure. Additionally, in patients with alcoholic hepatitis, the aspartate aminotransferase (AST) level is elevated but less than 300 IU/mL, and the ratio of AST to alanine aminotransferase (ALT) is usually more than 2.
CASE CONTINUES: FURTHER TESTING
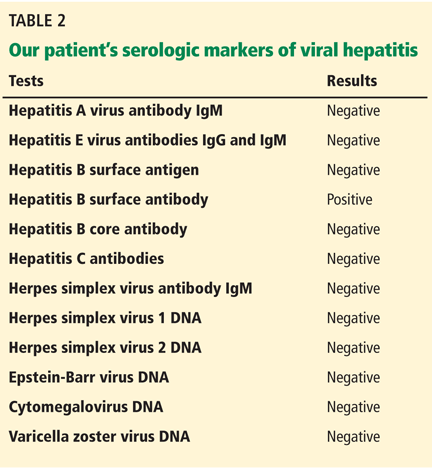
The results of our patient’s serologic tests are shown in Table 2. Other test results:
- Autoimmune markers including antinuclear antibodies, antimitochondrial antibodies, antismooth muscle antibodies, and liver and kidney microsomal antibodies are negative; her immunoglobulin G (IgG) level is normal
- Serum ceruloplasmin 25 mg/dL (normal 21–45)
- Free serum copper 120 µg/dL (normal 8–12)
- Abdominal ultrasonography is unremarkable, with normal liver parenchyma and no intrahepatic or extrahepatic biliary dilatation
- Doppler ultrasonography of the liver shows patent blood vessels.
3. Based on the new data, which of the following statements is correct?
- Hepatitis B is the cause of acute liver failure in this patient
- Herpetic hepatitis cannot be excluded on the basis of the available data
- Wilson disease is most likely the diagnosis, given her elevated free serum copper
- A normal serum ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease
Hepatitis B surface antigen and hepatitis B core antibodies were negative in our patient, excluding hepatitis B virus infection. The positive hepatitis B surface antibody indicates prior immunization.
Herpetic hepatitis is an uncommon but important cause of acute liver failure because the mortality rate is high if the patient is not treated early with acyclovir. Fever, elevated aminotransferases, and leukopenia are common with herpetic hepatitis. Fewer than 50% of patients with herpetic hepatitis have vesicular rash.4,5 The value of antibody serologic testing is limited due to high rates of false-positive and false-negative results. The gold standard diagnostic tests are viral load (detection of viral RNA by polymerase chain reaction), viral staining on liver biopsy, or both. In our patient, herpes simplex virus polymerase chain reaction testing was negative, which makes herpetic hepatitis unlikely.
Wilson disease is a genetic condition in which the ability to excrete copper in the bile is impaired, resulting in accumulation of copper in the hepatocytes. Subsequently, copper is released into the bloodstream and eventually into the urine.
However, copper excretion into the bile is impaired in patients with acute liver failure regardless of the etiology. Therefore, elevated free serum copper and 24-hour urine copper levels are not specific for the diagnosis of acute liver failure secondary to Wilson disease. Moreover, Kayser-Fleischer rings, which represent copper deposition in the limbus of the cornea, may not be apparent in the early stages of Wilson disease.
Since it is challenging to diagnose Wilson disease in the context of acute liver failure, Korman et al6 compared patients with acute liver failure secondary to Wilson disease with patients with acute liver failure secondary to other conditions. They found that alkaline phosphatase levels are frequently decreased in patients with acute liver failure secondary to Wilson disease,6 and that a ratio of alkaline phosphatase to total bilirubin of less than 4 is 94% sensitive and 96% specific for the diagnosis.6
Hemolysis is common in acute liver failure due to Wilson disease. This leads to disproportionate elevation of AST compared with ALT, since AST is present in red blood cells. Consequently, the ratio of AST to ALT is usually greater than 2.2, which provides a sensitivity of 94% and a specificity of 86% for the diagnosis.6 These two ratios together provide 100% sensitivity and 100% specificity for the diagnosis of Wilson disease in the context of acute liver failure.6
Ceruloplasmin. Patients with Wilson disease typically have a low ceruloplasmin level. However, because it is an acute-phase reaction protein, ceruloplasmin can be normal or elevated in patients with acute liver failure from Wilson disease.6 Therefore, a normal ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease.
CASE CONTINUES: A DEFINITIVE DIAGNOSIS
Our patient undergoes further testing, which reveals the following:
- Her 24-hour urinary excretion of copper is 150 µg (reference value < 30)
- Slit-lamp examination is normal and shows no evidence of Kayser-Fleischer rings
- Her ratio of alkaline phosphatase to total bilirubin is 0.77 based on her initial laboratory results (Table 1)
- Her AST-ALT ratio is 3.4.
The diagnosis in our patient is acute liver failure secondary to Wilson disease.
4. What is the most appropriate next step?
- Liver biopsy
- d-penicillamine by mouth
- Trientine by mouth
- Liver transplant
- Plasmapheresis
Liver biopsy. Accumulation of copper in the liver parenchyma in patients with Wilson disease is sporadic. Therefore, qualitative copper staining on liver biopsy can be falsely negative. Quantitative copper measurement in liver tissue is the gold standard for the diagnosis of Wilson disease. However, the test is time-consuming and is not rapidly available in the context of acute liver failure.
Chelating agents such as d-pencillamine and trientine are used to treat the chronic manifestations of Wilson disease but are not useful for acute liver failure secondary to Wilson disease.
Acute liver failure secondary to Wilson disease is life-threatening, and liver transplant is considered the only definitive life-saving therapy.
Therapeutic plasmapheresis has been reported to be a successful adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant.7 However, liver transplant is still the only definitive treatment.
CASE CONTINUES: THE PATIENT’S SISTER SEEKS CARE
The patient undergoes liver transplantation, with no perioperative or postoperative complications.
The patient’s 18-year-old sister is now seeking medical attention in the outpatient clinic, concerned that she may have Wilson disease. She is otherwise healthy and denies any symptoms or complaints.
5. What is the next step for the patient’s sister?
- Reassurance
- Prophylaxis with trientine
- Check liver enzyme levels, serum ceruloplasmin level, and urine copper, and order a slit-lamp examination
- Genetic testing
Wilson disease can be asymptomatic in its early stages and may be diagnosed incidentally during routine blood tests that reveal abnormal liver enzyme levels. All patients with a confirmed family history of Wilson disease should be screened even if they are asymptomatic. The diagnosis of Wilson disease should be established in first-degree relatives before specific treatment for the relatives is prescribed.
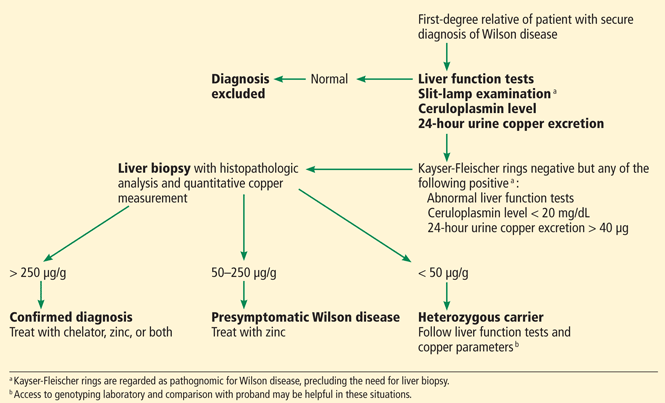
The first step in screening a first-degree relative for Wilson disease is to check liver enzyme levels (specifically aminotransferases, alkaline phosphatase, and bilirubin), serum ceruloplasmin level, and 24-hour urine copper, and order an ophthalmologic slit-lamp examination. If any of these tests is abnormal, liver biopsy should be performed for histopathologic evaluation and quantitative copper measurement. Kayser-Fleischer rings are seen in only 50% of patients with Wilson disease and hepatic involvement, but they are pathognomic. Guidelines8 for screening first-degree relatives of Wilson disease patients are shown in Figure 1.
Genetic analysis. ATP7B, the Wilson disease gene, is located on chromosome 13. At least 300 mutations of the gene have been described,2 and the most common mutation is present in only 15% to 30% of the Wilson disease population.8–10 Routine molecular testing of the ATP7B
CASE CONTINUES: WORKUP OF THE PATIENT’S SISTER
The patient’s sister has no symptoms and her physical examination is normal. Slit-lamp examination reveals no evidence of Kayser-Fleischer rings. Her laboratory values, including complete blood counts, complete metabolic panel, and INR, are within normal ranges. Other test results, however, are abnormal:
- Free serum copper level 27 µg/dL (normal 8–12)
- Serum ceruloplasmin 9.0 mg/dL (normal 20–50)
- 24-hour urinary copper excretion 135 µg (normal < 30).
She undergoes liver biopsy for quantitative copper measurement, and the result is very high at 1,118 µg/g dry weight (reference range 10–35). The diagnosis of Wilson disease is established.
TREATING CHRONIC WILSON DISEASE
6. Which of the following is not an appropriate next step for the patient’s sister?
- Tetrathiomolybdate
- d-penicillamine
- Trientine
- Zinc salts
- Prednisone
The goal of medical treatment of chronic Wilson disease is to improve symptoms and prevent progression of the disease.
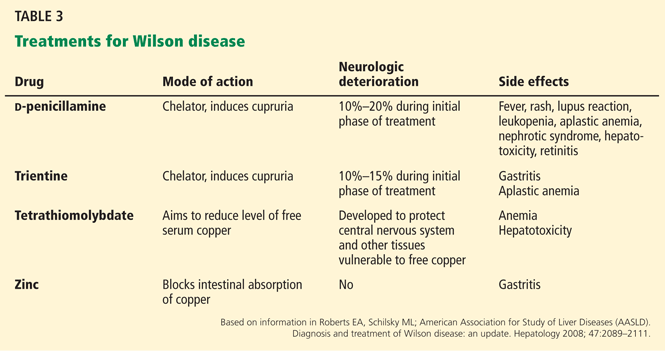
Chelating agents and zinc salts are the most commonly used medicines in the management of Wilson disease. Chelating agents remove copper from tissue, whereas zinc blocks the intestinal absorption of copper and stimulates the synthesis of endogenous chelators such as metallothioneins. Tetrathiomolybdate is an alternative agent developed to interfere with the distribution of excess body copper to susceptible target sites by reducing free serum copper (Table 3). There are no data to support the use of prednisone in the treatment of Wilson disease.
During treatment with chelating agents, 24-hour urinary excretion of copper is routinely monitored to determine the efficacy of therapy and adherence to treatment. Once de-coppering is achieved, as evidenced by a normalization of 24-hour urine copper excretion, the chelating agent can be switched to zinc salts to prevent intestinal absorption of copper.
Clinical and biochemical stabilization is achieved typically within 2 to 6 months of the initial treatment with chelating agents.8 Organ meats, nuts, shellfish, and chocolate are rich in copper and should be avoided.
The patient’s sister is started on trientine 250 mg orally three times daily on an empty stomach at least 1 hour before meals. Treatment is monitored by following 24-hour urine copper measurement. A 24-hour urine copper measurement at 3 months after starting treatment has increased from 54 at baseline to 350 µg, which indicates that the copper is being removed from tissues. The plan is for early substitution of zinc for long-term maintenance once de-coppering is completed.
KEY POINTS
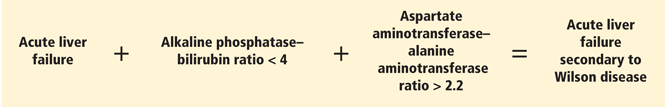
- Acute liver failure is severe acute liver injury characterized by coagulopathy (INR ≥ 1.5) and encephalopathy in a patient with no preexisting liver disease and with duration of symptoms less than 26 weeks.
- Acute liver failure secondary to Wilson disease is uncommon but should be excluded, particularly in young patients.
- The diagnosis of Wilson disease in the setting of acute liver failure is challenging because the serum ceruloplasmin level may be normal in acute liver failure secondary to Wilson disease, and free serum copper and 24-hour urine copper are usually elevated in all acute liver failure patients regardless of the etiology.
- A ratio of alkaline phosphatase to total bilirubin of less than 4 plus an AST-ALT ratio greater than 2.2 in a patient with acute liver failure should be regarded as Wilson disease until proven otherwise (Figure 2).
- Acute liver failure secondary to Wilson disease is usually fatal, and emergency liver transplant is a life-saving procedure.
- Screening of first-degree relatives of Wilson disease patients should include a history and physical examination, liver enzyme tests, complete blood cell count, serum ceruloplasmin level, serum free copper level, slit-lamp examination of the eyes, and 24-hour urinary copper measurement. Genetic tests are supplementary for screening but are not routinely available.
- Lee WM, Larson AM, Stravitz T. AASLD Position Paper: The management of acute liver failure: update 2011. www.aasld.org/sites/default/files/guideline_documents/alfenhanced.pdf. Accessed December 9, 2015.
- Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet 2010; 376:190–201.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Hanouneh IA, Khoriaty R, Zein NN. A 35-year-old Asian man with jaundice and markedly high aminotransferase levels. Cleve Clin J Med 2009; 76:449–456.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transpl 2007; 13:1428–1434.
- Korman JD, Volenberg I, Balko J, et al; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology 2008; 48:1167–1174.
- Morgan SM, Zantek ND. Therapeutic plasma exchange for fulminant hepatic failure secondary to Wilson's disease. J Clin Apher 2012; 27:282–286.
- Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47:2089–2111.
- Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet 1997; 61:317–328.
- Maier-Dobersberger T, Ferenci P, Polli C, et al. Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Ann Intern Med 1997; 127:21–26.
A 25-year-old woman presents to the emergency department with a 7-day history of fatigue and nausea. On presentation she denies having abdominal pain, headache, fever, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss. She recalls changes in the colors of her eyes and darkening urine over the last few days. Her medical history before this is unremarkable. She takes no prescription, over-the-counter, or herbal medications. She works as a librarian and has no occupational toxic exposures. She is single and has one sister with no prior medical history. She denies recent travel, sick contacts, smoking, recreational drug use, or pets at home.
On physical examination, her vital signs are temperature 37.3°C (99.1°F), heart rate 90 beats per minute, blood pressure 125/80 mm Hg, respiration rate 14 per minute, and oxygen saturation 97% on room air. She has icteric sclera and her skin is jaundiced. Cardiac examination is normal. Lungs are clear to auscultation and percussion bilaterally. Her abdomen is soft with no visceromegaly, masses, or tenderness. Extremities are normal with no edema. She is alert and oriented, but she has mild asterixis of the outstretched hands. The neurologic examination is otherwise unremarkable.
The patient’s basic laboratory values are listed in Table 1. Shortly after admission, she develops changes in her mental status, remaining alert but becoming agitated and oriented to person only. In view of her symptoms and laboratory findings, acute liver failure is suspected.
ACUTE LIVER FAILURE
1. The diagnostic criteria for acute liver failure include all of the following except which one?
- Acute elevation of liver biochemical tests
- Presence of preexisting liver disease
- Coagulopathy, defined by an international normalized ratio (INR) of 1.5 or greater
- Encephalopathy
- Duration of symptoms less than 26 weeks
Acute liver failure is defined by acute onset of worsening liver tests, coagulopathy (INR ≥ 1.5), and encephalopathy in patients with no preexisting liver disease and with symptom duration of less than 26 weeks.1 With a few exceptions, a history of preexisting liver disease negates the diagnosis of acute liver failure. Our patient meets the diagnostic criteria for acute liver failure.
Immediate management
Once acute liver failure is identified or suspected, the next step is to transfer the patient to the intensive care unit for close monitoring of mental status. Serial neurologic evaluations permit early detection of cerebral edema, which is considered the most common cause of death in patients with acute liver failure. Additionally, close monitoring of electrolytes and plasma glucose is necessary since these patients are susceptible to electrolyte disturbances and hypoglycemia.
Patients with acute liver failure are at increased risk of infections and should be routinely screened by obtaining urine and blood cultures.
Gastrointestinal bleeding is not uncommon in patients with acute liver failure and is usually due to gastric stress ulceration. Prophylaxis with a histamine 2 receptor antagonist or proton pump inhibitor should be considered in order to prevent gastrointestinal bleeding.
Treatment with N-acetylcysteine is beneficial, not only in patients with acute liver failure due to acetaminophen overdose, but also in those with acute liver failure from other causes.
CASE CONTINUES:
TRANSFER TO THE INTENSIVE CARE UNIT
The patient, now diagnosed with acute liver failure, is transferred to the intensive care unit. Arterial blood gas measurement shows:
- pH 7.38 (reference range 7.35–7.45)
- Pco2 40 mm Hg (36–46)
- Po2 97 mm Hg (85–95)
- Hco3 22 mmol/L (22–26).
A chest radiograph is obtained and is clear. Computed tomography (CT) of the brain reveals no edema. Transcranial Doppler ultrasonography does not show any intracranial fluid collections.
Blood and urine cultures are negative. Her hemoglobin level remains stable, and she does not develop signs of bleeding. She is started on a proton pump inhibitor for stress ulcer prophylaxis and is empirically given intravenous N-acetylcysteine until the cause of acute liver failure can be determined.
CAUSES OF ACUTE LIVER FAILURE
2. Which of the following can cause acute liver failure?
- Acetaminophen overdose
- Viral hepatitis
- Autoimmune hepatitis
- Wilson disease
- Alcoholic hepatitis
Drug-induced liver injury is the most common cause of acute liver failure in the United States,2,3 and of all drugs, acetaminophen overdose is the number-one cause. In acetaminophen-induced liver injury, serum aminotransferase levels are usually elevated to more than 1,000 U/L, while serum bilirubin remains normal in the early stages. Antimicrobial agents, antiepileptic drugs, and herbal supplements have also been implicated in acute liver failure. Our patient has denied taking herbal supplements or medications, including over-the-counter ones.
Acute viral hepatitis can explain the patient’s condition. It is a common cause of acute liver failure in the United States.2 Hepatitis A and E are more common in developing countries. Other viruses such as cytomegalovirus, Epstein-Barr virus, herpes simplex virus type 1 and 2, and varicella zoster virus can also cause acute liver failure. Serum aminotransferase levels may exceed 1,000 U/L in patients with viral hepatitis.
Autoimmune hepatitis is a rare cause of acute liver failure, but it should be considered in the differential diagnosis, particularly in middle-aged women with autoimmune disorders such as hypothyroidism. Autoimmune hepatitis can cause marked elevation in aminotransferase levels (> 1,000 U/L).
Wilson disease is an autosomal-recessive disease in which there is excessive accumulation of copper in the liver and other organs because of an inherited defect in the biliary excretion of copper. Wilson disease can cause acute liver failure and should be excluded in any patient, particularly if under age 40 with acute onset of unexplained hepatic, neurologic, or psychiatric disease.
Alcoholic hepatitis usually occurs in patients with a long-standing history of heavy alcohol use. As a result, most patients with alcoholic hepatitis have manifestations of chronic liver disease due to alcohol use. Therefore, by definition, it is not a cause of acute liver failure. Additionally, in patients with alcoholic hepatitis, the aspartate aminotransferase (AST) level is elevated but less than 300 IU/mL, and the ratio of AST to alanine aminotransferase (ALT) is usually more than 2.
CASE CONTINUES: FURTHER TESTING
The results of our patient’s serologic tests are shown in Table 2. Other test results:
- Autoimmune markers including antinuclear antibodies, antimitochondrial antibodies, antismooth muscle antibodies, and liver and kidney microsomal antibodies are negative; her immunoglobulin G (IgG) level is normal
- Serum ceruloplasmin 25 mg/dL (normal 21–45)
- Free serum copper 120 µg/dL (normal 8–12)
- Abdominal ultrasonography is unremarkable, with normal liver parenchyma and no intrahepatic or extrahepatic biliary dilatation
- Doppler ultrasonography of the liver shows patent blood vessels.
3. Based on the new data, which of the following statements is correct?
- Hepatitis B is the cause of acute liver failure in this patient
- Herpetic hepatitis cannot be excluded on the basis of the available data
- Wilson disease is most likely the diagnosis, given her elevated free serum copper
- A normal serum ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease
Hepatitis B surface antigen and hepatitis B core antibodies were negative in our patient, excluding hepatitis B virus infection. The positive hepatitis B surface antibody indicates prior immunization.
Herpetic hepatitis is an uncommon but important cause of acute liver failure because the mortality rate is high if the patient is not treated early with acyclovir. Fever, elevated aminotransferases, and leukopenia are common with herpetic hepatitis. Fewer than 50% of patients with herpetic hepatitis have vesicular rash.4,5 The value of antibody serologic testing is limited due to high rates of false-positive and false-negative results. The gold standard diagnostic tests are viral load (detection of viral RNA by polymerase chain reaction), viral staining on liver biopsy, or both. In our patient, herpes simplex virus polymerase chain reaction testing was negative, which makes herpetic hepatitis unlikely.
Wilson disease is a genetic condition in which the ability to excrete copper in the bile is impaired, resulting in accumulation of copper in the hepatocytes. Subsequently, copper is released into the bloodstream and eventually into the urine.
However, copper excretion into the bile is impaired in patients with acute liver failure regardless of the etiology. Therefore, elevated free serum copper and 24-hour urine copper levels are not specific for the diagnosis of acute liver failure secondary to Wilson disease. Moreover, Kayser-Fleischer rings, which represent copper deposition in the limbus of the cornea, may not be apparent in the early stages of Wilson disease.
Since it is challenging to diagnose Wilson disease in the context of acute liver failure, Korman et al6 compared patients with acute liver failure secondary to Wilson disease with patients with acute liver failure secondary to other conditions. They found that alkaline phosphatase levels are frequently decreased in patients with acute liver failure secondary to Wilson disease,6 and that a ratio of alkaline phosphatase to total bilirubin of less than 4 is 94% sensitive and 96% specific for the diagnosis.6
Hemolysis is common in acute liver failure due to Wilson disease. This leads to disproportionate elevation of AST compared with ALT, since AST is present in red blood cells. Consequently, the ratio of AST to ALT is usually greater than 2.2, which provides a sensitivity of 94% and a specificity of 86% for the diagnosis.6 These two ratios together provide 100% sensitivity and 100% specificity for the diagnosis of Wilson disease in the context of acute liver failure.6
Ceruloplasmin. Patients with Wilson disease typically have a low ceruloplasmin level. However, because it is an acute-phase reaction protein, ceruloplasmin can be normal or elevated in patients with acute liver failure from Wilson disease.6 Therefore, a normal ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease.
CASE CONTINUES: A DEFINITIVE DIAGNOSIS
Our patient undergoes further testing, which reveals the following:
- Her 24-hour urinary excretion of copper is 150 µg (reference value < 30)
- Slit-lamp examination is normal and shows no evidence of Kayser-Fleischer rings
- Her ratio of alkaline phosphatase to total bilirubin is 0.77 based on her initial laboratory results (Table 1)
- Her AST-ALT ratio is 3.4.
The diagnosis in our patient is acute liver failure secondary to Wilson disease.
4. What is the most appropriate next step?
- Liver biopsy
- d-penicillamine by mouth
- Trientine by mouth
- Liver transplant
- Plasmapheresis
Liver biopsy. Accumulation of copper in the liver parenchyma in patients with Wilson disease is sporadic. Therefore, qualitative copper staining on liver biopsy can be falsely negative. Quantitative copper measurement in liver tissue is the gold standard for the diagnosis of Wilson disease. However, the test is time-consuming and is not rapidly available in the context of acute liver failure.
Chelating agents such as d-pencillamine and trientine are used to treat the chronic manifestations of Wilson disease but are not useful for acute liver failure secondary to Wilson disease.
Acute liver failure secondary to Wilson disease is life-threatening, and liver transplant is considered the only definitive life-saving therapy.
Therapeutic plasmapheresis has been reported to be a successful adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant.7 However, liver transplant is still the only definitive treatment.
CASE CONTINUES: THE PATIENT’S SISTER SEEKS CARE
The patient undergoes liver transplantation, with no perioperative or postoperative complications.
The patient’s 18-year-old sister is now seeking medical attention in the outpatient clinic, concerned that she may have Wilson disease. She is otherwise healthy and denies any symptoms or complaints.
5. What is the next step for the patient’s sister?
- Reassurance
- Prophylaxis with trientine
- Check liver enzyme levels, serum ceruloplasmin level, and urine copper, and order a slit-lamp examination
- Genetic testing
Wilson disease can be asymptomatic in its early stages and may be diagnosed incidentally during routine blood tests that reveal abnormal liver enzyme levels. All patients with a confirmed family history of Wilson disease should be screened even if they are asymptomatic. The diagnosis of Wilson disease should be established in first-degree relatives before specific treatment for the relatives is prescribed.
The first step in screening a first-degree relative for Wilson disease is to check liver enzyme levels (specifically aminotransferases, alkaline phosphatase, and bilirubin), serum ceruloplasmin level, and 24-hour urine copper, and order an ophthalmologic slit-lamp examination. If any of these tests is abnormal, liver biopsy should be performed for histopathologic evaluation and quantitative copper measurement. Kayser-Fleischer rings are seen in only 50% of patients with Wilson disease and hepatic involvement, but they are pathognomic. Guidelines8 for screening first-degree relatives of Wilson disease patients are shown in Figure 1.
Genetic analysis. ATP7B, the Wilson disease gene, is located on chromosome 13. At least 300 mutations of the gene have been described,2 and the most common mutation is present in only 15% to 30% of the Wilson disease population.8–10 Routine molecular testing of the ATP7B
CASE CONTINUES: WORKUP OF THE PATIENT’S SISTER
The patient’s sister has no symptoms and her physical examination is normal. Slit-lamp examination reveals no evidence of Kayser-Fleischer rings. Her laboratory values, including complete blood counts, complete metabolic panel, and INR, are within normal ranges. Other test results, however, are abnormal:
- Free serum copper level 27 µg/dL (normal 8–12)
- Serum ceruloplasmin 9.0 mg/dL (normal 20–50)
- 24-hour urinary copper excretion 135 µg (normal < 30).
She undergoes liver biopsy for quantitative copper measurement, and the result is very high at 1,118 µg/g dry weight (reference range 10–35). The diagnosis of Wilson disease is established.
TREATING CHRONIC WILSON DISEASE
6. Which of the following is not an appropriate next step for the patient’s sister?
- Tetrathiomolybdate
- d-penicillamine
- Trientine
- Zinc salts
- Prednisone
The goal of medical treatment of chronic Wilson disease is to improve symptoms and prevent progression of the disease.
Chelating agents and zinc salts are the most commonly used medicines in the management of Wilson disease. Chelating agents remove copper from tissue, whereas zinc blocks the intestinal absorption of copper and stimulates the synthesis of endogenous chelators such as metallothioneins. Tetrathiomolybdate is an alternative agent developed to interfere with the distribution of excess body copper to susceptible target sites by reducing free serum copper (Table 3). There are no data to support the use of prednisone in the treatment of Wilson disease.
During treatment with chelating agents, 24-hour urinary excretion of copper is routinely monitored to determine the efficacy of therapy and adherence to treatment. Once de-coppering is achieved, as evidenced by a normalization of 24-hour urine copper excretion, the chelating agent can be switched to zinc salts to prevent intestinal absorption of copper.
Clinical and biochemical stabilization is achieved typically within 2 to 6 months of the initial treatment with chelating agents.8 Organ meats, nuts, shellfish, and chocolate are rich in copper and should be avoided.
The patient’s sister is started on trientine 250 mg orally three times daily on an empty stomach at least 1 hour before meals. Treatment is monitored by following 24-hour urine copper measurement. A 24-hour urine copper measurement at 3 months after starting treatment has increased from 54 at baseline to 350 µg, which indicates that the copper is being removed from tissues. The plan is for early substitution of zinc for long-term maintenance once de-coppering is completed.
KEY POINTS
- Acute liver failure is severe acute liver injury characterized by coagulopathy (INR ≥ 1.5) and encephalopathy in a patient with no preexisting liver disease and with duration of symptoms less than 26 weeks.
- Acute liver failure secondary to Wilson disease is uncommon but should be excluded, particularly in young patients.
- The diagnosis of Wilson disease in the setting of acute liver failure is challenging because the serum ceruloplasmin level may be normal in acute liver failure secondary to Wilson disease, and free serum copper and 24-hour urine copper are usually elevated in all acute liver failure patients regardless of the etiology.
- A ratio of alkaline phosphatase to total bilirubin of less than 4 plus an AST-ALT ratio greater than 2.2 in a patient with acute liver failure should be regarded as Wilson disease until proven otherwise (Figure 2).
- Acute liver failure secondary to Wilson disease is usually fatal, and emergency liver transplant is a life-saving procedure.
- Screening of first-degree relatives of Wilson disease patients should include a history and physical examination, liver enzyme tests, complete blood cell count, serum ceruloplasmin level, serum free copper level, slit-lamp examination of the eyes, and 24-hour urinary copper measurement. Genetic tests are supplementary for screening but are not routinely available.
A 25-year-old woman presents to the emergency department with a 7-day history of fatigue and nausea. On presentation she denies having abdominal pain, headache, fever, chills, night sweats, vomiting, diarrhea, melena, hematochezia, or weight loss. She recalls changes in the colors of her eyes and darkening urine over the last few days. Her medical history before this is unremarkable. She takes no prescription, over-the-counter, or herbal medications. She works as a librarian and has no occupational toxic exposures. She is single and has one sister with no prior medical history. She denies recent travel, sick contacts, smoking, recreational drug use, or pets at home.
On physical examination, her vital signs are temperature 37.3°C (99.1°F), heart rate 90 beats per minute, blood pressure 125/80 mm Hg, respiration rate 14 per minute, and oxygen saturation 97% on room air. She has icteric sclera and her skin is jaundiced. Cardiac examination is normal. Lungs are clear to auscultation and percussion bilaterally. Her abdomen is soft with no visceromegaly, masses, or tenderness. Extremities are normal with no edema. She is alert and oriented, but she has mild asterixis of the outstretched hands. The neurologic examination is otherwise unremarkable.
The patient’s basic laboratory values are listed in Table 1. Shortly after admission, she develops changes in her mental status, remaining alert but becoming agitated and oriented to person only. In view of her symptoms and laboratory findings, acute liver failure is suspected.
ACUTE LIVER FAILURE
1. The diagnostic criteria for acute liver failure include all of the following except which one?
- Acute elevation of liver biochemical tests
- Presence of preexisting liver disease
- Coagulopathy, defined by an international normalized ratio (INR) of 1.5 or greater
- Encephalopathy
- Duration of symptoms less than 26 weeks
Acute liver failure is defined by acute onset of worsening liver tests, coagulopathy (INR ≥ 1.5), and encephalopathy in patients with no preexisting liver disease and with symptom duration of less than 26 weeks.1 With a few exceptions, a history of preexisting liver disease negates the diagnosis of acute liver failure. Our patient meets the diagnostic criteria for acute liver failure.
Immediate management
Once acute liver failure is identified or suspected, the next step is to transfer the patient to the intensive care unit for close monitoring of mental status. Serial neurologic evaluations permit early detection of cerebral edema, which is considered the most common cause of death in patients with acute liver failure. Additionally, close monitoring of electrolytes and plasma glucose is necessary since these patients are susceptible to electrolyte disturbances and hypoglycemia.
Patients with acute liver failure are at increased risk of infections and should be routinely screened by obtaining urine and blood cultures.
Gastrointestinal bleeding is not uncommon in patients with acute liver failure and is usually due to gastric stress ulceration. Prophylaxis with a histamine 2 receptor antagonist or proton pump inhibitor should be considered in order to prevent gastrointestinal bleeding.
Treatment with N-acetylcysteine is beneficial, not only in patients with acute liver failure due to acetaminophen overdose, but also in those with acute liver failure from other causes.
CASE CONTINUES:
TRANSFER TO THE INTENSIVE CARE UNIT
The patient, now diagnosed with acute liver failure, is transferred to the intensive care unit. Arterial blood gas measurement shows:
- pH 7.38 (reference range 7.35–7.45)
- Pco2 40 mm Hg (36–46)
- Po2 97 mm Hg (85–95)
- Hco3 22 mmol/L (22–26).
A chest radiograph is obtained and is clear. Computed tomography (CT) of the brain reveals no edema. Transcranial Doppler ultrasonography does not show any intracranial fluid collections.
Blood and urine cultures are negative. Her hemoglobin level remains stable, and she does not develop signs of bleeding. She is started on a proton pump inhibitor for stress ulcer prophylaxis and is empirically given intravenous N-acetylcysteine until the cause of acute liver failure can be determined.
CAUSES OF ACUTE LIVER FAILURE
2. Which of the following can cause acute liver failure?
- Acetaminophen overdose
- Viral hepatitis
- Autoimmune hepatitis
- Wilson disease
- Alcoholic hepatitis
Drug-induced liver injury is the most common cause of acute liver failure in the United States,2,3 and of all drugs, acetaminophen overdose is the number-one cause. In acetaminophen-induced liver injury, serum aminotransferase levels are usually elevated to more than 1,000 U/L, while serum bilirubin remains normal in the early stages. Antimicrobial agents, antiepileptic drugs, and herbal supplements have also been implicated in acute liver failure. Our patient has denied taking herbal supplements or medications, including over-the-counter ones.
Acute viral hepatitis can explain the patient’s condition. It is a common cause of acute liver failure in the United States.2 Hepatitis A and E are more common in developing countries. Other viruses such as cytomegalovirus, Epstein-Barr virus, herpes simplex virus type 1 and 2, and varicella zoster virus can also cause acute liver failure. Serum aminotransferase levels may exceed 1,000 U/L in patients with viral hepatitis.
Autoimmune hepatitis is a rare cause of acute liver failure, but it should be considered in the differential diagnosis, particularly in middle-aged women with autoimmune disorders such as hypothyroidism. Autoimmune hepatitis can cause marked elevation in aminotransferase levels (> 1,000 U/L).
Wilson disease is an autosomal-recessive disease in which there is excessive accumulation of copper in the liver and other organs because of an inherited defect in the biliary excretion of copper. Wilson disease can cause acute liver failure and should be excluded in any patient, particularly if under age 40 with acute onset of unexplained hepatic, neurologic, or psychiatric disease.
Alcoholic hepatitis usually occurs in patients with a long-standing history of heavy alcohol use. As a result, most patients with alcoholic hepatitis have manifestations of chronic liver disease due to alcohol use. Therefore, by definition, it is not a cause of acute liver failure. Additionally, in patients with alcoholic hepatitis, the aspartate aminotransferase (AST) level is elevated but less than 300 IU/mL, and the ratio of AST to alanine aminotransferase (ALT) is usually more than 2.
CASE CONTINUES: FURTHER TESTING
The results of our patient’s serologic tests are shown in Table 2. Other test results:
- Autoimmune markers including antinuclear antibodies, antimitochondrial antibodies, antismooth muscle antibodies, and liver and kidney microsomal antibodies are negative; her immunoglobulin G (IgG) level is normal
- Serum ceruloplasmin 25 mg/dL (normal 21–45)
- Free serum copper 120 µg/dL (normal 8–12)
- Abdominal ultrasonography is unremarkable, with normal liver parenchyma and no intrahepatic or extrahepatic biliary dilatation
- Doppler ultrasonography of the liver shows patent blood vessels.
3. Based on the new data, which of the following statements is correct?
- Hepatitis B is the cause of acute liver failure in this patient
- Herpetic hepatitis cannot be excluded on the basis of the available data
- Wilson disease is most likely the diagnosis, given her elevated free serum copper
- A normal serum ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease
Hepatitis B surface antigen and hepatitis B core antibodies were negative in our patient, excluding hepatitis B virus infection. The positive hepatitis B surface antibody indicates prior immunization.
Herpetic hepatitis is an uncommon but important cause of acute liver failure because the mortality rate is high if the patient is not treated early with acyclovir. Fever, elevated aminotransferases, and leukopenia are common with herpetic hepatitis. Fewer than 50% of patients with herpetic hepatitis have vesicular rash.4,5 The value of antibody serologic testing is limited due to high rates of false-positive and false-negative results. The gold standard diagnostic tests are viral load (detection of viral RNA by polymerase chain reaction), viral staining on liver biopsy, or both. In our patient, herpes simplex virus polymerase chain reaction testing was negative, which makes herpetic hepatitis unlikely.
Wilson disease is a genetic condition in which the ability to excrete copper in the bile is impaired, resulting in accumulation of copper in the hepatocytes. Subsequently, copper is released into the bloodstream and eventually into the urine.
However, copper excretion into the bile is impaired in patients with acute liver failure regardless of the etiology. Therefore, elevated free serum copper and 24-hour urine copper levels are not specific for the diagnosis of acute liver failure secondary to Wilson disease. Moreover, Kayser-Fleischer rings, which represent copper deposition in the limbus of the cornea, may not be apparent in the early stages of Wilson disease.
Since it is challenging to diagnose Wilson disease in the context of acute liver failure, Korman et al6 compared patients with acute liver failure secondary to Wilson disease with patients with acute liver failure secondary to other conditions. They found that alkaline phosphatase levels are frequently decreased in patients with acute liver failure secondary to Wilson disease,6 and that a ratio of alkaline phosphatase to total bilirubin of less than 4 is 94% sensitive and 96% specific for the diagnosis.6
Hemolysis is common in acute liver failure due to Wilson disease. This leads to disproportionate elevation of AST compared with ALT, since AST is present in red blood cells. Consequently, the ratio of AST to ALT is usually greater than 2.2, which provides a sensitivity of 94% and a specificity of 86% for the diagnosis.6 These two ratios together provide 100% sensitivity and 100% specificity for the diagnosis of Wilson disease in the context of acute liver failure.6
Ceruloplasmin. Patients with Wilson disease typically have a low ceruloplasmin level. However, because it is an acute-phase reaction protein, ceruloplasmin can be normal or elevated in patients with acute liver failure from Wilson disease.6 Therefore, a normal ceruloplasmin level is not sufficient to rule out acute liver failure secondary to Wilson disease.
CASE CONTINUES: A DEFINITIVE DIAGNOSIS
Our patient undergoes further testing, which reveals the following:
- Her 24-hour urinary excretion of copper is 150 µg (reference value < 30)
- Slit-lamp examination is normal and shows no evidence of Kayser-Fleischer rings
- Her ratio of alkaline phosphatase to total bilirubin is 0.77 based on her initial laboratory results (Table 1)
- Her AST-ALT ratio is 3.4.
The diagnosis in our patient is acute liver failure secondary to Wilson disease.
4. What is the most appropriate next step?
- Liver biopsy
- d-penicillamine by mouth
- Trientine by mouth
- Liver transplant
- Plasmapheresis
Liver biopsy. Accumulation of copper in the liver parenchyma in patients with Wilson disease is sporadic. Therefore, qualitative copper staining on liver biopsy can be falsely negative. Quantitative copper measurement in liver tissue is the gold standard for the diagnosis of Wilson disease. However, the test is time-consuming and is not rapidly available in the context of acute liver failure.
Chelating agents such as d-pencillamine and trientine are used to treat the chronic manifestations of Wilson disease but are not useful for acute liver failure secondary to Wilson disease.
Acute liver failure secondary to Wilson disease is life-threatening, and liver transplant is considered the only definitive life-saving therapy.
Therapeutic plasmapheresis has been reported to be a successful adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant.7 However, liver transplant is still the only definitive treatment.
CASE CONTINUES: THE PATIENT’S SISTER SEEKS CARE
The patient undergoes liver transplantation, with no perioperative or postoperative complications.
The patient’s 18-year-old sister is now seeking medical attention in the outpatient clinic, concerned that she may have Wilson disease. She is otherwise healthy and denies any symptoms or complaints.
5. What is the next step for the patient’s sister?
- Reassurance
- Prophylaxis with trientine
- Check liver enzyme levels, serum ceruloplasmin level, and urine copper, and order a slit-lamp examination
- Genetic testing
Wilson disease can be asymptomatic in its early stages and may be diagnosed incidentally during routine blood tests that reveal abnormal liver enzyme levels. All patients with a confirmed family history of Wilson disease should be screened even if they are asymptomatic. The diagnosis of Wilson disease should be established in first-degree relatives before specific treatment for the relatives is prescribed.
The first step in screening a first-degree relative for Wilson disease is to check liver enzyme levels (specifically aminotransferases, alkaline phosphatase, and bilirubin), serum ceruloplasmin level, and 24-hour urine copper, and order an ophthalmologic slit-lamp examination. If any of these tests is abnormal, liver biopsy should be performed for histopathologic evaluation and quantitative copper measurement. Kayser-Fleischer rings are seen in only 50% of patients with Wilson disease and hepatic involvement, but they are pathognomic. Guidelines8 for screening first-degree relatives of Wilson disease patients are shown in Figure 1.
Genetic analysis. ATP7B, the Wilson disease gene, is located on chromosome 13. At least 300 mutations of the gene have been described,2 and the most common mutation is present in only 15% to 30% of the Wilson disease population.8–10 Routine molecular testing of the ATP7B
CASE CONTINUES: WORKUP OF THE PATIENT’S SISTER
The patient’s sister has no symptoms and her physical examination is normal. Slit-lamp examination reveals no evidence of Kayser-Fleischer rings. Her laboratory values, including complete blood counts, complete metabolic panel, and INR, are within normal ranges. Other test results, however, are abnormal:
- Free serum copper level 27 µg/dL (normal 8–12)
- Serum ceruloplasmin 9.0 mg/dL (normal 20–50)
- 24-hour urinary copper excretion 135 µg (normal < 30).
She undergoes liver biopsy for quantitative copper measurement, and the result is very high at 1,118 µg/g dry weight (reference range 10–35). The diagnosis of Wilson disease is established.
TREATING CHRONIC WILSON DISEASE
6. Which of the following is not an appropriate next step for the patient’s sister?
- Tetrathiomolybdate
- d-penicillamine
- Trientine
- Zinc salts
- Prednisone
The goal of medical treatment of chronic Wilson disease is to improve symptoms and prevent progression of the disease.
Chelating agents and zinc salts are the most commonly used medicines in the management of Wilson disease. Chelating agents remove copper from tissue, whereas zinc blocks the intestinal absorption of copper and stimulates the synthesis of endogenous chelators such as metallothioneins. Tetrathiomolybdate is an alternative agent developed to interfere with the distribution of excess body copper to susceptible target sites by reducing free serum copper (Table 3). There are no data to support the use of prednisone in the treatment of Wilson disease.
During treatment with chelating agents, 24-hour urinary excretion of copper is routinely monitored to determine the efficacy of therapy and adherence to treatment. Once de-coppering is achieved, as evidenced by a normalization of 24-hour urine copper excretion, the chelating agent can be switched to zinc salts to prevent intestinal absorption of copper.
Clinical and biochemical stabilization is achieved typically within 2 to 6 months of the initial treatment with chelating agents.8 Organ meats, nuts, shellfish, and chocolate are rich in copper and should be avoided.
The patient’s sister is started on trientine 250 mg orally three times daily on an empty stomach at least 1 hour before meals. Treatment is monitored by following 24-hour urine copper measurement. A 24-hour urine copper measurement at 3 months after starting treatment has increased from 54 at baseline to 350 µg, which indicates that the copper is being removed from tissues. The plan is for early substitution of zinc for long-term maintenance once de-coppering is completed.
KEY POINTS
- Acute liver failure is severe acute liver injury characterized by coagulopathy (INR ≥ 1.5) and encephalopathy in a patient with no preexisting liver disease and with duration of symptoms less than 26 weeks.
- Acute liver failure secondary to Wilson disease is uncommon but should be excluded, particularly in young patients.
- The diagnosis of Wilson disease in the setting of acute liver failure is challenging because the serum ceruloplasmin level may be normal in acute liver failure secondary to Wilson disease, and free serum copper and 24-hour urine copper are usually elevated in all acute liver failure patients regardless of the etiology.
- A ratio of alkaline phosphatase to total bilirubin of less than 4 plus an AST-ALT ratio greater than 2.2 in a patient with acute liver failure should be regarded as Wilson disease until proven otherwise (Figure 2).
- Acute liver failure secondary to Wilson disease is usually fatal, and emergency liver transplant is a life-saving procedure.
- Screening of first-degree relatives of Wilson disease patients should include a history and physical examination, liver enzyme tests, complete blood cell count, serum ceruloplasmin level, serum free copper level, slit-lamp examination of the eyes, and 24-hour urinary copper measurement. Genetic tests are supplementary for screening but are not routinely available.
- Lee WM, Larson AM, Stravitz T. AASLD Position Paper: The management of acute liver failure: update 2011. www.aasld.org/sites/default/files/guideline_documents/alfenhanced.pdf. Accessed December 9, 2015.
- Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet 2010; 376:190–201.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Hanouneh IA, Khoriaty R, Zein NN. A 35-year-old Asian man with jaundice and markedly high aminotransferase levels. Cleve Clin J Med 2009; 76:449–456.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transpl 2007; 13:1428–1434.
- Korman JD, Volenberg I, Balko J, et al; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology 2008; 48:1167–1174.
- Morgan SM, Zantek ND. Therapeutic plasma exchange for fulminant hepatic failure secondary to Wilson's disease. J Clin Apher 2012; 27:282–286.
- Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47:2089–2111.
- Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet 1997; 61:317–328.
- Maier-Dobersberger T, Ferenci P, Polli C, et al. Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Ann Intern Med 1997; 127:21–26.
- Lee WM, Larson AM, Stravitz T. AASLD Position Paper: The management of acute liver failure: update 2011. www.aasld.org/sites/default/files/guideline_documents/alfenhanced.pdf. Accessed December 9, 2015.
- Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet 2010; 376:190–201.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Hanouneh IA, Khoriaty R, Zein NN. A 35-year-old Asian man with jaundice and markedly high aminotransferase levels. Cleve Clin J Med 2009; 76:449–456.
- Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transpl 2007; 13:1428–1434.
- Korman JD, Volenberg I, Balko J, et al; Pediatric and Adult Acute Liver Failure Study Groups. Screening for Wilson disease in acute liver failure: a comparison of currently available diagnostic tests. Hepatology 2008; 48:1167–1174.
- Morgan SM, Zantek ND. Therapeutic plasma exchange for fulminant hepatic failure secondary to Wilson's disease. J Clin Apher 2012; 27:282–286.
- Roberts EA, Schilsky ML; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47:2089–2111.
- Shah AB, Chernov I, Zhang HT, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype-phenotype correlation, and functional analyses. Am J Hum Genet 1997; 61:317–328.
- Maier-Dobersberger T, Ferenci P, Polli C, et al. Detection of the His1069Gln mutation in Wilson disease by rapid polymerase chain reaction. Ann Intern Med 1997; 127:21–26.
