User login
Clinical features and diagnosis of small-vessel vasculitis
Vasculitis refers to inflammation of the blood vessel. This inflammation can cause vessel wall thickening that compromises or occludes the vessel lumen, ultimately resulting in organ ischemia. It also can cause vessel wall attenuation that predisposes to aneurysm formation or breach of the vessel integrity with resultant hemorrhage into the tissue.
Vasculitis can be thought of as a primary or secondary process. Primary vasculitides are unique disease entities without a currently identified underlying cause in which vasculitis forms the pathologic basis of tissue injury. Vasculitis can occur secondary to medication exposure or an underlying illness, including infections, malignancy, cryoglobulinemia, and rheumatic diseases (such as systemic lupus erythematosus, rheumatoid arthritis, Sjögren syndrome, or myositis).
Primary vasculitides may differ in epidemiology, such as the age at which they occur and the gender most likely to be affected, their clinical manifestations (including signs, symptoms, and patterns of organ involvement), the diagnostic approach (biopsy, arteriography, and laboratory investigation), treatment (supportive care, glucocorticoids alone, or in combination with other immunosuppressants), and the size of the vessels predominantly affected (large, medium, or small).
Small-vessel vasculitis affects the arteriole, capillary, and venule. An excellent example of small-vessel vasculitis and the one most commonly encountered in clinical practice is cutaneous vasculitis, in which extravasation of erythrocytes from disrupted small vessels is observed histologically, with the clinical sequelae of palpable purpura. Although categorization based on the predominant vessel size that is affected is a helpful way to view these diseases, this is not absolute and each disease has the potential to affect a diverse range of vessels.
This article explores the clinical features and diagnosis of three forms of vasculitis that predominantly affect the small vessels: granulomatosis with polyangiitis (GPA [Wegener’s granulomatosis]), microscopic polyangiitis (MPA), and eosinophilic GPA (Churg-Strauss syndrome, EGPA).
GRANULOMATOSIS WITH POLYANGIITIS
Granulomatosis with polyangiitis is characterized by granulomatous inflammation involving the respiratory tract and by vasculitis affecting small- to medium-sized vessels in which necrotizing glomerulonephritis is common.
Wide range of presentations, manifestations
Approximately 90% of patients with GPA have upper or lower airway involvement or both.1 Upper airway or ear symptoms affect 73% of patients initially and 92% overall.1 Direct inspection of the nasal membranes shows a cobblestoned or ulcerated appearance, and computed tomography reveals mucosal thickening of the sinuses. In some instances, sinus disease can compromise blood supply to the cartilaginous portion of the nasal septum, leading to nasal septum perforations or collapse of the nasal bridge. Another manifestation of upper airway disease and GPA is subglottic stenosis, a narrowing in the subglottic region located just below the vocal cords. The narrowing typically spans about 1 cm and rarely extends or involves the remainder of the trachea.
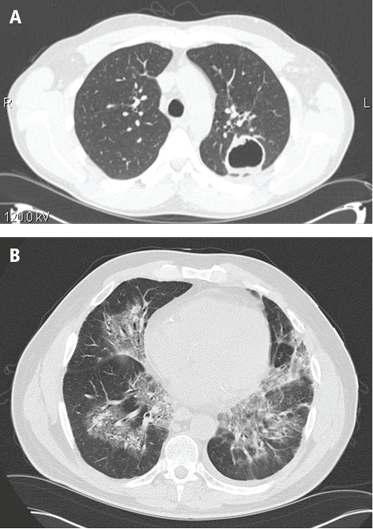
The lungs are involved in 85% of patients.1 Radiographic abnormalities can be diverse and include bilateral pulmonary nodular infiltrates, single or multiple cavities, and bilateral ground glass infiltrates as can be seen in pulmonary hemorrhage (Figure). Bronchoscopy may reveal endobronchial stenosis, and pleural disease can occur rarely.
Approximately 20% of patients with GPA may have glomerulonephritis when they first present for medical attention, but it eventually develops in nearly 80% of patients during the disease course.1 Despite its potential for rapid progression, glomerulonephritis presents a diagnostic challenge because it is asymptomatic. It is detected by evidence of proteinuria and an active urine sediment with dysmorphic red blood cells and red blood cell casts.
Ocular involvement occurs eventually in 52% of patients with GPA.1 Any ocular structure can be affected and ocular involvement can be visually threatening. The more prominent ocular manifestations include scleritis/episcleritis or orbital disease.
Cutaneous manifestations, observed in 46% of patients, include verrucous-appearing lesions on the elbow and infarctions in the skin and nail folds.1 Other rare manifestations can occur, such as pericarditis and cerebral vasculitis.
Although nearly all patients present with upper or lower airway symptoms, the multisystem nature of GPA explains the wide range of presentations and the varying degrees of disease severity.
Differential diagnosis
The differential diagnosis in GPA is varied. Particularly in the setting of isolated lung or sinus disease, infection is foremost in the differential diagnosis. Even in the nonimmunosuppressed host, unusual infections such as mycobacteria, histoplasmosis, and other fungal infections should be considered. Lymphadenopathy, rarely seen in GPA, should raise concern for other causes of disease. Lymphoproliferative processes and other neoplasms, other rheumatic diseases, granulomatous disease (ie, sarcoidosis), and other causes of glomerulonephritis (when present) also merit consideration. Differentiation of these entities from GPA is essential because the treatment differs in many instances.
The differential diagnosis for patients who present with midline destructive lesions must include other causes of collapse of the nasal bridge, nasal septum perforation, and possibly palate destruction. Erosions of the hard palate in particular should raise an immediate red flag for entities other than GPA, such as lymphoproliferative diseases; rare infections, particularly if the patient has studied or worked abroad; and cocaine exposure.
Diagnostic evaluation
A diagnosis of GPA is typically based on the presence of histologic features in a clinically compatible setting. Diagnostic features include necrosis, granulomatous inflammation, vasculitis, and special stains and cultures negative for microorganisms.
Biopsy sites are determined by evidence of clinical disease affecting a target organ and the likelihood of obtaining diagnostically meaningful findings from that site. One challenge is that biopsies are not always diagnostic. The changes tend to be patchy and the likelihood of a positive yield is associated with the amount of tissue that can be obtained. Tissues from the ear, nose, and throat have a yield of about 20%, depending upon the site and the biopsy size. The highest yield comes from radiographically abnormal pulmonary parenchyma. Although transbronchial biopsies are attractive because they are less invasive than open lung biopsy, they are also far less diagnostic, with fewer than 10% having a positive yield. Because cutaneous vasculitis is observed in many settings, its presence is usually insufficient evidence for diagnosis. The renal histologic appearance is a focal, segmental, crescentic, and necrotizing glomerulonephritis that has few to no immune complexes (pauciimmune glomerulonephritis).1–3
Chest imaging should be performed in any patient in whom GPA is part of the differential diagnosis, since up to one-third of patients may be asymptomatic yet have pulmonary radiographic findings.
Laboratory assessment should include serum chemistries to evaluate renal and hepatic function, complete blood count, erythrocyte sedimentation rate, measurement of C-reactive protein, and urinalysis. If the urinalysis is positive for blood, microscopy should be performed on fresh urine to look for casts. In the setting of pulmonary-renal manifestations, testing for other causes, such as antiglomerular basement antibodies and antinuclear antibodies, should be considered.
Serologic testing for antineutrophil cytoplasmic antibodies (ANCA) has provided a useful tool in suggesting the diagnosis of GPA. Two forms of ANCA have been identified in patients with vasculitis: ANCA directed against the neutrophil serine protease proteinase-3 (PR3), which results in a cytoplasmic immunofluorescence (cANCA) pattern; and ANCA directed against the neutrophil enzyme myeloperoxidase (MPO), which causes a perinuclear immunofluorescence (pANCA) pattern.4 Approximately 80% to 95% of ANCA found in patients with active severe GPA are detectable PR3-cANCA, while 5% to 20% are MPO-pANCA.5 The predictive value of ANCA for the diagnosis depends on the spectrum of clinical features. As ANCA can be seen in other settings, ANCA as the basis for diagnosis in place of tissue biopsy should be used with caution and only in selected instances where their predictive value would equal that of biopsy. The presence of ANCA is not necessary to establish the diagnosis, as up to 20% of patients with GPA may be ANCA-negative.6
MICROSCOPIC POLYANGIITIS
The history of MPA dates to 1866, with the description of periarteritis nodosa.7 The term “microscopic polyarteritis” was introduced in 1948, when glomerular disease was recognized in some patients.8 In 1994, the Chapel Hill Consensus Conference defined MPA as a necrotizing vasculitis with few or no immune deposits that affects small vessels (ie, capillaries, venules, or arterioles). Necrotizing arteritis of small- and medium-sized arteries may be present. Necrotizing glomerulonephritis and pulmonary capillaritis commonly occur.9 MPA shares many clinical features with GPA and is currently said to be distinguished by the absence of granulomatous inflammation.9
Presentations and manifestations
In one assessment of organ system involvement in 85 patients with MPA, investigators observed glomerular syndrome in 82% of patients.10 They also found a high predilection for involvement of the skin, joints, and lungs. Pulmonary hemorrhage is a particularly important manifestation of MPA because it can be immediately life-threatening.
Differential diagnosis
The differential diagnosis for MPA is similar to GPA in the inclusion of other causes of classic pulmonary-renal syndromes, such as antiglomerular basement membrane disease and systemic lupus erythematosus. Poststreptococcal glomerulonephritis should be considered when the kidney is the predominant organ involved in the absence of lung disease. In the setting of pulmonary infiltrates, infections and neoplasms remain significant in the differential diagnosis.
Diagnostic evaluation
The diagnosis of MPA is based on consistent clinical features and compatible histologic findings. The histologic renal lesion is identical to that seen in GPA. Pulmonary disease typically includes capillaritis and is notable for the absence of evidence of immune deposition, in contrast to antiglomerular basement membrane disease.
Chest imaging is indicated when MPA is part of the differential diagnosis. Computed tomography is the preferred technique, as early alveolar hemorrhage that can occur in MPA may not be visualized on a chest radiograph.
Laboratory assessment should include serum chemistries, complete blood count, erythrocyte sedimentation rate, measurement of C-reactive protein, and urinalysis. Additional testing should be pursued for other diseases as indicated by the clinical features.
Approximately 40% to 80% of patients with MPA have MPO-pANCA.5 Approximately 15% of patients are MPO-pANCA positive,6 and 0% to 20% are ANCA-negative. As with GPA, ANCA is useful to suggest—but not diagnose—disease in many instances. The absence of ANCA does not rule out MPA.
EOSINOPHILIC GPA
Eosinophilic GPA is a unique entity characterized by eosinophil-rich and granulomatous inflammation involving the respiratory tract and necrotizing vasculitis of small- to medium-size vessels. It is also associated with asthma and eosinophilia.
Different disease phases
Eosinophilic GPA is often thought of as having three phases: prodromal, eosinophilic, and vasculitic.11,12 Although helpful conceptually, these phases may not always be present and may not occur in sequence.
The prodromal phase is characterized by asthma associated with allergic rhinitis with or without polyposis. The eosinophilic phase is characterized by the presence of eosinophilia in the blood and tissue. Eosinophilia is a prominent feature, although accurate detection and assessment can be challenging in the setting of glucocorticoid use for asthma as this normalizes the eosinophil count.
The vasculitic phase distinguishes EGPA from other eosinophilic disorders. Features of vasculitis may occur in multiple organ sites, including the nerves, lungs, heart, gastrointestinal tract, and kidneys. In one series of 96 patients, nearly 100% had asthma, and peripheral nervous system involvement in the form of mononeuritis multiplex was present in 72%.12 Cardiac involvement is of particular importance as it is a prominent cause of disease-related mortality. Cardiac manifestations include myocarditis, pericarditis, endocarditis, valvulitis, and coronary vasculitis.
Differential diagnosis
The differential diagnosis of EGPA shares similarities with GPA and MPA, but also includes eosino philic disorders such as hypereosinophilic syndrome, eosinophilic leukemia, and parasitic diseases.
Diagnostic evaluation
Diagnosis is often based on the presence of asthma, a finding of peripheral eosinophilia (> 1,500 cells/mm3), and the presence of systemic vasculitis involving, ideally, two or more extrapulmonary organs. While histologic confirmation remains ideal, demonstration of characteristic findings on biopsy can be difficult. Glomerular involvement is far less common than in GPA and MPA, but, when present, the renal lesion is identical. Pulmonary histologic findings can be diverse and include the classic “allergic-granuloma” as originally described by Churg and Strauss, as well as isolated granulomatous inflammation, eosinophilic inflammation, or small-vessel vasculitis. Tissue eosinophilia is a prominent finding that typically is seen on biopsies of skin, nerve, and gastrointestinal tissues.
Chest imaging should be performed when EGPA is part of the differential diagnosis. Because of the potential for cardiac involvement, a baseline echocardiogram should be obtained. Pulmonary function tests may be useful, particularly in patients who have a strong asthmatic component.
Similar to GPA and MPA, laboratory assessment includes serum chemistries, complete blood count with differential to determine the eosinophil count, erythrocyte sedimentation rate, measurement of C-reactive protein, and urinalysis. With the allergic and asthmatic components, immunoglobulin E levels are frequently elevated. Additional testing for other eosinophilic diseases should be pursued as indicated by the clinical features.
Only about 40% of patients are ANCA-positive.13 Most of these are MPO-pANCA, with PR3-cANCA occurring less commonly. Although some reports have suggested differing clinical patterns of EGPA based on ANCA positivity, the presence or absence of ANCA is less helpful in the diagnosis.13
DIFFERENTIATION
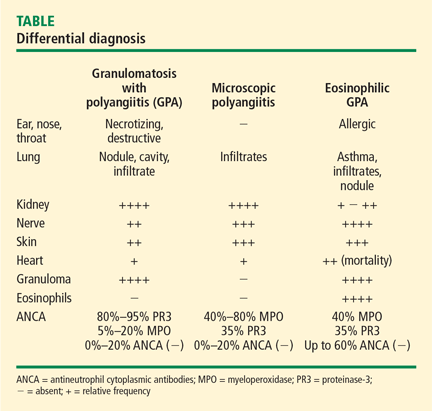
A key histologic difference between GPA and MPA is the presence of granulomatous inflammation in GPA and its absence in MPA under the current nomenclature system.9 Granulomatous inflammation can be seen in EGPA, but it is usually accompanied by eosinophils, which are less likely to be present in GPA and MPA.
The predominance of the ANCA immunofluorescence pattern and target antigen differs between GPA and MPA, with ANCA positivity occurring in 38% of patients with EGPA.13
SUMMARY
Conceptualizing vasculitic disease based on vessel size can be useful, but it is not an absolute definition. Although GPA, MPA, and EGPA predominantly affect small- to medium-sized vessels, these disease entities are phenotypically unique, with both shared features and differences. Common to all three entities is the potential for organ- and life-threatening manifestations, particularly involving the lungs, kidneys, nerves, gastrointestinal tract, and heart. All three entities need aggressive immuno suppression for severe disease. Recognition of these entities and the distinctions among them can guide the approach to diagnosis, treatment, and future outcomes.
- Hoffman GS, Kerr GS, Leavitt RY, et al Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Travis WD, Hoffman GS, Leavitt RY, Pass HI, Fauci AS. Surgical pathology of the lung in Wegener’s granulomatosis: review of 87 open lung biopsies from 67 patients. Am J Surg Pathol 1991; 15:315–333.
- Devaney KO, Travis WD, Hoffman G, Leavitt R, Lebovics R, Fauci AS. Interpretation of head and neck biopsies in Wegener’s granulomatosis: a pathologic study of 126 biopsies in 70 patients. Am J Surg Pathol 1990; 14:555–564.
- Bosch X, Guilabert A, Font J. Antineutrophil cytoplasmic antibodies. Lancet 2006; 368:404–418.
- Hoffman GS, Specks U. Antineutrophil cytoplasmic antibodies. Arthritis Rheum 1998;1521–1537.
- Wiik A. What you should know about PR3-ANCA. An introduction. Arthritis Res 2000; 2:252–254.
- Kussmaul A, Maier R. Über eine bisher nicht beschriebene eigenthümliche Arterienerkrankung (Periarteritis nodosa), die mit Morbus Brightii und rapid fortschreitender allgemeiner Muskellähmung einhergeht. Dtsch Arch Klin Med 1866; 1:484–518.
- Davson J, Ball J, Platt R. The kidney in periarteritis nodosa. Q J Med 1948; 17:175–202.
- Jennette C, Falk RJ, Andrassy K, et al Nomenclature of systemic vasculitides: proposal of an international consensus conference. Arthritis Rheum 1994; 37:187–192.
- Guillevin L, Durand-Gasselin B, Cevallos R, et al Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum 1999; 42:421–430.
- Keogh KA, Specks U. Churg-Strauss syndrome. Semin Respir Crit Care Med 2006; 27:148–157.
- Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome: clinical study and long-term follow-up of 96 patients. Medicine 1999; 78:26–37.
- Sablé-Fourtassou R, Cohen P, Mahr A, et al., for the French Vasculitis Study Group. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Intern Med 2005; 143:632–638.
Vasculitis refers to inflammation of the blood vessel. This inflammation can cause vessel wall thickening that compromises or occludes the vessel lumen, ultimately resulting in organ ischemia. It also can cause vessel wall attenuation that predisposes to aneurysm formation or breach of the vessel integrity with resultant hemorrhage into the tissue.
Vasculitis can be thought of as a primary or secondary process. Primary vasculitides are unique disease entities without a currently identified underlying cause in which vasculitis forms the pathologic basis of tissue injury. Vasculitis can occur secondary to medication exposure or an underlying illness, including infections, malignancy, cryoglobulinemia, and rheumatic diseases (such as systemic lupus erythematosus, rheumatoid arthritis, Sjögren syndrome, or myositis).
Primary vasculitides may differ in epidemiology, such as the age at which they occur and the gender most likely to be affected, their clinical manifestations (including signs, symptoms, and patterns of organ involvement), the diagnostic approach (biopsy, arteriography, and laboratory investigation), treatment (supportive care, glucocorticoids alone, or in combination with other immunosuppressants), and the size of the vessels predominantly affected (large, medium, or small).
Small-vessel vasculitis affects the arteriole, capillary, and venule. An excellent example of small-vessel vasculitis and the one most commonly encountered in clinical practice is cutaneous vasculitis, in which extravasation of erythrocytes from disrupted small vessels is observed histologically, with the clinical sequelae of palpable purpura. Although categorization based on the predominant vessel size that is affected is a helpful way to view these diseases, this is not absolute and each disease has the potential to affect a diverse range of vessels.
This article explores the clinical features and diagnosis of three forms of vasculitis that predominantly affect the small vessels: granulomatosis with polyangiitis (GPA [Wegener’s granulomatosis]), microscopic polyangiitis (MPA), and eosinophilic GPA (Churg-Strauss syndrome, EGPA).
GRANULOMATOSIS WITH POLYANGIITIS
Granulomatosis with polyangiitis is characterized by granulomatous inflammation involving the respiratory tract and by vasculitis affecting small- to medium-sized vessels in which necrotizing glomerulonephritis is common.
Wide range of presentations, manifestations
Approximately 90% of patients with GPA have upper or lower airway involvement or both.1 Upper airway or ear symptoms affect 73% of patients initially and 92% overall.1 Direct inspection of the nasal membranes shows a cobblestoned or ulcerated appearance, and computed tomography reveals mucosal thickening of the sinuses. In some instances, sinus disease can compromise blood supply to the cartilaginous portion of the nasal septum, leading to nasal septum perforations or collapse of the nasal bridge. Another manifestation of upper airway disease and GPA is subglottic stenosis, a narrowing in the subglottic region located just below the vocal cords. The narrowing typically spans about 1 cm and rarely extends or involves the remainder of the trachea.
The lungs are involved in 85% of patients.1 Radiographic abnormalities can be diverse and include bilateral pulmonary nodular infiltrates, single or multiple cavities, and bilateral ground glass infiltrates as can be seen in pulmonary hemorrhage (Figure). Bronchoscopy may reveal endobronchial stenosis, and pleural disease can occur rarely.
Approximately 20% of patients with GPA may have glomerulonephritis when they first present for medical attention, but it eventually develops in nearly 80% of patients during the disease course.1 Despite its potential for rapid progression, glomerulonephritis presents a diagnostic challenge because it is asymptomatic. It is detected by evidence of proteinuria and an active urine sediment with dysmorphic red blood cells and red blood cell casts.
Ocular involvement occurs eventually in 52% of patients with GPA.1 Any ocular structure can be affected and ocular involvement can be visually threatening. The more prominent ocular manifestations include scleritis/episcleritis or orbital disease.
Cutaneous manifestations, observed in 46% of patients, include verrucous-appearing lesions on the elbow and infarctions in the skin and nail folds.1 Other rare manifestations can occur, such as pericarditis and cerebral vasculitis.
Although nearly all patients present with upper or lower airway symptoms, the multisystem nature of GPA explains the wide range of presentations and the varying degrees of disease severity.
Differential diagnosis
The differential diagnosis in GPA is varied. Particularly in the setting of isolated lung or sinus disease, infection is foremost in the differential diagnosis. Even in the nonimmunosuppressed host, unusual infections such as mycobacteria, histoplasmosis, and other fungal infections should be considered. Lymphadenopathy, rarely seen in GPA, should raise concern for other causes of disease. Lymphoproliferative processes and other neoplasms, other rheumatic diseases, granulomatous disease (ie, sarcoidosis), and other causes of glomerulonephritis (when present) also merit consideration. Differentiation of these entities from GPA is essential because the treatment differs in many instances.
The differential diagnosis for patients who present with midline destructive lesions must include other causes of collapse of the nasal bridge, nasal septum perforation, and possibly palate destruction. Erosions of the hard palate in particular should raise an immediate red flag for entities other than GPA, such as lymphoproliferative diseases; rare infections, particularly if the patient has studied or worked abroad; and cocaine exposure.
Diagnostic evaluation
A diagnosis of GPA is typically based on the presence of histologic features in a clinically compatible setting. Diagnostic features include necrosis, granulomatous inflammation, vasculitis, and special stains and cultures negative for microorganisms.
Biopsy sites are determined by evidence of clinical disease affecting a target organ and the likelihood of obtaining diagnostically meaningful findings from that site. One challenge is that biopsies are not always diagnostic. The changes tend to be patchy and the likelihood of a positive yield is associated with the amount of tissue that can be obtained. Tissues from the ear, nose, and throat have a yield of about 20%, depending upon the site and the biopsy size. The highest yield comes from radiographically abnormal pulmonary parenchyma. Although transbronchial biopsies are attractive because they are less invasive than open lung biopsy, they are also far less diagnostic, with fewer than 10% having a positive yield. Because cutaneous vasculitis is observed in many settings, its presence is usually insufficient evidence for diagnosis. The renal histologic appearance is a focal, segmental, crescentic, and necrotizing glomerulonephritis that has few to no immune complexes (pauciimmune glomerulonephritis).1–3
Chest imaging should be performed in any patient in whom GPA is part of the differential diagnosis, since up to one-third of patients may be asymptomatic yet have pulmonary radiographic findings.
Laboratory assessment should include serum chemistries to evaluate renal and hepatic function, complete blood count, erythrocyte sedimentation rate, measurement of C-reactive protein, and urinalysis. If the urinalysis is positive for blood, microscopy should be performed on fresh urine to look for casts. In the setting of pulmonary-renal manifestations, testing for other causes, such as antiglomerular basement antibodies and antinuclear antibodies, should be considered.
Serologic testing for antineutrophil cytoplasmic antibodies (ANCA) has provided a useful tool in suggesting the diagnosis of GPA. Two forms of ANCA have been identified in patients with vasculitis: ANCA directed against the neutrophil serine protease proteinase-3 (PR3), which results in a cytoplasmic immunofluorescence (cANCA) pattern; and ANCA directed against the neutrophil enzyme myeloperoxidase (MPO), which causes a perinuclear immunofluorescence (pANCA) pattern.4 Approximately 80% to 95% of ANCA found in patients with active severe GPA are detectable PR3-cANCA, while 5% to 20% are MPO-pANCA.5 The predictive value of ANCA for the diagnosis depends on the spectrum of clinical features. As ANCA can be seen in other settings, ANCA as the basis for diagnosis in place of tissue biopsy should be used with caution and only in selected instances where their predictive value would equal that of biopsy. The presence of ANCA is not necessary to establish the diagnosis, as up to 20% of patients with GPA may be ANCA-negative.6
MICROSCOPIC POLYANGIITIS
The history of MPA dates to 1866, with the description of periarteritis nodosa.7 The term “microscopic polyarteritis” was introduced in 1948, when glomerular disease was recognized in some patients.8 In 1994, the Chapel Hill Consensus Conference defined MPA as a necrotizing vasculitis with few or no immune deposits that affects small vessels (ie, capillaries, venules, or arterioles). Necrotizing arteritis of small- and medium-sized arteries may be present. Necrotizing glomerulonephritis and pulmonary capillaritis commonly occur.9 MPA shares many clinical features with GPA and is currently said to be distinguished by the absence of granulomatous inflammation.9
Presentations and manifestations
In one assessment of organ system involvement in 85 patients with MPA, investigators observed glomerular syndrome in 82% of patients.10 They also found a high predilection for involvement of the skin, joints, and lungs. Pulmonary hemorrhage is a particularly important manifestation of MPA because it can be immediately life-threatening.
Differential diagnosis
The differential diagnosis for MPA is similar to GPA in the inclusion of other causes of classic pulmonary-renal syndromes, such as antiglomerular basement membrane disease and systemic lupus erythematosus. Poststreptococcal glomerulonephritis should be considered when the kidney is the predominant organ involved in the absence of lung disease. In the setting of pulmonary infiltrates, infections and neoplasms remain significant in the differential diagnosis.
Diagnostic evaluation
The diagnosis of MPA is based on consistent clinical features and compatible histologic findings. The histologic renal lesion is identical to that seen in GPA. Pulmonary disease typically includes capillaritis and is notable for the absence of evidence of immune deposition, in contrast to antiglomerular basement membrane disease.
Chest imaging is indicated when MPA is part of the differential diagnosis. Computed tomography is the preferred technique, as early alveolar hemorrhage that can occur in MPA may not be visualized on a chest radiograph.
Laboratory assessment should include serum chemistries, complete blood count, erythrocyte sedimentation rate, measurement of C-reactive protein, and urinalysis. Additional testing should be pursued for other diseases as indicated by the clinical features.
Approximately 40% to 80% of patients with MPA have MPO-pANCA.5 Approximately 15% of patients are MPO-pANCA positive,6 and 0% to 20% are ANCA-negative. As with GPA, ANCA is useful to suggest—but not diagnose—disease in many instances. The absence of ANCA does not rule out MPA.
EOSINOPHILIC GPA
Eosinophilic GPA is a unique entity characterized by eosinophil-rich and granulomatous inflammation involving the respiratory tract and necrotizing vasculitis of small- to medium-size vessels. It is also associated with asthma and eosinophilia.
Different disease phases
Eosinophilic GPA is often thought of as having three phases: prodromal, eosinophilic, and vasculitic.11,12 Although helpful conceptually, these phases may not always be present and may not occur in sequence.
The prodromal phase is characterized by asthma associated with allergic rhinitis with or without polyposis. The eosinophilic phase is characterized by the presence of eosinophilia in the blood and tissue. Eosinophilia is a prominent feature, although accurate detection and assessment can be challenging in the setting of glucocorticoid use for asthma as this normalizes the eosinophil count.
The vasculitic phase distinguishes EGPA from other eosinophilic disorders. Features of vasculitis may occur in multiple organ sites, including the nerves, lungs, heart, gastrointestinal tract, and kidneys. In one series of 96 patients, nearly 100% had asthma, and peripheral nervous system involvement in the form of mononeuritis multiplex was present in 72%.12 Cardiac involvement is of particular importance as it is a prominent cause of disease-related mortality. Cardiac manifestations include myocarditis, pericarditis, endocarditis, valvulitis, and coronary vasculitis.
Differential diagnosis
The differential diagnosis of EGPA shares similarities with GPA and MPA, but also includes eosino philic disorders such as hypereosinophilic syndrome, eosinophilic leukemia, and parasitic diseases.
Diagnostic evaluation
Diagnosis is often based on the presence of asthma, a finding of peripheral eosinophilia (> 1,500 cells/mm3), and the presence of systemic vasculitis involving, ideally, two or more extrapulmonary organs. While histologic confirmation remains ideal, demonstration of characteristic findings on biopsy can be difficult. Glomerular involvement is far less common than in GPA and MPA, but, when present, the renal lesion is identical. Pulmonary histologic findings can be diverse and include the classic “allergic-granuloma” as originally described by Churg and Strauss, as well as isolated granulomatous inflammation, eosinophilic inflammation, or small-vessel vasculitis. Tissue eosinophilia is a prominent finding that typically is seen on biopsies of skin, nerve, and gastrointestinal tissues.
Chest imaging should be performed when EGPA is part of the differential diagnosis. Because of the potential for cardiac involvement, a baseline echocardiogram should be obtained. Pulmonary function tests may be useful, particularly in patients who have a strong asthmatic component.
Similar to GPA and MPA, laboratory assessment includes serum chemistries, complete blood count with differential to determine the eosinophil count, erythrocyte sedimentation rate, measurement of C-reactive protein, and urinalysis. With the allergic and asthmatic components, immunoglobulin E levels are frequently elevated. Additional testing for other eosinophilic diseases should be pursued as indicated by the clinical features.
Only about 40% of patients are ANCA-positive.13 Most of these are MPO-pANCA, with PR3-cANCA occurring less commonly. Although some reports have suggested differing clinical patterns of EGPA based on ANCA positivity, the presence or absence of ANCA is less helpful in the diagnosis.13
DIFFERENTIATION
A key histologic difference between GPA and MPA is the presence of granulomatous inflammation in GPA and its absence in MPA under the current nomenclature system.9 Granulomatous inflammation can be seen in EGPA, but it is usually accompanied by eosinophils, which are less likely to be present in GPA and MPA.
The predominance of the ANCA immunofluorescence pattern and target antigen differs between GPA and MPA, with ANCA positivity occurring in 38% of patients with EGPA.13
SUMMARY
Conceptualizing vasculitic disease based on vessel size can be useful, but it is not an absolute definition. Although GPA, MPA, and EGPA predominantly affect small- to medium-sized vessels, these disease entities are phenotypically unique, with both shared features and differences. Common to all three entities is the potential for organ- and life-threatening manifestations, particularly involving the lungs, kidneys, nerves, gastrointestinal tract, and heart. All three entities need aggressive immuno suppression for severe disease. Recognition of these entities and the distinctions among them can guide the approach to diagnosis, treatment, and future outcomes.
Vasculitis refers to inflammation of the blood vessel. This inflammation can cause vessel wall thickening that compromises or occludes the vessel lumen, ultimately resulting in organ ischemia. It also can cause vessel wall attenuation that predisposes to aneurysm formation or breach of the vessel integrity with resultant hemorrhage into the tissue.
Vasculitis can be thought of as a primary or secondary process. Primary vasculitides are unique disease entities without a currently identified underlying cause in which vasculitis forms the pathologic basis of tissue injury. Vasculitis can occur secondary to medication exposure or an underlying illness, including infections, malignancy, cryoglobulinemia, and rheumatic diseases (such as systemic lupus erythematosus, rheumatoid arthritis, Sjögren syndrome, or myositis).
Primary vasculitides may differ in epidemiology, such as the age at which they occur and the gender most likely to be affected, their clinical manifestations (including signs, symptoms, and patterns of organ involvement), the diagnostic approach (biopsy, arteriography, and laboratory investigation), treatment (supportive care, glucocorticoids alone, or in combination with other immunosuppressants), and the size of the vessels predominantly affected (large, medium, or small).
Small-vessel vasculitis affects the arteriole, capillary, and venule. An excellent example of small-vessel vasculitis and the one most commonly encountered in clinical practice is cutaneous vasculitis, in which extravasation of erythrocytes from disrupted small vessels is observed histologically, with the clinical sequelae of palpable purpura. Although categorization based on the predominant vessel size that is affected is a helpful way to view these diseases, this is not absolute and each disease has the potential to affect a diverse range of vessels.
This article explores the clinical features and diagnosis of three forms of vasculitis that predominantly affect the small vessels: granulomatosis with polyangiitis (GPA [Wegener’s granulomatosis]), microscopic polyangiitis (MPA), and eosinophilic GPA (Churg-Strauss syndrome, EGPA).
GRANULOMATOSIS WITH POLYANGIITIS
Granulomatosis with polyangiitis is characterized by granulomatous inflammation involving the respiratory tract and by vasculitis affecting small- to medium-sized vessels in which necrotizing glomerulonephritis is common.
Wide range of presentations, manifestations
Approximately 90% of patients with GPA have upper or lower airway involvement or both.1 Upper airway or ear symptoms affect 73% of patients initially and 92% overall.1 Direct inspection of the nasal membranes shows a cobblestoned or ulcerated appearance, and computed tomography reveals mucosal thickening of the sinuses. In some instances, sinus disease can compromise blood supply to the cartilaginous portion of the nasal septum, leading to nasal septum perforations or collapse of the nasal bridge. Another manifestation of upper airway disease and GPA is subglottic stenosis, a narrowing in the subglottic region located just below the vocal cords. The narrowing typically spans about 1 cm and rarely extends or involves the remainder of the trachea.
The lungs are involved in 85% of patients.1 Radiographic abnormalities can be diverse and include bilateral pulmonary nodular infiltrates, single or multiple cavities, and bilateral ground glass infiltrates as can be seen in pulmonary hemorrhage (Figure). Bronchoscopy may reveal endobronchial stenosis, and pleural disease can occur rarely.
Approximately 20% of patients with GPA may have glomerulonephritis when they first present for medical attention, but it eventually develops in nearly 80% of patients during the disease course.1 Despite its potential for rapid progression, glomerulonephritis presents a diagnostic challenge because it is asymptomatic. It is detected by evidence of proteinuria and an active urine sediment with dysmorphic red blood cells and red blood cell casts.
Ocular involvement occurs eventually in 52% of patients with GPA.1 Any ocular structure can be affected and ocular involvement can be visually threatening. The more prominent ocular manifestations include scleritis/episcleritis or orbital disease.
Cutaneous manifestations, observed in 46% of patients, include verrucous-appearing lesions on the elbow and infarctions in the skin and nail folds.1 Other rare manifestations can occur, such as pericarditis and cerebral vasculitis.
Although nearly all patients present with upper or lower airway symptoms, the multisystem nature of GPA explains the wide range of presentations and the varying degrees of disease severity.
Differential diagnosis
The differential diagnosis in GPA is varied. Particularly in the setting of isolated lung or sinus disease, infection is foremost in the differential diagnosis. Even in the nonimmunosuppressed host, unusual infections such as mycobacteria, histoplasmosis, and other fungal infections should be considered. Lymphadenopathy, rarely seen in GPA, should raise concern for other causes of disease. Lymphoproliferative processes and other neoplasms, other rheumatic diseases, granulomatous disease (ie, sarcoidosis), and other causes of glomerulonephritis (when present) also merit consideration. Differentiation of these entities from GPA is essential because the treatment differs in many instances.
The differential diagnosis for patients who present with midline destructive lesions must include other causes of collapse of the nasal bridge, nasal septum perforation, and possibly palate destruction. Erosions of the hard palate in particular should raise an immediate red flag for entities other than GPA, such as lymphoproliferative diseases; rare infections, particularly if the patient has studied or worked abroad; and cocaine exposure.
Diagnostic evaluation
A diagnosis of GPA is typically based on the presence of histologic features in a clinically compatible setting. Diagnostic features include necrosis, granulomatous inflammation, vasculitis, and special stains and cultures negative for microorganisms.
Biopsy sites are determined by evidence of clinical disease affecting a target organ and the likelihood of obtaining diagnostically meaningful findings from that site. One challenge is that biopsies are not always diagnostic. The changes tend to be patchy and the likelihood of a positive yield is associated with the amount of tissue that can be obtained. Tissues from the ear, nose, and throat have a yield of about 20%, depending upon the site and the biopsy size. The highest yield comes from radiographically abnormal pulmonary parenchyma. Although transbronchial biopsies are attractive because they are less invasive than open lung biopsy, they are also far less diagnostic, with fewer than 10% having a positive yield. Because cutaneous vasculitis is observed in many settings, its presence is usually insufficient evidence for diagnosis. The renal histologic appearance is a focal, segmental, crescentic, and necrotizing glomerulonephritis that has few to no immune complexes (pauciimmune glomerulonephritis).1–3
Chest imaging should be performed in any patient in whom GPA is part of the differential diagnosis, since up to one-third of patients may be asymptomatic yet have pulmonary radiographic findings.
Laboratory assessment should include serum chemistries to evaluate renal and hepatic function, complete blood count, erythrocyte sedimentation rate, measurement of C-reactive protein, and urinalysis. If the urinalysis is positive for blood, microscopy should be performed on fresh urine to look for casts. In the setting of pulmonary-renal manifestations, testing for other causes, such as antiglomerular basement antibodies and antinuclear antibodies, should be considered.
Serologic testing for antineutrophil cytoplasmic antibodies (ANCA) has provided a useful tool in suggesting the diagnosis of GPA. Two forms of ANCA have been identified in patients with vasculitis: ANCA directed against the neutrophil serine protease proteinase-3 (PR3), which results in a cytoplasmic immunofluorescence (cANCA) pattern; and ANCA directed against the neutrophil enzyme myeloperoxidase (MPO), which causes a perinuclear immunofluorescence (pANCA) pattern.4 Approximately 80% to 95% of ANCA found in patients with active severe GPA are detectable PR3-cANCA, while 5% to 20% are MPO-pANCA.5 The predictive value of ANCA for the diagnosis depends on the spectrum of clinical features. As ANCA can be seen in other settings, ANCA as the basis for diagnosis in place of tissue biopsy should be used with caution and only in selected instances where their predictive value would equal that of biopsy. The presence of ANCA is not necessary to establish the diagnosis, as up to 20% of patients with GPA may be ANCA-negative.6
MICROSCOPIC POLYANGIITIS
The history of MPA dates to 1866, with the description of periarteritis nodosa.7 The term “microscopic polyarteritis” was introduced in 1948, when glomerular disease was recognized in some patients.8 In 1994, the Chapel Hill Consensus Conference defined MPA as a necrotizing vasculitis with few or no immune deposits that affects small vessels (ie, capillaries, venules, or arterioles). Necrotizing arteritis of small- and medium-sized arteries may be present. Necrotizing glomerulonephritis and pulmonary capillaritis commonly occur.9 MPA shares many clinical features with GPA and is currently said to be distinguished by the absence of granulomatous inflammation.9
Presentations and manifestations
In one assessment of organ system involvement in 85 patients with MPA, investigators observed glomerular syndrome in 82% of patients.10 They also found a high predilection for involvement of the skin, joints, and lungs. Pulmonary hemorrhage is a particularly important manifestation of MPA because it can be immediately life-threatening.
Differential diagnosis
The differential diagnosis for MPA is similar to GPA in the inclusion of other causes of classic pulmonary-renal syndromes, such as antiglomerular basement membrane disease and systemic lupus erythematosus. Poststreptococcal glomerulonephritis should be considered when the kidney is the predominant organ involved in the absence of lung disease. In the setting of pulmonary infiltrates, infections and neoplasms remain significant in the differential diagnosis.
Diagnostic evaluation
The diagnosis of MPA is based on consistent clinical features and compatible histologic findings. The histologic renal lesion is identical to that seen in GPA. Pulmonary disease typically includes capillaritis and is notable for the absence of evidence of immune deposition, in contrast to antiglomerular basement membrane disease.
Chest imaging is indicated when MPA is part of the differential diagnosis. Computed tomography is the preferred technique, as early alveolar hemorrhage that can occur in MPA may not be visualized on a chest radiograph.
Laboratory assessment should include serum chemistries, complete blood count, erythrocyte sedimentation rate, measurement of C-reactive protein, and urinalysis. Additional testing should be pursued for other diseases as indicated by the clinical features.
Approximately 40% to 80% of patients with MPA have MPO-pANCA.5 Approximately 15% of patients are MPO-pANCA positive,6 and 0% to 20% are ANCA-negative. As with GPA, ANCA is useful to suggest—but not diagnose—disease in many instances. The absence of ANCA does not rule out MPA.
EOSINOPHILIC GPA
Eosinophilic GPA is a unique entity characterized by eosinophil-rich and granulomatous inflammation involving the respiratory tract and necrotizing vasculitis of small- to medium-size vessels. It is also associated with asthma and eosinophilia.
Different disease phases
Eosinophilic GPA is often thought of as having three phases: prodromal, eosinophilic, and vasculitic.11,12 Although helpful conceptually, these phases may not always be present and may not occur in sequence.
The prodromal phase is characterized by asthma associated with allergic rhinitis with or without polyposis. The eosinophilic phase is characterized by the presence of eosinophilia in the blood and tissue. Eosinophilia is a prominent feature, although accurate detection and assessment can be challenging in the setting of glucocorticoid use for asthma as this normalizes the eosinophil count.
The vasculitic phase distinguishes EGPA from other eosinophilic disorders. Features of vasculitis may occur in multiple organ sites, including the nerves, lungs, heart, gastrointestinal tract, and kidneys. In one series of 96 patients, nearly 100% had asthma, and peripheral nervous system involvement in the form of mononeuritis multiplex was present in 72%.12 Cardiac involvement is of particular importance as it is a prominent cause of disease-related mortality. Cardiac manifestations include myocarditis, pericarditis, endocarditis, valvulitis, and coronary vasculitis.
Differential diagnosis
The differential diagnosis of EGPA shares similarities with GPA and MPA, but also includes eosino philic disorders such as hypereosinophilic syndrome, eosinophilic leukemia, and parasitic diseases.
Diagnostic evaluation
Diagnosis is often based on the presence of asthma, a finding of peripheral eosinophilia (> 1,500 cells/mm3), and the presence of systemic vasculitis involving, ideally, two or more extrapulmonary organs. While histologic confirmation remains ideal, demonstration of characteristic findings on biopsy can be difficult. Glomerular involvement is far less common than in GPA and MPA, but, when present, the renal lesion is identical. Pulmonary histologic findings can be diverse and include the classic “allergic-granuloma” as originally described by Churg and Strauss, as well as isolated granulomatous inflammation, eosinophilic inflammation, or small-vessel vasculitis. Tissue eosinophilia is a prominent finding that typically is seen on biopsies of skin, nerve, and gastrointestinal tissues.
Chest imaging should be performed when EGPA is part of the differential diagnosis. Because of the potential for cardiac involvement, a baseline echocardiogram should be obtained. Pulmonary function tests may be useful, particularly in patients who have a strong asthmatic component.
Similar to GPA and MPA, laboratory assessment includes serum chemistries, complete blood count with differential to determine the eosinophil count, erythrocyte sedimentation rate, measurement of C-reactive protein, and urinalysis. With the allergic and asthmatic components, immunoglobulin E levels are frequently elevated. Additional testing for other eosinophilic diseases should be pursued as indicated by the clinical features.
Only about 40% of patients are ANCA-positive.13 Most of these are MPO-pANCA, with PR3-cANCA occurring less commonly. Although some reports have suggested differing clinical patterns of EGPA based on ANCA positivity, the presence or absence of ANCA is less helpful in the diagnosis.13
DIFFERENTIATION
A key histologic difference between GPA and MPA is the presence of granulomatous inflammation in GPA and its absence in MPA under the current nomenclature system.9 Granulomatous inflammation can be seen in EGPA, but it is usually accompanied by eosinophils, which are less likely to be present in GPA and MPA.
The predominance of the ANCA immunofluorescence pattern and target antigen differs between GPA and MPA, with ANCA positivity occurring in 38% of patients with EGPA.13
SUMMARY
Conceptualizing vasculitic disease based on vessel size can be useful, but it is not an absolute definition. Although GPA, MPA, and EGPA predominantly affect small- to medium-sized vessels, these disease entities are phenotypically unique, with both shared features and differences. Common to all three entities is the potential for organ- and life-threatening manifestations, particularly involving the lungs, kidneys, nerves, gastrointestinal tract, and heart. All three entities need aggressive immuno suppression for severe disease. Recognition of these entities and the distinctions among them can guide the approach to diagnosis, treatment, and future outcomes.
- Hoffman GS, Kerr GS, Leavitt RY, et al Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Travis WD, Hoffman GS, Leavitt RY, Pass HI, Fauci AS. Surgical pathology of the lung in Wegener’s granulomatosis: review of 87 open lung biopsies from 67 patients. Am J Surg Pathol 1991; 15:315–333.
- Devaney KO, Travis WD, Hoffman G, Leavitt R, Lebovics R, Fauci AS. Interpretation of head and neck biopsies in Wegener’s granulomatosis: a pathologic study of 126 biopsies in 70 patients. Am J Surg Pathol 1990; 14:555–564.
- Bosch X, Guilabert A, Font J. Antineutrophil cytoplasmic antibodies. Lancet 2006; 368:404–418.
- Hoffman GS, Specks U. Antineutrophil cytoplasmic antibodies. Arthritis Rheum 1998;1521–1537.
- Wiik A. What you should know about PR3-ANCA. An introduction. Arthritis Res 2000; 2:252–254.
- Kussmaul A, Maier R. Über eine bisher nicht beschriebene eigenthümliche Arterienerkrankung (Periarteritis nodosa), die mit Morbus Brightii und rapid fortschreitender allgemeiner Muskellähmung einhergeht. Dtsch Arch Klin Med 1866; 1:484–518.
- Davson J, Ball J, Platt R. The kidney in periarteritis nodosa. Q J Med 1948; 17:175–202.
- Jennette C, Falk RJ, Andrassy K, et al Nomenclature of systemic vasculitides: proposal of an international consensus conference. Arthritis Rheum 1994; 37:187–192.
- Guillevin L, Durand-Gasselin B, Cevallos R, et al Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum 1999; 42:421–430.
- Keogh KA, Specks U. Churg-Strauss syndrome. Semin Respir Crit Care Med 2006; 27:148–157.
- Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome: clinical study and long-term follow-up of 96 patients. Medicine 1999; 78:26–37.
- Sablé-Fourtassou R, Cohen P, Mahr A, et al., for the French Vasculitis Study Group. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Intern Med 2005; 143:632–638.
- Hoffman GS, Kerr GS, Leavitt RY, et al Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Travis WD, Hoffman GS, Leavitt RY, Pass HI, Fauci AS. Surgical pathology of the lung in Wegener’s granulomatosis: review of 87 open lung biopsies from 67 patients. Am J Surg Pathol 1991; 15:315–333.
- Devaney KO, Travis WD, Hoffman G, Leavitt R, Lebovics R, Fauci AS. Interpretation of head and neck biopsies in Wegener’s granulomatosis: a pathologic study of 126 biopsies in 70 patients. Am J Surg Pathol 1990; 14:555–564.
- Bosch X, Guilabert A, Font J. Antineutrophil cytoplasmic antibodies. Lancet 2006; 368:404–418.
- Hoffman GS, Specks U. Antineutrophil cytoplasmic antibodies. Arthritis Rheum 1998;1521–1537.
- Wiik A. What you should know about PR3-ANCA. An introduction. Arthritis Res 2000; 2:252–254.
- Kussmaul A, Maier R. Über eine bisher nicht beschriebene eigenthümliche Arterienerkrankung (Periarteritis nodosa), die mit Morbus Brightii und rapid fortschreitender allgemeiner Muskellähmung einhergeht. Dtsch Arch Klin Med 1866; 1:484–518.
- Davson J, Ball J, Platt R. The kidney in periarteritis nodosa. Q J Med 1948; 17:175–202.
- Jennette C, Falk RJ, Andrassy K, et al Nomenclature of systemic vasculitides: proposal of an international consensus conference. Arthritis Rheum 1994; 37:187–192.
- Guillevin L, Durand-Gasselin B, Cevallos R, et al Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum 1999; 42:421–430.
- Keogh KA, Specks U. Churg-Strauss syndrome. Semin Respir Crit Care Med 2006; 27:148–157.
- Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome: clinical study and long-term follow-up of 96 patients. Medicine 1999; 78:26–37.
- Sablé-Fourtassou R, Cohen P, Mahr A, et al., for the French Vasculitis Study Group. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Intern Med 2005; 143:632–638.