User login
Do Probiotics Reduce C diff Risk in Hospitalized Patients?

A 68-year-old woman is admitted to the hospital with a diagnosis of community-acquired pneumonia. Should you add probiotics to her antibiotic regimen to prevent infection with Clostridium difficile?
Clostridium difficile infection (CDI) leads to significant morbidity, mortality, and treatment failures. In 2011, it culminated in a cost of $4.8 billion and 29,000 deaths.2,3 Risk factors for infection include antibiotic use, hospitalization, older age, and medical comorbidities.2 Probiotics have been proposed as one way to prevent CDI.
Several systematic reviews have demonstrated efficacy for probiotics in the prevention of CDI, although not all of them followed Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines or focused specifically on hospitalized patients, who are at increased risk.4-6 The largest high-quality randomized controlled trial (RCT) on the use of probiotics to prevent CDI, the PLACIDE trial, found no difference in CDI incidence between inpatients (ages 65 and older) who did and those who did not receive probiotics in addition to their oral or parenteral antibiotics; however, this trial had a lower incidence of CDI than was assumed in the power calculations.7 Guidelines from the American College of Gastroenterology and the Society for Healthcare Epidemiology of America do not include a recommendation for the use of probiotics in CDI prevention.8,9
Given the conflicting and poor-quality evidence and lack of recommendations, an additional systematic review and meta-analysis was performed, following PRISMA guidelines and focusing on studies conducted only in hospitalized adults.
STUDY SUMMARY
Probiotics prevent CDI in this population
This meta-analysis of 19 RCTs evaluated the efficacy of probiotics for the prevention of CDI in 6261 hospitalized adults taking antibiotics. All patients were 18 or older (mean age, 68-69) and received antibiotics orally, intravenously, or via both routes, for any medical indication.
Trials were included if the intervention was for CDI prevention and if the probiotic strains used were Lactobacillus, Saccharomyces, Bifidobacterium, or Streptococcus (alone or in combination). Probiotic doses ranged from 4 billion to 900 billion colony-forming U/d and were started from 1 to 7 days after the first antibiotic dose. Duration of probiotic use was either fixed at 14 to 21 days or varied based on the duration of antibiotics (extending 3-14 d after the last antibiotic dose).
Control groups received matching placebo in all but 2 trials; those 2 used usual care of no probiotics as the control. Exclusion criteria included pregnancy, immunocompromise, intensive care, a prosthetic heart valve, and pre-existing gastrointestinal disorders.
[polldaddy:10452484]
Continue to: The risk for CDI...
The risk for CDI was lower in the probiotic group (range 0%-11%) than in the control group (0%-40%), with no heterogeneity when the data from all 19 studies were pooled (relative risk [RR], 0.42). The median incidence of CDI in the control groups from all studies was 4%, which yielded a number needed to treat (NNT) of 43.
The researchers examined the NNT at varying incidence rates. If the CDI incidence was 1.2%, the NNT to prevent 1 case of CDI was 144; if the incidence was 7.4%, the NNT was 23. Compared with control groups, there was a significant reduction in CDI if probiotics were started within 1 to 2 days of antibiotic initiation (RR, 0.32), but not if they were started at 3 to 7 days (RR, 0.70). There was no significant difference in adverse events (ie, cramping, nausea, fever, soft stools, flatulence, taste disturbance) between probiotic and control groups (14% vs 16%).
WHAT’S NEW
Added benefit if probiotics taken sooner
This high-quality meta-analysis shows that administration of probiotics to hospitalized patients—particularly when started within 1 to 2 days of initiating antibiotic therapy—can prevent CDI.
CAVEATS
Limited applicability, lack of recommendations
Findings from this meta-analysis do not apply to patients who are pregnant; who have an immunocompromising condition, a prosthetic heart valve, or a pre-existing gastrointestinal disorder (eg, irritable bowel disease, pancreatitis); or who require intensive care. In addition, specific recommendations as to the optimal probiotic species, dose, formulation, and duration of use cannot be made based on this meta-analysis. Lastly, findings from this study do not apply to patients treated with antibiotics in the ambulatory care setting.
CHALLENGES TO IMPLEMENTATION
Limited availability in hospitals
The largest barrier to giving probiotics to hospitalized adults is their availability on local hospital formularies. Probiotics are not technically a medication; t
Continue to: ACKNOWLEDGMENT
ACKNOWLEDGMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2019. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2019;68[6]:351-352,354).
1. Shen NT, Maw A, Tmanova LL, et al. Timely use of probiotics in hospitalized adults prevents Clostridium difficile infection: a systematic review with meta-regression analysis. Gastroenterology. 2017;152(8):1889-1900.e9.
2. Evans CT, Safdar N. Current trends in the epidemiology and outcomes of Clostridium difficile infection. Clin Infect Dis. 2015;60(suppl 2):S66-S71.
3. Lessa FC, Winston LG, McDonald LC, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372(24):2369-2370.
4. Goldenberg JZ, Yap C, Lytvyn L, et al. Probiotics for the prevention of Clostridium difficile-associated diarrhea in adults and children. Cochrane Database Syst Rev. 2017;12:CD006095.
5. Lau CS, Chamberlain RS. Probiotics are effective at preventing Clostridium difficile–associated diarrhea: a systematic review and meta-analysis. Int J Gen Med. 2016:22:27-37.
6. Johnston BC, Goldenberg JZ, Guyatt GH. Probiotics for the prevention of Clostridium difficile–associated diarrhea. In response. Ann Intern Med. 2013;158(12):706-707.
7. Allen SJ, Wareham K, Wang D, et al. Lactobacilli and bifidobacteria in the prevention of antibiotic-associated diarrhoea and Clostridium difficile diarrhoea in older inpatients (PLACIDE): a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2013;382(9900):1249-1257.
8. Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol. 2013;108(4):478-498.
9. Cohen SH, Gerding DN, Johnson S, et al; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010;31(5):431-455.
A 68-year-old woman is admitted to the hospital with a diagnosis of community-acquired pneumonia. Should you add probiotics to her antibiotic regimen to prevent infection with Clostridium difficile?
Clostridium difficile infection (CDI) leads to significant morbidity, mortality, and treatment failures. In 2011, it culminated in a cost of $4.8 billion and 29,000 deaths.2,3 Risk factors for infection include antibiotic use, hospitalization, older age, and medical comorbidities.2 Probiotics have been proposed as one way to prevent CDI.
Several systematic reviews have demonstrated efficacy for probiotics in the prevention of CDI, although not all of them followed Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines or focused specifically on hospitalized patients, who are at increased risk.4-6 The largest high-quality randomized controlled trial (RCT) on the use of probiotics to prevent CDI, the PLACIDE trial, found no difference in CDI incidence between inpatients (ages 65 and older) who did and those who did not receive probiotics in addition to their oral or parenteral antibiotics; however, this trial had a lower incidence of CDI than was assumed in the power calculations.7 Guidelines from the American College of Gastroenterology and the Society for Healthcare Epidemiology of America do not include a recommendation for the use of probiotics in CDI prevention.8,9
Given the conflicting and poor-quality evidence and lack of recommendations, an additional systematic review and meta-analysis was performed, following PRISMA guidelines and focusing on studies conducted only in hospitalized adults.
STUDY SUMMARY
Probiotics prevent CDI in this population
This meta-analysis of 19 RCTs evaluated the efficacy of probiotics for the prevention of CDI in 6261 hospitalized adults taking antibiotics. All patients were 18 or older (mean age, 68-69) and received antibiotics orally, intravenously, or via both routes, for any medical indication.
Trials were included if the intervention was for CDI prevention and if the probiotic strains used were Lactobacillus, Saccharomyces, Bifidobacterium, or Streptococcus (alone or in combination). Probiotic doses ranged from 4 billion to 900 billion colony-forming U/d and were started from 1 to 7 days after the first antibiotic dose. Duration of probiotic use was either fixed at 14 to 21 days or varied based on the duration of antibiotics (extending 3-14 d after the last antibiotic dose).
Control groups received matching placebo in all but 2 trials; those 2 used usual care of no probiotics as the control. Exclusion criteria included pregnancy, immunocompromise, intensive care, a prosthetic heart valve, and pre-existing gastrointestinal disorders.
[polldaddy:10452484]
Continue to: The risk for CDI...
The risk for CDI was lower in the probiotic group (range 0%-11%) than in the control group (0%-40%), with no heterogeneity when the data from all 19 studies were pooled (relative risk [RR], 0.42). The median incidence of CDI in the control groups from all studies was 4%, which yielded a number needed to treat (NNT) of 43.
The researchers examined the NNT at varying incidence rates. If the CDI incidence was 1.2%, the NNT to prevent 1 case of CDI was 144; if the incidence was 7.4%, the NNT was 23. Compared with control groups, there was a significant reduction in CDI if probiotics were started within 1 to 2 days of antibiotic initiation (RR, 0.32), but not if they were started at 3 to 7 days (RR, 0.70). There was no significant difference in adverse events (ie, cramping, nausea, fever, soft stools, flatulence, taste disturbance) between probiotic and control groups (14% vs 16%).
WHAT’S NEW
Added benefit if probiotics taken sooner
This high-quality meta-analysis shows that administration of probiotics to hospitalized patients—particularly when started within 1 to 2 days of initiating antibiotic therapy—can prevent CDI.
CAVEATS
Limited applicability, lack of recommendations
Findings from this meta-analysis do not apply to patients who are pregnant; who have an immunocompromising condition, a prosthetic heart valve, or a pre-existing gastrointestinal disorder (eg, irritable bowel disease, pancreatitis); or who require intensive care. In addition, specific recommendations as to the optimal probiotic species, dose, formulation, and duration of use cannot be made based on this meta-analysis. Lastly, findings from this study do not apply to patients treated with antibiotics in the ambulatory care setting.
CHALLENGES TO IMPLEMENTATION
Limited availability in hospitals
The largest barrier to giving probiotics to hospitalized adults is their availability on local hospital formularies. Probiotics are not technically a medication; t
Continue to: ACKNOWLEDGMENT
ACKNOWLEDGMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2019. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2019;68[6]:351-352,354).
A 68-year-old woman is admitted to the hospital with a diagnosis of community-acquired pneumonia. Should you add probiotics to her antibiotic regimen to prevent infection with Clostridium difficile?
Clostridium difficile infection (CDI) leads to significant morbidity, mortality, and treatment failures. In 2011, it culminated in a cost of $4.8 billion and 29,000 deaths.2,3 Risk factors for infection include antibiotic use, hospitalization, older age, and medical comorbidities.2 Probiotics have been proposed as one way to prevent CDI.
Several systematic reviews have demonstrated efficacy for probiotics in the prevention of CDI, although not all of them followed Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines or focused specifically on hospitalized patients, who are at increased risk.4-6 The largest high-quality randomized controlled trial (RCT) on the use of probiotics to prevent CDI, the PLACIDE trial, found no difference in CDI incidence between inpatients (ages 65 and older) who did and those who did not receive probiotics in addition to their oral or parenteral antibiotics; however, this trial had a lower incidence of CDI than was assumed in the power calculations.7 Guidelines from the American College of Gastroenterology and the Society for Healthcare Epidemiology of America do not include a recommendation for the use of probiotics in CDI prevention.8,9
Given the conflicting and poor-quality evidence and lack of recommendations, an additional systematic review and meta-analysis was performed, following PRISMA guidelines and focusing on studies conducted only in hospitalized adults.
STUDY SUMMARY
Probiotics prevent CDI in this population
This meta-analysis of 19 RCTs evaluated the efficacy of probiotics for the prevention of CDI in 6261 hospitalized adults taking antibiotics. All patients were 18 or older (mean age, 68-69) and received antibiotics orally, intravenously, or via both routes, for any medical indication.
Trials were included if the intervention was for CDI prevention and if the probiotic strains used were Lactobacillus, Saccharomyces, Bifidobacterium, or Streptococcus (alone or in combination). Probiotic doses ranged from 4 billion to 900 billion colony-forming U/d and were started from 1 to 7 days after the first antibiotic dose. Duration of probiotic use was either fixed at 14 to 21 days or varied based on the duration of antibiotics (extending 3-14 d after the last antibiotic dose).
Control groups received matching placebo in all but 2 trials; those 2 used usual care of no probiotics as the control. Exclusion criteria included pregnancy, immunocompromise, intensive care, a prosthetic heart valve, and pre-existing gastrointestinal disorders.
[polldaddy:10452484]
Continue to: The risk for CDI...
The risk for CDI was lower in the probiotic group (range 0%-11%) than in the control group (0%-40%), with no heterogeneity when the data from all 19 studies were pooled (relative risk [RR], 0.42). The median incidence of CDI in the control groups from all studies was 4%, which yielded a number needed to treat (NNT) of 43.
The researchers examined the NNT at varying incidence rates. If the CDI incidence was 1.2%, the NNT to prevent 1 case of CDI was 144; if the incidence was 7.4%, the NNT was 23. Compared with control groups, there was a significant reduction in CDI if probiotics were started within 1 to 2 days of antibiotic initiation (RR, 0.32), but not if they were started at 3 to 7 days (RR, 0.70). There was no significant difference in adverse events (ie, cramping, nausea, fever, soft stools, flatulence, taste disturbance) between probiotic and control groups (14% vs 16%).
WHAT’S NEW
Added benefit if probiotics taken sooner
This high-quality meta-analysis shows that administration of probiotics to hospitalized patients—particularly when started within 1 to 2 days of initiating antibiotic therapy—can prevent CDI.
CAVEATS
Limited applicability, lack of recommendations
Findings from this meta-analysis do not apply to patients who are pregnant; who have an immunocompromising condition, a prosthetic heart valve, or a pre-existing gastrointestinal disorder (eg, irritable bowel disease, pancreatitis); or who require intensive care. In addition, specific recommendations as to the optimal probiotic species, dose, formulation, and duration of use cannot be made based on this meta-analysis. Lastly, findings from this study do not apply to patients treated with antibiotics in the ambulatory care setting.
CHALLENGES TO IMPLEMENTATION
Limited availability in hospitals
The largest barrier to giving probiotics to hospitalized adults is their availability on local hospital formularies. Probiotics are not technically a medication; t
Continue to: ACKNOWLEDGMENT
ACKNOWLEDGMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2019. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2019;68[6]:351-352,354).
1. Shen NT, Maw A, Tmanova LL, et al. Timely use of probiotics in hospitalized adults prevents Clostridium difficile infection: a systematic review with meta-regression analysis. Gastroenterology. 2017;152(8):1889-1900.e9.
2. Evans CT, Safdar N. Current trends in the epidemiology and outcomes of Clostridium difficile infection. Clin Infect Dis. 2015;60(suppl 2):S66-S71.
3. Lessa FC, Winston LG, McDonald LC, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372(24):2369-2370.
4. Goldenberg JZ, Yap C, Lytvyn L, et al. Probiotics for the prevention of Clostridium difficile-associated diarrhea in adults and children. Cochrane Database Syst Rev. 2017;12:CD006095.
5. Lau CS, Chamberlain RS. Probiotics are effective at preventing Clostridium difficile–associated diarrhea: a systematic review and meta-analysis. Int J Gen Med. 2016:22:27-37.
6. Johnston BC, Goldenberg JZ, Guyatt GH. Probiotics for the prevention of Clostridium difficile–associated diarrhea. In response. Ann Intern Med. 2013;158(12):706-707.
7. Allen SJ, Wareham K, Wang D, et al. Lactobacilli and bifidobacteria in the prevention of antibiotic-associated diarrhoea and Clostridium difficile diarrhoea in older inpatients (PLACIDE): a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2013;382(9900):1249-1257.
8. Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol. 2013;108(4):478-498.
9. Cohen SH, Gerding DN, Johnson S, et al; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010;31(5):431-455.
1. Shen NT, Maw A, Tmanova LL, et al. Timely use of probiotics in hospitalized adults prevents Clostridium difficile infection: a systematic review with meta-regression analysis. Gastroenterology. 2017;152(8):1889-1900.e9.
2. Evans CT, Safdar N. Current trends in the epidemiology and outcomes of Clostridium difficile infection. Clin Infect Dis. 2015;60(suppl 2):S66-S71.
3. Lessa FC, Winston LG, McDonald LC, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372(24):2369-2370.
4. Goldenberg JZ, Yap C, Lytvyn L, et al. Probiotics for the prevention of Clostridium difficile-associated diarrhea in adults and children. Cochrane Database Syst Rev. 2017;12:CD006095.
5. Lau CS, Chamberlain RS. Probiotics are effective at preventing Clostridium difficile–associated diarrhea: a systematic review and meta-analysis. Int J Gen Med. 2016:22:27-37.
6. Johnston BC, Goldenberg JZ, Guyatt GH. Probiotics for the prevention of Clostridium difficile–associated diarrhea. In response. Ann Intern Med. 2013;158(12):706-707.
7. Allen SJ, Wareham K, Wang D, et al. Lactobacilli and bifidobacteria in the prevention of antibiotic-associated diarrhoea and Clostridium difficile diarrhoea in older inpatients (PLACIDE): a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2013;382(9900):1249-1257.
8. Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol. 2013;108(4):478-498.
9. Cohen SH, Gerding DN, Johnson S, et al; Society for Healthcare Epidemiology of America; Infectious Diseases Society of America. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010;31(5):431-455.

Should You Switch the DAPT Agent a Month After ACS?

A 60-year-old man visits your clinic 30 days after he was hospitalized for acute coronary syndrome (ACS) due to ST-elevation myocardial infarction (STEMI). The patient underwent percutaneous coronary intervention (PCI) with placement of a stent and received aspirin and a loading dose of ticagrelor for antiplatelet therapy. He was discharged on dual antiplatelet therapy (DAPT) consisting of daily aspirin and ticagrelor. He asks about the risk for bleeding associated with these medications. Should you recommend any changes?
Platelet inhibition during and after ACS to prevent recurrent ischemic events is a cornerstone of treatment for patients after a myocardial infarction (MI).2 Current American College of Cardiology/American Heart Association and European Society of Cardiology guidelines recommend that patients with coronary artery disease who recently had an MI continue DAPT with aspirin and a P2Y12 blocker (clopidogrel, ticlopidine, ticagrelor, prasugrel, or cangrelor) for 12 months following ACS to reduce recurrent ischemia.2-4
Studies have shown that using the newer P2Y12 inhibitors (prasugrel and ticagrelor) after PCI leads to a significant reduction in recurrent ischemic events, compared with clopidogrel.5-7 These data prompted a guideline change recommending the use of the newer agents over clopidogrel for 12 months following PCI.2 Follow-up studies show strong evidence for the use of the newer P2Y12 agents in the first month following PCI, but they also demonstrate an increased bleeding risk in the maintenance phase (from 30 days to 12 months post-PCI).6,7 This increased risk is the basis for the study by Cuisset et al, which examined switching from a newer P2Y12 agent to clopidogrel after the initial 30-day period following PCI.
STUDY SUMMARY
Switched DAPT is superior
This open-label RCT (N = 646) evaluated changing DAPT from aspirin plus a newer P2Y12 blocker (prasugrel or ticagrelor) to a combination of aspirin and clopidogrel after the first month of DAPT post-ACS.1 Prior to PCI, patients received a loading dose of ticagrelor (180 mg) or prasugrel (60 mg). Subsequently, all patients took aspirin (75 mg/d) and either prasugrel (10 mg/d) or ticagrelor (90 mg bid) for 1 month. After 30 days, participants who had no adverse events were randomly assigned in a 1:1 ratio to continue the aspirin and newer P2Y12 blocker regimen or switch to aspirin and clopidogrel (75 mg/d). In the following year, researchers examined the composite outcome of cardiovascular death, urgent revascularization, stroke, and major bleeding (defined by a Bleeding Academic Research Consortium [BARC] classification ≥ Type 2 at 1-year post-ACS).
Of the participants (average age, 60), 40% had a STEMI and 60% had a non-STEMI. Overall, 43% of patients were prescribed ticagrelor and 57% prasugrel. At 1 year, 86% of the switched-DAPT group and 75% of the unchanged-DAPT group were still taking their medication. The composite outcome at 1-year follow-up was lower in the switched group compared with the unchanged group (13.4% vs 26.3%; hazard ratio [HR], 0.48; 95% confidence interval [CI], 0.34-0.68; number needed to treat [NNT], 8).
Bleeding events (ranging from minimal to fatal) were lower in the switched group (9.3% vs 23.5%; HR, 0.39; 95% CI, 0.27-0.57; NNT, 7) and events identified as BARC ≥ Type 2 (defined as needing medical treatment) were also lower in this group (4% vs 14.9%; HR, 0.30, 95% CI, 0.18-0.50; NNT, 9). There were no significant differences in reported recurrent cardiovascular ischemic events (9.3% vs 11.5%; HR, 0.80, 95% CI, 0.50-1.29).
WHAT’S NEW
Less bleeding, no increase in ischemic events
Cardiology guidelines recommend the newer P2Y12 blockers as part of DAPT after ACS, but this trial showed switching to clopidogrel for DAPT after 30 days of treatment lowers bleeding events with no difference in recurrent ischemic events.2-4
Continue to: CAVEATS
CAVEATS
Less-than-ideal study methods
In this open-label and unblinded study, the investigators adjudicating critical events were blinded to the treatment allocation. However, patients could self-report minor bleeding and medication discontinuation for which no consultation was sought. In addition, the investigators used opaque envelopes—a less-than-ideal method—to conceal allocation at enrollment.
CHALLENGES TO IMPLEMENTATION
PCP may not change cardiologist’s prescription
Implementing this practice is facilitated by the comparatively lower cost of clopidogrel versus the newer P2Y12 blockers. However, after ACS and PCI treatment, cardiologists usually initiate antiplatelet therapy and may continue to manage patients after discharge. The primary care provider (PCP) may not be responsible for the DAPT switch initially; furthermore, ordering a switch may require coordination if the PCP is hesitant to change the cardiologist’s prescription. Lastly, guidelines currently recommend using the newer P2Y12 blockers for 12 months.2 CR
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2019. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2019;68[3]:162,164).
1. Cuisset T, Deharo P, Quilici J, et al. Benefit of switching dual antiplatelet therapy after acute coronary syndrome: the TOPIC (timing of platelet inhibition after acute coronary syndrome) randomized study. Eur Heart J. 2017;38(41):3070-3078.
2. Levine GN, Bates ER, Bittl JA, et al. 2016 ACC/AHA guideline focused update on duration of dual antiplatelet therapy in patients with coronary artery disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2016;68(10):1082-1115.
3. Steg PG, James SK, Atar D, et al; Task Force on the management of ST-segment elevation acute myocardial infarction of the European Society of Cardiology (ESC). ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur Heart J. 2012;33(20):2569-2619.
4. Roffi M, Patrono C, Collet J-P, et al; ESC Scientific Document Group. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: task force for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J. 2015;37(3):267-315.
5. Antman EM, Wiviott SD, Murphy SA, et al. Early and late benefits of prasugrel in patients with acute coronary syndromes undergoing percutaneous coronary intervention: a TRITON-TIMI 38 (TRial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet InhibitioN with Prasugrel-Thrombolysis In Myocardial Infarction) analysis. J Am Coll Cardiol. 2008;51(21): 2028-2033.
6. Wallentin L, Becker RC, Budaj A, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045-1057.
7. Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357(20):2001-2015.
A 60-year-old man visits your clinic 30 days after he was hospitalized for acute coronary syndrome (ACS) due to ST-elevation myocardial infarction (STEMI). The patient underwent percutaneous coronary intervention (PCI) with placement of a stent and received aspirin and a loading dose of ticagrelor for antiplatelet therapy. He was discharged on dual antiplatelet therapy (DAPT) consisting of daily aspirin and ticagrelor. He asks about the risk for bleeding associated with these medications. Should you recommend any changes?
Platelet inhibition during and after ACS to prevent recurrent ischemic events is a cornerstone of treatment for patients after a myocardial infarction (MI).2 Current American College of Cardiology/American Heart Association and European Society of Cardiology guidelines recommend that patients with coronary artery disease who recently had an MI continue DAPT with aspirin and a P2Y12 blocker (clopidogrel, ticlopidine, ticagrelor, prasugrel, or cangrelor) for 12 months following ACS to reduce recurrent ischemia.2-4
Studies have shown that using the newer P2Y12 inhibitors (prasugrel and ticagrelor) after PCI leads to a significant reduction in recurrent ischemic events, compared with clopidogrel.5-7 These data prompted a guideline change recommending the use of the newer agents over clopidogrel for 12 months following PCI.2 Follow-up studies show strong evidence for the use of the newer P2Y12 agents in the first month following PCI, but they also demonstrate an increased bleeding risk in the maintenance phase (from 30 days to 12 months post-PCI).6,7 This increased risk is the basis for the study by Cuisset et al, which examined switching from a newer P2Y12 agent to clopidogrel after the initial 30-day period following PCI.
STUDY SUMMARY
Switched DAPT is superior
This open-label RCT (N = 646) evaluated changing DAPT from aspirin plus a newer P2Y12 blocker (prasugrel or ticagrelor) to a combination of aspirin and clopidogrel after the first month of DAPT post-ACS.1 Prior to PCI, patients received a loading dose of ticagrelor (180 mg) or prasugrel (60 mg). Subsequently, all patients took aspirin (75 mg/d) and either prasugrel (10 mg/d) or ticagrelor (90 mg bid) for 1 month. After 30 days, participants who had no adverse events were randomly assigned in a 1:1 ratio to continue the aspirin and newer P2Y12 blocker regimen or switch to aspirin and clopidogrel (75 mg/d). In the following year, researchers examined the composite outcome of cardiovascular death, urgent revascularization, stroke, and major bleeding (defined by a Bleeding Academic Research Consortium [BARC] classification ≥ Type 2 at 1-year post-ACS).
Of the participants (average age, 60), 40% had a STEMI and 60% had a non-STEMI. Overall, 43% of patients were prescribed ticagrelor and 57% prasugrel. At 1 year, 86% of the switched-DAPT group and 75% of the unchanged-DAPT group were still taking their medication. The composite outcome at 1-year follow-up was lower in the switched group compared with the unchanged group (13.4% vs 26.3%; hazard ratio [HR], 0.48; 95% confidence interval [CI], 0.34-0.68; number needed to treat [NNT], 8).
Bleeding events (ranging from minimal to fatal) were lower in the switched group (9.3% vs 23.5%; HR, 0.39; 95% CI, 0.27-0.57; NNT, 7) and events identified as BARC ≥ Type 2 (defined as needing medical treatment) were also lower in this group (4% vs 14.9%; HR, 0.30, 95% CI, 0.18-0.50; NNT, 9). There were no significant differences in reported recurrent cardiovascular ischemic events (9.3% vs 11.5%; HR, 0.80, 95% CI, 0.50-1.29).
WHAT’S NEW
Less bleeding, no increase in ischemic events
Cardiology guidelines recommend the newer P2Y12 blockers as part of DAPT after ACS, but this trial showed switching to clopidogrel for DAPT after 30 days of treatment lowers bleeding events with no difference in recurrent ischemic events.2-4
Continue to: CAVEATS
CAVEATS
Less-than-ideal study methods
In this open-label and unblinded study, the investigators adjudicating critical events were blinded to the treatment allocation. However, patients could self-report minor bleeding and medication discontinuation for which no consultation was sought. In addition, the investigators used opaque envelopes—a less-than-ideal method—to conceal allocation at enrollment.
CHALLENGES TO IMPLEMENTATION
PCP may not change cardiologist’s prescription
Implementing this practice is facilitated by the comparatively lower cost of clopidogrel versus the newer P2Y12 blockers. However, after ACS and PCI treatment, cardiologists usually initiate antiplatelet therapy and may continue to manage patients after discharge. The primary care provider (PCP) may not be responsible for the DAPT switch initially; furthermore, ordering a switch may require coordination if the PCP is hesitant to change the cardiologist’s prescription. Lastly, guidelines currently recommend using the newer P2Y12 blockers for 12 months.2 CR
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2019. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2019;68[3]:162,164).
A 60-year-old man visits your clinic 30 days after he was hospitalized for acute coronary syndrome (ACS) due to ST-elevation myocardial infarction (STEMI). The patient underwent percutaneous coronary intervention (PCI) with placement of a stent and received aspirin and a loading dose of ticagrelor for antiplatelet therapy. He was discharged on dual antiplatelet therapy (DAPT) consisting of daily aspirin and ticagrelor. He asks about the risk for bleeding associated with these medications. Should you recommend any changes?
Platelet inhibition during and after ACS to prevent recurrent ischemic events is a cornerstone of treatment for patients after a myocardial infarction (MI).2 Current American College of Cardiology/American Heart Association and European Society of Cardiology guidelines recommend that patients with coronary artery disease who recently had an MI continue DAPT with aspirin and a P2Y12 blocker (clopidogrel, ticlopidine, ticagrelor, prasugrel, or cangrelor) for 12 months following ACS to reduce recurrent ischemia.2-4
Studies have shown that using the newer P2Y12 inhibitors (prasugrel and ticagrelor) after PCI leads to a significant reduction in recurrent ischemic events, compared with clopidogrel.5-7 These data prompted a guideline change recommending the use of the newer agents over clopidogrel for 12 months following PCI.2 Follow-up studies show strong evidence for the use of the newer P2Y12 agents in the first month following PCI, but they also demonstrate an increased bleeding risk in the maintenance phase (from 30 days to 12 months post-PCI).6,7 This increased risk is the basis for the study by Cuisset et al, which examined switching from a newer P2Y12 agent to clopidogrel after the initial 30-day period following PCI.
STUDY SUMMARY
Switched DAPT is superior
This open-label RCT (N = 646) evaluated changing DAPT from aspirin plus a newer P2Y12 blocker (prasugrel or ticagrelor) to a combination of aspirin and clopidogrel after the first month of DAPT post-ACS.1 Prior to PCI, patients received a loading dose of ticagrelor (180 mg) or prasugrel (60 mg). Subsequently, all patients took aspirin (75 mg/d) and either prasugrel (10 mg/d) or ticagrelor (90 mg bid) for 1 month. After 30 days, participants who had no adverse events were randomly assigned in a 1:1 ratio to continue the aspirin and newer P2Y12 blocker regimen or switch to aspirin and clopidogrel (75 mg/d). In the following year, researchers examined the composite outcome of cardiovascular death, urgent revascularization, stroke, and major bleeding (defined by a Bleeding Academic Research Consortium [BARC] classification ≥ Type 2 at 1-year post-ACS).
Of the participants (average age, 60), 40% had a STEMI and 60% had a non-STEMI. Overall, 43% of patients were prescribed ticagrelor and 57% prasugrel. At 1 year, 86% of the switched-DAPT group and 75% of the unchanged-DAPT group were still taking their medication. The composite outcome at 1-year follow-up was lower in the switched group compared with the unchanged group (13.4% vs 26.3%; hazard ratio [HR], 0.48; 95% confidence interval [CI], 0.34-0.68; number needed to treat [NNT], 8).
Bleeding events (ranging from minimal to fatal) were lower in the switched group (9.3% vs 23.5%; HR, 0.39; 95% CI, 0.27-0.57; NNT, 7) and events identified as BARC ≥ Type 2 (defined as needing medical treatment) were also lower in this group (4% vs 14.9%; HR, 0.30, 95% CI, 0.18-0.50; NNT, 9). There were no significant differences in reported recurrent cardiovascular ischemic events (9.3% vs 11.5%; HR, 0.80, 95% CI, 0.50-1.29).
WHAT’S NEW
Less bleeding, no increase in ischemic events
Cardiology guidelines recommend the newer P2Y12 blockers as part of DAPT after ACS, but this trial showed switching to clopidogrel for DAPT after 30 days of treatment lowers bleeding events with no difference in recurrent ischemic events.2-4
Continue to: CAVEATS
CAVEATS
Less-than-ideal study methods
In this open-label and unblinded study, the investigators adjudicating critical events were blinded to the treatment allocation. However, patients could self-report minor bleeding and medication discontinuation for which no consultation was sought. In addition, the investigators used opaque envelopes—a less-than-ideal method—to conceal allocation at enrollment.
CHALLENGES TO IMPLEMENTATION
PCP may not change cardiologist’s prescription
Implementing this practice is facilitated by the comparatively lower cost of clopidogrel versus the newer P2Y12 blockers. However, after ACS and PCI treatment, cardiologists usually initiate antiplatelet therapy and may continue to manage patients after discharge. The primary care provider (PCP) may not be responsible for the DAPT switch initially; furthermore, ordering a switch may require coordination if the PCP is hesitant to change the cardiologist’s prescription. Lastly, guidelines currently recommend using the newer P2Y12 blockers for 12 months.2 CR
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2019. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2019;68[3]:162,164).
1. Cuisset T, Deharo P, Quilici J, et al. Benefit of switching dual antiplatelet therapy after acute coronary syndrome: the TOPIC (timing of platelet inhibition after acute coronary syndrome) randomized study. Eur Heart J. 2017;38(41):3070-3078.
2. Levine GN, Bates ER, Bittl JA, et al. 2016 ACC/AHA guideline focused update on duration of dual antiplatelet therapy in patients with coronary artery disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2016;68(10):1082-1115.
3. Steg PG, James SK, Atar D, et al; Task Force on the management of ST-segment elevation acute myocardial infarction of the European Society of Cardiology (ESC). ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur Heart J. 2012;33(20):2569-2619.
4. Roffi M, Patrono C, Collet J-P, et al; ESC Scientific Document Group. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: task force for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J. 2015;37(3):267-315.
5. Antman EM, Wiviott SD, Murphy SA, et al. Early and late benefits of prasugrel in patients with acute coronary syndromes undergoing percutaneous coronary intervention: a TRITON-TIMI 38 (TRial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet InhibitioN with Prasugrel-Thrombolysis In Myocardial Infarction) analysis. J Am Coll Cardiol. 2008;51(21): 2028-2033.
6. Wallentin L, Becker RC, Budaj A, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045-1057.
7. Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357(20):2001-2015.
1. Cuisset T, Deharo P, Quilici J, et al. Benefit of switching dual antiplatelet therapy after acute coronary syndrome: the TOPIC (timing of platelet inhibition after acute coronary syndrome) randomized study. Eur Heart J. 2017;38(41):3070-3078.
2. Levine GN, Bates ER, Bittl JA, et al. 2016 ACC/AHA guideline focused update on duration of dual antiplatelet therapy in patients with coronary artery disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2016;68(10):1082-1115.
3. Steg PG, James SK, Atar D, et al; Task Force on the management of ST-segment elevation acute myocardial infarction of the European Society of Cardiology (ESC). ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur Heart J. 2012;33(20):2569-2619.
4. Roffi M, Patrono C, Collet J-P, et al; ESC Scientific Document Group. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: task force for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J. 2015;37(3):267-315.
5. Antman EM, Wiviott SD, Murphy SA, et al. Early and late benefits of prasugrel in patients with acute coronary syndromes undergoing percutaneous coronary intervention: a TRITON-TIMI 38 (TRial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet InhibitioN with Prasugrel-Thrombolysis In Myocardial Infarction) analysis. J Am Coll Cardiol. 2008;51(21): 2028-2033.
6. Wallentin L, Becker RC, Budaj A, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045-1057.
7. Wiviott SD, Braunwald E, McCabe CH, et al; TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357(20):2001-2015.

Do probiotics reduce C diff risk in hospitalized patients?
ILLUSTRATIVE CASE
A 68-year-old woman is admitted to the hospital with a diagnosis of community-acquired pneumonia. Should you add probiotics to her antibiotic regimen to prevent infection with Clostridium difficile?
Clostridium difficile infection (CDI) leads to significant morbidity, mortality, and treatment failures. In 2011, it culminated in a cost of $4.8 billion and 29,000 deaths.2,3 Risk factors for infection include antibiotic use, hospitalization, older age, and medical comorbidities.2 Probiotics have been proposed as one way to prevent CDI.
While several systematic reviews have demonstrated efficacy for probiotics in the prevention of CDI,4-6 guidelines from the American College of Gastroenterology and the Society for Healthcare Epidemiology of America did not incorporate a recommendation for the use of probiotics in their CDI prevention strategy.7,8
The PLACIDE trial studied the use of probiotics in inpatients ages ≥ 65 years receiving either oral or parenteral antibiotics and found no difference in the incidence of CDI in those who received probiotics vs those who did not.9 Even though the PLACIDE trial was the largest, high-quality, randomized controlled trial (RCT) on the use of probiotics to prevent CDI, it had a lower incidence of CDI than was assumed in the power calculations. Additionally, previous systematic reviews did not always follow the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines, and did not focus specifically on hospitalized patients, who are at higher risk for CDI.
Given the conflicting and poor evidence and recommendations, an additional systematic review and meta-analysis was performed following PRISMA guidelines and focusing on studies conducted only on hospitalized adults.
STUDY SUMMARY
Probiotics prevent CDI in hospitalized patients receiving antibiotics
This meta-analysis of 19 RCTs evaluated the efficacy of probiotics for the prevention of CDI in 6261 adult hospitalized patients taking antibiotics. All patients were ≥ 18 years (mean age 68-69 years) and received antibiotics orally, intravenously, or via both routes for any medical indication.
Trials were included if the intervention was for CDI prevention and if the probiotics used were 1 or a combination of 4 strains (Lactobacillus, Saccharomyces, Bifidobacterium, Streptococcus). Probiotic doses ranged from 4 billion to 900 billion colony-forming u/day and were started from 1 to 7 days after first antibiotic dose. Duration of probiotic use was either fixed at between 14 and 21 days or varied based on the duration of antibiotics (extending 3-14 days after the last antibiotic dose).
Continue to: Control groups received...
Control groups received matching placebo in all trials but 2; those 2 used usual care of no probiotics as the control. Common patient exclusions were pregnancy, immune system compromise, intensive care, a prosthetic heart valve, and pre-existing gastrointestinal disorders.
The risk for CDI was lower in the probiotic group (range 0%-11%) than in the control group (0%-40%) with no heterogeneity (I2 = 0.0%; P = .56) when the data were pooled from all 19 studies (relative risk [RR] = 0.42; 95% confidence interval [CI], 0.30-0.57). The median incidence of CDI in the control groups from all studies was 4%, which yielded a number needed to treat (NNT) of 43 (95% CI, 36-58).
The researchers examined the NNT at varying incidence rates. If the incidence of CDI was 1.2%, the NNT to prevent 1 case of CDI was 144, and if the incidence was 7.4%, the NNT was 23. Compared with control groups, there was a significant reduction in CDI if probiotics were started within 1 to 2 days of antibiotic initiation (RR = 0.32; 95% CI, 0.22-0.48), but not if they were started at 3 to 7 days (RR = 0.70; 95% CI, 0.40-1.2). There was no significant difference in adverse events (ie, cramping, nausea, fever, soft stools, flatulence, taste disturbance) between probiotic and control groups (14% vs 16%; P = .35).
WHAT’S NEW
Probiotics provide added benefit if taken sooner rather than later
This high-quality meta-analysis shows that administration of probiotics to hospitalized patients—particularly when started within 1 to 2 days of initiating antibiotic therapy—can prevent CDI.
CAVEATS
Findings do not apply to all patients; specific recommendations are lacking
Findings from this meta-analysis do not apply to patients who have an immunocompromising condition, are pregnant, have a prosthetic heart valve, have a pre-existing gastrointestinal disorder (eg, irritable bowel disease, pancreatitis), or require intensive care. In addition, specific recommendations as to the optimal probiotic species, dose, formulation, and duration of use cannot be made based on this meta-analysis. Lastly, findings from this study do not apply to patients treated with antibiotics in the ambulatory care setting.
Continue to: CHALLENGES TO IMPLEMENTATION
CHALLENGES TO IMPLEMENTATION
Lack of “medication” status leads to limited availability in hospitals
The largest barrier to giving probiotics to hospitalized adult patients is the availability of probiotics on local hospital formularies. Probiotics are not technically a medication; they are not regulated or approved by the US Food and Drug Administration and thus, insurance coverage and availability for inpatient use are limited. Lastly, US cost-effectiveness data are lacking, although such data would likely be favorable given the high costs associated with treatment of CDI.
ACKNOWLEDGMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Shen NT, Maw A, Tmanova LL, et al. Timely use of probiotics in hospitalized adults prevents Clostridium difficile infection: a systematic review with meta-regression analysis. Gastroenterology. 2017;152:1889-1900.e9.
2. Evans CT, Safdar N. Current trends in the epidemiology and outcomes of Clostridium difficile infection. Clin Infect Dis. 2015;60(Suppl 2):S66-S71.
3. Lessa FC, Winston LG, McDonald LC, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372:2369-2370.
4. Goldenberg JZ, Yap C, Lytvyn L. Probiotics for the prevention of Clostridium difficile-associated diarrhea in adults and children. Cochrane Database Syst Rev. 2017;12:CD006095.
5. Lau CS, Chamberlain RS. Probiotics are effective at preventing Clostridium difficile–associated diarrhea: a systematic review and meta-analysis. Int J Gen Med. 2016:22:27-37.
6. Johnston BC, Goldenberg JZ, Guyatt GH. Probiotics for the prevention of Clostridium difficile–associated diarrhea. In response. Ann Intern Med. 2013;158:706-707.
7. Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol. 2013;108:478-498.
8. Cohen SH, Gerding DN, Johnson S, et al. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010;31:431-455.
9. Allen SJ, Wareham K, Wang D, et al. Lactobacilli and bifidobacteria in the prevention of antibiotic-associated diarrhoea and Clostridium difficile diarrhoea in older inpatients (PLACIDE): a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2013;382:1249-1257.
ILLUSTRATIVE CASE
A 68-year-old woman is admitted to the hospital with a diagnosis of community-acquired pneumonia. Should you add probiotics to her antibiotic regimen to prevent infection with Clostridium difficile?
Clostridium difficile infection (CDI) leads to significant morbidity, mortality, and treatment failures. In 2011, it culminated in a cost of $4.8 billion and 29,000 deaths.2,3 Risk factors for infection include antibiotic use, hospitalization, older age, and medical comorbidities.2 Probiotics have been proposed as one way to prevent CDI.
While several systematic reviews have demonstrated efficacy for probiotics in the prevention of CDI,4-6 guidelines from the American College of Gastroenterology and the Society for Healthcare Epidemiology of America did not incorporate a recommendation for the use of probiotics in their CDI prevention strategy.7,8
The PLACIDE trial studied the use of probiotics in inpatients ages ≥ 65 years receiving either oral or parenteral antibiotics and found no difference in the incidence of CDI in those who received probiotics vs those who did not.9 Even though the PLACIDE trial was the largest, high-quality, randomized controlled trial (RCT) on the use of probiotics to prevent CDI, it had a lower incidence of CDI than was assumed in the power calculations. Additionally, previous systematic reviews did not always follow the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines, and did not focus specifically on hospitalized patients, who are at higher risk for CDI.
Given the conflicting and poor evidence and recommendations, an additional systematic review and meta-analysis was performed following PRISMA guidelines and focusing on studies conducted only on hospitalized adults.
STUDY SUMMARY
Probiotics prevent CDI in hospitalized patients receiving antibiotics
This meta-analysis of 19 RCTs evaluated the efficacy of probiotics for the prevention of CDI in 6261 adult hospitalized patients taking antibiotics. All patients were ≥ 18 years (mean age 68-69 years) and received antibiotics orally, intravenously, or via both routes for any medical indication.
Trials were included if the intervention was for CDI prevention and if the probiotics used were 1 or a combination of 4 strains (Lactobacillus, Saccharomyces, Bifidobacterium, Streptococcus). Probiotic doses ranged from 4 billion to 900 billion colony-forming u/day and were started from 1 to 7 days after first antibiotic dose. Duration of probiotic use was either fixed at between 14 and 21 days or varied based on the duration of antibiotics (extending 3-14 days after the last antibiotic dose).
Continue to: Control groups received...
Control groups received matching placebo in all trials but 2; those 2 used usual care of no probiotics as the control. Common patient exclusions were pregnancy, immune system compromise, intensive care, a prosthetic heart valve, and pre-existing gastrointestinal disorders.
The risk for CDI was lower in the probiotic group (range 0%-11%) than in the control group (0%-40%) with no heterogeneity (I2 = 0.0%; P = .56) when the data were pooled from all 19 studies (relative risk [RR] = 0.42; 95% confidence interval [CI], 0.30-0.57). The median incidence of CDI in the control groups from all studies was 4%, which yielded a number needed to treat (NNT) of 43 (95% CI, 36-58).
The researchers examined the NNT at varying incidence rates. If the incidence of CDI was 1.2%, the NNT to prevent 1 case of CDI was 144, and if the incidence was 7.4%, the NNT was 23. Compared with control groups, there was a significant reduction in CDI if probiotics were started within 1 to 2 days of antibiotic initiation (RR = 0.32; 95% CI, 0.22-0.48), but not if they were started at 3 to 7 days (RR = 0.70; 95% CI, 0.40-1.2). There was no significant difference in adverse events (ie, cramping, nausea, fever, soft stools, flatulence, taste disturbance) between probiotic and control groups (14% vs 16%; P = .35).
WHAT’S NEW
Probiotics provide added benefit if taken sooner rather than later
This high-quality meta-analysis shows that administration of probiotics to hospitalized patients—particularly when started within 1 to 2 days of initiating antibiotic therapy—can prevent CDI.
CAVEATS
Findings do not apply to all patients; specific recommendations are lacking
Findings from this meta-analysis do not apply to patients who have an immunocompromising condition, are pregnant, have a prosthetic heart valve, have a pre-existing gastrointestinal disorder (eg, irritable bowel disease, pancreatitis), or require intensive care. In addition, specific recommendations as to the optimal probiotic species, dose, formulation, and duration of use cannot be made based on this meta-analysis. Lastly, findings from this study do not apply to patients treated with antibiotics in the ambulatory care setting.
Continue to: CHALLENGES TO IMPLEMENTATION
CHALLENGES TO IMPLEMENTATION
Lack of “medication” status leads to limited availability in hospitals
The largest barrier to giving probiotics to hospitalized adult patients is the availability of probiotics on local hospital formularies. Probiotics are not technically a medication; they are not regulated or approved by the US Food and Drug Administration and thus, insurance coverage and availability for inpatient use are limited. Lastly, US cost-effectiveness data are lacking, although such data would likely be favorable given the high costs associated with treatment of CDI.
ACKNOWLEDGMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
ILLUSTRATIVE CASE
A 68-year-old woman is admitted to the hospital with a diagnosis of community-acquired pneumonia. Should you add probiotics to her antibiotic regimen to prevent infection with Clostridium difficile?
Clostridium difficile infection (CDI) leads to significant morbidity, mortality, and treatment failures. In 2011, it culminated in a cost of $4.8 billion and 29,000 deaths.2,3 Risk factors for infection include antibiotic use, hospitalization, older age, and medical comorbidities.2 Probiotics have been proposed as one way to prevent CDI.
While several systematic reviews have demonstrated efficacy for probiotics in the prevention of CDI,4-6 guidelines from the American College of Gastroenterology and the Society for Healthcare Epidemiology of America did not incorporate a recommendation for the use of probiotics in their CDI prevention strategy.7,8
The PLACIDE trial studied the use of probiotics in inpatients ages ≥ 65 years receiving either oral or parenteral antibiotics and found no difference in the incidence of CDI in those who received probiotics vs those who did not.9 Even though the PLACIDE trial was the largest, high-quality, randomized controlled trial (RCT) on the use of probiotics to prevent CDI, it had a lower incidence of CDI than was assumed in the power calculations. Additionally, previous systematic reviews did not always follow the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines, and did not focus specifically on hospitalized patients, who are at higher risk for CDI.
Given the conflicting and poor evidence and recommendations, an additional systematic review and meta-analysis was performed following PRISMA guidelines and focusing on studies conducted only on hospitalized adults.
STUDY SUMMARY
Probiotics prevent CDI in hospitalized patients receiving antibiotics
This meta-analysis of 19 RCTs evaluated the efficacy of probiotics for the prevention of CDI in 6261 adult hospitalized patients taking antibiotics. All patients were ≥ 18 years (mean age 68-69 years) and received antibiotics orally, intravenously, or via both routes for any medical indication.
Trials were included if the intervention was for CDI prevention and if the probiotics used were 1 or a combination of 4 strains (Lactobacillus, Saccharomyces, Bifidobacterium, Streptococcus). Probiotic doses ranged from 4 billion to 900 billion colony-forming u/day and were started from 1 to 7 days after first antibiotic dose. Duration of probiotic use was either fixed at between 14 and 21 days or varied based on the duration of antibiotics (extending 3-14 days after the last antibiotic dose).
Continue to: Control groups received...
Control groups received matching placebo in all trials but 2; those 2 used usual care of no probiotics as the control. Common patient exclusions were pregnancy, immune system compromise, intensive care, a prosthetic heart valve, and pre-existing gastrointestinal disorders.
The risk for CDI was lower in the probiotic group (range 0%-11%) than in the control group (0%-40%) with no heterogeneity (I2 = 0.0%; P = .56) when the data were pooled from all 19 studies (relative risk [RR] = 0.42; 95% confidence interval [CI], 0.30-0.57). The median incidence of CDI in the control groups from all studies was 4%, which yielded a number needed to treat (NNT) of 43 (95% CI, 36-58).
The researchers examined the NNT at varying incidence rates. If the incidence of CDI was 1.2%, the NNT to prevent 1 case of CDI was 144, and if the incidence was 7.4%, the NNT was 23. Compared with control groups, there was a significant reduction in CDI if probiotics were started within 1 to 2 days of antibiotic initiation (RR = 0.32; 95% CI, 0.22-0.48), but not if they were started at 3 to 7 days (RR = 0.70; 95% CI, 0.40-1.2). There was no significant difference in adverse events (ie, cramping, nausea, fever, soft stools, flatulence, taste disturbance) between probiotic and control groups (14% vs 16%; P = .35).
WHAT’S NEW
Probiotics provide added benefit if taken sooner rather than later
This high-quality meta-analysis shows that administration of probiotics to hospitalized patients—particularly when started within 1 to 2 days of initiating antibiotic therapy—can prevent CDI.
CAVEATS
Findings do not apply to all patients; specific recommendations are lacking
Findings from this meta-analysis do not apply to patients who have an immunocompromising condition, are pregnant, have a prosthetic heart valve, have a pre-existing gastrointestinal disorder (eg, irritable bowel disease, pancreatitis), or require intensive care. In addition, specific recommendations as to the optimal probiotic species, dose, formulation, and duration of use cannot be made based on this meta-analysis. Lastly, findings from this study do not apply to patients treated with antibiotics in the ambulatory care setting.
Continue to: CHALLENGES TO IMPLEMENTATION
CHALLENGES TO IMPLEMENTATION
Lack of “medication” status leads to limited availability in hospitals
The largest barrier to giving probiotics to hospitalized adult patients is the availability of probiotics on local hospital formularies. Probiotics are not technically a medication; they are not regulated or approved by the US Food and Drug Administration and thus, insurance coverage and availability for inpatient use are limited. Lastly, US cost-effectiveness data are lacking, although such data would likely be favorable given the high costs associated with treatment of CDI.
ACKNOWLEDGMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Shen NT, Maw A, Tmanova LL, et al. Timely use of probiotics in hospitalized adults prevents Clostridium difficile infection: a systematic review with meta-regression analysis. Gastroenterology. 2017;152:1889-1900.e9.
2. Evans CT, Safdar N. Current trends in the epidemiology and outcomes of Clostridium difficile infection. Clin Infect Dis. 2015;60(Suppl 2):S66-S71.
3. Lessa FC, Winston LG, McDonald LC, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372:2369-2370.
4. Goldenberg JZ, Yap C, Lytvyn L. Probiotics for the prevention of Clostridium difficile-associated diarrhea in adults and children. Cochrane Database Syst Rev. 2017;12:CD006095.
5. Lau CS, Chamberlain RS. Probiotics are effective at preventing Clostridium difficile–associated diarrhea: a systematic review and meta-analysis. Int J Gen Med. 2016:22:27-37.
6. Johnston BC, Goldenberg JZ, Guyatt GH. Probiotics for the prevention of Clostridium difficile–associated diarrhea. In response. Ann Intern Med. 2013;158:706-707.
7. Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol. 2013;108:478-498.
8. Cohen SH, Gerding DN, Johnson S, et al. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010;31:431-455.
9. Allen SJ, Wareham K, Wang D, et al. Lactobacilli and bifidobacteria in the prevention of antibiotic-associated diarrhoea and Clostridium difficile diarrhoea in older inpatients (PLACIDE): a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2013;382:1249-1257.
1. Shen NT, Maw A, Tmanova LL, et al. Timely use of probiotics in hospitalized adults prevents Clostridium difficile infection: a systematic review with meta-regression analysis. Gastroenterology. 2017;152:1889-1900.e9.
2. Evans CT, Safdar N. Current trends in the epidemiology and outcomes of Clostridium difficile infection. Clin Infect Dis. 2015;60(Suppl 2):S66-S71.
3. Lessa FC, Winston LG, McDonald LC, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372:2369-2370.
4. Goldenberg JZ, Yap C, Lytvyn L. Probiotics for the prevention of Clostridium difficile-associated diarrhea in adults and children. Cochrane Database Syst Rev. 2017;12:CD006095.
5. Lau CS, Chamberlain RS. Probiotics are effective at preventing Clostridium difficile–associated diarrhea: a systematic review and meta-analysis. Int J Gen Med. 2016:22:27-37.
6. Johnston BC, Goldenberg JZ, Guyatt GH. Probiotics for the prevention of Clostridium difficile–associated diarrhea. In response. Ann Intern Med. 2013;158:706-707.
7. Surawicz CM, Brandt LJ, Binion DG, et al. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am J Gastroenterol. 2013;108:478-498.
8. Cohen SH, Gerding DN, Johnson S, et al. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol. 2010;31:431-455.
9. Allen SJ, Wareham K, Wang D, et al. Lactobacilli and bifidobacteria in the prevention of antibiotic-associated diarrhoea and Clostridium difficile diarrhoea in older inpatients (PLACIDE): a randomised, double-blind, placebo-controlled, multicentre trial. Lancet. 2013;382:1249-1257.
PRACTICE CHANGER
Start probiotics within 1 to 2 days of starting antibiotics in hospitalized patients to reduce the risk of Clostridium difficile infection.1
STRENGTH OF RECOMMENDATION
A: Based on a meta-analysis of randomized controlled trials.
Shen NT, Maw A, Tmanova LL, et al. Timely use of probiotics in hospitalized adults prevents Clostridium difficile infection: a systematic review with meta-regression analysis. Gastroenterology. 2017;152:1889-1900. e9.

How effective is spironolactone for treating resistant hypertension?
EVIDENCE SUMMARY
A 2017 meta-analysis of 4 RCTs (869 patients) evaluated the effectiveness of prescribing spironolactone for patients with resistant hypertension, defined as above-goal blood pressure (BP) despite treatment with at least 3 BP-lowering drugs (at least 1 of which was a diuretic).1 All 4 trials compared spironolactone 25 to 50 mg/d with placebo. Follow-up periods ranged from 8 to 16 weeks. The primary outcomes were systolic and diastolic BPs, which were evaluated in the office, at home, or with an ambulatory monitor.
Spironolactone markedly lowers systolic and diastolic BP
A statistically significant reduction in SBP occurred in the spironolactone group compared with the placebo group (weighted mean difference [WMD] = −16.7 mm Hg; 95% confidence interval [CI], −27.5 to −5.8 mm Hg). DBP also decreased (WMD = −6.11 mm Hg; 95% CI, −9.34 to −2.88 mm Hg).
Because significant heterogeneity was found in the initial pooled results (I2 = 96% for SBP; I2 = 85% for DBP), investigators performed an analysis that excluded a single study with a small sample size. The re-analysis continued to show significant reductions in SBP and DBP for spironolactone compared with placebo (SBP: WMD = −10.8 mm Hg; 95% CI, −13.16 to −8.43 mm Hg; DBP: WMD = −4.62 mm Hg; 95% CI, −6.05 to −3.2 mm Hg; I2 = 35%), confirming that the excluded trial was the source of heterogeneity in the initial analysis and that spironolactone continued to significantly lower BP for the treatment group compared with controls.
Add-on treatment with spironolactone also reduces BP
A 2016 meta-analysis of 5 RCTs with a total of 553 patients examined the effectiveness of add-on treatment with spironolactone (25-50 mg/d) for patients with resistant hypertension, defined as failure to achieve BP < 140/90 mm Hg despite treatment with 3 or more BP-lowering drugs, including one diuretic.2 Spironolactone was compared with placebo in 4 trials and with ramipril in the remaining study. The follow-up periods were 8 to 16 weeks. Researchers separated BP outcomes into 24-hour ambulatory systolic/diastolic BPs and office systolic/diastolic BPs.
The 24-hour ambulatory BPs were significantly lower in the spironolactone group compared with the control group (24-hour SBP: WMD = −10.5 mm Hg; 95% CI, −12.3 to −8.71 mm Hg; 24-hour DBP: WMD = −4.09 mm Hg; 95% CI, −5.28 to −2.91 mm Hg). No significant heterogeneity was noted in these analyses.
Office-based BPs also were markedly reduced in spironolactone groups compared with controls (office SBP: WMD = −17 mm Hg; 95% CI, −25 to −8.95 mm Hg); office DBP: WMD = −6.18 mm Hg; 95% CI, −9.3 to −3.05 mm Hg). Because the office-based BP data showed significant heterogeneity (I2 = 94% for SBP and 84.2% for DBP), 2 studies determined to be of lower quality caused by lack of detailed methodology were excluded from analysis, yielding continued statistically significant reductions in SBP (WMD = −11.7 mm Hg; 95% CI, −14.4 to −8.95 mm Hg) and DBP (WMD = −4.07 mm Hg; 95% CI, −5.6 to −2.54 mm Hg) compared with controls. Heterogeneity also decreased when the 2 studies were excluded (I2 = 21% for SBP and I2 = 59% for DBP).
How spironolactone compares with alternative drugs
A 2017 meta-analysis of 5 RCTs with 662 patients evaluated the effectiveness of spironolactone (25-50 mg/d) on resistant hypertension in patients taking 3 medications compared with a control group—placebo in 3 trials, placebo or bisoprolol (5-10 mg) in 1 trial, and an alternative treatment (candesartan 8 mg, atenolol 100 mg, or alpha methyldopa 750 mg) in 1 trial.3 Follow-up periods ranged from 4 to 16 weeks. Researchers evaluated changes in office and 24-hour ambulatory or home BP and completed separate analyses of pooled data for spironolactone compared with placebo groups, and spironolactone compared with alternative treatment groups.
Continue to: Investigators found a statistically significant...
Investigators found a statistically significant reduction in office SBP and DBP among patients taking spironolactone compared with control groups (SBP: WMD = −15.7 mm Hg; 95% CI, −20.5 to −11 mm Hg; DBP: WMD = −6.21 mm Hg; 95% CI, −8.33 to −4.1 mm Hg). A significant decrease also occurred in 24-hour ambulatory home SBP and DBP (SBP: MD = −8.7 mm Hg; 95% CI, −8.79 to −8.62 mm Hg; DBP: WMD = −4.12 mm Hg; 95% CI, −4.48 to −3.75 mm Hg).
Patients treated with spironolactone showed a marked decrease in home SBP compared with alternative drug groups (WMD = −4.5 mm Hg; 95% CI, −4.63 to −4.37 mm Hg), but alternative drugs reduced home DBP significantly more than spironolactone (WMD = 0.6 mm Hg; 95% CI, 0.55-0.65 mm Hg). Marked heterogeneity was found in these analyses, and the authors also noted that reductions in SBP are more clinically relevant than decreases in DBP.
RECOMMENDATIONS
The 2017 American Heart Association/American College of Cardiology evidence-based guideline recommends considering adding a mineralocorticoid receptor agonist to treatment regimens for resistant hypertension when: office BP remains ≥ 130/80 mm Hg; the patient is prescribed at least 3 antihypertensive agents at optimal doses including a diuretic; pseudoresistance (nonadherence, inaccurate measurements) is excluded; reversible lifestyle factors have been addressed; substances that interfere with BP treatment (such as nonsteroidal anti-inflammatory drugs and oral contraceptive pills) are excluded; and screening for secondary causes of hypertension is complete.4
The United Kingdom’s National Institute for Health and Care Excellence (NICE) evidence-based guideline recommends considering spironolactone 25 mg/d to treat resistant hypertension if the patient’s potassium level is 4.5 mmol/L or lower and BP is higher than 140/90 mm Hg despite treatment with an optimal or best-tolerated dose of an angiotensin-converting enzyme inhibitor or angiotensin II receptor blocker plus a calcium-channel blocker and diuretic.5
Editor’s takeaway
The evidence from multiple RCTs convincingly shows the effectiveness of spironolactone. Despite the SOR of C because of a disease-oriented outcome, we do treat to blood pressure goals, and therefore, spironolactone is a good option.
1. Zhao D, Liu H, Dong P, et al. A meta-analysis of add-on use of spironolactone in patients with resistant hypertension. Int J Cardiol. 2017;233:113-117.
2. Wang C, Xiong B, Huang J. Efficacy and safety of spironolactone in patients with resistant hypertension: a meta-analysis of randomised controlled trials. Heart Lung Circ. 2016;25:1021-1030.
3. Liu L, Xu B, Ju Y. Addition of spironolactone in patients with resistant hypertension: a meta-analysis of randomized controlled trials. Clin Exp Hypertens. 2017;39:257-263.
4. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2017. https://doi.org/10.1161/HYP.0000000000000065. Accessed June 6, 2019.
5. National Institute for Health and Care Excellence. Hypertension in adults: diagnosis and management. Clinical guideline [CG127]. August 2011. https://www.nice.org.uk/guidance/cg127/chapter/1-guidance#initiating-and-monitoring-antihypertensive-drug-treatment-including-blood-pressure-targets-2. Accessed June 6, 2019.
EVIDENCE SUMMARY
A 2017 meta-analysis of 4 RCTs (869 patients) evaluated the effectiveness of prescribing spironolactone for patients with resistant hypertension, defined as above-goal blood pressure (BP) despite treatment with at least 3 BP-lowering drugs (at least 1 of which was a diuretic).1 All 4 trials compared spironolactone 25 to 50 mg/d with placebo. Follow-up periods ranged from 8 to 16 weeks. The primary outcomes were systolic and diastolic BPs, which were evaluated in the office, at home, or with an ambulatory monitor.
Spironolactone markedly lowers systolic and diastolic BP
A statistically significant reduction in SBP occurred in the spironolactone group compared with the placebo group (weighted mean difference [WMD] = −16.7 mm Hg; 95% confidence interval [CI], −27.5 to −5.8 mm Hg). DBP also decreased (WMD = −6.11 mm Hg; 95% CI, −9.34 to −2.88 mm Hg).
Because significant heterogeneity was found in the initial pooled results (I2 = 96% for SBP; I2 = 85% for DBP), investigators performed an analysis that excluded a single study with a small sample size. The re-analysis continued to show significant reductions in SBP and DBP for spironolactone compared with placebo (SBP: WMD = −10.8 mm Hg; 95% CI, −13.16 to −8.43 mm Hg; DBP: WMD = −4.62 mm Hg; 95% CI, −6.05 to −3.2 mm Hg; I2 = 35%), confirming that the excluded trial was the source of heterogeneity in the initial analysis and that spironolactone continued to significantly lower BP for the treatment group compared with controls.
Add-on treatment with spironolactone also reduces BP
A 2016 meta-analysis of 5 RCTs with a total of 553 patients examined the effectiveness of add-on treatment with spironolactone (25-50 mg/d) for patients with resistant hypertension, defined as failure to achieve BP < 140/90 mm Hg despite treatment with 3 or more BP-lowering drugs, including one diuretic.2 Spironolactone was compared with placebo in 4 trials and with ramipril in the remaining study. The follow-up periods were 8 to 16 weeks. Researchers separated BP outcomes into 24-hour ambulatory systolic/diastolic BPs and office systolic/diastolic BPs.
The 24-hour ambulatory BPs were significantly lower in the spironolactone group compared with the control group (24-hour SBP: WMD = −10.5 mm Hg; 95% CI, −12.3 to −8.71 mm Hg; 24-hour DBP: WMD = −4.09 mm Hg; 95% CI, −5.28 to −2.91 mm Hg). No significant heterogeneity was noted in these analyses.
Office-based BPs also were markedly reduced in spironolactone groups compared with controls (office SBP: WMD = −17 mm Hg; 95% CI, −25 to −8.95 mm Hg); office DBP: WMD = −6.18 mm Hg; 95% CI, −9.3 to −3.05 mm Hg). Because the office-based BP data showed significant heterogeneity (I2 = 94% for SBP and 84.2% for DBP), 2 studies determined to be of lower quality caused by lack of detailed methodology were excluded from analysis, yielding continued statistically significant reductions in SBP (WMD = −11.7 mm Hg; 95% CI, −14.4 to −8.95 mm Hg) and DBP (WMD = −4.07 mm Hg; 95% CI, −5.6 to −2.54 mm Hg) compared with controls. Heterogeneity also decreased when the 2 studies were excluded (I2 = 21% for SBP and I2 = 59% for DBP).
How spironolactone compares with alternative drugs
A 2017 meta-analysis of 5 RCTs with 662 patients evaluated the effectiveness of spironolactone (25-50 mg/d) on resistant hypertension in patients taking 3 medications compared with a control group—placebo in 3 trials, placebo or bisoprolol (5-10 mg) in 1 trial, and an alternative treatment (candesartan 8 mg, atenolol 100 mg, or alpha methyldopa 750 mg) in 1 trial.3 Follow-up periods ranged from 4 to 16 weeks. Researchers evaluated changes in office and 24-hour ambulatory or home BP and completed separate analyses of pooled data for spironolactone compared with placebo groups, and spironolactone compared with alternative treatment groups.
Continue to: Investigators found a statistically significant...
Investigators found a statistically significant reduction in office SBP and DBP among patients taking spironolactone compared with control groups (SBP: WMD = −15.7 mm Hg; 95% CI, −20.5 to −11 mm Hg; DBP: WMD = −6.21 mm Hg; 95% CI, −8.33 to −4.1 mm Hg). A significant decrease also occurred in 24-hour ambulatory home SBP and DBP (SBP: MD = −8.7 mm Hg; 95% CI, −8.79 to −8.62 mm Hg; DBP: WMD = −4.12 mm Hg; 95% CI, −4.48 to −3.75 mm Hg).
Patients treated with spironolactone showed a marked decrease in home SBP compared with alternative drug groups (WMD = −4.5 mm Hg; 95% CI, −4.63 to −4.37 mm Hg), but alternative drugs reduced home DBP significantly more than spironolactone (WMD = 0.6 mm Hg; 95% CI, 0.55-0.65 mm Hg). Marked heterogeneity was found in these analyses, and the authors also noted that reductions in SBP are more clinically relevant than decreases in DBP.
RECOMMENDATIONS
The 2017 American Heart Association/American College of Cardiology evidence-based guideline recommends considering adding a mineralocorticoid receptor agonist to treatment regimens for resistant hypertension when: office BP remains ≥ 130/80 mm Hg; the patient is prescribed at least 3 antihypertensive agents at optimal doses including a diuretic; pseudoresistance (nonadherence, inaccurate measurements) is excluded; reversible lifestyle factors have been addressed; substances that interfere with BP treatment (such as nonsteroidal anti-inflammatory drugs and oral contraceptive pills) are excluded; and screening for secondary causes of hypertension is complete.4
The United Kingdom’s National Institute for Health and Care Excellence (NICE) evidence-based guideline recommends considering spironolactone 25 mg/d to treat resistant hypertension if the patient’s potassium level is 4.5 mmol/L or lower and BP is higher than 140/90 mm Hg despite treatment with an optimal or best-tolerated dose of an angiotensin-converting enzyme inhibitor or angiotensin II receptor blocker plus a calcium-channel blocker and diuretic.5
Editor’s takeaway
The evidence from multiple RCTs convincingly shows the effectiveness of spironolactone. Despite the SOR of C because of a disease-oriented outcome, we do treat to blood pressure goals, and therefore, spironolactone is a good option.
EVIDENCE SUMMARY
A 2017 meta-analysis of 4 RCTs (869 patients) evaluated the effectiveness of prescribing spironolactone for patients with resistant hypertension, defined as above-goal blood pressure (BP) despite treatment with at least 3 BP-lowering drugs (at least 1 of which was a diuretic).1 All 4 trials compared spironolactone 25 to 50 mg/d with placebo. Follow-up periods ranged from 8 to 16 weeks. The primary outcomes were systolic and diastolic BPs, which were evaluated in the office, at home, or with an ambulatory monitor.
Spironolactone markedly lowers systolic and diastolic BP
A statistically significant reduction in SBP occurred in the spironolactone group compared with the placebo group (weighted mean difference [WMD] = −16.7 mm Hg; 95% confidence interval [CI], −27.5 to −5.8 mm Hg). DBP also decreased (WMD = −6.11 mm Hg; 95% CI, −9.34 to −2.88 mm Hg).
Because significant heterogeneity was found in the initial pooled results (I2 = 96% for SBP; I2 = 85% for DBP), investigators performed an analysis that excluded a single study with a small sample size. The re-analysis continued to show significant reductions in SBP and DBP for spironolactone compared with placebo (SBP: WMD = −10.8 mm Hg; 95% CI, −13.16 to −8.43 mm Hg; DBP: WMD = −4.62 mm Hg; 95% CI, −6.05 to −3.2 mm Hg; I2 = 35%), confirming that the excluded trial was the source of heterogeneity in the initial analysis and that spironolactone continued to significantly lower BP for the treatment group compared with controls.
Add-on treatment with spironolactone also reduces BP
A 2016 meta-analysis of 5 RCTs with a total of 553 patients examined the effectiveness of add-on treatment with spironolactone (25-50 mg/d) for patients with resistant hypertension, defined as failure to achieve BP < 140/90 mm Hg despite treatment with 3 or more BP-lowering drugs, including one diuretic.2 Spironolactone was compared with placebo in 4 trials and with ramipril in the remaining study. The follow-up periods were 8 to 16 weeks. Researchers separated BP outcomes into 24-hour ambulatory systolic/diastolic BPs and office systolic/diastolic BPs.
The 24-hour ambulatory BPs were significantly lower in the spironolactone group compared with the control group (24-hour SBP: WMD = −10.5 mm Hg; 95% CI, −12.3 to −8.71 mm Hg; 24-hour DBP: WMD = −4.09 mm Hg; 95% CI, −5.28 to −2.91 mm Hg). No significant heterogeneity was noted in these analyses.
Office-based BPs also were markedly reduced in spironolactone groups compared with controls (office SBP: WMD = −17 mm Hg; 95% CI, −25 to −8.95 mm Hg); office DBP: WMD = −6.18 mm Hg; 95% CI, −9.3 to −3.05 mm Hg). Because the office-based BP data showed significant heterogeneity (I2 = 94% for SBP and 84.2% for DBP), 2 studies determined to be of lower quality caused by lack of detailed methodology were excluded from analysis, yielding continued statistically significant reductions in SBP (WMD = −11.7 mm Hg; 95% CI, −14.4 to −8.95 mm Hg) and DBP (WMD = −4.07 mm Hg; 95% CI, −5.6 to −2.54 mm Hg) compared with controls. Heterogeneity also decreased when the 2 studies were excluded (I2 = 21% for SBP and I2 = 59% for DBP).
How spironolactone compares with alternative drugs
A 2017 meta-analysis of 5 RCTs with 662 patients evaluated the effectiveness of spironolactone (25-50 mg/d) on resistant hypertension in patients taking 3 medications compared with a control group—placebo in 3 trials, placebo or bisoprolol (5-10 mg) in 1 trial, and an alternative treatment (candesartan 8 mg, atenolol 100 mg, or alpha methyldopa 750 mg) in 1 trial.3 Follow-up periods ranged from 4 to 16 weeks. Researchers evaluated changes in office and 24-hour ambulatory or home BP and completed separate analyses of pooled data for spironolactone compared with placebo groups, and spironolactone compared with alternative treatment groups.
Continue to: Investigators found a statistically significant...
Investigators found a statistically significant reduction in office SBP and DBP among patients taking spironolactone compared with control groups (SBP: WMD = −15.7 mm Hg; 95% CI, −20.5 to −11 mm Hg; DBP: WMD = −6.21 mm Hg; 95% CI, −8.33 to −4.1 mm Hg). A significant decrease also occurred in 24-hour ambulatory home SBP and DBP (SBP: MD = −8.7 mm Hg; 95% CI, −8.79 to −8.62 mm Hg; DBP: WMD = −4.12 mm Hg; 95% CI, −4.48 to −3.75 mm Hg).
Patients treated with spironolactone showed a marked decrease in home SBP compared with alternative drug groups (WMD = −4.5 mm Hg; 95% CI, −4.63 to −4.37 mm Hg), but alternative drugs reduced home DBP significantly more than spironolactone (WMD = 0.6 mm Hg; 95% CI, 0.55-0.65 mm Hg). Marked heterogeneity was found in these analyses, and the authors also noted that reductions in SBP are more clinically relevant than decreases in DBP.
RECOMMENDATIONS
The 2017 American Heart Association/American College of Cardiology evidence-based guideline recommends considering adding a mineralocorticoid receptor agonist to treatment regimens for resistant hypertension when: office BP remains ≥ 130/80 mm Hg; the patient is prescribed at least 3 antihypertensive agents at optimal doses including a diuretic; pseudoresistance (nonadherence, inaccurate measurements) is excluded; reversible lifestyle factors have been addressed; substances that interfere with BP treatment (such as nonsteroidal anti-inflammatory drugs and oral contraceptive pills) are excluded; and screening for secondary causes of hypertension is complete.4
The United Kingdom’s National Institute for Health and Care Excellence (NICE) evidence-based guideline recommends considering spironolactone 25 mg/d to treat resistant hypertension if the patient’s potassium level is 4.5 mmol/L or lower and BP is higher than 140/90 mm Hg despite treatment with an optimal or best-tolerated dose of an angiotensin-converting enzyme inhibitor or angiotensin II receptor blocker plus a calcium-channel blocker and diuretic.5
Editor’s takeaway
The evidence from multiple RCTs convincingly shows the effectiveness of spironolactone. Despite the SOR of C because of a disease-oriented outcome, we do treat to blood pressure goals, and therefore, spironolactone is a good option.
1. Zhao D, Liu H, Dong P, et al. A meta-analysis of add-on use of spironolactone in patients with resistant hypertension. Int J Cardiol. 2017;233:113-117.
2. Wang C, Xiong B, Huang J. Efficacy and safety of spironolactone in patients with resistant hypertension: a meta-analysis of randomised controlled trials. Heart Lung Circ. 2016;25:1021-1030.
3. Liu L, Xu B, Ju Y. Addition of spironolactone in patients with resistant hypertension: a meta-analysis of randomized controlled trials. Clin Exp Hypertens. 2017;39:257-263.
4. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2017. https://doi.org/10.1161/HYP.0000000000000065. Accessed June 6, 2019.
5. National Institute for Health and Care Excellence. Hypertension in adults: diagnosis and management. Clinical guideline [CG127]. August 2011. https://www.nice.org.uk/guidance/cg127/chapter/1-guidance#initiating-and-monitoring-antihypertensive-drug-treatment-including-blood-pressure-targets-2. Accessed June 6, 2019.
1. Zhao D, Liu H, Dong P, et al. A meta-analysis of add-on use of spironolactone in patients with resistant hypertension. Int J Cardiol. 2017;233:113-117.
2. Wang C, Xiong B, Huang J. Efficacy and safety of spironolactone in patients with resistant hypertension: a meta-analysis of randomised controlled trials. Heart Lung Circ. 2016;25:1021-1030.
3. Liu L, Xu B, Ju Y. Addition of spironolactone in patients with resistant hypertension: a meta-analysis of randomized controlled trials. Clin Exp Hypertens. 2017;39:257-263.
4. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2017. https://doi.org/10.1161/HYP.0000000000000065. Accessed June 6, 2019.
5. National Institute for Health and Care Excellence. Hypertension in adults: diagnosis and management. Clinical guideline [CG127]. August 2011. https://www.nice.org.uk/guidance/cg127/chapter/1-guidance#initiating-and-monitoring-antihypertensive-drug-treatment-including-blood-pressure-targets-2. Accessed June 6, 2019.
EVIDENCE-BASED ANSWER:
Very effective. Spironolactone reduces systolic blood pressure (SBP) by 11 to 17 mm Hg and diastolic blood pressure (DBP) by up to 6 mm Hg in patients with resistant hypertension taking 3 or more medications (strength of recommendation [SOR]: C, meta-analysis of randomized controlled trials [RCTs] of disease-oriented evidence).

Less Is More When It Comes to Ketorolac for Pain

A 46-year-old man with no significant medical history presents to the emergency department (ED) with right flank pain and nausea. CT reveals a 5-mm ureteral stone with no obstruction or hydronephrosis. You are planning to start him on IV ketorolac for pain. What is the most appropriate dose?
Ketorolac tromethamine is a highly effective NSAID. As a nonopiate analgesic, it is often the first choice for the treatment of acute pain in the flank, abdomen, musculoskeletal system, or head.2 While it is not associated with euphoria, withdrawal effects, or respiratory depression (like its opiate analgesic counterparts), ketorolac carries an FDA black-box warning for gastrointestinal, cardiovascular, renal, and bleeding risks.3
NSAIDs are known to have a “ceiling dose” at which maximum analgesic benefit is achieved; higher doses will not provide further pain relief. Higher doses of ketorolac may be used when the anti-inflammatory effects of NSAIDs are desired, but they are likely to cause more adverse effects.4 Available data describe the ceiling dose of ketorolac as 10 mg across dosage forms—yet the majority of research and most health care providers in current practice use higher doses (20 to 60 mg).4,5 The FDA-approved labeling provides for a maximum dose of 60 mg/d.3
In one recent study, ketorolac was prescribed above its ceiling dose in at least 97% of patients who received IV doses and at least 96% of those who received intramuscular (IM) doses in a US ED.6 If 10 mg of ketorolac is an effective analgesic dose, current practice exceeds the label recommendation to use the lowest effective dose. This study sought to determine the comparative efficacy of 3 different doses of IV ketorolac for acute pain management in an ED.
STUDY SUMMARY
10 mg of ketorolac is enough for pain
This randomized double-blind trial evaluated the effectiveness of ketorolac in 240 adult patients (ages 18 to 65) presenting to an ED with acute flank, abdominal, musculoskeletal, or headache pain.1 Acute pain was defined as onset within the past 30 days.
Patients were randomly assigned to receive either 10, 15, or 30 mg of IV ketorolac in 10 mL of normal saline. A pharmacist prepared the medication in identical syringes, which were delivered in a blinded manner to the nurses caring for the patients. Pain (measured using a 0-to-10 scale), vital signs, and adverse effects were assessed at baseline and at 15, 30, 60, 90, and 120 minutes. If patients were still in pain at 30 minutes, IV morphine (0.1 mg/kg) was offered. The primary outcome was a numerical pain score at 30 minutes after ketorolac administration; secondary outcomes included the occurrence of adverse events and the use of rescue
The treatment groups were similar in terms of demographics and baseline vital signs. Mean age was 39 to 42. Across the 3 groups, 36% to 40% of patients had abdominal pain, 26% to 39% had flank pain, 20% to 26% had musculoskeletal pain, and 1% to 11% had headache pain. Patients had experienced pain for an average of 1.5 to 3.5 days.
Continue to: Baseline pain scores...
Baseline pain scores were similar for all 3 groups (7.5-7.8 on a 10-point scale). In the intention-to-treat analysis, all 3 doses of ketorolac decreased pain significantly at 30 minutes, with no difference between the groups: mean pain scores postintervention were 5.1 for the 10- and 15-mg group and 4.8 for the 30-mg group. There was no difference between the groups at any other time intervals. There was also no difference between groups in the number of patients who needed rescue medication at 30 minutes (4 patients in the 10-mg group, 3 patients in the 15-mg group, and 4 patients in the 30-mg group). In addition, adverse events (eg, dizziness, nausea, headache, itching, flushing) did not differ between the groups.
WHAT’S NEW
10 mg is just as effective as 30 mg
This trial confirms that a low dose of IV ketorolac is just as effective as higher doses for acute pain control.
CAVEATS
2-hour limit; no look at long-term effects
It isn’t known whether the higher dose would have provided greater pain relief beyond the 120 minutes evaluated in this trial, or if alternative dosage forms (oral or IM) would result in different outcomes. This study was not designed to compare serious long-term adverse effects such as bleeding, renal impairment, or cardiovascular events. Additionally, this study was not powered to look at specific therapeutic indications or anti-inflammatory response.
CHALLENGES TO IMPLEMENTATION
10-mg single-dose vial not readily available
Ketorolac tromethamine for injection is available in the United States in 15-, 30-, and 60-mg single-dose vials. Because a 10-mg dose is not available as a single-dose vial, it would need to be specially prepared (as it was in this study). However, this study should reassure providers that using the lowest available dose (eg, 15 mg IV if that is what is available) will relieve acute pain as well as higher doses will. CR
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2019. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2019;68[1]:41-42).
1. Motov S, Yasavolian M, Likourezos A, et al. Comparison of intravenous ketorolac at three single-dose regimens for treating acute pain in the emergency department: a randomized controlled trial. Ann Emerg Med. 2017; 70:177-184.
2. Buckley MM, Brogden RN. Ketorolac: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential. Drugs. 1990;39: 86-109.
3. Ketorolac tromethamine [package insert]. Bedford, OH: Bedford Laboratories; 2009.
4. Catapano MS. The analgesic efficacy of ketorolac for acute pain. J Emerg Med. 1996;14:67-75.
5. García Rodríguez LA, Cattaruzzi C, Troncon MG, et al. Risk of hospitalization for upper gastrointestinal tract bleeding associated with ketorolac, other nonsteroidal anti-inflammatory drugs, calcium antagonists, and other antihypertensive drugs. Arch Intern Med. 1998;158:33-39.
6. Soleyman-Zomalan E, Motov S, Likourezos A, et al. Patterns of ketorolac dosing by emergency physicians. World J Emerg Med. 2017;8:43-46.
A 46-year-old man with no significant medical history presents to the emergency department (ED) with right flank pain and nausea. CT reveals a 5-mm ureteral stone with no obstruction or hydronephrosis. You are planning to start him on IV ketorolac for pain. What is the most appropriate dose?
Ketorolac tromethamine is a highly effective NSAID. As a nonopiate analgesic, it is often the first choice for the treatment of acute pain in the flank, abdomen, musculoskeletal system, or head.2 While it is not associated with euphoria, withdrawal effects, or respiratory depression (like its opiate analgesic counterparts), ketorolac carries an FDA black-box warning for gastrointestinal, cardiovascular, renal, and bleeding risks.3
NSAIDs are known to have a “ceiling dose” at which maximum analgesic benefit is achieved; higher doses will not provide further pain relief. Higher doses of ketorolac may be used when the anti-inflammatory effects of NSAIDs are desired, but they are likely to cause more adverse effects.4 Available data describe the ceiling dose of ketorolac as 10 mg across dosage forms—yet the majority of research and most health care providers in current practice use higher doses (20 to 60 mg).4,5 The FDA-approved labeling provides for a maximum dose of 60 mg/d.3
In one recent study, ketorolac was prescribed above its ceiling dose in at least 97% of patients who received IV doses and at least 96% of those who received intramuscular (IM) doses in a US ED.6 If 10 mg of ketorolac is an effective analgesic dose, current practice exceeds the label recommendation to use the lowest effective dose. This study sought to determine the comparative efficacy of 3 different doses of IV ketorolac for acute pain management in an ED.
STUDY SUMMARY
10 mg of ketorolac is enough for pain
This randomized double-blind trial evaluated the effectiveness of ketorolac in 240 adult patients (ages 18 to 65) presenting to an ED with acute flank, abdominal, musculoskeletal, or headache pain.1 Acute pain was defined as onset within the past 30 days.
Patients were randomly assigned to receive either 10, 15, or 30 mg of IV ketorolac in 10 mL of normal saline. A pharmacist prepared the medication in identical syringes, which were delivered in a blinded manner to the nurses caring for the patients. Pain (measured using a 0-to-10 scale), vital signs, and adverse effects were assessed at baseline and at 15, 30, 60, 90, and 120 minutes. If patients were still in pain at 30 minutes, IV morphine (0.1 mg/kg) was offered. The primary outcome was a numerical pain score at 30 minutes after ketorolac administration; secondary outcomes included the occurrence of adverse events and the use of rescue
The treatment groups were similar in terms of demographics and baseline vital signs. Mean age was 39 to 42. Across the 3 groups, 36% to 40% of patients had abdominal pain, 26% to 39% had flank pain, 20% to 26% had musculoskeletal pain, and 1% to 11% had headache pain. Patients had experienced pain for an average of 1.5 to 3.5 days.
Continue to: Baseline pain scores...
Baseline pain scores were similar for all 3 groups (7.5-7.8 on a 10-point scale). In the intention-to-treat analysis, all 3 doses of ketorolac decreased pain significantly at 30 minutes, with no difference between the groups: mean pain scores postintervention were 5.1 for the 10- and 15-mg group and 4.8 for the 30-mg group. There was no difference between the groups at any other time intervals. There was also no difference between groups in the number of patients who needed rescue medication at 30 minutes (4 patients in the 10-mg group, 3 patients in the 15-mg group, and 4 patients in the 30-mg group). In addition, adverse events (eg, dizziness, nausea, headache, itching, flushing) did not differ between the groups.
WHAT’S NEW
10 mg is just as effective as 30 mg
This trial confirms that a low dose of IV ketorolac is just as effective as higher doses for acute pain control.
CAVEATS
2-hour limit; no look at long-term effects
It isn’t known whether the higher dose would have provided greater pain relief beyond the 120 minutes evaluated in this trial, or if alternative dosage forms (oral or IM) would result in different outcomes. This study was not designed to compare serious long-term adverse effects such as bleeding, renal impairment, or cardiovascular events. Additionally, this study was not powered to look at specific therapeutic indications or anti-inflammatory response.
CHALLENGES TO IMPLEMENTATION
10-mg single-dose vial not readily available
Ketorolac tromethamine for injection is available in the United States in 15-, 30-, and 60-mg single-dose vials. Because a 10-mg dose is not available as a single-dose vial, it would need to be specially prepared (as it was in this study). However, this study should reassure providers that using the lowest available dose (eg, 15 mg IV if that is what is available) will relieve acute pain as well as higher doses will. CR
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2019. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2019;68[1]:41-42).
A 46-year-old man with no significant medical history presents to the emergency department (ED) with right flank pain and nausea. CT reveals a 5-mm ureteral stone with no obstruction or hydronephrosis. You are planning to start him on IV ketorolac for pain. What is the most appropriate dose?
Ketorolac tromethamine is a highly effective NSAID. As a nonopiate analgesic, it is often the first choice for the treatment of acute pain in the flank, abdomen, musculoskeletal system, or head.2 While it is not associated with euphoria, withdrawal effects, or respiratory depression (like its opiate analgesic counterparts), ketorolac carries an FDA black-box warning for gastrointestinal, cardiovascular, renal, and bleeding risks.3
NSAIDs are known to have a “ceiling dose” at which maximum analgesic benefit is achieved; higher doses will not provide further pain relief. Higher doses of ketorolac may be used when the anti-inflammatory effects of NSAIDs are desired, but they are likely to cause more adverse effects.4 Available data describe the ceiling dose of ketorolac as 10 mg across dosage forms—yet the majority of research and most health care providers in current practice use higher doses (20 to 60 mg).4,5 The FDA-approved labeling provides for a maximum dose of 60 mg/d.3
In one recent study, ketorolac was prescribed above its ceiling dose in at least 97% of patients who received IV doses and at least 96% of those who received intramuscular (IM) doses in a US ED.6 If 10 mg of ketorolac is an effective analgesic dose, current practice exceeds the label recommendation to use the lowest effective dose. This study sought to determine the comparative efficacy of 3 different doses of IV ketorolac for acute pain management in an ED.
STUDY SUMMARY
10 mg of ketorolac is enough for pain
This randomized double-blind trial evaluated the effectiveness of ketorolac in 240 adult patients (ages 18 to 65) presenting to an ED with acute flank, abdominal, musculoskeletal, or headache pain.1 Acute pain was defined as onset within the past 30 days.
Patients were randomly assigned to receive either 10, 15, or 30 mg of IV ketorolac in 10 mL of normal saline. A pharmacist prepared the medication in identical syringes, which were delivered in a blinded manner to the nurses caring for the patients. Pain (measured using a 0-to-10 scale), vital signs, and adverse effects were assessed at baseline and at 15, 30, 60, 90, and 120 minutes. If patients were still in pain at 30 minutes, IV morphine (0.1 mg/kg) was offered. The primary outcome was a numerical pain score at 30 minutes after ketorolac administration; secondary outcomes included the occurrence of adverse events and the use of rescue
The treatment groups were similar in terms of demographics and baseline vital signs. Mean age was 39 to 42. Across the 3 groups, 36% to 40% of patients had abdominal pain, 26% to 39% had flank pain, 20% to 26% had musculoskeletal pain, and 1% to 11% had headache pain. Patients had experienced pain for an average of 1.5 to 3.5 days.
Continue to: Baseline pain scores...
Baseline pain scores were similar for all 3 groups (7.5-7.8 on a 10-point scale). In the intention-to-treat analysis, all 3 doses of ketorolac decreased pain significantly at 30 minutes, with no difference between the groups: mean pain scores postintervention were 5.1 for the 10- and 15-mg group and 4.8 for the 30-mg group. There was no difference between the groups at any other time intervals. There was also no difference between groups in the number of patients who needed rescue medication at 30 minutes (4 patients in the 10-mg group, 3 patients in the 15-mg group, and 4 patients in the 30-mg group). In addition, adverse events (eg, dizziness, nausea, headache, itching, flushing) did not differ between the groups.
WHAT’S NEW
10 mg is just as effective as 30 mg
This trial confirms that a low dose of IV ketorolac is just as effective as higher doses for acute pain control.
CAVEATS
2-hour limit; no look at long-term effects
It isn’t known whether the higher dose would have provided greater pain relief beyond the 120 minutes evaluated in this trial, or if alternative dosage forms (oral or IM) would result in different outcomes. This study was not designed to compare serious long-term adverse effects such as bleeding, renal impairment, or cardiovascular events. Additionally, this study was not powered to look at specific therapeutic indications or anti-inflammatory response.
CHALLENGES TO IMPLEMENTATION
10-mg single-dose vial not readily available
Ketorolac tromethamine for injection is available in the United States in 15-, 30-, and 60-mg single-dose vials. Because a 10-mg dose is not available as a single-dose vial, it would need to be specially prepared (as it was in this study). However, this study should reassure providers that using the lowest available dose (eg, 15 mg IV if that is what is available) will relieve acute pain as well as higher doses will. CR
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2019. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2019;68[1]:41-42).
1. Motov S, Yasavolian M, Likourezos A, et al. Comparison of intravenous ketorolac at three single-dose regimens for treating acute pain in the emergency department: a randomized controlled trial. Ann Emerg Med. 2017; 70:177-184.
2. Buckley MM, Brogden RN. Ketorolac: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential. Drugs. 1990;39: 86-109.
3. Ketorolac tromethamine [package insert]. Bedford, OH: Bedford Laboratories; 2009.
4. Catapano MS. The analgesic efficacy of ketorolac for acute pain. J Emerg Med. 1996;14:67-75.
5. García Rodríguez LA, Cattaruzzi C, Troncon MG, et al. Risk of hospitalization for upper gastrointestinal tract bleeding associated with ketorolac, other nonsteroidal anti-inflammatory drugs, calcium antagonists, and other antihypertensive drugs. Arch Intern Med. 1998;158:33-39.
6. Soleyman-Zomalan E, Motov S, Likourezos A, et al. Patterns of ketorolac dosing by emergency physicians. World J Emerg Med. 2017;8:43-46.
1. Motov S, Yasavolian M, Likourezos A, et al. Comparison of intravenous ketorolac at three single-dose regimens for treating acute pain in the emergency department: a randomized controlled trial. Ann Emerg Med. 2017; 70:177-184.
2. Buckley MM, Brogden RN. Ketorolac: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential. Drugs. 1990;39: 86-109.
3. Ketorolac tromethamine [package insert]. Bedford, OH: Bedford Laboratories; 2009.
4. Catapano MS. The analgesic efficacy of ketorolac for acute pain. J Emerg Med. 1996;14:67-75.
5. García Rodríguez LA, Cattaruzzi C, Troncon MG, et al. Risk of hospitalization for upper gastrointestinal tract bleeding associated with ketorolac, other nonsteroidal anti-inflammatory drugs, calcium antagonists, and other antihypertensive drugs. Arch Intern Med. 1998;158:33-39.
6. Soleyman-Zomalan E, Motov S, Likourezos A, et al. Patterns of ketorolac dosing by emergency physicians. World J Emerg Med. 2017;8:43-46.

What are the risks of long-term PPI use for GERD symptoms in patients > 65 years?
EVIDENCE SUMMARY
A 2017 meta-analysis of 16 RCTs examined the risk of cardiovascular events in 7540 adult patients taking PPIs for GERD (mean ages 45-55 years).1 The primary outcome was cardiovascular events—including acute myocardial infarction, myocardial ischemia, angina pectoris, cardiac failure, and coronary artery stenosis—and cardiac disorders.
Analysis of pooled data found that PPI use was associated with a 70% increase in cardiovascular risk (relative risk [RR] = 1.7; 95% confidence interval [CI], 1.13-2.56; number needed to harm [NNH] = 241) when compared with controls (placebo, H2 blocker, or surgery). A subgroup analysis found that PPI use for longer than 8 weeks was associated with an even higher risk of adverse cardiovascular events (6 trials, 2296 patients; RR = 2.33; 95% CI, 1.33-4.08; NNH = 67) when compared with controls. The meta-analysis wasn’t limited by heterogeneity (I2 = 0).
C difficile infection risk is higherfor PPI users
A 2016 meta-analysis of 23 observational studies (19 case-control, 4 retrospective cohort; 186,033 patients) examined the risk of hospital-acquired C difficile infections in adults prescribed PPI for any indication.2 PPI exposure varied from use at time of diagnosis or hospitalization to any use within 90 days. Of the 23 studies, 16 reported sufficient data to calculate the mean age for the patients which was 69.9 years.
The risk of C difficile infection was found to be higher with PPI use than no use (pooled odds ratio [OR] = 1.81; 95% CI, 1.52-2.14). Although a significant association was found across a large group, the results were limited by considerable heterogeneity (I2 = 82%).
Risk of community-acquired pneumonia also increases with PPI use
A 2015 systematic review and meta-analysis of 33 trials (18 case-control, 10 cohort, 4 RCTs, and 1 case-crossover study) examined the risk of CAP in adult patients prescribed PPI for any indication for durations ranging from less than 1 month to > 6 months.3 The systematic review was distilled to 26 studies because of overlapping study populations. These 26 studies included 226,769 cases of CAP among 6,351,656 patients. The primary outcome was development of CAP, the secondary outcome was hospitalization for CAP.
PPI use, compared with no use, was associated with an increased risk of developing CAP (pooled OR = 1.49; 95% CI, 1.16-1.92) and an increased risk of hospitalization for CAP (pooled OR = 1.61; 95% CI, 1.12-2.31).
In a subgroup analysis for age, patients older than 65 years were also found to have an increased risk of developing CAP with PPI use (11 trials, total number of patients not provided; OR = 1.33; 95% CI, 1.13-1.58). Despite the significant associations of PPI use with risk revealed in the primary, secondary, and subgroup analyses, the results were limited by marked heterogeneity, with an I2 > 99%.
Continue to: Hip and vertebral fracture risks associated with PPIs
Hip and vertebral fracture riskis associated with PPIs
A 2011 systematic review and meta-analysis investigated the risk of fracture in adult patients taking PPIs for any indication.4 The analysis included 10 observational studies (4 cohort, 6 case-control) with a total of 223,210 fracture cases. The authors examined the incidence of hip, vertebral, and wrist or forearm fractures.
No significant association was found between PPI use and wrist or forearm fracture (3 studies; pooled OR = 1.09; 95% CI, 0.95-1.24). A modest association was noted between PPI use and both hip fractures (9 trials; OR = 1.25; 95% CI, 1.14-1.37) and vertebral fractures (4 trials; OR = 1.5; 95% CI, 1.32-1.72).
Subgroup analysis didn’t reveal evidence of an effect of duration of PPI use on fracture. Investigators didn’t conduct subgroup analysis of different patient ages. Final results were limited by significant heterogeneity with an I2 of 86%.
RECOMMENDATIONS
A 2015 American Geriatrics Society Beers Criteria update recommends limiting PPI use because of increased risk of C difficile infections and fractures. It also recommends against using PPIs for longer than 8 weeks except for high-risk patients (such as patients taking oral corticosteroids or chronic nonsteroidal anti-inflammatory drug users), patients with Barrett’s esophagitis, or patients who need maintenance after failure of a drug discontinuation trial or H2 blockers (quality of evidence, high; SOR, strong).5
Editor’s take
1. Sun S, Cui Z, Zhou M, et al. Proton pump inhibitor monotherapy and the risk of cardiovascular events in patients with gastro-esophageal reflux disease: a meta-analysis. Neurogastroenterol Motil. 2017;29:e12926.
2. Arriola V, Tischendorf J, Musuuza J, et al. Assessing the risk of hospital-acquired clostridium difficile infection with proton pump inhibitor use: a meta-analysis. Infect Control Hosp Epidemiol. 2016;37:1408-1417.
3. Lambert AA, Lam JO, Paik JJ, et al. Risk of community-acquired pneumonia with outpatient proton-pump inhibitor therapy: a systematic review and meta-analysis. PLoS One. 2015;10:e0128004.
4. Ngamruengphong S, Leontiadis GI, Radhi S, et al. Proton pump inhibitors and risk of fracture: a systematic review and meta-analysis of observational studies. Am J Gastroenterol. 2011;106:1209-1218.
5. American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 Updated Beers Criteria for Potentially Inappropriate Medication Use in Older Adults. J Am Geriatr Soc. 2015;63:2227-2246.
EVIDENCE SUMMARY
A 2017 meta-analysis of 16 RCTs examined the risk of cardiovascular events in 7540 adult patients taking PPIs for GERD (mean ages 45-55 years).1 The primary outcome was cardiovascular events—including acute myocardial infarction, myocardial ischemia, angina pectoris, cardiac failure, and coronary artery stenosis—and cardiac disorders.
Analysis of pooled data found that PPI use was associated with a 70% increase in cardiovascular risk (relative risk [RR] = 1.7; 95% confidence interval [CI], 1.13-2.56; number needed to harm [NNH] = 241) when compared with controls (placebo, H2 blocker, or surgery). A subgroup analysis found that PPI use for longer than 8 weeks was associated with an even higher risk of adverse cardiovascular events (6 trials, 2296 patients; RR = 2.33; 95% CI, 1.33-4.08; NNH = 67) when compared with controls. The meta-analysis wasn’t limited by heterogeneity (I2 = 0).
C difficile infection risk is higherfor PPI users
A 2016 meta-analysis of 23 observational studies (19 case-control, 4 retrospective cohort; 186,033 patients) examined the risk of hospital-acquired C difficile infections in adults prescribed PPI for any indication.2 PPI exposure varied from use at time of diagnosis or hospitalization to any use within 90 days. Of the 23 studies, 16 reported sufficient data to calculate the mean age for the patients which was 69.9 years.
The risk of C difficile infection was found to be higher with PPI use than no use (pooled odds ratio [OR] = 1.81; 95% CI, 1.52-2.14). Although a significant association was found across a large group, the results were limited by considerable heterogeneity (I2 = 82%).
Risk of community-acquired pneumonia also increases with PPI use
A 2015 systematic review and meta-analysis of 33 trials (18 case-control, 10 cohort, 4 RCTs, and 1 case-crossover study) examined the risk of CAP in adult patients prescribed PPI for any indication for durations ranging from less than 1 month to > 6 months.3 The systematic review was distilled to 26 studies because of overlapping study populations. These 26 studies included 226,769 cases of CAP among 6,351,656 patients. The primary outcome was development of CAP, the secondary outcome was hospitalization for CAP.
PPI use, compared with no use, was associated with an increased risk of developing CAP (pooled OR = 1.49; 95% CI, 1.16-1.92) and an increased risk of hospitalization for CAP (pooled OR = 1.61; 95% CI, 1.12-2.31).
In a subgroup analysis for age, patients older than 65 years were also found to have an increased risk of developing CAP with PPI use (11 trials, total number of patients not provided; OR = 1.33; 95% CI, 1.13-1.58). Despite the significant associations of PPI use with risk revealed in the primary, secondary, and subgroup analyses, the results were limited by marked heterogeneity, with an I2 > 99%.
Continue to: Hip and vertebral fracture risks associated with PPIs
Hip and vertebral fracture riskis associated with PPIs
A 2011 systematic review and meta-analysis investigated the risk of fracture in adult patients taking PPIs for any indication.4 The analysis included 10 observational studies (4 cohort, 6 case-control) with a total of 223,210 fracture cases. The authors examined the incidence of hip, vertebral, and wrist or forearm fractures.
No significant association was found between PPI use and wrist or forearm fracture (3 studies; pooled OR = 1.09; 95% CI, 0.95-1.24). A modest association was noted between PPI use and both hip fractures (9 trials; OR = 1.25; 95% CI, 1.14-1.37) and vertebral fractures (4 trials; OR = 1.5; 95% CI, 1.32-1.72).
Subgroup analysis didn’t reveal evidence of an effect of duration of PPI use on fracture. Investigators didn’t conduct subgroup analysis of different patient ages. Final results were limited by significant heterogeneity with an I2 of 86%.
RECOMMENDATIONS
A 2015 American Geriatrics Society Beers Criteria update recommends limiting PPI use because of increased risk of C difficile infections and fractures. It also recommends against using PPIs for longer than 8 weeks except for high-risk patients (such as patients taking oral corticosteroids or chronic nonsteroidal anti-inflammatory drug users), patients with Barrett’s esophagitis, or patients who need maintenance after failure of a drug discontinuation trial or H2 blockers (quality of evidence, high; SOR, strong).5
Editor’s take
EVIDENCE SUMMARY
A 2017 meta-analysis of 16 RCTs examined the risk of cardiovascular events in 7540 adult patients taking PPIs for GERD (mean ages 45-55 years).1 The primary outcome was cardiovascular events—including acute myocardial infarction, myocardial ischemia, angina pectoris, cardiac failure, and coronary artery stenosis—and cardiac disorders.
Analysis of pooled data found that PPI use was associated with a 70% increase in cardiovascular risk (relative risk [RR] = 1.7; 95% confidence interval [CI], 1.13-2.56; number needed to harm [NNH] = 241) when compared with controls (placebo, H2 blocker, or surgery). A subgroup analysis found that PPI use for longer than 8 weeks was associated with an even higher risk of adverse cardiovascular events (6 trials, 2296 patients; RR = 2.33; 95% CI, 1.33-4.08; NNH = 67) when compared with controls. The meta-analysis wasn’t limited by heterogeneity (I2 = 0).
C difficile infection risk is higherfor PPI users
A 2016 meta-analysis of 23 observational studies (19 case-control, 4 retrospective cohort; 186,033 patients) examined the risk of hospital-acquired C difficile infections in adults prescribed PPI for any indication.2 PPI exposure varied from use at time of diagnosis or hospitalization to any use within 90 days. Of the 23 studies, 16 reported sufficient data to calculate the mean age for the patients which was 69.9 years.
The risk of C difficile infection was found to be higher with PPI use than no use (pooled odds ratio [OR] = 1.81; 95% CI, 1.52-2.14). Although a significant association was found across a large group, the results were limited by considerable heterogeneity (I2 = 82%).
Risk of community-acquired pneumonia also increases with PPI use
A 2015 systematic review and meta-analysis of 33 trials (18 case-control, 10 cohort, 4 RCTs, and 1 case-crossover study) examined the risk of CAP in adult patients prescribed PPI for any indication for durations ranging from less than 1 month to > 6 months.3 The systematic review was distilled to 26 studies because of overlapping study populations. These 26 studies included 226,769 cases of CAP among 6,351,656 patients. The primary outcome was development of CAP, the secondary outcome was hospitalization for CAP.
PPI use, compared with no use, was associated with an increased risk of developing CAP (pooled OR = 1.49; 95% CI, 1.16-1.92) and an increased risk of hospitalization for CAP (pooled OR = 1.61; 95% CI, 1.12-2.31).
In a subgroup analysis for age, patients older than 65 years were also found to have an increased risk of developing CAP with PPI use (11 trials, total number of patients not provided; OR = 1.33; 95% CI, 1.13-1.58). Despite the significant associations of PPI use with risk revealed in the primary, secondary, and subgroup analyses, the results were limited by marked heterogeneity, with an I2 > 99%.
Continue to: Hip and vertebral fracture risks associated with PPIs
Hip and vertebral fracture riskis associated with PPIs
A 2011 systematic review and meta-analysis investigated the risk of fracture in adult patients taking PPIs for any indication.4 The analysis included 10 observational studies (4 cohort, 6 case-control) with a total of 223,210 fracture cases. The authors examined the incidence of hip, vertebral, and wrist or forearm fractures.
No significant association was found between PPI use and wrist or forearm fracture (3 studies; pooled OR = 1.09; 95% CI, 0.95-1.24). A modest association was noted between PPI use and both hip fractures (9 trials; OR = 1.25; 95% CI, 1.14-1.37) and vertebral fractures (4 trials; OR = 1.5; 95% CI, 1.32-1.72).
Subgroup analysis didn’t reveal evidence of an effect of duration of PPI use on fracture. Investigators didn’t conduct subgroup analysis of different patient ages. Final results were limited by significant heterogeneity with an I2 of 86%.
RECOMMENDATIONS
A 2015 American Geriatrics Society Beers Criteria update recommends limiting PPI use because of increased risk of C difficile infections and fractures. It also recommends against using PPIs for longer than 8 weeks except for high-risk patients (such as patients taking oral corticosteroids or chronic nonsteroidal anti-inflammatory drug users), patients with Barrett’s esophagitis, or patients who need maintenance after failure of a drug discontinuation trial or H2 blockers (quality of evidence, high; SOR, strong).5
Editor’s take
1. Sun S, Cui Z, Zhou M, et al. Proton pump inhibitor monotherapy and the risk of cardiovascular events in patients with gastro-esophageal reflux disease: a meta-analysis. Neurogastroenterol Motil. 2017;29:e12926.
2. Arriola V, Tischendorf J, Musuuza J, et al. Assessing the risk of hospital-acquired clostridium difficile infection with proton pump inhibitor use: a meta-analysis. Infect Control Hosp Epidemiol. 2016;37:1408-1417.
3. Lambert AA, Lam JO, Paik JJ, et al. Risk of community-acquired pneumonia with outpatient proton-pump inhibitor therapy: a systematic review and meta-analysis. PLoS One. 2015;10:e0128004.
4. Ngamruengphong S, Leontiadis GI, Radhi S, et al. Proton pump inhibitors and risk of fracture: a systematic review and meta-analysis of observational studies. Am J Gastroenterol. 2011;106:1209-1218.
5. American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 Updated Beers Criteria for Potentially Inappropriate Medication Use in Older Adults. J Am Geriatr Soc. 2015;63:2227-2246.
1. Sun S, Cui Z, Zhou M, et al. Proton pump inhibitor monotherapy and the risk of cardiovascular events in patients with gastro-esophageal reflux disease: a meta-analysis. Neurogastroenterol Motil. 2017;29:e12926.
2. Arriola V, Tischendorf J, Musuuza J, et al. Assessing the risk of hospital-acquired clostridium difficile infection with proton pump inhibitor use: a meta-analysis. Infect Control Hosp Epidemiol. 2016;37:1408-1417.
3. Lambert AA, Lam JO, Paik JJ, et al. Risk of community-acquired pneumonia with outpatient proton-pump inhibitor therapy: a systematic review and meta-analysis. PLoS One. 2015;10:e0128004.
4. Ngamruengphong S, Leontiadis GI, Radhi S, et al. Proton pump inhibitors and risk of fracture: a systematic review and meta-analysis of observational studies. Am J Gastroenterol. 2011;106:1209-1218.
5. American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 Updated Beers Criteria for Potentially Inappropriate Medication Use in Older Adults. J Am Geriatr Soc. 2015;63:2227-2246.
EVIDENCE-BASED ANSWER:
The use of proton pump inhibitors (PPIs) to control gastroesophageal reflux disease (GERD) is significantly associated with an increased risk of cardiovascular events such as acute myocardial infarction and myocardial ischemia, especially with treatment longer than 8 weeks (strength of recommendation [SOR]: A, systematic review of randomized, controlled trials [RCTs]). This summary is based on data extrapolated from studies on all adults because there is limited evidence that specifically addresses patients older than 65 years.
Adults taking PPIs also appear to be at increased risk of Clostridium difficile infection, community-acquired pneumonia (CAP; with use for < 30 days), and fracture (SOR: B, systematic reviews of heterogeneous prospective and retrospective observational studies).
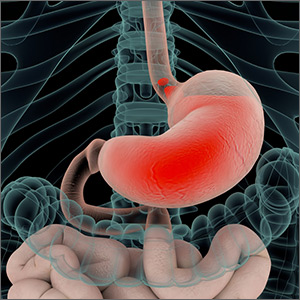
Should you switch the DAPT agent one month after ACS?
ILLUSTRATIVE CASE
A 60-year-old man is seen in your clinic 30 days after he was hospitalized for acute coronary syndrome (ACS) due to ST-elevation myocardial infarction (STEMI). He underwent percutaneous coronary intervention (PCI) with placement of one stent. He received aspirin and a loading dose of ticagrelor for antiplatelet therapy. He was discharged on dual antiplatelet therapy (DAPT) consisting of daily aspirin and ticagrelor. He asks about the risk of bleeding associated with these medications. Should you recommend any changes?
Platelet inhibition during and after ACS to prevent recurrent ischemic events is a cornerstone of treatment for patients after myocardial infarction (MI).2 Current American Cardiology Association and European Society of Cardiology guidelines recommend patients with coronary artery disease who have had a recent MI continue DAPT with aspirin and a P2Y12 blocker (ie, clopidogrel, ticlopidine, ticagrelor, prasugrel, or cangrelor) for 12 months following ACSto reduce recurrent ischemia.2-4
Studies have shown that using the newer P2Y12 inhibitors (ie, prasugrel and ticagrelor) after PCI leads to a significant reduction in recurrent ischemic events when compared to clopidogrel.5-7 These data led to a guideline change recommending the use of the newer agents over clopidogrel for 12 months following PCI.2 Follow-up studies evaluating the newer P2Y12 agents continue to show strong evidence for their use in the first month following PCI, while also demonstrating an increased bleeding risk in the maintenance phase (from 30 days to 12 months post-PCI).6,7 This increased risk is the basis for the current study, which tested switching from a newer P2Y12 agent to clopidogrel after the initial 30-day period following PCI.
STUDY SUMMARY
Switched DAPT is superior to unchanged DAPT
This open-label RCT (N = 646) examined changing DAPT from aspirin plus a newer P2Y12 blocker (prasugrel or ticagrelor) to a combination of aspirin and clopidogrel after the first month of DAPT post-ACS.1 Prior to PCI, all patients received a loading dose of ticagrelor 180 mg or prasugrel 60 mg. Subsequently, all patients in the trial took aspirin (75 mg/d) and one of the newer P2Y12 inhibitors (prasugrel 10 mg/d or ticagrelor 90 mg BID) for 1 month. For those enrollees who had no adverse events after 30 days, half were randomly switched to aspirin and clopidogrel 75 mg/d and the other half remained on aspirin and their newer P2Y12 blocker in a 1:1 ratio. For the next year, researchers examined the composite outcome of cardiovascular death, urgent revascularization, stroke, and major bleeding (as defined by the Bleeding Academic Research Consortium [BARC] classification ≥ Type 2 at 1 year post-ACS).
The average age of the participants was 60 years; 40% had experienced a STEMI and 60% had a non–STEMI. Overall, 43% of patients were prescribed ticagrelor and 57% prasugrel. At 1 year, 86% of the switched DAPT group and 75% of the unchanged DAPT group were still taking their medication. At the 1-year follow-up, the composite outcome was lower in the switched group, compared with the unchanged group (13% vs 26%; hazard ratio [HR] = 0.48; 95% confidence interval [CI], 0.34-0.68; number needed to treat [NNT] = 8).
All bleeding events (ranging from minimal to fatal) were lower in the switched group (9% vs 24%; HR = 0.39; 95% CI, 0.27-0.57; NNT = 7), and bleeding events identified as BARC ≥ Type 2 (defined as needing medical treatment) were also lower in the switched group (4% vs 15%; HR = 0.30, 95% CI, 0.18-0.50; NNT = 9). There were no significant differences in reported recurrent cardiovascular ischemic events (9.3% vs 11.5%; HR = 0.80, 95% CI, 0.50-1.29).
WHAT’S NEW
Fewer bleeding events without an increase in ischemic events
Cardiology guidelines recommend the newer P2Y12 blockers as part of DAPT after ACS, but this trial showed switching to clopidogrel for DAPT after 30 days of treatment lowers bleeding events with no difference in recurrent ischemic events.2-4
Continue to: CAVEATS
CAVEATS
Less-than-ideal study methods
This trial was an open-label, unblinded study. The investigators who adjudicated critical events were blinded to the treatment allocation, but some events, such as minor bleeding and medication discontinuation, could be self-reported by patients. In addition, the investigators used a less-than-ideal method (opaque envelopes) to conceal allocation at enrollment.
CHALLENGES TO IMPLEMENTATION
Implementation may require changing a cardiologist’s prescription
Implementing this practice change is facilitated by the fact that clopidogrel is currently less expensive than the newer P2Y12 blockers. However, after ACS and PCI treatment, cardiologists usually initiate antiplatelet therapy and may continue to manage patients after discharge. So the family physician (FP) may not be responsible for the DAPT switch initially. Further, switching may necessitate coordination with the cardiologist, as FPs may be hesitant to change cardiologists’ prescriptions. Lastly, guidelines currently recommend using the newer P2Y12 blockers for 12 months.2
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Cuisset T, Deharo P, Quilici J, et al. Benefit of switching dual antiplatelet therapy after acute coronary syndrome: the TOPIC (timing of platelet inhibition after acute coronary syndrome) randomized study. Eur Heart J. 2017;38:3070-3078.
2. Levine GN, Bates ER, Bittl JA, et al. 2016 ACC/AHA guideline focused update on duration of dual antiplatelet therapy in patients with coronary artery disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2016;68:1082-1115.
3. Steg PG, James SK, Atar D, et al. ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur Heart J. 2012;33:2569-2619.
4. Roffi M, Patrono C, Collet J-P, et al. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur Heart J. 2015;37:267-315.
5. Antman EM, Wiviott SD, Murphy SA, et al. Early and late benefits of prasugrel in patients with acute coronary syndromes undergoing percutaneous coronary intervention. J Am Coll Cardiol. 2008;51:2028-2033.
6. Wallentin L, Becker RC, Budaj A, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361:1045-1057.
7. Wiviott SD, Braunwald E, McCabe CH, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001-2015.
ILLUSTRATIVE CASE
A 60-year-old man is seen in your clinic 30 days after he was hospitalized for acute coronary syndrome (ACS) due to ST-elevation myocardial infarction (STEMI). He underwent percutaneous coronary intervention (PCI) with placement of one stent. He received aspirin and a loading dose of ticagrelor for antiplatelet therapy. He was discharged on dual antiplatelet therapy (DAPT) consisting of daily aspirin and ticagrelor. He asks about the risk of bleeding associated with these medications. Should you recommend any changes?
Platelet inhibition during and after ACS to prevent recurrent ischemic events is a cornerstone of treatment for patients after myocardial infarction (MI).2 Current American Cardiology Association and European Society of Cardiology guidelines recommend patients with coronary artery disease who have had a recent MI continue DAPT with aspirin and a P2Y12 blocker (ie, clopidogrel, ticlopidine, ticagrelor, prasugrel, or cangrelor) for 12 months following ACSto reduce recurrent ischemia.2-4
Studies have shown that using the newer P2Y12 inhibitors (ie, prasugrel and ticagrelor) after PCI leads to a significant reduction in recurrent ischemic events when compared to clopidogrel.5-7 These data led to a guideline change recommending the use of the newer agents over clopidogrel for 12 months following PCI.2 Follow-up studies evaluating the newer P2Y12 agents continue to show strong evidence for their use in the first month following PCI, while also demonstrating an increased bleeding risk in the maintenance phase (from 30 days to 12 months post-PCI).6,7 This increased risk is the basis for the current study, which tested switching from a newer P2Y12 agent to clopidogrel after the initial 30-day period following PCI.
STUDY SUMMARY
Switched DAPT is superior to unchanged DAPT
This open-label RCT (N = 646) examined changing DAPT from aspirin plus a newer P2Y12 blocker (prasugrel or ticagrelor) to a combination of aspirin and clopidogrel after the first month of DAPT post-ACS.1 Prior to PCI, all patients received a loading dose of ticagrelor 180 mg or prasugrel 60 mg. Subsequently, all patients in the trial took aspirin (75 mg/d) and one of the newer P2Y12 inhibitors (prasugrel 10 mg/d or ticagrelor 90 mg BID) for 1 month. For those enrollees who had no adverse events after 30 days, half were randomly switched to aspirin and clopidogrel 75 mg/d and the other half remained on aspirin and their newer P2Y12 blocker in a 1:1 ratio. For the next year, researchers examined the composite outcome of cardiovascular death, urgent revascularization, stroke, and major bleeding (as defined by the Bleeding Academic Research Consortium [BARC] classification ≥ Type 2 at 1 year post-ACS).
The average age of the participants was 60 years; 40% had experienced a STEMI and 60% had a non–STEMI. Overall, 43% of patients were prescribed ticagrelor and 57% prasugrel. At 1 year, 86% of the switched DAPT group and 75% of the unchanged DAPT group were still taking their medication. At the 1-year follow-up, the composite outcome was lower in the switched group, compared with the unchanged group (13% vs 26%; hazard ratio [HR] = 0.48; 95% confidence interval [CI], 0.34-0.68; number needed to treat [NNT] = 8).
All bleeding events (ranging from minimal to fatal) were lower in the switched group (9% vs 24%; HR = 0.39; 95% CI, 0.27-0.57; NNT = 7), and bleeding events identified as BARC ≥ Type 2 (defined as needing medical treatment) were also lower in the switched group (4% vs 15%; HR = 0.30, 95% CI, 0.18-0.50; NNT = 9). There were no significant differences in reported recurrent cardiovascular ischemic events (9.3% vs 11.5%; HR = 0.80, 95% CI, 0.50-1.29).
WHAT’S NEW
Fewer bleeding events without an increase in ischemic events
Cardiology guidelines recommend the newer P2Y12 blockers as part of DAPT after ACS, but this trial showed switching to clopidogrel for DAPT after 30 days of treatment lowers bleeding events with no difference in recurrent ischemic events.2-4
Continue to: CAVEATS
CAVEATS
Less-than-ideal study methods
This trial was an open-label, unblinded study. The investigators who adjudicated critical events were blinded to the treatment allocation, but some events, such as minor bleeding and medication discontinuation, could be self-reported by patients. In addition, the investigators used a less-than-ideal method (opaque envelopes) to conceal allocation at enrollment.
CHALLENGES TO IMPLEMENTATION
Implementation may require changing a cardiologist’s prescription
Implementing this practice change is facilitated by the fact that clopidogrel is currently less expensive than the newer P2Y12 blockers. However, after ACS and PCI treatment, cardiologists usually initiate antiplatelet therapy and may continue to manage patients after discharge. So the family physician (FP) may not be responsible for the DAPT switch initially. Further, switching may necessitate coordination with the cardiologist, as FPs may be hesitant to change cardiologists’ prescriptions. Lastly, guidelines currently recommend using the newer P2Y12 blockers for 12 months.2
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
ILLUSTRATIVE CASE
A 60-year-old man is seen in your clinic 30 days after he was hospitalized for acute coronary syndrome (ACS) due to ST-elevation myocardial infarction (STEMI). He underwent percutaneous coronary intervention (PCI) with placement of one stent. He received aspirin and a loading dose of ticagrelor for antiplatelet therapy. He was discharged on dual antiplatelet therapy (DAPT) consisting of daily aspirin and ticagrelor. He asks about the risk of bleeding associated with these medications. Should you recommend any changes?
Platelet inhibition during and after ACS to prevent recurrent ischemic events is a cornerstone of treatment for patients after myocardial infarction (MI).2 Current American Cardiology Association and European Society of Cardiology guidelines recommend patients with coronary artery disease who have had a recent MI continue DAPT with aspirin and a P2Y12 blocker (ie, clopidogrel, ticlopidine, ticagrelor, prasugrel, or cangrelor) for 12 months following ACSto reduce recurrent ischemia.2-4
Studies have shown that using the newer P2Y12 inhibitors (ie, prasugrel and ticagrelor) after PCI leads to a significant reduction in recurrent ischemic events when compared to clopidogrel.5-7 These data led to a guideline change recommending the use of the newer agents over clopidogrel for 12 months following PCI.2 Follow-up studies evaluating the newer P2Y12 agents continue to show strong evidence for their use in the first month following PCI, while also demonstrating an increased bleeding risk in the maintenance phase (from 30 days to 12 months post-PCI).6,7 This increased risk is the basis for the current study, which tested switching from a newer P2Y12 agent to clopidogrel after the initial 30-day period following PCI.
STUDY SUMMARY
Switched DAPT is superior to unchanged DAPT
This open-label RCT (N = 646) examined changing DAPT from aspirin plus a newer P2Y12 blocker (prasugrel or ticagrelor) to a combination of aspirin and clopidogrel after the first month of DAPT post-ACS.1 Prior to PCI, all patients received a loading dose of ticagrelor 180 mg or prasugrel 60 mg. Subsequently, all patients in the trial took aspirin (75 mg/d) and one of the newer P2Y12 inhibitors (prasugrel 10 mg/d or ticagrelor 90 mg BID) for 1 month. For those enrollees who had no adverse events after 30 days, half were randomly switched to aspirin and clopidogrel 75 mg/d and the other half remained on aspirin and their newer P2Y12 blocker in a 1:1 ratio. For the next year, researchers examined the composite outcome of cardiovascular death, urgent revascularization, stroke, and major bleeding (as defined by the Bleeding Academic Research Consortium [BARC] classification ≥ Type 2 at 1 year post-ACS).
The average age of the participants was 60 years; 40% had experienced a STEMI and 60% had a non–STEMI. Overall, 43% of patients were prescribed ticagrelor and 57% prasugrel. At 1 year, 86% of the switched DAPT group and 75% of the unchanged DAPT group were still taking their medication. At the 1-year follow-up, the composite outcome was lower in the switched group, compared with the unchanged group (13% vs 26%; hazard ratio [HR] = 0.48; 95% confidence interval [CI], 0.34-0.68; number needed to treat [NNT] = 8).
All bleeding events (ranging from minimal to fatal) were lower in the switched group (9% vs 24%; HR = 0.39; 95% CI, 0.27-0.57; NNT = 7), and bleeding events identified as BARC ≥ Type 2 (defined as needing medical treatment) were also lower in the switched group (4% vs 15%; HR = 0.30, 95% CI, 0.18-0.50; NNT = 9). There were no significant differences in reported recurrent cardiovascular ischemic events (9.3% vs 11.5%; HR = 0.80, 95% CI, 0.50-1.29).
WHAT’S NEW
Fewer bleeding events without an increase in ischemic events
Cardiology guidelines recommend the newer P2Y12 blockers as part of DAPT after ACS, but this trial showed switching to clopidogrel for DAPT after 30 days of treatment lowers bleeding events with no difference in recurrent ischemic events.2-4
Continue to: CAVEATS
CAVEATS
Less-than-ideal study methods
This trial was an open-label, unblinded study. The investigators who adjudicated critical events were blinded to the treatment allocation, but some events, such as minor bleeding and medication discontinuation, could be self-reported by patients. In addition, the investigators used a less-than-ideal method (opaque envelopes) to conceal allocation at enrollment.
CHALLENGES TO IMPLEMENTATION
Implementation may require changing a cardiologist’s prescription
Implementing this practice change is facilitated by the fact that clopidogrel is currently less expensive than the newer P2Y12 blockers. However, after ACS and PCI treatment, cardiologists usually initiate antiplatelet therapy and may continue to manage patients after discharge. So the family physician (FP) may not be responsible for the DAPT switch initially. Further, switching may necessitate coordination with the cardiologist, as FPs may be hesitant to change cardiologists’ prescriptions. Lastly, guidelines currently recommend using the newer P2Y12 blockers for 12 months.2
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Cuisset T, Deharo P, Quilici J, et al. Benefit of switching dual antiplatelet therapy after acute coronary syndrome: the TOPIC (timing of platelet inhibition after acute coronary syndrome) randomized study. Eur Heart J. 2017;38:3070-3078.
2. Levine GN, Bates ER, Bittl JA, et al. 2016 ACC/AHA guideline focused update on duration of dual antiplatelet therapy in patients with coronary artery disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2016;68:1082-1115.
3. Steg PG, James SK, Atar D, et al. ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur Heart J. 2012;33:2569-2619.
4. Roffi M, Patrono C, Collet J-P, et al. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur Heart J. 2015;37:267-315.
5. Antman EM, Wiviott SD, Murphy SA, et al. Early and late benefits of prasugrel in patients with acute coronary syndromes undergoing percutaneous coronary intervention. J Am Coll Cardiol. 2008;51:2028-2033.
6. Wallentin L, Becker RC, Budaj A, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361:1045-1057.
7. Wiviott SD, Braunwald E, McCabe CH, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001-2015.
1. Cuisset T, Deharo P, Quilici J, et al. Benefit of switching dual antiplatelet therapy after acute coronary syndrome: the TOPIC (timing of platelet inhibition after acute coronary syndrome) randomized study. Eur Heart J. 2017;38:3070-3078.
2. Levine GN, Bates ER, Bittl JA, et al. 2016 ACC/AHA guideline focused update on duration of dual antiplatelet therapy in patients with coronary artery disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2016;68:1082-1115.
3. Steg PG, James SK, Atar D, et al. ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur Heart J. 2012;33:2569-2619.
4. Roffi M, Patrono C, Collet J-P, et al. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur Heart J. 2015;37:267-315.
5. Antman EM, Wiviott SD, Murphy SA, et al. Early and late benefits of prasugrel in patients with acute coronary syndromes undergoing percutaneous coronary intervention. J Am Coll Cardiol. 2008;51:2028-2033.
6. Wallentin L, Becker RC, Budaj A, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361:1045-1057.
7. Wiviott SD, Braunwald E, McCabe CH, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001-2015.
PRACTICE CHANGER
Switch to clopidogrel from one of the newer P2Y12 blockers 1 month after an acute coronary event, while continuing aspirin, to decrease bleeding events without increasing the risk of ischemic events.1
STRENGTH OF RECOMMENDATION
B: Based on a single randomized controlled trial (RCT).
Cuisset T, Deharo P, Quilici J, et al. Benefit of switching dual antiplatelet therapy after acute coronary syndrome: the TOPIC (timing of platelet inhibition after acute coronary syndrome) randomized study. Eur Heart J. 2017;38:3070-3078.

Less is more when it comes to ketorolac for pain
ILLUSTRATIVE CASE
A 46-year-old man with no significant past medical history presents to the emergency department (ED) with right flank pain and nausea. A computed tomography scan reveals a 5-mm ureteral stone with no obstruction or hydronephrosis. You are planning on starting him on intravenous (IV) ketorolac for pain. What is the most appropriate dose?
Ketorolac tromethamine is a highly effective nonsteroidal anti-inflammatory drug (NSAID). As a non-opiate analgesic, it is often the optimal first choice for the treatment of acute conditions such as flank, abdominal, musculoskeletal, and headache pains.2 While it is not associated with euphoria, withdrawal effects, or respiratory depression (like its opiate analgesic counterparts), ketorolac carries a US Food and Drug Administration black box warning for gastrointestinal, cardiovascular, renal, and bleeding risks.3
NSAIDs are known to have a “ceiling dose,” a dose at which maximum analgesic benefit is achieved; higher doses will not provide further pain relief. Higher doses of ketorolac may be used when anti-inflammatory effects of NSAIDs are desired, but they are likely to cause more adverse effects.4 Available data describe the analgesic ceiling dose of ketorolac as 10 mg across dosage forms.4,5 Yet, the majority of research and the majority of health care providers in current practice use higher doses of 20 to 60 mg. The US Food and Drug Administration label provides for a maximum dose of 60 mg/d.3
In one recent study, ketorolac was prescribed above its ceiling dose of 10 mg in atleast 97% of patients who received IV doses and in at least 96% of patients receiving intramuscular (IM) doses in a US emergency department.6 If ketorolac 10 mg is an effective analgesic dose, current practice exceeds the label recommendation to use the lowest effective dose. This study sought to determine the comparative efficacy of 3 different doses of IV ketorolac for acute pain management in an ED.
STUDY SUMMARY
Though often used at higher doses, 10 mg of ketorolac is enough for pain
This randomized double-blind trial evaluated the effectiveness of 3 different doses of IV ketorolac for acute pain in 240 adult patients, ages 18 to 65 years, presenting to an ED with acute flank, abdominal, musculoskeletal, or headache pain.1 Acute pain was defined as onset ≤30 days.
Patients were randomized to receive either 10, 15, or 30 mg of IV ketorolac in 10 mL of normal saline. A pharmacist prepared the medication in identical syringes, and the syringes were delivered in a blinded manner to the nurses caring for the patients. Pain (measured using a 0 to 10 scale), vital signs, and adverse effects were assessed at baseline and at 15, 30, 60, 90, and 120 minutes. If patients were still in pain at 30 minutes, IV morphine 0.1 mg/kg was offered. The primary outcome was numerical pain score at 30 minutes after ketorolac administration; secondary outcomes included the occurrence of adverse events and the use of rescue medication.
The groups were similar in terms of demographics and baseline vital signs. The mean age of the participants was 39 to 42 years. Across the 3 groups, 36% to 40% of patients had abdominal pain, 26% to 39% had flank pain, 20% to 26% had musculoskeletal pain, and 1% to 11% had headache pain. Patients had pain for an average of 1.5 to 3.5 days.
Continue to: Baseline pain scores were similar...
Baseline pain scores were similar for all 3 groups (7.5-7.8 on a 10-point scale). In the intention-to-treat analysis, all 3 doses of ketorolac decreased pain significantly at 30 minutes, but there was no difference between the groups; for the 10- and 15-mg groups, the mean pain scores post-intervention were 5.1 (95% confidence interval [CI] 4.5-5.7 and 4.5-5.6, respectively); and for the 30-mg group, the mean pain score was 4.8 (95% CI, 4.2-5.5). No P values were provided. There was no difference between the groups at any other time intervals. There was also no difference in the number of patients who needed rescue medication (morphine) at 30 minutes between the groups (4 patients in the 10-mg group, 3 patients in the 15-mg group, and 4 patients in the 30-mg group; no P values were provided). In addition, adverse events (eg, dizziness, nausea, headache, itching, flushing) did not differ between the groups.
WHAT’S NEW
10 mg is just as effective as 30 mg
This trial confirms that a low dose (10 mg) of IV ketorolac is just as effective for acute pain control as higher 15- and 30-mg doses.
CAVEATS
A 2-hour time limit and no look at long-term effects
The ketorolac dose of 10 mg IV was specially prepared by the study pharmacist; it is unlikely this will be readily available in clinical settings. However, the 15-mg IV dose is also as effective as the higher 30-mg dose based on study results and is readily available.
It isn’t known whether the higher dose would have provided greater pain relief beyond the 120 minutes evaluated in this trial, or if alternative dosage forms (oral or IM) would result in different outcomes. This study was not designed to compare serious long-term adverse effects like bleeding, renal impairment, or cardiovascular events. Additionally, this study was not powered to look at specific therapeutic indications or anti-inflammatory response.
CHALLENGES TO IMPLEMENTATION
A 10-mg single-dose vial is not readily available
Ketorolac tromethamine for injection is available in the United States in 15-, 30-, and 60-mg single-dose vials. Because a 10-mg dose is not available as a single-dose vial, it would need to be specially prepared. However, this study should reassure providers that using the lowest available dose (eg, 15 mg IV if that is what is available) will relieve acute pain as well as higher doses.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Motov S, Yasavolian M, Likourezos A, et al. Comparison of intravenous ketorolac at three single-dose regimens for treating acute pain in the emergency department: a randomized controlled trial. Ann Emerg Med. 2017;70:177-184.
2. Buckley MM, Brogden RN. Ketorolac. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential. Drugs. 1990;39:86-109.
3. Ketorolac tromethamine [package insert]. Bedford, OH: Bedford Labratories; 2009.
4. Catapano MS. The analgesic efficacy of ketorolac for acute pain. J Emerg Med. 1996;14:67-75.
5. García Rodríguez LA, Cattaruzzi C, Troncon MG, et al. Risk of hospitalization for upper gastrointestinal tract bleeding associated with ketorolac, other nonsteroidal anti-inflammatory drugs, calcium antagonists, and other antihypertensive drugs. Arch Intern Med. 1998;158:33-39.
6. Soleyman-Zomalan E, Motov S, Likourezos A, et al. Patterns of ketorolac dosing by emergency physicians. World J Emerg Med. 2017;8:43-46.
ILLUSTRATIVE CASE
A 46-year-old man with no significant past medical history presents to the emergency department (ED) with right flank pain and nausea. A computed tomography scan reveals a 5-mm ureteral stone with no obstruction or hydronephrosis. You are planning on starting him on intravenous (IV) ketorolac for pain. What is the most appropriate dose?
Ketorolac tromethamine is a highly effective nonsteroidal anti-inflammatory drug (NSAID). As a non-opiate analgesic, it is often the optimal first choice for the treatment of acute conditions such as flank, abdominal, musculoskeletal, and headache pains.2 While it is not associated with euphoria, withdrawal effects, or respiratory depression (like its opiate analgesic counterparts), ketorolac carries a US Food and Drug Administration black box warning for gastrointestinal, cardiovascular, renal, and bleeding risks.3
NSAIDs are known to have a “ceiling dose,” a dose at which maximum analgesic benefit is achieved; higher doses will not provide further pain relief. Higher doses of ketorolac may be used when anti-inflammatory effects of NSAIDs are desired, but they are likely to cause more adverse effects.4 Available data describe the analgesic ceiling dose of ketorolac as 10 mg across dosage forms.4,5 Yet, the majority of research and the majority of health care providers in current practice use higher doses of 20 to 60 mg. The US Food and Drug Administration label provides for a maximum dose of 60 mg/d.3
In one recent study, ketorolac was prescribed above its ceiling dose of 10 mg in atleast 97% of patients who received IV doses and in at least 96% of patients receiving intramuscular (IM) doses in a US emergency department.6 If ketorolac 10 mg is an effective analgesic dose, current practice exceeds the label recommendation to use the lowest effective dose. This study sought to determine the comparative efficacy of 3 different doses of IV ketorolac for acute pain management in an ED.
STUDY SUMMARY
Though often used at higher doses, 10 mg of ketorolac is enough for pain
This randomized double-blind trial evaluated the effectiveness of 3 different doses of IV ketorolac for acute pain in 240 adult patients, ages 18 to 65 years, presenting to an ED with acute flank, abdominal, musculoskeletal, or headache pain.1 Acute pain was defined as onset ≤30 days.
Patients were randomized to receive either 10, 15, or 30 mg of IV ketorolac in 10 mL of normal saline. A pharmacist prepared the medication in identical syringes, and the syringes were delivered in a blinded manner to the nurses caring for the patients. Pain (measured using a 0 to 10 scale), vital signs, and adverse effects were assessed at baseline and at 15, 30, 60, 90, and 120 minutes. If patients were still in pain at 30 minutes, IV morphine 0.1 mg/kg was offered. The primary outcome was numerical pain score at 30 minutes after ketorolac administration; secondary outcomes included the occurrence of adverse events and the use of rescue medication.
The groups were similar in terms of demographics and baseline vital signs. The mean age of the participants was 39 to 42 years. Across the 3 groups, 36% to 40% of patients had abdominal pain, 26% to 39% had flank pain, 20% to 26% had musculoskeletal pain, and 1% to 11% had headache pain. Patients had pain for an average of 1.5 to 3.5 days.
Continue to: Baseline pain scores were similar...
Baseline pain scores were similar for all 3 groups (7.5-7.8 on a 10-point scale). In the intention-to-treat analysis, all 3 doses of ketorolac decreased pain significantly at 30 minutes, but there was no difference between the groups; for the 10- and 15-mg groups, the mean pain scores post-intervention were 5.1 (95% confidence interval [CI] 4.5-5.7 and 4.5-5.6, respectively); and for the 30-mg group, the mean pain score was 4.8 (95% CI, 4.2-5.5). No P values were provided. There was no difference between the groups at any other time intervals. There was also no difference in the number of patients who needed rescue medication (morphine) at 30 minutes between the groups (4 patients in the 10-mg group, 3 patients in the 15-mg group, and 4 patients in the 30-mg group; no P values were provided). In addition, adverse events (eg, dizziness, nausea, headache, itching, flushing) did not differ between the groups.
WHAT’S NEW
10 mg is just as effective as 30 mg
This trial confirms that a low dose (10 mg) of IV ketorolac is just as effective for acute pain control as higher 15- and 30-mg doses.
CAVEATS
A 2-hour time limit and no look at long-term effects
The ketorolac dose of 10 mg IV was specially prepared by the study pharmacist; it is unlikely this will be readily available in clinical settings. However, the 15-mg IV dose is also as effective as the higher 30-mg dose based on study results and is readily available.
It isn’t known whether the higher dose would have provided greater pain relief beyond the 120 minutes evaluated in this trial, or if alternative dosage forms (oral or IM) would result in different outcomes. This study was not designed to compare serious long-term adverse effects like bleeding, renal impairment, or cardiovascular events. Additionally, this study was not powered to look at specific therapeutic indications or anti-inflammatory response.
CHALLENGES TO IMPLEMENTATION
A 10-mg single-dose vial is not readily available
Ketorolac tromethamine for injection is available in the United States in 15-, 30-, and 60-mg single-dose vials. Because a 10-mg dose is not available as a single-dose vial, it would need to be specially prepared. However, this study should reassure providers that using the lowest available dose (eg, 15 mg IV if that is what is available) will relieve acute pain as well as higher doses.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
ILLUSTRATIVE CASE
A 46-year-old man with no significant past medical history presents to the emergency department (ED) with right flank pain and nausea. A computed tomography scan reveals a 5-mm ureteral stone with no obstruction or hydronephrosis. You are planning on starting him on intravenous (IV) ketorolac for pain. What is the most appropriate dose?
Ketorolac tromethamine is a highly effective nonsteroidal anti-inflammatory drug (NSAID). As a non-opiate analgesic, it is often the optimal first choice for the treatment of acute conditions such as flank, abdominal, musculoskeletal, and headache pains.2 While it is not associated with euphoria, withdrawal effects, or respiratory depression (like its opiate analgesic counterparts), ketorolac carries a US Food and Drug Administration black box warning for gastrointestinal, cardiovascular, renal, and bleeding risks.3
NSAIDs are known to have a “ceiling dose,” a dose at which maximum analgesic benefit is achieved; higher doses will not provide further pain relief. Higher doses of ketorolac may be used when anti-inflammatory effects of NSAIDs are desired, but they are likely to cause more adverse effects.4 Available data describe the analgesic ceiling dose of ketorolac as 10 mg across dosage forms.4,5 Yet, the majority of research and the majority of health care providers in current practice use higher doses of 20 to 60 mg. The US Food and Drug Administration label provides for a maximum dose of 60 mg/d.3
In one recent study, ketorolac was prescribed above its ceiling dose of 10 mg in atleast 97% of patients who received IV doses and in at least 96% of patients receiving intramuscular (IM) doses in a US emergency department.6 If ketorolac 10 mg is an effective analgesic dose, current practice exceeds the label recommendation to use the lowest effective dose. This study sought to determine the comparative efficacy of 3 different doses of IV ketorolac for acute pain management in an ED.
STUDY SUMMARY
Though often used at higher doses, 10 mg of ketorolac is enough for pain
This randomized double-blind trial evaluated the effectiveness of 3 different doses of IV ketorolac for acute pain in 240 adult patients, ages 18 to 65 years, presenting to an ED with acute flank, abdominal, musculoskeletal, or headache pain.1 Acute pain was defined as onset ≤30 days.
Patients were randomized to receive either 10, 15, or 30 mg of IV ketorolac in 10 mL of normal saline. A pharmacist prepared the medication in identical syringes, and the syringes were delivered in a blinded manner to the nurses caring for the patients. Pain (measured using a 0 to 10 scale), vital signs, and adverse effects were assessed at baseline and at 15, 30, 60, 90, and 120 minutes. If patients were still in pain at 30 minutes, IV morphine 0.1 mg/kg was offered. The primary outcome was numerical pain score at 30 minutes after ketorolac administration; secondary outcomes included the occurrence of adverse events and the use of rescue medication.
The groups were similar in terms of demographics and baseline vital signs. The mean age of the participants was 39 to 42 years. Across the 3 groups, 36% to 40% of patients had abdominal pain, 26% to 39% had flank pain, 20% to 26% had musculoskeletal pain, and 1% to 11% had headache pain. Patients had pain for an average of 1.5 to 3.5 days.
Continue to: Baseline pain scores were similar...
Baseline pain scores were similar for all 3 groups (7.5-7.8 on a 10-point scale). In the intention-to-treat analysis, all 3 doses of ketorolac decreased pain significantly at 30 minutes, but there was no difference between the groups; for the 10- and 15-mg groups, the mean pain scores post-intervention were 5.1 (95% confidence interval [CI] 4.5-5.7 and 4.5-5.6, respectively); and for the 30-mg group, the mean pain score was 4.8 (95% CI, 4.2-5.5). No P values were provided. There was no difference between the groups at any other time intervals. There was also no difference in the number of patients who needed rescue medication (morphine) at 30 minutes between the groups (4 patients in the 10-mg group, 3 patients in the 15-mg group, and 4 patients in the 30-mg group; no P values were provided). In addition, adverse events (eg, dizziness, nausea, headache, itching, flushing) did not differ between the groups.
WHAT’S NEW
10 mg is just as effective as 30 mg
This trial confirms that a low dose (10 mg) of IV ketorolac is just as effective for acute pain control as higher 15- and 30-mg doses.
CAVEATS
A 2-hour time limit and no look at long-term effects
The ketorolac dose of 10 mg IV was specially prepared by the study pharmacist; it is unlikely this will be readily available in clinical settings. However, the 15-mg IV dose is also as effective as the higher 30-mg dose based on study results and is readily available.
It isn’t known whether the higher dose would have provided greater pain relief beyond the 120 minutes evaluated in this trial, or if alternative dosage forms (oral or IM) would result in different outcomes. This study was not designed to compare serious long-term adverse effects like bleeding, renal impairment, or cardiovascular events. Additionally, this study was not powered to look at specific therapeutic indications or anti-inflammatory response.
CHALLENGES TO IMPLEMENTATION
A 10-mg single-dose vial is not readily available
Ketorolac tromethamine for injection is available in the United States in 15-, 30-, and 60-mg single-dose vials. Because a 10-mg dose is not available as a single-dose vial, it would need to be specially prepared. However, this study should reassure providers that using the lowest available dose (eg, 15 mg IV if that is what is available) will relieve acute pain as well as higher doses.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Motov S, Yasavolian M, Likourezos A, et al. Comparison of intravenous ketorolac at three single-dose regimens for treating acute pain in the emergency department: a randomized controlled trial. Ann Emerg Med. 2017;70:177-184.
2. Buckley MM, Brogden RN. Ketorolac. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential. Drugs. 1990;39:86-109.
3. Ketorolac tromethamine [package insert]. Bedford, OH: Bedford Labratories; 2009.
4. Catapano MS. The analgesic efficacy of ketorolac for acute pain. J Emerg Med. 1996;14:67-75.
5. García Rodríguez LA, Cattaruzzi C, Troncon MG, et al. Risk of hospitalization for upper gastrointestinal tract bleeding associated with ketorolac, other nonsteroidal anti-inflammatory drugs, calcium antagonists, and other antihypertensive drugs. Arch Intern Med. 1998;158:33-39.
6. Soleyman-Zomalan E, Motov S, Likourezos A, et al. Patterns of ketorolac dosing by emergency physicians. World J Emerg Med. 2017;8:43-46.
1. Motov S, Yasavolian M, Likourezos A, et al. Comparison of intravenous ketorolac at three single-dose regimens for treating acute pain in the emergency department: a randomized controlled trial. Ann Emerg Med. 2017;70:177-184.
2. Buckley MM, Brogden RN. Ketorolac. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential. Drugs. 1990;39:86-109.
3. Ketorolac tromethamine [package insert]. Bedford, OH: Bedford Labratories; 2009.
4. Catapano MS. The analgesic efficacy of ketorolac for acute pain. J Emerg Med. 1996;14:67-75.
5. García Rodríguez LA, Cattaruzzi C, Troncon MG, et al. Risk of hospitalization for upper gastrointestinal tract bleeding associated with ketorolac, other nonsteroidal anti-inflammatory drugs, calcium antagonists, and other antihypertensive drugs. Arch Intern Med. 1998;158:33-39.
6. Soleyman-Zomalan E, Motov S, Likourezos A, et al. Patterns of ketorolac dosing by emergency physicians. World J Emerg Med. 2017;8:43-46.
PRACTICE CHANGER
Use a low dose (10 mg) of intravenous ketorolac for moderate to severe acute pain in adults because it is as effective as higher doses (15-30 mg).1
STRENGTH OF RECOMMENDATION
B: Based on a single, good-quality randomized controlled trial.
Motov S, Yasavolian M, Likourezos A, et al. Comparison of intravenous ketorolac at three single-dose regimens for treating acute pain in the emergency department: a randomized controlled trial. Ann Emerg Med. 2017;70:177-184.

Time to Stop Glucosamine and Chondroitin for Knee OA?

A 65-year-old man with moderately severe osteoarthritis (OA) of the knee presents to your office for his annual exam. During the medication review, the patient mentions he is using glucosamine and chondroitin for his knee pain, which was recommended by a family member. Should you tell the patient to continue taking the medication?
Knee OA is a common condition in the United States, affecting an estimated 12% of adults ages 60 and older and 16% of those ages 70 and older.2 The primary goals of OA therapy are to minimize pain and improve function. The American Academy of Orthopedic Surgeons (AAOS) and the American College of Rheumatology (ACR) agree that firstline treatment recommendations include aerobic exercise, resistance training, and weight loss.
Initial pharmacologic therapies include full-strength acetaminophen or oral/topical NSAIDs; the latter are also used if pain is unresponsive to acetaminophen.3,4 If initial therapy is inadequate to control pain, tramadol, other opioids, duloxetine, or intra-articular injections with corticosteroids or hyaluronate are alternatives.3,4 Total knee replacement may be indicated in moderate or severe knee OA with radiographic evidence.5 Vitamin D, lateral wedge insoles, and antioxidants are not currently recommended.6
Prior studies evaluating glucosamine and/or chondroitin have provided conflicting results regarding evidence on pain reduction, function, and quality of life. Therefore, guidelines on OA management do not recommend their use (AAOS, strong; ACR, conditional).3,4 However, consumption remains high, with 6.5 million US adults reporting use of glucosamine and/or chondroitin in the prior 30 days.7
A 2015 systematic review of 43 randomized trials evaluating oral chondroitin sulfate for OA of varying severity suggested there may be a significant decrease in short-term and long-term pain with doses ≥ 800 mg/d compared with placebo (level of evidence, low; risk for bias, high).8 However, no significant difference was noted in short- or long-term function, and the trials were highly heterogeneous.
Studies included in the 2015 systematic review found that glucosamine plus chondroitin did not have a significant effect on short- or long-term pain or physical function compared with placebo. Although glucosamine plus chondroitin led to significantly decreased pain compared with other medication, sensitivity analyses conducted for larger studies (N > 200) with adequate methods of blinding and allocation concealment found no difference in pain.8 There was no statistically significant difference in adverse events for glucosamine plus chondroitin vs placebo, based on data from three studies included in the review.8
This RCT from Roman-Blas et al evaluated chondroitin and glucosamine vs placebo in patients with more severe OA. The study was supported by Tedec-Meiji Farma (Madrid), maker of the combination of chondroitin plus glucosamine used in the study.1
Continue to: STUDY SUMMARY
STUDY SUMMARY
Chondroitin + glucosamine not better than placebo
This multicenter, randomized, double-blind, placebo-controlled trial was conducted in nine rheumatology referral centers and one orthopedic center in Spain. The trial evaluated the efficacy of chondroitin sulfate (1,200 mg) plus glucosamine sulfate (1,500 mg) (CS/GS) compared with placebo in 164 patients with Grade 2 or 3 knee OA and moderate-to-severe knee pain. OA grade was ascertained using the Kellgren-Lawrence scale, corresponding to osteophytes and either possible (Grade 2) or definite (Grade 3) joint space narrowing. Knee pain severity was defined by a self-reported global pain score of 40 to 80 mm on a 100-mm visual analog scale (VAS).
No significant difference was noted in group characteristics; average age in the CS/GS group was 67 and in the placebo group, 65. Exclusion criteria included BMI ≥ 35, concurrent arthritic conditions, and any coexisting chronic disease that would prevent successful completion of the trial.1
The primary endpoint was mean reduction in global pain score on a 0- to 100-mm VAS at six months. Secondary outcomes included mean reduction in total and subscale scores in pain and function on the Western Ontario and McMaster Universities Osteoarthritis (WOMAC) index (0–100-mm VAS for each) and the use of rescue medication.
Baseline global pain scores were 62 mm in both groups. Acetaminophen, up to 3 g/d, was the only allowed rescue medication. Clinic visits occurred at 4, 12, and 24 weeks. A statistically significant difference between groups was defined as P < .03.1
Results. In the intention-to-treat analysis at six months, patients in the placebo group had a greater reduction in pain than the CS/GC group (–20 mm vs –12 mm; P = .029). No other difference was noted between the placebo and CS/GS groups in the total or subscales of the WOMAC index, and no difference was noted in use of acetaminophen. More patients in the placebo group had at least a 50% improvement in pain or function compared with the CS/GS group (47.4% vs 27.5%; P = .01).
Continue to: In the CS/GS group...
In the CS/GS group, 31% did not complete the six-month treatment period, compared with 18% in the placebo group. More patients dropped out because of adverse effects (diarrhea, upper abdominal pain, and constipation) in the CS/GS group than the placebo group (33 vs 19; P = .018).1
WHAT’S NEW
Pharma-sponsored study finds treatment ineffective
The effectiveness of CS/GS for the treatment of knee OA has been in question for years, but this RCT is the first trial sponsored by a pharmaceutical company to evaluate CS/GS efficacy. This trial found evidence of a lack of efficacy. In patients with more severe OA of the knee, placebo was more effective than CS/GS, and CS/GS had significantly more adverse events. Therefore, it may be time to advise patients to stop taking their CS/GS supplement.
CAVEATS
Cannot generalize findings
The study compared only one medication dosing regimen using a combination of CS and GS. Whether either agent alone, or different dosing, would lead to the same outcome is unknown.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2018. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2018; 67[9]:566-568).
1. Roman-Blas JA, Castañeda S, Sánchez-Pernaute O, et al. Combined treatment with chondroitin sulfate and glucosamine sulfate shows no superiority over placebo for reduction of joint pain and functional impairment in patients with knee osteoarthritis: a six-month multicenter, randomized, double-blind, placebo-controlled clinical trial. Arthritis Rheumatol. 2017;69:77-85.
2. Dillon CF, Rasch EK, Gu Q, et al. Prevalence of knee osteoarthritis in the United States: arthritis data from the Third National Health and Nutrition Examination Survey 1991-94. J Rheumatol. 2006;33:2271-2279.
3. Hochberg MC, Altman RD, April KT, et al. American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken). 2012;64:465-474.
4. Brown GA. AAOS clinical practice guideline: treatment of osteoarthritis of the knee: evidence-based guideline, 2nd ed. J Am Acad Orthop Surg. 2013;21:577-579.
5. Jordan KM, Arden NK, Doherty M, et al. EULAR Recommendations 2003: an evidence based approach to the management of knee osteoarthritis: report of a Task Force of the Standing Committee for International Clinical Studies Including Therapeutic Trials (ESCISIT). Ann Rheum Dis. 2003;62:1145-1155.
6. Ebell MH. Osteoarthritis: rapid evidence review. Am Fam Physician. 2018;97:523-526.
7. Clarke TC, Black LI, Stussman BJ, et al. Trends in the use of complementary health approaches among adults: United States, 2002-2012. Natl Health Stat Rep. 2015;(79):1-16.
8. Singh JA, Noorbaloochi S, MacDonald R, et al. Chondroitin for osteoarthritis. Cochrane Database Syst Rev. 2015;(1):CD005614.
A 65-year-old man with moderately severe osteoarthritis (OA) of the knee presents to your office for his annual exam. During the medication review, the patient mentions he is using glucosamine and chondroitin for his knee pain, which was recommended by a family member. Should you tell the patient to continue taking the medication?
Knee OA is a common condition in the United States, affecting an estimated 12% of adults ages 60 and older and 16% of those ages 70 and older.2 The primary goals of OA therapy are to minimize pain and improve function. The American Academy of Orthopedic Surgeons (AAOS) and the American College of Rheumatology (ACR) agree that firstline treatment recommendations include aerobic exercise, resistance training, and weight loss.
Initial pharmacologic therapies include full-strength acetaminophen or oral/topical NSAIDs; the latter are also used if pain is unresponsive to acetaminophen.3,4 If initial therapy is inadequate to control pain, tramadol, other opioids, duloxetine, or intra-articular injections with corticosteroids or hyaluronate are alternatives.3,4 Total knee replacement may be indicated in moderate or severe knee OA with radiographic evidence.5 Vitamin D, lateral wedge insoles, and antioxidants are not currently recommended.6
Prior studies evaluating glucosamine and/or chondroitin have provided conflicting results regarding evidence on pain reduction, function, and quality of life. Therefore, guidelines on OA management do not recommend their use (AAOS, strong; ACR, conditional).3,4 However, consumption remains high, with 6.5 million US adults reporting use of glucosamine and/or chondroitin in the prior 30 days.7
A 2015 systematic review of 43 randomized trials evaluating oral chondroitin sulfate for OA of varying severity suggested there may be a significant decrease in short-term and long-term pain with doses ≥ 800 mg/d compared with placebo (level of evidence, low; risk for bias, high).8 However, no significant difference was noted in short- or long-term function, and the trials were highly heterogeneous.
Studies included in the 2015 systematic review found that glucosamine plus chondroitin did not have a significant effect on short- or long-term pain or physical function compared with placebo. Although glucosamine plus chondroitin led to significantly decreased pain compared with other medication, sensitivity analyses conducted for larger studies (N > 200) with adequate methods of blinding and allocation concealment found no difference in pain.8 There was no statistically significant difference in adverse events for glucosamine plus chondroitin vs placebo, based on data from three studies included in the review.8
This RCT from Roman-Blas et al evaluated chondroitin and glucosamine vs placebo in patients with more severe OA. The study was supported by Tedec-Meiji Farma (Madrid), maker of the combination of chondroitin plus glucosamine used in the study.1
Continue to: STUDY SUMMARY
STUDY SUMMARY
Chondroitin + glucosamine not better than placebo
This multicenter, randomized, double-blind, placebo-controlled trial was conducted in nine rheumatology referral centers and one orthopedic center in Spain. The trial evaluated the efficacy of chondroitin sulfate (1,200 mg) plus glucosamine sulfate (1,500 mg) (CS/GS) compared with placebo in 164 patients with Grade 2 or 3 knee OA and moderate-to-severe knee pain. OA grade was ascertained using the Kellgren-Lawrence scale, corresponding to osteophytes and either possible (Grade 2) or definite (Grade 3) joint space narrowing. Knee pain severity was defined by a self-reported global pain score of 40 to 80 mm on a 100-mm visual analog scale (VAS).
No significant difference was noted in group characteristics; average age in the CS/GS group was 67 and in the placebo group, 65. Exclusion criteria included BMI ≥ 35, concurrent arthritic conditions, and any coexisting chronic disease that would prevent successful completion of the trial.1
The primary endpoint was mean reduction in global pain score on a 0- to 100-mm VAS at six months. Secondary outcomes included mean reduction in total and subscale scores in pain and function on the Western Ontario and McMaster Universities Osteoarthritis (WOMAC) index (0–100-mm VAS for each) and the use of rescue medication.
Baseline global pain scores were 62 mm in both groups. Acetaminophen, up to 3 g/d, was the only allowed rescue medication. Clinic visits occurred at 4, 12, and 24 weeks. A statistically significant difference between groups was defined as P < .03.1
Results. In the intention-to-treat analysis at six months, patients in the placebo group had a greater reduction in pain than the CS/GC group (–20 mm vs –12 mm; P = .029). No other difference was noted between the placebo and CS/GS groups in the total or subscales of the WOMAC index, and no difference was noted in use of acetaminophen. More patients in the placebo group had at least a 50% improvement in pain or function compared with the CS/GS group (47.4% vs 27.5%; P = .01).
Continue to: In the CS/GS group...
In the CS/GS group, 31% did not complete the six-month treatment period, compared with 18% in the placebo group. More patients dropped out because of adverse effects (diarrhea, upper abdominal pain, and constipation) in the CS/GS group than the placebo group (33 vs 19; P = .018).1
WHAT’S NEW
Pharma-sponsored study finds treatment ineffective
The effectiveness of CS/GS for the treatment of knee OA has been in question for years, but this RCT is the first trial sponsored by a pharmaceutical company to evaluate CS/GS efficacy. This trial found evidence of a lack of efficacy. In patients with more severe OA of the knee, placebo was more effective than CS/GS, and CS/GS had significantly more adverse events. Therefore, it may be time to advise patients to stop taking their CS/GS supplement.
CAVEATS
Cannot generalize findings
The study compared only one medication dosing regimen using a combination of CS and GS. Whether either agent alone, or different dosing, would lead to the same outcome is unknown.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2018. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2018; 67[9]:566-568).
A 65-year-old man with moderately severe osteoarthritis (OA) of the knee presents to your office for his annual exam. During the medication review, the patient mentions he is using glucosamine and chondroitin for his knee pain, which was recommended by a family member. Should you tell the patient to continue taking the medication?
Knee OA is a common condition in the United States, affecting an estimated 12% of adults ages 60 and older and 16% of those ages 70 and older.2 The primary goals of OA therapy are to minimize pain and improve function. The American Academy of Orthopedic Surgeons (AAOS) and the American College of Rheumatology (ACR) agree that firstline treatment recommendations include aerobic exercise, resistance training, and weight loss.
Initial pharmacologic therapies include full-strength acetaminophen or oral/topical NSAIDs; the latter are also used if pain is unresponsive to acetaminophen.3,4 If initial therapy is inadequate to control pain, tramadol, other opioids, duloxetine, or intra-articular injections with corticosteroids or hyaluronate are alternatives.3,4 Total knee replacement may be indicated in moderate or severe knee OA with radiographic evidence.5 Vitamin D, lateral wedge insoles, and antioxidants are not currently recommended.6
Prior studies evaluating glucosamine and/or chondroitin have provided conflicting results regarding evidence on pain reduction, function, and quality of life. Therefore, guidelines on OA management do not recommend their use (AAOS, strong; ACR, conditional).3,4 However, consumption remains high, with 6.5 million US adults reporting use of glucosamine and/or chondroitin in the prior 30 days.7
A 2015 systematic review of 43 randomized trials evaluating oral chondroitin sulfate for OA of varying severity suggested there may be a significant decrease in short-term and long-term pain with doses ≥ 800 mg/d compared with placebo (level of evidence, low; risk for bias, high).8 However, no significant difference was noted in short- or long-term function, and the trials were highly heterogeneous.
Studies included in the 2015 systematic review found that glucosamine plus chondroitin did not have a significant effect on short- or long-term pain or physical function compared with placebo. Although glucosamine plus chondroitin led to significantly decreased pain compared with other medication, sensitivity analyses conducted for larger studies (N > 200) with adequate methods of blinding and allocation concealment found no difference in pain.8 There was no statistically significant difference in adverse events for glucosamine plus chondroitin vs placebo, based on data from three studies included in the review.8
This RCT from Roman-Blas et al evaluated chondroitin and glucosamine vs placebo in patients with more severe OA. The study was supported by Tedec-Meiji Farma (Madrid), maker of the combination of chondroitin plus glucosamine used in the study.1
Continue to: STUDY SUMMARY
STUDY SUMMARY
Chondroitin + glucosamine not better than placebo
This multicenter, randomized, double-blind, placebo-controlled trial was conducted in nine rheumatology referral centers and one orthopedic center in Spain. The trial evaluated the efficacy of chondroitin sulfate (1,200 mg) plus glucosamine sulfate (1,500 mg) (CS/GS) compared with placebo in 164 patients with Grade 2 or 3 knee OA and moderate-to-severe knee pain. OA grade was ascertained using the Kellgren-Lawrence scale, corresponding to osteophytes and either possible (Grade 2) or definite (Grade 3) joint space narrowing. Knee pain severity was defined by a self-reported global pain score of 40 to 80 mm on a 100-mm visual analog scale (VAS).
No significant difference was noted in group characteristics; average age in the CS/GS group was 67 and in the placebo group, 65. Exclusion criteria included BMI ≥ 35, concurrent arthritic conditions, and any coexisting chronic disease that would prevent successful completion of the trial.1
The primary endpoint was mean reduction in global pain score on a 0- to 100-mm VAS at six months. Secondary outcomes included mean reduction in total and subscale scores in pain and function on the Western Ontario and McMaster Universities Osteoarthritis (WOMAC) index (0–100-mm VAS for each) and the use of rescue medication.
Baseline global pain scores were 62 mm in both groups. Acetaminophen, up to 3 g/d, was the only allowed rescue medication. Clinic visits occurred at 4, 12, and 24 weeks. A statistically significant difference between groups was defined as P < .03.1
Results. In the intention-to-treat analysis at six months, patients in the placebo group had a greater reduction in pain than the CS/GC group (–20 mm vs –12 mm; P = .029). No other difference was noted between the placebo and CS/GS groups in the total or subscales of the WOMAC index, and no difference was noted in use of acetaminophen. More patients in the placebo group had at least a 50% improvement in pain or function compared with the CS/GS group (47.4% vs 27.5%; P = .01).
Continue to: In the CS/GS group...
In the CS/GS group, 31% did not complete the six-month treatment period, compared with 18% in the placebo group. More patients dropped out because of adverse effects (diarrhea, upper abdominal pain, and constipation) in the CS/GS group than the placebo group (33 vs 19; P = .018).1
WHAT’S NEW
Pharma-sponsored study finds treatment ineffective
The effectiveness of CS/GS for the treatment of knee OA has been in question for years, but this RCT is the first trial sponsored by a pharmaceutical company to evaluate CS/GS efficacy. This trial found evidence of a lack of efficacy. In patients with more severe OA of the knee, placebo was more effective than CS/GS, and CS/GS had significantly more adverse events. Therefore, it may be time to advise patients to stop taking their CS/GS supplement.
CAVEATS
Cannot generalize findings
The study compared only one medication dosing regimen using a combination of CS and GS. Whether either agent alone, or different dosing, would lead to the same outcome is unknown.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
Copyright © 2018. The Family Physicians Inquiries Network. All rights reserved.
Reprinted with permission from the Family Physicians Inquiries Network and The Journal of Family Practice (2018; 67[9]:566-568).
1. Roman-Blas JA, Castañeda S, Sánchez-Pernaute O, et al. Combined treatment with chondroitin sulfate and glucosamine sulfate shows no superiority over placebo for reduction of joint pain and functional impairment in patients with knee osteoarthritis: a six-month multicenter, randomized, double-blind, placebo-controlled clinical trial. Arthritis Rheumatol. 2017;69:77-85.
2. Dillon CF, Rasch EK, Gu Q, et al. Prevalence of knee osteoarthritis in the United States: arthritis data from the Third National Health and Nutrition Examination Survey 1991-94. J Rheumatol. 2006;33:2271-2279.
3. Hochberg MC, Altman RD, April KT, et al. American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken). 2012;64:465-474.
4. Brown GA. AAOS clinical practice guideline: treatment of osteoarthritis of the knee: evidence-based guideline, 2nd ed. J Am Acad Orthop Surg. 2013;21:577-579.
5. Jordan KM, Arden NK, Doherty M, et al. EULAR Recommendations 2003: an evidence based approach to the management of knee osteoarthritis: report of a Task Force of the Standing Committee for International Clinical Studies Including Therapeutic Trials (ESCISIT). Ann Rheum Dis. 2003;62:1145-1155.
6. Ebell MH. Osteoarthritis: rapid evidence review. Am Fam Physician. 2018;97:523-526.
7. Clarke TC, Black LI, Stussman BJ, et al. Trends in the use of complementary health approaches among adults: United States, 2002-2012. Natl Health Stat Rep. 2015;(79):1-16.
8. Singh JA, Noorbaloochi S, MacDonald R, et al. Chondroitin for osteoarthritis. Cochrane Database Syst Rev. 2015;(1):CD005614.
1. Roman-Blas JA, Castañeda S, Sánchez-Pernaute O, et al. Combined treatment with chondroitin sulfate and glucosamine sulfate shows no superiority over placebo for reduction of joint pain and functional impairment in patients with knee osteoarthritis: a six-month multicenter, randomized, double-blind, placebo-controlled clinical trial. Arthritis Rheumatol. 2017;69:77-85.
2. Dillon CF, Rasch EK, Gu Q, et al. Prevalence of knee osteoarthritis in the United States: arthritis data from the Third National Health and Nutrition Examination Survey 1991-94. J Rheumatol. 2006;33:2271-2279.
3. Hochberg MC, Altman RD, April KT, et al. American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken). 2012;64:465-474.
4. Brown GA. AAOS clinical practice guideline: treatment of osteoarthritis of the knee: evidence-based guideline, 2nd ed. J Am Acad Orthop Surg. 2013;21:577-579.
5. Jordan KM, Arden NK, Doherty M, et al. EULAR Recommendations 2003: an evidence based approach to the management of knee osteoarthritis: report of a Task Force of the Standing Committee for International Clinical Studies Including Therapeutic Trials (ESCISIT). Ann Rheum Dis. 2003;62:1145-1155.
6. Ebell MH. Osteoarthritis: rapid evidence review. Am Fam Physician. 2018;97:523-526.
7. Clarke TC, Black LI, Stussman BJ, et al. Trends in the use of complementary health approaches among adults: United States, 2002-2012. Natl Health Stat Rep. 2015;(79):1-16.
8. Singh JA, Noorbaloochi S, MacDonald R, et al. Chondroitin for osteoarthritis. Cochrane Database Syst Rev. 2015;(1):CD005614.

Time to stop glucosamine and chondroitin for knee OA?
ILLUSTRATIVE CASE
A 65-year-old man with moderately severe osteoarthritis (OA) of the knee presents to your office for his annual exam. During the medication review, the patient mentions he is using glucosamine and chondroitin for his knee pain, which was recommended by a family member.
Should you tell the patient it’s okay to continue the medication?
Knee OA in the United States is a common condition and affects an estimated 12% of adults 60 years and older and 16% of adults 70 years and older.2 The primary goals of OA therapy are to minimize pain and improve function. The American Academy of Orthopedic Surgeons (AAOS) and the American College of Rheumatology (ACR) agree that first-line treatment recommendations include aerobic exercise, resistance training, and weight loss.
Initial pharmacologic therapies include full-strength acetaminophen or oral/topical nonsteroidal anti-inflammatory drugs (either initially or if unresponsive to acetaminophen).3,4 Alternative medication options for patients with an inadequate response to initial therapy include tramadol, other opioids, duloxetine, or intra-articular injections with corticosteroids or hyaluronate.3,4 Total knee replacement may be indicated in moderate or severe knee OA with radiographic evidence of OA.5 Vitamin D, lateral wedge insoles, and antioxidants are not currently recommended.6
Prior studies evaluating glucosamine and/or chondroitin have provided conflicting results regarding evidence on pain reduction, function, and quality of life. Therefore, guidelines on OA management do not recommend their use (AAOS, strong; ACR, conditional recommendation).3,4 However, consumption remains high, with 6.5 million US adults reporting use of glucosamine and/or chondroitin in the prior 30 days.7
A 2015 systematic review of 43 randomized trials evaluating oral chondroitin sulfate for OA of varying severity suggested there may be a significant decrease in short-term and long-term pain with doses of ≥800 mg/d compared with placebo (level of evidence, low; risk of bias, high).8 However, no significant difference was noted in short- or long-term function, and the trials were highly heterogeneous.
[polldaddy:10097537]
Studies included in the 2015 systematic review found that glucosamine plus chondroitin did not have a significant effect on short- or long-term pain or physical function compared with placebo. Although glucosamine plus chondroitin led to significantly decreased pain compared with other medication, sensitivity analyses conducted for larger studies (N>200) with adequate methods of blinding and allocation concealment found no difference in pain.8
Continue to: Three studies included...
Three studies included in the 2015 systematic review provided data on adverse events when comparing glucosamine plus chondroitin vs placebo, and found no statistically significant difference.8
This randomized controlled trial (RCT) from Roman-Blas et al1 evaluated chondroitin and glucosamine vs placebo in patients with more severe OA. The study was supported by Tedec-Meiji Farma (Madrid, Spain) maker of the combination of chondroitin plus glucosamine used in the study.
STUDY SUMMARY
Chondroitin + glucosamine was not better than placebo for pain
This multicenter, randomized, double-blind, placebo-controlled trial was conducted in 9 rheumatology referral centers and one orthopedic center in Spain. The trial evaluated the efficacy of chondroitin sulfate 1200 mg plus glucosamine sulfate 1500 mg (CS/GS) compared with placebo in 164 patients with Grade 2 or 3 knee OA and moderate to severe knee pain. OA grade was ascertained using the Kellgren-Lawrence scale, corresponding to osteophytes and either possible (Grade 2) or definite (Grade 3) joint space narrowing. Level of knee pain was defined by a self-reported global pain score of 40-80 mm on a 100-mm visual analog scale (VAS).
No significant difference was noted in group characteristics, and the average age in the CS/GS group was 67 years vs 65 years in the placebo group. Exclusion criteria included body mass index of ≥35 kg/m2, concurrent arthritic conditions, and any coexisting chronic disease that would prevent successful completion of the trial.1
The primary end point was mean reduction in global pain score on a 0- to 100-mm VAS at 6 months. Secondary outcomes included mean reduction in total and subscale scores in pain and function on the Western Ontario and McMaster Universities Osteoarthritis (WOMAC) index (0–100-mm VAS for each) and the use of rescue medication.
Continue to: Baseline global pain scores were...
Baseline global pain scores were 62 mm in both groups. Acetaminophen, up to 3 g/d, was the only allowed rescue medication. Clinic visits occurred at 4, 12, and 24 weeks. A statistically significant difference between groups was defined as P<.03.1
Results. In the intention-to-treat analysis at 6 months, patients in the placebo group had a greater reduction in pain than the CS/GC group (-20 mm vs -12 mm; P=.029). No other difference was noted between the placebo and CS/GS groups in the total or subscales of the WOMAC index, and no difference was noted in use of acetaminophen. More patients in the placebo group had at least a 50% improvement in pain or function compared with the CS/GS group (47.4% vs 27.5%; P=.01).
In the CS/GS group, 31% did not complete the 6-month treatment period, compared with 18% in the placebo group. More patients dropped out because of adverse effects (diarrhea, upper abdominal pain, and constipation) in the CS/GS group than the placebo group (33 vs 19; P=.018).1
WHAT’S NEW
A pharma-sponsored study finds treatment ineffective
The effectiveness of CS/GS for the treatment of knee OA has been in question for years, but this RCT is the first trial sponsored by a pharmaceutical company to evaluate CS/GS efficacy. This trial found evidence of a lack of efficacy. In patients with more severe OA of the knee, placebo was more effective than CS/GS, and CS/GS had significantly more adverse events. Therefore, it may be time to advise patients to stop taking their CS/GS supplement.
CAVEATS
Cannot generalize findings to CS or GS alone, or different dosages
The study compared only one medication dosing regimen using a combination of CS and GS. Whether either agent alone or different dosing would lead to the same outcome is unknown.
Continue to: CHALLENGES TO IMPLEMENTATION
CHALLENGES TO IMPLEMENTATION
An all-too-common product presents challenges
CS/GC is available over the counter and advertised directly to consumers. With this medication so readily available, identifying patients who are taking the supplement and encouraging discontinuation can be a challenge.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Roman-Blas JA, Castañeda S, Sánchez-Pernaute O, et al. Combined treatment with chondroitin sulfate and glucosamine sulfate shows no superiority over placebo for reduction of joint pain and functional impairment in patients with knee osteoarthritis: a six-month multicenter, randomized, double-blind, placebo-controlled clinical trial. Arthritis Rheumatol. 2017;69:77-85.
2. Dillon CF, Rasch EK, Gu Q, et al. Prevalence of knee osteoarthritis in the United States: arthritis data from the Third National Health and Nutrition Examination Survey 1991-94. J Rheumatol. 2006;33:2271-2279.
3. Hochberg MC, Altman RD, April KT, et al. American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken). 2012;64:465-474.
4. Brown GA. AAOS clinical practice guideline: treatment of osteoarthritis of the knee: evidence-based guideline, 2nd ed. J Am Acad Orthop Surg. 2013;21:577-579.
5. Jordan KM, Arden NK, Doherty M, et al. EULAR Recommendations 2003: an evidence based approach to the management of knee osteoarthritis: report of a Task Force of the Standing Committee for International Clinical Studies Including Therapeutic Trials (ESCISIT). Ann Rheum Dis. 2003;62:1145-1155.
6. Ebell MH. Osteoarthritis: rapid evidence review. Am Fam Physician. 2018;97:523-526.
7. Clarke TC, Black LI, Stussman BJ, et al. Trends in the use of complementary health approaches among adults: United States, 2002-2012. Natl Health Stat Rep. 2015;(79):1-16.
8. Singh JA, Noorbaloochi S, MacDonald R, et al. Chondroitin for osteoarthritis. Cochrane Database Syst Rev. 2015;(1):CD005614.
ILLUSTRATIVE CASE
A 65-year-old man with moderately severe osteoarthritis (OA) of the knee presents to your office for his annual exam. During the medication review, the patient mentions he is using glucosamine and chondroitin for his knee pain, which was recommended by a family member.
Should you tell the patient it’s okay to continue the medication?
Knee OA in the United States is a common condition and affects an estimated 12% of adults 60 years and older and 16% of adults 70 years and older.2 The primary goals of OA therapy are to minimize pain and improve function. The American Academy of Orthopedic Surgeons (AAOS) and the American College of Rheumatology (ACR) agree that first-line treatment recommendations include aerobic exercise, resistance training, and weight loss.
Initial pharmacologic therapies include full-strength acetaminophen or oral/topical nonsteroidal anti-inflammatory drugs (either initially or if unresponsive to acetaminophen).3,4 Alternative medication options for patients with an inadequate response to initial therapy include tramadol, other opioids, duloxetine, or intra-articular injections with corticosteroids or hyaluronate.3,4 Total knee replacement may be indicated in moderate or severe knee OA with radiographic evidence of OA.5 Vitamin D, lateral wedge insoles, and antioxidants are not currently recommended.6
Prior studies evaluating glucosamine and/or chondroitin have provided conflicting results regarding evidence on pain reduction, function, and quality of life. Therefore, guidelines on OA management do not recommend their use (AAOS, strong; ACR, conditional recommendation).3,4 However, consumption remains high, with 6.5 million US adults reporting use of glucosamine and/or chondroitin in the prior 30 days.7
A 2015 systematic review of 43 randomized trials evaluating oral chondroitin sulfate for OA of varying severity suggested there may be a significant decrease in short-term and long-term pain with doses of ≥800 mg/d compared with placebo (level of evidence, low; risk of bias, high).8 However, no significant difference was noted in short- or long-term function, and the trials were highly heterogeneous.
[polldaddy:10097537]
Studies included in the 2015 systematic review found that glucosamine plus chondroitin did not have a significant effect on short- or long-term pain or physical function compared with placebo. Although glucosamine plus chondroitin led to significantly decreased pain compared with other medication, sensitivity analyses conducted for larger studies (N>200) with adequate methods of blinding and allocation concealment found no difference in pain.8
Continue to: Three studies included...
Three studies included in the 2015 systematic review provided data on adverse events when comparing glucosamine plus chondroitin vs placebo, and found no statistically significant difference.8
This randomized controlled trial (RCT) from Roman-Blas et al1 evaluated chondroitin and glucosamine vs placebo in patients with more severe OA. The study was supported by Tedec-Meiji Farma (Madrid, Spain) maker of the combination of chondroitin plus glucosamine used in the study.
STUDY SUMMARY
Chondroitin + glucosamine was not better than placebo for pain
This multicenter, randomized, double-blind, placebo-controlled trial was conducted in 9 rheumatology referral centers and one orthopedic center in Spain. The trial evaluated the efficacy of chondroitin sulfate 1200 mg plus glucosamine sulfate 1500 mg (CS/GS) compared with placebo in 164 patients with Grade 2 or 3 knee OA and moderate to severe knee pain. OA grade was ascertained using the Kellgren-Lawrence scale, corresponding to osteophytes and either possible (Grade 2) or definite (Grade 3) joint space narrowing. Level of knee pain was defined by a self-reported global pain score of 40-80 mm on a 100-mm visual analog scale (VAS).
No significant difference was noted in group characteristics, and the average age in the CS/GS group was 67 years vs 65 years in the placebo group. Exclusion criteria included body mass index of ≥35 kg/m2, concurrent arthritic conditions, and any coexisting chronic disease that would prevent successful completion of the trial.1
The primary end point was mean reduction in global pain score on a 0- to 100-mm VAS at 6 months. Secondary outcomes included mean reduction in total and subscale scores in pain and function on the Western Ontario and McMaster Universities Osteoarthritis (WOMAC) index (0–100-mm VAS for each) and the use of rescue medication.
Continue to: Baseline global pain scores were...
Baseline global pain scores were 62 mm in both groups. Acetaminophen, up to 3 g/d, was the only allowed rescue medication. Clinic visits occurred at 4, 12, and 24 weeks. A statistically significant difference between groups was defined as P<.03.1
Results. In the intention-to-treat analysis at 6 months, patients in the placebo group had a greater reduction in pain than the CS/GC group (-20 mm vs -12 mm; P=.029). No other difference was noted between the placebo and CS/GS groups in the total or subscales of the WOMAC index, and no difference was noted in use of acetaminophen. More patients in the placebo group had at least a 50% improvement in pain or function compared with the CS/GS group (47.4% vs 27.5%; P=.01).
In the CS/GS group, 31% did not complete the 6-month treatment period, compared with 18% in the placebo group. More patients dropped out because of adverse effects (diarrhea, upper abdominal pain, and constipation) in the CS/GS group than the placebo group (33 vs 19; P=.018).1
WHAT’S NEW
A pharma-sponsored study finds treatment ineffective
The effectiveness of CS/GS for the treatment of knee OA has been in question for years, but this RCT is the first trial sponsored by a pharmaceutical company to evaluate CS/GS efficacy. This trial found evidence of a lack of efficacy. In patients with more severe OA of the knee, placebo was more effective than CS/GS, and CS/GS had significantly more adverse events. Therefore, it may be time to advise patients to stop taking their CS/GS supplement.
CAVEATS
Cannot generalize findings to CS or GS alone, or different dosages
The study compared only one medication dosing regimen using a combination of CS and GS. Whether either agent alone or different dosing would lead to the same outcome is unknown.
Continue to: CHALLENGES TO IMPLEMENTATION
CHALLENGES TO IMPLEMENTATION
An all-too-common product presents challenges
CS/GC is available over the counter and advertised directly to consumers. With this medication so readily available, identifying patients who are taking the supplement and encouraging discontinuation can be a challenge.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
ILLUSTRATIVE CASE
A 65-year-old man with moderately severe osteoarthritis (OA) of the knee presents to your office for his annual exam. During the medication review, the patient mentions he is using glucosamine and chondroitin for his knee pain, which was recommended by a family member.
Should you tell the patient it’s okay to continue the medication?
Knee OA in the United States is a common condition and affects an estimated 12% of adults 60 years and older and 16% of adults 70 years and older.2 The primary goals of OA therapy are to minimize pain and improve function. The American Academy of Orthopedic Surgeons (AAOS) and the American College of Rheumatology (ACR) agree that first-line treatment recommendations include aerobic exercise, resistance training, and weight loss.
Initial pharmacologic therapies include full-strength acetaminophen or oral/topical nonsteroidal anti-inflammatory drugs (either initially or if unresponsive to acetaminophen).3,4 Alternative medication options for patients with an inadequate response to initial therapy include tramadol, other opioids, duloxetine, or intra-articular injections with corticosteroids or hyaluronate.3,4 Total knee replacement may be indicated in moderate or severe knee OA with radiographic evidence of OA.5 Vitamin D, lateral wedge insoles, and antioxidants are not currently recommended.6
Prior studies evaluating glucosamine and/or chondroitin have provided conflicting results regarding evidence on pain reduction, function, and quality of life. Therefore, guidelines on OA management do not recommend their use (AAOS, strong; ACR, conditional recommendation).3,4 However, consumption remains high, with 6.5 million US adults reporting use of glucosamine and/or chondroitin in the prior 30 days.7
A 2015 systematic review of 43 randomized trials evaluating oral chondroitin sulfate for OA of varying severity suggested there may be a significant decrease in short-term and long-term pain with doses of ≥800 mg/d compared with placebo (level of evidence, low; risk of bias, high).8 However, no significant difference was noted in short- or long-term function, and the trials were highly heterogeneous.
[polldaddy:10097537]
Studies included in the 2015 systematic review found that glucosamine plus chondroitin did not have a significant effect on short- or long-term pain or physical function compared with placebo. Although glucosamine plus chondroitin led to significantly decreased pain compared with other medication, sensitivity analyses conducted for larger studies (N>200) with adequate methods of blinding and allocation concealment found no difference in pain.8
Continue to: Three studies included...
Three studies included in the 2015 systematic review provided data on adverse events when comparing glucosamine plus chondroitin vs placebo, and found no statistically significant difference.8
This randomized controlled trial (RCT) from Roman-Blas et al1 evaluated chondroitin and glucosamine vs placebo in patients with more severe OA. The study was supported by Tedec-Meiji Farma (Madrid, Spain) maker of the combination of chondroitin plus glucosamine used in the study.
STUDY SUMMARY
Chondroitin + glucosamine was not better than placebo for pain
This multicenter, randomized, double-blind, placebo-controlled trial was conducted in 9 rheumatology referral centers and one orthopedic center in Spain. The trial evaluated the efficacy of chondroitin sulfate 1200 mg plus glucosamine sulfate 1500 mg (CS/GS) compared with placebo in 164 patients with Grade 2 or 3 knee OA and moderate to severe knee pain. OA grade was ascertained using the Kellgren-Lawrence scale, corresponding to osteophytes and either possible (Grade 2) or definite (Grade 3) joint space narrowing. Level of knee pain was defined by a self-reported global pain score of 40-80 mm on a 100-mm visual analog scale (VAS).
No significant difference was noted in group characteristics, and the average age in the CS/GS group was 67 years vs 65 years in the placebo group. Exclusion criteria included body mass index of ≥35 kg/m2, concurrent arthritic conditions, and any coexisting chronic disease that would prevent successful completion of the trial.1
The primary end point was mean reduction in global pain score on a 0- to 100-mm VAS at 6 months. Secondary outcomes included mean reduction in total and subscale scores in pain and function on the Western Ontario and McMaster Universities Osteoarthritis (WOMAC) index (0–100-mm VAS for each) and the use of rescue medication.
Continue to: Baseline global pain scores were...
Baseline global pain scores were 62 mm in both groups. Acetaminophen, up to 3 g/d, was the only allowed rescue medication. Clinic visits occurred at 4, 12, and 24 weeks. A statistically significant difference between groups was defined as P<.03.1
Results. In the intention-to-treat analysis at 6 months, patients in the placebo group had a greater reduction in pain than the CS/GC group (-20 mm vs -12 mm; P=.029). No other difference was noted between the placebo and CS/GS groups in the total or subscales of the WOMAC index, and no difference was noted in use of acetaminophen. More patients in the placebo group had at least a 50% improvement in pain or function compared with the CS/GS group (47.4% vs 27.5%; P=.01).
In the CS/GS group, 31% did not complete the 6-month treatment period, compared with 18% in the placebo group. More patients dropped out because of adverse effects (diarrhea, upper abdominal pain, and constipation) in the CS/GS group than the placebo group (33 vs 19; P=.018).1
WHAT’S NEW
A pharma-sponsored study finds treatment ineffective
The effectiveness of CS/GS for the treatment of knee OA has been in question for years, but this RCT is the first trial sponsored by a pharmaceutical company to evaluate CS/GS efficacy. This trial found evidence of a lack of efficacy. In patients with more severe OA of the knee, placebo was more effective than CS/GS, and CS/GS had significantly more adverse events. Therefore, it may be time to advise patients to stop taking their CS/GS supplement.
CAVEATS
Cannot generalize findings to CS or GS alone, or different dosages
The study compared only one medication dosing regimen using a combination of CS and GS. Whether either agent alone or different dosing would lead to the same outcome is unknown.
Continue to: CHALLENGES TO IMPLEMENTATION
CHALLENGES TO IMPLEMENTATION
An all-too-common product presents challenges
CS/GC is available over the counter and advertised directly to consumers. With this medication so readily available, identifying patients who are taking the supplement and encouraging discontinuation can be a challenge.
ACKNOWLEDGEMENT
The PURLs Surveillance System was supported in part by Grant Number UL1RR024999 from the National Center For Research Resources, a Clinical Translational Science Award to the University of Chicago. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Research Resources or the National Institutes of Health.
1. Roman-Blas JA, Castañeda S, Sánchez-Pernaute O, et al. Combined treatment with chondroitin sulfate and glucosamine sulfate shows no superiority over placebo for reduction of joint pain and functional impairment in patients with knee osteoarthritis: a six-month multicenter, randomized, double-blind, placebo-controlled clinical trial. Arthritis Rheumatol. 2017;69:77-85.
2. Dillon CF, Rasch EK, Gu Q, et al. Prevalence of knee osteoarthritis in the United States: arthritis data from the Third National Health and Nutrition Examination Survey 1991-94. J Rheumatol. 2006;33:2271-2279.
3. Hochberg MC, Altman RD, April KT, et al. American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken). 2012;64:465-474.
4. Brown GA. AAOS clinical practice guideline: treatment of osteoarthritis of the knee: evidence-based guideline, 2nd ed. J Am Acad Orthop Surg. 2013;21:577-579.
5. Jordan KM, Arden NK, Doherty M, et al. EULAR Recommendations 2003: an evidence based approach to the management of knee osteoarthritis: report of a Task Force of the Standing Committee for International Clinical Studies Including Therapeutic Trials (ESCISIT). Ann Rheum Dis. 2003;62:1145-1155.
6. Ebell MH. Osteoarthritis: rapid evidence review. Am Fam Physician. 2018;97:523-526.
7. Clarke TC, Black LI, Stussman BJ, et al. Trends in the use of complementary health approaches among adults: United States, 2002-2012. Natl Health Stat Rep. 2015;(79):1-16.
8. Singh JA, Noorbaloochi S, MacDonald R, et al. Chondroitin for osteoarthritis. Cochrane Database Syst Rev. 2015;(1):CD005614.
1. Roman-Blas JA, Castañeda S, Sánchez-Pernaute O, et al. Combined treatment with chondroitin sulfate and glucosamine sulfate shows no superiority over placebo for reduction of joint pain and functional impairment in patients with knee osteoarthritis: a six-month multicenter, randomized, double-blind, placebo-controlled clinical trial. Arthritis Rheumatol. 2017;69:77-85.
2. Dillon CF, Rasch EK, Gu Q, et al. Prevalence of knee osteoarthritis in the United States: arthritis data from the Third National Health and Nutrition Examination Survey 1991-94. J Rheumatol. 2006;33:2271-2279.
3. Hochberg MC, Altman RD, April KT, et al. American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken). 2012;64:465-474.
4. Brown GA. AAOS clinical practice guideline: treatment of osteoarthritis of the knee: evidence-based guideline, 2nd ed. J Am Acad Orthop Surg. 2013;21:577-579.
5. Jordan KM, Arden NK, Doherty M, et al. EULAR Recommendations 2003: an evidence based approach to the management of knee osteoarthritis: report of a Task Force of the Standing Committee for International Clinical Studies Including Therapeutic Trials (ESCISIT). Ann Rheum Dis. 2003;62:1145-1155.
6. Ebell MH. Osteoarthritis: rapid evidence review. Am Fam Physician. 2018;97:523-526.
7. Clarke TC, Black LI, Stussman BJ, et al. Trends in the use of complementary health approaches among adults: United States, 2002-2012. Natl Health Stat Rep. 2015;(79):1-16.
8. Singh JA, Noorbaloochi S, MacDonald R, et al. Chondroitin for osteoarthritis. Cochrane Database Syst Rev. 2015;(1):CD005614.
PRACTICE CHANGER
Tell patients with moderately severe osteoarthritis to stop taking their glucosamine and chondroitin as it is less effective than placebo.1
STRENGTH OF RECOMMENDATION
B: Based on single, good-quality randomized controlled trial.
Roman-Blas JA, Castañeda S, Sánchez-Pernaute O, et al. Combined treatment with chondroitin sulfate and glucosamine sulfate shows no superiority over placebo for reduction of joint pain and functional impairment in patients with knee osteoarthritis: a six-month multicenter, randomized, double-blind, placebo-controlled clinical trial. Arthritis Rheumatol. 2017;69:77-85.
