User login
Role of the incretin pathway in the pathogenesis of type 2 diabetes mellitus
It has long been understood that the pathophysiology of type 2 diabetes mellitus (T2DM) is based on the triad of progressive decline in insulin-producing pancreatic beta cells, an increase in insulin resistance, and increased hepatic glucose production.1,2 It is now evident that other factors, including defective actions of the gastrointestinal (GI) incretin hormones glucagon-like peptide–1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), also play significant roles.2–5 The uncontrolled hyperglycemia resulting from such defects may lead to microvascular complications, including retinopathy, neuropathy, microangiopathy, and nephropathy, and macrovascular complications, such as coronary artery disease and peripheral vascular disease.
This review explores the growing understanding of the role of the incretins in normal insulin secretion, as well as in the pathogenesis of T2DM, and examines the pathophysiologic basis for the benefits and therapeutic application of incretin-based therapies in T2DM.1,2
THE GI SYSTEM AND GLUCOSE HOMEOSTASIS IN THE HEALTHY STATE
The GI system plays an integral role in glucose homeostasis.6 The observation that orally administered glucose provides a stronger insulinotropic stimulus than an intravenous glucose challenge provided insight into the regulation of plasma glucose by the GI system of healthy individuals.7 The incretin effect, as this is termed, may be responsible for 50% to 70% of the total insulin secreted following oral glucose intake.8
Two GI peptide hormones (the incretins)—GLP-1 and GIP—were found to exert major glucoregulatory actions.3,9,10 Within minutes of nutrient ingestion, GLP-1 is secreted from intestinal L cells in the distal ileum and colon, while GIP is released by intestinal K cells in the duodenum and jejunum.3 GLP-1 and GIP trigger their insulinotropic actions by binding beta-cell receptors.3 GLP-1 receptors are expressed on pancreatic glucagon-containing alpha and delta cells as well as on beta cells, whereas GIP receptors are expressed primarily on beta cells.3,8 GLP-1 receptors are also expressed in the central nervous system (CNS), peripheral nervous system, lung, heart, and GI tract, while GIP receptors are expressed in adipose tissue and the CNS.3 GLP-1 inhibits glucose-dependent glucagon secretion from alpha cells.3 In healthy individuals, fasting glucose is managed by tonic insulin/glucagon secretion, but excursions of postprandial glucose (PPG) are controlled by insulin and the incretin hormones.11
Additionally, in animal studies, GLP-1 has been shown to induce the transcriptional activation of the insulin gene and insulin biosynthesis, thus increasing beta-cell proliferation and decreasing beta-cell apoptosis.12 GLP-1 stimulates a CNS-mediated pathway of insulin secretion, slows gastric emptying, increases CNS-mediated satiety leading to reduced food intake, indirectly increases insulin sensitivity and nutrient uptake in skeletal muscle and adipose tissue, and exerts neuroprotective effects.8
Both GLP-1 and GIP are rapidly degraded by the serine protease dipeptidyl peptidase–4 (DPP-4), which is widely expressed in bound and free forms.14 A recent study in healthy adults showed that GLP-1 concentration declined even during maximal DPP-4 inhibition, suggesting that there may be pathways of GLP-1 elimination other than DPP-4 enzymatic degradation.15
INCRETINS AND THE PATHOGENESIS OF T2DM
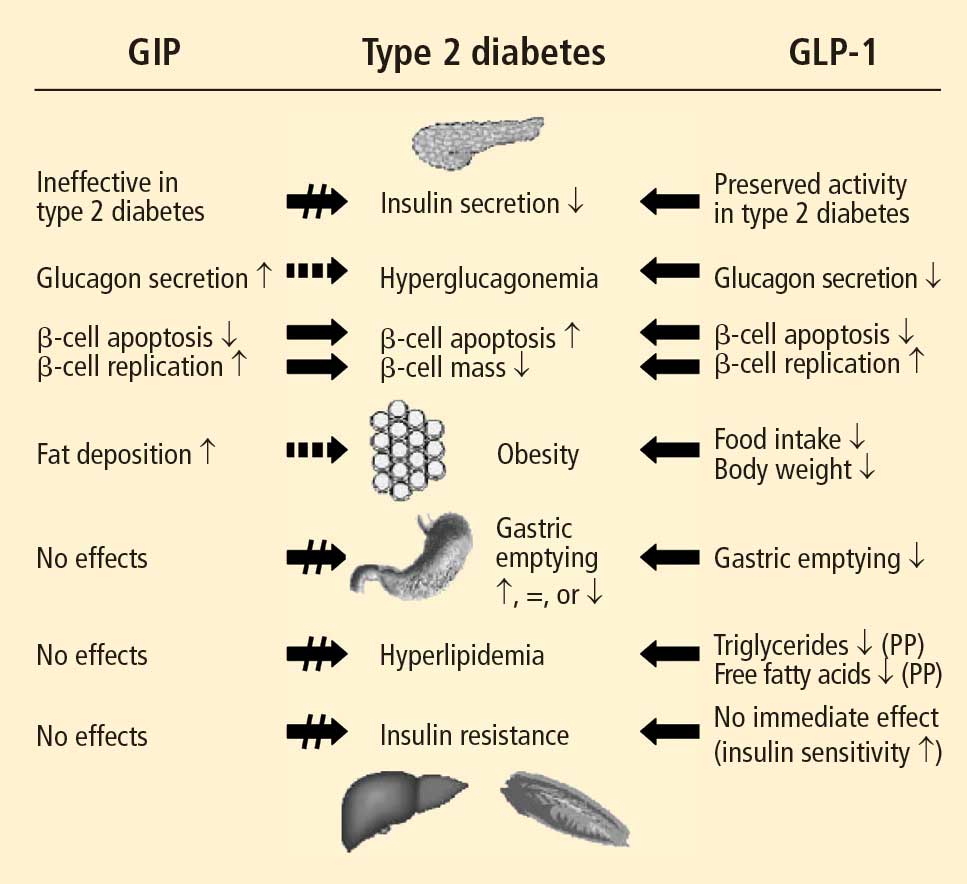
Studies have shown that incretin pathways play a role in the progression of T2DM.3,16 The significant reduction in the incretin effect seen in patients with T2DM has been attributed to several factors, including impaired secretion of GLP-1, accelerated metabolism of GLP-1 and GIP, and defective responsiveness to both hormones.16 Many patients with T2DM also have accelerated gastric emptying that may contribute to deterioration of their glycemic control.17
While GIP concentration is normal or modestly increased in patients with T2DM,16,18 the insulinotropic actions of GIP are significantly diminished.19 Thus, patients with T2DM have an impaired responsiveness to GIP with a possible link to GIP-receptor downregulation or desensitization.20
Are secretory defects a cause or result of T2DM?
In contrast to GIP, the secretion of GLP-1 has been shown to be deficient in patients with T2DM.18 As with GIP, it is unknown to what degree this defect is a cause or consequence of T2DM. In a study of identical twins, defective GLP-1 secretion was observed only in the one sibling with T2DM, suggesting that GLP-1 secretory deficits may be secondary to the development of T2DM.21 Despite the diminished secretion of GLP-1 in patients with T2DM, the insulinotropic actions of GLP-1 are preserved.19 It has also been shown that the effects of GLP-1 on gastric emptying and glucagon secretion are maintained in patients with T2DM.19,22,23
Whether this incretin dysregulation is responsible for or is the end result of hyperglycemia remains a subject of continued investigation. A recent study confirmed that the incretin effect is reduced in patients with T2DM, but advanced the concept that it may be a consequence of the diabetic state.16,24 Notably, impaired actions of GLP-1 and GIP and diminished concentrations of GLP-1 may be partially restored by improved glycemic control.24
Recent preclinical and clinical studies continue to clarify the roles of incretin hormones in T2DM. The findings from a study of obese diabetic mice suggest that the effect of GLP-1 therapy on the long-term remission of diabetes may be caused by improvements in beta-cell function and insulin sensitivity, as well as by a reduction in gluconeogenesis in the liver.25
Incretin effect and glucose tolerance, body mass index
Another study was conducted to evaluate quantitatively the separate impacts of obesity and hyperglycemia on the incretin effect in patients with T2DM, patients with impaired glucose tolerance, and patients with normal glucose tolerance.26 There was a significant (P ≤ .05) reduction in the incretin effect in terms of total insulin secretion, beta-cell glucose sensitivity, and the GLP-1 response to oral glucose in patients with T2DM compared with individuals whose glucose tolerance was normal or impaired. Each manifestation of the incretin effect was inversely related to both glucose tolerance and body mass index in an independent, additive manner (P ≤ .05); thus, glucose tolerance and obesity attenuate the incretin effect on beta-cell function and GLP-1 response independently of each other.
Exogenous GLP-1 has been shown to restore the regulation of blood glucose to near-normal concentrations in patients with T2DM.27 Several studies of patients with T2DM have shown that synthetic GLP-1 administration induces insulin secretion,19,27 slows gastric emptying (which is accelerated in patients with T2DM), and decreases inappropriately elevated glucagon secretion.19,23,28 Acute GLP-1 infusion studies showed that GLP-1 improved fasting plasma glucose (FPG) and PPG concentrations23,27; long-term studies showed that this hormone exerts euglycemic effects, leading to improvements in glycosylated hemoglobin (HbA1c), and induces weight loss.29
TARGETING FUNDAMENTAL DEFECTS OF T2DM WITH INCRETIN-BASED THERAPIES
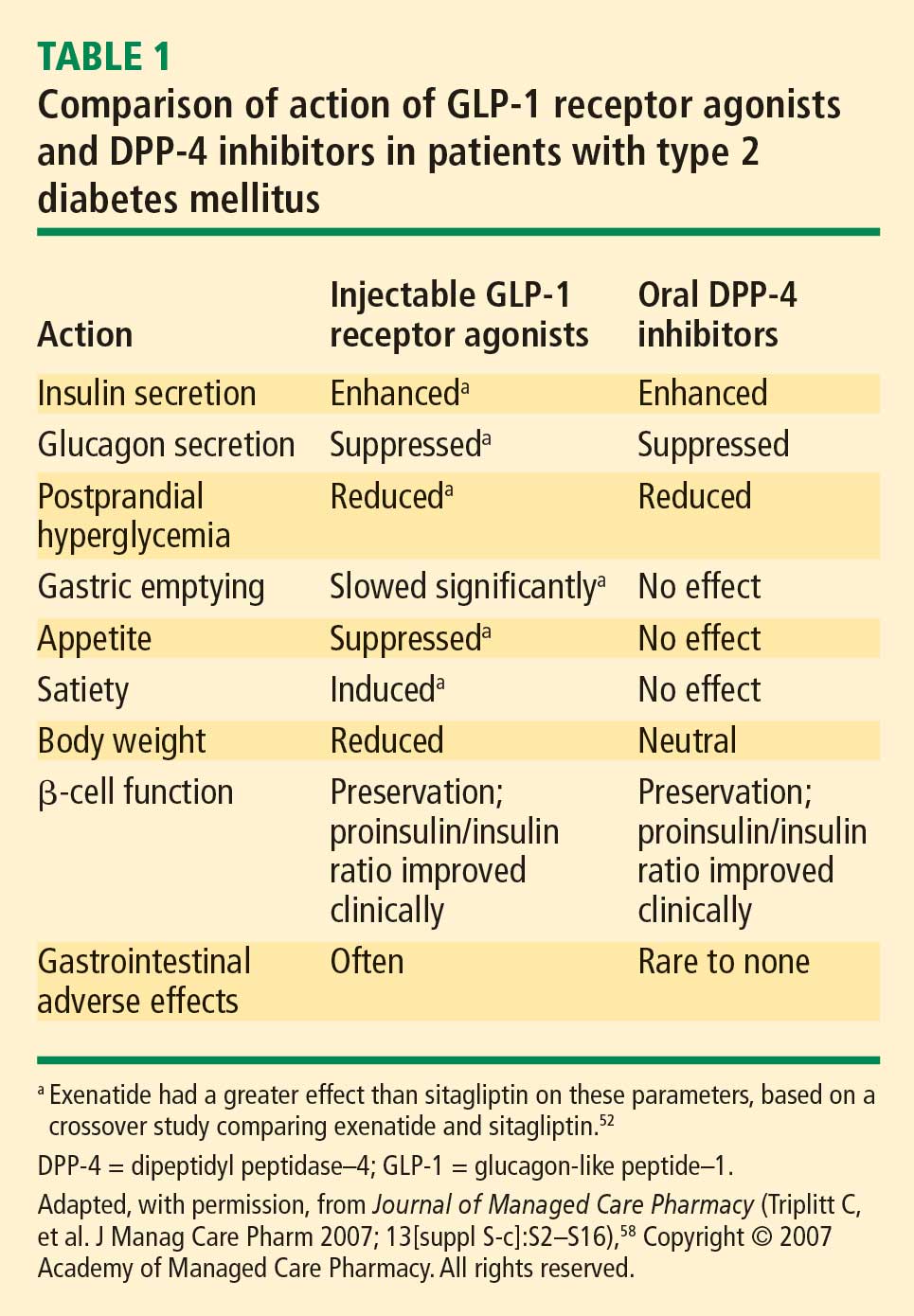
Recognition and a better understanding of the role of the incretins and the enzyme involved in their degradation have led to the development of two incretin-based treatments: the GLP-1 receptor agonists, which possess many of the glucoregulatory actions of incretin peptides, and the DPP-4 inhibitors.5 Both the GLP-1 receptor agonists and the DPP-4 inhibitors have demonstrated safety and efficacy in the management of hyperglycemia in patients with T2DM.
GLP-1 receptor agonists
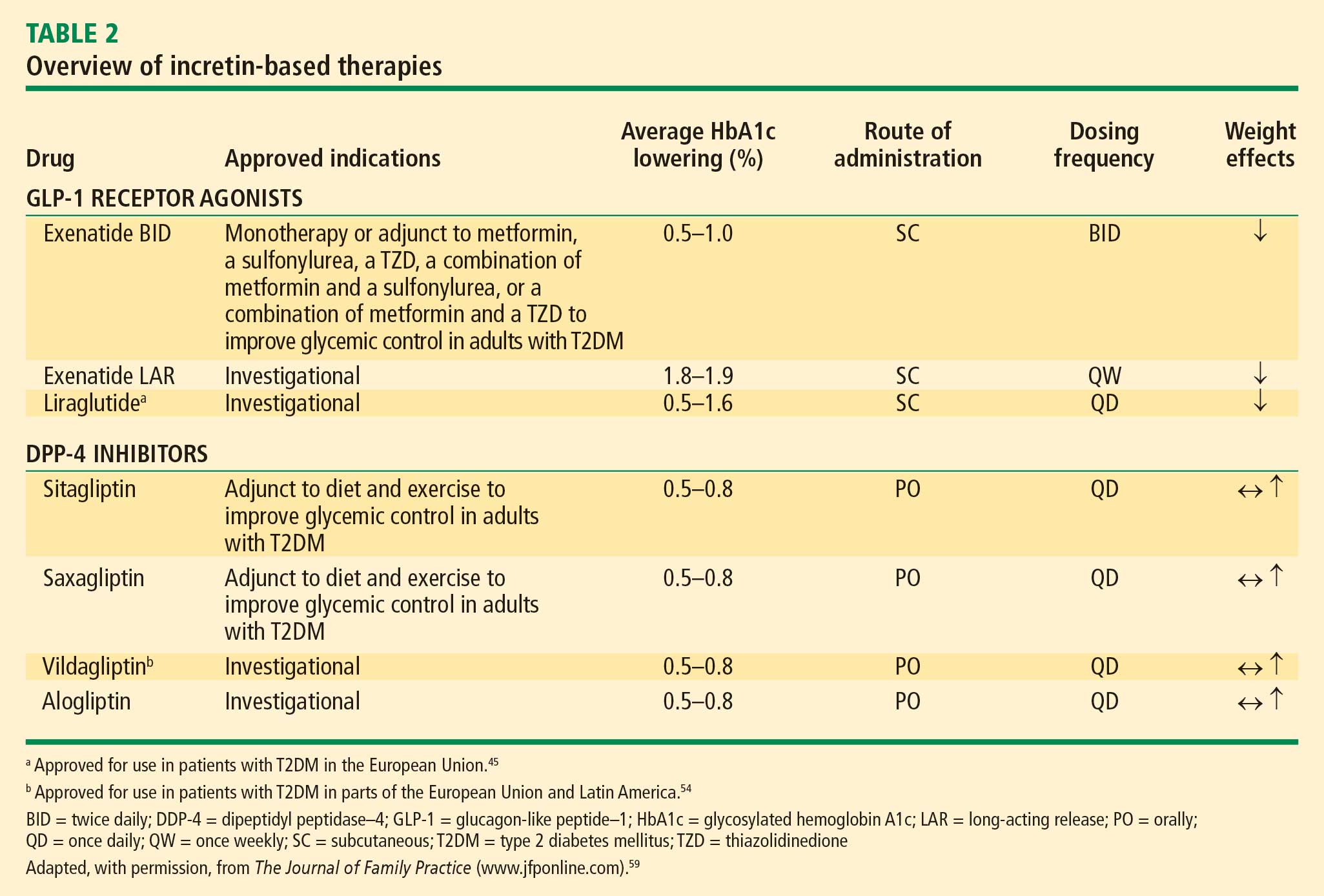
The GLP-1 receptor agonist exenatide is a synthetic form of exendin-4 and has a unique amino acid sequence that renders it resistant to degradation by DPP-4, making its actions longer lasting than endogenous GLP-1.5,30 Exenatide has a half-life of 2.4 hours and is detectable for up to 10 hours after subcutaneous (SC) injection.5,30 It is administered BID and has been approved as monotherapy or an adjunct therapy in patients with T2DM who have inadequate glycemic control following treatment with metformin, a sulfonylurea, a thiazolidinedione (TZD), or metformin in combination with a sulfonylurea or a TZD.31–35
In both human and animal studies, exenatide has been shown to enhance glucose-dependent insulin secretion and suppress inappropriate glucagon secretion in a glucose-dependent manner, reduce food intake and body weight, and acutely improve beta-cell function by enhancing first- and second-phase insulin secretion.5,36,37
In a small study involving 17 patients with T2DM, exenatide was shown to slow gastric emptying, which could be an important mechanism contributing to its beneficial effects on PPG concentration.38 Exenatide also has been shown to attenuate postprandial hyperglycemia, a risk factor for cardiovascular disease (CVD), by reducing endogenous glucose production by about 50% in patients with T2DM.39 Another mechanism for glycemic control may exist, as a recent animal study has shown that exenatide, similar to endogenous GLP-1, lowers blood glucose concentration independent of changes in pancreatic islet hormone secretion or delayed gastric emptying.40
A formulation of exenatide that is administered once weekly—exenatide long-acting release (LAR)—is in clinical evaluation and under review by the US Food and Drug Administration (FDA). In a short-term study, exenatide-LAR (0.8 mg or 2.0 mg) was administered once weekly for 15 weeks to patients with T2DM whose glycemia was suboptimally controlled with metformin alone or in combination with diet and exercise. Compared with placebo, treatment with exenatide once weekly was associated with markedly reduced HbA1c, FPG, PPG and body weight.41 In a larger, 30-week, phase 3 trial, Diabetes Therapy Utilization: Researching Changes in A1C, Weight and Other Factors Through Intervention with Exenatide ONce Weekly (DURATION-1), exenatide-LAR 2 mg once weekly was compared with exenatide 10 mg BID in patients with T2DM. Exenatide-LAR once weekly was associated with a significantly greater reduction in HbA1c (–1.9% vs –1.5%, P = .0023), and with a similar low risk of hypoglycemia and reduction in body weight (–3.7 kg vs –3.6 kg, P = .89) compared with the BID formulation.42
Liraglutide, recently approved in the European Union for T2DM and also under regulatory review in the United States, is a DPP-4–resistant human analogue GLP-1 receptor agonist in clinical development that has a 97% homology to native GLP-1.43–45 In contrast to exenatide, the acetylated liraglutide molecule allows binding to serum albumin and provides resistance to DPP-4 degradation, thus prolonging the half-life of liraglutide to approximately 12 hours. Liraglutide is administered SC QD as monotherapy or in combination with other antidiabetes agents such as metformin or sulfonylurea to patients with T2DM.44–47 Liraglutide has been shown to reduce HbA1c, decrease body weight, and lead to a lower incidence of hypoglycemia compared with the sulfonylurea glimepiride.
DPP-4 inhibitors
Sitagliptin is a DPP-4 inhibitor indicated as monotherapy or in combination with metformin or a TZD in patients with T2DM with inadequate glycemic control.48–51 Given orally, sitagliptin does not bind to the GLP-1 receptor agonist and has been shown to inhibit circulating DPP-4 activity by about 80%.52,53 Sitagliptin has been associated with an approximate twofold increase in postprandial GLP-1 plasma concentrations compared with placebo in healthy human subjects and in patients with T2DM.53 Saxagliptin, another potent DPP-4 inhibitor, significantly reduced HbA1c and FPG concentrations in patients with T2DM54 with a neutral effect on weight; it was recently approved by the FDA for treatment of T2DM.55
The DPP-4 inhibitor vildagliptin is currently being used in the European Union and Latin America but has yet to receive regulatory approval in the United States.54 Alogliptin, a novel, high-affinity, high-specificity DPP-4 inhibitor currently in development, provides rapid and sustained DPP-4 inhibition and significantly reduces HbA1c, FPG, and PPG concentrations with no change in body weight in patients with T2DM.56,57
Incretin-based therapies compared
Effects of incretin-based therapies
The number of people with T2DM, overweight/obesity, or CVD, alone or in combination, is approaching epidemic proportions, with the mechanisms of these conditions interrelated. Approximately 24 million Americans have diabetes, and T2DM accounts for more than 90% of these cases.61 Most patients with T2DM are not achieving HbA1c targets.62–64 About 60% of deaths among patients with T2DM are caused by CVD.65 Compounding the problem, overweight/obesity enhances the risk for CV-related morbidities in patients with diabetes.66 A cluster of metabolic disorders referred to as the metabolic syndrome (which includes hyperglycemia, measures of central obesity, and a series of significant CV risk factors) is common in patients with T2DM and CVD.67 Unfortunately, many antidiabetes drugs that successfully manage glycemic control also cause weight gain, which in theory may increase CV risk in patients with T2DM.68
Data from studies of patients with T2DM show that exenatide improves glycemic control and reduces body weight. Exenatide administered BID significantly reduced HbA1c (–0.40% to –0.86%) and weight (–1.6 kg to –2.8 kg) relative to baseline in three 30-week, placebo-controlled clinical trials.31,33,34 In subsequent 2-year, open-label extension studies, exenatide produced significant reductions from baseline in HbA1c (–20.9% at 30 weeks) and weight (–2.1 kg at 30 weeks). Both decreases were sustained through 2 years (HbA1c –1.1%, weight –4.7 kg) with a low incidence of hypoglycemia.31 Further post hoc analysis of the open-label extension of the 30-week trials followed patients treated with exenatide BID for 3 years or longer.69 In addition to markedly decreasing HbA1c from baseline levels (–1.1% at 3 years and –0.8% at up to 3.5 years; P < .0001), adjunctive exenatide produced significant reductions in body weight—up to –5.3 kg after 3.5 years of therapy.31,69 At 3.5 years, continued exenatide therapy resulted in a –6% reduction in low-density lipoprotein cholesterol, a 24% mean increase in high-density lipoprotein cholesterol, and a mean reduction in blood pressure of –2% to –4% from baseline levels. Improvements in hepatic biomarkers and homeostasis model assessment-B, a measure of beta-cell function, were seen after 2 and 3 years of exenatide treatment.31 Hypoglycemia was generally mild and transient.
In comparative head-to-head studies, exenatide BID and insulin analogues reduced HbA1c by similar magnitudes; yet exenatide treatment resulted in better control in terms of PPG and weight loss, while insulin glargine and insulin aspart produced weight gain.70–73
Mechanisms of cardioprotective effects
Although the mechanisms for the potential cardioprotective effects of GLP-1 and its receptor agonists remain to be fully elucidated, a recent study suggested that two novel pathways could be involved—one that is dependent on the known GLP-1 receptor pathway, and one that is independent of the GLP-1 receptor pathway.74 Correlating with observations of a potential cardioprotective effect, an infusion of recombinant GLP-1 in patients with acute myocardial infarction, when added to standard therapy, resulted in improved left ventricular function and was associated with reduced mortality.75 Evidence continues to accumulate for potential cardioprotective effects of the GLP-1 receptor agonists, indicating that they may have a positive impact on macrovascular complications in patients with T2DM.
CONCLUSION
T2DM, which is often associated with overweight and obesity, remains a significant challenge worldwide. The broad spectrum of glucoregulatory actions of the incretin hormones GLP-1 and GIP, and their importance in maintaining glucose homeostasis, have been recognized and correlated with the pathogenesis of T2DM. An improved understanding of the roles played by GLP-1 and GIP in the pathogenesis of T2DM may provide clinicians with important details regarding the therapeutic application of incretin-based therapies, including the GLP-1 receptor agonist exenatide and the DPP-4 inhibitors sitagliptin and saxagliptin. Antidiabetes agents whose development is based on the multiple pharmacologic effects of incretin hormones can address the multifaceted nature of T2DM and overcome some current limitations of traditional therapies, especially those related to weight. This becomes more compelling given the close link among T2DM, obesity, and increased CV risk.
- Boyle PJ, Freeman JS. Application of incretin mimetics and dipeptidyl peptidase IV inhibitors in managing type 2 diabetes mellitus. J Am Osteopath Assoc 2007; 107(suppl 3):S10–S16.
- Freeman JS. The pathophysiologic role of incretins. J Am Osteopath Assoc 2007; 107(suppl 3):S6–S9.
- Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006; 368:1696–1705.
- Nauck MA, Baller B, Meier JJ. Gastric inhibitory polypeptide and glucagon-like peptide-1 in the pathogenesis of type 2 diabetes. Diabetes 2004; 53(suppl 3):S190–S196.
- Stonehouse A, Okerson T, Kendall D, Maggs D. Emerging incretin based therapies for type 2 diabetes: incretin mimetics and DPP-4 inhibitors. Curr Diabetes Rev 2008; 4:101–109.
- Huda MS, Wilding JP, Pinkney JH. Gut peptides and the regulation of appetite. Obes Rev 2006; 7:163–182.
- Elrick H, Stimmler L, Hlad CJ Jr, Arai Y. Plasma insulin response to oral and intravenous glucose administration. J Clin Endocrinol Metab 1964; 24:1076–1082.
- Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007; 132:2131–2157.
- Brown JC, Dryburgh JR, Ross SA, Dupré J. Identification and actions of gastric inhibitory polypeptide. Recent Prog Horm Res 1975; 31:487–532.
- Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet 1987; 2:1300–1304.
- Nauck MA, Homberger E, Siegel EG, et al. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J Clin Endocrinol Metab 1986; 63:492–498.
- Farilla L, Bulotta A, Hirshberg B, et al. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology 2003; 144:5149–5158.
- Van Gaal LF, Gutkin SW, Nauck MA. Exploiting the antidiabetic properties of incretins to treat type 2 diabetes mellitus: glucagon-like peptide 1 receptor agonists or insulin for patients with inadequate glycemic control? Eur J Endocrinol 2008; 158:773–784.
- Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes 1995; 44:1126–1131.
- Dai H, Gustavson SM, Preston GM, Eskra JD, Calle R, Hirshberg B. Non-linear increase in GLP-1 levels in response to DPP-IV inhibition in healthy adult subjects. Diabetes Obes Metab 2008; 10:506–513.
- Nauck MA, Stöckmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 1986; 29:46–52.
- Phillips WT, Schwartz JG, McMahan CA. Rapid gastric emptying of an oral glucose solution in type 2 diabetic patients. J Nucl Med 1992; 33:1496–1500.
- Toft-Nielsen MB, Damholt MB, Madsbad S, et al. Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J Clin Endocrinol Metab 2001; 86:3717–3723.
- Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest 1993; 91:301–307.
- Lynn FC, Thompson SA, Pospisilik JA, et al. A novel pathway for regulation of glucose-dependent insulinotropic polypeptide (GIP) receptor expression in beta cells. FASEB J 2003; 17:91–93.
- Vaag AA, Holst JJ, Vølund A, Beck-Nielsen HB. Gut incretin hormones in identical twins discordant for non-insulin-dependent diabetes mellitus (NIDDM)—evidence for decreased glucagon-like peptide-1 secretion during oral glucose ingestion in NIDDM twins. Eur J Endocrinol 1996; 135:425–432.
- Meier JJ, Gallwitz B, Salmen S, et al. Normalization of glucose concentrations and deceleration of gastric emptying after solid meals during intravenous glucagon-like peptide 1 in patients with type 2 diabetes. J Clin Endocrinol Metab 2003; 88:2719–2725.
- Nauck MA, Kleine N, Orskov C, Holst JJ, Willms B, Creutzfeldt W. Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7-36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 1993; 36:741–744.
- Knop FK, Vilsbøll T, Højberg PV, et al. Reduced incretin effect in type 2 diabetes: cause or consequence of the diabetic state? Diabetes 2007; 56:1951–1959.
- Lee YS, Shin S, Shigihara T, et al. Glucagon-like peptide-1 gene therapy in obese diabetic mice results in long-term cure of diabetes by improving insulin sensitivity and reducing hepatic gluconeogenesis. Diabetes 2007; 56:1671–1679.
- Muscelli E, Mari A, Casolaro A, et al. Separate impact of obesity and glucose tolerance on the incretin effect in normal subjects and type 2 diabetic patients. Diabetes 2008; 57:1340–1348.
- Nathan DM, Schreiber E, Fogel H, Mojsov S, Habener JF. Insulinotropic action of glucagonlike peptide-I-(7-37) in diabetic and nondiabetic subjects. Diabetes Care 1992; 15:270–276.
- Kolterman OG, Buse JB, Fineman MS, et al. Synthetic exendin-4 (exenatide) significantly reduces postprandial and fasting plasma glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab 2003; 88:3082–3089.
- Zander M, Madsbad S, Madsen JL, Holst JJ. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet 2002; 359:824–830.
- Kolterman OG, Kim DD, Shen L, et al. Pharmacokinetics, pharmacodynamics, and safety of exenatide in patients with type 2 diabetes mellitus. Am J Health Syst Pharm 2005; 62:173–181.
- Buse JB, Henry RR, Han J, et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care 2004; 27:2628–2635.
- Byetta [package insert]. San Diego, CA: Amylin Pharmaceuticals, Inc.; 2009.
- DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 2005; 28:1092–1100.
- Kendall DM, Riddle MC, Rosenstock J, et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in patients with type 2 diabetes treated with metformin and a sulfonylurea. Diabetes Care 2005; 28:1083–1091.
- Zinman B, Hoogwerf BJ, Durán García S, et al. The effect of adding exenatide to a thiazolidinedione in suboptimally controlled type 2 diabetes: a randomized trial. Ann Intern Med 2007; 146:477–485.
- Fehse F, Trautmann M, Holst JJ, et al. Exenatide augments first- and second-phase insulin secretion in response to intravenous glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab 2005; 90:5991–5997.
- Parkes DG, Pittner R, Jodka C, Smith P, Young A. Insulinotropic actions of exendin-4 and glucagon-like peptide-1 in vivo and in vitro. Metabolism 2001; 50:583–589.
- Linnebjerg H, Park S, Kothare PA, et al. Effect of exenatide on gastric emptying and relationship to postprandial glycemia in type 2 diabetes. Regul Pept 2008; 151:123–129.
- Cervera A, Wajcberg E, Sriwijitkamol A, et al. Mechanism of action of exenatide to reduce postprandial hyperglycemia in type 2 diabetes. Am J Physiol Endocrinol Metab 2008; 294:E846–E852.
- Ionut V, Zheng D, Stefanovski D, Bergman RN. Exenatide can reduce glucose independent of islet hormones or gastric emptying. Am J Physiol Endocrinol Metab 2008; 295:E269–E277.
- Kim D, MacConell L, Zhuang D, et al. Effects of once-weekly dosing of a long-acting release formulation of exenatide on glucose control and body weight in subjects with type 2 diabetes. Diabetes Care 2007; 30:1487–1493.
- Drucker DJ, Buse JB, Taylor K, et al. Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: a randomised, open-label, non-inferiority study. Lancet 2008; 372:1240–1250.
- Knudsen LB, Nielsen PF, Huusfeldt PO, et al. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem 2000; 43:1664–1669.
- Nauck M, Frid A, Hermansen K, et al; for the LEAD-2 Study Group. Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: the LEAD (liraglutide effect and action in diabetes)-2 study. Diabetes Care 2009; 32:84–90.
- Committee for Medicinal Products for Human Use: Summary of Positive Opinion for Victoza. European Medicines Agency Web site. http://www.emea.europa.eu/pdfs/human/opinion/Victoza_14168909en.pdf. Published April 23, 2009. Accessed September 21, 2009.
- Garber A, Henry R, Ratner R, et al. Liraglutide versus glimepiride monotherapy for type 2 diabetes (LEAD-3 Mono): a randomised, 52-week, phase III, double-blind, parallel-treatment trial. Lancet 2009; 373:473–481.
- Marre M, Shaw J, Brändle M, et al. Liraglutide, a once-daily human GLP-1 analogue, added to a sulphonylurea over 26 weeks produces greater improvements in glycaemic and weight control compared with adding rosiglitazone or placebo in subjects with type 2 diabetes (LEAD-1 SU). Diabet Med 2009; 26:268–278.
- Aschner P, Kipnes MS, Lunceford JK, et al; for the Sitagliptin 021 Study Group. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care 2006; 29:2632–2637.
- Charbonnel B, Karasik A, Liu J, Wu M, Meininger G; for the Sitagliptin Study 020 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006; 29:2638–2643.
- Raz I, Hanefeld M, Xu L, et al. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia 2006; 49:2564–2571.
- Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P; for the Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2006; 28:1556–1568.
- DeFronzo RA, Okerson T, Viswanathan P, Guan X, Holcombe JH, MacConell L. Effects of exenatide versus sitagliptin on postprandial glucose, insulin and glucagon secretion, gastric emptying, and caloric intake: a randomized, cross-over study. Curr Med Res Opin 2008; 24:2943–2952.
- Herman GA, Stevens C, Van Dyck K, et al. Pharmacokinetics and pharmacodynamics of sitagliptin, an inhibitor of dipeptidyl peptidase IV, in healthy subjects: results from two randomized, double-blind, placebo-controlled studies with single oral doses. Clin Pharmacol Ther 2005; 78:675–688.
- Baggio LL, Drucker DJ, Maida A, Lamont BJ. ADA 2008: incretin-based therapeutics. MedscapeCME Web site. http://www.medscape.com/viewprogram/15786. Accessed September 18, 2009.
- US Department of Health and Human Services. FDA approves new drug treatment for type 2 diabetes. US Food and Drug Administration Web site. http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm174780.htm. Published July 31, 2009. Accessed September 18, 2009.
- Covington P, Christopher R, Davenport M, et al. Pharmacokinetic, pharmacodynamic, and tolerability profiles of the dipeptidyl peptidase-4 inhibitor alogliptin: a randomized, double-blind, placebo-controlled, multiple-dose study in adult patients with type 2 diabetes. Clin Ther 2008; 30:499–512.
- DeFronzo RA, Fleck PR, Wilson CA, Mekki Q; on behalf of the Alogliptin Study 010 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor alogliptin in patients with type 2 diabetes and inadequate glycemic control: a randomized, double-blind, placebo-controlled study. Diabetes Care 2008; 31:2315–2317.
- Triplitt CL, McGill JB, Porte D Jr, Conner CS. The changing landscape of type 2 diabetes: the role of incretin-based therapies in managed care outcomes. J Manag Care Pharm 2007; 13(9 suppl C):S2–S16.
- Garber AJ, Spann SJ. An overview of incretin clinical trials. J Fam Pract 2008; 57(9 suppl):S10–S18.
- Henry RR. Evolving concepts of type 2 diabetes management with oral medications: new approaches to an old disease. Curr Med Res Opin 2008; 24:2189–2202.
- Centers for Disease Control and Prevention. National diabetes fact sheet: general information and national estimates on diabetes in the United States, 2007. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/diabetes/pubs/pdf/ndfs_2007.pdf. Published 2008. Accessed September 21, 2009.
- Ong KL, Cheung BM, Wong LY, Wt NM, Tan KC, Lam KS. Prevalence, treatment, and control of diagnosed diabetes in the U.S. National Health and Nutrition Examination Survey 1999–2004. Ann Epidemiol 2008; 18:222–229.
- Sanders CL, Yesupriya AJ, Curtin LR. Analysis of population structure and stratification in NHANES III self-reported race/ethnicities. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/genomics/events/file/print/10year/08_pop_struct_ab.pdf. Accessed September 21, 2009.
- Koro CE, Bowlin SJ, Bourgeois N, Fedder DO. Glycemic control from 1988 to 2000 among US adults diagnosed with type 2 diabetes: a preliminary report. Diabetes Care 2004; 27:17–20.
- Haffner SM, Lehto S, Rönnemaa T, Pyörälä K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229–234.
- Lloyd-Jones D, Adams R, Carnethon M, et al. Heart disease and stroke statistics—2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2009; 119:480–486.
- Stonehouse AH, Holcombe JH, Kendall DM. GLP-1 analogues, DPP-IV inhibitors and the metabolic syndrome. In: Fonseca V, ed. Therapeutic Strategies in Metabolic Syndrome. Oxford, UK: Atlas Medical Publishing Ltd; 2008: 137–157.
- Purnell JQ, Weyer C. Weight effect of current and experimental drugs for diabetes mellitus: from promotion to alleviation of obesity. Treat Endocrinol 2003; 2:33–47.
- Klonoff DC, Buse JB, Nielsen LL, et al. Exenatide effects on diabetes, obesity, cardiovascular risk factors and hepatic biomarkers in patients with type 2 diabetes treated for at least 3 years. Curr Med Res Opin 2008; 24:275–286.
- Barnett AH, Burger J, Johns D, et al. Tolerability and efficacy of exenatide and titrated insulin glargine in adult patients with type 2 diabetes previously uncontrolled with metformin or a sulfonylurea: a multinational, randomized, open-label, two-period, crossover noninferiority trial. Clin Ther 2007; 29:2333–2348.
- Glass LC, Qu Y, Lenox S, et al. Effects of exenatide versus insulin analogues on weight change in subjects with type 2 diabetes: a pooled post-hoc analysis. Curr Med Res Opin 2008; 24:639–644.
- Heine RJ, Van Gaal LF, Johns D, Mihm MJ, Widel MH, Brodows RG; for the GWAA Study Group. Exenatide versus insulin glargine in patients with suboptimally controlled type 2 diabetes: a randomized trial. Ann Intern Med 2005; 143:559–569.
- Nauck MA, Duran S, Kim D, et al. A comparison of twice-daily exenatide and biphasic insulin aspart in patients with type 2 diabetes who were suboptimally controlled with sulfonylurea and metformin: a non-inferiority study. Diabetologia 2007; 50:259–267.
- Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation 2008; 117:2340–2350.
- Nikolaidis LA, Mankad S, Sokos GG, et al. Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation 2004; 109:962–965.
It has long been understood that the pathophysiology of type 2 diabetes mellitus (T2DM) is based on the triad of progressive decline in insulin-producing pancreatic beta cells, an increase in insulin resistance, and increased hepatic glucose production.1,2 It is now evident that other factors, including defective actions of the gastrointestinal (GI) incretin hormones glucagon-like peptide–1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), also play significant roles.2–5 The uncontrolled hyperglycemia resulting from such defects may lead to microvascular complications, including retinopathy, neuropathy, microangiopathy, and nephropathy, and macrovascular complications, such as coronary artery disease and peripheral vascular disease.
This review explores the growing understanding of the role of the incretins in normal insulin secretion, as well as in the pathogenesis of T2DM, and examines the pathophysiologic basis for the benefits and therapeutic application of incretin-based therapies in T2DM.1,2
THE GI SYSTEM AND GLUCOSE HOMEOSTASIS IN THE HEALTHY STATE
The GI system plays an integral role in glucose homeostasis.6 The observation that orally administered glucose provides a stronger insulinotropic stimulus than an intravenous glucose challenge provided insight into the regulation of plasma glucose by the GI system of healthy individuals.7 The incretin effect, as this is termed, may be responsible for 50% to 70% of the total insulin secreted following oral glucose intake.8
Two GI peptide hormones (the incretins)—GLP-1 and GIP—were found to exert major glucoregulatory actions.3,9,10 Within minutes of nutrient ingestion, GLP-1 is secreted from intestinal L cells in the distal ileum and colon, while GIP is released by intestinal K cells in the duodenum and jejunum.3 GLP-1 and GIP trigger their insulinotropic actions by binding beta-cell receptors.3 GLP-1 receptors are expressed on pancreatic glucagon-containing alpha and delta cells as well as on beta cells, whereas GIP receptors are expressed primarily on beta cells.3,8 GLP-1 receptors are also expressed in the central nervous system (CNS), peripheral nervous system, lung, heart, and GI tract, while GIP receptors are expressed in adipose tissue and the CNS.3 GLP-1 inhibits glucose-dependent glucagon secretion from alpha cells.3 In healthy individuals, fasting glucose is managed by tonic insulin/glucagon secretion, but excursions of postprandial glucose (PPG) are controlled by insulin and the incretin hormones.11
Additionally, in animal studies, GLP-1 has been shown to induce the transcriptional activation of the insulin gene and insulin biosynthesis, thus increasing beta-cell proliferation and decreasing beta-cell apoptosis.12 GLP-1 stimulates a CNS-mediated pathway of insulin secretion, slows gastric emptying, increases CNS-mediated satiety leading to reduced food intake, indirectly increases insulin sensitivity and nutrient uptake in skeletal muscle and adipose tissue, and exerts neuroprotective effects.8
Both GLP-1 and GIP are rapidly degraded by the serine protease dipeptidyl peptidase–4 (DPP-4), which is widely expressed in bound and free forms.14 A recent study in healthy adults showed that GLP-1 concentration declined even during maximal DPP-4 inhibition, suggesting that there may be pathways of GLP-1 elimination other than DPP-4 enzymatic degradation.15
INCRETINS AND THE PATHOGENESIS OF T2DM
Studies have shown that incretin pathways play a role in the progression of T2DM.3,16 The significant reduction in the incretin effect seen in patients with T2DM has been attributed to several factors, including impaired secretion of GLP-1, accelerated metabolism of GLP-1 and GIP, and defective responsiveness to both hormones.16 Many patients with T2DM also have accelerated gastric emptying that may contribute to deterioration of their glycemic control.17
While GIP concentration is normal or modestly increased in patients with T2DM,16,18 the insulinotropic actions of GIP are significantly diminished.19 Thus, patients with T2DM have an impaired responsiveness to GIP with a possible link to GIP-receptor downregulation or desensitization.20
Are secretory defects a cause or result of T2DM?
In contrast to GIP, the secretion of GLP-1 has been shown to be deficient in patients with T2DM.18 As with GIP, it is unknown to what degree this defect is a cause or consequence of T2DM. In a study of identical twins, defective GLP-1 secretion was observed only in the one sibling with T2DM, suggesting that GLP-1 secretory deficits may be secondary to the development of T2DM.21 Despite the diminished secretion of GLP-1 in patients with T2DM, the insulinotropic actions of GLP-1 are preserved.19 It has also been shown that the effects of GLP-1 on gastric emptying and glucagon secretion are maintained in patients with T2DM.19,22,23
Whether this incretin dysregulation is responsible for or is the end result of hyperglycemia remains a subject of continued investigation. A recent study confirmed that the incretin effect is reduced in patients with T2DM, but advanced the concept that it may be a consequence of the diabetic state.16,24 Notably, impaired actions of GLP-1 and GIP and diminished concentrations of GLP-1 may be partially restored by improved glycemic control.24
Recent preclinical and clinical studies continue to clarify the roles of incretin hormones in T2DM. The findings from a study of obese diabetic mice suggest that the effect of GLP-1 therapy on the long-term remission of diabetes may be caused by improvements in beta-cell function and insulin sensitivity, as well as by a reduction in gluconeogenesis in the liver.25
Incretin effect and glucose tolerance, body mass index
Another study was conducted to evaluate quantitatively the separate impacts of obesity and hyperglycemia on the incretin effect in patients with T2DM, patients with impaired glucose tolerance, and patients with normal glucose tolerance.26 There was a significant (P ≤ .05) reduction in the incretin effect in terms of total insulin secretion, beta-cell glucose sensitivity, and the GLP-1 response to oral glucose in patients with T2DM compared with individuals whose glucose tolerance was normal or impaired. Each manifestation of the incretin effect was inversely related to both glucose tolerance and body mass index in an independent, additive manner (P ≤ .05); thus, glucose tolerance and obesity attenuate the incretin effect on beta-cell function and GLP-1 response independently of each other.
Exogenous GLP-1 has been shown to restore the regulation of blood glucose to near-normal concentrations in patients with T2DM.27 Several studies of patients with T2DM have shown that synthetic GLP-1 administration induces insulin secretion,19,27 slows gastric emptying (which is accelerated in patients with T2DM), and decreases inappropriately elevated glucagon secretion.19,23,28 Acute GLP-1 infusion studies showed that GLP-1 improved fasting plasma glucose (FPG) and PPG concentrations23,27; long-term studies showed that this hormone exerts euglycemic effects, leading to improvements in glycosylated hemoglobin (HbA1c), and induces weight loss.29
TARGETING FUNDAMENTAL DEFECTS OF T2DM WITH INCRETIN-BASED THERAPIES
Recognition and a better understanding of the role of the incretins and the enzyme involved in their degradation have led to the development of two incretin-based treatments: the GLP-1 receptor agonists, which possess many of the glucoregulatory actions of incretin peptides, and the DPP-4 inhibitors.5 Both the GLP-1 receptor agonists and the DPP-4 inhibitors have demonstrated safety and efficacy in the management of hyperglycemia in patients with T2DM.
GLP-1 receptor agonists
The GLP-1 receptor agonist exenatide is a synthetic form of exendin-4 and has a unique amino acid sequence that renders it resistant to degradation by DPP-4, making its actions longer lasting than endogenous GLP-1.5,30 Exenatide has a half-life of 2.4 hours and is detectable for up to 10 hours after subcutaneous (SC) injection.5,30 It is administered BID and has been approved as monotherapy or an adjunct therapy in patients with T2DM who have inadequate glycemic control following treatment with metformin, a sulfonylurea, a thiazolidinedione (TZD), or metformin in combination with a sulfonylurea or a TZD.31–35
In both human and animal studies, exenatide has been shown to enhance glucose-dependent insulin secretion and suppress inappropriate glucagon secretion in a glucose-dependent manner, reduce food intake and body weight, and acutely improve beta-cell function by enhancing first- and second-phase insulin secretion.5,36,37
In a small study involving 17 patients with T2DM, exenatide was shown to slow gastric emptying, which could be an important mechanism contributing to its beneficial effects on PPG concentration.38 Exenatide also has been shown to attenuate postprandial hyperglycemia, a risk factor for cardiovascular disease (CVD), by reducing endogenous glucose production by about 50% in patients with T2DM.39 Another mechanism for glycemic control may exist, as a recent animal study has shown that exenatide, similar to endogenous GLP-1, lowers blood glucose concentration independent of changes in pancreatic islet hormone secretion or delayed gastric emptying.40
A formulation of exenatide that is administered once weekly—exenatide long-acting release (LAR)—is in clinical evaluation and under review by the US Food and Drug Administration (FDA). In a short-term study, exenatide-LAR (0.8 mg or 2.0 mg) was administered once weekly for 15 weeks to patients with T2DM whose glycemia was suboptimally controlled with metformin alone or in combination with diet and exercise. Compared with placebo, treatment with exenatide once weekly was associated with markedly reduced HbA1c, FPG, PPG and body weight.41 In a larger, 30-week, phase 3 trial, Diabetes Therapy Utilization: Researching Changes in A1C, Weight and Other Factors Through Intervention with Exenatide ONce Weekly (DURATION-1), exenatide-LAR 2 mg once weekly was compared with exenatide 10 mg BID in patients with T2DM. Exenatide-LAR once weekly was associated with a significantly greater reduction in HbA1c (–1.9% vs –1.5%, P = .0023), and with a similar low risk of hypoglycemia and reduction in body weight (–3.7 kg vs –3.6 kg, P = .89) compared with the BID formulation.42
Liraglutide, recently approved in the European Union for T2DM and also under regulatory review in the United States, is a DPP-4–resistant human analogue GLP-1 receptor agonist in clinical development that has a 97% homology to native GLP-1.43–45 In contrast to exenatide, the acetylated liraglutide molecule allows binding to serum albumin and provides resistance to DPP-4 degradation, thus prolonging the half-life of liraglutide to approximately 12 hours. Liraglutide is administered SC QD as monotherapy or in combination with other antidiabetes agents such as metformin or sulfonylurea to patients with T2DM.44–47 Liraglutide has been shown to reduce HbA1c, decrease body weight, and lead to a lower incidence of hypoglycemia compared with the sulfonylurea glimepiride.
DPP-4 inhibitors
Sitagliptin is a DPP-4 inhibitor indicated as monotherapy or in combination with metformin or a TZD in patients with T2DM with inadequate glycemic control.48–51 Given orally, sitagliptin does not bind to the GLP-1 receptor agonist and has been shown to inhibit circulating DPP-4 activity by about 80%.52,53 Sitagliptin has been associated with an approximate twofold increase in postprandial GLP-1 plasma concentrations compared with placebo in healthy human subjects and in patients with T2DM.53 Saxagliptin, another potent DPP-4 inhibitor, significantly reduced HbA1c and FPG concentrations in patients with T2DM54 with a neutral effect on weight; it was recently approved by the FDA for treatment of T2DM.55
The DPP-4 inhibitor vildagliptin is currently being used in the European Union and Latin America but has yet to receive regulatory approval in the United States.54 Alogliptin, a novel, high-affinity, high-specificity DPP-4 inhibitor currently in development, provides rapid and sustained DPP-4 inhibition and significantly reduces HbA1c, FPG, and PPG concentrations with no change in body weight in patients with T2DM.56,57
Incretin-based therapies compared
Effects of incretin-based therapies
The number of people with T2DM, overweight/obesity, or CVD, alone or in combination, is approaching epidemic proportions, with the mechanisms of these conditions interrelated. Approximately 24 million Americans have diabetes, and T2DM accounts for more than 90% of these cases.61 Most patients with T2DM are not achieving HbA1c targets.62–64 About 60% of deaths among patients with T2DM are caused by CVD.65 Compounding the problem, overweight/obesity enhances the risk for CV-related morbidities in patients with diabetes.66 A cluster of metabolic disorders referred to as the metabolic syndrome (which includes hyperglycemia, measures of central obesity, and a series of significant CV risk factors) is common in patients with T2DM and CVD.67 Unfortunately, many antidiabetes drugs that successfully manage glycemic control also cause weight gain, which in theory may increase CV risk in patients with T2DM.68
Data from studies of patients with T2DM show that exenatide improves glycemic control and reduces body weight. Exenatide administered BID significantly reduced HbA1c (–0.40% to –0.86%) and weight (–1.6 kg to –2.8 kg) relative to baseline in three 30-week, placebo-controlled clinical trials.31,33,34 In subsequent 2-year, open-label extension studies, exenatide produced significant reductions from baseline in HbA1c (–20.9% at 30 weeks) and weight (–2.1 kg at 30 weeks). Both decreases were sustained through 2 years (HbA1c –1.1%, weight –4.7 kg) with a low incidence of hypoglycemia.31 Further post hoc analysis of the open-label extension of the 30-week trials followed patients treated with exenatide BID for 3 years or longer.69 In addition to markedly decreasing HbA1c from baseline levels (–1.1% at 3 years and –0.8% at up to 3.5 years; P < .0001), adjunctive exenatide produced significant reductions in body weight—up to –5.3 kg after 3.5 years of therapy.31,69 At 3.5 years, continued exenatide therapy resulted in a –6% reduction in low-density lipoprotein cholesterol, a 24% mean increase in high-density lipoprotein cholesterol, and a mean reduction in blood pressure of –2% to –4% from baseline levels. Improvements in hepatic biomarkers and homeostasis model assessment-B, a measure of beta-cell function, were seen after 2 and 3 years of exenatide treatment.31 Hypoglycemia was generally mild and transient.
In comparative head-to-head studies, exenatide BID and insulin analogues reduced HbA1c by similar magnitudes; yet exenatide treatment resulted in better control in terms of PPG and weight loss, while insulin glargine and insulin aspart produced weight gain.70–73
Mechanisms of cardioprotective effects
Although the mechanisms for the potential cardioprotective effects of GLP-1 and its receptor agonists remain to be fully elucidated, a recent study suggested that two novel pathways could be involved—one that is dependent on the known GLP-1 receptor pathway, and one that is independent of the GLP-1 receptor pathway.74 Correlating with observations of a potential cardioprotective effect, an infusion of recombinant GLP-1 in patients with acute myocardial infarction, when added to standard therapy, resulted in improved left ventricular function and was associated with reduced mortality.75 Evidence continues to accumulate for potential cardioprotective effects of the GLP-1 receptor agonists, indicating that they may have a positive impact on macrovascular complications in patients with T2DM.
CONCLUSION
T2DM, which is often associated with overweight and obesity, remains a significant challenge worldwide. The broad spectrum of glucoregulatory actions of the incretin hormones GLP-1 and GIP, and their importance in maintaining glucose homeostasis, have been recognized and correlated with the pathogenesis of T2DM. An improved understanding of the roles played by GLP-1 and GIP in the pathogenesis of T2DM may provide clinicians with important details regarding the therapeutic application of incretin-based therapies, including the GLP-1 receptor agonist exenatide and the DPP-4 inhibitors sitagliptin and saxagliptin. Antidiabetes agents whose development is based on the multiple pharmacologic effects of incretin hormones can address the multifaceted nature of T2DM and overcome some current limitations of traditional therapies, especially those related to weight. This becomes more compelling given the close link among T2DM, obesity, and increased CV risk.
It has long been understood that the pathophysiology of type 2 diabetes mellitus (T2DM) is based on the triad of progressive decline in insulin-producing pancreatic beta cells, an increase in insulin resistance, and increased hepatic glucose production.1,2 It is now evident that other factors, including defective actions of the gastrointestinal (GI) incretin hormones glucagon-like peptide–1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), also play significant roles.2–5 The uncontrolled hyperglycemia resulting from such defects may lead to microvascular complications, including retinopathy, neuropathy, microangiopathy, and nephropathy, and macrovascular complications, such as coronary artery disease and peripheral vascular disease.
This review explores the growing understanding of the role of the incretins in normal insulin secretion, as well as in the pathogenesis of T2DM, and examines the pathophysiologic basis for the benefits and therapeutic application of incretin-based therapies in T2DM.1,2
THE GI SYSTEM AND GLUCOSE HOMEOSTASIS IN THE HEALTHY STATE
The GI system plays an integral role in glucose homeostasis.6 The observation that orally administered glucose provides a stronger insulinotropic stimulus than an intravenous glucose challenge provided insight into the regulation of plasma glucose by the GI system of healthy individuals.7 The incretin effect, as this is termed, may be responsible for 50% to 70% of the total insulin secreted following oral glucose intake.8
Two GI peptide hormones (the incretins)—GLP-1 and GIP—were found to exert major glucoregulatory actions.3,9,10 Within minutes of nutrient ingestion, GLP-1 is secreted from intestinal L cells in the distal ileum and colon, while GIP is released by intestinal K cells in the duodenum and jejunum.3 GLP-1 and GIP trigger their insulinotropic actions by binding beta-cell receptors.3 GLP-1 receptors are expressed on pancreatic glucagon-containing alpha and delta cells as well as on beta cells, whereas GIP receptors are expressed primarily on beta cells.3,8 GLP-1 receptors are also expressed in the central nervous system (CNS), peripheral nervous system, lung, heart, and GI tract, while GIP receptors are expressed in adipose tissue and the CNS.3 GLP-1 inhibits glucose-dependent glucagon secretion from alpha cells.3 In healthy individuals, fasting glucose is managed by tonic insulin/glucagon secretion, but excursions of postprandial glucose (PPG) are controlled by insulin and the incretin hormones.11
Additionally, in animal studies, GLP-1 has been shown to induce the transcriptional activation of the insulin gene and insulin biosynthesis, thus increasing beta-cell proliferation and decreasing beta-cell apoptosis.12 GLP-1 stimulates a CNS-mediated pathway of insulin secretion, slows gastric emptying, increases CNS-mediated satiety leading to reduced food intake, indirectly increases insulin sensitivity and nutrient uptake in skeletal muscle and adipose tissue, and exerts neuroprotective effects.8
Both GLP-1 and GIP are rapidly degraded by the serine protease dipeptidyl peptidase–4 (DPP-4), which is widely expressed in bound and free forms.14 A recent study in healthy adults showed that GLP-1 concentration declined even during maximal DPP-4 inhibition, suggesting that there may be pathways of GLP-1 elimination other than DPP-4 enzymatic degradation.15
INCRETINS AND THE PATHOGENESIS OF T2DM
Studies have shown that incretin pathways play a role in the progression of T2DM.3,16 The significant reduction in the incretin effect seen in patients with T2DM has been attributed to several factors, including impaired secretion of GLP-1, accelerated metabolism of GLP-1 and GIP, and defective responsiveness to both hormones.16 Many patients with T2DM also have accelerated gastric emptying that may contribute to deterioration of their glycemic control.17
While GIP concentration is normal or modestly increased in patients with T2DM,16,18 the insulinotropic actions of GIP are significantly diminished.19 Thus, patients with T2DM have an impaired responsiveness to GIP with a possible link to GIP-receptor downregulation or desensitization.20
Are secretory defects a cause or result of T2DM?
In contrast to GIP, the secretion of GLP-1 has been shown to be deficient in patients with T2DM.18 As with GIP, it is unknown to what degree this defect is a cause or consequence of T2DM. In a study of identical twins, defective GLP-1 secretion was observed only in the one sibling with T2DM, suggesting that GLP-1 secretory deficits may be secondary to the development of T2DM.21 Despite the diminished secretion of GLP-1 in patients with T2DM, the insulinotropic actions of GLP-1 are preserved.19 It has also been shown that the effects of GLP-1 on gastric emptying and glucagon secretion are maintained in patients with T2DM.19,22,23
Whether this incretin dysregulation is responsible for or is the end result of hyperglycemia remains a subject of continued investigation. A recent study confirmed that the incretin effect is reduced in patients with T2DM, but advanced the concept that it may be a consequence of the diabetic state.16,24 Notably, impaired actions of GLP-1 and GIP and diminished concentrations of GLP-1 may be partially restored by improved glycemic control.24
Recent preclinical and clinical studies continue to clarify the roles of incretin hormones in T2DM. The findings from a study of obese diabetic mice suggest that the effect of GLP-1 therapy on the long-term remission of diabetes may be caused by improvements in beta-cell function and insulin sensitivity, as well as by a reduction in gluconeogenesis in the liver.25
Incretin effect and glucose tolerance, body mass index
Another study was conducted to evaluate quantitatively the separate impacts of obesity and hyperglycemia on the incretin effect in patients with T2DM, patients with impaired glucose tolerance, and patients with normal glucose tolerance.26 There was a significant (P ≤ .05) reduction in the incretin effect in terms of total insulin secretion, beta-cell glucose sensitivity, and the GLP-1 response to oral glucose in patients with T2DM compared with individuals whose glucose tolerance was normal or impaired. Each manifestation of the incretin effect was inversely related to both glucose tolerance and body mass index in an independent, additive manner (P ≤ .05); thus, glucose tolerance and obesity attenuate the incretin effect on beta-cell function and GLP-1 response independently of each other.
Exogenous GLP-1 has been shown to restore the regulation of blood glucose to near-normal concentrations in patients with T2DM.27 Several studies of patients with T2DM have shown that synthetic GLP-1 administration induces insulin secretion,19,27 slows gastric emptying (which is accelerated in patients with T2DM), and decreases inappropriately elevated glucagon secretion.19,23,28 Acute GLP-1 infusion studies showed that GLP-1 improved fasting plasma glucose (FPG) and PPG concentrations23,27; long-term studies showed that this hormone exerts euglycemic effects, leading to improvements in glycosylated hemoglobin (HbA1c), and induces weight loss.29
TARGETING FUNDAMENTAL DEFECTS OF T2DM WITH INCRETIN-BASED THERAPIES
Recognition and a better understanding of the role of the incretins and the enzyme involved in their degradation have led to the development of two incretin-based treatments: the GLP-1 receptor agonists, which possess many of the glucoregulatory actions of incretin peptides, and the DPP-4 inhibitors.5 Both the GLP-1 receptor agonists and the DPP-4 inhibitors have demonstrated safety and efficacy in the management of hyperglycemia in patients with T2DM.
GLP-1 receptor agonists
The GLP-1 receptor agonist exenatide is a synthetic form of exendin-4 and has a unique amino acid sequence that renders it resistant to degradation by DPP-4, making its actions longer lasting than endogenous GLP-1.5,30 Exenatide has a half-life of 2.4 hours and is detectable for up to 10 hours after subcutaneous (SC) injection.5,30 It is administered BID and has been approved as monotherapy or an adjunct therapy in patients with T2DM who have inadequate glycemic control following treatment with metformin, a sulfonylurea, a thiazolidinedione (TZD), or metformin in combination with a sulfonylurea or a TZD.31–35
In both human and animal studies, exenatide has been shown to enhance glucose-dependent insulin secretion and suppress inappropriate glucagon secretion in a glucose-dependent manner, reduce food intake and body weight, and acutely improve beta-cell function by enhancing first- and second-phase insulin secretion.5,36,37
In a small study involving 17 patients with T2DM, exenatide was shown to slow gastric emptying, which could be an important mechanism contributing to its beneficial effects on PPG concentration.38 Exenatide also has been shown to attenuate postprandial hyperglycemia, a risk factor for cardiovascular disease (CVD), by reducing endogenous glucose production by about 50% in patients with T2DM.39 Another mechanism for glycemic control may exist, as a recent animal study has shown that exenatide, similar to endogenous GLP-1, lowers blood glucose concentration independent of changes in pancreatic islet hormone secretion or delayed gastric emptying.40
A formulation of exenatide that is administered once weekly—exenatide long-acting release (LAR)—is in clinical evaluation and under review by the US Food and Drug Administration (FDA). In a short-term study, exenatide-LAR (0.8 mg or 2.0 mg) was administered once weekly for 15 weeks to patients with T2DM whose glycemia was suboptimally controlled with metformin alone or in combination with diet and exercise. Compared with placebo, treatment with exenatide once weekly was associated with markedly reduced HbA1c, FPG, PPG and body weight.41 In a larger, 30-week, phase 3 trial, Diabetes Therapy Utilization: Researching Changes in A1C, Weight and Other Factors Through Intervention with Exenatide ONce Weekly (DURATION-1), exenatide-LAR 2 mg once weekly was compared with exenatide 10 mg BID in patients with T2DM. Exenatide-LAR once weekly was associated with a significantly greater reduction in HbA1c (–1.9% vs –1.5%, P = .0023), and with a similar low risk of hypoglycemia and reduction in body weight (–3.7 kg vs –3.6 kg, P = .89) compared with the BID formulation.42
Liraglutide, recently approved in the European Union for T2DM and also under regulatory review in the United States, is a DPP-4–resistant human analogue GLP-1 receptor agonist in clinical development that has a 97% homology to native GLP-1.43–45 In contrast to exenatide, the acetylated liraglutide molecule allows binding to serum albumin and provides resistance to DPP-4 degradation, thus prolonging the half-life of liraglutide to approximately 12 hours. Liraglutide is administered SC QD as monotherapy or in combination with other antidiabetes agents such as metformin or sulfonylurea to patients with T2DM.44–47 Liraglutide has been shown to reduce HbA1c, decrease body weight, and lead to a lower incidence of hypoglycemia compared with the sulfonylurea glimepiride.
DPP-4 inhibitors
Sitagliptin is a DPP-4 inhibitor indicated as monotherapy or in combination with metformin or a TZD in patients with T2DM with inadequate glycemic control.48–51 Given orally, sitagliptin does not bind to the GLP-1 receptor agonist and has been shown to inhibit circulating DPP-4 activity by about 80%.52,53 Sitagliptin has been associated with an approximate twofold increase in postprandial GLP-1 plasma concentrations compared with placebo in healthy human subjects and in patients with T2DM.53 Saxagliptin, another potent DPP-4 inhibitor, significantly reduced HbA1c and FPG concentrations in patients with T2DM54 with a neutral effect on weight; it was recently approved by the FDA for treatment of T2DM.55
The DPP-4 inhibitor vildagliptin is currently being used in the European Union and Latin America but has yet to receive regulatory approval in the United States.54 Alogliptin, a novel, high-affinity, high-specificity DPP-4 inhibitor currently in development, provides rapid and sustained DPP-4 inhibition and significantly reduces HbA1c, FPG, and PPG concentrations with no change in body weight in patients with T2DM.56,57
Incretin-based therapies compared
Effects of incretin-based therapies
The number of people with T2DM, overweight/obesity, or CVD, alone or in combination, is approaching epidemic proportions, with the mechanisms of these conditions interrelated. Approximately 24 million Americans have diabetes, and T2DM accounts for more than 90% of these cases.61 Most patients with T2DM are not achieving HbA1c targets.62–64 About 60% of deaths among patients with T2DM are caused by CVD.65 Compounding the problem, overweight/obesity enhances the risk for CV-related morbidities in patients with diabetes.66 A cluster of metabolic disorders referred to as the metabolic syndrome (which includes hyperglycemia, measures of central obesity, and a series of significant CV risk factors) is common in patients with T2DM and CVD.67 Unfortunately, many antidiabetes drugs that successfully manage glycemic control also cause weight gain, which in theory may increase CV risk in patients with T2DM.68
Data from studies of patients with T2DM show that exenatide improves glycemic control and reduces body weight. Exenatide administered BID significantly reduced HbA1c (–0.40% to –0.86%) and weight (–1.6 kg to –2.8 kg) relative to baseline in three 30-week, placebo-controlled clinical trials.31,33,34 In subsequent 2-year, open-label extension studies, exenatide produced significant reductions from baseline in HbA1c (–20.9% at 30 weeks) and weight (–2.1 kg at 30 weeks). Both decreases were sustained through 2 years (HbA1c –1.1%, weight –4.7 kg) with a low incidence of hypoglycemia.31 Further post hoc analysis of the open-label extension of the 30-week trials followed patients treated with exenatide BID for 3 years or longer.69 In addition to markedly decreasing HbA1c from baseline levels (–1.1% at 3 years and –0.8% at up to 3.5 years; P < .0001), adjunctive exenatide produced significant reductions in body weight—up to –5.3 kg after 3.5 years of therapy.31,69 At 3.5 years, continued exenatide therapy resulted in a –6% reduction in low-density lipoprotein cholesterol, a 24% mean increase in high-density lipoprotein cholesterol, and a mean reduction in blood pressure of –2% to –4% from baseline levels. Improvements in hepatic biomarkers and homeostasis model assessment-B, a measure of beta-cell function, were seen after 2 and 3 years of exenatide treatment.31 Hypoglycemia was generally mild and transient.
In comparative head-to-head studies, exenatide BID and insulin analogues reduced HbA1c by similar magnitudes; yet exenatide treatment resulted in better control in terms of PPG and weight loss, while insulin glargine and insulin aspart produced weight gain.70–73
Mechanisms of cardioprotective effects
Although the mechanisms for the potential cardioprotective effects of GLP-1 and its receptor agonists remain to be fully elucidated, a recent study suggested that two novel pathways could be involved—one that is dependent on the known GLP-1 receptor pathway, and one that is independent of the GLP-1 receptor pathway.74 Correlating with observations of a potential cardioprotective effect, an infusion of recombinant GLP-1 in patients with acute myocardial infarction, when added to standard therapy, resulted in improved left ventricular function and was associated with reduced mortality.75 Evidence continues to accumulate for potential cardioprotective effects of the GLP-1 receptor agonists, indicating that they may have a positive impact on macrovascular complications in patients with T2DM.
CONCLUSION
T2DM, which is often associated with overweight and obesity, remains a significant challenge worldwide. The broad spectrum of glucoregulatory actions of the incretin hormones GLP-1 and GIP, and their importance in maintaining glucose homeostasis, have been recognized and correlated with the pathogenesis of T2DM. An improved understanding of the roles played by GLP-1 and GIP in the pathogenesis of T2DM may provide clinicians with important details regarding the therapeutic application of incretin-based therapies, including the GLP-1 receptor agonist exenatide and the DPP-4 inhibitors sitagliptin and saxagliptin. Antidiabetes agents whose development is based on the multiple pharmacologic effects of incretin hormones can address the multifaceted nature of T2DM and overcome some current limitations of traditional therapies, especially those related to weight. This becomes more compelling given the close link among T2DM, obesity, and increased CV risk.
- Boyle PJ, Freeman JS. Application of incretin mimetics and dipeptidyl peptidase IV inhibitors in managing type 2 diabetes mellitus. J Am Osteopath Assoc 2007; 107(suppl 3):S10–S16.
- Freeman JS. The pathophysiologic role of incretins. J Am Osteopath Assoc 2007; 107(suppl 3):S6–S9.
- Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006; 368:1696–1705.
- Nauck MA, Baller B, Meier JJ. Gastric inhibitory polypeptide and glucagon-like peptide-1 in the pathogenesis of type 2 diabetes. Diabetes 2004; 53(suppl 3):S190–S196.
- Stonehouse A, Okerson T, Kendall D, Maggs D. Emerging incretin based therapies for type 2 diabetes: incretin mimetics and DPP-4 inhibitors. Curr Diabetes Rev 2008; 4:101–109.
- Huda MS, Wilding JP, Pinkney JH. Gut peptides and the regulation of appetite. Obes Rev 2006; 7:163–182.
- Elrick H, Stimmler L, Hlad CJ Jr, Arai Y. Plasma insulin response to oral and intravenous glucose administration. J Clin Endocrinol Metab 1964; 24:1076–1082.
- Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007; 132:2131–2157.
- Brown JC, Dryburgh JR, Ross SA, Dupré J. Identification and actions of gastric inhibitory polypeptide. Recent Prog Horm Res 1975; 31:487–532.
- Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet 1987; 2:1300–1304.
- Nauck MA, Homberger E, Siegel EG, et al. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J Clin Endocrinol Metab 1986; 63:492–498.
- Farilla L, Bulotta A, Hirshberg B, et al. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology 2003; 144:5149–5158.
- Van Gaal LF, Gutkin SW, Nauck MA. Exploiting the antidiabetic properties of incretins to treat type 2 diabetes mellitus: glucagon-like peptide 1 receptor agonists or insulin for patients with inadequate glycemic control? Eur J Endocrinol 2008; 158:773–784.
- Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes 1995; 44:1126–1131.
- Dai H, Gustavson SM, Preston GM, Eskra JD, Calle R, Hirshberg B. Non-linear increase in GLP-1 levels in response to DPP-IV inhibition in healthy adult subjects. Diabetes Obes Metab 2008; 10:506–513.
- Nauck MA, Stöckmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 1986; 29:46–52.
- Phillips WT, Schwartz JG, McMahan CA. Rapid gastric emptying of an oral glucose solution in type 2 diabetic patients. J Nucl Med 1992; 33:1496–1500.
- Toft-Nielsen MB, Damholt MB, Madsbad S, et al. Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J Clin Endocrinol Metab 2001; 86:3717–3723.
- Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest 1993; 91:301–307.
- Lynn FC, Thompson SA, Pospisilik JA, et al. A novel pathway for regulation of glucose-dependent insulinotropic polypeptide (GIP) receptor expression in beta cells. FASEB J 2003; 17:91–93.
- Vaag AA, Holst JJ, Vølund A, Beck-Nielsen HB. Gut incretin hormones in identical twins discordant for non-insulin-dependent diabetes mellitus (NIDDM)—evidence for decreased glucagon-like peptide-1 secretion during oral glucose ingestion in NIDDM twins. Eur J Endocrinol 1996; 135:425–432.
- Meier JJ, Gallwitz B, Salmen S, et al. Normalization of glucose concentrations and deceleration of gastric emptying after solid meals during intravenous glucagon-like peptide 1 in patients with type 2 diabetes. J Clin Endocrinol Metab 2003; 88:2719–2725.
- Nauck MA, Kleine N, Orskov C, Holst JJ, Willms B, Creutzfeldt W. Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7-36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 1993; 36:741–744.
- Knop FK, Vilsbøll T, Højberg PV, et al. Reduced incretin effect in type 2 diabetes: cause or consequence of the diabetic state? Diabetes 2007; 56:1951–1959.
- Lee YS, Shin S, Shigihara T, et al. Glucagon-like peptide-1 gene therapy in obese diabetic mice results in long-term cure of diabetes by improving insulin sensitivity and reducing hepatic gluconeogenesis. Diabetes 2007; 56:1671–1679.
- Muscelli E, Mari A, Casolaro A, et al. Separate impact of obesity and glucose tolerance on the incretin effect in normal subjects and type 2 diabetic patients. Diabetes 2008; 57:1340–1348.
- Nathan DM, Schreiber E, Fogel H, Mojsov S, Habener JF. Insulinotropic action of glucagonlike peptide-I-(7-37) in diabetic and nondiabetic subjects. Diabetes Care 1992; 15:270–276.
- Kolterman OG, Buse JB, Fineman MS, et al. Synthetic exendin-4 (exenatide) significantly reduces postprandial and fasting plasma glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab 2003; 88:3082–3089.
- Zander M, Madsbad S, Madsen JL, Holst JJ. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet 2002; 359:824–830.
- Kolterman OG, Kim DD, Shen L, et al. Pharmacokinetics, pharmacodynamics, and safety of exenatide in patients with type 2 diabetes mellitus. Am J Health Syst Pharm 2005; 62:173–181.
- Buse JB, Henry RR, Han J, et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care 2004; 27:2628–2635.
- Byetta [package insert]. San Diego, CA: Amylin Pharmaceuticals, Inc.; 2009.
- DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 2005; 28:1092–1100.
- Kendall DM, Riddle MC, Rosenstock J, et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in patients with type 2 diabetes treated with metformin and a sulfonylurea. Diabetes Care 2005; 28:1083–1091.
- Zinman B, Hoogwerf BJ, Durán García S, et al. The effect of adding exenatide to a thiazolidinedione in suboptimally controlled type 2 diabetes: a randomized trial. Ann Intern Med 2007; 146:477–485.
- Fehse F, Trautmann M, Holst JJ, et al. Exenatide augments first- and second-phase insulin secretion in response to intravenous glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab 2005; 90:5991–5997.
- Parkes DG, Pittner R, Jodka C, Smith P, Young A. Insulinotropic actions of exendin-4 and glucagon-like peptide-1 in vivo and in vitro. Metabolism 2001; 50:583–589.
- Linnebjerg H, Park S, Kothare PA, et al. Effect of exenatide on gastric emptying and relationship to postprandial glycemia in type 2 diabetes. Regul Pept 2008; 151:123–129.
- Cervera A, Wajcberg E, Sriwijitkamol A, et al. Mechanism of action of exenatide to reduce postprandial hyperglycemia in type 2 diabetes. Am J Physiol Endocrinol Metab 2008; 294:E846–E852.
- Ionut V, Zheng D, Stefanovski D, Bergman RN. Exenatide can reduce glucose independent of islet hormones or gastric emptying. Am J Physiol Endocrinol Metab 2008; 295:E269–E277.
- Kim D, MacConell L, Zhuang D, et al. Effects of once-weekly dosing of a long-acting release formulation of exenatide on glucose control and body weight in subjects with type 2 diabetes. Diabetes Care 2007; 30:1487–1493.
- Drucker DJ, Buse JB, Taylor K, et al. Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: a randomised, open-label, non-inferiority study. Lancet 2008; 372:1240–1250.
- Knudsen LB, Nielsen PF, Huusfeldt PO, et al. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem 2000; 43:1664–1669.
- Nauck M, Frid A, Hermansen K, et al; for the LEAD-2 Study Group. Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: the LEAD (liraglutide effect and action in diabetes)-2 study. Diabetes Care 2009; 32:84–90.
- Committee for Medicinal Products for Human Use: Summary of Positive Opinion for Victoza. European Medicines Agency Web site. http://www.emea.europa.eu/pdfs/human/opinion/Victoza_14168909en.pdf. Published April 23, 2009. Accessed September 21, 2009.
- Garber A, Henry R, Ratner R, et al. Liraglutide versus glimepiride monotherapy for type 2 diabetes (LEAD-3 Mono): a randomised, 52-week, phase III, double-blind, parallel-treatment trial. Lancet 2009; 373:473–481.
- Marre M, Shaw J, Brändle M, et al. Liraglutide, a once-daily human GLP-1 analogue, added to a sulphonylurea over 26 weeks produces greater improvements in glycaemic and weight control compared with adding rosiglitazone or placebo in subjects with type 2 diabetes (LEAD-1 SU). Diabet Med 2009; 26:268–278.
- Aschner P, Kipnes MS, Lunceford JK, et al; for the Sitagliptin 021 Study Group. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care 2006; 29:2632–2637.
- Charbonnel B, Karasik A, Liu J, Wu M, Meininger G; for the Sitagliptin Study 020 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006; 29:2638–2643.
- Raz I, Hanefeld M, Xu L, et al. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia 2006; 49:2564–2571.
- Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P; for the Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2006; 28:1556–1568.
- DeFronzo RA, Okerson T, Viswanathan P, Guan X, Holcombe JH, MacConell L. Effects of exenatide versus sitagliptin on postprandial glucose, insulin and glucagon secretion, gastric emptying, and caloric intake: a randomized, cross-over study. Curr Med Res Opin 2008; 24:2943–2952.
- Herman GA, Stevens C, Van Dyck K, et al. Pharmacokinetics and pharmacodynamics of sitagliptin, an inhibitor of dipeptidyl peptidase IV, in healthy subjects: results from two randomized, double-blind, placebo-controlled studies with single oral doses. Clin Pharmacol Ther 2005; 78:675–688.
- Baggio LL, Drucker DJ, Maida A, Lamont BJ. ADA 2008: incretin-based therapeutics. MedscapeCME Web site. http://www.medscape.com/viewprogram/15786. Accessed September 18, 2009.
- US Department of Health and Human Services. FDA approves new drug treatment for type 2 diabetes. US Food and Drug Administration Web site. http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm174780.htm. Published July 31, 2009. Accessed September 18, 2009.
- Covington P, Christopher R, Davenport M, et al. Pharmacokinetic, pharmacodynamic, and tolerability profiles of the dipeptidyl peptidase-4 inhibitor alogliptin: a randomized, double-blind, placebo-controlled, multiple-dose study in adult patients with type 2 diabetes. Clin Ther 2008; 30:499–512.
- DeFronzo RA, Fleck PR, Wilson CA, Mekki Q; on behalf of the Alogliptin Study 010 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor alogliptin in patients with type 2 diabetes and inadequate glycemic control: a randomized, double-blind, placebo-controlled study. Diabetes Care 2008; 31:2315–2317.
- Triplitt CL, McGill JB, Porte D Jr, Conner CS. The changing landscape of type 2 diabetes: the role of incretin-based therapies in managed care outcomes. J Manag Care Pharm 2007; 13(9 suppl C):S2–S16.
- Garber AJ, Spann SJ. An overview of incretin clinical trials. J Fam Pract 2008; 57(9 suppl):S10–S18.
- Henry RR. Evolving concepts of type 2 diabetes management with oral medications: new approaches to an old disease. Curr Med Res Opin 2008; 24:2189–2202.
- Centers for Disease Control and Prevention. National diabetes fact sheet: general information and national estimates on diabetes in the United States, 2007. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/diabetes/pubs/pdf/ndfs_2007.pdf. Published 2008. Accessed September 21, 2009.
- Ong KL, Cheung BM, Wong LY, Wt NM, Tan KC, Lam KS. Prevalence, treatment, and control of diagnosed diabetes in the U.S. National Health and Nutrition Examination Survey 1999–2004. Ann Epidemiol 2008; 18:222–229.
- Sanders CL, Yesupriya AJ, Curtin LR. Analysis of population structure and stratification in NHANES III self-reported race/ethnicities. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/genomics/events/file/print/10year/08_pop_struct_ab.pdf. Accessed September 21, 2009.
- Koro CE, Bowlin SJ, Bourgeois N, Fedder DO. Glycemic control from 1988 to 2000 among US adults diagnosed with type 2 diabetes: a preliminary report. Diabetes Care 2004; 27:17–20.
- Haffner SM, Lehto S, Rönnemaa T, Pyörälä K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229–234.
- Lloyd-Jones D, Adams R, Carnethon M, et al. Heart disease and stroke statistics—2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2009; 119:480–486.
- Stonehouse AH, Holcombe JH, Kendall DM. GLP-1 analogues, DPP-IV inhibitors and the metabolic syndrome. In: Fonseca V, ed. Therapeutic Strategies in Metabolic Syndrome. Oxford, UK: Atlas Medical Publishing Ltd; 2008: 137–157.
- Purnell JQ, Weyer C. Weight effect of current and experimental drugs for diabetes mellitus: from promotion to alleviation of obesity. Treat Endocrinol 2003; 2:33–47.
- Klonoff DC, Buse JB, Nielsen LL, et al. Exenatide effects on diabetes, obesity, cardiovascular risk factors and hepatic biomarkers in patients with type 2 diabetes treated for at least 3 years. Curr Med Res Opin 2008; 24:275–286.
- Barnett AH, Burger J, Johns D, et al. Tolerability and efficacy of exenatide and titrated insulin glargine in adult patients with type 2 diabetes previously uncontrolled with metformin or a sulfonylurea: a multinational, randomized, open-label, two-period, crossover noninferiority trial. Clin Ther 2007; 29:2333–2348.
- Glass LC, Qu Y, Lenox S, et al. Effects of exenatide versus insulin analogues on weight change in subjects with type 2 diabetes: a pooled post-hoc analysis. Curr Med Res Opin 2008; 24:639–644.
- Heine RJ, Van Gaal LF, Johns D, Mihm MJ, Widel MH, Brodows RG; for the GWAA Study Group. Exenatide versus insulin glargine in patients with suboptimally controlled type 2 diabetes: a randomized trial. Ann Intern Med 2005; 143:559–569.
- Nauck MA, Duran S, Kim D, et al. A comparison of twice-daily exenatide and biphasic insulin aspart in patients with type 2 diabetes who were suboptimally controlled with sulfonylurea and metformin: a non-inferiority study. Diabetologia 2007; 50:259–267.
- Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation 2008; 117:2340–2350.
- Nikolaidis LA, Mankad S, Sokos GG, et al. Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation 2004; 109:962–965.
- Boyle PJ, Freeman JS. Application of incretin mimetics and dipeptidyl peptidase IV inhibitors in managing type 2 diabetes mellitus. J Am Osteopath Assoc 2007; 107(suppl 3):S10–S16.
- Freeman JS. The pathophysiologic role of incretins. J Am Osteopath Assoc 2007; 107(suppl 3):S6–S9.
- Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006; 368:1696–1705.
- Nauck MA, Baller B, Meier JJ. Gastric inhibitory polypeptide and glucagon-like peptide-1 in the pathogenesis of type 2 diabetes. Diabetes 2004; 53(suppl 3):S190–S196.
- Stonehouse A, Okerson T, Kendall D, Maggs D. Emerging incretin based therapies for type 2 diabetes: incretin mimetics and DPP-4 inhibitors. Curr Diabetes Rev 2008; 4:101–109.
- Huda MS, Wilding JP, Pinkney JH. Gut peptides and the regulation of appetite. Obes Rev 2006; 7:163–182.
- Elrick H, Stimmler L, Hlad CJ Jr, Arai Y. Plasma insulin response to oral and intravenous glucose administration. J Clin Endocrinol Metab 1964; 24:1076–1082.
- Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007; 132:2131–2157.
- Brown JC, Dryburgh JR, Ross SA, Dupré J. Identification and actions of gastric inhibitory polypeptide. Recent Prog Horm Res 1975; 31:487–532.
- Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet 1987; 2:1300–1304.
- Nauck MA, Homberger E, Siegel EG, et al. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J Clin Endocrinol Metab 1986; 63:492–498.
- Farilla L, Bulotta A, Hirshberg B, et al. Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology 2003; 144:5149–5158.
- Van Gaal LF, Gutkin SW, Nauck MA. Exploiting the antidiabetic properties of incretins to treat type 2 diabetes mellitus: glucagon-like peptide 1 receptor agonists or insulin for patients with inadequate glycemic control? Eur J Endocrinol 2008; 158:773–784.
- Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes 1995; 44:1126–1131.
- Dai H, Gustavson SM, Preston GM, Eskra JD, Calle R, Hirshberg B. Non-linear increase in GLP-1 levels in response to DPP-IV inhibition in healthy adult subjects. Diabetes Obes Metab 2008; 10:506–513.
- Nauck MA, Stöckmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 1986; 29:46–52.
- Phillips WT, Schwartz JG, McMahan CA. Rapid gastric emptying of an oral glucose solution in type 2 diabetic patients. J Nucl Med 1992; 33:1496–1500.
- Toft-Nielsen MB, Damholt MB, Madsbad S, et al. Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J Clin Endocrinol Metab 2001; 86:3717–3723.
- Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest 1993; 91:301–307.
- Lynn FC, Thompson SA, Pospisilik JA, et al. A novel pathway for regulation of glucose-dependent insulinotropic polypeptide (GIP) receptor expression in beta cells. FASEB J 2003; 17:91–93.
- Vaag AA, Holst JJ, Vølund A, Beck-Nielsen HB. Gut incretin hormones in identical twins discordant for non-insulin-dependent diabetes mellitus (NIDDM)—evidence for decreased glucagon-like peptide-1 secretion during oral glucose ingestion in NIDDM twins. Eur J Endocrinol 1996; 135:425–432.
- Meier JJ, Gallwitz B, Salmen S, et al. Normalization of glucose concentrations and deceleration of gastric emptying after solid meals during intravenous glucagon-like peptide 1 in patients with type 2 diabetes. J Clin Endocrinol Metab 2003; 88:2719–2725.
- Nauck MA, Kleine N, Orskov C, Holst JJ, Willms B, Creutzfeldt W. Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7-36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 1993; 36:741–744.
- Knop FK, Vilsbøll T, Højberg PV, et al. Reduced incretin effect in type 2 diabetes: cause or consequence of the diabetic state? Diabetes 2007; 56:1951–1959.
- Lee YS, Shin S, Shigihara T, et al. Glucagon-like peptide-1 gene therapy in obese diabetic mice results in long-term cure of diabetes by improving insulin sensitivity and reducing hepatic gluconeogenesis. Diabetes 2007; 56:1671–1679.
- Muscelli E, Mari A, Casolaro A, et al. Separate impact of obesity and glucose tolerance on the incretin effect in normal subjects and type 2 diabetic patients. Diabetes 2008; 57:1340–1348.
- Nathan DM, Schreiber E, Fogel H, Mojsov S, Habener JF. Insulinotropic action of glucagonlike peptide-I-(7-37) in diabetic and nondiabetic subjects. Diabetes Care 1992; 15:270–276.
- Kolterman OG, Buse JB, Fineman MS, et al. Synthetic exendin-4 (exenatide) significantly reduces postprandial and fasting plasma glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab 2003; 88:3082–3089.
- Zander M, Madsbad S, Madsen JL, Holst JJ. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet 2002; 359:824–830.
- Kolterman OG, Kim DD, Shen L, et al. Pharmacokinetics, pharmacodynamics, and safety of exenatide in patients with type 2 diabetes mellitus. Am J Health Syst Pharm 2005; 62:173–181.
- Buse JB, Henry RR, Han J, et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care 2004; 27:2628–2635.
- Byetta [package insert]. San Diego, CA: Amylin Pharmaceuticals, Inc.; 2009.
- DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 2005; 28:1092–1100.
- Kendall DM, Riddle MC, Rosenstock J, et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in patients with type 2 diabetes treated with metformin and a sulfonylurea. Diabetes Care 2005; 28:1083–1091.
- Zinman B, Hoogwerf BJ, Durán García S, et al. The effect of adding exenatide to a thiazolidinedione in suboptimally controlled type 2 diabetes: a randomized trial. Ann Intern Med 2007; 146:477–485.
- Fehse F, Trautmann M, Holst JJ, et al. Exenatide augments first- and second-phase insulin secretion in response to intravenous glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab 2005; 90:5991–5997.
- Parkes DG, Pittner R, Jodka C, Smith P, Young A. Insulinotropic actions of exendin-4 and glucagon-like peptide-1 in vivo and in vitro. Metabolism 2001; 50:583–589.
- Linnebjerg H, Park S, Kothare PA, et al. Effect of exenatide on gastric emptying and relationship to postprandial glycemia in type 2 diabetes. Regul Pept 2008; 151:123–129.
- Cervera A, Wajcberg E, Sriwijitkamol A, et al. Mechanism of action of exenatide to reduce postprandial hyperglycemia in type 2 diabetes. Am J Physiol Endocrinol Metab 2008; 294:E846–E852.
- Ionut V, Zheng D, Stefanovski D, Bergman RN. Exenatide can reduce glucose independent of islet hormones or gastric emptying. Am J Physiol Endocrinol Metab 2008; 295:E269–E277.
- Kim D, MacConell L, Zhuang D, et al. Effects of once-weekly dosing of a long-acting release formulation of exenatide on glucose control and body weight in subjects with type 2 diabetes. Diabetes Care 2007; 30:1487–1493.
- Drucker DJ, Buse JB, Taylor K, et al. Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: a randomised, open-label, non-inferiority study. Lancet 2008; 372:1240–1250.
- Knudsen LB, Nielsen PF, Huusfeldt PO, et al. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem 2000; 43:1664–1669.
- Nauck M, Frid A, Hermansen K, et al; for the LEAD-2 Study Group. Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: the LEAD (liraglutide effect and action in diabetes)-2 study. Diabetes Care 2009; 32:84–90.
- Committee for Medicinal Products for Human Use: Summary of Positive Opinion for Victoza. European Medicines Agency Web site. http://www.emea.europa.eu/pdfs/human/opinion/Victoza_14168909en.pdf. Published April 23, 2009. Accessed September 21, 2009.
- Garber A, Henry R, Ratner R, et al. Liraglutide versus glimepiride monotherapy for type 2 diabetes (LEAD-3 Mono): a randomised, 52-week, phase III, double-blind, parallel-treatment trial. Lancet 2009; 373:473–481.
- Marre M, Shaw J, Brändle M, et al. Liraglutide, a once-daily human GLP-1 analogue, added to a sulphonylurea over 26 weeks produces greater improvements in glycaemic and weight control compared with adding rosiglitazone or placebo in subjects with type 2 diabetes (LEAD-1 SU). Diabet Med 2009; 26:268–278.
- Aschner P, Kipnes MS, Lunceford JK, et al; for the Sitagliptin 021 Study Group. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care 2006; 29:2632–2637.
- Charbonnel B, Karasik A, Liu J, Wu M, Meininger G; for the Sitagliptin Study 020 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing metformin therapy in patients with type 2 diabetes inadequately controlled with metformin alone. Diabetes Care 2006; 29:2638–2643.
- Raz I, Hanefeld M, Xu L, et al. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia 2006; 49:2564–2571.
- Rosenstock J, Brazg R, Andryuk PJ, Lu K, Stein P; for the Sitagliptin Study 019 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin Ther 2006; 28:1556–1568.
- DeFronzo RA, Okerson T, Viswanathan P, Guan X, Holcombe JH, MacConell L. Effects of exenatide versus sitagliptin on postprandial glucose, insulin and glucagon secretion, gastric emptying, and caloric intake: a randomized, cross-over study. Curr Med Res Opin 2008; 24:2943–2952.
- Herman GA, Stevens C, Van Dyck K, et al. Pharmacokinetics and pharmacodynamics of sitagliptin, an inhibitor of dipeptidyl peptidase IV, in healthy subjects: results from two randomized, double-blind, placebo-controlled studies with single oral doses. Clin Pharmacol Ther 2005; 78:675–688.
- Baggio LL, Drucker DJ, Maida A, Lamont BJ. ADA 2008: incretin-based therapeutics. MedscapeCME Web site. http://www.medscape.com/viewprogram/15786. Accessed September 18, 2009.
- US Department of Health and Human Services. FDA approves new drug treatment for type 2 diabetes. US Food and Drug Administration Web site. http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm174780.htm. Published July 31, 2009. Accessed September 18, 2009.
- Covington P, Christopher R, Davenport M, et al. Pharmacokinetic, pharmacodynamic, and tolerability profiles of the dipeptidyl peptidase-4 inhibitor alogliptin: a randomized, double-blind, placebo-controlled, multiple-dose study in adult patients with type 2 diabetes. Clin Ther 2008; 30:499–512.
- DeFronzo RA, Fleck PR, Wilson CA, Mekki Q; on behalf of the Alogliptin Study 010 Group. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor alogliptin in patients with type 2 diabetes and inadequate glycemic control: a randomized, double-blind, placebo-controlled study. Diabetes Care 2008; 31:2315–2317.
- Triplitt CL, McGill JB, Porte D Jr, Conner CS. The changing landscape of type 2 diabetes: the role of incretin-based therapies in managed care outcomes. J Manag Care Pharm 2007; 13(9 suppl C):S2–S16.
- Garber AJ, Spann SJ. An overview of incretin clinical trials. J Fam Pract 2008; 57(9 suppl):S10–S18.
- Henry RR. Evolving concepts of type 2 diabetes management with oral medications: new approaches to an old disease. Curr Med Res Opin 2008; 24:2189–2202.
- Centers for Disease Control and Prevention. National diabetes fact sheet: general information and national estimates on diabetes in the United States, 2007. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/diabetes/pubs/pdf/ndfs_2007.pdf. Published 2008. Accessed September 21, 2009.
- Ong KL, Cheung BM, Wong LY, Wt NM, Tan KC, Lam KS. Prevalence, treatment, and control of diagnosed diabetes in the U.S. National Health and Nutrition Examination Survey 1999–2004. Ann Epidemiol 2008; 18:222–229.
- Sanders CL, Yesupriya AJ, Curtin LR. Analysis of population structure and stratification in NHANES III self-reported race/ethnicities. Centers for Disease Control and Prevention Web site. http://www.cdc.gov/genomics/events/file/print/10year/08_pop_struct_ab.pdf. Accessed September 21, 2009.
- Koro CE, Bowlin SJ, Bourgeois N, Fedder DO. Glycemic control from 1988 to 2000 among US adults diagnosed with type 2 diabetes: a preliminary report. Diabetes Care 2004; 27:17–20.
- Haffner SM, Lehto S, Rönnemaa T, Pyörälä K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229–234.
- Lloyd-Jones D, Adams R, Carnethon M, et al. Heart disease and stroke statistics—2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2009; 119:480–486.
- Stonehouse AH, Holcombe JH, Kendall DM. GLP-1 analogues, DPP-IV inhibitors and the metabolic syndrome. In: Fonseca V, ed. Therapeutic Strategies in Metabolic Syndrome. Oxford, UK: Atlas Medical Publishing Ltd; 2008: 137–157.
- Purnell JQ, Weyer C. Weight effect of current and experimental drugs for diabetes mellitus: from promotion to alleviation of obesity. Treat Endocrinol 2003; 2:33–47.
- Klonoff DC, Buse JB, Nielsen LL, et al. Exenatide effects on diabetes, obesity, cardiovascular risk factors and hepatic biomarkers in patients with type 2 diabetes treated for at least 3 years. Curr Med Res Opin 2008; 24:275–286.
- Barnett AH, Burger J, Johns D, et al. Tolerability and efficacy of exenatide and titrated insulin glargine in adult patients with type 2 diabetes previously uncontrolled with metformin or a sulfonylurea: a multinational, randomized, open-label, two-period, crossover noninferiority trial. Clin Ther 2007; 29:2333–2348.
- Glass LC, Qu Y, Lenox S, et al. Effects of exenatide versus insulin analogues on weight change in subjects with type 2 diabetes: a pooled post-hoc analysis. Curr Med Res Opin 2008; 24:639–644.
- Heine RJ, Van Gaal LF, Johns D, Mihm MJ, Widel MH, Brodows RG; for the GWAA Study Group. Exenatide versus insulin glargine in patients with suboptimally controlled type 2 diabetes: a randomized trial. Ann Intern Med 2005; 143:559–569.
- Nauck MA, Duran S, Kim D, et al. A comparison of twice-daily exenatide and biphasic insulin aspart in patients with type 2 diabetes who were suboptimally controlled with sulfonylurea and metformin: a non-inferiority study. Diabetologia 2007; 50:259–267.
- Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation 2008; 117:2340–2350.
- Nikolaidis LA, Mankad S, Sokos GG, et al. Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation 2004; 109:962–965.
KEY POINTS
- The incretin effect may be responsible for up to 70% of insulin secretion following oral glucose ingestion; reduction of the incretin effect contributes to T2DM pathophysiology.
- It is unknown whether incretin defects are a cause or consequence of T2DM.
- Incretin therapies effectively lower glucose with concomitant favorable effects on body weight. GLP-1 receptor agonists reduce weight, while DPP-4 inhibitors are weight neutral.