User login
Pimavanserin for psychosis in patients with Parkinson’s disease
Pimavanserin is a potent 5-HT2A inverse agonist and 5-HT2C inverse agonist, with 5-fold greater affinity for the 5-HT2A receptor.1 Although antagonists block agonist actions at the receptor site, inverse agonists reduce the level of baseline constitutive activity seen in many G protein-coupled receptors. This medication is FDA approved for treating hallucinations and delusions associated with Parkinson’s disease (PD) psychosis (Table 1).1
In the pivotal 6-week clinical trial, pimavanserin significantly reduced positive symptoms seen in PD patients with psychosis (effect size = 0.50), with no evident impairment of motor function.2 Only 2 adverse effects occurred in ≥5% of pimavanserin-treated patients and at ≥2 times the rate of placebo: peripheral edema (7% vs 3% for placebo) and confusion (6% vs 3% for placebo). There was a mean increase in the QTc of 7.3 milliseconds compared with placebo in the pivotal phase III study.
Clinical implications
Despite numerous developments in the pharmacotherapeutics of psychotic disorders, patients with psychosis related to PD previously responded in a robust manner to only 1 antipsychotic, low-dosage clozapine (mean effect size, 0.80),2 with numerous failed trials for other atypical antipsychotics, including quetiapine.3,4 The pathophysiology of psychosis in PD patients is not related to dopamine agonist treatment, but is caused by the accumulation of cortical Lewy body burden, which results in loss of serotonergic signaling from dorsal raphe neurons. The net effect is up-regulation of postsynaptic 5-HT2A receptors.5 Psychosis is the most common cause of nursing home placement among PD patients without dementia.6
Receptor blocking. Based on the finding that clozapine in low dosages acts at 5-HT2A receptors,7 pimavanserin was designed to be a potent 5-HT2A inverse agonist, with more than 5-fold higher selectivity over 5-HT2C receptors, and no appreciable affinity for other serotonergic, adrenergic, dopaminergic, muscarinic, or histaminergic receptors8 (Table 2). The concept that 5-HT2A receptor stimulation can cause psychosis with prominent visual hallucinations is known from studies of LSD and other hallucinogenic compounds whose activity is blocked by 5-HT2A antagonists.

As an agent devoid of dopamine D2 antagonism, pimavanserin carries no risk of exacerbating motor symptoms, which was commonly seen with most atypical antipsychotics studied for psychosis in PD patients, except for clozapine and quetiapine.3 Although quetiapine did not cause motor effects, it proved ineffective in multiple studies (n = 153), likely because of the near absence of potent 5-HT2A binding.4
Pimavanserin also lacks:
- the hematologic monitoring requirement of clozapine
- clozapine’s risks of sedation, orthostasis, and anticholinergic and metabolic adverse effects.
Pimavanserin is significantly more potent than other non-antipsychotic psychotropics at the 5-HT2Areceptor, including doxepin (26 nM), trazodone (36 nM), and mirtazapine (60 nM).
Use in psychosis associated with PD. Recommended dosage is 34 mg once daily without titration (with or without food), based on results from a phase III clinical trial2 (because of the FDA breakthrough therapy designation for this compound, only 1 phase III trial was required). Pimavanserin produced significant improvement on the PD-adapted Scale for the Assessment of Positive Symptoms (SAPS-PD), a 9-item instrument extracted from the larger SAPS used in schizophrenia research. Specifically, pimavanserin was effective for both the hallucinations and delusions components of the SAPS-PD.
Pharmacologic profile, adverse effects. Pimavanserin lacks affinity for receptors other than 5-HT2A and 5-HT2C, leading to an absence of significant anticholinergic effects, orthostasis, or sedation in clinical trials.2 In all short-term clinical trials, the only common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were peripheral edema (7% vs 2% placebo) and confusional state (6% vs 3% placebo).2 More than 300 patients have been treated for >6 months, >270 have been treated for at least 12 months, and >150 have been treated for at least 24 months with no adverse effects other than those seen in the short-term trials.1
There is a measurable impact on cardiac conduction seen in phase III data and in the thorough QT study. In the thorough QT study, 252 healthy participants received multiple dosages in a randomized, double-blind manner with positive controls.1 The maximum mean change from baseline was 13.5 milliseconds at dosages twice the recommended dosage, and the upper limit of the 90% CI was only slightly greater at 16.6 milliseconds. Subsequent kinetic analyses suggested concentration-dependent QTc interval prolongation in the therapeutic range, with a recommendation to halve the daily dosage in patients taking potent cytochrome P450 (CYP) 3A4 inhibitors.
In the 6-week, placebo-controlled effectiveness studies, mean increases in QTc interval were in the range of 5 to 8 milliseconds. There were sporadic reports of QTcF values ≥500 milliseconds, or changes from baseline QTc values ≥60 milliseconds in pimavanserin-treated participants, although the incidence generally was the same for pimavanserin and placebo groups. There were no reports of torsades de pointes or any differences from placebo in the incidence of adverse reactions associated with delayed ventricular repolarization.
How it works
The theory behind development of pimavanserin rests in the finding that low-dosage clozapine (6.25 to 50 mg/d) was effective for PD patients with psychosis (effect size 0.80).8 Although clozapine has high affinity for multiple sites, including histamine H1 receptors (Ki = 1.13 nM), α-1A and a α-2C adrenergic receptors (Ki = 1.62 nM and 6 nM, respectively), 5-HT2A receptors (Ki = 5.35 nM), and muscarinic M1 receptors (Ki = 6 nM), the hypothesized primary mechanism of clozapine’s effectiveness for PD psychosis at low dosages focused on the 5-HT2Areceptor. This idea was based on the knowledge that hallucinogens such as mescaline, psilocybin, and LSD are 5-HT2A agonists.9 This hallucinogenic activity can be blocked with 5-HT2A antagonists. Because of pimavanserin’s binding profile, the compound was studied as a treatment for psychosis in PD patients.
Pharmacokinetics
Pimavanserin demonstrates dose-proportional pharmacokinetics after a single oral dose as much as 7.5 times the recommended dosage. The pharmacokinetics of pimavanserin were similar in study participants (mean age, 72.4) and healthy controls, and a high-fat meal had no impact on the maximum blood levels (Cmax) or total drug exposure (area under the curve [AUC]).
The mean plasma half-lives for pimavanserin and its metabolite N-desmethyl-pimavanserin (AC-279) are 57 hours and 200 hours, respectively. Although the metabolite appears active in in vitro assays, it does not cross the blood-brain barrier to any appreciable extent, therefore contributing little to the clinical effect. The median time to maximum concentration (Tmax) of pimavanserin is 6 hours with a range of 4 to 24 hours, while the median Tmax of the primary metabolite AC-279 is 6 hours. The bioavailability of pimavanserin in an oral tablet or solution essentially is identical.
Pimavanserin is primarily metabolized via CYP3A4 to AC-279, and strong CYP3A4 inhibitors (eg, ketoconazole, itraconazole, clarithromycin, indinavir) increase pimavanserin Cmax by 1.5-fold, and AUC by 3-fold. In patients taking strong CYP3A4 inhibitors, the dosage of pimavanserin should be reduced by 50% to 17 mg/d. Conversely, patients on CYP3A4 inducers (eg, rifampin, carbamazepine, phenytoin) should be monitored for lack of efficacy; consider a dosage increase as necessary. Neither pimavanserin nor its metabolite, AC-279, are inhibitors or inducers of major CYP enzymes or drug transporters.
Efficacy in PD psychosis
Study 1. This 6-week, fixed dosage, double-blind, placebo-controlled trial was performed in adult PD patients age ≥40 with PD psychosis.2 Participants had to have (1) a PD diagnosis for at least 1 year and (2) psychotic symptoms that developed after diagnosis. Psychotic symptoms had to be present for at least 1 month, occurring at least weekly in the month before screening, and severe enough to warrant antipsychotic treatment. Baseline Mini-Mental State Examination score had to be ≥21 out of 30, with no evidence of delirium. Patients with dementia preceding or concurrent with the PD diagnosis were excluded. Antipsychotic treatments were not permitted during the trial.
After a 2-week nonpharmacotherapeutic lead-in phase that included a brief, daily psychosocial intervention by a caregiver, 199 patients who still met severity criteria were randomly allocated in a 1:1 manner to pimavanserin (34 mg of active drug, reported in the paper as 40 mg of pimavanserin tartrate) or matched placebo. Based on kinetic modeling and earlier clinical data, lower dosages (ie, 17 mg) were not explored, because they achieved only 50% of the steady state plasma levels thought to be required for efficacy.
The primary outcome was assessed by central, independent raters using the PD-adapted SAPS-PD. The efficacy analysis included 95 pimavanserin-treated individuals and 90 taking placebo. Baseline SAPS-PD scores were 14.7 ± 5.55 in the placebo group, and 15.9 ± 6.12 in the pimavanserin arm. Participants had a mean age of 72.4 and 94% white ethnicity across both cohorts; 42% of the placebo group and 33% of the pimavanserin group were female. Antipsychotic exposure in the 21 days prior to study entry were reported in 17% (n = 15) and 19% (n = 18) of the placebo and pimavanserin groups, respectively, with the most common agent being quetiapine (13 of 15, placebo, 16 of 18, pimavanserin). Approximately one-third of all participants were taking a cholinesterase inhibitor throughout the study.
Efficacy outcome. Pimavanserin was associated with a 5.79-point decrease in SAPS-PD scores compared with 2.73-point decrease for placebo (difference −3.06, 95% CI −4.91 to −1.20; P = .001). The effect size for this difference (Cohen’s d) was 0.50. The significant effect of pimavanserin vs placebo also was seen in separate analyses of the SAPS-PD subscore for hallucinations and delusions (effect size 0.50), and individually for hallucinations (effect size 0.45) and delusions (effect size 0.33). Separation from placebo appeared after the second week of pimavanserin treatment, and continued through the end of the study. There is unpublished data showing efficacy through week 10, and longer term, uncontrolled data consistent with sustained response. An exploratory analysis of caregiver burden demonstrated an effect size of 0.50.
Tolerability
The discontinuation rate because of adverse events for pimavanserin and placebo-treated patients was 10 patients in the pimavanserin group (4 due to psychotic symptoms within 10 days of starting the study drug) compared with 2 in the placebo group. There was no evidence of motor worsening in either group, demonstrated by the score on part II of the Unified Parkinson’s Disease Rating Scale (UPDRS) that captures self-reported activities of daily living, or on UPDRS part III (motor examination). Pimavanserin has no contraindications.
Unique clinical issues
Binding properties. Pimavanserin possesses potent 5-HT2A inverse agonist properties required to manage psychosis in PD patients, but lacks clozapine’s affinities for α-1 adrenergic, muscarinic, or histaminergic receptors that contribute to clozapine’s poor tolerability. Moreover, pimavanserin has no appreciable affinity for dopaminergic receptors, and therefore does not induce motor adverse effects.
Clozapine aside, all available atypical antipsychotics have proved ineffective for psychosis in PD patients, and most caused significant motor worsening.3 Although quetiapine does not cause motor effects, it has been shown to be ineffective for psychosis in PD patients in multiple trials.4
The effect size for clozapine response is large (0.80) in PD patients with psychosis, but tolerability issues and administrative burdens regarding patient and prescriber registration and routine hematological monitoring pose significant clinical barriers. Clozapine also lacks an FDA indication for this purpose, which may pose a hurdle to its use in certain treatment settings.
Why Rx? The reasons to prescribe pimavanserin for PD patients with psychosis likely include:
- absence of tolerability issues seen with the only other effective agent, clozapine
- lack of motor effects
- lack of administrative and monitoring burden related to clozapine prescribing
- only agent with FDA approval for hallucinations and delusions in PD patients with psychosis.
Dosing
The recommended dosage of pimavanserin is 34 mg/d administered as a single dose with or without food. There is no need for titration, and none was performed in the pivotal clinical trial. Given the long half-life (57 hours), steady state is not achieved until day 12, therefore initiation with a lower dosage might prolong the time to efficacy. There is no dosage adjustment required in patients with mild or moderate renal impairment, but pimavanserin treatment is not recommended in patients with severe renal impairment. Pimavanserin has not been evaluated in patients with hepatic impairment (using Child-Pugh criteria), and is not recommended for these patients.
Other key aspects of dosing to keep in mind.
- Because pimavanserin is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a strong CYP3A4 inhibitor; the recommended dosage is 17 mg/d when administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of pimavanserin with CYP3A4 inducers, patients should be monitored for lack of efficacy during concomitant use with a CYP3A4 inducer, and consideration given to a dosage increase.
Use in pregnancy and lactation. There are no data on the use of pimavanserin in pregnant women, but no developmental effects were seen when the drug was administered orally at 10 or 12 times the maximum recommended human dosage to rats or rabbits during organogenesis. Pimavanserin was not teratogenic in pregnant rats and rabbits. There is no information regarding the presence of pimavanserin in human breast milk.
Geriatric patients. No dosage adjustment is required for older patients. The study population in the pivotal trial was mean age 72.4 years.
Summing up
Before development of pimavanserin, clozapine was the only effective treatment for psychosis in PD patients. Despite clozapine’s robust effects across several trials, patients often were given ineffective medications, such as quetiapine, because of the administrative and tolerability barriers posed by clozapine use. Because psychosis is the most common cause of nursing home placement in non-demented PD patients, an agent with demonstrated efficacy and without the adverse effect profile of clozapine or monitoring requirements represents an enormous advance in the treatment of psychosis in PD patients.
1. Nuplazid [package insert]. San Diego, CA: Acadia Pharmaceuticals Inc.; 2016.
2. Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. [Erratum in Lancet. 2014;384(9937):28]. Lancet. 2014;383(9916):533-540.
3. Borek LL, Friedman JH. Treating psychosis in movement disorder patients: a review. Expert Opin Pharmacother. 2014;15(11):1553-1564.
4. Desmarais P, Massoud F, Filion J, et al. Quetiapine for psychosis in Parkinson disease and neurodegenerative parkinsonian disorders: a systematic review. J Geriatr Psychiatry Neurol. 2016;29(4):227-236.
5. Ballanger B, Strafella AP, van Eimeren T, et al. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch Neurol. 2010;67(4):416-421.
6. Ravina B, Marder K, Fernandez HH, et al. Diagnostic criteria for psychosis in Parkinson’s disease: report of an NINDS, NIMH work group. Mov Disord. 2007;22(8):1061-1068.
7. Nordström AL, Farde L, Nyberg S, et al. D1, D2, and 5-HT2 receptor occupancy in relation to clozapine serum concentration: a PET study of schizophrenic patients. Am J Psychiatry. 1995;152(10):1444-1449.
8. Hacksell U, Burstein ES, McFarland K, et al. On the discovery and development of pimavanserin: a novel drug candidate for Parkinson’s psychosis. Neurochem Res. 2014;39(10):2008-2017.
9. Moreno JL, Holloway T, Albizu L, et al. Metabotropic glutamate mGlu2 receptor is necessary for the pharmacological and behavioral effects induced by hallucinogenic 5-HT2A receptor agonists. Neurosci Lett. 2011;493(3):76-79.
Pimavanserin is a potent 5-HT2A inverse agonist and 5-HT2C inverse agonist, with 5-fold greater affinity for the 5-HT2A receptor.1 Although antagonists block agonist actions at the receptor site, inverse agonists reduce the level of baseline constitutive activity seen in many G protein-coupled receptors. This medication is FDA approved for treating hallucinations and delusions associated with Parkinson’s disease (PD) psychosis (Table 1).1
In the pivotal 6-week clinical trial, pimavanserin significantly reduced positive symptoms seen in PD patients with psychosis (effect size = 0.50), with no evident impairment of motor function.2 Only 2 adverse effects occurred in ≥5% of pimavanserin-treated patients and at ≥2 times the rate of placebo: peripheral edema (7% vs 3% for placebo) and confusion (6% vs 3% for placebo). There was a mean increase in the QTc of 7.3 milliseconds compared with placebo in the pivotal phase III study.
Clinical implications
Despite numerous developments in the pharmacotherapeutics of psychotic disorders, patients with psychosis related to PD previously responded in a robust manner to only 1 antipsychotic, low-dosage clozapine (mean effect size, 0.80),2 with numerous failed trials for other atypical antipsychotics, including quetiapine.3,4 The pathophysiology of psychosis in PD patients is not related to dopamine agonist treatment, but is caused by the accumulation of cortical Lewy body burden, which results in loss of serotonergic signaling from dorsal raphe neurons. The net effect is up-regulation of postsynaptic 5-HT2A receptors.5 Psychosis is the most common cause of nursing home placement among PD patients without dementia.6
Receptor blocking. Based on the finding that clozapine in low dosages acts at 5-HT2A receptors,7 pimavanserin was designed to be a potent 5-HT2A inverse agonist, with more than 5-fold higher selectivity over 5-HT2C receptors, and no appreciable affinity for other serotonergic, adrenergic, dopaminergic, muscarinic, or histaminergic receptors8 (Table 2). The concept that 5-HT2A receptor stimulation can cause psychosis with prominent visual hallucinations is known from studies of LSD and other hallucinogenic compounds whose activity is blocked by 5-HT2A antagonists.
As an agent devoid of dopamine D2 antagonism, pimavanserin carries no risk of exacerbating motor symptoms, which was commonly seen with most atypical antipsychotics studied for psychosis in PD patients, except for clozapine and quetiapine.3 Although quetiapine did not cause motor effects, it proved ineffective in multiple studies (n = 153), likely because of the near absence of potent 5-HT2A binding.4
Pimavanserin also lacks:
- the hematologic monitoring requirement of clozapine
- clozapine’s risks of sedation, orthostasis, and anticholinergic and metabolic adverse effects.
Pimavanserin is significantly more potent than other non-antipsychotic psychotropics at the 5-HT2Areceptor, including doxepin (26 nM), trazodone (36 nM), and mirtazapine (60 nM).
Use in psychosis associated with PD. Recommended dosage is 34 mg once daily without titration (with or without food), based on results from a phase III clinical trial2 (because of the FDA breakthrough therapy designation for this compound, only 1 phase III trial was required). Pimavanserin produced significant improvement on the PD-adapted Scale for the Assessment of Positive Symptoms (SAPS-PD), a 9-item instrument extracted from the larger SAPS used in schizophrenia research. Specifically, pimavanserin was effective for both the hallucinations and delusions components of the SAPS-PD.
Pharmacologic profile, adverse effects. Pimavanserin lacks affinity for receptors other than 5-HT2A and 5-HT2C, leading to an absence of significant anticholinergic effects, orthostasis, or sedation in clinical trials.2 In all short-term clinical trials, the only common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were peripheral edema (7% vs 2% placebo) and confusional state (6% vs 3% placebo).2 More than 300 patients have been treated for >6 months, >270 have been treated for at least 12 months, and >150 have been treated for at least 24 months with no adverse effects other than those seen in the short-term trials.1
There is a measurable impact on cardiac conduction seen in phase III data and in the thorough QT study. In the thorough QT study, 252 healthy participants received multiple dosages in a randomized, double-blind manner with positive controls.1 The maximum mean change from baseline was 13.5 milliseconds at dosages twice the recommended dosage, and the upper limit of the 90% CI was only slightly greater at 16.6 milliseconds. Subsequent kinetic analyses suggested concentration-dependent QTc interval prolongation in the therapeutic range, with a recommendation to halve the daily dosage in patients taking potent cytochrome P450 (CYP) 3A4 inhibitors.
In the 6-week, placebo-controlled effectiveness studies, mean increases in QTc interval were in the range of 5 to 8 milliseconds. There were sporadic reports of QTcF values ≥500 milliseconds, or changes from baseline QTc values ≥60 milliseconds in pimavanserin-treated participants, although the incidence generally was the same for pimavanserin and placebo groups. There were no reports of torsades de pointes or any differences from placebo in the incidence of adverse reactions associated with delayed ventricular repolarization.
How it works
The theory behind development of pimavanserin rests in the finding that low-dosage clozapine (6.25 to 50 mg/d) was effective for PD patients with psychosis (effect size 0.80).8 Although clozapine has high affinity for multiple sites, including histamine H1 receptors (Ki = 1.13 nM), α-1A and a α-2C adrenergic receptors (Ki = 1.62 nM and 6 nM, respectively), 5-HT2A receptors (Ki = 5.35 nM), and muscarinic M1 receptors (Ki = 6 nM), the hypothesized primary mechanism of clozapine’s effectiveness for PD psychosis at low dosages focused on the 5-HT2Areceptor. This idea was based on the knowledge that hallucinogens such as mescaline, psilocybin, and LSD are 5-HT2A agonists.9 This hallucinogenic activity can be blocked with 5-HT2A antagonists. Because of pimavanserin’s binding profile, the compound was studied as a treatment for psychosis in PD patients.
Pharmacokinetics
Pimavanserin demonstrates dose-proportional pharmacokinetics after a single oral dose as much as 7.5 times the recommended dosage. The pharmacokinetics of pimavanserin were similar in study participants (mean age, 72.4) and healthy controls, and a high-fat meal had no impact on the maximum blood levels (Cmax) or total drug exposure (area under the curve [AUC]).
The mean plasma half-lives for pimavanserin and its metabolite N-desmethyl-pimavanserin (AC-279) are 57 hours and 200 hours, respectively. Although the metabolite appears active in in vitro assays, it does not cross the blood-brain barrier to any appreciable extent, therefore contributing little to the clinical effect. The median time to maximum concentration (Tmax) of pimavanserin is 6 hours with a range of 4 to 24 hours, while the median Tmax of the primary metabolite AC-279 is 6 hours. The bioavailability of pimavanserin in an oral tablet or solution essentially is identical.
Pimavanserin is primarily metabolized via CYP3A4 to AC-279, and strong CYP3A4 inhibitors (eg, ketoconazole, itraconazole, clarithromycin, indinavir) increase pimavanserin Cmax by 1.5-fold, and AUC by 3-fold. In patients taking strong CYP3A4 inhibitors, the dosage of pimavanserin should be reduced by 50% to 17 mg/d. Conversely, patients on CYP3A4 inducers (eg, rifampin, carbamazepine, phenytoin) should be monitored for lack of efficacy; consider a dosage increase as necessary. Neither pimavanserin nor its metabolite, AC-279, are inhibitors or inducers of major CYP enzymes or drug transporters.
Efficacy in PD psychosis
Study 1. This 6-week, fixed dosage, double-blind, placebo-controlled trial was performed in adult PD patients age ≥40 with PD psychosis.2 Participants had to have (1) a PD diagnosis for at least 1 year and (2) psychotic symptoms that developed after diagnosis. Psychotic symptoms had to be present for at least 1 month, occurring at least weekly in the month before screening, and severe enough to warrant antipsychotic treatment. Baseline Mini-Mental State Examination score had to be ≥21 out of 30, with no evidence of delirium. Patients with dementia preceding or concurrent with the PD diagnosis were excluded. Antipsychotic treatments were not permitted during the trial.
After a 2-week nonpharmacotherapeutic lead-in phase that included a brief, daily psychosocial intervention by a caregiver, 199 patients who still met severity criteria were randomly allocated in a 1:1 manner to pimavanserin (34 mg of active drug, reported in the paper as 40 mg of pimavanserin tartrate) or matched placebo. Based on kinetic modeling and earlier clinical data, lower dosages (ie, 17 mg) were not explored, because they achieved only 50% of the steady state plasma levels thought to be required for efficacy.
The primary outcome was assessed by central, independent raters using the PD-adapted SAPS-PD. The efficacy analysis included 95 pimavanserin-treated individuals and 90 taking placebo. Baseline SAPS-PD scores were 14.7 ± 5.55 in the placebo group, and 15.9 ± 6.12 in the pimavanserin arm. Participants had a mean age of 72.4 and 94% white ethnicity across both cohorts; 42% of the placebo group and 33% of the pimavanserin group were female. Antipsychotic exposure in the 21 days prior to study entry were reported in 17% (n = 15) and 19% (n = 18) of the placebo and pimavanserin groups, respectively, with the most common agent being quetiapine (13 of 15, placebo, 16 of 18, pimavanserin). Approximately one-third of all participants were taking a cholinesterase inhibitor throughout the study.
Efficacy outcome. Pimavanserin was associated with a 5.79-point decrease in SAPS-PD scores compared with 2.73-point decrease for placebo (difference −3.06, 95% CI −4.91 to −1.20; P = .001). The effect size for this difference (Cohen’s d) was 0.50. The significant effect of pimavanserin vs placebo also was seen in separate analyses of the SAPS-PD subscore for hallucinations and delusions (effect size 0.50), and individually for hallucinations (effect size 0.45) and delusions (effect size 0.33). Separation from placebo appeared after the second week of pimavanserin treatment, and continued through the end of the study. There is unpublished data showing efficacy through week 10, and longer term, uncontrolled data consistent with sustained response. An exploratory analysis of caregiver burden demonstrated an effect size of 0.50.
Tolerability
The discontinuation rate because of adverse events for pimavanserin and placebo-treated patients was 10 patients in the pimavanserin group (4 due to psychotic symptoms within 10 days of starting the study drug) compared with 2 in the placebo group. There was no evidence of motor worsening in either group, demonstrated by the score on part II of the Unified Parkinson’s Disease Rating Scale (UPDRS) that captures self-reported activities of daily living, or on UPDRS part III (motor examination). Pimavanserin has no contraindications.
Unique clinical issues
Binding properties. Pimavanserin possesses potent 5-HT2A inverse agonist properties required to manage psychosis in PD patients, but lacks clozapine’s affinities for α-1 adrenergic, muscarinic, or histaminergic receptors that contribute to clozapine’s poor tolerability. Moreover, pimavanserin has no appreciable affinity for dopaminergic receptors, and therefore does not induce motor adverse effects.
Clozapine aside, all available atypical antipsychotics have proved ineffective for psychosis in PD patients, and most caused significant motor worsening.3 Although quetiapine does not cause motor effects, it has been shown to be ineffective for psychosis in PD patients in multiple trials.4
The effect size for clozapine response is large (0.80) in PD patients with psychosis, but tolerability issues and administrative burdens regarding patient and prescriber registration and routine hematological monitoring pose significant clinical barriers. Clozapine also lacks an FDA indication for this purpose, which may pose a hurdle to its use in certain treatment settings.
Why Rx? The reasons to prescribe pimavanserin for PD patients with psychosis likely include:
- absence of tolerability issues seen with the only other effective agent, clozapine
- lack of motor effects
- lack of administrative and monitoring burden related to clozapine prescribing
- only agent with FDA approval for hallucinations and delusions in PD patients with psychosis.
Dosing
The recommended dosage of pimavanserin is 34 mg/d administered as a single dose with or without food. There is no need for titration, and none was performed in the pivotal clinical trial. Given the long half-life (57 hours), steady state is not achieved until day 12, therefore initiation with a lower dosage might prolong the time to efficacy. There is no dosage adjustment required in patients with mild or moderate renal impairment, but pimavanserin treatment is not recommended in patients with severe renal impairment. Pimavanserin has not been evaluated in patients with hepatic impairment (using Child-Pugh criteria), and is not recommended for these patients.
Other key aspects of dosing to keep in mind.
- Because pimavanserin is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a strong CYP3A4 inhibitor; the recommended dosage is 17 mg/d when administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of pimavanserin with CYP3A4 inducers, patients should be monitored for lack of efficacy during concomitant use with a CYP3A4 inducer, and consideration given to a dosage increase.
Use in pregnancy and lactation. There are no data on the use of pimavanserin in pregnant women, but no developmental effects were seen when the drug was administered orally at 10 or 12 times the maximum recommended human dosage to rats or rabbits during organogenesis. Pimavanserin was not teratogenic in pregnant rats and rabbits. There is no information regarding the presence of pimavanserin in human breast milk.
Geriatric patients. No dosage adjustment is required for older patients. The study population in the pivotal trial was mean age 72.4 years.
Summing up
Before development of pimavanserin, clozapine was the only effective treatment for psychosis in PD patients. Despite clozapine’s robust effects across several trials, patients often were given ineffective medications, such as quetiapine, because of the administrative and tolerability barriers posed by clozapine use. Because psychosis is the most common cause of nursing home placement in non-demented PD patients, an agent with demonstrated efficacy and without the adverse effect profile of clozapine or monitoring requirements represents an enormous advance in the treatment of psychosis in PD patients.
Pimavanserin is a potent 5-HT2A inverse agonist and 5-HT2C inverse agonist, with 5-fold greater affinity for the 5-HT2A receptor.1 Although antagonists block agonist actions at the receptor site, inverse agonists reduce the level of baseline constitutive activity seen in many G protein-coupled receptors. This medication is FDA approved for treating hallucinations and delusions associated with Parkinson’s disease (PD) psychosis (Table 1).1
In the pivotal 6-week clinical trial, pimavanserin significantly reduced positive symptoms seen in PD patients with psychosis (effect size = 0.50), with no evident impairment of motor function.2 Only 2 adverse effects occurred in ≥5% of pimavanserin-treated patients and at ≥2 times the rate of placebo: peripheral edema (7% vs 3% for placebo) and confusion (6% vs 3% for placebo). There was a mean increase in the QTc of 7.3 milliseconds compared with placebo in the pivotal phase III study.
Clinical implications
Despite numerous developments in the pharmacotherapeutics of psychotic disorders, patients with psychosis related to PD previously responded in a robust manner to only 1 antipsychotic, low-dosage clozapine (mean effect size, 0.80),2 with numerous failed trials for other atypical antipsychotics, including quetiapine.3,4 The pathophysiology of psychosis in PD patients is not related to dopamine agonist treatment, but is caused by the accumulation of cortical Lewy body burden, which results in loss of serotonergic signaling from dorsal raphe neurons. The net effect is up-regulation of postsynaptic 5-HT2A receptors.5 Psychosis is the most common cause of nursing home placement among PD patients without dementia.6
Receptor blocking. Based on the finding that clozapine in low dosages acts at 5-HT2A receptors,7 pimavanserin was designed to be a potent 5-HT2A inverse agonist, with more than 5-fold higher selectivity over 5-HT2C receptors, and no appreciable affinity for other serotonergic, adrenergic, dopaminergic, muscarinic, or histaminergic receptors8 (Table 2). The concept that 5-HT2A receptor stimulation can cause psychosis with prominent visual hallucinations is known from studies of LSD and other hallucinogenic compounds whose activity is blocked by 5-HT2A antagonists.
As an agent devoid of dopamine D2 antagonism, pimavanserin carries no risk of exacerbating motor symptoms, which was commonly seen with most atypical antipsychotics studied for psychosis in PD patients, except for clozapine and quetiapine.3 Although quetiapine did not cause motor effects, it proved ineffective in multiple studies (n = 153), likely because of the near absence of potent 5-HT2A binding.4
Pimavanserin also lacks:
- the hematologic monitoring requirement of clozapine
- clozapine’s risks of sedation, orthostasis, and anticholinergic and metabolic adverse effects.
Pimavanserin is significantly more potent than other non-antipsychotic psychotropics at the 5-HT2Areceptor, including doxepin (26 nM), trazodone (36 nM), and mirtazapine (60 nM).
Use in psychosis associated with PD. Recommended dosage is 34 mg once daily without titration (with or without food), based on results from a phase III clinical trial2 (because of the FDA breakthrough therapy designation for this compound, only 1 phase III trial was required). Pimavanserin produced significant improvement on the PD-adapted Scale for the Assessment of Positive Symptoms (SAPS-PD), a 9-item instrument extracted from the larger SAPS used in schizophrenia research. Specifically, pimavanserin was effective for both the hallucinations and delusions components of the SAPS-PD.
Pharmacologic profile, adverse effects. Pimavanserin lacks affinity for receptors other than 5-HT2A and 5-HT2C, leading to an absence of significant anticholinergic effects, orthostasis, or sedation in clinical trials.2 In all short-term clinical trials, the only common adverse reactions (incidence ≥5% and at least twice the rate of placebo) were peripheral edema (7% vs 2% placebo) and confusional state (6% vs 3% placebo).2 More than 300 patients have been treated for >6 months, >270 have been treated for at least 12 months, and >150 have been treated for at least 24 months with no adverse effects other than those seen in the short-term trials.1
There is a measurable impact on cardiac conduction seen in phase III data and in the thorough QT study. In the thorough QT study, 252 healthy participants received multiple dosages in a randomized, double-blind manner with positive controls.1 The maximum mean change from baseline was 13.5 milliseconds at dosages twice the recommended dosage, and the upper limit of the 90% CI was only slightly greater at 16.6 milliseconds. Subsequent kinetic analyses suggested concentration-dependent QTc interval prolongation in the therapeutic range, with a recommendation to halve the daily dosage in patients taking potent cytochrome P450 (CYP) 3A4 inhibitors.
In the 6-week, placebo-controlled effectiveness studies, mean increases in QTc interval were in the range of 5 to 8 milliseconds. There were sporadic reports of QTcF values ≥500 milliseconds, or changes from baseline QTc values ≥60 milliseconds in pimavanserin-treated participants, although the incidence generally was the same for pimavanserin and placebo groups. There were no reports of torsades de pointes or any differences from placebo in the incidence of adverse reactions associated with delayed ventricular repolarization.
How it works
The theory behind development of pimavanserin rests in the finding that low-dosage clozapine (6.25 to 50 mg/d) was effective for PD patients with psychosis (effect size 0.80).8 Although clozapine has high affinity for multiple sites, including histamine H1 receptors (Ki = 1.13 nM), α-1A and a α-2C adrenergic receptors (Ki = 1.62 nM and 6 nM, respectively), 5-HT2A receptors (Ki = 5.35 nM), and muscarinic M1 receptors (Ki = 6 nM), the hypothesized primary mechanism of clozapine’s effectiveness for PD psychosis at low dosages focused on the 5-HT2Areceptor. This idea was based on the knowledge that hallucinogens such as mescaline, psilocybin, and LSD are 5-HT2A agonists.9 This hallucinogenic activity can be blocked with 5-HT2A antagonists. Because of pimavanserin’s binding profile, the compound was studied as a treatment for psychosis in PD patients.
Pharmacokinetics
Pimavanserin demonstrates dose-proportional pharmacokinetics after a single oral dose as much as 7.5 times the recommended dosage. The pharmacokinetics of pimavanserin were similar in study participants (mean age, 72.4) and healthy controls, and a high-fat meal had no impact on the maximum blood levels (Cmax) or total drug exposure (area under the curve [AUC]).
The mean plasma half-lives for pimavanserin and its metabolite N-desmethyl-pimavanserin (AC-279) are 57 hours and 200 hours, respectively. Although the metabolite appears active in in vitro assays, it does not cross the blood-brain barrier to any appreciable extent, therefore contributing little to the clinical effect. The median time to maximum concentration (Tmax) of pimavanserin is 6 hours with a range of 4 to 24 hours, while the median Tmax of the primary metabolite AC-279 is 6 hours. The bioavailability of pimavanserin in an oral tablet or solution essentially is identical.
Pimavanserin is primarily metabolized via CYP3A4 to AC-279, and strong CYP3A4 inhibitors (eg, ketoconazole, itraconazole, clarithromycin, indinavir) increase pimavanserin Cmax by 1.5-fold, and AUC by 3-fold. In patients taking strong CYP3A4 inhibitors, the dosage of pimavanserin should be reduced by 50% to 17 mg/d. Conversely, patients on CYP3A4 inducers (eg, rifampin, carbamazepine, phenytoin) should be monitored for lack of efficacy; consider a dosage increase as necessary. Neither pimavanserin nor its metabolite, AC-279, are inhibitors or inducers of major CYP enzymes or drug transporters.
Efficacy in PD psychosis
Study 1. This 6-week, fixed dosage, double-blind, placebo-controlled trial was performed in adult PD patients age ≥40 with PD psychosis.2 Participants had to have (1) a PD diagnosis for at least 1 year and (2) psychotic symptoms that developed after diagnosis. Psychotic symptoms had to be present for at least 1 month, occurring at least weekly in the month before screening, and severe enough to warrant antipsychotic treatment. Baseline Mini-Mental State Examination score had to be ≥21 out of 30, with no evidence of delirium. Patients with dementia preceding or concurrent with the PD diagnosis were excluded. Antipsychotic treatments were not permitted during the trial.
After a 2-week nonpharmacotherapeutic lead-in phase that included a brief, daily psychosocial intervention by a caregiver, 199 patients who still met severity criteria were randomly allocated in a 1:1 manner to pimavanserin (34 mg of active drug, reported in the paper as 40 mg of pimavanserin tartrate) or matched placebo. Based on kinetic modeling and earlier clinical data, lower dosages (ie, 17 mg) were not explored, because they achieved only 50% of the steady state plasma levels thought to be required for efficacy.
The primary outcome was assessed by central, independent raters using the PD-adapted SAPS-PD. The efficacy analysis included 95 pimavanserin-treated individuals and 90 taking placebo. Baseline SAPS-PD scores were 14.7 ± 5.55 in the placebo group, and 15.9 ± 6.12 in the pimavanserin arm. Participants had a mean age of 72.4 and 94% white ethnicity across both cohorts; 42% of the placebo group and 33% of the pimavanserin group were female. Antipsychotic exposure in the 21 days prior to study entry were reported in 17% (n = 15) and 19% (n = 18) of the placebo and pimavanserin groups, respectively, with the most common agent being quetiapine (13 of 15, placebo, 16 of 18, pimavanserin). Approximately one-third of all participants were taking a cholinesterase inhibitor throughout the study.
Efficacy outcome. Pimavanserin was associated with a 5.79-point decrease in SAPS-PD scores compared with 2.73-point decrease for placebo (difference −3.06, 95% CI −4.91 to −1.20; P = .001). The effect size for this difference (Cohen’s d) was 0.50. The significant effect of pimavanserin vs placebo also was seen in separate analyses of the SAPS-PD subscore for hallucinations and delusions (effect size 0.50), and individually for hallucinations (effect size 0.45) and delusions (effect size 0.33). Separation from placebo appeared after the second week of pimavanserin treatment, and continued through the end of the study. There is unpublished data showing efficacy through week 10, and longer term, uncontrolled data consistent with sustained response. An exploratory analysis of caregiver burden demonstrated an effect size of 0.50.
Tolerability
The discontinuation rate because of adverse events for pimavanserin and placebo-treated patients was 10 patients in the pimavanserin group (4 due to psychotic symptoms within 10 days of starting the study drug) compared with 2 in the placebo group. There was no evidence of motor worsening in either group, demonstrated by the score on part II of the Unified Parkinson’s Disease Rating Scale (UPDRS) that captures self-reported activities of daily living, or on UPDRS part III (motor examination). Pimavanserin has no contraindications.
Unique clinical issues
Binding properties. Pimavanserin possesses potent 5-HT2A inverse agonist properties required to manage psychosis in PD patients, but lacks clozapine’s affinities for α-1 adrenergic, muscarinic, or histaminergic receptors that contribute to clozapine’s poor tolerability. Moreover, pimavanserin has no appreciable affinity for dopaminergic receptors, and therefore does not induce motor adverse effects.
Clozapine aside, all available atypical antipsychotics have proved ineffective for psychosis in PD patients, and most caused significant motor worsening.3 Although quetiapine does not cause motor effects, it has been shown to be ineffective for psychosis in PD patients in multiple trials.4
The effect size for clozapine response is large (0.80) in PD patients with psychosis, but tolerability issues and administrative burdens regarding patient and prescriber registration and routine hematological monitoring pose significant clinical barriers. Clozapine also lacks an FDA indication for this purpose, which may pose a hurdle to its use in certain treatment settings.
Why Rx? The reasons to prescribe pimavanserin for PD patients with psychosis likely include:
- absence of tolerability issues seen with the only other effective agent, clozapine
- lack of motor effects
- lack of administrative and monitoring burden related to clozapine prescribing
- only agent with FDA approval for hallucinations and delusions in PD patients with psychosis.
Dosing
The recommended dosage of pimavanserin is 34 mg/d administered as a single dose with or without food. There is no need for titration, and none was performed in the pivotal clinical trial. Given the long half-life (57 hours), steady state is not achieved until day 12, therefore initiation with a lower dosage might prolong the time to efficacy. There is no dosage adjustment required in patients with mild or moderate renal impairment, but pimavanserin treatment is not recommended in patients with severe renal impairment. Pimavanserin has not been evaluated in patients with hepatic impairment (using Child-Pugh criteria), and is not recommended for these patients.
Other key aspects of dosing to keep in mind.
- Because pimavanserin is metabolized primarily by CYP3A4, dosage adjustment is required in the presence of a strong CYP3A4 inhibitor; the recommended dosage is 17 mg/d when administered concomitantly with a strong CYP3A4 inhibitor.
- Because data are not available regarding concomitant use of pimavanserin with CYP3A4 inducers, patients should be monitored for lack of efficacy during concomitant use with a CYP3A4 inducer, and consideration given to a dosage increase.
Use in pregnancy and lactation. There are no data on the use of pimavanserin in pregnant women, but no developmental effects were seen when the drug was administered orally at 10 or 12 times the maximum recommended human dosage to rats or rabbits during organogenesis. Pimavanserin was not teratogenic in pregnant rats and rabbits. There is no information regarding the presence of pimavanserin in human breast milk.
Geriatric patients. No dosage adjustment is required for older patients. The study population in the pivotal trial was mean age 72.4 years.
Summing up
Before development of pimavanserin, clozapine was the only effective treatment for psychosis in PD patients. Despite clozapine’s robust effects across several trials, patients often were given ineffective medications, such as quetiapine, because of the administrative and tolerability barriers posed by clozapine use. Because psychosis is the most common cause of nursing home placement in non-demented PD patients, an agent with demonstrated efficacy and without the adverse effect profile of clozapine or monitoring requirements represents an enormous advance in the treatment of psychosis in PD patients.
1. Nuplazid [package insert]. San Diego, CA: Acadia Pharmaceuticals Inc.; 2016.
2. Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. [Erratum in Lancet. 2014;384(9937):28]. Lancet. 2014;383(9916):533-540.
3. Borek LL, Friedman JH. Treating psychosis in movement disorder patients: a review. Expert Opin Pharmacother. 2014;15(11):1553-1564.
4. Desmarais P, Massoud F, Filion J, et al. Quetiapine for psychosis in Parkinson disease and neurodegenerative parkinsonian disorders: a systematic review. J Geriatr Psychiatry Neurol. 2016;29(4):227-236.
5. Ballanger B, Strafella AP, van Eimeren T, et al. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch Neurol. 2010;67(4):416-421.
6. Ravina B, Marder K, Fernandez HH, et al. Diagnostic criteria for psychosis in Parkinson’s disease: report of an NINDS, NIMH work group. Mov Disord. 2007;22(8):1061-1068.
7. Nordström AL, Farde L, Nyberg S, et al. D1, D2, and 5-HT2 receptor occupancy in relation to clozapine serum concentration: a PET study of schizophrenic patients. Am J Psychiatry. 1995;152(10):1444-1449.
8. Hacksell U, Burstein ES, McFarland K, et al. On the discovery and development of pimavanserin: a novel drug candidate for Parkinson’s psychosis. Neurochem Res. 2014;39(10):2008-2017.
9. Moreno JL, Holloway T, Albizu L, et al. Metabotropic glutamate mGlu2 receptor is necessary for the pharmacological and behavioral effects induced by hallucinogenic 5-HT2A receptor agonists. Neurosci Lett. 2011;493(3):76-79.
1. Nuplazid [package insert]. San Diego, CA: Acadia Pharmaceuticals Inc.; 2016.
2. Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. [Erratum in Lancet. 2014;384(9937):28]. Lancet. 2014;383(9916):533-540.
3. Borek LL, Friedman JH. Treating psychosis in movement disorder patients: a review. Expert Opin Pharmacother. 2014;15(11):1553-1564.
4. Desmarais P, Massoud F, Filion J, et al. Quetiapine for psychosis in Parkinson disease and neurodegenerative parkinsonian disorders: a systematic review. J Geriatr Psychiatry Neurol. 2016;29(4):227-236.
5. Ballanger B, Strafella AP, van Eimeren T, et al. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch Neurol. 2010;67(4):416-421.
6. Ravina B, Marder K, Fernandez HH, et al. Diagnostic criteria for psychosis in Parkinson’s disease: report of an NINDS, NIMH work group. Mov Disord. 2007;22(8):1061-1068.
7. Nordström AL, Farde L, Nyberg S, et al. D1, D2, and 5-HT2 receptor occupancy in relation to clozapine serum concentration: a PET study of schizophrenic patients. Am J Psychiatry. 1995;152(10):1444-1449.
8. Hacksell U, Burstein ES, McFarland K, et al. On the discovery and development of pimavanserin: a novel drug candidate for Parkinson’s psychosis. Neurochem Res. 2014;39(10):2008-2017.
9. Moreno JL, Holloway T, Albizu L, et al. Metabotropic glutamate mGlu2 receptor is necessary for the pharmacological and behavioral effects induced by hallucinogenic 5-HT2A receptor agonists. Neurosci Lett. 2011;493(3):76-79.
Ketamine for treatment-resistant depression

Is monitoring of plasma antipsychotic levels useful?
Although there has been success in defining the minimum therapeutic response threshold for certain antipsychotics—for example, clozapine (350 to 450 ng/mL), haloperidol (3 to 5 ng/mL), and fluphenazine (0.8 ng/mL)—one aspect of antipsychotic plasma levels not widely discussed is their value as a marker of adherence.
Many schizophrenia patients achieve an optimal response to agents for which there is no depot formulation. For them, maintenance of symptom control depends wholly on oral medication adherence.1 Regrettably, nonadherence with oral antipsychotic treatment is prevalent among patients with schizophrenia; yet, in routine clinical practice, the extent of nonadherence rarely is measured.
Studies have been able to quantify oral medication nonadherence using monitoring devices, such as the Medication Event Monitoring System (MEMS) that electronically records opening of the medication container and strongly correlates with pill count. Although patients knew they were participating in a trial using MEMS technology, only 48% were able to take their medication at least 80% of the time in a 4-week study,2 and only 43% met the 70% adherence threshold in a 6-month trial.3
Clinicians, patients: Unreliable indicators of adherence
Neither clinician rating nor patient self-reporting is a reliable predictor of adherence with an oral medication regimen. In the 6-month adherence trial,3 clinicians estimated that 95% of their patients met the 70% adherence threshold (the actual percentage was 43%); in a 12-week study, clinician ratings correlated weakly with adherence (r = 0.32; P = .001), but patient self-reporting showed no significant correlation (r = 0.18; P = .08) with pill count.4
Clinicians underestimate not only the extent of nonadherence but also the impact that even a brief period of modest nonadherence has on the risk of relapse. In an 18-month prospective study of patients who recently had been given a diagnosis of schizophrenia, and in whom clinician and patient reports were supplemented with a pill count every 1 to 2 weeks and plasma antipsychotic levels every 4 weeks, any period of at least mild nonadherence was significantly predictive of symptom exacerbation or relapse (hazard ratio [HR], 3.4; 95% CI, 1.4–8.4; P < .002).1 Moreover, 50% to 75% adherence for ≥ 2 consecutive weeks increased the HR to 5.8, and moderate nonadherence (<50% for 2.0 to 3.9 weeks) to an HR of 28.5.
There might be a better method already available
Given the poor correlation between a clinician’s judgment and a patient’s actual pill-taking, it is clear that more accurate methods of tracking adherence must be devised. Because MEMS technology is not widely available, and because pill counts require a home visit or a cooperative patient who brings medications to office visits, plasma antipsychotic monitoring potentially is an appealing method of tracking adherence.
Correlation between the plasma antipsychotic level and relapse is not consistently seen in the literature,4 but plasma levels obtained during periods of clinical stability offer the opportunity to define, for the individual patient, a range of drug exposure that is associated with clinical response. The ideal plasma level baseline is obtained at steady state during a presumed period of observed adherence, such as during a hospital stay. Although patients can be nonadherent in the hospital, this setting offers the best proxy for an acute clinical response to a given plasma level. The alternative is to obtain several plasma levels during a period of outpatient clinical stability.
Clinicians must be mindful that changes in the plasma antipsychotic level after hospital discharge might not reflect poor adherence; environmental factors (eg, exposure to cytochrome P450 or P-glycoprotein inducers) can have a significant impact on results. Resumption of smoking is a classic example, and routinely is associated with a 50% reduction in plasma clozapine levels.5
There also is expected variability in plasma antipsychotic levels based on (1) the timing of prior doses with regard to trough levels, and (2) the effects of an occasional missed dose. Nevertheless, in a sample of adherent clozapine-treated patients, investigators found that 98% of patients had a coefficient of variability (CV) of 30% for sequential plasma concentrations (mean CV, 14%).6
Clinicians should inform patients that the plasma antipsychotic level is a tool for helping track treatment engagement before relapse—the same way metabolic monitoring helps track abnormalities that can be associated with future cardiovascular events. (Clinicians also must be charitable with their patients when discussing a significant drop in the plasma antipsychotic level [eg, >30%], acknowledging that many patients often miss doses.)
Using the patient’s input about specific difficulties with a medication regimen, clinicians should strive to find ways to improve oral medication adherence. In many cases, the clinician can assist through medication simplification, consolidation of multiple daily doses, provision of pill boxes, and discussions about long-acting injectable (LAI) antipsychotics.
In short, plasma antipsychotic levels offer an opportunity to have a richer, evidence-based discussion about adherence, beyond the trite, ineffective question “Did you take your medication?” Use of an objective measure can (1) serve as a benchmark for the patient (eg, “You seemed to do better when your clozapine level was above 400 ng/mL”), and (2) remind clinicians of the variable adherence inherent with oral medication regimens.
A note about long-acting injectable antipsychotics
Because nonadherence is seen throughout the course of schizophrenia, discussion of LAI therapy should not be limited to patients with chronic illness. Results of recent naturalistic7 and randomized studies8 show significant reduction in the rate of psychotic relapse and improved symptom control8 among first-episode patients who are taking an LAI. Moreover, compelling data show that most first-episode patients who are taking an oral antipsychotic will accept a recommendation for treatment with an LAI.9
Summing up: 2 Tools for achieving therapeutic success
Monitoring plasma levels of antipsychotics offers a method for quantifying the problem of nonadherence. For many patients, an LAI antipsychotic provides a solution to nonadherence, and the increasing variety of LAI preparations means more options with which to match individual patients.
Clinicians have a limited amount of time to spend with patients in the office, but time spent discussing LAIs is an investment in the patient’s stability and functional outcome. Minutes once spent managing nonadherence can be devoted to understanding the patient’s aspirations and to developing strategies to achieve those goals.
In the end, isn’t that what we’d rather be talking about with our patients?
1. Subotnik KL, Nuechterlein KH, Ventura J, et al. Risperidone nonadherence and return of positive symptoms in the early course of schizophrenia. Am J Psychiatry. 2011;168(3):286-292.
2. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
3. Byerly MJ, Thompson A, Carmody T, et al. Validity of electronically monitored medication adherence and conventional adherence measures in schizophrenia. Psychiatr Serv. 2007;58(6):844-847.
4. Velligan DI, Wang M, Diamond P, et al. Relationships among subjective and objective measures of adherence to oral antipsychotic medications. Psychiatr Serv. 2007;58(9):1187-1192.
5. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21(6):569-574.
6. Dugan DJ, Ereshefsky L, Toney GB, et al. Dose and interval adherence among stabilized clozapine-treated patients measured by medication event monitoring. Presented at: Meeting of the New Clinical Drug Evaluation Unit; May 30-June 2, 2000; Boca Raton, FL.
7. Tiihonen J, Haukka J, Taylor M, et al. A nationwide cohort study of oral and depot antipsychotics after first hospitalization for schizophrenia. [Erratum in Am J Psychiatry. 2012;169(2):223]. Am J Psychiatry. 2011;168(6):603-609.
8. Subotnik KL, Casaus LR, Ventura J, et al. Long-acting injectable risperidone for relapse prevention and control of breakthrough symptoms after a recent first episode of schizophrenia: a randomized clinical trial. JAMA Psychiatry. 2015;72(8):822-829.
9. Weiden PJ, Schooler NR, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406.
Although there has been success in defining the minimum therapeutic response threshold for certain antipsychotics—for example, clozapine (350 to 450 ng/mL), haloperidol (3 to 5 ng/mL), and fluphenazine (0.8 ng/mL)—one aspect of antipsychotic plasma levels not widely discussed is their value as a marker of adherence.
Many schizophrenia patients achieve an optimal response to agents for which there is no depot formulation. For them, maintenance of symptom control depends wholly on oral medication adherence.1 Regrettably, nonadherence with oral antipsychotic treatment is prevalent among patients with schizophrenia; yet, in routine clinical practice, the extent of nonadherence rarely is measured.
Studies have been able to quantify oral medication nonadherence using monitoring devices, such as the Medication Event Monitoring System (MEMS) that electronically records opening of the medication container and strongly correlates with pill count. Although patients knew they were participating in a trial using MEMS technology, only 48% were able to take their medication at least 80% of the time in a 4-week study,2 and only 43% met the 70% adherence threshold in a 6-month trial.3
Clinicians, patients: Unreliable indicators of adherence
Neither clinician rating nor patient self-reporting is a reliable predictor of adherence with an oral medication regimen. In the 6-month adherence trial,3 clinicians estimated that 95% of their patients met the 70% adherence threshold (the actual percentage was 43%); in a 12-week study, clinician ratings correlated weakly with adherence (r = 0.32; P = .001), but patient self-reporting showed no significant correlation (r = 0.18; P = .08) with pill count.4
Clinicians underestimate not only the extent of nonadherence but also the impact that even a brief period of modest nonadherence has on the risk of relapse. In an 18-month prospective study of patients who recently had been given a diagnosis of schizophrenia, and in whom clinician and patient reports were supplemented with a pill count every 1 to 2 weeks and plasma antipsychotic levels every 4 weeks, any period of at least mild nonadherence was significantly predictive of symptom exacerbation or relapse (hazard ratio [HR], 3.4; 95% CI, 1.4–8.4; P < .002).1 Moreover, 50% to 75% adherence for ≥ 2 consecutive weeks increased the HR to 5.8, and moderate nonadherence (<50% for 2.0 to 3.9 weeks) to an HR of 28.5.
There might be a better method already available
Given the poor correlation between a clinician’s judgment and a patient’s actual pill-taking, it is clear that more accurate methods of tracking adherence must be devised. Because MEMS technology is not widely available, and because pill counts require a home visit or a cooperative patient who brings medications to office visits, plasma antipsychotic monitoring potentially is an appealing method of tracking adherence.
Correlation between the plasma antipsychotic level and relapse is not consistently seen in the literature,4 but plasma levels obtained during periods of clinical stability offer the opportunity to define, for the individual patient, a range of drug exposure that is associated with clinical response. The ideal plasma level baseline is obtained at steady state during a presumed period of observed adherence, such as during a hospital stay. Although patients can be nonadherent in the hospital, this setting offers the best proxy for an acute clinical response to a given plasma level. The alternative is to obtain several plasma levels during a period of outpatient clinical stability.
Clinicians must be mindful that changes in the plasma antipsychotic level after hospital discharge might not reflect poor adherence; environmental factors (eg, exposure to cytochrome P450 or P-glycoprotein inducers) can have a significant impact on results. Resumption of smoking is a classic example, and routinely is associated with a 50% reduction in plasma clozapine levels.5
There also is expected variability in plasma antipsychotic levels based on (1) the timing of prior doses with regard to trough levels, and (2) the effects of an occasional missed dose. Nevertheless, in a sample of adherent clozapine-treated patients, investigators found that 98% of patients had a coefficient of variability (CV) of 30% for sequential plasma concentrations (mean CV, 14%).6
Clinicians should inform patients that the plasma antipsychotic level is a tool for helping track treatment engagement before relapse—the same way metabolic monitoring helps track abnormalities that can be associated with future cardiovascular events. (Clinicians also must be charitable with their patients when discussing a significant drop in the plasma antipsychotic level [eg, >30%], acknowledging that many patients often miss doses.)
Using the patient’s input about specific difficulties with a medication regimen, clinicians should strive to find ways to improve oral medication adherence. In many cases, the clinician can assist through medication simplification, consolidation of multiple daily doses, provision of pill boxes, and discussions about long-acting injectable (LAI) antipsychotics.
In short, plasma antipsychotic levels offer an opportunity to have a richer, evidence-based discussion about adherence, beyond the trite, ineffective question “Did you take your medication?” Use of an objective measure can (1) serve as a benchmark for the patient (eg, “You seemed to do better when your clozapine level was above 400 ng/mL”), and (2) remind clinicians of the variable adherence inherent with oral medication regimens.
A note about long-acting injectable antipsychotics
Because nonadherence is seen throughout the course of schizophrenia, discussion of LAI therapy should not be limited to patients with chronic illness. Results of recent naturalistic7 and randomized studies8 show significant reduction in the rate of psychotic relapse and improved symptom control8 among first-episode patients who are taking an LAI. Moreover, compelling data show that most first-episode patients who are taking an oral antipsychotic will accept a recommendation for treatment with an LAI.9
Summing up: 2 Tools for achieving therapeutic success
Monitoring plasma levels of antipsychotics offers a method for quantifying the problem of nonadherence. For many patients, an LAI antipsychotic provides a solution to nonadherence, and the increasing variety of LAI preparations means more options with which to match individual patients.
Clinicians have a limited amount of time to spend with patients in the office, but time spent discussing LAIs is an investment in the patient’s stability and functional outcome. Minutes once spent managing nonadherence can be devoted to understanding the patient’s aspirations and to developing strategies to achieve those goals.
In the end, isn’t that what we’d rather be talking about with our patients?
Although there has been success in defining the minimum therapeutic response threshold for certain antipsychotics—for example, clozapine (350 to 450 ng/mL), haloperidol (3 to 5 ng/mL), and fluphenazine (0.8 ng/mL)—one aspect of antipsychotic plasma levels not widely discussed is their value as a marker of adherence.
Many schizophrenia patients achieve an optimal response to agents for which there is no depot formulation. For them, maintenance of symptom control depends wholly on oral medication adherence.1 Regrettably, nonadherence with oral antipsychotic treatment is prevalent among patients with schizophrenia; yet, in routine clinical practice, the extent of nonadherence rarely is measured.
Studies have been able to quantify oral medication nonadherence using monitoring devices, such as the Medication Event Monitoring System (MEMS) that electronically records opening of the medication container and strongly correlates with pill count. Although patients knew they were participating in a trial using MEMS technology, only 48% were able to take their medication at least 80% of the time in a 4-week study,2 and only 43% met the 70% adherence threshold in a 6-month trial.3
Clinicians, patients: Unreliable indicators of adherence
Neither clinician rating nor patient self-reporting is a reliable predictor of adherence with an oral medication regimen. In the 6-month adherence trial,3 clinicians estimated that 95% of their patients met the 70% adherence threshold (the actual percentage was 43%); in a 12-week study, clinician ratings correlated weakly with adherence (r = 0.32; P = .001), but patient self-reporting showed no significant correlation (r = 0.18; P = .08) with pill count.4
Clinicians underestimate not only the extent of nonadherence but also the impact that even a brief period of modest nonadherence has on the risk of relapse. In an 18-month prospective study of patients who recently had been given a diagnosis of schizophrenia, and in whom clinician and patient reports were supplemented with a pill count every 1 to 2 weeks and plasma antipsychotic levels every 4 weeks, any period of at least mild nonadherence was significantly predictive of symptom exacerbation or relapse (hazard ratio [HR], 3.4; 95% CI, 1.4–8.4; P < .002).1 Moreover, 50% to 75% adherence for ≥ 2 consecutive weeks increased the HR to 5.8, and moderate nonadherence (<50% for 2.0 to 3.9 weeks) to an HR of 28.5.
There might be a better method already available
Given the poor correlation between a clinician’s judgment and a patient’s actual pill-taking, it is clear that more accurate methods of tracking adherence must be devised. Because MEMS technology is not widely available, and because pill counts require a home visit or a cooperative patient who brings medications to office visits, plasma antipsychotic monitoring potentially is an appealing method of tracking adherence.
Correlation between the plasma antipsychotic level and relapse is not consistently seen in the literature,4 but plasma levels obtained during periods of clinical stability offer the opportunity to define, for the individual patient, a range of drug exposure that is associated with clinical response. The ideal plasma level baseline is obtained at steady state during a presumed period of observed adherence, such as during a hospital stay. Although patients can be nonadherent in the hospital, this setting offers the best proxy for an acute clinical response to a given plasma level. The alternative is to obtain several plasma levels during a period of outpatient clinical stability.
Clinicians must be mindful that changes in the plasma antipsychotic level after hospital discharge might not reflect poor adherence; environmental factors (eg, exposure to cytochrome P450 or P-glycoprotein inducers) can have a significant impact on results. Resumption of smoking is a classic example, and routinely is associated with a 50% reduction in plasma clozapine levels.5
There also is expected variability in plasma antipsychotic levels based on (1) the timing of prior doses with regard to trough levels, and (2) the effects of an occasional missed dose. Nevertheless, in a sample of adherent clozapine-treated patients, investigators found that 98% of patients had a coefficient of variability (CV) of 30% for sequential plasma concentrations (mean CV, 14%).6
Clinicians should inform patients that the plasma antipsychotic level is a tool for helping track treatment engagement before relapse—the same way metabolic monitoring helps track abnormalities that can be associated with future cardiovascular events. (Clinicians also must be charitable with their patients when discussing a significant drop in the plasma antipsychotic level [eg, >30%], acknowledging that many patients often miss doses.)
Using the patient’s input about specific difficulties with a medication regimen, clinicians should strive to find ways to improve oral medication adherence. In many cases, the clinician can assist through medication simplification, consolidation of multiple daily doses, provision of pill boxes, and discussions about long-acting injectable (LAI) antipsychotics.
In short, plasma antipsychotic levels offer an opportunity to have a richer, evidence-based discussion about adherence, beyond the trite, ineffective question “Did you take your medication?” Use of an objective measure can (1) serve as a benchmark for the patient (eg, “You seemed to do better when your clozapine level was above 400 ng/mL”), and (2) remind clinicians of the variable adherence inherent with oral medication regimens.
A note about long-acting injectable antipsychotics
Because nonadherence is seen throughout the course of schizophrenia, discussion of LAI therapy should not be limited to patients with chronic illness. Results of recent naturalistic7 and randomized studies8 show significant reduction in the rate of psychotic relapse and improved symptom control8 among first-episode patients who are taking an LAI. Moreover, compelling data show that most first-episode patients who are taking an oral antipsychotic will accept a recommendation for treatment with an LAI.9
Summing up: 2 Tools for achieving therapeutic success
Monitoring plasma levels of antipsychotics offers a method for quantifying the problem of nonadherence. For many patients, an LAI antipsychotic provides a solution to nonadherence, and the increasing variety of LAI preparations means more options with which to match individual patients.
Clinicians have a limited amount of time to spend with patients in the office, but time spent discussing LAIs is an investment in the patient’s stability and functional outcome. Minutes once spent managing nonadherence can be devoted to understanding the patient’s aspirations and to developing strategies to achieve those goals.
In the end, isn’t that what we’d rather be talking about with our patients?
1. Subotnik KL, Nuechterlein KH, Ventura J, et al. Risperidone nonadherence and return of positive symptoms in the early course of schizophrenia. Am J Psychiatry. 2011;168(3):286-292.
2. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
3. Byerly MJ, Thompson A, Carmody T, et al. Validity of electronically monitored medication adherence and conventional adherence measures in schizophrenia. Psychiatr Serv. 2007;58(6):844-847.
4. Velligan DI, Wang M, Diamond P, et al. Relationships among subjective and objective measures of adherence to oral antipsychotic medications. Psychiatr Serv. 2007;58(9):1187-1192.
5. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21(6):569-574.
6. Dugan DJ, Ereshefsky L, Toney GB, et al. Dose and interval adherence among stabilized clozapine-treated patients measured by medication event monitoring. Presented at: Meeting of the New Clinical Drug Evaluation Unit; May 30-June 2, 2000; Boca Raton, FL.
7. Tiihonen J, Haukka J, Taylor M, et al. A nationwide cohort study of oral and depot antipsychotics after first hospitalization for schizophrenia. [Erratum in Am J Psychiatry. 2012;169(2):223]. Am J Psychiatry. 2011;168(6):603-609.
8. Subotnik KL, Casaus LR, Ventura J, et al. Long-acting injectable risperidone for relapse prevention and control of breakthrough symptoms after a recent first episode of schizophrenia: a randomized clinical trial. JAMA Psychiatry. 2015;72(8):822-829.
9. Weiden PJ, Schooler NR, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406.
1. Subotnik KL, Nuechterlein KH, Ventura J, et al. Risperidone nonadherence and return of positive symptoms in the early course of schizophrenia. Am J Psychiatry. 2011;168(3):286-292.
2. Remington G, Teo C, Mann S, et al. Examining levels of antipsychotic adherence to better understand nonadherence. J Clin Psychopharmacol. 2013;33(2):261-263.
3. Byerly MJ, Thompson A, Carmody T, et al. Validity of electronically monitored medication adherence and conventional adherence measures in schizophrenia. Psychiatr Serv. 2007;58(6):844-847.
4. Velligan DI, Wang M, Diamond P, et al. Relationships among subjective and objective measures of adherence to oral antipsychotic medications. Psychiatr Serv. 2007;58(9):1187-1192.
5. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21(6):569-574.
6. Dugan DJ, Ereshefsky L, Toney GB, et al. Dose and interval adherence among stabilized clozapine-treated patients measured by medication event monitoring. Presented at: Meeting of the New Clinical Drug Evaluation Unit; May 30-June 2, 2000; Boca Raton, FL.
7. Tiihonen J, Haukka J, Taylor M, et al. A nationwide cohort study of oral and depot antipsychotics after first hospitalization for schizophrenia. [Erratum in Am J Psychiatry. 2012;169(2):223]. Am J Psychiatry. 2011;168(6):603-609.
8. Subotnik KL, Casaus LR, Ventura J, et al. Long-acting injectable risperidone for relapse prevention and control of breakthrough symptoms after a recent first episode of schizophrenia: a randomized clinical trial. JAMA Psychiatry. 2015;72(8):822-829.
9. Weiden PJ, Schooler NR, Weedon JC, et al. A randomized controlled trial of long-acting injectable risperidone vs continuation on oral atypical antipsychotics for first-episode schizophrenia patients: initial adherence outcome. J Clin Psychiatry. 2009;70(10):1397-1406.

Second of 2 parts: The mysteries of psychiatry maintenance of certification, further unraveled
To recap what I discussed in Part 1 of this article (December 2014): As part of a trend across all medical specialty boards, the American Board of Psychiatry and Neurology (ABPN) instituted a recertification process for all new general psychiatry certifications, starting October 1, 1994.1 In 2000, the specialties that comprise the American Board of Medical Specialties (ABMS) agreed to develop a comprehensive maintenance of certification (MOC) process to demonstrate ongoing learning and competency beyond what can be captured by a recertification examination. All ABMS member boards now use a 4-part process for recertification.
A great deal of professional and personal importance has been attached to maintaining one’s general and subspecialty certifications. To that end, the 2 parts of this article highlight current ABPN MOC requirements and provide resources for understanding, tracking, and completing the self-assessment (SA) and performance-in-practice (PIP) components.
In this installment, I examine 3 components of MOC:
• continuing medical education (CME), including SA requirements
• improvement in medical practice (PIP)
• continuous maintenance of certification (C-MOC)
In addition to this review, all physicians who are subject to MOC should download and read the 20-page revised MOC Program booklet v. 2.1 (May 2014).2
Continuing medical education
The CME requirement is clear: All diplomate physicians must accrue, on average, 30 Category-1 CME credits a year; the CME must be relevant to the specialty or subspecialty in which the diplomate practices.3 For physicians who hold >1 ABPN certificates, the total CME requirement is the same; CME credits can be applied across each specialty and subspecialty.
The May 2014 MOC revision states that, for physicians who certified or recertified between 2005 and 2011 and who applied for the 2015 examination in 2014, the required CME credit total is 270.2 For all subsequent years of certification or recertification, including 2012, diplomates are enrolled in C-MOC, which is described below.2
To even out the accrual of CME credits across the prior 10 years, ABPN mandates that, for diplomates who certified or recertified between 2005 and 2011, one hundred fifty of the CME credits be accrued in the 5 years before they apply for the examination. Diplomates in C-MOC should accrue, on average, 30 CME credits a year in each of the 3-year blocks (ie, 90 units in each block).2
Self-assessment
SA is a specific form of CME that is designed to provide comprehensive test-based feedback on knowledge acquired, to enhance the learning process.4 SA CME feedback must include:
• the correct answer to each test question
• recommended literature resources for each question
• performance compared to peers on each question.
Given the structured nature of SA activities, beginning January 1, 2014, one must use only ABPN-approved SA products (see Related Resources for a list of APBN-approved SA products).5
Table 1 and Table 2 outline SA requirements for, respectively, physicians who certified or recertified from 2005 through 2011, and those who certified or recertified in 2012 (and later). The SA requirement increases after 2011 to 24 credits in each 3-year block (8 credits a year, on average).2 Multiple SA activities can be used to fulfill the credit requirement of each 3-year block.
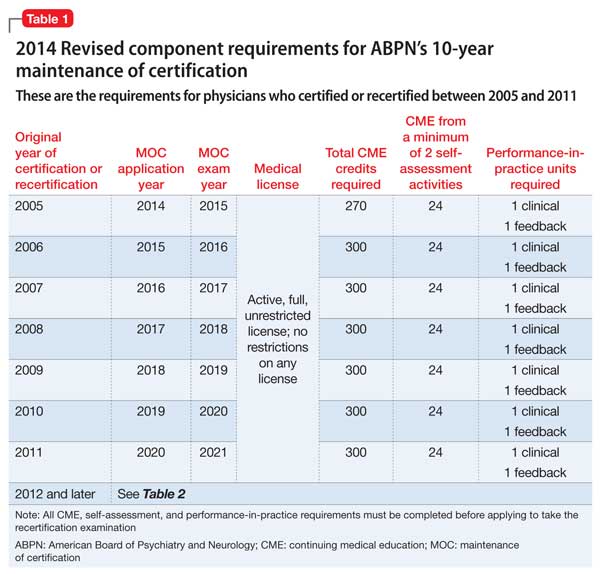
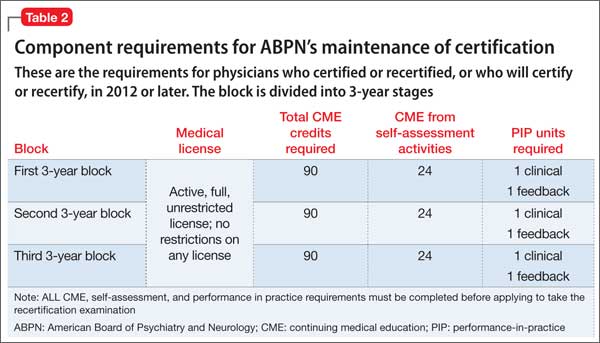
Note: Credits accrued by performing SA activities count toward the CME credit total.
Improvement in medical practice, or PIP
Physicians who are active clinically must complete PIP modules. Each module comprises peer or patient feedback plus a clinical aspect. The May 2014 MOC revision simplified the feedback process to mandate peer or patient feedback—but not both, as required previously.2 For the feedback PIP module, the physician selects 5 peers or patients to complete review forms, examines the results, and creates a plan of improvement. An exception to this “rule of 5” applies to diplomates who have a supervisor capable of evaluating all general competencies, defined below.
Related Resources provides a link to ABPN-created forms.
Within 24 months, but not sooner than 1 month, 5 peers or patients (or 1 applicable supervisor) are selected to complete review forms; changes in practice are noted. The same peers or patients might be selected for a second review. As noted in Table 1 and Table 2, the number of PIP modules is fewer for physicians who certified or recertified between 2005 and 2011; from 2012 onward, 1 PIP clinical module is required in each 3-year block.2
There are 6 ABPN-approved feedback module options, of which the diplomate must choose 1 in any given block2:
• 5 patient surveys
• 5 peer evaluations of general competenciesa
• 5 resident evaluations of general competenciesa
• 360° evaluation of general competencies,a with 5 respondents
• institutional peer review of general competencies,a with 5 respondents
• 1 supervisor evaluation of general competencies.a
aGeneral competencies include patient care; practice-based learning and improvement; professionalism; medical knowledge; interpersonal and communication skills; and system-based practices.
Although many institutions have a quality improvement (QI) program, that program must be approved by the Multi-Specialty MOC Portfolio Approval Program sponsored by ABMS for a clinician to receive credit for 1 PIP clinical module. If the approved QI program includes patient or peer feedback (eg, a survey), the diplo mate can receive credit for 1 PIP feedback module.2
For the clinical PIP module, the physician selects 5 charts for review and examines them based on criteria found in an ABPN-approved (starting in 2014) PIP product. (Related Resources provides a link to this list.) After reviewing the initial 5 charts, a plan for improvement is created. Within 24 months, but no sooner than 1 month, 5 charts are again selected and reviewed, and changes in practice are noted. The same charts can be selected for the second review.
As noted in Table 1 and Table 2, the number of PIP modules is fewer for those who initially certified or recertified between 2005 and 2011; from 2012 onward, 1 PIP clinical module is required in each 3-year block.2
The C-MOC process
Physicians who certified or recertified in 2012, or who will certify or recertify after that year, are enrolled automatically in C-MOC.6,7 The purpose of C-MOC is to keep diplomates on track to fulfill the higher level of SA requirements that began with this group; this is done by mandating use of the ABPN Physician Folios system. As shown in Table 2, there is no longer a 10-year cycle; instead, there are continuous 3-year stages, within which each diplomate must accrue 90 CME credits (on average, 30 credits a year), 24 SA credits (on average, 8 a year), 1 PIP clinical module, and 1 PIP feedback module.6,7
The first 3-year block of C-MOC requirements will be waived for physicians who complete Accreditation Council on Graduate Medical Education–accredited or ABPN-approved subspecialty training in 2012 or later—if they pass the corresponding ABPN subspecialty examination during the first 3-year block of enrollment in C-MOC.2 For diplomates enrolled in C-MOC, failure to track progress of each 3-year block, via the ABPN Physician Folios system, has significant consequences: Those who do not complete the first stage of the program by the end of 3 years will be listed on the ABPN Web site as “certified— not meeting MOC requirements.” Those who do not complete 2 stages by the end of 6 years will be listed as “not certified.”2
Cognitive exam still in place. The only remnant of the old 10-year cycle is the requirement to pass the cognitive examination every 10 years, although the exam can be taken earlier if the diplomate wishes. If all requirements are met and one does not sit for, or fails, the exam, the ABPN Web site will report the diplomate as “not meeting MOC requirements.” One can retake the exam within 1 year of the failed or missed exam, but a subsequent failure or missed exam will result in being listed as “not certified.”2
Fee structure. Instead of a single fee paid at the time of the exam(s), physicians in the C-MOC program pay an annual fee that covers participation in ABPN Physician Folios and 1 exam in a 10-year period. Fewer than 10 years of participation, or applying for a combined examination (for diplomates who hold multiple certifications), requires an additional fee.7
Bottom Line
Maintenance of certification (MOC) is manageable, although it requires you to be familiar with its various elements. Those elements include continuing medical education (CME requirements); the additional self-assessment component of CME; performance-in-practice modules; and continuous maintenance of certification. The MOC program booklet of the American Board of Psychiatry and Neurology provides all necessary details.
Disclosure
Dr. Meyer reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Faulkner LR, Tivnan PW, Winstead DK, et al. The ABPN Maintenance of Certification Program for psychiatrists: past history, current status, and future directions. Acad Psychiatry. 2008;32(3):241-248.
2. Maintenance of Certification Program. American Board of Psychiatry and Neurology Inc. http://www.abpn.com/ downloads/moc/moc_web_doc.pdf. Published May 2014. Accessed August 25, 2014.
3. Faulkner LR, Vondrak PA. Frequently asked questions about maintenance of certification (MOC). J Clin Psychiatry. 2010;71(5):632-633.
4. Ebert MH, Faulkner L, Stubbe DE, et al. Maintenance of certification in psychiatry. J Clin Psychiatry. 2009;70(10):e39.
5. Approved MOC Products. American Board of Psychiatry and Neurology Inc. http://www.abpn.com/moc_products. asp. Accessed August 25, 2014.
6. Continuous MOC (C-MOC). American Board of Psychiatry and Neurology Inc. http://www.abpn.com/downloads/ moc/ContinuousCertificationApproach_0311.pdf. Accessed August 25, 2014.
7. C-MOC Program Overview. American Board of Psychiatry and Neurology Inc. http://www.abpn.com/downloads/ moc/moc-handouts-CMOC-051314.pdf. Published May 13, 2014. Accessed August 25, 2014.
To recap what I discussed in Part 1 of this article (December 2014): As part of a trend across all medical specialty boards, the American Board of Psychiatry and Neurology (ABPN) instituted a recertification process for all new general psychiatry certifications, starting October 1, 1994.1 In 2000, the specialties that comprise the American Board of Medical Specialties (ABMS) agreed to develop a comprehensive maintenance of certification (MOC) process to demonstrate ongoing learning and competency beyond what can be captured by a recertification examination. All ABMS member boards now use a 4-part process for recertification.
A great deal of professional and personal importance has been attached to maintaining one’s general and subspecialty certifications. To that end, the 2 parts of this article highlight current ABPN MOC requirements and provide resources for understanding, tracking, and completing the self-assessment (SA) and performance-in-practice (PIP) components.
In this installment, I examine 3 components of MOC:
• continuing medical education (CME), including SA requirements
• improvement in medical practice (PIP)
• continuous maintenance of certification (C-MOC)
In addition to this review, all physicians who are subject to MOC should download and read the 20-page revised MOC Program booklet v. 2.1 (May 2014).2
Continuing medical education
The CME requirement is clear: All diplomate physicians must accrue, on average, 30 Category-1 CME credits a year; the CME must be relevant to the specialty or subspecialty in which the diplomate practices.3 For physicians who hold >1 ABPN certificates, the total CME requirement is the same; CME credits can be applied across each specialty and subspecialty.
The May 2014 MOC revision states that, for physicians who certified or recertified between 2005 and 2011 and who applied for the 2015 examination in 2014, the required CME credit total is 270.2 For all subsequent years of certification or recertification, including 2012, diplomates are enrolled in C-MOC, which is described below.2
To even out the accrual of CME credits across the prior 10 years, ABPN mandates that, for diplomates who certified or recertified between 2005 and 2011, one hundred fifty of the CME credits be accrued in the 5 years before they apply for the examination. Diplomates in C-MOC should accrue, on average, 30 CME credits a year in each of the 3-year blocks (ie, 90 units in each block).2
Self-assessment
SA is a specific form of CME that is designed to provide comprehensive test-based feedback on knowledge acquired, to enhance the learning process.4 SA CME feedback must include:
• the correct answer to each test question
• recommended literature resources for each question
• performance compared to peers on each question.
Given the structured nature of SA activities, beginning January 1, 2014, one must use only ABPN-approved SA products (see Related Resources for a list of APBN-approved SA products).5
Table 1 and Table 2 outline SA requirements for, respectively, physicians who certified or recertified from 2005 through 2011, and those who certified or recertified in 2012 (and later). The SA requirement increases after 2011 to 24 credits in each 3-year block (8 credits a year, on average).2 Multiple SA activities can be used to fulfill the credit requirement of each 3-year block.
Note: Credits accrued by performing SA activities count toward the CME credit total.
Improvement in medical practice, or PIP
Physicians who are active clinically must complete PIP modules. Each module comprises peer or patient feedback plus a clinical aspect. The May 2014 MOC revision simplified the feedback process to mandate peer or patient feedback—but not both, as required previously.2 For the feedback PIP module, the physician selects 5 peers or patients to complete review forms, examines the results, and creates a plan of improvement. An exception to this “rule of 5” applies to diplomates who have a supervisor capable of evaluating all general competencies, defined below.
Related Resources provides a link to ABPN-created forms.
Within 24 months, but not sooner than 1 month, 5 peers or patients (or 1 applicable supervisor) are selected to complete review forms; changes in practice are noted. The same peers or patients might be selected for a second review. As noted in Table 1 and Table 2, the number of PIP modules is fewer for physicians who certified or recertified between 2005 and 2011; from 2012 onward, 1 PIP clinical module is required in each 3-year block.2
There are 6 ABPN-approved feedback module options, of which the diplomate must choose 1 in any given block2:
• 5 patient surveys
• 5 peer evaluations of general competenciesa
• 5 resident evaluations of general competenciesa
• 360° evaluation of general competencies,a with 5 respondents
• institutional peer review of general competencies,a with 5 respondents
• 1 supervisor evaluation of general competencies.a
aGeneral competencies include patient care; practice-based learning and improvement; professionalism; medical knowledge; interpersonal and communication skills; and system-based practices.
Although many institutions have a quality improvement (QI) program, that program must be approved by the Multi-Specialty MOC Portfolio Approval Program sponsored by ABMS for a clinician to receive credit for 1 PIP clinical module. If the approved QI program includes patient or peer feedback (eg, a survey), the diplo mate can receive credit for 1 PIP feedback module.2
For the clinical PIP module, the physician selects 5 charts for review and examines them based on criteria found in an ABPN-approved (starting in 2014) PIP product. (Related Resources provides a link to this list.) After reviewing the initial 5 charts, a plan for improvement is created. Within 24 months, but no sooner than 1 month, 5 charts are again selected and reviewed, and changes in practice are noted. The same charts can be selected for the second review.
As noted in Table 1 and Table 2, the number of PIP modules is fewer for those who initially certified or recertified between 2005 and 2011; from 2012 onward, 1 PIP clinical module is required in each 3-year block.2
The C-MOC process
Physicians who certified or recertified in 2012, or who will certify or recertify after that year, are enrolled automatically in C-MOC.6,7 The purpose of C-MOC is to keep diplomates on track to fulfill the higher level of SA requirements that began with this group; this is done by mandating use of the ABPN Physician Folios system. As shown in Table 2, there is no longer a 10-year cycle; instead, there are continuous 3-year stages, within which each diplomate must accrue 90 CME credits (on average, 30 credits a year), 24 SA credits (on average, 8 a year), 1 PIP clinical module, and 1 PIP feedback module.6,7
The first 3-year block of C-MOC requirements will be waived for physicians who complete Accreditation Council on Graduate Medical Education–accredited or ABPN-approved subspecialty training in 2012 or later—if they pass the corresponding ABPN subspecialty examination during the first 3-year block of enrollment in C-MOC.2 For diplomates enrolled in C-MOC, failure to track progress of each 3-year block, via the ABPN Physician Folios system, has significant consequences: Those who do not complete the first stage of the program by the end of 3 years will be listed on the ABPN Web site as “certified— not meeting MOC requirements.” Those who do not complete 2 stages by the end of 6 years will be listed as “not certified.”2
Cognitive exam still in place. The only remnant of the old 10-year cycle is the requirement to pass the cognitive examination every 10 years, although the exam can be taken earlier if the diplomate wishes. If all requirements are met and one does not sit for, or fails, the exam, the ABPN Web site will report the diplomate as “not meeting MOC requirements.” One can retake the exam within 1 year of the failed or missed exam, but a subsequent failure or missed exam will result in being listed as “not certified.”2
Fee structure. Instead of a single fee paid at the time of the exam(s), physicians in the C-MOC program pay an annual fee that covers participation in ABPN Physician Folios and 1 exam in a 10-year period. Fewer than 10 years of participation, or applying for a combined examination (for diplomates who hold multiple certifications), requires an additional fee.7
Bottom Line
Maintenance of certification (MOC) is manageable, although it requires you to be familiar with its various elements. Those elements include continuing medical education (CME requirements); the additional self-assessment component of CME; performance-in-practice modules; and continuous maintenance of certification. The MOC program booklet of the American Board of Psychiatry and Neurology provides all necessary details.
Disclosure
Dr. Meyer reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
To recap what I discussed in Part 1 of this article (December 2014): As part of a trend across all medical specialty boards, the American Board of Psychiatry and Neurology (ABPN) instituted a recertification process for all new general psychiatry certifications, starting October 1, 1994.1 In 2000, the specialties that comprise the American Board of Medical Specialties (ABMS) agreed to develop a comprehensive maintenance of certification (MOC) process to demonstrate ongoing learning and competency beyond what can be captured by a recertification examination. All ABMS member boards now use a 4-part process for recertification.
A great deal of professional and personal importance has been attached to maintaining one’s general and subspecialty certifications. To that end, the 2 parts of this article highlight current ABPN MOC requirements and provide resources for understanding, tracking, and completing the self-assessment (SA) and performance-in-practice (PIP) components.
In this installment, I examine 3 components of MOC:
• continuing medical education (CME), including SA requirements
• improvement in medical practice (PIP)
• continuous maintenance of certification (C-MOC)
In addition to this review, all physicians who are subject to MOC should download and read the 20-page revised MOC Program booklet v. 2.1 (May 2014).2
Continuing medical education
The CME requirement is clear: All diplomate physicians must accrue, on average, 30 Category-1 CME credits a year; the CME must be relevant to the specialty or subspecialty in which the diplomate practices.3 For physicians who hold >1 ABPN certificates, the total CME requirement is the same; CME credits can be applied across each specialty and subspecialty.
The May 2014 MOC revision states that, for physicians who certified or recertified between 2005 and 2011 and who applied for the 2015 examination in 2014, the required CME credit total is 270.2 For all subsequent years of certification or recertification, including 2012, diplomates are enrolled in C-MOC, which is described below.2
To even out the accrual of CME credits across the prior 10 years, ABPN mandates that, for diplomates who certified or recertified between 2005 and 2011, one hundred fifty of the CME credits be accrued in the 5 years before they apply for the examination. Diplomates in C-MOC should accrue, on average, 30 CME credits a year in each of the 3-year blocks (ie, 90 units in each block).2
Self-assessment
SA is a specific form of CME that is designed to provide comprehensive test-based feedback on knowledge acquired, to enhance the learning process.4 SA CME feedback must include:
• the correct answer to each test question
• recommended literature resources for each question
• performance compared to peers on each question.
Given the structured nature of SA activities, beginning January 1, 2014, one must use only ABPN-approved SA products (see Related Resources for a list of APBN-approved SA products).5
Table 1 and Table 2 outline SA requirements for, respectively, physicians who certified or recertified from 2005 through 2011, and those who certified or recertified in 2012 (and later). The SA requirement increases after 2011 to 24 credits in each 3-year block (8 credits a year, on average).2 Multiple SA activities can be used to fulfill the credit requirement of each 3-year block.
Note: Credits accrued by performing SA activities count toward the CME credit total.
Improvement in medical practice, or PIP
Physicians who are active clinically must complete PIP modules. Each module comprises peer or patient feedback plus a clinical aspect. The May 2014 MOC revision simplified the feedback process to mandate peer or patient feedback—but not both, as required previously.2 For the feedback PIP module, the physician selects 5 peers or patients to complete review forms, examines the results, and creates a plan of improvement. An exception to this “rule of 5” applies to diplomates who have a supervisor capable of evaluating all general competencies, defined below.
Related Resources provides a link to ABPN-created forms.
Within 24 months, but not sooner than 1 month, 5 peers or patients (or 1 applicable supervisor) are selected to complete review forms; changes in practice are noted. The same peers or patients might be selected for a second review. As noted in Table 1 and Table 2, the number of PIP modules is fewer for physicians who certified or recertified between 2005 and 2011; from 2012 onward, 1 PIP clinical module is required in each 3-year block.2
There are 6 ABPN-approved feedback module options, of which the diplomate must choose 1 in any given block2:
• 5 patient surveys
• 5 peer evaluations of general competenciesa
• 5 resident evaluations of general competenciesa
• 360° evaluation of general competencies,a with 5 respondents
• institutional peer review of general competencies,a with 5 respondents
• 1 supervisor evaluation of general competencies.a
aGeneral competencies include patient care; practice-based learning and improvement; professionalism; medical knowledge; interpersonal and communication skills; and system-based practices.
Although many institutions have a quality improvement (QI) program, that program must be approved by the Multi-Specialty MOC Portfolio Approval Program sponsored by ABMS for a clinician to receive credit for 1 PIP clinical module. If the approved QI program includes patient or peer feedback (eg, a survey), the diplo mate can receive credit for 1 PIP feedback module.2
For the clinical PIP module, the physician selects 5 charts for review and examines them based on criteria found in an ABPN-approved (starting in 2014) PIP product. (Related Resources provides a link to this list.) After reviewing the initial 5 charts, a plan for improvement is created. Within 24 months, but no sooner than 1 month, 5 charts are again selected and reviewed, and changes in practice are noted. The same charts can be selected for the second review.
As noted in Table 1 and Table 2, the number of PIP modules is fewer for those who initially certified or recertified between 2005 and 2011; from 2012 onward, 1 PIP clinical module is required in each 3-year block.2
The C-MOC process
Physicians who certified or recertified in 2012, or who will certify or recertify after that year, are enrolled automatically in C-MOC.6,7 The purpose of C-MOC is to keep diplomates on track to fulfill the higher level of SA requirements that began with this group; this is done by mandating use of the ABPN Physician Folios system. As shown in Table 2, there is no longer a 10-year cycle; instead, there are continuous 3-year stages, within which each diplomate must accrue 90 CME credits (on average, 30 credits a year), 24 SA credits (on average, 8 a year), 1 PIP clinical module, and 1 PIP feedback module.6,7
The first 3-year block of C-MOC requirements will be waived for physicians who complete Accreditation Council on Graduate Medical Education–accredited or ABPN-approved subspecialty training in 2012 or later—if they pass the corresponding ABPN subspecialty examination during the first 3-year block of enrollment in C-MOC.2 For diplomates enrolled in C-MOC, failure to track progress of each 3-year block, via the ABPN Physician Folios system, has significant consequences: Those who do not complete the first stage of the program by the end of 3 years will be listed on the ABPN Web site as “certified— not meeting MOC requirements.” Those who do not complete 2 stages by the end of 6 years will be listed as “not certified.”2
Cognitive exam still in place. The only remnant of the old 10-year cycle is the requirement to pass the cognitive examination every 10 years, although the exam can be taken earlier if the diplomate wishes. If all requirements are met and one does not sit for, or fails, the exam, the ABPN Web site will report the diplomate as “not meeting MOC requirements.” One can retake the exam within 1 year of the failed or missed exam, but a subsequent failure or missed exam will result in being listed as “not certified.”2
Fee structure. Instead of a single fee paid at the time of the exam(s), physicians in the C-MOC program pay an annual fee that covers participation in ABPN Physician Folios and 1 exam in a 10-year period. Fewer than 10 years of participation, or applying for a combined examination (for diplomates who hold multiple certifications), requires an additional fee.7
Bottom Line
Maintenance of certification (MOC) is manageable, although it requires you to be familiar with its various elements. Those elements include continuing medical education (CME requirements); the additional self-assessment component of CME; performance-in-practice modules; and continuous maintenance of certification. The MOC program booklet of the American Board of Psychiatry and Neurology provides all necessary details.
Disclosure
Dr. Meyer reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Faulkner LR, Tivnan PW, Winstead DK, et al. The ABPN Maintenance of Certification Program for psychiatrists: past history, current status, and future directions. Acad Psychiatry. 2008;32(3):241-248.
2. Maintenance of Certification Program. American Board of Psychiatry and Neurology Inc. http://www.abpn.com/ downloads/moc/moc_web_doc.pdf. Published May 2014. Accessed August 25, 2014.
3. Faulkner LR, Vondrak PA. Frequently asked questions about maintenance of certification (MOC). J Clin Psychiatry. 2010;71(5):632-633.
4. Ebert MH, Faulkner L, Stubbe DE, et al. Maintenance of certification in psychiatry. J Clin Psychiatry. 2009;70(10):e39.
5. Approved MOC Products. American Board of Psychiatry and Neurology Inc. http://www.abpn.com/moc_products. asp. Accessed August 25, 2014.
6. Continuous MOC (C-MOC). American Board of Psychiatry and Neurology Inc. http://www.abpn.com/downloads/ moc/ContinuousCertificationApproach_0311.pdf. Accessed August 25, 2014.
7. C-MOC Program Overview. American Board of Psychiatry and Neurology Inc. http://www.abpn.com/downloads/ moc/moc-handouts-CMOC-051314.pdf. Published May 13, 2014. Accessed August 25, 2014.
1. Faulkner LR, Tivnan PW, Winstead DK, et al. The ABPN Maintenance of Certification Program for psychiatrists: past history, current status, and future directions. Acad Psychiatry. 2008;32(3):241-248.
2. Maintenance of Certification Program. American Board of Psychiatry and Neurology Inc. http://www.abpn.com/ downloads/moc/moc_web_doc.pdf. Published May 2014. Accessed August 25, 2014.
3. Faulkner LR, Vondrak PA. Frequently asked questions about maintenance of certification (MOC). J Clin Psychiatry. 2010;71(5):632-633.
4. Ebert MH, Faulkner L, Stubbe DE, et al. Maintenance of certification in psychiatry. J Clin Psychiatry. 2009;70(10):e39.
5. Approved MOC Products. American Board of Psychiatry and Neurology Inc. http://www.abpn.com/moc_products. asp. Accessed August 25, 2014.
6. Continuous MOC (C-MOC). American Board of Psychiatry and Neurology Inc. http://www.abpn.com/downloads/ moc/ContinuousCertificationApproach_0311.pdf. Accessed August 25, 2014.
7. C-MOC Program Overview. American Board of Psychiatry and Neurology Inc. http://www.abpn.com/downloads/ moc/moc-handouts-CMOC-051314.pdf. Published May 13, 2014. Accessed August 25, 2014.

A guide to the mysteries of maintenance of certification
As part of a general trend among all medical specialty boards, the American Board of Psychiatry and Neurology (ABPN) instituted a recertification process for all new general psychiatry certifications on October 1, 1994.1 In 2000, the individual specialties that constitute the American Board of Medical Specialties (ABMS) subsequently agreed to develop a comprehensive maintenance of certification (MOC) process to demonstrate ongoing learning and competency beyond what could be captured by a recertification examination alone.
All ABMS member boards now use a 4-part process for recertification. For ABPN, those 4 core components are listed in the Table.1,2
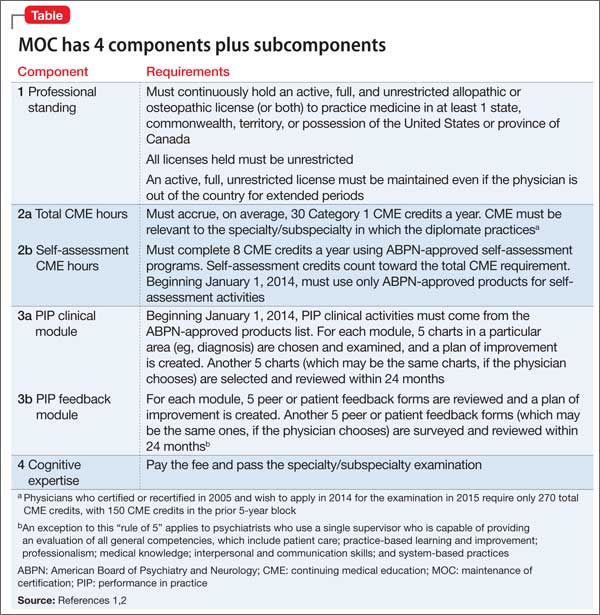
ABPN component 1 (maintaining an unrestricted medical license) and component 4 (passing the recertification examination) are straightforward; however, requirements for continuing medical education (CME), including the specific need to accrue ABPN-approved self-assessment (SA) CME hours, and the Improvement in Medical Practice (performance in practice, or PIP) module, have stoked significant commentary and confusion.
Based on feedback,3,4 ABPN in 2014:
• modified the SA and PIP requirements for physicians who certified or recertified between 2005 and 2011
• changed the specific requirement for the PIP feedback component.
These modifications only added to feelings of uncertainty about the MOC process among many psychiatrists.5
Given the professional and personal importance attached to maintaining one’s general and subspecialty certifications, the 2 parts of this article—here and in the January 2015 issue—have been constructed to highlight current ABPN MOC requirements and provide resources for understanding, tracking, and completing the SA and PIP portions.
In addition to this review, I urge all physicians who are subject to MOC to read the 20-page revised MOC Program bookleta (version 2.1, May 2014).5
aDownload the booklet at www.abpn.com/downloads/moc/ moc_web_doc.pdf.
Who must recertify?
As of October 1, 1994, all physicians who achieve ABPN certifications in general psychiatry are issued a 10-year, time-limited certificate that expires on December 31 of the 10th year.3 Note that the 10-year, time-limited certificate in child and adolescent psychiatry began in 1995 and expires 10 years later on December 31.
Certificates in the subspecialties (addiction psychiatry, forensic psychiatry, geriatric psychiatry, etc.), including those issued before October 1, 1994, are 10-year, time-limited certificates that expire on December 31.3 This expiration date often is overlooked by physicians who are exempt from the MOC process for their general psychiatry, or child and adolescent psychiatry certification. There is no exemption for any subspecialty certificate (aside from child and adolescent psychiatry before 1995), regardless of the date of issue.
Moreover, physicians who hold a certificate in a subspecialty also must maintain certification in their specialty (general psychiatry) to apply for recertification in their subspecialization. One exception: Diplomates in child and adolescent psychiatry do not need to maintain current certification in general psychiatry for their subspecialty certification to remain valid or to recertify in child and adolescent psychiatry.
The need to maintain multiple certifications can seem onerous, but note that CME, SA, and PIP activities that have been completed in one area of specialization or subspecialization accrue and count for multiple certifications for diplomates certified in 2 or more areas.5
Get started!
Tracking your progress is critical to keeping up with MOC requirements. You can do this with a personal spreadsheet or by using online resources. Although it is not required, ABPN has established a system that allows diplomates to create and maintain, at no cost, a physician folio on the ABPN server that facilitates documentation of CME hours, including specific SA hours, and PIP module completion.6 All diplomates are required to maintain records of SA activities, CME activities, and PIP units; the ABPN will audit approximately 5% of examination applications.5
Regardless of what documentation method you choose, you should establish an active profile on the ABPN site (www.abpn. com/folios), confirm your contact information, and, if you are not active clinically, update your clinical status. ABPN requires that diplomates self-report their clinical status every 24 months—information that is available to the public. Clinical status also identifies to ABPN those PIP modules that you must complete.
ABPN recognizes 3 categories of clinical status5:
1. Clinically active. Provided any amount of direct or consultative care, or both, in the preceding 24 months, including supervision of residents.
a) Engaged in direct or consultative care, or both, sufficient to complete Improvement in Medical Practice (PIP) units.
b) Engaged in direct or consultative care, or both, that is insufficient to complete PIP units.
2. Clinically inactive. Did not provide direct or consultative care in the preceding 24 months.
3. Status unknown. No information is available on clinical activity.
Based on these definitions, physicians in Category 1a are required to complete all components of the MOC program, including PIP units; physicians in Category 1b or Category 2 are required to complete all components of the MOC program except PIP units.
A change in status from Category 1b or 2 to Category 1a (eg, moving from a purely administrative position to one with clinical duties) requires completion of ≥1 PIP unit.
The easy parts
Licenses. Maintaining your unrestricted professional license(s) is mandatory; the language of this requirement is unambiguous (Table).5 The plural form of license is intentional: Some physicians have medical licenses in multiple states and, in some jurisdictions, licenses are required to supervise physician assistants and other personnel or to prescribe controlled substances. Any restriction on a professional license should be discussed with ABPN and resolved to prevent rejection of the examination application.5
Examinations. For physicians who are not yet enrolled in the continuous-MOC (C-MOC) process (to be discussed in Part 2 of this article), an application to take the examination in Year 10 can be filed in Year 9 of the cycle—after the CME, SA, and PIP requirements are completed. Once a diplomate becomes subject to the C-MOC process by certifying or recertifying from 2012 onwards, completion of each 3-year module of CME, SA, and PIP will not coincide with the 10-year time frame of the examination.
The application deadline for all MOC examinations typically is the year before the examination; the examination should be taken in the year the certificate expires, although it can be taken earlier if desired.7 The examinations are computer-based and administered at a certified testing center. For diplomates who have more than 1 ABPN certificate and want to combine multiple examinations into 1 test session, a reduced fee structure applies.
The general psychiatry examination comprises 220 single-answer, multiple-choice questions that must be completed within 290 minutes, with 10 extra minutes allotted to read on-screen instructions, sign in, and complete a post-examination survey.8 The combined examinations comprise 100 questions from each ABPN specialty or subspecialty area.5
The content of the 2015 general psychiatry examinationb is available on the ABPN Web site.7 Note that the recertification examination in general psychiatry does not cover neurology topics.
bDownload the outline of the examination at www.abpn.com/ downloads/content_outlines/MOC/2015-MOC-Psych-blueprint-060314-EWM-MR.pdf.
Examinations administered in 2015 and 2016 will use only diagnostic criteria that have not changed from DSM-IV-TR9: Neither obsolete diagnoses or subtypes from DSM-IV-TR nor new diagnoses or subtypes in DSM-5 (eg, hoarding disorder) will be tested.9 Diagnoses that are exactly or substantially the same will be tested; these include diagnoses:
• with a name change only (eg, “phonological disorder” in DSM-IV-TR is “speech sound disorder” in DSM-5)
• expanded into >1 new diagnosis (eg, hypochondriasis was expanded to 2 new diagnoses: somatic symptom disorder and illness anxiety disorder)
• subsumed or combined into a new diagnosis (eg, substance use and dependence are now combined into substance use disorder in DSM-5).9
For these diagnoses, both DSM-IV-TR and DSM-5 diagnoses will be provided on examinations.
Beginning in 2017, all examinations will use DSM-5 classifications and diagnostic criteria.9
Part 2 of this article in the January 2015 issue reviews other key aspects of MOC: continuing medical education (CME), including self-assessment requirements; performance in practice (PIP); and continuous maintenance of certification (C-MOC).
BOTTOM LINE
Maintenance of certification (MOC) is a manageable process, although it requires you to be familiar with its various elements, including the duration of certification, licensing requirements, and the examination. Start the process by (1) establishing a login on the ABPN Web site and (2) reviewing the MOC program booklet.
Related Resources
• ABPN MOC home page. www.abpn.com/moc.html
• ABPN-approved products for SA, CME, and PIP modules. www.abpn.com/moc_products.asp
• Peer and patient feedback forms– Peer feedback form v1. www.abpn.com/downloads/moc/PIP-peer-feedback-v1-051914.pdf
– Patient feedback form v1. www.abpn.com/downloads/moc/PIP-patient-feedback-v1-051914.pdf
– Patient feedback form v2. www.abpn.com/downloads/moc/PIP-patient-feedback-v2-051914.pdf
• ABPN physician folio page. https://application.abpn.com/webclient/folios.aspx
Disclosure
Dr. Meyer reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Faulkner LR, Tivnan PW, Winstead DK, et al. The ABPN Maintenance of Certification Program for psychiatrists: past history, current status, and future directions. Acad Psychiatry. 2008;32(3):241-248.
2. Ebert MH, Faulkner L, Stubbe DE, et al. Maintenance of certification in psychiatry. J Clin Psychiatry. 2009;70(10):e39.
3. Faulkner LR, Vondrak PA. Frequently asked questions about maintenance of certification (MOC). J Clin Psychiatry. 2010;71(5):632-633.
4. Faulkner LR, Juul D, Andrade NN, et al. Recent trends in american board of psychiatry and neurology psychiatric subspecialties. Acad Psychiatry. 2011;35(1):35-39.
5. Maintenance of certification program. American Board of Psychiatry and Neurology, Inc. http://abpn.com/ downloads/moc/moc_web_doc.pdf. Published May 2014. Accessed August 25, 2014.
6. Physician folios. American Board of Psychiatry and Neurology, Inc. https://application.abpn.com/webclient/ folios.aspx. Accessed August 25, 2014.
7. Maintenance of certification examination in psychiatry 2015 content blueprint. American Board of Psychiatry and Neurology, Inc. http://www.abpn.com/downloads/ content_outlines/MOC/2015-MOC-Psych-blueprint- 060314-EWM-MR.pdf. Published June 2, 2014. Accessed August 25, 2014.
8. Instructions for the 2015 psychiatry maintenance of certification examination. American Board of Psychiatry and Neurology, Inc. http://www.abpn.com/downloads/ content_outlines/MOC/2015-MOC-Psych-Format-and- Scoring-060214-RL-MR.pdf. Published June 2, 2014. Accessed August 25, 2014.
9. DSM-5 conversion. American Board of Psychiatry and Neurology, Inc. http://www.abpn.com/ifas.html. Accessed August 25, 2014.
As part of a general trend among all medical specialty boards, the American Board of Psychiatry and Neurology (ABPN) instituted a recertification process for all new general psychiatry certifications on October 1, 1994.1 In 2000, the individual specialties that constitute the American Board of Medical Specialties (ABMS) subsequently agreed to develop a comprehensive maintenance of certification (MOC) process to demonstrate ongoing learning and competency beyond what could be captured by a recertification examination alone.
All ABMS member boards now use a 4-part process for recertification. For ABPN, those 4 core components are listed in the Table.1,2
ABPN component 1 (maintaining an unrestricted medical license) and component 4 (passing the recertification examination) are straightforward; however, requirements for continuing medical education (CME), including the specific need to accrue ABPN-approved self-assessment (SA) CME hours, and the Improvement in Medical Practice (performance in practice, or PIP) module, have stoked significant commentary and confusion.
Based on feedback,3,4 ABPN in 2014:
• modified the SA and PIP requirements for physicians who certified or recertified between 2005 and 2011
• changed the specific requirement for the PIP feedback component.
These modifications only added to feelings of uncertainty about the MOC process among many psychiatrists.5
Given the professional and personal importance attached to maintaining one’s general and subspecialty certifications, the 2 parts of this article—here and in the January 2015 issue—have been constructed to highlight current ABPN MOC requirements and provide resources for understanding, tracking, and completing the SA and PIP portions.
In addition to this review, I urge all physicians who are subject to MOC to read the 20-page revised MOC Program bookleta (version 2.1, May 2014).5
aDownload the booklet at www.abpn.com/downloads/moc/ moc_web_doc.pdf.
Who must recertify?
As of October 1, 1994, all physicians who achieve ABPN certifications in general psychiatry are issued a 10-year, time-limited certificate that expires on December 31 of the 10th year.3 Note that the 10-year, time-limited certificate in child and adolescent psychiatry began in 1995 and expires 10 years later on December 31.
Certificates in the subspecialties (addiction psychiatry, forensic psychiatry, geriatric psychiatry, etc.), including those issued before October 1, 1994, are 10-year, time-limited certificates that expire on December 31.3 This expiration date often is overlooked by physicians who are exempt from the MOC process for their general psychiatry, or child and adolescent psychiatry certification. There is no exemption for any subspecialty certificate (aside from child and adolescent psychiatry before 1995), regardless of the date of issue.
Moreover, physicians who hold a certificate in a subspecialty also must maintain certification in their specialty (general psychiatry) to apply for recertification in their subspecialization. One exception: Diplomates in child and adolescent psychiatry do not need to maintain current certification in general psychiatry for their subspecialty certification to remain valid or to recertify in child and adolescent psychiatry.
The need to maintain multiple certifications can seem onerous, but note that CME, SA, and PIP activities that have been completed in one area of specialization or subspecialization accrue and count for multiple certifications for diplomates certified in 2 or more areas.5
Get started!
Tracking your progress is critical to keeping up with MOC requirements. You can do this with a personal spreadsheet or by using online resources. Although it is not required, ABPN has established a system that allows diplomates to create and maintain, at no cost, a physician folio on the ABPN server that facilitates documentation of CME hours, including specific SA hours, and PIP module completion.6 All diplomates are required to maintain records of SA activities, CME activities, and PIP units; the ABPN will audit approximately 5% of examination applications.5
Regardless of what documentation method you choose, you should establish an active profile on the ABPN site (www.abpn. com/folios), confirm your contact information, and, if you are not active clinically, update your clinical status. ABPN requires that diplomates self-report their clinical status every 24 months—information that is available to the public. Clinical status also identifies to ABPN those PIP modules that you must complete.
ABPN recognizes 3 categories of clinical status5:
1. Clinically active. Provided any amount of direct or consultative care, or both, in the preceding 24 months, including supervision of residents.
a) Engaged in direct or consultative care, or both, sufficient to complete Improvement in Medical Practice (PIP) units.
b) Engaged in direct or consultative care, or both, that is insufficient to complete PIP units.
2. Clinically inactive. Did not provide direct or consultative care in the preceding 24 months.
3. Status unknown. No information is available on clinical activity.
Based on these definitions, physicians in Category 1a are required to complete all components of the MOC program, including PIP units; physicians in Category 1b or Category 2 are required to complete all components of the MOC program except PIP units.
A change in status from Category 1b or 2 to Category 1a (eg, moving from a purely administrative position to one with clinical duties) requires completion of ≥1 PIP unit.
The easy parts
Licenses. Maintaining your unrestricted professional license(s) is mandatory; the language of this requirement is unambiguous (Table).5 The plural form of license is intentional: Some physicians have medical licenses in multiple states and, in some jurisdictions, licenses are required to supervise physician assistants and other personnel or to prescribe controlled substances. Any restriction on a professional license should be discussed with ABPN and resolved to prevent rejection of the examination application.5
Examinations. For physicians who are not yet enrolled in the continuous-MOC (C-MOC) process (to be discussed in Part 2 of this article), an application to take the examination in Year 10 can be filed in Year 9 of the cycle—after the CME, SA, and PIP requirements are completed. Once a diplomate becomes subject to the C-MOC process by certifying or recertifying from 2012 onwards, completion of each 3-year module of CME, SA, and PIP will not coincide with the 10-year time frame of the examination.
The application deadline for all MOC examinations typically is the year before the examination; the examination should be taken in the year the certificate expires, although it can be taken earlier if desired.7 The examinations are computer-based and administered at a certified testing center. For diplomates who have more than 1 ABPN certificate and want to combine multiple examinations into 1 test session, a reduced fee structure applies.
The general psychiatry examination comprises 220 single-answer, multiple-choice questions that must be completed within 290 minutes, with 10 extra minutes allotted to read on-screen instructions, sign in, and complete a post-examination survey.8 The combined examinations comprise 100 questions from each ABPN specialty or subspecialty area.5
The content of the 2015 general psychiatry examinationb is available on the ABPN Web site.7 Note that the recertification examination in general psychiatry does not cover neurology topics.
bDownload the outline of the examination at www.abpn.com/ downloads/content_outlines/MOC/2015-MOC-Psych-blueprint-060314-EWM-MR.pdf.
Examinations administered in 2015 and 2016 will use only diagnostic criteria that have not changed from DSM-IV-TR9: Neither obsolete diagnoses or subtypes from DSM-IV-TR nor new diagnoses or subtypes in DSM-5 (eg, hoarding disorder) will be tested.9 Diagnoses that are exactly or substantially the same will be tested; these include diagnoses:
• with a name change only (eg, “phonological disorder” in DSM-IV-TR is “speech sound disorder” in DSM-5)
• expanded into >1 new diagnosis (eg, hypochondriasis was expanded to 2 new diagnoses: somatic symptom disorder and illness anxiety disorder)
• subsumed or combined into a new diagnosis (eg, substance use and dependence are now combined into substance use disorder in DSM-5).9
For these diagnoses, both DSM-IV-TR and DSM-5 diagnoses will be provided on examinations.
Beginning in 2017, all examinations will use DSM-5 classifications and diagnostic criteria.9
Part 2 of this article in the January 2015 issue reviews other key aspects of MOC: continuing medical education (CME), including self-assessment requirements; performance in practice (PIP); and continuous maintenance of certification (C-MOC).
BOTTOM LINE
Maintenance of certification (MOC) is a manageable process, although it requires you to be familiar with its various elements, including the duration of certification, licensing requirements, and the examination. Start the process by (1) establishing a login on the ABPN Web site and (2) reviewing the MOC program booklet.
Related Resources
• ABPN MOC home page. www.abpn.com/moc.html
• ABPN-approved products for SA, CME, and PIP modules. www.abpn.com/moc_products.asp
• Peer and patient feedback forms– Peer feedback form v1. www.abpn.com/downloads/moc/PIP-peer-feedback-v1-051914.pdf
– Patient feedback form v1. www.abpn.com/downloads/moc/PIP-patient-feedback-v1-051914.pdf
– Patient feedback form v2. www.abpn.com/downloads/moc/PIP-patient-feedback-v2-051914.pdf
• ABPN physician folio page. https://application.abpn.com/webclient/folios.aspx
Disclosure
Dr. Meyer reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
As part of a general trend among all medical specialty boards, the American Board of Psychiatry and Neurology (ABPN) instituted a recertification process for all new general psychiatry certifications on October 1, 1994.1 In 2000, the individual specialties that constitute the American Board of Medical Specialties (ABMS) subsequently agreed to develop a comprehensive maintenance of certification (MOC) process to demonstrate ongoing learning and competency beyond what could be captured by a recertification examination alone.
All ABMS member boards now use a 4-part process for recertification. For ABPN, those 4 core components are listed in the Table.1,2
ABPN component 1 (maintaining an unrestricted medical license) and component 4 (passing the recertification examination) are straightforward; however, requirements for continuing medical education (CME), including the specific need to accrue ABPN-approved self-assessment (SA) CME hours, and the Improvement in Medical Practice (performance in practice, or PIP) module, have stoked significant commentary and confusion.
Based on feedback,3,4 ABPN in 2014:
• modified the SA and PIP requirements for physicians who certified or recertified between 2005 and 2011
• changed the specific requirement for the PIP feedback component.
These modifications only added to feelings of uncertainty about the MOC process among many psychiatrists.5
Given the professional and personal importance attached to maintaining one’s general and subspecialty certifications, the 2 parts of this article—here and in the January 2015 issue—have been constructed to highlight current ABPN MOC requirements and provide resources for understanding, tracking, and completing the SA and PIP portions.
In addition to this review, I urge all physicians who are subject to MOC to read the 20-page revised MOC Program bookleta (version 2.1, May 2014).5
aDownload the booklet at www.abpn.com/downloads/moc/ moc_web_doc.pdf.
Who must recertify?
As of October 1, 1994, all physicians who achieve ABPN certifications in general psychiatry are issued a 10-year, time-limited certificate that expires on December 31 of the 10th year.3 Note that the 10-year, time-limited certificate in child and adolescent psychiatry began in 1995 and expires 10 years later on December 31.
Certificates in the subspecialties (addiction psychiatry, forensic psychiatry, geriatric psychiatry, etc.), including those issued before October 1, 1994, are 10-year, time-limited certificates that expire on December 31.3 This expiration date often is overlooked by physicians who are exempt from the MOC process for their general psychiatry, or child and adolescent psychiatry certification. There is no exemption for any subspecialty certificate (aside from child and adolescent psychiatry before 1995), regardless of the date of issue.
Moreover, physicians who hold a certificate in a subspecialty also must maintain certification in their specialty (general psychiatry) to apply for recertification in their subspecialization. One exception: Diplomates in child and adolescent psychiatry do not need to maintain current certification in general psychiatry for their subspecialty certification to remain valid or to recertify in child and adolescent psychiatry.
The need to maintain multiple certifications can seem onerous, but note that CME, SA, and PIP activities that have been completed in one area of specialization or subspecialization accrue and count for multiple certifications for diplomates certified in 2 or more areas.5
Get started!
Tracking your progress is critical to keeping up with MOC requirements. You can do this with a personal spreadsheet or by using online resources. Although it is not required, ABPN has established a system that allows diplomates to create and maintain, at no cost, a physician folio on the ABPN server that facilitates documentation of CME hours, including specific SA hours, and PIP module completion.6 All diplomates are required to maintain records of SA activities, CME activities, and PIP units; the ABPN will audit approximately 5% of examination applications.5
Regardless of what documentation method you choose, you should establish an active profile on the ABPN site (www.abpn. com/folios), confirm your contact information, and, if you are not active clinically, update your clinical status. ABPN requires that diplomates self-report their clinical status every 24 months—information that is available to the public. Clinical status also identifies to ABPN those PIP modules that you must complete.
ABPN recognizes 3 categories of clinical status5:
1. Clinically active. Provided any amount of direct or consultative care, or both, in the preceding 24 months, including supervision of residents.
a) Engaged in direct or consultative care, or both, sufficient to complete Improvement in Medical Practice (PIP) units.
b) Engaged in direct or consultative care, or both, that is insufficient to complete PIP units.
2. Clinically inactive. Did not provide direct or consultative care in the preceding 24 months.
3. Status unknown. No information is available on clinical activity.
Based on these definitions, physicians in Category 1a are required to complete all components of the MOC program, including PIP units; physicians in Category 1b or Category 2 are required to complete all components of the MOC program except PIP units.
A change in status from Category 1b or 2 to Category 1a (eg, moving from a purely administrative position to one with clinical duties) requires completion of ≥1 PIP unit.
The easy parts
Licenses. Maintaining your unrestricted professional license(s) is mandatory; the language of this requirement is unambiguous (Table).5 The plural form of license is intentional: Some physicians have medical licenses in multiple states and, in some jurisdictions, licenses are required to supervise physician assistants and other personnel or to prescribe controlled substances. Any restriction on a professional license should be discussed with ABPN and resolved to prevent rejection of the examination application.5
Examinations. For physicians who are not yet enrolled in the continuous-MOC (C-MOC) process (to be discussed in Part 2 of this article), an application to take the examination in Year 10 can be filed in Year 9 of the cycle—after the CME, SA, and PIP requirements are completed. Once a diplomate becomes subject to the C-MOC process by certifying or recertifying from 2012 onwards, completion of each 3-year module of CME, SA, and PIP will not coincide with the 10-year time frame of the examination.
The application deadline for all MOC examinations typically is the year before the examination; the examination should be taken in the year the certificate expires, although it can be taken earlier if desired.7 The examinations are computer-based and administered at a certified testing center. For diplomates who have more than 1 ABPN certificate and want to combine multiple examinations into 1 test session, a reduced fee structure applies.
The general psychiatry examination comprises 220 single-answer, multiple-choice questions that must be completed within 290 minutes, with 10 extra minutes allotted to read on-screen instructions, sign in, and complete a post-examination survey.8 The combined examinations comprise 100 questions from each ABPN specialty or subspecialty area.5
The content of the 2015 general psychiatry examinationb is available on the ABPN Web site.7 Note that the recertification examination in general psychiatry does not cover neurology topics.
bDownload the outline of the examination at www.abpn.com/ downloads/content_outlines/MOC/2015-MOC-Psych-blueprint-060314-EWM-MR.pdf.
Examinations administered in 2015 and 2016 will use only diagnostic criteria that have not changed from DSM-IV-TR9: Neither obsolete diagnoses or subtypes from DSM-IV-TR nor new diagnoses or subtypes in DSM-5 (eg, hoarding disorder) will be tested.9 Diagnoses that are exactly or substantially the same will be tested; these include diagnoses:
• with a name change only (eg, “phonological disorder” in DSM-IV-TR is “speech sound disorder” in DSM-5)
• expanded into >1 new diagnosis (eg, hypochondriasis was expanded to 2 new diagnoses: somatic symptom disorder and illness anxiety disorder)
• subsumed or combined into a new diagnosis (eg, substance use and dependence are now combined into substance use disorder in DSM-5).9
For these diagnoses, both DSM-IV-TR and DSM-5 diagnoses will be provided on examinations.
Beginning in 2017, all examinations will use DSM-5 classifications and diagnostic criteria.9
Part 2 of this article in the January 2015 issue reviews other key aspects of MOC: continuing medical education (CME), including self-assessment requirements; performance in practice (PIP); and continuous maintenance of certification (C-MOC).
BOTTOM LINE
Maintenance of certification (MOC) is a manageable process, although it requires you to be familiar with its various elements, including the duration of certification, licensing requirements, and the examination. Start the process by (1) establishing a login on the ABPN Web site and (2) reviewing the MOC program booklet.
Related Resources
• ABPN MOC home page. www.abpn.com/moc.html
• ABPN-approved products for SA, CME, and PIP modules. www.abpn.com/moc_products.asp
• Peer and patient feedback forms– Peer feedback form v1. www.abpn.com/downloads/moc/PIP-peer-feedback-v1-051914.pdf
– Patient feedback form v1. www.abpn.com/downloads/moc/PIP-patient-feedback-v1-051914.pdf
– Patient feedback form v2. www.abpn.com/downloads/moc/PIP-patient-feedback-v2-051914.pdf
• ABPN physician folio page. https://application.abpn.com/webclient/folios.aspx
Disclosure
Dr. Meyer reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Faulkner LR, Tivnan PW, Winstead DK, et al. The ABPN Maintenance of Certification Program for psychiatrists: past history, current status, and future directions. Acad Psychiatry. 2008;32(3):241-248.
2. Ebert MH, Faulkner L, Stubbe DE, et al. Maintenance of certification in psychiatry. J Clin Psychiatry. 2009;70(10):e39.
3. Faulkner LR, Vondrak PA. Frequently asked questions about maintenance of certification (MOC). J Clin Psychiatry. 2010;71(5):632-633.
4. Faulkner LR, Juul D, Andrade NN, et al. Recent trends in american board of psychiatry and neurology psychiatric subspecialties. Acad Psychiatry. 2011;35(1):35-39.
5. Maintenance of certification program. American Board of Psychiatry and Neurology, Inc. http://abpn.com/ downloads/moc/moc_web_doc.pdf. Published May 2014. Accessed August 25, 2014.
6. Physician folios. American Board of Psychiatry and Neurology, Inc. https://application.abpn.com/webclient/ folios.aspx. Accessed August 25, 2014.
7. Maintenance of certification examination in psychiatry 2015 content blueprint. American Board of Psychiatry and Neurology, Inc. http://www.abpn.com/downloads/ content_outlines/MOC/2015-MOC-Psych-blueprint- 060314-EWM-MR.pdf. Published June 2, 2014. Accessed August 25, 2014.
8. Instructions for the 2015 psychiatry maintenance of certification examination. American Board of Psychiatry and Neurology, Inc. http://www.abpn.com/downloads/ content_outlines/MOC/2015-MOC-Psych-Format-and- Scoring-060214-RL-MR.pdf. Published June 2, 2014. Accessed August 25, 2014.
9. DSM-5 conversion. American Board of Psychiatry and Neurology, Inc. http://www.abpn.com/ifas.html. Accessed August 25, 2014.
1. Faulkner LR, Tivnan PW, Winstead DK, et al. The ABPN Maintenance of Certification Program for psychiatrists: past history, current status, and future directions. Acad Psychiatry. 2008;32(3):241-248.
2. Ebert MH, Faulkner L, Stubbe DE, et al. Maintenance of certification in psychiatry. J Clin Psychiatry. 2009;70(10):e39.
3. Faulkner LR, Vondrak PA. Frequently asked questions about maintenance of certification (MOC). J Clin Psychiatry. 2010;71(5):632-633.
4. Faulkner LR, Juul D, Andrade NN, et al. Recent trends in american board of psychiatry and neurology psychiatric subspecialties. Acad Psychiatry. 2011;35(1):35-39.
5. Maintenance of certification program. American Board of Psychiatry and Neurology, Inc. http://abpn.com/ downloads/moc/moc_web_doc.pdf. Published May 2014. Accessed August 25, 2014.
6. Physician folios. American Board of Psychiatry and Neurology, Inc. https://application.abpn.com/webclient/ folios.aspx. Accessed August 25, 2014.
7. Maintenance of certification examination in psychiatry 2015 content blueprint. American Board of Psychiatry and Neurology, Inc. http://www.abpn.com/downloads/ content_outlines/MOC/2015-MOC-Psych-blueprint- 060314-EWM-MR.pdf. Published June 2, 2014. Accessed August 25, 2014.
8. Instructions for the 2015 psychiatry maintenance of certification examination. American Board of Psychiatry and Neurology, Inc. http://www.abpn.com/downloads/ content_outlines/MOC/2015-MOC-Psych-Format-and- Scoring-060214-RL-MR.pdf. Published June 2, 2014. Accessed August 25, 2014.
9. DSM-5 conversion. American Board of Psychiatry and Neurology, Inc. http://www.abpn.com/ifas.html. Accessed August 25, 2014.

Loss of enzyme induction: Ups and downs of a hidden drug-drug interaction
Mr. P, age 35 with schizophrenia and seizure disorder, has been maintained on risperidone, 6 mg qhs, and phenytoin, 300 mg qhs. For clinical reasons, the treating neurologist changes the anticonvulsant to divalproex. One week later, Mr. P presents to the emergency room complaining of jaw and neck stiffness.
Ms. K, age 43 with a history of schizoaffective disorder, bipolar type, and erratic medication adherence, is being treated with quetiapine, 600 mg at bedtime, and carbamazepine, 1,000 mg/d. Between appointments she stops taking carbamazepine, believing it is causing her to hear voices from her television. Two weeks later, the manager of Ms. K’s independent living facility tells the psychiatrist that the patient appears excessively sedated and has fallen twice in the past few days.
Mrs. T, a 39-year-old state hospital resident with schizoaffective disorder, bipolar type, has been treated with clozapine, 250 mg bid for 6 months; her most recent trough serum level was 492 ng/mL. She smokes 15 cigarettes/d. Two weeks after the hospital institutes a no-smoking policy, Mrs. T complains of excessive drooling and lightheadedness. Her trough clozapine level is now 875 ng/mL.
Discontinuing a medication that has enzyme-inducing effects presents a hidden problem for patients receiving antipsychotic pharmacotherapy. Certain hepatic enzymes responsible for antipsychotic metabolism—as well those involved in intercellular drug transport—are induced by medication or environmental exposures.1,2 Adding a medication that induces these enzymes to the regimen of a patient receiving antipsychotic therapy can result in markedly reduced serum antipsychotic levels, and discontinuing an inducing agent can result in increased antipsychotic levels.
Drug-drug interactions (DDIs) are a substantial contributor to adverse drug reactions (Box).3-7 Antipsychotic prescribing information highlights potential DDIs from the use of enzyme inhibitors and inducers but identifies only effects caused by adding a second agent. The prescriber remains the sole line of defense for monitoring for DDIs when discontinuing a medication that has inducing or inhibiting effects.
Most psychiatrists are aware that certain medications have clinically significant effects on cytochrome P450 (CYP) activity and of the potential for CYP inhibitors to generate DDIs. Clinicians often are aware of antidepressant medications’ CYP-inhibiting effects, know that levels of other medications will change when discontinuing a potent P450 inhibitor, and understand the need to increase dosages of medications influenced by such agents.8
However, few studies have evaluated the effects of enzyme induction on antipsychotic drug levels,9,10 and the literature rarely discusses changes in serum drug levels after loss of enzyme or drug transport induction.11 If unrecognized, these changes may have significant clinical consequences.
Drug-drug interactions (DDIs) are a common and often preventable cause of morbidity and mortality. National surveillance data showed 700,000 emergency room visits related to adverse drug reactions (ADRs) in the 2 years from January 2004 through 2005.3 ADRs are particularly concerning for psychiatrists managing polypharmacy regimens for patients with severe mental disorders such as schizophrenia.
Literature on DDIs with antipsychotics focuses primarily on kinetic interactions that generate supratherapeutic drug levels.4,5 Because development of side effects is associated with reduced adherence, these kinetic interactions may increase the risk of adverse effects and lead to patients stopping the antipsychotic treatment.6,7
Two induction pathways
The primary mechanism underlying clinically significant DDIs occurs during CYP-mediated phase I metabolism. Molecules undergo oxidative conversion into metabolites that can be conjugated by phase II enzymes, generating more soluble forms that facilitate excretion.
The workhorse of human CYP metabolism is 3A4 (Table 1),12,13 which comprises 30% of hepatic activity and 70% of gut cytochrome activity.14 CYP 1A2 is responsible for 10% to 15% of CYP activity.
Both CYP 3A4 and 1A2 are inducible. A wide variety of medications induce 3A4 activity. The list of 1A2 inducers is shorter; the most common are aryl hydrocarbons from cigarette smoke and proton pump inhibitors.
CYP 2D6 accounts for 20% of hepatic cytochrome activity but is not inducible. CYP 2D6 is well known to psychiatrists because some selective serotonin reuptake inhibitors (SSRIs) and the non-SSRI antidepressant bupropion are potent inhibitors of this enzyme.15,16
P-glycoprotein (PGP) induction. Transmembrane shuttles such as P-glycoprotein (PGP) are an important component of drug disposition. PGP belongs to the family of ATP binding cassette (ABC) transporters that bring molecules across cellular barriers.17,18 It was first described in cancer cells that developed multiple drug resistance (MDR) and is often referred to as MDR1.19 PGP is encoded on human chromosome 7 and expressed in normal tissues, particularly in areas where cells seek to limit drug influx, such as those lining the luminal surface of the small and large intestine and those lining the blood-brain barrier and blood-testis barrier. The expression of PGP in hepatic cells promotes drug clearance by enhancing biliary drug excretion.
PGP is encoded on the same chromosome as CYP 3A4, and these 2 proteins frequently are expressed in the same cells, particularly in the intestinal lining and liver. Moreover, PGP is inducible, and there is substantial overlap between medications that are substrates for—or inducers of—PGP and CYP 3A4 activity. This makes it challenging to determine whether the kinetic effects of a second medication are the result of interference of 3A4, PGP, or both.
Polymorphisms in PGP activity may influence the penetration of psychotropic medications into the CNS. Studies indicate an association between certain PGP polymorphisms and treatment outcomes.17,18
Table 1
What induces CYP 1A2 and 3A4?
| Enzyme | Description | Inducers* |
|---|---|---|
| CYP 1A2 |
| Aryl hydrocarbons (smoking), protonpump inhibitors (omeprazole > lansoprazole > pantoprazole), modafinil, St. John’s wort, chargrilled meat, cruciferous vegetables such as broccoli and cabbage, flavones, protein supplements |
| CYP 3A4 |
| Carbamazepine, phenytoin, phenobarbital, rifampin, oxcarbazepine, efavirenz, glucocorticoids, modafinil, nevirapine, pioglitazone, St. John’s wort |
| * Listed in order from strongest to weakest induction | ||
| CYP: cytochrome P450; PGP: P-glycoprotein | ||
| Source: References 12,13 | ||
Stopping an inducer
In general, inducers of CYP enzymes stimulate gene transcription within hours of exposure; maximum transcriptional activity occurs after 10 to 12 hours of exposure. As transcription increases, the concentration of the CYP mRNA transcript steadily accumulates, as does concentration of CYP protein.
After an inducer is discontinued, transcription returns to basal levels within 18 hours; however, the degradation of CYP proteins is a first-order process, with a half-life of 8 to 30 hours. As a result, the decrease in cellular CYP concentration—and the level of activity—lags behind the decreased synthesis from reduced mRNA levels.
Interactions with antipsychotics
Effects on serum antipsychotic levels caused by discontinuing a CYP or PGP inducer can be predicted from data on decreases in antipsychotic levels following inducer exposure. Except for ziprasidone and paliperidone, most atypical antipsychotics are prone to substantial decreases during concomitant inducer use (Table 2).21
The effect of enzyme inducers on risperidone is particularly interesting. Conversion of risperidone to its active metabolite 9-OH risperidone (paliperidone) occurs primarily via 2D6,22 yet concurrent use of carbamazepine—a potent CYP 3A4 inducer—results in a 50% decrease in the concentration of the active moiety (risperidone plus 9-OH risperidone). This finding and other early investigations suggested that CYP 3A had a role in risperidone metabolism,23,24 but these early studies and case series often involved molecules that had activity at both 3A4 and PGP. Further research clarified that effects on PGP—and not 3A4—are responsible for the changes in risperidone metabolism observed with the use of carbamazepine and other medications.25,26
Induction in case patients: Follow-up. Regardless of whether induction is mediated by ≥1 metabolic pathways, the loss of the inducer will result in serum antipsychotic increases that are proportional to the initial decrease.20 For example, with risperidone, the expected decrease is 50%. Therefore, after Mr. P stopped taking phenytoin, his serum risperidone level would be expected to double, which resulted in extrapyramidal side effects.
Quetiapine clearance is increased 5-fold by inducer exposure, so a clinician treating Ms. K would expect a marked increase in somnolence—and possibly orthostasis—as serum quetiapine levels peak 1 to 2 weeks following carbamazepine discontinuation.
The effects of smoking cessation on serum clozapine levels have been well-documented.1,27 Clinicians should anticipate median increases in serum clozapine levels of 55% after a patient discontinues smoking (aryl hydrocarbon exposure), but changes vary substantially among individuals. Mrs. T’s serum clozapine increased approximately 78%.
Careful clinical monitoring and slow downward adjustment of antipsychotic doses could have prevented the adverse effects these 3 patients experienced after loss of CYP/PGP induction and the consequences those side effects present for future medication adherence. When loss of induction is unplanned—as when Ms. K stopped taking carbamazepine but continued quetiapine—clinicians need to be alert to the fact that the patient was prescribed an inducer and include the loss of induction as a hypothesis for the patient’s somnolence.
Table 2
Effects of CYP/PGP induction on atypical antipsychotics
| Antipsychotic | Metabolic pathways | Effect of induction |
|---|---|---|
| Aripiprazole | 2D6 and 3A4 convert aripiprazole to active metabolite dehydro-aripiprazole | 3A4 induction decreases maximum concentration of aripiprazole and metabolite by 70% |
| Clozapine | Multiple enzymes convert clozapine to N-desmethylclozapine; mean contributions of CYP 1A2, 2C19, 3A4, 2C9, and 2D6 are 30%, 24%, 22%, 12%, and 6%, respectively, with CYP 1A2 predominantly involved at low concentrations | Loss of smoking-related 1A2 induction results in 50% increase in serum levels |
| Olanzapine | Direct glucuronidation or 1A2-mediated oxidation to N-desmethlyolanzapine | Carbamazepine use increases clearance by 50%. Olanzapine concentration:dose ratio is about 5-fold lower in smokers (7.9 +/- 2.6) than in nonsmokers (1.56 +/- 1.1; P |
| Paliperidone | 59% excreted unchanged in urine; phase I metabolism accounts for ≤10% of drug clearance | Unlikely to significantly impact levels, but impact of PGP induction is unknown |
| Quetiapine | 3A4-mediated sulfoxidation to inactive metabolite is primary pathway, but numerous metabolites noted, with 1 active metabolite (norquetiapine) | Phenytoin increases clearance 5-fold |
| Risperidone | 2D6 converts risperidone to active metabolite 9-OH risperidone | In a drug interaction study of risperidone, 6 mg/d for 3 weeks, followed by 3 weeks of carbamazepine, active moiety concentration was decreased by about 50% |
| Ziprasidone | 3A4 (~1/3); aldehyde oxidase (~2/3) | Approximately 35% decrease in ziprasidone exposure by carbamazepine |
| CYP: cytochrome P450; PGP: P-glycoprotein | ||
| Source: Reference 21 | ||
Clinical considerations
In the absence of detailed data on antipsychotic metabolism, clinicians can make intelligent decisions regarding potential DDIs by:
- knowing the extent of induction by common offenders (such as carbamazepine or phenytoin) documented in the medication’s prescribing information or demonstrated through convincing case reports or case series
- memorizing the list of CYP 1A2 and CYP 3A4/PGP inducers.
Patients who may be susceptible to effects from loss of enzyme induction (including smokers receiving olanzapine or clozapine or others taking 3A4/PGP inducers) must be identified, and plans made for dosage adjustments if inducing agents are discontinued for a sufficient time (≥1 week) to result in downregulation of CYP or PGP activity. A slow taper of the antipsychotic over 1 to 2 weeks to the new target dose should compensate for loss of enzyme or PGP induction.
For newer antipsychotic medications with limited data, the proposed discontinuation of an inducer should, at the minimum, prompt a discussion between the psychiatrist and patient regarding the expected increase in serum antipsychotic levels and potential adverse effects that may result. Clinicians also must make every attempt to stay apprised of a patient’s current medications, bearing in mind that another provider may prescribe an inducer. Patients with schizophrenia always should be educated to contact the psychiatrist following any change in medication regimen, placing particular emphasis on the 1 or 2 medications that are known to be implicated in DDIs with the patient’s current antipsychotic.
- Flockhart DA. Drug interactions: Cytochrome P450 drug interaction table. Indiana University School of Medicine. 2007. http://medicine.iupui.edu/flockhart/table.htm.
- Cozza KL, Armstrong SC, Oesterheld JR. Concise guide to drug interaction principles for medical practice: Cytochrome P450s, UGTS, p-glycoproteins. Washington, DC: American Psychiatric Press, Inc; 2003.
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Carbamazepine • Carbatrol, Tegretol
- Clozapine • Clozaril
- Divalproex • Depakote
- Efavirenz • Sustiva
- Lansoprazole • Prevacid
- Modafinil • Provigil
- Nevirapine • Viramune
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Oxcarbazepine • Trileptal
- Paliperidone • Invega
- Pantoprazole • Protonix
- Phenobarbital • Barbita, Luminal, others
- Phenytoin • Dilantin
- Pioglitazone • Actos
- Quetiapine • Seroquel
- Rifampin • Rifadin, Rimactane
- Risperidone • Risperdal
- Ziprasidone • Geodon
Dr. Meyer receives grant/research support from the National Institute of Mental Health, Pfizer Inc., and the University of California. He is a consultant to Bristol-Myers Squibb, Organon, Vanda Pharmaceuticals, and Wyeth, and a speaker for AstraZeneca, Bristol-Myers Squibb, Dainippon Sumitomo Pharma, and Pfizer Inc.
Ms. Leckband reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21:569-574.
2. Wong YW, Yeh C, Thyrum PT. The effects of concomitant phenytoin administration on the steady-state pharmacokinetics of quetiapine. J Clin Psychopharmacol. 2001;21:89-93.
3. Budnitz DS, Pollock DA, Weidenbach KN, et al. National surveillance of emergency department visits for outpatient adverse drug events. JAMA. 2006;296:1858-1866.
4. Prior TI, Baker GB. Interactions between the cytochrome P450 system and the second-generation antipsychotics. J Psychiatry Neurosci. 2003;28:99-112.
5. Spina E, de Leon J. Metabolic drug interactions with newer antipsychotics: a comparative review. Basic Clin Pharmacol Toxicol. 2007;100:4-22.
6. Preskorn SH. Drug-drug interactions: proof of relevance (part II): cause of tolerability problems or noncompliance. J Psychiatr Pract. 2005;11:397-401.
7. Weiden PJ, Mackell JA, McDonnell D. Obesity as a risk factor for antipsychotic noncompliance. Schizophr Res. 2004;66:51-7.
8. Preskorn SH, Flockhart D. 2006 guide to psychiatric drug interactions. Prim Psychiatry. 2006;13:35-64.
9. Spina E, Perucca E. Clinical significance of pharmacokinetic interactions between antiepileptic and psychotropic drugs. Epilepsia. 2002;43(suppl 2):37-44.
10. Meyer JM. Drug-drug interactions with antipsychotics. CNS Spectr. 2007;12:6-9.
11. Takahashi H, Yoshida K, Higuchi H, et al. Development of parkinsonian symptoms after discontinuation of carbamazepine in patients concurrently treated with risperidone: two case reports. Clin Neuropharmacol. 2001;24:358-360.
12. Rendic S. Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab Rev. 2002;34:83-448.
13. Hong CC, Tang BK, Hammond GL, et al. Cytochrome P450 1A2 (CYP1A2) activity and risk factors for breast cancer: a cross-sectional study. Breast Cancer Res. 2004;6:R352-365.
14. Cozza KL, Armstrong SC, Oesterheld JR. Concise guide to drug interaction principles for medical practice: cytochrome P450s, UGTS, p-glycoproteins. Washington, DC: American Psychiatric Press, Inc; 2003.
15. Kirchheiner J, Seeringer A. Clinical implications of pharmacogenetics of cytochrome P450 drug metabolizing enzymes. Biochim Biophys Acta. 2007;1770:489-494.
16. Kotlyar M, Brauer LH, Tracy TS, et al. Inhibition of CYP2D6 activity by bupropion. J Clin Psychopharmacol. 2005;25:226-229.
17. Uhr M, Tontsch A, Namendorf C, et al. Polymorphisms in the drug transporter gene ABCB1 predict antidepressant treatment response in depression. Neuron. 2008;57:203-209.
18. Bozina N, Kuzman MR, Medved V, et al. Associations between MDR1 gene polymorphisms and schizophrenia and therapeutic response to olanzapine in female schizophrenic patients. J Psychiatr Res. 2008;42:89-97.
19. Kim RB. Drugs as p-glycoprotein substrates, inhibitors, and inducers. Drug Metab Rev. 2002;34:47-54.
20. Hollenberg PF. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab Rev. 2002;34:17-35.
21. Physicians’ desk reference. 62nd ed. Montvale, NJ: Thomson Healthcare Inc.; 2007.
22. Heykants J, Huang ML, Mannens G, et al. The pharmacokinetics of risperidone in humans: a summary. J Clin Psychiatry. 1994;55 (suppl):13-7.
23. de Leon J, Bork J. Risperidone and cytochrome P450 3A. J Clin Psychiatry. 1997;58:450.-
24. Lane HY, Chang WH. Risperidone-carbamazepine interactions: is cytochrome P450 3A involved? J Clin Psychiatry. 1998;59:430-431.
25. Ejsing TB, Pedersen AD, Linnet K. P-glycoprotein interaction with risperidone and 9-OH-risperidone studied in vitro, in knock-out mice and in drug-drug interaction experiments. Hum Psychopharmacol. 2005;20:493-500.
26. Cousein E, Barthelemy C, Poullain S, et al. P-glycoprotein and cytochrome P450 3A4 involvement in risperidone transport using an in vitro Caco-2/TC7 model and an in vivo model. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:878-886.
27. Rostami-Hodjegan A, Amin AM, et al. Influence of dose, cigarette smoking, age, sex, and metabolic activity on plasma clozapine concentrations: a predictive model and nomograms to aid clozapine dose adjustment and to assess compliance in individual patients. J Clin Psychopharmacol. 2004;24:70-78.
28. Flockhart DA. Drug interactions: cytochrome P450 drug interaction table. Indiana University School of Medicine. 2007. Available at: http://medicine.iupui.edu/flockhart/table.htm. Accessed October 22, 2008.
Mr. P, age 35 with schizophrenia and seizure disorder, has been maintained on risperidone, 6 mg qhs, and phenytoin, 300 mg qhs. For clinical reasons, the treating neurologist changes the anticonvulsant to divalproex. One week later, Mr. P presents to the emergency room complaining of jaw and neck stiffness.
Ms. K, age 43 with a history of schizoaffective disorder, bipolar type, and erratic medication adherence, is being treated with quetiapine, 600 mg at bedtime, and carbamazepine, 1,000 mg/d. Between appointments she stops taking carbamazepine, believing it is causing her to hear voices from her television. Two weeks later, the manager of Ms. K’s independent living facility tells the psychiatrist that the patient appears excessively sedated and has fallen twice in the past few days.
Mrs. T, a 39-year-old state hospital resident with schizoaffective disorder, bipolar type, has been treated with clozapine, 250 mg bid for 6 months; her most recent trough serum level was 492 ng/mL. She smokes 15 cigarettes/d. Two weeks after the hospital institutes a no-smoking policy, Mrs. T complains of excessive drooling and lightheadedness. Her trough clozapine level is now 875 ng/mL.
Discontinuing a medication that has enzyme-inducing effects presents a hidden problem for patients receiving antipsychotic pharmacotherapy. Certain hepatic enzymes responsible for antipsychotic metabolism—as well those involved in intercellular drug transport—are induced by medication or environmental exposures.1,2 Adding a medication that induces these enzymes to the regimen of a patient receiving antipsychotic therapy can result in markedly reduced serum antipsychotic levels, and discontinuing an inducing agent can result in increased antipsychotic levels.
Drug-drug interactions (DDIs) are a substantial contributor to adverse drug reactions (Box).3-7 Antipsychotic prescribing information highlights potential DDIs from the use of enzyme inhibitors and inducers but identifies only effects caused by adding a second agent. The prescriber remains the sole line of defense for monitoring for DDIs when discontinuing a medication that has inducing or inhibiting effects.
Most psychiatrists are aware that certain medications have clinically significant effects on cytochrome P450 (CYP) activity and of the potential for CYP inhibitors to generate DDIs. Clinicians often are aware of antidepressant medications’ CYP-inhibiting effects, know that levels of other medications will change when discontinuing a potent P450 inhibitor, and understand the need to increase dosages of medications influenced by such agents.8
However, few studies have evaluated the effects of enzyme induction on antipsychotic drug levels,9,10 and the literature rarely discusses changes in serum drug levels after loss of enzyme or drug transport induction.11 If unrecognized, these changes may have significant clinical consequences.
Drug-drug interactions (DDIs) are a common and often preventable cause of morbidity and mortality. National surveillance data showed 700,000 emergency room visits related to adverse drug reactions (ADRs) in the 2 years from January 2004 through 2005.3 ADRs are particularly concerning for psychiatrists managing polypharmacy regimens for patients with severe mental disorders such as schizophrenia.
Literature on DDIs with antipsychotics focuses primarily on kinetic interactions that generate supratherapeutic drug levels.4,5 Because development of side effects is associated with reduced adherence, these kinetic interactions may increase the risk of adverse effects and lead to patients stopping the antipsychotic treatment.6,7
Two induction pathways
The primary mechanism underlying clinically significant DDIs occurs during CYP-mediated phase I metabolism. Molecules undergo oxidative conversion into metabolites that can be conjugated by phase II enzymes, generating more soluble forms that facilitate excretion.
The workhorse of human CYP metabolism is 3A4 (Table 1),12,13 which comprises 30% of hepatic activity and 70% of gut cytochrome activity.14 CYP 1A2 is responsible for 10% to 15% of CYP activity.
Both CYP 3A4 and 1A2 are inducible. A wide variety of medications induce 3A4 activity. The list of 1A2 inducers is shorter; the most common are aryl hydrocarbons from cigarette smoke and proton pump inhibitors.
CYP 2D6 accounts for 20% of hepatic cytochrome activity but is not inducible. CYP 2D6 is well known to psychiatrists because some selective serotonin reuptake inhibitors (SSRIs) and the non-SSRI antidepressant bupropion are potent inhibitors of this enzyme.15,16
P-glycoprotein (PGP) induction. Transmembrane shuttles such as P-glycoprotein (PGP) are an important component of drug disposition. PGP belongs to the family of ATP binding cassette (ABC) transporters that bring molecules across cellular barriers.17,18 It was first described in cancer cells that developed multiple drug resistance (MDR) and is often referred to as MDR1.19 PGP is encoded on human chromosome 7 and expressed in normal tissues, particularly in areas where cells seek to limit drug influx, such as those lining the luminal surface of the small and large intestine and those lining the blood-brain barrier and blood-testis barrier. The expression of PGP in hepatic cells promotes drug clearance by enhancing biliary drug excretion.
PGP is encoded on the same chromosome as CYP 3A4, and these 2 proteins frequently are expressed in the same cells, particularly in the intestinal lining and liver. Moreover, PGP is inducible, and there is substantial overlap between medications that are substrates for—or inducers of—PGP and CYP 3A4 activity. This makes it challenging to determine whether the kinetic effects of a second medication are the result of interference of 3A4, PGP, or both.
Polymorphisms in PGP activity may influence the penetration of psychotropic medications into the CNS. Studies indicate an association between certain PGP polymorphisms and treatment outcomes.17,18
Table 1
What induces CYP 1A2 and 3A4?
| Enzyme | Description | Inducers* |
|---|---|---|
| CYP 1A2 |
| Aryl hydrocarbons (smoking), protonpump inhibitors (omeprazole > lansoprazole > pantoprazole), modafinil, St. John’s wort, chargrilled meat, cruciferous vegetables such as broccoli and cabbage, flavones, protein supplements |
| CYP 3A4 |
| Carbamazepine, phenytoin, phenobarbital, rifampin, oxcarbazepine, efavirenz, glucocorticoids, modafinil, nevirapine, pioglitazone, St. John’s wort |
| * Listed in order from strongest to weakest induction | ||
| CYP: cytochrome P450; PGP: P-glycoprotein | ||
| Source: References 12,13 | ||
Stopping an inducer
In general, inducers of CYP enzymes stimulate gene transcription within hours of exposure; maximum transcriptional activity occurs after 10 to 12 hours of exposure. As transcription increases, the concentration of the CYP mRNA transcript steadily accumulates, as does concentration of CYP protein.
After an inducer is discontinued, transcription returns to basal levels within 18 hours; however, the degradation of CYP proteins is a first-order process, with a half-life of 8 to 30 hours. As a result, the decrease in cellular CYP concentration—and the level of activity—lags behind the decreased synthesis from reduced mRNA levels.
Interactions with antipsychotics
Effects on serum antipsychotic levels caused by discontinuing a CYP or PGP inducer can be predicted from data on decreases in antipsychotic levels following inducer exposure. Except for ziprasidone and paliperidone, most atypical antipsychotics are prone to substantial decreases during concomitant inducer use (Table 2).21
The effect of enzyme inducers on risperidone is particularly interesting. Conversion of risperidone to its active metabolite 9-OH risperidone (paliperidone) occurs primarily via 2D6,22 yet concurrent use of carbamazepine—a potent CYP 3A4 inducer—results in a 50% decrease in the concentration of the active moiety (risperidone plus 9-OH risperidone). This finding and other early investigations suggested that CYP 3A had a role in risperidone metabolism,23,24 but these early studies and case series often involved molecules that had activity at both 3A4 and PGP. Further research clarified that effects on PGP—and not 3A4—are responsible for the changes in risperidone metabolism observed with the use of carbamazepine and other medications.25,26
Induction in case patients: Follow-up. Regardless of whether induction is mediated by ≥1 metabolic pathways, the loss of the inducer will result in serum antipsychotic increases that are proportional to the initial decrease.20 For example, with risperidone, the expected decrease is 50%. Therefore, after Mr. P stopped taking phenytoin, his serum risperidone level would be expected to double, which resulted in extrapyramidal side effects.
Quetiapine clearance is increased 5-fold by inducer exposure, so a clinician treating Ms. K would expect a marked increase in somnolence—and possibly orthostasis—as serum quetiapine levels peak 1 to 2 weeks following carbamazepine discontinuation.
The effects of smoking cessation on serum clozapine levels have been well-documented.1,27 Clinicians should anticipate median increases in serum clozapine levels of 55% after a patient discontinues smoking (aryl hydrocarbon exposure), but changes vary substantially among individuals. Mrs. T’s serum clozapine increased approximately 78%.
Careful clinical monitoring and slow downward adjustment of antipsychotic doses could have prevented the adverse effects these 3 patients experienced after loss of CYP/PGP induction and the consequences those side effects present for future medication adherence. When loss of induction is unplanned—as when Ms. K stopped taking carbamazepine but continued quetiapine—clinicians need to be alert to the fact that the patient was prescribed an inducer and include the loss of induction as a hypothesis for the patient’s somnolence.
Table 2
Effects of CYP/PGP induction on atypical antipsychotics
| Antipsychotic | Metabolic pathways | Effect of induction |
|---|---|---|
| Aripiprazole | 2D6 and 3A4 convert aripiprazole to active metabolite dehydro-aripiprazole | 3A4 induction decreases maximum concentration of aripiprazole and metabolite by 70% |
| Clozapine | Multiple enzymes convert clozapine to N-desmethylclozapine; mean contributions of CYP 1A2, 2C19, 3A4, 2C9, and 2D6 are 30%, 24%, 22%, 12%, and 6%, respectively, with CYP 1A2 predominantly involved at low concentrations | Loss of smoking-related 1A2 induction results in 50% increase in serum levels |
| Olanzapine | Direct glucuronidation or 1A2-mediated oxidation to N-desmethlyolanzapine | Carbamazepine use increases clearance by 50%. Olanzapine concentration:dose ratio is about 5-fold lower in smokers (7.9 +/- 2.6) than in nonsmokers (1.56 +/- 1.1; P |
| Paliperidone | 59% excreted unchanged in urine; phase I metabolism accounts for ≤10% of drug clearance | Unlikely to significantly impact levels, but impact of PGP induction is unknown |
| Quetiapine | 3A4-mediated sulfoxidation to inactive metabolite is primary pathway, but numerous metabolites noted, with 1 active metabolite (norquetiapine) | Phenytoin increases clearance 5-fold |
| Risperidone | 2D6 converts risperidone to active metabolite 9-OH risperidone | In a drug interaction study of risperidone, 6 mg/d for 3 weeks, followed by 3 weeks of carbamazepine, active moiety concentration was decreased by about 50% |
| Ziprasidone | 3A4 (~1/3); aldehyde oxidase (~2/3) | Approximately 35% decrease in ziprasidone exposure by carbamazepine |
| CYP: cytochrome P450; PGP: P-glycoprotein | ||
| Source: Reference 21 | ||
Clinical considerations
In the absence of detailed data on antipsychotic metabolism, clinicians can make intelligent decisions regarding potential DDIs by:
- knowing the extent of induction by common offenders (such as carbamazepine or phenytoin) documented in the medication’s prescribing information or demonstrated through convincing case reports or case series
- memorizing the list of CYP 1A2 and CYP 3A4/PGP inducers.
Patients who may be susceptible to effects from loss of enzyme induction (including smokers receiving olanzapine or clozapine or others taking 3A4/PGP inducers) must be identified, and plans made for dosage adjustments if inducing agents are discontinued for a sufficient time (≥1 week) to result in downregulation of CYP or PGP activity. A slow taper of the antipsychotic over 1 to 2 weeks to the new target dose should compensate for loss of enzyme or PGP induction.
For newer antipsychotic medications with limited data, the proposed discontinuation of an inducer should, at the minimum, prompt a discussion between the psychiatrist and patient regarding the expected increase in serum antipsychotic levels and potential adverse effects that may result. Clinicians also must make every attempt to stay apprised of a patient’s current medications, bearing in mind that another provider may prescribe an inducer. Patients with schizophrenia always should be educated to contact the psychiatrist following any change in medication regimen, placing particular emphasis on the 1 or 2 medications that are known to be implicated in DDIs with the patient’s current antipsychotic.
- Flockhart DA. Drug interactions: Cytochrome P450 drug interaction table. Indiana University School of Medicine. 2007. http://medicine.iupui.edu/flockhart/table.htm.
- Cozza KL, Armstrong SC, Oesterheld JR. Concise guide to drug interaction principles for medical practice: Cytochrome P450s, UGTS, p-glycoproteins. Washington, DC: American Psychiatric Press, Inc; 2003.
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Carbamazepine • Carbatrol, Tegretol
- Clozapine • Clozaril
- Divalproex • Depakote
- Efavirenz • Sustiva
- Lansoprazole • Prevacid
- Modafinil • Provigil
- Nevirapine • Viramune
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Oxcarbazepine • Trileptal
- Paliperidone • Invega
- Pantoprazole • Protonix
- Phenobarbital • Barbita, Luminal, others
- Phenytoin • Dilantin
- Pioglitazone • Actos
- Quetiapine • Seroquel
- Rifampin • Rifadin, Rimactane
- Risperidone • Risperdal
- Ziprasidone • Geodon
Dr. Meyer receives grant/research support from the National Institute of Mental Health, Pfizer Inc., and the University of California. He is a consultant to Bristol-Myers Squibb, Organon, Vanda Pharmaceuticals, and Wyeth, and a speaker for AstraZeneca, Bristol-Myers Squibb, Dainippon Sumitomo Pharma, and Pfizer Inc.
Ms. Leckband reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Mr. P, age 35 with schizophrenia and seizure disorder, has been maintained on risperidone, 6 mg qhs, and phenytoin, 300 mg qhs. For clinical reasons, the treating neurologist changes the anticonvulsant to divalproex. One week later, Mr. P presents to the emergency room complaining of jaw and neck stiffness.
Ms. K, age 43 with a history of schizoaffective disorder, bipolar type, and erratic medication adherence, is being treated with quetiapine, 600 mg at bedtime, and carbamazepine, 1,000 mg/d. Between appointments she stops taking carbamazepine, believing it is causing her to hear voices from her television. Two weeks later, the manager of Ms. K’s independent living facility tells the psychiatrist that the patient appears excessively sedated and has fallen twice in the past few days.
Mrs. T, a 39-year-old state hospital resident with schizoaffective disorder, bipolar type, has been treated with clozapine, 250 mg bid for 6 months; her most recent trough serum level was 492 ng/mL. She smokes 15 cigarettes/d. Two weeks after the hospital institutes a no-smoking policy, Mrs. T complains of excessive drooling and lightheadedness. Her trough clozapine level is now 875 ng/mL.
Discontinuing a medication that has enzyme-inducing effects presents a hidden problem for patients receiving antipsychotic pharmacotherapy. Certain hepatic enzymes responsible for antipsychotic metabolism—as well those involved in intercellular drug transport—are induced by medication or environmental exposures.1,2 Adding a medication that induces these enzymes to the regimen of a patient receiving antipsychotic therapy can result in markedly reduced serum antipsychotic levels, and discontinuing an inducing agent can result in increased antipsychotic levels.
Drug-drug interactions (DDIs) are a substantial contributor to adverse drug reactions (Box).3-7 Antipsychotic prescribing information highlights potential DDIs from the use of enzyme inhibitors and inducers but identifies only effects caused by adding a second agent. The prescriber remains the sole line of defense for monitoring for DDIs when discontinuing a medication that has inducing or inhibiting effects.
Most psychiatrists are aware that certain medications have clinically significant effects on cytochrome P450 (CYP) activity and of the potential for CYP inhibitors to generate DDIs. Clinicians often are aware of antidepressant medications’ CYP-inhibiting effects, know that levels of other medications will change when discontinuing a potent P450 inhibitor, and understand the need to increase dosages of medications influenced by such agents.8
However, few studies have evaluated the effects of enzyme induction on antipsychotic drug levels,9,10 and the literature rarely discusses changes in serum drug levels after loss of enzyme or drug transport induction.11 If unrecognized, these changes may have significant clinical consequences.
Drug-drug interactions (DDIs) are a common and often preventable cause of morbidity and mortality. National surveillance data showed 700,000 emergency room visits related to adverse drug reactions (ADRs) in the 2 years from January 2004 through 2005.3 ADRs are particularly concerning for psychiatrists managing polypharmacy regimens for patients with severe mental disorders such as schizophrenia.
Literature on DDIs with antipsychotics focuses primarily on kinetic interactions that generate supratherapeutic drug levels.4,5 Because development of side effects is associated with reduced adherence, these kinetic interactions may increase the risk of adverse effects and lead to patients stopping the antipsychotic treatment.6,7
Two induction pathways
The primary mechanism underlying clinically significant DDIs occurs during CYP-mediated phase I metabolism. Molecules undergo oxidative conversion into metabolites that can be conjugated by phase II enzymes, generating more soluble forms that facilitate excretion.
The workhorse of human CYP metabolism is 3A4 (Table 1),12,13 which comprises 30% of hepatic activity and 70% of gut cytochrome activity.14 CYP 1A2 is responsible for 10% to 15% of CYP activity.
Both CYP 3A4 and 1A2 are inducible. A wide variety of medications induce 3A4 activity. The list of 1A2 inducers is shorter; the most common are aryl hydrocarbons from cigarette smoke and proton pump inhibitors.
CYP 2D6 accounts for 20% of hepatic cytochrome activity but is not inducible. CYP 2D6 is well known to psychiatrists because some selective serotonin reuptake inhibitors (SSRIs) and the non-SSRI antidepressant bupropion are potent inhibitors of this enzyme.15,16
P-glycoprotein (PGP) induction. Transmembrane shuttles such as P-glycoprotein (PGP) are an important component of drug disposition. PGP belongs to the family of ATP binding cassette (ABC) transporters that bring molecules across cellular barriers.17,18 It was first described in cancer cells that developed multiple drug resistance (MDR) and is often referred to as MDR1.19 PGP is encoded on human chromosome 7 and expressed in normal tissues, particularly in areas where cells seek to limit drug influx, such as those lining the luminal surface of the small and large intestine and those lining the blood-brain barrier and blood-testis barrier. The expression of PGP in hepatic cells promotes drug clearance by enhancing biliary drug excretion.
PGP is encoded on the same chromosome as CYP 3A4, and these 2 proteins frequently are expressed in the same cells, particularly in the intestinal lining and liver. Moreover, PGP is inducible, and there is substantial overlap between medications that are substrates for—or inducers of—PGP and CYP 3A4 activity. This makes it challenging to determine whether the kinetic effects of a second medication are the result of interference of 3A4, PGP, or both.
Polymorphisms in PGP activity may influence the penetration of psychotropic medications into the CNS. Studies indicate an association between certain PGP polymorphisms and treatment outcomes.17,18
Table 1
What induces CYP 1A2 and 3A4?
| Enzyme | Description | Inducers* |
|---|---|---|
| CYP 1A2 |
| Aryl hydrocarbons (smoking), protonpump inhibitors (omeprazole > lansoprazole > pantoprazole), modafinil, St. John’s wort, chargrilled meat, cruciferous vegetables such as broccoli and cabbage, flavones, protein supplements |
| CYP 3A4 |
| Carbamazepine, phenytoin, phenobarbital, rifampin, oxcarbazepine, efavirenz, glucocorticoids, modafinil, nevirapine, pioglitazone, St. John’s wort |
| * Listed in order from strongest to weakest induction | ||
| CYP: cytochrome P450; PGP: P-glycoprotein | ||
| Source: References 12,13 | ||
Stopping an inducer
In general, inducers of CYP enzymes stimulate gene transcription within hours of exposure; maximum transcriptional activity occurs after 10 to 12 hours of exposure. As transcription increases, the concentration of the CYP mRNA transcript steadily accumulates, as does concentration of CYP protein.
After an inducer is discontinued, transcription returns to basal levels within 18 hours; however, the degradation of CYP proteins is a first-order process, with a half-life of 8 to 30 hours. As a result, the decrease in cellular CYP concentration—and the level of activity—lags behind the decreased synthesis from reduced mRNA levels.
Interactions with antipsychotics
Effects on serum antipsychotic levels caused by discontinuing a CYP or PGP inducer can be predicted from data on decreases in antipsychotic levels following inducer exposure. Except for ziprasidone and paliperidone, most atypical antipsychotics are prone to substantial decreases during concomitant inducer use (Table 2).21
The effect of enzyme inducers on risperidone is particularly interesting. Conversion of risperidone to its active metabolite 9-OH risperidone (paliperidone) occurs primarily via 2D6,22 yet concurrent use of carbamazepine—a potent CYP 3A4 inducer—results in a 50% decrease in the concentration of the active moiety (risperidone plus 9-OH risperidone). This finding and other early investigations suggested that CYP 3A had a role in risperidone metabolism,23,24 but these early studies and case series often involved molecules that had activity at both 3A4 and PGP. Further research clarified that effects on PGP—and not 3A4—are responsible for the changes in risperidone metabolism observed with the use of carbamazepine and other medications.25,26
Induction in case patients: Follow-up. Regardless of whether induction is mediated by ≥1 metabolic pathways, the loss of the inducer will result in serum antipsychotic increases that are proportional to the initial decrease.20 For example, with risperidone, the expected decrease is 50%. Therefore, after Mr. P stopped taking phenytoin, his serum risperidone level would be expected to double, which resulted in extrapyramidal side effects.
Quetiapine clearance is increased 5-fold by inducer exposure, so a clinician treating Ms. K would expect a marked increase in somnolence—and possibly orthostasis—as serum quetiapine levels peak 1 to 2 weeks following carbamazepine discontinuation.
The effects of smoking cessation on serum clozapine levels have been well-documented.1,27 Clinicians should anticipate median increases in serum clozapine levels of 55% after a patient discontinues smoking (aryl hydrocarbon exposure), but changes vary substantially among individuals. Mrs. T’s serum clozapine increased approximately 78%.
Careful clinical monitoring and slow downward adjustment of antipsychotic doses could have prevented the adverse effects these 3 patients experienced after loss of CYP/PGP induction and the consequences those side effects present for future medication adherence. When loss of induction is unplanned—as when Ms. K stopped taking carbamazepine but continued quetiapine—clinicians need to be alert to the fact that the patient was prescribed an inducer and include the loss of induction as a hypothesis for the patient’s somnolence.
Table 2
Effects of CYP/PGP induction on atypical antipsychotics
| Antipsychotic | Metabolic pathways | Effect of induction |
|---|---|---|
| Aripiprazole | 2D6 and 3A4 convert aripiprazole to active metabolite dehydro-aripiprazole | 3A4 induction decreases maximum concentration of aripiprazole and metabolite by 70% |
| Clozapine | Multiple enzymes convert clozapine to N-desmethylclozapine; mean contributions of CYP 1A2, 2C19, 3A4, 2C9, and 2D6 are 30%, 24%, 22%, 12%, and 6%, respectively, with CYP 1A2 predominantly involved at low concentrations | Loss of smoking-related 1A2 induction results in 50% increase in serum levels |
| Olanzapine | Direct glucuronidation or 1A2-mediated oxidation to N-desmethlyolanzapine | Carbamazepine use increases clearance by 50%. Olanzapine concentration:dose ratio is about 5-fold lower in smokers (7.9 +/- 2.6) than in nonsmokers (1.56 +/- 1.1; P |
| Paliperidone | 59% excreted unchanged in urine; phase I metabolism accounts for ≤10% of drug clearance | Unlikely to significantly impact levels, but impact of PGP induction is unknown |
| Quetiapine | 3A4-mediated sulfoxidation to inactive metabolite is primary pathway, but numerous metabolites noted, with 1 active metabolite (norquetiapine) | Phenytoin increases clearance 5-fold |
| Risperidone | 2D6 converts risperidone to active metabolite 9-OH risperidone | In a drug interaction study of risperidone, 6 mg/d for 3 weeks, followed by 3 weeks of carbamazepine, active moiety concentration was decreased by about 50% |
| Ziprasidone | 3A4 (~1/3); aldehyde oxidase (~2/3) | Approximately 35% decrease in ziprasidone exposure by carbamazepine |
| CYP: cytochrome P450; PGP: P-glycoprotein | ||
| Source: Reference 21 | ||
Clinical considerations
In the absence of detailed data on antipsychotic metabolism, clinicians can make intelligent decisions regarding potential DDIs by:
- knowing the extent of induction by common offenders (such as carbamazepine or phenytoin) documented in the medication’s prescribing information or demonstrated through convincing case reports or case series
- memorizing the list of CYP 1A2 and CYP 3A4/PGP inducers.
Patients who may be susceptible to effects from loss of enzyme induction (including smokers receiving olanzapine or clozapine or others taking 3A4/PGP inducers) must be identified, and plans made for dosage adjustments if inducing agents are discontinued for a sufficient time (≥1 week) to result in downregulation of CYP or PGP activity. A slow taper of the antipsychotic over 1 to 2 weeks to the new target dose should compensate for loss of enzyme or PGP induction.
For newer antipsychotic medications with limited data, the proposed discontinuation of an inducer should, at the minimum, prompt a discussion between the psychiatrist and patient regarding the expected increase in serum antipsychotic levels and potential adverse effects that may result. Clinicians also must make every attempt to stay apprised of a patient’s current medications, bearing in mind that another provider may prescribe an inducer. Patients with schizophrenia always should be educated to contact the psychiatrist following any change in medication regimen, placing particular emphasis on the 1 or 2 medications that are known to be implicated in DDIs with the patient’s current antipsychotic.
- Flockhart DA. Drug interactions: Cytochrome P450 drug interaction table. Indiana University School of Medicine. 2007. http://medicine.iupui.edu/flockhart/table.htm.
- Cozza KL, Armstrong SC, Oesterheld JR. Concise guide to drug interaction principles for medical practice: Cytochrome P450s, UGTS, p-glycoproteins. Washington, DC: American Psychiatric Press, Inc; 2003.
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Carbamazepine • Carbatrol, Tegretol
- Clozapine • Clozaril
- Divalproex • Depakote
- Efavirenz • Sustiva
- Lansoprazole • Prevacid
- Modafinil • Provigil
- Nevirapine • Viramune
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Oxcarbazepine • Trileptal
- Paliperidone • Invega
- Pantoprazole • Protonix
- Phenobarbital • Barbita, Luminal, others
- Phenytoin • Dilantin
- Pioglitazone • Actos
- Quetiapine • Seroquel
- Rifampin • Rifadin, Rimactane
- Risperidone • Risperdal
- Ziprasidone • Geodon
Dr. Meyer receives grant/research support from the National Institute of Mental Health, Pfizer Inc., and the University of California. He is a consultant to Bristol-Myers Squibb, Organon, Vanda Pharmaceuticals, and Wyeth, and a speaker for AstraZeneca, Bristol-Myers Squibb, Dainippon Sumitomo Pharma, and Pfizer Inc.
Ms. Leckband reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21:569-574.
2. Wong YW, Yeh C, Thyrum PT. The effects of concomitant phenytoin administration on the steady-state pharmacokinetics of quetiapine. J Clin Psychopharmacol. 2001;21:89-93.
3. Budnitz DS, Pollock DA, Weidenbach KN, et al. National surveillance of emergency department visits for outpatient adverse drug events. JAMA. 2006;296:1858-1866.
4. Prior TI, Baker GB. Interactions between the cytochrome P450 system and the second-generation antipsychotics. J Psychiatry Neurosci. 2003;28:99-112.
5. Spina E, de Leon J. Metabolic drug interactions with newer antipsychotics: a comparative review. Basic Clin Pharmacol Toxicol. 2007;100:4-22.
6. Preskorn SH. Drug-drug interactions: proof of relevance (part II): cause of tolerability problems or noncompliance. J Psychiatr Pract. 2005;11:397-401.
7. Weiden PJ, Mackell JA, McDonnell D. Obesity as a risk factor for antipsychotic noncompliance. Schizophr Res. 2004;66:51-7.
8. Preskorn SH, Flockhart D. 2006 guide to psychiatric drug interactions. Prim Psychiatry. 2006;13:35-64.
9. Spina E, Perucca E. Clinical significance of pharmacokinetic interactions between antiepileptic and psychotropic drugs. Epilepsia. 2002;43(suppl 2):37-44.
10. Meyer JM. Drug-drug interactions with antipsychotics. CNS Spectr. 2007;12:6-9.
11. Takahashi H, Yoshida K, Higuchi H, et al. Development of parkinsonian symptoms after discontinuation of carbamazepine in patients concurrently treated with risperidone: two case reports. Clin Neuropharmacol. 2001;24:358-360.
12. Rendic S. Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab Rev. 2002;34:83-448.
13. Hong CC, Tang BK, Hammond GL, et al. Cytochrome P450 1A2 (CYP1A2) activity and risk factors for breast cancer: a cross-sectional study. Breast Cancer Res. 2004;6:R352-365.
14. Cozza KL, Armstrong SC, Oesterheld JR. Concise guide to drug interaction principles for medical practice: cytochrome P450s, UGTS, p-glycoproteins. Washington, DC: American Psychiatric Press, Inc; 2003.
15. Kirchheiner J, Seeringer A. Clinical implications of pharmacogenetics of cytochrome P450 drug metabolizing enzymes. Biochim Biophys Acta. 2007;1770:489-494.
16. Kotlyar M, Brauer LH, Tracy TS, et al. Inhibition of CYP2D6 activity by bupropion. J Clin Psychopharmacol. 2005;25:226-229.
17. Uhr M, Tontsch A, Namendorf C, et al. Polymorphisms in the drug transporter gene ABCB1 predict antidepressant treatment response in depression. Neuron. 2008;57:203-209.
18. Bozina N, Kuzman MR, Medved V, et al. Associations between MDR1 gene polymorphisms and schizophrenia and therapeutic response to olanzapine in female schizophrenic patients. J Psychiatr Res. 2008;42:89-97.
19. Kim RB. Drugs as p-glycoprotein substrates, inhibitors, and inducers. Drug Metab Rev. 2002;34:47-54.
20. Hollenberg PF. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab Rev. 2002;34:17-35.
21. Physicians’ desk reference. 62nd ed. Montvale, NJ: Thomson Healthcare Inc.; 2007.
22. Heykants J, Huang ML, Mannens G, et al. The pharmacokinetics of risperidone in humans: a summary. J Clin Psychiatry. 1994;55 (suppl):13-7.
23. de Leon J, Bork J. Risperidone and cytochrome P450 3A. J Clin Psychiatry. 1997;58:450.-
24. Lane HY, Chang WH. Risperidone-carbamazepine interactions: is cytochrome P450 3A involved? J Clin Psychiatry. 1998;59:430-431.
25. Ejsing TB, Pedersen AD, Linnet K. P-glycoprotein interaction with risperidone and 9-OH-risperidone studied in vitro, in knock-out mice and in drug-drug interaction experiments. Hum Psychopharmacol. 2005;20:493-500.
26. Cousein E, Barthelemy C, Poullain S, et al. P-glycoprotein and cytochrome P450 3A4 involvement in risperidone transport using an in vitro Caco-2/TC7 model and an in vivo model. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:878-886.
27. Rostami-Hodjegan A, Amin AM, et al. Influence of dose, cigarette smoking, age, sex, and metabolic activity on plasma clozapine concentrations: a predictive model and nomograms to aid clozapine dose adjustment and to assess compliance in individual patients. J Clin Psychopharmacol. 2004;24:70-78.
28. Flockhart DA. Drug interactions: cytochrome P450 drug interaction table. Indiana University School of Medicine. 2007. Available at: http://medicine.iupui.edu/flockhart/table.htm. Accessed October 22, 2008.
1. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21:569-574.
2. Wong YW, Yeh C, Thyrum PT. The effects of concomitant phenytoin administration on the steady-state pharmacokinetics of quetiapine. J Clin Psychopharmacol. 2001;21:89-93.
3. Budnitz DS, Pollock DA, Weidenbach KN, et al. National surveillance of emergency department visits for outpatient adverse drug events. JAMA. 2006;296:1858-1866.
4. Prior TI, Baker GB. Interactions between the cytochrome P450 system and the second-generation antipsychotics. J Psychiatry Neurosci. 2003;28:99-112.
5. Spina E, de Leon J. Metabolic drug interactions with newer antipsychotics: a comparative review. Basic Clin Pharmacol Toxicol. 2007;100:4-22.
6. Preskorn SH. Drug-drug interactions: proof of relevance (part II): cause of tolerability problems or noncompliance. J Psychiatr Pract. 2005;11:397-401.
7. Weiden PJ, Mackell JA, McDonnell D. Obesity as a risk factor for antipsychotic noncompliance. Schizophr Res. 2004;66:51-7.
8. Preskorn SH, Flockhart D. 2006 guide to psychiatric drug interactions. Prim Psychiatry. 2006;13:35-64.
9. Spina E, Perucca E. Clinical significance of pharmacokinetic interactions between antiepileptic and psychotropic drugs. Epilepsia. 2002;43(suppl 2):37-44.
10. Meyer JM. Drug-drug interactions with antipsychotics. CNS Spectr. 2007;12:6-9.
11. Takahashi H, Yoshida K, Higuchi H, et al. Development of parkinsonian symptoms after discontinuation of carbamazepine in patients concurrently treated with risperidone: two case reports. Clin Neuropharmacol. 2001;24:358-360.
12. Rendic S. Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab Rev. 2002;34:83-448.
13. Hong CC, Tang BK, Hammond GL, et al. Cytochrome P450 1A2 (CYP1A2) activity and risk factors for breast cancer: a cross-sectional study. Breast Cancer Res. 2004;6:R352-365.
14. Cozza KL, Armstrong SC, Oesterheld JR. Concise guide to drug interaction principles for medical practice: cytochrome P450s, UGTS, p-glycoproteins. Washington, DC: American Psychiatric Press, Inc; 2003.
15. Kirchheiner J, Seeringer A. Clinical implications of pharmacogenetics of cytochrome P450 drug metabolizing enzymes. Biochim Biophys Acta. 2007;1770:489-494.
16. Kotlyar M, Brauer LH, Tracy TS, et al. Inhibition of CYP2D6 activity by bupropion. J Clin Psychopharmacol. 2005;25:226-229.
17. Uhr M, Tontsch A, Namendorf C, et al. Polymorphisms in the drug transporter gene ABCB1 predict antidepressant treatment response in depression. Neuron. 2008;57:203-209.
18. Bozina N, Kuzman MR, Medved V, et al. Associations between MDR1 gene polymorphisms and schizophrenia and therapeutic response to olanzapine in female schizophrenic patients. J Psychiatr Res. 2008;42:89-97.
19. Kim RB. Drugs as p-glycoprotein substrates, inhibitors, and inducers. Drug Metab Rev. 2002;34:47-54.
20. Hollenberg PF. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab Rev. 2002;34:17-35.
21. Physicians’ desk reference. 62nd ed. Montvale, NJ: Thomson Healthcare Inc.; 2007.
22. Heykants J, Huang ML, Mannens G, et al. The pharmacokinetics of risperidone in humans: a summary. J Clin Psychiatry. 1994;55 (suppl):13-7.
23. de Leon J, Bork J. Risperidone and cytochrome P450 3A. J Clin Psychiatry. 1997;58:450.-
24. Lane HY, Chang WH. Risperidone-carbamazepine interactions: is cytochrome P450 3A involved? J Clin Psychiatry. 1998;59:430-431.
25. Ejsing TB, Pedersen AD, Linnet K. P-glycoprotein interaction with risperidone and 9-OH-risperidone studied in vitro, in knock-out mice and in drug-drug interaction experiments. Hum Psychopharmacol. 2005;20:493-500.
26. Cousein E, Barthelemy C, Poullain S, et al. P-glycoprotein and cytochrome P450 3A4 involvement in risperidone transport using an in vitro Caco-2/TC7 model and an in vivo model. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:878-886.
27. Rostami-Hodjegan A, Amin AM, et al. Influence of dose, cigarette smoking, age, sex, and metabolic activity on plasma clozapine concentrations: a predictive model and nomograms to aid clozapine dose adjustment and to assess compliance in individual patients. J Clin Psychopharmacol. 2004;24:70-78.
28. Flockhart DA. Drug interactions: cytochrome P450 drug interaction table. Indiana University School of Medicine. 2007. Available at: http://medicine.iupui.edu/flockhart/table.htm. Accessed October 22, 2008.