User login
Nausea, vomiting, and panic attacks in a 50-year-old woman
A 50-year-old woman presents to the emergency department because of repeated episodes of vomiting over the past 12 hours. She reports eight episodes of non-bloody, nonbilious emesis associated with palpitations and feelings of anxiety, but with no fever or diarrhea. She has not traveled recently and does not have any sick contacts.
She reports that she never had health problems until 6 months ago, when she began having panic attacks that woke her from sleep. The episodes first occurred once or twice per week, usually at night, and involved palpitations and feelings of anxiety that lasted 2 to 4 hours, but no other associated symptoms. After a month, the episodes began to occur more regularly during the day and were accompanied by a pounding headache that began in the back of her neck and extended up and over her head. Her primary care physician prescribed sertraline (Zoloft) and referred her to a neurologist to evaluate the headaches. The neurologic workup included brain magnetic resonance imaging and electroencephalography, both of which were normal.
After 8 weeks on sertraline, the episodes continued to increase in frequency and severity, and her physician switched her to paroxetine (Paxil) and added lorazepam (Ativan), which did not improve her symptoms. Over the past 2 months, during which time she has not been taking any medications, the episodes began to involve nausea and, more recently, vomiting, with episodes occurring as often as once or twice daily, and with intermittent symptom-free days. None of the prior episodes was accompanied by symptoms as severe as those she is currently experiencing.
She is otherwise healthy with no chronic diseases. Her surgical history includes resection of an angiolipoma from her right arm and dilation and curettage for endometrial polyps. She has no personal or family history of psychiatric illness.
PHYSICAL EXAMINATION
The patient is slender and tremulous but does not appear diaphoretic. Her blood pressure is 176/92 mm Hg, pulse 98, temperature 36.5°C (97.7°F), and respiratory rate 20 per minute. Oxygen saturation by pulse oximetry is 98% on room air. She has dry mucus membranes and orthostatic hypotension, but her physical examination is otherwise normal. Electrocardiography (ECG) shows a normal sinus rhythm with a prolonged QTc of 571 ms and peaked P and T waves.
LABORATORY VALUES
- Hemoglobin 15.6 g/dL (reference range 11.5–15.5)
- Hematocrit 47.2% (36.0–46.0)
- Platelet count 448 × 109/L (150–400)
- White cell count 18.65 × 109/L (3.70–11.00)
- Potassium 2.5 mmol/L (3.5–4.0)
- Chloride 97 mmol/L (98–110)
- Bicarbonate 21 mmol/L (23–32)
- Anion gap 20 mmol/L (0–15)
- Glucose 233 mg/dL (65–100).
Sodium, blood urea nitrogen, and creatinine levels are all within normal limits. Urinalysis suggests a urinary tract infection.
IS THIS A PANIC ATTACK?
1. Which of the following is not characteristic of a panic attack?
- Nausea and vomiting
- Onset during sleep
- Palpitations
- Chest pain or discomfort
- Headache
- Trembling or shaking
According to the Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition) (DSM-IV), the diagnosis of panic attack requires the presence of intense fear or discomfort and four or more other symptoms that may come from any of six domains:
- Cardiovascular: palpitations, pounding heart, tachycardia, and chest pain or discomfort
- Autonomic: sweating, chills or hot flushes, and trembling or shaking
- Pulmonary: shortness of breath or a smothering sensation
- Neurologic: dizziness or light-headedness and paresthesias
- Gastrointestinal: choking and nausea or abdominal distress
- Psychological: compass derealization, depersonalization, and the fear of losing control or “going crazy.”1
Two aspects of the patient’s history may be misinterpreted by those unfamiliar with the symptomatology of panic attack. First, although panic disorder carries an increased risk of many comorbidities, including migraine, headache is not typically associated with the panic attacks themselves.2 Second, while not a part of the diagnostic criteria, sleep disturbances are common in patients with panic disorder, and 30% to 45% of patients with the disorder experience recurrent nocturnal panic attacks.3 Therefore, the correct answer is headache.
THE DIFFERENTIAL DIAGNOSIS
When considering a diagnosis of panic attack or panic disorder, the DSM-IV mandates that medical causes of the symptoms must be excluded. Common conditions causing a similar spectrum of symptoms include hyperthyroidism, caffeine and stimulant use or abuse, asthma, cardiac arrhythmias, alcohol withdrawal, and, more rarely, complex partial seizures and pheochromocytoma.2,4 Many of these conditions can be ruled out by the history alone in a reliable patient.
Our patient’s electrocardiogram showed no evidence of ischemia or arrhythmias. Also, her recent negative neurologic workup makes seizure activity less likely.
Many of this patient’s laboratory abnormalities are easily explained by her repeated bouts of vomiting. Specifically, her elevated hemoglobin level and hematocrit are likely secondary to volume contraction, while hypochloremia is seen following losses of HCl with emesis. Typically, however, patients with vomiting have a hypochloremic metabolic alkalosis, and her low serum bicarbonate level is inconsistent with the history.
Three factors might be contributing to this patient’s hypokalemia. First, in a volume-depleted state, the cortical collecting tubules secrete potassium in exchange for increased sodium reabsorption in an attempt to correct volume status. Second, the alkalotic state caused by losses of acid with vomiting results in a transcellular shift of potassium ions into cells in exchange for hydrogen ions. Third, increased levels of epinephrine also cause a shift of potassium ions into cells.5 Potassium is not lost directly through nausea and vomiting.
A state of catecholamine excess, such as during a severe panic attack or in the presence of a catecholamine-secreting tumor, could explain many of her abnormalities. In addition to causing hypokalemia, epinephrine has a gluconeogenic effect, whereas norepinephrine inhibits insulin release, providing a potential explanation for hyperglycemia in a patient with no risk factors for diabetes. Finally, catecholamine excess contributes to lactic acidosis, which could help to explain the low serum bicarbonate level and the elevated anion gap, but unless we take arterial blood gas measurements, the patient’s acid-base status cannot be determined.
While panic attacks do stimulate the sympathetic nervous system, certain elements of her history raise the clinical suspicion for another process. First, the severity of the electrolyte abnormalities is suspicious. Second, a typical panic attack peaks at 10 minutes and begins to subside, whereas this woman’s symptoms have persisted for 12 hours. Finally, the clinical history, in particular the prominence of headaches associated with the symptoms, is inconsistent with classic panic attack. Consequently, an alternative diagnosis, such as pheochromocytoma, deserves more careful evaluation.
Whenever laboratory results do not fit with the clinical scenario or patient, however, one final possibility should always be considered—laboratory error. Errors can be preanalytical (eg, patient misidentification), analytical, or postanalytical. In aggregate, the frequency of errors in laboratory results is 1 in 214 to 8,316.6 Given that even the more conservative estimates show an incidence higher than that of many of the rare diseases for which clinicians may be testing, laboratory error always deserves consideration.
COULD THIS BE PHEOCHROMOCYTOMA?
Pheochromocytoma is a neuroendocrine tumor most commonly arising from the chromaffin cells of the adrenal medulla. However, extra-adrenal pheochromocytoma, generally paraganglioma, accounts for 15% to 20% of these tumors. Although the condition is generally considered very rare, autopsy studies have demonstrated a prevalence of 0.05%, suggesting that many tumors are either missed or are not clinically significant.
The diagnosis is most often sought in hypertensive patients, a population in which pheochromocytoma has a prevalence of 0.1% to 0.6%.7
2. What is the most common presenting symptom of pheochromocytoma?
- Paroxysmal hypertension
- Sustained hypertension
- Nausea
- Cardiomyopathy
- Headache
- Hemorrhagic shock
- Psychological symptoms such as anxiety or panic
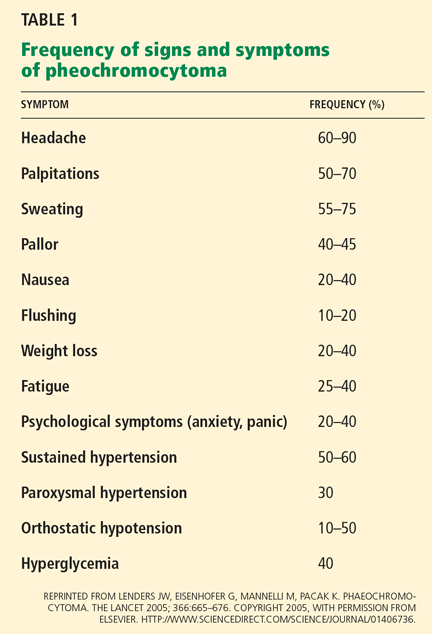
Although 50% to 60% of patients with pheochromocytoma have sustained hypertension, it may be absent in patients with primarily epinephrine-secreting tumors or large tumors that degrade catecholamines, leading to normal or low blood pressure.
Cardiomyopathy is a rare consequence of untreated pheochromocytoma, caused by the effects of excess circulating catecholamines over a long period of time.8 As seen in this patient, a prolonged QTc on ECG associated with elevated levels of norepinephrine and normetanephrine may be the only red flag.9
Pheochromocytoma is typically an extremely well-vascularized tumor, and rupture or hemorrhage is a rare but often fatal complication.
IMPORTANT FAMILY HISTORY
The classic “rule of 10s” suggests that 10% of pheochromocytomas are hereditary, but in fact the number may be higher. In a large cohort of patients with apparently sporadic pheochromocytoma, 25% were found to have germ-line mutations.10 This finding highlights the importance not only of obtaining a thorough family history, but also of genetic testing and counseling once the diagnosis has been made.
3. Which hereditary syndrome is not associated with pheochromocytoma?
- Von Hippel-Lindau syndrome
- Neurofibromatosis type 1
- Neurofibromatosis type 2
- Multiple endocrine neoplasia type 2
- Paraganglioma syndromes
Germ-line mutations in five genes related to three hereditary syndromes (von Hippel-Lindau, neurofibromatosis type 1, and multiple endocrine neoplasia type 2) and in two genes related to paraganglioma syndromes are known to be associated with pheochromocytoma.7
Von Hippel-Lindau syndrome
Von Hippel-Lindau syndrome affects 1 in 36,000 live births. It is caused by a mutation of the von Hippel-Lindau gene on chromosome 3, and 10% to 20% of patients with the syndrome have pheochromocytoma. Other associated problems include renal clear-cell carcinomas and cysts, central nervous system and retinal hemangioblastomas, pancreatic tumors and cysts, endolymphatic tumors, and epididymal cysts.
Neurofibromatosis type 1
Neurofibromatosis type 1 affects 1 in 2,500 to 3,000 individuals and is caused by a mutation of the neurofibromatosis type 1 gene on chromosome 17. The disease is diagnosed by the presence of café-au-lait macules, axillary or inguinal freckling (or both), dermal or plexiform neurofibromas, Lisch nodules, or osseous lesions, but the condition is associated with many other pathologic findings, including optic pathway gliomas, cardiovascular abnormalities, and, in up to 5.7% of patients, pheochromocytoma.11
Neurofibromatosis type 2
Neurofibromatosis type 2 affects 1 in 25,000 live births and is caused by a mutation of the neurofibromatosis type 2 gene on chromosome 22. Patients often develop nervous system tumors, ophthalmologic pathology, and cutaneous lesions, but the condition is not associated with pheochromocytoma.12
Multiple endocrine neoplasia type 2
Multiple endocrine neoplasia type 2 affects 1 in 35,000 individuals and is caused by an activating mutation of the RET proto-oncogene on chromosome 21. The syndrome is most worrisome because of the 95% lifetime risk of medullary thyroid carcinoma in affected patients, but it is also associated with a 50% risk of pheochromocytoma and a 20% to 30% risk of primary hyperparathyroidism. Pheochromocytoma is the presenting clinical problem in 10% to 30% of patients.13
Paraganglioma syndromes
Paraganglioma syndromes are caused by mutations in the three genes encoding subunits of the succinate dehydrogenase enzyme. These mutations affect 1 in 30,000 to 100,000 individuals and incur a 70% lifetime risk of developing pheochromocytoma or paraganglioma.14
TESTING FOR AND MANAGING PHEOCHROMOCYTOMA
The consequences of untreated pheochromocytoma are potentially devastating and include progression to metastatic disease, hypertensive crises, cardiomyopathy, and adrenal hemorrhage. Nevertheless, the average patient goes 3 years before receiving the correct diagnosis.7 Consequently, heightened suspicion and tests with both high sensitivity and specificity are needed.
4. Which test for pheochromocytoma has the highest sensitivity?
- Plasma free metanephrines
- Plasma catecholamines
- Urine total metanephrines
- Urine fractionated metanephrines
- Urine catecholamines
- Urine vanillylmandelic acid

Some researchers have also examined plasma total metanephrines and found that any one of these three biochemical markers at a value two times greater than the upper limit of normal provides specificity of around 95%.16
Further laboratory tests in our patient
- Serum dopamine 70 pg/mL (reference range 0–20)
- Norepinephrine 2,018 pg/mL (80–520)
- Epinephrine 2,479 pg/mL (10–200)
- Free normetanephrine 12 pg/mL (< 0.9)
- Free metanephrine 17.8 pg/mL (< 0.5).
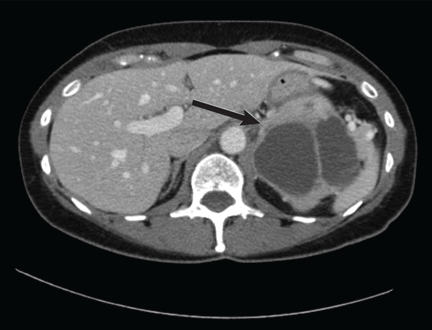
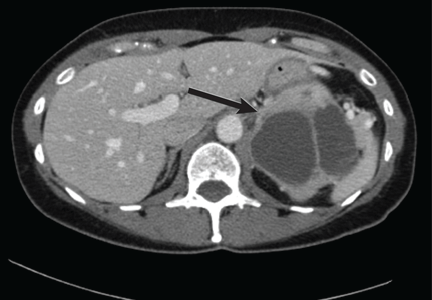
VALUE OF IMAGING STUDIES
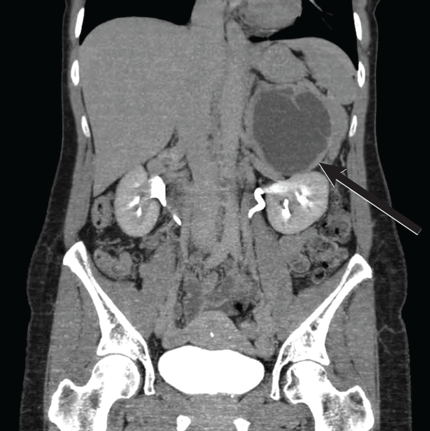
Computed tomography has a sensitivity of up to 95% for detecting adrenal tumors and is able to detect tumors larger than 0.5 cm, but its specificity may be as low as 50%.17 Studies utilizing modern imaging equipment report a prevalence of adrenal incidentaloma of 4%, of which only 1.5% to 11% are pheochromocytoma.18 Thus, while the simultaneous occurrence of pheochromocytoma-like symptoms and an incidentaloma is not common, the potential for unnecessary surgery precludes diagnosis and treatment based on symptoms and imaging alone.
Magnetic resonance imaging has similar sensitivity and specificity but can better characterize the tumor’s blood supply and relationship to other structures.
Iodine 131 metaiodobenzylguanidine (MIBG) scanning is a physiologic study that uses a radiolabeled amine. Since it can identify pheochromocytoma regardless of location, MIBG scanning is typically used when pheochromocytoma is diagnosed by biochemical testing but CT and MRI fail to locate the lesion, or as a follow-up test in patients in whom recurrence or metastasis is suspected or documented.
The specificity of MIBG scanning is 95% to 100%, but the need to protect the thyroid from ablation and the potential need to repeat scans for up to 72 hours make it a poor choice for the initial evaluation.17
5. What is the next best step in our patient’s management?
- Treat her hypertension with a beta-blocker
- Begin a course of alpha-blockade
- Urgent surgery
- Observation
Because of the high concentration of circulating catecholamines and the instability of the tumor to physical manipulation, appropriate medical management before surgical resection is of paramount importance.
Beta-blockade can lead to malignant hypertension due to the unopposed alpha stimulation and must not be begun until alpha-blockade has been started. The standard of care is to give an alpha-blocker or calcium channel blocker 10 to 14 days before surgery. Typically, oral phenoxybenzamine (Dibenzyline) 10 mg twice daily is started and titrated upward daily by 10 to 20 mg until a target seated blood pressure of 120/80 mm Hg is obtained. Selective alpha-1 blockers such as prazosin (Minipress) and terazosin (Hytrin) have also been used and have the benefit of a preserved alpha-2 catecholamine reuptake mechanism.17
After several days, a beta-blocker may be added, particularly for patients with arrhythmias.7 In patients with refractory hypertension, metyrosine (Demser) can be useful.
During surgery, the patient’s hemodynamic stability and glucose levels can fluctuate rapidly from sudden releases of catecholamines during manipulation of the tumor, as well as from the sudden loss of catecholamines after ligation of draining vessels. Advances in medical care have reduced the perioperative death rate from 50% to less than 3%.7,19
CASE CONCLUSION AND FOLLOW-UP
Two months after her initial presentation, the patient underwent open surgery and had the mass removed without complications. She reports that the “panic attacks” have ceased completely.
The recurrence rate of pheochromocytoma is 13% in patients with sporadic disease and 33% in patients with familial syndromes. The overall recurrence rate with long-term follow-up is 17%, half of recurrences being malignant disease. All patients should therefore be followed in the clinic annually for at least 10 years to identify and treat recurrences early,7 and many experts recommend lifelong follow-up, even for patients without hereditary syndromes.17
Nearly every diagnosis in the DSM-IV includes the caveat that medical causes of disease must be excluded before psychiatric labels can be applied. Although panic disorder and panic attack are far more common than pheochromocytoma, just as essential hypertension is far more common than pheochromocytoma, physicians need to remember that pheochromocytoma can cause symptoms common to both illnesses. Thus, while rare conditions are rare, atypical presentations of common conditions may deserve a second glance.
- Yates WR. Phenomenology and epidemiology of panic disorder. Ann Clin Psychiatry 2009; 21:95–102.
- Katon WJ. Clinical practice. Panic disorder. N Engl J Med 2006; 354:2360–2367.
- Craske MG, Tsao JC. Assessment and treatment of nocturnal panic attacks. Sleep Med Rev 2005; 9:173–184.
- Roy-Byrne PP, Craske MG, Stein MB. Panic disorder. Lancet 2006; 368:1023–1032.
- Beal AL, Deuser WE, Beilman GJ. A role for epinephrine in post-traumatic hypokalemia. Shock 2007; 27:358–363.
- Kalra J. Medical errors: impact on clinical laboratories and other critical areas. Clin Biochem 2004; 37:1052–1062.
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet 2005; 366:665–675.
- Leissner KB, Mahmood F, Aragam JR, Amouzgar A, Ortega R. Catecholamine-induced cardiomyopathy and pheochromocytoma. Anesth Analg 2008; 107:410–412.
- Yu R, Furmark L, Wong C. Cardiac abnormalities associated with pheochromocytoma and other adrenal tumors. Endocr Pract 2009; 15:10–16.
- Neumann HP, Bausch B, McWhinney SR, et al; Freiburg-Warsaw-Columbus Pheochromocytoma Study Group. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346:1459–1466.
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics 2009; 123:124–133.
- Asthagiri AR, Parry DM, Butman JA, et al. Neurofibromatosis type 2. Lancet 2009; 373:1974–1986.
- Callender GG, Rich TA, Perrier ND. Multiple endocrine neoplasia syndromes. Surg Clin North Am 2008; 88:863–895.
- Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. J Intern Med 2009; 266:19–42.
- Yu R, Nissen NN, Chopra P, Dhall D, Phillips E, Wei M. Diagnosis and treatment of pheochromocytoma in an academic hospital from 1997 to 2007. Am J Med 2009; 122:85–95.
- Grouzmann E, Drouard-Troalen L, Baudin E, et al. Diagnostic accuracy of free and total metanephrines in plasma and fractionated metanephrines in urine of patients with pheochromocytoma. Eur J Endocrinol 2010; 162:951–960.
- Mittendorf EA, Evans DB, Lee JE, Perrier ND. Pheochromocytoma: advances in genetics, diagnosis, localization, and treatment. Hematol Oncol Clin North Am 2007; 21:509–525.
- Singh PK, Buch HN. Adrenal incidentaloma: evaluation and management. J Clin Pathol 2008; 61:1168–1173.
- Kasturi S, Kutikov A, Guzzo TJ, Smith AL, Wein AJ. Modern management of pheochromocytoma. Nat Clin Pract Urol 2007; 4:630–633.
A 50-year-old woman presents to the emergency department because of repeated episodes of vomiting over the past 12 hours. She reports eight episodes of non-bloody, nonbilious emesis associated with palpitations and feelings of anxiety, but with no fever or diarrhea. She has not traveled recently and does not have any sick contacts.
She reports that she never had health problems until 6 months ago, when she began having panic attacks that woke her from sleep. The episodes first occurred once or twice per week, usually at night, and involved palpitations and feelings of anxiety that lasted 2 to 4 hours, but no other associated symptoms. After a month, the episodes began to occur more regularly during the day and were accompanied by a pounding headache that began in the back of her neck and extended up and over her head. Her primary care physician prescribed sertraline (Zoloft) and referred her to a neurologist to evaluate the headaches. The neurologic workup included brain magnetic resonance imaging and electroencephalography, both of which were normal.
After 8 weeks on sertraline, the episodes continued to increase in frequency and severity, and her physician switched her to paroxetine (Paxil) and added lorazepam (Ativan), which did not improve her symptoms. Over the past 2 months, during which time she has not been taking any medications, the episodes began to involve nausea and, more recently, vomiting, with episodes occurring as often as once or twice daily, and with intermittent symptom-free days. None of the prior episodes was accompanied by symptoms as severe as those she is currently experiencing.
She is otherwise healthy with no chronic diseases. Her surgical history includes resection of an angiolipoma from her right arm and dilation and curettage for endometrial polyps. She has no personal or family history of psychiatric illness.
PHYSICAL EXAMINATION
The patient is slender and tremulous but does not appear diaphoretic. Her blood pressure is 176/92 mm Hg, pulse 98, temperature 36.5°C (97.7°F), and respiratory rate 20 per minute. Oxygen saturation by pulse oximetry is 98% on room air. She has dry mucus membranes and orthostatic hypotension, but her physical examination is otherwise normal. Electrocardiography (ECG) shows a normal sinus rhythm with a prolonged QTc of 571 ms and peaked P and T waves.
LABORATORY VALUES
- Hemoglobin 15.6 g/dL (reference range 11.5–15.5)
- Hematocrit 47.2% (36.0–46.0)
- Platelet count 448 × 109/L (150–400)
- White cell count 18.65 × 109/L (3.70–11.00)
- Potassium 2.5 mmol/L (3.5–4.0)
- Chloride 97 mmol/L (98–110)
- Bicarbonate 21 mmol/L (23–32)
- Anion gap 20 mmol/L (0–15)
- Glucose 233 mg/dL (65–100).
Sodium, blood urea nitrogen, and creatinine levels are all within normal limits. Urinalysis suggests a urinary tract infection.
IS THIS A PANIC ATTACK?
1. Which of the following is not characteristic of a panic attack?
- Nausea and vomiting
- Onset during sleep
- Palpitations
- Chest pain or discomfort
- Headache
- Trembling or shaking
According to the Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition) (DSM-IV), the diagnosis of panic attack requires the presence of intense fear or discomfort and four or more other symptoms that may come from any of six domains:
- Cardiovascular: palpitations, pounding heart, tachycardia, and chest pain or discomfort
- Autonomic: sweating, chills or hot flushes, and trembling or shaking
- Pulmonary: shortness of breath or a smothering sensation
- Neurologic: dizziness or light-headedness and paresthesias
- Gastrointestinal: choking and nausea or abdominal distress
- Psychological: compass derealization, depersonalization, and the fear of losing control or “going crazy.”1
Two aspects of the patient’s history may be misinterpreted by those unfamiliar with the symptomatology of panic attack. First, although panic disorder carries an increased risk of many comorbidities, including migraine, headache is not typically associated with the panic attacks themselves.2 Second, while not a part of the diagnostic criteria, sleep disturbances are common in patients with panic disorder, and 30% to 45% of patients with the disorder experience recurrent nocturnal panic attacks.3 Therefore, the correct answer is headache.
THE DIFFERENTIAL DIAGNOSIS
When considering a diagnosis of panic attack or panic disorder, the DSM-IV mandates that medical causes of the symptoms must be excluded. Common conditions causing a similar spectrum of symptoms include hyperthyroidism, caffeine and stimulant use or abuse, asthma, cardiac arrhythmias, alcohol withdrawal, and, more rarely, complex partial seizures and pheochromocytoma.2,4 Many of these conditions can be ruled out by the history alone in a reliable patient.
Our patient’s electrocardiogram showed no evidence of ischemia or arrhythmias. Also, her recent negative neurologic workup makes seizure activity less likely.
Many of this patient’s laboratory abnormalities are easily explained by her repeated bouts of vomiting. Specifically, her elevated hemoglobin level and hematocrit are likely secondary to volume contraction, while hypochloremia is seen following losses of HCl with emesis. Typically, however, patients with vomiting have a hypochloremic metabolic alkalosis, and her low serum bicarbonate level is inconsistent with the history.
Three factors might be contributing to this patient’s hypokalemia. First, in a volume-depleted state, the cortical collecting tubules secrete potassium in exchange for increased sodium reabsorption in an attempt to correct volume status. Second, the alkalotic state caused by losses of acid with vomiting results in a transcellular shift of potassium ions into cells in exchange for hydrogen ions. Third, increased levels of epinephrine also cause a shift of potassium ions into cells.5 Potassium is not lost directly through nausea and vomiting.
A state of catecholamine excess, such as during a severe panic attack or in the presence of a catecholamine-secreting tumor, could explain many of her abnormalities. In addition to causing hypokalemia, epinephrine has a gluconeogenic effect, whereas norepinephrine inhibits insulin release, providing a potential explanation for hyperglycemia in a patient with no risk factors for diabetes. Finally, catecholamine excess contributes to lactic acidosis, which could help to explain the low serum bicarbonate level and the elevated anion gap, but unless we take arterial blood gas measurements, the patient’s acid-base status cannot be determined.
While panic attacks do stimulate the sympathetic nervous system, certain elements of her history raise the clinical suspicion for another process. First, the severity of the electrolyte abnormalities is suspicious. Second, a typical panic attack peaks at 10 minutes and begins to subside, whereas this woman’s symptoms have persisted for 12 hours. Finally, the clinical history, in particular the prominence of headaches associated with the symptoms, is inconsistent with classic panic attack. Consequently, an alternative diagnosis, such as pheochromocytoma, deserves more careful evaluation.
Whenever laboratory results do not fit with the clinical scenario or patient, however, one final possibility should always be considered—laboratory error. Errors can be preanalytical (eg, patient misidentification), analytical, or postanalytical. In aggregate, the frequency of errors in laboratory results is 1 in 214 to 8,316.6 Given that even the more conservative estimates show an incidence higher than that of many of the rare diseases for which clinicians may be testing, laboratory error always deserves consideration.
COULD THIS BE PHEOCHROMOCYTOMA?
Pheochromocytoma is a neuroendocrine tumor most commonly arising from the chromaffin cells of the adrenal medulla. However, extra-adrenal pheochromocytoma, generally paraganglioma, accounts for 15% to 20% of these tumors. Although the condition is generally considered very rare, autopsy studies have demonstrated a prevalence of 0.05%, suggesting that many tumors are either missed or are not clinically significant.
The diagnosis is most often sought in hypertensive patients, a population in which pheochromocytoma has a prevalence of 0.1% to 0.6%.7
2. What is the most common presenting symptom of pheochromocytoma?
- Paroxysmal hypertension
- Sustained hypertension
- Nausea
- Cardiomyopathy
- Headache
- Hemorrhagic shock
- Psychological symptoms such as anxiety or panic
Although 50% to 60% of patients with pheochromocytoma have sustained hypertension, it may be absent in patients with primarily epinephrine-secreting tumors or large tumors that degrade catecholamines, leading to normal or low blood pressure.
Cardiomyopathy is a rare consequence of untreated pheochromocytoma, caused by the effects of excess circulating catecholamines over a long period of time.8 As seen in this patient, a prolonged QTc on ECG associated with elevated levels of norepinephrine and normetanephrine may be the only red flag.9
Pheochromocytoma is typically an extremely well-vascularized tumor, and rupture or hemorrhage is a rare but often fatal complication.
IMPORTANT FAMILY HISTORY
The classic “rule of 10s” suggests that 10% of pheochromocytomas are hereditary, but in fact the number may be higher. In a large cohort of patients with apparently sporadic pheochromocytoma, 25% were found to have germ-line mutations.10 This finding highlights the importance not only of obtaining a thorough family history, but also of genetic testing and counseling once the diagnosis has been made.
3. Which hereditary syndrome is not associated with pheochromocytoma?
- Von Hippel-Lindau syndrome
- Neurofibromatosis type 1
- Neurofibromatosis type 2
- Multiple endocrine neoplasia type 2
- Paraganglioma syndromes
Germ-line mutations in five genes related to three hereditary syndromes (von Hippel-Lindau, neurofibromatosis type 1, and multiple endocrine neoplasia type 2) and in two genes related to paraganglioma syndromes are known to be associated with pheochromocytoma.7
Von Hippel-Lindau syndrome
Von Hippel-Lindau syndrome affects 1 in 36,000 live births. It is caused by a mutation of the von Hippel-Lindau gene on chromosome 3, and 10% to 20% of patients with the syndrome have pheochromocytoma. Other associated problems include renal clear-cell carcinomas and cysts, central nervous system and retinal hemangioblastomas, pancreatic tumors and cysts, endolymphatic tumors, and epididymal cysts.
Neurofibromatosis type 1
Neurofibromatosis type 1 affects 1 in 2,500 to 3,000 individuals and is caused by a mutation of the neurofibromatosis type 1 gene on chromosome 17. The disease is diagnosed by the presence of café-au-lait macules, axillary or inguinal freckling (or both), dermal or plexiform neurofibromas, Lisch nodules, or osseous lesions, but the condition is associated with many other pathologic findings, including optic pathway gliomas, cardiovascular abnormalities, and, in up to 5.7% of patients, pheochromocytoma.11
Neurofibromatosis type 2
Neurofibromatosis type 2 affects 1 in 25,000 live births and is caused by a mutation of the neurofibromatosis type 2 gene on chromosome 22. Patients often develop nervous system tumors, ophthalmologic pathology, and cutaneous lesions, but the condition is not associated with pheochromocytoma.12
Multiple endocrine neoplasia type 2
Multiple endocrine neoplasia type 2 affects 1 in 35,000 individuals and is caused by an activating mutation of the RET proto-oncogene on chromosome 21. The syndrome is most worrisome because of the 95% lifetime risk of medullary thyroid carcinoma in affected patients, but it is also associated with a 50% risk of pheochromocytoma and a 20% to 30% risk of primary hyperparathyroidism. Pheochromocytoma is the presenting clinical problem in 10% to 30% of patients.13
Paraganglioma syndromes
Paraganglioma syndromes are caused by mutations in the three genes encoding subunits of the succinate dehydrogenase enzyme. These mutations affect 1 in 30,000 to 100,000 individuals and incur a 70% lifetime risk of developing pheochromocytoma or paraganglioma.14
TESTING FOR AND MANAGING PHEOCHROMOCYTOMA
The consequences of untreated pheochromocytoma are potentially devastating and include progression to metastatic disease, hypertensive crises, cardiomyopathy, and adrenal hemorrhage. Nevertheless, the average patient goes 3 years before receiving the correct diagnosis.7 Consequently, heightened suspicion and tests with both high sensitivity and specificity are needed.
4. Which test for pheochromocytoma has the highest sensitivity?
- Plasma free metanephrines
- Plasma catecholamines
- Urine total metanephrines
- Urine fractionated metanephrines
- Urine catecholamines
- Urine vanillylmandelic acid
Some researchers have also examined plasma total metanephrines and found that any one of these three biochemical markers at a value two times greater than the upper limit of normal provides specificity of around 95%.16
Further laboratory tests in our patient
- Serum dopamine 70 pg/mL (reference range 0–20)
- Norepinephrine 2,018 pg/mL (80–520)
- Epinephrine 2,479 pg/mL (10–200)
- Free normetanephrine 12 pg/mL (< 0.9)
- Free metanephrine 17.8 pg/mL (< 0.5).
VALUE OF IMAGING STUDIES
Computed tomography has a sensitivity of up to 95% for detecting adrenal tumors and is able to detect tumors larger than 0.5 cm, but its specificity may be as low as 50%.17 Studies utilizing modern imaging equipment report a prevalence of adrenal incidentaloma of 4%, of which only 1.5% to 11% are pheochromocytoma.18 Thus, while the simultaneous occurrence of pheochromocytoma-like symptoms and an incidentaloma is not common, the potential for unnecessary surgery precludes diagnosis and treatment based on symptoms and imaging alone.
Magnetic resonance imaging has similar sensitivity and specificity but can better characterize the tumor’s blood supply and relationship to other structures.
Iodine 131 metaiodobenzylguanidine (MIBG) scanning is a physiologic study that uses a radiolabeled amine. Since it can identify pheochromocytoma regardless of location, MIBG scanning is typically used when pheochromocytoma is diagnosed by biochemical testing but CT and MRI fail to locate the lesion, or as a follow-up test in patients in whom recurrence or metastasis is suspected or documented.
The specificity of MIBG scanning is 95% to 100%, but the need to protect the thyroid from ablation and the potential need to repeat scans for up to 72 hours make it a poor choice for the initial evaluation.17
5. What is the next best step in our patient’s management?
- Treat her hypertension with a beta-blocker
- Begin a course of alpha-blockade
- Urgent surgery
- Observation
Because of the high concentration of circulating catecholamines and the instability of the tumor to physical manipulation, appropriate medical management before surgical resection is of paramount importance.
Beta-blockade can lead to malignant hypertension due to the unopposed alpha stimulation and must not be begun until alpha-blockade has been started. The standard of care is to give an alpha-blocker or calcium channel blocker 10 to 14 days before surgery. Typically, oral phenoxybenzamine (Dibenzyline) 10 mg twice daily is started and titrated upward daily by 10 to 20 mg until a target seated blood pressure of 120/80 mm Hg is obtained. Selective alpha-1 blockers such as prazosin (Minipress) and terazosin (Hytrin) have also been used and have the benefit of a preserved alpha-2 catecholamine reuptake mechanism.17
After several days, a beta-blocker may be added, particularly for patients with arrhythmias.7 In patients with refractory hypertension, metyrosine (Demser) can be useful.
During surgery, the patient’s hemodynamic stability and glucose levels can fluctuate rapidly from sudden releases of catecholamines during manipulation of the tumor, as well as from the sudden loss of catecholamines after ligation of draining vessels. Advances in medical care have reduced the perioperative death rate from 50% to less than 3%.7,19
CASE CONCLUSION AND FOLLOW-UP
Two months after her initial presentation, the patient underwent open surgery and had the mass removed without complications. She reports that the “panic attacks” have ceased completely.
The recurrence rate of pheochromocytoma is 13% in patients with sporadic disease and 33% in patients with familial syndromes. The overall recurrence rate with long-term follow-up is 17%, half of recurrences being malignant disease. All patients should therefore be followed in the clinic annually for at least 10 years to identify and treat recurrences early,7 and many experts recommend lifelong follow-up, even for patients without hereditary syndromes.17
Nearly every diagnosis in the DSM-IV includes the caveat that medical causes of disease must be excluded before psychiatric labels can be applied. Although panic disorder and panic attack are far more common than pheochromocytoma, just as essential hypertension is far more common than pheochromocytoma, physicians need to remember that pheochromocytoma can cause symptoms common to both illnesses. Thus, while rare conditions are rare, atypical presentations of common conditions may deserve a second glance.
A 50-year-old woman presents to the emergency department because of repeated episodes of vomiting over the past 12 hours. She reports eight episodes of non-bloody, nonbilious emesis associated with palpitations and feelings of anxiety, but with no fever or diarrhea. She has not traveled recently and does not have any sick contacts.
She reports that she never had health problems until 6 months ago, when she began having panic attacks that woke her from sleep. The episodes first occurred once or twice per week, usually at night, and involved palpitations and feelings of anxiety that lasted 2 to 4 hours, but no other associated symptoms. After a month, the episodes began to occur more regularly during the day and were accompanied by a pounding headache that began in the back of her neck and extended up and over her head. Her primary care physician prescribed sertraline (Zoloft) and referred her to a neurologist to evaluate the headaches. The neurologic workup included brain magnetic resonance imaging and electroencephalography, both of which were normal.
After 8 weeks on sertraline, the episodes continued to increase in frequency and severity, and her physician switched her to paroxetine (Paxil) and added lorazepam (Ativan), which did not improve her symptoms. Over the past 2 months, during which time she has not been taking any medications, the episodes began to involve nausea and, more recently, vomiting, with episodes occurring as often as once or twice daily, and with intermittent symptom-free days. None of the prior episodes was accompanied by symptoms as severe as those she is currently experiencing.
She is otherwise healthy with no chronic diseases. Her surgical history includes resection of an angiolipoma from her right arm and dilation and curettage for endometrial polyps. She has no personal or family history of psychiatric illness.
PHYSICAL EXAMINATION
The patient is slender and tremulous but does not appear diaphoretic. Her blood pressure is 176/92 mm Hg, pulse 98, temperature 36.5°C (97.7°F), and respiratory rate 20 per minute. Oxygen saturation by pulse oximetry is 98% on room air. She has dry mucus membranes and orthostatic hypotension, but her physical examination is otherwise normal. Electrocardiography (ECG) shows a normal sinus rhythm with a prolonged QTc of 571 ms and peaked P and T waves.
LABORATORY VALUES
- Hemoglobin 15.6 g/dL (reference range 11.5–15.5)
- Hematocrit 47.2% (36.0–46.0)
- Platelet count 448 × 109/L (150–400)
- White cell count 18.65 × 109/L (3.70–11.00)
- Potassium 2.5 mmol/L (3.5–4.0)
- Chloride 97 mmol/L (98–110)
- Bicarbonate 21 mmol/L (23–32)
- Anion gap 20 mmol/L (0–15)
- Glucose 233 mg/dL (65–100).
Sodium, blood urea nitrogen, and creatinine levels are all within normal limits. Urinalysis suggests a urinary tract infection.
IS THIS A PANIC ATTACK?
1. Which of the following is not characteristic of a panic attack?
- Nausea and vomiting
- Onset during sleep
- Palpitations
- Chest pain or discomfort
- Headache
- Trembling or shaking
According to the Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition) (DSM-IV), the diagnosis of panic attack requires the presence of intense fear or discomfort and four or more other symptoms that may come from any of six domains:
- Cardiovascular: palpitations, pounding heart, tachycardia, and chest pain or discomfort
- Autonomic: sweating, chills or hot flushes, and trembling or shaking
- Pulmonary: shortness of breath or a smothering sensation
- Neurologic: dizziness or light-headedness and paresthesias
- Gastrointestinal: choking and nausea or abdominal distress
- Psychological: compass derealization, depersonalization, and the fear of losing control or “going crazy.”1
Two aspects of the patient’s history may be misinterpreted by those unfamiliar with the symptomatology of panic attack. First, although panic disorder carries an increased risk of many comorbidities, including migraine, headache is not typically associated with the panic attacks themselves.2 Second, while not a part of the diagnostic criteria, sleep disturbances are common in patients with panic disorder, and 30% to 45% of patients with the disorder experience recurrent nocturnal panic attacks.3 Therefore, the correct answer is headache.
THE DIFFERENTIAL DIAGNOSIS
When considering a diagnosis of panic attack or panic disorder, the DSM-IV mandates that medical causes of the symptoms must be excluded. Common conditions causing a similar spectrum of symptoms include hyperthyroidism, caffeine and stimulant use or abuse, asthma, cardiac arrhythmias, alcohol withdrawal, and, more rarely, complex partial seizures and pheochromocytoma.2,4 Many of these conditions can be ruled out by the history alone in a reliable patient.
Our patient’s electrocardiogram showed no evidence of ischemia or arrhythmias. Also, her recent negative neurologic workup makes seizure activity less likely.
Many of this patient’s laboratory abnormalities are easily explained by her repeated bouts of vomiting. Specifically, her elevated hemoglobin level and hematocrit are likely secondary to volume contraction, while hypochloremia is seen following losses of HCl with emesis. Typically, however, patients with vomiting have a hypochloremic metabolic alkalosis, and her low serum bicarbonate level is inconsistent with the history.
Three factors might be contributing to this patient’s hypokalemia. First, in a volume-depleted state, the cortical collecting tubules secrete potassium in exchange for increased sodium reabsorption in an attempt to correct volume status. Second, the alkalotic state caused by losses of acid with vomiting results in a transcellular shift of potassium ions into cells in exchange for hydrogen ions. Third, increased levels of epinephrine also cause a shift of potassium ions into cells.5 Potassium is not lost directly through nausea and vomiting.
A state of catecholamine excess, such as during a severe panic attack or in the presence of a catecholamine-secreting tumor, could explain many of her abnormalities. In addition to causing hypokalemia, epinephrine has a gluconeogenic effect, whereas norepinephrine inhibits insulin release, providing a potential explanation for hyperglycemia in a patient with no risk factors for diabetes. Finally, catecholamine excess contributes to lactic acidosis, which could help to explain the low serum bicarbonate level and the elevated anion gap, but unless we take arterial blood gas measurements, the patient’s acid-base status cannot be determined.
While panic attacks do stimulate the sympathetic nervous system, certain elements of her history raise the clinical suspicion for another process. First, the severity of the electrolyte abnormalities is suspicious. Second, a typical panic attack peaks at 10 minutes and begins to subside, whereas this woman’s symptoms have persisted for 12 hours. Finally, the clinical history, in particular the prominence of headaches associated with the symptoms, is inconsistent with classic panic attack. Consequently, an alternative diagnosis, such as pheochromocytoma, deserves more careful evaluation.
Whenever laboratory results do not fit with the clinical scenario or patient, however, one final possibility should always be considered—laboratory error. Errors can be preanalytical (eg, patient misidentification), analytical, or postanalytical. In aggregate, the frequency of errors in laboratory results is 1 in 214 to 8,316.6 Given that even the more conservative estimates show an incidence higher than that of many of the rare diseases for which clinicians may be testing, laboratory error always deserves consideration.
COULD THIS BE PHEOCHROMOCYTOMA?
Pheochromocytoma is a neuroendocrine tumor most commonly arising from the chromaffin cells of the adrenal medulla. However, extra-adrenal pheochromocytoma, generally paraganglioma, accounts for 15% to 20% of these tumors. Although the condition is generally considered very rare, autopsy studies have demonstrated a prevalence of 0.05%, suggesting that many tumors are either missed or are not clinically significant.
The diagnosis is most often sought in hypertensive patients, a population in which pheochromocytoma has a prevalence of 0.1% to 0.6%.7
2. What is the most common presenting symptom of pheochromocytoma?
- Paroxysmal hypertension
- Sustained hypertension
- Nausea
- Cardiomyopathy
- Headache
- Hemorrhagic shock
- Psychological symptoms such as anxiety or panic
Although 50% to 60% of patients with pheochromocytoma have sustained hypertension, it may be absent in patients with primarily epinephrine-secreting tumors or large tumors that degrade catecholamines, leading to normal or low blood pressure.
Cardiomyopathy is a rare consequence of untreated pheochromocytoma, caused by the effects of excess circulating catecholamines over a long period of time.8 As seen in this patient, a prolonged QTc on ECG associated with elevated levels of norepinephrine and normetanephrine may be the only red flag.9
Pheochromocytoma is typically an extremely well-vascularized tumor, and rupture or hemorrhage is a rare but often fatal complication.
IMPORTANT FAMILY HISTORY
The classic “rule of 10s” suggests that 10% of pheochromocytomas are hereditary, but in fact the number may be higher. In a large cohort of patients with apparently sporadic pheochromocytoma, 25% were found to have germ-line mutations.10 This finding highlights the importance not only of obtaining a thorough family history, but also of genetic testing and counseling once the diagnosis has been made.
3. Which hereditary syndrome is not associated with pheochromocytoma?
- Von Hippel-Lindau syndrome
- Neurofibromatosis type 1
- Neurofibromatosis type 2
- Multiple endocrine neoplasia type 2
- Paraganglioma syndromes
Germ-line mutations in five genes related to three hereditary syndromes (von Hippel-Lindau, neurofibromatosis type 1, and multiple endocrine neoplasia type 2) and in two genes related to paraganglioma syndromes are known to be associated with pheochromocytoma.7
Von Hippel-Lindau syndrome
Von Hippel-Lindau syndrome affects 1 in 36,000 live births. It is caused by a mutation of the von Hippel-Lindau gene on chromosome 3, and 10% to 20% of patients with the syndrome have pheochromocytoma. Other associated problems include renal clear-cell carcinomas and cysts, central nervous system and retinal hemangioblastomas, pancreatic tumors and cysts, endolymphatic tumors, and epididymal cysts.
Neurofibromatosis type 1
Neurofibromatosis type 1 affects 1 in 2,500 to 3,000 individuals and is caused by a mutation of the neurofibromatosis type 1 gene on chromosome 17. The disease is diagnosed by the presence of café-au-lait macules, axillary or inguinal freckling (or both), dermal or plexiform neurofibromas, Lisch nodules, or osseous lesions, but the condition is associated with many other pathologic findings, including optic pathway gliomas, cardiovascular abnormalities, and, in up to 5.7% of patients, pheochromocytoma.11
Neurofibromatosis type 2
Neurofibromatosis type 2 affects 1 in 25,000 live births and is caused by a mutation of the neurofibromatosis type 2 gene on chromosome 22. Patients often develop nervous system tumors, ophthalmologic pathology, and cutaneous lesions, but the condition is not associated with pheochromocytoma.12
Multiple endocrine neoplasia type 2
Multiple endocrine neoplasia type 2 affects 1 in 35,000 individuals and is caused by an activating mutation of the RET proto-oncogene on chromosome 21. The syndrome is most worrisome because of the 95% lifetime risk of medullary thyroid carcinoma in affected patients, but it is also associated with a 50% risk of pheochromocytoma and a 20% to 30% risk of primary hyperparathyroidism. Pheochromocytoma is the presenting clinical problem in 10% to 30% of patients.13
Paraganglioma syndromes
Paraganglioma syndromes are caused by mutations in the three genes encoding subunits of the succinate dehydrogenase enzyme. These mutations affect 1 in 30,000 to 100,000 individuals and incur a 70% lifetime risk of developing pheochromocytoma or paraganglioma.14
TESTING FOR AND MANAGING PHEOCHROMOCYTOMA
The consequences of untreated pheochromocytoma are potentially devastating and include progression to metastatic disease, hypertensive crises, cardiomyopathy, and adrenal hemorrhage. Nevertheless, the average patient goes 3 years before receiving the correct diagnosis.7 Consequently, heightened suspicion and tests with both high sensitivity and specificity are needed.
4. Which test for pheochromocytoma has the highest sensitivity?
- Plasma free metanephrines
- Plasma catecholamines
- Urine total metanephrines
- Urine fractionated metanephrines
- Urine catecholamines
- Urine vanillylmandelic acid
Some researchers have also examined plasma total metanephrines and found that any one of these three biochemical markers at a value two times greater than the upper limit of normal provides specificity of around 95%.16
Further laboratory tests in our patient
- Serum dopamine 70 pg/mL (reference range 0–20)
- Norepinephrine 2,018 pg/mL (80–520)
- Epinephrine 2,479 pg/mL (10–200)
- Free normetanephrine 12 pg/mL (< 0.9)
- Free metanephrine 17.8 pg/mL (< 0.5).
VALUE OF IMAGING STUDIES
Computed tomography has a sensitivity of up to 95% for detecting adrenal tumors and is able to detect tumors larger than 0.5 cm, but its specificity may be as low as 50%.17 Studies utilizing modern imaging equipment report a prevalence of adrenal incidentaloma of 4%, of which only 1.5% to 11% are pheochromocytoma.18 Thus, while the simultaneous occurrence of pheochromocytoma-like symptoms and an incidentaloma is not common, the potential for unnecessary surgery precludes diagnosis and treatment based on symptoms and imaging alone.
Magnetic resonance imaging has similar sensitivity and specificity but can better characterize the tumor’s blood supply and relationship to other structures.
Iodine 131 metaiodobenzylguanidine (MIBG) scanning is a physiologic study that uses a radiolabeled amine. Since it can identify pheochromocytoma regardless of location, MIBG scanning is typically used when pheochromocytoma is diagnosed by biochemical testing but CT and MRI fail to locate the lesion, or as a follow-up test in patients in whom recurrence or metastasis is suspected or documented.
The specificity of MIBG scanning is 95% to 100%, but the need to protect the thyroid from ablation and the potential need to repeat scans for up to 72 hours make it a poor choice for the initial evaluation.17
5. What is the next best step in our patient’s management?
- Treat her hypertension with a beta-blocker
- Begin a course of alpha-blockade
- Urgent surgery
- Observation
Because of the high concentration of circulating catecholamines and the instability of the tumor to physical manipulation, appropriate medical management before surgical resection is of paramount importance.
Beta-blockade can lead to malignant hypertension due to the unopposed alpha stimulation and must not be begun until alpha-blockade has been started. The standard of care is to give an alpha-blocker or calcium channel blocker 10 to 14 days before surgery. Typically, oral phenoxybenzamine (Dibenzyline) 10 mg twice daily is started and titrated upward daily by 10 to 20 mg until a target seated blood pressure of 120/80 mm Hg is obtained. Selective alpha-1 blockers such as prazosin (Minipress) and terazosin (Hytrin) have also been used and have the benefit of a preserved alpha-2 catecholamine reuptake mechanism.17
After several days, a beta-blocker may be added, particularly for patients with arrhythmias.7 In patients with refractory hypertension, metyrosine (Demser) can be useful.
During surgery, the patient’s hemodynamic stability and glucose levels can fluctuate rapidly from sudden releases of catecholamines during manipulation of the tumor, as well as from the sudden loss of catecholamines after ligation of draining vessels. Advances in medical care have reduced the perioperative death rate from 50% to less than 3%.7,19
CASE CONCLUSION AND FOLLOW-UP
Two months after her initial presentation, the patient underwent open surgery and had the mass removed without complications. She reports that the “panic attacks” have ceased completely.
The recurrence rate of pheochromocytoma is 13% in patients with sporadic disease and 33% in patients with familial syndromes. The overall recurrence rate with long-term follow-up is 17%, half of recurrences being malignant disease. All patients should therefore be followed in the clinic annually for at least 10 years to identify and treat recurrences early,7 and many experts recommend lifelong follow-up, even for patients without hereditary syndromes.17
Nearly every diagnosis in the DSM-IV includes the caveat that medical causes of disease must be excluded before psychiatric labels can be applied. Although panic disorder and panic attack are far more common than pheochromocytoma, just as essential hypertension is far more common than pheochromocytoma, physicians need to remember that pheochromocytoma can cause symptoms common to both illnesses. Thus, while rare conditions are rare, atypical presentations of common conditions may deserve a second glance.
- Yates WR. Phenomenology and epidemiology of panic disorder. Ann Clin Psychiatry 2009; 21:95–102.
- Katon WJ. Clinical practice. Panic disorder. N Engl J Med 2006; 354:2360–2367.
- Craske MG, Tsao JC. Assessment and treatment of nocturnal panic attacks. Sleep Med Rev 2005; 9:173–184.
- Roy-Byrne PP, Craske MG, Stein MB. Panic disorder. Lancet 2006; 368:1023–1032.
- Beal AL, Deuser WE, Beilman GJ. A role for epinephrine in post-traumatic hypokalemia. Shock 2007; 27:358–363.
- Kalra J. Medical errors: impact on clinical laboratories and other critical areas. Clin Biochem 2004; 37:1052–1062.
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet 2005; 366:665–675.
- Leissner KB, Mahmood F, Aragam JR, Amouzgar A, Ortega R. Catecholamine-induced cardiomyopathy and pheochromocytoma. Anesth Analg 2008; 107:410–412.
- Yu R, Furmark L, Wong C. Cardiac abnormalities associated with pheochromocytoma and other adrenal tumors. Endocr Pract 2009; 15:10–16.
- Neumann HP, Bausch B, McWhinney SR, et al; Freiburg-Warsaw-Columbus Pheochromocytoma Study Group. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346:1459–1466.
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics 2009; 123:124–133.
- Asthagiri AR, Parry DM, Butman JA, et al. Neurofibromatosis type 2. Lancet 2009; 373:1974–1986.
- Callender GG, Rich TA, Perrier ND. Multiple endocrine neoplasia syndromes. Surg Clin North Am 2008; 88:863–895.
- Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. J Intern Med 2009; 266:19–42.
- Yu R, Nissen NN, Chopra P, Dhall D, Phillips E, Wei M. Diagnosis and treatment of pheochromocytoma in an academic hospital from 1997 to 2007. Am J Med 2009; 122:85–95.
- Grouzmann E, Drouard-Troalen L, Baudin E, et al. Diagnostic accuracy of free and total metanephrines in plasma and fractionated metanephrines in urine of patients with pheochromocytoma. Eur J Endocrinol 2010; 162:951–960.
- Mittendorf EA, Evans DB, Lee JE, Perrier ND. Pheochromocytoma: advances in genetics, diagnosis, localization, and treatment. Hematol Oncol Clin North Am 2007; 21:509–525.
- Singh PK, Buch HN. Adrenal incidentaloma: evaluation and management. J Clin Pathol 2008; 61:1168–1173.
- Kasturi S, Kutikov A, Guzzo TJ, Smith AL, Wein AJ. Modern management of pheochromocytoma. Nat Clin Pract Urol 2007; 4:630–633.
- Yates WR. Phenomenology and epidemiology of panic disorder. Ann Clin Psychiatry 2009; 21:95–102.
- Katon WJ. Clinical practice. Panic disorder. N Engl J Med 2006; 354:2360–2367.
- Craske MG, Tsao JC. Assessment and treatment of nocturnal panic attacks. Sleep Med Rev 2005; 9:173–184.
- Roy-Byrne PP, Craske MG, Stein MB. Panic disorder. Lancet 2006; 368:1023–1032.
- Beal AL, Deuser WE, Beilman GJ. A role for epinephrine in post-traumatic hypokalemia. Shock 2007; 27:358–363.
- Kalra J. Medical errors: impact on clinical laboratories and other critical areas. Clin Biochem 2004; 37:1052–1062.
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet 2005; 366:665–675.
- Leissner KB, Mahmood F, Aragam JR, Amouzgar A, Ortega R. Catecholamine-induced cardiomyopathy and pheochromocytoma. Anesth Analg 2008; 107:410–412.
- Yu R, Furmark L, Wong C. Cardiac abnormalities associated with pheochromocytoma and other adrenal tumors. Endocr Pract 2009; 15:10–16.
- Neumann HP, Bausch B, McWhinney SR, et al; Freiburg-Warsaw-Columbus Pheochromocytoma Study Group. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346:1459–1466.
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics 2009; 123:124–133.
- Asthagiri AR, Parry DM, Butman JA, et al. Neurofibromatosis type 2. Lancet 2009; 373:1974–1986.
- Callender GG, Rich TA, Perrier ND. Multiple endocrine neoplasia syndromes. Surg Clin North Am 2008; 88:863–895.
- Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. J Intern Med 2009; 266:19–42.
- Yu R, Nissen NN, Chopra P, Dhall D, Phillips E, Wei M. Diagnosis and treatment of pheochromocytoma in an academic hospital from 1997 to 2007. Am J Med 2009; 122:85–95.
- Grouzmann E, Drouard-Troalen L, Baudin E, et al. Diagnostic accuracy of free and total metanephrines in plasma and fractionated metanephrines in urine of patients with pheochromocytoma. Eur J Endocrinol 2010; 162:951–960.
- Mittendorf EA, Evans DB, Lee JE, Perrier ND. Pheochromocytoma: advances in genetics, diagnosis, localization, and treatment. Hematol Oncol Clin North Am 2007; 21:509–525.
- Singh PK, Buch HN. Adrenal incidentaloma: evaluation and management. J Clin Pathol 2008; 61:1168–1173.
- Kasturi S, Kutikov A, Guzzo TJ, Smith AL, Wein AJ. Modern management of pheochromocytoma. Nat Clin Pract Urol 2007; 4:630–633.