User login
Targeted therapies have revolutionized the treatment of numerous different cancer types and ushered in an era of personalized medicine, yet they can be prohibitively costly. As patent protection expires on many of the first FDA-approved monoclonal antibodies developed for oncologic indications, the doors are opened for other companies to develop their own version of these drugs, known as biosimilars. The price of biosimilars is expected to be considerably lower than the original drugs upon which they are based.
Bevacizumab-awwb, marketed as Mvasi by Amgen and Allergen, became the first such drug to receive approval by the US Food and Drug Administration for the treatment of cancer in fall last year.1 It is a biosimilar of Genentech’s anti-angiogenesis drug, bevacizumab (Avastin), a monoclonal antibody that targets vascular endothelial growth factor-A (VEGF-A).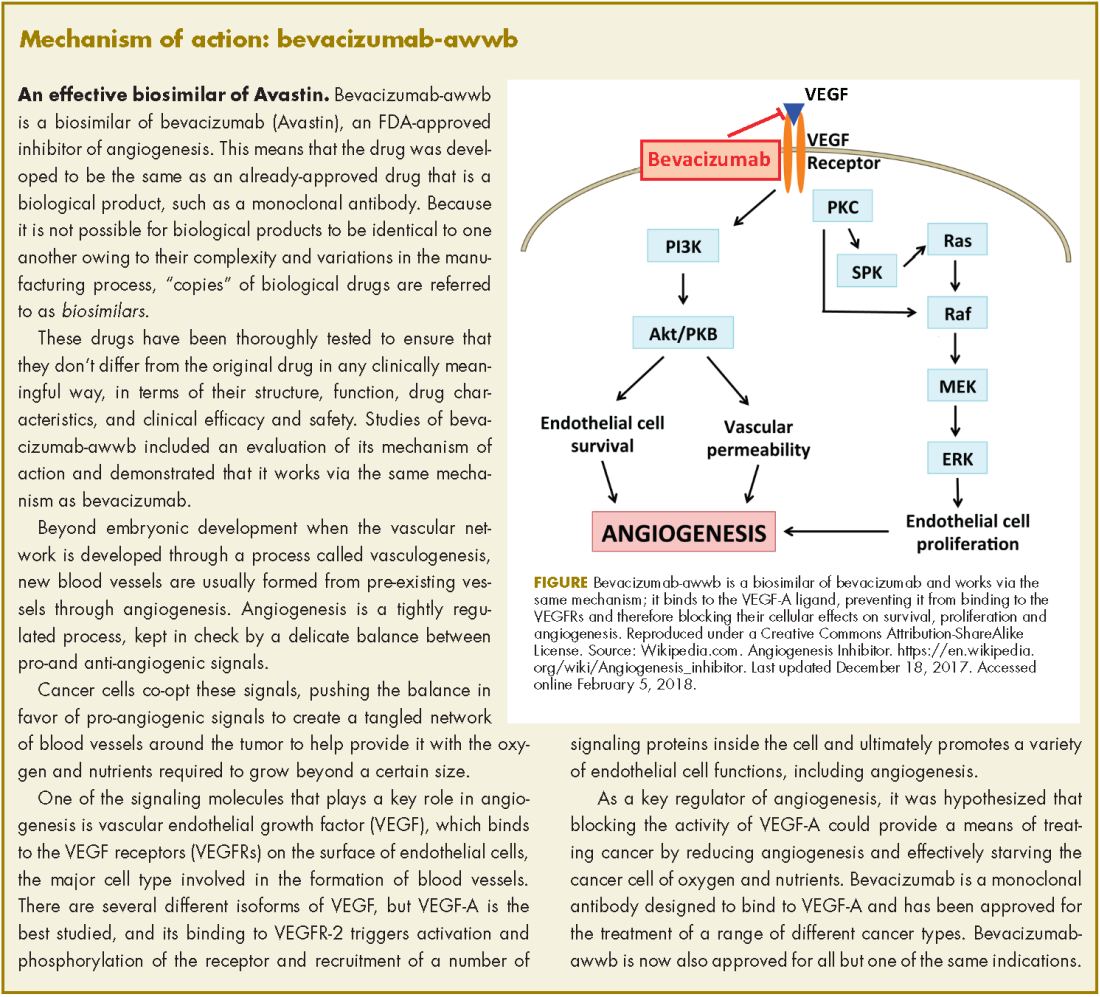
The approval of biosimilars is based on rigorous demonstration of a high level of similarity between the biosimilar and the already-approved reference drug, in terms of structure, function, pharmacokinetics, pharmacodynamics, and clinical efficacy and safety.
Bevacizumab-awwb was approved for the first- or second-line treatment of metastatic colorectal cancer (mCRC) in combination with 5-fluorouracil-based chemotherapy; the second-line treatment of mCRC in combination with fluoropyrimidine-oxaliplatin chemotherapy in patients who progressed on first-line bevacizumab; the first-line treatment of unresectable, locally advanced, recurrent or metastatic nonsquamous non-small cell lung cancer (NSCLC) in combination with carboplatin and paclitaxel; the second-line treatment of glioblastoma (GBM) as monotherapy; and in patients with persistent, recurrent, or metastatic cervical cancer in combination with paclitaxel and cisplatin or paclitaxel and topotecan. It was not approved for the treatment of ovarian cancer, for which bevacizumab is indicated.
The majority of the data used to support approval came from 2 studies – a 3-arm, single-dose pharmacokinetics study, and a comparative clinical study in patients with advanced/metastatic NSCLC. In the pharmacokinetics study, 202 healthy men received an infusion of 3 mg/kg of bevacizumab-awwb, US-approved bevacizumab, or EU-approved bevacizumab. Bevacizumab-awwb was shown to have pharmacokinetic similarity to both approved forms of bevacizumab, and safety and tolerability were comparable, with none of the participants developing binding or neutralizing antidrug antibodies.2
In the clinical study, 648 patients received an infusion of bevacizumab-awwb or EU-approved bevacizumab at a dose of 15 mg/kg every 3 weeks in combination with 6 AUC carboplatin and 200 mg/m2 paclitaxel for 6 cycles. The overall response rate was 39% for bevacizumab-awwb, compared with 41.7% for EU-bevacizumab, and there were 2 complete responses in each group. The median duration of response for bevacizumab-awwb compared with EU-bevacizumab was 5.8 months versus 5.6 months, respectively, and median progression-free survival was 6.6 months versus 7.9 months.3
In terms of safety, the rates of grade 3/4 adverse events (AEs) were 42.9% in the biosimilar arm, compared with 44.3% for the reference drug. Overall, there were no clinically meaningful differences in AEs, serious AEs, deaths, or treatment discontinuations.
The recommended dose for bevacizumab-awwb in patients with mCRC is a 5 mg/kg intravenous dose administered every 2 weeks with bolus-IFL, a 10 mg/kg IV dose administered every 2 weeks with FOLFOX4, or a 5 mg/kg IV dose administered every 2 weeks or 7.5 mg/kg IV dose administered every 3 weeks with fluoropyrimidine-irinotecan or fluoropyrimidine-oxaliplatin-based chemotherapy.
For patients with NSCLC, bevacizumab-awwb should be administered at a 15 mg/kg IV dose every 3 weeks with the carboplatin–paclitaxel combination; for GBM patients, a 10 mg/kg IV dose should be administered every 3 weeks; and for patients with cervical cancer, an IV dose of 15 mg/kg every 3 weeks in combination with paclitaxel–cisplatin or paclitaxel–topotecan is recommended.
The prescribing information outlines warnings and precautions to advise clinicians administering the new biosimilar of the risks of gastrointestinal (GI) perforations, surgery and wound healing complications, and severe and potentially fatal pulmonary, GI, central nervous system, and vaginal bleeding.4
Treatment should be discontinued if GI perforation occurs. Patients should not take bevacizumab-awwb in the 28 days before elective surgery and after surgery until the wound is healed, and treatment should be discontinued if the surgical wound breaks open. Bevacizumab-awwb should not be administered to patients with severe hemorrhage or those with hemoptysis.
Blood pressure should be monitored every 2-3 weeks during treatment and hypertension treated with antihypertensive therapy. Treatment should be temporarily suspended in patients with severe hypertension that is not controlled with antihypertensive therapy and discontinued in patients who experience hypertensive crisis or hypertensive encephalopathy.
Proteinuria should be monitored by dipstick urine analysis during treatment, and patients with a 2+ or greater reading (concentration, 100 mg/dL) should undergo further assessment with 24-hour urine collection. Treatment should be suspended if proteinuria levels are ≥2 g/24h and can be resumed when they fall below that level, but should be discontinued in patients with nephrotic syndrome. Treatment should also be discontinued in patients who develop posterior reversible encephalopathy syndrome, and patients should be advised of the potential for fetal harm
1. FDA approves first biosimilar for the treatment of cancer. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm576112.htm. September 14, 2017. Accessed January 31, 2018.
2. Markus R, Chow V, Pan X, and Hanes V. A phase I, randomized, single-dose study evaluating the pharmacokinetic equivalence of biosimilar ABP 215 and bevacizumab in healthy adult men. Cancer Chemother. Pharmacol. 2017;80:755-763.
3. Thatcher N, Thomas M, Ostoros G, et al. Randomized, double-blind, phase 3 study comparing biosimilar candidate ABP-215 with bevacizumab in patients with non-squamous NSCLC. J Thorac Oncol. 2017;12(1):S902-S903.
4. Mvasi (bevacizumab-awwb) solution, for intravenous infusion. Prescribing information. Amgen Inc, https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761028s000lbl.pdf. September 2017. Accessed January 31, 2018.
Targeted therapies have revolutionized the treatment of numerous different cancer types and ushered in an era of personalized medicine, yet they can be prohibitively costly. As patent protection expires on many of the first FDA-approved monoclonal antibodies developed for oncologic indications, the doors are opened for other companies to develop their own version of these drugs, known as biosimilars. The price of biosimilars is expected to be considerably lower than the original drugs upon which they are based.
Bevacizumab-awwb, marketed as Mvasi by Amgen and Allergen, became the first such drug to receive approval by the US Food and Drug Administration for the treatment of cancer in fall last year.1 It is a biosimilar of Genentech’s anti-angiogenesis drug, bevacizumab (Avastin), a monoclonal antibody that targets vascular endothelial growth factor-A (VEGF-A).
The approval of biosimilars is based on rigorous demonstration of a high level of similarity between the biosimilar and the already-approved reference drug, in terms of structure, function, pharmacokinetics, pharmacodynamics, and clinical efficacy and safety.
Bevacizumab-awwb was approved for the first- or second-line treatment of metastatic colorectal cancer (mCRC) in combination with 5-fluorouracil-based chemotherapy; the second-line treatment of mCRC in combination with fluoropyrimidine-oxaliplatin chemotherapy in patients who progressed on first-line bevacizumab; the first-line treatment of unresectable, locally advanced, recurrent or metastatic nonsquamous non-small cell lung cancer (NSCLC) in combination with carboplatin and paclitaxel; the second-line treatment of glioblastoma (GBM) as monotherapy; and in patients with persistent, recurrent, or metastatic cervical cancer in combination with paclitaxel and cisplatin or paclitaxel and topotecan. It was not approved for the treatment of ovarian cancer, for which bevacizumab is indicated.
The majority of the data used to support approval came from 2 studies – a 3-arm, single-dose pharmacokinetics study, and a comparative clinical study in patients with advanced/metastatic NSCLC. In the pharmacokinetics study, 202 healthy men received an infusion of 3 mg/kg of bevacizumab-awwb, US-approved bevacizumab, or EU-approved bevacizumab. Bevacizumab-awwb was shown to have pharmacokinetic similarity to both approved forms of bevacizumab, and safety and tolerability were comparable, with none of the participants developing binding or neutralizing antidrug antibodies.2
In the clinical study, 648 patients received an infusion of bevacizumab-awwb or EU-approved bevacizumab at a dose of 15 mg/kg every 3 weeks in combination with 6 AUC carboplatin and 200 mg/m2 paclitaxel for 6 cycles. The overall response rate was 39% for bevacizumab-awwb, compared with 41.7% for EU-bevacizumab, and there were 2 complete responses in each group. The median duration of response for bevacizumab-awwb compared with EU-bevacizumab was 5.8 months versus 5.6 months, respectively, and median progression-free survival was 6.6 months versus 7.9 months.3
In terms of safety, the rates of grade 3/4 adverse events (AEs) were 42.9% in the biosimilar arm, compared with 44.3% for the reference drug. Overall, there were no clinically meaningful differences in AEs, serious AEs, deaths, or treatment discontinuations.
The recommended dose for bevacizumab-awwb in patients with mCRC is a 5 mg/kg intravenous dose administered every 2 weeks with bolus-IFL, a 10 mg/kg IV dose administered every 2 weeks with FOLFOX4, or a 5 mg/kg IV dose administered every 2 weeks or 7.5 mg/kg IV dose administered every 3 weeks with fluoropyrimidine-irinotecan or fluoropyrimidine-oxaliplatin-based chemotherapy.
For patients with NSCLC, bevacizumab-awwb should be administered at a 15 mg/kg IV dose every 3 weeks with the carboplatin–paclitaxel combination; for GBM patients, a 10 mg/kg IV dose should be administered every 3 weeks; and for patients with cervical cancer, an IV dose of 15 mg/kg every 3 weeks in combination with paclitaxel–cisplatin or paclitaxel–topotecan is recommended.
The prescribing information outlines warnings and precautions to advise clinicians administering the new biosimilar of the risks of gastrointestinal (GI) perforations, surgery and wound healing complications, and severe and potentially fatal pulmonary, GI, central nervous system, and vaginal bleeding.4
Treatment should be discontinued if GI perforation occurs. Patients should not take bevacizumab-awwb in the 28 days before elective surgery and after surgery until the wound is healed, and treatment should be discontinued if the surgical wound breaks open. Bevacizumab-awwb should not be administered to patients with severe hemorrhage or those with hemoptysis.
Blood pressure should be monitored every 2-3 weeks during treatment and hypertension treated with antihypertensive therapy. Treatment should be temporarily suspended in patients with severe hypertension that is not controlled with antihypertensive therapy and discontinued in patients who experience hypertensive crisis or hypertensive encephalopathy.
Proteinuria should be monitored by dipstick urine analysis during treatment, and patients with a 2+ or greater reading (concentration, 100 mg/dL) should undergo further assessment with 24-hour urine collection. Treatment should be suspended if proteinuria levels are ≥2 g/24h and can be resumed when they fall below that level, but should be discontinued in patients with nephrotic syndrome. Treatment should also be discontinued in patients who develop posterior reversible encephalopathy syndrome, and patients should be advised of the potential for fetal harm
Targeted therapies have revolutionized the treatment of numerous different cancer types and ushered in an era of personalized medicine, yet they can be prohibitively costly. As patent protection expires on many of the first FDA-approved monoclonal antibodies developed for oncologic indications, the doors are opened for other companies to develop their own version of these drugs, known as biosimilars. The price of biosimilars is expected to be considerably lower than the original drugs upon which they are based.
Bevacizumab-awwb, marketed as Mvasi by Amgen and Allergen, became the first such drug to receive approval by the US Food and Drug Administration for the treatment of cancer in fall last year.1 It is a biosimilar of Genentech’s anti-angiogenesis drug, bevacizumab (Avastin), a monoclonal antibody that targets vascular endothelial growth factor-A (VEGF-A).
The approval of biosimilars is based on rigorous demonstration of a high level of similarity between the biosimilar and the already-approved reference drug, in terms of structure, function, pharmacokinetics, pharmacodynamics, and clinical efficacy and safety.
Bevacizumab-awwb was approved for the first- or second-line treatment of metastatic colorectal cancer (mCRC) in combination with 5-fluorouracil-based chemotherapy; the second-line treatment of mCRC in combination with fluoropyrimidine-oxaliplatin chemotherapy in patients who progressed on first-line bevacizumab; the first-line treatment of unresectable, locally advanced, recurrent or metastatic nonsquamous non-small cell lung cancer (NSCLC) in combination with carboplatin and paclitaxel; the second-line treatment of glioblastoma (GBM) as monotherapy; and in patients with persistent, recurrent, or metastatic cervical cancer in combination with paclitaxel and cisplatin or paclitaxel and topotecan. It was not approved for the treatment of ovarian cancer, for which bevacizumab is indicated.
The majority of the data used to support approval came from 2 studies – a 3-arm, single-dose pharmacokinetics study, and a comparative clinical study in patients with advanced/metastatic NSCLC. In the pharmacokinetics study, 202 healthy men received an infusion of 3 mg/kg of bevacizumab-awwb, US-approved bevacizumab, or EU-approved bevacizumab. Bevacizumab-awwb was shown to have pharmacokinetic similarity to both approved forms of bevacizumab, and safety and tolerability were comparable, with none of the participants developing binding or neutralizing antidrug antibodies.2
In the clinical study, 648 patients received an infusion of bevacizumab-awwb or EU-approved bevacizumab at a dose of 15 mg/kg every 3 weeks in combination with 6 AUC carboplatin and 200 mg/m2 paclitaxel for 6 cycles. The overall response rate was 39% for bevacizumab-awwb, compared with 41.7% for EU-bevacizumab, and there were 2 complete responses in each group. The median duration of response for bevacizumab-awwb compared with EU-bevacizumab was 5.8 months versus 5.6 months, respectively, and median progression-free survival was 6.6 months versus 7.9 months.3
In terms of safety, the rates of grade 3/4 adverse events (AEs) were 42.9% in the biosimilar arm, compared with 44.3% for the reference drug. Overall, there were no clinically meaningful differences in AEs, serious AEs, deaths, or treatment discontinuations.
The recommended dose for bevacizumab-awwb in patients with mCRC is a 5 mg/kg intravenous dose administered every 2 weeks with bolus-IFL, a 10 mg/kg IV dose administered every 2 weeks with FOLFOX4, or a 5 mg/kg IV dose administered every 2 weeks or 7.5 mg/kg IV dose administered every 3 weeks with fluoropyrimidine-irinotecan or fluoropyrimidine-oxaliplatin-based chemotherapy.
For patients with NSCLC, bevacizumab-awwb should be administered at a 15 mg/kg IV dose every 3 weeks with the carboplatin–paclitaxel combination; for GBM patients, a 10 mg/kg IV dose should be administered every 3 weeks; and for patients with cervical cancer, an IV dose of 15 mg/kg every 3 weeks in combination with paclitaxel–cisplatin or paclitaxel–topotecan is recommended.
The prescribing information outlines warnings and precautions to advise clinicians administering the new biosimilar of the risks of gastrointestinal (GI) perforations, surgery and wound healing complications, and severe and potentially fatal pulmonary, GI, central nervous system, and vaginal bleeding.4
Treatment should be discontinued if GI perforation occurs. Patients should not take bevacizumab-awwb in the 28 days before elective surgery and after surgery until the wound is healed, and treatment should be discontinued if the surgical wound breaks open. Bevacizumab-awwb should not be administered to patients with severe hemorrhage or those with hemoptysis.
Blood pressure should be monitored every 2-3 weeks during treatment and hypertension treated with antihypertensive therapy. Treatment should be temporarily suspended in patients with severe hypertension that is not controlled with antihypertensive therapy and discontinued in patients who experience hypertensive crisis or hypertensive encephalopathy.
Proteinuria should be monitored by dipstick urine analysis during treatment, and patients with a 2+ or greater reading (concentration, 100 mg/dL) should undergo further assessment with 24-hour urine collection. Treatment should be suspended if proteinuria levels are ≥2 g/24h and can be resumed when they fall below that level, but should be discontinued in patients with nephrotic syndrome. Treatment should also be discontinued in patients who develop posterior reversible encephalopathy syndrome, and patients should be advised of the potential for fetal harm
1. FDA approves first biosimilar for the treatment of cancer. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm576112.htm. September 14, 2017. Accessed January 31, 2018.
2. Markus R, Chow V, Pan X, and Hanes V. A phase I, randomized, single-dose study evaluating the pharmacokinetic equivalence of biosimilar ABP 215 and bevacizumab in healthy adult men. Cancer Chemother. Pharmacol. 2017;80:755-763.
3. Thatcher N, Thomas M, Ostoros G, et al. Randomized, double-blind, phase 3 study comparing biosimilar candidate ABP-215 with bevacizumab in patients with non-squamous NSCLC. J Thorac Oncol. 2017;12(1):S902-S903.
4. Mvasi (bevacizumab-awwb) solution, for intravenous infusion. Prescribing information. Amgen Inc, https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761028s000lbl.pdf. September 2017. Accessed January 31, 2018.
1. FDA approves first biosimilar for the treatment of cancer. FDA News Release. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm576112.htm. September 14, 2017. Accessed January 31, 2018.
2. Markus R, Chow V, Pan X, and Hanes V. A phase I, randomized, single-dose study evaluating the pharmacokinetic equivalence of biosimilar ABP 215 and bevacizumab in healthy adult men. Cancer Chemother. Pharmacol. 2017;80:755-763.
3. Thatcher N, Thomas M, Ostoros G, et al. Randomized, double-blind, phase 3 study comparing biosimilar candidate ABP-215 with bevacizumab in patients with non-squamous NSCLC. J Thorac Oncol. 2017;12(1):S902-S903.
4. Mvasi (bevacizumab-awwb) solution, for intravenous infusion. Prescribing information. Amgen Inc, https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761028s000lbl.pdf. September 2017. Accessed January 31, 2018.
