User login
According to the US Food and Drug Administration (FDA), a biosimilar is “highly similar to an FDA-approved biological product, . . . and has no clinically meaningful differences in terms of safety and effectiveness.”1 The Biologics Price Competition and Innovation (BPCI) Act of 2009 created an expedited pathway for the approval of products shown to be biosimilar to FDA-licensed reference products.2 In 2013, the European Medicines Agency approved the first biosimilar modeled on infliximab (Remsima [formerly known as CT-P13], Celltrion Healthcare Co, Ltd) for the same indications as its reference product.3 In 2016, the FDA approved Inflectra (Hospira, a Pfizer Company), an infliximab biosimilar; Erelzi (Sandoz, a Novartis Division), an etanercept biosimilar; and Amjevita (Amgen Inc), an adalimumab biosimilar, all for numerous clinical indications including plaque psoriasis and psoriatic arthritis.4-6
There has been a substantial amount of distrust surrounding the biosimilars; however, as the patents for the biologic agents expire, new biosimilars will undoubtedly flood the market. In this article, we provide information that will help dermatologists understand the need for and use of these agents.
Biosimilars Versus Generic Drugs
Small-molecule generics can be made in a process that is relatively inexpensive, reproducible, and able to yield identical products with each lot.7 In contrast, biosimilars are large complex proteins made in living cells. They differ from their reference product because of changes that occur during manufacturing (eg, purification system, posttranslational modifications).7-9 Glycosylation is particularly sensitive to manufacturing and can affect the immunogenicity of the product.9 The impact of manufacturing can be substantial; for example, during phase 3 trials for efalizumab, a change in the manufacturing facility affected pharmacokinetic properties to such a degree that the FDA required a repeat of the trials.10
FDA Guidelines on Biosimilarity
The FDA outlines the following approach to demonstrate biosimilarity.2 The first step is structural characterization to evaluate the primary, secondary, tertiary, and quaternary structures and posttranslational modifications. The next step utilizes in vivo and/or in vitro functional assays to compare the biosimilar and reference product. The third step is a focus on toxicity and immunogenicity. The fourth step involves clinical studies to study pharmacokinetic and pharmacodynamic data, immunogenicity, safety, and efficacy. After the biosimilar has been approved, there must be a system in place to monitor postmarketing safety. If a biosimilar is tested in one patient population (eg, patients with plaque psoriasis), a request can be made to approve the drug for all the conditions that the reference product was approved for, such as plaque psoriasis, rheumatoid arthritis, and inflammatory bowel disease, even though clinical trials were not performed in all of these patient populations.2 The BPCI Act leaves it up to the FDA to determine how much and what type of data (eg, in vitro, in vivo, clinical) are required.11
Extrapolation and Interchangeability
Once a biosimilar has been approved, 2 questions must be answered: First, can its use be extrapolated to all indications for the reference product? The infliximab biosimilar approved by the European Medicines Agency and the FDA had only been studied in patients with ankylosing spondylitis12 and rheumatoid arthritis,13 yet it was granted all the indications for infliximab, including severe plaque psoriasis.14 As of now, the various regulatory agencies differ on their policies regarding extrapolation. Extrapolation is not automatically bestowed on a biosimilar in the United States but can be requested by the manufacturer.2
Second, can the biosimilar be seamlessly switched with its reference product at the pharmacy level? The BPCI Act allows for the substitution of biosimilars that are deemed interchangeable without notifying the provider, yet individual states ultimately can pass laws regarding this issue.15,16 An interchangeable agent would “produce the same clinical result as the reference product,” and “the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the reference product is not greater than the risk of using the reference product.”15 Generic drugs are allowed to be substituted without notifying the patient or prescriber16; however, biosimilars that are not deemed interchangeable would require permission from the prescriber before substitution.11
Biosimilars for Psoriasis
In April 2016, an infliximab biosimilar (Inflectra) became the second biosimilar approved by the FDA.4 Inflectra was studied in clinical trials for patients with ankylosing spondylitis17 and rheumatoid arthritis,18 and in both trials the biosimilar was found to have similar efficacy and safety profiles to that of the reference product. In August 2016, an etanercept biosimilar (Erelzi) was approved,5 and in September 2016, an adalimumab biosimilar (Amjevita) was approved.6
The Table summarizes clinical trials (both completed and ongoing) evaluating biosimilars in adults with plaque psoriasis; thus far, there are 2464 participants enrolled across 5 different studies of adalimumab biosimilars (registered at www.clinicaltrials.gov with the identifiers NCT01970488, NCT02016105, NCT02489227, NCT02714322, NCT02581345) and 531 participants in an etanercept biosimilar study (NCT01891864).
A phase 3 double-blind study compared adalimumab to an adalimumab biosimilar (ABP 501) in 350 adults with plaque psoriasis (NCT01970488). Participants received an initial loading dose of adalimumab (n=175) or ABP 501 (n=175) 80 mg subcutaneously on week 1/day 1, followed by 40 mg at week 2 every 2 weeks thereafter. At week 16, participants with psoriasis area and severity index (PASI) 50 or greater remained in the study for up to 52 weeks; those who were receiving adalimumab were re-randomized to receive either ABP 501 or adalimumab. Participants receiving ABP 501 continued to receive the biosimilar. The mean PASI improvement at weeks 16, 32, and 50 was 86.6, 87.6, and 87.2, respectively, in the ABP 501/ABP 501 group (A/A) compared to 88.0, 88.2, and 88.1, respectively, in the adalimumab/adalimumab group (B/B).19 Autoantibodies developed in 68.4% of participants in the A/A group compared to 74.7% in the B/B group. The incidence of treatment-emergent adverse events (TEAEs) was 86.2% in the A/A group and 78.5% in the B/B group. The most common TEAEs were nasopharyngitis, headache, and upper respiratory tract infection. The incidence of serious TEAEs was 4.6% in the A/A group compared to 5.1% in the B/B group. Overall, the efficacy, safety, and immunogenicity of the adalimumab biosimilar was comparable to the reference product.19
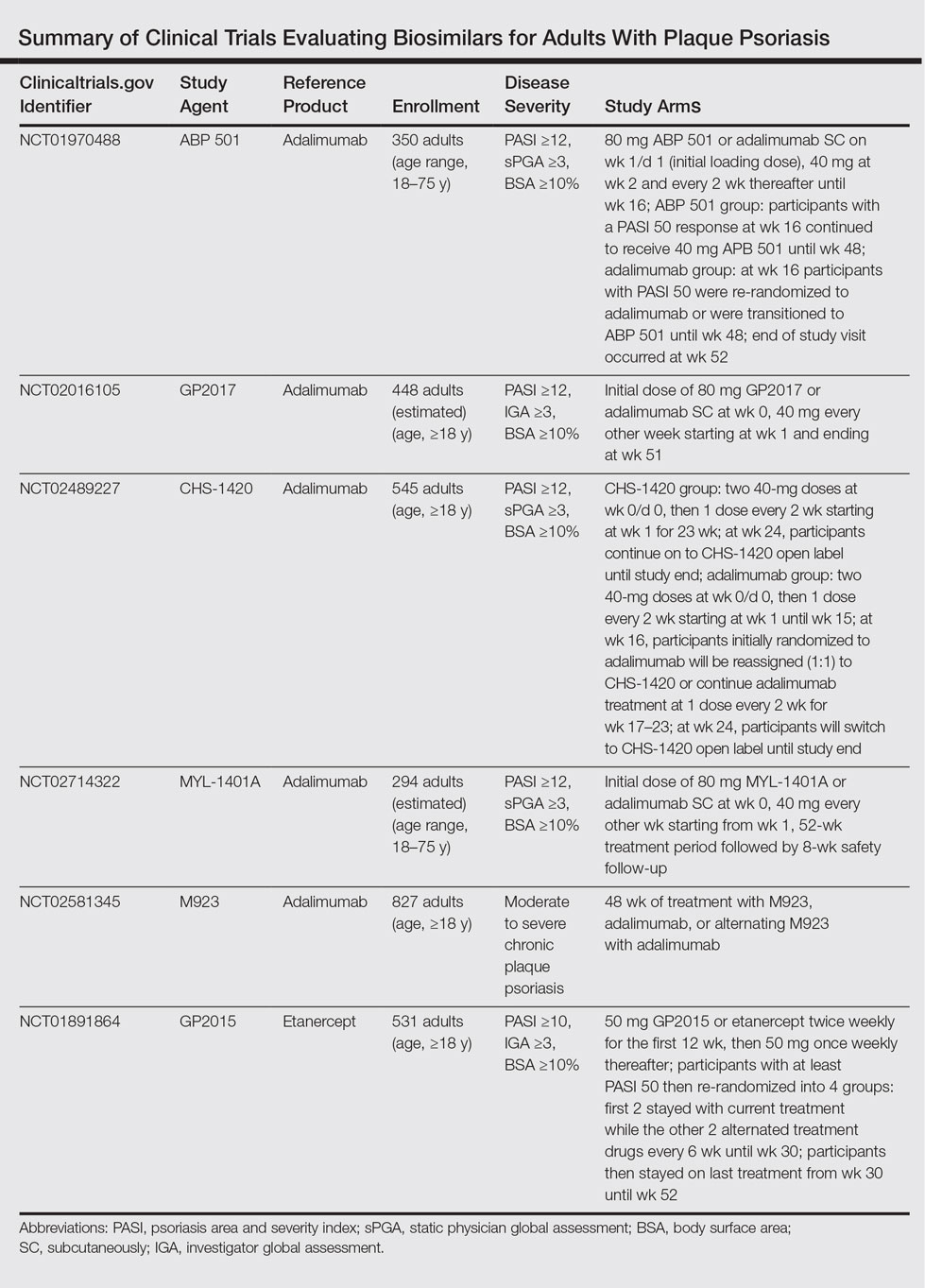
A second phase 3 trial (ADACCESS) evaluated the adalimumab biosimilar GP2017 (NCT02016105). Participants received an initial dose of 80 mg subcutaneously of either GP2017 or adalimumab at week 0, followed by 40 mg every other week starting at week 1 and ending at week 51. The study has been completed but results are not yet available.
The third trial is evaluating the adalimumab biosimilar CHS-1420 (NCT02489227). Participants in the experimental arm receive two 40-mg doses of CHS-1420 at week 0/day 0, and then 1 dose every 2 weeks from week 1 for 23 weeks. At week 24, participants continue with an open-label study. Participants in the adalimumab group receive two 40-mg doses at week 0/day 0, and then 1 dose every 2 weeks from week 1 to week 15. At week 16, participants will be re-randomized (1:1) to continue adalimumab or start CHS-1420 at one 40-mg dose every 2 weeks during weeks 17 to 23. At week 24, participants will switch to CHS-1420 open label until the end of the study. Study results are not yet available; the study is ongoing but not recruiting.
The fourth ongoing trial is evaluating the adalimumab biosimilar MYL-1401A (NCT02714322). Participants receive an initial dose of 80 mg subcutaneously of either MYL-1401A or adalimumab (2:1), followed by 40 mg every other week starting 1 week after the initial dose. After the 52-week treatment period, there is an 8-week safety follow-up period. Study results are not yet available; the study is ongoing but not recruiting.
A fifth adalimumab biosimilar, M923, also is currently being tested in clinical trials (NCT02581345). Participants receive either M923, adalimumab, or alternate between the 2 agents. Although the study is still ongoing, data released from the manufacturer state that the proportion of participants who achieved PASI 75 after 16 weeks of treatment was equivalent in the 2 treatment groups. The proportion of participants who achieved PASI 90, as well as the type, frequency, and severity of adverse events, also were comparable.20
The EGALITY trial, completed in March 2015, compared the etanercept biosimilar GP2015 to etanercept over a 52-week period (NCT01891864). Participants received either GP2015 or etanercept 50 mg twice weekly for the first 12 weeks. Participants with at least PASI 50 were then re-randomized into 4 groups: the first 2 groups stayed with their current treatments while the other 2 groups alternated treatments every 6 weeks until week 30. Participants then stayed on their last treatment from week 30 to week 52. The adjusted PASI 75 response rate at week 12 was 73.4% in the group receiving GP2015 and 75.7% in the group receiving etanercept.21 The percentage change in PASI score at all time points was found to be comparable from baseline until week 52. Importantly, the incidence of TEAEs up to week 52 was comparable and no new safety issues were reported. Additionally, switching participants from etanercept to the biosimilar during the subsequent treatment periods did not cause an increase in formation of antidrug antibodies.21
There are 2 upcoming studies involving biosimilars that are not yet recruiting patients. The first (NCT02925338) will analyze the characteristics of patients treated with Inflectra as well as their response to treatment. The second (NCT02762955) will be comparing the efficacy and safety of an adalimumab biosimilar (BCD-057, BIOCAD) to adalimumab.
Economic Advantages of Biosimilars
The annual economic burden of psoriasis in the United States is substantial, with estimates between $35.2 billion22 and $112 billion.23 Biosimilars can be 25% to 30% cheaper than their reference products9,11,24 and have the potential to save the US health care system billions of dollars.25 Furthermore, the developers of biosimilars could offer patient assistance programs.11 That being said, drug developers can extend patents for their branded drugs; for instance, 2 patents for Enbrel (Amgen Inc) could protect the drug until 2029.26,27
Although cost is an important factor in deciding which medications to prescribe for patients, it should never take precedence over safety and efficacy. Manufacturers can develop new drugs with greater efficacy, fewer side effects, or more convenient dosing schedules,26,27 or they could offer co-payment assistance programs.26,28 Physicians also must consider how the biosimilars will be integrated into drug formularies. Would patients be required to use a biosimilar before a branded drug?11,29 Will patients already taking a branded drug be grandfathered in?11 Would they have to pay a premium to continue taking their drug? And finally, could changes in formularies and employer-payer relationships destabilize patient regimens?30
Conclusion
Preliminary results suggest that biosimilars can have similar safety, efficacy, and immunogenicity data compared to their reference products.19,21 Biosimilars have the potential to greatly reduce the cost burden associated with psoriasis. However, how similar is “highly similar”? Although cost is an important consideration in selecting drug therapies, the reason for using a biosimilar should never be based on cost alone.
- Information on biosimilars. US Food and Drug Administration website. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/. Updated May 10, 2016. Accessed July 5, 2016.
- US Department of Health and Human Services. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product: Guidance for Industry. Silver Spring, MD: US Food and Drug Administration; 2015.
- McKeage K. A review of CT-P13: an infliximab biosimilar. BioDrugs. 2014;28:313-321.
- FDA approves Inflectra, a biosimilar to Remicade [news release]. Silver Spring, MD: US Food and Drug Administration; April 5, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm494227.htm. Updated April 20, 2016. Accessed January 23, 2017.
- FDA approves Erelzi, a biosimilar to Enbrel [news release]. Silver Spring, MD: US Food and Drug Administration; August 30, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm518639.htm. Accessed January 23, 2017.
- FDA approves Amjevita, a biosimilar to Humira [news release]. Silver Spring, MD: US Food and Drug Administration; September 23, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm522243.htm. Accessed January 23, 2017.
- Scott BJ, Klein AV, Wang J. Biosimilar monoclonal antibodies: a Canadian regulatory perspective on the assessment of clinically relevant differences and indication extrapolation [published online June 26, 2014]. J Clin Pharmacol. 2015;55(suppl 3):S123-S132.
- Mellstedt H, Niederwieser D, Ludwig H. The challenge of biosimilars [published online September 14, 2007]. Ann Oncol. 2008;19:411-419.
- Puig L. Biosimilars and reference biologics: decisions on biosimilar interchangeability require the involvement of dermatologists [published online October 2, 2013]. Actas Dermosifiliogr. 2014;105:435-437.
- Strober BE, Armour K, Romiti R, et al. Biopharmaceuticals and biosimilars in psoriasis: what the dermatologist needs to know. J Am Acad Dermatol. 2012;66:317-322.
- Falit BP, Singh SC, Brennan TA. Biosimilar competition in the United States: statutory incentives, payers, and pharmacy benefit managers. Health Aff (Millwood). 2015;34:294-301.
- Park W, Hrycaj P, Jeka S, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72:1605-1612.
- Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72:1613-1620.
- Carretero Hernandez G, Puig L. The use of biosimilar drugs in psoriasis: a position paper. Actas Dermosifiliogr. 2015;106:249-251.
- Regulation of Biological Products, 42 USC §262 (2013).
- Ventola CL. Evaluation of biosimilars for formulary inclusion: factors for consideration by P&T committees. P T. 2015;40:680-689.
- Park W, Yoo DH, Jaworski J, et al. Comparable long-term efficacy, as assessed by patient-reported outcomes, safety and pharmacokinetics, of CT-P13 and reference infliximab in patients with ankylosing spondylitis: 54-week results from the randomized, parallel-group PLANETAS study. Arthritis Res Ther. 2016;18:25.
- Yoo DH, Racewicz A, Brzezicki J, et al. A phase III randomized study to evaluate the efficacy and safety of CT-P13 compared with reference infliximab in patients with active rheumatoid arthritis: 54-week results from the PLANETRA study. Arthritis Res Ther. 2015;18:82.
- Strober B, Foley P, Philipp S, et al. Evaluation of efficacy and safety of ABP 501 in a phase 3 study in subjects with moderate to severe plaque psoriasis: 52-week results. J Am Acad Dermatol. 2016;74(5, suppl 1):AB249.
- Momenta Pharmaceuticals announces positive top-line phase 3 results for M923, a proposed Humira (adalimumab) biosimilar [news release]. Cambridge, MA: Momenta Pharmaceuticals, Inc; November 29, 2016. http://ir.momentapharma.com/releasedetail.cfm?ReleaseID=1001255. Accessed January 25, 2017.
- Griffiths CE, Thaci D, Gerdes S, et al. The EGALITY study: a confirmatory, randomised, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, versus the originator product in patients with moderate to severe chronic plaque-type psoriasis [published online October 27, 2016]. Br J Dermatol. doi:10.1111/bjd.15152.
- Vanderpuye-Orgle J, Zhao Y, Lu J, et al. Evaluating the economic burden of psoriasis in the United States [published online April 14, 2015]. J Am Acad Dermatol. 2015;72:961-967.
- Brezinski EA, Dhillon JS, Armstrong AW. Economic burden of psoriasis in the United States: a systematic review. JAMA Dermatol. 2015;151:651-658.
- Menter MA, Griffiths CE. Psoriasis: the future. Dermatol Clin. 2015;33:161-166.
- Hackbarth GM, Crosson FJ, Miller ME. Report to the Congress: improving incentives in the Medicare program. Medicare Payment Advisory Commission, Washington, DC; 2009.
- Lovenworth SJ. The new biosimilar era: the basics, the landscape, and the future. Bloomberg website. http://about.bloomberglaw.com/practitioner-contributions/the-new-biosimilar-era-the-basics-the-landscape-and-the-future. Published September 21, 2012. Accessed July 6, 2016.
- Blackstone EA, Joseph PF. The economics of biosimilars. Am Health Drug Benefits. 2013;6:469-478.
- Calvo B, Zuniga L. The US approach to biosimilars: the long-awaited FDA approval pathway. BioDrugs. 2012;26:357-361.
- Lucio SD, Stevenson JG, Hoffman JM. Biosimilars: implications for health-system pharmacists. Am J Health Syst Pharm. 2013;70:2004-2017.
- Barriers to access attributed to formulary changes. Manag Care. 2012;21:41.
According to the US Food and Drug Administration (FDA), a biosimilar is “highly similar to an FDA-approved biological product, . . . and has no clinically meaningful differences in terms of safety and effectiveness.”1 The Biologics Price Competition and Innovation (BPCI) Act of 2009 created an expedited pathway for the approval of products shown to be biosimilar to FDA-licensed reference products.2 In 2013, the European Medicines Agency approved the first biosimilar modeled on infliximab (Remsima [formerly known as CT-P13], Celltrion Healthcare Co, Ltd) for the same indications as its reference product.3 In 2016, the FDA approved Inflectra (Hospira, a Pfizer Company), an infliximab biosimilar; Erelzi (Sandoz, a Novartis Division), an etanercept biosimilar; and Amjevita (Amgen Inc), an adalimumab biosimilar, all for numerous clinical indications including plaque psoriasis and psoriatic arthritis.4-6
There has been a substantial amount of distrust surrounding the biosimilars; however, as the patents for the biologic agents expire, new biosimilars will undoubtedly flood the market. In this article, we provide information that will help dermatologists understand the need for and use of these agents.
Biosimilars Versus Generic Drugs
Small-molecule generics can be made in a process that is relatively inexpensive, reproducible, and able to yield identical products with each lot.7 In contrast, biosimilars are large complex proteins made in living cells. They differ from their reference product because of changes that occur during manufacturing (eg, purification system, posttranslational modifications).7-9 Glycosylation is particularly sensitive to manufacturing and can affect the immunogenicity of the product.9 The impact of manufacturing can be substantial; for example, during phase 3 trials for efalizumab, a change in the manufacturing facility affected pharmacokinetic properties to such a degree that the FDA required a repeat of the trials.10
FDA Guidelines on Biosimilarity
The FDA outlines the following approach to demonstrate biosimilarity.2 The first step is structural characterization to evaluate the primary, secondary, tertiary, and quaternary structures and posttranslational modifications. The next step utilizes in vivo and/or in vitro functional assays to compare the biosimilar and reference product. The third step is a focus on toxicity and immunogenicity. The fourth step involves clinical studies to study pharmacokinetic and pharmacodynamic data, immunogenicity, safety, and efficacy. After the biosimilar has been approved, there must be a system in place to monitor postmarketing safety. If a biosimilar is tested in one patient population (eg, patients with plaque psoriasis), a request can be made to approve the drug for all the conditions that the reference product was approved for, such as plaque psoriasis, rheumatoid arthritis, and inflammatory bowel disease, even though clinical trials were not performed in all of these patient populations.2 The BPCI Act leaves it up to the FDA to determine how much and what type of data (eg, in vitro, in vivo, clinical) are required.11
Extrapolation and Interchangeability
Once a biosimilar has been approved, 2 questions must be answered: First, can its use be extrapolated to all indications for the reference product? The infliximab biosimilar approved by the European Medicines Agency and the FDA had only been studied in patients with ankylosing spondylitis12 and rheumatoid arthritis,13 yet it was granted all the indications for infliximab, including severe plaque psoriasis.14 As of now, the various regulatory agencies differ on their policies regarding extrapolation. Extrapolation is not automatically bestowed on a biosimilar in the United States but can be requested by the manufacturer.2
Second, can the biosimilar be seamlessly switched with its reference product at the pharmacy level? The BPCI Act allows for the substitution of biosimilars that are deemed interchangeable without notifying the provider, yet individual states ultimately can pass laws regarding this issue.15,16 An interchangeable agent would “produce the same clinical result as the reference product,” and “the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the reference product is not greater than the risk of using the reference product.”15 Generic drugs are allowed to be substituted without notifying the patient or prescriber16; however, biosimilars that are not deemed interchangeable would require permission from the prescriber before substitution.11
Biosimilars for Psoriasis
In April 2016, an infliximab biosimilar (Inflectra) became the second biosimilar approved by the FDA.4 Inflectra was studied in clinical trials for patients with ankylosing spondylitis17 and rheumatoid arthritis,18 and in both trials the biosimilar was found to have similar efficacy and safety profiles to that of the reference product. In August 2016, an etanercept biosimilar (Erelzi) was approved,5 and in September 2016, an adalimumab biosimilar (Amjevita) was approved.6
The Table summarizes clinical trials (both completed and ongoing) evaluating biosimilars in adults with plaque psoriasis; thus far, there are 2464 participants enrolled across 5 different studies of adalimumab biosimilars (registered at www.clinicaltrials.gov with the identifiers NCT01970488, NCT02016105, NCT02489227, NCT02714322, NCT02581345) and 531 participants in an etanercept biosimilar study (NCT01891864).
A phase 3 double-blind study compared adalimumab to an adalimumab biosimilar (ABP 501) in 350 adults with plaque psoriasis (NCT01970488). Participants received an initial loading dose of adalimumab (n=175) or ABP 501 (n=175) 80 mg subcutaneously on week 1/day 1, followed by 40 mg at week 2 every 2 weeks thereafter. At week 16, participants with psoriasis area and severity index (PASI) 50 or greater remained in the study for up to 52 weeks; those who were receiving adalimumab were re-randomized to receive either ABP 501 or adalimumab. Participants receiving ABP 501 continued to receive the biosimilar. The mean PASI improvement at weeks 16, 32, and 50 was 86.6, 87.6, and 87.2, respectively, in the ABP 501/ABP 501 group (A/A) compared to 88.0, 88.2, and 88.1, respectively, in the adalimumab/adalimumab group (B/B).19 Autoantibodies developed in 68.4% of participants in the A/A group compared to 74.7% in the B/B group. The incidence of treatment-emergent adverse events (TEAEs) was 86.2% in the A/A group and 78.5% in the B/B group. The most common TEAEs were nasopharyngitis, headache, and upper respiratory tract infection. The incidence of serious TEAEs was 4.6% in the A/A group compared to 5.1% in the B/B group. Overall, the efficacy, safety, and immunogenicity of the adalimumab biosimilar was comparable to the reference product.19
A second phase 3 trial (ADACCESS) evaluated the adalimumab biosimilar GP2017 (NCT02016105). Participants received an initial dose of 80 mg subcutaneously of either GP2017 or adalimumab at week 0, followed by 40 mg every other week starting at week 1 and ending at week 51. The study has been completed but results are not yet available.
The third trial is evaluating the adalimumab biosimilar CHS-1420 (NCT02489227). Participants in the experimental arm receive two 40-mg doses of CHS-1420 at week 0/day 0, and then 1 dose every 2 weeks from week 1 for 23 weeks. At week 24, participants continue with an open-label study. Participants in the adalimumab group receive two 40-mg doses at week 0/day 0, and then 1 dose every 2 weeks from week 1 to week 15. At week 16, participants will be re-randomized (1:1) to continue adalimumab or start CHS-1420 at one 40-mg dose every 2 weeks during weeks 17 to 23. At week 24, participants will switch to CHS-1420 open label until the end of the study. Study results are not yet available; the study is ongoing but not recruiting.
The fourth ongoing trial is evaluating the adalimumab biosimilar MYL-1401A (NCT02714322). Participants receive an initial dose of 80 mg subcutaneously of either MYL-1401A or adalimumab (2:1), followed by 40 mg every other week starting 1 week after the initial dose. After the 52-week treatment period, there is an 8-week safety follow-up period. Study results are not yet available; the study is ongoing but not recruiting.
A fifth adalimumab biosimilar, M923, also is currently being tested in clinical trials (NCT02581345). Participants receive either M923, adalimumab, or alternate between the 2 agents. Although the study is still ongoing, data released from the manufacturer state that the proportion of participants who achieved PASI 75 after 16 weeks of treatment was equivalent in the 2 treatment groups. The proportion of participants who achieved PASI 90, as well as the type, frequency, and severity of adverse events, also were comparable.20
The EGALITY trial, completed in March 2015, compared the etanercept biosimilar GP2015 to etanercept over a 52-week period (NCT01891864). Participants received either GP2015 or etanercept 50 mg twice weekly for the first 12 weeks. Participants with at least PASI 50 were then re-randomized into 4 groups: the first 2 groups stayed with their current treatments while the other 2 groups alternated treatments every 6 weeks until week 30. Participants then stayed on their last treatment from week 30 to week 52. The adjusted PASI 75 response rate at week 12 was 73.4% in the group receiving GP2015 and 75.7% in the group receiving etanercept.21 The percentage change in PASI score at all time points was found to be comparable from baseline until week 52. Importantly, the incidence of TEAEs up to week 52 was comparable and no new safety issues were reported. Additionally, switching participants from etanercept to the biosimilar during the subsequent treatment periods did not cause an increase in formation of antidrug antibodies.21
There are 2 upcoming studies involving biosimilars that are not yet recruiting patients. The first (NCT02925338) will analyze the characteristics of patients treated with Inflectra as well as their response to treatment. The second (NCT02762955) will be comparing the efficacy and safety of an adalimumab biosimilar (BCD-057, BIOCAD) to adalimumab.
Economic Advantages of Biosimilars
The annual economic burden of psoriasis in the United States is substantial, with estimates between $35.2 billion22 and $112 billion.23 Biosimilars can be 25% to 30% cheaper than their reference products9,11,24 and have the potential to save the US health care system billions of dollars.25 Furthermore, the developers of biosimilars could offer patient assistance programs.11 That being said, drug developers can extend patents for their branded drugs; for instance, 2 patents for Enbrel (Amgen Inc) could protect the drug until 2029.26,27
Although cost is an important factor in deciding which medications to prescribe for patients, it should never take precedence over safety and efficacy. Manufacturers can develop new drugs with greater efficacy, fewer side effects, or more convenient dosing schedules,26,27 or they could offer co-payment assistance programs.26,28 Physicians also must consider how the biosimilars will be integrated into drug formularies. Would patients be required to use a biosimilar before a branded drug?11,29 Will patients already taking a branded drug be grandfathered in?11 Would they have to pay a premium to continue taking their drug? And finally, could changes in formularies and employer-payer relationships destabilize patient regimens?30
Conclusion
Preliminary results suggest that biosimilars can have similar safety, efficacy, and immunogenicity data compared to their reference products.19,21 Biosimilars have the potential to greatly reduce the cost burden associated with psoriasis. However, how similar is “highly similar”? Although cost is an important consideration in selecting drug therapies, the reason for using a biosimilar should never be based on cost alone.
According to the US Food and Drug Administration (FDA), a biosimilar is “highly similar to an FDA-approved biological product, . . . and has no clinically meaningful differences in terms of safety and effectiveness.”1 The Biologics Price Competition and Innovation (BPCI) Act of 2009 created an expedited pathway for the approval of products shown to be biosimilar to FDA-licensed reference products.2 In 2013, the European Medicines Agency approved the first biosimilar modeled on infliximab (Remsima [formerly known as CT-P13], Celltrion Healthcare Co, Ltd) for the same indications as its reference product.3 In 2016, the FDA approved Inflectra (Hospira, a Pfizer Company), an infliximab biosimilar; Erelzi (Sandoz, a Novartis Division), an etanercept biosimilar; and Amjevita (Amgen Inc), an adalimumab biosimilar, all for numerous clinical indications including plaque psoriasis and psoriatic arthritis.4-6
There has been a substantial amount of distrust surrounding the biosimilars; however, as the patents for the biologic agents expire, new biosimilars will undoubtedly flood the market. In this article, we provide information that will help dermatologists understand the need for and use of these agents.
Biosimilars Versus Generic Drugs
Small-molecule generics can be made in a process that is relatively inexpensive, reproducible, and able to yield identical products with each lot.7 In contrast, biosimilars are large complex proteins made in living cells. They differ from their reference product because of changes that occur during manufacturing (eg, purification system, posttranslational modifications).7-9 Glycosylation is particularly sensitive to manufacturing and can affect the immunogenicity of the product.9 The impact of manufacturing can be substantial; for example, during phase 3 trials for efalizumab, a change in the manufacturing facility affected pharmacokinetic properties to such a degree that the FDA required a repeat of the trials.10
FDA Guidelines on Biosimilarity
The FDA outlines the following approach to demonstrate biosimilarity.2 The first step is structural characterization to evaluate the primary, secondary, tertiary, and quaternary structures and posttranslational modifications. The next step utilizes in vivo and/or in vitro functional assays to compare the biosimilar and reference product. The third step is a focus on toxicity and immunogenicity. The fourth step involves clinical studies to study pharmacokinetic and pharmacodynamic data, immunogenicity, safety, and efficacy. After the biosimilar has been approved, there must be a system in place to monitor postmarketing safety. If a biosimilar is tested in one patient population (eg, patients with plaque psoriasis), a request can be made to approve the drug for all the conditions that the reference product was approved for, such as plaque psoriasis, rheumatoid arthritis, and inflammatory bowel disease, even though clinical trials were not performed in all of these patient populations.2 The BPCI Act leaves it up to the FDA to determine how much and what type of data (eg, in vitro, in vivo, clinical) are required.11
Extrapolation and Interchangeability
Once a biosimilar has been approved, 2 questions must be answered: First, can its use be extrapolated to all indications for the reference product? The infliximab biosimilar approved by the European Medicines Agency and the FDA had only been studied in patients with ankylosing spondylitis12 and rheumatoid arthritis,13 yet it was granted all the indications for infliximab, including severe plaque psoriasis.14 As of now, the various regulatory agencies differ on their policies regarding extrapolation. Extrapolation is not automatically bestowed on a biosimilar in the United States but can be requested by the manufacturer.2
Second, can the biosimilar be seamlessly switched with its reference product at the pharmacy level? The BPCI Act allows for the substitution of biosimilars that are deemed interchangeable without notifying the provider, yet individual states ultimately can pass laws regarding this issue.15,16 An interchangeable agent would “produce the same clinical result as the reference product,” and “the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the reference product is not greater than the risk of using the reference product.”15 Generic drugs are allowed to be substituted without notifying the patient or prescriber16; however, biosimilars that are not deemed interchangeable would require permission from the prescriber before substitution.11
Biosimilars for Psoriasis
In April 2016, an infliximab biosimilar (Inflectra) became the second biosimilar approved by the FDA.4 Inflectra was studied in clinical trials for patients with ankylosing spondylitis17 and rheumatoid arthritis,18 and in both trials the biosimilar was found to have similar efficacy and safety profiles to that of the reference product. In August 2016, an etanercept biosimilar (Erelzi) was approved,5 and in September 2016, an adalimumab biosimilar (Amjevita) was approved.6
The Table summarizes clinical trials (both completed and ongoing) evaluating biosimilars in adults with plaque psoriasis; thus far, there are 2464 participants enrolled across 5 different studies of adalimumab biosimilars (registered at www.clinicaltrials.gov with the identifiers NCT01970488, NCT02016105, NCT02489227, NCT02714322, NCT02581345) and 531 participants in an etanercept biosimilar study (NCT01891864).
A phase 3 double-blind study compared adalimumab to an adalimumab biosimilar (ABP 501) in 350 adults with plaque psoriasis (NCT01970488). Participants received an initial loading dose of adalimumab (n=175) or ABP 501 (n=175) 80 mg subcutaneously on week 1/day 1, followed by 40 mg at week 2 every 2 weeks thereafter. At week 16, participants with psoriasis area and severity index (PASI) 50 or greater remained in the study for up to 52 weeks; those who were receiving adalimumab were re-randomized to receive either ABP 501 or adalimumab. Participants receiving ABP 501 continued to receive the biosimilar. The mean PASI improvement at weeks 16, 32, and 50 was 86.6, 87.6, and 87.2, respectively, in the ABP 501/ABP 501 group (A/A) compared to 88.0, 88.2, and 88.1, respectively, in the adalimumab/adalimumab group (B/B).19 Autoantibodies developed in 68.4% of participants in the A/A group compared to 74.7% in the B/B group. The incidence of treatment-emergent adverse events (TEAEs) was 86.2% in the A/A group and 78.5% in the B/B group. The most common TEAEs were nasopharyngitis, headache, and upper respiratory tract infection. The incidence of serious TEAEs was 4.6% in the A/A group compared to 5.1% in the B/B group. Overall, the efficacy, safety, and immunogenicity of the adalimumab biosimilar was comparable to the reference product.19
A second phase 3 trial (ADACCESS) evaluated the adalimumab biosimilar GP2017 (NCT02016105). Participants received an initial dose of 80 mg subcutaneously of either GP2017 or adalimumab at week 0, followed by 40 mg every other week starting at week 1 and ending at week 51. The study has been completed but results are not yet available.
The third trial is evaluating the adalimumab biosimilar CHS-1420 (NCT02489227). Participants in the experimental arm receive two 40-mg doses of CHS-1420 at week 0/day 0, and then 1 dose every 2 weeks from week 1 for 23 weeks. At week 24, participants continue with an open-label study. Participants in the adalimumab group receive two 40-mg doses at week 0/day 0, and then 1 dose every 2 weeks from week 1 to week 15. At week 16, participants will be re-randomized (1:1) to continue adalimumab or start CHS-1420 at one 40-mg dose every 2 weeks during weeks 17 to 23. At week 24, participants will switch to CHS-1420 open label until the end of the study. Study results are not yet available; the study is ongoing but not recruiting.
The fourth ongoing trial is evaluating the adalimumab biosimilar MYL-1401A (NCT02714322). Participants receive an initial dose of 80 mg subcutaneously of either MYL-1401A or adalimumab (2:1), followed by 40 mg every other week starting 1 week after the initial dose. After the 52-week treatment period, there is an 8-week safety follow-up period. Study results are not yet available; the study is ongoing but not recruiting.
A fifth adalimumab biosimilar, M923, also is currently being tested in clinical trials (NCT02581345). Participants receive either M923, adalimumab, or alternate between the 2 agents. Although the study is still ongoing, data released from the manufacturer state that the proportion of participants who achieved PASI 75 after 16 weeks of treatment was equivalent in the 2 treatment groups. The proportion of participants who achieved PASI 90, as well as the type, frequency, and severity of adverse events, also were comparable.20
The EGALITY trial, completed in March 2015, compared the etanercept biosimilar GP2015 to etanercept over a 52-week period (NCT01891864). Participants received either GP2015 or etanercept 50 mg twice weekly for the first 12 weeks. Participants with at least PASI 50 were then re-randomized into 4 groups: the first 2 groups stayed with their current treatments while the other 2 groups alternated treatments every 6 weeks until week 30. Participants then stayed on their last treatment from week 30 to week 52. The adjusted PASI 75 response rate at week 12 was 73.4% in the group receiving GP2015 and 75.7% in the group receiving etanercept.21 The percentage change in PASI score at all time points was found to be comparable from baseline until week 52. Importantly, the incidence of TEAEs up to week 52 was comparable and no new safety issues were reported. Additionally, switching participants from etanercept to the biosimilar during the subsequent treatment periods did not cause an increase in formation of antidrug antibodies.21
There are 2 upcoming studies involving biosimilars that are not yet recruiting patients. The first (NCT02925338) will analyze the characteristics of patients treated with Inflectra as well as their response to treatment. The second (NCT02762955) will be comparing the efficacy and safety of an adalimumab biosimilar (BCD-057, BIOCAD) to adalimumab.
Economic Advantages of Biosimilars
The annual economic burden of psoriasis in the United States is substantial, with estimates between $35.2 billion22 and $112 billion.23 Biosimilars can be 25% to 30% cheaper than their reference products9,11,24 and have the potential to save the US health care system billions of dollars.25 Furthermore, the developers of biosimilars could offer patient assistance programs.11 That being said, drug developers can extend patents for their branded drugs; for instance, 2 patents for Enbrel (Amgen Inc) could protect the drug until 2029.26,27
Although cost is an important factor in deciding which medications to prescribe for patients, it should never take precedence over safety and efficacy. Manufacturers can develop new drugs with greater efficacy, fewer side effects, or more convenient dosing schedules,26,27 or they could offer co-payment assistance programs.26,28 Physicians also must consider how the biosimilars will be integrated into drug formularies. Would patients be required to use a biosimilar before a branded drug?11,29 Will patients already taking a branded drug be grandfathered in?11 Would they have to pay a premium to continue taking their drug? And finally, could changes in formularies and employer-payer relationships destabilize patient regimens?30
Conclusion
Preliminary results suggest that biosimilars can have similar safety, efficacy, and immunogenicity data compared to their reference products.19,21 Biosimilars have the potential to greatly reduce the cost burden associated with psoriasis. However, how similar is “highly similar”? Although cost is an important consideration in selecting drug therapies, the reason for using a biosimilar should never be based on cost alone.
- Information on biosimilars. US Food and Drug Administration website. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/. Updated May 10, 2016. Accessed July 5, 2016.
- US Department of Health and Human Services. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product: Guidance for Industry. Silver Spring, MD: US Food and Drug Administration; 2015.
- McKeage K. A review of CT-P13: an infliximab biosimilar. BioDrugs. 2014;28:313-321.
- FDA approves Inflectra, a biosimilar to Remicade [news release]. Silver Spring, MD: US Food and Drug Administration; April 5, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm494227.htm. Updated April 20, 2016. Accessed January 23, 2017.
- FDA approves Erelzi, a biosimilar to Enbrel [news release]. Silver Spring, MD: US Food and Drug Administration; August 30, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm518639.htm. Accessed January 23, 2017.
- FDA approves Amjevita, a biosimilar to Humira [news release]. Silver Spring, MD: US Food and Drug Administration; September 23, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm522243.htm. Accessed January 23, 2017.
- Scott BJ, Klein AV, Wang J. Biosimilar monoclonal antibodies: a Canadian regulatory perspective on the assessment of clinically relevant differences and indication extrapolation [published online June 26, 2014]. J Clin Pharmacol. 2015;55(suppl 3):S123-S132.
- Mellstedt H, Niederwieser D, Ludwig H. The challenge of biosimilars [published online September 14, 2007]. Ann Oncol. 2008;19:411-419.
- Puig L. Biosimilars and reference biologics: decisions on biosimilar interchangeability require the involvement of dermatologists [published online October 2, 2013]. Actas Dermosifiliogr. 2014;105:435-437.
- Strober BE, Armour K, Romiti R, et al. Biopharmaceuticals and biosimilars in psoriasis: what the dermatologist needs to know. J Am Acad Dermatol. 2012;66:317-322.
- Falit BP, Singh SC, Brennan TA. Biosimilar competition in the United States: statutory incentives, payers, and pharmacy benefit managers. Health Aff (Millwood). 2015;34:294-301.
- Park W, Hrycaj P, Jeka S, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72:1605-1612.
- Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72:1613-1620.
- Carretero Hernandez G, Puig L. The use of biosimilar drugs in psoriasis: a position paper. Actas Dermosifiliogr. 2015;106:249-251.
- Regulation of Biological Products, 42 USC §262 (2013).
- Ventola CL. Evaluation of biosimilars for formulary inclusion: factors for consideration by P&T committees. P T. 2015;40:680-689.
- Park W, Yoo DH, Jaworski J, et al. Comparable long-term efficacy, as assessed by patient-reported outcomes, safety and pharmacokinetics, of CT-P13 and reference infliximab in patients with ankylosing spondylitis: 54-week results from the randomized, parallel-group PLANETAS study. Arthritis Res Ther. 2016;18:25.
- Yoo DH, Racewicz A, Brzezicki J, et al. A phase III randomized study to evaluate the efficacy and safety of CT-P13 compared with reference infliximab in patients with active rheumatoid arthritis: 54-week results from the PLANETRA study. Arthritis Res Ther. 2015;18:82.
- Strober B, Foley P, Philipp S, et al. Evaluation of efficacy and safety of ABP 501 in a phase 3 study in subjects with moderate to severe plaque psoriasis: 52-week results. J Am Acad Dermatol. 2016;74(5, suppl 1):AB249.
- Momenta Pharmaceuticals announces positive top-line phase 3 results for M923, a proposed Humira (adalimumab) biosimilar [news release]. Cambridge, MA: Momenta Pharmaceuticals, Inc; November 29, 2016. http://ir.momentapharma.com/releasedetail.cfm?ReleaseID=1001255. Accessed January 25, 2017.
- Griffiths CE, Thaci D, Gerdes S, et al. The EGALITY study: a confirmatory, randomised, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, versus the originator product in patients with moderate to severe chronic plaque-type psoriasis [published online October 27, 2016]. Br J Dermatol. doi:10.1111/bjd.15152.
- Vanderpuye-Orgle J, Zhao Y, Lu J, et al. Evaluating the economic burden of psoriasis in the United States [published online April 14, 2015]. J Am Acad Dermatol. 2015;72:961-967.
- Brezinski EA, Dhillon JS, Armstrong AW. Economic burden of psoriasis in the United States: a systematic review. JAMA Dermatol. 2015;151:651-658.
- Menter MA, Griffiths CE. Psoriasis: the future. Dermatol Clin. 2015;33:161-166.
- Hackbarth GM, Crosson FJ, Miller ME. Report to the Congress: improving incentives in the Medicare program. Medicare Payment Advisory Commission, Washington, DC; 2009.
- Lovenworth SJ. The new biosimilar era: the basics, the landscape, and the future. Bloomberg website. http://about.bloomberglaw.com/practitioner-contributions/the-new-biosimilar-era-the-basics-the-landscape-and-the-future. Published September 21, 2012. Accessed July 6, 2016.
- Blackstone EA, Joseph PF. The economics of biosimilars. Am Health Drug Benefits. 2013;6:469-478.
- Calvo B, Zuniga L. The US approach to biosimilars: the long-awaited FDA approval pathway. BioDrugs. 2012;26:357-361.
- Lucio SD, Stevenson JG, Hoffman JM. Biosimilars: implications for health-system pharmacists. Am J Health Syst Pharm. 2013;70:2004-2017.
- Barriers to access attributed to formulary changes. Manag Care. 2012;21:41.
- Information on biosimilars. US Food and Drug Administration website. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/. Updated May 10, 2016. Accessed July 5, 2016.
- US Department of Health and Human Services. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product: Guidance for Industry. Silver Spring, MD: US Food and Drug Administration; 2015.
- McKeage K. A review of CT-P13: an infliximab biosimilar. BioDrugs. 2014;28:313-321.
- FDA approves Inflectra, a biosimilar to Remicade [news release]. Silver Spring, MD: US Food and Drug Administration; April 5, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm494227.htm. Updated April 20, 2016. Accessed January 23, 2017.
- FDA approves Erelzi, a biosimilar to Enbrel [news release]. Silver Spring, MD: US Food and Drug Administration; August 30, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm518639.htm. Accessed January 23, 2017.
- FDA approves Amjevita, a biosimilar to Humira [news release]. Silver Spring, MD: US Food and Drug Administration; September 23, 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm522243.htm. Accessed January 23, 2017.
- Scott BJ, Klein AV, Wang J. Biosimilar monoclonal antibodies: a Canadian regulatory perspective on the assessment of clinically relevant differences and indication extrapolation [published online June 26, 2014]. J Clin Pharmacol. 2015;55(suppl 3):S123-S132.
- Mellstedt H, Niederwieser D, Ludwig H. The challenge of biosimilars [published online September 14, 2007]. Ann Oncol. 2008;19:411-419.
- Puig L. Biosimilars and reference biologics: decisions on biosimilar interchangeability require the involvement of dermatologists [published online October 2, 2013]. Actas Dermosifiliogr. 2014;105:435-437.
- Strober BE, Armour K, Romiti R, et al. Biopharmaceuticals and biosimilars in psoriasis: what the dermatologist needs to know. J Am Acad Dermatol. 2012;66:317-322.
- Falit BP, Singh SC, Brennan TA. Biosimilar competition in the United States: statutory incentives, payers, and pharmacy benefit managers. Health Aff (Millwood). 2015;34:294-301.
- Park W, Hrycaj P, Jeka S, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72:1605-1612.
- Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72:1613-1620.
- Carretero Hernandez G, Puig L. The use of biosimilar drugs in psoriasis: a position paper. Actas Dermosifiliogr. 2015;106:249-251.
- Regulation of Biological Products, 42 USC §262 (2013).
- Ventola CL. Evaluation of biosimilars for formulary inclusion: factors for consideration by P&T committees. P T. 2015;40:680-689.
- Park W, Yoo DH, Jaworski J, et al. Comparable long-term efficacy, as assessed by patient-reported outcomes, safety and pharmacokinetics, of CT-P13 and reference infliximab in patients with ankylosing spondylitis: 54-week results from the randomized, parallel-group PLANETAS study. Arthritis Res Ther. 2016;18:25.
- Yoo DH, Racewicz A, Brzezicki J, et al. A phase III randomized study to evaluate the efficacy and safety of CT-P13 compared with reference infliximab in patients with active rheumatoid arthritis: 54-week results from the PLANETRA study. Arthritis Res Ther. 2015;18:82.
- Strober B, Foley P, Philipp S, et al. Evaluation of efficacy and safety of ABP 501 in a phase 3 study in subjects with moderate to severe plaque psoriasis: 52-week results. J Am Acad Dermatol. 2016;74(5, suppl 1):AB249.
- Momenta Pharmaceuticals announces positive top-line phase 3 results for M923, a proposed Humira (adalimumab) biosimilar [news release]. Cambridge, MA: Momenta Pharmaceuticals, Inc; November 29, 2016. http://ir.momentapharma.com/releasedetail.cfm?ReleaseID=1001255. Accessed January 25, 2017.
- Griffiths CE, Thaci D, Gerdes S, et al. The EGALITY study: a confirmatory, randomised, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, versus the originator product in patients with moderate to severe chronic plaque-type psoriasis [published online October 27, 2016]. Br J Dermatol. doi:10.1111/bjd.15152.
- Vanderpuye-Orgle J, Zhao Y, Lu J, et al. Evaluating the economic burden of psoriasis in the United States [published online April 14, 2015]. J Am Acad Dermatol. 2015;72:961-967.
- Brezinski EA, Dhillon JS, Armstrong AW. Economic burden of psoriasis in the United States: a systematic review. JAMA Dermatol. 2015;151:651-658.
- Menter MA, Griffiths CE. Psoriasis: the future. Dermatol Clin. 2015;33:161-166.
- Hackbarth GM, Crosson FJ, Miller ME. Report to the Congress: improving incentives in the Medicare program. Medicare Payment Advisory Commission, Washington, DC; 2009.
- Lovenworth SJ. The new biosimilar era: the basics, the landscape, and the future. Bloomberg website. http://about.bloomberglaw.com/practitioner-contributions/the-new-biosimilar-era-the-basics-the-landscape-and-the-future. Published September 21, 2012. Accessed July 6, 2016.
- Blackstone EA, Joseph PF. The economics of biosimilars. Am Health Drug Benefits. 2013;6:469-478.
- Calvo B, Zuniga L. The US approach to biosimilars: the long-awaited FDA approval pathway. BioDrugs. 2012;26:357-361.
- Lucio SD, Stevenson JG, Hoffman JM. Biosimilars: implications for health-system pharmacists. Am J Health Syst Pharm. 2013;70:2004-2017.
- Barriers to access attributed to formulary changes. Manag Care. 2012;21:41.
Practice Points
- Three biosimilars have been approved by the US Food and Drug Administration to treat adult patients with plaque psoriasis and psoriatic arthritis.
- By virtue of their production, biosimilars are not identical to their reference products, and we must ensure that their safety is comparable.