User login
The Diagnosis: Lymphomatoid Papulosis
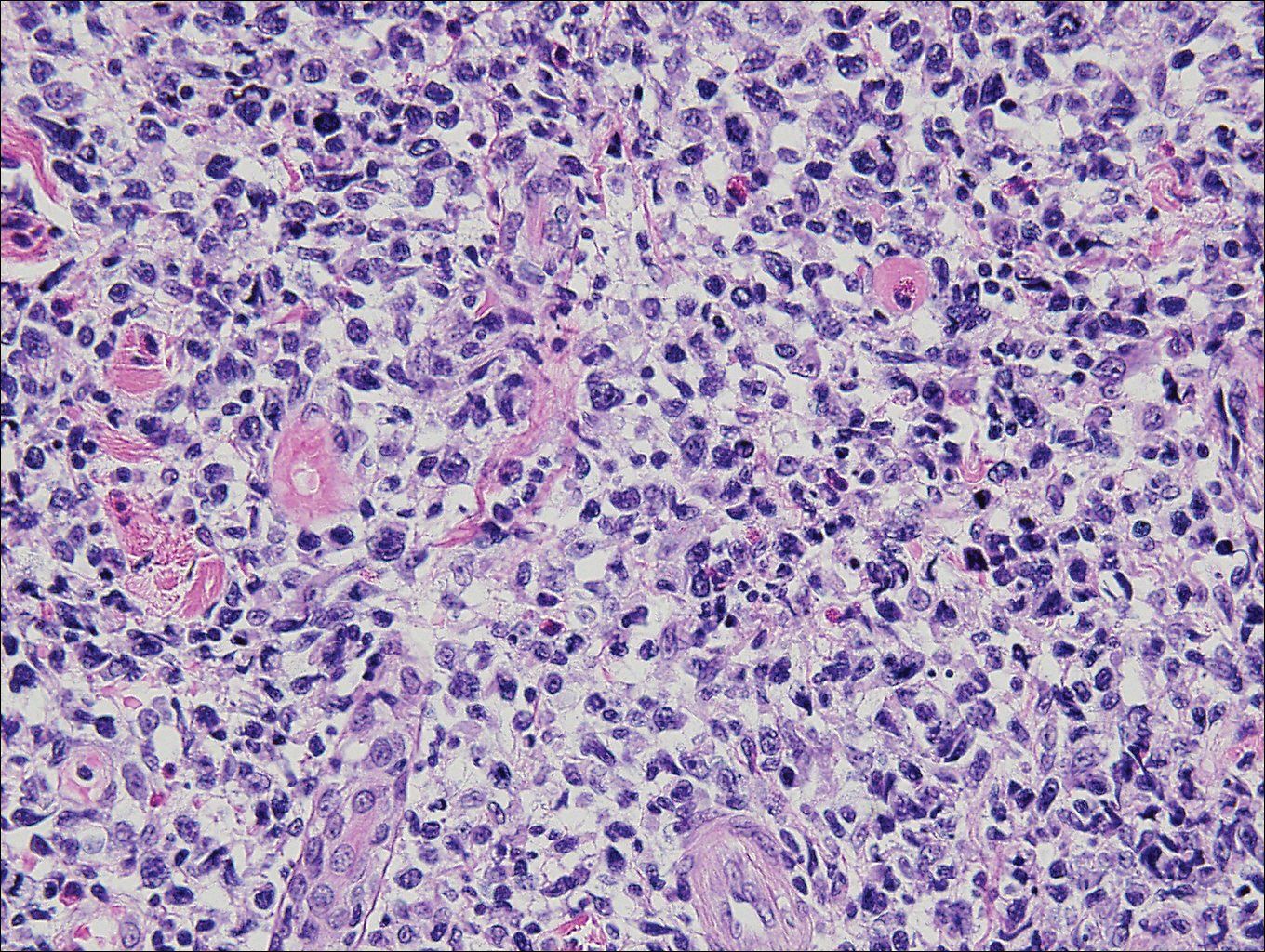
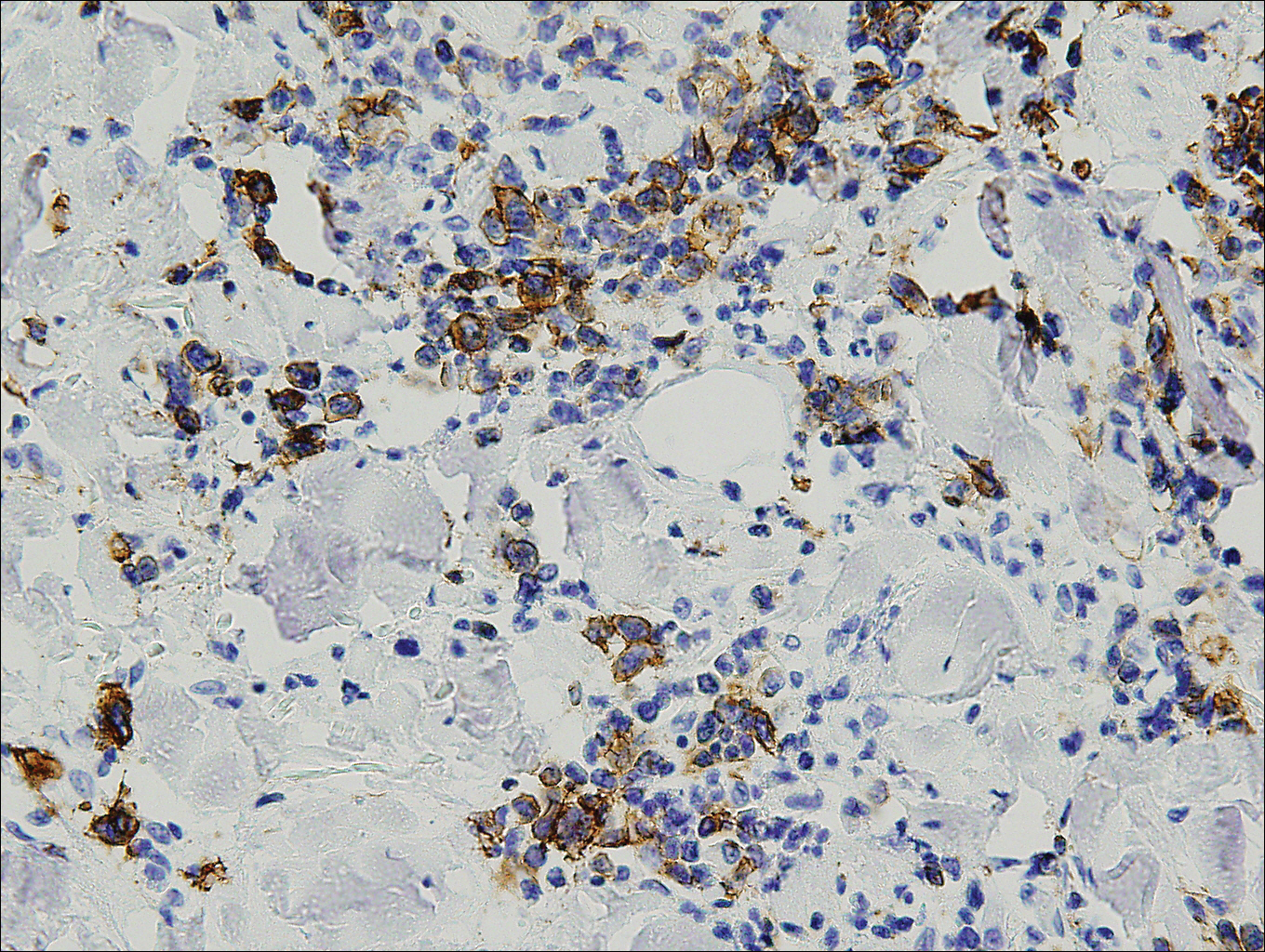
Histopathologic and immunohistochemical examination of the ulcer revealed a dense nodular and diffuse infiltrate in the papillary and reticular dermis comprised predominantly of atypical, CD30+, small T cells and large lymphoid cells admixed with neutrophils and eosinophils (Figures 1 and 2). Tissue cultures and infectious stains were negative. The complete blood cell count, metabolic panel, serum lactate dehydrogenase level, and peripheral blood flow cytometry were normal. Correlation of the lesions' self-healing nature with the histopathologic and immunohistochemical findings led to a diagnosis of lymphomatoid papulosis (LyP). In light of this diagnosis, a shave biopsy was obtained of one of the patient's poikilodermatous patches and was found to be consistent with poikilodermatous mycosis fungoides (MF).
At 4-month follow-up, the patient reported that she continued to develop crops of 1 to 3 LyP lesions each month. She continued to deny systemic concerns, and the poikilodermatous MF appeared unchanged. As part of a hematologic workup, a positron emission tomography-computed tomography scan revealed glucose-avid lymph nodes in the axillary, supraclavicular, abdominal, and inguinal regions. These findings raised concern for possible lymphomatous involvement of the patient's MF. Systemic therapy may be required pending further surveillance.
Lymphomatoid papulosis is a chronic papulonecrotic disease characterized clinically by recurrent crops of self-healing papules. Histopathologically, LyP features a perivascular infiltrate with atypical dermal T cells. Macaulay1 first described LyP in 1968 in a 41-year-old woman with a several-year history of continuously self-resolving crops of necrotic papules, noting the paradox between the patient's benign clinical course and malignant-seeming histology featuring "an alarming infiltrate of anaplastic cells." Since this report, LyP has continued to spur debate regarding its malignant potential but is now recognized as an indolent cutaneous T-cell lymphoma with an excellent prognosis.2
There are several histopathologic subtypes of LyP, the most common of which are type A, resembling Hodgkin lymphoma; type B, resembling MF; type C, resembling primary cutaneous anaplastic large cell lymphoma (C-ALCL); and type D, resembling aggressive epidermotropic CD8+ cutaneous T-cell lymphoma.2
The multifocal ulcers and eschars of LyP may appropriately raise suspicion for an infectious process, as in the present case. Numerous reports show that LyP may be initially misdiagnosed as an infection, such as cellulitis,3 furunculosis,4 parapoxvirus Orf,5 and ecthyma.6 Furthermore, several cutaneous infections have histopathologic features indistinguishable from LyP.7 For example, herpes simplex virus infection, molluscum contagiosum, Milker nodule, syphilis, and leishmaniasis may contain an appreciable number of large CD30+ T cells, which is compatible with both LyP type C and C-ALCL.7 As in the present case, the final diagnosis rests on clinicopathologic correlation, with LyP often distinguished by its invariable self-resolution, unlike its numerous infectious mimickers. The self-regressing nature of LyP also helps differentiate LyP occurring in the setting of MF from MF that has underwent CD30+ large cell transformation. In addition, the diagnosis of MF-associated LyP is favored over transformed MF when, as in the present case, CD30+ lesions develop on skin distinct from MF-affected skin.
Although isolated LyP is benign, 18% (11/61) of patients will subsequently develop lymphoma. More commonly, lymphomas may precede or occur concomitantly with the onset of LyP. In a retrospective study of 84 LyP patients, for example, 40% (34/84) had prior or concomitant lymphoma.8 Owing to the well-established link between LyP and lymphoma, there is appropriate emphasis on close monitoring of these patients. In addition, a careful history and physical examination are necessary to evaluate for a preceding, previously undiagnosed lymphoma. In point of fact, our patient had undiagnosed poikilodermatous MF prior to developing LyP, which was proven by biopsy at the time of LyP diagnosis. A distinct clinical variant of MF, poikilodermatous MF is characterized by hyperpigmented and hypopigmented patches, atrophy, and telangiectasia. A study of 49 patients with poikilodermatous MF found that this variant had an earlier age of onset compared with other types of MF. The study also showed that 18% (9/49) of patients had coexistent LyP, suggesting that poikilodermatous MF and LyP may be more frequently associated than previously believed.9
Treatment of LyP is unnecessary beyond basic wound care to avoid bacterial superinfection.2,10 Therapy for poikilodermatous MF, similar to other types of MF, is based on disease stage. Topical therapy may be utilized for localized disease, while systemic therapies are reserved for recalcitrant cases and internal involvement.9
Acknowledgments
We thank David L. Ramsay, MD, for obtaining aspects of the patient's history, and Shane A. Meehan, MD, and Adnan Mir, MD, PhD, as well as Cynthia M. Magro, MD, (all from New York, New York) for performing the histopathologic and immunohistochemical analyses.
- Macaulay WL. Lymphomatoid papulosis. a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Meena M, Martin PA, Abouseif C, et al. Lymphomatoid papulosis type C of the eyelid in a young girl: a case report and review of literature. Orbit. 2014;3:395-398.
- Dinotta F, Lacarrubba F, Micali G. Sixteen-year-old girl with papules and nodules on the face and upper limbs. Pediatr Dermatol. 2014;31:103-104.
- Eminger LA, Shinohara MM, Kim EJ, et al. Clinicopathologic challenge: acral lymphomatoid papulosis. Int J Dermatol. 2012;51:531-534.
- Harder D, Kuhn A, Mahrle G. Lymphomatoid papulosis resembling ecthyma. a case report. Z Hautkr. 1989;64:593-595.
- Werner B, Massone C, Kerl H, et al. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J Cutan Pathol. 2008;35:1100-1107.
- Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-581.
- Abbott RA, Sahni D, Robson A, et al. Poikilodermatous mycosis fungoides: a study of its clinicopathological, immunophenotypic, and prognostic features. J Am Acad Dermatol. 2011;65:313-319.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
The Diagnosis: Lymphomatoid Papulosis
Histopathologic and immunohistochemical examination of the ulcer revealed a dense nodular and diffuse infiltrate in the papillary and reticular dermis comprised predominantly of atypical, CD30+, small T cells and large lymphoid cells admixed with neutrophils and eosinophils (Figures 1 and 2). Tissue cultures and infectious stains were negative. The complete blood cell count, metabolic panel, serum lactate dehydrogenase level, and peripheral blood flow cytometry were normal. Correlation of the lesions' self-healing nature with the histopathologic and immunohistochemical findings led to a diagnosis of lymphomatoid papulosis (LyP). In light of this diagnosis, a shave biopsy was obtained of one of the patient's poikilodermatous patches and was found to be consistent with poikilodermatous mycosis fungoides (MF).
At 4-month follow-up, the patient reported that she continued to develop crops of 1 to 3 LyP lesions each month. She continued to deny systemic concerns, and the poikilodermatous MF appeared unchanged. As part of a hematologic workup, a positron emission tomography-computed tomography scan revealed glucose-avid lymph nodes in the axillary, supraclavicular, abdominal, and inguinal regions. These findings raised concern for possible lymphomatous involvement of the patient's MF. Systemic therapy may be required pending further surveillance.
Lymphomatoid papulosis is a chronic papulonecrotic disease characterized clinically by recurrent crops of self-healing papules. Histopathologically, LyP features a perivascular infiltrate with atypical dermal T cells. Macaulay1 first described LyP in 1968 in a 41-year-old woman with a several-year history of continuously self-resolving crops of necrotic papules, noting the paradox between the patient's benign clinical course and malignant-seeming histology featuring "an alarming infiltrate of anaplastic cells." Since this report, LyP has continued to spur debate regarding its malignant potential but is now recognized as an indolent cutaneous T-cell lymphoma with an excellent prognosis.2
There are several histopathologic subtypes of LyP, the most common of which are type A, resembling Hodgkin lymphoma; type B, resembling MF; type C, resembling primary cutaneous anaplastic large cell lymphoma (C-ALCL); and type D, resembling aggressive epidermotropic CD8+ cutaneous T-cell lymphoma.2
The multifocal ulcers and eschars of LyP may appropriately raise suspicion for an infectious process, as in the present case. Numerous reports show that LyP may be initially misdiagnosed as an infection, such as cellulitis,3 furunculosis,4 parapoxvirus Orf,5 and ecthyma.6 Furthermore, several cutaneous infections have histopathologic features indistinguishable from LyP.7 For example, herpes simplex virus infection, molluscum contagiosum, Milker nodule, syphilis, and leishmaniasis may contain an appreciable number of large CD30+ T cells, which is compatible with both LyP type C and C-ALCL.7 As in the present case, the final diagnosis rests on clinicopathologic correlation, with LyP often distinguished by its invariable self-resolution, unlike its numerous infectious mimickers. The self-regressing nature of LyP also helps differentiate LyP occurring in the setting of MF from MF that has underwent CD30+ large cell transformation. In addition, the diagnosis of MF-associated LyP is favored over transformed MF when, as in the present case, CD30+ lesions develop on skin distinct from MF-affected skin.
Although isolated LyP is benign, 18% (11/61) of patients will subsequently develop lymphoma. More commonly, lymphomas may precede or occur concomitantly with the onset of LyP. In a retrospective study of 84 LyP patients, for example, 40% (34/84) had prior or concomitant lymphoma.8 Owing to the well-established link between LyP and lymphoma, there is appropriate emphasis on close monitoring of these patients. In addition, a careful history and physical examination are necessary to evaluate for a preceding, previously undiagnosed lymphoma. In point of fact, our patient had undiagnosed poikilodermatous MF prior to developing LyP, which was proven by biopsy at the time of LyP diagnosis. A distinct clinical variant of MF, poikilodermatous MF is characterized by hyperpigmented and hypopigmented patches, atrophy, and telangiectasia. A study of 49 patients with poikilodermatous MF found that this variant had an earlier age of onset compared with other types of MF. The study also showed that 18% (9/49) of patients had coexistent LyP, suggesting that poikilodermatous MF and LyP may be more frequently associated than previously believed.9
Treatment of LyP is unnecessary beyond basic wound care to avoid bacterial superinfection.2,10 Therapy for poikilodermatous MF, similar to other types of MF, is based on disease stage. Topical therapy may be utilized for localized disease, while systemic therapies are reserved for recalcitrant cases and internal involvement.9
Acknowledgments
We thank David L. Ramsay, MD, for obtaining aspects of the patient's history, and Shane A. Meehan, MD, and Adnan Mir, MD, PhD, as well as Cynthia M. Magro, MD, (all from New York, New York) for performing the histopathologic and immunohistochemical analyses.
The Diagnosis: Lymphomatoid Papulosis
Histopathologic and immunohistochemical examination of the ulcer revealed a dense nodular and diffuse infiltrate in the papillary and reticular dermis comprised predominantly of atypical, CD30+, small T cells and large lymphoid cells admixed with neutrophils and eosinophils (Figures 1 and 2). Tissue cultures and infectious stains were negative. The complete blood cell count, metabolic panel, serum lactate dehydrogenase level, and peripheral blood flow cytometry were normal. Correlation of the lesions' self-healing nature with the histopathologic and immunohistochemical findings led to a diagnosis of lymphomatoid papulosis (LyP). In light of this diagnosis, a shave biopsy was obtained of one of the patient's poikilodermatous patches and was found to be consistent with poikilodermatous mycosis fungoides (MF).
At 4-month follow-up, the patient reported that she continued to develop crops of 1 to 3 LyP lesions each month. She continued to deny systemic concerns, and the poikilodermatous MF appeared unchanged. As part of a hematologic workup, a positron emission tomography-computed tomography scan revealed glucose-avid lymph nodes in the axillary, supraclavicular, abdominal, and inguinal regions. These findings raised concern for possible lymphomatous involvement of the patient's MF. Systemic therapy may be required pending further surveillance.
Lymphomatoid papulosis is a chronic papulonecrotic disease characterized clinically by recurrent crops of self-healing papules. Histopathologically, LyP features a perivascular infiltrate with atypical dermal T cells. Macaulay1 first described LyP in 1968 in a 41-year-old woman with a several-year history of continuously self-resolving crops of necrotic papules, noting the paradox between the patient's benign clinical course and malignant-seeming histology featuring "an alarming infiltrate of anaplastic cells." Since this report, LyP has continued to spur debate regarding its malignant potential but is now recognized as an indolent cutaneous T-cell lymphoma with an excellent prognosis.2
There are several histopathologic subtypes of LyP, the most common of which are type A, resembling Hodgkin lymphoma; type B, resembling MF; type C, resembling primary cutaneous anaplastic large cell lymphoma (C-ALCL); and type D, resembling aggressive epidermotropic CD8+ cutaneous T-cell lymphoma.2
The multifocal ulcers and eschars of LyP may appropriately raise suspicion for an infectious process, as in the present case. Numerous reports show that LyP may be initially misdiagnosed as an infection, such as cellulitis,3 furunculosis,4 parapoxvirus Orf,5 and ecthyma.6 Furthermore, several cutaneous infections have histopathologic features indistinguishable from LyP.7 For example, herpes simplex virus infection, molluscum contagiosum, Milker nodule, syphilis, and leishmaniasis may contain an appreciable number of large CD30+ T cells, which is compatible with both LyP type C and C-ALCL.7 As in the present case, the final diagnosis rests on clinicopathologic correlation, with LyP often distinguished by its invariable self-resolution, unlike its numerous infectious mimickers. The self-regressing nature of LyP also helps differentiate LyP occurring in the setting of MF from MF that has underwent CD30+ large cell transformation. In addition, the diagnosis of MF-associated LyP is favored over transformed MF when, as in the present case, CD30+ lesions develop on skin distinct from MF-affected skin.
Although isolated LyP is benign, 18% (11/61) of patients will subsequently develop lymphoma. More commonly, lymphomas may precede or occur concomitantly with the onset of LyP. In a retrospective study of 84 LyP patients, for example, 40% (34/84) had prior or concomitant lymphoma.8 Owing to the well-established link between LyP and lymphoma, there is appropriate emphasis on close monitoring of these patients. In addition, a careful history and physical examination are necessary to evaluate for a preceding, previously undiagnosed lymphoma. In point of fact, our patient had undiagnosed poikilodermatous MF prior to developing LyP, which was proven by biopsy at the time of LyP diagnosis. A distinct clinical variant of MF, poikilodermatous MF is characterized by hyperpigmented and hypopigmented patches, atrophy, and telangiectasia. A study of 49 patients with poikilodermatous MF found that this variant had an earlier age of onset compared with other types of MF. The study also showed that 18% (9/49) of patients had coexistent LyP, suggesting that poikilodermatous MF and LyP may be more frequently associated than previously believed.9
Treatment of LyP is unnecessary beyond basic wound care to avoid bacterial superinfection.2,10 Therapy for poikilodermatous MF, similar to other types of MF, is based on disease stage. Topical therapy may be utilized for localized disease, while systemic therapies are reserved for recalcitrant cases and internal involvement.9
Acknowledgments
We thank David L. Ramsay, MD, for obtaining aspects of the patient's history, and Shane A. Meehan, MD, and Adnan Mir, MD, PhD, as well as Cynthia M. Magro, MD, (all from New York, New York) for performing the histopathologic and immunohistochemical analyses.
- Macaulay WL. Lymphomatoid papulosis. a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Meena M, Martin PA, Abouseif C, et al. Lymphomatoid papulosis type C of the eyelid in a young girl: a case report and review of literature. Orbit. 2014;3:395-398.
- Dinotta F, Lacarrubba F, Micali G. Sixteen-year-old girl with papules and nodules on the face and upper limbs. Pediatr Dermatol. 2014;31:103-104.
- Eminger LA, Shinohara MM, Kim EJ, et al. Clinicopathologic challenge: acral lymphomatoid papulosis. Int J Dermatol. 2012;51:531-534.
- Harder D, Kuhn A, Mahrle G. Lymphomatoid papulosis resembling ecthyma. a case report. Z Hautkr. 1989;64:593-595.
- Werner B, Massone C, Kerl H, et al. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J Cutan Pathol. 2008;35:1100-1107.
- Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-581.
- Abbott RA, Sahni D, Robson A, et al. Poikilodermatous mycosis fungoides: a study of its clinicopathological, immunophenotypic, and prognostic features. J Am Acad Dermatol. 2011;65:313-319.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
- Macaulay WL. Lymphomatoid papulosis. a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Meena M, Martin PA, Abouseif C, et al. Lymphomatoid papulosis type C of the eyelid in a young girl: a case report and review of literature. Orbit. 2014;3:395-398.
- Dinotta F, Lacarrubba F, Micali G. Sixteen-year-old girl with papules and nodules on the face and upper limbs. Pediatr Dermatol. 2014;31:103-104.
- Eminger LA, Shinohara MM, Kim EJ, et al. Clinicopathologic challenge: acral lymphomatoid papulosis. Int J Dermatol. 2012;51:531-534.
- Harder D, Kuhn A, Mahrle G. Lymphomatoid papulosis resembling ecthyma. a case report. Z Hautkr. 1989;64:593-595.
- Werner B, Massone C, Kerl H, et al. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J Cutan Pathol. 2008;35:1100-1107.
- Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-581.
- Abbott RA, Sahni D, Robson A, et al. Poikilodermatous mycosis fungoides: a study of its clinicopathological, immunophenotypic, and prognostic features. J Am Acad Dermatol. 2011;65:313-319.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
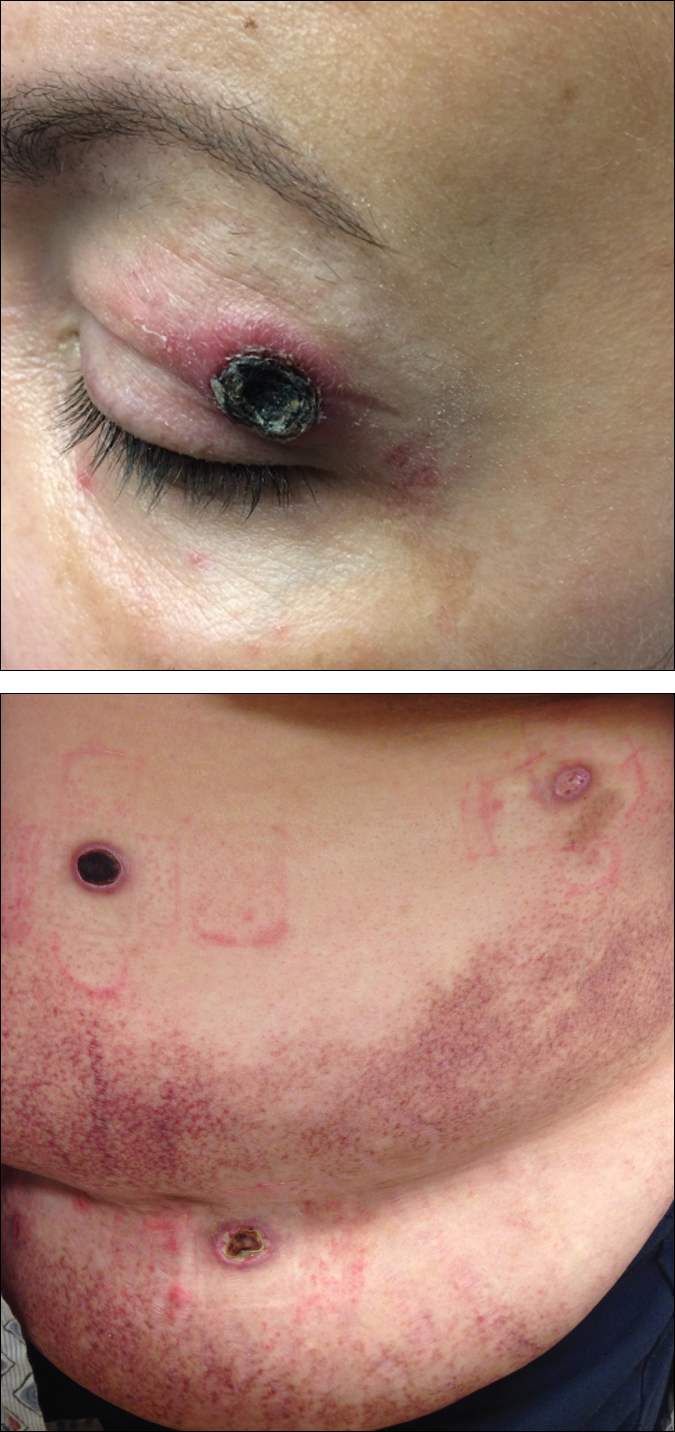
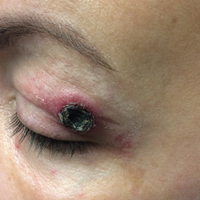
A 50-year-old woman presented for evaluation of black eschars on the face and body. Over the preceding 8 weeks she had developed several asymptomatic papules that gradually enlarged, ulcerated, and formed a black eschar, prior to gradually self-resolving over the course of several weeks. During this time, new lesions were forming. The resulting skin revealed dyspigmentation and scar formation. Prior to presentation, antimicrobial therapy had been initiated for a presumed infectious etiology; however, the eruption continued to progress. The patient denied sick contacts, livestock exposure, or recent travel. A complete review of systems, including fever, chills, or lymphadenopathy, was negative. Physical examination revealed 6 circular necrotic ulcers with an overlying black eschar on the face (top), trunk (bottom), hands, and thighs, all in various stages of healing. In addition, large, reticulated, poikilodermatous patches were incidentally noted in areas free of ulcers and eschars on the trunk (bottom) and bilateral arms and legs. Upon questioning, the patient said these patches had been present for more than 30 years. A punch biopsy from an ulcer on the chest was obtained and sent for histopathologic and immunohistochemical examination.