User login
The Diagnosis: Kikuchi-Fujimoto Disease
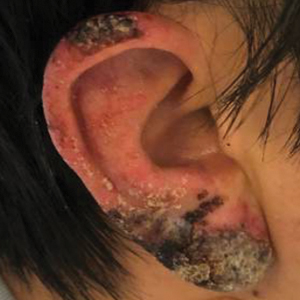
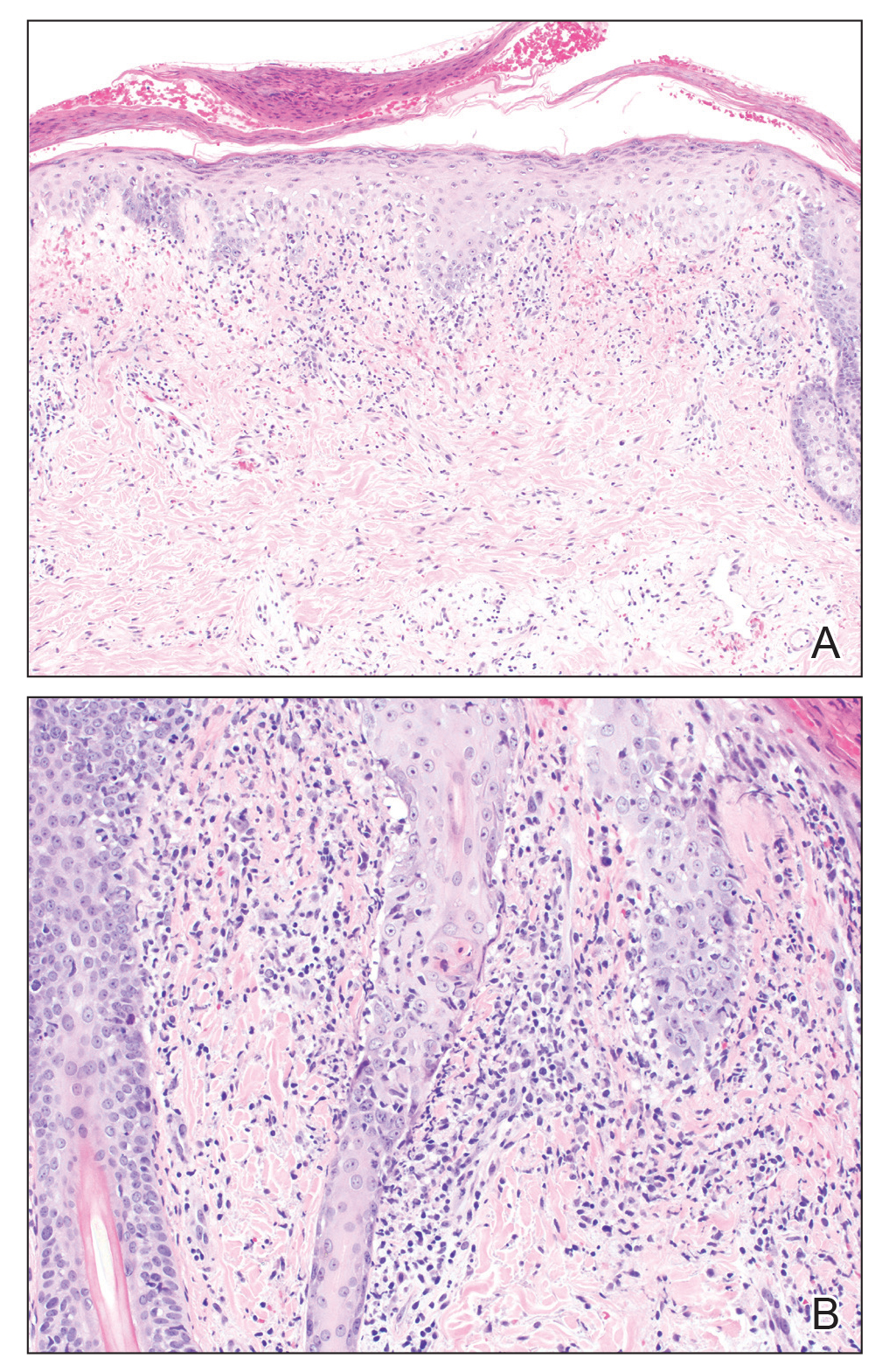
A skin biopsy from the left helix was obtained. Histopathologic examination revealed a vacuolar interface reaction with marked papillary dermal edema and a patchy perijunctional lymphocytic infiltrate. The dermis was free of increased mucin (Figure 1). Immunohistochemical staining for CD56 and Epstein-Barr virus (EBV)–encoded small nuclear RNA chromogenic in situ hybridization were negative. Laboratory workup was remarkable for elevated transaminases and inflammatory markers (eg, C-reactive protein, erythrocyte sedimentation rate) but negative for rheumatologic markers (eg, antinuclear antibodies, antineutrophil cytoplasmic antibodies, myeloperoxidase antibodies, serine protease IgG). An extensive infectious workup was unrevealing. Computed tomography highlighted prominent lymphadenopathy throughout the cervical and supraclavicular chains and a large necrotic lymph node in the porta hepatis (Figure 2). Right neck lymph node aspiration revealed necrotizing lymphadenitis in a background of histiocytes and mixed lymphocytes. Coupling the clinical presentation and histomorphology with imaging, a diagnosis of Kikuchi-Fujimoto disease (KD) was rendered.

Kikuchi-Fujimoto disease is a rare illness of unknown etiology characterized by cervical lymphadenopathy and fever. Originally described in Japan, KD affects all racial and ethnic groups1,2 but more commonly is seen in women and patients younger than 40 years.3 It can be associated with systemic lupus erythematosus (SLE) and other autoimmune diseases (eg, relapsing polychondritis, adult-onset Still disease),3 and lymphoma.4 Multiple infections have been implicated in the pathogenesis of KD, including EBV and other human herpesviruses; HIV; human T-cell leukemia virus type 1; dengue virus; parvovirus B19; and Yersinia enterocolitica, Bartonella, Brucella, and Toxoplasma infections.3,5,6
Kikuchi-Fujimoto disease classically presents with fever and cervical lymphadenopathy. In a retrospective review of 244 patients with KD, the 3 most common manifestations included lymphadenopathy, fever, and rash.7 A diagnosis of KD is rendered based on clinical presentation and lymph node histopathologic findings of paracortical necrosis and florid histiocytic infiltrate.1
The cutaneous manifestations of KD are heterogeneous yet mostly transient. Cutaneous involvement is reported in 16.6% to 40% of patients.3,5,6 Common cutaneous manifestations include erythematous macules, papules, patches, and plaques; erosions, nodules, and bullae less commonly can occur.6 A variety of cutaneous manifestations have been reported in KD, including lesions mimicking pigmented purpuric dermatoses, vasculitis, Sweet syndrome, drug eruptions, and viral exanthems.6 Signs and symptoms of KD usually resolve within 1 to 4 months. Although there are no established treatments for this disease, patients with severe or persistent symptoms can be treated with steroids or hydroxychloroquine. Recurrences after treatment have been reported.8
Systemic lupus erythematosus is a multiorgan disease with protean manifestations. Cutaneous manifestations of SLE include malar erythema and discoid, annular, and papulosquamous lesions. Histopathologic patterns frequently observed in cutaneous lesions associated with SLE include interface dermatitis with perivascular infiltrates, dermal mucin, and plasmacytoid dendritic cells (marked by CD123 staining); these findings were notably absent in our case.6
Lupus vulgaris is a form of cutaneous tuberculosis that results from reactivation of Mycobacterium tuberculosis in tubercles formed during preceding hematogenous dissemination. The head and neck region is the most common location, particularly the nose, cheeks, and earlobes. Small, brown-red, soft papules coalesce into gelatinous plaques, demonstrating a characteristic apple jelly appearance on diascopy. Other clinical manifestations include the plaque/plane, hypertrophic/tumorlike, and ulcerative/scarring forms.9 Delayed-type hypersensitivity testing by tuberculin skin test, interferon-gamma release assay, or polymerase chain reaction–based assays can detect Mycobacterium tuberculosis. Histopathology shows well-formed granulomas surrounded by chronic inflammatory cells and central necrosis.
Hydroa vacciniforme–like (HV-like) eruption is a rare photosensitive disorder characterized by vesiculopapules on sun-exposed areas. Hydroa vacciniforme–like eruptions rarely have been reported to progress to EBVassociated malignant lymphoma.10 Unlike typical hydroa vacciniforme, which resolves by early adulthood, HV-like eruptions can become more severe with age and are associated with systemic manifestations, including fevers, lymphadenopathy, and liver damage. Histopathologic examination reveals a dense infiltrate of atypical T lymphocytes or natural killer cells (CD56+), which stain positive for EBV-encoded small nuclear RNA,10 in contrast to the patchy perijunctional lymphocytic infiltrate seen in KD.
This case highlights the protean cutaneous manifestations of a rare rheumatologic entity. It demonstrates the importance of a full systemic workup when considering an enigmatic disease. Our patient was started on prednisone 20 mg and hydroxychloroquine 200 mg daily. Within 24 hours, the fevers and rash both improved.
- Turner RR, Martin J, Dorfman RF. Necrotizing lymphadenitis. a study of 30 cases. Am J Surg Pathol. 1983;7:115-123.
- Dorfman RF, Berry GJ. Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol. 1988;5:329-345.
- Atwater AR, Longley BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yoshino T, Mannami T, Ichimura K, et al. Two cases of histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto’s disease) following diffuse large B-cell lymphoma. Hum Pathol. 2000;31:1328-1331.
- Yen A, Fearneyhough P, Raimer SS, et al. EBV-associated Kikuchi’s histiocytic necrotizing lymphadenitis with cutaneous manifestations. J Am Acad Dermatol. 1997;36:342-346.
- Kim JH, Kim YB, In SI, et al. The cutaneous lesions of Kikuchi’s disease: a comprehensive analysis of 16 cases based on the clinicopathologic, immunohistochemical, and immunofluorescence studies with an emphasis on the differential diagnosis. Hum Pathol. 2010;41:1245-1254.
- Kucukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto Disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Smith KG, Becker GJ, Busmanis I. Recurrent Kikuchi’s disease. Lancet. 1992;340:124.
- Macgregor R. Cutaneous tuberculosis. Clin Dermatol. 1995;13:245-255.
- Iwatsuki K, Ohtsuka M, Harada H, et al. Clinicopathologic manifestations of Epstein-Barr virus–associated cutaneous lymphoproliferative disorders. Arch Dermatol. 1997;133:1081-1086.
The Diagnosis: Kikuchi-Fujimoto Disease
A skin biopsy from the left helix was obtained. Histopathologic examination revealed a vacuolar interface reaction with marked papillary dermal edema and a patchy perijunctional lymphocytic infiltrate. The dermis was free of increased mucin (Figure 1). Immunohistochemical staining for CD56 and Epstein-Barr virus (EBV)–encoded small nuclear RNA chromogenic in situ hybridization were negative. Laboratory workup was remarkable for elevated transaminases and inflammatory markers (eg, C-reactive protein, erythrocyte sedimentation rate) but negative for rheumatologic markers (eg, antinuclear antibodies, antineutrophil cytoplasmic antibodies, myeloperoxidase antibodies, serine protease IgG). An extensive infectious workup was unrevealing. Computed tomography highlighted prominent lymphadenopathy throughout the cervical and supraclavicular chains and a large necrotic lymph node in the porta hepatis (Figure 2). Right neck lymph node aspiration revealed necrotizing lymphadenitis in a background of histiocytes and mixed lymphocytes. Coupling the clinical presentation and histomorphology with imaging, a diagnosis of Kikuchi-Fujimoto disease (KD) was rendered.
Kikuchi-Fujimoto disease is a rare illness of unknown etiology characterized by cervical lymphadenopathy and fever. Originally described in Japan, KD affects all racial and ethnic groups1,2 but more commonly is seen in women and patients younger than 40 years.3 It can be associated with systemic lupus erythematosus (SLE) and other autoimmune diseases (eg, relapsing polychondritis, adult-onset Still disease),3 and lymphoma.4 Multiple infections have been implicated in the pathogenesis of KD, including EBV and other human herpesviruses; HIV; human T-cell leukemia virus type 1; dengue virus; parvovirus B19; and Yersinia enterocolitica, Bartonella, Brucella, and Toxoplasma infections.3,5,6
Kikuchi-Fujimoto disease classically presents with fever and cervical lymphadenopathy. In a retrospective review of 244 patients with KD, the 3 most common manifestations included lymphadenopathy, fever, and rash.7 A diagnosis of KD is rendered based on clinical presentation and lymph node histopathologic findings of paracortical necrosis and florid histiocytic infiltrate.1
The cutaneous manifestations of KD are heterogeneous yet mostly transient. Cutaneous involvement is reported in 16.6% to 40% of patients.3,5,6 Common cutaneous manifestations include erythematous macules, papules, patches, and plaques; erosions, nodules, and bullae less commonly can occur.6 A variety of cutaneous manifestations have been reported in KD, including lesions mimicking pigmented purpuric dermatoses, vasculitis, Sweet syndrome, drug eruptions, and viral exanthems.6 Signs and symptoms of KD usually resolve within 1 to 4 months. Although there are no established treatments for this disease, patients with severe or persistent symptoms can be treated with steroids or hydroxychloroquine. Recurrences after treatment have been reported.8
Systemic lupus erythematosus is a multiorgan disease with protean manifestations. Cutaneous manifestations of SLE include malar erythema and discoid, annular, and papulosquamous lesions. Histopathologic patterns frequently observed in cutaneous lesions associated with SLE include interface dermatitis with perivascular infiltrates, dermal mucin, and plasmacytoid dendritic cells (marked by CD123 staining); these findings were notably absent in our case.6
Lupus vulgaris is a form of cutaneous tuberculosis that results from reactivation of Mycobacterium tuberculosis in tubercles formed during preceding hematogenous dissemination. The head and neck region is the most common location, particularly the nose, cheeks, and earlobes. Small, brown-red, soft papules coalesce into gelatinous plaques, demonstrating a characteristic apple jelly appearance on diascopy. Other clinical manifestations include the plaque/plane, hypertrophic/tumorlike, and ulcerative/scarring forms.9 Delayed-type hypersensitivity testing by tuberculin skin test, interferon-gamma release assay, or polymerase chain reaction–based assays can detect Mycobacterium tuberculosis. Histopathology shows well-formed granulomas surrounded by chronic inflammatory cells and central necrosis.
Hydroa vacciniforme–like (HV-like) eruption is a rare photosensitive disorder characterized by vesiculopapules on sun-exposed areas. Hydroa vacciniforme–like eruptions rarely have been reported to progress to EBVassociated malignant lymphoma.10 Unlike typical hydroa vacciniforme, which resolves by early adulthood, HV-like eruptions can become more severe with age and are associated with systemic manifestations, including fevers, lymphadenopathy, and liver damage. Histopathologic examination reveals a dense infiltrate of atypical T lymphocytes or natural killer cells (CD56+), which stain positive for EBV-encoded small nuclear RNA,10 in contrast to the patchy perijunctional lymphocytic infiltrate seen in KD.
This case highlights the protean cutaneous manifestations of a rare rheumatologic entity. It demonstrates the importance of a full systemic workup when considering an enigmatic disease. Our patient was started on prednisone 20 mg and hydroxychloroquine 200 mg daily. Within 24 hours, the fevers and rash both improved.
The Diagnosis: Kikuchi-Fujimoto Disease
A skin biopsy from the left helix was obtained. Histopathologic examination revealed a vacuolar interface reaction with marked papillary dermal edema and a patchy perijunctional lymphocytic infiltrate. The dermis was free of increased mucin (Figure 1). Immunohistochemical staining for CD56 and Epstein-Barr virus (EBV)–encoded small nuclear RNA chromogenic in situ hybridization were negative. Laboratory workup was remarkable for elevated transaminases and inflammatory markers (eg, C-reactive protein, erythrocyte sedimentation rate) but negative for rheumatologic markers (eg, antinuclear antibodies, antineutrophil cytoplasmic antibodies, myeloperoxidase antibodies, serine protease IgG). An extensive infectious workup was unrevealing. Computed tomography highlighted prominent lymphadenopathy throughout the cervical and supraclavicular chains and a large necrotic lymph node in the porta hepatis (Figure 2). Right neck lymph node aspiration revealed necrotizing lymphadenitis in a background of histiocytes and mixed lymphocytes. Coupling the clinical presentation and histomorphology with imaging, a diagnosis of Kikuchi-Fujimoto disease (KD) was rendered.
Kikuchi-Fujimoto disease is a rare illness of unknown etiology characterized by cervical lymphadenopathy and fever. Originally described in Japan, KD affects all racial and ethnic groups1,2 but more commonly is seen in women and patients younger than 40 years.3 It can be associated with systemic lupus erythematosus (SLE) and other autoimmune diseases (eg, relapsing polychondritis, adult-onset Still disease),3 and lymphoma.4 Multiple infections have been implicated in the pathogenesis of KD, including EBV and other human herpesviruses; HIV; human T-cell leukemia virus type 1; dengue virus; parvovirus B19; and Yersinia enterocolitica, Bartonella, Brucella, and Toxoplasma infections.3,5,6
Kikuchi-Fujimoto disease classically presents with fever and cervical lymphadenopathy. In a retrospective review of 244 patients with KD, the 3 most common manifestations included lymphadenopathy, fever, and rash.7 A diagnosis of KD is rendered based on clinical presentation and lymph node histopathologic findings of paracortical necrosis and florid histiocytic infiltrate.1
The cutaneous manifestations of KD are heterogeneous yet mostly transient. Cutaneous involvement is reported in 16.6% to 40% of patients.3,5,6 Common cutaneous manifestations include erythematous macules, papules, patches, and plaques; erosions, nodules, and bullae less commonly can occur.6 A variety of cutaneous manifestations have been reported in KD, including lesions mimicking pigmented purpuric dermatoses, vasculitis, Sweet syndrome, drug eruptions, and viral exanthems.6 Signs and symptoms of KD usually resolve within 1 to 4 months. Although there are no established treatments for this disease, patients with severe or persistent symptoms can be treated with steroids or hydroxychloroquine. Recurrences after treatment have been reported.8
Systemic lupus erythematosus is a multiorgan disease with protean manifestations. Cutaneous manifestations of SLE include malar erythema and discoid, annular, and papulosquamous lesions. Histopathologic patterns frequently observed in cutaneous lesions associated with SLE include interface dermatitis with perivascular infiltrates, dermal mucin, and plasmacytoid dendritic cells (marked by CD123 staining); these findings were notably absent in our case.6
Lupus vulgaris is a form of cutaneous tuberculosis that results from reactivation of Mycobacterium tuberculosis in tubercles formed during preceding hematogenous dissemination. The head and neck region is the most common location, particularly the nose, cheeks, and earlobes. Small, brown-red, soft papules coalesce into gelatinous plaques, demonstrating a characteristic apple jelly appearance on diascopy. Other clinical manifestations include the plaque/plane, hypertrophic/tumorlike, and ulcerative/scarring forms.9 Delayed-type hypersensitivity testing by tuberculin skin test, interferon-gamma release assay, or polymerase chain reaction–based assays can detect Mycobacterium tuberculosis. Histopathology shows well-formed granulomas surrounded by chronic inflammatory cells and central necrosis.
Hydroa vacciniforme–like (HV-like) eruption is a rare photosensitive disorder characterized by vesiculopapules on sun-exposed areas. Hydroa vacciniforme–like eruptions rarely have been reported to progress to EBVassociated malignant lymphoma.10 Unlike typical hydroa vacciniforme, which resolves by early adulthood, HV-like eruptions can become more severe with age and are associated with systemic manifestations, including fevers, lymphadenopathy, and liver damage. Histopathologic examination reveals a dense infiltrate of atypical T lymphocytes or natural killer cells (CD56+), which stain positive for EBV-encoded small nuclear RNA,10 in contrast to the patchy perijunctional lymphocytic infiltrate seen in KD.
This case highlights the protean cutaneous manifestations of a rare rheumatologic entity. It demonstrates the importance of a full systemic workup when considering an enigmatic disease. Our patient was started on prednisone 20 mg and hydroxychloroquine 200 mg daily. Within 24 hours, the fevers and rash both improved.
- Turner RR, Martin J, Dorfman RF. Necrotizing lymphadenitis. a study of 30 cases. Am J Surg Pathol. 1983;7:115-123.
- Dorfman RF, Berry GJ. Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol. 1988;5:329-345.
- Atwater AR, Longley BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yoshino T, Mannami T, Ichimura K, et al. Two cases of histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto’s disease) following diffuse large B-cell lymphoma. Hum Pathol. 2000;31:1328-1331.
- Yen A, Fearneyhough P, Raimer SS, et al. EBV-associated Kikuchi’s histiocytic necrotizing lymphadenitis with cutaneous manifestations. J Am Acad Dermatol. 1997;36:342-346.
- Kim JH, Kim YB, In SI, et al. The cutaneous lesions of Kikuchi’s disease: a comprehensive analysis of 16 cases based on the clinicopathologic, immunohistochemical, and immunofluorescence studies with an emphasis on the differential diagnosis. Hum Pathol. 2010;41:1245-1254.
- Kucukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto Disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Smith KG, Becker GJ, Busmanis I. Recurrent Kikuchi’s disease. Lancet. 1992;340:124.
- Macgregor R. Cutaneous tuberculosis. Clin Dermatol. 1995;13:245-255.
- Iwatsuki K, Ohtsuka M, Harada H, et al. Clinicopathologic manifestations of Epstein-Barr virus–associated cutaneous lymphoproliferative disorders. Arch Dermatol. 1997;133:1081-1086.
- Turner RR, Martin J, Dorfman RF. Necrotizing lymphadenitis. a study of 30 cases. Am J Surg Pathol. 1983;7:115-123.
- Dorfman RF, Berry GJ. Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol. 1988;5:329-345.
- Atwater AR, Longley BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yoshino T, Mannami T, Ichimura K, et al. Two cases of histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto’s disease) following diffuse large B-cell lymphoma. Hum Pathol. 2000;31:1328-1331.
- Yen A, Fearneyhough P, Raimer SS, et al. EBV-associated Kikuchi’s histiocytic necrotizing lymphadenitis with cutaneous manifestations. J Am Acad Dermatol. 1997;36:342-346.
- Kim JH, Kim YB, In SI, et al. The cutaneous lesions of Kikuchi’s disease: a comprehensive analysis of 16 cases based on the clinicopathologic, immunohistochemical, and immunofluorescence studies with an emphasis on the differential diagnosis. Hum Pathol. 2010;41:1245-1254.
- Kucukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto Disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Smith KG, Becker GJ, Busmanis I. Recurrent Kikuchi’s disease. Lancet. 1992;340:124.
- Macgregor R. Cutaneous tuberculosis. Clin Dermatol. 1995;13:245-255.
- Iwatsuki K, Ohtsuka M, Harada H, et al. Clinicopathologic manifestations of Epstein-Barr virus–associated cutaneous lymphoproliferative disorders. Arch Dermatol. 1997;133:1081-1086.
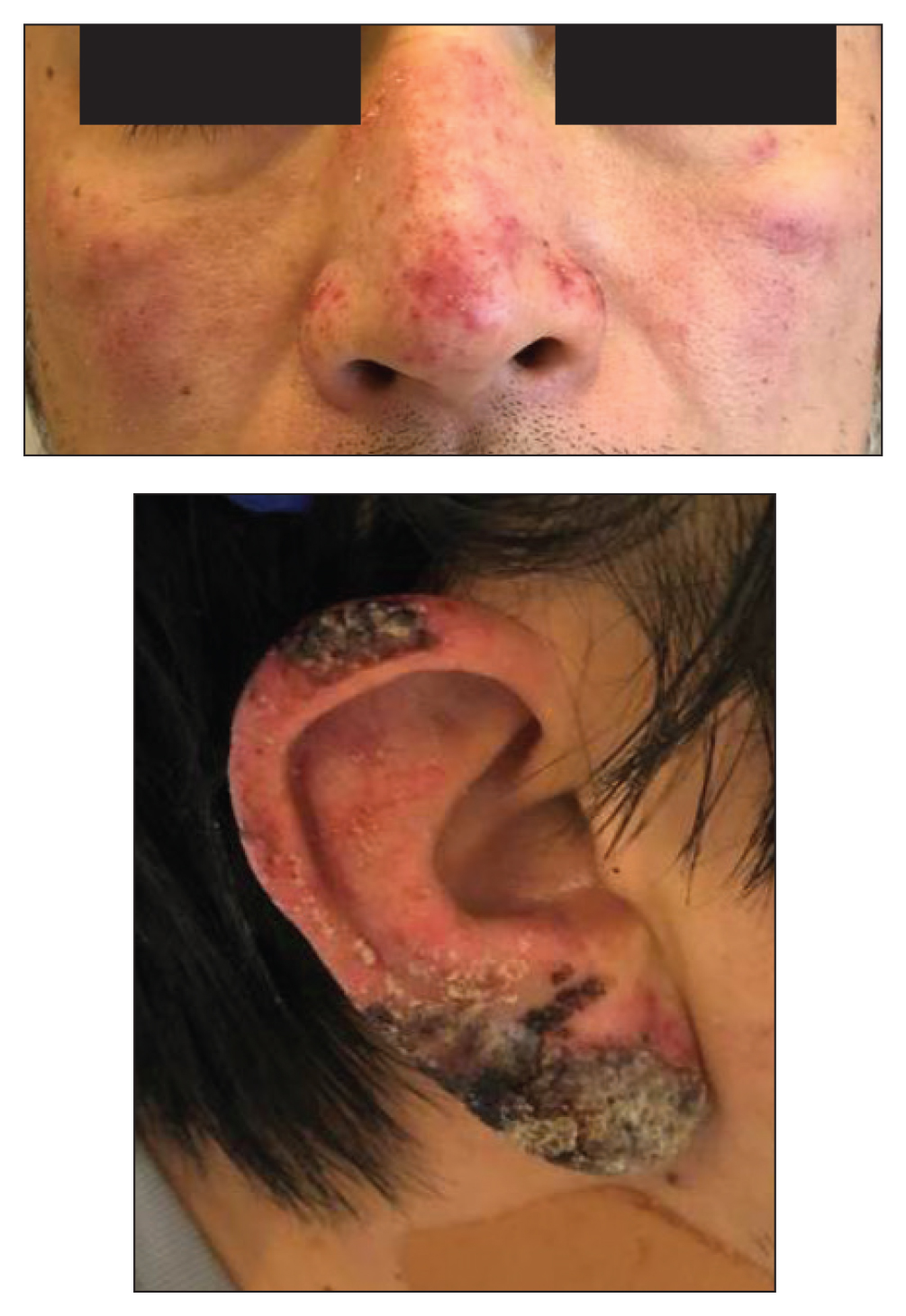
A healthy 42-year-old Japanese man presented with painful lymphadenopathy and fevers of 1 month’s duration as well as a pruritic rash and bilateral ear redness and crusting of 1 week’s duration. He initially was seen at an outside facility and was treated with antibiotics and supportive care for cervical adenitis. During clinical evaluation, he denied joint pain, photosensitivity, and oral lesions. His medical and family history were noncontributory. Although he reported recent travel to multiple countries, he denied exposure to animals, ticks, or sick individuals. Physical examination revealed erythematous blanching papules on the nose and cheeks (top) as well as crusted papules coalescing into plaques on the bilateral helices and lobules (bottom).