User login
Compared with first-generation antipsychotics, second-generation antipsychotics (SGAs) have a lower risk for extrapyramidal symptoms. Yet tardive dyskinesia (TD) remains a concern because of the widespread use of SGAs for multiple indications.1 Prior to April 2017, clinicians had no FDA-approved TD treatment options. The most widely used agent worldwide, tetrabenazine, had positive efficacy data in TD trials over the past 45 years but was not available in the United States until 2008, and its sole indication was for chorea associated with Huntington’s disease.2 Moreover, the use of tetrabenazine involved slow titration, multiple daily dosing, cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, and tolerability issues.
Tetrabenazine is an inhibitor of vesicular monoamine transport type 2 (VMAT2), a transport protein located almost exclusively in the CNS whose role is to place monoamine neurotransmitters (dopamine, serotonin, norepinephrine) into presynaptic vesicles. By decreasing dopamine transport into these presynaptic vesicles, synaptic dopamine release is lessened, thus reducing postsynaptic dopamine D2 receptor activity and the severity of dyskinetic movements.1
In 2 pivotal 12-week clinical trials, deutetrabenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) scores (see Efficacy).6,7
Clinical implications
TD remains a substantial public health concern due to the increasing use of antipsychotics for mood and other disorders beyond the initial indications for schizophrenia.1 Although exposure to dopamine D2antagonism results in postsynaptic receptor upregulation and supersensitivity that underlies the development of dyskinesia, this process is often rapidly reversible in animal models.1 The persistence of TD symptoms in up to 80% of patients after dopamine receptor blocking agents (DRBAs) are stopped has led to hypotheses that the underlying pathophysiology of TD is also a problem with neuroplasticity. Aside from DRBA exposure, environmental factors (eg, oxidative stress) and genetic predisposition might contribute to TD risk.1
Before 2017, only 1 medication (branched-chain amino acids) had been FDA-approved for treating TD in the United States, and only a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]) had positive results from controlled trials, most with small effect sizes.8 Moreover, there was only 1 controlled trial each for clonazepam and EGb-761.1 A branched-chain amino acid preparation received FDA approval for managing TD in male patients, but is no longer commercially available, except from compounding pharmacies.9
Tetrabenazine was developed in the mid-1950s to avoid orthostasis and sedation associated with reserpine.10 Both reserpine and tetrabenazine proved effective for TD,11 but tetrabenazine lacked reserpine’s peripheral adverse effects. However, the kinetics of tetrabenazine necessitated multiple daily doses, and CYP2D6 genotyping was required for doses >50 mg/d.2
Receptor blocking. The mechanism that distinguishes the clinical profiles of reserpine and tetrabenazine relates to their differential properties at VMAT.12 VMAT exists in 2 forms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the CNS.13 Tetrabenazine is a specific and reversible VMAT2 inhibitor, whereas reserpine is an irreversible and nonselective antagonist of VMAT1 and VMAT2. It is reserpine’s VMAT1 inhibition that results in peripheral adverse effects such as orthostasis. Tetrabenazine is rapidly and extensively converted into 2 isomers, alpha-dihydrotetrabenazine (α-DHTBZ) and beta-dihydrotetrabenazine (β-DHTBZ), both of which are metabolized by CYP2D6, with a role for CYP3A4 in α-DHTBZ metabolism.1 These DHTBZ metabolites have a short half-life when generated from oral tetrabenazine, a feature that necessitates multiple daily dosing; moreover, the existence of 2D6 polymorphisms led to FDA-mandated CYP2D6 genotyping for tetrabenazine doses >50 mg/d when it was approved for Huntington’s chorea. The concern is that 2D6 poor metabolizers will have excessive exposure to the VMAT2 effects of DHTBZ, resulting in sedation, akathisia, parkinsonism, and mood symptoms.2
How deuterium impacts medication kinetics. Deuterium is a naturally occurring, stable, nontoxic isotope of hydrogen. Humans have 5 g of deuterium in their body at any time, mostly in the form of heavy water (D2O).14 When deuterium is used to replace selected hydrogen atoms, the resulting molecule will have similar configuration and receptor-binding properties but markedly different kinetics. Because the carbon–deuterium covalent bond requires 8 times more energy to break than a carbon–hydrogen bond, the half-life is prolonged.15 Utilizing this knowledge, a deuterated form of tetrabenazine, deutetrabenazine, was synthesized with such a purpose in mind. While the active metabolites of deutetrabenazine retain the VMAT2 affinity of non-deuterated tetrabenazine, the substitution of deuterium for hydrogen at specific positions slows the breakdown of metabolites, resulting in sustained duration of action, greater active drug exposure, and less impact of 2D6 genotype on drug exposure, thus eliminating the need for genotyping, unless one wants to exceed 36 mg/d.
Deutetrabenazine was first studied in Huntington’s chorea in a 13-week, double-blind, placebo-controlled, parallel-group study (N = 90).4 The maximum daily deutetrabenazine dose was 48 mg, but reduced to 36 mg in those taking strong CYP2D6 inhibitors (bupropion, fluoxetine, or paroxetine). Blinded 2D6 genotyping was performed, but there was no dose modification required based on 2D6 genotype. There was a 36.4% reduction in total maximal chorea score for deutetrabenazine compared with 14.4% for placebo (P < .001).4 Importantly, adverse effects were comparable between both groups, with 1 drop-out in the deutetrabenazine arm vs 2 in the placebo arm. The only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo was somnolence: 11.1% for deutetrabenazine vs 4.4% for placebo.4 The mean deutetrabenazine daily dose at the end of the treatment period was 39.7 ± 9.3 mg, and for those with impaired CYP2D6 function (poor metabolizers or those taking strong CYP2D6 inhibiting medications), the mean daily dose was 34.8 mg ± 3.8 mg.4
Use in tardive dyskinesia. The recommended starting dosage for TD treatment is 6 mg, twice daily with food. The dose may be increased at weekly intervals in increments of 6 mg/d to a maximum recommended daily dosage of 48 mg.5 The maximum daily dose is 36 mg (18 mg, twice daily) in patients receiving strong CYP2D6 inhibitors or who are 2D6 poor metabolizers.5
Deutetrabenazine has not been studied in those with moderate or severe hepatic impairment, and its use is contraindicated in these patients.5 No clinical studies have been conducted to assess the effect of renal impairment on the pharmacokinetics of deutetrabenazine.5
Pharmacologic profile, adverse reactions
When the data from the two 12-week, phase 3 placebo-controlled studies were pooled, the most common adverse reactions occurring in >3% of deutetrabenazine patients and greater than placebo were nasopharyngeal symptoms (4% vs 2% placebo) and insomnia (4% vs 1% placebo).5 Importantly, in neither TD study were there clinically significant changes in rating scales for depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms. The mean QT prolongation for a single 24 mg dose of deutetrabenazine in healthy volunteers was 4.5 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 6.5 milliseconds.5 For tetrabenazine, single 50 mg doses administered to volunteers resulted in mean QT prolongation of 8 milliseconds.5 In patients requiring deutetrabenazine doses >24 mg/d who are taking other medications known to prolong QTc, assess the QTc interval before and after increasing the dose of deutetrabenazine or other medications that are known to prolong QTc.5
How it works
Tetrabenazine is the only agent that has demonstrated significant efficacy for TD management, but its use involves slow titration, multiple daily dosing, CYP2D6 genotyping for doses >50 mg/d, and tolerability issues. For example, the most common adverse effects in the pivotal tetrabenazine Huntington’s disease trial were sedation/somnolence (tetrabenazine 31% vs 3% for placebo), insomnia (tetrabenazine 22% vs 0% for placebo), depression (tetrabenazine 19% vs 0% for placebo), fatigue (tetrabenazine 22% vs 13% for placebo), and akathisia (tetrabenazine 19% vs 0% for placebo).2 For comparison, the only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo in the pivotal Huntington’s disease trial was somnolence (11.1% for deutetrabenazine vs 4.4% for placebo).4
Pharmacokinetics
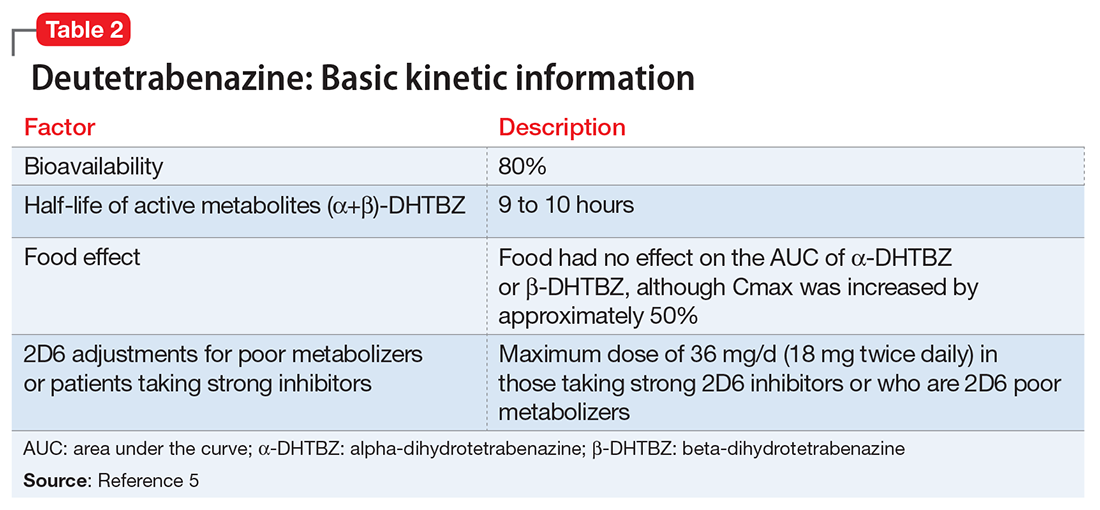
Deutetrabenazine has 80% oral bioavailability, and is rapidly converted to its active metabolites after oral dosing (Table 2).5 Linear dose dependence of Cmax and area under the curve (AUC) was observed for the active metabolites following single or multiple doses of deutetrabenazine (6 to 24 mg and 7.5 to 22.5 mg, twice daily).15 Cmax of deuterated α-DHTBZ and β-DHTBZ is reached within 3 to 4 hours after dosing, with a steady state ratio of 3:1 for the α-DHTBZ vs the β-DHTBZ form. Food had no effect on AUC, but did increase Cmax by 50%.5
Deutetrabenazine is metabolized through carbonyl reductase enzymes to its active metabolites, and these are further metabolized through multiple CYP pathways, predominantly 2D6 and to a lesser extent 3A4. The effect of CYP2D6 inhibition on the pharmacokinetics of deutetrabenazine and its α-DHTBZ and β-DHTBZ metabolites was studied in 24 healthy participants following a single 22.5 mg dose of deutetrabenazine given after 8 days of administration of the strong CYP2D6 inhibitor paroxetine, 20 mg/d. In the presence of paroxetine, systemic exposure (AUC) of α-DHTBZ was 1.9-fold higher and β-DHTBZ was 6.5-fold higher, resulting in an approximately 3-fold increase in AUC for total (α+β)-DHTBZ, with corresponding increases in mean half-life of approximately 1.5-fold and 2.7-fold, respectively.5 Neither deutetrabenazine or its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that deutetrabenazine and its active metabolites are unlikely to inhibit most major drug transporters at clinically relevant concentrations.
Efficacy
Efficacy was established in two 12-week, double-blind, placebo-controlled trials of adult patients with TD (ages 18 to 80).6,7 Eligible participants had:
- TD diagnosis for ≥3 months before screening and a history of DRBA treatment for ≥3 months (≥1 month if age ≥60)
- Total AIMS motor score ≥6 (items 1 to 7) at both screening and baseline, verified by a blinded central rater at screening via central video rating
- Patients with an underlying psychiatric illness had to be stable. Psychoactive medication use, including antipsychotics, was allowed if stable for ≥30 days before screening (antidepressants, ≥45 days).
Exclusion criteria included treatment with tetrabenazine, reserpine, α-methyl-p-tyrosine, strong anticholinergic medications, dopamine antagonizing antiemetics (eg, metoclopramide, prochlorperazine, promethazine), dopamine agonists, levodopa, stimulants, or a monoamine oxidase inhibitor (MAOI) within 30 days of screening or baseline, or treatment with botulinum toxin within 3 months of screening; and presence of a neurologic condition that could confound TD assessments, serious untreated or undertreated psychiatric illness, or unstable medical illness. Patients with a history of or active suicidal ideation or behavior within 6 months of screening or score ≥11 on the depression subscale of the Hospital Anxiety and Depression Scale were excluded. Those participants with Fridericia-corrected QT interval values >450 milliseconds in men, >460 milliseconds in women, or >480 milliseconds in patients with a right bundle branch block on electrocardiography at screening also were excluded.
The flexible-dose TD study was performed in 117 participants randomized in a 1:1 manner to deutetrabenazine or placebo, both administered twice daily, titrated to optimal dosage (12 to 48 mg/d) over 6 weeks, and then administered at that dose for another 6 weeks.7 The population demographics were: mean age, 54.6 ± 10.3 years, 52.1% female, 69.2% white, and 80.3% receiving ongoing dopamine antagonists, with a mean TD duration of 74.7 ± 81.5 months. Sixty-eight percent had schizophrenia spectrum disorders, and 30% had mood disorders. The primary outcome was change in total AIMS score (items 1 to 7) assessed by central, independent raters. The mean baseline AIMS score for items 1 to 7 was 9.6 ± 3.9, with 82.9% of participants with baseline AIMS scores ≥6. Study treatment retention was high: placebo 88.1%, deutetrabenazine 89.7%.7 There was a mean 3 point decrease in AIMS score for deutetrabenazine compared with 1.4 for placebo (P = .019). Among those with baseline AIMS scores ≥6, there was a 3.4 point decrease in AIMS scores for deutetrabenazine compared with a 1.9 point decrease for placebo (P = .027). The only adverse effects that occurred in ≥5% of deutetrabenazine participants and at a rate ≥2 times the rate in placebo were insomnia (deutetrabenazine 6.9% vs placebo 1.7%) and akathisia (deutetrabenazine 5.2% vs placebo 0%).
The fixed-dose TD study was performed in 293 participants randomized in 1:1:1:1 manner to 1 of 3 fixed doses of deutetrabenazine (12 mg/d, 24 mg/d, or 36 mg/d) or placebo, both administered twice daily.6 The starting dose of deutetrabenazine was 6 mg twice daily. During the dose escalation period (through Week 4), the dose of study drug was increased weekly in increments of 6 mg/d until the randomized dose was achieved. Patients continued to receive the dose they were assigned to over a maintenance period of 8 weeks.6 The population demographics were: mean age, 56.4 ± 11.3 years, 55% female, 79% white, 76% receiving ongoing dopamine antagonists, with a mean TD duration of 67.2 ± 66 months. Sixty percent had schizophrenia spectrum disorders, and 36% had mood disorders. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. The mean AIMS score at baseline was 9.5 ± 2.7 in the placebo group, and for deutetrabenazine: 9.6 ± 2.4 in the 12 mg/d group, 9.4 ± 2.9 in the 24 mg/d group, and 10.1 ± 3.2 in the 36 mg/d group. The 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). Study treatment retention rates were high: placebo 90.5%, deutetrabenazine 88%. Across all doses, only 1 adverse effect occurred in ≥5% of deutetrabenazine participants: headache (5% deutetrabenazine vs 6% placebo). At the highest dose, 36 mg/d, the only adverse effects that occurred in ≥5% of participants were diarrhea (7% deutetrabenazine vs 3% placebo) and headache (7% deutetrabenazine vs 6% placebo).
Outcome. In the flexible-dose study (mean dose 38.8 ± 7.92 mg/d), the deutetrabenazine arm experienced a mean 30% reduction in AIMS scores from baseline at the Week 12 endpoint. Compared with placebo, the mean reduction in AIMS scores (standard error) was: −3.0 (0.45) deutetrabenazine vs −1.6 (0.46) placebo (P = .019).7 For the fixed-dose study, the 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). In addition to these mean changes from baseline, 35% of the 24 mg/d group and 33% of the 36 mg/d group demonstrated ≥50% reduction in AIMS scores.6
Tolerability
In the 2 phase 3 trials, there were no adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo.5 Discontinuations because of adverse events were low in both pivotal studies across all treatment groups: 3.4% for placebo vs 1.7% for deutetrabenazine in the flexible-dose trial,7 and 3% for placebo vs 4% for deutetrabenazine in the fixed-dose study.6 In neither trial were there clinically significant changes in ratings of depression, suicidality, parkinsonism, or schizophrenia symptoms. The mean QT prolongation in healthy volunteers is described above.
Clinical considerations
Unique properties. Deutetrabenazine utilizes the greater bond strength of the carbon–deuterium bond to slow CYP metabolism, resulting in prolonged duration of action that is well tolerated, and provides significant efficacy.
Why Rx? The reasons to prescribe deutetrabenazine for TD patients include:
- only 1 of 2 agents with FDA approval for TD
- fewer tolerability issues than with tetrabenazine
- lower sedation rates in TD trials than with valbenazine
- no signal for effects on mood parameters or rates of parkinsonism when used for TD.
Dosing
The recommended starting dosage of deutetrabenazine is 6 mg twice daily taken with food, increasing by 6 mg/d weekly as needed, with a maximum dose of 48 mg/d or 36 mg/d in those taking strong CYP2D6 inhibitors or who are 2D6 poor metabolizers. Deutetrabenazine is contraindicated in patients with hepatic impairment (as determined by Child-Pugh criteria16). There are no data in patients with renal impairment. The combined efficacy and tolerability of dosages >48 mg/d has not been evaluated. Overdoses of tetrabenazine ranging from 100 to 1,000 mg have been reported in the literature and were associated with acute dystonia, oculogyric crisis, nausea and vomiting, sweating, sedation, hypotension, confusion, diarrhea, hallucinations, rubor, and tremor.5
Contraindications
When used for TD, deutetrabenazine is contraindicated for patients taking reserpine, tetrabenazine, valbenazine, or MAOIs, and for patients with hepatic impairment. As with most medications, there are no data on deutetrabenazine use in pregnant women; however, oral administration of deutetrabenazine (5, 10, or 30 mg/kg/d) or tetrabenazine (30 mg/kg/d) to pregnant rats during organogenesis had no clear effect on embryofetal development. The highest dose tested was 6 times the maximum recommended human dose of 48 mg/d on a body surface area (mg/m2) basis. There are no data on the presence of deutetrabenazine or its metabolites in human milk, the effects on the breastfed infant, or the effects of the drug on milk production.
1. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
2. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
3. Meyer JM. Valbenazine for tardive dyskinesia. Current Psychiatry. 2017;16(5):40-46.
4. Huntington Study Group; Frank S, Testa CM, Stamler D, et al. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA. 2016;316(1):40-50.
5. Austedo [package insert]. North Wales, PA: Teva Pharmaceuticals USA, Inc.; 2017.
6. Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595-604.
7. Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88(21):2003-2010.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Richardson MA, Small AM, Read LL, et al. Branched chain amino acid treatment of tardive dyskinesia in children and adolescents. J Clin Psychiatry. 2004;65(1):92-96.
10. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
11. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
12. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
13. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
14. Kushner DJ, Baker A, Dunstall TG. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can J Physiol Pharmacol. 1999;77(2):79-88.
15. United States Securities and Exchange Commission. Form S-1 Registration Statement of Auspex Pharmaceuticals, Inc. https://www.sec.gov/Archives/edgar/data/1454189/000119312513481239/d627086ds1.htm. Published December 20, 2013. Accessed July 1, 2016.
16. Cholongitas E, Papatheodoridis GV, Vangeli M, et al. Systematic review: the model for end-stage liver disease—should it replace Child-Pugh’s classification for assessing prognosis in cirrhosis? Aliment Pharmacol Ther. 2005;22(11-12):1079-1089.
Compared with first-generation antipsychotics, second-generation antipsychotics (SGAs) have a lower risk for extrapyramidal symptoms. Yet tardive dyskinesia (TD) remains a concern because of the widespread use of SGAs for multiple indications.1 Prior to April 2017, clinicians had no FDA-approved TD treatment options. The most widely used agent worldwide, tetrabenazine, had positive efficacy data in TD trials over the past 45 years but was not available in the United States until 2008, and its sole indication was for chorea associated with Huntington’s disease.2 Moreover, the use of tetrabenazine involved slow titration, multiple daily dosing, cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, and tolerability issues.
Tetrabenazine is an inhibitor of vesicular monoamine transport type 2 (VMAT2), a transport protein located almost exclusively in the CNS whose role is to place monoamine neurotransmitters (dopamine, serotonin, norepinephrine) into presynaptic vesicles. By decreasing dopamine transport into these presynaptic vesicles, synaptic dopamine release is lessened, thus reducing postsynaptic dopamine D2 receptor activity and the severity of dyskinetic movements.1
In 2 pivotal 12-week clinical trials, deutetrabenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) scores (see Efficacy).6,7
Clinical implications
TD remains a substantial public health concern due to the increasing use of antipsychotics for mood and other disorders beyond the initial indications for schizophrenia.1 Although exposure to dopamine D2antagonism results in postsynaptic receptor upregulation and supersensitivity that underlies the development of dyskinesia, this process is often rapidly reversible in animal models.1 The persistence of TD symptoms in up to 80% of patients after dopamine receptor blocking agents (DRBAs) are stopped has led to hypotheses that the underlying pathophysiology of TD is also a problem with neuroplasticity. Aside from DRBA exposure, environmental factors (eg, oxidative stress) and genetic predisposition might contribute to TD risk.1
Before 2017, only 1 medication (branched-chain amino acids) had been FDA-approved for treating TD in the United States, and only a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]) had positive results from controlled trials, most with small effect sizes.8 Moreover, there was only 1 controlled trial each for clonazepam and EGb-761.1 A branched-chain amino acid preparation received FDA approval for managing TD in male patients, but is no longer commercially available, except from compounding pharmacies.9
Tetrabenazine was developed in the mid-1950s to avoid orthostasis and sedation associated with reserpine.10 Both reserpine and tetrabenazine proved effective for TD,11 but tetrabenazine lacked reserpine’s peripheral adverse effects. However, the kinetics of tetrabenazine necessitated multiple daily doses, and CYP2D6 genotyping was required for doses >50 mg/d.2
Receptor blocking. The mechanism that distinguishes the clinical profiles of reserpine and tetrabenazine relates to their differential properties at VMAT.12 VMAT exists in 2 forms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the CNS.13 Tetrabenazine is a specific and reversible VMAT2 inhibitor, whereas reserpine is an irreversible and nonselective antagonist of VMAT1 and VMAT2. It is reserpine’s VMAT1 inhibition that results in peripheral adverse effects such as orthostasis. Tetrabenazine is rapidly and extensively converted into 2 isomers, alpha-dihydrotetrabenazine (α-DHTBZ) and beta-dihydrotetrabenazine (β-DHTBZ), both of which are metabolized by CYP2D6, with a role for CYP3A4 in α-DHTBZ metabolism.1 These DHTBZ metabolites have a short half-life when generated from oral tetrabenazine, a feature that necessitates multiple daily dosing; moreover, the existence of 2D6 polymorphisms led to FDA-mandated CYP2D6 genotyping for tetrabenazine doses >50 mg/d when it was approved for Huntington’s chorea. The concern is that 2D6 poor metabolizers will have excessive exposure to the VMAT2 effects of DHTBZ, resulting in sedation, akathisia, parkinsonism, and mood symptoms.2
How deuterium impacts medication kinetics. Deuterium is a naturally occurring, stable, nontoxic isotope of hydrogen. Humans have 5 g of deuterium in their body at any time, mostly in the form of heavy water (D2O).14 When deuterium is used to replace selected hydrogen atoms, the resulting molecule will have similar configuration and receptor-binding properties but markedly different kinetics. Because the carbon–deuterium covalent bond requires 8 times more energy to break than a carbon–hydrogen bond, the half-life is prolonged.15 Utilizing this knowledge, a deuterated form of tetrabenazine, deutetrabenazine, was synthesized with such a purpose in mind. While the active metabolites of deutetrabenazine retain the VMAT2 affinity of non-deuterated tetrabenazine, the substitution of deuterium for hydrogen at specific positions slows the breakdown of metabolites, resulting in sustained duration of action, greater active drug exposure, and less impact of 2D6 genotype on drug exposure, thus eliminating the need for genotyping, unless one wants to exceed 36 mg/d.
Deutetrabenazine was first studied in Huntington’s chorea in a 13-week, double-blind, placebo-controlled, parallel-group study (N = 90).4 The maximum daily deutetrabenazine dose was 48 mg, but reduced to 36 mg in those taking strong CYP2D6 inhibitors (bupropion, fluoxetine, or paroxetine). Blinded 2D6 genotyping was performed, but there was no dose modification required based on 2D6 genotype. There was a 36.4% reduction in total maximal chorea score for deutetrabenazine compared with 14.4% for placebo (P < .001).4 Importantly, adverse effects were comparable between both groups, with 1 drop-out in the deutetrabenazine arm vs 2 in the placebo arm. The only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo was somnolence: 11.1% for deutetrabenazine vs 4.4% for placebo.4 The mean deutetrabenazine daily dose at the end of the treatment period was 39.7 ± 9.3 mg, and for those with impaired CYP2D6 function (poor metabolizers or those taking strong CYP2D6 inhibiting medications), the mean daily dose was 34.8 mg ± 3.8 mg.4
Use in tardive dyskinesia. The recommended starting dosage for TD treatment is 6 mg, twice daily with food. The dose may be increased at weekly intervals in increments of 6 mg/d to a maximum recommended daily dosage of 48 mg.5 The maximum daily dose is 36 mg (18 mg, twice daily) in patients receiving strong CYP2D6 inhibitors or who are 2D6 poor metabolizers.5
Deutetrabenazine has not been studied in those with moderate or severe hepatic impairment, and its use is contraindicated in these patients.5 No clinical studies have been conducted to assess the effect of renal impairment on the pharmacokinetics of deutetrabenazine.5
Pharmacologic profile, adverse reactions
When the data from the two 12-week, phase 3 placebo-controlled studies were pooled, the most common adverse reactions occurring in >3% of deutetrabenazine patients and greater than placebo were nasopharyngeal symptoms (4% vs 2% placebo) and insomnia (4% vs 1% placebo).5 Importantly, in neither TD study were there clinically significant changes in rating scales for depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms. The mean QT prolongation for a single 24 mg dose of deutetrabenazine in healthy volunteers was 4.5 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 6.5 milliseconds.5 For tetrabenazine, single 50 mg doses administered to volunteers resulted in mean QT prolongation of 8 milliseconds.5 In patients requiring deutetrabenazine doses >24 mg/d who are taking other medications known to prolong QTc, assess the QTc interval before and after increasing the dose of deutetrabenazine or other medications that are known to prolong QTc.5
How it works
Tetrabenazine is the only agent that has demonstrated significant efficacy for TD management, but its use involves slow titration, multiple daily dosing, CYP2D6 genotyping for doses >50 mg/d, and tolerability issues. For example, the most common adverse effects in the pivotal tetrabenazine Huntington’s disease trial were sedation/somnolence (tetrabenazine 31% vs 3% for placebo), insomnia (tetrabenazine 22% vs 0% for placebo), depression (tetrabenazine 19% vs 0% for placebo), fatigue (tetrabenazine 22% vs 13% for placebo), and akathisia (tetrabenazine 19% vs 0% for placebo).2 For comparison, the only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo in the pivotal Huntington’s disease trial was somnolence (11.1% for deutetrabenazine vs 4.4% for placebo).4
Pharmacokinetics
Deutetrabenazine has 80% oral bioavailability, and is rapidly converted to its active metabolites after oral dosing (Table 2).5 Linear dose dependence of Cmax and area under the curve (AUC) was observed for the active metabolites following single or multiple doses of deutetrabenazine (6 to 24 mg and 7.5 to 22.5 mg, twice daily).15 Cmax of deuterated α-DHTBZ and β-DHTBZ is reached within 3 to 4 hours after dosing, with a steady state ratio of 3:1 for the α-DHTBZ vs the β-DHTBZ form. Food had no effect on AUC, but did increase Cmax by 50%.5
Deutetrabenazine is metabolized through carbonyl reductase enzymes to its active metabolites, and these are further metabolized through multiple CYP pathways, predominantly 2D6 and to a lesser extent 3A4. The effect of CYP2D6 inhibition on the pharmacokinetics of deutetrabenazine and its α-DHTBZ and β-DHTBZ metabolites was studied in 24 healthy participants following a single 22.5 mg dose of deutetrabenazine given after 8 days of administration of the strong CYP2D6 inhibitor paroxetine, 20 mg/d. In the presence of paroxetine, systemic exposure (AUC) of α-DHTBZ was 1.9-fold higher and β-DHTBZ was 6.5-fold higher, resulting in an approximately 3-fold increase in AUC for total (α+β)-DHTBZ, with corresponding increases in mean half-life of approximately 1.5-fold and 2.7-fold, respectively.5 Neither deutetrabenazine or its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that deutetrabenazine and its active metabolites are unlikely to inhibit most major drug transporters at clinically relevant concentrations.
Efficacy
Efficacy was established in two 12-week, double-blind, placebo-controlled trials of adult patients with TD (ages 18 to 80).6,7 Eligible participants had:
- TD diagnosis for ≥3 months before screening and a history of DRBA treatment for ≥3 months (≥1 month if age ≥60)
- Total AIMS motor score ≥6 (items 1 to 7) at both screening and baseline, verified by a blinded central rater at screening via central video rating
- Patients with an underlying psychiatric illness had to be stable. Psychoactive medication use, including antipsychotics, was allowed if stable for ≥30 days before screening (antidepressants, ≥45 days).
Exclusion criteria included treatment with tetrabenazine, reserpine, α-methyl-p-tyrosine, strong anticholinergic medications, dopamine antagonizing antiemetics (eg, metoclopramide, prochlorperazine, promethazine), dopamine agonists, levodopa, stimulants, or a monoamine oxidase inhibitor (MAOI) within 30 days of screening or baseline, or treatment with botulinum toxin within 3 months of screening; and presence of a neurologic condition that could confound TD assessments, serious untreated or undertreated psychiatric illness, or unstable medical illness. Patients with a history of or active suicidal ideation or behavior within 6 months of screening or score ≥11 on the depression subscale of the Hospital Anxiety and Depression Scale were excluded. Those participants with Fridericia-corrected QT interval values >450 milliseconds in men, >460 milliseconds in women, or >480 milliseconds in patients with a right bundle branch block on electrocardiography at screening also were excluded.
The flexible-dose TD study was performed in 117 participants randomized in a 1:1 manner to deutetrabenazine or placebo, both administered twice daily, titrated to optimal dosage (12 to 48 mg/d) over 6 weeks, and then administered at that dose for another 6 weeks.7 The population demographics were: mean age, 54.6 ± 10.3 years, 52.1% female, 69.2% white, and 80.3% receiving ongoing dopamine antagonists, with a mean TD duration of 74.7 ± 81.5 months. Sixty-eight percent had schizophrenia spectrum disorders, and 30% had mood disorders. The primary outcome was change in total AIMS score (items 1 to 7) assessed by central, independent raters. The mean baseline AIMS score for items 1 to 7 was 9.6 ± 3.9, with 82.9% of participants with baseline AIMS scores ≥6. Study treatment retention was high: placebo 88.1%, deutetrabenazine 89.7%.7 There was a mean 3 point decrease in AIMS score for deutetrabenazine compared with 1.4 for placebo (P = .019). Among those with baseline AIMS scores ≥6, there was a 3.4 point decrease in AIMS scores for deutetrabenazine compared with a 1.9 point decrease for placebo (P = .027). The only adverse effects that occurred in ≥5% of deutetrabenazine participants and at a rate ≥2 times the rate in placebo were insomnia (deutetrabenazine 6.9% vs placebo 1.7%) and akathisia (deutetrabenazine 5.2% vs placebo 0%).
The fixed-dose TD study was performed in 293 participants randomized in 1:1:1:1 manner to 1 of 3 fixed doses of deutetrabenazine (12 mg/d, 24 mg/d, or 36 mg/d) or placebo, both administered twice daily.6 The starting dose of deutetrabenazine was 6 mg twice daily. During the dose escalation period (through Week 4), the dose of study drug was increased weekly in increments of 6 mg/d until the randomized dose was achieved. Patients continued to receive the dose they were assigned to over a maintenance period of 8 weeks.6 The population demographics were: mean age, 56.4 ± 11.3 years, 55% female, 79% white, 76% receiving ongoing dopamine antagonists, with a mean TD duration of 67.2 ± 66 months. Sixty percent had schizophrenia spectrum disorders, and 36% had mood disorders. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. The mean AIMS score at baseline was 9.5 ± 2.7 in the placebo group, and for deutetrabenazine: 9.6 ± 2.4 in the 12 mg/d group, 9.4 ± 2.9 in the 24 mg/d group, and 10.1 ± 3.2 in the 36 mg/d group. The 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). Study treatment retention rates were high: placebo 90.5%, deutetrabenazine 88%. Across all doses, only 1 adverse effect occurred in ≥5% of deutetrabenazine participants: headache (5% deutetrabenazine vs 6% placebo). At the highest dose, 36 mg/d, the only adverse effects that occurred in ≥5% of participants were diarrhea (7% deutetrabenazine vs 3% placebo) and headache (7% deutetrabenazine vs 6% placebo).
Outcome. In the flexible-dose study (mean dose 38.8 ± 7.92 mg/d), the deutetrabenazine arm experienced a mean 30% reduction in AIMS scores from baseline at the Week 12 endpoint. Compared with placebo, the mean reduction in AIMS scores (standard error) was: −3.0 (0.45) deutetrabenazine vs −1.6 (0.46) placebo (P = .019).7 For the fixed-dose study, the 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). In addition to these mean changes from baseline, 35% of the 24 mg/d group and 33% of the 36 mg/d group demonstrated ≥50% reduction in AIMS scores.6
Tolerability
In the 2 phase 3 trials, there were no adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo.5 Discontinuations because of adverse events were low in both pivotal studies across all treatment groups: 3.4% for placebo vs 1.7% for deutetrabenazine in the flexible-dose trial,7 and 3% for placebo vs 4% for deutetrabenazine in the fixed-dose study.6 In neither trial were there clinically significant changes in ratings of depression, suicidality, parkinsonism, or schizophrenia symptoms. The mean QT prolongation in healthy volunteers is described above.
Clinical considerations
Unique properties. Deutetrabenazine utilizes the greater bond strength of the carbon–deuterium bond to slow CYP metabolism, resulting in prolonged duration of action that is well tolerated, and provides significant efficacy.
Why Rx? The reasons to prescribe deutetrabenazine for TD patients include:
- only 1 of 2 agents with FDA approval for TD
- fewer tolerability issues than with tetrabenazine
- lower sedation rates in TD trials than with valbenazine
- no signal for effects on mood parameters or rates of parkinsonism when used for TD.
Dosing
The recommended starting dosage of deutetrabenazine is 6 mg twice daily taken with food, increasing by 6 mg/d weekly as needed, with a maximum dose of 48 mg/d or 36 mg/d in those taking strong CYP2D6 inhibitors or who are 2D6 poor metabolizers. Deutetrabenazine is contraindicated in patients with hepatic impairment (as determined by Child-Pugh criteria16). There are no data in patients with renal impairment. The combined efficacy and tolerability of dosages >48 mg/d has not been evaluated. Overdoses of tetrabenazine ranging from 100 to 1,000 mg have been reported in the literature and were associated with acute dystonia, oculogyric crisis, nausea and vomiting, sweating, sedation, hypotension, confusion, diarrhea, hallucinations, rubor, and tremor.5
Contraindications
When used for TD, deutetrabenazine is contraindicated for patients taking reserpine, tetrabenazine, valbenazine, or MAOIs, and for patients with hepatic impairment. As with most medications, there are no data on deutetrabenazine use in pregnant women; however, oral administration of deutetrabenazine (5, 10, or 30 mg/kg/d) or tetrabenazine (30 mg/kg/d) to pregnant rats during organogenesis had no clear effect on embryofetal development. The highest dose tested was 6 times the maximum recommended human dose of 48 mg/d on a body surface area (mg/m2) basis. There are no data on the presence of deutetrabenazine or its metabolites in human milk, the effects on the breastfed infant, or the effects of the drug on milk production.
Compared with first-generation antipsychotics, second-generation antipsychotics (SGAs) have a lower risk for extrapyramidal symptoms. Yet tardive dyskinesia (TD) remains a concern because of the widespread use of SGAs for multiple indications.1 Prior to April 2017, clinicians had no FDA-approved TD treatment options. The most widely used agent worldwide, tetrabenazine, had positive efficacy data in TD trials over the past 45 years but was not available in the United States until 2008, and its sole indication was for chorea associated with Huntington’s disease.2 Moreover, the use of tetrabenazine involved slow titration, multiple daily dosing, cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, and tolerability issues.
Tetrabenazine is an inhibitor of vesicular monoamine transport type 2 (VMAT2), a transport protein located almost exclusively in the CNS whose role is to place monoamine neurotransmitters (dopamine, serotonin, norepinephrine) into presynaptic vesicles. By decreasing dopamine transport into these presynaptic vesicles, synaptic dopamine release is lessened, thus reducing postsynaptic dopamine D2 receptor activity and the severity of dyskinetic movements.1
In 2 pivotal 12-week clinical trials, deutetrabenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) scores (see Efficacy).6,7
Clinical implications
TD remains a substantial public health concern due to the increasing use of antipsychotics for mood and other disorders beyond the initial indications for schizophrenia.1 Although exposure to dopamine D2antagonism results in postsynaptic receptor upregulation and supersensitivity that underlies the development of dyskinesia, this process is often rapidly reversible in animal models.1 The persistence of TD symptoms in up to 80% of patients after dopamine receptor blocking agents (DRBAs) are stopped has led to hypotheses that the underlying pathophysiology of TD is also a problem with neuroplasticity. Aside from DRBA exposure, environmental factors (eg, oxidative stress) and genetic predisposition might contribute to TD risk.1
Before 2017, only 1 medication (branched-chain amino acids) had been FDA-approved for treating TD in the United States, and only a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]) had positive results from controlled trials, most with small effect sizes.8 Moreover, there was only 1 controlled trial each for clonazepam and EGb-761.1 A branched-chain amino acid preparation received FDA approval for managing TD in male patients, but is no longer commercially available, except from compounding pharmacies.9
Tetrabenazine was developed in the mid-1950s to avoid orthostasis and sedation associated with reserpine.10 Both reserpine and tetrabenazine proved effective for TD,11 but tetrabenazine lacked reserpine’s peripheral adverse effects. However, the kinetics of tetrabenazine necessitated multiple daily doses, and CYP2D6 genotyping was required for doses >50 mg/d.2
Receptor blocking. The mechanism that distinguishes the clinical profiles of reserpine and tetrabenazine relates to their differential properties at VMAT.12 VMAT exists in 2 forms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the CNS.13 Tetrabenazine is a specific and reversible VMAT2 inhibitor, whereas reserpine is an irreversible and nonselective antagonist of VMAT1 and VMAT2. It is reserpine’s VMAT1 inhibition that results in peripheral adverse effects such as orthostasis. Tetrabenazine is rapidly and extensively converted into 2 isomers, alpha-dihydrotetrabenazine (α-DHTBZ) and beta-dihydrotetrabenazine (β-DHTBZ), both of which are metabolized by CYP2D6, with a role for CYP3A4 in α-DHTBZ metabolism.1 These DHTBZ metabolites have a short half-life when generated from oral tetrabenazine, a feature that necessitates multiple daily dosing; moreover, the existence of 2D6 polymorphisms led to FDA-mandated CYP2D6 genotyping for tetrabenazine doses >50 mg/d when it was approved for Huntington’s chorea. The concern is that 2D6 poor metabolizers will have excessive exposure to the VMAT2 effects of DHTBZ, resulting in sedation, akathisia, parkinsonism, and mood symptoms.2
How deuterium impacts medication kinetics. Deuterium is a naturally occurring, stable, nontoxic isotope of hydrogen. Humans have 5 g of deuterium in their body at any time, mostly in the form of heavy water (D2O).14 When deuterium is used to replace selected hydrogen atoms, the resulting molecule will have similar configuration and receptor-binding properties but markedly different kinetics. Because the carbon–deuterium covalent bond requires 8 times more energy to break than a carbon–hydrogen bond, the half-life is prolonged.15 Utilizing this knowledge, a deuterated form of tetrabenazine, deutetrabenazine, was synthesized with such a purpose in mind. While the active metabolites of deutetrabenazine retain the VMAT2 affinity of non-deuterated tetrabenazine, the substitution of deuterium for hydrogen at specific positions slows the breakdown of metabolites, resulting in sustained duration of action, greater active drug exposure, and less impact of 2D6 genotype on drug exposure, thus eliminating the need for genotyping, unless one wants to exceed 36 mg/d.
Deutetrabenazine was first studied in Huntington’s chorea in a 13-week, double-blind, placebo-controlled, parallel-group study (N = 90).4 The maximum daily deutetrabenazine dose was 48 mg, but reduced to 36 mg in those taking strong CYP2D6 inhibitors (bupropion, fluoxetine, or paroxetine). Blinded 2D6 genotyping was performed, but there was no dose modification required based on 2D6 genotype. There was a 36.4% reduction in total maximal chorea score for deutetrabenazine compared with 14.4% for placebo (P < .001).4 Importantly, adverse effects were comparable between both groups, with 1 drop-out in the deutetrabenazine arm vs 2 in the placebo arm. The only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo was somnolence: 11.1% for deutetrabenazine vs 4.4% for placebo.4 The mean deutetrabenazine daily dose at the end of the treatment period was 39.7 ± 9.3 mg, and for those with impaired CYP2D6 function (poor metabolizers or those taking strong CYP2D6 inhibiting medications), the mean daily dose was 34.8 mg ± 3.8 mg.4
Use in tardive dyskinesia. The recommended starting dosage for TD treatment is 6 mg, twice daily with food. The dose may be increased at weekly intervals in increments of 6 mg/d to a maximum recommended daily dosage of 48 mg.5 The maximum daily dose is 36 mg (18 mg, twice daily) in patients receiving strong CYP2D6 inhibitors or who are 2D6 poor metabolizers.5
Deutetrabenazine has not been studied in those with moderate or severe hepatic impairment, and its use is contraindicated in these patients.5 No clinical studies have been conducted to assess the effect of renal impairment on the pharmacokinetics of deutetrabenazine.5
Pharmacologic profile, adverse reactions
When the data from the two 12-week, phase 3 placebo-controlled studies were pooled, the most common adverse reactions occurring in >3% of deutetrabenazine patients and greater than placebo were nasopharyngeal symptoms (4% vs 2% placebo) and insomnia (4% vs 1% placebo).5 Importantly, in neither TD study were there clinically significant changes in rating scales for depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms. The mean QT prolongation for a single 24 mg dose of deutetrabenazine in healthy volunteers was 4.5 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 6.5 milliseconds.5 For tetrabenazine, single 50 mg doses administered to volunteers resulted in mean QT prolongation of 8 milliseconds.5 In patients requiring deutetrabenazine doses >24 mg/d who are taking other medications known to prolong QTc, assess the QTc interval before and after increasing the dose of deutetrabenazine or other medications that are known to prolong QTc.5
How it works
Tetrabenazine is the only agent that has demonstrated significant efficacy for TD management, but its use involves slow titration, multiple daily dosing, CYP2D6 genotyping for doses >50 mg/d, and tolerability issues. For example, the most common adverse effects in the pivotal tetrabenazine Huntington’s disease trial were sedation/somnolence (tetrabenazine 31% vs 3% for placebo), insomnia (tetrabenazine 22% vs 0% for placebo), depression (tetrabenazine 19% vs 0% for placebo), fatigue (tetrabenazine 22% vs 13% for placebo), and akathisia (tetrabenazine 19% vs 0% for placebo).2 For comparison, the only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo in the pivotal Huntington’s disease trial was somnolence (11.1% for deutetrabenazine vs 4.4% for placebo).4
Pharmacokinetics
Deutetrabenazine has 80% oral bioavailability, and is rapidly converted to its active metabolites after oral dosing (Table 2).5 Linear dose dependence of Cmax and area under the curve (AUC) was observed for the active metabolites following single or multiple doses of deutetrabenazine (6 to 24 mg and 7.5 to 22.5 mg, twice daily).15 Cmax of deuterated α-DHTBZ and β-DHTBZ is reached within 3 to 4 hours after dosing, with a steady state ratio of 3:1 for the α-DHTBZ vs the β-DHTBZ form. Food had no effect on AUC, but did increase Cmax by 50%.5
Deutetrabenazine is metabolized through carbonyl reductase enzymes to its active metabolites, and these are further metabolized through multiple CYP pathways, predominantly 2D6 and to a lesser extent 3A4. The effect of CYP2D6 inhibition on the pharmacokinetics of deutetrabenazine and its α-DHTBZ and β-DHTBZ metabolites was studied in 24 healthy participants following a single 22.5 mg dose of deutetrabenazine given after 8 days of administration of the strong CYP2D6 inhibitor paroxetine, 20 mg/d. In the presence of paroxetine, systemic exposure (AUC) of α-DHTBZ was 1.9-fold higher and β-DHTBZ was 6.5-fold higher, resulting in an approximately 3-fold increase in AUC for total (α+β)-DHTBZ, with corresponding increases in mean half-life of approximately 1.5-fold and 2.7-fold, respectively.5 Neither deutetrabenazine or its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that deutetrabenazine and its active metabolites are unlikely to inhibit most major drug transporters at clinically relevant concentrations.
Efficacy
Efficacy was established in two 12-week, double-blind, placebo-controlled trials of adult patients with TD (ages 18 to 80).6,7 Eligible participants had:
- TD diagnosis for ≥3 months before screening and a history of DRBA treatment for ≥3 months (≥1 month if age ≥60)
- Total AIMS motor score ≥6 (items 1 to 7) at both screening and baseline, verified by a blinded central rater at screening via central video rating
- Patients with an underlying psychiatric illness had to be stable. Psychoactive medication use, including antipsychotics, was allowed if stable for ≥30 days before screening (antidepressants, ≥45 days).
Exclusion criteria included treatment with tetrabenazine, reserpine, α-methyl-p-tyrosine, strong anticholinergic medications, dopamine antagonizing antiemetics (eg, metoclopramide, prochlorperazine, promethazine), dopamine agonists, levodopa, stimulants, or a monoamine oxidase inhibitor (MAOI) within 30 days of screening or baseline, or treatment with botulinum toxin within 3 months of screening; and presence of a neurologic condition that could confound TD assessments, serious untreated or undertreated psychiatric illness, or unstable medical illness. Patients with a history of or active suicidal ideation or behavior within 6 months of screening or score ≥11 on the depression subscale of the Hospital Anxiety and Depression Scale were excluded. Those participants with Fridericia-corrected QT interval values >450 milliseconds in men, >460 milliseconds in women, or >480 milliseconds in patients with a right bundle branch block on electrocardiography at screening also were excluded.
The flexible-dose TD study was performed in 117 participants randomized in a 1:1 manner to deutetrabenazine or placebo, both administered twice daily, titrated to optimal dosage (12 to 48 mg/d) over 6 weeks, and then administered at that dose for another 6 weeks.7 The population demographics were: mean age, 54.6 ± 10.3 years, 52.1% female, 69.2% white, and 80.3% receiving ongoing dopamine antagonists, with a mean TD duration of 74.7 ± 81.5 months. Sixty-eight percent had schizophrenia spectrum disorders, and 30% had mood disorders. The primary outcome was change in total AIMS score (items 1 to 7) assessed by central, independent raters. The mean baseline AIMS score for items 1 to 7 was 9.6 ± 3.9, with 82.9% of participants with baseline AIMS scores ≥6. Study treatment retention was high: placebo 88.1%, deutetrabenazine 89.7%.7 There was a mean 3 point decrease in AIMS score for deutetrabenazine compared with 1.4 for placebo (P = .019). Among those with baseline AIMS scores ≥6, there was a 3.4 point decrease in AIMS scores for deutetrabenazine compared with a 1.9 point decrease for placebo (P = .027). The only adverse effects that occurred in ≥5% of deutetrabenazine participants and at a rate ≥2 times the rate in placebo were insomnia (deutetrabenazine 6.9% vs placebo 1.7%) and akathisia (deutetrabenazine 5.2% vs placebo 0%).
The fixed-dose TD study was performed in 293 participants randomized in 1:1:1:1 manner to 1 of 3 fixed doses of deutetrabenazine (12 mg/d, 24 mg/d, or 36 mg/d) or placebo, both administered twice daily.6 The starting dose of deutetrabenazine was 6 mg twice daily. During the dose escalation period (through Week 4), the dose of study drug was increased weekly in increments of 6 mg/d until the randomized dose was achieved. Patients continued to receive the dose they were assigned to over a maintenance period of 8 weeks.6 The population demographics were: mean age, 56.4 ± 11.3 years, 55% female, 79% white, 76% receiving ongoing dopamine antagonists, with a mean TD duration of 67.2 ± 66 months. Sixty percent had schizophrenia spectrum disorders, and 36% had mood disorders. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. The mean AIMS score at baseline was 9.5 ± 2.7 in the placebo group, and for deutetrabenazine: 9.6 ± 2.4 in the 12 mg/d group, 9.4 ± 2.9 in the 24 mg/d group, and 10.1 ± 3.2 in the 36 mg/d group. The 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). Study treatment retention rates were high: placebo 90.5%, deutetrabenazine 88%. Across all doses, only 1 adverse effect occurred in ≥5% of deutetrabenazine participants: headache (5% deutetrabenazine vs 6% placebo). At the highest dose, 36 mg/d, the only adverse effects that occurred in ≥5% of participants were diarrhea (7% deutetrabenazine vs 3% placebo) and headache (7% deutetrabenazine vs 6% placebo).
Outcome. In the flexible-dose study (mean dose 38.8 ± 7.92 mg/d), the deutetrabenazine arm experienced a mean 30% reduction in AIMS scores from baseline at the Week 12 endpoint. Compared with placebo, the mean reduction in AIMS scores (standard error) was: −3.0 (0.45) deutetrabenazine vs −1.6 (0.46) placebo (P = .019).7 For the fixed-dose study, the 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). In addition to these mean changes from baseline, 35% of the 24 mg/d group and 33% of the 36 mg/d group demonstrated ≥50% reduction in AIMS scores.6
Tolerability
In the 2 phase 3 trials, there were no adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo.5 Discontinuations because of adverse events were low in both pivotal studies across all treatment groups: 3.4% for placebo vs 1.7% for deutetrabenazine in the flexible-dose trial,7 and 3% for placebo vs 4% for deutetrabenazine in the fixed-dose study.6 In neither trial were there clinically significant changes in ratings of depression, suicidality, parkinsonism, or schizophrenia symptoms. The mean QT prolongation in healthy volunteers is described above.
Clinical considerations
Unique properties. Deutetrabenazine utilizes the greater bond strength of the carbon–deuterium bond to slow CYP metabolism, resulting in prolonged duration of action that is well tolerated, and provides significant efficacy.
Why Rx? The reasons to prescribe deutetrabenazine for TD patients include:
- only 1 of 2 agents with FDA approval for TD
- fewer tolerability issues than with tetrabenazine
- lower sedation rates in TD trials than with valbenazine
- no signal for effects on mood parameters or rates of parkinsonism when used for TD.
Dosing
The recommended starting dosage of deutetrabenazine is 6 mg twice daily taken with food, increasing by 6 mg/d weekly as needed, with a maximum dose of 48 mg/d or 36 mg/d in those taking strong CYP2D6 inhibitors or who are 2D6 poor metabolizers. Deutetrabenazine is contraindicated in patients with hepatic impairment (as determined by Child-Pugh criteria16). There are no data in patients with renal impairment. The combined efficacy and tolerability of dosages >48 mg/d has not been evaluated. Overdoses of tetrabenazine ranging from 100 to 1,000 mg have been reported in the literature and were associated with acute dystonia, oculogyric crisis, nausea and vomiting, sweating, sedation, hypotension, confusion, diarrhea, hallucinations, rubor, and tremor.5
Contraindications
When used for TD, deutetrabenazine is contraindicated for patients taking reserpine, tetrabenazine, valbenazine, or MAOIs, and for patients with hepatic impairment. As with most medications, there are no data on deutetrabenazine use in pregnant women; however, oral administration of deutetrabenazine (5, 10, or 30 mg/kg/d) or tetrabenazine (30 mg/kg/d) to pregnant rats during organogenesis had no clear effect on embryofetal development. The highest dose tested was 6 times the maximum recommended human dose of 48 mg/d on a body surface area (mg/m2) basis. There are no data on the presence of deutetrabenazine or its metabolites in human milk, the effects on the breastfed infant, or the effects of the drug on milk production.
1. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
2. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
3. Meyer JM. Valbenazine for tardive dyskinesia. Current Psychiatry. 2017;16(5):40-46.
4. Huntington Study Group; Frank S, Testa CM, Stamler D, et al. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA. 2016;316(1):40-50.
5. Austedo [package insert]. North Wales, PA: Teva Pharmaceuticals USA, Inc.; 2017.
6. Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595-604.
7. Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88(21):2003-2010.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Richardson MA, Small AM, Read LL, et al. Branched chain amino acid treatment of tardive dyskinesia in children and adolescents. J Clin Psychiatry. 2004;65(1):92-96.
10. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
11. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
12. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
13. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
14. Kushner DJ, Baker A, Dunstall TG. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can J Physiol Pharmacol. 1999;77(2):79-88.
15. United States Securities and Exchange Commission. Form S-1 Registration Statement of Auspex Pharmaceuticals, Inc. https://www.sec.gov/Archives/edgar/data/1454189/000119312513481239/d627086ds1.htm. Published December 20, 2013. Accessed July 1, 2016.
16. Cholongitas E, Papatheodoridis GV, Vangeli M, et al. Systematic review: the model for end-stage liver disease—should it replace Child-Pugh’s classification for assessing prognosis in cirrhosis? Aliment Pharmacol Ther. 2005;22(11-12):1079-1089.
1. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
2. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
3. Meyer JM. Valbenazine for tardive dyskinesia. Current Psychiatry. 2017;16(5):40-46.
4. Huntington Study Group; Frank S, Testa CM, Stamler D, et al. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA. 2016;316(1):40-50.
5. Austedo [package insert]. North Wales, PA: Teva Pharmaceuticals USA, Inc.; 2017.
6. Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595-604.
7. Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88(21):2003-2010.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Richardson MA, Small AM, Read LL, et al. Branched chain amino acid treatment of tardive dyskinesia in children and adolescents. J Clin Psychiatry. 2004;65(1):92-96.
10. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
11. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
12. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
13. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
14. Kushner DJ, Baker A, Dunstall TG. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can J Physiol Pharmacol. 1999;77(2):79-88.
15. United States Securities and Exchange Commission. Form S-1 Registration Statement of Auspex Pharmaceuticals, Inc. https://www.sec.gov/Archives/edgar/data/1454189/000119312513481239/d627086ds1.htm. Published December 20, 2013. Accessed July 1, 2016.
16. Cholongitas E, Papatheodoridis GV, Vangeli M, et al. Systematic review: the model for end-stage liver disease—should it replace Child-Pugh’s classification for assessing prognosis in cirrhosis? Aliment Pharmacol Ther. 2005;22(11-12):1079-1089.