User login
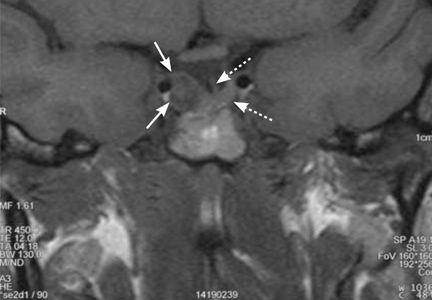
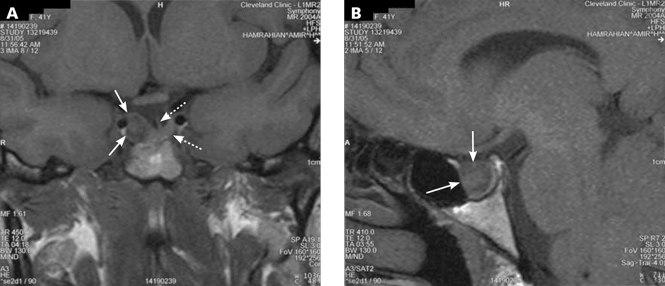
A 39-year-old woman is referred for evaluation of a pituitary mass, which was found on magnetic resonance imaging (MRI) performed because of persistent vertigo. The mass, measuring 1.1 by 1.0 cm, arises from the right portion of the sella turcica and does not reach the optic chiasm (Figure 1). It appears hypointense on MRI and enhances after contrast is given, suggesting it is a pituitary adenoma.
On physical examination she does not have any stigmata of Cushing syndrome or of acromegaly. Her blood pressure is 116/72 mm Hg and her heart rate is regular at 68 beats per minute. Her visual fields are normal as assessed by confrontation, and she has no galactorrhea.
How should this patient be evaluated?
BY DEFINITION, INCIDENTALOMAS ARE UNSUSPECTED
Pituitary “incidentalomas” are, by definition, masses that are discovered by computed tomography (CT) or MRI performed to evaluate unrelated disorders (such as head trauma), for cancer staging, or because of nonspecific symptoms such as dizziness and headache. In some series, headache was the most common reason for imaging studies that led to the discovery of pituitary incidentalomas.1
With more patients undergoing computed tomography (CT) and MRI, more incidentalomas are being discovered. Incidentally discovered pituitary adenomas accounted for 12% of the pituitary tumors in a series of 353 consecutive patients with a presumptive diagnosis of pituitary tumor at one institution over a 14-year period.2 Pituitary masses other than adenomas are discussed later in this paper.
Microadenomas are common, macroadenomas less so
Autopsy studies have revealed pituitary microadenomas (ie, < 10 mm in greatest dimension) in 3% to 27% of patients with no history of pituitary disorders. Macroadenomas (10 mm or larger), on the other hand, are found in fewer than 0.5% of people.3,4 Recently, a study of MRI in 2,000 healthy adult volunteers, age 45 to 97 years, found pituitary macroadenomas in 0.3%.5
Hall et al6 found that 10% of relatively young (< 60 years old) healthy volunteers harbored a pituitary microadenoma on pituitary MRI, but none had a macroadenoma. In a meta-analysis by Ezzat and colleagues,3 adenomas of all sizes were found in 1% to 40% of imaging or postmortem studies (for an average of 16.7%), but macroadenomas were found in only 0.16% to 0.2% of the population.
Although the natural history of pituitary incidentalomas is not well characterized, the numbers suggest that microadenomas rarely grow into macroadenomas.7 Another possibility is that most macroadenomas cause symptoms and therefore come to clinical attention, and thus are not incidentalomas per se.
THE INITIAL EVALUATION: TWO QUESTIONS
The initial approach to a patient with a pituitary incidentaloma should be guided by two questions:
- Is the tumor hormonally active?
- Is it causing a mass effect (ie, is it exerting pressure on adjacent structures)?
IS THE TUMOR HORMONALLY ACTIVE?
A careful history and physical examination may reveal overlooked symptoms or signs of hypersecretion of a specific hormone, which can be evaluated in detail to establish the diagnosis. However, most patients with pituitary incidentalomas have no symptoms, and for them there is no real consensus about the optimal workup strategy.
Prolactin excess
King et al8 calculated that the serum prolactin level is the single most cost-effective screening test for hormonal activity in patients with incidentally discovered pituitary microadenomas. They also suggested, however, that it may be cost-effective to measure multiple hormones in very anxious patients, since a negative test may provide reassurance and improve quality of life.
One should be careful in interpreting elevated prolactin levels in patients with pituitary incidentalomas, since a number of medications (eg, metoclopramide [Reglan], verapamil [Calan], phenothiazines) and disorders (eg, hypothyroidism, cirrhosis, renal failure) can cause mild to moderate elevations of prolactin. In general, a prolactin level of more than 200 ng/mL is almost always diagnostic of prolactinomas. In our experience, a prolactin level above 100 ng/mL is almost always due to a prolactin-secreting pituitary adenoma, except during pregnancy and in some patients who receive antipsychotics or metoclopramide. For these patients, if it is clinically safe to hold or switch medications, retesting after a drug holiday may prove useful and diagnostic.
Growth hormone excess
Growth hormone hypersecretion has been reported in patients with pituitary tumors who have no clinical stigmata of acromegaly.9,10 Moreover, acral changes may not correlate with the metabolic consequences of growth hormone excess.11 In a study by Reincke et al,12 one of 18 patients with pituitary incidentalomas and no apparent acromegalic features had a growth hormone-secreting pituitary adenoma. For this reason, looking for so-called silent growth hormone hypersecretion may be warranted in patients with pituitary tumors, especially in those with macroadenomas.9
The best initial test for growth hormone hypersecretion is the measurement of insulin-like growth factor-1 (IGF-1).13 A normal age- and sex-adjusted IGF-1 level almost always rules out acromegaly.
Further hormonal evaluation
Further hormonal evaluation should be guided by the clinical picture.
Cortisol. In a patient with excess weight gain, central obesity, proximal myopathy, and skin manifestations that suggest hypercortisolism, appropriate initial tests would be a midnight salivary cortisol level, an overnight 1-mg (low-dose) dexamethasone suppression test, or a 24-hour urinary free cortisol level.
Thyroid hormones. Patients with symptoms that suggest hyperthyroidism should have their thyroid-stimulating hormone (TSH; thyrotropin) and free thyroxine (T4) levels measured to rule out a TSH-secreting pituitary adenoma, a very rare tumor.
Gonadotropins. Screening for a gonadotropin-secreting pituitary adenoma by measuring follicle-stimulating hormone, luteinizing hormone, and gonadotropin alpha subunit is not routinely indicated, since almost all of such tumors are clinically silent and generally come to clinical attention only because of a mass effect (see below).
IS THERE A MASS EFFECT?
Pituitary macroadenomas can also cause problems via a mass effect. Examples: hypopituitarism, visual field defects (by compressing the optic chiasm), cranial neuropathy (eg, diplopia, eyelid ptosis secondary to lateral extension of the tumor into a cavernous sinus), and headache.
Hypopituitarism
Hypopituitarism can range from deficiency of one pituitary hormone to the loss of all anterior pituitary hormones (panhypopituitarism).
Hypopituitarism from a mass effect is rare in patients with microadenomas, but one or more anterior pituitary hormone deficiencies are found in more than 30% of patients with a pituitary macroadenoma.3,12,14 With some exceptions, including pituitary apoplexy, the loss of pituitary hormone secretion is slowly progressive; symptoms tend to be nonspecific and often are not noticed at first.
Increased intrasellar pressure may play a role in the pathogenesis of hypopituitarism in patients with pituitary masses.15 Blood flow through the portal vessels is decreased, possibly resulting in diminished delivery of hypo-thalamic hormones to pituitary cells or leading to variable ischemia or necrosis of the normal gland, or both.
All patients with a pituitary macroadenoma should undergo a hormonal evaluation to look for pituitary hormone deficiency.
Growth hormone, gonadotropin deficiencies. In general, pituitary hormone deficiencies from an expanding pituitary tumor tend to begin with growth hormone or the gonadotropins (luteinizing hormone and follicle-stimulating hormone), or both.
Low serum testosterone levels in men (estradiol in women) along with normal or low follicle-stimulating hormone and luteinizing hormone levels are consistent with gonadotropin deficiency in men and amenorrheic premenopausal women.
Failure of the follicle-stimulating hormone and luteinizing hormone levels to rise after menopause is also consistent with gonadotropin deficiency. The presence of regular menses almost always indicates a normal gonadotropin axis. In women with irregular menstruation, hormonal evaluation can be challenging for evaluation of the gonadotropin axis and usually is not indicated.
Patients with deficiencies of two or more pituitary axes and low IGF-1 levels can be presumed to have growth hormone deficiency and usually do not need dynamic testing. But when testing is indicated, the growth hormone axis is best evaluated by dynamic testing, using either a growth hormone-releasing hormone/ arginine stimulation test or the insulin tolerance test.
Thyroid deficiencies. As the tumor expands, deficiencies of thyrotropin and adrenocorticotropic hormone (ACTH) secretion may follow those of growth hormone and gonadotropins. In our experience, the thyrotropin axis is usually affected before the corticotropin axis.
To evaluate the thyrotropin axis, the serum thyrotropin level should be measured along with the free thyroxine level or the free thyroxine index. A low free thyroxine level with a low or normal thyrotropin level is consistent with secondary hypothyroidism. It is inappropriate to measure thyrotropin without also measuring thyroxine in a patient with pituitary disorder, since a normal thyrotropin level in a patient with hypopituitarism is not uncommon.
Adrenal insufficiency. The ACTH stimulation test or an early morning (8 am) plasma cortisol level are both reasonable initial tests to evaluate the hypothalamic-pituitary-adrenal axis. An early morning cortisol level lower than 3 μg/dL confirms adrenal insufficiency, while a value higher than 15 μg/dL makes the diagnosis highly unlikely. Cortisol levels in the range of 3 to 15 μg/dL are indeterminate and should be further evaluated by an ACTH stimulation test, which can be performed anytime during the day.
The standard-dose ACTH stimulation test uses an intravenous or intramuscular injection of 250 μg of cosyntropin (Cortrosyn; ACTH 1–24). A normal response is a plasma cortisol concentration higher than 18 μg/dL at 30 minutes.
The sensitivity of the ACTH stimulation test in detecting mild, partial adrenal insufficiency is higher if a lower dose of cosyntropin is used (1 μg intravenously). However, the low-dose test has a higher false-positive rate. In most clinical situations, the 30-minute cortisol value during a standard-dose ACTH stimulation test has a diagnostic accuracy close to that of the low-dose ACTH stimulation test.16 Patients with recent-onset ACTH deficiency (eg, in pituitary apoplexy or within 2 to 4 weeks following pituitary surgery) may have a normal response to the ACTH stimulation test, since their adrenal glands have not undergone sufficient atrophy and still respond to ACTH stimulation.
The insulin tolerance test is considered the gold standard for evaluating the hypothalamic-pituitary-adrenal axis, but it needs to be performed by an experienced clinician and is usually not needed for everyday clinical practice.
Visual field defects

Visual field loss generally begins in the superior temporal fields, which explains why the patient may not notice it at first. Then, with continued growth and compression, vision loss extends into the inferior temporal fields, then into the nasal fields as a late effect.
Because the patient may not notice the visual field defect, formal visual field testing is warranted if the tumor compresses or abuts the optic chiasm. While bitemporal hemianopia is the classic manifestation of chiasmal compression, variable visual field defects may occur depending on which portion of the optic apparatus is involved.
Cranial neuropathy
Abnormal eye movements, which may cause diplopia, result from extension of a pituitary tumor into one or both cavernous sinuses. Compression of the third (occulomotor, the cranial nerve most often affected), fourth (trochlear), and sixth (abducens) cranial nerves leads to eye movement deviations as well as eyelid ptosis due to third nerve dysfunction. Cranial neuropathy most commonly occurs in the setting of pituitary apoplexy (see below) but may occur without it.
Headache
Headache can be associated with pituitary tumors, but the underlying pathophysiology remains uncertain. Possible mechanisms include structural causes such as dural stretching or cavernous sinus invasion.17 Other possible mechanisms are an increase in the intrasellar pressure and tumor activity.15,18 The link between headache and tumor activity is supported by the observation that headaches resolve in some patients with acromegaly shortly after they start taking somatostatin analogues.19
Migraine may be the most common type of headache reported in patients with pituitary adenomas; however, short-lasting unilateral neuralgiform headache with conjunctival injection and tearing (SUNCT) has also been reported.19
Of interest, there seems to be a strong association between pituitary-associated headache and a family history of headache.19 That said, headache is a common symptom in the general population, and establishing a cause-and-effect relationship prior to surgical removal of a pituitary tumor can be challenging. Approximately 50% of patients with headache who undergo an operation for a pituitary tumor have relief after surgery; however, 35% may not have relief, and up to 15% have a worsening of their headaches.19
OUR PATIENT’S HORMONAL EVALUATION
In the patient we described earlier, hormonal evaluation revealed the following:
- Prolactin 12.2 ng/mL (reference range 2–17.4)
- IGF-1 189 ng/mL (114–492)
- Thyrotropin 1.63 μU/mL (0.4–5.5)
- Free thyroxine index 9.5 μg/L (6–11)
- Maximum cortisol during a low-dose ACTH stimulation test 18.4 μg/dL. In short, all her test results were normal.
A formal visual field test was not performed, since the pituitary mass did not reach the optic chiasm (Figure 1).
ADENOMAS VS OTHER SELLAR MASSES

In some cases, it may be difficult to distinguish a nonadenomatous lesion from a nonfunctioning pituitary adenoma. However, several endocrine, radiographic, and neurologic features may help to differentiate pituitary tumors from other, less common sellar disorders.20
For instance, diabetes insipidus is extremely rare in patients with pituitary adenomas at presentation without significant suprasellar extension of the tumor. Therefore, its presence strongly suggests a nonpituitary cause such as hypophysitis, sarcoidosis, or a meta-static lesion.21
Some radiographic features that suggest sellar masses other than pituitary tumors include calcifications on CT in patients with craniopharyngiomas and meningiomas or a rapidly enlarging mass with lack of sellar enlargement (sellar remodeling), which suggests a metastatic lesion. While a dural tail sign (a linear enhanced structure or “tail” extending away from the tumor mass along the dural surface) may be seen with some meningiomas, peripheral enhancement of the dura is not specific for meningioma and may be seen with pituitary apoplexy as well.22,23
Cranial neuropathy is less common in patients with pituitary adenomas than in those with nonadenomatous masses (for example a metastasis or a meningioma), although the acute onset of cranial neuropathy often accompanies a hemorrhagic infarction of a preexisting pituitary adenoma (pituitary apoplexy).20
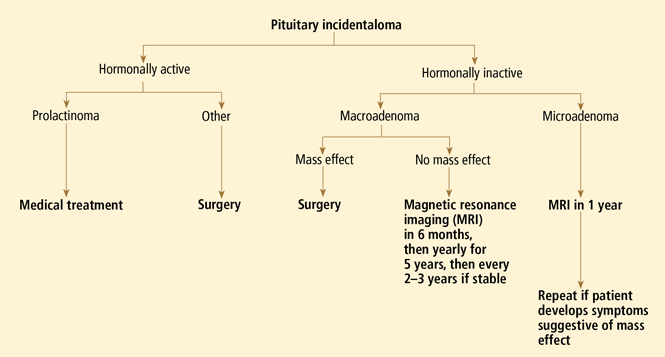
OUR RECOMMENDATIONS
Our approach to a patient with a pituitary incidentaloma is summarized in Figure 4.
If the tumor is hormonally active
Prolactinoma is the exception. For this tumor, dopamine agonists can resolve symptoms and shrink the tumor in most cases. Even in patients with a visual field defect associated with a macroprolactinoma, vision usually improves within days after starting a dopamine agonist, before the tumor has observably shrunk. However, a follow-up visual field test is necessary 2 to 6 weeks after starting therapy to establish that the tumor is responding to therapy; if the tumor does not respond, surgery may be necessary.
If the tumor is hormonally inactive
If the tumor is hormonally inactive, its further evaluation depends on its size and whether there is a mass effect. In patients with a nonfunctioning pituitary macroadenoma, a comprehensive hormonal evaluation for hypopituitarism should be done. Patients with a visual field defect or cranial neuropathy should undergo surgical tumor resection. If there is no mass effect, observation may be an acceptable strategy. We, and others,1,25 recommend surgery for most patients with pituitary macroadenomas abutting the optic chiasm.
If the tumor is small
If the tumor is small (ie, a microadenoma), the risk of its growing is low. Three small studies followed such patients prospectively and found a 0 to 14% risk of tumor enlargement over a mean follow-up period of 1.8 to 6.7 years.12,25,26 While there is no consensus about how soon to follow up patients with nonfunctioning pituitary microadenomas, we obtain a follow-up MRI study in 1 year, with no further routine imaging if the tumor has remained stable, unless the patient develops symptoms or signs suggesting a mass effect.
If the tumor is large
If the tumor is large (ie, a macroadenoma), the risk of further growth is expected to be higher, since the tumor has already shown the propensity to grow. In the same three series discussed above, the risk of tumor growth for a pituitary macroadenomas was about 30% over the mean follow-up of 1.8 to 6.7 years.12,25,26
Furthermore, several recent studies have suggested a higher propensity to grow and to cause symptoms and signs than previously thought. For example, Karavitaki et al7 studied 24 patients who had nonfunctional macroadenomas and found that the 48-month probability of enlargement was 44%; of this group, 57% showed new or worsening visual field defects, and an additional 21% showed new chiasmatic compression without vision loss. Similarly, Arita and colleagues27 found that 21 (50%) of 42 nonfunctional adenomas (mean size 18.3 ± 7 mm) increased by at least 10% over an average of 32 months after the initial evaluation. Ten patients became symptomatic over a mean of about 5 years, with 4 of these 10 (9.5% of the entire cohort) suffering symptomatic pituitary apoplexy. Therefore, one may argue for surgery (especially in young patients) for pituitary macroadenomas even in the absence of mass effect.
We would obtain a follow-up MRI study at 6 months, then yearly for 5 years, and then every 2 to 3 years if the tumor is stable. Surgery would be indicated if there is evidence of tumor growth or a mass effect.
While tumor growth has been found to be independent of age in some studies,27 others have found longer tumor doubling time in patients older than 60 years.28
The risk of pituitary apoplexy
Pituitary apoplexy results from a hemorrhagic infarction of the tumor and manifests clinically as the sudden onset of severe headache, nausea, vomiting, vision loss, and cranial nerve palsies. While most cases of pituitary apoplexy are spontaneous, precipitating factors may include head injury, anticoagulant therapy, dopamine agonists, radiation therapy, or dynamic endocrine tests.29
It is important to educate patients and their families about the symptoms of pituitary apoplexy, especially patients with pituitary macroadenomas. If the condition is unrecognized and untreated, patients can develop hypotension and shock secondary to adrenal insufficiency, as well as irreversible vision loss or diplopia.
Surgery is generally recommended in cases of progressive vision loss or cranial neuropathy, preferably within 24 or 48 hours of onset if feasible, to minimize the risk of a permanent neurologic deficit.
Clinically significant pituitary apoplexy is rare in patients with pituitary microadenomas. In the study by Arita et al,27 the risk of pituitary apoplexy during 5 years of follow-up was 9.5%, and all of the tumors involved were macroadenomas. This rate is higher than in some other studies, in which the risk of apoplexy ranged from 0.4% to 7% during a mean follow-up of 2 to 6 years.1,25,30
CASE FOLLOW-UP
Since our patient had no evidence of hormonal hypersecretion or mass effect and no hypopituitarism, we asked her to return in 6 months. A repeat MRI study showed the tumor to be stable, with no evidence of growth. The patient was scheduled for a return visit in 1 year.
- Sanno N, Oyama K, Tahara S, Teramoto A, Kato Y. A survey of pituitary incidentaloma in Japan. Eur J Endocrinol 2003; 149:123–127.
- Gsponer J, De Tribolet N, Déruaz JP, et al. Diagnosis, treatment, and outcome of pituitary tumors and other abnormal intrasellar masses. Retrospective analysis of 353 patients. Medicine (Baltimore) 1999; 78:236–269.
- Ezzat S, Asa SL, Couldwell WT, et al. The prevalence of pituitary adenomas: a systematic review. Cancer 2004; 101:613–619.
- Molitch ME, Russell EJ. The pituitary “incidentaloma.” Ann Intern Med 1990; 112:925–931.
- Vernooij MW, Ikram MA, Tanghe HL, et al. Incidental findings on brain MRI in the general population. N Engl J Med 2007; 357:1821–1828.
- Hall WA, Luciano MG, Doppman JL, Patronas NJ, Oldfield EH. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Intern Med 1994; 120:817–820.
- Karavitaki N, Collison K, Halliday J, et al. What is the natural history of nonoperated nonfunctioning pituitary adenomas? Clin Endocrinol (Oxf) 2007; 67:938–943.
- King JT, Justice AC, Aron DC. Management of incidental pituitary microadenomas: a cost-effectiveness analysis. J Clin Endocrinol Metab 1997; 82:3625–3632.
- Klibanski A, Zervas NT, Kovacs K, Ridgway EC. Clinically silent hypersecretion of growth hormone in patients with pituitary tumors. J Neurosurg 1987; 66:806–811.
- Trouillas J, Sassolas G, Loras B, et al. Somatotropic adenomas without acromegaly. Pathol Res Pract 1991; 187:943–949.
- Cryer PE, Daughaday WH. Regulation of growth hormone secretion in acromegaly. J Clin Endocrinol Metab 1969; 29:386–393.
- Reincke M, Allolio B, Saeger W, Menzel J, Winkelmann W. The ‘incidentaloma’ of the pituitary gland. Is neurosurgery required? JAMA 1990; 263:2772–2776.
- Giustina A, Barkan A, Casanueva FF, et al. Criteria for cure of acromegaly: a consensus statement. J Clin Endocrinol Metab 2000; 85:526–529.
- Nammour GM, Ybarra J, Naheedy MH, Romeo JH, Aron DC. Incidental pituitary macroadenoma: a population-based study. Am J Med Sci 1997; 314:287–291.
- Arafah BM, Prunty D, Ybarra J, Hlavin ML, Selman WR. The dominant role of increased intrasellar pressure in the pathogenesis of hypopituitarism, hyperprolactinemia, and headaches in patients with pituitary adenomas. J Clin Endocrinol Metab 2000; 85:1789–1793.
- Mayenknecht J, Diederich S, Bahr V, Plockinger U, Oelkers W. Comparison of low and high dose corticotropin stimulation tests in patients with pituitary disease. J Clin Endocrinol Metab 1998; 83:1558–1562.
- Forsyth PA, Posner JB. Headaches in patients with brain tumors: a study of 111 patients. Neurology 1993; 43:1678–1683.
- Abe T, Matsumoto K, Kuwazawa J, Toyoda I, Sasaki K. Headache associated with pituitary adenomas. Headache 1998; 38:782–786.
- Levy MJ, Matharu MS, Meeran K, Powell M, Goadsby PJ. The clinical characteristics of headache in patients with pituitary tumours. Brain 2005; 128:1921–1930.
- Freda PU, Post KD. Differential diagnosis of sellar masses. Endocrinol Metab Clin North Am 1999; 28:81–117.
- Gopan T, Toms SA, Prayson RA, Suh JH, Hamrahian AH, Weil RJ. Symptomatic pituitary metastases from renal cell carcinoma. Pituitary 2007; 10:251–259.
- Moore AF, Grinspoon SK. A dural tale. J Clin Endocrinol Metab 2007; 92:3367–3368.
- Smirniotopoulos JG, Murphy FM, Rushing EJ, Rees JH, Schroeder JW. Patterns of contrast enhancement in the brain and meninges. Radiographics 2007; 27:525–551.
- Chanson P, Daujat F, Young J, et al. Normal pituitary hypertrophy as a frequent cause of pituitary incidentaloma: a follow-up study. J Clin Endocrinol Metab 2001; 86:3009–3015.
- Donovan LE, Corenblum B. The natural history of the pituitary incidentaloma. Arch Intern Med 1995; 155:181–183.
- Feldkamp J, Santen R, Harms E, Aulich A, Modder U, Scherbaum WA. Incidentally discovered pituitary lesions: high frequency of macroadenomas and hormone-secreting adenomas—results of a prospective study. Clin Endocrinol (Oxf) 1999; 51:109–113.
- Arita K, Tominaga A, Sugiyama K, et al. Natural course of incidentally found nonfunctioning pituitary adenoma, with special reference to pituitary apoplexy during follow-up examination. J Neurosurg 2006; 104:884–891.
- Tanaka Y, Hongo K, Tada T, Sakai K, Kakizawa Y, Kobayashi S. Growth pattern and rate in residual nonfunctioning pituitary adenomas: correlations among tumor volume doubling time, patient age, and MIB-1 index. J Neurosurg 2003; 98:359–365.
- Biousse V, Newman NJ, Oyesiku NM. Precipitating factors in pituitary apoplexy. J Neurol Neurosurg Psychiatry 2001; 71:542–545.
- Nishizawa S, Ohta S, Yokoyama T, Uemura K. Therapeutic strategy for incidentally found pituitary tumors (“pituitary incidentalomas”). Neurosurgery 1998; 43:1344–1348.
A 39-year-old woman is referred for evaluation of a pituitary mass, which was found on magnetic resonance imaging (MRI) performed because of persistent vertigo. The mass, measuring 1.1 by 1.0 cm, arises from the right portion of the sella turcica and does not reach the optic chiasm (Figure 1). It appears hypointense on MRI and enhances after contrast is given, suggesting it is a pituitary adenoma.
On physical examination she does not have any stigmata of Cushing syndrome or of acromegaly. Her blood pressure is 116/72 mm Hg and her heart rate is regular at 68 beats per minute. Her visual fields are normal as assessed by confrontation, and she has no galactorrhea.
How should this patient be evaluated?
BY DEFINITION, INCIDENTALOMAS ARE UNSUSPECTED
Pituitary “incidentalomas” are, by definition, masses that are discovered by computed tomography (CT) or MRI performed to evaluate unrelated disorders (such as head trauma), for cancer staging, or because of nonspecific symptoms such as dizziness and headache. In some series, headache was the most common reason for imaging studies that led to the discovery of pituitary incidentalomas.1
With more patients undergoing computed tomography (CT) and MRI, more incidentalomas are being discovered. Incidentally discovered pituitary adenomas accounted for 12% of the pituitary tumors in a series of 353 consecutive patients with a presumptive diagnosis of pituitary tumor at one institution over a 14-year period.2 Pituitary masses other than adenomas are discussed later in this paper.
Microadenomas are common, macroadenomas less so
Autopsy studies have revealed pituitary microadenomas (ie, < 10 mm in greatest dimension) in 3% to 27% of patients with no history of pituitary disorders. Macroadenomas (10 mm or larger), on the other hand, are found in fewer than 0.5% of people.3,4 Recently, a study of MRI in 2,000 healthy adult volunteers, age 45 to 97 years, found pituitary macroadenomas in 0.3%.5
Hall et al6 found that 10% of relatively young (< 60 years old) healthy volunteers harbored a pituitary microadenoma on pituitary MRI, but none had a macroadenoma. In a meta-analysis by Ezzat and colleagues,3 adenomas of all sizes were found in 1% to 40% of imaging or postmortem studies (for an average of 16.7%), but macroadenomas were found in only 0.16% to 0.2% of the population.
Although the natural history of pituitary incidentalomas is not well characterized, the numbers suggest that microadenomas rarely grow into macroadenomas.7 Another possibility is that most macroadenomas cause symptoms and therefore come to clinical attention, and thus are not incidentalomas per se.
THE INITIAL EVALUATION: TWO QUESTIONS
The initial approach to a patient with a pituitary incidentaloma should be guided by two questions:
- Is the tumor hormonally active?
- Is it causing a mass effect (ie, is it exerting pressure on adjacent structures)?
IS THE TUMOR HORMONALLY ACTIVE?
A careful history and physical examination may reveal overlooked symptoms or signs of hypersecretion of a specific hormone, which can be evaluated in detail to establish the diagnosis. However, most patients with pituitary incidentalomas have no symptoms, and for them there is no real consensus about the optimal workup strategy.
Prolactin excess
King et al8 calculated that the serum prolactin level is the single most cost-effective screening test for hormonal activity in patients with incidentally discovered pituitary microadenomas. They also suggested, however, that it may be cost-effective to measure multiple hormones in very anxious patients, since a negative test may provide reassurance and improve quality of life.
One should be careful in interpreting elevated prolactin levels in patients with pituitary incidentalomas, since a number of medications (eg, metoclopramide [Reglan], verapamil [Calan], phenothiazines) and disorders (eg, hypothyroidism, cirrhosis, renal failure) can cause mild to moderate elevations of prolactin. In general, a prolactin level of more than 200 ng/mL is almost always diagnostic of prolactinomas. In our experience, a prolactin level above 100 ng/mL is almost always due to a prolactin-secreting pituitary adenoma, except during pregnancy and in some patients who receive antipsychotics or metoclopramide. For these patients, if it is clinically safe to hold or switch medications, retesting after a drug holiday may prove useful and diagnostic.
Growth hormone excess
Growth hormone hypersecretion has been reported in patients with pituitary tumors who have no clinical stigmata of acromegaly.9,10 Moreover, acral changes may not correlate with the metabolic consequences of growth hormone excess.11 In a study by Reincke et al,12 one of 18 patients with pituitary incidentalomas and no apparent acromegalic features had a growth hormone-secreting pituitary adenoma. For this reason, looking for so-called silent growth hormone hypersecretion may be warranted in patients with pituitary tumors, especially in those with macroadenomas.9
The best initial test for growth hormone hypersecretion is the measurement of insulin-like growth factor-1 (IGF-1).13 A normal age- and sex-adjusted IGF-1 level almost always rules out acromegaly.
Further hormonal evaluation
Further hormonal evaluation should be guided by the clinical picture.
Cortisol. In a patient with excess weight gain, central obesity, proximal myopathy, and skin manifestations that suggest hypercortisolism, appropriate initial tests would be a midnight salivary cortisol level, an overnight 1-mg (low-dose) dexamethasone suppression test, or a 24-hour urinary free cortisol level.
Thyroid hormones. Patients with symptoms that suggest hyperthyroidism should have their thyroid-stimulating hormone (TSH; thyrotropin) and free thyroxine (T4) levels measured to rule out a TSH-secreting pituitary adenoma, a very rare tumor.
Gonadotropins. Screening for a gonadotropin-secreting pituitary adenoma by measuring follicle-stimulating hormone, luteinizing hormone, and gonadotropin alpha subunit is not routinely indicated, since almost all of such tumors are clinically silent and generally come to clinical attention only because of a mass effect (see below).
IS THERE A MASS EFFECT?
Pituitary macroadenomas can also cause problems via a mass effect. Examples: hypopituitarism, visual field defects (by compressing the optic chiasm), cranial neuropathy (eg, diplopia, eyelid ptosis secondary to lateral extension of the tumor into a cavernous sinus), and headache.
Hypopituitarism
Hypopituitarism can range from deficiency of one pituitary hormone to the loss of all anterior pituitary hormones (panhypopituitarism).
Hypopituitarism from a mass effect is rare in patients with microadenomas, but one or more anterior pituitary hormone deficiencies are found in more than 30% of patients with a pituitary macroadenoma.3,12,14 With some exceptions, including pituitary apoplexy, the loss of pituitary hormone secretion is slowly progressive; symptoms tend to be nonspecific and often are not noticed at first.
Increased intrasellar pressure may play a role in the pathogenesis of hypopituitarism in patients with pituitary masses.15 Blood flow through the portal vessels is decreased, possibly resulting in diminished delivery of hypo-thalamic hormones to pituitary cells or leading to variable ischemia or necrosis of the normal gland, or both.
All patients with a pituitary macroadenoma should undergo a hormonal evaluation to look for pituitary hormone deficiency.
Growth hormone, gonadotropin deficiencies. In general, pituitary hormone deficiencies from an expanding pituitary tumor tend to begin with growth hormone or the gonadotropins (luteinizing hormone and follicle-stimulating hormone), or both.
Low serum testosterone levels in men (estradiol in women) along with normal or low follicle-stimulating hormone and luteinizing hormone levels are consistent with gonadotropin deficiency in men and amenorrheic premenopausal women.
Failure of the follicle-stimulating hormone and luteinizing hormone levels to rise after menopause is also consistent with gonadotropin deficiency. The presence of regular menses almost always indicates a normal gonadotropin axis. In women with irregular menstruation, hormonal evaluation can be challenging for evaluation of the gonadotropin axis and usually is not indicated.
Patients with deficiencies of two or more pituitary axes and low IGF-1 levels can be presumed to have growth hormone deficiency and usually do not need dynamic testing. But when testing is indicated, the growth hormone axis is best evaluated by dynamic testing, using either a growth hormone-releasing hormone/ arginine stimulation test or the insulin tolerance test.
Thyroid deficiencies. As the tumor expands, deficiencies of thyrotropin and adrenocorticotropic hormone (ACTH) secretion may follow those of growth hormone and gonadotropins. In our experience, the thyrotropin axis is usually affected before the corticotropin axis.
To evaluate the thyrotropin axis, the serum thyrotropin level should be measured along with the free thyroxine level or the free thyroxine index. A low free thyroxine level with a low or normal thyrotropin level is consistent with secondary hypothyroidism. It is inappropriate to measure thyrotropin without also measuring thyroxine in a patient with pituitary disorder, since a normal thyrotropin level in a patient with hypopituitarism is not uncommon.
Adrenal insufficiency. The ACTH stimulation test or an early morning (8 am) plasma cortisol level are both reasonable initial tests to evaluate the hypothalamic-pituitary-adrenal axis. An early morning cortisol level lower than 3 μg/dL confirms adrenal insufficiency, while a value higher than 15 μg/dL makes the diagnosis highly unlikely. Cortisol levels in the range of 3 to 15 μg/dL are indeterminate and should be further evaluated by an ACTH stimulation test, which can be performed anytime during the day.
The standard-dose ACTH stimulation test uses an intravenous or intramuscular injection of 250 μg of cosyntropin (Cortrosyn; ACTH 1–24). A normal response is a plasma cortisol concentration higher than 18 μg/dL at 30 minutes.
The sensitivity of the ACTH stimulation test in detecting mild, partial adrenal insufficiency is higher if a lower dose of cosyntropin is used (1 μg intravenously). However, the low-dose test has a higher false-positive rate. In most clinical situations, the 30-minute cortisol value during a standard-dose ACTH stimulation test has a diagnostic accuracy close to that of the low-dose ACTH stimulation test.16 Patients with recent-onset ACTH deficiency (eg, in pituitary apoplexy or within 2 to 4 weeks following pituitary surgery) may have a normal response to the ACTH stimulation test, since their adrenal glands have not undergone sufficient atrophy and still respond to ACTH stimulation.
The insulin tolerance test is considered the gold standard for evaluating the hypothalamic-pituitary-adrenal axis, but it needs to be performed by an experienced clinician and is usually not needed for everyday clinical practice.
Visual field defects
Visual field loss generally begins in the superior temporal fields, which explains why the patient may not notice it at first. Then, with continued growth and compression, vision loss extends into the inferior temporal fields, then into the nasal fields as a late effect.
Because the patient may not notice the visual field defect, formal visual field testing is warranted if the tumor compresses or abuts the optic chiasm. While bitemporal hemianopia is the classic manifestation of chiasmal compression, variable visual field defects may occur depending on which portion of the optic apparatus is involved.
Cranial neuropathy
Abnormal eye movements, which may cause diplopia, result from extension of a pituitary tumor into one or both cavernous sinuses. Compression of the third (occulomotor, the cranial nerve most often affected), fourth (trochlear), and sixth (abducens) cranial nerves leads to eye movement deviations as well as eyelid ptosis due to third nerve dysfunction. Cranial neuropathy most commonly occurs in the setting of pituitary apoplexy (see below) but may occur without it.
Headache
Headache can be associated with pituitary tumors, but the underlying pathophysiology remains uncertain. Possible mechanisms include structural causes such as dural stretching or cavernous sinus invasion.17 Other possible mechanisms are an increase in the intrasellar pressure and tumor activity.15,18 The link between headache and tumor activity is supported by the observation that headaches resolve in some patients with acromegaly shortly after they start taking somatostatin analogues.19
Migraine may be the most common type of headache reported in patients with pituitary adenomas; however, short-lasting unilateral neuralgiform headache with conjunctival injection and tearing (SUNCT) has also been reported.19
Of interest, there seems to be a strong association between pituitary-associated headache and a family history of headache.19 That said, headache is a common symptom in the general population, and establishing a cause-and-effect relationship prior to surgical removal of a pituitary tumor can be challenging. Approximately 50% of patients with headache who undergo an operation for a pituitary tumor have relief after surgery; however, 35% may not have relief, and up to 15% have a worsening of their headaches.19
OUR PATIENT’S HORMONAL EVALUATION
In the patient we described earlier, hormonal evaluation revealed the following:
- Prolactin 12.2 ng/mL (reference range 2–17.4)
- IGF-1 189 ng/mL (114–492)
- Thyrotropin 1.63 μU/mL (0.4–5.5)
- Free thyroxine index 9.5 μg/L (6–11)
- Maximum cortisol during a low-dose ACTH stimulation test 18.4 μg/dL. In short, all her test results were normal.
A formal visual field test was not performed, since the pituitary mass did not reach the optic chiasm (Figure 1).
ADENOMAS VS OTHER SELLAR MASSES
In some cases, it may be difficult to distinguish a nonadenomatous lesion from a nonfunctioning pituitary adenoma. However, several endocrine, radiographic, and neurologic features may help to differentiate pituitary tumors from other, less common sellar disorders.20
For instance, diabetes insipidus is extremely rare in patients with pituitary adenomas at presentation without significant suprasellar extension of the tumor. Therefore, its presence strongly suggests a nonpituitary cause such as hypophysitis, sarcoidosis, or a meta-static lesion.21
Some radiographic features that suggest sellar masses other than pituitary tumors include calcifications on CT in patients with craniopharyngiomas and meningiomas or a rapidly enlarging mass with lack of sellar enlargement (sellar remodeling), which suggests a metastatic lesion. While a dural tail sign (a linear enhanced structure or “tail” extending away from the tumor mass along the dural surface) may be seen with some meningiomas, peripheral enhancement of the dura is not specific for meningioma and may be seen with pituitary apoplexy as well.22,23
Cranial neuropathy is less common in patients with pituitary adenomas than in those with nonadenomatous masses (for example a metastasis or a meningioma), although the acute onset of cranial neuropathy often accompanies a hemorrhagic infarction of a preexisting pituitary adenoma (pituitary apoplexy).20
OUR RECOMMENDATIONS
Our approach to a patient with a pituitary incidentaloma is summarized in Figure 4.
If the tumor is hormonally active
Prolactinoma is the exception. For this tumor, dopamine agonists can resolve symptoms and shrink the tumor in most cases. Even in patients with a visual field defect associated with a macroprolactinoma, vision usually improves within days after starting a dopamine agonist, before the tumor has observably shrunk. However, a follow-up visual field test is necessary 2 to 6 weeks after starting therapy to establish that the tumor is responding to therapy; if the tumor does not respond, surgery may be necessary.
If the tumor is hormonally inactive
If the tumor is hormonally inactive, its further evaluation depends on its size and whether there is a mass effect. In patients with a nonfunctioning pituitary macroadenoma, a comprehensive hormonal evaluation for hypopituitarism should be done. Patients with a visual field defect or cranial neuropathy should undergo surgical tumor resection. If there is no mass effect, observation may be an acceptable strategy. We, and others,1,25 recommend surgery for most patients with pituitary macroadenomas abutting the optic chiasm.
If the tumor is small
If the tumor is small (ie, a microadenoma), the risk of its growing is low. Three small studies followed such patients prospectively and found a 0 to 14% risk of tumor enlargement over a mean follow-up period of 1.8 to 6.7 years.12,25,26 While there is no consensus about how soon to follow up patients with nonfunctioning pituitary microadenomas, we obtain a follow-up MRI study in 1 year, with no further routine imaging if the tumor has remained stable, unless the patient develops symptoms or signs suggesting a mass effect.
If the tumor is large
If the tumor is large (ie, a macroadenoma), the risk of further growth is expected to be higher, since the tumor has already shown the propensity to grow. In the same three series discussed above, the risk of tumor growth for a pituitary macroadenomas was about 30% over the mean follow-up of 1.8 to 6.7 years.12,25,26
Furthermore, several recent studies have suggested a higher propensity to grow and to cause symptoms and signs than previously thought. For example, Karavitaki et al7 studied 24 patients who had nonfunctional macroadenomas and found that the 48-month probability of enlargement was 44%; of this group, 57% showed new or worsening visual field defects, and an additional 21% showed new chiasmatic compression without vision loss. Similarly, Arita and colleagues27 found that 21 (50%) of 42 nonfunctional adenomas (mean size 18.3 ± 7 mm) increased by at least 10% over an average of 32 months after the initial evaluation. Ten patients became symptomatic over a mean of about 5 years, with 4 of these 10 (9.5% of the entire cohort) suffering symptomatic pituitary apoplexy. Therefore, one may argue for surgery (especially in young patients) for pituitary macroadenomas even in the absence of mass effect.
We would obtain a follow-up MRI study at 6 months, then yearly for 5 years, and then every 2 to 3 years if the tumor is stable. Surgery would be indicated if there is evidence of tumor growth or a mass effect.
While tumor growth has been found to be independent of age in some studies,27 others have found longer tumor doubling time in patients older than 60 years.28
The risk of pituitary apoplexy
Pituitary apoplexy results from a hemorrhagic infarction of the tumor and manifests clinically as the sudden onset of severe headache, nausea, vomiting, vision loss, and cranial nerve palsies. While most cases of pituitary apoplexy are spontaneous, precipitating factors may include head injury, anticoagulant therapy, dopamine agonists, radiation therapy, or dynamic endocrine tests.29
It is important to educate patients and their families about the symptoms of pituitary apoplexy, especially patients with pituitary macroadenomas. If the condition is unrecognized and untreated, patients can develop hypotension and shock secondary to adrenal insufficiency, as well as irreversible vision loss or diplopia.
Surgery is generally recommended in cases of progressive vision loss or cranial neuropathy, preferably within 24 or 48 hours of onset if feasible, to minimize the risk of a permanent neurologic deficit.
Clinically significant pituitary apoplexy is rare in patients with pituitary microadenomas. In the study by Arita et al,27 the risk of pituitary apoplexy during 5 years of follow-up was 9.5%, and all of the tumors involved were macroadenomas. This rate is higher than in some other studies, in which the risk of apoplexy ranged from 0.4% to 7% during a mean follow-up of 2 to 6 years.1,25,30
CASE FOLLOW-UP
Since our patient had no evidence of hormonal hypersecretion or mass effect and no hypopituitarism, we asked her to return in 6 months. A repeat MRI study showed the tumor to be stable, with no evidence of growth. The patient was scheduled for a return visit in 1 year.
A 39-year-old woman is referred for evaluation of a pituitary mass, which was found on magnetic resonance imaging (MRI) performed because of persistent vertigo. The mass, measuring 1.1 by 1.0 cm, arises from the right portion of the sella turcica and does not reach the optic chiasm (Figure 1). It appears hypointense on MRI and enhances after contrast is given, suggesting it is a pituitary adenoma.
On physical examination she does not have any stigmata of Cushing syndrome or of acromegaly. Her blood pressure is 116/72 mm Hg and her heart rate is regular at 68 beats per minute. Her visual fields are normal as assessed by confrontation, and she has no galactorrhea.
How should this patient be evaluated?
BY DEFINITION, INCIDENTALOMAS ARE UNSUSPECTED
Pituitary “incidentalomas” are, by definition, masses that are discovered by computed tomography (CT) or MRI performed to evaluate unrelated disorders (such as head trauma), for cancer staging, or because of nonspecific symptoms such as dizziness and headache. In some series, headache was the most common reason for imaging studies that led to the discovery of pituitary incidentalomas.1
With more patients undergoing computed tomography (CT) and MRI, more incidentalomas are being discovered. Incidentally discovered pituitary adenomas accounted for 12% of the pituitary tumors in a series of 353 consecutive patients with a presumptive diagnosis of pituitary tumor at one institution over a 14-year period.2 Pituitary masses other than adenomas are discussed later in this paper.
Microadenomas are common, macroadenomas less so
Autopsy studies have revealed pituitary microadenomas (ie, < 10 mm in greatest dimension) in 3% to 27% of patients with no history of pituitary disorders. Macroadenomas (10 mm or larger), on the other hand, are found in fewer than 0.5% of people.3,4 Recently, a study of MRI in 2,000 healthy adult volunteers, age 45 to 97 years, found pituitary macroadenomas in 0.3%.5
Hall et al6 found that 10% of relatively young (< 60 years old) healthy volunteers harbored a pituitary microadenoma on pituitary MRI, but none had a macroadenoma. In a meta-analysis by Ezzat and colleagues,3 adenomas of all sizes were found in 1% to 40% of imaging or postmortem studies (for an average of 16.7%), but macroadenomas were found in only 0.16% to 0.2% of the population.
Although the natural history of pituitary incidentalomas is not well characterized, the numbers suggest that microadenomas rarely grow into macroadenomas.7 Another possibility is that most macroadenomas cause symptoms and therefore come to clinical attention, and thus are not incidentalomas per se.
THE INITIAL EVALUATION: TWO QUESTIONS
The initial approach to a patient with a pituitary incidentaloma should be guided by two questions:
- Is the tumor hormonally active?
- Is it causing a mass effect (ie, is it exerting pressure on adjacent structures)?
IS THE TUMOR HORMONALLY ACTIVE?
A careful history and physical examination may reveal overlooked symptoms or signs of hypersecretion of a specific hormone, which can be evaluated in detail to establish the diagnosis. However, most patients with pituitary incidentalomas have no symptoms, and for them there is no real consensus about the optimal workup strategy.
Prolactin excess
King et al8 calculated that the serum prolactin level is the single most cost-effective screening test for hormonal activity in patients with incidentally discovered pituitary microadenomas. They also suggested, however, that it may be cost-effective to measure multiple hormones in very anxious patients, since a negative test may provide reassurance and improve quality of life.
One should be careful in interpreting elevated prolactin levels in patients with pituitary incidentalomas, since a number of medications (eg, metoclopramide [Reglan], verapamil [Calan], phenothiazines) and disorders (eg, hypothyroidism, cirrhosis, renal failure) can cause mild to moderate elevations of prolactin. In general, a prolactin level of more than 200 ng/mL is almost always diagnostic of prolactinomas. In our experience, a prolactin level above 100 ng/mL is almost always due to a prolactin-secreting pituitary adenoma, except during pregnancy and in some patients who receive antipsychotics or metoclopramide. For these patients, if it is clinically safe to hold or switch medications, retesting after a drug holiday may prove useful and diagnostic.
Growth hormone excess
Growth hormone hypersecretion has been reported in patients with pituitary tumors who have no clinical stigmata of acromegaly.9,10 Moreover, acral changes may not correlate with the metabolic consequences of growth hormone excess.11 In a study by Reincke et al,12 one of 18 patients with pituitary incidentalomas and no apparent acromegalic features had a growth hormone-secreting pituitary adenoma. For this reason, looking for so-called silent growth hormone hypersecretion may be warranted in patients with pituitary tumors, especially in those with macroadenomas.9
The best initial test for growth hormone hypersecretion is the measurement of insulin-like growth factor-1 (IGF-1).13 A normal age- and sex-adjusted IGF-1 level almost always rules out acromegaly.
Further hormonal evaluation
Further hormonal evaluation should be guided by the clinical picture.
Cortisol. In a patient with excess weight gain, central obesity, proximal myopathy, and skin manifestations that suggest hypercortisolism, appropriate initial tests would be a midnight salivary cortisol level, an overnight 1-mg (low-dose) dexamethasone suppression test, or a 24-hour urinary free cortisol level.
Thyroid hormones. Patients with symptoms that suggest hyperthyroidism should have their thyroid-stimulating hormone (TSH; thyrotropin) and free thyroxine (T4) levels measured to rule out a TSH-secreting pituitary adenoma, a very rare tumor.
Gonadotropins. Screening for a gonadotropin-secreting pituitary adenoma by measuring follicle-stimulating hormone, luteinizing hormone, and gonadotropin alpha subunit is not routinely indicated, since almost all of such tumors are clinically silent and generally come to clinical attention only because of a mass effect (see below).
IS THERE A MASS EFFECT?
Pituitary macroadenomas can also cause problems via a mass effect. Examples: hypopituitarism, visual field defects (by compressing the optic chiasm), cranial neuropathy (eg, diplopia, eyelid ptosis secondary to lateral extension of the tumor into a cavernous sinus), and headache.
Hypopituitarism
Hypopituitarism can range from deficiency of one pituitary hormone to the loss of all anterior pituitary hormones (panhypopituitarism).
Hypopituitarism from a mass effect is rare in patients with microadenomas, but one or more anterior pituitary hormone deficiencies are found in more than 30% of patients with a pituitary macroadenoma.3,12,14 With some exceptions, including pituitary apoplexy, the loss of pituitary hormone secretion is slowly progressive; symptoms tend to be nonspecific and often are not noticed at first.
Increased intrasellar pressure may play a role in the pathogenesis of hypopituitarism in patients with pituitary masses.15 Blood flow through the portal vessels is decreased, possibly resulting in diminished delivery of hypo-thalamic hormones to pituitary cells or leading to variable ischemia or necrosis of the normal gland, or both.
All patients with a pituitary macroadenoma should undergo a hormonal evaluation to look for pituitary hormone deficiency.
Growth hormone, gonadotropin deficiencies. In general, pituitary hormone deficiencies from an expanding pituitary tumor tend to begin with growth hormone or the gonadotropins (luteinizing hormone and follicle-stimulating hormone), or both.
Low serum testosterone levels in men (estradiol in women) along with normal or low follicle-stimulating hormone and luteinizing hormone levels are consistent with gonadotropin deficiency in men and amenorrheic premenopausal women.
Failure of the follicle-stimulating hormone and luteinizing hormone levels to rise after menopause is also consistent with gonadotropin deficiency. The presence of regular menses almost always indicates a normal gonadotropin axis. In women with irregular menstruation, hormonal evaluation can be challenging for evaluation of the gonadotropin axis and usually is not indicated.
Patients with deficiencies of two or more pituitary axes and low IGF-1 levels can be presumed to have growth hormone deficiency and usually do not need dynamic testing. But when testing is indicated, the growth hormone axis is best evaluated by dynamic testing, using either a growth hormone-releasing hormone/ arginine stimulation test or the insulin tolerance test.
Thyroid deficiencies. As the tumor expands, deficiencies of thyrotropin and adrenocorticotropic hormone (ACTH) secretion may follow those of growth hormone and gonadotropins. In our experience, the thyrotropin axis is usually affected before the corticotropin axis.
To evaluate the thyrotropin axis, the serum thyrotropin level should be measured along with the free thyroxine level or the free thyroxine index. A low free thyroxine level with a low or normal thyrotropin level is consistent with secondary hypothyroidism. It is inappropriate to measure thyrotropin without also measuring thyroxine in a patient with pituitary disorder, since a normal thyrotropin level in a patient with hypopituitarism is not uncommon.
Adrenal insufficiency. The ACTH stimulation test or an early morning (8 am) plasma cortisol level are both reasonable initial tests to evaluate the hypothalamic-pituitary-adrenal axis. An early morning cortisol level lower than 3 μg/dL confirms adrenal insufficiency, while a value higher than 15 μg/dL makes the diagnosis highly unlikely. Cortisol levels in the range of 3 to 15 μg/dL are indeterminate and should be further evaluated by an ACTH stimulation test, which can be performed anytime during the day.
The standard-dose ACTH stimulation test uses an intravenous or intramuscular injection of 250 μg of cosyntropin (Cortrosyn; ACTH 1–24). A normal response is a plasma cortisol concentration higher than 18 μg/dL at 30 minutes.
The sensitivity of the ACTH stimulation test in detecting mild, partial adrenal insufficiency is higher if a lower dose of cosyntropin is used (1 μg intravenously). However, the low-dose test has a higher false-positive rate. In most clinical situations, the 30-minute cortisol value during a standard-dose ACTH stimulation test has a diagnostic accuracy close to that of the low-dose ACTH stimulation test.16 Patients with recent-onset ACTH deficiency (eg, in pituitary apoplexy or within 2 to 4 weeks following pituitary surgery) may have a normal response to the ACTH stimulation test, since their adrenal glands have not undergone sufficient atrophy and still respond to ACTH stimulation.
The insulin tolerance test is considered the gold standard for evaluating the hypothalamic-pituitary-adrenal axis, but it needs to be performed by an experienced clinician and is usually not needed for everyday clinical practice.
Visual field defects
Visual field loss generally begins in the superior temporal fields, which explains why the patient may not notice it at first. Then, with continued growth and compression, vision loss extends into the inferior temporal fields, then into the nasal fields as a late effect.
Because the patient may not notice the visual field defect, formal visual field testing is warranted if the tumor compresses or abuts the optic chiasm. While bitemporal hemianopia is the classic manifestation of chiasmal compression, variable visual field defects may occur depending on which portion of the optic apparatus is involved.
Cranial neuropathy
Abnormal eye movements, which may cause diplopia, result from extension of a pituitary tumor into one or both cavernous sinuses. Compression of the third (occulomotor, the cranial nerve most often affected), fourth (trochlear), and sixth (abducens) cranial nerves leads to eye movement deviations as well as eyelid ptosis due to third nerve dysfunction. Cranial neuropathy most commonly occurs in the setting of pituitary apoplexy (see below) but may occur without it.
Headache
Headache can be associated with pituitary tumors, but the underlying pathophysiology remains uncertain. Possible mechanisms include structural causes such as dural stretching or cavernous sinus invasion.17 Other possible mechanisms are an increase in the intrasellar pressure and tumor activity.15,18 The link between headache and tumor activity is supported by the observation that headaches resolve in some patients with acromegaly shortly after they start taking somatostatin analogues.19
Migraine may be the most common type of headache reported in patients with pituitary adenomas; however, short-lasting unilateral neuralgiform headache with conjunctival injection and tearing (SUNCT) has also been reported.19
Of interest, there seems to be a strong association between pituitary-associated headache and a family history of headache.19 That said, headache is a common symptom in the general population, and establishing a cause-and-effect relationship prior to surgical removal of a pituitary tumor can be challenging. Approximately 50% of patients with headache who undergo an operation for a pituitary tumor have relief after surgery; however, 35% may not have relief, and up to 15% have a worsening of their headaches.19
OUR PATIENT’S HORMONAL EVALUATION
In the patient we described earlier, hormonal evaluation revealed the following:
- Prolactin 12.2 ng/mL (reference range 2–17.4)
- IGF-1 189 ng/mL (114–492)
- Thyrotropin 1.63 μU/mL (0.4–5.5)
- Free thyroxine index 9.5 μg/L (6–11)
- Maximum cortisol during a low-dose ACTH stimulation test 18.4 μg/dL. In short, all her test results were normal.
A formal visual field test was not performed, since the pituitary mass did not reach the optic chiasm (Figure 1).
ADENOMAS VS OTHER SELLAR MASSES
In some cases, it may be difficult to distinguish a nonadenomatous lesion from a nonfunctioning pituitary adenoma. However, several endocrine, radiographic, and neurologic features may help to differentiate pituitary tumors from other, less common sellar disorders.20
For instance, diabetes insipidus is extremely rare in patients with pituitary adenomas at presentation without significant suprasellar extension of the tumor. Therefore, its presence strongly suggests a nonpituitary cause such as hypophysitis, sarcoidosis, or a meta-static lesion.21
Some radiographic features that suggest sellar masses other than pituitary tumors include calcifications on CT in patients with craniopharyngiomas and meningiomas or a rapidly enlarging mass with lack of sellar enlargement (sellar remodeling), which suggests a metastatic lesion. While a dural tail sign (a linear enhanced structure or “tail” extending away from the tumor mass along the dural surface) may be seen with some meningiomas, peripheral enhancement of the dura is not specific for meningioma and may be seen with pituitary apoplexy as well.22,23
Cranial neuropathy is less common in patients with pituitary adenomas than in those with nonadenomatous masses (for example a metastasis or a meningioma), although the acute onset of cranial neuropathy often accompanies a hemorrhagic infarction of a preexisting pituitary adenoma (pituitary apoplexy).20
OUR RECOMMENDATIONS
Our approach to a patient with a pituitary incidentaloma is summarized in Figure 4.
If the tumor is hormonally active
Prolactinoma is the exception. For this tumor, dopamine agonists can resolve symptoms and shrink the tumor in most cases. Even in patients with a visual field defect associated with a macroprolactinoma, vision usually improves within days after starting a dopamine agonist, before the tumor has observably shrunk. However, a follow-up visual field test is necessary 2 to 6 weeks after starting therapy to establish that the tumor is responding to therapy; if the tumor does not respond, surgery may be necessary.
If the tumor is hormonally inactive
If the tumor is hormonally inactive, its further evaluation depends on its size and whether there is a mass effect. In patients with a nonfunctioning pituitary macroadenoma, a comprehensive hormonal evaluation for hypopituitarism should be done. Patients with a visual field defect or cranial neuropathy should undergo surgical tumor resection. If there is no mass effect, observation may be an acceptable strategy. We, and others,1,25 recommend surgery for most patients with pituitary macroadenomas abutting the optic chiasm.
If the tumor is small
If the tumor is small (ie, a microadenoma), the risk of its growing is low. Three small studies followed such patients prospectively and found a 0 to 14% risk of tumor enlargement over a mean follow-up period of 1.8 to 6.7 years.12,25,26 While there is no consensus about how soon to follow up patients with nonfunctioning pituitary microadenomas, we obtain a follow-up MRI study in 1 year, with no further routine imaging if the tumor has remained stable, unless the patient develops symptoms or signs suggesting a mass effect.
If the tumor is large
If the tumor is large (ie, a macroadenoma), the risk of further growth is expected to be higher, since the tumor has already shown the propensity to grow. In the same three series discussed above, the risk of tumor growth for a pituitary macroadenomas was about 30% over the mean follow-up of 1.8 to 6.7 years.12,25,26
Furthermore, several recent studies have suggested a higher propensity to grow and to cause symptoms and signs than previously thought. For example, Karavitaki et al7 studied 24 patients who had nonfunctional macroadenomas and found that the 48-month probability of enlargement was 44%; of this group, 57% showed new or worsening visual field defects, and an additional 21% showed new chiasmatic compression without vision loss. Similarly, Arita and colleagues27 found that 21 (50%) of 42 nonfunctional adenomas (mean size 18.3 ± 7 mm) increased by at least 10% over an average of 32 months after the initial evaluation. Ten patients became symptomatic over a mean of about 5 years, with 4 of these 10 (9.5% of the entire cohort) suffering symptomatic pituitary apoplexy. Therefore, one may argue for surgery (especially in young patients) for pituitary macroadenomas even in the absence of mass effect.
We would obtain a follow-up MRI study at 6 months, then yearly for 5 years, and then every 2 to 3 years if the tumor is stable. Surgery would be indicated if there is evidence of tumor growth or a mass effect.
While tumor growth has been found to be independent of age in some studies,27 others have found longer tumor doubling time in patients older than 60 years.28
The risk of pituitary apoplexy
Pituitary apoplexy results from a hemorrhagic infarction of the tumor and manifests clinically as the sudden onset of severe headache, nausea, vomiting, vision loss, and cranial nerve palsies. While most cases of pituitary apoplexy are spontaneous, precipitating factors may include head injury, anticoagulant therapy, dopamine agonists, radiation therapy, or dynamic endocrine tests.29
It is important to educate patients and their families about the symptoms of pituitary apoplexy, especially patients with pituitary macroadenomas. If the condition is unrecognized and untreated, patients can develop hypotension and shock secondary to adrenal insufficiency, as well as irreversible vision loss or diplopia.
Surgery is generally recommended in cases of progressive vision loss or cranial neuropathy, preferably within 24 or 48 hours of onset if feasible, to minimize the risk of a permanent neurologic deficit.
Clinically significant pituitary apoplexy is rare in patients with pituitary microadenomas. In the study by Arita et al,27 the risk of pituitary apoplexy during 5 years of follow-up was 9.5%, and all of the tumors involved were macroadenomas. This rate is higher than in some other studies, in which the risk of apoplexy ranged from 0.4% to 7% during a mean follow-up of 2 to 6 years.1,25,30
CASE FOLLOW-UP
Since our patient had no evidence of hormonal hypersecretion or mass effect and no hypopituitarism, we asked her to return in 6 months. A repeat MRI study showed the tumor to be stable, with no evidence of growth. The patient was scheduled for a return visit in 1 year.
- Sanno N, Oyama K, Tahara S, Teramoto A, Kato Y. A survey of pituitary incidentaloma in Japan. Eur J Endocrinol 2003; 149:123–127.
- Gsponer J, De Tribolet N, Déruaz JP, et al. Diagnosis, treatment, and outcome of pituitary tumors and other abnormal intrasellar masses. Retrospective analysis of 353 patients. Medicine (Baltimore) 1999; 78:236–269.
- Ezzat S, Asa SL, Couldwell WT, et al. The prevalence of pituitary adenomas: a systematic review. Cancer 2004; 101:613–619.
- Molitch ME, Russell EJ. The pituitary “incidentaloma.” Ann Intern Med 1990; 112:925–931.
- Vernooij MW, Ikram MA, Tanghe HL, et al. Incidental findings on brain MRI in the general population. N Engl J Med 2007; 357:1821–1828.
- Hall WA, Luciano MG, Doppman JL, Patronas NJ, Oldfield EH. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Intern Med 1994; 120:817–820.
- Karavitaki N, Collison K, Halliday J, et al. What is the natural history of nonoperated nonfunctioning pituitary adenomas? Clin Endocrinol (Oxf) 2007; 67:938–943.
- King JT, Justice AC, Aron DC. Management of incidental pituitary microadenomas: a cost-effectiveness analysis. J Clin Endocrinol Metab 1997; 82:3625–3632.
- Klibanski A, Zervas NT, Kovacs K, Ridgway EC. Clinically silent hypersecretion of growth hormone in patients with pituitary tumors. J Neurosurg 1987; 66:806–811.
- Trouillas J, Sassolas G, Loras B, et al. Somatotropic adenomas without acromegaly. Pathol Res Pract 1991; 187:943–949.
- Cryer PE, Daughaday WH. Regulation of growth hormone secretion in acromegaly. J Clin Endocrinol Metab 1969; 29:386–393.
- Reincke M, Allolio B, Saeger W, Menzel J, Winkelmann W. The ‘incidentaloma’ of the pituitary gland. Is neurosurgery required? JAMA 1990; 263:2772–2776.
- Giustina A, Barkan A, Casanueva FF, et al. Criteria for cure of acromegaly: a consensus statement. J Clin Endocrinol Metab 2000; 85:526–529.
- Nammour GM, Ybarra J, Naheedy MH, Romeo JH, Aron DC. Incidental pituitary macroadenoma: a population-based study. Am J Med Sci 1997; 314:287–291.
- Arafah BM, Prunty D, Ybarra J, Hlavin ML, Selman WR. The dominant role of increased intrasellar pressure in the pathogenesis of hypopituitarism, hyperprolactinemia, and headaches in patients with pituitary adenomas. J Clin Endocrinol Metab 2000; 85:1789–1793.
- Mayenknecht J, Diederich S, Bahr V, Plockinger U, Oelkers W. Comparison of low and high dose corticotropin stimulation tests in patients with pituitary disease. J Clin Endocrinol Metab 1998; 83:1558–1562.
- Forsyth PA, Posner JB. Headaches in patients with brain tumors: a study of 111 patients. Neurology 1993; 43:1678–1683.
- Abe T, Matsumoto K, Kuwazawa J, Toyoda I, Sasaki K. Headache associated with pituitary adenomas. Headache 1998; 38:782–786.
- Levy MJ, Matharu MS, Meeran K, Powell M, Goadsby PJ. The clinical characteristics of headache in patients with pituitary tumours. Brain 2005; 128:1921–1930.
- Freda PU, Post KD. Differential diagnosis of sellar masses. Endocrinol Metab Clin North Am 1999; 28:81–117.
- Gopan T, Toms SA, Prayson RA, Suh JH, Hamrahian AH, Weil RJ. Symptomatic pituitary metastases from renal cell carcinoma. Pituitary 2007; 10:251–259.
- Moore AF, Grinspoon SK. A dural tale. J Clin Endocrinol Metab 2007; 92:3367–3368.
- Smirniotopoulos JG, Murphy FM, Rushing EJ, Rees JH, Schroeder JW. Patterns of contrast enhancement in the brain and meninges. Radiographics 2007; 27:525–551.
- Chanson P, Daujat F, Young J, et al. Normal pituitary hypertrophy as a frequent cause of pituitary incidentaloma: a follow-up study. J Clin Endocrinol Metab 2001; 86:3009–3015.
- Donovan LE, Corenblum B. The natural history of the pituitary incidentaloma. Arch Intern Med 1995; 155:181–183.
- Feldkamp J, Santen R, Harms E, Aulich A, Modder U, Scherbaum WA. Incidentally discovered pituitary lesions: high frequency of macroadenomas and hormone-secreting adenomas—results of a prospective study. Clin Endocrinol (Oxf) 1999; 51:109–113.
- Arita K, Tominaga A, Sugiyama K, et al. Natural course of incidentally found nonfunctioning pituitary adenoma, with special reference to pituitary apoplexy during follow-up examination. J Neurosurg 2006; 104:884–891.
- Tanaka Y, Hongo K, Tada T, Sakai K, Kakizawa Y, Kobayashi S. Growth pattern and rate in residual nonfunctioning pituitary adenomas: correlations among tumor volume doubling time, patient age, and MIB-1 index. J Neurosurg 2003; 98:359–365.
- Biousse V, Newman NJ, Oyesiku NM. Precipitating factors in pituitary apoplexy. J Neurol Neurosurg Psychiatry 2001; 71:542–545.
- Nishizawa S, Ohta S, Yokoyama T, Uemura K. Therapeutic strategy for incidentally found pituitary tumors (“pituitary incidentalomas”). Neurosurgery 1998; 43:1344–1348.
- Sanno N, Oyama K, Tahara S, Teramoto A, Kato Y. A survey of pituitary incidentaloma in Japan. Eur J Endocrinol 2003; 149:123–127.
- Gsponer J, De Tribolet N, Déruaz JP, et al. Diagnosis, treatment, and outcome of pituitary tumors and other abnormal intrasellar masses. Retrospective analysis of 353 patients. Medicine (Baltimore) 1999; 78:236–269.
- Ezzat S, Asa SL, Couldwell WT, et al. The prevalence of pituitary adenomas: a systematic review. Cancer 2004; 101:613–619.
- Molitch ME, Russell EJ. The pituitary “incidentaloma.” Ann Intern Med 1990; 112:925–931.
- Vernooij MW, Ikram MA, Tanghe HL, et al. Incidental findings on brain MRI in the general population. N Engl J Med 2007; 357:1821–1828.
- Hall WA, Luciano MG, Doppman JL, Patronas NJ, Oldfield EH. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Intern Med 1994; 120:817–820.
- Karavitaki N, Collison K, Halliday J, et al. What is the natural history of nonoperated nonfunctioning pituitary adenomas? Clin Endocrinol (Oxf) 2007; 67:938–943.
- King JT, Justice AC, Aron DC. Management of incidental pituitary microadenomas: a cost-effectiveness analysis. J Clin Endocrinol Metab 1997; 82:3625–3632.
- Klibanski A, Zervas NT, Kovacs K, Ridgway EC. Clinically silent hypersecretion of growth hormone in patients with pituitary tumors. J Neurosurg 1987; 66:806–811.
- Trouillas J, Sassolas G, Loras B, et al. Somatotropic adenomas without acromegaly. Pathol Res Pract 1991; 187:943–949.
- Cryer PE, Daughaday WH. Regulation of growth hormone secretion in acromegaly. J Clin Endocrinol Metab 1969; 29:386–393.
- Reincke M, Allolio B, Saeger W, Menzel J, Winkelmann W. The ‘incidentaloma’ of the pituitary gland. Is neurosurgery required? JAMA 1990; 263:2772–2776.
- Giustina A, Barkan A, Casanueva FF, et al. Criteria for cure of acromegaly: a consensus statement. J Clin Endocrinol Metab 2000; 85:526–529.
- Nammour GM, Ybarra J, Naheedy MH, Romeo JH, Aron DC. Incidental pituitary macroadenoma: a population-based study. Am J Med Sci 1997; 314:287–291.
- Arafah BM, Prunty D, Ybarra J, Hlavin ML, Selman WR. The dominant role of increased intrasellar pressure in the pathogenesis of hypopituitarism, hyperprolactinemia, and headaches in patients with pituitary adenomas. J Clin Endocrinol Metab 2000; 85:1789–1793.
- Mayenknecht J, Diederich S, Bahr V, Plockinger U, Oelkers W. Comparison of low and high dose corticotropin stimulation tests in patients with pituitary disease. J Clin Endocrinol Metab 1998; 83:1558–1562.
- Forsyth PA, Posner JB. Headaches in patients with brain tumors: a study of 111 patients. Neurology 1993; 43:1678–1683.
- Abe T, Matsumoto K, Kuwazawa J, Toyoda I, Sasaki K. Headache associated with pituitary adenomas. Headache 1998; 38:782–786.
- Levy MJ, Matharu MS, Meeran K, Powell M, Goadsby PJ. The clinical characteristics of headache in patients with pituitary tumours. Brain 2005; 128:1921–1930.
- Freda PU, Post KD. Differential diagnosis of sellar masses. Endocrinol Metab Clin North Am 1999; 28:81–117.
- Gopan T, Toms SA, Prayson RA, Suh JH, Hamrahian AH, Weil RJ. Symptomatic pituitary metastases from renal cell carcinoma. Pituitary 2007; 10:251–259.
- Moore AF, Grinspoon SK. A dural tale. J Clin Endocrinol Metab 2007; 92:3367–3368.
- Smirniotopoulos JG, Murphy FM, Rushing EJ, Rees JH, Schroeder JW. Patterns of contrast enhancement in the brain and meninges. Radiographics 2007; 27:525–551.
- Chanson P, Daujat F, Young J, et al. Normal pituitary hypertrophy as a frequent cause of pituitary incidentaloma: a follow-up study. J Clin Endocrinol Metab 2001; 86:3009–3015.
- Donovan LE, Corenblum B. The natural history of the pituitary incidentaloma. Arch Intern Med 1995; 155:181–183.
- Feldkamp J, Santen R, Harms E, Aulich A, Modder U, Scherbaum WA. Incidentally discovered pituitary lesions: high frequency of macroadenomas and hormone-secreting adenomas—results of a prospective study. Clin Endocrinol (Oxf) 1999; 51:109–113.
- Arita K, Tominaga A, Sugiyama K, et al. Natural course of incidentally found nonfunctioning pituitary adenoma, with special reference to pituitary apoplexy during follow-up examination. J Neurosurg 2006; 104:884–891.
- Tanaka Y, Hongo K, Tada T, Sakai K, Kakizawa Y, Kobayashi S. Growth pattern and rate in residual nonfunctioning pituitary adenomas: correlations among tumor volume doubling time, patient age, and MIB-1 index. J Neurosurg 2003; 98:359–365.
- Biousse V, Newman NJ, Oyesiku NM. Precipitating factors in pituitary apoplexy. J Neurol Neurosurg Psychiatry 2001; 71:542–545.
- Nishizawa S, Ohta S, Yokoyama T, Uemura K. Therapeutic strategy for incidentally found pituitary tumors (“pituitary incidentalomas”). Neurosurgery 1998; 43:1344–1348.
KEY POINTS
- Two key questions that must be answered when a pituitary incidentaloma is discovered are whether it is hormonally active and whether it is causing a mass effect (eg, a visual field defect due to pressure on the optic chiasm).
- Incidentalomas that are not hormonally active and that are not causing a mass effect can generally be managed by watchful waiting.
- Hormonally active prolactin-secreting tumors can be treated with dopamine agonists. Other hormonally active tumors and those that are causing a mass effect should be surgically removed.
- The risks of further tumor growth and of pituitary apoplexy are higher in tumors that are larger when discovered.