This review highlights important recent publications in the area of neuroscience and heart-brain medicine. Abnormalities of regulation of the circulation by catecholamine systems figure as a general theme of the topics highlighted. These topics, which are reviewed in turn below, are (1) mechanisms of cardiac sympathetic denervation in Parkinson disease (PD), (2) cytoplasmic monoamine metabolites as autotoxins, and (3) the validity of power spectral analysis of heart rate variability to indicate cardiac sympathetic tone.
MECHANISMS OF CARDIAC SYMPATHETIC DENERVATION IN PARKINSON DISEASE
The movement disorder component of PD is well recognized as resulting from loss of dopaminergic neurons in the nigrostriatal system of the brain. The finding of low myocardial 6-[18F]fluorodopamine–derived radioactivity by positron emission tomography provided the first neuroimaging evidence for loss of catecholaminergic neurons outside the brain in PD.1 Many reports using 123I-metaiodobenzylguanidine scanning have concurred with this finding. Beginning in the early 2000s, post-mortem neuropathologic studies demonstrated virtually absent immunoreactivity for tyrosine hydroxylase, the rate-limiting enzyme in norepinephrine biosynthesis, in epicardial nerves in PD.2,3 These results provided clues to the mechanism of autonomic dysfunction in PD, a prominent nonmotor manifestation of the disease.
With kind permission from Springer Science+Business Media: Acta Neuropathologica,
Figure 1. Tyrosine hydroxylase immunoreactivity (THir) in epicardial nerve from (A) a control subject and (B) a patient with familial Parkinson disease due to duplication of the gene encoding alpha-synuclein (PARK4).6Alpha-synuclein is a key protein in the pathogenesis of PD. It is abundant in Lewy bodies and Lewy neurites, and mutations or multiplications of the gene that encodes it cause rare inherited forms of PD. In 2001 we reported evidence for cardiac sympathetic denervation, neurogenic orthostatic hypotension, and baroreflex failure in familial PD from mutation of the gene encoding alpha-synuclein.4 Subsequently we reported analogous denervation in familial PD from triplication of the normal gene.5 This past year Orimo’s group in Tokyo provided the first pathological confirmation of cardiac sympathetic denervation in familial PD from inherited alpha-synucleinopathy, based on severely decreased epicardial neuronal tyrosine hydroxylase immunoreactivity (Figure 1).6 In contrast, patients with familial PD from parkin gene mutation, which is not thought to be a Lewy body disease, have been found to have normal cardiac 123I-metaiodobenzyl-guanidine–derived radioactivity and normal epicardial neuronal tyrosine hydroxylase immunoreactivity.7 These findings establish a link between alpha-synucleinopathy and cardiac sympathetic denervation.
Some individuals who die without clinical parkinsonism have Lewy bodies detected pathologically. Growing evidence shows that incidental Lewy body disease represents early, presymptomatic PD.8 Orimo’s group therefore studied cardiac tissues and paravertebral sympathetic ganglia from patients with incidental Lewy body disease.9 Postmortem tissues were likewise obtained from comparison subjects with multiple system atrophy and from control subjects. Immunohistochemical analyses were performed using antibodies against tyrosine hydroxylase, phosphorylated neurofilament as a marker of axons, and phosphorylated alpha-synuclein as a marker of abnormal alpha-synuclein deposits. Key findings from this study9 were as follows:
Reprinted from Brain (Orimo S, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008; 131:642–650) by permission of Oxford University Press.
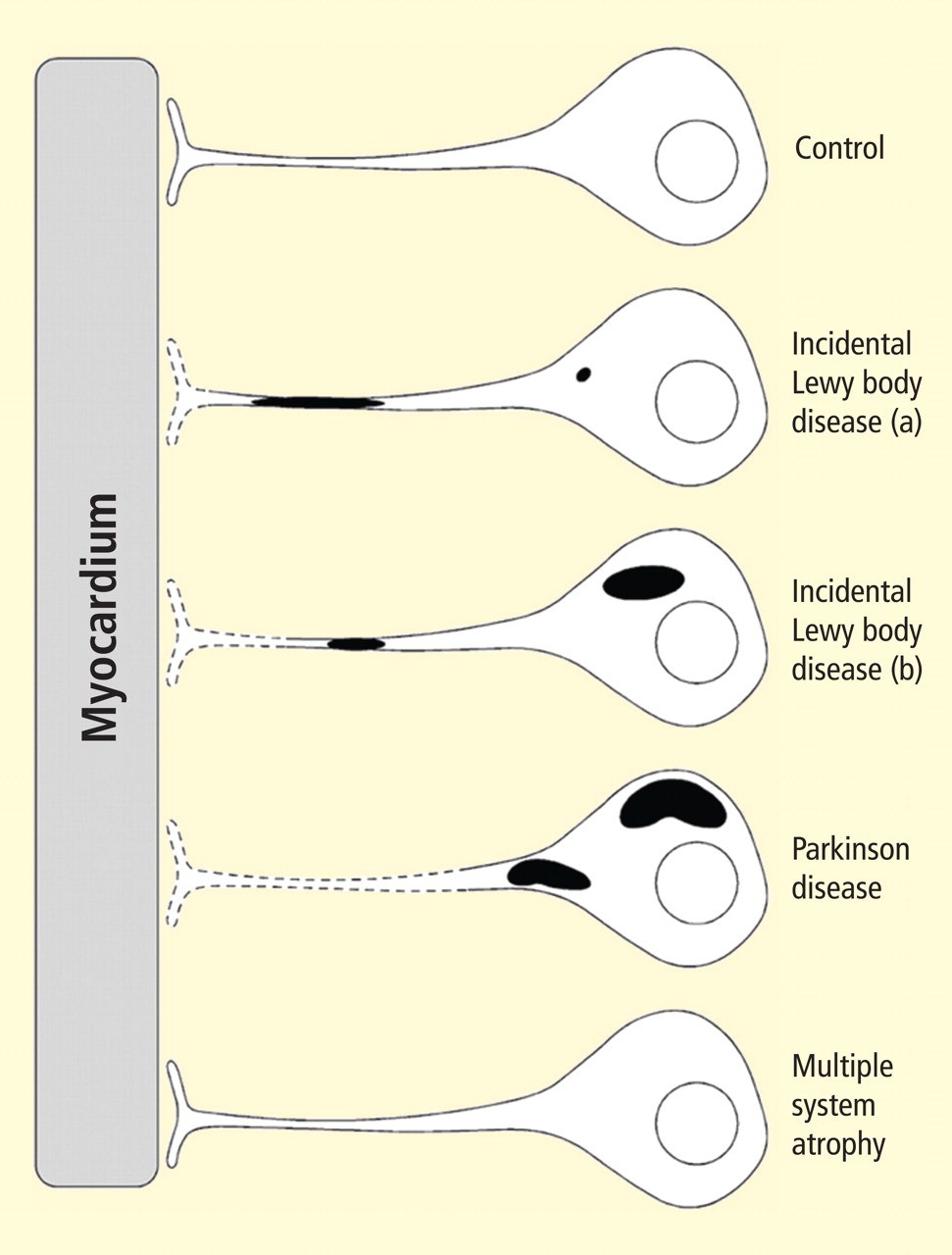
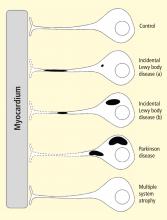
Figure 2. Concept diagram of the pathogenetic sequence of cardiac sympathetic denervation. In incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons (a), alpha-synuclein aggregates (black shading) accumulate abundantly in the distal axons but sparsely in the paravertebral sympathetic ganglia. In contrast, in incidental Lewy body disease with decreased THir axons (b), alpha-synuclein aggregates diminish in the distal axons but increase in the paravertebral sympathetic ganglia. In Parkinson disease, alpha-synuclein aggregates disappear in the distal axons and accumulate much more abundantly in the paravertebral sympathetic ganglia. In multiple system atrophy, alpha-synuclein aggregates are generally not observed (as in controls), with a few exceptions. Dotted lines indicate degeneration of THir axons.9Alpha-synuclein aggregates in distal epicardial nerve fascicles were more abundant in incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons than in incidental Lewy body disease with decreased THir axons (Figure 2).
Alpha-synuclein aggregates in the epicardial nerve fibers were closely related to the disappearance of THir axons.
In incidental Lewy body disease with preserved THir axons, alpha-synuclein aggregates were consistently more abundant in the epicardial nerves than in the paravertebral sympathetic ganglia (Figure 2).
Distally dominant accumulation of alpha-synuclein aggregates was reversed in incidental Lewy body disease with decreased THir axons and in PD, because both conditions involve fewer alpha-synuclein aggregates in axons and more abundant aggregates in the paravertebral sympathetic ganglia (Figure 2).
Thus, accumulation of alpha-synuclein aggregates in distal cardiac sympathetic axons precedes aggregation in neuronal somata or ganglionic neurites, heralding centripetal degeneration of cardiac sympathetic nerves in PD. This chronological and dynamic relationship between alpha-synuclein aggregation and distally dominant degeneration of cardiac noradrenergic nerves may represent the pathological mechanism behind a common degenerative process in PD.
In conclusion, cardiac noradrenergic denervation in Lewy body diseases, even in early stages, accounts for reduced cardiac uptake of 123I-metaiodobenzylguanidine and 6-[18F]fluorodopamine in PD. Alpha-synuclein aggregation appears to be intimately involved in the cardiac noradrenergic denervation that attends Lewy body diseases. The pathogenetic process seems to proceed in a centripetal, retrograde direction.
CYTOPLASMIC MONOAMINE METABOLITES AS AUTOTOXINS
Current concepts about mechanisms of PD emphasize pathologic alpha-synuclein accumulation, oxidative injury, impaired proteasomal or mitochondrial functions, neuroinflammation, or abnormal kinase signaling. These concepts do not explain relatively selective nigrostriatal dopaminergic and cardiac noradrenergic denervation in PD.
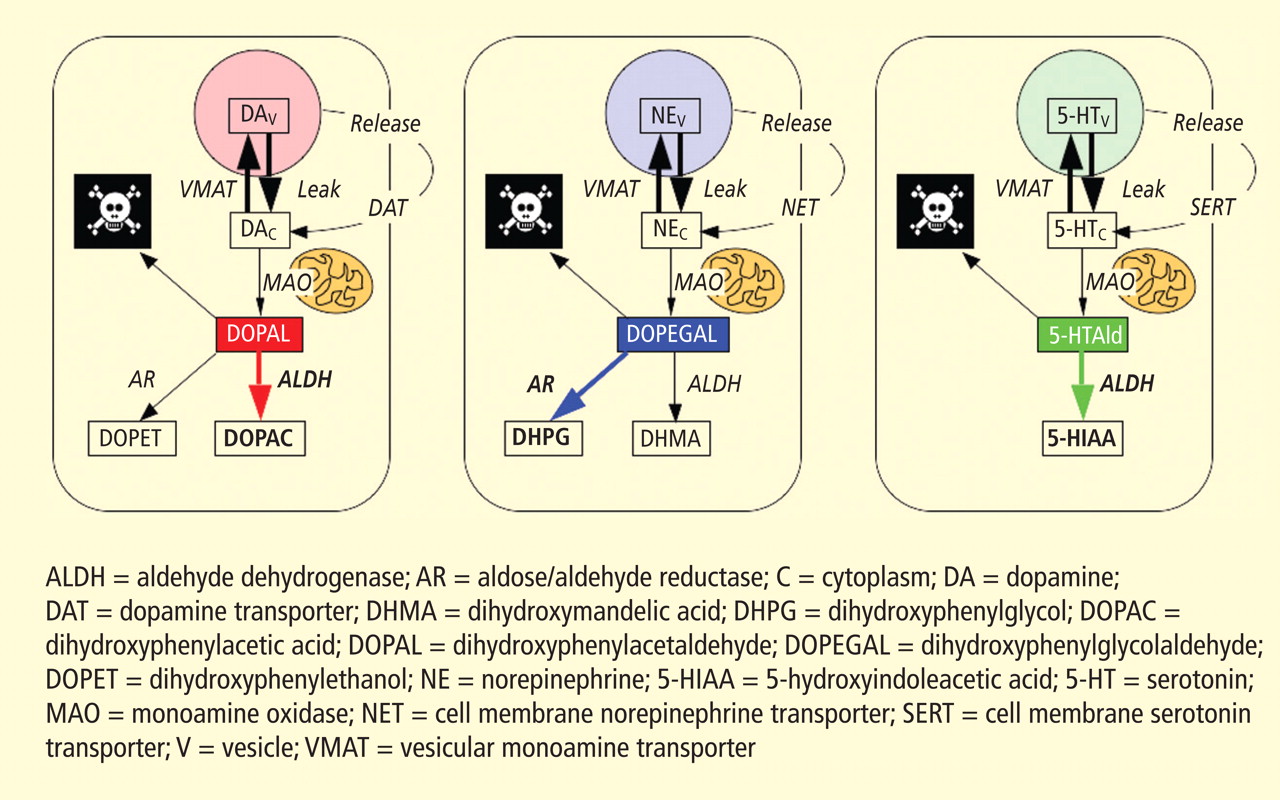
Figure 3. According to the monoamine aldehyde hypothesis, interference with the vesicular recycling of cytoplasmic monoamines (dopamine [DA], norepinephrine [NE], and serotonin [5-HT]) augments formation of toxic aldehydes. For instance, DA that leaks from vesicles (V) into the cytoplasm (C) or that is taken up via the cell membrane DA transporter (DAT) and escapes vesicular reuptake via the vesicular monoamine transporter (VMAT) is subject to oxidative deamination catalyzed by monoamine oxidase (MAO) to form the catecholaldehyde DOPAL, which is toxic. DOPAL is detoxified by ALDH to form DOPAC, the major metabolic route, or by AR to form DOPET, the minor metabolic route. Analogously, NE is converted to DOPEGAL, and 5-HT is converted to 5-HT-aldehyde (5-HTAld).
A potential explanation is that cytoplasmic catecholamine metabolites are autotoxins (Figure 3). The mechanisms of autotoxicity include spontaneous auto-oxidation, to form quinones and chromes leading to increased production of reactive oxygen species, and enzymatic oxidation.
Catecholamines in the neuronal cytoplasm undergo enzymatic oxidative deamination to form catecholaldehydes (dihydroxyphenylacetaldehyde [DOPAL] from dopamine), which are cytotoxic, as predicted by Blaschko more than a half century ago.10 DOPAL is detoxified mainly by aldehyde dehydrogenase (ALDH). In the substantia nigra, aldehyde dehydrogenase 1A1 (ALDH1A1) is the main isoform of ALDH, and postmortem studies have noted decreased nigral ALDH1A1 gene expression11,12 and protein content13 in PD patients.
All neurons express alpha-synuclein. Current concepts about mechanisms also do not explain the relatively selective aggregation of alpha-synuclein in catecholaminergic neurons. Alpha-synuclein appears to play a role in the cycling of catecholamines across vesicular and cell membranes.14
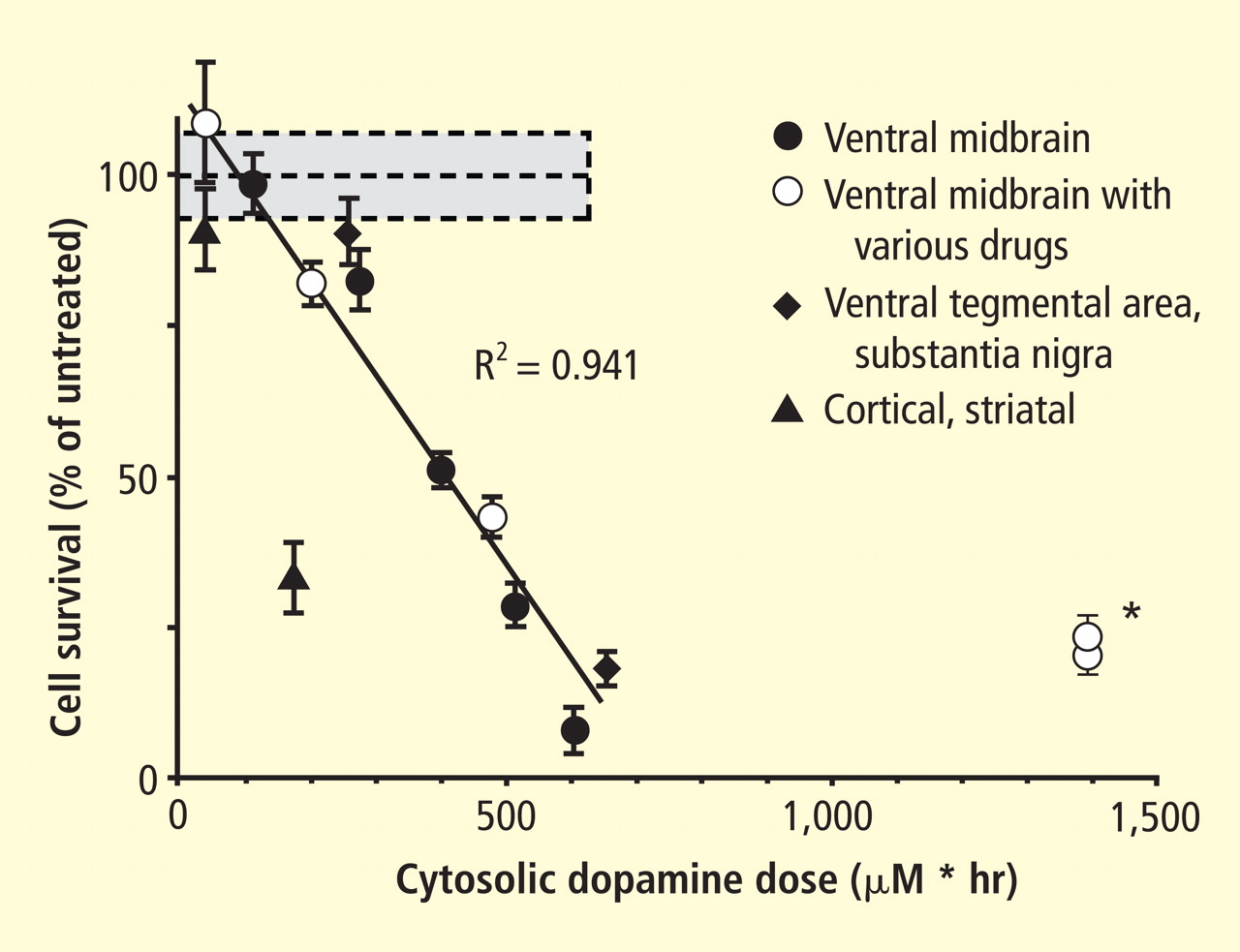
Figure 4. Cell survival and cytoplasmic dopamine are inversely related, according to a murine model by Mosharov et al.16 Graph shows the dependence of cell survival under l-dopa–induced stress on the cytoplasmic dopamine (DAcyt) dose in mouse neurons. The DAcyt dose was estimated as: [DAcyt]×TExposure = [DAcyt]×Ln([L-dopa]/K0.5)/k,where [DAcyt] is the concentration of cytosolic DA in cells treated with a saturating level (> 50 μM) of l-dopa for 1 hour, where [l-dopa] is the initial drug concentration, and where K0.5 = 9.7 μM and k = 0.15 hr−1 are the kinetic constants. TExposure approximates the time during which extracellular l-dopa remained higher than K0.5. The data points are (from left to right): filled circles—ventral midbrain cultures treated with 25, 100, 250, 500, and 1,000 μM l-dopa alone; open circles—ventral midbrain neurons treated with 250 μM l-dopa in the presence of benserazide, methamphetamine, reserpine, pargyline, and pargyline reserpine; diamonds—ventral tegmental area and substantia nigra neurons; triangles—striatal and cortical neurons treated with 250 μM l-dopa. Dotted lines and shaded boxes represent mean ± SEM in untreated cells. The solid line is the linear fit of all data points, excluding striatal and cortical neurons and the two data points indicated by the asterisk. Treatments to the right of this line are neuroprotective, as the same level of cell death is achieved with higher DAcyt doses; treatments to the left of this line are more susceptible to DAcyt stress.In the past year, a few important studies have been published related to autotoxicity of cytoplasmic catecholamine metabolites and to pathogenic interactions with alpha-synuclein. In 2006, Mosharov et al reported that alpha-synuclein overexpression increases cytoplasmic dopamine concentrations in rat pheochromocytoma PC-12 cells.15 Recently, the same group, using intracellular patch electrochemistry, directly measured cytoplasmic dopamine in cultured midbrain neurons and found that increases in dopamine and its metabolites are neurotoxic, whereas manipulations that reduce cytoplasmic dopamine are neuroprotective (Figure 4).16 Levodopa (l-dopa) increased cytoplasmic dopamine more in substantia nigra neurons than in ventral tegmental neurons, suggesting that this difference might help explain the greater susceptibility of nigral neurons to the pathogenetic process. The greater buildup of cytoplasmic dopamine seemed to depend on dihydropyridine-sensitive calcium (Ca2+) channels. Finally, dopaminergic neurons lacking alpha-synuclein were resistant to l-dopa–induced cell death. These findings led the authors to propose a “multiple-hit” model (Figure 5) in which interactions between intracellular ionized calcium, cytoplasmic dopamine, and alpha-synuclein underlie susceptibility of nigral neurons in PD.16
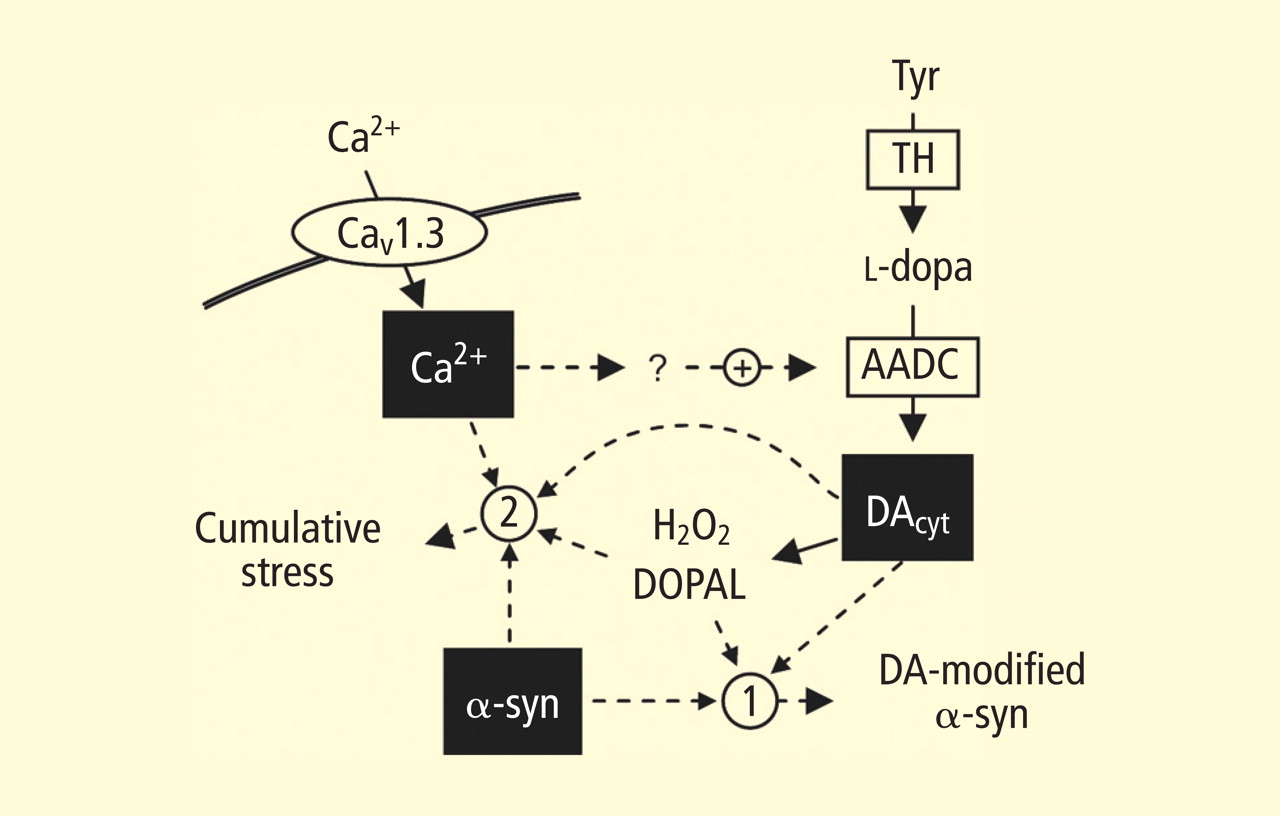
Figure 5. The “multiple-hit” model of Parkinson disease pathogenesis,16 which holds that neurotoxicity is a result of multiple factors, including the presence of alpha-synuclein (α-syn), elevation of cytoplasmic calcium (Ca2+), and buildup of cytoplasmic dopamine (DAcyt) and its metabolites. Nonexclusive toxic steps may result from (1) mechanisms that require direct interaction between DA or its metabolites with α-syn, such as DA-modified stabilization of α-syn protofibrils or inhibition of chaperone-mediated autophagy, or (2) cumulative damage from multiple independent sources. Reducing the levels of any of the three players provides neuroprotection. (AADC = aromatic l-amino acid decarboxylase; DOPAL = dihydroxyphenylacetaldehyde; TH = tyrosine hydroxylase)Burke et al added a potentially important clue, demonstrating that DOPAL potently oligomerizes and aggregates alpha-synuclein.17 This finding introduces the possibility of multiple pathogenetic positive feedback loops.
Under resting conditions, most catecholamine turnover results from leakage from vesicular stores into the cytoplasm and subsequent oxidative deamination by monoamine oxidase. Ordinarily, however, catecholamines in the cytoplasm are efficiently recycled back into the vesicles via the type 2 vesicular monoamine transporter (VMAT-2). Accordingly, interference with VMAT functions would be expected to tend to build up cytoplasmic catecholamines, with potentially cytotoxic consequences. In 2007, Caudle et al reported that mice with severely decreased VMAT-2 have aging-associated decreases in striatal dopamine that begin in the terminal fields, alpha-synuclein deposition in substantia nigra neurons, and l-dopa–responsive behavioral deficits.18 More recently the same group noted nonmotor signs associated with PD in VMAT-2–deficient mice, such as anosmia, gastrointestinal hypomotility, sleep disturbances, anxiety, and depression.19 Since VMAT-2 serves to recycle not only dopamine but also norepinephrine and serotonin, this single abnormality could help explain loss of all three types of monoaminergic neurons in PD.
Finally, Pena-Silva et al recently tested whether serotonin induces oxidative stress in human heart valves.20 They showed that in heart valves from explanted human hearts not used for transplantation, incubation of homogenates of cardiac valves and blood vessels with serotonin increased generation of the superoxide free radical. Inhibitors of monoamine oxidase prevented this effect. Dopamine also increased superoxide levels in heart valves, and this effect was also attenuated by monoamine oxidase inhibition. These findings fit with the concept that the aldehydes produced by the action of monoamine oxidase on cytoplasmic monoamines generate toxic free radicals.
VALIDITY OF POWER SPECTRAL ANALYSIS OF HEART RATE VARIABILITY TO INDICATE CARDIAC SYMPATHETIC TONE
Power spectral analysis of heart rate variability is simple, relatively inexpensive, noninvasive, and widely used to indicate cardiac sympathetic “tone” or sympathovagal “balance.” Almost 2,000 studies to date have used this modality. Relatively increased cardiac sympathetic tone, reflected by low-frequency (LF) power or the ratio of LF power to high-frequency (HF) power, is an adverse prognostic sign in a variety of conditions. Nevertheless, the validity of LF power, or the LF:HF ratio, as an index of cardiac sympathetic tone remains unsettled.
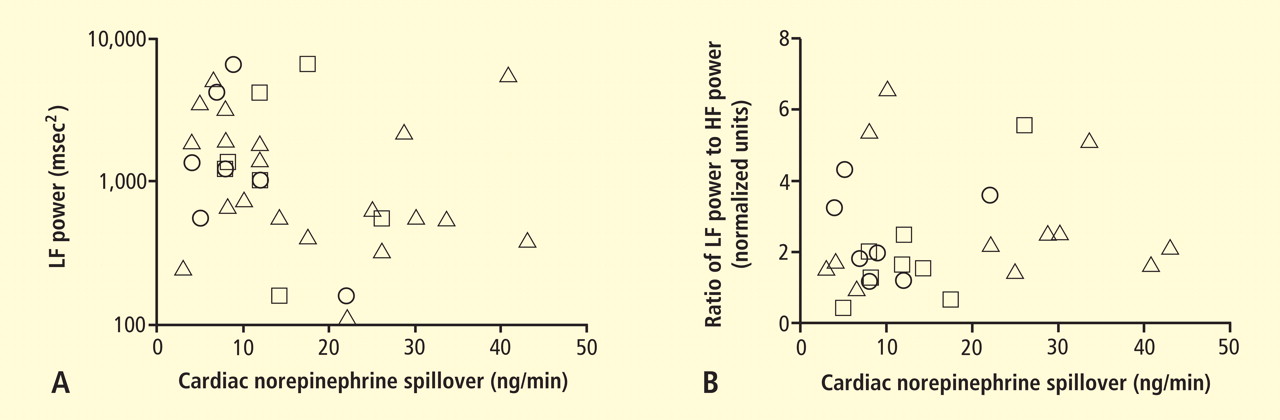
In 2007 we assessed the validity of power spectral analysis rather directly, by taking advantage of our ability to delineate cardiac sympathetic innervation. We compared LF power in patients with cardiac sympathetic denervation, indicated by low myocardial levels of 6-[18F]fluorodopamine–derived radioactivity or low rates of norepinephrine entry into coronary sinus plasma (cardiac norepinephrine spillover), with values in patients with intact innervation. LF power was unrelated to myocardial 6-[18F]fluorodopamine–derived radioactivity or cardiac norepinephrine spillover, but it was related to baroreflex-cardiovagal gain. Patients with a low baroreflex-cardiovagal gain had low LF power, regardless of cardiac innervation. From these findings we concluded that LF power reflects baroreflex function, not cardiac sympathetic innervation.21
Figure 6. Relationships of heart rate variability indices with cardiac norepinephrine spillover. Graphs show time and frequency domain heart rate variability measures (LF = low frequency; HF = high frequency) versus cardiac norepinephrine in healthy subjects (squares) and patients with major depression (triangles) and panic disorder (circles).22 Recently Baumert et al also examined the relationship between indices from power spectral analysis of heart rate variability and cardiac norepinephrine spillover.22 They found, as we did, that none of the standard heart rate variability parameters was correlated with cardiac norepinephrine spillover (Figure 6). The same group reported a positive correlation between the heart rate–corrected QT interval and cardiac norepinephrine spillover.23 Among patients with major depression, the distribution of cardiac norepinephrine spillover seemed bimodal. Overall, cardiac norepinephrine spillover was not increased, although a subgroup had clearly increased spillover.
In congestive heart failure, baroreflex-cardiovagal gain tends to be low and cardiac sympathetic outflow markedly increased, yet the LF:HF ratio is not increased during supine rest.24,25 It therefore appears that power spectral analysis of heart rate variability may provide a measure of baroreflexive modulation of autonomic outflows to the heart but not a measure of those outflows themselves. The search continues for a valid, noninvasive means to assess cardiac sympathetic function.
References
Goldstein DS, Holmes C, Cannon RO, Eisenhofer G, Kopin IJ. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med1997; 336:696–702.
Orimo S, Ozawa E, Oka T, et al Different histopathology accounting for a decrease in myocardial MIBG uptake in PD and MSA. Neurology2001; 57:1140–1141.
Amino T, Orimo S, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Path2005; 15:29–34.
Goldstein DS, Li S-T, Kopin IJ. Sympathetic neurocirculatory failure in Parkinson disease: evidence for an etiologic role of α-synuclein. Ann Intern Med2001; 135:1010–1011.
Singleton A, Gwinn-Hardy K, Sharabi Y, et al Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain2004; 127:768–772.
Orimo S, Uchihara T, Nakamura A, et al Cardiac sympathetic denervation in Parkinson’s disease linked to SNCA duplication. Acta Neuropathol2008; 116:575–577.
Orimo S, Amino T, Yokochi M, et al Preserved cardiac sympathetic nerve accounts for normal cardiac uptake of MIBG in PARK2. Mov Disord2005; 20:1350–1353.
Dickson DW, Fujishiro H, DelleDonne A, et al Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol2008; 115:437–444.
Orimo S, Uchihara T, Nakamura A, et al Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain2008; 131:642–650.
Blaschko H. Amine oxidase and amine metabolism. Pharmacol Rev1952; 4:415–458.
Galter D, Buervenich S, Carmine A, Anvret M, Olson L. ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson’s disease and in the ventral tegmental area in schizophrenia. Neurobiol Dis2003; 14:637–647.
Mandel S, Grunblatt E, Riederer P, et al Gene expression profiling of sporadic Parkinson’s disease substantia nigra pars compacta reveals impairment of ubiquitin-proteasome subunits, SKP1A, aldehyde dehydrogenase, and chaperone HSC-70. Ann N Y Acad Sci2005; 1053:356–375.
Werner CJ, Heyny-von Haussen R, Mall G, Wolf S. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome Sci2008; 6:8.
Larsen KE, Schmitz Y, Troyer MD, et al Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci2006; 26:11915–11922.
Mosharov EV, Staal RG, Bove J, et al Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J Neurosci2006; 26:9304–9311.
Mosharov EV, Larsen KE, Kanter E, et al Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron2009; 62:218–229.
Burke WJ, Kumar VB, Pandey N, et al Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol2008; 115:193–203.
Caudle WM, Richardson JR, Wang MZ, et al Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci2007; 27:8138–8148.
Taylor TN, Caudle WM, Shepherd KR, et al Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci2009; 29:8103–8113.
Pena-Silva RA, Miller JD, Chu Y, Heistad DD. Serotonin produces monoamine oxidase-dependent oxidative stress in human heart valves. Am J Physiol Heart Circ Physiol2009; 297:H1354–H1360.
Moak JP, Goldstein DS, Eldadah BA, et al Supine low-frequency power of heart rate variability reflects baroreflex function, not cardiac sympathetic innervation. Heart Rhythm2007; 4:1523–1529.
Baumert M, Lambert GW, Dawood T, et al Short-term heart rate variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol2009; 297:H674–H679.
Baumert M, Lambert GW, Dawood T, et al QT interval variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol2008; 295:H962–H968.
Tygesen H, Rundqvist B, Waagstein F, Wennerblom B. Heart rate variability measurement correlates with cardiac norepinephrine spillover in congestive heart failure. Am J Cardiol2001; 87:1308–1311.
Kingwell BA, Thompson JM, Kaye DM, McPherson GA, Jennings GL, Esler MD. Heart rate spectral analysis, cardiac norepinephrine spillover, and muscle sympathetic nerve activity during human sympathetic nervous activation and failure. Circulation1994; 90:234–240.
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, Clinical Neurosciences Program, Division of Intramural Research, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Building 10, Room 5N220, 10 Center Drive, MSC-1620, Bethesda, MD 20892–1620; goldsteind@ninds.nih.gov
Dr. Goldstein reported that he has no financial relationships that pose a potential conflict of interest with this article.
Some of the research summarized here was supported by the intramural research program of the National Institutes of Health.
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, Clinical Neurosciences Program, Division of Intramural Research, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Building 10, Room 5N220, 10 Center Drive, MSC-1620, Bethesda, MD 20892–1620; goldsteind@ninds.nih.gov
Dr. Goldstein reported that he has no financial relationships that pose a potential conflict of interest with this article.
Some of the research summarized here was supported by the intramural research program of the National Institutes of Health.
Author and Disclosure Information
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, Clinical Neurosciences Program, Division of Intramural Research, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Building 10, Room 5N220, 10 Center Drive, MSC-1620, Bethesda, MD 20892–1620; goldsteind@ninds.nih.gov
Dr. Goldstein reported that he has no financial relationships that pose a potential conflict of interest with this article.
Some of the research summarized here was supported by the intramural research program of the National Institutes of Health.
This review highlights important recent publications in the area of neuroscience and heart-brain medicine. Abnormalities of regulation of the circulation by catecholamine systems figure as a general theme of the topics highlighted. These topics, which are reviewed in turn below, are (1) mechanisms of cardiac sympathetic denervation in Parkinson disease (PD), (2) cytoplasmic monoamine metabolites as autotoxins, and (3) the validity of power spectral analysis of heart rate variability to indicate cardiac sympathetic tone.
MECHANISMS OF CARDIAC SYMPATHETIC DENERVATION IN PARKINSON DISEASE
The movement disorder component of PD is well recognized as resulting from loss of dopaminergic neurons in the nigrostriatal system of the brain. The finding of low myocardial 6-[18F]fluorodopamine–derived radioactivity by positron emission tomography provided the first neuroimaging evidence for loss of catecholaminergic neurons outside the brain in PD.1 Many reports using 123I-metaiodobenzylguanidine scanning have concurred with this finding. Beginning in the early 2000s, post-mortem neuropathologic studies demonstrated virtually absent immunoreactivity for tyrosine hydroxylase, the rate-limiting enzyme in norepinephrine biosynthesis, in epicardial nerves in PD.2,3 These results provided clues to the mechanism of autonomic dysfunction in PD, a prominent nonmotor manifestation of the disease.
With kind permission from Springer Science+Business Media: Acta Neuropathologica,
Figure 1. Tyrosine hydroxylase immunoreactivity (THir) in epicardial nerve from (A) a control subject and (B) a patient with familial Parkinson disease due to duplication of the gene encoding alpha-synuclein (PARK4).6Alpha-synuclein is a key protein in the pathogenesis of PD. It is abundant in Lewy bodies and Lewy neurites, and mutations or multiplications of the gene that encodes it cause rare inherited forms of PD. In 2001 we reported evidence for cardiac sympathetic denervation, neurogenic orthostatic hypotension, and baroreflex failure in familial PD from mutation of the gene encoding alpha-synuclein.4 Subsequently we reported analogous denervation in familial PD from triplication of the normal gene.5 This past year Orimo’s group in Tokyo provided the first pathological confirmation of cardiac sympathetic denervation in familial PD from inherited alpha-synucleinopathy, based on severely decreased epicardial neuronal tyrosine hydroxylase immunoreactivity (Figure 1).6 In contrast, patients with familial PD from parkin gene mutation, which is not thought to be a Lewy body disease, have been found to have normal cardiac 123I-metaiodobenzyl-guanidine–derived radioactivity and normal epicardial neuronal tyrosine hydroxylase immunoreactivity.7 These findings establish a link between alpha-synucleinopathy and cardiac sympathetic denervation.
Some individuals who die without clinical parkinsonism have Lewy bodies detected pathologically. Growing evidence shows that incidental Lewy body disease represents early, presymptomatic PD.8 Orimo’s group therefore studied cardiac tissues and paravertebral sympathetic ganglia from patients with incidental Lewy body disease.9 Postmortem tissues were likewise obtained from comparison subjects with multiple system atrophy and from control subjects. Immunohistochemical analyses were performed using antibodies against tyrosine hydroxylase, phosphorylated neurofilament as a marker of axons, and phosphorylated alpha-synuclein as a marker of abnormal alpha-synuclein deposits. Key findings from this study9 were as follows:
Reprinted from Brain (Orimo S, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008; 131:642–650) by permission of Oxford University Press.
Figure 2. Concept diagram of the pathogenetic sequence of cardiac sympathetic denervation. In incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons (a), alpha-synuclein aggregates (black shading) accumulate abundantly in the distal axons but sparsely in the paravertebral sympathetic ganglia. In contrast, in incidental Lewy body disease with decreased THir axons (b), alpha-synuclein aggregates diminish in the distal axons but increase in the paravertebral sympathetic ganglia. In Parkinson disease, alpha-synuclein aggregates disappear in the distal axons and accumulate much more abundantly in the paravertebral sympathetic ganglia. In multiple system atrophy, alpha-synuclein aggregates are generally not observed (as in controls), with a few exceptions. Dotted lines indicate degeneration of THir axons.9Alpha-synuclein aggregates in distal epicardial nerve fascicles were more abundant in incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons than in incidental Lewy body disease with decreased THir axons (Figure 2).
Alpha-synuclein aggregates in the epicardial nerve fibers were closely related to the disappearance of THir axons.
In incidental Lewy body disease with preserved THir axons, alpha-synuclein aggregates were consistently more abundant in the epicardial nerves than in the paravertebral sympathetic ganglia (Figure 2).
Distally dominant accumulation of alpha-synuclein aggregates was reversed in incidental Lewy body disease with decreased THir axons and in PD, because both conditions involve fewer alpha-synuclein aggregates in axons and more abundant aggregates in the paravertebral sympathetic ganglia (Figure 2).
Thus, accumulation of alpha-synuclein aggregates in distal cardiac sympathetic axons precedes aggregation in neuronal somata or ganglionic neurites, heralding centripetal degeneration of cardiac sympathetic nerves in PD. This chronological and dynamic relationship between alpha-synuclein aggregation and distally dominant degeneration of cardiac noradrenergic nerves may represent the pathological mechanism behind a common degenerative process in PD.
In conclusion, cardiac noradrenergic denervation in Lewy body diseases, even in early stages, accounts for reduced cardiac uptake of 123I-metaiodobenzylguanidine and 6-[18F]fluorodopamine in PD. Alpha-synuclein aggregation appears to be intimately involved in the cardiac noradrenergic denervation that attends Lewy body diseases. The pathogenetic process seems to proceed in a centripetal, retrograde direction.
CYTOPLASMIC MONOAMINE METABOLITES AS AUTOTOXINS
Current concepts about mechanisms of PD emphasize pathologic alpha-synuclein accumulation, oxidative injury, impaired proteasomal or mitochondrial functions, neuroinflammation, or abnormal kinase signaling. These concepts do not explain relatively selective nigrostriatal dopaminergic and cardiac noradrenergic denervation in PD.
Figure 3. According to the monoamine aldehyde hypothesis, interference with the vesicular recycling of cytoplasmic monoamines (dopamine [DA], norepinephrine [NE], and serotonin [5-HT]) augments formation of toxic aldehydes. For instance, DA that leaks from vesicles (V) into the cytoplasm (C) or that is taken up via the cell membrane DA transporter (DAT) and escapes vesicular reuptake via the vesicular monoamine transporter (VMAT) is subject to oxidative deamination catalyzed by monoamine oxidase (MAO) to form the catecholaldehyde DOPAL, which is toxic. DOPAL is detoxified by ALDH to form DOPAC, the major metabolic route, or by AR to form DOPET, the minor metabolic route. Analogously, NE is converted to DOPEGAL, and 5-HT is converted to 5-HT-aldehyde (5-HTAld).
A potential explanation is that cytoplasmic catecholamine metabolites are autotoxins (Figure 3). The mechanisms of autotoxicity include spontaneous auto-oxidation, to form quinones and chromes leading to increased production of reactive oxygen species, and enzymatic oxidation.
Catecholamines in the neuronal cytoplasm undergo enzymatic oxidative deamination to form catecholaldehydes (dihydroxyphenylacetaldehyde [DOPAL] from dopamine), which are cytotoxic, as predicted by Blaschko more than a half century ago.10 DOPAL is detoxified mainly by aldehyde dehydrogenase (ALDH). In the substantia nigra, aldehyde dehydrogenase 1A1 (ALDH1A1) is the main isoform of ALDH, and postmortem studies have noted decreased nigral ALDH1A1 gene expression11,12 and protein content13 in PD patients.
All neurons express alpha-synuclein. Current concepts about mechanisms also do not explain the relatively selective aggregation of alpha-synuclein in catecholaminergic neurons. Alpha-synuclein appears to play a role in the cycling of catecholamines across vesicular and cell membranes.14
Figure 4. Cell survival and cytoplasmic dopamine are inversely related, according to a murine model by Mosharov et al.16 Graph shows the dependence of cell survival under l-dopa–induced stress on the cytoplasmic dopamine (DAcyt) dose in mouse neurons. The DAcyt dose was estimated as: [DAcyt]×TExposure = [DAcyt]×Ln([L-dopa]/K0.5)/k,where [DAcyt] is the concentration of cytosolic DA in cells treated with a saturating level (> 50 μM) of l-dopa for 1 hour, where [l-dopa] is the initial drug concentration, and where K0.5 = 9.7 μM and k = 0.15 hr−1 are the kinetic constants. TExposure approximates the time during which extracellular l-dopa remained higher than K0.5. The data points are (from left to right): filled circles—ventral midbrain cultures treated with 25, 100, 250, 500, and 1,000 μM l-dopa alone; open circles—ventral midbrain neurons treated with 250 μM l-dopa in the presence of benserazide, methamphetamine, reserpine, pargyline, and pargyline reserpine; diamonds—ventral tegmental area and substantia nigra neurons; triangles—striatal and cortical neurons treated with 250 μM l-dopa. Dotted lines and shaded boxes represent mean ± SEM in untreated cells. The solid line is the linear fit of all data points, excluding striatal and cortical neurons and the two data points indicated by the asterisk. Treatments to the right of this line are neuroprotective, as the same level of cell death is achieved with higher DAcyt doses; treatments to the left of this line are more susceptible to DAcyt stress.In the past year, a few important studies have been published related to autotoxicity of cytoplasmic catecholamine metabolites and to pathogenic interactions with alpha-synuclein. In 2006, Mosharov et al reported that alpha-synuclein overexpression increases cytoplasmic dopamine concentrations in rat pheochromocytoma PC-12 cells.15 Recently, the same group, using intracellular patch electrochemistry, directly measured cytoplasmic dopamine in cultured midbrain neurons and found that increases in dopamine and its metabolites are neurotoxic, whereas manipulations that reduce cytoplasmic dopamine are neuroprotective (Figure 4).16 Levodopa (l-dopa) increased cytoplasmic dopamine more in substantia nigra neurons than in ventral tegmental neurons, suggesting that this difference might help explain the greater susceptibility of nigral neurons to the pathogenetic process. The greater buildup of cytoplasmic dopamine seemed to depend on dihydropyridine-sensitive calcium (Ca2+) channels. Finally, dopaminergic neurons lacking alpha-synuclein were resistant to l-dopa–induced cell death. These findings led the authors to propose a “multiple-hit” model (Figure 5) in which interactions between intracellular ionized calcium, cytoplasmic dopamine, and alpha-synuclein underlie susceptibility of nigral neurons in PD.16
Figure 5. The “multiple-hit” model of Parkinson disease pathogenesis,16 which holds that neurotoxicity is a result of multiple factors, including the presence of alpha-synuclein (α-syn), elevation of cytoplasmic calcium (Ca2+), and buildup of cytoplasmic dopamine (DAcyt) and its metabolites. Nonexclusive toxic steps may result from (1) mechanisms that require direct interaction between DA or its metabolites with α-syn, such as DA-modified stabilization of α-syn protofibrils or inhibition of chaperone-mediated autophagy, or (2) cumulative damage from multiple independent sources. Reducing the levels of any of the three players provides neuroprotection. (AADC = aromatic l-amino acid decarboxylase; DOPAL = dihydroxyphenylacetaldehyde; TH = tyrosine hydroxylase)Burke et al added a potentially important clue, demonstrating that DOPAL potently oligomerizes and aggregates alpha-synuclein.17 This finding introduces the possibility of multiple pathogenetic positive feedback loops.
Under resting conditions, most catecholamine turnover results from leakage from vesicular stores into the cytoplasm and subsequent oxidative deamination by monoamine oxidase. Ordinarily, however, catecholamines in the cytoplasm are efficiently recycled back into the vesicles via the type 2 vesicular monoamine transporter (VMAT-2). Accordingly, interference with VMAT functions would be expected to tend to build up cytoplasmic catecholamines, with potentially cytotoxic consequences. In 2007, Caudle et al reported that mice with severely decreased VMAT-2 have aging-associated decreases in striatal dopamine that begin in the terminal fields, alpha-synuclein deposition in substantia nigra neurons, and l-dopa–responsive behavioral deficits.18 More recently the same group noted nonmotor signs associated with PD in VMAT-2–deficient mice, such as anosmia, gastrointestinal hypomotility, sleep disturbances, anxiety, and depression.19 Since VMAT-2 serves to recycle not only dopamine but also norepinephrine and serotonin, this single abnormality could help explain loss of all three types of monoaminergic neurons in PD.
Finally, Pena-Silva et al recently tested whether serotonin induces oxidative stress in human heart valves.20 They showed that in heart valves from explanted human hearts not used for transplantation, incubation of homogenates of cardiac valves and blood vessels with serotonin increased generation of the superoxide free radical. Inhibitors of monoamine oxidase prevented this effect. Dopamine also increased superoxide levels in heart valves, and this effect was also attenuated by monoamine oxidase inhibition. These findings fit with the concept that the aldehydes produced by the action of monoamine oxidase on cytoplasmic monoamines generate toxic free radicals.
VALIDITY OF POWER SPECTRAL ANALYSIS OF HEART RATE VARIABILITY TO INDICATE CARDIAC SYMPATHETIC TONE
Power spectral analysis of heart rate variability is simple, relatively inexpensive, noninvasive, and widely used to indicate cardiac sympathetic “tone” or sympathovagal “balance.” Almost 2,000 studies to date have used this modality. Relatively increased cardiac sympathetic tone, reflected by low-frequency (LF) power or the ratio of LF power to high-frequency (HF) power, is an adverse prognostic sign in a variety of conditions. Nevertheless, the validity of LF power, or the LF:HF ratio, as an index of cardiac sympathetic tone remains unsettled.
In 2007 we assessed the validity of power spectral analysis rather directly, by taking advantage of our ability to delineate cardiac sympathetic innervation. We compared LF power in patients with cardiac sympathetic denervation, indicated by low myocardial levels of 6-[18F]fluorodopamine–derived radioactivity or low rates of norepinephrine entry into coronary sinus plasma (cardiac norepinephrine spillover), with values in patients with intact innervation. LF power was unrelated to myocardial 6-[18F]fluorodopamine–derived radioactivity or cardiac norepinephrine spillover, but it was related to baroreflex-cardiovagal gain. Patients with a low baroreflex-cardiovagal gain had low LF power, regardless of cardiac innervation. From these findings we concluded that LF power reflects baroreflex function, not cardiac sympathetic innervation.21
Figure 6. Relationships of heart rate variability indices with cardiac norepinephrine spillover. Graphs show time and frequency domain heart rate variability measures (LF = low frequency; HF = high frequency) versus cardiac norepinephrine in healthy subjects (squares) and patients with major depression (triangles) and panic disorder (circles).22 Recently Baumert et al also examined the relationship between indices from power spectral analysis of heart rate variability and cardiac norepinephrine spillover.22 They found, as we did, that none of the standard heart rate variability parameters was correlated with cardiac norepinephrine spillover (Figure 6). The same group reported a positive correlation between the heart rate–corrected QT interval and cardiac norepinephrine spillover.23 Among patients with major depression, the distribution of cardiac norepinephrine spillover seemed bimodal. Overall, cardiac norepinephrine spillover was not increased, although a subgroup had clearly increased spillover.
In congestive heart failure, baroreflex-cardiovagal gain tends to be low and cardiac sympathetic outflow markedly increased, yet the LF:HF ratio is not increased during supine rest.24,25 It therefore appears that power spectral analysis of heart rate variability may provide a measure of baroreflexive modulation of autonomic outflows to the heart but not a measure of those outflows themselves. The search continues for a valid, noninvasive means to assess cardiac sympathetic function.
This review highlights important recent publications in the area of neuroscience and heart-brain medicine. Abnormalities of regulation of the circulation by catecholamine systems figure as a general theme of the topics highlighted. These topics, which are reviewed in turn below, are (1) mechanisms of cardiac sympathetic denervation in Parkinson disease (PD), (2) cytoplasmic monoamine metabolites as autotoxins, and (3) the validity of power spectral analysis of heart rate variability to indicate cardiac sympathetic tone.
MECHANISMS OF CARDIAC SYMPATHETIC DENERVATION IN PARKINSON DISEASE
The movement disorder component of PD is well recognized as resulting from loss of dopaminergic neurons in the nigrostriatal system of the brain. The finding of low myocardial 6-[18F]fluorodopamine–derived radioactivity by positron emission tomography provided the first neuroimaging evidence for loss of catecholaminergic neurons outside the brain in PD.1 Many reports using 123I-metaiodobenzylguanidine scanning have concurred with this finding. Beginning in the early 2000s, post-mortem neuropathologic studies demonstrated virtually absent immunoreactivity for tyrosine hydroxylase, the rate-limiting enzyme in norepinephrine biosynthesis, in epicardial nerves in PD.2,3 These results provided clues to the mechanism of autonomic dysfunction in PD, a prominent nonmotor manifestation of the disease.
With kind permission from Springer Science+Business Media: Acta Neuropathologica,
Figure 1. Tyrosine hydroxylase immunoreactivity (THir) in epicardial nerve from (A) a control subject and (B) a patient with familial Parkinson disease due to duplication of the gene encoding alpha-synuclein (PARK4).6Alpha-synuclein is a key protein in the pathogenesis of PD. It is abundant in Lewy bodies and Lewy neurites, and mutations or multiplications of the gene that encodes it cause rare inherited forms of PD. In 2001 we reported evidence for cardiac sympathetic denervation, neurogenic orthostatic hypotension, and baroreflex failure in familial PD from mutation of the gene encoding alpha-synuclein.4 Subsequently we reported analogous denervation in familial PD from triplication of the normal gene.5 This past year Orimo’s group in Tokyo provided the first pathological confirmation of cardiac sympathetic denervation in familial PD from inherited alpha-synucleinopathy, based on severely decreased epicardial neuronal tyrosine hydroxylase immunoreactivity (Figure 1).6 In contrast, patients with familial PD from parkin gene mutation, which is not thought to be a Lewy body disease, have been found to have normal cardiac 123I-metaiodobenzyl-guanidine–derived radioactivity and normal epicardial neuronal tyrosine hydroxylase immunoreactivity.7 These findings establish a link between alpha-synucleinopathy and cardiac sympathetic denervation.
Some individuals who die without clinical parkinsonism have Lewy bodies detected pathologically. Growing evidence shows that incidental Lewy body disease represents early, presymptomatic PD.8 Orimo’s group therefore studied cardiac tissues and paravertebral sympathetic ganglia from patients with incidental Lewy body disease.9 Postmortem tissues were likewise obtained from comparison subjects with multiple system atrophy and from control subjects. Immunohistochemical analyses were performed using antibodies against tyrosine hydroxylase, phosphorylated neurofilament as a marker of axons, and phosphorylated alpha-synuclein as a marker of abnormal alpha-synuclein deposits. Key findings from this study9 were as follows:
Reprinted from Brain (Orimo S, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008; 131:642–650) by permission of Oxford University Press.
Figure 2. Concept diagram of the pathogenetic sequence of cardiac sympathetic denervation. In incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons (a), alpha-synuclein aggregates (black shading) accumulate abundantly in the distal axons but sparsely in the paravertebral sympathetic ganglia. In contrast, in incidental Lewy body disease with decreased THir axons (b), alpha-synuclein aggregates diminish in the distal axons but increase in the paravertebral sympathetic ganglia. In Parkinson disease, alpha-synuclein aggregates disappear in the distal axons and accumulate much more abundantly in the paravertebral sympathetic ganglia. In multiple system atrophy, alpha-synuclein aggregates are generally not observed (as in controls), with a few exceptions. Dotted lines indicate degeneration of THir axons.9Alpha-synuclein aggregates in distal epicardial nerve fascicles were more abundant in incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons than in incidental Lewy body disease with decreased THir axons (Figure 2).
Alpha-synuclein aggregates in the epicardial nerve fibers were closely related to the disappearance of THir axons.
In incidental Lewy body disease with preserved THir axons, alpha-synuclein aggregates were consistently more abundant in the epicardial nerves than in the paravertebral sympathetic ganglia (Figure 2).
Distally dominant accumulation of alpha-synuclein aggregates was reversed in incidental Lewy body disease with decreased THir axons and in PD, because both conditions involve fewer alpha-synuclein aggregates in axons and more abundant aggregates in the paravertebral sympathetic ganglia (Figure 2).
Thus, accumulation of alpha-synuclein aggregates in distal cardiac sympathetic axons precedes aggregation in neuronal somata or ganglionic neurites, heralding centripetal degeneration of cardiac sympathetic nerves in PD. This chronological and dynamic relationship between alpha-synuclein aggregation and distally dominant degeneration of cardiac noradrenergic nerves may represent the pathological mechanism behind a common degenerative process in PD.
In conclusion, cardiac noradrenergic denervation in Lewy body diseases, even in early stages, accounts for reduced cardiac uptake of 123I-metaiodobenzylguanidine and 6-[18F]fluorodopamine in PD. Alpha-synuclein aggregation appears to be intimately involved in the cardiac noradrenergic denervation that attends Lewy body diseases. The pathogenetic process seems to proceed in a centripetal, retrograde direction.
CYTOPLASMIC MONOAMINE METABOLITES AS AUTOTOXINS
Current concepts about mechanisms of PD emphasize pathologic alpha-synuclein accumulation, oxidative injury, impaired proteasomal or mitochondrial functions, neuroinflammation, or abnormal kinase signaling. These concepts do not explain relatively selective nigrostriatal dopaminergic and cardiac noradrenergic denervation in PD.
Figure 3. According to the monoamine aldehyde hypothesis, interference with the vesicular recycling of cytoplasmic monoamines (dopamine [DA], norepinephrine [NE], and serotonin [5-HT]) augments formation of toxic aldehydes. For instance, DA that leaks from vesicles (V) into the cytoplasm (C) or that is taken up via the cell membrane DA transporter (DAT) and escapes vesicular reuptake via the vesicular monoamine transporter (VMAT) is subject to oxidative deamination catalyzed by monoamine oxidase (MAO) to form the catecholaldehyde DOPAL, which is toxic. DOPAL is detoxified by ALDH to form DOPAC, the major metabolic route, or by AR to form DOPET, the minor metabolic route. Analogously, NE is converted to DOPEGAL, and 5-HT is converted to 5-HT-aldehyde (5-HTAld).
A potential explanation is that cytoplasmic catecholamine metabolites are autotoxins (Figure 3). The mechanisms of autotoxicity include spontaneous auto-oxidation, to form quinones and chromes leading to increased production of reactive oxygen species, and enzymatic oxidation.
Catecholamines in the neuronal cytoplasm undergo enzymatic oxidative deamination to form catecholaldehydes (dihydroxyphenylacetaldehyde [DOPAL] from dopamine), which are cytotoxic, as predicted by Blaschko more than a half century ago.10 DOPAL is detoxified mainly by aldehyde dehydrogenase (ALDH). In the substantia nigra, aldehyde dehydrogenase 1A1 (ALDH1A1) is the main isoform of ALDH, and postmortem studies have noted decreased nigral ALDH1A1 gene expression11,12 and protein content13 in PD patients.
All neurons express alpha-synuclein. Current concepts about mechanisms also do not explain the relatively selective aggregation of alpha-synuclein in catecholaminergic neurons. Alpha-synuclein appears to play a role in the cycling of catecholamines across vesicular and cell membranes.14
Figure 4. Cell survival and cytoplasmic dopamine are inversely related, according to a murine model by Mosharov et al.16 Graph shows the dependence of cell survival under l-dopa–induced stress on the cytoplasmic dopamine (DAcyt) dose in mouse neurons. The DAcyt dose was estimated as: [DAcyt]×TExposure = [DAcyt]×Ln([L-dopa]/K0.5)/k,where [DAcyt] is the concentration of cytosolic DA in cells treated with a saturating level (> 50 μM) of l-dopa for 1 hour, where [l-dopa] is the initial drug concentration, and where K0.5 = 9.7 μM and k = 0.15 hr−1 are the kinetic constants. TExposure approximates the time during which extracellular l-dopa remained higher than K0.5. The data points are (from left to right): filled circles—ventral midbrain cultures treated with 25, 100, 250, 500, and 1,000 μM l-dopa alone; open circles—ventral midbrain neurons treated with 250 μM l-dopa in the presence of benserazide, methamphetamine, reserpine, pargyline, and pargyline reserpine; diamonds—ventral tegmental area and substantia nigra neurons; triangles—striatal and cortical neurons treated with 250 μM l-dopa. Dotted lines and shaded boxes represent mean ± SEM in untreated cells. The solid line is the linear fit of all data points, excluding striatal and cortical neurons and the two data points indicated by the asterisk. Treatments to the right of this line are neuroprotective, as the same level of cell death is achieved with higher DAcyt doses; treatments to the left of this line are more susceptible to DAcyt stress.In the past year, a few important studies have been published related to autotoxicity of cytoplasmic catecholamine metabolites and to pathogenic interactions with alpha-synuclein. In 2006, Mosharov et al reported that alpha-synuclein overexpression increases cytoplasmic dopamine concentrations in rat pheochromocytoma PC-12 cells.15 Recently, the same group, using intracellular patch electrochemistry, directly measured cytoplasmic dopamine in cultured midbrain neurons and found that increases in dopamine and its metabolites are neurotoxic, whereas manipulations that reduce cytoplasmic dopamine are neuroprotective (Figure 4).16 Levodopa (l-dopa) increased cytoplasmic dopamine more in substantia nigra neurons than in ventral tegmental neurons, suggesting that this difference might help explain the greater susceptibility of nigral neurons to the pathogenetic process. The greater buildup of cytoplasmic dopamine seemed to depend on dihydropyridine-sensitive calcium (Ca2+) channels. Finally, dopaminergic neurons lacking alpha-synuclein were resistant to l-dopa–induced cell death. These findings led the authors to propose a “multiple-hit” model (Figure 5) in which interactions between intracellular ionized calcium, cytoplasmic dopamine, and alpha-synuclein underlie susceptibility of nigral neurons in PD.16
Figure 5. The “multiple-hit” model of Parkinson disease pathogenesis,16 which holds that neurotoxicity is a result of multiple factors, including the presence of alpha-synuclein (α-syn), elevation of cytoplasmic calcium (Ca2+), and buildup of cytoplasmic dopamine (DAcyt) and its metabolites. Nonexclusive toxic steps may result from (1) mechanisms that require direct interaction between DA or its metabolites with α-syn, such as DA-modified stabilization of α-syn protofibrils or inhibition of chaperone-mediated autophagy, or (2) cumulative damage from multiple independent sources. Reducing the levels of any of the three players provides neuroprotection. (AADC = aromatic l-amino acid decarboxylase; DOPAL = dihydroxyphenylacetaldehyde; TH = tyrosine hydroxylase)Burke et al added a potentially important clue, demonstrating that DOPAL potently oligomerizes and aggregates alpha-synuclein.17 This finding introduces the possibility of multiple pathogenetic positive feedback loops.
Under resting conditions, most catecholamine turnover results from leakage from vesicular stores into the cytoplasm and subsequent oxidative deamination by monoamine oxidase. Ordinarily, however, catecholamines in the cytoplasm are efficiently recycled back into the vesicles via the type 2 vesicular monoamine transporter (VMAT-2). Accordingly, interference with VMAT functions would be expected to tend to build up cytoplasmic catecholamines, with potentially cytotoxic consequences. In 2007, Caudle et al reported that mice with severely decreased VMAT-2 have aging-associated decreases in striatal dopamine that begin in the terminal fields, alpha-synuclein deposition in substantia nigra neurons, and l-dopa–responsive behavioral deficits.18 More recently the same group noted nonmotor signs associated with PD in VMAT-2–deficient mice, such as anosmia, gastrointestinal hypomotility, sleep disturbances, anxiety, and depression.19 Since VMAT-2 serves to recycle not only dopamine but also norepinephrine and serotonin, this single abnormality could help explain loss of all three types of monoaminergic neurons in PD.
Finally, Pena-Silva et al recently tested whether serotonin induces oxidative stress in human heart valves.20 They showed that in heart valves from explanted human hearts not used for transplantation, incubation of homogenates of cardiac valves and blood vessels with serotonin increased generation of the superoxide free radical. Inhibitors of monoamine oxidase prevented this effect. Dopamine also increased superoxide levels in heart valves, and this effect was also attenuated by monoamine oxidase inhibition. These findings fit with the concept that the aldehydes produced by the action of monoamine oxidase on cytoplasmic monoamines generate toxic free radicals.
VALIDITY OF POWER SPECTRAL ANALYSIS OF HEART RATE VARIABILITY TO INDICATE CARDIAC SYMPATHETIC TONE
Power spectral analysis of heart rate variability is simple, relatively inexpensive, noninvasive, and widely used to indicate cardiac sympathetic “tone” or sympathovagal “balance.” Almost 2,000 studies to date have used this modality. Relatively increased cardiac sympathetic tone, reflected by low-frequency (LF) power or the ratio of LF power to high-frequency (HF) power, is an adverse prognostic sign in a variety of conditions. Nevertheless, the validity of LF power, or the LF:HF ratio, as an index of cardiac sympathetic tone remains unsettled.
In 2007 we assessed the validity of power spectral analysis rather directly, by taking advantage of our ability to delineate cardiac sympathetic innervation. We compared LF power in patients with cardiac sympathetic denervation, indicated by low myocardial levels of 6-[18F]fluorodopamine–derived radioactivity or low rates of norepinephrine entry into coronary sinus plasma (cardiac norepinephrine spillover), with values in patients with intact innervation. LF power was unrelated to myocardial 6-[18F]fluorodopamine–derived radioactivity or cardiac norepinephrine spillover, but it was related to baroreflex-cardiovagal gain. Patients with a low baroreflex-cardiovagal gain had low LF power, regardless of cardiac innervation. From these findings we concluded that LF power reflects baroreflex function, not cardiac sympathetic innervation.21
Figure 6. Relationships of heart rate variability indices with cardiac norepinephrine spillover. Graphs show time and frequency domain heart rate variability measures (LF = low frequency; HF = high frequency) versus cardiac norepinephrine in healthy subjects (squares) and patients with major depression (triangles) and panic disorder (circles).22 Recently Baumert et al also examined the relationship between indices from power spectral analysis of heart rate variability and cardiac norepinephrine spillover.22 They found, as we did, that none of the standard heart rate variability parameters was correlated with cardiac norepinephrine spillover (Figure 6). The same group reported a positive correlation between the heart rate–corrected QT interval and cardiac norepinephrine spillover.23 Among patients with major depression, the distribution of cardiac norepinephrine spillover seemed bimodal. Overall, cardiac norepinephrine spillover was not increased, although a subgroup had clearly increased spillover.
In congestive heart failure, baroreflex-cardiovagal gain tends to be low and cardiac sympathetic outflow markedly increased, yet the LF:HF ratio is not increased during supine rest.24,25 It therefore appears that power spectral analysis of heart rate variability may provide a measure of baroreflexive modulation of autonomic outflows to the heart but not a measure of those outflows themselves. The search continues for a valid, noninvasive means to assess cardiac sympathetic function.
References
Goldstein DS, Holmes C, Cannon RO, Eisenhofer G, Kopin IJ. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med1997; 336:696–702.
Orimo S, Ozawa E, Oka T, et al Different histopathology accounting for a decrease in myocardial MIBG uptake in PD and MSA. Neurology2001; 57:1140–1141.
Amino T, Orimo S, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Path2005; 15:29–34.
Goldstein DS, Li S-T, Kopin IJ. Sympathetic neurocirculatory failure in Parkinson disease: evidence for an etiologic role of α-synuclein. Ann Intern Med2001; 135:1010–1011.
Singleton A, Gwinn-Hardy K, Sharabi Y, et al Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain2004; 127:768–772.
Orimo S, Uchihara T, Nakamura A, et al Cardiac sympathetic denervation in Parkinson’s disease linked to SNCA duplication. Acta Neuropathol2008; 116:575–577.
Orimo S, Amino T, Yokochi M, et al Preserved cardiac sympathetic nerve accounts for normal cardiac uptake of MIBG in PARK2. Mov Disord2005; 20:1350–1353.
Dickson DW, Fujishiro H, DelleDonne A, et al Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol2008; 115:437–444.
Orimo S, Uchihara T, Nakamura A, et al Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain2008; 131:642–650.
Blaschko H. Amine oxidase and amine metabolism. Pharmacol Rev1952; 4:415–458.
Galter D, Buervenich S, Carmine A, Anvret M, Olson L. ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson’s disease and in the ventral tegmental area in schizophrenia. Neurobiol Dis2003; 14:637–647.
Mandel S, Grunblatt E, Riederer P, et al Gene expression profiling of sporadic Parkinson’s disease substantia nigra pars compacta reveals impairment of ubiquitin-proteasome subunits, SKP1A, aldehyde dehydrogenase, and chaperone HSC-70. Ann N Y Acad Sci2005; 1053:356–375.
Werner CJ, Heyny-von Haussen R, Mall G, Wolf S. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome Sci2008; 6:8.
Larsen KE, Schmitz Y, Troyer MD, et al Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci2006; 26:11915–11922.
Mosharov EV, Staal RG, Bove J, et al Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J Neurosci2006; 26:9304–9311.
Mosharov EV, Larsen KE, Kanter E, et al Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron2009; 62:218–229.
Burke WJ, Kumar VB, Pandey N, et al Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol2008; 115:193–203.
Caudle WM, Richardson JR, Wang MZ, et al Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci2007; 27:8138–8148.
Taylor TN, Caudle WM, Shepherd KR, et al Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci2009; 29:8103–8113.
Pena-Silva RA, Miller JD, Chu Y, Heistad DD. Serotonin produces monoamine oxidase-dependent oxidative stress in human heart valves. Am J Physiol Heart Circ Physiol2009; 297:H1354–H1360.
Moak JP, Goldstein DS, Eldadah BA, et al Supine low-frequency power of heart rate variability reflects baroreflex function, not cardiac sympathetic innervation. Heart Rhythm2007; 4:1523–1529.
Baumert M, Lambert GW, Dawood T, et al Short-term heart rate variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol2009; 297:H674–H679.
Baumert M, Lambert GW, Dawood T, et al QT interval variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol2008; 295:H962–H968.
Tygesen H, Rundqvist B, Waagstein F, Wennerblom B. Heart rate variability measurement correlates with cardiac norepinephrine spillover in congestive heart failure. Am J Cardiol2001; 87:1308–1311.
Kingwell BA, Thompson JM, Kaye DM, McPherson GA, Jennings GL, Esler MD. Heart rate spectral analysis, cardiac norepinephrine spillover, and muscle sympathetic nerve activity during human sympathetic nervous activation and failure. Circulation1994; 90:234–240.
References
Goldstein DS, Holmes C, Cannon RO, Eisenhofer G, Kopin IJ. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med1997; 336:696–702.
Orimo S, Ozawa E, Oka T, et al Different histopathology accounting for a decrease in myocardial MIBG uptake in PD and MSA. Neurology2001; 57:1140–1141.
Amino T, Orimo S, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Path2005; 15:29–34.
Goldstein DS, Li S-T, Kopin IJ. Sympathetic neurocirculatory failure in Parkinson disease: evidence for an etiologic role of α-synuclein. Ann Intern Med2001; 135:1010–1011.
Singleton A, Gwinn-Hardy K, Sharabi Y, et al Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain2004; 127:768–772.
Orimo S, Uchihara T, Nakamura A, et al Cardiac sympathetic denervation in Parkinson’s disease linked to SNCA duplication. Acta Neuropathol2008; 116:575–577.
Orimo S, Amino T, Yokochi M, et al Preserved cardiac sympathetic nerve accounts for normal cardiac uptake of MIBG in PARK2. Mov Disord2005; 20:1350–1353.
Dickson DW, Fujishiro H, DelleDonne A, et al Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol2008; 115:437–444.
Orimo S, Uchihara T, Nakamura A, et al Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain2008; 131:642–650.
Blaschko H. Amine oxidase and amine metabolism. Pharmacol Rev1952; 4:415–458.
Galter D, Buervenich S, Carmine A, Anvret M, Olson L. ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson’s disease and in the ventral tegmental area in schizophrenia. Neurobiol Dis2003; 14:637–647.
Mandel S, Grunblatt E, Riederer P, et al Gene expression profiling of sporadic Parkinson’s disease substantia nigra pars compacta reveals impairment of ubiquitin-proteasome subunits, SKP1A, aldehyde dehydrogenase, and chaperone HSC-70. Ann N Y Acad Sci2005; 1053:356–375.
Werner CJ, Heyny-von Haussen R, Mall G, Wolf S. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome Sci2008; 6:8.
Larsen KE, Schmitz Y, Troyer MD, et al Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci2006; 26:11915–11922.
Mosharov EV, Staal RG, Bove J, et al Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J Neurosci2006; 26:9304–9311.
Mosharov EV, Larsen KE, Kanter E, et al Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron2009; 62:218–229.
Burke WJ, Kumar VB, Pandey N, et al Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol2008; 115:193–203.
Caudle WM, Richardson JR, Wang MZ, et al Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci2007; 27:8138–8148.
Taylor TN, Caudle WM, Shepherd KR, et al Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci2009; 29:8103–8113.
Pena-Silva RA, Miller JD, Chu Y, Heistad DD. Serotonin produces monoamine oxidase-dependent oxidative stress in human heart valves. Am J Physiol Heart Circ Physiol2009; 297:H1354–H1360.
Moak JP, Goldstein DS, Eldadah BA, et al Supine low-frequency power of heart rate variability reflects baroreflex function, not cardiac sympathetic innervation. Heart Rhythm2007; 4:1523–1529.
Baumert M, Lambert GW, Dawood T, et al Short-term heart rate variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol2009; 297:H674–H679.
Baumert M, Lambert GW, Dawood T, et al QT interval variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol2008; 295:H962–H968.
Tygesen H, Rundqvist B, Waagstein F, Wennerblom B. Heart rate variability measurement correlates with cardiac norepinephrine spillover in congestive heart failure. Am J Cardiol2001; 87:1308–1311.
Kingwell BA, Thompson JM, Kaye DM, McPherson GA, Jennings GL, Esler MD. Heart rate spectral analysis, cardiac norepinephrine spillover, and muscle sympathetic nerve activity during human sympathetic nervous activation and failure. Circulation1994; 90:234–240.

 Reprinted from Brain (Orimo S, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008; 131:642–650) by permission of Oxford University Press.
Reprinted from Brain (Orimo S, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008; 131:642–650) by permission of Oxford University Press.