This review highlights important recent publications in the area of neuroscience and heart-brain medicine. Abnormalities of regulation of the circulation by catecholamine systems figure as a general theme of the topics highlighted. These topics, which are reviewed in turn below, are (1) mechanisms of cardiac sympathetic denervation in Parkinson disease (PD), (2) cytoplasmic monoamine metabolites as autotoxins, and (3) the validity of power spectral analysis of heart rate variability to indicate cardiac sympathetic tone.
MECHANISMS OF CARDIAC SYMPATHETIC DENERVATION IN PARKINSON DISEASE
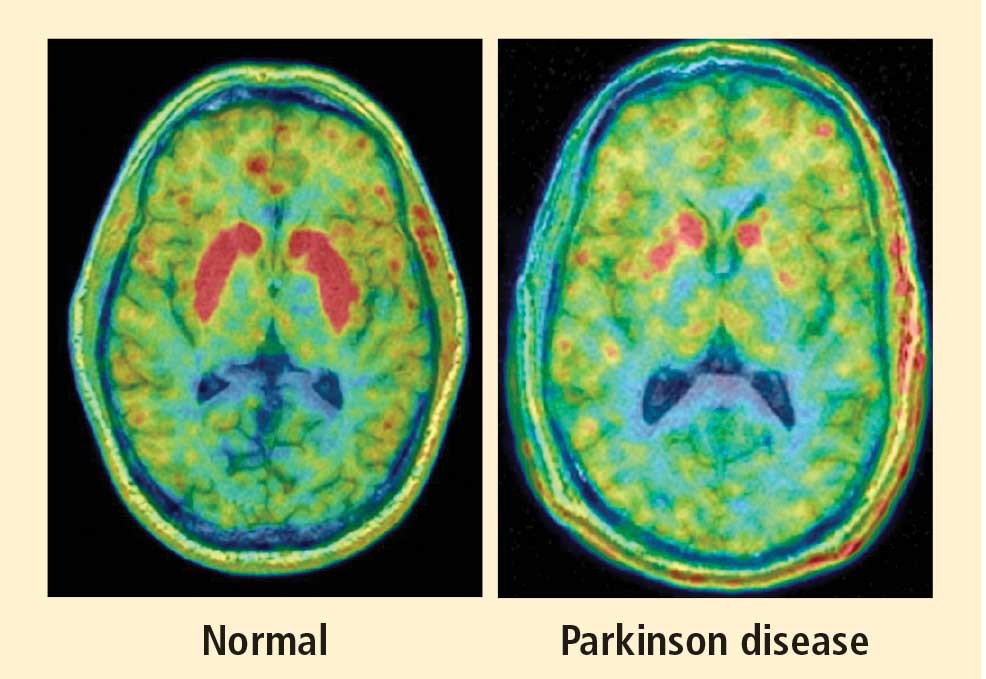
The movement disorder component of PD is well recognized as resulting from loss of dopaminergic neurons in the nigrostriatal system of the brain. The finding of low myocardial 6-[18F]fluorodopamine–derived radioactivity by positron emission tomography provided the first neuroimaging evidence for loss of catecholaminergic neurons outside the brain in PD.1 Many reports using 123I-metaiodobenzylguanidine scanning have concurred with this finding. Beginning in the early 2000s, post-mortem neuropathologic studies demonstrated virtually absent immunoreactivity for tyrosine hydroxylase, the rate-limiting enzyme in norepinephrine biosynthesis, in epicardial nerves in PD.2,3 These results provided clues to the mechanism of autonomic dysfunction in PD, a prominent nonmotor manifestation of the disease.
With kind permission from Springer Science+Business Media: Acta Neuropathologica,
Figure 1. Tyrosine hydroxylase immunoreactivity (THir) in epicardial nerve from (A) a control subject and (B) a patient with familial Parkinson disease due to duplication of the gene encoding alpha-synuclein (PARK4).6Alpha-synuclein is a key protein in the pathogenesis of PD. It is abundant in Lewy bodies and Lewy neurites, and mutations or multiplications of the gene that encodes it cause rare inherited forms of PD. In 2001 we reported evidence for cardiac sympathetic denervation, neurogenic orthostatic hypotension, and baroreflex failure in familial PD from mutation of the gene encoding alpha-synuclein.4 Subsequently we reported analogous denervation in familial PD from triplication of the normal gene.5 This past year Orimo’s group in Tokyo provided the first pathological confirmation of cardiac sympathetic denervation in familial PD from inherited alpha-synucleinopathy, based on severely decreased epicardial neuronal tyrosine hydroxylase immunoreactivity (Figure 1).6 In contrast, patients with familial PD from parkin gene mutation, which is not thought to be a Lewy body disease, have been found to have normal cardiac 123I-metaiodobenzyl-guanidine–derived radioactivity and normal epicardial neuronal tyrosine hydroxylase immunoreactivity.7 These findings establish a link between alpha-synucleinopathy and cardiac sympathetic denervation.
Some individuals who die without clinical parkinsonism have Lewy bodies detected pathologically. Growing evidence shows that incidental Lewy body disease represents early, presymptomatic PD.8 Orimo’s group therefore studied cardiac tissues and paravertebral sympathetic ganglia from patients with incidental Lewy body disease.9 Postmortem tissues were likewise obtained from comparison subjects with multiple system atrophy and from control subjects. Immunohistochemical analyses were performed using antibodies against tyrosine hydroxylase, phosphorylated neurofilament as a marker of axons, and phosphorylated alpha-synuclein as a marker of abnormal alpha-synuclein deposits. Key findings from this study9 were as follows:
Reprinted from Brain (Orimo S, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008; 131:642–650) by permission of Oxford University Press.
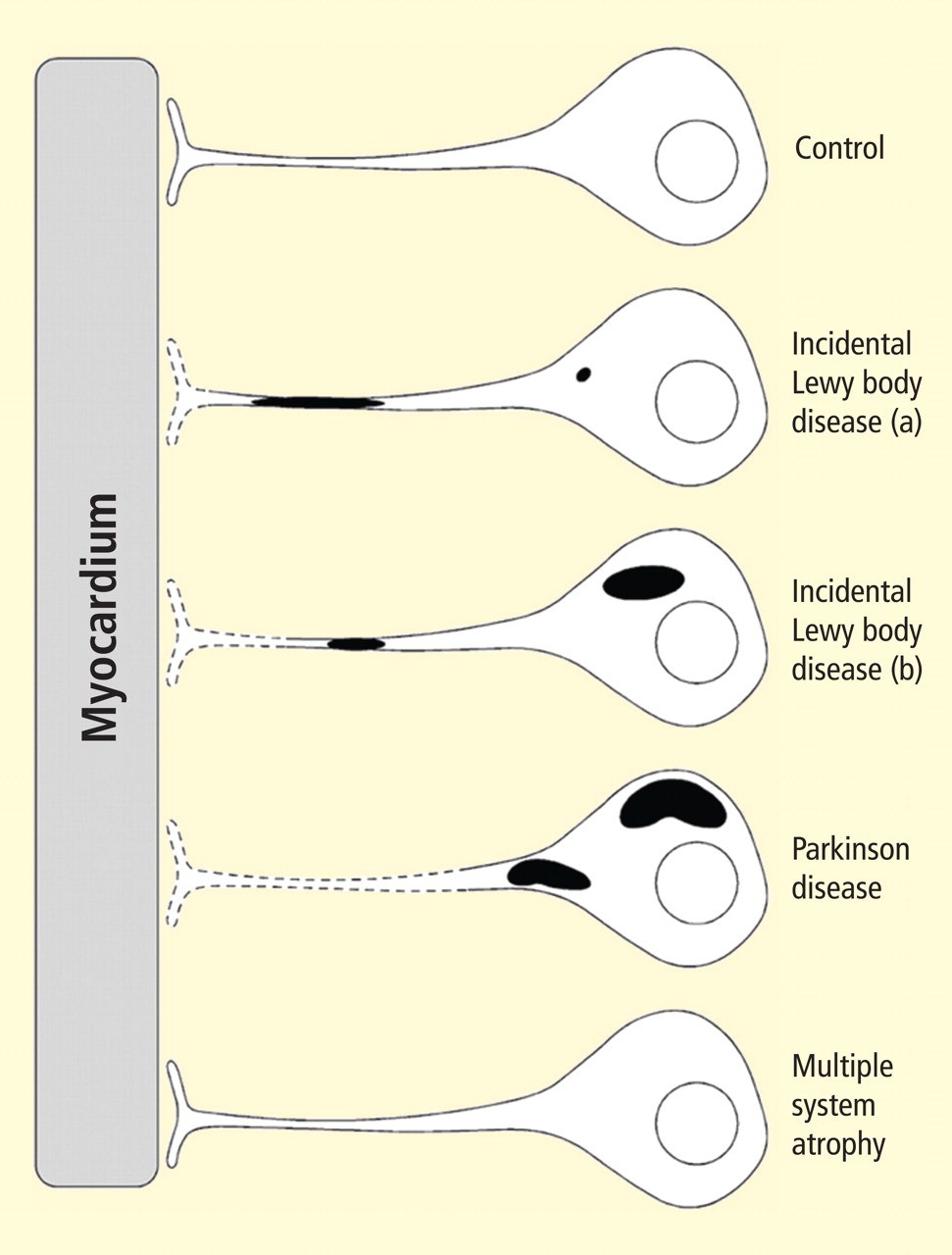
Figure 2. Concept diagram of the pathogenetic sequence of cardiac sympathetic denervation. In incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons (a), alpha-synuclein aggregates (black shading) accumulate abundantly in the distal axons but sparsely in the paravertebral sympathetic ganglia. In contrast, in incidental Lewy body disease with decreased THir axons (b), alpha-synuclein aggregates diminish in the distal axons but increase in the paravertebral sympathetic ganglia. In Parkinson disease, alpha-synuclein aggregates disappear in the distal axons and accumulate much more abundantly in the paravertebral sympathetic ganglia. In multiple system atrophy, alpha-synuclein aggregates are generally not observed (as in controls), with a few exceptions. Dotted lines indicate degeneration of THir axons.9Alpha-synuclein aggregates in distal epicardial nerve fascicles were more abundant in incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons than in incidental Lewy body disease with decreased THir axons (Figure 2).
Alpha-synuclein aggregates in the epicardial nerve fibers were closely related to the disappearance of THir axons.
In incidental Lewy body disease with preserved THir axons, alpha-synuclein aggregates were consistently more abundant in the epicardial nerves than in the paravertebral sympathetic ganglia (Figure 2).
Distally dominant accumulation of alpha-synuclein aggregates was reversed in incidental Lewy body disease with decreased THir axons and in PD, because both conditions involve fewer alpha-synuclein aggregates in axons and more abundant aggregates in the paravertebral sympathetic ganglia (Figure 2).
Thus, accumulation of alpha-synuclein aggregates in distal cardiac sympathetic axons precedes aggregation in neuronal somata or ganglionic neurites, heralding centripetal degeneration of cardiac sympathetic nerves in PD. This chronological and dynamic relationship between alpha-synuclein aggregation and distally dominant degeneration of cardiac noradrenergic nerves may represent the pathological mechanism behind a common degenerative process in PD.
In conclusion, cardiac noradrenergic denervation in Lewy body diseases, even in early stages, accounts for reduced cardiac uptake of 123I-metaiodobenzylguanidine and 6-[18F]fluorodopamine in PD. Alpha-synuclein aggregation appears to be intimately involved in the cardiac noradrenergic denervation that attends Lewy body diseases. The pathogenetic process seems to proceed in a centripetal, retrograde direction.
CYTOPLASMIC MONOAMINE METABOLITES AS AUTOTOXINS
Current concepts about mechanisms of PD emphasize pathologic alpha-synuclein accumulation, oxidative injury, impaired proteasomal or mitochondrial functions, neuroinflammation, or abnormal kinase signaling. These concepts do not explain relatively selective nigrostriatal dopaminergic and cardiac noradrenergic denervation in PD.
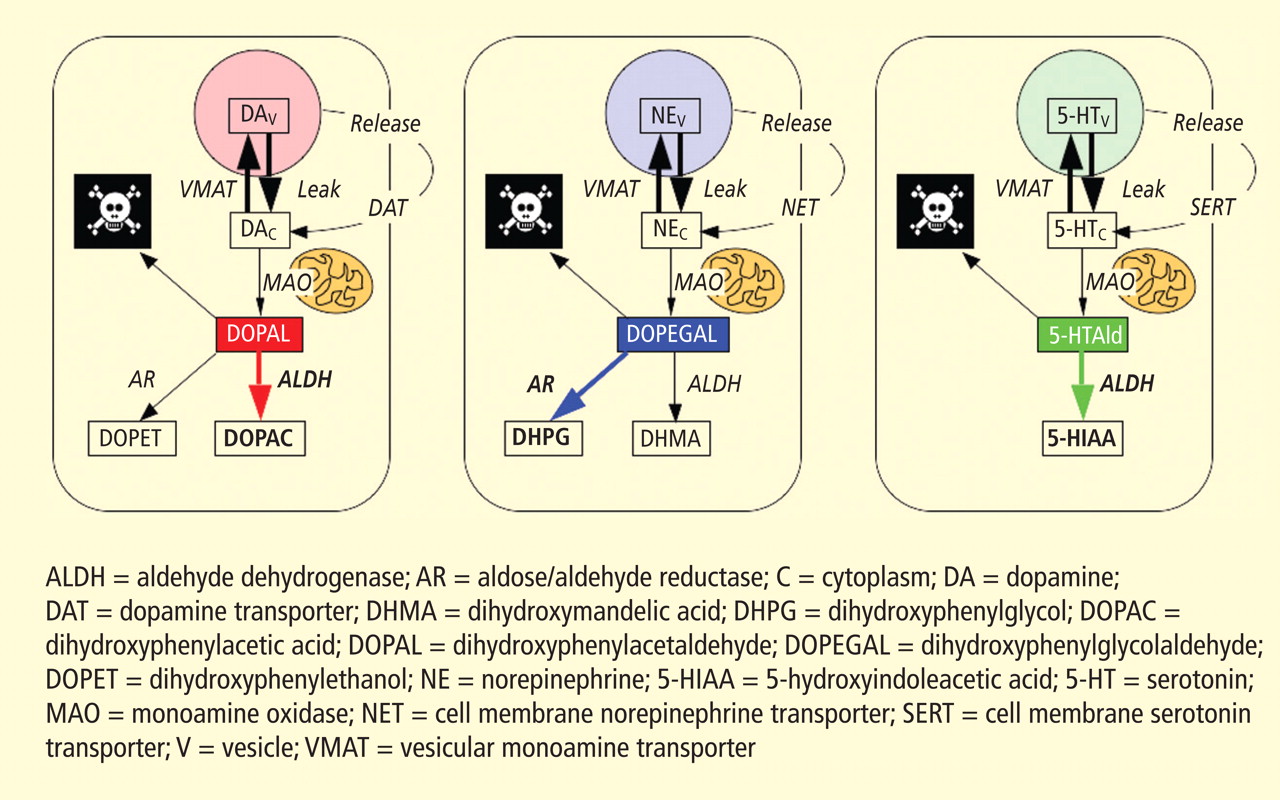
Figure 3. According to the monoamine aldehyde hypothesis, interference with the vesicular recycling of cytoplasmic monoamines (dopamine [DA], norepinephrine [NE], and serotonin [5-HT]) augments formation of toxic aldehydes. For instance, DA that leaks from vesicles (V) into the cytoplasm (C) or that is taken up via the cell membrane DA transporter (DAT) and escapes vesicular reuptake via the vesicular monoamine transporter (VMAT) is subject to oxidative deamination catalyzed by monoamine oxidase (MAO) to form the catecholaldehyde DOPAL, which is toxic. DOPAL is detoxified by ALDH to form DOPAC, the major metabolic route, or by AR to form DOPET, the minor metabolic route. Analogously, NE is converted to DOPEGAL, and 5-HT is converted to 5-HT-aldehyde (5-HTAld).
A potential explanation is that cytoplasmic catecholamine metabolites are autotoxins (Figure 3). The mechanisms of autotoxicity include spontaneous auto-oxidation, to form quinones and chromes leading to increased production of reactive oxygen species, and enzymatic oxidation.
Catecholamines in the neuronal cytoplasm undergo enzymatic oxidative deamination to form catecholaldehydes (dihydroxyphenylacetaldehyde [DOPAL] from dopamine), which are cytotoxic, as predicted by Blaschko more than a half century ago.10 DOPAL is detoxified mainly by aldehyde dehydrogenase (ALDH). In the substantia nigra, aldehyde dehydrogenase 1A1 (ALDH1A1) is the main isoform of ALDH, and postmortem studies have noted decreased nigral ALDH1A1 gene expression11,12 and protein content13 in PD patients.
All neurons express alpha-synuclein. Current concepts about mechanisms also do not explain the relatively selective aggregation of alpha-synuclein in catecholaminergic neurons. Alpha-synuclein appears to play a role in the cycling of catecholamines across vesicular and cell membranes.14
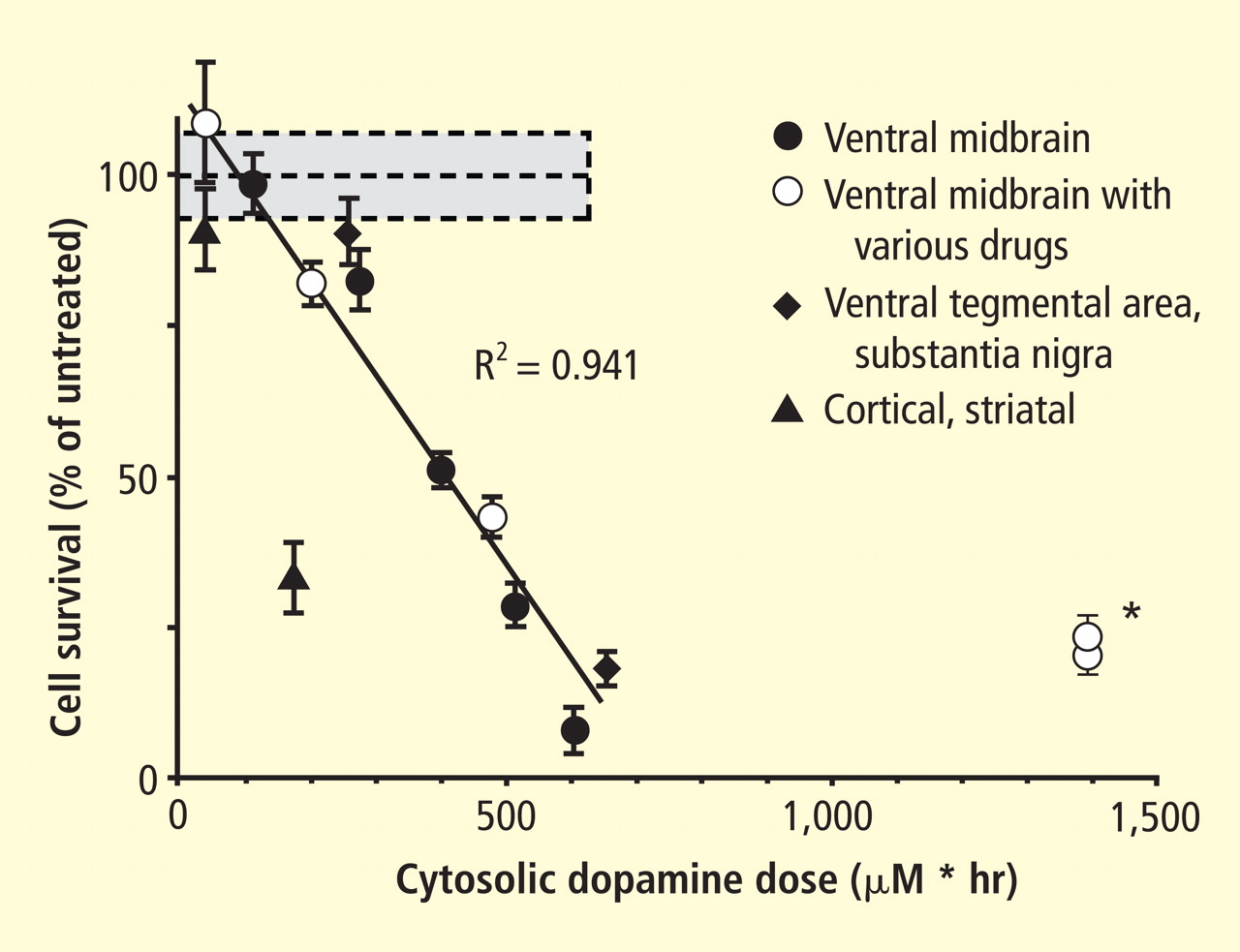
Figure 4. Cell survival and cytoplasmic dopamine are inversely related, according to a murine model by Mosharov et al.16 Graph shows the dependence of cell survival under l-dopa–induced stress on the cytoplasmic dopamine (DAcyt) dose in mouse neurons. The DAcyt dose was estimated as: [DAcyt]×TExposure = [DAcyt]×Ln([L-dopa]/K0.5)/k,where [DAcyt] is the concentration of cytosolic DA in cells treated with a saturating level (> 50 μM) of l-dopa for 1 hour, where [l-dopa] is the initial drug concentration, and where K0.5 = 9.7 μM and k = 0.15 hr−1 are the kinetic constants. TExposure approximates the time during which extracellular l-dopa remained higher than K0.5. The data points are (from left to right): filled circles—ventral midbrain cultures treated with 25, 100, 250, 500, and 1,000 μM l-dopa alone; open circles—ventral midbrain neurons treated with 250 μM l-dopa in the presence of benserazide, methamphetamine, reserpine, pargyline, and pargyline reserpine; diamonds—ventral tegmental area and substantia nigra neurons; triangles—striatal and cortical neurons treated with 250 μM l-dopa. Dotted lines and shaded boxes represent mean ± SEM in untreated cells. The solid line is the linear fit of all data points, excluding striatal and cortical neurons and the two data points indicated by the asterisk. Treatments to the right of this line are neuroprotective, as the same level of cell death is achieved with higher DAcyt doses; treatments to the left of this line are more susceptible to DAcyt stress.In the past year, a few important studies have been published related to autotoxicity of cytoplasmic catecholamine metabolites and to pathogenic interactions with alpha-synuclein. In 2006, Mosharov et al reported that alpha-synuclein overexpression increases cytoplasmic dopamine concentrations in rat pheochromocytoma PC-12 cells.15 Recently, the same group, using intracellular patch electrochemistry, directly measured cytoplasmic dopamine in cultured midbrain neurons and found that increases in dopamine and its metabolites are neurotoxic, whereas manipulations that reduce cytoplasmic dopamine are neuroprotective (Figure 4).16 Levodopa (l-dopa) increased cytoplasmic dopamine more in substantia nigra neurons than in ventral tegmental neurons, suggesting that this difference might help explain the greater susceptibility of nigral neurons to the pathogenetic process. The greater buildup of cytoplasmic dopamine seemed to depend on dihydropyridine-sensitive calcium (Ca2+) channels. Finally, dopaminergic neurons lacking alpha-synuclein were resistant to l-dopa–induced cell death. These findings led the authors to propose a “multiple-hit” model (Figure 5) in which interactions between intracellular ionized calcium, cytoplasmic dopamine, and alpha-synuclein underlie susceptibility of nigral neurons in PD.16
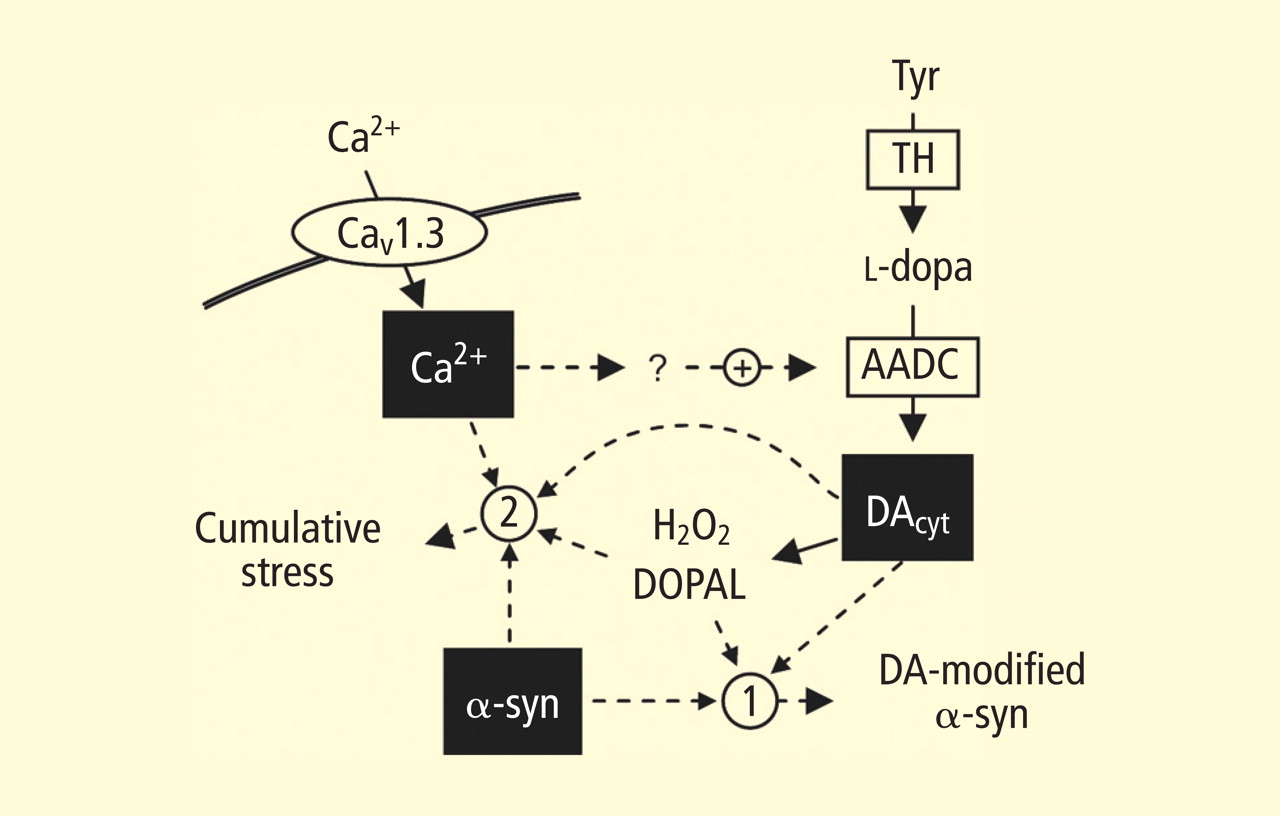
Figure 5. The “multiple-hit” model of Parkinson disease pathogenesis,16 which holds that neurotoxicity is a result of multiple factors, including the presence of alpha-synuclein (α-syn), elevation of cytoplasmic calcium (Ca2+), and buildup of cytoplasmic dopamine (DAcyt) and its metabolites. Nonexclusive toxic steps may result from (1) mechanisms that require direct interaction between DA or its metabolites with α-syn, such as DA-modified stabilization of α-syn protofibrils or inhibition of chaperone-mediated autophagy, or (2) cumulative damage from multiple independent sources. Reducing the levels of any of the three players provides neuroprotection. (AADC = aromatic l-amino acid decarboxylase; DOPAL = dihydroxyphenylacetaldehyde; TH = tyrosine hydroxylase)Burke et al added a potentially important clue, demonstrating that DOPAL potently oligomerizes and aggregates alpha-synuclein.17 This finding introduces the possibility of multiple pathogenetic positive feedback loops.
Under resting conditions, most catecholamine turnover results from leakage from vesicular stores into the cytoplasm and subsequent oxidative deamination by monoamine oxidase. Ordinarily, however, catecholamines in the cytoplasm are efficiently recycled back into the vesicles via the type 2 vesicular monoamine transporter (VMAT-2). Accordingly, interference with VMAT functions would be expected to tend to build up cytoplasmic catecholamines, with potentially cytotoxic consequences. In 2007, Caudle et al reported that mice with severely decreased VMAT-2 have aging-associated decreases in striatal dopamine that begin in the terminal fields, alpha-synuclein deposition in substantia nigra neurons, and l-dopa–responsive behavioral deficits.18 More recently the same group noted nonmotor signs associated with PD in VMAT-2–deficient mice, such as anosmia, gastrointestinal hypomotility, sleep disturbances, anxiety, and depression.19 Since VMAT-2 serves to recycle not only dopamine but also norepinephrine and serotonin, this single abnormality could help explain loss of all three types of monoaminergic neurons in PD.
Finally, Pena-Silva et al recently tested whether serotonin induces oxidative stress in human heart valves.20 They showed that in heart valves from explanted human hearts not used for transplantation, incubation of homogenates of cardiac valves and blood vessels with serotonin increased generation of the superoxide free radical. Inhibitors of monoamine oxidase prevented this effect. Dopamine also increased superoxide levels in heart valves, and this effect was also attenuated by monoamine oxidase inhibition. These findings fit with the concept that the aldehydes produced by the action of monoamine oxidase on cytoplasmic monoamines generate toxic free radicals.
VALIDITY OF POWER SPECTRAL ANALYSIS OF HEART RATE VARIABILITY TO INDICATE CARDIAC SYMPATHETIC TONE
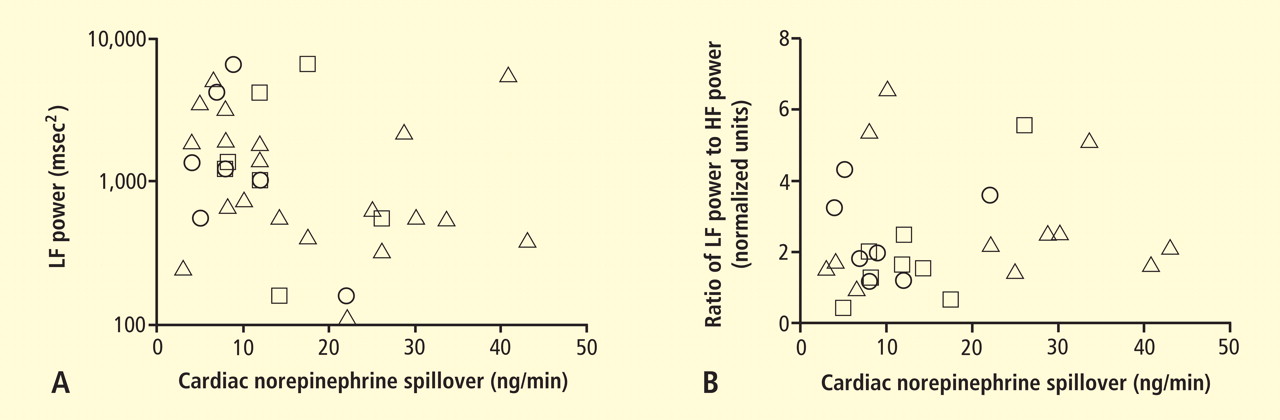
Power spectral analysis of heart rate variability is simple, relatively inexpensive, noninvasive, and widely used to indicate cardiac sympathetic “tone” or sympathovagal “balance.” Almost 2,000 studies to date have used this modality. Relatively increased cardiac sympathetic tone, reflected by low-frequency (LF) power or the ratio of LF power to high-frequency (HF) power, is an adverse prognostic sign in a variety of conditions. Nevertheless, the validity of LF power, or the LF:HF ratio, as an index of cardiac sympathetic tone remains unsettled.
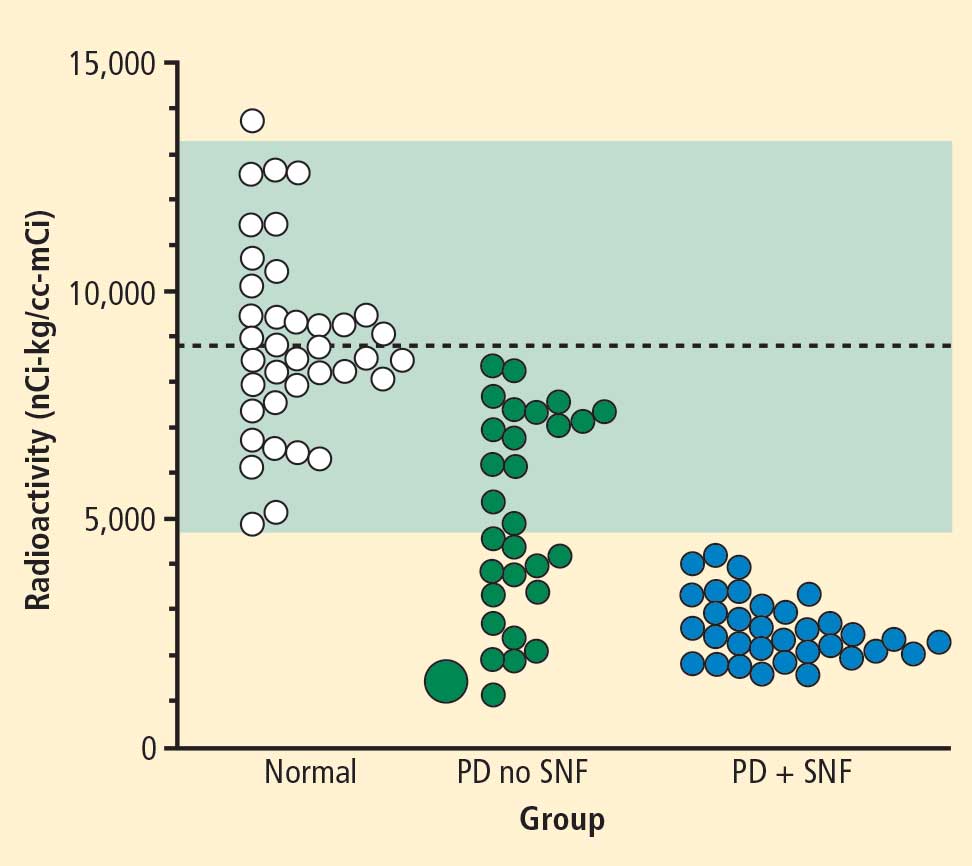
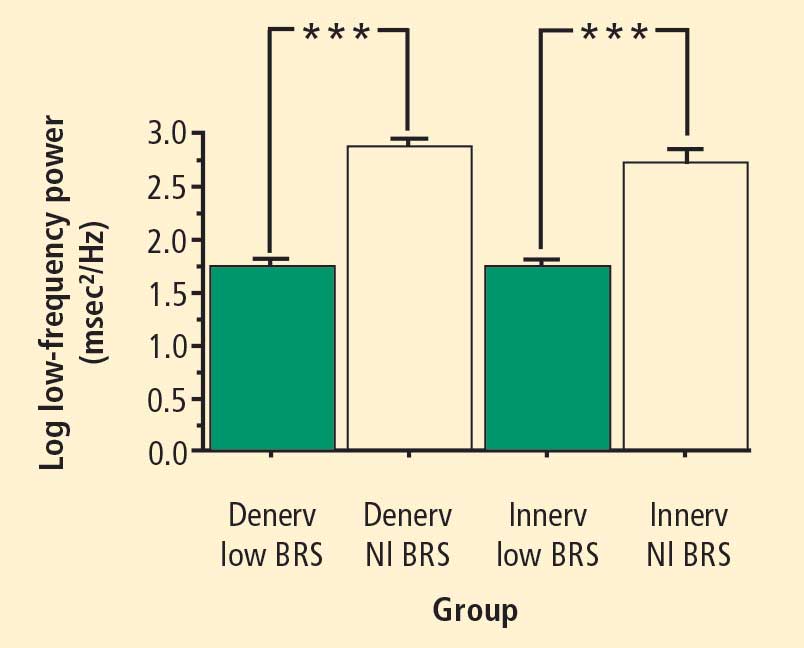
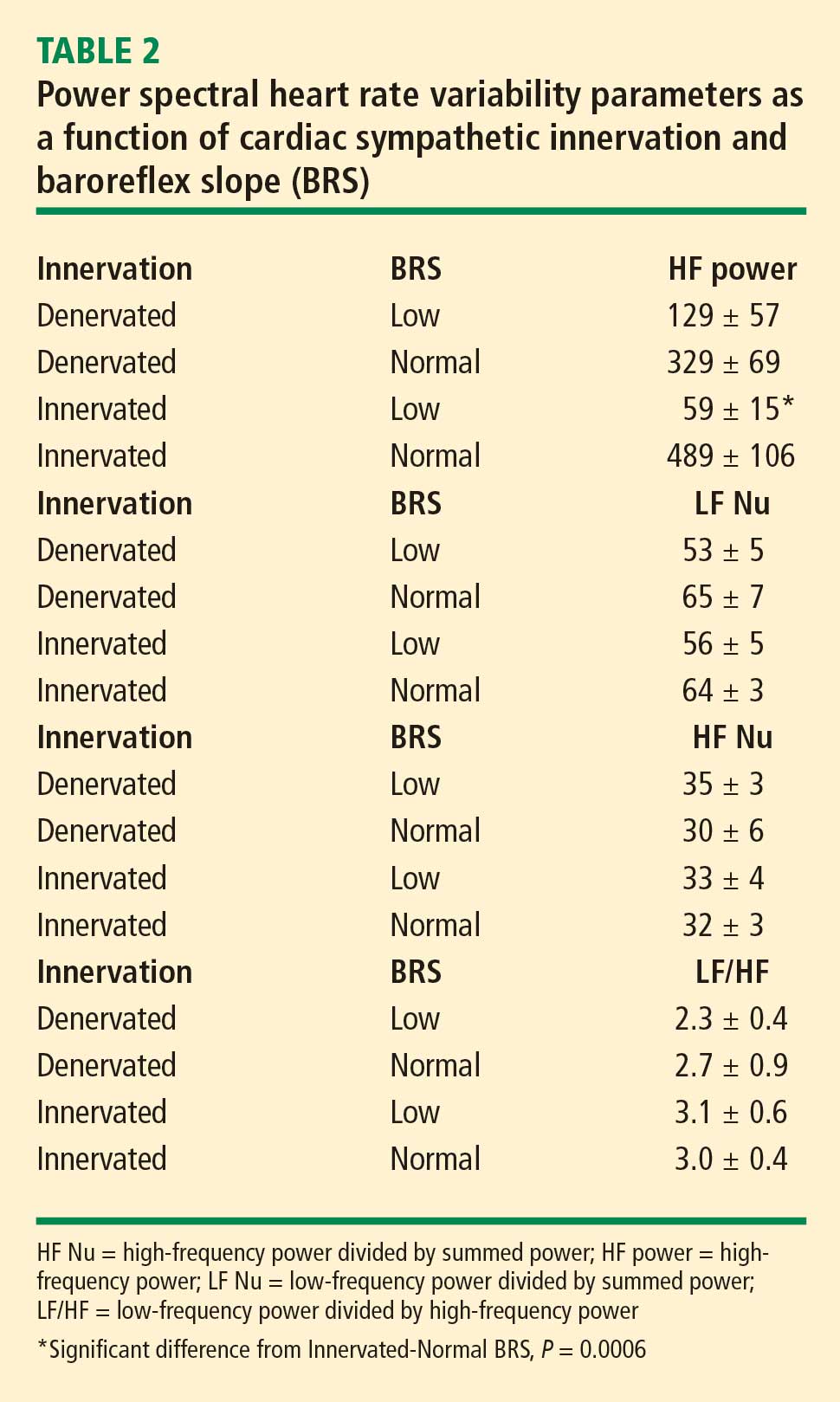
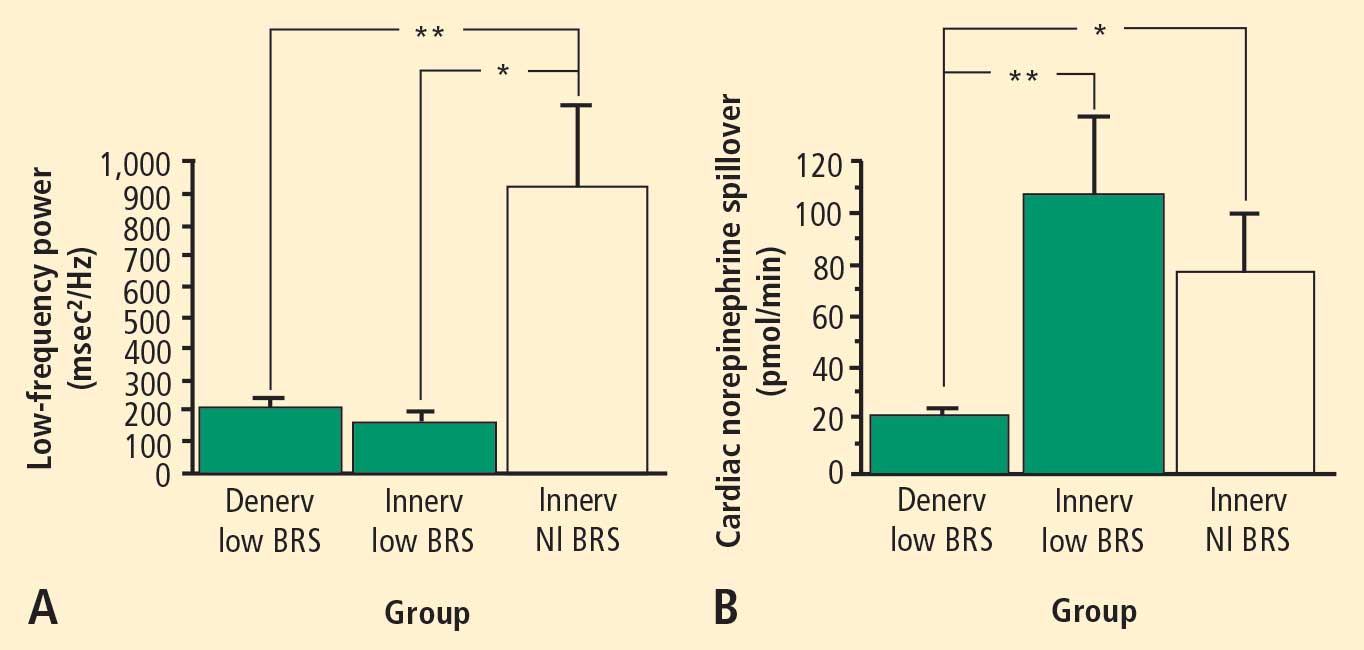
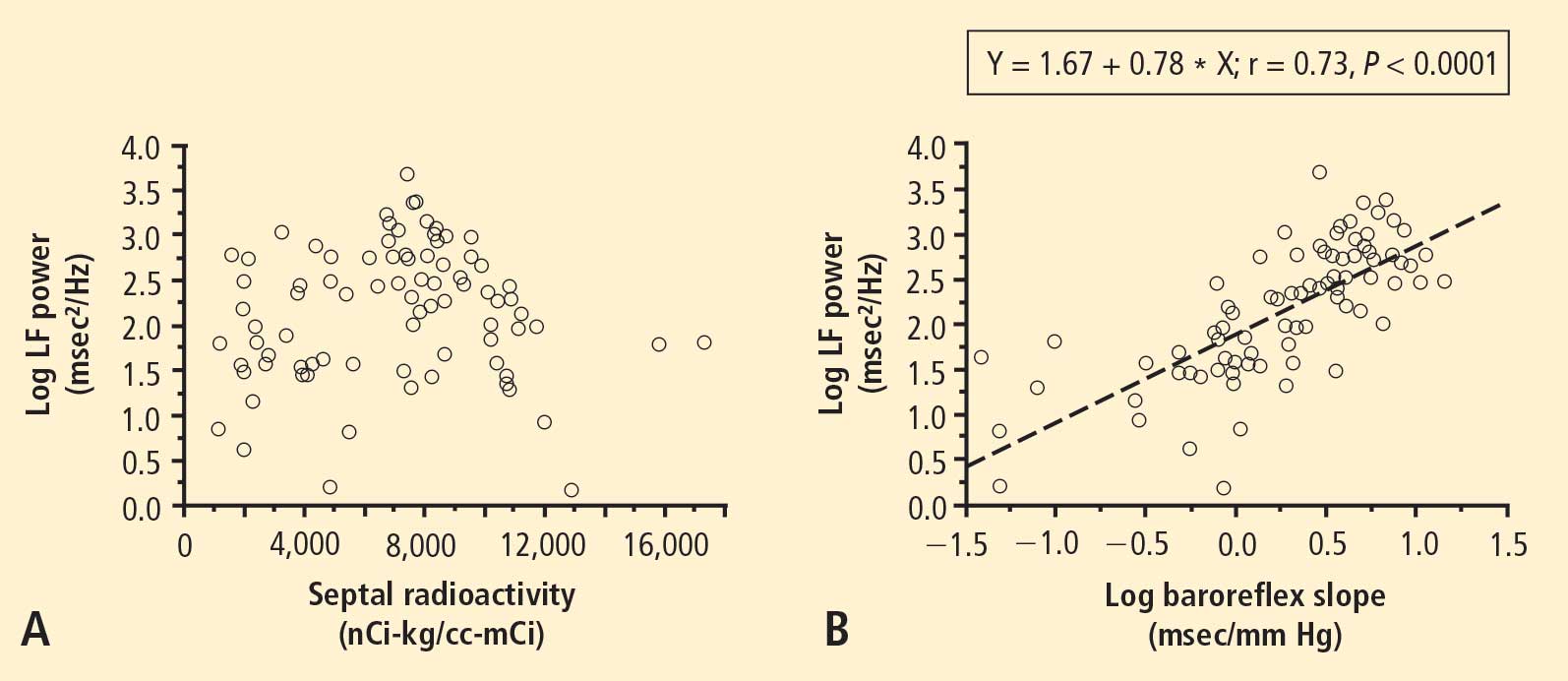
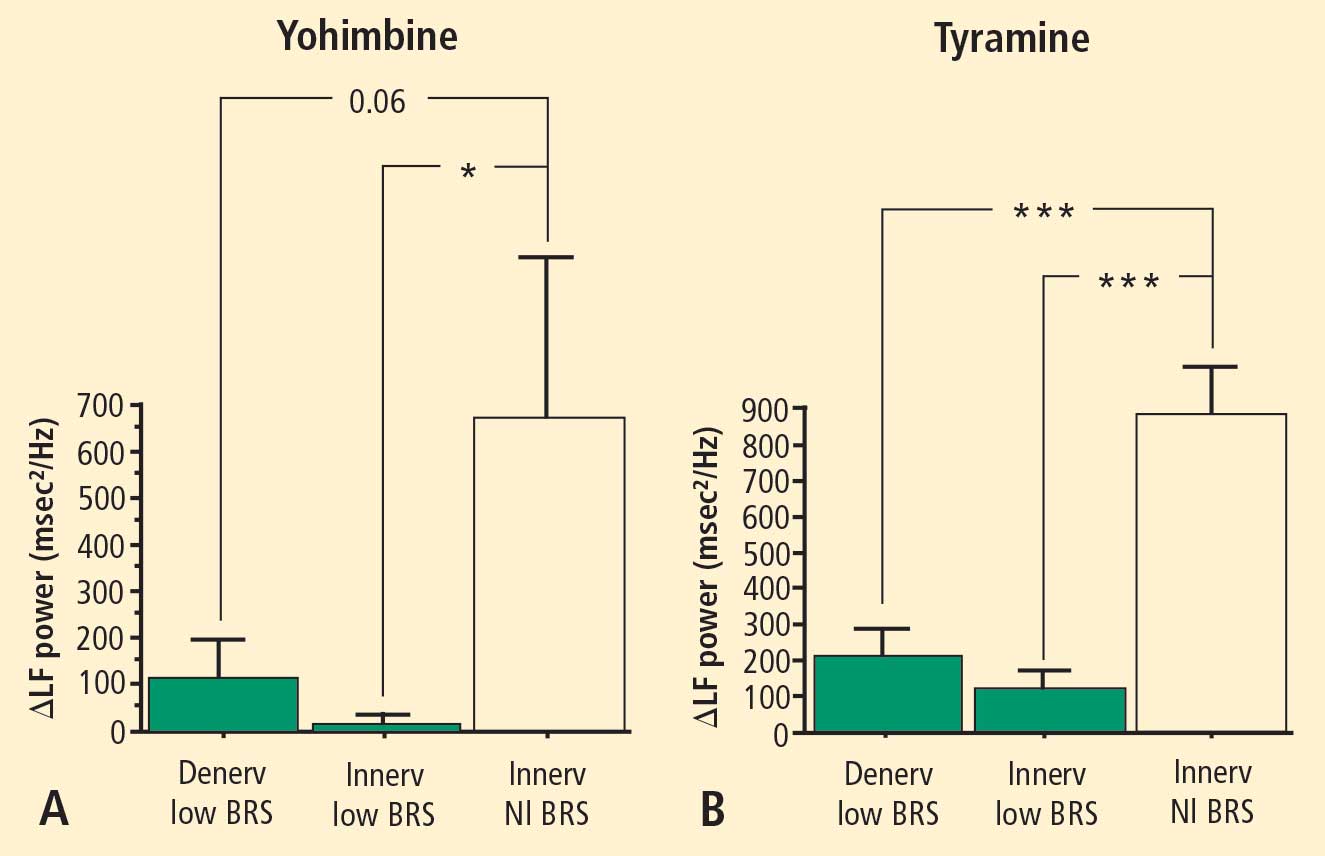
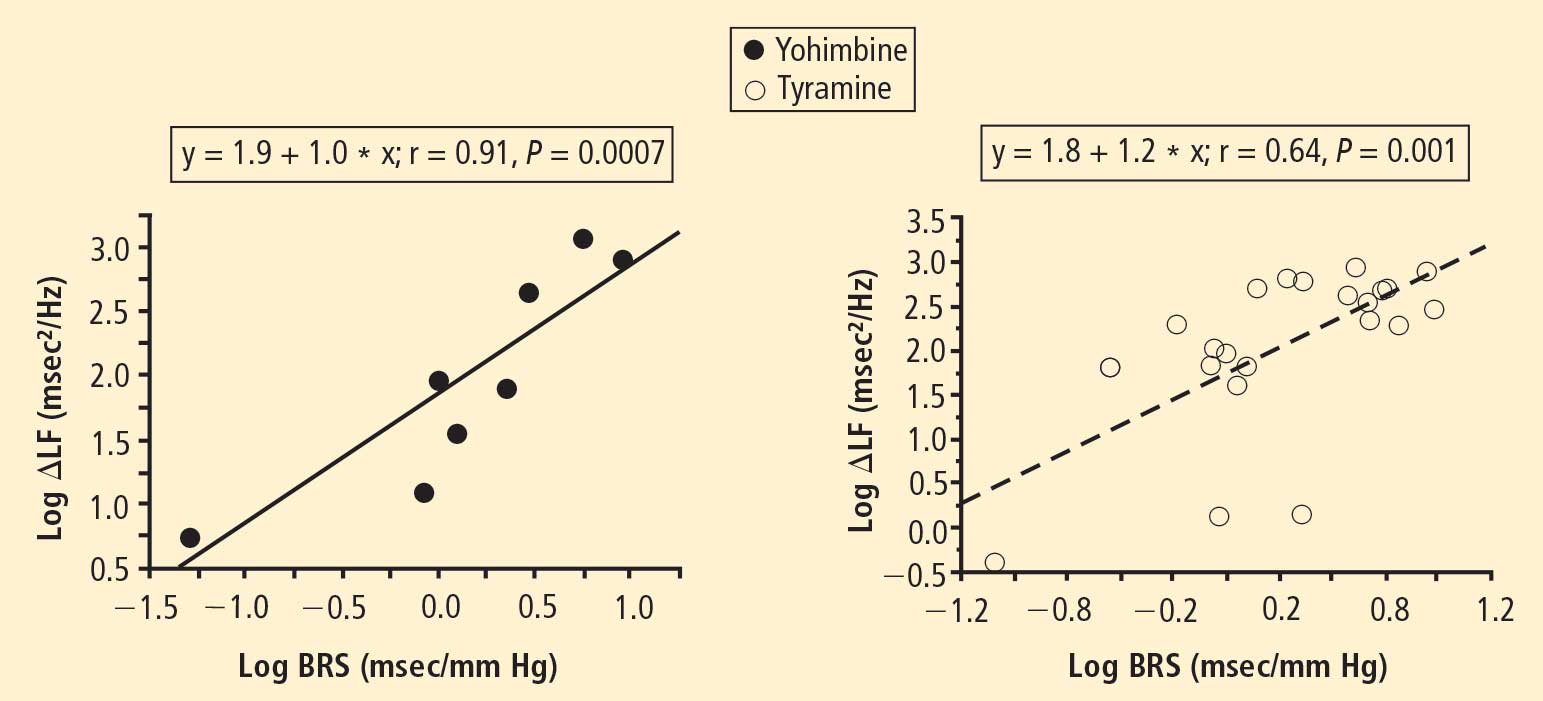
In 2007 we assessed the validity of power spectral analysis rather directly, by taking advantage of our ability to delineate cardiac sympathetic innervation. We compared LF power in patients with cardiac sympathetic denervation, indicated by low myocardial levels of 6-[18F]fluorodopamine–derived radioactivity or low rates of norepinephrine entry into coronary sinus plasma (cardiac norepinephrine spillover), with values in patients with intact innervation. LF power was unrelated to myocardial 6-[18F]fluorodopamine–derived radioactivity or cardiac norepinephrine spillover, but it was related to baroreflex-cardiovagal gain. Patients with a low baroreflex-cardiovagal gain had low LF power, regardless of cardiac innervation. From these findings we concluded that LF power reflects baroreflex function, not cardiac sympathetic innervation.21
Figure 6. Relationships of heart rate variability indices with cardiac norepinephrine spillover. Graphs show time and frequency domain heart rate variability measures (LF = low frequency; HF = high frequency) versus cardiac norepinephrine in healthy subjects (squares) and patients with major depression (triangles) and panic disorder (circles).22 Recently Baumert et al also examined the relationship between indices from power spectral analysis of heart rate variability and cardiac norepinephrine spillover.22 They found, as we did, that none of the standard heart rate variability parameters was correlated with cardiac norepinephrine spillover (Figure 6). The same group reported a positive correlation between the heart rate–corrected QT interval and cardiac norepinephrine spillover.23 Among patients with major depression, the distribution of cardiac norepinephrine spillover seemed bimodal. Overall, cardiac norepinephrine spillover was not increased, although a subgroup had clearly increased spillover.
In congestive heart failure, baroreflex-cardiovagal gain tends to be low and cardiac sympathetic outflow markedly increased, yet the LF:HF ratio is not increased during supine rest.24,25 It therefore appears that power spectral analysis of heart rate variability may provide a measure of baroreflexive modulation of autonomic outflows to the heart but not a measure of those outflows themselves. The search continues for a valid, noninvasive means to assess cardiac sympathetic function.
References
Goldstein DS, Holmes C, Cannon RO, Eisenhofer G, Kopin IJ. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med1997; 336:696–702.
Orimo S, Ozawa E, Oka T, et al Different histopathology accounting for a decrease in myocardial MIBG uptake in PD and MSA. Neurology2001; 57:1140–1141.
Amino T, Orimo S, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Path2005; 15:29–34.
Goldstein DS, Li S-T, Kopin IJ. Sympathetic neurocirculatory failure in Parkinson disease: evidence for an etiologic role of α-synuclein. Ann Intern Med2001; 135:1010–1011.
Singleton A, Gwinn-Hardy K, Sharabi Y, et al Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain2004; 127:768–772.
Orimo S, Uchihara T, Nakamura A, et al Cardiac sympathetic denervation in Parkinson’s disease linked to SNCA duplication. Acta Neuropathol2008; 116:575–577.
Orimo S, Amino T, Yokochi M, et al Preserved cardiac sympathetic nerve accounts for normal cardiac uptake of MIBG in PARK2. Mov Disord2005; 20:1350–1353.
Dickson DW, Fujishiro H, DelleDonne A, et al Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol2008; 115:437–444.
Orimo S, Uchihara T, Nakamura A, et al Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain2008; 131:642–650.
Blaschko H. Amine oxidase and amine metabolism. Pharmacol Rev1952; 4:415–458.
Galter D, Buervenich S, Carmine A, Anvret M, Olson L. ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson’s disease and in the ventral tegmental area in schizophrenia. Neurobiol Dis2003; 14:637–647.
Mandel S, Grunblatt E, Riederer P, et al Gene expression profiling of sporadic Parkinson’s disease substantia nigra pars compacta reveals impairment of ubiquitin-proteasome subunits, SKP1A, aldehyde dehydrogenase, and chaperone HSC-70. Ann N Y Acad Sci2005; 1053:356–375.
Werner CJ, Heyny-von Haussen R, Mall G, Wolf S. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome Sci2008; 6:8.
Larsen KE, Schmitz Y, Troyer MD, et al Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci2006; 26:11915–11922.
Mosharov EV, Staal RG, Bove J, et al Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J Neurosci2006; 26:9304–9311.
Mosharov EV, Larsen KE, Kanter E, et al Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron2009; 62:218–229.
Burke WJ, Kumar VB, Pandey N, et al Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol2008; 115:193–203.
Caudle WM, Richardson JR, Wang MZ, et al Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci2007; 27:8138–8148.
Taylor TN, Caudle WM, Shepherd KR, et al Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci2009; 29:8103–8113.
Pena-Silva RA, Miller JD, Chu Y, Heistad DD. Serotonin produces monoamine oxidase-dependent oxidative stress in human heart valves. Am J Physiol Heart Circ Physiol2009; 297:H1354–H1360.
Moak JP, Goldstein DS, Eldadah BA, et al Supine low-frequency power of heart rate variability reflects baroreflex function, not cardiac sympathetic innervation. Heart Rhythm2007; 4:1523–1529.
Baumert M, Lambert GW, Dawood T, et al Short-term heart rate variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol2009; 297:H674–H679.
Baumert M, Lambert GW, Dawood T, et al QT interval variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol2008; 295:H962–H968.
Tygesen H, Rundqvist B, Waagstein F, Wennerblom B. Heart rate variability measurement correlates with cardiac norepinephrine spillover in congestive heart failure. Am J Cardiol2001; 87:1308–1311.
Kingwell BA, Thompson JM, Kaye DM, McPherson GA, Jennings GL, Esler MD. Heart rate spectral analysis, cardiac norepinephrine spillover, and muscle sympathetic nerve activity during human sympathetic nervous activation and failure. Circulation1994; 90:234–240.
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, Clinical Neurosciences Program, Division of Intramural Research, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Building 10, Room 5N220, 10 Center Drive, MSC-1620, Bethesda, MD 20892–1620; goldsteind@ninds.nih.gov
Dr. Goldstein reported that he has no financial relationships that pose a potential conflict of interest with this article.
Some of the research summarized here was supported by the intramural research program of the National Institutes of Health.
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, Clinical Neurosciences Program, Division of Intramural Research, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Building 10, Room 5N220, 10 Center Drive, MSC-1620, Bethesda, MD 20892–1620; goldsteind@ninds.nih.gov
Dr. Goldstein reported that he has no financial relationships that pose a potential conflict of interest with this article.
Some of the research summarized here was supported by the intramural research program of the National Institutes of Health.
Author and Disclosure Information
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, Clinical Neurosciences Program, Division of Intramural Research, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Building 10, Room 5N220, 10 Center Drive, MSC-1620, Bethesda, MD 20892–1620; goldsteind@ninds.nih.gov
Dr. Goldstein reported that he has no financial relationships that pose a potential conflict of interest with this article.
Some of the research summarized here was supported by the intramural research program of the National Institutes of Health.
This review highlights important recent publications in the area of neuroscience and heart-brain medicine. Abnormalities of regulation of the circulation by catecholamine systems figure as a general theme of the topics highlighted. These topics, which are reviewed in turn below, are (1) mechanisms of cardiac sympathetic denervation in Parkinson disease (PD), (2) cytoplasmic monoamine metabolites as autotoxins, and (3) the validity of power spectral analysis of heart rate variability to indicate cardiac sympathetic tone.
MECHANISMS OF CARDIAC SYMPATHETIC DENERVATION IN PARKINSON DISEASE
The movement disorder component of PD is well recognized as resulting from loss of dopaminergic neurons in the nigrostriatal system of the brain. The finding of low myocardial 6-[18F]fluorodopamine–derived radioactivity by positron emission tomography provided the first neuroimaging evidence for loss of catecholaminergic neurons outside the brain in PD.1 Many reports using 123I-metaiodobenzylguanidine scanning have concurred with this finding. Beginning in the early 2000s, post-mortem neuropathologic studies demonstrated virtually absent immunoreactivity for tyrosine hydroxylase, the rate-limiting enzyme in norepinephrine biosynthesis, in epicardial nerves in PD.2,3 These results provided clues to the mechanism of autonomic dysfunction in PD, a prominent nonmotor manifestation of the disease.
With kind permission from Springer Science+Business Media: Acta Neuropathologica,
Figure 1. Tyrosine hydroxylase immunoreactivity (THir) in epicardial nerve from (A) a control subject and (B) a patient with familial Parkinson disease due to duplication of the gene encoding alpha-synuclein (PARK4).6Alpha-synuclein is a key protein in the pathogenesis of PD. It is abundant in Lewy bodies and Lewy neurites, and mutations or multiplications of the gene that encodes it cause rare inherited forms of PD. In 2001 we reported evidence for cardiac sympathetic denervation, neurogenic orthostatic hypotension, and baroreflex failure in familial PD from mutation of the gene encoding alpha-synuclein.4 Subsequently we reported analogous denervation in familial PD from triplication of the normal gene.5 This past year Orimo’s group in Tokyo provided the first pathological confirmation of cardiac sympathetic denervation in familial PD from inherited alpha-synucleinopathy, based on severely decreased epicardial neuronal tyrosine hydroxylase immunoreactivity (Figure 1).6 In contrast, patients with familial PD from parkin gene mutation, which is not thought to be a Lewy body disease, have been found to have normal cardiac 123I-metaiodobenzyl-guanidine–derived radioactivity and normal epicardial neuronal tyrosine hydroxylase immunoreactivity.7 These findings establish a link between alpha-synucleinopathy and cardiac sympathetic denervation.
Some individuals who die without clinical parkinsonism have Lewy bodies detected pathologically. Growing evidence shows that incidental Lewy body disease represents early, presymptomatic PD.8 Orimo’s group therefore studied cardiac tissues and paravertebral sympathetic ganglia from patients with incidental Lewy body disease.9 Postmortem tissues were likewise obtained from comparison subjects with multiple system atrophy and from control subjects. Immunohistochemical analyses were performed using antibodies against tyrosine hydroxylase, phosphorylated neurofilament as a marker of axons, and phosphorylated alpha-synuclein as a marker of abnormal alpha-synuclein deposits. Key findings from this study9 were as follows:
Reprinted from Brain (Orimo S, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008; 131:642–650) by permission of Oxford University Press.
Figure 2. Concept diagram of the pathogenetic sequence of cardiac sympathetic denervation. In incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons (a), alpha-synuclein aggregates (black shading) accumulate abundantly in the distal axons but sparsely in the paravertebral sympathetic ganglia. In contrast, in incidental Lewy body disease with decreased THir axons (b), alpha-synuclein aggregates diminish in the distal axons but increase in the paravertebral sympathetic ganglia. In Parkinson disease, alpha-synuclein aggregates disappear in the distal axons and accumulate much more abundantly in the paravertebral sympathetic ganglia. In multiple system atrophy, alpha-synuclein aggregates are generally not observed (as in controls), with a few exceptions. Dotted lines indicate degeneration of THir axons.9Alpha-synuclein aggregates in distal epicardial nerve fascicles were more abundant in incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons than in incidental Lewy body disease with decreased THir axons (Figure 2).
Alpha-synuclein aggregates in the epicardial nerve fibers were closely related to the disappearance of THir axons.
In incidental Lewy body disease with preserved THir axons, alpha-synuclein aggregates were consistently more abundant in the epicardial nerves than in the paravertebral sympathetic ganglia (Figure 2).
Distally dominant accumulation of alpha-synuclein aggregates was reversed in incidental Lewy body disease with decreased THir axons and in PD, because both conditions involve fewer alpha-synuclein aggregates in axons and more abundant aggregates in the paravertebral sympathetic ganglia (Figure 2).
Thus, accumulation of alpha-synuclein aggregates in distal cardiac sympathetic axons precedes aggregation in neuronal somata or ganglionic neurites, heralding centripetal degeneration of cardiac sympathetic nerves in PD. This chronological and dynamic relationship between alpha-synuclein aggregation and distally dominant degeneration of cardiac noradrenergic nerves may represent the pathological mechanism behind a common degenerative process in PD.
In conclusion, cardiac noradrenergic denervation in Lewy body diseases, even in early stages, accounts for reduced cardiac uptake of 123I-metaiodobenzylguanidine and 6-[18F]fluorodopamine in PD. Alpha-synuclein aggregation appears to be intimately involved in the cardiac noradrenergic denervation that attends Lewy body diseases. The pathogenetic process seems to proceed in a centripetal, retrograde direction.
CYTOPLASMIC MONOAMINE METABOLITES AS AUTOTOXINS
Current concepts about mechanisms of PD emphasize pathologic alpha-synuclein accumulation, oxidative injury, impaired proteasomal or mitochondrial functions, neuroinflammation, or abnormal kinase signaling. These concepts do not explain relatively selective nigrostriatal dopaminergic and cardiac noradrenergic denervation in PD.
Figure 3. According to the monoamine aldehyde hypothesis, interference with the vesicular recycling of cytoplasmic monoamines (dopamine [DA], norepinephrine [NE], and serotonin [5-HT]) augments formation of toxic aldehydes. For instance, DA that leaks from vesicles (V) into the cytoplasm (C) or that is taken up via the cell membrane DA transporter (DAT) and escapes vesicular reuptake via the vesicular monoamine transporter (VMAT) is subject to oxidative deamination catalyzed by monoamine oxidase (MAO) to form the catecholaldehyde DOPAL, which is toxic. DOPAL is detoxified by ALDH to form DOPAC, the major metabolic route, or by AR to form DOPET, the minor metabolic route. Analogously, NE is converted to DOPEGAL, and 5-HT is converted to 5-HT-aldehyde (5-HTAld).
A potential explanation is that cytoplasmic catecholamine metabolites are autotoxins (Figure 3). The mechanisms of autotoxicity include spontaneous auto-oxidation, to form quinones and chromes leading to increased production of reactive oxygen species, and enzymatic oxidation.
Catecholamines in the neuronal cytoplasm undergo enzymatic oxidative deamination to form catecholaldehydes (dihydroxyphenylacetaldehyde [DOPAL] from dopamine), which are cytotoxic, as predicted by Blaschko more than a half century ago.10 DOPAL is detoxified mainly by aldehyde dehydrogenase (ALDH). In the substantia nigra, aldehyde dehydrogenase 1A1 (ALDH1A1) is the main isoform of ALDH, and postmortem studies have noted decreased nigral ALDH1A1 gene expression11,12 and protein content13 in PD patients.
All neurons express alpha-synuclein. Current concepts about mechanisms also do not explain the relatively selective aggregation of alpha-synuclein in catecholaminergic neurons. Alpha-synuclein appears to play a role in the cycling of catecholamines across vesicular and cell membranes.14
Figure 4. Cell survival and cytoplasmic dopamine are inversely related, according to a murine model by Mosharov et al.16 Graph shows the dependence of cell survival under l-dopa–induced stress on the cytoplasmic dopamine (DAcyt) dose in mouse neurons. The DAcyt dose was estimated as: [DAcyt]×TExposure = [DAcyt]×Ln([L-dopa]/K0.5)/k,where [DAcyt] is the concentration of cytosolic DA in cells treated with a saturating level (> 50 μM) of l-dopa for 1 hour, where [l-dopa] is the initial drug concentration, and where K0.5 = 9.7 μM and k = 0.15 hr−1 are the kinetic constants. TExposure approximates the time during which extracellular l-dopa remained higher than K0.5. The data points are (from left to right): filled circles—ventral midbrain cultures treated with 25, 100, 250, 500, and 1,000 μM l-dopa alone; open circles—ventral midbrain neurons treated with 250 μM l-dopa in the presence of benserazide, methamphetamine, reserpine, pargyline, and pargyline reserpine; diamonds—ventral tegmental area and substantia nigra neurons; triangles—striatal and cortical neurons treated with 250 μM l-dopa. Dotted lines and shaded boxes represent mean ± SEM in untreated cells. The solid line is the linear fit of all data points, excluding striatal and cortical neurons and the two data points indicated by the asterisk. Treatments to the right of this line are neuroprotective, as the same level of cell death is achieved with higher DAcyt doses; treatments to the left of this line are more susceptible to DAcyt stress.In the past year, a few important studies have been published related to autotoxicity of cytoplasmic catecholamine metabolites and to pathogenic interactions with alpha-synuclein. In 2006, Mosharov et al reported that alpha-synuclein overexpression increases cytoplasmic dopamine concentrations in rat pheochromocytoma PC-12 cells.15 Recently, the same group, using intracellular patch electrochemistry, directly measured cytoplasmic dopamine in cultured midbrain neurons and found that increases in dopamine and its metabolites are neurotoxic, whereas manipulations that reduce cytoplasmic dopamine are neuroprotective (Figure 4).16 Levodopa (l-dopa) increased cytoplasmic dopamine more in substantia nigra neurons than in ventral tegmental neurons, suggesting that this difference might help explain the greater susceptibility of nigral neurons to the pathogenetic process. The greater buildup of cytoplasmic dopamine seemed to depend on dihydropyridine-sensitive calcium (Ca2+) channels. Finally, dopaminergic neurons lacking alpha-synuclein were resistant to l-dopa–induced cell death. These findings led the authors to propose a “multiple-hit” model (Figure 5) in which interactions between intracellular ionized calcium, cytoplasmic dopamine, and alpha-synuclein underlie susceptibility of nigral neurons in PD.16
Figure 5. The “multiple-hit” model of Parkinson disease pathogenesis,16 which holds that neurotoxicity is a result of multiple factors, including the presence of alpha-synuclein (α-syn), elevation of cytoplasmic calcium (Ca2+), and buildup of cytoplasmic dopamine (DAcyt) and its metabolites. Nonexclusive toxic steps may result from (1) mechanisms that require direct interaction between DA or its metabolites with α-syn, such as DA-modified stabilization of α-syn protofibrils or inhibition of chaperone-mediated autophagy, or (2) cumulative damage from multiple independent sources. Reducing the levels of any of the three players provides neuroprotection. (AADC = aromatic l-amino acid decarboxylase; DOPAL = dihydroxyphenylacetaldehyde; TH = tyrosine hydroxylase)Burke et al added a potentially important clue, demonstrating that DOPAL potently oligomerizes and aggregates alpha-synuclein.17 This finding introduces the possibility of multiple pathogenetic positive feedback loops.
Under resting conditions, most catecholamine turnover results from leakage from vesicular stores into the cytoplasm and subsequent oxidative deamination by monoamine oxidase. Ordinarily, however, catecholamines in the cytoplasm are efficiently recycled back into the vesicles via the type 2 vesicular monoamine transporter (VMAT-2). Accordingly, interference with VMAT functions would be expected to tend to build up cytoplasmic catecholamines, with potentially cytotoxic consequences. In 2007, Caudle et al reported that mice with severely decreased VMAT-2 have aging-associated decreases in striatal dopamine that begin in the terminal fields, alpha-synuclein deposition in substantia nigra neurons, and l-dopa–responsive behavioral deficits.18 More recently the same group noted nonmotor signs associated with PD in VMAT-2–deficient mice, such as anosmia, gastrointestinal hypomotility, sleep disturbances, anxiety, and depression.19 Since VMAT-2 serves to recycle not only dopamine but also norepinephrine and serotonin, this single abnormality could help explain loss of all three types of monoaminergic neurons in PD.
Finally, Pena-Silva et al recently tested whether serotonin induces oxidative stress in human heart valves.20 They showed that in heart valves from explanted human hearts not used for transplantation, incubation of homogenates of cardiac valves and blood vessels with serotonin increased generation of the superoxide free radical. Inhibitors of monoamine oxidase prevented this effect. Dopamine also increased superoxide levels in heart valves, and this effect was also attenuated by monoamine oxidase inhibition. These findings fit with the concept that the aldehydes produced by the action of monoamine oxidase on cytoplasmic monoamines generate toxic free radicals.
VALIDITY OF POWER SPECTRAL ANALYSIS OF HEART RATE VARIABILITY TO INDICATE CARDIAC SYMPATHETIC TONE
Power spectral analysis of heart rate variability is simple, relatively inexpensive, noninvasive, and widely used to indicate cardiac sympathetic “tone” or sympathovagal “balance.” Almost 2,000 studies to date have used this modality. Relatively increased cardiac sympathetic tone, reflected by low-frequency (LF) power or the ratio of LF power to high-frequency (HF) power, is an adverse prognostic sign in a variety of conditions. Nevertheless, the validity of LF power, or the LF:HF ratio, as an index of cardiac sympathetic tone remains unsettled.
In 2007 we assessed the validity of power spectral analysis rather directly, by taking advantage of our ability to delineate cardiac sympathetic innervation. We compared LF power in patients with cardiac sympathetic denervation, indicated by low myocardial levels of 6-[18F]fluorodopamine–derived radioactivity or low rates of norepinephrine entry into coronary sinus plasma (cardiac norepinephrine spillover), with values in patients with intact innervation. LF power was unrelated to myocardial 6-[18F]fluorodopamine–derived radioactivity or cardiac norepinephrine spillover, but it was related to baroreflex-cardiovagal gain. Patients with a low baroreflex-cardiovagal gain had low LF power, regardless of cardiac innervation. From these findings we concluded that LF power reflects baroreflex function, not cardiac sympathetic innervation.21
Figure 6. Relationships of heart rate variability indices with cardiac norepinephrine spillover. Graphs show time and frequency domain heart rate variability measures (LF = low frequency; HF = high frequency) versus cardiac norepinephrine in healthy subjects (squares) and patients with major depression (triangles) and panic disorder (circles).22 Recently Baumert et al also examined the relationship between indices from power spectral analysis of heart rate variability and cardiac norepinephrine spillover.22 They found, as we did, that none of the standard heart rate variability parameters was correlated with cardiac norepinephrine spillover (Figure 6). The same group reported a positive correlation between the heart rate–corrected QT interval and cardiac norepinephrine spillover.23 Among patients with major depression, the distribution of cardiac norepinephrine spillover seemed bimodal. Overall, cardiac norepinephrine spillover was not increased, although a subgroup had clearly increased spillover.
In congestive heart failure, baroreflex-cardiovagal gain tends to be low and cardiac sympathetic outflow markedly increased, yet the LF:HF ratio is not increased during supine rest.24,25 It therefore appears that power spectral analysis of heart rate variability may provide a measure of baroreflexive modulation of autonomic outflows to the heart but not a measure of those outflows themselves. The search continues for a valid, noninvasive means to assess cardiac sympathetic function.
This review highlights important recent publications in the area of neuroscience and heart-brain medicine. Abnormalities of regulation of the circulation by catecholamine systems figure as a general theme of the topics highlighted. These topics, which are reviewed in turn below, are (1) mechanisms of cardiac sympathetic denervation in Parkinson disease (PD), (2) cytoplasmic monoamine metabolites as autotoxins, and (3) the validity of power spectral analysis of heart rate variability to indicate cardiac sympathetic tone.
MECHANISMS OF CARDIAC SYMPATHETIC DENERVATION IN PARKINSON DISEASE
The movement disorder component of PD is well recognized as resulting from loss of dopaminergic neurons in the nigrostriatal system of the brain. The finding of low myocardial 6-[18F]fluorodopamine–derived radioactivity by positron emission tomography provided the first neuroimaging evidence for loss of catecholaminergic neurons outside the brain in PD.1 Many reports using 123I-metaiodobenzylguanidine scanning have concurred with this finding. Beginning in the early 2000s, post-mortem neuropathologic studies demonstrated virtually absent immunoreactivity for tyrosine hydroxylase, the rate-limiting enzyme in norepinephrine biosynthesis, in epicardial nerves in PD.2,3 These results provided clues to the mechanism of autonomic dysfunction in PD, a prominent nonmotor manifestation of the disease.
With kind permission from Springer Science+Business Media: Acta Neuropathologica,
Figure 1. Tyrosine hydroxylase immunoreactivity (THir) in epicardial nerve from (A) a control subject and (B) a patient with familial Parkinson disease due to duplication of the gene encoding alpha-synuclein (PARK4).6Alpha-synuclein is a key protein in the pathogenesis of PD. It is abundant in Lewy bodies and Lewy neurites, and mutations or multiplications of the gene that encodes it cause rare inherited forms of PD. In 2001 we reported evidence for cardiac sympathetic denervation, neurogenic orthostatic hypotension, and baroreflex failure in familial PD from mutation of the gene encoding alpha-synuclein.4 Subsequently we reported analogous denervation in familial PD from triplication of the normal gene.5 This past year Orimo’s group in Tokyo provided the first pathological confirmation of cardiac sympathetic denervation in familial PD from inherited alpha-synucleinopathy, based on severely decreased epicardial neuronal tyrosine hydroxylase immunoreactivity (Figure 1).6 In contrast, patients with familial PD from parkin gene mutation, which is not thought to be a Lewy body disease, have been found to have normal cardiac 123I-metaiodobenzyl-guanidine–derived radioactivity and normal epicardial neuronal tyrosine hydroxylase immunoreactivity.7 These findings establish a link between alpha-synucleinopathy and cardiac sympathetic denervation.
Some individuals who die without clinical parkinsonism have Lewy bodies detected pathologically. Growing evidence shows that incidental Lewy body disease represents early, presymptomatic PD.8 Orimo’s group therefore studied cardiac tissues and paravertebral sympathetic ganglia from patients with incidental Lewy body disease.9 Postmortem tissues were likewise obtained from comparison subjects with multiple system atrophy and from control subjects. Immunohistochemical analyses were performed using antibodies against tyrosine hydroxylase, phosphorylated neurofilament as a marker of axons, and phosphorylated alpha-synuclein as a marker of abnormal alpha-synuclein deposits. Key findings from this study9 were as follows:
Reprinted from Brain (Orimo S, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008; 131:642–650) by permission of Oxford University Press.
Figure 2. Concept diagram of the pathogenetic sequence of cardiac sympathetic denervation. In incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons (a), alpha-synuclein aggregates (black shading) accumulate abundantly in the distal axons but sparsely in the paravertebral sympathetic ganglia. In contrast, in incidental Lewy body disease with decreased THir axons (b), alpha-synuclein aggregates diminish in the distal axons but increase in the paravertebral sympathetic ganglia. In Parkinson disease, alpha-synuclein aggregates disappear in the distal axons and accumulate much more abundantly in the paravertebral sympathetic ganglia. In multiple system atrophy, alpha-synuclein aggregates are generally not observed (as in controls), with a few exceptions. Dotted lines indicate degeneration of THir axons.9Alpha-synuclein aggregates in distal epicardial nerve fascicles were more abundant in incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons than in incidental Lewy body disease with decreased THir axons (Figure 2).
Alpha-synuclein aggregates in the epicardial nerve fibers were closely related to the disappearance of THir axons.
In incidental Lewy body disease with preserved THir axons, alpha-synuclein aggregates were consistently more abundant in the epicardial nerves than in the paravertebral sympathetic ganglia (Figure 2).
Distally dominant accumulation of alpha-synuclein aggregates was reversed in incidental Lewy body disease with decreased THir axons and in PD, because both conditions involve fewer alpha-synuclein aggregates in axons and more abundant aggregates in the paravertebral sympathetic ganglia (Figure 2).
Thus, accumulation of alpha-synuclein aggregates in distal cardiac sympathetic axons precedes aggregation in neuronal somata or ganglionic neurites, heralding centripetal degeneration of cardiac sympathetic nerves in PD. This chronological and dynamic relationship between alpha-synuclein aggregation and distally dominant degeneration of cardiac noradrenergic nerves may represent the pathological mechanism behind a common degenerative process in PD.
In conclusion, cardiac noradrenergic denervation in Lewy body diseases, even in early stages, accounts for reduced cardiac uptake of 123I-metaiodobenzylguanidine and 6-[18F]fluorodopamine in PD. Alpha-synuclein aggregation appears to be intimately involved in the cardiac noradrenergic denervation that attends Lewy body diseases. The pathogenetic process seems to proceed in a centripetal, retrograde direction.
CYTOPLASMIC MONOAMINE METABOLITES AS AUTOTOXINS
Current concepts about mechanisms of PD emphasize pathologic alpha-synuclein accumulation, oxidative injury, impaired proteasomal or mitochondrial functions, neuroinflammation, or abnormal kinase signaling. These concepts do not explain relatively selective nigrostriatal dopaminergic and cardiac noradrenergic denervation in PD.
Figure 3. According to the monoamine aldehyde hypothesis, interference with the vesicular recycling of cytoplasmic monoamines (dopamine [DA], norepinephrine [NE], and serotonin [5-HT]) augments formation of toxic aldehydes. For instance, DA that leaks from vesicles (V) into the cytoplasm (C) or that is taken up via the cell membrane DA transporter (DAT) and escapes vesicular reuptake via the vesicular monoamine transporter (VMAT) is subject to oxidative deamination catalyzed by monoamine oxidase (MAO) to form the catecholaldehyde DOPAL, which is toxic. DOPAL is detoxified by ALDH to form DOPAC, the major metabolic route, or by AR to form DOPET, the minor metabolic route. Analogously, NE is converted to DOPEGAL, and 5-HT is converted to 5-HT-aldehyde (5-HTAld).
A potential explanation is that cytoplasmic catecholamine metabolites are autotoxins (Figure 3). The mechanisms of autotoxicity include spontaneous auto-oxidation, to form quinones and chromes leading to increased production of reactive oxygen species, and enzymatic oxidation.
Catecholamines in the neuronal cytoplasm undergo enzymatic oxidative deamination to form catecholaldehydes (dihydroxyphenylacetaldehyde [DOPAL] from dopamine), which are cytotoxic, as predicted by Blaschko more than a half century ago.10 DOPAL is detoxified mainly by aldehyde dehydrogenase (ALDH). In the substantia nigra, aldehyde dehydrogenase 1A1 (ALDH1A1) is the main isoform of ALDH, and postmortem studies have noted decreased nigral ALDH1A1 gene expression11,12 and protein content13 in PD patients.
All neurons express alpha-synuclein. Current concepts about mechanisms also do not explain the relatively selective aggregation of alpha-synuclein in catecholaminergic neurons. Alpha-synuclein appears to play a role in the cycling of catecholamines across vesicular and cell membranes.14
Figure 4. Cell survival and cytoplasmic dopamine are inversely related, according to a murine model by Mosharov et al.16 Graph shows the dependence of cell survival under l-dopa–induced stress on the cytoplasmic dopamine (DAcyt) dose in mouse neurons. The DAcyt dose was estimated as: [DAcyt]×TExposure = [DAcyt]×Ln([L-dopa]/K0.5)/k,where [DAcyt] is the concentration of cytosolic DA in cells treated with a saturating level (> 50 μM) of l-dopa for 1 hour, where [l-dopa] is the initial drug concentration, and where K0.5 = 9.7 μM and k = 0.15 hr−1 are the kinetic constants. TExposure approximates the time during which extracellular l-dopa remained higher than K0.5. The data points are (from left to right): filled circles—ventral midbrain cultures treated with 25, 100, 250, 500, and 1,000 μM l-dopa alone; open circles—ventral midbrain neurons treated with 250 μM l-dopa in the presence of benserazide, methamphetamine, reserpine, pargyline, and pargyline reserpine; diamonds—ventral tegmental area and substantia nigra neurons; triangles—striatal and cortical neurons treated with 250 μM l-dopa. Dotted lines and shaded boxes represent mean ± SEM in untreated cells. The solid line is the linear fit of all data points, excluding striatal and cortical neurons and the two data points indicated by the asterisk. Treatments to the right of this line are neuroprotective, as the same level of cell death is achieved with higher DAcyt doses; treatments to the left of this line are more susceptible to DAcyt stress.In the past year, a few important studies have been published related to autotoxicity of cytoplasmic catecholamine metabolites and to pathogenic interactions with alpha-synuclein. In 2006, Mosharov et al reported that alpha-synuclein overexpression increases cytoplasmic dopamine concentrations in rat pheochromocytoma PC-12 cells.15 Recently, the same group, using intracellular patch electrochemistry, directly measured cytoplasmic dopamine in cultured midbrain neurons and found that increases in dopamine and its metabolites are neurotoxic, whereas manipulations that reduce cytoplasmic dopamine are neuroprotective (Figure 4).16 Levodopa (l-dopa) increased cytoplasmic dopamine more in substantia nigra neurons than in ventral tegmental neurons, suggesting that this difference might help explain the greater susceptibility of nigral neurons to the pathogenetic process. The greater buildup of cytoplasmic dopamine seemed to depend on dihydropyridine-sensitive calcium (Ca2+) channels. Finally, dopaminergic neurons lacking alpha-synuclein were resistant to l-dopa–induced cell death. These findings led the authors to propose a “multiple-hit” model (Figure 5) in which interactions between intracellular ionized calcium, cytoplasmic dopamine, and alpha-synuclein underlie susceptibility of nigral neurons in PD.16
Figure 5. The “multiple-hit” model of Parkinson disease pathogenesis,16 which holds that neurotoxicity is a result of multiple factors, including the presence of alpha-synuclein (α-syn), elevation of cytoplasmic calcium (Ca2+), and buildup of cytoplasmic dopamine (DAcyt) and its metabolites. Nonexclusive toxic steps may result from (1) mechanisms that require direct interaction between DA or its metabolites with α-syn, such as DA-modified stabilization of α-syn protofibrils or inhibition of chaperone-mediated autophagy, or (2) cumulative damage from multiple independent sources. Reducing the levels of any of the three players provides neuroprotection. (AADC = aromatic l-amino acid decarboxylase; DOPAL = dihydroxyphenylacetaldehyde; TH = tyrosine hydroxylase)Burke et al added a potentially important clue, demonstrating that DOPAL potently oligomerizes and aggregates alpha-synuclein.17 This finding introduces the possibility of multiple pathogenetic positive feedback loops.
Under resting conditions, most catecholamine turnover results from leakage from vesicular stores into the cytoplasm and subsequent oxidative deamination by monoamine oxidase. Ordinarily, however, catecholamines in the cytoplasm are efficiently recycled back into the vesicles via the type 2 vesicular monoamine transporter (VMAT-2). Accordingly, interference with VMAT functions would be expected to tend to build up cytoplasmic catecholamines, with potentially cytotoxic consequences. In 2007, Caudle et al reported that mice with severely decreased VMAT-2 have aging-associated decreases in striatal dopamine that begin in the terminal fields, alpha-synuclein deposition in substantia nigra neurons, and l-dopa–responsive behavioral deficits.18 More recently the same group noted nonmotor signs associated with PD in VMAT-2–deficient mice, such as anosmia, gastrointestinal hypomotility, sleep disturbances, anxiety, and depression.19 Since VMAT-2 serves to recycle not only dopamine but also norepinephrine and serotonin, this single abnormality could help explain loss of all three types of monoaminergic neurons in PD.
Finally, Pena-Silva et al recently tested whether serotonin induces oxidative stress in human heart valves.20 They showed that in heart valves from explanted human hearts not used for transplantation, incubation of homogenates of cardiac valves and blood vessels with serotonin increased generation of the superoxide free radical. Inhibitors of monoamine oxidase prevented this effect. Dopamine also increased superoxide levels in heart valves, and this effect was also attenuated by monoamine oxidase inhibition. These findings fit with the concept that the aldehydes produced by the action of monoamine oxidase on cytoplasmic monoamines generate toxic free radicals.
VALIDITY OF POWER SPECTRAL ANALYSIS OF HEART RATE VARIABILITY TO INDICATE CARDIAC SYMPATHETIC TONE
Power spectral analysis of heart rate variability is simple, relatively inexpensive, noninvasive, and widely used to indicate cardiac sympathetic “tone” or sympathovagal “balance.” Almost 2,000 studies to date have used this modality. Relatively increased cardiac sympathetic tone, reflected by low-frequency (LF) power or the ratio of LF power to high-frequency (HF) power, is an adverse prognostic sign in a variety of conditions. Nevertheless, the validity of LF power, or the LF:HF ratio, as an index of cardiac sympathetic tone remains unsettled.
In 2007 we assessed the validity of power spectral analysis rather directly, by taking advantage of our ability to delineate cardiac sympathetic innervation. We compared LF power in patients with cardiac sympathetic denervation, indicated by low myocardial levels of 6-[18F]fluorodopamine–derived radioactivity or low rates of norepinephrine entry into coronary sinus plasma (cardiac norepinephrine spillover), with values in patients with intact innervation. LF power was unrelated to myocardial 6-[18F]fluorodopamine–derived radioactivity or cardiac norepinephrine spillover, but it was related to baroreflex-cardiovagal gain. Patients with a low baroreflex-cardiovagal gain had low LF power, regardless of cardiac innervation. From these findings we concluded that LF power reflects baroreflex function, not cardiac sympathetic innervation.21
Figure 6. Relationships of heart rate variability indices with cardiac norepinephrine spillover. Graphs show time and frequency domain heart rate variability measures (LF = low frequency; HF = high frequency) versus cardiac norepinephrine in healthy subjects (squares) and patients with major depression (triangles) and panic disorder (circles).22 Recently Baumert et al also examined the relationship between indices from power spectral analysis of heart rate variability and cardiac norepinephrine spillover.22 They found, as we did, that none of the standard heart rate variability parameters was correlated with cardiac norepinephrine spillover (Figure 6). The same group reported a positive correlation between the heart rate–corrected QT interval and cardiac norepinephrine spillover.23 Among patients with major depression, the distribution of cardiac norepinephrine spillover seemed bimodal. Overall, cardiac norepinephrine spillover was not increased, although a subgroup had clearly increased spillover.
In congestive heart failure, baroreflex-cardiovagal gain tends to be low and cardiac sympathetic outflow markedly increased, yet the LF:HF ratio is not increased during supine rest.24,25 It therefore appears that power spectral analysis of heart rate variability may provide a measure of baroreflexive modulation of autonomic outflows to the heart but not a measure of those outflows themselves. The search continues for a valid, noninvasive means to assess cardiac sympathetic function.
References
Goldstein DS, Holmes C, Cannon RO, Eisenhofer G, Kopin IJ. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med1997; 336:696–702.
Orimo S, Ozawa E, Oka T, et al Different histopathology accounting for a decrease in myocardial MIBG uptake in PD and MSA. Neurology2001; 57:1140–1141.
Amino T, Orimo S, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Path2005; 15:29–34.
Goldstein DS, Li S-T, Kopin IJ. Sympathetic neurocirculatory failure in Parkinson disease: evidence for an etiologic role of α-synuclein. Ann Intern Med2001; 135:1010–1011.
Singleton A, Gwinn-Hardy K, Sharabi Y, et al Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain2004; 127:768–772.
Orimo S, Uchihara T, Nakamura A, et al Cardiac sympathetic denervation in Parkinson’s disease linked to SNCA duplication. Acta Neuropathol2008; 116:575–577.
Orimo S, Amino T, Yokochi M, et al Preserved cardiac sympathetic nerve accounts for normal cardiac uptake of MIBG in PARK2. Mov Disord2005; 20:1350–1353.
Dickson DW, Fujishiro H, DelleDonne A, et al Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol2008; 115:437–444.
Orimo S, Uchihara T, Nakamura A, et al Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain2008; 131:642–650.
Blaschko H. Amine oxidase and amine metabolism. Pharmacol Rev1952; 4:415–458.
Galter D, Buervenich S, Carmine A, Anvret M, Olson L. ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson’s disease and in the ventral tegmental area in schizophrenia. Neurobiol Dis2003; 14:637–647.
Mandel S, Grunblatt E, Riederer P, et al Gene expression profiling of sporadic Parkinson’s disease substantia nigra pars compacta reveals impairment of ubiquitin-proteasome subunits, SKP1A, aldehyde dehydrogenase, and chaperone HSC-70. Ann N Y Acad Sci2005; 1053:356–375.
Werner CJ, Heyny-von Haussen R, Mall G, Wolf S. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome Sci2008; 6:8.
Larsen KE, Schmitz Y, Troyer MD, et al Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci2006; 26:11915–11922.
Mosharov EV, Staal RG, Bove J, et al Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J Neurosci2006; 26:9304–9311.
Mosharov EV, Larsen KE, Kanter E, et al Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron2009; 62:218–229.
Burke WJ, Kumar VB, Pandey N, et al Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol2008; 115:193–203.
Caudle WM, Richardson JR, Wang MZ, et al Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci2007; 27:8138–8148.
Taylor TN, Caudle WM, Shepherd KR, et al Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci2009; 29:8103–8113.
Pena-Silva RA, Miller JD, Chu Y, Heistad DD. Serotonin produces monoamine oxidase-dependent oxidative stress in human heart valves. Am J Physiol Heart Circ Physiol2009; 297:H1354–H1360.
Moak JP, Goldstein DS, Eldadah BA, et al Supine low-frequency power of heart rate variability reflects baroreflex function, not cardiac sympathetic innervation. Heart Rhythm2007; 4:1523–1529.
Baumert M, Lambert GW, Dawood T, et al Short-term heart rate variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol2009; 297:H674–H679.
Baumert M, Lambert GW, Dawood T, et al QT interval variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol2008; 295:H962–H968.
Tygesen H, Rundqvist B, Waagstein F, Wennerblom B. Heart rate variability measurement correlates with cardiac norepinephrine spillover in congestive heart failure. Am J Cardiol2001; 87:1308–1311.
Kingwell BA, Thompson JM, Kaye DM, McPherson GA, Jennings GL, Esler MD. Heart rate spectral analysis, cardiac norepinephrine spillover, and muscle sympathetic nerve activity during human sympathetic nervous activation and failure. Circulation1994; 90:234–240.
References
Goldstein DS, Holmes C, Cannon RO, Eisenhofer G, Kopin IJ. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med1997; 336:696–702.
Orimo S, Ozawa E, Oka T, et al Different histopathology accounting for a decrease in myocardial MIBG uptake in PD and MSA. Neurology2001; 57:1140–1141.
Amino T, Orimo S, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Path2005; 15:29–34.
Goldstein DS, Li S-T, Kopin IJ. Sympathetic neurocirculatory failure in Parkinson disease: evidence for an etiologic role of α-synuclein. Ann Intern Med2001; 135:1010–1011.
Singleton A, Gwinn-Hardy K, Sharabi Y, et al Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain2004; 127:768–772.
Orimo S, Uchihara T, Nakamura A, et al Cardiac sympathetic denervation in Parkinson’s disease linked to SNCA duplication. Acta Neuropathol2008; 116:575–577.
Orimo S, Amino T, Yokochi M, et al Preserved cardiac sympathetic nerve accounts for normal cardiac uptake of MIBG in PARK2. Mov Disord2005; 20:1350–1353.
Dickson DW, Fujishiro H, DelleDonne A, et al Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol2008; 115:437–444.
Orimo S, Uchihara T, Nakamura A, et al Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain2008; 131:642–650.
Blaschko H. Amine oxidase and amine metabolism. Pharmacol Rev1952; 4:415–458.
Galter D, Buervenich S, Carmine A, Anvret M, Olson L. ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson’s disease and in the ventral tegmental area in schizophrenia. Neurobiol Dis2003; 14:637–647.
Mandel S, Grunblatt E, Riederer P, et al Gene expression profiling of sporadic Parkinson’s disease substantia nigra pars compacta reveals impairment of ubiquitin-proteasome subunits, SKP1A, aldehyde dehydrogenase, and chaperone HSC-70. Ann N Y Acad Sci2005; 1053:356–375.
Werner CJ, Heyny-von Haussen R, Mall G, Wolf S. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome Sci2008; 6:8.
Larsen KE, Schmitz Y, Troyer MD, et al Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci2006; 26:11915–11922.
Mosharov EV, Staal RG, Bove J, et al Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J Neurosci2006; 26:9304–9311.
Mosharov EV, Larsen KE, Kanter E, et al Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron2009; 62:218–229.
Burke WJ, Kumar VB, Pandey N, et al Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol2008; 115:193–203.
Caudle WM, Richardson JR, Wang MZ, et al Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci2007; 27:8138–8148.
Taylor TN, Caudle WM, Shepherd KR, et al Nonmotor symptoms of Parkinson’s disease revealed in an animal model with reduced monoamine storage capacity. J Neurosci2009; 29:8103–8113.
Pena-Silva RA, Miller JD, Chu Y, Heistad DD. Serotonin produces monoamine oxidase-dependent oxidative stress in human heart valves. Am J Physiol Heart Circ Physiol2009; 297:H1354–H1360.
Moak JP, Goldstein DS, Eldadah BA, et al Supine low-frequency power of heart rate variability reflects baroreflex function, not cardiac sympathetic innervation. Heart Rhythm2007; 4:1523–1529.
Baumert M, Lambert GW, Dawood T, et al Short-term heart rate variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol2009; 297:H674–H679.
Baumert M, Lambert GW, Dawood T, et al QT interval variability and cardiac norepinephrine spillover in patients with depression and panic disorder. Am J Physiol Heart Circ Physiol2008; 295:H962–H968.
Tygesen H, Rundqvist B, Waagstein F, Wennerblom B. Heart rate variability measurement correlates with cardiac norepinephrine spillover in congestive heart failure. Am J Cardiol2001; 87:1308–1311.
Kingwell BA, Thompson JM, Kaye DM, McPherson GA, Jennings GL, Esler MD. Heart rate spectral analysis, cardiac norepinephrine spillover, and muscle sympathetic nerve activity during human sympathetic nervous activation and failure. Circulation1994; 90:234–240.
This is a momentous occasion for me, for the extraordinary people in the Clinical Neurocardiology Section at the National Institutes of Health (NIH), and for my family—my wife Minka and son Joey drove all the way from Maryland late last night and early this morning to be here. I thank them publicly here.
THE ‘SPARKLE OF INSIGHT’ FROM ENLIGHTENED INDUCTION
In these brief comments, as I look back on the road I have taken over the past 40 years carrying out patient-oriented research in heart-brain medicine, I would like to convey a viewpoint instead of dwelling on the presentation of research data.
The idea I wish to convey is that ignorance isn’t biased. If you have a hypothesis you want to test, you are inherently biased to find something positive—and, if you are in academic medicine, publishable—in the data you obtain. But if you have the technical capability to measure something no one else can measure, and you have sufficient mastery of the topic to know what is not yet known, then if you make an observation that you did not predict and if you recognize its significance, you have made a discovery. You have revealed a bit of the truth. You experience the highest joy and thrill a scientist can feel—a “sparkle of insight.” When this happens, if you have sense, you stop what you have been doing to pursue that discovery.
Hardly anyone has received a Nobel Prize for testing a theory, but many Nobel Prizes have been awarded for technological advances and for discoveries based on those advances. In my view, discoverers use an enlightened inductive approach at least as much as deduction. They develop new technology that enables key novel measurements, and they keep in mind gaps in knowledge, so that they are ready to appreciate the significance of their observations.
A PERSONAL EXAMPLE
‘You have to measure something’
Let me share an example of this process by relating a sparkle of insight I had several years ago. When I began working at the NIH, I met with the chief of the Hypertension-Endocrine Branch of the National Heart, Lung, and Blood Institute about the research program I would pursue. After listening patiently to me for many minutes as I spouted about how I was going to test hypotheses derived from the concepts that people with hypertension are “hyper-tense,” and that stress causes heart disease, the chief responded, “Well, these ideas are all well and good. But what are you going to measure? You can measure whatever you want, but you have to measure something.”
Measure something. I wanted to see if there was hyperactivity of the sympathetic nervous system or excessive sympathetic innervation in hypertension, and I started working on ways to measure sympathetic activity.
The sympathetic nervous system at a glance
First I should introduce you to the sympathetic nervous system, which is one of the main effectors by which the brain regulates the heart and blood vessels. It is a key link between the brain and heart. The sympathetic nerves to the heart and other organs do not come directly from the brain but from ganglia, which are clumps of nerve cell bodies strung like pearls on a necklace on each side of the spinal column. This origin outside the central nervous system will be an important fact to keep in mind.
In the heart, the sympathetic nerves travel with the coronary arteries and then dive into the heart muscle from the outside. Sympathetic nerves also enmesh the walls of arteries and arterioles. The arterioles constitute the main determinant of total peripheral resistance to blood flow in the body and therefore figure prominently in the control of blood pressure. The architectural association between sympathetic nerves and the muscle in the heart and arteriolar walls has enticed hypertension researchers for many decades.
A false start with plasma norepinephrine measurement
I developed novel methods for measuring plasma levels of norepinephrine, which is the chemical messenger that the sympathetic nervous system uses in regulation of the circulation, and of adrenaline (epinephrine), which is the well-known and potent “fight-or-flight” hormone.1 Applying this technology to patients with high blood pressure led to several publications2–9 but actually shed more heat than light on the hypothesis of sympathetic hyperactivity as a cause of or contributor to hypertension. In the face of negative data, the theory was qualified—sympathetic hyperactivity might be apparent only in the young, or the thin, or the Caucasian, or the male—but not abandoned.
Insights from visualizing sympathetic nerves in the heart
Then I embarked on a project to visualize sympathetic nerves in the heart, by a new technology called positron emission tomographic (PET) scanning. With several colleagues—including Irwin J. Kopin, Graeme Eisenhofer, Peter Chang, David Hovevey-Zion, Ehud Grossman, and Courtney Holmes—to whom I will always be grateful, I developed a PET imaging agent called 6-[18F]fluorodopamine.10–13
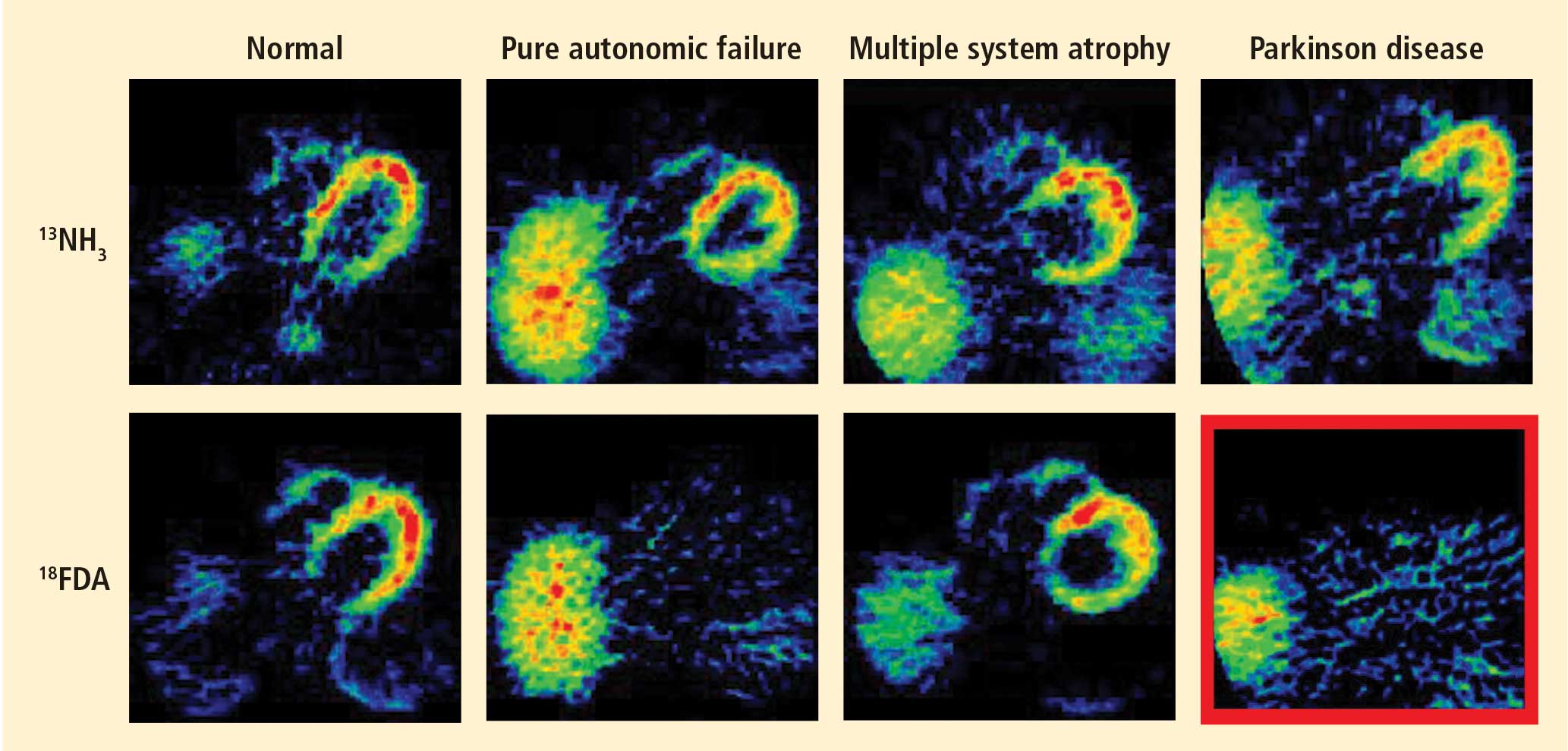
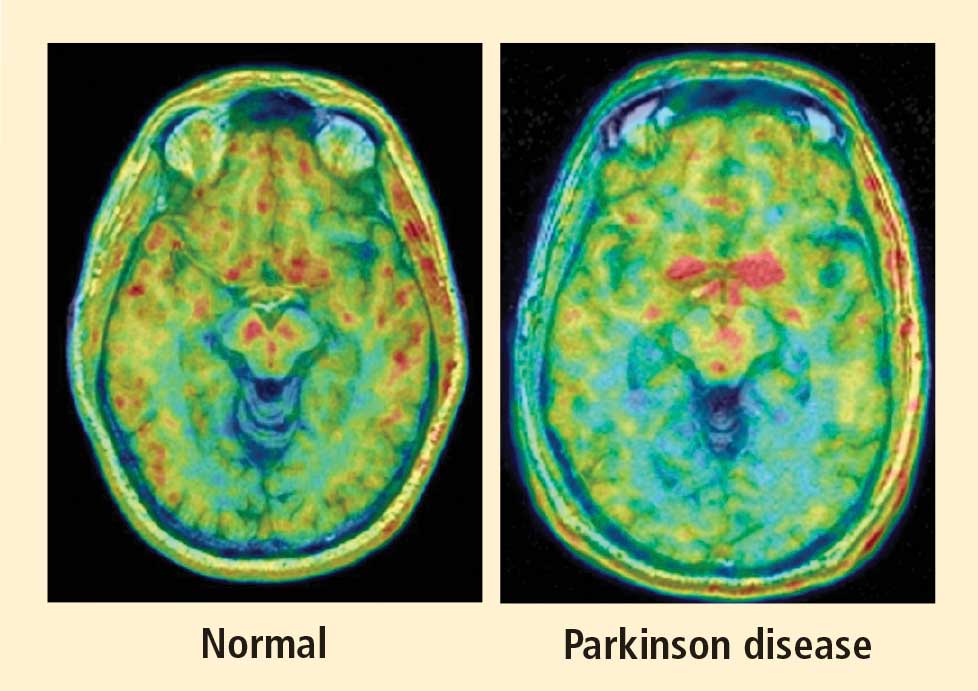
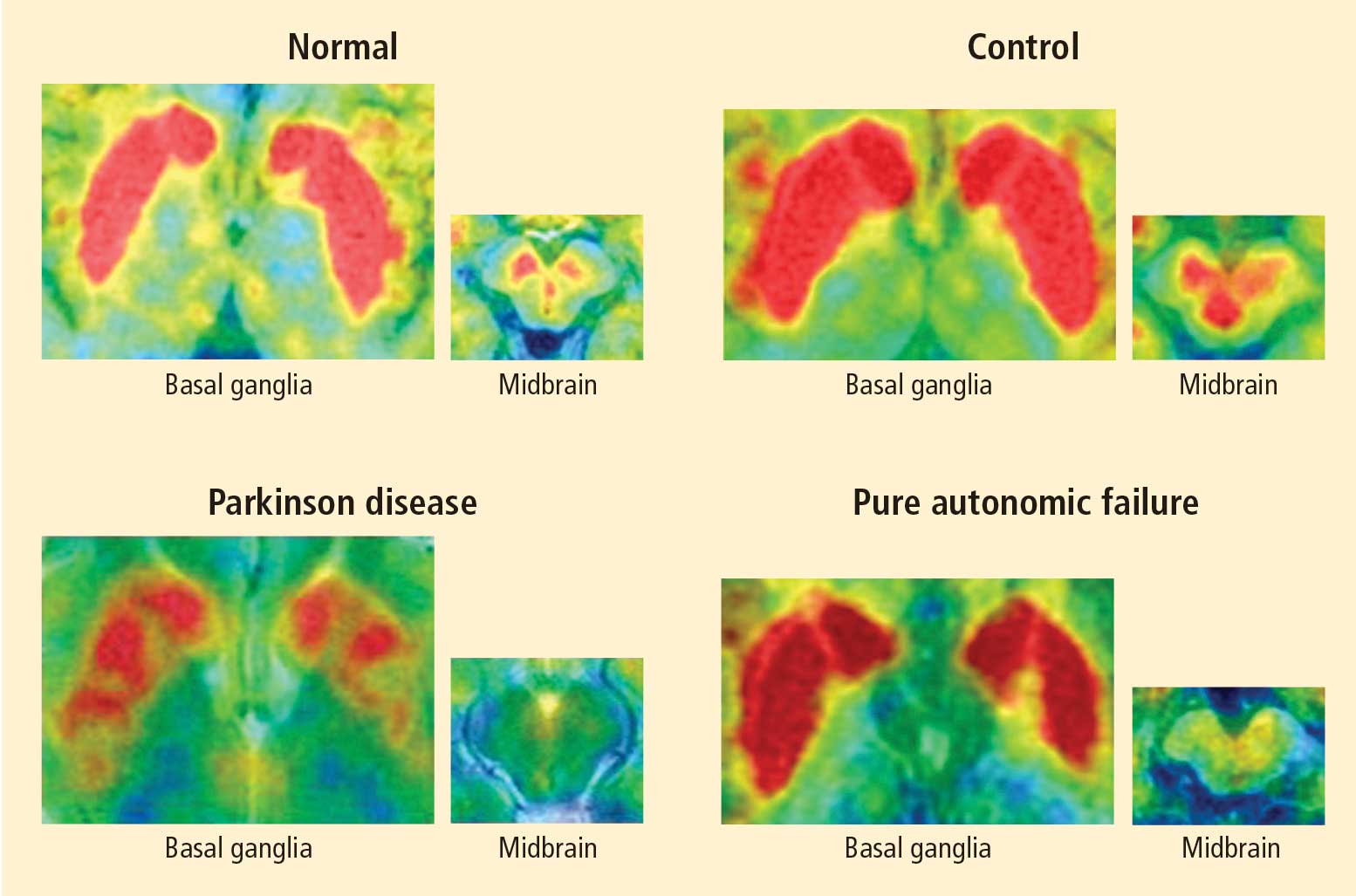
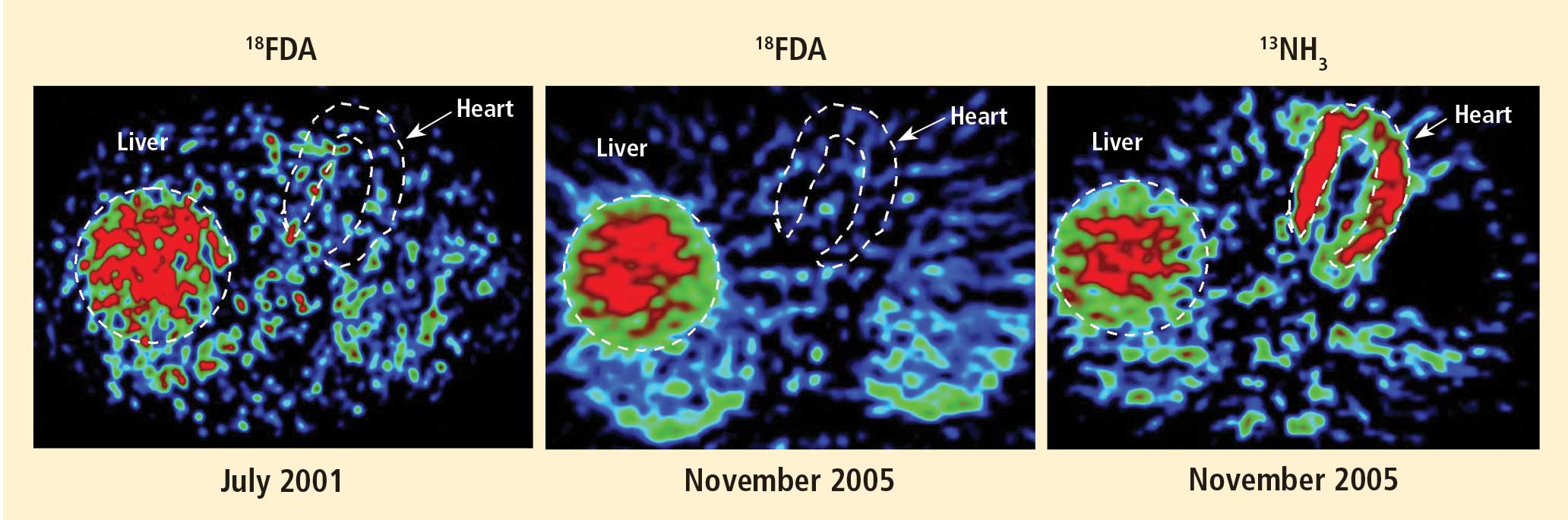
Figure 1. Thoracic positron emission tomographic scans in a healthy control subject and in patients with pure autonomic failure, multiple system atrophy, and Parkinson disease. The top row shows 13N-labeled ammonia perfusion scans and the bottom row shows 6-[18F]fluoro dopamine sympathoneural scans in each subject. Note the absence of cardiac 6-[18F]fluorodopamine-derived radioactivity in the subjects with pure autonomic failure and Parkinson disease in contrast with the normal radioactivity in the patient with multiple system atrophy; the decrease in radioactivity is particularly severe in the patient with Parkinson disease (red border). Adapted from Goldstein et al.14After injection of 6-[18F]fluorodopamine into a person’s vein, PET scan slices of the chest reveal the sympathetic nerves in the heart (Figure 1). The top row of Figure 1 shows where the blood is going—perfusion—in four people, and the bottom row shows the 6-[18F]fluorodopamine scans in the same people. The horseshoe-shaped structure is the main pumping muscle of the heart, the left ventricular myocardium. The “blob” on the patient’s right is the liver.
Normally, PET scans using 6-[18F]fluorodopamine look remarkably similar to scans using 13N-labeled ammonia, a perfusion imaging agent. The first patient I studied with this new technology was a patient with a rare disease called pure autonomic failure (PAF). In PAF, there was already good evidence for a loss of sympathetic nerves throughout the body. Myocardial perfusion in this patient was normal, but there was much less than normal 6-[18F]fluorodopamine-derived radioactivity in the heart muscle. In another uncommon disease, multiple system atrophy (MSA), the perfusion was also normal, and the cardiac sympathetic nerves seemed intact, in line with what was already known about this progressive neurodegenerative disease.
Then I tested a patient who had been thought to have MSA but actually had Parkinson disease (PD) with orthostatic hypotension (a fall in blood pressure each time the person stands up). PD with orthostatic hypotension can be very difficult to distinguish from the parkinsonian form of MSA. To my complete surprise, the patient with PD had a remarkable decrease in 6-[18F]fluorodopa mine-derived radioactivity in the heart muscle. There was normal blood flow to the heart muscle, so the 6-[18F]fluorodopamine was being delivered, but there was no evidence of sympathetic nerves in the heart. The scans resembled those in the PAF patient, not the MSA patient.
This finding did not arise from a prediction to test a hypothesis. It wasn’t long before I tested additional PD patients and found the same unexpected results.14,15 Because I was ignorant, I wasn’t biased. I felt I had put my finger on a piece of the truth, and I had to stop and think about the implications of this discovery. I never did come to test the hypotheses that I had sought out originally to test. Instead, I followed a totally new path, based on the discovery of cardiac sympathetic denervation in PD.
Beyond a brain disease: Seeing PD as a heart-brain disorder
More than 50 neuroimaging studies since our original report have agreed remarkably consistently on the association between PD and loss of sympathetic nerves in the heart; moreover, postmortem pathology studies have amply confirmed that a profound loss of cardiac sympathetic nerves is characteristic of PD.16 I have yet to come across a single patient with PD and orthostatic hypotension who has not had cardiac sympathetic denervation, and virtually all patients with PD who do not have orthostatic hypotension seem to have at least partial loss of cardiac sympathetic nerves.
Considering that the source of those nerves is the ganglia, which lie outside the central nervous system, PD must be more than a brain disease and more than a movement disorder. It must also be a disease of the sympathetic nerves in the heart, a form of a dysautonomia, and a heart-brain disorder.
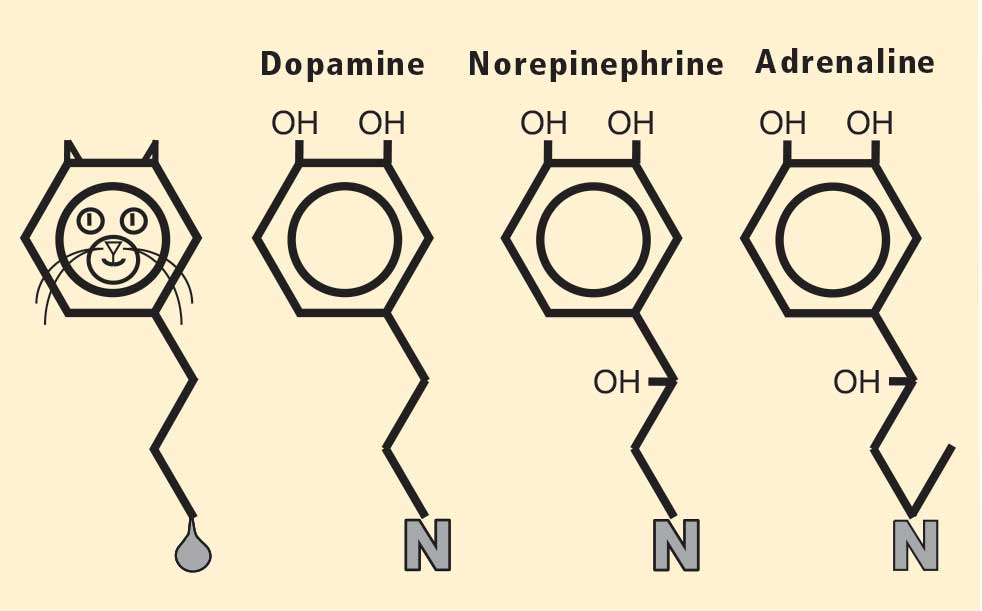
The role of catecholamines: Another discovery born of unbiased ignorance
Figure 2. The chemical structure of each of the catecholamines —dopamine, norepinephrine, and adrenaline (epinephrine)—resembles a cat. The head of the cat is the catechol nucleus, with the two pointy ears corresponding to the adjacent hydroxyl groups on the benzene ring. At the other end of the cat is a hydrocarbon tail, ending in a “uriniferous” amine group. Adapted from Goldstein.17To appreciate fully the significance of this finding, I must mention my favorite chemical family, the catecholamines, whose chemical structures resemble cats (Figure 2).17 PD results from a loss of a particular chemical, dopamine, in a particular pathway in the brain; dopa mine is a catecholamine. The other catecholamines in humans are norepinephrine and adrenaline. As noted above, norepinephrine is the chemical messenger of the sympathetic nerves, and adrenaline is the well-known hormone that produces many of the signs of emotional distress.
Almost a half century ago, Hornykiewicz and colleagues made the pivotal discovery that PD features loss of dopamine in the nigrostriatal system in the brain.18 Given the cardiac sympathetic denervation, PD might be a disease of catecholamine systems both inside and outside the central nervous system—dopamine in the nigrostriatal system, and norepinephrine in the sympathetic nerves of the heart.
Then what of the third catecholamine, adrenaline, in PD? Plasma levels of adrenaline and of its metabolite, metanephrine, are normal in PD, even in patients who have PD and orthostatic hypotension, which involves loss of norepinephrine-producing nerves not only in the heart but in other organs.19 What is different about the adrenaline-producing cells in the medulla (from the Latin for “marrow”) of the adrenal glands atop each kidney? Why aren’t these catecholamine-producing cells also lost in PD?
I have some ideas in mind but won’t go into them here. The point is that the discovery of normal adrenaline-producing cells in PD, despite loss of cells producing the other catecholamines, was not based on my testing a hypothesis. It was a discovery born of ignorance, and because ignorance isn’t biased, that discovery points to the truth. Whatever the eventual explanation for the specific pattern of catecholamine cell loss in PD, it cannot refute the discovery itself.
HOW DISCOVERIES ARISE: AN APPLIED EXERCISE FOR READERS
Figure 3. High-resolution positron emission tomographic scans, superimposed over magnetic resonance images, at the level of the basal ganglia, after intravenous administration of 6-[18F]fluorodopa in a control subject (left) and a patient with Parkinson disease (PD) (right). Red indicates the maximum amount of radioactivity. Note the severely decreased 6-[18F]fluorodopa-derived radioactivity in the striatum—epecially the putamen—in the PD patient.Now let’s have you, the reader, make a discovery and induce its significance based on what I have tried to teach so far, that discoveries arise from the application of relevant technology and from insights of the prepared mind. Take a look at the scans in the left panels of Figures 3 and 4. The large red structures in Figure 3, which look like a sad clown’s eyes, correspond to the striatum. The striatum is made up of the putamen, which is like the mascara on the side of the sad clown’s eyes, and the caudate, which is like the beady eyes themselves. In the left panel of Figure 4, the small spots in the midbrain correspond to the substantia nigra, a major site of dopamine-producing neurons in the human brain.
Figure 4. High-resolution positron emission tomographic scans, superimposed over magnetic resonance images, at the level of the midbrain, after intravenous administration of 6-[18F]fluorodopa in a control subject (left) and a patient with Parkinson disease (PD) (right). Red indicates the maximum amount of radioactivity. Note the severely decreased 6-[18F]fluorodopa-derived radioactivity bilaterally in the region corresponding to the substantia nigra in the PD patient.We can see in the right panel of Figure 3 that in PD there is a loss of the ability to store dopamine in the striatum—especially in the putamen, the mascara of the sad clown’s eyes. In the right panel of Figure 4 we see that in the brainstem there is a loss of the dopamine-containing nerve cells in the substantia nigra. These scans therefore demonstrate graphically the nigrostriatal lesion characteristic of PD. There is a loss of the nerve cells in the substantia nigra in the midbrain and a loss of the dopamine-containing terminals in the striatum.
Figure 5. High-resolution positron emission tomographic scans, superimposed over magnetic resonance images, at the levels of the basal ganglia and midbrain, after intravenous administration of 6-[18F]fluorodopa in four subjects: a normal volunteer (upper left) a control patient without parkinsonism or autonomic failure (upper right) a patient with Parkinson disease (PD) (lower left) a patient with pure autonomic failure (PAF) (lower right). Red indicates the maximum amount of radioactivity. Note the severely decreased 6-[18F] fluorodopa-derived radioactivity bilaterally in the region corresponding to the substantia nigra in both PD and PAF. Adapted from Goldstein et al.20Now take a look at the scans of these areas in a patient with PAF in Figure 5. Remember that PAF involves a loss of sympathetic nerves in the heart, just like in PD, but that PAF does not involve parkinsonism. Look at the sad clown’s eyes. The mascara is there, of course, because the patient does not have parkinsonism. But now look for the spots in the substantia nigra—they are missing, just as in PD.
PAF is a rare disease, and I have only studied several cases with high-resolution PET scanning of the brain, but so far they have all had this unexpected, unpredicted finding of loss of dopaminergic neurons in the substantia nigra.20
What does this pattern mean? If PAF patients have just as much loss of nigral neurons as PD patients do, and if PAF patients do not have parkinsonism, then the movement disorder in PD cannot result from loss of the dopamine neurons in the substantia nigra per se. Instead, the movement disorder in PD seems to come from loss of the dopaminergic terminals in the striatum.
How can PAF patients have normal dopamine terminals in the putamen when the number of dopaminergic cell bodies is severely reduced? Somehow, PAF patients must be able to sprout new terminals, even as they lose the cell bodies. Maybe if we knew how PAF patients do this, we would have a way to treat or even prevent PD.
How do PAF patients maintain normal dopamine terminals as the cell bodies die off? No one knows. Until now, no one thought of asking such a question. No one hypothesized that this discovery would be made, but it was. And because ignorance isn’t biased, we have put our finger on the truth. By keeping in mind what isn’t known, we could see what wasn’t there. Now we can begin to think of what to look for next.
SUMMARY AND CONCLUSIONS
Because ignorance isn’t biased, if you have the tools to make relevant measurements, if you have sufficient mastery of the subject to know what isn’t known, and if you have access to patients with rare but informative disorders, you can make important discoveries based on inductions from observations.
The discoveries that cardiac sympathetic denervation characterizes PD and that parkinsonism does not result from loss of dopamine neurons per se depended crucially on studying patients with a rare disease, PAF. In 1657, William Harvey—the same William Harvey who first described the circulation of the blood and who first pointed out the effects of emotions on the heart—wrote eloquently about the extraordinary power of studying patients with rare diseases:Nature is nowhere accustomed more openly to display her secret mysteries than in cases where she shows traces of her workings apart from the beaten path; nor is there any better way to advance the proper practice of medicine than to give our minds to the discovery of the usual law of nature, by the careful investigation of cases of rarer forms of disease. For it has been found in almost all things, that what they contain of use or of application, is hardly perceived unless we are deprived of them, or they become deranged in some way.21
I hope I have convinced you of the importance of seeing what isn’t there. My thanks go out again to the Earl and Doris Bakken Heart-Brain Institute for this prestigious award, to my family, to my colleagues and friends, and to my patients. As I have written in Adrenaline and the Inner World: An Introduction to Scientific Integrative Medicine,17 patients serve as a unique scientific resource. They report what is wrong; they tell us the truth. We have to make sense of what they teach.
References
Goldstein DS, Feuerstein G, Izzo JL, Kopin IJ, Keiser HR. Validity and reliability of liquid chromatography with electrochemical detection for measuring plasma levels of norepinephrine and epinephrine in man. Life Sci1981; 28:467–475.
Goldstein DS. Plasma norepinephrine during stress in essential hypertension. Hypertension1981; 3:551–556.
Goldstein DS. Plasma norepinephrine in essential hypertension: a study of the studies. Hypertension1981; 3:48–52.
Goldstein D, Horwitz D, Keiser HR, Polinsky RJ, Kopin IJ. Plasma l-[3H]norepinephrine, d-[14C]norepinephrine, and d,l-[3H] isoproterenol kinetics in essential hypertension. J Clin Invest1983; 72:1748–1758.
Goldstein DS. Arterial baroreflex sensitivity, plasma catecholamines, and pressor responsiveness in essential hypertension. Circulation1983; 68:234–240.
Goldstein DS. Plasma catecholamines and essential hypertension: an analytical review. Hypertension1983; 5:86–99.
Goldstein DS, Lake CR, Chernow B, et al. Age-dependence of hypertensive-normotensive differences in plasma norepinephrine. Hypertension1983; 5:100–104.
Goldstein DS, McCarty R, Polinsky RJ, Kopin IJ. Relationship between plasma norepinephrine and sympathetic neural activity. Hypertension1983; 5:552–559.
Goldstein DS, Lake CR. Plasma norepinephrine and epinephrine levels in essential hypertension. Fed Proc1984; 43:57–61.
Eisenhofer G, Hovevey-Sion D, Kopin IJ, et al. Neuronal uptake and metabolism of 2- and 6-fluorodopamine: false neurotransmitters for positron emission tomographic imaging of sympathetically innervated tissues. J Pharmacol Exp Ther1989; 248:419–427.
Chang PC, Szemeredi K, Grossman E, Kopin IJ, Goldstein DS. Fate of tritiated 6-fluorodopamine in rats: a false neurotransmitter for positron emission tomographic imaging of sympathetic innervation and function. J Pharmacol Exp Ther1990; 255:809–817.
Goldstein DS, Holmes C. Metabolic fate of the sympathoneural imaging agent 6-[18F]fluorodopamine in humans. Clin Exp Hypertens1997; 19:155–161.
Goldstein DS, Eisenhofer G, Dunn BB, et al. Positron emission tomographic imaging of cardiac sympathetic innervation using 6-[18F] fluorodopamine: initial findings in humans. J Am Coll Cardiol1993; 22:1961–1971.
Goldstein DS, Holmes C, Cannon RO, Eisenhofer G, Kopin IJ. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med1997; 336:696–702.
Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO. Cardiac sympathetic denervation in Parkinson disease. Ann Intern Med2000; 133:338–347.
Amino T, Orimo S, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Pathol2005; 15:29–34.
Goldstein DS. Adrenaline and the Inner World: An Introduction to Scientific Integrative Medicine. Baltimore, MD: Johns Hopkins University Press; 2006.
Ehringer H, Hornykiewicz O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system [in German]. Wien Klin Wochenschr1960; 38:1236–1239.
Goldstein DS, Holmes C, Sharabi Y, Brentzel S, Eisenhofer G. Plasma levels of catechols and metanephrines in neurogenic orthostatic hypotension. Neurology2003; 60:1327–1332.
Goldstein DS, Holmes C, Sato T, et al. Central dopamine deficiency in pure autonomic failure. Clin Auton Res2008; 18:58–65.
Hervey Wyatt RBWilliam Harvey 1578 to 1657Whitefish, MTKessinger Publishing2005:161–162.
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, Clinical Neurosciences Program, Division of Intramural Research, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Building 10, Room 6N252, 10 Center Drive, MSC-1620, Bethesda, MD 20892-1620; goldsteind@ninds.nih.gov
Dr. Goldstein reported that he has no financial interests or relationships that pose a potential conflict of interest with this article.
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, Clinical Neurosciences Program, Division of Intramural Research, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Building 10, Room 6N252, 10 Center Drive, MSC-1620, Bethesda, MD 20892-1620; goldsteind@ninds.nih.gov
Dr. Goldstein reported that he has no financial interests or relationships that pose a potential conflict of interest with this article.
Author and Disclosure Information
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, Clinical Neurosciences Program, Division of Intramural Research, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Building 10, Room 6N252, 10 Center Drive, MSC-1620, Bethesda, MD 20892-1620; goldsteind@ninds.nih.gov
Dr. Goldstein reported that he has no financial interests or relationships that pose a potential conflict of interest with this article.
This is a momentous occasion for me, for the extraordinary people in the Clinical Neurocardiology Section at the National Institutes of Health (NIH), and for my family—my wife Minka and son Joey drove all the way from Maryland late last night and early this morning to be here. I thank them publicly here.
THE ‘SPARKLE OF INSIGHT’ FROM ENLIGHTENED INDUCTION
In these brief comments, as I look back on the road I have taken over the past 40 years carrying out patient-oriented research in heart-brain medicine, I would like to convey a viewpoint instead of dwelling on the presentation of research data.
The idea I wish to convey is that ignorance isn’t biased. If you have a hypothesis you want to test, you are inherently biased to find something positive—and, if you are in academic medicine, publishable—in the data you obtain. But if you have the technical capability to measure something no one else can measure, and you have sufficient mastery of the topic to know what is not yet known, then if you make an observation that you did not predict and if you recognize its significance, you have made a discovery. You have revealed a bit of the truth. You experience the highest joy and thrill a scientist can feel—a “sparkle of insight.” When this happens, if you have sense, you stop what you have been doing to pursue that discovery.
Hardly anyone has received a Nobel Prize for testing a theory, but many Nobel Prizes have been awarded for technological advances and for discoveries based on those advances. In my view, discoverers use an enlightened inductive approach at least as much as deduction. They develop new technology that enables key novel measurements, and they keep in mind gaps in knowledge, so that they are ready to appreciate the significance of their observations.
A PERSONAL EXAMPLE
‘You have to measure something’
Let me share an example of this process by relating a sparkle of insight I had several years ago. When I began working at the NIH, I met with the chief of the Hypertension-Endocrine Branch of the National Heart, Lung, and Blood Institute about the research program I would pursue. After listening patiently to me for many minutes as I spouted about how I was going to test hypotheses derived from the concepts that people with hypertension are “hyper-tense,” and that stress causes heart disease, the chief responded, “Well, these ideas are all well and good. But what are you going to measure? You can measure whatever you want, but you have to measure something.”
Measure something. I wanted to see if there was hyperactivity of the sympathetic nervous system or excessive sympathetic innervation in hypertension, and I started working on ways to measure sympathetic activity.
The sympathetic nervous system at a glance
First I should introduce you to the sympathetic nervous system, which is one of the main effectors by which the brain regulates the heart and blood vessels. It is a key link between the brain and heart. The sympathetic nerves to the heart and other organs do not come directly from the brain but from ganglia, which are clumps of nerve cell bodies strung like pearls on a necklace on each side of the spinal column. This origin outside the central nervous system will be an important fact to keep in mind.
In the heart, the sympathetic nerves travel with the coronary arteries and then dive into the heart muscle from the outside. Sympathetic nerves also enmesh the walls of arteries and arterioles. The arterioles constitute the main determinant of total peripheral resistance to blood flow in the body and therefore figure prominently in the control of blood pressure. The architectural association between sympathetic nerves and the muscle in the heart and arteriolar walls has enticed hypertension researchers for many decades.
A false start with plasma norepinephrine measurement
I developed novel methods for measuring plasma levels of norepinephrine, which is the chemical messenger that the sympathetic nervous system uses in regulation of the circulation, and of adrenaline (epinephrine), which is the well-known and potent “fight-or-flight” hormone.1 Applying this technology to patients with high blood pressure led to several publications2–9 but actually shed more heat than light on the hypothesis of sympathetic hyperactivity as a cause of or contributor to hypertension. In the face of negative data, the theory was qualified—sympathetic hyperactivity might be apparent only in the young, or the thin, or the Caucasian, or the male—but not abandoned.
Insights from visualizing sympathetic nerves in the heart
Then I embarked on a project to visualize sympathetic nerves in the heart, by a new technology called positron emission tomographic (PET) scanning. With several colleagues—including Irwin J. Kopin, Graeme Eisenhofer, Peter Chang, David Hovevey-Zion, Ehud Grossman, and Courtney Holmes—to whom I will always be grateful, I developed a PET imaging agent called 6-[18F]fluorodopamine.10–13
Figure 1. Thoracic positron emission tomographic scans in a healthy control subject and in patients with pure autonomic failure, multiple system atrophy, and Parkinson disease. The top row shows 13N-labeled ammonia perfusion scans and the bottom row shows 6-[18F]fluoro dopamine sympathoneural scans in each subject. Note the absence of cardiac 6-[18F]fluorodopamine-derived radioactivity in the subjects with pure autonomic failure and Parkinson disease in contrast with the normal radioactivity in the patient with multiple system atrophy; the decrease in radioactivity is particularly severe in the patient with Parkinson disease (red border). Adapted from Goldstein et al.14After injection of 6-[18F]fluorodopamine into a person’s vein, PET scan slices of the chest reveal the sympathetic nerves in the heart (Figure 1). The top row of Figure 1 shows where the blood is going—perfusion—in four people, and the bottom row shows the 6-[18F]fluorodopamine scans in the same people. The horseshoe-shaped structure is the main pumping muscle of the heart, the left ventricular myocardium. The “blob” on the patient’s right is the liver.
Normally, PET scans using 6-[18F]fluorodopamine look remarkably similar to scans using 13N-labeled ammonia, a perfusion imaging agent. The first patient I studied with this new technology was a patient with a rare disease called pure autonomic failure (PAF). In PAF, there was already good evidence for a loss of sympathetic nerves throughout the body. Myocardial perfusion in this patient was normal, but there was much less than normal 6-[18F]fluorodopamine-derived radioactivity in the heart muscle. In another uncommon disease, multiple system atrophy (MSA), the perfusion was also normal, and the cardiac sympathetic nerves seemed intact, in line with what was already known about this progressive neurodegenerative disease.
Then I tested a patient who had been thought to have MSA but actually had Parkinson disease (PD) with orthostatic hypotension (a fall in blood pressure each time the person stands up). PD with orthostatic hypotension can be very difficult to distinguish from the parkinsonian form of MSA. To my complete surprise, the patient with PD had a remarkable decrease in 6-[18F]fluorodopa mine-derived radioactivity in the heart muscle. There was normal blood flow to the heart muscle, so the 6-[18F]fluorodopamine was being delivered, but there was no evidence of sympathetic nerves in the heart. The scans resembled those in the PAF patient, not the MSA patient.
This finding did not arise from a prediction to test a hypothesis. It wasn’t long before I tested additional PD patients and found the same unexpected results.14,15 Because I was ignorant, I wasn’t biased. I felt I had put my finger on a piece of the truth, and I had to stop and think about the implications of this discovery. I never did come to test the hypotheses that I had sought out originally to test. Instead, I followed a totally new path, based on the discovery of cardiac sympathetic denervation in PD.
Beyond a brain disease: Seeing PD as a heart-brain disorder
More than 50 neuroimaging studies since our original report have agreed remarkably consistently on the association between PD and loss of sympathetic nerves in the heart; moreover, postmortem pathology studies have amply confirmed that a profound loss of cardiac sympathetic nerves is characteristic of PD.16 I have yet to come across a single patient with PD and orthostatic hypotension who has not had cardiac sympathetic denervation, and virtually all patients with PD who do not have orthostatic hypotension seem to have at least partial loss of cardiac sympathetic nerves.
Considering that the source of those nerves is the ganglia, which lie outside the central nervous system, PD must be more than a brain disease and more than a movement disorder. It must also be a disease of the sympathetic nerves in the heart, a form of a dysautonomia, and a heart-brain disorder.
The role of catecholamines: Another discovery born of unbiased ignorance
Figure 2. The chemical structure of each of the catecholamines —dopamine, norepinephrine, and adrenaline (epinephrine)—resembles a cat. The head of the cat is the catechol nucleus, with the two pointy ears corresponding to the adjacent hydroxyl groups on the benzene ring. At the other end of the cat is a hydrocarbon tail, ending in a “uriniferous” amine group. Adapted from Goldstein.17To appreciate fully the significance of this finding, I must mention my favorite chemical family, the catecholamines, whose chemical structures resemble cats (Figure 2).17 PD results from a loss of a particular chemical, dopamine, in a particular pathway in the brain; dopa mine is a catecholamine. The other catecholamines in humans are norepinephrine and adrenaline. As noted above, norepinephrine is the chemical messenger of the sympathetic nerves, and adrenaline is the well-known hormone that produces many of the signs of emotional distress.
Almost a half century ago, Hornykiewicz and colleagues made the pivotal discovery that PD features loss of dopamine in the nigrostriatal system in the brain.18 Given the cardiac sympathetic denervation, PD might be a disease of catecholamine systems both inside and outside the central nervous system—dopamine in the nigrostriatal system, and norepinephrine in the sympathetic nerves of the heart.
Then what of the third catecholamine, adrenaline, in PD? Plasma levels of adrenaline and of its metabolite, metanephrine, are normal in PD, even in patients who have PD and orthostatic hypotension, which involves loss of norepinephrine-producing nerves not only in the heart but in other organs.19 What is different about the adrenaline-producing cells in the medulla (from the Latin for “marrow”) of the adrenal glands atop each kidney? Why aren’t these catecholamine-producing cells also lost in PD?
I have some ideas in mind but won’t go into them here. The point is that the discovery of normal adrenaline-producing cells in PD, despite loss of cells producing the other catecholamines, was not based on my testing a hypothesis. It was a discovery born of ignorance, and because ignorance isn’t biased, that discovery points to the truth. Whatever the eventual explanation for the specific pattern of catecholamine cell loss in PD, it cannot refute the discovery itself.
HOW DISCOVERIES ARISE: AN APPLIED EXERCISE FOR READERS
Figure 3. High-resolution positron emission tomographic scans, superimposed over magnetic resonance images, at the level of the basal ganglia, after intravenous administration of 6-[18F]fluorodopa in a control subject (left) and a patient with Parkinson disease (PD) (right). Red indicates the maximum amount of radioactivity. Note the severely decreased 6-[18F]fluorodopa-derived radioactivity in the striatum—epecially the putamen—in the PD patient.Now let’s have you, the reader, make a discovery and induce its significance based on what I have tried to teach so far, that discoveries arise from the application of relevant technology and from insights of the prepared mind. Take a look at the scans in the left panels of Figures 3 and 4. The large red structures in Figure 3, which look like a sad clown’s eyes, correspond to the striatum. The striatum is made up of the putamen, which is like the mascara on the side of the sad clown’s eyes, and the caudate, which is like the beady eyes themselves. In the left panel of Figure 4, the small spots in the midbrain correspond to the substantia nigra, a major site of dopamine-producing neurons in the human brain.
Figure 4. High-resolution positron emission tomographic scans, superimposed over magnetic resonance images, at the level of the midbrain, after intravenous administration of 6-[18F]fluorodopa in a control subject (left) and a patient with Parkinson disease (PD) (right). Red indicates the maximum amount of radioactivity. Note the severely decreased 6-[18F]fluorodopa-derived radioactivity bilaterally in the region corresponding to the substantia nigra in the PD patient.We can see in the right panel of Figure 3 that in PD there is a loss of the ability to store dopamine in the striatum—especially in the putamen, the mascara of the sad clown’s eyes. In the right panel of Figure 4 we see that in the brainstem there is a loss of the dopamine-containing nerve cells in the substantia nigra. These scans therefore demonstrate graphically the nigrostriatal lesion characteristic of PD. There is a loss of the nerve cells in the substantia nigra in the midbrain and a loss of the dopamine-containing terminals in the striatum.
Figure 5. High-resolution positron emission tomographic scans, superimposed over magnetic resonance images, at the levels of the basal ganglia and midbrain, after intravenous administration of 6-[18F]fluorodopa in four subjects: a normal volunteer (upper left) a control patient without parkinsonism or autonomic failure (upper right) a patient with Parkinson disease (PD) (lower left) a patient with pure autonomic failure (PAF) (lower right). Red indicates the maximum amount of radioactivity. Note the severely decreased 6-[18F] fluorodopa-derived radioactivity bilaterally in the region corresponding to the substantia nigra in both PD and PAF. Adapted from Goldstein et al.20Now take a look at the scans of these areas in a patient with PAF in Figure 5. Remember that PAF involves a loss of sympathetic nerves in the heart, just like in PD, but that PAF does not involve parkinsonism. Look at the sad clown’s eyes. The mascara is there, of course, because the patient does not have parkinsonism. But now look for the spots in the substantia nigra—they are missing, just as in PD.
PAF is a rare disease, and I have only studied several cases with high-resolution PET scanning of the brain, but so far they have all had this unexpected, unpredicted finding of loss of dopaminergic neurons in the substantia nigra.20
What does this pattern mean? If PAF patients have just as much loss of nigral neurons as PD patients do, and if PAF patients do not have parkinsonism, then the movement disorder in PD cannot result from loss of the dopamine neurons in the substantia nigra per se. Instead, the movement disorder in PD seems to come from loss of the dopaminergic terminals in the striatum.
How can PAF patients have normal dopamine terminals in the putamen when the number of dopaminergic cell bodies is severely reduced? Somehow, PAF patients must be able to sprout new terminals, even as they lose the cell bodies. Maybe if we knew how PAF patients do this, we would have a way to treat or even prevent PD.
How do PAF patients maintain normal dopamine terminals as the cell bodies die off? No one knows. Until now, no one thought of asking such a question. No one hypothesized that this discovery would be made, but it was. And because ignorance isn’t biased, we have put our finger on the truth. By keeping in mind what isn’t known, we could see what wasn’t there. Now we can begin to think of what to look for next.
SUMMARY AND CONCLUSIONS
Because ignorance isn’t biased, if you have the tools to make relevant measurements, if you have sufficient mastery of the subject to know what isn’t known, and if you have access to patients with rare but informative disorders, you can make important discoveries based on inductions from observations.
The discoveries that cardiac sympathetic denervation characterizes PD and that parkinsonism does not result from loss of dopamine neurons per se depended crucially on studying patients with a rare disease, PAF. In 1657, William Harvey—the same William Harvey who first described the circulation of the blood and who first pointed out the effects of emotions on the heart—wrote eloquently about the extraordinary power of studying patients with rare diseases:Nature is nowhere accustomed more openly to display her secret mysteries than in cases where she shows traces of her workings apart from the beaten path; nor is there any better way to advance the proper practice of medicine than to give our minds to the discovery of the usual law of nature, by the careful investigation of cases of rarer forms of disease. For it has been found in almost all things, that what they contain of use or of application, is hardly perceived unless we are deprived of them, or they become deranged in some way.21
I hope I have convinced you of the importance of seeing what isn’t there. My thanks go out again to the Earl and Doris Bakken Heart-Brain Institute for this prestigious award, to my family, to my colleagues and friends, and to my patients. As I have written in Adrenaline and the Inner World: An Introduction to Scientific Integrative Medicine,17 patients serve as a unique scientific resource. They report what is wrong; they tell us the truth. We have to make sense of what they teach.
This is a momentous occasion for me, for the extraordinary people in the Clinical Neurocardiology Section at the National Institutes of Health (NIH), and for my family—my wife Minka and son Joey drove all the way from Maryland late last night and early this morning to be here. I thank them publicly here.
THE ‘SPARKLE OF INSIGHT’ FROM ENLIGHTENED INDUCTION
In these brief comments, as I look back on the road I have taken over the past 40 years carrying out patient-oriented research in heart-brain medicine, I would like to convey a viewpoint instead of dwelling on the presentation of research data.
The idea I wish to convey is that ignorance isn’t biased. If you have a hypothesis you want to test, you are inherently biased to find something positive—and, if you are in academic medicine, publishable—in the data you obtain. But if you have the technical capability to measure something no one else can measure, and you have sufficient mastery of the topic to know what is not yet known, then if you make an observation that you did not predict and if you recognize its significance, you have made a discovery. You have revealed a bit of the truth. You experience the highest joy and thrill a scientist can feel—a “sparkle of insight.” When this happens, if you have sense, you stop what you have been doing to pursue that discovery.
Hardly anyone has received a Nobel Prize for testing a theory, but many Nobel Prizes have been awarded for technological advances and for discoveries based on those advances. In my view, discoverers use an enlightened inductive approach at least as much as deduction. They develop new technology that enables key novel measurements, and they keep in mind gaps in knowledge, so that they are ready to appreciate the significance of their observations.
A PERSONAL EXAMPLE
‘You have to measure something’
Let me share an example of this process by relating a sparkle of insight I had several years ago. When I began working at the NIH, I met with the chief of the Hypertension-Endocrine Branch of the National Heart, Lung, and Blood Institute about the research program I would pursue. After listening patiently to me for many minutes as I spouted about how I was going to test hypotheses derived from the concepts that people with hypertension are “hyper-tense,” and that stress causes heart disease, the chief responded, “Well, these ideas are all well and good. But what are you going to measure? You can measure whatever you want, but you have to measure something.”
Measure something. I wanted to see if there was hyperactivity of the sympathetic nervous system or excessive sympathetic innervation in hypertension, and I started working on ways to measure sympathetic activity.
The sympathetic nervous system at a glance
First I should introduce you to the sympathetic nervous system, which is one of the main effectors by which the brain regulates the heart and blood vessels. It is a key link between the brain and heart. The sympathetic nerves to the heart and other organs do not come directly from the brain but from ganglia, which are clumps of nerve cell bodies strung like pearls on a necklace on each side of the spinal column. This origin outside the central nervous system will be an important fact to keep in mind.
In the heart, the sympathetic nerves travel with the coronary arteries and then dive into the heart muscle from the outside. Sympathetic nerves also enmesh the walls of arteries and arterioles. The arterioles constitute the main determinant of total peripheral resistance to blood flow in the body and therefore figure prominently in the control of blood pressure. The architectural association between sympathetic nerves and the muscle in the heart and arteriolar walls has enticed hypertension researchers for many decades.
A false start with plasma norepinephrine measurement
I developed novel methods for measuring plasma levels of norepinephrine, which is the chemical messenger that the sympathetic nervous system uses in regulation of the circulation, and of adrenaline (epinephrine), which is the well-known and potent “fight-or-flight” hormone.1 Applying this technology to patients with high blood pressure led to several publications2–9 but actually shed more heat than light on the hypothesis of sympathetic hyperactivity as a cause of or contributor to hypertension. In the face of negative data, the theory was qualified—sympathetic hyperactivity might be apparent only in the young, or the thin, or the Caucasian, or the male—but not abandoned.
Insights from visualizing sympathetic nerves in the heart
Then I embarked on a project to visualize sympathetic nerves in the heart, by a new technology called positron emission tomographic (PET) scanning. With several colleagues—including Irwin J. Kopin, Graeme Eisenhofer, Peter Chang, David Hovevey-Zion, Ehud Grossman, and Courtney Holmes—to whom I will always be grateful, I developed a PET imaging agent called 6-[18F]fluorodopamine.10–13
Figure 1. Thoracic positron emission tomographic scans in a healthy control subject and in patients with pure autonomic failure, multiple system atrophy, and Parkinson disease. The top row shows 13N-labeled ammonia perfusion scans and the bottom row shows 6-[18F]fluoro dopamine sympathoneural scans in each subject. Note the absence of cardiac 6-[18F]fluorodopamine-derived radioactivity in the subjects with pure autonomic failure and Parkinson disease in contrast with the normal radioactivity in the patient with multiple system atrophy; the decrease in radioactivity is particularly severe in the patient with Parkinson disease (red border). Adapted from Goldstein et al.14After injection of 6-[18F]fluorodopamine into a person’s vein, PET scan slices of the chest reveal the sympathetic nerves in the heart (Figure 1). The top row of Figure 1 shows where the blood is going—perfusion—in four people, and the bottom row shows the 6-[18F]fluorodopamine scans in the same people. The horseshoe-shaped structure is the main pumping muscle of the heart, the left ventricular myocardium. The “blob” on the patient’s right is the liver.
Normally, PET scans using 6-[18F]fluorodopamine look remarkably similar to scans using 13N-labeled ammonia, a perfusion imaging agent. The first patient I studied with this new technology was a patient with a rare disease called pure autonomic failure (PAF). In PAF, there was already good evidence for a loss of sympathetic nerves throughout the body. Myocardial perfusion in this patient was normal, but there was much less than normal 6-[18F]fluorodopamine-derived radioactivity in the heart muscle. In another uncommon disease, multiple system atrophy (MSA), the perfusion was also normal, and the cardiac sympathetic nerves seemed intact, in line with what was already known about this progressive neurodegenerative disease.
Then I tested a patient who had been thought to have MSA but actually had Parkinson disease (PD) with orthostatic hypotension (a fall in blood pressure each time the person stands up). PD with orthostatic hypotension can be very difficult to distinguish from the parkinsonian form of MSA. To my complete surprise, the patient with PD had a remarkable decrease in 6-[18F]fluorodopa mine-derived radioactivity in the heart muscle. There was normal blood flow to the heart muscle, so the 6-[18F]fluorodopamine was being delivered, but there was no evidence of sympathetic nerves in the heart. The scans resembled those in the PAF patient, not the MSA patient.
This finding did not arise from a prediction to test a hypothesis. It wasn’t long before I tested additional PD patients and found the same unexpected results.14,15 Because I was ignorant, I wasn’t biased. I felt I had put my finger on a piece of the truth, and I had to stop and think about the implications of this discovery. I never did come to test the hypotheses that I had sought out originally to test. Instead, I followed a totally new path, based on the discovery of cardiac sympathetic denervation in PD.
Beyond a brain disease: Seeing PD as a heart-brain disorder
More than 50 neuroimaging studies since our original report have agreed remarkably consistently on the association between PD and loss of sympathetic nerves in the heart; moreover, postmortem pathology studies have amply confirmed that a profound loss of cardiac sympathetic nerves is characteristic of PD.16 I have yet to come across a single patient with PD and orthostatic hypotension who has not had cardiac sympathetic denervation, and virtually all patients with PD who do not have orthostatic hypotension seem to have at least partial loss of cardiac sympathetic nerves.
Considering that the source of those nerves is the ganglia, which lie outside the central nervous system, PD must be more than a brain disease and more than a movement disorder. It must also be a disease of the sympathetic nerves in the heart, a form of a dysautonomia, and a heart-brain disorder.
The role of catecholamines: Another discovery born of unbiased ignorance
Figure 2. The chemical structure of each of the catecholamines —dopamine, norepinephrine, and adrenaline (epinephrine)—resembles a cat. The head of the cat is the catechol nucleus, with the two pointy ears corresponding to the adjacent hydroxyl groups on the benzene ring. At the other end of the cat is a hydrocarbon tail, ending in a “uriniferous” amine group. Adapted from Goldstein.17To appreciate fully the significance of this finding, I must mention my favorite chemical family, the catecholamines, whose chemical structures resemble cats (Figure 2).17 PD results from a loss of a particular chemical, dopamine, in a particular pathway in the brain; dopa mine is a catecholamine. The other catecholamines in humans are norepinephrine and adrenaline. As noted above, norepinephrine is the chemical messenger of the sympathetic nerves, and adrenaline is the well-known hormone that produces many of the signs of emotional distress.
Almost a half century ago, Hornykiewicz and colleagues made the pivotal discovery that PD features loss of dopamine in the nigrostriatal system in the brain.18 Given the cardiac sympathetic denervation, PD might be a disease of catecholamine systems both inside and outside the central nervous system—dopamine in the nigrostriatal system, and norepinephrine in the sympathetic nerves of the heart.
Then what of the third catecholamine, adrenaline, in PD? Plasma levels of adrenaline and of its metabolite, metanephrine, are normal in PD, even in patients who have PD and orthostatic hypotension, which involves loss of norepinephrine-producing nerves not only in the heart but in other organs.19 What is different about the adrenaline-producing cells in the medulla (from the Latin for “marrow”) of the adrenal glands atop each kidney? Why aren’t these catecholamine-producing cells also lost in PD?
I have some ideas in mind but won’t go into them here. The point is that the discovery of normal adrenaline-producing cells in PD, despite loss of cells producing the other catecholamines, was not based on my testing a hypothesis. It was a discovery born of ignorance, and because ignorance isn’t biased, that discovery points to the truth. Whatever the eventual explanation for the specific pattern of catecholamine cell loss in PD, it cannot refute the discovery itself.
HOW DISCOVERIES ARISE: AN APPLIED EXERCISE FOR READERS
Figure 3. High-resolution positron emission tomographic scans, superimposed over magnetic resonance images, at the level of the basal ganglia, after intravenous administration of 6-[18F]fluorodopa in a control subject (left) and a patient with Parkinson disease (PD) (right). Red indicates the maximum amount of radioactivity. Note the severely decreased 6-[18F]fluorodopa-derived radioactivity in the striatum—epecially the putamen—in the PD patient.Now let’s have you, the reader, make a discovery and induce its significance based on what I have tried to teach so far, that discoveries arise from the application of relevant technology and from insights of the prepared mind. Take a look at the scans in the left panels of Figures 3 and 4. The large red structures in Figure 3, which look like a sad clown’s eyes, correspond to the striatum. The striatum is made up of the putamen, which is like the mascara on the side of the sad clown’s eyes, and the caudate, which is like the beady eyes themselves. In the left panel of Figure 4, the small spots in the midbrain correspond to the substantia nigra, a major site of dopamine-producing neurons in the human brain.
Figure 4. High-resolution positron emission tomographic scans, superimposed over magnetic resonance images, at the level of the midbrain, after intravenous administration of 6-[18F]fluorodopa in a control subject (left) and a patient with Parkinson disease (PD) (right). Red indicates the maximum amount of radioactivity. Note the severely decreased 6-[18F]fluorodopa-derived radioactivity bilaterally in the region corresponding to the substantia nigra in the PD patient.We can see in the right panel of Figure 3 that in PD there is a loss of the ability to store dopamine in the striatum—especially in the putamen, the mascara of the sad clown’s eyes. In the right panel of Figure 4 we see that in the brainstem there is a loss of the dopamine-containing nerve cells in the substantia nigra. These scans therefore demonstrate graphically the nigrostriatal lesion characteristic of PD. There is a loss of the nerve cells in the substantia nigra in the midbrain and a loss of the dopamine-containing terminals in the striatum.
Figure 5. High-resolution positron emission tomographic scans, superimposed over magnetic resonance images, at the levels of the basal ganglia and midbrain, after intravenous administration of 6-[18F]fluorodopa in four subjects: a normal volunteer (upper left) a control patient without parkinsonism or autonomic failure (upper right) a patient with Parkinson disease (PD) (lower left) a patient with pure autonomic failure (PAF) (lower right). Red indicates the maximum amount of radioactivity. Note the severely decreased 6-[18F] fluorodopa-derived radioactivity bilaterally in the region corresponding to the substantia nigra in both PD and PAF. Adapted from Goldstein et al.20Now take a look at the scans of these areas in a patient with PAF in Figure 5. Remember that PAF involves a loss of sympathetic nerves in the heart, just like in PD, but that PAF does not involve parkinsonism. Look at the sad clown’s eyes. The mascara is there, of course, because the patient does not have parkinsonism. But now look for the spots in the substantia nigra—they are missing, just as in PD.
PAF is a rare disease, and I have only studied several cases with high-resolution PET scanning of the brain, but so far they have all had this unexpected, unpredicted finding of loss of dopaminergic neurons in the substantia nigra.20
What does this pattern mean? If PAF patients have just as much loss of nigral neurons as PD patients do, and if PAF patients do not have parkinsonism, then the movement disorder in PD cannot result from loss of the dopamine neurons in the substantia nigra per se. Instead, the movement disorder in PD seems to come from loss of the dopaminergic terminals in the striatum.
How can PAF patients have normal dopamine terminals in the putamen when the number of dopaminergic cell bodies is severely reduced? Somehow, PAF patients must be able to sprout new terminals, even as they lose the cell bodies. Maybe if we knew how PAF patients do this, we would have a way to treat or even prevent PD.
How do PAF patients maintain normal dopamine terminals as the cell bodies die off? No one knows. Until now, no one thought of asking such a question. No one hypothesized that this discovery would be made, but it was. And because ignorance isn’t biased, we have put our finger on the truth. By keeping in mind what isn’t known, we could see what wasn’t there. Now we can begin to think of what to look for next.
SUMMARY AND CONCLUSIONS
Because ignorance isn’t biased, if you have the tools to make relevant measurements, if you have sufficient mastery of the subject to know what isn’t known, and if you have access to patients with rare but informative disorders, you can make important discoveries based on inductions from observations.
The discoveries that cardiac sympathetic denervation characterizes PD and that parkinsonism does not result from loss of dopamine neurons per se depended crucially on studying patients with a rare disease, PAF. In 1657, William Harvey—the same William Harvey who first described the circulation of the blood and who first pointed out the effects of emotions on the heart—wrote eloquently about the extraordinary power of studying patients with rare diseases:Nature is nowhere accustomed more openly to display her secret mysteries than in cases where she shows traces of her workings apart from the beaten path; nor is there any better way to advance the proper practice of medicine than to give our minds to the discovery of the usual law of nature, by the careful investigation of cases of rarer forms of disease. For it has been found in almost all things, that what they contain of use or of application, is hardly perceived unless we are deprived of them, or they become deranged in some way.21
I hope I have convinced you of the importance of seeing what isn’t there. My thanks go out again to the Earl and Doris Bakken Heart-Brain Institute for this prestigious award, to my family, to my colleagues and friends, and to my patients. As I have written in Adrenaline and the Inner World: An Introduction to Scientific Integrative Medicine,17 patients serve as a unique scientific resource. They report what is wrong; they tell us the truth. We have to make sense of what they teach.
References
Goldstein DS, Feuerstein G, Izzo JL, Kopin IJ, Keiser HR. Validity and reliability of liquid chromatography with electrochemical detection for measuring plasma levels of norepinephrine and epinephrine in man. Life Sci1981; 28:467–475.
Goldstein DS. Plasma norepinephrine during stress in essential hypertension. Hypertension1981; 3:551–556.
Goldstein DS. Plasma norepinephrine in essential hypertension: a study of the studies. Hypertension1981; 3:48–52.
Goldstein D, Horwitz D, Keiser HR, Polinsky RJ, Kopin IJ. Plasma l-[3H]norepinephrine, d-[14C]norepinephrine, and d,l-[3H] isoproterenol kinetics in essential hypertension. J Clin Invest1983; 72:1748–1758.
Goldstein DS. Arterial baroreflex sensitivity, plasma catecholamines, and pressor responsiveness in essential hypertension. Circulation1983; 68:234–240.
Goldstein DS. Plasma catecholamines and essential hypertension: an analytical review. Hypertension1983; 5:86–99.
Goldstein DS, Lake CR, Chernow B, et al. Age-dependence of hypertensive-normotensive differences in plasma norepinephrine. Hypertension1983; 5:100–104.
Goldstein DS, McCarty R, Polinsky RJ, Kopin IJ. Relationship between plasma norepinephrine and sympathetic neural activity. Hypertension1983; 5:552–559.
Goldstein DS, Lake CR. Plasma norepinephrine and epinephrine levels in essential hypertension. Fed Proc1984; 43:57–61.
Eisenhofer G, Hovevey-Sion D, Kopin IJ, et al. Neuronal uptake and metabolism of 2- and 6-fluorodopamine: false neurotransmitters for positron emission tomographic imaging of sympathetically innervated tissues. J Pharmacol Exp Ther1989; 248:419–427.
Chang PC, Szemeredi K, Grossman E, Kopin IJ, Goldstein DS. Fate of tritiated 6-fluorodopamine in rats: a false neurotransmitter for positron emission tomographic imaging of sympathetic innervation and function. J Pharmacol Exp Ther1990; 255:809–817.
Goldstein DS, Holmes C. Metabolic fate of the sympathoneural imaging agent 6-[18F]fluorodopamine in humans. Clin Exp Hypertens1997; 19:155–161.
Goldstein DS, Eisenhofer G, Dunn BB, et al. Positron emission tomographic imaging of cardiac sympathetic innervation using 6-[18F] fluorodopamine: initial findings in humans. J Am Coll Cardiol1993; 22:1961–1971.
Goldstein DS, Holmes C, Cannon RO, Eisenhofer G, Kopin IJ. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med1997; 336:696–702.
Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO. Cardiac sympathetic denervation in Parkinson disease. Ann Intern Med2000; 133:338–347.
Amino T, Orimo S, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Pathol2005; 15:29–34.
Goldstein DS. Adrenaline and the Inner World: An Introduction to Scientific Integrative Medicine. Baltimore, MD: Johns Hopkins University Press; 2006.
Ehringer H, Hornykiewicz O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system [in German]. Wien Klin Wochenschr1960; 38:1236–1239.
Goldstein DS, Holmes C, Sharabi Y, Brentzel S, Eisenhofer G. Plasma levels of catechols and metanephrines in neurogenic orthostatic hypotension. Neurology2003; 60:1327–1332.
Goldstein DS, Holmes C, Sato T, et al. Central dopamine deficiency in pure autonomic failure. Clin Auton Res2008; 18:58–65.
Hervey Wyatt RBWilliam Harvey 1578 to 1657Whitefish, MTKessinger Publishing2005:161–162.
References
Goldstein DS, Feuerstein G, Izzo JL, Kopin IJ, Keiser HR. Validity and reliability of liquid chromatography with electrochemical detection for measuring plasma levels of norepinephrine and epinephrine in man. Life Sci1981; 28:467–475.
Goldstein DS. Plasma norepinephrine during stress in essential hypertension. Hypertension1981; 3:551–556.
Goldstein DS. Plasma norepinephrine in essential hypertension: a study of the studies. Hypertension1981; 3:48–52.
Goldstein D, Horwitz D, Keiser HR, Polinsky RJ, Kopin IJ. Plasma l-[3H]norepinephrine, d-[14C]norepinephrine, and d,l-[3H] isoproterenol kinetics in essential hypertension. J Clin Invest1983; 72:1748–1758.
Goldstein DS. Arterial baroreflex sensitivity, plasma catecholamines, and pressor responsiveness in essential hypertension. Circulation1983; 68:234–240.
Goldstein DS. Plasma catecholamines and essential hypertension: an analytical review. Hypertension1983; 5:86–99.
Goldstein DS, Lake CR, Chernow B, et al. Age-dependence of hypertensive-normotensive differences in plasma norepinephrine. Hypertension1983; 5:100–104.
Goldstein DS, McCarty R, Polinsky RJ, Kopin IJ. Relationship between plasma norepinephrine and sympathetic neural activity. Hypertension1983; 5:552–559.
Goldstein DS, Lake CR. Plasma norepinephrine and epinephrine levels in essential hypertension. Fed Proc1984; 43:57–61.
Eisenhofer G, Hovevey-Sion D, Kopin IJ, et al. Neuronal uptake and metabolism of 2- and 6-fluorodopamine: false neurotransmitters for positron emission tomographic imaging of sympathetically innervated tissues. J Pharmacol Exp Ther1989; 248:419–427.
Chang PC, Szemeredi K, Grossman E, Kopin IJ, Goldstein DS. Fate of tritiated 6-fluorodopamine in rats: a false neurotransmitter for positron emission tomographic imaging of sympathetic innervation and function. J Pharmacol Exp Ther1990; 255:809–817.
Goldstein DS, Holmes C. Metabolic fate of the sympathoneural imaging agent 6-[18F]fluorodopamine in humans. Clin Exp Hypertens1997; 19:155–161.
Goldstein DS, Eisenhofer G, Dunn BB, et al. Positron emission tomographic imaging of cardiac sympathetic innervation using 6-[18F] fluorodopamine: initial findings in humans. J Am Coll Cardiol1993; 22:1961–1971.
Goldstein DS, Holmes C, Cannon RO, Eisenhofer G, Kopin IJ. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med1997; 336:696–702.
Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO. Cardiac sympathetic denervation in Parkinson disease. Ann Intern Med2000; 133:338–347.
Amino T, Orimo S, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Pathol2005; 15:29–34.
Goldstein DS. Adrenaline and the Inner World: An Introduction to Scientific Integrative Medicine. Baltimore, MD: Johns Hopkins University Press; 2006.
Ehringer H, Hornykiewicz O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system [in German]. Wien Klin Wochenschr1960; 38:1236–1239.
Goldstein DS, Holmes C, Sharabi Y, Brentzel S, Eisenhofer G. Plasma levels of catechols and metanephrines in neurogenic orthostatic hypotension. Neurology2003; 60:1327–1332.
Goldstein DS, Holmes C, Sato T, et al. Central dopamine deficiency in pure autonomic failure. Clin Auton Res2008; 18:58–65.
Hervey Wyatt RBWilliam Harvey 1578 to 1657Whitefish, MTKessinger Publishing2005:161–162.
In Parkinson disease (PD), by the time the movement disorder develops, most of the nigrostriatal dopamine terminals have been lost. Identification of biomarkers of PD should improve early diagnosis and spur development of effective treatments.
Braak has proposed a pathogenetic sequence beginning outside the brain, with invasion of peripheral, vulnerable autonomic neurons, followed by alpha-synucleinopathy in lower brainstem nuclei and then by alpha-synucleinopathy in the midbrain substantia nigra and then finally in the cerebral cortex.3,4 Consistent with early involvement of peripheral autonomic or lower brainstem centers, several studies of de novo PD have reported evidence of cardiac noradrenergic denervation5,8,14,22 or of decreased baroreflex-cardiovagal function.1,2,6,14,18
Whether these abnormalities can actually precede symptomatic PD has been unknown. Here we report the case of a patient who had cardiac noradrenergic denervation, detected by 6-[18F]fluorodopamine positron emission tomography, and decreased baroreflex-cardiovagal gain, detected by abnormal beat-to-beat blood pressure and heart rate responses to the Valsalva maneuver, 4 years before the clinical onset of PD.
CASE REPORT
A 56-year-old man was referred for possible pheochromocytoma, based on episodic hypertensive episodes and symptoms suggesting excessive catecholamine effects.
He had no serious health problems until about 1998, when he began to experience malaise and exercise intolerance and episodes of hypertension or hypotension, palpitations, and chest tightness. He also had a long history of constipation and dyspepsia, a tendency to urinary retention, and complained of a sense of fullness in the left neck. The patient’s career was in marketing and business development, until he quit work due to his symptoms. His mother had died of PD. Cardiac catheterization showed normal coronary arteries. Gastrointestinal endoscopy was unrevealing. Biochemical testing showed elevated plasma levels and urinary excretion of epinephrine. Thyroid function was normal.
Figure 1. Thoracic 6-[18F]fluorodopamine (18FDA) and 13N-ammonia (13NH3) images in July 2001 and November 2005. Note absence of left ventricular myocardial 6-[18F]fluorodopamine-derived radioactivity at both times, indicating cardiac sympathetic denervation. Myocardial perfusion, as indicated by 13NH3-derived radioactivity, was normal.Because of the hypertensive paroxysms, pheochromocytoma was suspected. In April 2000, the patient had a plasma epinephrine level about twice the upper limit of normal and a plasma metanephrine level about 50% above normal. In July 2001, he was evaluated at the National Institutes of Health (NIH). Normal follow-up plasma metanephrine, and failure of 6-[18F]fluorodopamine PET to detect an adrenal or extra-adrenal focus of radioactivity, excluded pheochromocytoma.17 At that time the concentration of 6-[18F]fluorodopamine-derived radioactivity was found to be markedly decreased in the left ventricular myocardium (Figure 1).
Figure 2. Beat-to-beat heart rate and blood pressure responses to the Valsalva maneuver (12-second duration, 30 mm Hg) in July 2001 and November 2005. In the latter recording, note progressive decline in blood pressure during Phase II, smaller pressure overshoot, and delayed return of pressure toward baseline in Phase IV, consistent with worsening baroreflex-sympathoneural function. Heart rate responses during and after the maneuver were also smaller in 2005 than in 2001, despite larger changes in blood pressure, consistent with worsening baroreflex-cardiovagal function.Autonomic function testing included measurements of beat-to-beat blood pressure and heart rate during and after performance of the Valsalva maneuver. Blood pressure decreased early in Phase II and then leveled off, and there was an overshoot in pressure during Phase IV (dashed line in Figure 2), which are normal findings. Baroreflex-cardiovagal gain, calculated from the slope of the relationship between cardiac interbeat interval (with one beat delay) and systolic blood pressure during Phase II of the maneuver, was decreased at 3.2 msec/mm Hg; baroreflex-cardiovagal gain calculated from the data in Phase IV after release of the maneuver was also decreased at 3.1 msec/mm Hg).11,14,15
Over several months in 2005 the patient noted progressive slowing of movement and inability to relax the arms, small handwriting, decreased facial expression, and decreased voice volume. The patient returned to the NIH in November 2005, to participate in a protocol on pseudopheochromocytoma, the evaluation again including 6-[18F]fluorodopamine positron emission tomographic scanning and beat-to-beat blood pressure and heart rate associated with the Valsalva maneuver. 6-[18F]fluorodopamine PET again revealed severely decreased 6-[18F]fluorodopaminederived radioactivity throughout the left ventricular myocardium (Figure 1). In the interventricular septum, radioactivity at the midpoint of the scanning frame between 5 and 10 minutes after initiation of injection of 6-[18F]fluorodopamine was 1,286 nCi-kg/cc-mCi, more than 2 standard deviations below the normal mean and one of the lowest values we have recorded so far (Figure 3). Blood pressure decreased progressively in Phase II of the Valsalva maneuver, to a greater extent than in 2001, there was no overshoot of pressure after release of the maneuver, and the return of pressure toward baseline was prolonged, findings pointing to failure of sympathetically mediated reflexive vasoconstriction.12,23 Baroreflex-cardiovagal gain was also lower than in 2001 (1.2 msec/mm Hg from the results in Phase II, 2.6 msec/mm Hg from those in Phase IV), both because the range of heart was smaller and the extent of change in systolic pressure larger in 2005 than in 2001.
Figure 3. Individual values for septal myocardial 6-[18F]fluorodopamine-derived radioactivity, in normal control subjects (white circles), patients with Parkinson disease without sympathetic neurocirculatory failure (PD no SNF, green circles), patients with Parkinson disease and sympathetic neurocirculatory failure (PD SNF, blue circles), and the case reported here (large green circle). Dashed line shows the normal mean value and light green shaded area 2 standard deviations from the normal mean. Note markedly decreased 6-[18F]fluorodopamine-derived radioactivity in the current case.As a test of the status of the adrenomedullary hormonal system, blood was obtained via an indwelling arm catheter during supine rest and after bolus i.v. injection of 1 mg of glucagon and assayed for plasma catecholamines in our laboratory. Both in July 2001 and November 2005, the ratio of plasma epinephrine (in pg/mL) to norepinephrine (in pg/mL) was relatively high during supine rest (76:99 and 101:234), and the patient had large increases of plasma epinephrine levels in response to glucagon (peak values more than 250 pg/mL, more than six times the normal peak value).
Neurological consultation in November 2005 noted stooped posture and axial instability, cogwheel rigidity in all four extremities, paucity of spontaneous movements, masked face with infrequent blinking, and monotone voice, but with normal speed of gait and no resting tremor. The patient was diagnosed with mild PD.
DISCUSSION
In this patient, results of 6-[18F]fluorodopamine PET scanning indicated cardiac sympathetic denervation 4 years before the clinical onset of PD. Considering that in PD loss of cardiac noradrenergic innervation progresses slowly over years,13 and that the patient already had evidence for markedly decreased cardiac noradrenergic innervation at the time of initial evaluation, loss of cardiac sympathetic nerves probably preceded the movement disorder by many more than the 4 years between initial testing and the onset of PD.
The findings in this case fit with previous reports of cardiac noradrenergic denervation in de novo PD and with the concept of a peripheral-to-central and caudal-to-rostral pathogenetic sequence. Orimo and co-workers have noted loss of noradrenergic terminal innervation of the myocardium before loss of cell bodies in sympathetic ganglia in PD.16
Our patient also had evidence for decreased baroreflex-cardiovagal function 4 years before the movement disorder. The baroreflex is a homeostatic arc, and abnormalities of afferent neurotransmission, central integration by brainstem centers, or vagal efferent pre-ganglionic or post-ganglionic fibers could result in the same clinical laboratory finding of low baroreflex-cardiovagal gain. In particular, the extent to which baroreflex-cardiovagal failure in PD reflects a brainstem lesion, as opposed to an afferent lesion or loss of parasympathetic cholinergic efferents, remains unknown. The results in our patient are consistent with the view that baroreflex-cardiovagal function worsens over years before the onset of PD.
Chronic constipation, which also preceded parkinsonism in our case, would be consistent with early dysregulation of gastrointestinal autonomic function. Accumulations of alpha-synuclein in enteric neurons and in the dorsal motor nucleus of the vagus nerve, the central neural site of origin of parasympathetic innervation of much of the gastrointestinal tract, has been reported to be an early pathological finding.3 As noted above, however, the occurrence of central neural pathology would not exclude a concurrent afferent or efferent lesion, and studies have found Lewy bodies in the myenteric plexus of both the esophagus and colon,9 as well as loss of enteric dopaminergic neurons in PD with chronic constipation.19
Evidence for abnormalities of the sympathetic norad renergic and parasympathetic cholinergic components of the autonomic nervous system in our patient occurred without evidence for compromised adrenomedullary function. On the contrary, the patient had augmented plasma epinephrine responses to glucagon injection, both upon initial evaluation and at follow-up. The patient therefore did not appear to have diffuse loss of catecholaminergic cells. Although studies have noted decreased adrenomedullary catecholamine concentrations in patients with severe PD,7,20,21 plasma levels of epinephrine and its O-methylated metabolite, metanephrine, have been reported to be normal.10
Combined cardiac sympathetic denervation (with attendant denervation supersensitivity), baroreflex-cardiovagal hypofunction, and adrenomedullary hyper-responsiveness might explain the symptoms and signs of cardiovascular instability, such as episodic hypertensive paroxysms, tachycardia, palpitations, and chest pain despite normal coronary arteries, that led to clinical suspicion of pheochromocytoma in this patient.
The results in this case lead us to propose that cardiac sympathetic denervation and decreased baroreflex-cardiovagal gain may be biomarkers of early autonomic involvement in PD. Studies in progress about autonomic function in relatives of patients with familial PD should help test this hypothesis.
References
Bonuccelli U, Lucetti C, Del Dotto P, et al. Orthostatic hypotension in de novo Parkinson disease. Arch Neurol2003; 60:1400–1404.
Bouhaddi M, Vuillier F, Fortrat JO, et al. Impaired cardiovascular autonomic control in newly and long-term-treated patients with Parkinson’s disease: involvement of L-dopa therapy. Auton Neurosci2004; 116:30–38.
Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett2006; 396:67–72.
Braak H, Rub U, Gai WP, Del Tredici K. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm2003; 110:517–536.
Braune S. The role of cardiac metaiodobenzylguanidine uptake in the differential diagnosis of parkinsonian syndromes. Clin Auton Res2001; 11:351–355.
Camerlingo M, Aillon C, Bottacchi E, et al. Parasympathetic assessment in Parkinson’s disease. Adv Neurol1987; 45:267–269.
Carmichael SW, Wilson RJ, Brimijoin WS, et al. Decreased catecholamines in the adrenal medulla of patients with parkinsonism. N Engl J Med1988; 318:254.
Druschky A, Hilz MJ, Platsch G, et al. Differentiation of Parkinson’s disease and multiple system atrophy in early disease stages by means of I-123-MIBG-SPECT. J Neurol Sci2000; 175:3–12.
Edwards LL, Quigley EM, Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease: frequency and pathophysiology. Neurology1992; 42:726–732.
Goldstein DS, Holmes C, Sharabi Y, Brentzel S, Eisenhofer G. Plasma levels of catechols and metanephrines in neurogenic orthostatic hypotension. Neurology2003; 60:1327–1332.
Goldstein DS, Horwitz D, Keiser HR. Comparison of techniques for measuring baroreflex sensitivity in man. Circulation1982; 66:432–439.
Goldstein DS, Tack C. Noninvasive detection of sympathetic neurocirculatory failure. Clin Auton Res2000; 10:285–291.
Li ST, Dendi R, Holmes C, Goldstein DS. Progressive loss of cardiac sympathetic innervation in Parkinson’s disease. Ann Neurol2002; 52:220–223.
Oka H, Mochio S, Onouchi K, Morita M, Yoshioka M, Inoue K. Cardiovascular dysautonomia in de novo Parkinson’s disease. J Neurol Sci2006; 241:59–65.
Oka H, Mochio S, Yoshioka M, Morita M, Onouchi K, Inoue K. Cardiovascular dysautonomia in Parkinson’s disease and multiple system atrophy. Acta Neurol Scand2006; 113:221–227.
Orimo S, Amino T, Itoh Y, et al. Cardiac sympathetic denervation precedes neuronal loss in the sympathetic ganglia in Lewy body disease. Acta Neuropathol (Berl)2005; 109:583–588.
Pacak K, Eisenhofer G, Carrasquillo JA, Chen CC, Li ST, Goldstein DS. 6-[18F]fluorodopamine positron emission tomographic (PET) scanning for diagnostic localization of pheochromocytoma. Hypertension2001; 38:6–8.
Quadri R, Comino I, Scarzella L, et al. Autonomic nervous function in de novo parkinsonian patients in basal condition and after acute levodopa administration. Funct Neurol2000; 15:81–86.
Singaram C, Ashraf W, Gaumnitz EA, et al. Dopaminergic defect of enteric nervous system in Parkinson’s disease patients with chronic constipation. Lancet1995; 346:861–864.
Stoddard SL, Ahlskog JE, Kelly PJ, et al. Decreased adrenal medullary catecholamines in adrenal transplanted parkinsonian patients compared to nephrectomy patients. Exp Neurol1989; 104:218–222.
Stoddard SL, Tyce GM, Ahlskog JE, Zinsmeister AR, Carmichael SW. Decreased catecholamine content in parkinsonian adrenal medullae. Exp Neurol1989; 104:22–27.
Takatsu H, Nishida H, Matsuo H, et al. Cardiac sympathetic denervation from the early stage of Parkinson’s disease: clinical and experimental studies with radiolabeled MIBG. J Nucl Med2000; 41:71–77.
Vogel ER, Sandroni P, Low PA. Blood pressure recovery from Valsalva maneuver in patients with autonomic failure. Neurology2005; 65:1533–1537.
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, National Institute of Neurological Disorders and Stroke (NINDS), National Institutes of Health (NIH), Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Clinical Neurocardiology Section, NINDS, NIH, 10 Center Drive MSC-1620, Building 10, Room 6N252, Bethesda, MD 20892-1620;goldsteind@ninds.nih.gov
*This article is reprinted, with permission, from Clinical Autonomic Research (Goldstein DS, et al. Clin Auton Res 2007; 17:118–121). The original publication is available at www.springerlink.com.
No author conflicts of interest were reported in the original publication of this article.
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, National Institute of Neurological Disorders and Stroke (NINDS), National Institutes of Health (NIH), Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Clinical Neurocardiology Section, NINDS, NIH, 10 Center Drive MSC-1620, Building 10, Room 6N252, Bethesda, MD 20892-1620;goldsteind@ninds.nih.gov
*This article is reprinted, with permission, from Clinical Autonomic Research (Goldstein DS, et al. Clin Auton Res 2007; 17:118–121). The original publication is available at www.springerlink.com.
No author conflicts of interest were reported in the original publication of this article.
Author and Disclosure Information
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, National Institute of Neurological Disorders and Stroke (NINDS), National Institutes of Health (NIH), Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Clinical Neurocardiology Section, NINDS, NIH, 10 Center Drive MSC-1620, Building 10, Room 6N252, Bethesda, MD 20892-1620;goldsteind@ninds.nih.gov
*This article is reprinted, with permission, from Clinical Autonomic Research (Goldstein DS, et al. Clin Auton Res 2007; 17:118–121). The original publication is available at www.springerlink.com.
No author conflicts of interest were reported in the original publication of this article.
In Parkinson disease (PD), by the time the movement disorder develops, most of the nigrostriatal dopamine terminals have been lost. Identification of biomarkers of PD should improve early diagnosis and spur development of effective treatments.
Braak has proposed a pathogenetic sequence beginning outside the brain, with invasion of peripheral, vulnerable autonomic neurons, followed by alpha-synucleinopathy in lower brainstem nuclei and then by alpha-synucleinopathy in the midbrain substantia nigra and then finally in the cerebral cortex.3,4 Consistent with early involvement of peripheral autonomic or lower brainstem centers, several studies of de novo PD have reported evidence of cardiac noradrenergic denervation5,8,14,22 or of decreased baroreflex-cardiovagal function.1,2,6,14,18
Whether these abnormalities can actually precede symptomatic PD has been unknown. Here we report the case of a patient who had cardiac noradrenergic denervation, detected by 6-[18F]fluorodopamine positron emission tomography, and decreased baroreflex-cardiovagal gain, detected by abnormal beat-to-beat blood pressure and heart rate responses to the Valsalva maneuver, 4 years before the clinical onset of PD.
CASE REPORT
A 56-year-old man was referred for possible pheochromocytoma, based on episodic hypertensive episodes and symptoms suggesting excessive catecholamine effects.
He had no serious health problems until about 1998, when he began to experience malaise and exercise intolerance and episodes of hypertension or hypotension, palpitations, and chest tightness. He also had a long history of constipation and dyspepsia, a tendency to urinary retention, and complained of a sense of fullness in the left neck. The patient’s career was in marketing and business development, until he quit work due to his symptoms. His mother had died of PD. Cardiac catheterization showed normal coronary arteries. Gastrointestinal endoscopy was unrevealing. Biochemical testing showed elevated plasma levels and urinary excretion of epinephrine. Thyroid function was normal.
Figure 1. Thoracic 6-[18F]fluorodopamine (18FDA) and 13N-ammonia (13NH3) images in July 2001 and November 2005. Note absence of left ventricular myocardial 6-[18F]fluorodopamine-derived radioactivity at both times, indicating cardiac sympathetic denervation. Myocardial perfusion, as indicated by 13NH3-derived radioactivity, was normal.Because of the hypertensive paroxysms, pheochromocytoma was suspected. In April 2000, the patient had a plasma epinephrine level about twice the upper limit of normal and a plasma metanephrine level about 50% above normal. In July 2001, he was evaluated at the National Institutes of Health (NIH). Normal follow-up plasma metanephrine, and failure of 6-[18F]fluorodopamine PET to detect an adrenal or extra-adrenal focus of radioactivity, excluded pheochromocytoma.17 At that time the concentration of 6-[18F]fluorodopamine-derived radioactivity was found to be markedly decreased in the left ventricular myocardium (Figure 1).
Figure 2. Beat-to-beat heart rate and blood pressure responses to the Valsalva maneuver (12-second duration, 30 mm Hg) in July 2001 and November 2005. In the latter recording, note progressive decline in blood pressure during Phase II, smaller pressure overshoot, and delayed return of pressure toward baseline in Phase IV, consistent with worsening baroreflex-sympathoneural function. Heart rate responses during and after the maneuver were also smaller in 2005 than in 2001, despite larger changes in blood pressure, consistent with worsening baroreflex-cardiovagal function.Autonomic function testing included measurements of beat-to-beat blood pressure and heart rate during and after performance of the Valsalva maneuver. Blood pressure decreased early in Phase II and then leveled off, and there was an overshoot in pressure during Phase IV (dashed line in Figure 2), which are normal findings. Baroreflex-cardiovagal gain, calculated from the slope of the relationship between cardiac interbeat interval (with one beat delay) and systolic blood pressure during Phase II of the maneuver, was decreased at 3.2 msec/mm Hg; baroreflex-cardiovagal gain calculated from the data in Phase IV after release of the maneuver was also decreased at 3.1 msec/mm Hg).11,14,15
Over several months in 2005 the patient noted progressive slowing of movement and inability to relax the arms, small handwriting, decreased facial expression, and decreased voice volume. The patient returned to the NIH in November 2005, to participate in a protocol on pseudopheochromocytoma, the evaluation again including 6-[18F]fluorodopamine positron emission tomographic scanning and beat-to-beat blood pressure and heart rate associated with the Valsalva maneuver. 6-[18F]fluorodopamine PET again revealed severely decreased 6-[18F]fluorodopaminederived radioactivity throughout the left ventricular myocardium (Figure 1). In the interventricular septum, radioactivity at the midpoint of the scanning frame between 5 and 10 minutes after initiation of injection of 6-[18F]fluorodopamine was 1,286 nCi-kg/cc-mCi, more than 2 standard deviations below the normal mean and one of the lowest values we have recorded so far (Figure 3). Blood pressure decreased progressively in Phase II of the Valsalva maneuver, to a greater extent than in 2001, there was no overshoot of pressure after release of the maneuver, and the return of pressure toward baseline was prolonged, findings pointing to failure of sympathetically mediated reflexive vasoconstriction.12,23 Baroreflex-cardiovagal gain was also lower than in 2001 (1.2 msec/mm Hg from the results in Phase II, 2.6 msec/mm Hg from those in Phase IV), both because the range of heart was smaller and the extent of change in systolic pressure larger in 2005 than in 2001.
Figure 3. Individual values for septal myocardial 6-[18F]fluorodopamine-derived radioactivity, in normal control subjects (white circles), patients with Parkinson disease without sympathetic neurocirculatory failure (PD no SNF, green circles), patients with Parkinson disease and sympathetic neurocirculatory failure (PD SNF, blue circles), and the case reported here (large green circle). Dashed line shows the normal mean value and light green shaded area 2 standard deviations from the normal mean. Note markedly decreased 6-[18F]fluorodopamine-derived radioactivity in the current case.As a test of the status of the adrenomedullary hormonal system, blood was obtained via an indwelling arm catheter during supine rest and after bolus i.v. injection of 1 mg of glucagon and assayed for plasma catecholamines in our laboratory. Both in July 2001 and November 2005, the ratio of plasma epinephrine (in pg/mL) to norepinephrine (in pg/mL) was relatively high during supine rest (76:99 and 101:234), and the patient had large increases of plasma epinephrine levels in response to glucagon (peak values more than 250 pg/mL, more than six times the normal peak value).
Neurological consultation in November 2005 noted stooped posture and axial instability, cogwheel rigidity in all four extremities, paucity of spontaneous movements, masked face with infrequent blinking, and monotone voice, but with normal speed of gait and no resting tremor. The patient was diagnosed with mild PD.
DISCUSSION
In this patient, results of 6-[18F]fluorodopamine PET scanning indicated cardiac sympathetic denervation 4 years before the clinical onset of PD. Considering that in PD loss of cardiac noradrenergic innervation progresses slowly over years,13 and that the patient already had evidence for markedly decreased cardiac noradrenergic innervation at the time of initial evaluation, loss of cardiac sympathetic nerves probably preceded the movement disorder by many more than the 4 years between initial testing and the onset of PD.
The findings in this case fit with previous reports of cardiac noradrenergic denervation in de novo PD and with the concept of a peripheral-to-central and caudal-to-rostral pathogenetic sequence. Orimo and co-workers have noted loss of noradrenergic terminal innervation of the myocardium before loss of cell bodies in sympathetic ganglia in PD.16
Our patient also had evidence for decreased baroreflex-cardiovagal function 4 years before the movement disorder. The baroreflex is a homeostatic arc, and abnormalities of afferent neurotransmission, central integration by brainstem centers, or vagal efferent pre-ganglionic or post-ganglionic fibers could result in the same clinical laboratory finding of low baroreflex-cardiovagal gain. In particular, the extent to which baroreflex-cardiovagal failure in PD reflects a brainstem lesion, as opposed to an afferent lesion or loss of parasympathetic cholinergic efferents, remains unknown. The results in our patient are consistent with the view that baroreflex-cardiovagal function worsens over years before the onset of PD.
Chronic constipation, which also preceded parkinsonism in our case, would be consistent with early dysregulation of gastrointestinal autonomic function. Accumulations of alpha-synuclein in enteric neurons and in the dorsal motor nucleus of the vagus nerve, the central neural site of origin of parasympathetic innervation of much of the gastrointestinal tract, has been reported to be an early pathological finding.3 As noted above, however, the occurrence of central neural pathology would not exclude a concurrent afferent or efferent lesion, and studies have found Lewy bodies in the myenteric plexus of both the esophagus and colon,9 as well as loss of enteric dopaminergic neurons in PD with chronic constipation.19
Evidence for abnormalities of the sympathetic norad renergic and parasympathetic cholinergic components of the autonomic nervous system in our patient occurred without evidence for compromised adrenomedullary function. On the contrary, the patient had augmented plasma epinephrine responses to glucagon injection, both upon initial evaluation and at follow-up. The patient therefore did not appear to have diffuse loss of catecholaminergic cells. Although studies have noted decreased adrenomedullary catecholamine concentrations in patients with severe PD,7,20,21 plasma levels of epinephrine and its O-methylated metabolite, metanephrine, have been reported to be normal.10
Combined cardiac sympathetic denervation (with attendant denervation supersensitivity), baroreflex-cardiovagal hypofunction, and adrenomedullary hyper-responsiveness might explain the symptoms and signs of cardiovascular instability, such as episodic hypertensive paroxysms, tachycardia, palpitations, and chest pain despite normal coronary arteries, that led to clinical suspicion of pheochromocytoma in this patient.
The results in this case lead us to propose that cardiac sympathetic denervation and decreased baroreflex-cardiovagal gain may be biomarkers of early autonomic involvement in PD. Studies in progress about autonomic function in relatives of patients with familial PD should help test this hypothesis.
In Parkinson disease (PD), by the time the movement disorder develops, most of the nigrostriatal dopamine terminals have been lost. Identification of biomarkers of PD should improve early diagnosis and spur development of effective treatments.
Braak has proposed a pathogenetic sequence beginning outside the brain, with invasion of peripheral, vulnerable autonomic neurons, followed by alpha-synucleinopathy in lower brainstem nuclei and then by alpha-synucleinopathy in the midbrain substantia nigra and then finally in the cerebral cortex.3,4 Consistent with early involvement of peripheral autonomic or lower brainstem centers, several studies of de novo PD have reported evidence of cardiac noradrenergic denervation5,8,14,22 or of decreased baroreflex-cardiovagal function.1,2,6,14,18
Whether these abnormalities can actually precede symptomatic PD has been unknown. Here we report the case of a patient who had cardiac noradrenergic denervation, detected by 6-[18F]fluorodopamine positron emission tomography, and decreased baroreflex-cardiovagal gain, detected by abnormal beat-to-beat blood pressure and heart rate responses to the Valsalva maneuver, 4 years before the clinical onset of PD.
CASE REPORT
A 56-year-old man was referred for possible pheochromocytoma, based on episodic hypertensive episodes and symptoms suggesting excessive catecholamine effects.
He had no serious health problems until about 1998, when he began to experience malaise and exercise intolerance and episodes of hypertension or hypotension, palpitations, and chest tightness. He also had a long history of constipation and dyspepsia, a tendency to urinary retention, and complained of a sense of fullness in the left neck. The patient’s career was in marketing and business development, until he quit work due to his symptoms. His mother had died of PD. Cardiac catheterization showed normal coronary arteries. Gastrointestinal endoscopy was unrevealing. Biochemical testing showed elevated plasma levels and urinary excretion of epinephrine. Thyroid function was normal.
Figure 1. Thoracic 6-[18F]fluorodopamine (18FDA) and 13N-ammonia (13NH3) images in July 2001 and November 2005. Note absence of left ventricular myocardial 6-[18F]fluorodopamine-derived radioactivity at both times, indicating cardiac sympathetic denervation. Myocardial perfusion, as indicated by 13NH3-derived radioactivity, was normal.Because of the hypertensive paroxysms, pheochromocytoma was suspected. In April 2000, the patient had a plasma epinephrine level about twice the upper limit of normal and a plasma metanephrine level about 50% above normal. In July 2001, he was evaluated at the National Institutes of Health (NIH). Normal follow-up plasma metanephrine, and failure of 6-[18F]fluorodopamine PET to detect an adrenal or extra-adrenal focus of radioactivity, excluded pheochromocytoma.17 At that time the concentration of 6-[18F]fluorodopamine-derived radioactivity was found to be markedly decreased in the left ventricular myocardium (Figure 1).
Figure 2. Beat-to-beat heart rate and blood pressure responses to the Valsalva maneuver (12-second duration, 30 mm Hg) in July 2001 and November 2005. In the latter recording, note progressive decline in blood pressure during Phase II, smaller pressure overshoot, and delayed return of pressure toward baseline in Phase IV, consistent with worsening baroreflex-sympathoneural function. Heart rate responses during and after the maneuver were also smaller in 2005 than in 2001, despite larger changes in blood pressure, consistent with worsening baroreflex-cardiovagal function.Autonomic function testing included measurements of beat-to-beat blood pressure and heart rate during and after performance of the Valsalva maneuver. Blood pressure decreased early in Phase II and then leveled off, and there was an overshoot in pressure during Phase IV (dashed line in Figure 2), which are normal findings. Baroreflex-cardiovagal gain, calculated from the slope of the relationship between cardiac interbeat interval (with one beat delay) and systolic blood pressure during Phase II of the maneuver, was decreased at 3.2 msec/mm Hg; baroreflex-cardiovagal gain calculated from the data in Phase IV after release of the maneuver was also decreased at 3.1 msec/mm Hg).11,14,15
Over several months in 2005 the patient noted progressive slowing of movement and inability to relax the arms, small handwriting, decreased facial expression, and decreased voice volume. The patient returned to the NIH in November 2005, to participate in a protocol on pseudopheochromocytoma, the evaluation again including 6-[18F]fluorodopamine positron emission tomographic scanning and beat-to-beat blood pressure and heart rate associated with the Valsalva maneuver. 6-[18F]fluorodopamine PET again revealed severely decreased 6-[18F]fluorodopaminederived radioactivity throughout the left ventricular myocardium (Figure 1). In the interventricular septum, radioactivity at the midpoint of the scanning frame between 5 and 10 minutes after initiation of injection of 6-[18F]fluorodopamine was 1,286 nCi-kg/cc-mCi, more than 2 standard deviations below the normal mean and one of the lowest values we have recorded so far (Figure 3). Blood pressure decreased progressively in Phase II of the Valsalva maneuver, to a greater extent than in 2001, there was no overshoot of pressure after release of the maneuver, and the return of pressure toward baseline was prolonged, findings pointing to failure of sympathetically mediated reflexive vasoconstriction.12,23 Baroreflex-cardiovagal gain was also lower than in 2001 (1.2 msec/mm Hg from the results in Phase II, 2.6 msec/mm Hg from those in Phase IV), both because the range of heart was smaller and the extent of change in systolic pressure larger in 2005 than in 2001.
Figure 3. Individual values for septal myocardial 6-[18F]fluorodopamine-derived radioactivity, in normal control subjects (white circles), patients with Parkinson disease without sympathetic neurocirculatory failure (PD no SNF, green circles), patients with Parkinson disease and sympathetic neurocirculatory failure (PD SNF, blue circles), and the case reported here (large green circle). Dashed line shows the normal mean value and light green shaded area 2 standard deviations from the normal mean. Note markedly decreased 6-[18F]fluorodopamine-derived radioactivity in the current case.As a test of the status of the adrenomedullary hormonal system, blood was obtained via an indwelling arm catheter during supine rest and after bolus i.v. injection of 1 mg of glucagon and assayed for plasma catecholamines in our laboratory. Both in July 2001 and November 2005, the ratio of plasma epinephrine (in pg/mL) to norepinephrine (in pg/mL) was relatively high during supine rest (76:99 and 101:234), and the patient had large increases of plasma epinephrine levels in response to glucagon (peak values more than 250 pg/mL, more than six times the normal peak value).
Neurological consultation in November 2005 noted stooped posture and axial instability, cogwheel rigidity in all four extremities, paucity of spontaneous movements, masked face with infrequent blinking, and monotone voice, but with normal speed of gait and no resting tremor. The patient was diagnosed with mild PD.
DISCUSSION
In this patient, results of 6-[18F]fluorodopamine PET scanning indicated cardiac sympathetic denervation 4 years before the clinical onset of PD. Considering that in PD loss of cardiac noradrenergic innervation progresses slowly over years,13 and that the patient already had evidence for markedly decreased cardiac noradrenergic innervation at the time of initial evaluation, loss of cardiac sympathetic nerves probably preceded the movement disorder by many more than the 4 years between initial testing and the onset of PD.
The findings in this case fit with previous reports of cardiac noradrenergic denervation in de novo PD and with the concept of a peripheral-to-central and caudal-to-rostral pathogenetic sequence. Orimo and co-workers have noted loss of noradrenergic terminal innervation of the myocardium before loss of cell bodies in sympathetic ganglia in PD.16
Our patient also had evidence for decreased baroreflex-cardiovagal function 4 years before the movement disorder. The baroreflex is a homeostatic arc, and abnormalities of afferent neurotransmission, central integration by brainstem centers, or vagal efferent pre-ganglionic or post-ganglionic fibers could result in the same clinical laboratory finding of low baroreflex-cardiovagal gain. In particular, the extent to which baroreflex-cardiovagal failure in PD reflects a brainstem lesion, as opposed to an afferent lesion or loss of parasympathetic cholinergic efferents, remains unknown. The results in our patient are consistent with the view that baroreflex-cardiovagal function worsens over years before the onset of PD.
Chronic constipation, which also preceded parkinsonism in our case, would be consistent with early dysregulation of gastrointestinal autonomic function. Accumulations of alpha-synuclein in enteric neurons and in the dorsal motor nucleus of the vagus nerve, the central neural site of origin of parasympathetic innervation of much of the gastrointestinal tract, has been reported to be an early pathological finding.3 As noted above, however, the occurrence of central neural pathology would not exclude a concurrent afferent or efferent lesion, and studies have found Lewy bodies in the myenteric plexus of both the esophagus and colon,9 as well as loss of enteric dopaminergic neurons in PD with chronic constipation.19
Evidence for abnormalities of the sympathetic norad renergic and parasympathetic cholinergic components of the autonomic nervous system in our patient occurred without evidence for compromised adrenomedullary function. On the contrary, the patient had augmented plasma epinephrine responses to glucagon injection, both upon initial evaluation and at follow-up. The patient therefore did not appear to have diffuse loss of catecholaminergic cells. Although studies have noted decreased adrenomedullary catecholamine concentrations in patients with severe PD,7,20,21 plasma levels of epinephrine and its O-methylated metabolite, metanephrine, have been reported to be normal.10
Combined cardiac sympathetic denervation (with attendant denervation supersensitivity), baroreflex-cardiovagal hypofunction, and adrenomedullary hyper-responsiveness might explain the symptoms and signs of cardiovascular instability, such as episodic hypertensive paroxysms, tachycardia, palpitations, and chest pain despite normal coronary arteries, that led to clinical suspicion of pheochromocytoma in this patient.
The results in this case lead us to propose that cardiac sympathetic denervation and decreased baroreflex-cardiovagal gain may be biomarkers of early autonomic involvement in PD. Studies in progress about autonomic function in relatives of patients with familial PD should help test this hypothesis.
References
Bonuccelli U, Lucetti C, Del Dotto P, et al. Orthostatic hypotension in de novo Parkinson disease. Arch Neurol2003; 60:1400–1404.
Bouhaddi M, Vuillier F, Fortrat JO, et al. Impaired cardiovascular autonomic control in newly and long-term-treated patients with Parkinson’s disease: involvement of L-dopa therapy. Auton Neurosci2004; 116:30–38.
Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett2006; 396:67–72.
Braak H, Rub U, Gai WP, Del Tredici K. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm2003; 110:517–536.
Braune S. The role of cardiac metaiodobenzylguanidine uptake in the differential diagnosis of parkinsonian syndromes. Clin Auton Res2001; 11:351–355.
Camerlingo M, Aillon C, Bottacchi E, et al. Parasympathetic assessment in Parkinson’s disease. Adv Neurol1987; 45:267–269.
Carmichael SW, Wilson RJ, Brimijoin WS, et al. Decreased catecholamines in the adrenal medulla of patients with parkinsonism. N Engl J Med1988; 318:254.
Druschky A, Hilz MJ, Platsch G, et al. Differentiation of Parkinson’s disease and multiple system atrophy in early disease stages by means of I-123-MIBG-SPECT. J Neurol Sci2000; 175:3–12.
Edwards LL, Quigley EM, Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease: frequency and pathophysiology. Neurology1992; 42:726–732.
Goldstein DS, Holmes C, Sharabi Y, Brentzel S, Eisenhofer G. Plasma levels of catechols and metanephrines in neurogenic orthostatic hypotension. Neurology2003; 60:1327–1332.
Goldstein DS, Horwitz D, Keiser HR. Comparison of techniques for measuring baroreflex sensitivity in man. Circulation1982; 66:432–439.
Goldstein DS, Tack C. Noninvasive detection of sympathetic neurocirculatory failure. Clin Auton Res2000; 10:285–291.
Li ST, Dendi R, Holmes C, Goldstein DS. Progressive loss of cardiac sympathetic innervation in Parkinson’s disease. Ann Neurol2002; 52:220–223.
Oka H, Mochio S, Onouchi K, Morita M, Yoshioka M, Inoue K. Cardiovascular dysautonomia in de novo Parkinson’s disease. J Neurol Sci2006; 241:59–65.
Oka H, Mochio S, Yoshioka M, Morita M, Onouchi K, Inoue K. Cardiovascular dysautonomia in Parkinson’s disease and multiple system atrophy. Acta Neurol Scand2006; 113:221–227.
Orimo S, Amino T, Itoh Y, et al. Cardiac sympathetic denervation precedes neuronal loss in the sympathetic ganglia in Lewy body disease. Acta Neuropathol (Berl)2005; 109:583–588.
Pacak K, Eisenhofer G, Carrasquillo JA, Chen CC, Li ST, Goldstein DS. 6-[18F]fluorodopamine positron emission tomographic (PET) scanning for diagnostic localization of pheochromocytoma. Hypertension2001; 38:6–8.
Quadri R, Comino I, Scarzella L, et al. Autonomic nervous function in de novo parkinsonian patients in basal condition and after acute levodopa administration. Funct Neurol2000; 15:81–86.
Singaram C, Ashraf W, Gaumnitz EA, et al. Dopaminergic defect of enteric nervous system in Parkinson’s disease patients with chronic constipation. Lancet1995; 346:861–864.
Stoddard SL, Ahlskog JE, Kelly PJ, et al. Decreased adrenal medullary catecholamines in adrenal transplanted parkinsonian patients compared to nephrectomy patients. Exp Neurol1989; 104:218–222.
Stoddard SL, Tyce GM, Ahlskog JE, Zinsmeister AR, Carmichael SW. Decreased catecholamine content in parkinsonian adrenal medullae. Exp Neurol1989; 104:22–27.
Takatsu H, Nishida H, Matsuo H, et al. Cardiac sympathetic denervation from the early stage of Parkinson’s disease: clinical and experimental studies with radiolabeled MIBG. J Nucl Med2000; 41:71–77.
Vogel ER, Sandroni P, Low PA. Blood pressure recovery from Valsalva maneuver in patients with autonomic failure. Neurology2005; 65:1533–1537.
References
Bonuccelli U, Lucetti C, Del Dotto P, et al. Orthostatic hypotension in de novo Parkinson disease. Arch Neurol2003; 60:1400–1404.
Bouhaddi M, Vuillier F, Fortrat JO, et al. Impaired cardiovascular autonomic control in newly and long-term-treated patients with Parkinson’s disease: involvement of L-dopa therapy. Auton Neurosci2004; 116:30–38.
Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett2006; 396:67–72.
Braak H, Rub U, Gai WP, Del Tredici K. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm2003; 110:517–536.
Braune S. The role of cardiac metaiodobenzylguanidine uptake in the differential diagnosis of parkinsonian syndromes. Clin Auton Res2001; 11:351–355.
Camerlingo M, Aillon C, Bottacchi E, et al. Parasympathetic assessment in Parkinson’s disease. Adv Neurol1987; 45:267–269.
Carmichael SW, Wilson RJ, Brimijoin WS, et al. Decreased catecholamines in the adrenal medulla of patients with parkinsonism. N Engl J Med1988; 318:254.
Druschky A, Hilz MJ, Platsch G, et al. Differentiation of Parkinson’s disease and multiple system atrophy in early disease stages by means of I-123-MIBG-SPECT. J Neurol Sci2000; 175:3–12.
Edwards LL, Quigley EM, Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease: frequency and pathophysiology. Neurology1992; 42:726–732.
Goldstein DS, Holmes C, Sharabi Y, Brentzel S, Eisenhofer G. Plasma levels of catechols and metanephrines in neurogenic orthostatic hypotension. Neurology2003; 60:1327–1332.
Goldstein DS, Horwitz D, Keiser HR. Comparison of techniques for measuring baroreflex sensitivity in man. Circulation1982; 66:432–439.
Goldstein DS, Tack C. Noninvasive detection of sympathetic neurocirculatory failure. Clin Auton Res2000; 10:285–291.
Li ST, Dendi R, Holmes C, Goldstein DS. Progressive loss of cardiac sympathetic innervation in Parkinson’s disease. Ann Neurol2002; 52:220–223.
Oka H, Mochio S, Onouchi K, Morita M, Yoshioka M, Inoue K. Cardiovascular dysautonomia in de novo Parkinson’s disease. J Neurol Sci2006; 241:59–65.
Oka H, Mochio S, Yoshioka M, Morita M, Onouchi K, Inoue K. Cardiovascular dysautonomia in Parkinson’s disease and multiple system atrophy. Acta Neurol Scand2006; 113:221–227.
Orimo S, Amino T, Itoh Y, et al. Cardiac sympathetic denervation precedes neuronal loss in the sympathetic ganglia in Lewy body disease. Acta Neuropathol (Berl)2005; 109:583–588.
Pacak K, Eisenhofer G, Carrasquillo JA, Chen CC, Li ST, Goldstein DS. 6-[18F]fluorodopamine positron emission tomographic (PET) scanning for diagnostic localization of pheochromocytoma. Hypertension2001; 38:6–8.
Quadri R, Comino I, Scarzella L, et al. Autonomic nervous function in de novo parkinsonian patients in basal condition and after acute levodopa administration. Funct Neurol2000; 15:81–86.
Singaram C, Ashraf W, Gaumnitz EA, et al. Dopaminergic defect of enteric nervous system in Parkinson’s disease patients with chronic constipation. Lancet1995; 346:861–864.
Stoddard SL, Ahlskog JE, Kelly PJ, et al. Decreased adrenal medullary catecholamines in adrenal transplanted parkinsonian patients compared to nephrectomy patients. Exp Neurol1989; 104:218–222.
Stoddard SL, Tyce GM, Ahlskog JE, Zinsmeister AR, Carmichael SW. Decreased catecholamine content in parkinsonian adrenal medullae. Exp Neurol1989; 104:22–27.
Takatsu H, Nishida H, Matsuo H, et al. Cardiac sympathetic denervation from the early stage of Parkinson’s disease: clinical and experimental studies with radiolabeled MIBG. J Nucl Med2000; 41:71–77.
Vogel ER, Sandroni P, Low PA. Blood pressure recovery from Valsalva maneuver in patients with autonomic failure. Neurology2005; 65:1533–1537.
Spectral analysis of heart rate variability (HRV) has been used widely as a noninvasive technique for examining sympathetic and parasympathetic nervous outflows to the heart. Low-frequency (LF) and high-frequency (HF) power have been used most commonly. Human and animal experiments have repeatedly confirmed the dependence of HF power on respiration-related alterations in parasympathetic cardiovagal outflow–respiratory sinus arrhythmia; however, whether LF power provides an indirect measure of cardiac sympathetic activity has been contentious. Pagani et al1 reported that LF power (normalized to total spectral power) increased during states associated with sympathetic noradrenergic activation and that bilateral stellectomy in dogs reduced LF power. Alvarenga et al,2 however, reported that LF power was unrelated to all measures of norepinephrine kinetics in the heart; and in congestive heart failure, which is associated with a high rate of entry of norepinephrine into coronary sinus plasma (cardiac norepinephrine spillover),3 LF power is decreased, not increased as might be expected if LF power reflected sympathetic activity.4–7
Sleight et al8 proposed an alternative explanation for the origin of LF power. In a small group of human subjects, power spectral analysis of HRV showed that the amplitude of LF power was related to baroreflex gain and not to the level of sympathetic activity. Carotid sinus stimulation increased LF power only in individuals with normal baroreflex sensitivity and did not do so in those with depressed baroreflex gain. Therefore, results of power spectral analysis of LF power might reflect baroreflex-cardiovagal function.9
Studies of patients with dysautonomias provide an unusual opportunity to examine neurocirculatory correlates of LF power. Some chronic autonomic failure syndromes feature cardiac sympathetic denervation, whereas others do not. Parkinson disease with neurogenic orthostatic hypotension and pure autonomic failure feature cardiac sympathetic denervation, whereas multiple system atrophy does not.10 All 3 diseases involve baroreflex-cardiovagal and baro-reflex-sympathoneural failure.11 Chronic orthostatic intolerance syndromes (postural tachycardia syndrome, neurocardiogenic syncope) do not entail either cardiac sympathetic denervation or baroreflex failure.12
For this article, we carried out power spectral analyses of HRV on digitized electrocardiographic recordings from dysautonomia patients and normal volunteers during supine rest, measurement of cardiac norepinephrine spillover, and intravenous infusion of yohimbine and tyramine, 2 drugs that are known to release norepinephrine from cardiac sympathetic nerves.13,14 Cardiac sympathetic innervation was assessed by 6-[18F]fluorodopamine positron emission tomographic scanning.15
We hypothesized that if LF power indicated cardiac sympathetic innervation and function, then patients with neuroimaging or neurochemical evidence of cardiac sympathetic denervation would have low LF power and attenuated increments in LF power in response to yohimbine and tyramine. Alternatively, if LF power was reflective of baroreflex function, alterations of LF power would be independent of cardiac sympathetic innervation status and correlate with changes in baroreflex gain.
METHODS
The study protocols were approved by the Intramural Research Board of the National Institute of Neurological Disorders and Stroke. All subjects were studied at the National Institutes of Health Clinical Center after giving informed, written consent.
Subjects
The study population consisted of a total of 98 subjects who participated in research protocols studying chronic orthostatic intolerance and chronic autonomic failure (Table 1). The subjects underwent autonomic function testing and had reviewable, digitized electrocardiographic data enabling retrospective power spectral analysis of HRV. ECG and blood pressure data were sampled at 1 kHz.
The study subjects were separated into 4 groups, depending on their state of cardiac sympathetic innervation and baroreflex-cardiovagal slope (BRS; see below). There were 40 subjects with intact sympathetic innervation and normal BRS (Innervated-Normal BRS), 24 with intact sympathetic innervation and low BRS (Innervated-Low BRS), 4 with sympathetic denervation and normal BRS (Denervated-Normal BRS), and 30 with sympathetic denervation and low BRS (Denervated-Low BRS).
Autonomic function testing
Each subject was studied while supine with head on pillow after an overnight fast. Each patient had monitoring of the electrocardiogram and beat-tobeat blood pressure using either noninvasive devices (Finometer, Finapres Medical Systems, Amsterdam, the Netherlands; Portapres, Finapres Medical Systems; or Colin tonometer, Colin Medical Instruments, San Antonio, TX) or a brachial intra-arterial catheter. We previously studied formally and reported excellent agreement between intra-arterial and these noninvasively obtained measures of beat-to-beat blood pressure.16 Continuous vital signs data were digitized and recorded using a PowerLab (AD Instruments Pty Ltd, Castle Hill, Australia) data acquisition system and stored for later analysis on an Apple PowerBook G4 computer (Apple, Cupertino, CA).
After about a 10-min baseline period, each subject performed a Valsalva maneuver (30 mm Hg for 12 sec) at least 3 times.
Baroreflex function
As an index of baroreflex function, we used the slope of the relationship between cardiac interbeat interval and systolic blood pressure during phase II of the Valsalva maneuver.17 BRS, in units of msec/mm Hg, was calculated from the linear regression equation for the relationship between interbeat interval (with 1-beat delay) and systolic pressure. A BRS value of ≤3 msec/mm Hg was considered low.11
Pharmacologic testing
Pharmacologic testing was performed on completion of the autonomic evaluation, using either tyramine or yohimbine. If a subject received both drugs, each drug administration was on a separate day. The durations of drug infusion were sufficient for heart rate and blood pressure to reach plateau values.
In a total of 22 subjects (Table 1), yohimbine was infused intravenously at 62.5 μg/kg over 3 min and then at 0.5 μg/kg/min for 12 min. In a total of 50 subjects, tyramine was infused at a rate of 1 mg/min for 10 min. In patients with severe supine hypertension (systolic pressure more than 200 mm Hg) and orthostatic hypotension, the test drugs were infused during head-up tilting (15° to 30°), to decrease baseline pressure, or else the drugs were not given.
HRV analysis
LF power (0.04 to 0.15 Hz), HF power (0.16 to 0.4 Hz), and total power (TP, 0.0 to 0.4 Hz) were calculated using Chart 5.4.2 and the HRV module version 1.03 (PowerLab, AD Instruments Pty Ltd, Castle Hill, Australia). Stable heart rate epochs 3 to 5 min in duration were chosen for analysis. One epoch was sampled immediately before initiation of drug testing; the second followed attainment of steady-state hemodynamic effects. Interbeat interval data were reviewed carefully to eliminate artifacts from noise and T waves, using segments with little to no premature beats. LF power and HF power were calculated as absolute power (msec2), with or without normalization for total power (0.04 to 0.4 Hz). Reported LF or HF power was integrated within their defined frequency bands.
Cardiac sympathetic neuroimaging
For cardiac sympathetic neuroimaging the subject was positioned supine, feet-first in a GE Advance scanner (General Electric, Milwaukee, WI), with the thorax in the gantry. After positioning the patient with the thorax in the scanner and transmission scanning for attenuation correction, 6-[18F]fluorodopamine (usual dose 1 mCi, specific activity 1.0 to 4.0 Ci/mmole, in about 10 mL normal saline) was infused intravenously at a constant rate for 3 min, and dynamic scanning data were obtained for thoracic radioactivity, with the midpoint of the scanning interval at 7.5 min after injection of the tracer (data collection interval between 5 and 10 min). Cardiac sympathetic denervation was defined by low concentrations of 6-[18F] fluorodopamine-derived radioactivity in the interventricular septum (< 5,000 nCi-kg/cc-mCi) or left ventricular free wall (< 4,000 nCi-kg/cc-mCi) corresponding to about 2 SD below the normal means.
Cardiac norepinephrine spillover
Subgroups of subjects (3 PD + NOH, 3 MSA, 3 PAF, 5 normal volunteers) underwent right heart catheterization for measurement of cardiac norepinephrine spillover. 3H-Norepinephrine was infused intravenously, and arterial and coronary sinus blood was sampled and coronary sinus blood flow was measured by thermodilution for measurements of cardiac norepinephrine spillover as described previously.18 In some subjects, yohimbine was infused during cardiac catheterization. Patients with chronic autonomic failure received the doses described above; normal volunteers and patients with chronic orthostatic intolerance received twice the doses described above.
Data analysis
Statistical analyses were performed using StatView version 5.0.1. (SAS Institute, Cary, NC). Mean values in the baseline condition for the several subject groups were compared using single-factor ANOVA. Responses to drugs were analyzed by dependent-means t tests. Differences in response to pharmacologic tests among subject groups were compared using repeated measures analyses of variance. Relationships between individual hemodynamic values were assessed by linear regression and calculation of Pearson correlation coefficients. Post-hoc testing consisted of the Fisher PLSD test. Multiple regression analysis was done on the individual data, with the log of LF power as the dependent measure and the log of baroreflex slope and septal 6-[18F] fluorodopamine-derived radioactivity as independent measures. Mean values were expressed ± SEM.
RESULTS
Baseline
Across the 7 subject groups (N = 98), LF power was unrelated to subject group (F = 1.2). When individual subjects were stratified in terms of cardiac sympathetic denervation or innervation, based on concentrations of 6-[18F]fluorodopamine-derived radioactivity more than 2 SD below the normal mean, then LF power was lower in the Denervated group (mean 221 ± 55 msec2/Hz, N = 34) than in the Innervated group (516 ± 93 msec2/Hz, N = 64, F = 4.8, P = 0.03). LF power normalized for total power, HF normalized for total power, and the ratio of LF:HF were not related to 6-[18F]fluorodopamine-derived radioactivity.
When subjects were stratified in terms of BRS, then LF power was lower in the Low BRS group (223 ± 105 msec2/Hz, N = 46) than in the Normal BRS group (617 ± 97 msec2/Hz, N = 25, F = 6.1, P = 0.02). The Low BRS group did not differ from the Normal BRS group in normalized LF power (F = 0.8).
Figure 1. Mean (± SEM) values for the log of low-frequency power of heart rate variability in subject groups with innervated (Innerv) or denervated (Denerv) hearts, as indicated by low 6-[18F] fluorodopamine-derived radioactivity, and normal (Nl) or low baroreflex-cardiovagal slope (BRS), as indicated by slope ≤3 msec/mm Hg during the Valsalva maneuver. ***Significant difference, P < 0.001.When individual subjects were stratified into 4 groups, based on both cardiac 6-[18F]fluorodopamine-derived radioactivity (Innervated or Denervated) and on baroreflex-cardiovagal slope (Normal BRS or Low BRS), then both LF power and the log of LF power varied highly significantly as a function of subject group (F = 9.5, P < 0.0001; F = 4.6, P = 0.0004). The Denervated-Low BRS group had lower LF power than did the Denervated-Normal BRS group (P = 0.05), and the Innervated-Low BRS group had lower LF power than did the Innervated-Normal BRS group (P < 0.0001). When level of baroreflex function was taken into account, the Innervated and Denervated groups did not differ in LF power (Figure 1).
Values for HF power also varied with subject group when individual subjects were stratified in terms of both cardiac sympathetic innervation and BRS (F = 4.9, P = 0.004; Table 2). The Innervated-Low BRS group had lower HF power than did the Innervated-Normal BRS group (P = 0.003); however, the Denervated-Low BRS group did not differ from the Denervated-Normal BRS group in HF power. Normalization of LF and HF power for total power, and the ratio of low-to-high frequency did not reveal additional group differences (Table 2). In particular, the LF:HF ratio did not vary with the subject group (F = 0.6).
Figure 2. Mean (± SEM) values for (A) low-frequency power of heart rate variability and (B) cardiac norepinephrine spillover during right heart catheterization in subject groups with innervated (Innerv) or denervated (Denerv) hearts, as indicated by low 6-[18F]fluorodopaminederived radioactivity, and normal (Nl) or low baroreflex-cardiovagal slope (BRS), as indicated by slope ≤3 msec/mm Hg during the Valsalva maneuver. *Significant difference, P < 0.05. **Significant difference, P < 0.01.Analysis of data from subjects during cardiac catheterization showed that LF power varied as a function of subject group (F = 5.3, P = 0.03, Figure 2). The Innervated-Low BRS group had lower LF power than did the Innervated-Normal BRS group (P = 0.04), whereas the Denervated-Low BRS and Innervated-Low BRS groups did not differ in LF power. As expected, the Denervated-Low BRS group had lower cardiac norepinephrine spillover than the Innervated-Low BRS group.
Figure 3. Individual values for the log of low-frequency (LF) power as a function of (A) septal 6-[18F]fluorodopamine-derived radioactivity and (B) the log of baroreflex-cardiovagal slope.Individual values for LF power were positively correlated with BRS. When values for both variables were log-transformed, the log of LF power correlated positively with the log of BRS slope (r = 0.72, P < 0.0001, Figure 3). Individual values for the log of LF power were also correlated with the magnitude of decrease in systolic pressure during performance of the Valsalva maneuver (r = −0.60, P < 0.0001) and with the orthostatic change in systolic pressure (r = 0.58, P < 0.0001). In contrast, the log of LF power was unrelated to the septal myocardial concentration of 6-[18F]fluorodopamine-derived radioactivity, the plasma norepinephrine concentration, or cardiac norepinephrine spillover.
From multiple regression analysis for the log of LF power as the dependent measure and the log of baroreflex slope and septal 6-[18F]fluorodopamine-derived radioactivity as independent measures, the regression coefficient for the log of baroreflex slope was 0.92 (P < 0.0001), whereas the regression coefficient for 6-[18F] fluorodopamine-derived radioactivity was 3 ×10−6.
At baseline, the log of HF power correlated positively with the log of LF power (r = 0.77, P < 0.0001). HF power varied with the subject group (F = 4.9, P = 0.004). As with LF power, HF power was greater in the Innervated-Normal BRS than in the Innervated-Low BRS (P = 0.001, Table 2). As expected, the log of HF power correlated positively with the log of BRS (r = 0.60, P < 0.0001). The log of HF power also correlated negatively with the magnitude of decrease in systolic pressure during the Valsalva maneuver (r = −0.24, P = 0.02) and positively with the orthostatic change in systolic pressure (r = 0.40, P = 0.004).
Yohimbine
Figure 4. Mean (± SEM) values for the change in low-frequency power (ΔLF power) of heart rate variability during (A) intravenous infusion of yohimbine or (B) tyramine in groups with innervated (Innerv) or denervated (Denerv) hearts, as indicated by low 6-[18F]fluorodopaminederived radioactivity, and normal (Nl) or low baroreflex-cardiovagal slope (BRS), as indicated by slope ≤3 msec/mm Hg during the Valsalva maneuver. *Significant difference, P < 0.05. ***Significant difference, P < 0.001.Yohimbine infusion increased LF power (t = 2.9, P = 0.007). The group with cardiac sympathetic denervation did not differ from the group with intact cardiac innervation in terms of the change in LF power during yohimbine infusion (F = 0.7). Yohimbine infusion increased LF power in the Innervated-Normal BRS group (t = 2.8, P = 0.01), but not in the innervated or denervated groups with low BRS (Figure 4). The Innervated-Normal BRS group had a larger increase in LF power during yohimbine infusion than did the Innervated-Low BRS group (P = 0.02). Too few patients with cardiac denervation and normal BRS were studied to include in the ANOVA. The log of the change in LF power during yohimbine administration was positively correlated with the log of BRS at baseline (Figure 5).
Figure 5. Individual values for the log of change in low-frequency power (log ΔLF power) as a function of baroreflex-cardiovagal slope at baseline. Left: yohimbine infusion. Right: tyramine infusion.Yohimbine increased HF power in the Innervated-Normal BRS group (t = 2.1, P = 0.05) but not in the innervated or denervated groups with low BRS.
The change in LF power in response to yohimbine during cardiac catheterization was unrelated to the change in cardiac norepinephrine spillover (r = −0.09, N = 12).
Tyramine
Overall, tyramine infusion increased LF power (t = 2.9, P = 0.008). The group with cardiac sympathetic denervation did not differ from the group with intact cardiac innervation in terms of the change in LF power during tyramine infusion (F = 1.7). Tyramine increased LF power in the Innervated-Normal BRS group but not in the Innervated-Low BRS or Denervated-Low BRS groups (Figure 4; data for 2 outliers excluded). The log of the change in LF power during tyramine administration was positively correlated with the log of BRS at baseline (Figure 5; data for 2 outliers excluded).
DISCUSSION
In this study, patients with neuroimaging evidence of cardiac sympathetic denervation had low LF power of heart rate variability. At first glance, this finding would seem to support the view that LF power can provide an index of cardiac sympathetic outflow. As explained below, several lines of evidence from the present study led us to infer that the association between low LF power and cardiac sympathetic innervation is determined mainly by concurrent baroreflex function.
Patients with low BRS had low LF power, and patients with normal BRS had normal LF power, regardless of the status of cardiac sympathetic innervation as assessed by 6-[18F]fluorodopamine scanning. Neither normalization of LF and HF power for total power nor use of the LF:HF ratio improved the association with indices of cardiac sympathetic innervation.
Neurochemical findings during cardiac catheterization supported the above results based on cardiac sympathetic neuroimaging. Among patients with innervated hearts who had normal cardiac norepinephrine spillover, LF power was decreased only in the group with low BRS and was normal in the group with normal BRS. As expected, cardiac norepinephrine spillover was decreased in patients with neuroimaging evidence of cardiac sympathetic denervation.
Effects of pharmacological manipulations that increase norepinephrine release from sympathetic nerves provided further support for an association between baroreflex failure and low LF power, independent of cardiac sympathetic function. Both tyramine and yohimbine increased LF power only in the subjects with normal BRS. In subjects with low BRS, neither drug increased LF power, even in the group with intact cardiac sympathetic innervation. Moreover, individual values for responses of the log of LF power to both drugs were correlated positively with the log of BRS at baseline.
The fact that HF power was positively correlated with LF power could also fit with the notion of baroreflex function acting as a common determinant of values of both variables. We cannot exclude concurrent parasympathetic cardiovagal and sympathetic denervation as an explanation for the association between HF and LF power. Inhibition of the effects of parasympathetic activity after atropine administration results in the almost complete absence of both LF and HF HRV, further suggesting a common determinant.19
Several previous investigations have cast doubt on the validity of LF power as a measure of sympathetic activity because of dissociations between LF power and cardiac norepinephrine spillover, directly recorded sympathetic nerve traffic, and plasma norepinephrine levels.4,6,20 Such dissociations are especially glaring in patients with congestive heart failure, which is characterized by decreased LF power9 despite marked cardiac sympathetic activation.3
Other pathophysiologic states do result in both decreased sympathetic nervous system activity and decreased LF power. In these pathophysiologic states, the possibility remains that low LF power might reflect failure of baroreflexive modulation of sympathetic neuronal outflows, rather than sympathoinhibition itself. For instance, Wiklund et al21 noted low LF power in patients with palmar hyperhidrosis undergoing bilateral transthoracic sympathectomy; however, baroreflex-cardiovagal sensitivity also declines after thoracic sympathectomy.22
Sleight et al8 suggested dependence of LF power on baroreflex function, based on effects of carotid baroreceptor stimulation in 3 patients: 1 with normal BRS; 1 with ischemic heart disease, congestive heart failure, and normal BRS; and 1 with ischemic heart disease, congestive heart failure, and initially low BRS who subsequently had an improved clinical state and BRS. In the baseline state, both congestive heart failure patients had low LF power, despite a presumably hypernoradrenergic state. Direct baroreceptor stimulation at 0.1 Hz increased LF power in the normal subject and in the patient with congestive heart failure and normal BRS. The congestive heart failure patient with low BRS did not have an increase in LF power until BRS normalized. These data revealed an initial dissociation between cardiac noradrenergic state in the patients with congestive heart failure and LF power. During carotid sinus stimulation, LF power increased only when BRS was normal. Low BRS obviated this effect.
Because congestive heart failure is well known to be associated with baroreflex-cardiovagal inhibition,23–25 the finding of low LF power in heart failure also supports an association between LF power and BRS, independently of increased tonic release of norepinephrine from sympathetic nerves in the heart. Cevese et al26 inhibited noradrenergic vasomotor tone using an alpha-adrenoceptor blocker in human subjects while maintaining mean blood pressure at control levels using angiotensin II. This drug combination, which would be expected to attenuate sympathetically mediated vasomotor tone and thereby decrease arterial baroreceptor input, markedly decreased or abolished LF power of HRV, suggesting that, at least under resting supine conditions, a baroreflex mechanism accounts almost entirely for LF power of HRV.
deBoer et al27 developed a beat-to-beat model of the human circulation using a set of differential equations and the following principles of operation: (1) the baroreflex regulates heart rate and peripheral vascular resistance; (2) Windkessel properties characterize the systemic arterial tree; (3) contractile properties of the ventricular myocardium follow the Starling law; and (4) respiration exerts mechanical effects on blood pressure. The model attributes LF power to a resonance in the circulatory control system, produced by a slow time constant for reflexive sympathetically mediated responses to beat-tobeat blood pressure changes. The resonance can be upregulated or downregulated by the state of baroreflex activity. The model of deBoer et al predicts that changes in blood pressure would lead heart rate changes at 0.1 Hz through a delayed sympathetic response. Changes in HR would depend on summed effects of sympathetic and vagal effects, with the sympathetic response delaying the overall response. At the respiratory frequency (0.2 to 0.3 Hz), blood pressure and HR changes would occur with little delay because of fast parasympathetic control. In essence, the response of the sympathetic nervous system behaves as a low band pass filter, with a peak response at 0.1 Hz and little response at frequencies above 0.2 Hz. Systolic blood pressure would lead to changes in heart rate via the baroreflex. In general the results of this study fit with the deBoer model.
In conclusion, LF power derived from the interbeat interval spectrogram predominantly reflects baroreflex-mediated, phasic changes in cardiovagal and sympathetic noradrenergic outflows. In the setting of baroreflex failure, baseline LF power is reduced, regardless of the status of cardiac sympathetic innervation.
LIMITATIONS
The combination of cardiac sympathetic denervation and normal baroreflex function seems quite rare. One must exercise caution in drawing inferences from the findings in the Denervated-Normal BRS group, which contained only 4 subjects, even though the difference in mean log-transformed LF power from the Denervated-Low BRS group was highly statistically significant.
All of the testing in our study was done with the subjects supine. LF power measured in other positions might have different sources and meaning.
References
Pagani M, Lombardi F, Guzzetti S, et al. Power spectral analysis of heart rate and arterial pressure variabilities as a marker of sympatho-vagal interaction in man and conscious dog. Circ Res1986; 59:178–193.
Alvarenga ME, Richards JC, Lambert G, Esler MD. Psychophysiological mechanisms in panic disorder: a correlative analysis of noradrenaline spillover, neuronal noradrenaline reuptake, power spectral analysis of heart rate variability, and psychological variables. Psychosom Med2006; 68:8–16.
Eisenhofer G, Friberg P, Rundqvist B, et al. Cardiac sympathetic nerve function in congestive heart failure. Circulation1996; 93:1667–1676.
Notarius CF, Butler GC, Ando S, Pollard MJ, Senn BL, Floras JS. Dissociation between microneurographic and heart rate variability estimates of sympathetic tone in normal subjects and patients with heart failure. Clin Sci (Lond)1999; 96:557–565.
Scalvini S, Volterrani M, Zanelli E, et al. Is heart rate variability a reliable method to assess autonomic modulation in left ventricular dysfunction and heart failure? Assessment of autonomic modulation with heart rate variability. Int J Cardiol1998; 67:9–17.
Kingwell BA, Thompson JM, Kaye DM, McPherson GA, Jennings GL, Esler MD. Heart rate spectral analysis, cardiac norepinephrine spillover, and muscle sympathetic nerve activity during human sympathetic nervous activation and failure. Circulation1994; 90:234–240.
van de Borne P, Montano N, Pagani M, Oren R, Somers VK. Absence of low-frequency variability of sympathetic nerve activity in severe heart failure. Circulation1997; 95:1449–1454.
Sleight P, La Rovere MT, Mortara A, et al. Physiology and pathophysiology of heart rate and blood pressure variability in humans: is power spectral analysis largely an index of baroreflex gain?Clin Sci (Lond)1995; 88:103–109.
Saul JP, Arai Y, Berger RD, Lilly LS, Colucci WS, Cohen RJ. Assessment of autonomic regulation in chronic congestive heart failure by heart rate spectral analysis. Am J Cardiol1988; 61:1292–1299.
Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO. Cardiac sympathetic denervation in Parkinson disease. Ann Intern Med2000; 133:338–347.
Goldstein DS, Eldadah BA, Holmes C, et al. Neurocirculatory abnormalities in Parkinson disease with orthostatic hypotension. Independence from levodopa treatment. Hypertension2005; 46:1333–1339.
Goldstein DS, Eldadah B, Holmes C, Pechnik S, Moak J, Sharabi Y. Neurocirculatory abnormalities in chronic orthostatic intolerance. Circulation2005; 111:839–845.
Goldstein DS, Holmes C, Frank SM, et al. Cardiac sympathetic dysautonomia in chronic orthostatic intolerance syndromes. Circulation2002; 106:2358–2365.
Lord SW, Clayton RH, Mitchell L, Dark JH, Murray A, McComb JM. Sympathetic reinnervation and heart rate variability after cardiac transplantation. Heart1997; 77:532–538.
Goldstein DS, Eisenhofer G, Dunn BB, et al. Positron emission tomographic imaging of cardiac sympathetic innervation using 6-[18F]fluorodopamine: initial findings in humans. J Am Coll Cardiol1993; 22:1961–1971.
Goldstein DS, Tack C. Non-invasive detection of sympathetic neurocirculatory failure. Clin Auton Res2000; 10:285–291.
Goldstein DS, Horwitz D, Keiser HR. Comparison of techniques for measuring baroreflex sensitivity in man. Circulation1982; 66:432–439.
Goldstein DS, Brush JE, Eisenhofer G, Stull R, Esler M. In vivo measurement of neuronal uptake of norepinephrine in the human heart. Circulation1988; 78:41–48.
Koh J, Brown TE, Beightol LA, Ha CY, Eckberg DL. Human autonomic rhythms: vagal cardiac mechanisms in tetraplegic subjects. J Physiol1994; 474:483–495.
Saul JP, Rea RF, Eckberg DL, Berger RD, Cohen RJ. Heart rate and muscle sympathetic nerve variability during reflex changes of autonomic activity. Am J Physiol1990; 258:H713–H721.
Wiklund U, Koskinen LO, Niklasson U, Bjerle P, Elfversson J. Endoscopic transthoracic sympathectomy affects the autonomic modulation of heart rate in patients with palmar hyperhidrosis. Acta Neurochir (Wien)2000; 142:691–696.
Kawamata YT, Kawamata T, Omote K, et al. Endoscopic thoracic sympathectomy suppresses baroreflex control of heart rate in patients with essential hyperhidrosis. Anesth Analg2004; 98:37–39.
Goldstein RE, Beiser GD, Stampfer M, Epstein SE. Impairment of autonomically mediated heart rate control in patients with cardiac dysfunction. Circ Res1975; 36:571–578.
Cody RJ, Franklin KW, Kluger J, Laragh JH. Mechanisms governing the postural response and baroreceptor abnormalities in chronic congestive heart failure: effects of acute and long-term convertingenzyme inhibition. Circulation1982; 66:135–142.
Creager MA. Baroreceptor reflex function in congestive heart failure. Am J Cardiol1992; 69:10G–15G; discussion 15G–16G.
Cevese A, Gulli G, Polati E, Gottin L, Grasso R. Baroreflex and oscillation of heart period at 0.1 Hz studied by alpha-blockade and crossspectral analysis in healthy humans. J Physiol2001; 531:235–244.
deBoer RW, Karemaker JM, Strackee J. Hemodynamic fluctuations and baroreflex sensitivity in humans: a beat-to-beat model. Am J Physiol1987; 253:H680–689.
Jeffrey P. Moak, MD Children’s National Medical Center, Washington, DC
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, National Institute of Neurological Disorders and Stroke (NINDS), National Institutes of Health (NIH), Bethesda, MD
Jeffrey P. Moak, MD Children’s National Medical Center, Washington, DC
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, National Institute of Neurological Disorders and Stroke (NINDS), National Institutes of Health (NIH), Bethesda, MD
No author conflicts of interest were reported in the original publication of this article.
Author and Disclosure Information
Jeffrey P. Moak, MD Children’s National Medical Center, Washington, DC
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, National Institute of Neurological Disorders and Stroke (NINDS), National Institutes of Health (NIH), Bethesda, MD
Spectral analysis of heart rate variability (HRV) has been used widely as a noninvasive technique for examining sympathetic and parasympathetic nervous outflows to the heart. Low-frequency (LF) and high-frequency (HF) power have been used most commonly. Human and animal experiments have repeatedly confirmed the dependence of HF power on respiration-related alterations in parasympathetic cardiovagal outflow–respiratory sinus arrhythmia; however, whether LF power provides an indirect measure of cardiac sympathetic activity has been contentious. Pagani et al1 reported that LF power (normalized to total spectral power) increased during states associated with sympathetic noradrenergic activation and that bilateral stellectomy in dogs reduced LF power. Alvarenga et al,2 however, reported that LF power was unrelated to all measures of norepinephrine kinetics in the heart; and in congestive heart failure, which is associated with a high rate of entry of norepinephrine into coronary sinus plasma (cardiac norepinephrine spillover),3 LF power is decreased, not increased as might be expected if LF power reflected sympathetic activity.4–7
Sleight et al8 proposed an alternative explanation for the origin of LF power. In a small group of human subjects, power spectral analysis of HRV showed that the amplitude of LF power was related to baroreflex gain and not to the level of sympathetic activity. Carotid sinus stimulation increased LF power only in individuals with normal baroreflex sensitivity and did not do so in those with depressed baroreflex gain. Therefore, results of power spectral analysis of LF power might reflect baroreflex-cardiovagal function.9
Studies of patients with dysautonomias provide an unusual opportunity to examine neurocirculatory correlates of LF power. Some chronic autonomic failure syndromes feature cardiac sympathetic denervation, whereas others do not. Parkinson disease with neurogenic orthostatic hypotension and pure autonomic failure feature cardiac sympathetic denervation, whereas multiple system atrophy does not.10 All 3 diseases involve baroreflex-cardiovagal and baro-reflex-sympathoneural failure.11 Chronic orthostatic intolerance syndromes (postural tachycardia syndrome, neurocardiogenic syncope) do not entail either cardiac sympathetic denervation or baroreflex failure.12
For this article, we carried out power spectral analyses of HRV on digitized electrocardiographic recordings from dysautonomia patients and normal volunteers during supine rest, measurement of cardiac norepinephrine spillover, and intravenous infusion of yohimbine and tyramine, 2 drugs that are known to release norepinephrine from cardiac sympathetic nerves.13,14 Cardiac sympathetic innervation was assessed by 6-[18F]fluorodopamine positron emission tomographic scanning.15
We hypothesized that if LF power indicated cardiac sympathetic innervation and function, then patients with neuroimaging or neurochemical evidence of cardiac sympathetic denervation would have low LF power and attenuated increments in LF power in response to yohimbine and tyramine. Alternatively, if LF power was reflective of baroreflex function, alterations of LF power would be independent of cardiac sympathetic innervation status and correlate with changes in baroreflex gain.
METHODS
The study protocols were approved by the Intramural Research Board of the National Institute of Neurological Disorders and Stroke. All subjects were studied at the National Institutes of Health Clinical Center after giving informed, written consent.
Subjects
The study population consisted of a total of 98 subjects who participated in research protocols studying chronic orthostatic intolerance and chronic autonomic failure (Table 1). The subjects underwent autonomic function testing and had reviewable, digitized electrocardiographic data enabling retrospective power spectral analysis of HRV. ECG and blood pressure data were sampled at 1 kHz.
The study subjects were separated into 4 groups, depending on their state of cardiac sympathetic innervation and baroreflex-cardiovagal slope (BRS; see below). There were 40 subjects with intact sympathetic innervation and normal BRS (Innervated-Normal BRS), 24 with intact sympathetic innervation and low BRS (Innervated-Low BRS), 4 with sympathetic denervation and normal BRS (Denervated-Normal BRS), and 30 with sympathetic denervation and low BRS (Denervated-Low BRS).
Autonomic function testing
Each subject was studied while supine with head on pillow after an overnight fast. Each patient had monitoring of the electrocardiogram and beat-tobeat blood pressure using either noninvasive devices (Finometer, Finapres Medical Systems, Amsterdam, the Netherlands; Portapres, Finapres Medical Systems; or Colin tonometer, Colin Medical Instruments, San Antonio, TX) or a brachial intra-arterial catheter. We previously studied formally and reported excellent agreement between intra-arterial and these noninvasively obtained measures of beat-to-beat blood pressure.16 Continuous vital signs data were digitized and recorded using a PowerLab (AD Instruments Pty Ltd, Castle Hill, Australia) data acquisition system and stored for later analysis on an Apple PowerBook G4 computer (Apple, Cupertino, CA).
After about a 10-min baseline period, each subject performed a Valsalva maneuver (30 mm Hg for 12 sec) at least 3 times.
Baroreflex function
As an index of baroreflex function, we used the slope of the relationship between cardiac interbeat interval and systolic blood pressure during phase II of the Valsalva maneuver.17 BRS, in units of msec/mm Hg, was calculated from the linear regression equation for the relationship between interbeat interval (with 1-beat delay) and systolic pressure. A BRS value of ≤3 msec/mm Hg was considered low.11
Pharmacologic testing
Pharmacologic testing was performed on completion of the autonomic evaluation, using either tyramine or yohimbine. If a subject received both drugs, each drug administration was on a separate day. The durations of drug infusion were sufficient for heart rate and blood pressure to reach plateau values.
In a total of 22 subjects (Table 1), yohimbine was infused intravenously at 62.5 μg/kg over 3 min and then at 0.5 μg/kg/min for 12 min. In a total of 50 subjects, tyramine was infused at a rate of 1 mg/min for 10 min. In patients with severe supine hypertension (systolic pressure more than 200 mm Hg) and orthostatic hypotension, the test drugs were infused during head-up tilting (15° to 30°), to decrease baseline pressure, or else the drugs were not given.
HRV analysis
LF power (0.04 to 0.15 Hz), HF power (0.16 to 0.4 Hz), and total power (TP, 0.0 to 0.4 Hz) were calculated using Chart 5.4.2 and the HRV module version 1.03 (PowerLab, AD Instruments Pty Ltd, Castle Hill, Australia). Stable heart rate epochs 3 to 5 min in duration were chosen for analysis. One epoch was sampled immediately before initiation of drug testing; the second followed attainment of steady-state hemodynamic effects. Interbeat interval data were reviewed carefully to eliminate artifacts from noise and T waves, using segments with little to no premature beats. LF power and HF power were calculated as absolute power (msec2), with or without normalization for total power (0.04 to 0.4 Hz). Reported LF or HF power was integrated within their defined frequency bands.
Cardiac sympathetic neuroimaging
For cardiac sympathetic neuroimaging the subject was positioned supine, feet-first in a GE Advance scanner (General Electric, Milwaukee, WI), with the thorax in the gantry. After positioning the patient with the thorax in the scanner and transmission scanning for attenuation correction, 6-[18F]fluorodopamine (usual dose 1 mCi, specific activity 1.0 to 4.0 Ci/mmole, in about 10 mL normal saline) was infused intravenously at a constant rate for 3 min, and dynamic scanning data were obtained for thoracic radioactivity, with the midpoint of the scanning interval at 7.5 min after injection of the tracer (data collection interval between 5 and 10 min). Cardiac sympathetic denervation was defined by low concentrations of 6-[18F] fluorodopamine-derived radioactivity in the interventricular septum (< 5,000 nCi-kg/cc-mCi) or left ventricular free wall (< 4,000 nCi-kg/cc-mCi) corresponding to about 2 SD below the normal means.
Cardiac norepinephrine spillover
Subgroups of subjects (3 PD + NOH, 3 MSA, 3 PAF, 5 normal volunteers) underwent right heart catheterization for measurement of cardiac norepinephrine spillover. 3H-Norepinephrine was infused intravenously, and arterial and coronary sinus blood was sampled and coronary sinus blood flow was measured by thermodilution for measurements of cardiac norepinephrine spillover as described previously.18 In some subjects, yohimbine was infused during cardiac catheterization. Patients with chronic autonomic failure received the doses described above; normal volunteers and patients with chronic orthostatic intolerance received twice the doses described above.
Data analysis
Statistical analyses were performed using StatView version 5.0.1. (SAS Institute, Cary, NC). Mean values in the baseline condition for the several subject groups were compared using single-factor ANOVA. Responses to drugs were analyzed by dependent-means t tests. Differences in response to pharmacologic tests among subject groups were compared using repeated measures analyses of variance. Relationships between individual hemodynamic values were assessed by linear regression and calculation of Pearson correlation coefficients. Post-hoc testing consisted of the Fisher PLSD test. Multiple regression analysis was done on the individual data, with the log of LF power as the dependent measure and the log of baroreflex slope and septal 6-[18F] fluorodopamine-derived radioactivity as independent measures. Mean values were expressed ± SEM.
RESULTS
Baseline
Across the 7 subject groups (N = 98), LF power was unrelated to subject group (F = 1.2). When individual subjects were stratified in terms of cardiac sympathetic denervation or innervation, based on concentrations of 6-[18F]fluorodopamine-derived radioactivity more than 2 SD below the normal mean, then LF power was lower in the Denervated group (mean 221 ± 55 msec2/Hz, N = 34) than in the Innervated group (516 ± 93 msec2/Hz, N = 64, F = 4.8, P = 0.03). LF power normalized for total power, HF normalized for total power, and the ratio of LF:HF were not related to 6-[18F]fluorodopamine-derived radioactivity.
When subjects were stratified in terms of BRS, then LF power was lower in the Low BRS group (223 ± 105 msec2/Hz, N = 46) than in the Normal BRS group (617 ± 97 msec2/Hz, N = 25, F = 6.1, P = 0.02). The Low BRS group did not differ from the Normal BRS group in normalized LF power (F = 0.8).
Figure 1. Mean (± SEM) values for the log of low-frequency power of heart rate variability in subject groups with innervated (Innerv) or denervated (Denerv) hearts, as indicated by low 6-[18F] fluorodopamine-derived radioactivity, and normal (Nl) or low baroreflex-cardiovagal slope (BRS), as indicated by slope ≤3 msec/mm Hg during the Valsalva maneuver. ***Significant difference, P < 0.001.When individual subjects were stratified into 4 groups, based on both cardiac 6-[18F]fluorodopamine-derived radioactivity (Innervated or Denervated) and on baroreflex-cardiovagal slope (Normal BRS or Low BRS), then both LF power and the log of LF power varied highly significantly as a function of subject group (F = 9.5, P < 0.0001; F = 4.6, P = 0.0004). The Denervated-Low BRS group had lower LF power than did the Denervated-Normal BRS group (P = 0.05), and the Innervated-Low BRS group had lower LF power than did the Innervated-Normal BRS group (P < 0.0001). When level of baroreflex function was taken into account, the Innervated and Denervated groups did not differ in LF power (Figure 1).
Values for HF power also varied with subject group when individual subjects were stratified in terms of both cardiac sympathetic innervation and BRS (F = 4.9, P = 0.004; Table 2). The Innervated-Low BRS group had lower HF power than did the Innervated-Normal BRS group (P = 0.003); however, the Denervated-Low BRS group did not differ from the Denervated-Normal BRS group in HF power. Normalization of LF and HF power for total power, and the ratio of low-to-high frequency did not reveal additional group differences (Table 2). In particular, the LF:HF ratio did not vary with the subject group (F = 0.6).
Figure 2. Mean (± SEM) values for (A) low-frequency power of heart rate variability and (B) cardiac norepinephrine spillover during right heart catheterization in subject groups with innervated (Innerv) or denervated (Denerv) hearts, as indicated by low 6-[18F]fluorodopaminederived radioactivity, and normal (Nl) or low baroreflex-cardiovagal slope (BRS), as indicated by slope ≤3 msec/mm Hg during the Valsalva maneuver. *Significant difference, P < 0.05. **Significant difference, P < 0.01.Analysis of data from subjects during cardiac catheterization showed that LF power varied as a function of subject group (F = 5.3, P = 0.03, Figure 2). The Innervated-Low BRS group had lower LF power than did the Innervated-Normal BRS group (P = 0.04), whereas the Denervated-Low BRS and Innervated-Low BRS groups did not differ in LF power. As expected, the Denervated-Low BRS group had lower cardiac norepinephrine spillover than the Innervated-Low BRS group.
Figure 3. Individual values for the log of low-frequency (LF) power as a function of (A) septal 6-[18F]fluorodopamine-derived radioactivity and (B) the log of baroreflex-cardiovagal slope.Individual values for LF power were positively correlated with BRS. When values for both variables were log-transformed, the log of LF power correlated positively with the log of BRS slope (r = 0.72, P < 0.0001, Figure 3). Individual values for the log of LF power were also correlated with the magnitude of decrease in systolic pressure during performance of the Valsalva maneuver (r = −0.60, P < 0.0001) and with the orthostatic change in systolic pressure (r = 0.58, P < 0.0001). In contrast, the log of LF power was unrelated to the septal myocardial concentration of 6-[18F]fluorodopamine-derived radioactivity, the plasma norepinephrine concentration, or cardiac norepinephrine spillover.
From multiple regression analysis for the log of LF power as the dependent measure and the log of baroreflex slope and septal 6-[18F]fluorodopamine-derived radioactivity as independent measures, the regression coefficient for the log of baroreflex slope was 0.92 (P < 0.0001), whereas the regression coefficient for 6-[18F] fluorodopamine-derived radioactivity was 3 ×10−6.
At baseline, the log of HF power correlated positively with the log of LF power (r = 0.77, P < 0.0001). HF power varied with the subject group (F = 4.9, P = 0.004). As with LF power, HF power was greater in the Innervated-Normal BRS than in the Innervated-Low BRS (P = 0.001, Table 2). As expected, the log of HF power correlated positively with the log of BRS (r = 0.60, P < 0.0001). The log of HF power also correlated negatively with the magnitude of decrease in systolic pressure during the Valsalva maneuver (r = −0.24, P = 0.02) and positively with the orthostatic change in systolic pressure (r = 0.40, P = 0.004).
Yohimbine
Figure 4. Mean (± SEM) values for the change in low-frequency power (ΔLF power) of heart rate variability during (A) intravenous infusion of yohimbine or (B) tyramine in groups with innervated (Innerv) or denervated (Denerv) hearts, as indicated by low 6-[18F]fluorodopaminederived radioactivity, and normal (Nl) or low baroreflex-cardiovagal slope (BRS), as indicated by slope ≤3 msec/mm Hg during the Valsalva maneuver. *Significant difference, P < 0.05. ***Significant difference, P < 0.001.Yohimbine infusion increased LF power (t = 2.9, P = 0.007). The group with cardiac sympathetic denervation did not differ from the group with intact cardiac innervation in terms of the change in LF power during yohimbine infusion (F = 0.7). Yohimbine infusion increased LF power in the Innervated-Normal BRS group (t = 2.8, P = 0.01), but not in the innervated or denervated groups with low BRS (Figure 4). The Innervated-Normal BRS group had a larger increase in LF power during yohimbine infusion than did the Innervated-Low BRS group (P = 0.02). Too few patients with cardiac denervation and normal BRS were studied to include in the ANOVA. The log of the change in LF power during yohimbine administration was positively correlated with the log of BRS at baseline (Figure 5).
Figure 5. Individual values for the log of change in low-frequency power (log ΔLF power) as a function of baroreflex-cardiovagal slope at baseline. Left: yohimbine infusion. Right: tyramine infusion.Yohimbine increased HF power in the Innervated-Normal BRS group (t = 2.1, P = 0.05) but not in the innervated or denervated groups with low BRS.
The change in LF power in response to yohimbine during cardiac catheterization was unrelated to the change in cardiac norepinephrine spillover (r = −0.09, N = 12).
Tyramine
Overall, tyramine infusion increased LF power (t = 2.9, P = 0.008). The group with cardiac sympathetic denervation did not differ from the group with intact cardiac innervation in terms of the change in LF power during tyramine infusion (F = 1.7). Tyramine increased LF power in the Innervated-Normal BRS group but not in the Innervated-Low BRS or Denervated-Low BRS groups (Figure 4; data for 2 outliers excluded). The log of the change in LF power during tyramine administration was positively correlated with the log of BRS at baseline (Figure 5; data for 2 outliers excluded).
DISCUSSION
In this study, patients with neuroimaging evidence of cardiac sympathetic denervation had low LF power of heart rate variability. At first glance, this finding would seem to support the view that LF power can provide an index of cardiac sympathetic outflow. As explained below, several lines of evidence from the present study led us to infer that the association between low LF power and cardiac sympathetic innervation is determined mainly by concurrent baroreflex function.
Patients with low BRS had low LF power, and patients with normal BRS had normal LF power, regardless of the status of cardiac sympathetic innervation as assessed by 6-[18F]fluorodopamine scanning. Neither normalization of LF and HF power for total power nor use of the LF:HF ratio improved the association with indices of cardiac sympathetic innervation.
Neurochemical findings during cardiac catheterization supported the above results based on cardiac sympathetic neuroimaging. Among patients with innervated hearts who had normal cardiac norepinephrine spillover, LF power was decreased only in the group with low BRS and was normal in the group with normal BRS. As expected, cardiac norepinephrine spillover was decreased in patients with neuroimaging evidence of cardiac sympathetic denervation.
Effects of pharmacological manipulations that increase norepinephrine release from sympathetic nerves provided further support for an association between baroreflex failure and low LF power, independent of cardiac sympathetic function. Both tyramine and yohimbine increased LF power only in the subjects with normal BRS. In subjects with low BRS, neither drug increased LF power, even in the group with intact cardiac sympathetic innervation. Moreover, individual values for responses of the log of LF power to both drugs were correlated positively with the log of BRS at baseline.
The fact that HF power was positively correlated with LF power could also fit with the notion of baroreflex function acting as a common determinant of values of both variables. We cannot exclude concurrent parasympathetic cardiovagal and sympathetic denervation as an explanation for the association between HF and LF power. Inhibition of the effects of parasympathetic activity after atropine administration results in the almost complete absence of both LF and HF HRV, further suggesting a common determinant.19
Several previous investigations have cast doubt on the validity of LF power as a measure of sympathetic activity because of dissociations between LF power and cardiac norepinephrine spillover, directly recorded sympathetic nerve traffic, and plasma norepinephrine levels.4,6,20 Such dissociations are especially glaring in patients with congestive heart failure, which is characterized by decreased LF power9 despite marked cardiac sympathetic activation.3
Other pathophysiologic states do result in both decreased sympathetic nervous system activity and decreased LF power. In these pathophysiologic states, the possibility remains that low LF power might reflect failure of baroreflexive modulation of sympathetic neuronal outflows, rather than sympathoinhibition itself. For instance, Wiklund et al21 noted low LF power in patients with palmar hyperhidrosis undergoing bilateral transthoracic sympathectomy; however, baroreflex-cardiovagal sensitivity also declines after thoracic sympathectomy.22
Sleight et al8 suggested dependence of LF power on baroreflex function, based on effects of carotid baroreceptor stimulation in 3 patients: 1 with normal BRS; 1 with ischemic heart disease, congestive heart failure, and normal BRS; and 1 with ischemic heart disease, congestive heart failure, and initially low BRS who subsequently had an improved clinical state and BRS. In the baseline state, both congestive heart failure patients had low LF power, despite a presumably hypernoradrenergic state. Direct baroreceptor stimulation at 0.1 Hz increased LF power in the normal subject and in the patient with congestive heart failure and normal BRS. The congestive heart failure patient with low BRS did not have an increase in LF power until BRS normalized. These data revealed an initial dissociation between cardiac noradrenergic state in the patients with congestive heart failure and LF power. During carotid sinus stimulation, LF power increased only when BRS was normal. Low BRS obviated this effect.
Because congestive heart failure is well known to be associated with baroreflex-cardiovagal inhibition,23–25 the finding of low LF power in heart failure also supports an association between LF power and BRS, independently of increased tonic release of norepinephrine from sympathetic nerves in the heart. Cevese et al26 inhibited noradrenergic vasomotor tone using an alpha-adrenoceptor blocker in human subjects while maintaining mean blood pressure at control levels using angiotensin II. This drug combination, which would be expected to attenuate sympathetically mediated vasomotor tone and thereby decrease arterial baroreceptor input, markedly decreased or abolished LF power of HRV, suggesting that, at least under resting supine conditions, a baroreflex mechanism accounts almost entirely for LF power of HRV.
deBoer et al27 developed a beat-to-beat model of the human circulation using a set of differential equations and the following principles of operation: (1) the baroreflex regulates heart rate and peripheral vascular resistance; (2) Windkessel properties characterize the systemic arterial tree; (3) contractile properties of the ventricular myocardium follow the Starling law; and (4) respiration exerts mechanical effects on blood pressure. The model attributes LF power to a resonance in the circulatory control system, produced by a slow time constant for reflexive sympathetically mediated responses to beat-tobeat blood pressure changes. The resonance can be upregulated or downregulated by the state of baroreflex activity. The model of deBoer et al predicts that changes in blood pressure would lead heart rate changes at 0.1 Hz through a delayed sympathetic response. Changes in HR would depend on summed effects of sympathetic and vagal effects, with the sympathetic response delaying the overall response. At the respiratory frequency (0.2 to 0.3 Hz), blood pressure and HR changes would occur with little delay because of fast parasympathetic control. In essence, the response of the sympathetic nervous system behaves as a low band pass filter, with a peak response at 0.1 Hz and little response at frequencies above 0.2 Hz. Systolic blood pressure would lead to changes in heart rate via the baroreflex. In general the results of this study fit with the deBoer model.
In conclusion, LF power derived from the interbeat interval spectrogram predominantly reflects baroreflex-mediated, phasic changes in cardiovagal and sympathetic noradrenergic outflows. In the setting of baroreflex failure, baseline LF power is reduced, regardless of the status of cardiac sympathetic innervation.
LIMITATIONS
The combination of cardiac sympathetic denervation and normal baroreflex function seems quite rare. One must exercise caution in drawing inferences from the findings in the Denervated-Normal BRS group, which contained only 4 subjects, even though the difference in mean log-transformed LF power from the Denervated-Low BRS group was highly statistically significant.
All of the testing in our study was done with the subjects supine. LF power measured in other positions might have different sources and meaning.
Spectral analysis of heart rate variability (HRV) has been used widely as a noninvasive technique for examining sympathetic and parasympathetic nervous outflows to the heart. Low-frequency (LF) and high-frequency (HF) power have been used most commonly. Human and animal experiments have repeatedly confirmed the dependence of HF power on respiration-related alterations in parasympathetic cardiovagal outflow–respiratory sinus arrhythmia; however, whether LF power provides an indirect measure of cardiac sympathetic activity has been contentious. Pagani et al1 reported that LF power (normalized to total spectral power) increased during states associated with sympathetic noradrenergic activation and that bilateral stellectomy in dogs reduced LF power. Alvarenga et al,2 however, reported that LF power was unrelated to all measures of norepinephrine kinetics in the heart; and in congestive heart failure, which is associated with a high rate of entry of norepinephrine into coronary sinus plasma (cardiac norepinephrine spillover),3 LF power is decreased, not increased as might be expected if LF power reflected sympathetic activity.4–7
Sleight et al8 proposed an alternative explanation for the origin of LF power. In a small group of human subjects, power spectral analysis of HRV showed that the amplitude of LF power was related to baroreflex gain and not to the level of sympathetic activity. Carotid sinus stimulation increased LF power only in individuals with normal baroreflex sensitivity and did not do so in those with depressed baroreflex gain. Therefore, results of power spectral analysis of LF power might reflect baroreflex-cardiovagal function.9
Studies of patients with dysautonomias provide an unusual opportunity to examine neurocirculatory correlates of LF power. Some chronic autonomic failure syndromes feature cardiac sympathetic denervation, whereas others do not. Parkinson disease with neurogenic orthostatic hypotension and pure autonomic failure feature cardiac sympathetic denervation, whereas multiple system atrophy does not.10 All 3 diseases involve baroreflex-cardiovagal and baro-reflex-sympathoneural failure.11 Chronic orthostatic intolerance syndromes (postural tachycardia syndrome, neurocardiogenic syncope) do not entail either cardiac sympathetic denervation or baroreflex failure.12
For this article, we carried out power spectral analyses of HRV on digitized electrocardiographic recordings from dysautonomia patients and normal volunteers during supine rest, measurement of cardiac norepinephrine spillover, and intravenous infusion of yohimbine and tyramine, 2 drugs that are known to release norepinephrine from cardiac sympathetic nerves.13,14 Cardiac sympathetic innervation was assessed by 6-[18F]fluorodopamine positron emission tomographic scanning.15
We hypothesized that if LF power indicated cardiac sympathetic innervation and function, then patients with neuroimaging or neurochemical evidence of cardiac sympathetic denervation would have low LF power and attenuated increments in LF power in response to yohimbine and tyramine. Alternatively, if LF power was reflective of baroreflex function, alterations of LF power would be independent of cardiac sympathetic innervation status and correlate with changes in baroreflex gain.
METHODS
The study protocols were approved by the Intramural Research Board of the National Institute of Neurological Disorders and Stroke. All subjects were studied at the National Institutes of Health Clinical Center after giving informed, written consent.
Subjects
The study population consisted of a total of 98 subjects who participated in research protocols studying chronic orthostatic intolerance and chronic autonomic failure (Table 1). The subjects underwent autonomic function testing and had reviewable, digitized electrocardiographic data enabling retrospective power spectral analysis of HRV. ECG and blood pressure data were sampled at 1 kHz.
The study subjects were separated into 4 groups, depending on their state of cardiac sympathetic innervation and baroreflex-cardiovagal slope (BRS; see below). There were 40 subjects with intact sympathetic innervation and normal BRS (Innervated-Normal BRS), 24 with intact sympathetic innervation and low BRS (Innervated-Low BRS), 4 with sympathetic denervation and normal BRS (Denervated-Normal BRS), and 30 with sympathetic denervation and low BRS (Denervated-Low BRS).
Autonomic function testing
Each subject was studied while supine with head on pillow after an overnight fast. Each patient had monitoring of the electrocardiogram and beat-tobeat blood pressure using either noninvasive devices (Finometer, Finapres Medical Systems, Amsterdam, the Netherlands; Portapres, Finapres Medical Systems; or Colin tonometer, Colin Medical Instruments, San Antonio, TX) or a brachial intra-arterial catheter. We previously studied formally and reported excellent agreement between intra-arterial and these noninvasively obtained measures of beat-to-beat blood pressure.16 Continuous vital signs data were digitized and recorded using a PowerLab (AD Instruments Pty Ltd, Castle Hill, Australia) data acquisition system and stored for later analysis on an Apple PowerBook G4 computer (Apple, Cupertino, CA).
After about a 10-min baseline period, each subject performed a Valsalva maneuver (30 mm Hg for 12 sec) at least 3 times.
Baroreflex function
As an index of baroreflex function, we used the slope of the relationship between cardiac interbeat interval and systolic blood pressure during phase II of the Valsalva maneuver.17 BRS, in units of msec/mm Hg, was calculated from the linear regression equation for the relationship between interbeat interval (with 1-beat delay) and systolic pressure. A BRS value of ≤3 msec/mm Hg was considered low.11
Pharmacologic testing
Pharmacologic testing was performed on completion of the autonomic evaluation, using either tyramine or yohimbine. If a subject received both drugs, each drug administration was on a separate day. The durations of drug infusion were sufficient for heart rate and blood pressure to reach plateau values.
In a total of 22 subjects (Table 1), yohimbine was infused intravenously at 62.5 μg/kg over 3 min and then at 0.5 μg/kg/min for 12 min. In a total of 50 subjects, tyramine was infused at a rate of 1 mg/min for 10 min. In patients with severe supine hypertension (systolic pressure more than 200 mm Hg) and orthostatic hypotension, the test drugs were infused during head-up tilting (15° to 30°), to decrease baseline pressure, or else the drugs were not given.
HRV analysis
LF power (0.04 to 0.15 Hz), HF power (0.16 to 0.4 Hz), and total power (TP, 0.0 to 0.4 Hz) were calculated using Chart 5.4.2 and the HRV module version 1.03 (PowerLab, AD Instruments Pty Ltd, Castle Hill, Australia). Stable heart rate epochs 3 to 5 min in duration were chosen for analysis. One epoch was sampled immediately before initiation of drug testing; the second followed attainment of steady-state hemodynamic effects. Interbeat interval data were reviewed carefully to eliminate artifacts from noise and T waves, using segments with little to no premature beats. LF power and HF power were calculated as absolute power (msec2), with or without normalization for total power (0.04 to 0.4 Hz). Reported LF or HF power was integrated within their defined frequency bands.
Cardiac sympathetic neuroimaging
For cardiac sympathetic neuroimaging the subject was positioned supine, feet-first in a GE Advance scanner (General Electric, Milwaukee, WI), with the thorax in the gantry. After positioning the patient with the thorax in the scanner and transmission scanning for attenuation correction, 6-[18F]fluorodopamine (usual dose 1 mCi, specific activity 1.0 to 4.0 Ci/mmole, in about 10 mL normal saline) was infused intravenously at a constant rate for 3 min, and dynamic scanning data were obtained for thoracic radioactivity, with the midpoint of the scanning interval at 7.5 min after injection of the tracer (data collection interval between 5 and 10 min). Cardiac sympathetic denervation was defined by low concentrations of 6-[18F] fluorodopamine-derived radioactivity in the interventricular septum (< 5,000 nCi-kg/cc-mCi) or left ventricular free wall (< 4,000 nCi-kg/cc-mCi) corresponding to about 2 SD below the normal means.
Cardiac norepinephrine spillover
Subgroups of subjects (3 PD + NOH, 3 MSA, 3 PAF, 5 normal volunteers) underwent right heart catheterization for measurement of cardiac norepinephrine spillover. 3H-Norepinephrine was infused intravenously, and arterial and coronary sinus blood was sampled and coronary sinus blood flow was measured by thermodilution for measurements of cardiac norepinephrine spillover as described previously.18 In some subjects, yohimbine was infused during cardiac catheterization. Patients with chronic autonomic failure received the doses described above; normal volunteers and patients with chronic orthostatic intolerance received twice the doses described above.
Data analysis
Statistical analyses were performed using StatView version 5.0.1. (SAS Institute, Cary, NC). Mean values in the baseline condition for the several subject groups were compared using single-factor ANOVA. Responses to drugs were analyzed by dependent-means t tests. Differences in response to pharmacologic tests among subject groups were compared using repeated measures analyses of variance. Relationships between individual hemodynamic values were assessed by linear regression and calculation of Pearson correlation coefficients. Post-hoc testing consisted of the Fisher PLSD test. Multiple regression analysis was done on the individual data, with the log of LF power as the dependent measure and the log of baroreflex slope and septal 6-[18F] fluorodopamine-derived radioactivity as independent measures. Mean values were expressed ± SEM.
RESULTS
Baseline
Across the 7 subject groups (N = 98), LF power was unrelated to subject group (F = 1.2). When individual subjects were stratified in terms of cardiac sympathetic denervation or innervation, based on concentrations of 6-[18F]fluorodopamine-derived radioactivity more than 2 SD below the normal mean, then LF power was lower in the Denervated group (mean 221 ± 55 msec2/Hz, N = 34) than in the Innervated group (516 ± 93 msec2/Hz, N = 64, F = 4.8, P = 0.03). LF power normalized for total power, HF normalized for total power, and the ratio of LF:HF were not related to 6-[18F]fluorodopamine-derived radioactivity.
When subjects were stratified in terms of BRS, then LF power was lower in the Low BRS group (223 ± 105 msec2/Hz, N = 46) than in the Normal BRS group (617 ± 97 msec2/Hz, N = 25, F = 6.1, P = 0.02). The Low BRS group did not differ from the Normal BRS group in normalized LF power (F = 0.8).
Figure 1. Mean (± SEM) values for the log of low-frequency power of heart rate variability in subject groups with innervated (Innerv) or denervated (Denerv) hearts, as indicated by low 6-[18F] fluorodopamine-derived radioactivity, and normal (Nl) or low baroreflex-cardiovagal slope (BRS), as indicated by slope ≤3 msec/mm Hg during the Valsalva maneuver. ***Significant difference, P < 0.001.When individual subjects were stratified into 4 groups, based on both cardiac 6-[18F]fluorodopamine-derived radioactivity (Innervated or Denervated) and on baroreflex-cardiovagal slope (Normal BRS or Low BRS), then both LF power and the log of LF power varied highly significantly as a function of subject group (F = 9.5, P < 0.0001; F = 4.6, P = 0.0004). The Denervated-Low BRS group had lower LF power than did the Denervated-Normal BRS group (P = 0.05), and the Innervated-Low BRS group had lower LF power than did the Innervated-Normal BRS group (P < 0.0001). When level of baroreflex function was taken into account, the Innervated and Denervated groups did not differ in LF power (Figure 1).
Values for HF power also varied with subject group when individual subjects were stratified in terms of both cardiac sympathetic innervation and BRS (F = 4.9, P = 0.004; Table 2). The Innervated-Low BRS group had lower HF power than did the Innervated-Normal BRS group (P = 0.003); however, the Denervated-Low BRS group did not differ from the Denervated-Normal BRS group in HF power. Normalization of LF and HF power for total power, and the ratio of low-to-high frequency did not reveal additional group differences (Table 2). In particular, the LF:HF ratio did not vary with the subject group (F = 0.6).
Figure 2. Mean (± SEM) values for (A) low-frequency power of heart rate variability and (B) cardiac norepinephrine spillover during right heart catheterization in subject groups with innervated (Innerv) or denervated (Denerv) hearts, as indicated by low 6-[18F]fluorodopaminederived radioactivity, and normal (Nl) or low baroreflex-cardiovagal slope (BRS), as indicated by slope ≤3 msec/mm Hg during the Valsalva maneuver. *Significant difference, P < 0.05. **Significant difference, P < 0.01.Analysis of data from subjects during cardiac catheterization showed that LF power varied as a function of subject group (F = 5.3, P = 0.03, Figure 2). The Innervated-Low BRS group had lower LF power than did the Innervated-Normal BRS group (P = 0.04), whereas the Denervated-Low BRS and Innervated-Low BRS groups did not differ in LF power. As expected, the Denervated-Low BRS group had lower cardiac norepinephrine spillover than the Innervated-Low BRS group.
Figure 3. Individual values for the log of low-frequency (LF) power as a function of (A) septal 6-[18F]fluorodopamine-derived radioactivity and (B) the log of baroreflex-cardiovagal slope.Individual values for LF power were positively correlated with BRS. When values for both variables were log-transformed, the log of LF power correlated positively with the log of BRS slope (r = 0.72, P < 0.0001, Figure 3). Individual values for the log of LF power were also correlated with the magnitude of decrease in systolic pressure during performance of the Valsalva maneuver (r = −0.60, P < 0.0001) and with the orthostatic change in systolic pressure (r = 0.58, P < 0.0001). In contrast, the log of LF power was unrelated to the septal myocardial concentration of 6-[18F]fluorodopamine-derived radioactivity, the plasma norepinephrine concentration, or cardiac norepinephrine spillover.
From multiple regression analysis for the log of LF power as the dependent measure and the log of baroreflex slope and septal 6-[18F]fluorodopamine-derived radioactivity as independent measures, the regression coefficient for the log of baroreflex slope was 0.92 (P < 0.0001), whereas the regression coefficient for 6-[18F] fluorodopamine-derived radioactivity was 3 ×10−6.
At baseline, the log of HF power correlated positively with the log of LF power (r = 0.77, P < 0.0001). HF power varied with the subject group (F = 4.9, P = 0.004). As with LF power, HF power was greater in the Innervated-Normal BRS than in the Innervated-Low BRS (P = 0.001, Table 2). As expected, the log of HF power correlated positively with the log of BRS (r = 0.60, P < 0.0001). The log of HF power also correlated negatively with the magnitude of decrease in systolic pressure during the Valsalva maneuver (r = −0.24, P = 0.02) and positively with the orthostatic change in systolic pressure (r = 0.40, P = 0.004).
Yohimbine
Figure 4. Mean (± SEM) values for the change in low-frequency power (ΔLF power) of heart rate variability during (A) intravenous infusion of yohimbine or (B) tyramine in groups with innervated (Innerv) or denervated (Denerv) hearts, as indicated by low 6-[18F]fluorodopaminederived radioactivity, and normal (Nl) or low baroreflex-cardiovagal slope (BRS), as indicated by slope ≤3 msec/mm Hg during the Valsalva maneuver. *Significant difference, P < 0.05. ***Significant difference, P < 0.001.Yohimbine infusion increased LF power (t = 2.9, P = 0.007). The group with cardiac sympathetic denervation did not differ from the group with intact cardiac innervation in terms of the change in LF power during yohimbine infusion (F = 0.7). Yohimbine infusion increased LF power in the Innervated-Normal BRS group (t = 2.8, P = 0.01), but not in the innervated or denervated groups with low BRS (Figure 4). The Innervated-Normal BRS group had a larger increase in LF power during yohimbine infusion than did the Innervated-Low BRS group (P = 0.02). Too few patients with cardiac denervation and normal BRS were studied to include in the ANOVA. The log of the change in LF power during yohimbine administration was positively correlated with the log of BRS at baseline (Figure 5).
Figure 5. Individual values for the log of change in low-frequency power (log ΔLF power) as a function of baroreflex-cardiovagal slope at baseline. Left: yohimbine infusion. Right: tyramine infusion.Yohimbine increased HF power in the Innervated-Normal BRS group (t = 2.1, P = 0.05) but not in the innervated or denervated groups with low BRS.
The change in LF power in response to yohimbine during cardiac catheterization was unrelated to the change in cardiac norepinephrine spillover (r = −0.09, N = 12).
Tyramine
Overall, tyramine infusion increased LF power (t = 2.9, P = 0.008). The group with cardiac sympathetic denervation did not differ from the group with intact cardiac innervation in terms of the change in LF power during tyramine infusion (F = 1.7). Tyramine increased LF power in the Innervated-Normal BRS group but not in the Innervated-Low BRS or Denervated-Low BRS groups (Figure 4; data for 2 outliers excluded). The log of the change in LF power during tyramine administration was positively correlated with the log of BRS at baseline (Figure 5; data for 2 outliers excluded).
DISCUSSION
In this study, patients with neuroimaging evidence of cardiac sympathetic denervation had low LF power of heart rate variability. At first glance, this finding would seem to support the view that LF power can provide an index of cardiac sympathetic outflow. As explained below, several lines of evidence from the present study led us to infer that the association between low LF power and cardiac sympathetic innervation is determined mainly by concurrent baroreflex function.
Patients with low BRS had low LF power, and patients with normal BRS had normal LF power, regardless of the status of cardiac sympathetic innervation as assessed by 6-[18F]fluorodopamine scanning. Neither normalization of LF and HF power for total power nor use of the LF:HF ratio improved the association with indices of cardiac sympathetic innervation.
Neurochemical findings during cardiac catheterization supported the above results based on cardiac sympathetic neuroimaging. Among patients with innervated hearts who had normal cardiac norepinephrine spillover, LF power was decreased only in the group with low BRS and was normal in the group with normal BRS. As expected, cardiac norepinephrine spillover was decreased in patients with neuroimaging evidence of cardiac sympathetic denervation.
Effects of pharmacological manipulations that increase norepinephrine release from sympathetic nerves provided further support for an association between baroreflex failure and low LF power, independent of cardiac sympathetic function. Both tyramine and yohimbine increased LF power only in the subjects with normal BRS. In subjects with low BRS, neither drug increased LF power, even in the group with intact cardiac sympathetic innervation. Moreover, individual values for responses of the log of LF power to both drugs were correlated positively with the log of BRS at baseline.
The fact that HF power was positively correlated with LF power could also fit with the notion of baroreflex function acting as a common determinant of values of both variables. We cannot exclude concurrent parasympathetic cardiovagal and sympathetic denervation as an explanation for the association between HF and LF power. Inhibition of the effects of parasympathetic activity after atropine administration results in the almost complete absence of both LF and HF HRV, further suggesting a common determinant.19
Several previous investigations have cast doubt on the validity of LF power as a measure of sympathetic activity because of dissociations between LF power and cardiac norepinephrine spillover, directly recorded sympathetic nerve traffic, and plasma norepinephrine levels.4,6,20 Such dissociations are especially glaring in patients with congestive heart failure, which is characterized by decreased LF power9 despite marked cardiac sympathetic activation.3
Other pathophysiologic states do result in both decreased sympathetic nervous system activity and decreased LF power. In these pathophysiologic states, the possibility remains that low LF power might reflect failure of baroreflexive modulation of sympathetic neuronal outflows, rather than sympathoinhibition itself. For instance, Wiklund et al21 noted low LF power in patients with palmar hyperhidrosis undergoing bilateral transthoracic sympathectomy; however, baroreflex-cardiovagal sensitivity also declines after thoracic sympathectomy.22
Sleight et al8 suggested dependence of LF power on baroreflex function, based on effects of carotid baroreceptor stimulation in 3 patients: 1 with normal BRS; 1 with ischemic heart disease, congestive heart failure, and normal BRS; and 1 with ischemic heart disease, congestive heart failure, and initially low BRS who subsequently had an improved clinical state and BRS. In the baseline state, both congestive heart failure patients had low LF power, despite a presumably hypernoradrenergic state. Direct baroreceptor stimulation at 0.1 Hz increased LF power in the normal subject and in the patient with congestive heart failure and normal BRS. The congestive heart failure patient with low BRS did not have an increase in LF power until BRS normalized. These data revealed an initial dissociation between cardiac noradrenergic state in the patients with congestive heart failure and LF power. During carotid sinus stimulation, LF power increased only when BRS was normal. Low BRS obviated this effect.
Because congestive heart failure is well known to be associated with baroreflex-cardiovagal inhibition,23–25 the finding of low LF power in heart failure also supports an association between LF power and BRS, independently of increased tonic release of norepinephrine from sympathetic nerves in the heart. Cevese et al26 inhibited noradrenergic vasomotor tone using an alpha-adrenoceptor blocker in human subjects while maintaining mean blood pressure at control levels using angiotensin II. This drug combination, which would be expected to attenuate sympathetically mediated vasomotor tone and thereby decrease arterial baroreceptor input, markedly decreased or abolished LF power of HRV, suggesting that, at least under resting supine conditions, a baroreflex mechanism accounts almost entirely for LF power of HRV.
deBoer et al27 developed a beat-to-beat model of the human circulation using a set of differential equations and the following principles of operation: (1) the baroreflex regulates heart rate and peripheral vascular resistance; (2) Windkessel properties characterize the systemic arterial tree; (3) contractile properties of the ventricular myocardium follow the Starling law; and (4) respiration exerts mechanical effects on blood pressure. The model attributes LF power to a resonance in the circulatory control system, produced by a slow time constant for reflexive sympathetically mediated responses to beat-tobeat blood pressure changes. The resonance can be upregulated or downregulated by the state of baroreflex activity. The model of deBoer et al predicts that changes in blood pressure would lead heart rate changes at 0.1 Hz through a delayed sympathetic response. Changes in HR would depend on summed effects of sympathetic and vagal effects, with the sympathetic response delaying the overall response. At the respiratory frequency (0.2 to 0.3 Hz), blood pressure and HR changes would occur with little delay because of fast parasympathetic control. In essence, the response of the sympathetic nervous system behaves as a low band pass filter, with a peak response at 0.1 Hz and little response at frequencies above 0.2 Hz. Systolic blood pressure would lead to changes in heart rate via the baroreflex. In general the results of this study fit with the deBoer model.
In conclusion, LF power derived from the interbeat interval spectrogram predominantly reflects baroreflex-mediated, phasic changes in cardiovagal and sympathetic noradrenergic outflows. In the setting of baroreflex failure, baseline LF power is reduced, regardless of the status of cardiac sympathetic innervation.
LIMITATIONS
The combination of cardiac sympathetic denervation and normal baroreflex function seems quite rare. One must exercise caution in drawing inferences from the findings in the Denervated-Normal BRS group, which contained only 4 subjects, even though the difference in mean log-transformed LF power from the Denervated-Low BRS group was highly statistically significant.
All of the testing in our study was done with the subjects supine. LF power measured in other positions might have different sources and meaning.
References
Pagani M, Lombardi F, Guzzetti S, et al. Power spectral analysis of heart rate and arterial pressure variabilities as a marker of sympatho-vagal interaction in man and conscious dog. Circ Res1986; 59:178–193.
Alvarenga ME, Richards JC, Lambert G, Esler MD. Psychophysiological mechanisms in panic disorder: a correlative analysis of noradrenaline spillover, neuronal noradrenaline reuptake, power spectral analysis of heart rate variability, and psychological variables. Psychosom Med2006; 68:8–16.
Eisenhofer G, Friberg P, Rundqvist B, et al. Cardiac sympathetic nerve function in congestive heart failure. Circulation1996; 93:1667–1676.
Notarius CF, Butler GC, Ando S, Pollard MJ, Senn BL, Floras JS. Dissociation between microneurographic and heart rate variability estimates of sympathetic tone in normal subjects and patients with heart failure. Clin Sci (Lond)1999; 96:557–565.
Scalvini S, Volterrani M, Zanelli E, et al. Is heart rate variability a reliable method to assess autonomic modulation in left ventricular dysfunction and heart failure? Assessment of autonomic modulation with heart rate variability. Int J Cardiol1998; 67:9–17.
Kingwell BA, Thompson JM, Kaye DM, McPherson GA, Jennings GL, Esler MD. Heart rate spectral analysis, cardiac norepinephrine spillover, and muscle sympathetic nerve activity during human sympathetic nervous activation and failure. Circulation1994; 90:234–240.
van de Borne P, Montano N, Pagani M, Oren R, Somers VK. Absence of low-frequency variability of sympathetic nerve activity in severe heart failure. Circulation1997; 95:1449–1454.
Sleight P, La Rovere MT, Mortara A, et al. Physiology and pathophysiology of heart rate and blood pressure variability in humans: is power spectral analysis largely an index of baroreflex gain?Clin Sci (Lond)1995; 88:103–109.
Saul JP, Arai Y, Berger RD, Lilly LS, Colucci WS, Cohen RJ. Assessment of autonomic regulation in chronic congestive heart failure by heart rate spectral analysis. Am J Cardiol1988; 61:1292–1299.
Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO. Cardiac sympathetic denervation in Parkinson disease. Ann Intern Med2000; 133:338–347.
Goldstein DS, Eldadah BA, Holmes C, et al. Neurocirculatory abnormalities in Parkinson disease with orthostatic hypotension. Independence from levodopa treatment. Hypertension2005; 46:1333–1339.
Goldstein DS, Eldadah B, Holmes C, Pechnik S, Moak J, Sharabi Y. Neurocirculatory abnormalities in chronic orthostatic intolerance. Circulation2005; 111:839–845.
Goldstein DS, Holmes C, Frank SM, et al. Cardiac sympathetic dysautonomia in chronic orthostatic intolerance syndromes. Circulation2002; 106:2358–2365.
Lord SW, Clayton RH, Mitchell L, Dark JH, Murray A, McComb JM. Sympathetic reinnervation and heart rate variability after cardiac transplantation. Heart1997; 77:532–538.
Goldstein DS, Eisenhofer G, Dunn BB, et al. Positron emission tomographic imaging of cardiac sympathetic innervation using 6-[18F]fluorodopamine: initial findings in humans. J Am Coll Cardiol1993; 22:1961–1971.
Goldstein DS, Tack C. Non-invasive detection of sympathetic neurocirculatory failure. Clin Auton Res2000; 10:285–291.
Goldstein DS, Horwitz D, Keiser HR. Comparison of techniques for measuring baroreflex sensitivity in man. Circulation1982; 66:432–439.
Goldstein DS, Brush JE, Eisenhofer G, Stull R, Esler M. In vivo measurement of neuronal uptake of norepinephrine in the human heart. Circulation1988; 78:41–48.
Koh J, Brown TE, Beightol LA, Ha CY, Eckberg DL. Human autonomic rhythms: vagal cardiac mechanisms in tetraplegic subjects. J Physiol1994; 474:483–495.
Saul JP, Rea RF, Eckberg DL, Berger RD, Cohen RJ. Heart rate and muscle sympathetic nerve variability during reflex changes of autonomic activity. Am J Physiol1990; 258:H713–H721.
Wiklund U, Koskinen LO, Niklasson U, Bjerle P, Elfversson J. Endoscopic transthoracic sympathectomy affects the autonomic modulation of heart rate in patients with palmar hyperhidrosis. Acta Neurochir (Wien)2000; 142:691–696.
Kawamata YT, Kawamata T, Omote K, et al. Endoscopic thoracic sympathectomy suppresses baroreflex control of heart rate in patients with essential hyperhidrosis. Anesth Analg2004; 98:37–39.
Goldstein RE, Beiser GD, Stampfer M, Epstein SE. Impairment of autonomically mediated heart rate control in patients with cardiac dysfunction. Circ Res1975; 36:571–578.
Cody RJ, Franklin KW, Kluger J, Laragh JH. Mechanisms governing the postural response and baroreceptor abnormalities in chronic congestive heart failure: effects of acute and long-term convertingenzyme inhibition. Circulation1982; 66:135–142.
Creager MA. Baroreceptor reflex function in congestive heart failure. Am J Cardiol1992; 69:10G–15G; discussion 15G–16G.
Cevese A, Gulli G, Polati E, Gottin L, Grasso R. Baroreflex and oscillation of heart period at 0.1 Hz studied by alpha-blockade and crossspectral analysis in healthy humans. J Physiol2001; 531:235–244.
deBoer RW, Karemaker JM, Strackee J. Hemodynamic fluctuations and baroreflex sensitivity in humans: a beat-to-beat model. Am J Physiol1987; 253:H680–689.
References
Pagani M, Lombardi F, Guzzetti S, et al. Power spectral analysis of heart rate and arterial pressure variabilities as a marker of sympatho-vagal interaction in man and conscious dog. Circ Res1986; 59:178–193.
Alvarenga ME, Richards JC, Lambert G, Esler MD. Psychophysiological mechanisms in panic disorder: a correlative analysis of noradrenaline spillover, neuronal noradrenaline reuptake, power spectral analysis of heart rate variability, and psychological variables. Psychosom Med2006; 68:8–16.
Eisenhofer G, Friberg P, Rundqvist B, et al. Cardiac sympathetic nerve function in congestive heart failure. Circulation1996; 93:1667–1676.
Notarius CF, Butler GC, Ando S, Pollard MJ, Senn BL, Floras JS. Dissociation between microneurographic and heart rate variability estimates of sympathetic tone in normal subjects and patients with heart failure. Clin Sci (Lond)1999; 96:557–565.
Scalvini S, Volterrani M, Zanelli E, et al. Is heart rate variability a reliable method to assess autonomic modulation in left ventricular dysfunction and heart failure? Assessment of autonomic modulation with heart rate variability. Int J Cardiol1998; 67:9–17.
Kingwell BA, Thompson JM, Kaye DM, McPherson GA, Jennings GL, Esler MD. Heart rate spectral analysis, cardiac norepinephrine spillover, and muscle sympathetic nerve activity during human sympathetic nervous activation and failure. Circulation1994; 90:234–240.
van de Borne P, Montano N, Pagani M, Oren R, Somers VK. Absence of low-frequency variability of sympathetic nerve activity in severe heart failure. Circulation1997; 95:1449–1454.
Sleight P, La Rovere MT, Mortara A, et al. Physiology and pathophysiology of heart rate and blood pressure variability in humans: is power spectral analysis largely an index of baroreflex gain?Clin Sci (Lond)1995; 88:103–109.
Saul JP, Arai Y, Berger RD, Lilly LS, Colucci WS, Cohen RJ. Assessment of autonomic regulation in chronic congestive heart failure by heart rate spectral analysis. Am J Cardiol1988; 61:1292–1299.
Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO. Cardiac sympathetic denervation in Parkinson disease. Ann Intern Med2000; 133:338–347.
Goldstein DS, Eldadah BA, Holmes C, et al. Neurocirculatory abnormalities in Parkinson disease with orthostatic hypotension. Independence from levodopa treatment. Hypertension2005; 46:1333–1339.
Goldstein DS, Eldadah B, Holmes C, Pechnik S, Moak J, Sharabi Y. Neurocirculatory abnormalities in chronic orthostatic intolerance. Circulation2005; 111:839–845.
Goldstein DS, Holmes C, Frank SM, et al. Cardiac sympathetic dysautonomia in chronic orthostatic intolerance syndromes. Circulation2002; 106:2358–2365.
Lord SW, Clayton RH, Mitchell L, Dark JH, Murray A, McComb JM. Sympathetic reinnervation and heart rate variability after cardiac transplantation. Heart1997; 77:532–538.
Goldstein DS, Eisenhofer G, Dunn BB, et al. Positron emission tomographic imaging of cardiac sympathetic innervation using 6-[18F]fluorodopamine: initial findings in humans. J Am Coll Cardiol1993; 22:1961–1971.
Goldstein DS, Tack C. Non-invasive detection of sympathetic neurocirculatory failure. Clin Auton Res2000; 10:285–291.
Goldstein DS, Horwitz D, Keiser HR. Comparison of techniques for measuring baroreflex sensitivity in man. Circulation1982; 66:432–439.
Goldstein DS, Brush JE, Eisenhofer G, Stull R, Esler M. In vivo measurement of neuronal uptake of norepinephrine in the human heart. Circulation1988; 78:41–48.
Koh J, Brown TE, Beightol LA, Ha CY, Eckberg DL. Human autonomic rhythms: vagal cardiac mechanisms in tetraplegic subjects. J Physiol1994; 474:483–495.
Saul JP, Rea RF, Eckberg DL, Berger RD, Cohen RJ. Heart rate and muscle sympathetic nerve variability during reflex changes of autonomic activity. Am J Physiol1990; 258:H713–H721.
Wiklund U, Koskinen LO, Niklasson U, Bjerle P, Elfversson J. Endoscopic transthoracic sympathectomy affects the autonomic modulation of heart rate in patients with palmar hyperhidrosis. Acta Neurochir (Wien)2000; 142:691–696.
Kawamata YT, Kawamata T, Omote K, et al. Endoscopic thoracic sympathectomy suppresses baroreflex control of heart rate in patients with essential hyperhidrosis. Anesth Analg2004; 98:37–39.
Goldstein RE, Beiser GD, Stampfer M, Epstein SE. Impairment of autonomically mediated heart rate control in patients with cardiac dysfunction. Circ Res1975; 36:571–578.
Cody RJ, Franklin KW, Kluger J, Laragh JH. Mechanisms governing the postural response and baroreceptor abnormalities in chronic congestive heart failure: effects of acute and long-term convertingenzyme inhibition. Circulation1982; 66:135–142.
Creager MA. Baroreceptor reflex function in congestive heart failure. Am J Cardiol1992; 69:10G–15G; discussion 15G–16G.
Cevese A, Gulli G, Polati E, Gottin L, Grasso R. Baroreflex and oscillation of heart period at 0.1 Hz studied by alpha-blockade and crossspectral analysis in healthy humans. J Physiol2001; 531:235–244.
deBoer RW, Karemaker JM, Strackee J. Hemodynamic fluctuations and baroreflex sensitivity in humans: a beat-to-beat model. Am J Physiol1987; 253:H680–689.
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Chief, Clinical Neurocardiology Section, CNP, DIR, NINDS, NIH, 10 Center Drive MSC-1620, Building 10, Room 6N252, Bethesda, MD 20892-1620; goldsteind@ninds.nih.gov
Dr. Goldstein reported that he has no financial relationships that pose a potential conflict of interest with this article.
This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Neurological Disorders and Stroke.
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Chief, Clinical Neurocardiology Section, CNP, DIR, NINDS, NIH, 10 Center Drive MSC-1620, Building 10, Room 6N252, Bethesda, MD 20892-1620; goldsteind@ninds.nih.gov
Dr. Goldstein reported that he has no financial relationships that pose a potential conflict of interest with this article.
This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Neurological Disorders and Stroke.
Author and Disclosure Information
David S. Goldstein, MD, PhD Clinical Neurocardiology Section, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Chief, Clinical Neurocardiology Section, CNP, DIR, NINDS, NIH, 10 Center Drive MSC-1620, Building 10, Room 6N252, Bethesda, MD 20892-1620; goldsteind@ninds.nih.gov
Dr. Goldstein reported that he has no financial relationships that pose a potential conflict of interest with this article.
This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Neurological Disorders and Stroke.

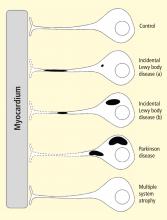 Reprinted from Brain (Orimo S, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008; 131:642–650) by permission of Oxford University Press.
Reprinted from Brain (Orimo S, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008; 131:642–650) by permission of Oxford University Press.