User login
This is a momentous occasion for me, for the extraordinary people in the Clinical Neurocardiology Section at the National Institutes of Health (NIH), and for my family—my wife Minka and son Joey drove all the way from Maryland late last night and early this morning to be here. I thank them publicly here.
THE ‘SPARKLE OF INSIGHT’ FROM ENLIGHTENED INDUCTION
In these brief comments, as I look back on the road I have taken over the past 40 years carrying out patient-oriented research in heart-brain medicine, I would like to convey a viewpoint instead of dwelling on the presentation of research data.
The idea I wish to convey is that ignorance isn’t biased. If you have a hypothesis you want to test, you are inherently biased to find something positive—and, if you are in academic medicine, publishable—in the data you obtain. But if you have the technical capability to measure something no one else can measure, and you have sufficient mastery of the topic to know what is not yet known, then if you make an observation that you did not predict and if you recognize its significance, you have made a discovery. You have revealed a bit of the truth. You experience the highest joy and thrill a scientist can feel—a “sparkle of insight.” When this happens, if you have sense, you stop what you have been doing to pursue that discovery.
Hardly anyone has received a Nobel Prize for testing a theory, but many Nobel Prizes have been awarded for technological advances and for discoveries based on those advances. In my view, discoverers use an enlightened inductive approach at least as much as deduction. They develop new technology that enables key novel measurements, and they keep in mind gaps in knowledge, so that they are ready to appreciate the significance of their observations.
A PERSONAL EXAMPLE
‘You have to measure something’
Let me share an example of this process by relating a sparkle of insight I had several years ago. When I began working at the NIH, I met with the chief of the Hypertension-Endocrine Branch of the National Heart, Lung, and Blood Institute about the research program I would pursue. After listening patiently to me for many minutes as I spouted about how I was going to test hypotheses derived from the concepts that people with hypertension are “hyper-tense,” and that stress causes heart disease, the chief responded, “Well, these ideas are all well and good. But what are you going to measure? You can measure whatever you want, but you have to measure something.”
Measure something. I wanted to see if there was hyperactivity of the sympathetic nervous system or excessive sympathetic innervation in hypertension, and I started working on ways to measure sympathetic activity.
The sympathetic nervous system at a glance
First I should introduce you to the sympathetic nervous system, which is one of the main effectors by which the brain regulates the heart and blood vessels. It is a key link between the brain and heart. The sympathetic nerves to the heart and other organs do not come directly from the brain but from ganglia, which are clumps of nerve cell bodies strung like pearls on a necklace on each side of the spinal column. This origin outside the central nervous system will be an important fact to keep in mind.
In the heart, the sympathetic nerves travel with the coronary arteries and then dive into the heart muscle from the outside. Sympathetic nerves also enmesh the walls of arteries and arterioles. The arterioles constitute the main determinant of total peripheral resistance to blood flow in the body and therefore figure prominently in the control of blood pressure. The architectural association between sympathetic nerves and the muscle in the heart and arteriolar walls has enticed hypertension researchers for many decades.
A false start with plasma norepinephrine measurement
I developed novel methods for measuring plasma levels of norepinephrine, which is the chemical messenger that the sympathetic nervous system uses in regulation of the circulation, and of adrenaline (epinephrine), which is the well-known and potent “fight-or-flight” hormone.1 Applying this technology to patients with high blood pressure led to several publications2–9 but actually shed more heat than light on the hypothesis of sympathetic hyperactivity as a cause of or contributor to hypertension. In the face of negative data, the theory was qualified—sympathetic hyperactivity might be apparent only in the young, or the thin, or the Caucasian, or the male—but not abandoned.
Insights from visualizing sympathetic nerves in the heart
Then I embarked on a project to visualize sympathetic nerves in the heart, by a new technology called positron emission tomographic (PET) scanning. With several colleagues—including Irwin J. Kopin, Graeme Eisenhofer, Peter Chang, David Hovevey-Zion, Ehud Grossman, and Courtney Holmes—to whom I will always be grateful, I developed a PET imaging agent called 6-[18F]fluorodopamine.10–13
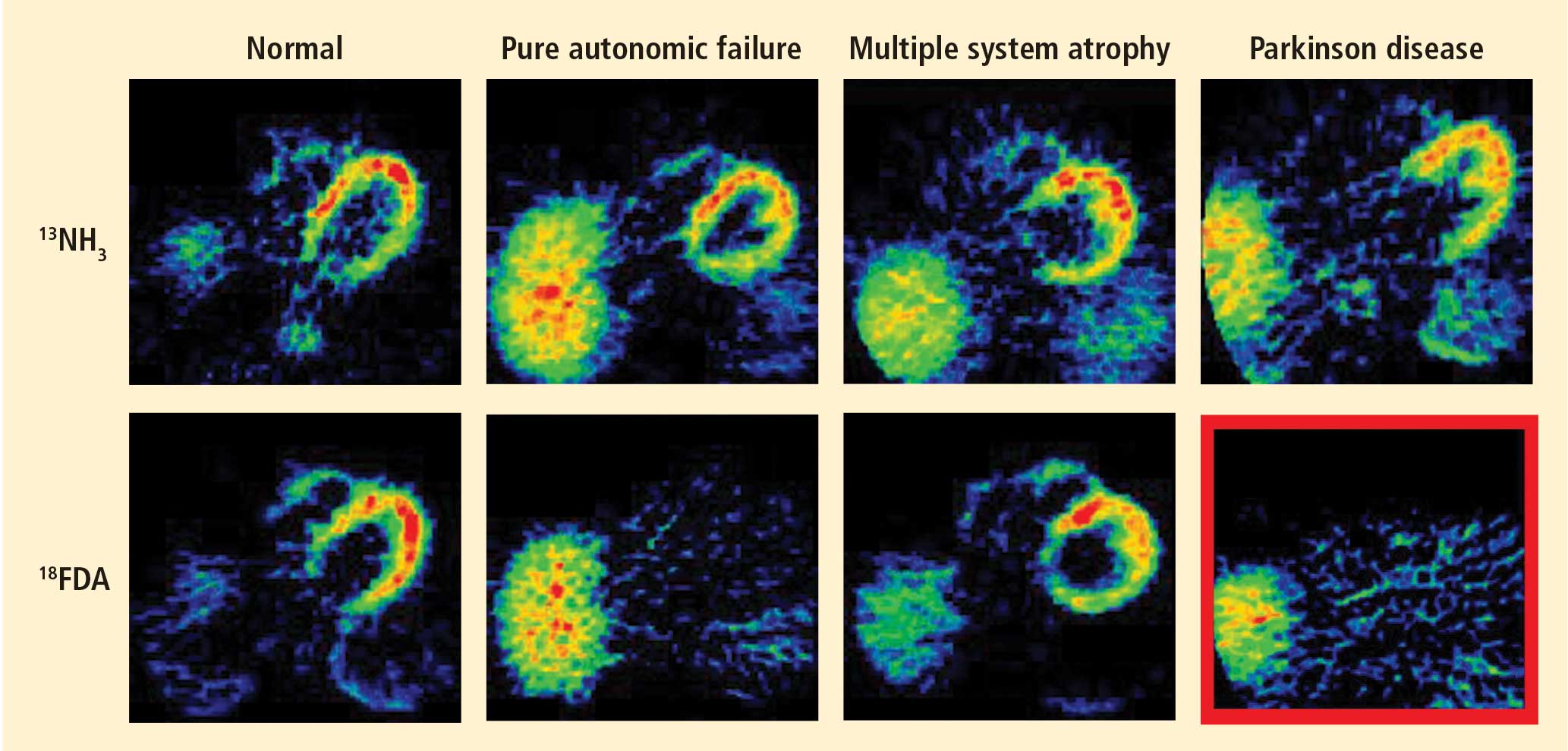
Normally, PET scans using 6-[18F]fluorodopamine look remarkably similar to scans using 13N-labeled ammonia, a perfusion imaging agent. The first patient I studied with this new technology was a patient with a rare disease called pure autonomic failure (PAF). In PAF, there was already good evidence for a loss of sympathetic nerves throughout the body. Myocardial perfusion in this patient was normal, but there was much less than normal 6-[18F]fluorodopamine-derived radioactivity in the heart muscle. In another uncommon disease, multiple system atrophy (MSA), the perfusion was also normal, and the cardiac sympathetic nerves seemed intact, in line with what was already known about this progressive neurodegenerative disease.
Then I tested a patient who had been thought to have MSA but actually had Parkinson disease (PD) with orthostatic hypotension (a fall in blood pressure each time the person stands up). PD with orthostatic hypotension can be very difficult to distinguish from the parkinsonian form of MSA. To my complete surprise, the patient with PD had a remarkable decrease in 6-[18F]fluorodopa mine-derived radioactivity in the heart muscle. There was normal blood flow to the heart muscle, so the 6-[18F]fluorodopamine was being delivered, but there was no evidence of sympathetic nerves in the heart. The scans resembled those in the PAF patient, not the MSA patient.
This finding did not arise from a prediction to test a hypothesis. It wasn’t long before I tested additional PD patients and found the same unexpected results.14,15 Because I was ignorant, I wasn’t biased. I felt I had put my finger on a piece of the truth, and I had to stop and think about the implications of this discovery. I never did come to test the hypotheses that I had sought out originally to test. Instead, I followed a totally new path, based on the discovery of cardiac sympathetic denervation in PD.
Beyond a brain disease: Seeing PD as a heart-brain disorder
More than 50 neuroimaging studies since our original report have agreed remarkably consistently on the association between PD and loss of sympathetic nerves in the heart; moreover, postmortem pathology studies have amply confirmed that a profound loss of cardiac sympathetic nerves is characteristic of PD.16 I have yet to come across a single patient with PD and orthostatic hypotension who has not had cardiac sympathetic denervation, and virtually all patients with PD who do not have orthostatic hypotension seem to have at least partial loss of cardiac sympathetic nerves.
Considering that the source of those nerves is the ganglia, which lie outside the central nervous system, PD must be more than a brain disease and more than a movement disorder. It must also be a disease of the sympathetic nerves in the heart, a form of a dysautonomia, and a heart-brain disorder.
The role of catecholamines: Another discovery born of unbiased ignorance
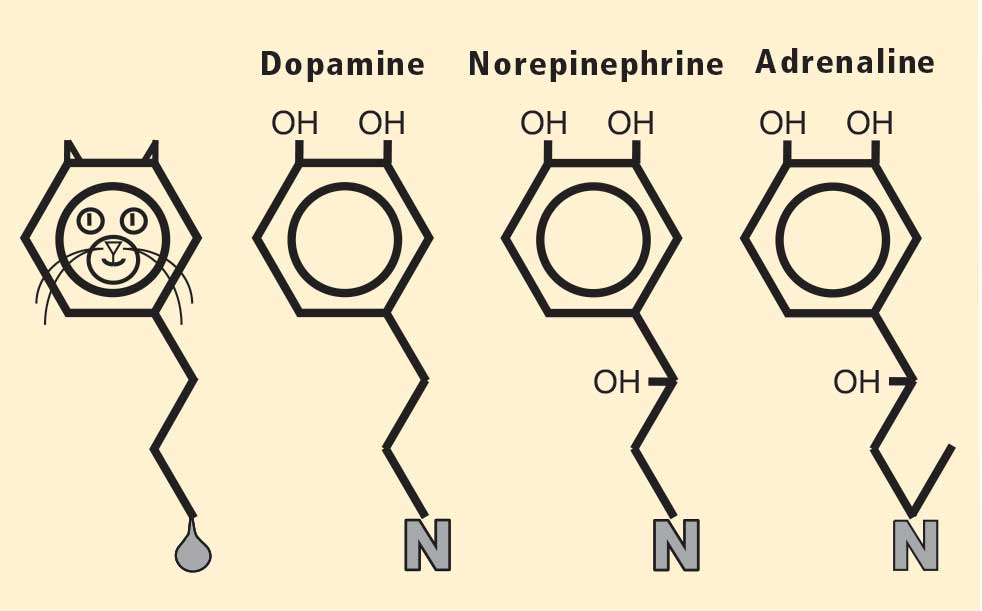
Almost a half century ago, Hornykiewicz and colleagues made the pivotal discovery that PD features loss of dopamine in the nigrostriatal system in the brain.18 Given the cardiac sympathetic denervation, PD might be a disease of catecholamine systems both inside and outside the central nervous system—dopamine in the nigrostriatal system, and norepinephrine in the sympathetic nerves of the heart.
Then what of the third catecholamine, adrenaline, in PD? Plasma levels of adrenaline and of its metabolite, metanephrine, are normal in PD, even in patients who have PD and orthostatic hypotension, which involves loss of norepinephrine-producing nerves not only in the heart but in other organs.19 What is different about the adrenaline-producing cells in the medulla (from the Latin for “marrow”) of the adrenal glands atop each kidney? Why aren’t these catecholamine-producing cells also lost in PD?
I have some ideas in mind but won’t go into them here. The point is that the discovery of normal adrenaline-producing cells in PD, despite loss of cells producing the other catecholamines, was not based on my testing a hypothesis. It was a discovery born of ignorance, and because ignorance isn’t biased, that discovery points to the truth. Whatever the eventual explanation for the specific pattern of catecholamine cell loss in PD, it cannot refute the discovery itself.
HOW DISCOVERIES ARISE: AN APPLIED EXERCISE FOR READERS
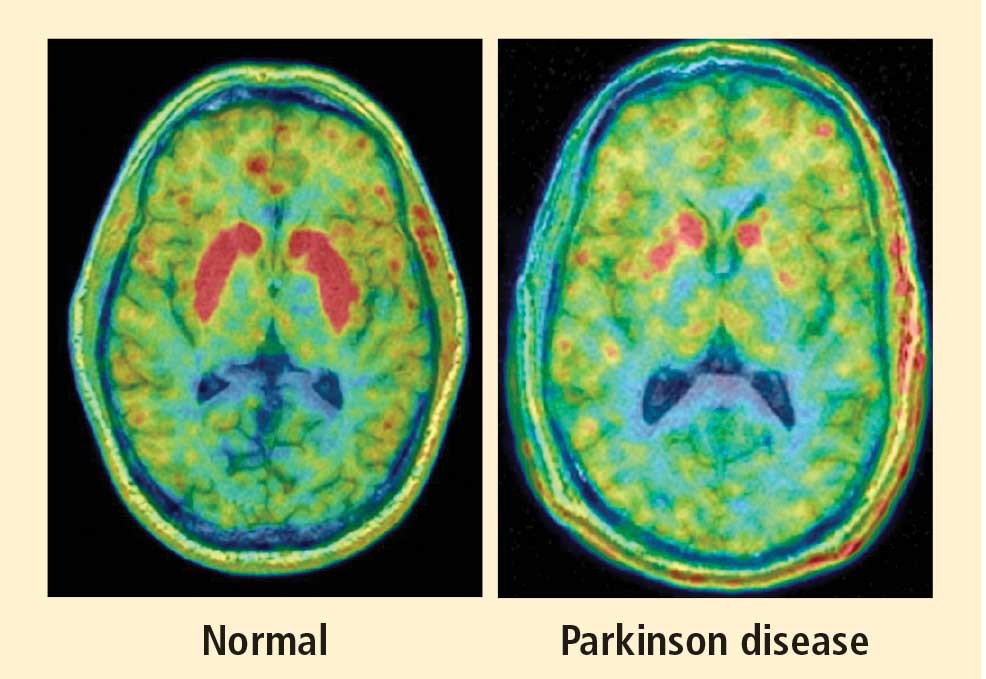
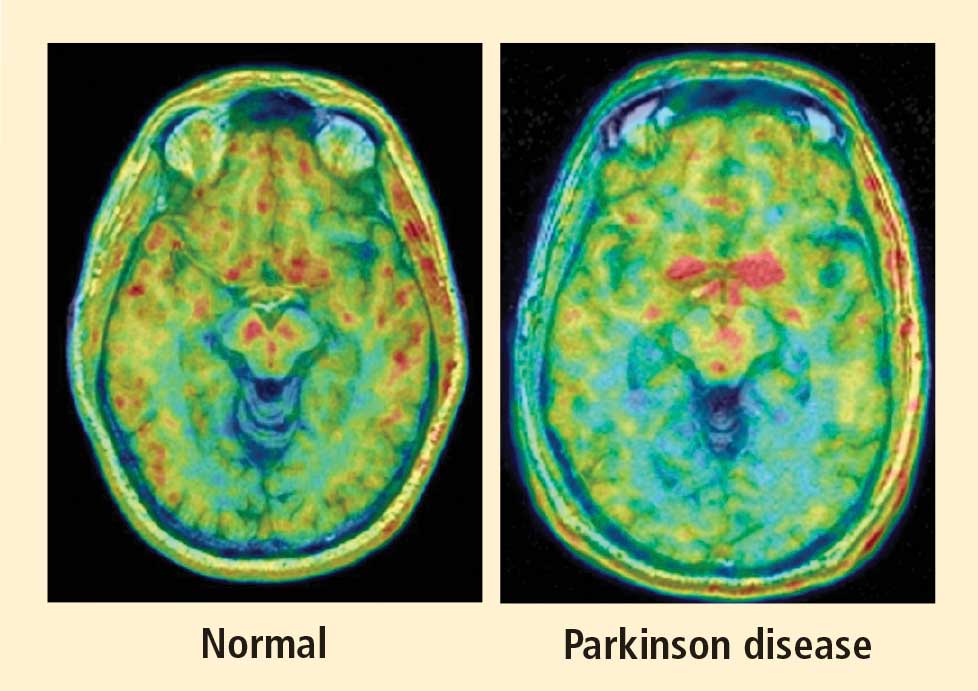
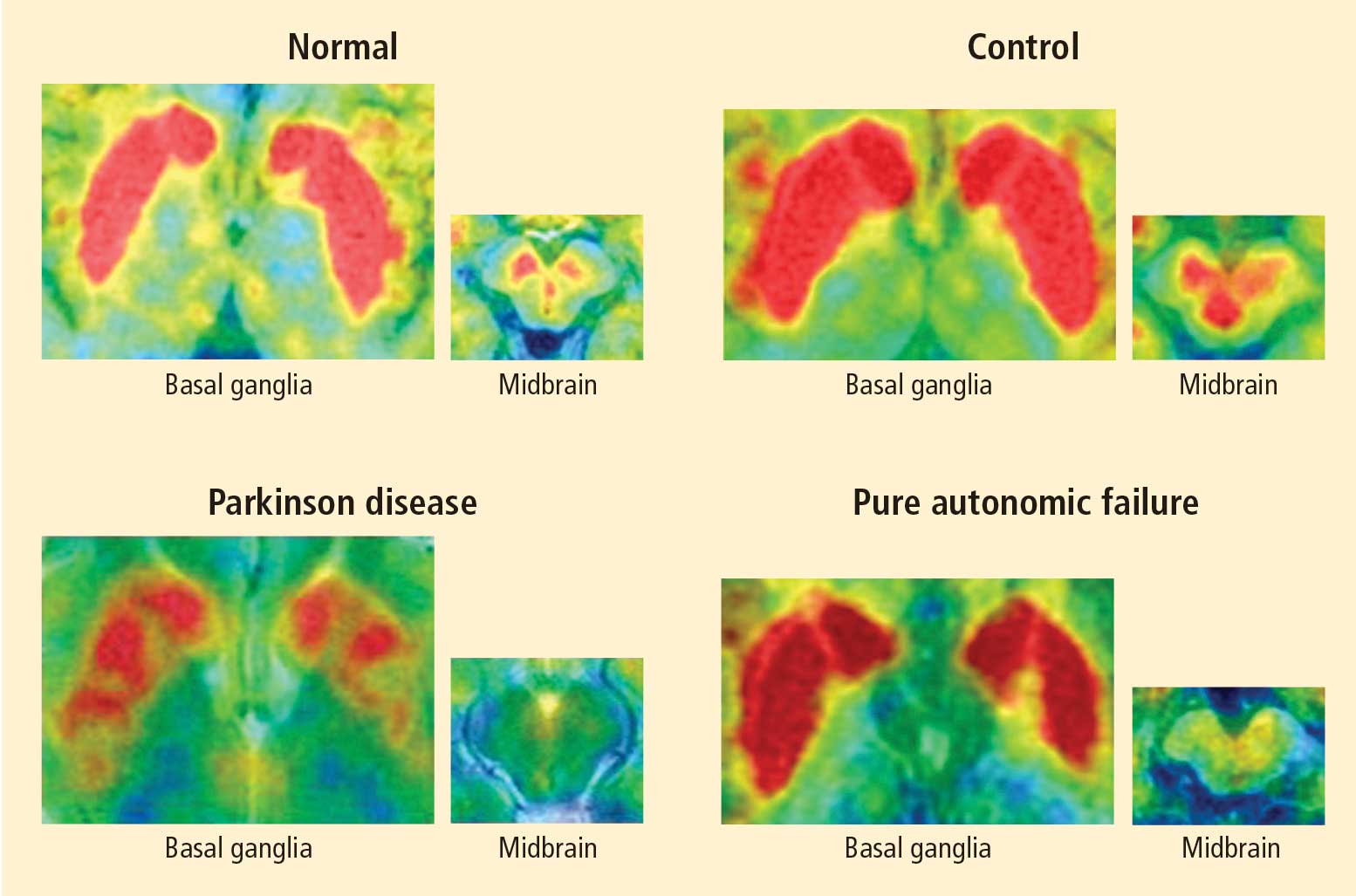
PAF is a rare disease, and I have only studied several cases with high-resolution PET scanning of the brain, but so far they have all had this unexpected, unpredicted finding of loss of dopaminergic neurons in the substantia nigra.20
What does this pattern mean? If PAF patients have just as much loss of nigral neurons as PD patients do, and if PAF patients do not have parkinsonism, then the movement disorder in PD cannot result from loss of the dopamine neurons in the substantia nigra per se. Instead, the movement disorder in PD seems to come from loss of the dopaminergic terminals in the striatum.
How can PAF patients have normal dopamine terminals in the putamen when the number of dopaminergic cell bodies is severely reduced? Somehow, PAF patients must be able to sprout new terminals, even as they lose the cell bodies. Maybe if we knew how PAF patients do this, we would have a way to treat or even prevent PD.
How do PAF patients maintain normal dopamine terminals as the cell bodies die off? No one knows. Until now, no one thought of asking such a question. No one hypothesized that this discovery would be made, but it was. And because ignorance isn’t biased, we have put our finger on the truth. By keeping in mind what isn’t known, we could see what wasn’t there. Now we can begin to think of what to look for next.
SUMMARY AND CONCLUSIONS
Because ignorance isn’t biased, if you have the tools to make relevant measurements, if you have sufficient mastery of the subject to know what isn’t known, and if you have access to patients with rare but informative disorders, you can make important discoveries based on inductions from observations.
The discoveries that cardiac sympathetic denervation characterizes PD and that parkinsonism does not result from loss of dopamine neurons per se depended crucially on studying patients with a rare disease, PAF. In 1657, William Harvey—the same William Harvey who first described the circulation of the blood and who first pointed out the effects of emotions on the heart—wrote eloquently about the extraordinary power of studying patients with rare diseases:
I hope I have convinced you of the importance of seeing what isn’t there. My thanks go out again to the Earl and Doris Bakken Heart-Brain Institute for this prestigious award, to my family, to my colleagues and friends, and to my patients. As I have written in Adrenaline and the Inner World: An Introduction to Scientific Integrative Medicine,17 patients serve as a unique scientific resource. They report what is wrong; they tell us the truth. We have to make sense of what they teach.
- Goldstein DS, Feuerstein G, Izzo JL, Kopin IJ, Keiser HR. Validity and reliability of liquid chromatography with electrochemical detection for measuring plasma levels of norepinephrine and epinephrine in man. Life Sci 1981; 28:467–475.
- Goldstein DS. Plasma norepinephrine during stress in essential hypertension. Hypertension 1981; 3:551–556.
- Goldstein DS. Plasma norepinephrine in essential hypertension: a study of the studies. Hypertension 1981; 3:48–52.
- Goldstein D, Horwitz D, Keiser HR, Polinsky RJ, Kopin IJ. Plasma l-[3H]norepinephrine, d-[14C]norepinephrine, and d,l-[3H] isoproterenol kinetics in essential hypertension. J Clin Invest 1983; 72:1748–1758.
- Goldstein DS. Arterial baroreflex sensitivity, plasma catecholamines, and pressor responsiveness in essential hypertension. Circulation 1983; 68:234–240.
- Goldstein DS. Plasma catecholamines and essential hypertension: an analytical review. Hypertension 1983; 5:86–99.
- Goldstein DS, Lake CR, Chernow B, et al. Age-dependence of hypertensive-normotensive differences in plasma norepinephrine. Hypertension 1983; 5:100–104.
- Goldstein DS, McCarty R, Polinsky RJ, Kopin IJ. Relationship between plasma norepinephrine and sympathetic neural activity. Hypertension 1983; 5:552–559.
- Goldstein DS, Lake CR. Plasma norepinephrine and epinephrine levels in essential hypertension. Fed Proc 1984; 43:57–61.
- Eisenhofer G, Hovevey-Sion D, Kopin IJ, et al. Neuronal uptake and metabolism of 2- and 6-fluorodopamine: false neurotransmitters for positron emission tomographic imaging of sympathetically innervated tissues. J Pharmacol Exp Ther 1989; 248:419–427.
- Chang PC, Szemeredi K, Grossman E, Kopin IJ, Goldstein DS. Fate of tritiated 6-fluorodopamine in rats: a false neurotransmitter for positron emission tomographic imaging of sympathetic innervation and function. J Pharmacol Exp Ther 1990; 255:809–817.
- Goldstein DS, Holmes C. Metabolic fate of the sympathoneural imaging agent 6-[18F]fluorodopamine in humans. Clin Exp Hypertens 1997; 19:155–161.
- Goldstein DS, Eisenhofer G, Dunn BB, et al. Positron emission tomographic imaging of cardiac sympathetic innervation using 6-[18F] fluorodopamine: initial findings in humans. J Am Coll Cardiol 1993; 22:1961–1971.
- Goldstein DS, Holmes C, Cannon RO, Eisenhofer G, Kopin IJ. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med 1997; 336:696–702.
- Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO. Cardiac sympathetic denervation in Parkinson disease. Ann Intern Med 2000; 133:338–347.
- Amino T, Orimo S, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Pathol 2005; 15:29–34.
- Goldstein DS. Adrenaline and the Inner World: An Introduction to Scientific Integrative Medicine. Baltimore, MD: Johns Hopkins University Press; 2006.
- Ehringer H, Hornykiewicz O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system [in German]. Wien Klin Wochenschr 1960; 38:1236–1239.
- Goldstein DS, Holmes C, Sharabi Y, Brentzel S, Eisenhofer G. Plasma levels of catechols and metanephrines in neurogenic orthostatic hypotension. Neurology 2003; 60:1327–1332.
- Goldstein DS, Holmes C, Sato T, et al. Central dopamine deficiency in pure autonomic failure. Clin Auton Res 2008; 18:58–65.
- Hervey Wyatt RB William Harvey 1578 to 1657 Whitefish, MT Kessinger Publishing 2005:161–162.
This is a momentous occasion for me, for the extraordinary people in the Clinical Neurocardiology Section at the National Institutes of Health (NIH), and for my family—my wife Minka and son Joey drove all the way from Maryland late last night and early this morning to be here. I thank them publicly here.
THE ‘SPARKLE OF INSIGHT’ FROM ENLIGHTENED INDUCTION
In these brief comments, as I look back on the road I have taken over the past 40 years carrying out patient-oriented research in heart-brain medicine, I would like to convey a viewpoint instead of dwelling on the presentation of research data.
The idea I wish to convey is that ignorance isn’t biased. If you have a hypothesis you want to test, you are inherently biased to find something positive—and, if you are in academic medicine, publishable—in the data you obtain. But if you have the technical capability to measure something no one else can measure, and you have sufficient mastery of the topic to know what is not yet known, then if you make an observation that you did not predict and if you recognize its significance, you have made a discovery. You have revealed a bit of the truth. You experience the highest joy and thrill a scientist can feel—a “sparkle of insight.” When this happens, if you have sense, you stop what you have been doing to pursue that discovery.
Hardly anyone has received a Nobel Prize for testing a theory, but many Nobel Prizes have been awarded for technological advances and for discoveries based on those advances. In my view, discoverers use an enlightened inductive approach at least as much as deduction. They develop new technology that enables key novel measurements, and they keep in mind gaps in knowledge, so that they are ready to appreciate the significance of their observations.
A PERSONAL EXAMPLE
‘You have to measure something’
Let me share an example of this process by relating a sparkle of insight I had several years ago. When I began working at the NIH, I met with the chief of the Hypertension-Endocrine Branch of the National Heart, Lung, and Blood Institute about the research program I would pursue. After listening patiently to me for many minutes as I spouted about how I was going to test hypotheses derived from the concepts that people with hypertension are “hyper-tense,” and that stress causes heart disease, the chief responded, “Well, these ideas are all well and good. But what are you going to measure? You can measure whatever you want, but you have to measure something.”
Measure something. I wanted to see if there was hyperactivity of the sympathetic nervous system or excessive sympathetic innervation in hypertension, and I started working on ways to measure sympathetic activity.
The sympathetic nervous system at a glance
First I should introduce you to the sympathetic nervous system, which is one of the main effectors by which the brain regulates the heart and blood vessels. It is a key link between the brain and heart. The sympathetic nerves to the heart and other organs do not come directly from the brain but from ganglia, which are clumps of nerve cell bodies strung like pearls on a necklace on each side of the spinal column. This origin outside the central nervous system will be an important fact to keep in mind.
In the heart, the sympathetic nerves travel with the coronary arteries and then dive into the heart muscle from the outside. Sympathetic nerves also enmesh the walls of arteries and arterioles. The arterioles constitute the main determinant of total peripheral resistance to blood flow in the body and therefore figure prominently in the control of blood pressure. The architectural association between sympathetic nerves and the muscle in the heart and arteriolar walls has enticed hypertension researchers for many decades.
A false start with plasma norepinephrine measurement
I developed novel methods for measuring plasma levels of norepinephrine, which is the chemical messenger that the sympathetic nervous system uses in regulation of the circulation, and of adrenaline (epinephrine), which is the well-known and potent “fight-or-flight” hormone.1 Applying this technology to patients with high blood pressure led to several publications2–9 but actually shed more heat than light on the hypothesis of sympathetic hyperactivity as a cause of or contributor to hypertension. In the face of negative data, the theory was qualified—sympathetic hyperactivity might be apparent only in the young, or the thin, or the Caucasian, or the male—but not abandoned.
Insights from visualizing sympathetic nerves in the heart
Then I embarked on a project to visualize sympathetic nerves in the heart, by a new technology called positron emission tomographic (PET) scanning. With several colleagues—including Irwin J. Kopin, Graeme Eisenhofer, Peter Chang, David Hovevey-Zion, Ehud Grossman, and Courtney Holmes—to whom I will always be grateful, I developed a PET imaging agent called 6-[18F]fluorodopamine.10–13
Normally, PET scans using 6-[18F]fluorodopamine look remarkably similar to scans using 13N-labeled ammonia, a perfusion imaging agent. The first patient I studied with this new technology was a patient with a rare disease called pure autonomic failure (PAF). In PAF, there was already good evidence for a loss of sympathetic nerves throughout the body. Myocardial perfusion in this patient was normal, but there was much less than normal 6-[18F]fluorodopamine-derived radioactivity in the heart muscle. In another uncommon disease, multiple system atrophy (MSA), the perfusion was also normal, and the cardiac sympathetic nerves seemed intact, in line with what was already known about this progressive neurodegenerative disease.
Then I tested a patient who had been thought to have MSA but actually had Parkinson disease (PD) with orthostatic hypotension (a fall in blood pressure each time the person stands up). PD with orthostatic hypotension can be very difficult to distinguish from the parkinsonian form of MSA. To my complete surprise, the patient with PD had a remarkable decrease in 6-[18F]fluorodopa mine-derived radioactivity in the heart muscle. There was normal blood flow to the heart muscle, so the 6-[18F]fluorodopamine was being delivered, but there was no evidence of sympathetic nerves in the heart. The scans resembled those in the PAF patient, not the MSA patient.
This finding did not arise from a prediction to test a hypothesis. It wasn’t long before I tested additional PD patients and found the same unexpected results.14,15 Because I was ignorant, I wasn’t biased. I felt I had put my finger on a piece of the truth, and I had to stop and think about the implications of this discovery. I never did come to test the hypotheses that I had sought out originally to test. Instead, I followed a totally new path, based on the discovery of cardiac sympathetic denervation in PD.
Beyond a brain disease: Seeing PD as a heart-brain disorder
More than 50 neuroimaging studies since our original report have agreed remarkably consistently on the association between PD and loss of sympathetic nerves in the heart; moreover, postmortem pathology studies have amply confirmed that a profound loss of cardiac sympathetic nerves is characteristic of PD.16 I have yet to come across a single patient with PD and orthostatic hypotension who has not had cardiac sympathetic denervation, and virtually all patients with PD who do not have orthostatic hypotension seem to have at least partial loss of cardiac sympathetic nerves.
Considering that the source of those nerves is the ganglia, which lie outside the central nervous system, PD must be more than a brain disease and more than a movement disorder. It must also be a disease of the sympathetic nerves in the heart, a form of a dysautonomia, and a heart-brain disorder.
The role of catecholamines: Another discovery born of unbiased ignorance
Almost a half century ago, Hornykiewicz and colleagues made the pivotal discovery that PD features loss of dopamine in the nigrostriatal system in the brain.18 Given the cardiac sympathetic denervation, PD might be a disease of catecholamine systems both inside and outside the central nervous system—dopamine in the nigrostriatal system, and norepinephrine in the sympathetic nerves of the heart.
Then what of the third catecholamine, adrenaline, in PD? Plasma levels of adrenaline and of its metabolite, metanephrine, are normal in PD, even in patients who have PD and orthostatic hypotension, which involves loss of norepinephrine-producing nerves not only in the heart but in other organs.19 What is different about the adrenaline-producing cells in the medulla (from the Latin for “marrow”) of the adrenal glands atop each kidney? Why aren’t these catecholamine-producing cells also lost in PD?
I have some ideas in mind but won’t go into them here. The point is that the discovery of normal adrenaline-producing cells in PD, despite loss of cells producing the other catecholamines, was not based on my testing a hypothesis. It was a discovery born of ignorance, and because ignorance isn’t biased, that discovery points to the truth. Whatever the eventual explanation for the specific pattern of catecholamine cell loss in PD, it cannot refute the discovery itself.
HOW DISCOVERIES ARISE: AN APPLIED EXERCISE FOR READERS
PAF is a rare disease, and I have only studied several cases with high-resolution PET scanning of the brain, but so far they have all had this unexpected, unpredicted finding of loss of dopaminergic neurons in the substantia nigra.20
What does this pattern mean? If PAF patients have just as much loss of nigral neurons as PD patients do, and if PAF patients do not have parkinsonism, then the movement disorder in PD cannot result from loss of the dopamine neurons in the substantia nigra per se. Instead, the movement disorder in PD seems to come from loss of the dopaminergic terminals in the striatum.
How can PAF patients have normal dopamine terminals in the putamen when the number of dopaminergic cell bodies is severely reduced? Somehow, PAF patients must be able to sprout new terminals, even as they lose the cell bodies. Maybe if we knew how PAF patients do this, we would have a way to treat or even prevent PD.
How do PAF patients maintain normal dopamine terminals as the cell bodies die off? No one knows. Until now, no one thought of asking such a question. No one hypothesized that this discovery would be made, but it was. And because ignorance isn’t biased, we have put our finger on the truth. By keeping in mind what isn’t known, we could see what wasn’t there. Now we can begin to think of what to look for next.
SUMMARY AND CONCLUSIONS
Because ignorance isn’t biased, if you have the tools to make relevant measurements, if you have sufficient mastery of the subject to know what isn’t known, and if you have access to patients with rare but informative disorders, you can make important discoveries based on inductions from observations.
The discoveries that cardiac sympathetic denervation characterizes PD and that parkinsonism does not result from loss of dopamine neurons per se depended crucially on studying patients with a rare disease, PAF. In 1657, William Harvey—the same William Harvey who first described the circulation of the blood and who first pointed out the effects of emotions on the heart—wrote eloquently about the extraordinary power of studying patients with rare diseases:
I hope I have convinced you of the importance of seeing what isn’t there. My thanks go out again to the Earl and Doris Bakken Heart-Brain Institute for this prestigious award, to my family, to my colleagues and friends, and to my patients. As I have written in Adrenaline and the Inner World: An Introduction to Scientific Integrative Medicine,17 patients serve as a unique scientific resource. They report what is wrong; they tell us the truth. We have to make sense of what they teach.
This is a momentous occasion for me, for the extraordinary people in the Clinical Neurocardiology Section at the National Institutes of Health (NIH), and for my family—my wife Minka and son Joey drove all the way from Maryland late last night and early this morning to be here. I thank them publicly here.
THE ‘SPARKLE OF INSIGHT’ FROM ENLIGHTENED INDUCTION
In these brief comments, as I look back on the road I have taken over the past 40 years carrying out patient-oriented research in heart-brain medicine, I would like to convey a viewpoint instead of dwelling on the presentation of research data.
The idea I wish to convey is that ignorance isn’t biased. If you have a hypothesis you want to test, you are inherently biased to find something positive—and, if you are in academic medicine, publishable—in the data you obtain. But if you have the technical capability to measure something no one else can measure, and you have sufficient mastery of the topic to know what is not yet known, then if you make an observation that you did not predict and if you recognize its significance, you have made a discovery. You have revealed a bit of the truth. You experience the highest joy and thrill a scientist can feel—a “sparkle of insight.” When this happens, if you have sense, you stop what you have been doing to pursue that discovery.
Hardly anyone has received a Nobel Prize for testing a theory, but many Nobel Prizes have been awarded for technological advances and for discoveries based on those advances. In my view, discoverers use an enlightened inductive approach at least as much as deduction. They develop new technology that enables key novel measurements, and they keep in mind gaps in knowledge, so that they are ready to appreciate the significance of their observations.
A PERSONAL EXAMPLE
‘You have to measure something’
Let me share an example of this process by relating a sparkle of insight I had several years ago. When I began working at the NIH, I met with the chief of the Hypertension-Endocrine Branch of the National Heart, Lung, and Blood Institute about the research program I would pursue. After listening patiently to me for many minutes as I spouted about how I was going to test hypotheses derived from the concepts that people with hypertension are “hyper-tense,” and that stress causes heart disease, the chief responded, “Well, these ideas are all well and good. But what are you going to measure? You can measure whatever you want, but you have to measure something.”
Measure something. I wanted to see if there was hyperactivity of the sympathetic nervous system or excessive sympathetic innervation in hypertension, and I started working on ways to measure sympathetic activity.
The sympathetic nervous system at a glance
First I should introduce you to the sympathetic nervous system, which is one of the main effectors by which the brain regulates the heart and blood vessels. It is a key link between the brain and heart. The sympathetic nerves to the heart and other organs do not come directly from the brain but from ganglia, which are clumps of nerve cell bodies strung like pearls on a necklace on each side of the spinal column. This origin outside the central nervous system will be an important fact to keep in mind.
In the heart, the sympathetic nerves travel with the coronary arteries and then dive into the heart muscle from the outside. Sympathetic nerves also enmesh the walls of arteries and arterioles. The arterioles constitute the main determinant of total peripheral resistance to blood flow in the body and therefore figure prominently in the control of blood pressure. The architectural association between sympathetic nerves and the muscle in the heart and arteriolar walls has enticed hypertension researchers for many decades.
A false start with plasma norepinephrine measurement
I developed novel methods for measuring plasma levels of norepinephrine, which is the chemical messenger that the sympathetic nervous system uses in regulation of the circulation, and of adrenaline (epinephrine), which is the well-known and potent “fight-or-flight” hormone.1 Applying this technology to patients with high blood pressure led to several publications2–9 but actually shed more heat than light on the hypothesis of sympathetic hyperactivity as a cause of or contributor to hypertension. In the face of negative data, the theory was qualified—sympathetic hyperactivity might be apparent only in the young, or the thin, or the Caucasian, or the male—but not abandoned.
Insights from visualizing sympathetic nerves in the heart
Then I embarked on a project to visualize sympathetic nerves in the heart, by a new technology called positron emission tomographic (PET) scanning. With several colleagues—including Irwin J. Kopin, Graeme Eisenhofer, Peter Chang, David Hovevey-Zion, Ehud Grossman, and Courtney Holmes—to whom I will always be grateful, I developed a PET imaging agent called 6-[18F]fluorodopamine.10–13
Normally, PET scans using 6-[18F]fluorodopamine look remarkably similar to scans using 13N-labeled ammonia, a perfusion imaging agent. The first patient I studied with this new technology was a patient with a rare disease called pure autonomic failure (PAF). In PAF, there was already good evidence for a loss of sympathetic nerves throughout the body. Myocardial perfusion in this patient was normal, but there was much less than normal 6-[18F]fluorodopamine-derived radioactivity in the heart muscle. In another uncommon disease, multiple system atrophy (MSA), the perfusion was also normal, and the cardiac sympathetic nerves seemed intact, in line with what was already known about this progressive neurodegenerative disease.
Then I tested a patient who had been thought to have MSA but actually had Parkinson disease (PD) with orthostatic hypotension (a fall in blood pressure each time the person stands up). PD with orthostatic hypotension can be very difficult to distinguish from the parkinsonian form of MSA. To my complete surprise, the patient with PD had a remarkable decrease in 6-[18F]fluorodopa mine-derived radioactivity in the heart muscle. There was normal blood flow to the heart muscle, so the 6-[18F]fluorodopamine was being delivered, but there was no evidence of sympathetic nerves in the heart. The scans resembled those in the PAF patient, not the MSA patient.
This finding did not arise from a prediction to test a hypothesis. It wasn’t long before I tested additional PD patients and found the same unexpected results.14,15 Because I was ignorant, I wasn’t biased. I felt I had put my finger on a piece of the truth, and I had to stop and think about the implications of this discovery. I never did come to test the hypotheses that I had sought out originally to test. Instead, I followed a totally new path, based on the discovery of cardiac sympathetic denervation in PD.
Beyond a brain disease: Seeing PD as a heart-brain disorder
More than 50 neuroimaging studies since our original report have agreed remarkably consistently on the association between PD and loss of sympathetic nerves in the heart; moreover, postmortem pathology studies have amply confirmed that a profound loss of cardiac sympathetic nerves is characteristic of PD.16 I have yet to come across a single patient with PD and orthostatic hypotension who has not had cardiac sympathetic denervation, and virtually all patients with PD who do not have orthostatic hypotension seem to have at least partial loss of cardiac sympathetic nerves.
Considering that the source of those nerves is the ganglia, which lie outside the central nervous system, PD must be more than a brain disease and more than a movement disorder. It must also be a disease of the sympathetic nerves in the heart, a form of a dysautonomia, and a heart-brain disorder.
The role of catecholamines: Another discovery born of unbiased ignorance
Almost a half century ago, Hornykiewicz and colleagues made the pivotal discovery that PD features loss of dopamine in the nigrostriatal system in the brain.18 Given the cardiac sympathetic denervation, PD might be a disease of catecholamine systems both inside and outside the central nervous system—dopamine in the nigrostriatal system, and norepinephrine in the sympathetic nerves of the heart.
Then what of the third catecholamine, adrenaline, in PD? Plasma levels of adrenaline and of its metabolite, metanephrine, are normal in PD, even in patients who have PD and orthostatic hypotension, which involves loss of norepinephrine-producing nerves not only in the heart but in other organs.19 What is different about the adrenaline-producing cells in the medulla (from the Latin for “marrow”) of the adrenal glands atop each kidney? Why aren’t these catecholamine-producing cells also lost in PD?
I have some ideas in mind but won’t go into them here. The point is that the discovery of normal adrenaline-producing cells in PD, despite loss of cells producing the other catecholamines, was not based on my testing a hypothesis. It was a discovery born of ignorance, and because ignorance isn’t biased, that discovery points to the truth. Whatever the eventual explanation for the specific pattern of catecholamine cell loss in PD, it cannot refute the discovery itself.
HOW DISCOVERIES ARISE: AN APPLIED EXERCISE FOR READERS
PAF is a rare disease, and I have only studied several cases with high-resolution PET scanning of the brain, but so far they have all had this unexpected, unpredicted finding of loss of dopaminergic neurons in the substantia nigra.20
What does this pattern mean? If PAF patients have just as much loss of nigral neurons as PD patients do, and if PAF patients do not have parkinsonism, then the movement disorder in PD cannot result from loss of the dopamine neurons in the substantia nigra per se. Instead, the movement disorder in PD seems to come from loss of the dopaminergic terminals in the striatum.
How can PAF patients have normal dopamine terminals in the putamen when the number of dopaminergic cell bodies is severely reduced? Somehow, PAF patients must be able to sprout new terminals, even as they lose the cell bodies. Maybe if we knew how PAF patients do this, we would have a way to treat or even prevent PD.
How do PAF patients maintain normal dopamine terminals as the cell bodies die off? No one knows. Until now, no one thought of asking such a question. No one hypothesized that this discovery would be made, but it was. And because ignorance isn’t biased, we have put our finger on the truth. By keeping in mind what isn’t known, we could see what wasn’t there. Now we can begin to think of what to look for next.
SUMMARY AND CONCLUSIONS
Because ignorance isn’t biased, if you have the tools to make relevant measurements, if you have sufficient mastery of the subject to know what isn’t known, and if you have access to patients with rare but informative disorders, you can make important discoveries based on inductions from observations.
The discoveries that cardiac sympathetic denervation characterizes PD and that parkinsonism does not result from loss of dopamine neurons per se depended crucially on studying patients with a rare disease, PAF. In 1657, William Harvey—the same William Harvey who first described the circulation of the blood and who first pointed out the effects of emotions on the heart—wrote eloquently about the extraordinary power of studying patients with rare diseases:
I hope I have convinced you of the importance of seeing what isn’t there. My thanks go out again to the Earl and Doris Bakken Heart-Brain Institute for this prestigious award, to my family, to my colleagues and friends, and to my patients. As I have written in Adrenaline and the Inner World: An Introduction to Scientific Integrative Medicine,17 patients serve as a unique scientific resource. They report what is wrong; they tell us the truth. We have to make sense of what they teach.
- Goldstein DS, Feuerstein G, Izzo JL, Kopin IJ, Keiser HR. Validity and reliability of liquid chromatography with electrochemical detection for measuring plasma levels of norepinephrine and epinephrine in man. Life Sci 1981; 28:467–475.
- Goldstein DS. Plasma norepinephrine during stress in essential hypertension. Hypertension 1981; 3:551–556.
- Goldstein DS. Plasma norepinephrine in essential hypertension: a study of the studies. Hypertension 1981; 3:48–52.
- Goldstein D, Horwitz D, Keiser HR, Polinsky RJ, Kopin IJ. Plasma l-[3H]norepinephrine, d-[14C]norepinephrine, and d,l-[3H] isoproterenol kinetics in essential hypertension. J Clin Invest 1983; 72:1748–1758.
- Goldstein DS. Arterial baroreflex sensitivity, plasma catecholamines, and pressor responsiveness in essential hypertension. Circulation 1983; 68:234–240.
- Goldstein DS. Plasma catecholamines and essential hypertension: an analytical review. Hypertension 1983; 5:86–99.
- Goldstein DS, Lake CR, Chernow B, et al. Age-dependence of hypertensive-normotensive differences in plasma norepinephrine. Hypertension 1983; 5:100–104.
- Goldstein DS, McCarty R, Polinsky RJ, Kopin IJ. Relationship between plasma norepinephrine and sympathetic neural activity. Hypertension 1983; 5:552–559.
- Goldstein DS, Lake CR. Plasma norepinephrine and epinephrine levels in essential hypertension. Fed Proc 1984; 43:57–61.
- Eisenhofer G, Hovevey-Sion D, Kopin IJ, et al. Neuronal uptake and metabolism of 2- and 6-fluorodopamine: false neurotransmitters for positron emission tomographic imaging of sympathetically innervated tissues. J Pharmacol Exp Ther 1989; 248:419–427.
- Chang PC, Szemeredi K, Grossman E, Kopin IJ, Goldstein DS. Fate of tritiated 6-fluorodopamine in rats: a false neurotransmitter for positron emission tomographic imaging of sympathetic innervation and function. J Pharmacol Exp Ther 1990; 255:809–817.
- Goldstein DS, Holmes C. Metabolic fate of the sympathoneural imaging agent 6-[18F]fluorodopamine in humans. Clin Exp Hypertens 1997; 19:155–161.
- Goldstein DS, Eisenhofer G, Dunn BB, et al. Positron emission tomographic imaging of cardiac sympathetic innervation using 6-[18F] fluorodopamine: initial findings in humans. J Am Coll Cardiol 1993; 22:1961–1971.
- Goldstein DS, Holmes C, Cannon RO, Eisenhofer G, Kopin IJ. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med 1997; 336:696–702.
- Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO. Cardiac sympathetic denervation in Parkinson disease. Ann Intern Med 2000; 133:338–347.
- Amino T, Orimo S, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Pathol 2005; 15:29–34.
- Goldstein DS. Adrenaline and the Inner World: An Introduction to Scientific Integrative Medicine. Baltimore, MD: Johns Hopkins University Press; 2006.
- Ehringer H, Hornykiewicz O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system [in German]. Wien Klin Wochenschr 1960; 38:1236–1239.
- Goldstein DS, Holmes C, Sharabi Y, Brentzel S, Eisenhofer G. Plasma levels of catechols and metanephrines in neurogenic orthostatic hypotension. Neurology 2003; 60:1327–1332.
- Goldstein DS, Holmes C, Sato T, et al. Central dopamine deficiency in pure autonomic failure. Clin Auton Res 2008; 18:58–65.
- Hervey Wyatt RB William Harvey 1578 to 1657 Whitefish, MT Kessinger Publishing 2005:161–162.
- Goldstein DS, Feuerstein G, Izzo JL, Kopin IJ, Keiser HR. Validity and reliability of liquid chromatography with electrochemical detection for measuring plasma levels of norepinephrine and epinephrine in man. Life Sci 1981; 28:467–475.
- Goldstein DS. Plasma norepinephrine during stress in essential hypertension. Hypertension 1981; 3:551–556.
- Goldstein DS. Plasma norepinephrine in essential hypertension: a study of the studies. Hypertension 1981; 3:48–52.
- Goldstein D, Horwitz D, Keiser HR, Polinsky RJ, Kopin IJ. Plasma l-[3H]norepinephrine, d-[14C]norepinephrine, and d,l-[3H] isoproterenol kinetics in essential hypertension. J Clin Invest 1983; 72:1748–1758.
- Goldstein DS. Arterial baroreflex sensitivity, plasma catecholamines, and pressor responsiveness in essential hypertension. Circulation 1983; 68:234–240.
- Goldstein DS. Plasma catecholamines and essential hypertension: an analytical review. Hypertension 1983; 5:86–99.
- Goldstein DS, Lake CR, Chernow B, et al. Age-dependence of hypertensive-normotensive differences in plasma norepinephrine. Hypertension 1983; 5:100–104.
- Goldstein DS, McCarty R, Polinsky RJ, Kopin IJ. Relationship between plasma norepinephrine and sympathetic neural activity. Hypertension 1983; 5:552–559.
- Goldstein DS, Lake CR. Plasma norepinephrine and epinephrine levels in essential hypertension. Fed Proc 1984; 43:57–61.
- Eisenhofer G, Hovevey-Sion D, Kopin IJ, et al. Neuronal uptake and metabolism of 2- and 6-fluorodopamine: false neurotransmitters for positron emission tomographic imaging of sympathetically innervated tissues. J Pharmacol Exp Ther 1989; 248:419–427.
- Chang PC, Szemeredi K, Grossman E, Kopin IJ, Goldstein DS. Fate of tritiated 6-fluorodopamine in rats: a false neurotransmitter for positron emission tomographic imaging of sympathetic innervation and function. J Pharmacol Exp Ther 1990; 255:809–817.
- Goldstein DS, Holmes C. Metabolic fate of the sympathoneural imaging agent 6-[18F]fluorodopamine in humans. Clin Exp Hypertens 1997; 19:155–161.
- Goldstein DS, Eisenhofer G, Dunn BB, et al. Positron emission tomographic imaging of cardiac sympathetic innervation using 6-[18F] fluorodopamine: initial findings in humans. J Am Coll Cardiol 1993; 22:1961–1971.
- Goldstein DS, Holmes C, Cannon RO, Eisenhofer G, Kopin IJ. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med 1997; 336:696–702.
- Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO. Cardiac sympathetic denervation in Parkinson disease. Ann Intern Med 2000; 133:338–347.
- Amino T, Orimo S, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Pathol 2005; 15:29–34.
- Goldstein DS. Adrenaline and the Inner World: An Introduction to Scientific Integrative Medicine. Baltimore, MD: Johns Hopkins University Press; 2006.
- Ehringer H, Hornykiewicz O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system [in German]. Wien Klin Wochenschr 1960; 38:1236–1239.
- Goldstein DS, Holmes C, Sharabi Y, Brentzel S, Eisenhofer G. Plasma levels of catechols and metanephrines in neurogenic orthostatic hypotension. Neurology 2003; 60:1327–1332.
- Goldstein DS, Holmes C, Sato T, et al. Central dopamine deficiency in pure autonomic failure. Clin Auton Res 2008; 18:58–65.
- Hervey Wyatt RB William Harvey 1578 to 1657 Whitefish, MT Kessinger Publishing 2005:161–162.