User login
Spectral analysis of heart rate variability (HRV) has been used widely as a noninvasive technique for examining sympathetic and parasympathetic nervous outflows to the heart. Low-frequency (LF) and high-frequency (HF) power have been used most commonly. Human and animal experiments have repeatedly confirmed the dependence of HF power on respiration-related alterations in parasympathetic cardiovagal outflow–respiratory sinus arrhythmia; however, whether LF power provides an indirect measure of cardiac sympathetic activity has been contentious. Pagani et al1 reported that LF power (normalized to total spectral power) increased during states associated with sympathetic noradrenergic activation and that bilateral stellectomy in dogs reduced LF power. Alvarenga et al,2 however, reported that LF power was unrelated to all measures of norepinephrine kinetics in the heart; and in congestive heart failure, which is associated with a high rate of entry of norepinephrine into coronary sinus plasma (cardiac norepinephrine spillover),3 LF power is decreased, not increased as might be expected if LF power reflected sympathetic activity.4–7
Sleight et al8 proposed an alternative explanation for the origin of LF power. In a small group of human subjects, power spectral analysis of HRV showed that the amplitude of LF power was related to baroreflex gain and not to the level of sympathetic activity. Carotid sinus stimulation increased LF power only in individuals with normal baroreflex sensitivity and did not do so in those with depressed baroreflex gain. Therefore, results of power spectral analysis of LF power might reflect baroreflex-cardiovagal function.9
Studies of patients with dysautonomias provide an unusual opportunity to examine neurocirculatory correlates of LF power. Some chronic autonomic failure syndromes feature cardiac sympathetic denervation, whereas others do not. Parkinson disease with neurogenic orthostatic hypotension and pure autonomic failure feature cardiac sympathetic denervation, whereas multiple system atrophy does not.10 All 3 diseases involve baroreflex-cardiovagal and baro-reflex-sympathoneural failure.11 Chronic orthostatic intolerance syndromes (postural tachycardia syndrome, neurocardiogenic syncope) do not entail either cardiac sympathetic denervation or baroreflex failure.12
For this article, we carried out power spectral analyses of HRV on digitized electrocardiographic recordings from dysautonomia patients and normal volunteers during supine rest, measurement of cardiac norepinephrine spillover, and intravenous infusion of yohimbine and tyramine, 2 drugs that are known to release norepinephrine from cardiac sympathetic nerves.13,14 Cardiac sympathetic innervation was assessed by 6-[18F]fluorodopamine positron emission tomographic scanning.15
We hypothesized that if LF power indicated cardiac sympathetic innervation and function, then patients with neuroimaging or neurochemical evidence of cardiac sympathetic denervation would have low LF power and attenuated increments in LF power in response to yohimbine and tyramine. Alternatively, if LF power was reflective of baroreflex function, alterations of LF power would be independent of cardiac sympathetic innervation status and correlate with changes in baroreflex gain.
METHODS
The study protocols were approved by the Intramural Research Board of the National Institute of Neurological Disorders and Stroke. All subjects were studied at the National Institutes of Health Clinical Center after giving informed, written consent.
Subjects
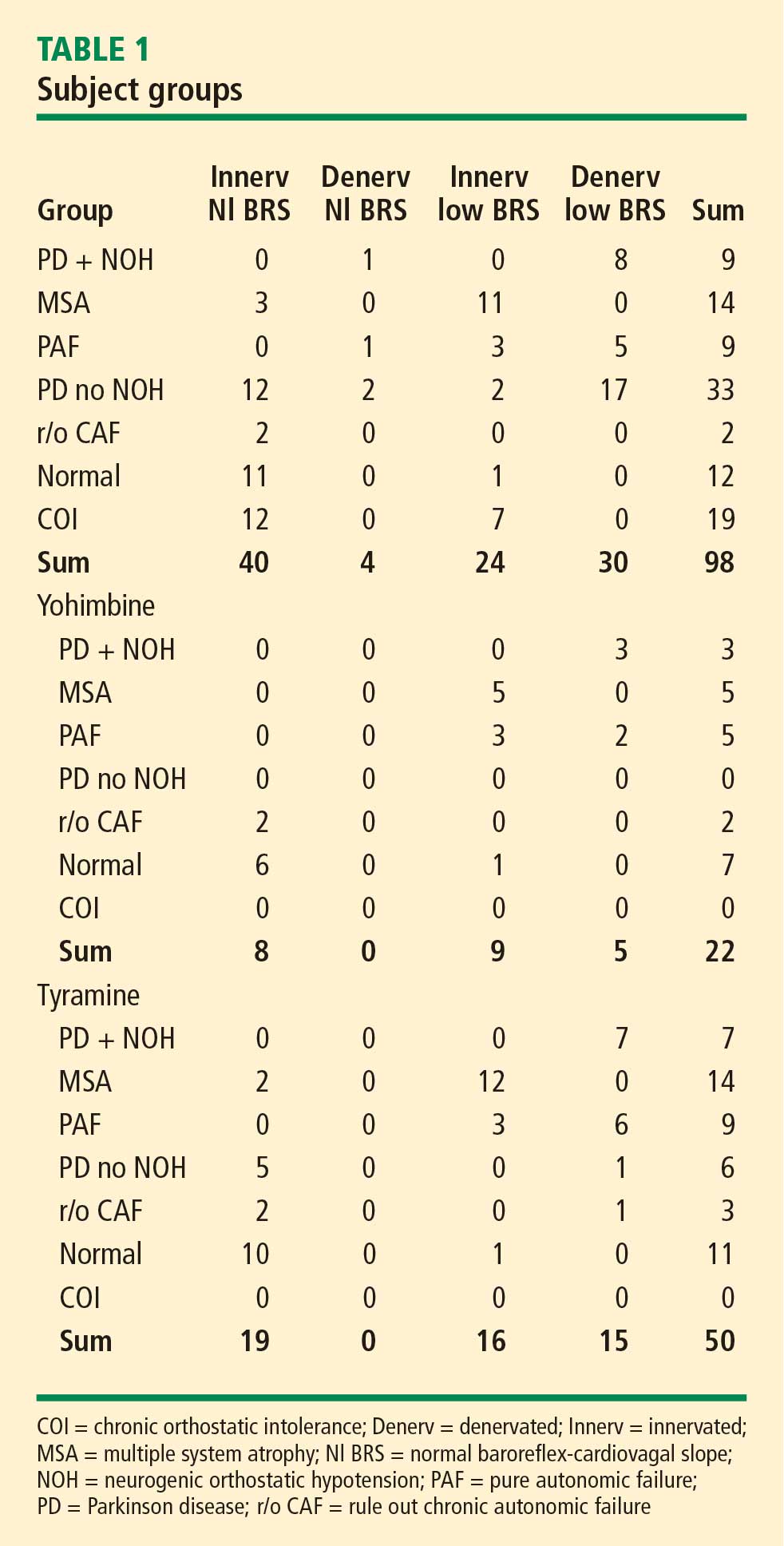
The study subjects were separated into 4 groups, depending on their state of cardiac sympathetic innervation and baroreflex-cardiovagal slope (BRS; see below). There were 40 subjects with intact sympathetic innervation and normal BRS (Innervated-Normal BRS), 24 with intact sympathetic innervation and low BRS (Innervated-Low BRS), 4 with sympathetic denervation and normal BRS (Denervated-Normal BRS), and 30 with sympathetic denervation and low BRS (Denervated-Low BRS).
Autonomic function testing
Each subject was studied while supine with head on pillow after an overnight fast. Each patient had monitoring of the electrocardiogram and beat-tobeat blood pressure using either noninvasive devices (Finometer, Finapres Medical Systems, Amsterdam, the Netherlands; Portapres, Finapres Medical Systems; or Colin tonometer, Colin Medical Instruments, San Antonio, TX) or a brachial intra-arterial catheter. We previously studied formally and reported excellent agreement between intra-arterial and these noninvasively obtained measures of beat-to-beat blood pressure.16 Continuous vital signs data were digitized and recorded using a PowerLab (AD Instruments Pty Ltd, Castle Hill, Australia) data acquisition system and stored for later analysis on an Apple PowerBook G4 computer (Apple, Cupertino, CA).
After about a 10-min baseline period, each subject performed a Valsalva maneuver (30 mm Hg for 12 sec) at least 3 times.
Baroreflex function
As an index of baroreflex function, we used the slope of the relationship between cardiac interbeat interval and systolic blood pressure during phase II of the Valsalva maneuver.17 BRS, in units of msec/mm Hg, was calculated from the linear regression equation for the relationship between interbeat interval (with 1-beat delay) and systolic pressure. A BRS value of ≤3 msec/mm Hg was considered low.11
Pharmacologic testing
Pharmacologic testing was performed on completion of the autonomic evaluation, using either tyramine or yohimbine. If a subject received both drugs, each drug administration was on a separate day. The durations of drug infusion were sufficient for heart rate and blood pressure to reach plateau values.
In a total of 22 subjects (Table 1), yohimbine was infused intravenously at 62.5 μg/kg over 3 min and then at 0.5 μg/kg/min for 12 min. In a total of 50 subjects, tyramine was infused at a rate of 1 mg/min for 10 min. In patients with severe supine hypertension (systolic pressure more than 200 mm Hg) and orthostatic hypotension, the test drugs were infused during head-up tilting (15° to 30°), to decrease baseline pressure, or else the drugs were not given.
HRV analysis
LF power (0.04 to 0.15 Hz), HF power (0.16 to 0.4 Hz), and total power (TP, 0.0 to 0.4 Hz) were calculated using Chart 5.4.2 and the HRV module version 1.03 (PowerLab, AD Instruments Pty Ltd, Castle Hill, Australia). Stable heart rate epochs 3 to 5 min in duration were chosen for analysis. One epoch was sampled immediately before initiation of drug testing; the second followed attainment of steady-state hemodynamic effects. Interbeat interval data were reviewed carefully to eliminate artifacts from noise and T waves, using segments with little to no premature beats. LF power and HF power were calculated as absolute power (msec2), with or without normalization for total power (0.04 to 0.4 Hz). Reported LF or HF power was integrated within their defined frequency bands.
Cardiac sympathetic neuroimaging
For cardiac sympathetic neuroimaging the subject was positioned supine, feet-first in a GE Advance scanner (General Electric, Milwaukee, WI), with the thorax in the gantry. After positioning the patient with the thorax in the scanner and transmission scanning for attenuation correction, 6-[18F]fluorodopamine (usual dose 1 mCi, specific activity 1.0 to 4.0 Ci/mmole, in about 10 mL normal saline) was infused intravenously at a constant rate for 3 min, and dynamic scanning data were obtained for thoracic radioactivity, with the midpoint of the scanning interval at 7.5 min after injection of the tracer (data collection interval between 5 and 10 min). Cardiac sympathetic denervation was defined by low concentrations of 6-[18F] fluorodopamine-derived radioactivity in the interventricular septum (< 5,000 nCi-kg/cc-mCi) or left ventricular free wall (< 4,000 nCi-kg/cc-mCi) corresponding to about 2 SD below the normal means.
Cardiac norepinephrine spillover
Subgroups of subjects (3 PD + NOH, 3 MSA, 3 PAF, 5 normal volunteers) underwent right heart catheterization for measurement of cardiac norepinephrine spillover. 3H-Norepinephrine was infused intravenously, and arterial and coronary sinus blood was sampled and coronary sinus blood flow was measured by thermodilution for measurements of cardiac norepinephrine spillover as described previously.18 In some subjects, yohimbine was infused during cardiac catheterization. Patients with chronic autonomic failure received the doses described above; normal volunteers and patients with chronic orthostatic intolerance received twice the doses described above.
Data analysis
Statistical analyses were performed using StatView version 5.0.1. (SAS Institute, Cary, NC). Mean values in the baseline condition for the several subject groups were compared using single-factor ANOVA. Responses to drugs were analyzed by dependent-means t tests. Differences in response to pharmacologic tests among subject groups were compared using repeated measures analyses of variance. Relationships between individual hemodynamic values were assessed by linear regression and calculation of Pearson correlation coefficients. Post-hoc testing consisted of the Fisher PLSD test. Multiple regression analysis was done on the individual data, with the log of LF power as the dependent measure and the log of baroreflex slope and septal 6-[18F] fluorodopamine-derived radioactivity as independent measures. Mean values were expressed ± SEM.
RESULTS
Baseline
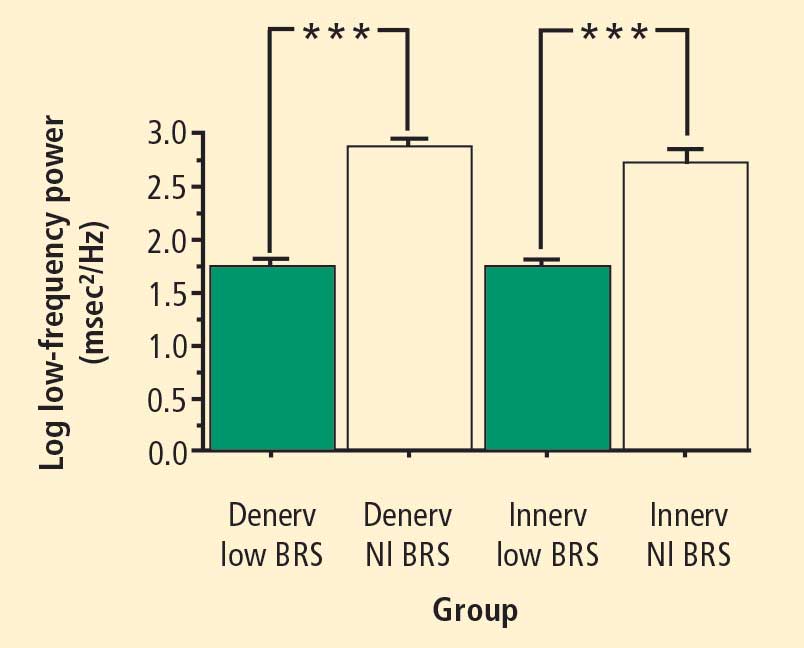
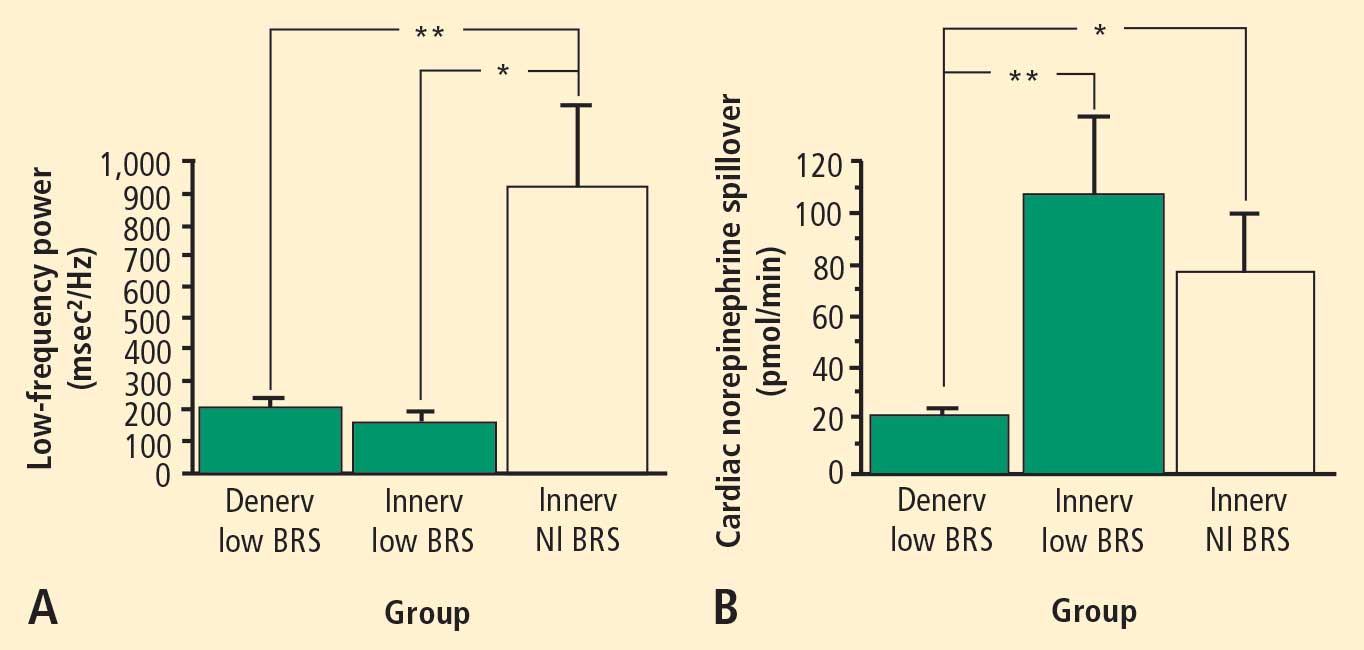
Across the 7 subject groups (N = 98), LF power was unrelated to subject group (F = 1.2). When individual subjects were stratified in terms of cardiac sympathetic denervation or innervation, based on concentrations of 6-[18F]fluorodopamine-derived radioactivity more than 2 SD below the normal mean, then LF power was lower in the Denervated group (mean 221 ± 55 msec2/Hz, N = 34) than in the Innervated group (516 ± 93 msec2/Hz, N = 64, F = 4.8, P = 0.03). LF power normalized for total power, HF normalized for total power, and the ratio of LF:HF were not related to 6-[18F]fluorodopamine-derived radioactivity.
When subjects were stratified in terms of BRS, then LF power was lower in the Low BRS group (223 ± 105 msec2/Hz, N = 46) than in the Normal BRS group (617 ± 97 msec2/Hz, N = 25, F = 6.1, P = 0.02). The Low BRS group did not differ from the Normal BRS group in normalized LF power (F = 0.8).
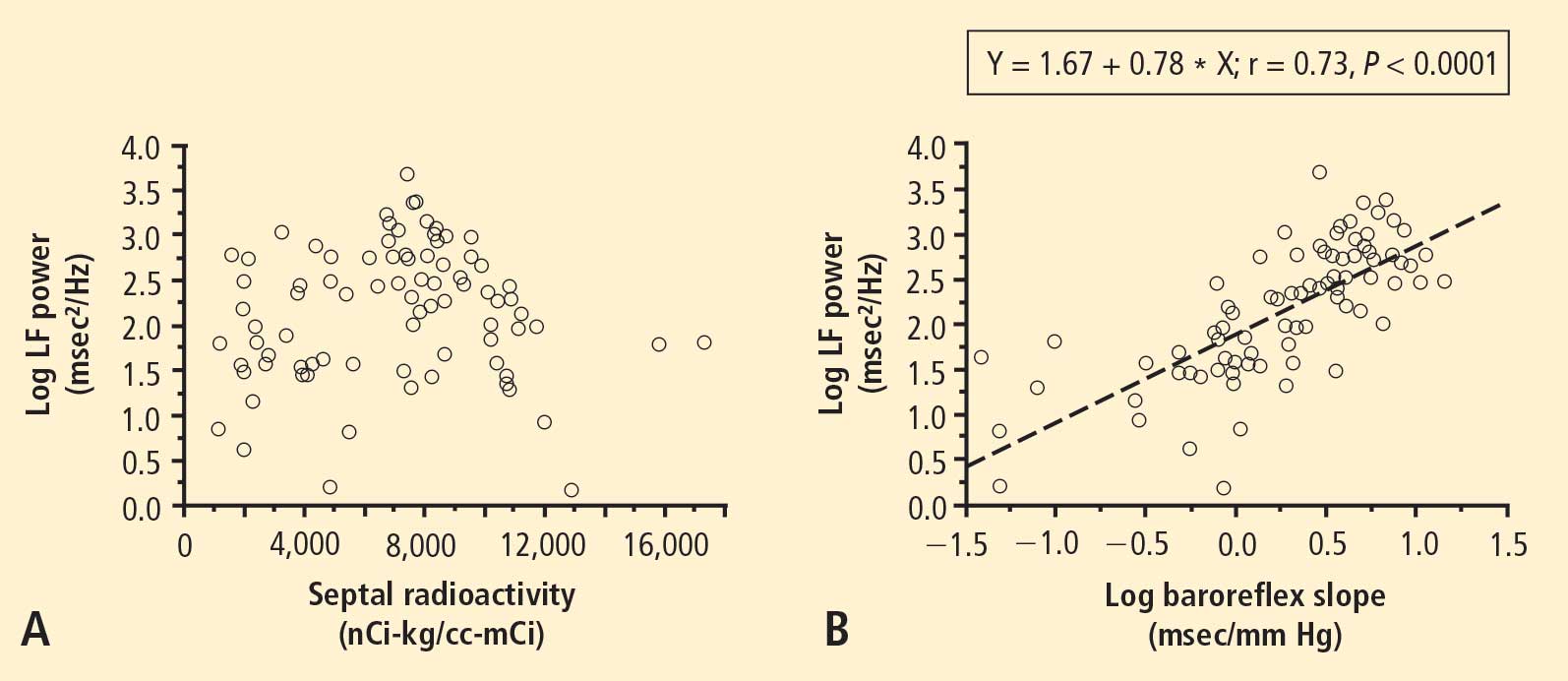
From multiple regression analysis for the log of LF power as the dependent measure and the log of baroreflex slope and septal 6-[18F]fluorodopamine-derived radioactivity as independent measures, the regression coefficient for the log of baroreflex slope was 0.92 (P < 0.0001), whereas the regression coefficient for 6-[18F] fluorodopamine-derived radioactivity was 3 ×10−6.
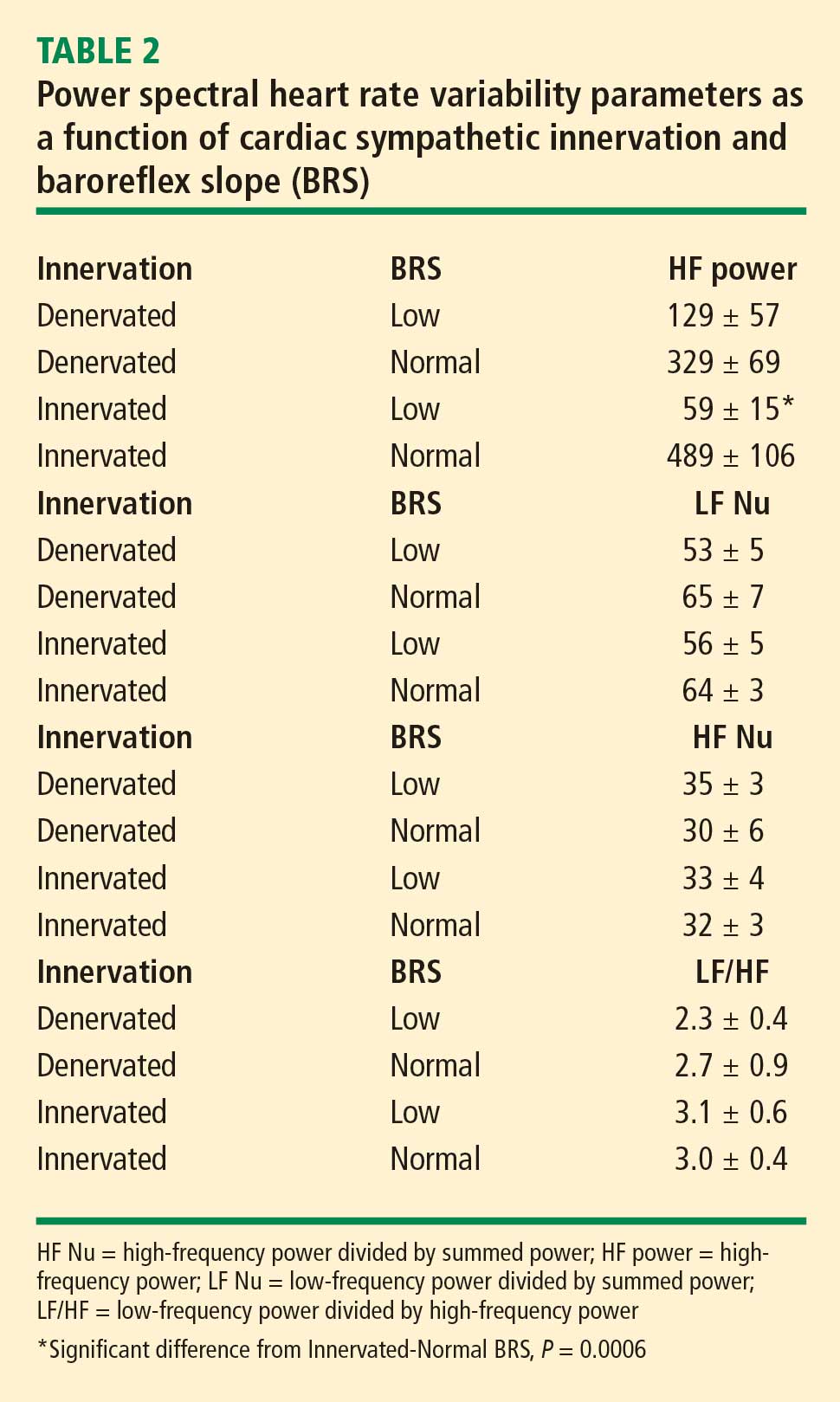
At baseline, the log of HF power correlated positively with the log of LF power (r = 0.77, P < 0.0001). HF power varied with the subject group (F = 4.9, P = 0.004). As with LF power, HF power was greater in the Innervated-Normal BRS than in the Innervated-Low BRS (P = 0.001, Table 2). As expected, the log of HF power correlated positively with the log of BRS (r = 0.60, P < 0.0001). The log of HF power also correlated negatively with the magnitude of decrease in systolic pressure during the Valsalva maneuver (r = −0.24, P = 0.02) and positively with the orthostatic change in systolic pressure (r = 0.40, P = 0.004).
Yohimbine
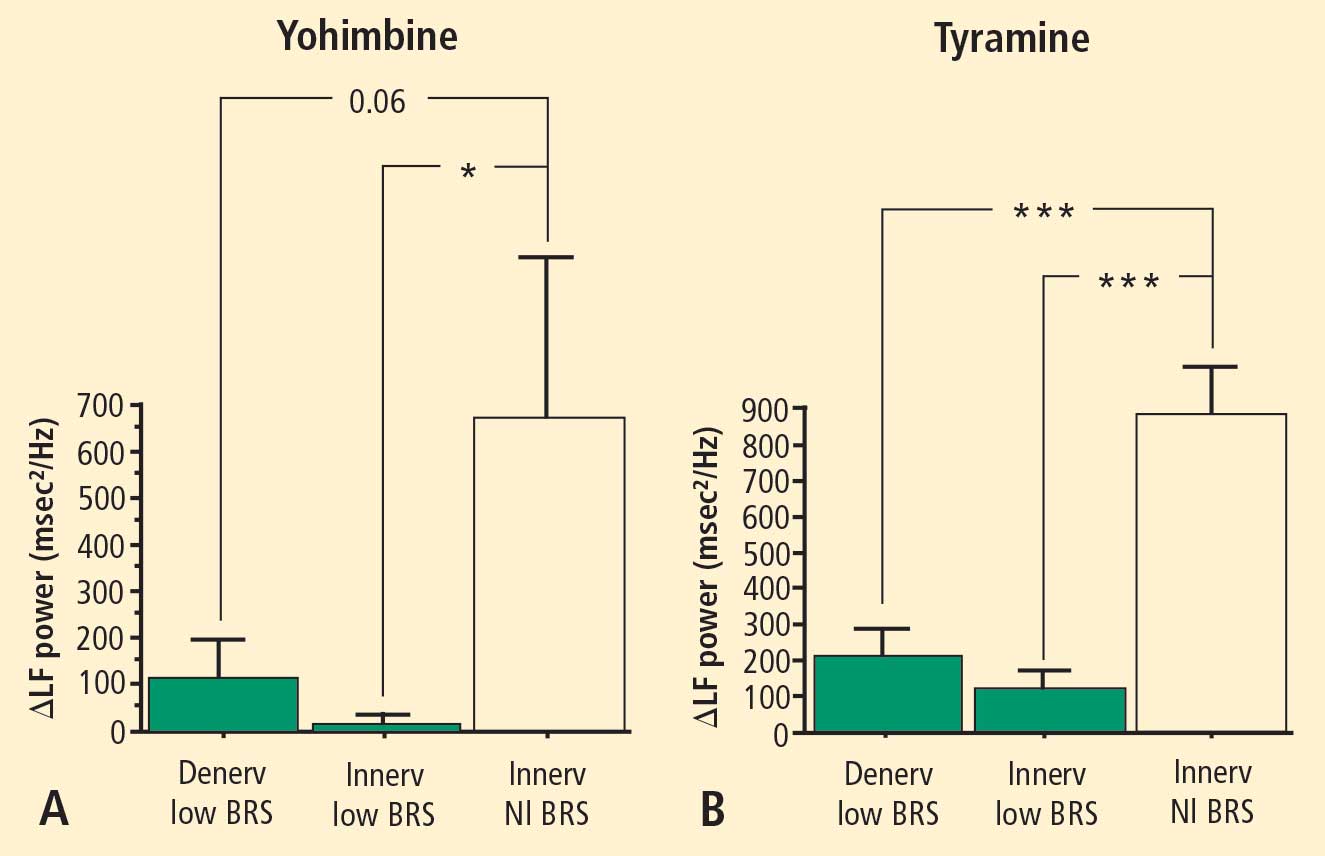
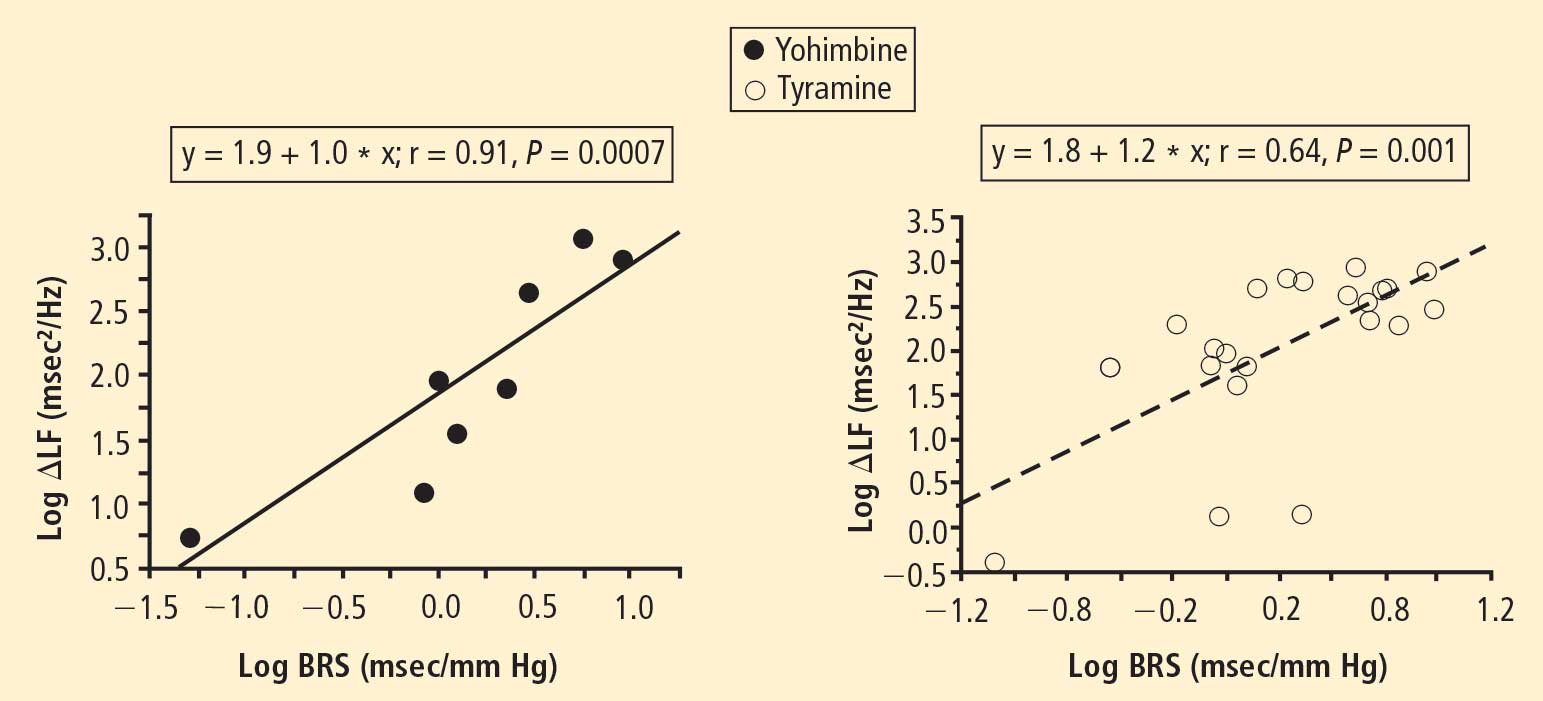
The change in LF power in response to yohimbine during cardiac catheterization was unrelated to the change in cardiac norepinephrine spillover (r = −0.09, N = 12).
Tyramine
Overall, tyramine infusion increased LF power (t = 2.9, P = 0.008). The group with cardiac sympathetic denervation did not differ from the group with intact cardiac innervation in terms of the change in LF power during tyramine infusion (F = 1.7). Tyramine increased LF power in the Innervated-Normal BRS group but not in the Innervated-Low BRS or Denervated-Low BRS groups (Figure 4; data for 2 outliers excluded). The log of the change in LF power during tyramine administration was positively correlated with the log of BRS at baseline (Figure 5; data for 2 outliers excluded).
DISCUSSION
In this study, patients with neuroimaging evidence of cardiac sympathetic denervation had low LF power of heart rate variability. At first glance, this finding would seem to support the view that LF power can provide an index of cardiac sympathetic outflow. As explained below, several lines of evidence from the present study led us to infer that the association between low LF power and cardiac sympathetic innervation is determined mainly by concurrent baroreflex function.
Patients with low BRS had low LF power, and patients with normal BRS had normal LF power, regardless of the status of cardiac sympathetic innervation as assessed by 6-[18F]fluorodopamine scanning. Neither normalization of LF and HF power for total power nor use of the LF:HF ratio improved the association with indices of cardiac sympathetic innervation.
Neurochemical findings during cardiac catheterization supported the above results based on cardiac sympathetic neuroimaging. Among patients with innervated hearts who had normal cardiac norepinephrine spillover, LF power was decreased only in the group with low BRS and was normal in the group with normal BRS. As expected, cardiac norepinephrine spillover was decreased in patients with neuroimaging evidence of cardiac sympathetic denervation.
Effects of pharmacological manipulations that increase norepinephrine release from sympathetic nerves provided further support for an association between baroreflex failure and low LF power, independent of cardiac sympathetic function. Both tyramine and yohimbine increased LF power only in the subjects with normal BRS. In subjects with low BRS, neither drug increased LF power, even in the group with intact cardiac sympathetic innervation. Moreover, individual values for responses of the log of LF power to both drugs were correlated positively with the log of BRS at baseline.
The fact that HF power was positively correlated with LF power could also fit with the notion of baroreflex function acting as a common determinant of values of both variables. We cannot exclude concurrent parasympathetic cardiovagal and sympathetic denervation as an explanation for the association between HF and LF power. Inhibition of the effects of parasympathetic activity after atropine administration results in the almost complete absence of both LF and HF HRV, further suggesting a common determinant.19
Several previous investigations have cast doubt on the validity of LF power as a measure of sympathetic activity because of dissociations between LF power and cardiac norepinephrine spillover, directly recorded sympathetic nerve traffic, and plasma norepinephrine levels.4,6,20 Such dissociations are especially glaring in patients with congestive heart failure, which is characterized by decreased LF power9 despite marked cardiac sympathetic activation.3
Other pathophysiologic states do result in both decreased sympathetic nervous system activity and decreased LF power. In these pathophysiologic states, the possibility remains that low LF power might reflect failure of baroreflexive modulation of sympathetic neuronal outflows, rather than sympathoinhibition itself. For instance, Wiklund et al21 noted low LF power in patients with palmar hyperhidrosis undergoing bilateral transthoracic sympathectomy; however, baroreflex-cardiovagal sensitivity also declines after thoracic sympathectomy.22
Sleight et al8 suggested dependence of LF power on baroreflex function, based on effects of carotid baroreceptor stimulation in 3 patients: 1 with normal BRS; 1 with ischemic heart disease, congestive heart failure, and normal BRS; and 1 with ischemic heart disease, congestive heart failure, and initially low BRS who subsequently had an improved clinical state and BRS. In the baseline state, both congestive heart failure patients had low LF power, despite a presumably hypernoradrenergic state. Direct baroreceptor stimulation at 0.1 Hz increased LF power in the normal subject and in the patient with congestive heart failure and normal BRS. The congestive heart failure patient with low BRS did not have an increase in LF power until BRS normalized. These data revealed an initial dissociation between cardiac noradrenergic state in the patients with congestive heart failure and LF power. During carotid sinus stimulation, LF power increased only when BRS was normal. Low BRS obviated this effect.
Because congestive heart failure is well known to be associated with baroreflex-cardiovagal inhibition,23–25 the finding of low LF power in heart failure also supports an association between LF power and BRS, independently of increased tonic release of norepinephrine from sympathetic nerves in the heart. Cevese et al26 inhibited noradrenergic vasomotor tone using an alpha-adrenoceptor blocker in human subjects while maintaining mean blood pressure at control levels using angiotensin II. This drug combination, which would be expected to attenuate sympathetically mediated vasomotor tone and thereby decrease arterial baroreceptor input, markedly decreased or abolished LF power of HRV, suggesting that, at least under resting supine conditions, a baroreflex mechanism accounts almost entirely for LF power of HRV.
deBoer et al27 developed a beat-to-beat model of the human circulation using a set of differential equations and the following principles of operation: (1) the baroreflex regulates heart rate and peripheral vascular resistance; (2) Windkessel properties characterize the systemic arterial tree; (3) contractile properties of the ventricular myocardium follow the Starling law; and (4) respiration exerts mechanical effects on blood pressure. The model attributes LF power to a resonance in the circulatory control system, produced by a slow time constant for reflexive sympathetically mediated responses to beat-tobeat blood pressure changes. The resonance can be upregulated or downregulated by the state of baroreflex activity. The model of deBoer et al predicts that changes in blood pressure would lead heart rate changes at 0.1 Hz through a delayed sympathetic response. Changes in HR would depend on summed effects of sympathetic and vagal effects, with the sympathetic response delaying the overall response. At the respiratory frequency (0.2 to 0.3 Hz), blood pressure and HR changes would occur with little delay because of fast parasympathetic control. In essence, the response of the sympathetic nervous system behaves as a low band pass filter, with a peak response at 0.1 Hz and little response at frequencies above 0.2 Hz. Systolic blood pressure would lead to changes in heart rate via the baroreflex. In general the results of this study fit with the deBoer model.
In conclusion, LF power derived from the interbeat interval spectrogram predominantly reflects baroreflex-mediated, phasic changes in cardiovagal and sympathetic noradrenergic outflows. In the setting of baroreflex failure, baseline LF power is reduced, regardless of the status of cardiac sympathetic innervation.
LIMITATIONS
The combination of cardiac sympathetic denervation and normal baroreflex function seems quite rare. One must exercise caution in drawing inferences from the findings in the Denervated-Normal BRS group, which contained only 4 subjects, even though the difference in mean log-transformed LF power from the Denervated-Low BRS group was highly statistically significant.
All of the testing in our study was done with the subjects supine. LF power measured in other positions might have different sources and meaning.
- Pagani M, Lombardi F, Guzzetti S, et al. Power spectral analysis of heart rate and arterial pressure variabilities as a marker of sympatho-vagal interaction in man and conscious dog. Circ Res 1986; 59:178–193.
- Alvarenga ME, Richards JC, Lambert G, Esler MD. Psychophysiological mechanisms in panic disorder: a correlative analysis of noradrenaline spillover, neuronal noradrenaline reuptake, power spectral analysis of heart rate variability, and psychological variables. Psychosom Med 2006; 68:8–16.
- Eisenhofer G, Friberg P, Rundqvist B, et al. Cardiac sympathetic nerve function in congestive heart failure. Circulation 1996; 93:1667–1676.
- Notarius CF, Butler GC, Ando S, Pollard MJ, Senn BL, Floras JS. Dissociation between microneurographic and heart rate variability estimates of sympathetic tone in normal subjects and patients with heart failure. Clin Sci (Lond) 1999; 96:557–565.
- Scalvini S, Volterrani M, Zanelli E, et al. Is heart rate variability a reliable method to assess autonomic modulation in left ventricular dysfunction and heart failure? Assessment of autonomic modulation with heart rate variability. Int J Cardiol 1998; 67:9–17.
- Kingwell BA, Thompson JM, Kaye DM, McPherson GA, Jennings GL, Esler MD. Heart rate spectral analysis, cardiac norepinephrine spillover, and muscle sympathetic nerve activity during human sympathetic nervous activation and failure. Circulation 1994; 90:234–240.
- van de Borne P, Montano N, Pagani M, Oren R, Somers VK. Absence of low-frequency variability of sympathetic nerve activity in severe heart failure. Circulation 1997; 95:1449–1454.
- Sleight P, La Rovere MT, Mortara A, et al. Physiology and pathophysiology of heart rate and blood pressure variability in humans: is power spectral analysis largely an index of baroreflex gain? Clin Sci (Lond) 1995; 88:103–109.
- Saul JP, Arai Y, Berger RD, Lilly LS, Colucci WS, Cohen RJ. Assessment of autonomic regulation in chronic congestive heart failure by heart rate spectral analysis. Am J Cardiol 1988; 61:1292–1299.
- Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO. Cardiac sympathetic denervation in Parkinson disease. Ann Intern Med 2000; 133:338–347.
- Goldstein DS, Eldadah BA, Holmes C, et al. Neurocirculatory abnormalities in Parkinson disease with orthostatic hypotension. Independence from levodopa treatment. Hypertension 2005; 46:1333–1339.
- Goldstein DS, Eldadah B, Holmes C, Pechnik S, Moak J, Sharabi Y. Neurocirculatory abnormalities in chronic orthostatic intolerance. Circulation 2005; 111:839–845.
- Goldstein DS, Holmes C, Frank SM, et al. Cardiac sympathetic dysautonomia in chronic orthostatic intolerance syndromes. Circulation 2002; 106:2358–2365.
- Lord SW, Clayton RH, Mitchell L, Dark JH, Murray A, McComb JM. Sympathetic reinnervation and heart rate variability after cardiac transplantation. Heart 1997; 77:532–538.
- Goldstein DS, Eisenhofer G, Dunn BB, et al. Positron emission tomographic imaging of cardiac sympathetic innervation using 6-[18F]fluorodopamine: initial findings in humans. J Am Coll Cardiol 1993; 22:1961–1971.
- Goldstein DS, Tack C. Non-invasive detection of sympathetic neurocirculatory failure. Clin Auton Res 2000; 10:285–291.
- Goldstein DS, Horwitz D, Keiser HR. Comparison of techniques for measuring baroreflex sensitivity in man. Circulation 1982; 66:432–439.
- Goldstein DS, Brush JE, Eisenhofer G, Stull R, Esler M. In vivo measurement of neuronal uptake of norepinephrine in the human heart. Circulation 1988; 78:41–48.
- Koh J, Brown TE, Beightol LA, Ha CY, Eckberg DL. Human autonomic rhythms: vagal cardiac mechanisms in tetraplegic subjects. J Physiol 1994; 474:483–495.
- Saul JP, Rea RF, Eckberg DL, Berger RD, Cohen RJ. Heart rate and muscle sympathetic nerve variability during reflex changes of autonomic activity. Am J Physiol 1990; 258:H713–H721.
- Wiklund U, Koskinen LO, Niklasson U, Bjerle P, Elfversson J. Endoscopic transthoracic sympathectomy affects the autonomic modulation of heart rate in patients with palmar hyperhidrosis. Acta Neurochir (Wien) 2000; 142:691–696.
- Kawamata YT, Kawamata T, Omote K, et al. Endoscopic thoracic sympathectomy suppresses baroreflex control of heart rate in patients with essential hyperhidrosis. Anesth Analg 2004; 98:37–39.
- Goldstein RE, Beiser GD, Stampfer M, Epstein SE. Impairment of autonomically mediated heart rate control in patients with cardiac dysfunction. Circ Res 1975; 36:571–578.
- Cody RJ, Franklin KW, Kluger J, Laragh JH. Mechanisms governing the postural response and baroreceptor abnormalities in chronic congestive heart failure: effects of acute and long-term convertingenzyme inhibition. Circulation 1982; 66:135–142.
- Creager MA. Baroreceptor reflex function in congestive heart failure. Am J Cardiol 1992; 69:10G–15G; discussion 15G–16G.
- Cevese A, Gulli G, Polati E, Gottin L, Grasso R. Baroreflex and oscillation of heart period at 0.1 Hz studied by alpha-blockade and crossspectral analysis in healthy humans. J Physiol 2001; 531:235–244.
- deBoer RW, Karemaker JM, Strackee J. Hemodynamic fluctuations and baroreflex sensitivity in humans: a beat-to-beat model. Am J Physiol 1987; 253:H680–689.
Spectral analysis of heart rate variability (HRV) has been used widely as a noninvasive technique for examining sympathetic and parasympathetic nervous outflows to the heart. Low-frequency (LF) and high-frequency (HF) power have been used most commonly. Human and animal experiments have repeatedly confirmed the dependence of HF power on respiration-related alterations in parasympathetic cardiovagal outflow–respiratory sinus arrhythmia; however, whether LF power provides an indirect measure of cardiac sympathetic activity has been contentious. Pagani et al1 reported that LF power (normalized to total spectral power) increased during states associated with sympathetic noradrenergic activation and that bilateral stellectomy in dogs reduced LF power. Alvarenga et al,2 however, reported that LF power was unrelated to all measures of norepinephrine kinetics in the heart; and in congestive heart failure, which is associated with a high rate of entry of norepinephrine into coronary sinus plasma (cardiac norepinephrine spillover),3 LF power is decreased, not increased as might be expected if LF power reflected sympathetic activity.4–7
Sleight et al8 proposed an alternative explanation for the origin of LF power. In a small group of human subjects, power spectral analysis of HRV showed that the amplitude of LF power was related to baroreflex gain and not to the level of sympathetic activity. Carotid sinus stimulation increased LF power only in individuals with normal baroreflex sensitivity and did not do so in those with depressed baroreflex gain. Therefore, results of power spectral analysis of LF power might reflect baroreflex-cardiovagal function.9
Studies of patients with dysautonomias provide an unusual opportunity to examine neurocirculatory correlates of LF power. Some chronic autonomic failure syndromes feature cardiac sympathetic denervation, whereas others do not. Parkinson disease with neurogenic orthostatic hypotension and pure autonomic failure feature cardiac sympathetic denervation, whereas multiple system atrophy does not.10 All 3 diseases involve baroreflex-cardiovagal and baro-reflex-sympathoneural failure.11 Chronic orthostatic intolerance syndromes (postural tachycardia syndrome, neurocardiogenic syncope) do not entail either cardiac sympathetic denervation or baroreflex failure.12
For this article, we carried out power spectral analyses of HRV on digitized electrocardiographic recordings from dysautonomia patients and normal volunteers during supine rest, measurement of cardiac norepinephrine spillover, and intravenous infusion of yohimbine and tyramine, 2 drugs that are known to release norepinephrine from cardiac sympathetic nerves.13,14 Cardiac sympathetic innervation was assessed by 6-[18F]fluorodopamine positron emission tomographic scanning.15
We hypothesized that if LF power indicated cardiac sympathetic innervation and function, then patients with neuroimaging or neurochemical evidence of cardiac sympathetic denervation would have low LF power and attenuated increments in LF power in response to yohimbine and tyramine. Alternatively, if LF power was reflective of baroreflex function, alterations of LF power would be independent of cardiac sympathetic innervation status and correlate with changes in baroreflex gain.
METHODS
The study protocols were approved by the Intramural Research Board of the National Institute of Neurological Disorders and Stroke. All subjects were studied at the National Institutes of Health Clinical Center after giving informed, written consent.
Subjects
The study subjects were separated into 4 groups, depending on their state of cardiac sympathetic innervation and baroreflex-cardiovagal slope (BRS; see below). There were 40 subjects with intact sympathetic innervation and normal BRS (Innervated-Normal BRS), 24 with intact sympathetic innervation and low BRS (Innervated-Low BRS), 4 with sympathetic denervation and normal BRS (Denervated-Normal BRS), and 30 with sympathetic denervation and low BRS (Denervated-Low BRS).
Autonomic function testing
Each subject was studied while supine with head on pillow after an overnight fast. Each patient had monitoring of the electrocardiogram and beat-tobeat blood pressure using either noninvasive devices (Finometer, Finapres Medical Systems, Amsterdam, the Netherlands; Portapres, Finapres Medical Systems; or Colin tonometer, Colin Medical Instruments, San Antonio, TX) or a brachial intra-arterial catheter. We previously studied formally and reported excellent agreement between intra-arterial and these noninvasively obtained measures of beat-to-beat blood pressure.16 Continuous vital signs data were digitized and recorded using a PowerLab (AD Instruments Pty Ltd, Castle Hill, Australia) data acquisition system and stored for later analysis on an Apple PowerBook G4 computer (Apple, Cupertino, CA).
After about a 10-min baseline period, each subject performed a Valsalva maneuver (30 mm Hg for 12 sec) at least 3 times.
Baroreflex function
As an index of baroreflex function, we used the slope of the relationship between cardiac interbeat interval and systolic blood pressure during phase II of the Valsalva maneuver.17 BRS, in units of msec/mm Hg, was calculated from the linear regression equation for the relationship between interbeat interval (with 1-beat delay) and systolic pressure. A BRS value of ≤3 msec/mm Hg was considered low.11
Pharmacologic testing
Pharmacologic testing was performed on completion of the autonomic evaluation, using either tyramine or yohimbine. If a subject received both drugs, each drug administration was on a separate day. The durations of drug infusion were sufficient for heart rate and blood pressure to reach plateau values.
In a total of 22 subjects (Table 1), yohimbine was infused intravenously at 62.5 μg/kg over 3 min and then at 0.5 μg/kg/min for 12 min. In a total of 50 subjects, tyramine was infused at a rate of 1 mg/min for 10 min. In patients with severe supine hypertension (systolic pressure more than 200 mm Hg) and orthostatic hypotension, the test drugs were infused during head-up tilting (15° to 30°), to decrease baseline pressure, or else the drugs were not given.
HRV analysis
LF power (0.04 to 0.15 Hz), HF power (0.16 to 0.4 Hz), and total power (TP, 0.0 to 0.4 Hz) were calculated using Chart 5.4.2 and the HRV module version 1.03 (PowerLab, AD Instruments Pty Ltd, Castle Hill, Australia). Stable heart rate epochs 3 to 5 min in duration were chosen for analysis. One epoch was sampled immediately before initiation of drug testing; the second followed attainment of steady-state hemodynamic effects. Interbeat interval data were reviewed carefully to eliminate artifacts from noise and T waves, using segments with little to no premature beats. LF power and HF power were calculated as absolute power (msec2), with or without normalization for total power (0.04 to 0.4 Hz). Reported LF or HF power was integrated within their defined frequency bands.
Cardiac sympathetic neuroimaging
For cardiac sympathetic neuroimaging the subject was positioned supine, feet-first in a GE Advance scanner (General Electric, Milwaukee, WI), with the thorax in the gantry. After positioning the patient with the thorax in the scanner and transmission scanning for attenuation correction, 6-[18F]fluorodopamine (usual dose 1 mCi, specific activity 1.0 to 4.0 Ci/mmole, in about 10 mL normal saline) was infused intravenously at a constant rate for 3 min, and dynamic scanning data were obtained for thoracic radioactivity, with the midpoint of the scanning interval at 7.5 min after injection of the tracer (data collection interval between 5 and 10 min). Cardiac sympathetic denervation was defined by low concentrations of 6-[18F] fluorodopamine-derived radioactivity in the interventricular septum (< 5,000 nCi-kg/cc-mCi) or left ventricular free wall (< 4,000 nCi-kg/cc-mCi) corresponding to about 2 SD below the normal means.
Cardiac norepinephrine spillover
Subgroups of subjects (3 PD + NOH, 3 MSA, 3 PAF, 5 normal volunteers) underwent right heart catheterization for measurement of cardiac norepinephrine spillover. 3H-Norepinephrine was infused intravenously, and arterial and coronary sinus blood was sampled and coronary sinus blood flow was measured by thermodilution for measurements of cardiac norepinephrine spillover as described previously.18 In some subjects, yohimbine was infused during cardiac catheterization. Patients with chronic autonomic failure received the doses described above; normal volunteers and patients with chronic orthostatic intolerance received twice the doses described above.
Data analysis
Statistical analyses were performed using StatView version 5.0.1. (SAS Institute, Cary, NC). Mean values in the baseline condition for the several subject groups were compared using single-factor ANOVA. Responses to drugs were analyzed by dependent-means t tests. Differences in response to pharmacologic tests among subject groups were compared using repeated measures analyses of variance. Relationships between individual hemodynamic values were assessed by linear regression and calculation of Pearson correlation coefficients. Post-hoc testing consisted of the Fisher PLSD test. Multiple regression analysis was done on the individual data, with the log of LF power as the dependent measure and the log of baroreflex slope and septal 6-[18F] fluorodopamine-derived radioactivity as independent measures. Mean values were expressed ± SEM.
RESULTS
Baseline
Across the 7 subject groups (N = 98), LF power was unrelated to subject group (F = 1.2). When individual subjects were stratified in terms of cardiac sympathetic denervation or innervation, based on concentrations of 6-[18F]fluorodopamine-derived radioactivity more than 2 SD below the normal mean, then LF power was lower in the Denervated group (mean 221 ± 55 msec2/Hz, N = 34) than in the Innervated group (516 ± 93 msec2/Hz, N = 64, F = 4.8, P = 0.03). LF power normalized for total power, HF normalized for total power, and the ratio of LF:HF were not related to 6-[18F]fluorodopamine-derived radioactivity.
When subjects were stratified in terms of BRS, then LF power was lower in the Low BRS group (223 ± 105 msec2/Hz, N = 46) than in the Normal BRS group (617 ± 97 msec2/Hz, N = 25, F = 6.1, P = 0.02). The Low BRS group did not differ from the Normal BRS group in normalized LF power (F = 0.8).
From multiple regression analysis for the log of LF power as the dependent measure and the log of baroreflex slope and septal 6-[18F]fluorodopamine-derived radioactivity as independent measures, the regression coefficient for the log of baroreflex slope was 0.92 (P < 0.0001), whereas the regression coefficient for 6-[18F] fluorodopamine-derived radioactivity was 3 ×10−6.
At baseline, the log of HF power correlated positively with the log of LF power (r = 0.77, P < 0.0001). HF power varied with the subject group (F = 4.9, P = 0.004). As with LF power, HF power was greater in the Innervated-Normal BRS than in the Innervated-Low BRS (P = 0.001, Table 2). As expected, the log of HF power correlated positively with the log of BRS (r = 0.60, P < 0.0001). The log of HF power also correlated negatively with the magnitude of decrease in systolic pressure during the Valsalva maneuver (r = −0.24, P = 0.02) and positively with the orthostatic change in systolic pressure (r = 0.40, P = 0.004).
Yohimbine
The change in LF power in response to yohimbine during cardiac catheterization was unrelated to the change in cardiac norepinephrine spillover (r = −0.09, N = 12).
Tyramine
Overall, tyramine infusion increased LF power (t = 2.9, P = 0.008). The group with cardiac sympathetic denervation did not differ from the group with intact cardiac innervation in terms of the change in LF power during tyramine infusion (F = 1.7). Tyramine increased LF power in the Innervated-Normal BRS group but not in the Innervated-Low BRS or Denervated-Low BRS groups (Figure 4; data for 2 outliers excluded). The log of the change in LF power during tyramine administration was positively correlated with the log of BRS at baseline (Figure 5; data for 2 outliers excluded).
DISCUSSION
In this study, patients with neuroimaging evidence of cardiac sympathetic denervation had low LF power of heart rate variability. At first glance, this finding would seem to support the view that LF power can provide an index of cardiac sympathetic outflow. As explained below, several lines of evidence from the present study led us to infer that the association between low LF power and cardiac sympathetic innervation is determined mainly by concurrent baroreflex function.
Patients with low BRS had low LF power, and patients with normal BRS had normal LF power, regardless of the status of cardiac sympathetic innervation as assessed by 6-[18F]fluorodopamine scanning. Neither normalization of LF and HF power for total power nor use of the LF:HF ratio improved the association with indices of cardiac sympathetic innervation.
Neurochemical findings during cardiac catheterization supported the above results based on cardiac sympathetic neuroimaging. Among patients with innervated hearts who had normal cardiac norepinephrine spillover, LF power was decreased only in the group with low BRS and was normal in the group with normal BRS. As expected, cardiac norepinephrine spillover was decreased in patients with neuroimaging evidence of cardiac sympathetic denervation.
Effects of pharmacological manipulations that increase norepinephrine release from sympathetic nerves provided further support for an association between baroreflex failure and low LF power, independent of cardiac sympathetic function. Both tyramine and yohimbine increased LF power only in the subjects with normal BRS. In subjects with low BRS, neither drug increased LF power, even in the group with intact cardiac sympathetic innervation. Moreover, individual values for responses of the log of LF power to both drugs were correlated positively with the log of BRS at baseline.
The fact that HF power was positively correlated with LF power could also fit with the notion of baroreflex function acting as a common determinant of values of both variables. We cannot exclude concurrent parasympathetic cardiovagal and sympathetic denervation as an explanation for the association between HF and LF power. Inhibition of the effects of parasympathetic activity after atropine administration results in the almost complete absence of both LF and HF HRV, further suggesting a common determinant.19
Several previous investigations have cast doubt on the validity of LF power as a measure of sympathetic activity because of dissociations between LF power and cardiac norepinephrine spillover, directly recorded sympathetic nerve traffic, and plasma norepinephrine levels.4,6,20 Such dissociations are especially glaring in patients with congestive heart failure, which is characterized by decreased LF power9 despite marked cardiac sympathetic activation.3
Other pathophysiologic states do result in both decreased sympathetic nervous system activity and decreased LF power. In these pathophysiologic states, the possibility remains that low LF power might reflect failure of baroreflexive modulation of sympathetic neuronal outflows, rather than sympathoinhibition itself. For instance, Wiklund et al21 noted low LF power in patients with palmar hyperhidrosis undergoing bilateral transthoracic sympathectomy; however, baroreflex-cardiovagal sensitivity also declines after thoracic sympathectomy.22
Sleight et al8 suggested dependence of LF power on baroreflex function, based on effects of carotid baroreceptor stimulation in 3 patients: 1 with normal BRS; 1 with ischemic heart disease, congestive heart failure, and normal BRS; and 1 with ischemic heart disease, congestive heart failure, and initially low BRS who subsequently had an improved clinical state and BRS. In the baseline state, both congestive heart failure patients had low LF power, despite a presumably hypernoradrenergic state. Direct baroreceptor stimulation at 0.1 Hz increased LF power in the normal subject and in the patient with congestive heart failure and normal BRS. The congestive heart failure patient with low BRS did not have an increase in LF power until BRS normalized. These data revealed an initial dissociation between cardiac noradrenergic state in the patients with congestive heart failure and LF power. During carotid sinus stimulation, LF power increased only when BRS was normal. Low BRS obviated this effect.
Because congestive heart failure is well known to be associated with baroreflex-cardiovagal inhibition,23–25 the finding of low LF power in heart failure also supports an association between LF power and BRS, independently of increased tonic release of norepinephrine from sympathetic nerves in the heart. Cevese et al26 inhibited noradrenergic vasomotor tone using an alpha-adrenoceptor blocker in human subjects while maintaining mean blood pressure at control levels using angiotensin II. This drug combination, which would be expected to attenuate sympathetically mediated vasomotor tone and thereby decrease arterial baroreceptor input, markedly decreased or abolished LF power of HRV, suggesting that, at least under resting supine conditions, a baroreflex mechanism accounts almost entirely for LF power of HRV.
deBoer et al27 developed a beat-to-beat model of the human circulation using a set of differential equations and the following principles of operation: (1) the baroreflex regulates heart rate and peripheral vascular resistance; (2) Windkessel properties characterize the systemic arterial tree; (3) contractile properties of the ventricular myocardium follow the Starling law; and (4) respiration exerts mechanical effects on blood pressure. The model attributes LF power to a resonance in the circulatory control system, produced by a slow time constant for reflexive sympathetically mediated responses to beat-tobeat blood pressure changes. The resonance can be upregulated or downregulated by the state of baroreflex activity. The model of deBoer et al predicts that changes in blood pressure would lead heart rate changes at 0.1 Hz through a delayed sympathetic response. Changes in HR would depend on summed effects of sympathetic and vagal effects, with the sympathetic response delaying the overall response. At the respiratory frequency (0.2 to 0.3 Hz), blood pressure and HR changes would occur with little delay because of fast parasympathetic control. In essence, the response of the sympathetic nervous system behaves as a low band pass filter, with a peak response at 0.1 Hz and little response at frequencies above 0.2 Hz. Systolic blood pressure would lead to changes in heart rate via the baroreflex. In general the results of this study fit with the deBoer model.
In conclusion, LF power derived from the interbeat interval spectrogram predominantly reflects baroreflex-mediated, phasic changes in cardiovagal and sympathetic noradrenergic outflows. In the setting of baroreflex failure, baseline LF power is reduced, regardless of the status of cardiac sympathetic innervation.
LIMITATIONS
The combination of cardiac sympathetic denervation and normal baroreflex function seems quite rare. One must exercise caution in drawing inferences from the findings in the Denervated-Normal BRS group, which contained only 4 subjects, even though the difference in mean log-transformed LF power from the Denervated-Low BRS group was highly statistically significant.
All of the testing in our study was done with the subjects supine. LF power measured in other positions might have different sources and meaning.
Spectral analysis of heart rate variability (HRV) has been used widely as a noninvasive technique for examining sympathetic and parasympathetic nervous outflows to the heart. Low-frequency (LF) and high-frequency (HF) power have been used most commonly. Human and animal experiments have repeatedly confirmed the dependence of HF power on respiration-related alterations in parasympathetic cardiovagal outflow–respiratory sinus arrhythmia; however, whether LF power provides an indirect measure of cardiac sympathetic activity has been contentious. Pagani et al1 reported that LF power (normalized to total spectral power) increased during states associated with sympathetic noradrenergic activation and that bilateral stellectomy in dogs reduced LF power. Alvarenga et al,2 however, reported that LF power was unrelated to all measures of norepinephrine kinetics in the heart; and in congestive heart failure, which is associated with a high rate of entry of norepinephrine into coronary sinus plasma (cardiac norepinephrine spillover),3 LF power is decreased, not increased as might be expected if LF power reflected sympathetic activity.4–7
Sleight et al8 proposed an alternative explanation for the origin of LF power. In a small group of human subjects, power spectral analysis of HRV showed that the amplitude of LF power was related to baroreflex gain and not to the level of sympathetic activity. Carotid sinus stimulation increased LF power only in individuals with normal baroreflex sensitivity and did not do so in those with depressed baroreflex gain. Therefore, results of power spectral analysis of LF power might reflect baroreflex-cardiovagal function.9
Studies of patients with dysautonomias provide an unusual opportunity to examine neurocirculatory correlates of LF power. Some chronic autonomic failure syndromes feature cardiac sympathetic denervation, whereas others do not. Parkinson disease with neurogenic orthostatic hypotension and pure autonomic failure feature cardiac sympathetic denervation, whereas multiple system atrophy does not.10 All 3 diseases involve baroreflex-cardiovagal and baro-reflex-sympathoneural failure.11 Chronic orthostatic intolerance syndromes (postural tachycardia syndrome, neurocardiogenic syncope) do not entail either cardiac sympathetic denervation or baroreflex failure.12
For this article, we carried out power spectral analyses of HRV on digitized electrocardiographic recordings from dysautonomia patients and normal volunteers during supine rest, measurement of cardiac norepinephrine spillover, and intravenous infusion of yohimbine and tyramine, 2 drugs that are known to release norepinephrine from cardiac sympathetic nerves.13,14 Cardiac sympathetic innervation was assessed by 6-[18F]fluorodopamine positron emission tomographic scanning.15
We hypothesized that if LF power indicated cardiac sympathetic innervation and function, then patients with neuroimaging or neurochemical evidence of cardiac sympathetic denervation would have low LF power and attenuated increments in LF power in response to yohimbine and tyramine. Alternatively, if LF power was reflective of baroreflex function, alterations of LF power would be independent of cardiac sympathetic innervation status and correlate with changes in baroreflex gain.
METHODS
The study protocols were approved by the Intramural Research Board of the National Institute of Neurological Disorders and Stroke. All subjects were studied at the National Institutes of Health Clinical Center after giving informed, written consent.
Subjects
The study subjects were separated into 4 groups, depending on their state of cardiac sympathetic innervation and baroreflex-cardiovagal slope (BRS; see below). There were 40 subjects with intact sympathetic innervation and normal BRS (Innervated-Normal BRS), 24 with intact sympathetic innervation and low BRS (Innervated-Low BRS), 4 with sympathetic denervation and normal BRS (Denervated-Normal BRS), and 30 with sympathetic denervation and low BRS (Denervated-Low BRS).
Autonomic function testing
Each subject was studied while supine with head on pillow after an overnight fast. Each patient had monitoring of the electrocardiogram and beat-tobeat blood pressure using either noninvasive devices (Finometer, Finapres Medical Systems, Amsterdam, the Netherlands; Portapres, Finapres Medical Systems; or Colin tonometer, Colin Medical Instruments, San Antonio, TX) or a brachial intra-arterial catheter. We previously studied formally and reported excellent agreement between intra-arterial and these noninvasively obtained measures of beat-to-beat blood pressure.16 Continuous vital signs data were digitized and recorded using a PowerLab (AD Instruments Pty Ltd, Castle Hill, Australia) data acquisition system and stored for later analysis on an Apple PowerBook G4 computer (Apple, Cupertino, CA).
After about a 10-min baseline period, each subject performed a Valsalva maneuver (30 mm Hg for 12 sec) at least 3 times.
Baroreflex function
As an index of baroreflex function, we used the slope of the relationship between cardiac interbeat interval and systolic blood pressure during phase II of the Valsalva maneuver.17 BRS, in units of msec/mm Hg, was calculated from the linear regression equation for the relationship between interbeat interval (with 1-beat delay) and systolic pressure. A BRS value of ≤3 msec/mm Hg was considered low.11
Pharmacologic testing
Pharmacologic testing was performed on completion of the autonomic evaluation, using either tyramine or yohimbine. If a subject received both drugs, each drug administration was on a separate day. The durations of drug infusion were sufficient for heart rate and blood pressure to reach plateau values.
In a total of 22 subjects (Table 1), yohimbine was infused intravenously at 62.5 μg/kg over 3 min and then at 0.5 μg/kg/min for 12 min. In a total of 50 subjects, tyramine was infused at a rate of 1 mg/min for 10 min. In patients with severe supine hypertension (systolic pressure more than 200 mm Hg) and orthostatic hypotension, the test drugs were infused during head-up tilting (15° to 30°), to decrease baseline pressure, or else the drugs were not given.
HRV analysis
LF power (0.04 to 0.15 Hz), HF power (0.16 to 0.4 Hz), and total power (TP, 0.0 to 0.4 Hz) were calculated using Chart 5.4.2 and the HRV module version 1.03 (PowerLab, AD Instruments Pty Ltd, Castle Hill, Australia). Stable heart rate epochs 3 to 5 min in duration were chosen for analysis. One epoch was sampled immediately before initiation of drug testing; the second followed attainment of steady-state hemodynamic effects. Interbeat interval data were reviewed carefully to eliminate artifacts from noise and T waves, using segments with little to no premature beats. LF power and HF power were calculated as absolute power (msec2), with or without normalization for total power (0.04 to 0.4 Hz). Reported LF or HF power was integrated within their defined frequency bands.
Cardiac sympathetic neuroimaging
For cardiac sympathetic neuroimaging the subject was positioned supine, feet-first in a GE Advance scanner (General Electric, Milwaukee, WI), with the thorax in the gantry. After positioning the patient with the thorax in the scanner and transmission scanning for attenuation correction, 6-[18F]fluorodopamine (usual dose 1 mCi, specific activity 1.0 to 4.0 Ci/mmole, in about 10 mL normal saline) was infused intravenously at a constant rate for 3 min, and dynamic scanning data were obtained for thoracic radioactivity, with the midpoint of the scanning interval at 7.5 min after injection of the tracer (data collection interval between 5 and 10 min). Cardiac sympathetic denervation was defined by low concentrations of 6-[18F] fluorodopamine-derived radioactivity in the interventricular septum (< 5,000 nCi-kg/cc-mCi) or left ventricular free wall (< 4,000 nCi-kg/cc-mCi) corresponding to about 2 SD below the normal means.
Cardiac norepinephrine spillover
Subgroups of subjects (3 PD + NOH, 3 MSA, 3 PAF, 5 normal volunteers) underwent right heart catheterization for measurement of cardiac norepinephrine spillover. 3H-Norepinephrine was infused intravenously, and arterial and coronary sinus blood was sampled and coronary sinus blood flow was measured by thermodilution for measurements of cardiac norepinephrine spillover as described previously.18 In some subjects, yohimbine was infused during cardiac catheterization. Patients with chronic autonomic failure received the doses described above; normal volunteers and patients with chronic orthostatic intolerance received twice the doses described above.
Data analysis
Statistical analyses were performed using StatView version 5.0.1. (SAS Institute, Cary, NC). Mean values in the baseline condition for the several subject groups were compared using single-factor ANOVA. Responses to drugs were analyzed by dependent-means t tests. Differences in response to pharmacologic tests among subject groups were compared using repeated measures analyses of variance. Relationships between individual hemodynamic values were assessed by linear regression and calculation of Pearson correlation coefficients. Post-hoc testing consisted of the Fisher PLSD test. Multiple regression analysis was done on the individual data, with the log of LF power as the dependent measure and the log of baroreflex slope and septal 6-[18F] fluorodopamine-derived radioactivity as independent measures. Mean values were expressed ± SEM.
RESULTS
Baseline
Across the 7 subject groups (N = 98), LF power was unrelated to subject group (F = 1.2). When individual subjects were stratified in terms of cardiac sympathetic denervation or innervation, based on concentrations of 6-[18F]fluorodopamine-derived radioactivity more than 2 SD below the normal mean, then LF power was lower in the Denervated group (mean 221 ± 55 msec2/Hz, N = 34) than in the Innervated group (516 ± 93 msec2/Hz, N = 64, F = 4.8, P = 0.03). LF power normalized for total power, HF normalized for total power, and the ratio of LF:HF were not related to 6-[18F]fluorodopamine-derived radioactivity.
When subjects were stratified in terms of BRS, then LF power was lower in the Low BRS group (223 ± 105 msec2/Hz, N = 46) than in the Normal BRS group (617 ± 97 msec2/Hz, N = 25, F = 6.1, P = 0.02). The Low BRS group did not differ from the Normal BRS group in normalized LF power (F = 0.8).
From multiple regression analysis for the log of LF power as the dependent measure and the log of baroreflex slope and septal 6-[18F]fluorodopamine-derived radioactivity as independent measures, the regression coefficient for the log of baroreflex slope was 0.92 (P < 0.0001), whereas the regression coefficient for 6-[18F] fluorodopamine-derived radioactivity was 3 ×10−6.
At baseline, the log of HF power correlated positively with the log of LF power (r = 0.77, P < 0.0001). HF power varied with the subject group (F = 4.9, P = 0.004). As with LF power, HF power was greater in the Innervated-Normal BRS than in the Innervated-Low BRS (P = 0.001, Table 2). As expected, the log of HF power correlated positively with the log of BRS (r = 0.60, P < 0.0001). The log of HF power also correlated negatively with the magnitude of decrease in systolic pressure during the Valsalva maneuver (r = −0.24, P = 0.02) and positively with the orthostatic change in systolic pressure (r = 0.40, P = 0.004).
Yohimbine
The change in LF power in response to yohimbine during cardiac catheterization was unrelated to the change in cardiac norepinephrine spillover (r = −0.09, N = 12).
Tyramine
Overall, tyramine infusion increased LF power (t = 2.9, P = 0.008). The group with cardiac sympathetic denervation did not differ from the group with intact cardiac innervation in terms of the change in LF power during tyramine infusion (F = 1.7). Tyramine increased LF power in the Innervated-Normal BRS group but not in the Innervated-Low BRS or Denervated-Low BRS groups (Figure 4; data for 2 outliers excluded). The log of the change in LF power during tyramine administration was positively correlated with the log of BRS at baseline (Figure 5; data for 2 outliers excluded).
DISCUSSION
In this study, patients with neuroimaging evidence of cardiac sympathetic denervation had low LF power of heart rate variability. At first glance, this finding would seem to support the view that LF power can provide an index of cardiac sympathetic outflow. As explained below, several lines of evidence from the present study led us to infer that the association between low LF power and cardiac sympathetic innervation is determined mainly by concurrent baroreflex function.
Patients with low BRS had low LF power, and patients with normal BRS had normal LF power, regardless of the status of cardiac sympathetic innervation as assessed by 6-[18F]fluorodopamine scanning. Neither normalization of LF and HF power for total power nor use of the LF:HF ratio improved the association with indices of cardiac sympathetic innervation.
Neurochemical findings during cardiac catheterization supported the above results based on cardiac sympathetic neuroimaging. Among patients with innervated hearts who had normal cardiac norepinephrine spillover, LF power was decreased only in the group with low BRS and was normal in the group with normal BRS. As expected, cardiac norepinephrine spillover was decreased in patients with neuroimaging evidence of cardiac sympathetic denervation.
Effects of pharmacological manipulations that increase norepinephrine release from sympathetic nerves provided further support for an association between baroreflex failure and low LF power, independent of cardiac sympathetic function. Both tyramine and yohimbine increased LF power only in the subjects with normal BRS. In subjects with low BRS, neither drug increased LF power, even in the group with intact cardiac sympathetic innervation. Moreover, individual values for responses of the log of LF power to both drugs were correlated positively with the log of BRS at baseline.
The fact that HF power was positively correlated with LF power could also fit with the notion of baroreflex function acting as a common determinant of values of both variables. We cannot exclude concurrent parasympathetic cardiovagal and sympathetic denervation as an explanation for the association between HF and LF power. Inhibition of the effects of parasympathetic activity after atropine administration results in the almost complete absence of both LF and HF HRV, further suggesting a common determinant.19
Several previous investigations have cast doubt on the validity of LF power as a measure of sympathetic activity because of dissociations between LF power and cardiac norepinephrine spillover, directly recorded sympathetic nerve traffic, and plasma norepinephrine levels.4,6,20 Such dissociations are especially glaring in patients with congestive heart failure, which is characterized by decreased LF power9 despite marked cardiac sympathetic activation.3
Other pathophysiologic states do result in both decreased sympathetic nervous system activity and decreased LF power. In these pathophysiologic states, the possibility remains that low LF power might reflect failure of baroreflexive modulation of sympathetic neuronal outflows, rather than sympathoinhibition itself. For instance, Wiklund et al21 noted low LF power in patients with palmar hyperhidrosis undergoing bilateral transthoracic sympathectomy; however, baroreflex-cardiovagal sensitivity also declines after thoracic sympathectomy.22
Sleight et al8 suggested dependence of LF power on baroreflex function, based on effects of carotid baroreceptor stimulation in 3 patients: 1 with normal BRS; 1 with ischemic heart disease, congestive heart failure, and normal BRS; and 1 with ischemic heart disease, congestive heart failure, and initially low BRS who subsequently had an improved clinical state and BRS. In the baseline state, both congestive heart failure patients had low LF power, despite a presumably hypernoradrenergic state. Direct baroreceptor stimulation at 0.1 Hz increased LF power in the normal subject and in the patient with congestive heart failure and normal BRS. The congestive heart failure patient with low BRS did not have an increase in LF power until BRS normalized. These data revealed an initial dissociation between cardiac noradrenergic state in the patients with congestive heart failure and LF power. During carotid sinus stimulation, LF power increased only when BRS was normal. Low BRS obviated this effect.
Because congestive heart failure is well known to be associated with baroreflex-cardiovagal inhibition,23–25 the finding of low LF power in heart failure also supports an association between LF power and BRS, independently of increased tonic release of norepinephrine from sympathetic nerves in the heart. Cevese et al26 inhibited noradrenergic vasomotor tone using an alpha-adrenoceptor blocker in human subjects while maintaining mean blood pressure at control levels using angiotensin II. This drug combination, which would be expected to attenuate sympathetically mediated vasomotor tone and thereby decrease arterial baroreceptor input, markedly decreased or abolished LF power of HRV, suggesting that, at least under resting supine conditions, a baroreflex mechanism accounts almost entirely for LF power of HRV.
deBoer et al27 developed a beat-to-beat model of the human circulation using a set of differential equations and the following principles of operation: (1) the baroreflex regulates heart rate and peripheral vascular resistance; (2) Windkessel properties characterize the systemic arterial tree; (3) contractile properties of the ventricular myocardium follow the Starling law; and (4) respiration exerts mechanical effects on blood pressure. The model attributes LF power to a resonance in the circulatory control system, produced by a slow time constant for reflexive sympathetically mediated responses to beat-tobeat blood pressure changes. The resonance can be upregulated or downregulated by the state of baroreflex activity. The model of deBoer et al predicts that changes in blood pressure would lead heart rate changes at 0.1 Hz through a delayed sympathetic response. Changes in HR would depend on summed effects of sympathetic and vagal effects, with the sympathetic response delaying the overall response. At the respiratory frequency (0.2 to 0.3 Hz), blood pressure and HR changes would occur with little delay because of fast parasympathetic control. In essence, the response of the sympathetic nervous system behaves as a low band pass filter, with a peak response at 0.1 Hz and little response at frequencies above 0.2 Hz. Systolic blood pressure would lead to changes in heart rate via the baroreflex. In general the results of this study fit with the deBoer model.
In conclusion, LF power derived from the interbeat interval spectrogram predominantly reflects baroreflex-mediated, phasic changes in cardiovagal and sympathetic noradrenergic outflows. In the setting of baroreflex failure, baseline LF power is reduced, regardless of the status of cardiac sympathetic innervation.
LIMITATIONS
The combination of cardiac sympathetic denervation and normal baroreflex function seems quite rare. One must exercise caution in drawing inferences from the findings in the Denervated-Normal BRS group, which contained only 4 subjects, even though the difference in mean log-transformed LF power from the Denervated-Low BRS group was highly statistically significant.
All of the testing in our study was done with the subjects supine. LF power measured in other positions might have different sources and meaning.
- Pagani M, Lombardi F, Guzzetti S, et al. Power spectral analysis of heart rate and arterial pressure variabilities as a marker of sympatho-vagal interaction in man and conscious dog. Circ Res 1986; 59:178–193.
- Alvarenga ME, Richards JC, Lambert G, Esler MD. Psychophysiological mechanisms in panic disorder: a correlative analysis of noradrenaline spillover, neuronal noradrenaline reuptake, power spectral analysis of heart rate variability, and psychological variables. Psychosom Med 2006; 68:8–16.
- Eisenhofer G, Friberg P, Rundqvist B, et al. Cardiac sympathetic nerve function in congestive heart failure. Circulation 1996; 93:1667–1676.
- Notarius CF, Butler GC, Ando S, Pollard MJ, Senn BL, Floras JS. Dissociation between microneurographic and heart rate variability estimates of sympathetic tone in normal subjects and patients with heart failure. Clin Sci (Lond) 1999; 96:557–565.
- Scalvini S, Volterrani M, Zanelli E, et al. Is heart rate variability a reliable method to assess autonomic modulation in left ventricular dysfunction and heart failure? Assessment of autonomic modulation with heart rate variability. Int J Cardiol 1998; 67:9–17.
- Kingwell BA, Thompson JM, Kaye DM, McPherson GA, Jennings GL, Esler MD. Heart rate spectral analysis, cardiac norepinephrine spillover, and muscle sympathetic nerve activity during human sympathetic nervous activation and failure. Circulation 1994; 90:234–240.
- van de Borne P, Montano N, Pagani M, Oren R, Somers VK. Absence of low-frequency variability of sympathetic nerve activity in severe heart failure. Circulation 1997; 95:1449–1454.
- Sleight P, La Rovere MT, Mortara A, et al. Physiology and pathophysiology of heart rate and blood pressure variability in humans: is power spectral analysis largely an index of baroreflex gain? Clin Sci (Lond) 1995; 88:103–109.
- Saul JP, Arai Y, Berger RD, Lilly LS, Colucci WS, Cohen RJ. Assessment of autonomic regulation in chronic congestive heart failure by heart rate spectral analysis. Am J Cardiol 1988; 61:1292–1299.
- Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO. Cardiac sympathetic denervation in Parkinson disease. Ann Intern Med 2000; 133:338–347.
- Goldstein DS, Eldadah BA, Holmes C, et al. Neurocirculatory abnormalities in Parkinson disease with orthostatic hypotension. Independence from levodopa treatment. Hypertension 2005; 46:1333–1339.
- Goldstein DS, Eldadah B, Holmes C, Pechnik S, Moak J, Sharabi Y. Neurocirculatory abnormalities in chronic orthostatic intolerance. Circulation 2005; 111:839–845.
- Goldstein DS, Holmes C, Frank SM, et al. Cardiac sympathetic dysautonomia in chronic orthostatic intolerance syndromes. Circulation 2002; 106:2358–2365.
- Lord SW, Clayton RH, Mitchell L, Dark JH, Murray A, McComb JM. Sympathetic reinnervation and heart rate variability after cardiac transplantation. Heart 1997; 77:532–538.
- Goldstein DS, Eisenhofer G, Dunn BB, et al. Positron emission tomographic imaging of cardiac sympathetic innervation using 6-[18F]fluorodopamine: initial findings in humans. J Am Coll Cardiol 1993; 22:1961–1971.
- Goldstein DS, Tack C. Non-invasive detection of sympathetic neurocirculatory failure. Clin Auton Res 2000; 10:285–291.
- Goldstein DS, Horwitz D, Keiser HR. Comparison of techniques for measuring baroreflex sensitivity in man. Circulation 1982; 66:432–439.
- Goldstein DS, Brush JE, Eisenhofer G, Stull R, Esler M. In vivo measurement of neuronal uptake of norepinephrine in the human heart. Circulation 1988; 78:41–48.
- Koh J, Brown TE, Beightol LA, Ha CY, Eckberg DL. Human autonomic rhythms: vagal cardiac mechanisms in tetraplegic subjects. J Physiol 1994; 474:483–495.
- Saul JP, Rea RF, Eckberg DL, Berger RD, Cohen RJ. Heart rate and muscle sympathetic nerve variability during reflex changes of autonomic activity. Am J Physiol 1990; 258:H713–H721.
- Wiklund U, Koskinen LO, Niklasson U, Bjerle P, Elfversson J. Endoscopic transthoracic sympathectomy affects the autonomic modulation of heart rate in patients with palmar hyperhidrosis. Acta Neurochir (Wien) 2000; 142:691–696.
- Kawamata YT, Kawamata T, Omote K, et al. Endoscopic thoracic sympathectomy suppresses baroreflex control of heart rate in patients with essential hyperhidrosis. Anesth Analg 2004; 98:37–39.
- Goldstein RE, Beiser GD, Stampfer M, Epstein SE. Impairment of autonomically mediated heart rate control in patients with cardiac dysfunction. Circ Res 1975; 36:571–578.
- Cody RJ, Franklin KW, Kluger J, Laragh JH. Mechanisms governing the postural response and baroreceptor abnormalities in chronic congestive heart failure: effects of acute and long-term convertingenzyme inhibition. Circulation 1982; 66:135–142.
- Creager MA. Baroreceptor reflex function in congestive heart failure. Am J Cardiol 1992; 69:10G–15G; discussion 15G–16G.
- Cevese A, Gulli G, Polati E, Gottin L, Grasso R. Baroreflex and oscillation of heart period at 0.1 Hz studied by alpha-blockade and crossspectral analysis in healthy humans. J Physiol 2001; 531:235–244.
- deBoer RW, Karemaker JM, Strackee J. Hemodynamic fluctuations and baroreflex sensitivity in humans: a beat-to-beat model. Am J Physiol 1987; 253:H680–689.
- Pagani M, Lombardi F, Guzzetti S, et al. Power spectral analysis of heart rate and arterial pressure variabilities as a marker of sympatho-vagal interaction in man and conscious dog. Circ Res 1986; 59:178–193.
- Alvarenga ME, Richards JC, Lambert G, Esler MD. Psychophysiological mechanisms in panic disorder: a correlative analysis of noradrenaline spillover, neuronal noradrenaline reuptake, power spectral analysis of heart rate variability, and psychological variables. Psychosom Med 2006; 68:8–16.
- Eisenhofer G, Friberg P, Rundqvist B, et al. Cardiac sympathetic nerve function in congestive heart failure. Circulation 1996; 93:1667–1676.
- Notarius CF, Butler GC, Ando S, Pollard MJ, Senn BL, Floras JS. Dissociation between microneurographic and heart rate variability estimates of sympathetic tone in normal subjects and patients with heart failure. Clin Sci (Lond) 1999; 96:557–565.
- Scalvini S, Volterrani M, Zanelli E, et al. Is heart rate variability a reliable method to assess autonomic modulation in left ventricular dysfunction and heart failure? Assessment of autonomic modulation with heart rate variability. Int J Cardiol 1998; 67:9–17.
- Kingwell BA, Thompson JM, Kaye DM, McPherson GA, Jennings GL, Esler MD. Heart rate spectral analysis, cardiac norepinephrine spillover, and muscle sympathetic nerve activity during human sympathetic nervous activation and failure. Circulation 1994; 90:234–240.
- van de Borne P, Montano N, Pagani M, Oren R, Somers VK. Absence of low-frequency variability of sympathetic nerve activity in severe heart failure. Circulation 1997; 95:1449–1454.
- Sleight P, La Rovere MT, Mortara A, et al. Physiology and pathophysiology of heart rate and blood pressure variability in humans: is power spectral analysis largely an index of baroreflex gain? Clin Sci (Lond) 1995; 88:103–109.
- Saul JP, Arai Y, Berger RD, Lilly LS, Colucci WS, Cohen RJ. Assessment of autonomic regulation in chronic congestive heart failure by heart rate spectral analysis. Am J Cardiol 1988; 61:1292–1299.
- Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO. Cardiac sympathetic denervation in Parkinson disease. Ann Intern Med 2000; 133:338–347.
- Goldstein DS, Eldadah BA, Holmes C, et al. Neurocirculatory abnormalities in Parkinson disease with orthostatic hypotension. Independence from levodopa treatment. Hypertension 2005; 46:1333–1339.
- Goldstein DS, Eldadah B, Holmes C, Pechnik S, Moak J, Sharabi Y. Neurocirculatory abnormalities in chronic orthostatic intolerance. Circulation 2005; 111:839–845.
- Goldstein DS, Holmes C, Frank SM, et al. Cardiac sympathetic dysautonomia in chronic orthostatic intolerance syndromes. Circulation 2002; 106:2358–2365.
- Lord SW, Clayton RH, Mitchell L, Dark JH, Murray A, McComb JM. Sympathetic reinnervation and heart rate variability after cardiac transplantation. Heart 1997; 77:532–538.
- Goldstein DS, Eisenhofer G, Dunn BB, et al. Positron emission tomographic imaging of cardiac sympathetic innervation using 6-[18F]fluorodopamine: initial findings in humans. J Am Coll Cardiol 1993; 22:1961–1971.
- Goldstein DS, Tack C. Non-invasive detection of sympathetic neurocirculatory failure. Clin Auton Res 2000; 10:285–291.
- Goldstein DS, Horwitz D, Keiser HR. Comparison of techniques for measuring baroreflex sensitivity in man. Circulation 1982; 66:432–439.
- Goldstein DS, Brush JE, Eisenhofer G, Stull R, Esler M. In vivo measurement of neuronal uptake of norepinephrine in the human heart. Circulation 1988; 78:41–48.
- Koh J, Brown TE, Beightol LA, Ha CY, Eckberg DL. Human autonomic rhythms: vagal cardiac mechanisms in tetraplegic subjects. J Physiol 1994; 474:483–495.
- Saul JP, Rea RF, Eckberg DL, Berger RD, Cohen RJ. Heart rate and muscle sympathetic nerve variability during reflex changes of autonomic activity. Am J Physiol 1990; 258:H713–H721.
- Wiklund U, Koskinen LO, Niklasson U, Bjerle P, Elfversson J. Endoscopic transthoracic sympathectomy affects the autonomic modulation of heart rate in patients with palmar hyperhidrosis. Acta Neurochir (Wien) 2000; 142:691–696.
- Kawamata YT, Kawamata T, Omote K, et al. Endoscopic thoracic sympathectomy suppresses baroreflex control of heart rate in patients with essential hyperhidrosis. Anesth Analg 2004; 98:37–39.
- Goldstein RE, Beiser GD, Stampfer M, Epstein SE. Impairment of autonomically mediated heart rate control in patients with cardiac dysfunction. Circ Res 1975; 36:571–578.
- Cody RJ, Franklin KW, Kluger J, Laragh JH. Mechanisms governing the postural response and baroreceptor abnormalities in chronic congestive heart failure: effects of acute and long-term convertingenzyme inhibition. Circulation 1982; 66:135–142.
- Creager MA. Baroreceptor reflex function in congestive heart failure. Am J Cardiol 1992; 69:10G–15G; discussion 15G–16G.
- Cevese A, Gulli G, Polati E, Gottin L, Grasso R. Baroreflex and oscillation of heart period at 0.1 Hz studied by alpha-blockade and crossspectral analysis in healthy humans. J Physiol 2001; 531:235–244.
- deBoer RW, Karemaker JM, Strackee J. Hemodynamic fluctuations and baroreflex sensitivity in humans: a beat-to-beat model. Am J Physiol 1987; 253:H680–689.