User login
Although the US Food and Drug Administration (FDA) has granted emergency use authorization of remdesivir (Gilead Sciences, Inc., Foster City, California) to treat COVID-19, the disease caused by SARS-CoV-2, the drug is considered an investigational agent, not yet formally approved by the FDA and whose efficacy and safety has not yet been fully characterized. Remdesivir has been shown to be effective in reducing the time to recovery of people with COVID-19 disease. It has not been tested in a large controlled clinical trial of pregnant women with COVID-19; however, remdesivir has been given to pregnant women infected with COVID-19 in a compassionate use protocol. For pregnant women, the drug should only be used if the potential benefit justifies the potential risk to the mother and fetus.1
Pharmacology. Remdesivir is a nucleoside RNA polymerase inhibitor. It has a molecular formula of
C27H35N6O8P and a molecular weight of 602.6 g/mol.1
Mechanism of action. From FDA’s fact sheet: “Remdesivir is an adenosine nucleotide prodrug that distributes into cells where it is metabolized to form the pharmacologically active nucleoside triphosphate metabolite. Metabolism of remdesivir to remdesivir triphosphate has been demonstrated in multiple cell types. Remdesivir triphosphate acts as an analog of adenosine triphosphate (ATP) and competes with the natural ATP substrate for incorporation into nascent RNA chains by the SARS-CoV-2 RNA-dependent RNA polymerase, which results in chain termination during replication of the viral RNA. Remdesivir triphosphate is a weak inhibitor of mammalian DNA and RNA polymerases with low potential for mitochondrial toxicity.”1
Treatment protocols
Remdesivir is authorized for treatment of hospitalized patients with severe COVID-19 disease, defined as patients with an oxygen saturation ≤ 94% on room air or requiring supplemental oxygen or requiring mechanical ventilation or requiring extracorporeal membrane oxygenation (ECMO). The optimal dose and duration of treatment of COVID-19 with remdesivir is unknown.1
Prior to initiating treatment, the estimated glomerular filtration rate should be documented to be ≥ 30 mL/min. An excipient used in the remdesivir formulation—sulfobutylether-β-cylcodextrin sodium salt—is renally cleared and accumulates in patients with decreased renal function.
Baseline liver function tests should be performed prior to treatment and daily during the course of treatment. Remdesivir should not be initiated in patients with an alanine aminotransferase (ALT) level ≥ 5 times the upper limit of normal at baseline. Remdesivir should be discontinued in patients who develop an ALT level ≥ 5 times the upper limit of normal or in patients who develop elevated ALT levels and have increased bilirubin, alkaline phosphatase, or international normalized ratio.1
In one open-label study (GS-US-540-5773), remdesivir treatment was discontinued due to an adverse event in 5% of patients on a 5-day regimen and in 10% of patients on a 10-day regimen.1
Under the emergency use authorization, two treatment protocols have been proposed depending on the clinical severity of the COVID-19 infection1:
- Protocol 1: For people with COVID-19 requiring mechanical ventilation and/or ECMO, the duration of therapy is 10 days, beginning with a loading dose of remdesivir 200 mg infused intravenously for 30 to 120 minutes on day 1 followed by a once-daily dose of 100 mg for 9 days.
- Protocol 2: For people with COVID-19 disease not requiring mechanical ventilation and/or ECMO, the duration of therapy is 5 days, beginning with a loading dose of remdesivir 200 mg infused intravenously for 30 to 120 minutes on day 1 followed by a once-daily dose of 100 mg for 4 days. If the patient does not show clinical improvement, treatment may be extended for an additional 5 days.
Continue to: Randomized placebo-controlled trial results...
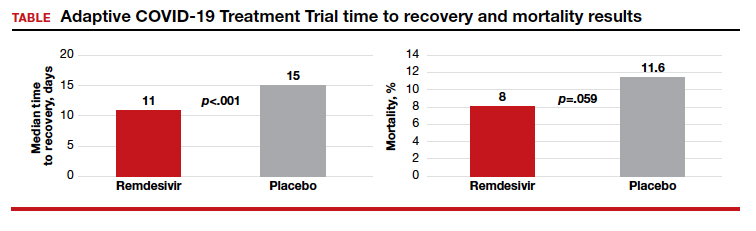
Randomized placebo-controlled trial results
The Adaptive COVID-19 Treatment Trial (ACTT), sponsored by the National Institute of Allergy and Infectious Diseases, is a randomized, double-blind, placebo-controlled trial conducted by Gilead Sciences. The study began in February and evaluated up to 10 days of remdesivir treatment—200 mg IV once daily for 1 day followed by 100 mg IV once daily for 9 days in hospitalized adult patients with COVID-19. Patients were enrolled in a 1:1 manner to remdesivir or placebo, and time to recovery within 28 days after randomization was the trial’s endpoint. According to preliminary analysis of 606 recovered patients, recovery took a median of 11 days in the remdesivir group and 15 days in the placebo group (hazard ratio, 1.31; 95% confidence interval (CI), 1.12‒1.54; P<.001). Mortality rates were 8.0% and 11.6% in the remdesivir and placebo groups, respectively (P=.059).1
5 vs 10 days of remdesivir treatment
The Gilead Sciences‒sponsored study GS-US-540-5773 was a randomized, open-label multicenter trial of patients with severe COVID-19. A total of 197 adult patients received 10-day remdesivir treatment (200 mg IV once daily for 1 day followed by 100 mg IV once daily for 9 days). An additional 200 adult patients received 5-day remdesivir treatment (200 mg IV once daily followed by 100 mg IV for 4 days). Both groups also received standard of care. Results suggested that patients receiving 10 days of remdesivir had similar improvement in clinical status compared with those receiving a 5-day treatment course (10-to-5 day odds ratio, 0.76; 95% CI, 0.51‒1.13] on day 14).1 Improvement in clinical status was defined as an improvement of 2 or more points from baseline on a predefined 7-point scale that ranged from hospital discharge to increasing levels of oxygen support to death. Clinical recovery was achieved if patients ceased the need for oxygen support or were discharged.1
The time to clinical improvement for 50% of patients was similar in each treatment group (10 days in the 5-day group versus 11 days in the 10-day group). By day 14, observed clinical improvement rates were 65% and 54% in the 5- and 10-day treatment groups, respectively. Clinical recovery rates were 70% and 59% in the 5- and 10-day treatment groups and mortality rates were 8% and 11%.1
Adverse events
The use of remdesivir is contraindicated in patients who are hypersensitive to the drug. Its infusion may cause hypotension, nausea, vomiting, diaphoresis, and shivering. If signs of a clinically significant infusion reaction are observed the infusion should be discontinued. As noted above, elevation in ALT levels occurs with remdesivir treatment.1
Reporting serious adverse events. If a serious and unexpected adverse event occurs and appears to be associated with the use of remdesivir, the prescribing health care provider and/or the provider’s designee should complete and submit a MedWatch form to the FDA using one of the following methods1:
- Complete and submit the report online: www.fda.gov/medwatch/report.htm
- Return form FDA 3500 (available at http://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Forms/UCM163919.pdf) to the FDA by mail (MedWatch, 5600 Fishers Lane, Rockville, MD 20852-9787) or fax (1-800-FDA-0178)
- Gilead requests that all FDA MedWatch forms also be returned to Gilead Pharmacovigilance and Epidemiology: fax: 1-650-522-5477 726; e-mail: Safety_fc@gilead.com
Continue to: Drug interactions...
Drug interactions
Remdesivir has not been evaluated for drug-drug interactions in humans. The clinical relevance of in vitro drug interactions also has not been established. According to the FDA, remdesivir is a substrate for the drug metabolizing enzymes CYP2C8, CYP2D6, and CYP3A4, and is a substrate for organic anion transporting polypeptides 1B1 (OAPT1B1) and P-glycoprotein (P-gp) transporters. In vitro, remdesivir inhibits CYP3A4, OATP1B1, OATP1B3, BSEP, MRP4, and NTCP.1
Pregnancy risk summary
Remdesivir has not been studied adequately in pregnant women and only should be used during pregnancy if the potential benefit of the drug justifies the potential risk to both mother and fetus.
Nonclinical animal studies that included systemic exposure of the predominant circulating metabolite of remdesivir in pregnant rats and rabbits (at 4 times the recommended dose of human exposure) demonstrated no adverse effect on embryofetal development.1
Breastfeeding
The only information regarding breastfeeding and remdesivir comes from animal studies. The drug and its metabolites were detected in the plasma of nursing rat pups whose mothers given intravenous remdesivir daily from gestation day 6 to lactation day 20. Measured on lactation day 10, remdesivir exposure in the pups was about 1% that of maternal exposure.1
“Because of the potential for viral transmission to SARS-CoV-2-negative infants and adverse reactions from the drug in breastfeeding infants, the developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for remdesivir and any potential adverse effects on the breastfed child from remdesivir or from the underlying maternal condition.”1
- US Food and Drug Administration. Fact Sheet for Health Care Providers Emergency Use Authorization (UA) of Remdesivir (GS-5734)TM. https://www.fda.gov/media/137566/download. Accessed May 19, 2020.
Although the US Food and Drug Administration (FDA) has granted emergency use authorization of remdesivir (Gilead Sciences, Inc., Foster City, California) to treat COVID-19, the disease caused by SARS-CoV-2, the drug is considered an investigational agent, not yet formally approved by the FDA and whose efficacy and safety has not yet been fully characterized. Remdesivir has been shown to be effective in reducing the time to recovery of people with COVID-19 disease. It has not been tested in a large controlled clinical trial of pregnant women with COVID-19; however, remdesivir has been given to pregnant women infected with COVID-19 in a compassionate use protocol. For pregnant women, the drug should only be used if the potential benefit justifies the potential risk to the mother and fetus.1
Pharmacology. Remdesivir is a nucleoside RNA polymerase inhibitor. It has a molecular formula of
C27H35N6O8P and a molecular weight of 602.6 g/mol.1
Mechanism of action. From FDA’s fact sheet: “Remdesivir is an adenosine nucleotide prodrug that distributes into cells where it is metabolized to form the pharmacologically active nucleoside triphosphate metabolite. Metabolism of remdesivir to remdesivir triphosphate has been demonstrated in multiple cell types. Remdesivir triphosphate acts as an analog of adenosine triphosphate (ATP) and competes with the natural ATP substrate for incorporation into nascent RNA chains by the SARS-CoV-2 RNA-dependent RNA polymerase, which results in chain termination during replication of the viral RNA. Remdesivir triphosphate is a weak inhibitor of mammalian DNA and RNA polymerases with low potential for mitochondrial toxicity.”1
Treatment protocols
Remdesivir is authorized for treatment of hospitalized patients with severe COVID-19 disease, defined as patients with an oxygen saturation ≤ 94% on room air or requiring supplemental oxygen or requiring mechanical ventilation or requiring extracorporeal membrane oxygenation (ECMO). The optimal dose and duration of treatment of COVID-19 with remdesivir is unknown.1
Prior to initiating treatment, the estimated glomerular filtration rate should be documented to be ≥ 30 mL/min. An excipient used in the remdesivir formulation—sulfobutylether-β-cylcodextrin sodium salt—is renally cleared and accumulates in patients with decreased renal function.
Baseline liver function tests should be performed prior to treatment and daily during the course of treatment. Remdesivir should not be initiated in patients with an alanine aminotransferase (ALT) level ≥ 5 times the upper limit of normal at baseline. Remdesivir should be discontinued in patients who develop an ALT level ≥ 5 times the upper limit of normal or in patients who develop elevated ALT levels and have increased bilirubin, alkaline phosphatase, or international normalized ratio.1
In one open-label study (GS-US-540-5773), remdesivir treatment was discontinued due to an adverse event in 5% of patients on a 5-day regimen and in 10% of patients on a 10-day regimen.1
Under the emergency use authorization, two treatment protocols have been proposed depending on the clinical severity of the COVID-19 infection1:
- Protocol 1: For people with COVID-19 requiring mechanical ventilation and/or ECMO, the duration of therapy is 10 days, beginning with a loading dose of remdesivir 200 mg infused intravenously for 30 to 120 minutes on day 1 followed by a once-daily dose of 100 mg for 9 days.
- Protocol 2: For people with COVID-19 disease not requiring mechanical ventilation and/or ECMO, the duration of therapy is 5 days, beginning with a loading dose of remdesivir 200 mg infused intravenously for 30 to 120 minutes on day 1 followed by a once-daily dose of 100 mg for 4 days. If the patient does not show clinical improvement, treatment may be extended for an additional 5 days.
Continue to: Randomized placebo-controlled trial results...
Randomized placebo-controlled trial results
The Adaptive COVID-19 Treatment Trial (ACTT), sponsored by the National Institute of Allergy and Infectious Diseases, is a randomized, double-blind, placebo-controlled trial conducted by Gilead Sciences. The study began in February and evaluated up to 10 days of remdesivir treatment—200 mg IV once daily for 1 day followed by 100 mg IV once daily for 9 days in hospitalized adult patients with COVID-19. Patients were enrolled in a 1:1 manner to remdesivir or placebo, and time to recovery within 28 days after randomization was the trial’s endpoint. According to preliminary analysis of 606 recovered patients, recovery took a median of 11 days in the remdesivir group and 15 days in the placebo group (hazard ratio, 1.31; 95% confidence interval (CI), 1.12‒1.54; P<.001). Mortality rates were 8.0% and 11.6% in the remdesivir and placebo groups, respectively (P=.059).1
5 vs 10 days of remdesivir treatment
The Gilead Sciences‒sponsored study GS-US-540-5773 was a randomized, open-label multicenter trial of patients with severe COVID-19. A total of 197 adult patients received 10-day remdesivir treatment (200 mg IV once daily for 1 day followed by 100 mg IV once daily for 9 days). An additional 200 adult patients received 5-day remdesivir treatment (200 mg IV once daily followed by 100 mg IV for 4 days). Both groups also received standard of care. Results suggested that patients receiving 10 days of remdesivir had similar improvement in clinical status compared with those receiving a 5-day treatment course (10-to-5 day odds ratio, 0.76; 95% CI, 0.51‒1.13] on day 14).1 Improvement in clinical status was defined as an improvement of 2 or more points from baseline on a predefined 7-point scale that ranged from hospital discharge to increasing levels of oxygen support to death. Clinical recovery was achieved if patients ceased the need for oxygen support or were discharged.1
The time to clinical improvement for 50% of patients was similar in each treatment group (10 days in the 5-day group versus 11 days in the 10-day group). By day 14, observed clinical improvement rates were 65% and 54% in the 5- and 10-day treatment groups, respectively. Clinical recovery rates were 70% and 59% in the 5- and 10-day treatment groups and mortality rates were 8% and 11%.1
Adverse events
The use of remdesivir is contraindicated in patients who are hypersensitive to the drug. Its infusion may cause hypotension, nausea, vomiting, diaphoresis, and shivering. If signs of a clinically significant infusion reaction are observed the infusion should be discontinued. As noted above, elevation in ALT levels occurs with remdesivir treatment.1
Reporting serious adverse events. If a serious and unexpected adverse event occurs and appears to be associated with the use of remdesivir, the prescribing health care provider and/or the provider’s designee should complete and submit a MedWatch form to the FDA using one of the following methods1:
- Complete and submit the report online: www.fda.gov/medwatch/report.htm
- Return form FDA 3500 (available at http://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Forms/UCM163919.pdf) to the FDA by mail (MedWatch, 5600 Fishers Lane, Rockville, MD 20852-9787) or fax (1-800-FDA-0178)
- Gilead requests that all FDA MedWatch forms also be returned to Gilead Pharmacovigilance and Epidemiology: fax: 1-650-522-5477 726; e-mail: Safety_fc@gilead.com
Continue to: Drug interactions...
Drug interactions
Remdesivir has not been evaluated for drug-drug interactions in humans. The clinical relevance of in vitro drug interactions also has not been established. According to the FDA, remdesivir is a substrate for the drug metabolizing enzymes CYP2C8, CYP2D6, and CYP3A4, and is a substrate for organic anion transporting polypeptides 1B1 (OAPT1B1) and P-glycoprotein (P-gp) transporters. In vitro, remdesivir inhibits CYP3A4, OATP1B1, OATP1B3, BSEP, MRP4, and NTCP.1
Pregnancy risk summary
Remdesivir has not been studied adequately in pregnant women and only should be used during pregnancy if the potential benefit of the drug justifies the potential risk to both mother and fetus.
Nonclinical animal studies that included systemic exposure of the predominant circulating metabolite of remdesivir in pregnant rats and rabbits (at 4 times the recommended dose of human exposure) demonstrated no adverse effect on embryofetal development.1
Breastfeeding
The only information regarding breastfeeding and remdesivir comes from animal studies. The drug and its metabolites were detected in the plasma of nursing rat pups whose mothers given intravenous remdesivir daily from gestation day 6 to lactation day 20. Measured on lactation day 10, remdesivir exposure in the pups was about 1% that of maternal exposure.1
“Because of the potential for viral transmission to SARS-CoV-2-negative infants and adverse reactions from the drug in breastfeeding infants, the developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for remdesivir and any potential adverse effects on the breastfed child from remdesivir or from the underlying maternal condition.”1
Although the US Food and Drug Administration (FDA) has granted emergency use authorization of remdesivir (Gilead Sciences, Inc., Foster City, California) to treat COVID-19, the disease caused by SARS-CoV-2, the drug is considered an investigational agent, not yet formally approved by the FDA and whose efficacy and safety has not yet been fully characterized. Remdesivir has been shown to be effective in reducing the time to recovery of people with COVID-19 disease. It has not been tested in a large controlled clinical trial of pregnant women with COVID-19; however, remdesivir has been given to pregnant women infected with COVID-19 in a compassionate use protocol. For pregnant women, the drug should only be used if the potential benefit justifies the potential risk to the mother and fetus.1
Pharmacology. Remdesivir is a nucleoside RNA polymerase inhibitor. It has a molecular formula of
C27H35N6O8P and a molecular weight of 602.6 g/mol.1
Mechanism of action. From FDA’s fact sheet: “Remdesivir is an adenosine nucleotide prodrug that distributes into cells where it is metabolized to form the pharmacologically active nucleoside triphosphate metabolite. Metabolism of remdesivir to remdesivir triphosphate has been demonstrated in multiple cell types. Remdesivir triphosphate acts as an analog of adenosine triphosphate (ATP) and competes with the natural ATP substrate for incorporation into nascent RNA chains by the SARS-CoV-2 RNA-dependent RNA polymerase, which results in chain termination during replication of the viral RNA. Remdesivir triphosphate is a weak inhibitor of mammalian DNA and RNA polymerases with low potential for mitochondrial toxicity.”1
Treatment protocols
Remdesivir is authorized for treatment of hospitalized patients with severe COVID-19 disease, defined as patients with an oxygen saturation ≤ 94% on room air or requiring supplemental oxygen or requiring mechanical ventilation or requiring extracorporeal membrane oxygenation (ECMO). The optimal dose and duration of treatment of COVID-19 with remdesivir is unknown.1
Prior to initiating treatment, the estimated glomerular filtration rate should be documented to be ≥ 30 mL/min. An excipient used in the remdesivir formulation—sulfobutylether-β-cylcodextrin sodium salt—is renally cleared and accumulates in patients with decreased renal function.
Baseline liver function tests should be performed prior to treatment and daily during the course of treatment. Remdesivir should not be initiated in patients with an alanine aminotransferase (ALT) level ≥ 5 times the upper limit of normal at baseline. Remdesivir should be discontinued in patients who develop an ALT level ≥ 5 times the upper limit of normal or in patients who develop elevated ALT levels and have increased bilirubin, alkaline phosphatase, or international normalized ratio.1
In one open-label study (GS-US-540-5773), remdesivir treatment was discontinued due to an adverse event in 5% of patients on a 5-day regimen and in 10% of patients on a 10-day regimen.1
Under the emergency use authorization, two treatment protocols have been proposed depending on the clinical severity of the COVID-19 infection1:
- Protocol 1: For people with COVID-19 requiring mechanical ventilation and/or ECMO, the duration of therapy is 10 days, beginning with a loading dose of remdesivir 200 mg infused intravenously for 30 to 120 minutes on day 1 followed by a once-daily dose of 100 mg for 9 days.
- Protocol 2: For people with COVID-19 disease not requiring mechanical ventilation and/or ECMO, the duration of therapy is 5 days, beginning with a loading dose of remdesivir 200 mg infused intravenously for 30 to 120 minutes on day 1 followed by a once-daily dose of 100 mg for 4 days. If the patient does not show clinical improvement, treatment may be extended for an additional 5 days.
Continue to: Randomized placebo-controlled trial results...
Randomized placebo-controlled trial results
The Adaptive COVID-19 Treatment Trial (ACTT), sponsored by the National Institute of Allergy and Infectious Diseases, is a randomized, double-blind, placebo-controlled trial conducted by Gilead Sciences. The study began in February and evaluated up to 10 days of remdesivir treatment—200 mg IV once daily for 1 day followed by 100 mg IV once daily for 9 days in hospitalized adult patients with COVID-19. Patients were enrolled in a 1:1 manner to remdesivir or placebo, and time to recovery within 28 days after randomization was the trial’s endpoint. According to preliminary analysis of 606 recovered patients, recovery took a median of 11 days in the remdesivir group and 15 days in the placebo group (hazard ratio, 1.31; 95% confidence interval (CI), 1.12‒1.54; P<.001). Mortality rates were 8.0% and 11.6% in the remdesivir and placebo groups, respectively (P=.059).1
5 vs 10 days of remdesivir treatment
The Gilead Sciences‒sponsored study GS-US-540-5773 was a randomized, open-label multicenter trial of patients with severe COVID-19. A total of 197 adult patients received 10-day remdesivir treatment (200 mg IV once daily for 1 day followed by 100 mg IV once daily for 9 days). An additional 200 adult patients received 5-day remdesivir treatment (200 mg IV once daily followed by 100 mg IV for 4 days). Both groups also received standard of care. Results suggested that patients receiving 10 days of remdesivir had similar improvement in clinical status compared with those receiving a 5-day treatment course (10-to-5 day odds ratio, 0.76; 95% CI, 0.51‒1.13] on day 14).1 Improvement in clinical status was defined as an improvement of 2 or more points from baseline on a predefined 7-point scale that ranged from hospital discharge to increasing levels of oxygen support to death. Clinical recovery was achieved if patients ceased the need for oxygen support or were discharged.1
The time to clinical improvement for 50% of patients was similar in each treatment group (10 days in the 5-day group versus 11 days in the 10-day group). By day 14, observed clinical improvement rates were 65% and 54% in the 5- and 10-day treatment groups, respectively. Clinical recovery rates were 70% and 59% in the 5- and 10-day treatment groups and mortality rates were 8% and 11%.1
Adverse events
The use of remdesivir is contraindicated in patients who are hypersensitive to the drug. Its infusion may cause hypotension, nausea, vomiting, diaphoresis, and shivering. If signs of a clinically significant infusion reaction are observed the infusion should be discontinued. As noted above, elevation in ALT levels occurs with remdesivir treatment.1
Reporting serious adverse events. If a serious and unexpected adverse event occurs and appears to be associated with the use of remdesivir, the prescribing health care provider and/or the provider’s designee should complete and submit a MedWatch form to the FDA using one of the following methods1:
- Complete and submit the report online: www.fda.gov/medwatch/report.htm
- Return form FDA 3500 (available at http://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Forms/UCM163919.pdf) to the FDA by mail (MedWatch, 5600 Fishers Lane, Rockville, MD 20852-9787) or fax (1-800-FDA-0178)
- Gilead requests that all FDA MedWatch forms also be returned to Gilead Pharmacovigilance and Epidemiology: fax: 1-650-522-5477 726; e-mail: Safety_fc@gilead.com
Continue to: Drug interactions...
Drug interactions
Remdesivir has not been evaluated for drug-drug interactions in humans. The clinical relevance of in vitro drug interactions also has not been established. According to the FDA, remdesivir is a substrate for the drug metabolizing enzymes CYP2C8, CYP2D6, and CYP3A4, and is a substrate for organic anion transporting polypeptides 1B1 (OAPT1B1) and P-glycoprotein (P-gp) transporters. In vitro, remdesivir inhibits CYP3A4, OATP1B1, OATP1B3, BSEP, MRP4, and NTCP.1
Pregnancy risk summary
Remdesivir has not been studied adequately in pregnant women and only should be used during pregnancy if the potential benefit of the drug justifies the potential risk to both mother and fetus.
Nonclinical animal studies that included systemic exposure of the predominant circulating metabolite of remdesivir in pregnant rats and rabbits (at 4 times the recommended dose of human exposure) demonstrated no adverse effect on embryofetal development.1
Breastfeeding
The only information regarding breastfeeding and remdesivir comes from animal studies. The drug and its metabolites were detected in the plasma of nursing rat pups whose mothers given intravenous remdesivir daily from gestation day 6 to lactation day 20. Measured on lactation day 10, remdesivir exposure in the pups was about 1% that of maternal exposure.1
“Because of the potential for viral transmission to SARS-CoV-2-negative infants and adverse reactions from the drug in breastfeeding infants, the developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for remdesivir and any potential adverse effects on the breastfed child from remdesivir or from the underlying maternal condition.”1
- US Food and Drug Administration. Fact Sheet for Health Care Providers Emergency Use Authorization (UA) of Remdesivir (GS-5734)TM. https://www.fda.gov/media/137566/download. Accessed May 19, 2020.
- US Food and Drug Administration. Fact Sheet for Health Care Providers Emergency Use Authorization (UA) of Remdesivir (GS-5734)TM. https://www.fda.gov/media/137566/download. Accessed May 19, 2020.