User login
The Diagnosis: Relapsing Polychondritis
Due to suspicion of relapsing polychondritis (RP), we also performed an audiometric evaluation, which demonstrated bilateral sensorineural hearing loss. Echocardiography highlighted mild to moderate mitralic and tricuspidal insufficiency without hemodynamic impairment (ejection fraction, 50%). Corticosteroid therapy was started (prednisone 0.5 mg/kg/d). After 7 days of treatment, inflammation was remarkably reduced, and the patient no longer reported pain.
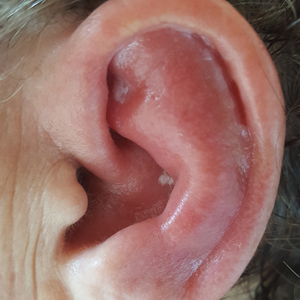
Relapsing polychondritis is a rare noninfective condition characterized by focal inflammatory destruction of ear cartilage, followed by fibroblastic regeneration. It often is associated with ocular inflammation, including conjunctivitis, scleritis, and episcleritis; cochlear or vestibular lesions; and seronegative nonerosive inflammatory arthritis.1 Clinical examination of the affected area shows swelling, redness, and tenderness of the ear, which could lead to a misdiagnosis of cellulitis. A typical and useful differentiating sign is the sparing of the noncartilaginous parts of the ear lobule. If not promptly diagnosed and treated, the destructive process can cause thinning of the cartilage, leading to deformities of the external ear.
The differential diagnosis includes erysipelas, which presents as a rapidly appearing inflammatory patch with sharply defined borders, accompanied by regional lymphadenopathy or skin streaking as well as fever. Sweet syndrome usually presents with tender erythematous or violaceous skin papules, plaques, or nodules, frequently with a pseudovesicular appearance; patients generally present with a classic fever and peripheral neutrophilia.2 The localized cutaneous form of leishmaniasis usually appears with a papule that generally develops into an ulcerative nodular lesion. Our patient did not have a history of exposure to topical substances that could point to photocontact dermatitis.
Dion et al3 proposed 3 distinct clinical phenotypes of RP: (1) patients with concomitant myelodysplastic syndrome or other hematologic malignancy (<10% of patients), mostly older men with a poor prognosis; (2) patients with tracheobronchial involvement (approximately 25% of patients); and (3) patients who do not have hematologic or tracheobronchial involvement (approximately 65% of patients) with a good prognosis.
Two sets of diagnostic criteria have been proposed. The criteria from McAdam et al4 required the presence of 3 or more of the following clinical features: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation (eg, conjunctivitis, keratitis, scleritis/episcleritis, uveitis), respiratory tract chondritis (laryngeal and/or tracheal cartilages), and cochlear and/or vestibular dysfunction (eg, neurosensory hearing loss, tinnitus, vertigo). These criteria were modified by Damiani and Levine.5 According to the latter, all patients were required to have one of the following: at least 4 of the McAdam et al4 diagnostic criteria; 1 or more of the clinical findings included in the McAdam et al4 criteria with histologic features suggestive for RP; or chondritis at 2 or more separate anatomic locations with a response to glucocorticoids and/or dapsone.
No laboratory findings are specific for RP, and nonspecific indicators of inflammation--elevated erythrocyte sedimentation rate and C-reactive protein--often are present.
The cause of RP is unknown. Familial clustering has not been observed. Terao et al6 found that HLA-DRB1*1602, -DQB1*0502, and -B*6701, in linkage disequilibrium with each other, are associated with susceptibility to RP.
There is no universal consensus about treatment, but a course of steroids leads to the resolution of the acute phase. Maintenance treatment can include dapsone, azathioprine, methotrexate, cyclophosphamide, and cyclosporine.7,8 Some studies have described the successful use of anti-tumor necrosis factor α inhibitors and rituximab.9,10
- Borgia F, Giuffrida R, Guarneri F, et al. Relapsing polychondritis: an updated review. Biomedicines. 2018;6:84.
- Rednic S, Damian L, Talarico R, et al. Relapsing polychondritis: state of the art on clinical practice guidelines. RMD Open. 2018:4(suppl 1)e000788.
- Dion J, Costedoat-Chalumeau N, Sène D, et al. Relapsing polychondritis can be characterized by three different clinical phenotypes: analysis of a recent series of 142 patients. Arthritis Rheumatol. 2016;68:2992-3001.
- McAdam LP, O'Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore). 1976;55:193.
- Damiani JM, Levine HL. Relapsing polychondritis--report of ten cases. Laryngoscope. 1979;89:929-946.
- Terao C, Yoshifuji H, Yamano Y, et al. Genotyping of relapsing polychondritis identified novel susceptibility HLA alleles and distinct genetic characteristics from other rheumatic diseases. Rheumatology (Oxford). 2016;55:1686.016-82
- Goldenberg G, Sangueza OP, Jorizzo JL. Successful treatment of relapsing polychondritis with mycophenolate mofetil. J Dermatolog Treat. 2006;17:158-159.
- Handler RP. Leflunomide for relapsing polychondritis: successful longterm treatment. J Rheumatol. 2006;33:1916; author reply 1916-1917.
- Carter JD. Treatment of relapsing polychondritis with a TNF antagonist. J Rheumatol. 2005;32:1413.
- Leroux G, Costedoat-Chalumeau N, Brihaye B, et al. Treatment of relapsing polychondritis with rituximab: a retrospective study of nine patients. Arthritis Rheum. 2009;61:577-582.
The Diagnosis: Relapsing Polychondritis
Due to suspicion of relapsing polychondritis (RP), we also performed an audiometric evaluation, which demonstrated bilateral sensorineural hearing loss. Echocardiography highlighted mild to moderate mitralic and tricuspidal insufficiency without hemodynamic impairment (ejection fraction, 50%). Corticosteroid therapy was started (prednisone 0.5 mg/kg/d). After 7 days of treatment, inflammation was remarkably reduced, and the patient no longer reported pain.
Relapsing polychondritis is a rare noninfective condition characterized by focal inflammatory destruction of ear cartilage, followed by fibroblastic regeneration. It often is associated with ocular inflammation, including conjunctivitis, scleritis, and episcleritis; cochlear or vestibular lesions; and seronegative nonerosive inflammatory arthritis.1 Clinical examination of the affected area shows swelling, redness, and tenderness of the ear, which could lead to a misdiagnosis of cellulitis. A typical and useful differentiating sign is the sparing of the noncartilaginous parts of the ear lobule. If not promptly diagnosed and treated, the destructive process can cause thinning of the cartilage, leading to deformities of the external ear.
The differential diagnosis includes erysipelas, which presents as a rapidly appearing inflammatory patch with sharply defined borders, accompanied by regional lymphadenopathy or skin streaking as well as fever. Sweet syndrome usually presents with tender erythematous or violaceous skin papules, plaques, or nodules, frequently with a pseudovesicular appearance; patients generally present with a classic fever and peripheral neutrophilia.2 The localized cutaneous form of leishmaniasis usually appears with a papule that generally develops into an ulcerative nodular lesion. Our patient did not have a history of exposure to topical substances that could point to photocontact dermatitis.
Dion et al3 proposed 3 distinct clinical phenotypes of RP: (1) patients with concomitant myelodysplastic syndrome or other hematologic malignancy (<10% of patients), mostly older men with a poor prognosis; (2) patients with tracheobronchial involvement (approximately 25% of patients); and (3) patients who do not have hematologic or tracheobronchial involvement (approximately 65% of patients) with a good prognosis.
Two sets of diagnostic criteria have been proposed. The criteria from McAdam et al4 required the presence of 3 or more of the following clinical features: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation (eg, conjunctivitis, keratitis, scleritis/episcleritis, uveitis), respiratory tract chondritis (laryngeal and/or tracheal cartilages), and cochlear and/or vestibular dysfunction (eg, neurosensory hearing loss, tinnitus, vertigo). These criteria were modified by Damiani and Levine.5 According to the latter, all patients were required to have one of the following: at least 4 of the McAdam et al4 diagnostic criteria; 1 or more of the clinical findings included in the McAdam et al4 criteria with histologic features suggestive for RP; or chondritis at 2 or more separate anatomic locations with a response to glucocorticoids and/or dapsone.
No laboratory findings are specific for RP, and nonspecific indicators of inflammation--elevated erythrocyte sedimentation rate and C-reactive protein--often are present.
The cause of RP is unknown. Familial clustering has not been observed. Terao et al6 found that HLA-DRB1*1602, -DQB1*0502, and -B*6701, in linkage disequilibrium with each other, are associated with susceptibility to RP.
There is no universal consensus about treatment, but a course of steroids leads to the resolution of the acute phase. Maintenance treatment can include dapsone, azathioprine, methotrexate, cyclophosphamide, and cyclosporine.7,8 Some studies have described the successful use of anti-tumor necrosis factor α inhibitors and rituximab.9,10
The Diagnosis: Relapsing Polychondritis
Due to suspicion of relapsing polychondritis (RP), we also performed an audiometric evaluation, which demonstrated bilateral sensorineural hearing loss. Echocardiography highlighted mild to moderate mitralic and tricuspidal insufficiency without hemodynamic impairment (ejection fraction, 50%). Corticosteroid therapy was started (prednisone 0.5 mg/kg/d). After 7 days of treatment, inflammation was remarkably reduced, and the patient no longer reported pain.
Relapsing polychondritis is a rare noninfective condition characterized by focal inflammatory destruction of ear cartilage, followed by fibroblastic regeneration. It often is associated with ocular inflammation, including conjunctivitis, scleritis, and episcleritis; cochlear or vestibular lesions; and seronegative nonerosive inflammatory arthritis.1 Clinical examination of the affected area shows swelling, redness, and tenderness of the ear, which could lead to a misdiagnosis of cellulitis. A typical and useful differentiating sign is the sparing of the noncartilaginous parts of the ear lobule. If not promptly diagnosed and treated, the destructive process can cause thinning of the cartilage, leading to deformities of the external ear.
The differential diagnosis includes erysipelas, which presents as a rapidly appearing inflammatory patch with sharply defined borders, accompanied by regional lymphadenopathy or skin streaking as well as fever. Sweet syndrome usually presents with tender erythematous or violaceous skin papules, plaques, or nodules, frequently with a pseudovesicular appearance; patients generally present with a classic fever and peripheral neutrophilia.2 The localized cutaneous form of leishmaniasis usually appears with a papule that generally develops into an ulcerative nodular lesion. Our patient did not have a history of exposure to topical substances that could point to photocontact dermatitis.
Dion et al3 proposed 3 distinct clinical phenotypes of RP: (1) patients with concomitant myelodysplastic syndrome or other hematologic malignancy (<10% of patients), mostly older men with a poor prognosis; (2) patients with tracheobronchial involvement (approximately 25% of patients); and (3) patients who do not have hematologic or tracheobronchial involvement (approximately 65% of patients) with a good prognosis.
Two sets of diagnostic criteria have been proposed. The criteria from McAdam et al4 required the presence of 3 or more of the following clinical features: bilateral auricular chondritis, nonerosive seronegative inflammatory polyarthritis, nasal chondritis, ocular inflammation (eg, conjunctivitis, keratitis, scleritis/episcleritis, uveitis), respiratory tract chondritis (laryngeal and/or tracheal cartilages), and cochlear and/or vestibular dysfunction (eg, neurosensory hearing loss, tinnitus, vertigo). These criteria were modified by Damiani and Levine.5 According to the latter, all patients were required to have one of the following: at least 4 of the McAdam et al4 diagnostic criteria; 1 or more of the clinical findings included in the McAdam et al4 criteria with histologic features suggestive for RP; or chondritis at 2 or more separate anatomic locations with a response to glucocorticoids and/or dapsone.
No laboratory findings are specific for RP, and nonspecific indicators of inflammation--elevated erythrocyte sedimentation rate and C-reactive protein--often are present.
The cause of RP is unknown. Familial clustering has not been observed. Terao et al6 found that HLA-DRB1*1602, -DQB1*0502, and -B*6701, in linkage disequilibrium with each other, are associated with susceptibility to RP.
There is no universal consensus about treatment, but a course of steroids leads to the resolution of the acute phase. Maintenance treatment can include dapsone, azathioprine, methotrexate, cyclophosphamide, and cyclosporine.7,8 Some studies have described the successful use of anti-tumor necrosis factor α inhibitors and rituximab.9,10
- Borgia F, Giuffrida R, Guarneri F, et al. Relapsing polychondritis: an updated review. Biomedicines. 2018;6:84.
- Rednic S, Damian L, Talarico R, et al. Relapsing polychondritis: state of the art on clinical practice guidelines. RMD Open. 2018:4(suppl 1)e000788.
- Dion J, Costedoat-Chalumeau N, Sène D, et al. Relapsing polychondritis can be characterized by three different clinical phenotypes: analysis of a recent series of 142 patients. Arthritis Rheumatol. 2016;68:2992-3001.
- McAdam LP, O'Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore). 1976;55:193.
- Damiani JM, Levine HL. Relapsing polychondritis--report of ten cases. Laryngoscope. 1979;89:929-946.
- Terao C, Yoshifuji H, Yamano Y, et al. Genotyping of relapsing polychondritis identified novel susceptibility HLA alleles and distinct genetic characteristics from other rheumatic diseases. Rheumatology (Oxford). 2016;55:1686.016-82
- Goldenberg G, Sangueza OP, Jorizzo JL. Successful treatment of relapsing polychondritis with mycophenolate mofetil. J Dermatolog Treat. 2006;17:158-159.
- Handler RP. Leflunomide for relapsing polychondritis: successful longterm treatment. J Rheumatol. 2006;33:1916; author reply 1916-1917.
- Carter JD. Treatment of relapsing polychondritis with a TNF antagonist. J Rheumatol. 2005;32:1413.
- Leroux G, Costedoat-Chalumeau N, Brihaye B, et al. Treatment of relapsing polychondritis with rituximab: a retrospective study of nine patients. Arthritis Rheum. 2009;61:577-582.
- Borgia F, Giuffrida R, Guarneri F, et al. Relapsing polychondritis: an updated review. Biomedicines. 2018;6:84.
- Rednic S, Damian L, Talarico R, et al. Relapsing polychondritis: state of the art on clinical practice guidelines. RMD Open. 2018:4(suppl 1)e000788.
- Dion J, Costedoat-Chalumeau N, Sène D, et al. Relapsing polychondritis can be characterized by three different clinical phenotypes: analysis of a recent series of 142 patients. Arthritis Rheumatol. 2016;68:2992-3001.
- McAdam LP, O'Hanlan MA, Bluestone R, et al. Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine (Baltimore). 1976;55:193.
- Damiani JM, Levine HL. Relapsing polychondritis--report of ten cases. Laryngoscope. 1979;89:929-946.
- Terao C, Yoshifuji H, Yamano Y, et al. Genotyping of relapsing polychondritis identified novel susceptibility HLA alleles and distinct genetic characteristics from other rheumatic diseases. Rheumatology (Oxford). 2016;55:1686.016-82
- Goldenberg G, Sangueza OP, Jorizzo JL. Successful treatment of relapsing polychondritis with mycophenolate mofetil. J Dermatolog Treat. 2006;17:158-159.
- Handler RP. Leflunomide for relapsing polychondritis: successful longterm treatment. J Rheumatol. 2006;33:1916; author reply 1916-1917.
- Carter JD. Treatment of relapsing polychondritis with a TNF antagonist. J Rheumatol. 2005;32:1413.
- Leroux G, Costedoat-Chalumeau N, Brihaye B, et al. Treatment of relapsing polychondritis with rituximab: a retrospective study of nine patients. Arthritis Rheum. 2009;61:577-582.
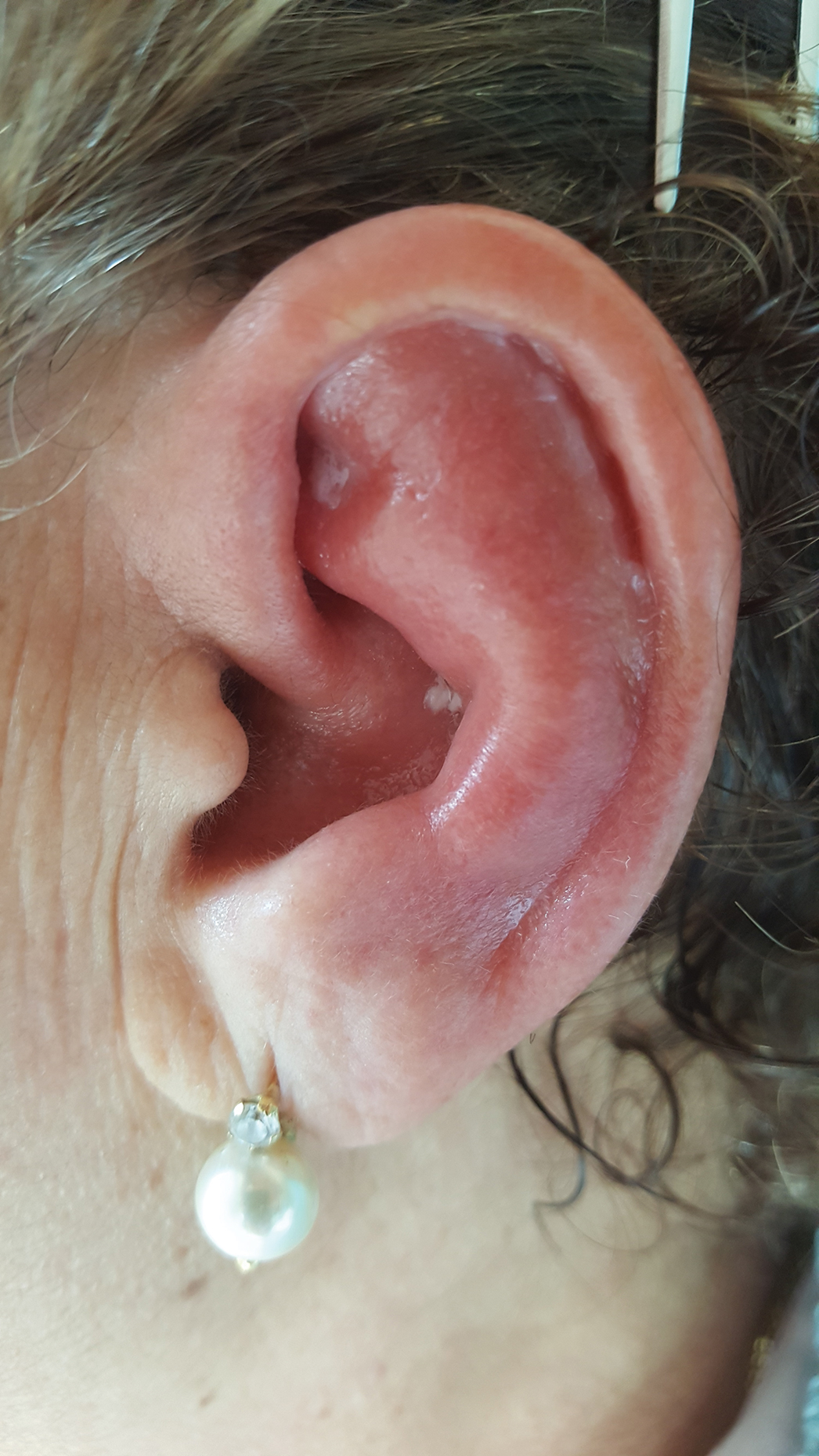
A 67-year-old woman presented with severe pain of the left external ear. She explained that similar episodes had occurred 2 years prior and affected the right ear and the nose. Her general practitioner prescribed topical and systemic antibiotic treatment, but there was no improvement. The patient also reported diffuse small joint pain without any radiologic sign of erosive arthritis. Physical examination revealed a red swollen external ear that was tender to the touch from the helix to the antitragus; conversely, the earlobe did not present any sign of inflammation. Redness of the left eye also was noticed, and a slit-lamp examination confirmed our suspect of scleritis. Results from routine blood tests, including an autoimmune panel, were within reference range, except for a nonspecific increase of inflammatory markers (erythrocyte sedimentation rate, 43 mm/h [reference range, 0–20 mm/h]; C-reactive protein, 5.65 mg/L [reference range, 0.08–3.1 mg/L]).