User login
Soft-tissue sarcomas are tumors of the mesenchymal system, and half develop in the extremities.1 Although patients with soft-tissue sarcomas have been treated with a combination of surgery, radiation therapy, and chemotherapy, it remains unclear whether either radiation or chemotherapy improves outcomes for these patients. Soft-tissue sarcomas are therefore currently treated with surgical resection when possible, with or without chemotherapy or radiation.
Even though multimodal therapy for patients with these tumors is controversial, a multidisciplinary conference among the many providers who may be involved in the management these patients—orthopedic, medical, and radiation oncologists, as well as the referring primary care physician, plastic and reconstructive surgeons, physical therapists, and radiologists and pathologists with expertise in these tumors—is helpful.2 This article presents an overview of the management of these patients, with a focus on the mainstay treatment, surgical resection. The roles of chemotherapy and radiation therapy for soft-tissue sarcomas, while touched upon here, are detailed in the final two articles in this supplement.
HISTOLOGIC GRADING AND THERAPY IMPLICATIONS
The prognosis of soft-tissue sarcomas correlates with histopathologic grade, and a three-grade system appears to be more accurate than a two-grade system.3 In general, low-grade lesions (grade 1) are unlikely to metastasize and are therefore less likely to need treatment with chemotherapy or radiation, as the risks of these therapies would most likely outweigh any benefit in terms of local control.
Specifically, the risk of radiation involves debilitation of local wound healing and the chance of dedifferentiation of low-grade lesions to higher-grade lesions with more metastatic potential. Grade 2 and 3 lesions are usually considered high-grade and are more likely to be treated with radiation and chemotherapy. Radiation is frequently used in patients with high-grade lesions when anticipated margins or actual margins are less than 1 cm.4–6
Chemotherapy’s lack of proven efficacy for soft-tissue sarcomas likely stems from poor understanding of the pathophysiology, molecular biology, and even some aspects of the natural history of these uncommon and heterogeneous tumors. There are more than 50 subtypes of soft-tissue sarcoma,7,8 and this heterogeneity has likely contributed to the difficulty of identifying chemotherapeutic agents that are highly active against these diseases.9
THE ROLE OF FAMILIAL GENETICS
Developing effective chemotherapeutic strategies may depend on grouping soft-tissue sarcomas more homogeneously. To compare like lesions with like lesions, molecular analysis and even molecular signatures may be of assistance. Along these lines, critical mutations and translocations have been described for several soft-tissue sarcoma subtypes.
Li-Fraumeni syndrome is an autosomal dominant cancer predisposition syndrome caused by germline mutations (ie, in every cell) in the p53 gene.10 Patients with Li-Fraumeni syndrome have an increased risk of developing soft-tissue sarcomas.1,11
Neurofibromatosis type 1 is caused by germline mutations in the NF1 gene, and malignant peripheral nerve sheath tumors occur within neurofibromas in neurofibromatosis patients and typically have additional mutations in CDKN2A or p53.9 INI1 loss is seen in all cases of extrarenal rhabdoid tumors and has been reported in a subset of epithelioid sarcomas (those occurring in proximal/axial regions).9,12 Delineation and greater understanding of these genetic abnormalities may lead to more effective medical therapy.
EVALUATION OF SUSPICIOUS SOFT-TISSUE MASSES
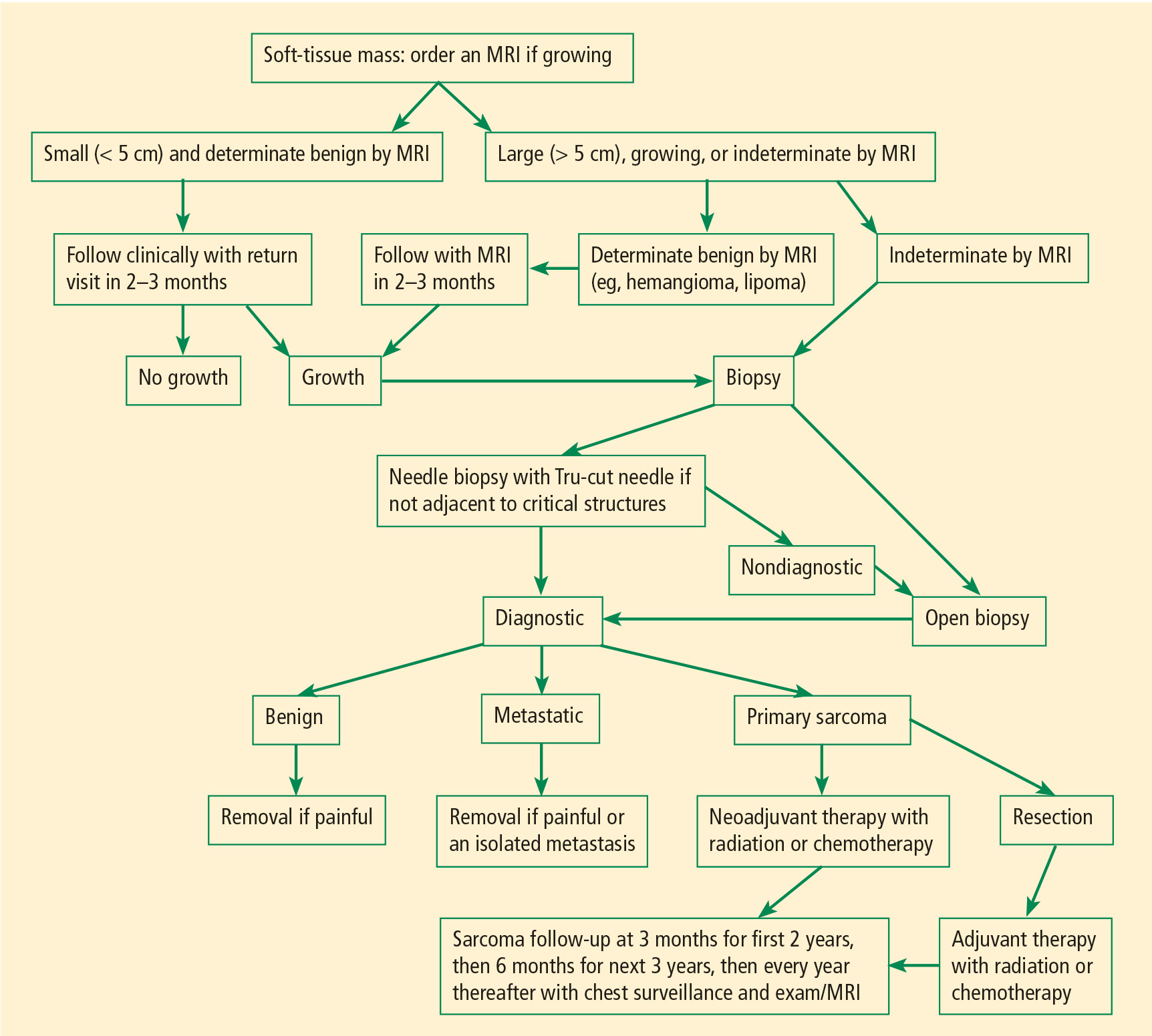
Figure 1 presents in flow chart form our general approach to the evaluation and management of patients with a soft-tissue mass suspicious for sarcoma—an approach detailed in the text below.
History and physical examination
Patients with soft-tissue sarcomas present with a mass that generally is increasing in size. The location and depth of the mass can be assessed on physical examination. In general, the deeper the mass, the more likely it is to be a sarcoma.13 Unlike bone sarcomas, soft-tissue sarcomas frequently are not associated with pain, so lack of pain does not make a mass more likely to be benign. In general, the only way to be sure that a mass is not malignant is to biopsy it. However, there are certain symptoms and signs that make a benign diagnosis much more likely. For example, very soft superficial masses that have not changed in size in years tend to be benign lipomas, and discolored lesions that go away with elevation of the affected body part tend to be hemangiomas.
Magnetic resonance imaging
Magnetic resonance imaging (MRI) is the primary imaging method for soft-tissue sarcomas. The benignity of a lesion such as a lipoma or hemangioma may be able to be determined with high certainty on MRI, in which case we call the imaging of the lesion “determinate.” Such lesions with determinate imaging (often referred to as “determinate lesions”) usually do not require a biopsy. However, the nature and identity of most lesions cannot be determined by MRI; although the MRI is still useful to help plan the biopsy in these cases, these lesions are termed “indeterminate” by MRI and should usually be biopsied.
Lesions that can be deemed determinate and usually be diagnosed as benign based on MRI findings include lipomas, hemangiomas, granuloma annulare, and ganglion cysts. However, most other soft-tissue lesions are indeterminate on MRI and, except in rare circumstances, require a biopsy to determine what they are and how they should be treated.
BIOPSY
The primary biopsy procedures for soft-tissue sarcomas are needle or open biopsy techniques and, in general, are similar to those for bone sarcomas, as reviewed in the previous article in this supplement. Regardless of the biopsy technique, hemostasis must be meticulous and patients are generally advised to not use the affected limb for at least several days after the biopsy to reduce the risk of a cancer cell–laden hematoma. It is preferable for the biopsy to be performed by or in consultation with the surgeon who will do the resection, if required.
Avoid transverse incisions
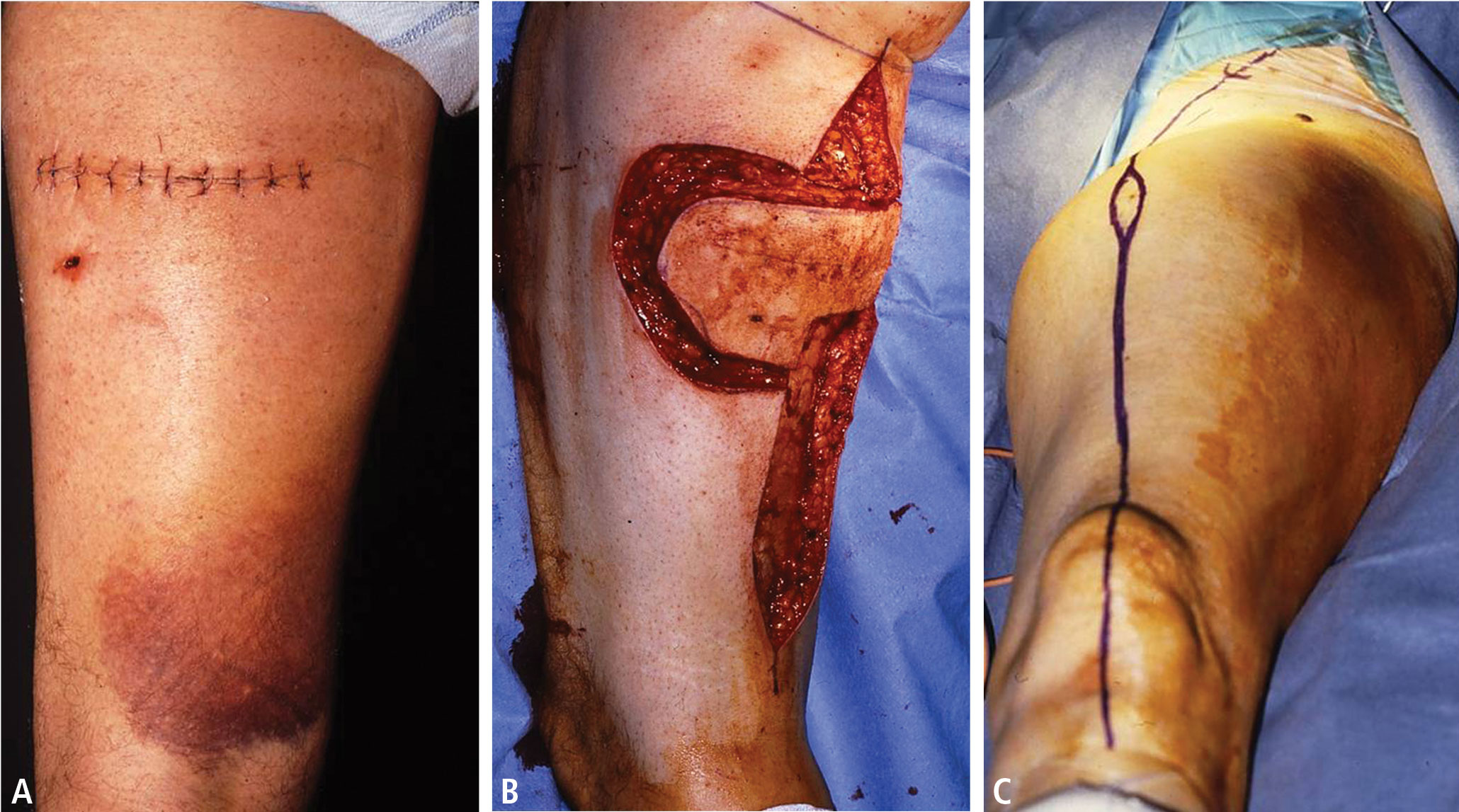
Lymph node biopsies
Lymph node biopsies are not generally indicated in patients with soft-tissue sarcoma. However, lymph node assessment and management should be considered in cases of clear cell sarcoma, epithelioid sarcoma, angiosarcoma, and embryonal/alveolar rhabdomyosarcoma, each of which has a greater than 10% incidence of lymph node metastasis.14 In this subset of soft-tissue sarcomas, a 5-year survival rate of 46% has been reported with therapeutic lymphadenectomy with curative intent versus nearly 0% with no lymphadenectomy or noncurative lymphadenectomy.14
Our approach in these sarcomas that go to the lymph nodes with increased relative frequency has been to first resect the sarcoma and then, after the margin is determined to be negative on the permanent pathology report, to schedule a nuclear medicine radiotracer study to analyze the drainage of the surgical bed. With this information we take the patient to the operating room and assess the location of the sentinel node (ie, node with the highest level of activity) through the skin using a radioactive counter with a sterile probe. We then make an incision in this area and find the lymph node. Upon removal of the “hot” lymph node, we reassess the radioactivity of the resected node and its node bed to be sure that we have the sentinel node. If this node or any node in the dissection has tumor in it, we do a therapeutic lymphadenectomy to remove all the lymph nodes in the area. For example, in the lower leg the lymphatic drainage is to the popliteal area, the inguinal area, or both. In the lower arm the lymphatic drainage is to the epitrochlear area and the axilla.
RESECTION
The resection surgery involves careful preoperative planning, almost always with an MRI and subsequent review by musculoskeletal tumor radiologists. In the operating room, general anesthesia is preferred to avoid ineffective blocks or overly effective blocks, which prevent neurologic examination immediately after the operation. If the functional loss is not too great, resection of the entire muscle or muscles involved is performed. If neurovascular structures are not encased (ie, not more than 50% surrounded in the case of arteries or motor nerves), then these structures are spared. If arteries are encased, the vessels are bypassed and the encased structure is left with the resection specimen. If the tumor is adjacent to but not encasing the neurovascular structures, the best course is to discuss with the radiation oncology team whether they prefer preoperative or postoperative radiation therapy. In general, for a high-grade lesion with adjacent neurovascular structues and no plane between the tumor and these structures, we ask our radiation oncologist colleagues to see the patient and discuss preoperative or perioperative (brachytherapy) radiation therapy. Postoperatively, where there is less than a 1-cm margin with no fascial boundary, we generally recommend radiation.
Margins
In our experience, margins of 1 cm or greater or resections with a fascial boundary are adequate and will leave patients with a much lower than 10% risk of recurrence. Others have postulated that margins that are smaller than this can have a very low rate of recurrence if perioperative (preoperative, intraoperative, or postoperative) radiation is given (personal communication from Drs. Jeffrey Eckardt and Dempsey Springfield). However, no well-controlled study has demonstrated how close the margin can be while still achieving an acceptable recurrence rate, and such a study would be very hard to perform given the rarity and heterogeneity of soft-tissue sarcomas and the variability in their assessment and surgical treatment.
Intralesional surgery leads to recurrence
Intralesional surgery will always lead to recurrence if the lesion is truly a soft-tissue sarcoma, even in spite of radiation therapy, chemotherapy, or both. Myomectomy and compartmental resections are frequently necessary to achieve a negative margin (normal tissue around the entire resection specimen). If intralesional surgery has been performed at an outside institution, we have generally recommended resection of the tumor bed, and in our experience this has reduced the recurrence rate after intralesional surgery to levels near those obtained when we perform the biopsy. In our experience, intralesional surgery without tumor bed resection will result in recurrence in nearly every case.
Reconstruction
Postoperative reconstruction of the defect involves closure of the fascia and skin with minimal tension, if possible. If there is tension, a vacuum-assisted closure dressing is placed on the wound and the patient returns for definitive closure, usually with a muscle flap. If the flap is a straightforward rotational flap, such as a medial gastrocnemius, or if only a split-thickness skin graft is required because there is healthy muscle in the floor of the open wound, this can be performed by experienced orthopedic surgeons. If these straightforward solutions are not possible, consultation with plastic surgeons is required, and they will cover the area with a complex rotational flap or, occasionally, with a free flap. For split-thickness skin grafts, it is prudent to make certain that the width of a #15 knife blade can pass between the blade and the housing of the Padgett dermatome and to take the skin from the extremity ipsilateral to the sarcoma (even with negative margins) to ensure that skin will not be contaminated with errant sarcoma cells.
Reconstruction following sarcoma resection is discussed in further detail in the next article in this supplement.
OUTCOMES AND FOLLOW-UP
The recurrence rate for soft-tissue sarcomas resected at Cleveland Clinic over the past 15 years has been less than 10%. This rate is comparable to the rates at other institutions that perform a high volume of sarcoma resections, but at institutions without a group dedicated to these procedures or without substantial experience in them, the recurrence rate is much higher, particularly with positive margins.15
Cure for soft-tissue sarcomas depends on being disease-free not only locally but also systemically. Most metastases from soft-tissue sarcomas are to the lung and, less commonly (as noted above), the lymph nodes. We assess local recurrence and metastatic disease at 3-month intervals for the first 2 years. Among patients who are disease-free at 2 years after the definitive surgery, the cure rate is 80% to 85%. After 2 years, we assess patients for presence of disease at 6-month intervals for the next 3 years and at yearly intervals thereafter.
Patients who have a recurrence are at increased risk for metastatic disease, and it is often very hard to achieve local control, as these patients frequently have had tumor contamination of the wound. At that point, unless the entire wound is excised or an amputation is performed, recurrences will continue. A nomogram has been validated for evaluating 10-year soft-tissue sarcoma–specific survival16 and is freely available at www.nomograms.org.
FUTURE DIRECTIONS
Future research challenges in this area include breaking down soft-tissue sarcoma subgroups more homogeneously, possibly with genetic markers, to better determine which lesions might benefit from chemotherapy. The goal of improved subtyping is to decrease the metastatic rate of soft-tissue sarcomas in much the same manner that directed chemotherapy has improved the metastasis and cure rates for patients with Ewing sarcoma and osteosarcoma.
- Simon MA, Springfield D, eds. Surgery for Bone and Soft-tissue Tumors. Philadelphia, PA: Lippincott-Raven; 1998.
- Glencross J, Balasubramanian SP, Bacon J, Robinson MH, Reed MW. An audit of the management of soft tissue sarcoma within a health region in the UK. Eur J Surg Oncol 2003; 29:670–675.
- Kandel RA, Bell RS, Wunder JS, et al. Comparison between a 2- and 3-grade system in predicting metastatic-free survival in extremity soft-tissue sarcoma. J Surg Oncol 1999; 72:77–82.
- Suit HD, Spiro I. Role of radiation in the management of adult patients with sarcoma of soft tissue. Semin Surg Oncol 1994; 10:347–356.
- Suit H, Spiro I. Preoperative radiation therapy for patients with sarcoma of the soft tissues. Cancer Treat Res 1993; 67:99–105.
- Suit HD, Mankin HJ, Wood WC, et al. Treatment of the patient with stage M0 soft tissue sarcoma. J Clin Oncol 1988; 6:854–862.
- Fletcher CDM, Unni KK, Mertens F. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002.
- Brennan MF, Singer S, Maki RG, O’Sullivan B. Sarcomas of the soft tissue and bone. In: DeVita Jr VT, Lawrence TS, Rosenbert SA, eds. Cancer: Principles & Practice of Oncology. 8th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008.
- Weiss SW, Goldblum JR. Enzinger and Weiss’s Soft Tissue Tumors. 5th ed. Philadelphia, PA: Mosby Elsevier; 2008.
- Gonzalez KD, Buzin CH, Noltner KA, et al. High frequency of de novo mutations in Li-Fraumeni syndrome. J Med Genet 2009; 46:689–693.
- Gonzalez KD, Noltner KA, Buzin CH, et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol 2009; 27:1250–1256.
- Modena P, Lualdi E, Facchinetti F, et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res 2005; 65:4012–4019.
- Peabody TD, Simon MA. Principles of staging of soft-tissue sarcomas. Clin Orthop Relat Res 1993; 289:19–31.
- Fong Y, Coit DG, Woodruff JM, Brennan MF. Lymph node metastasis from soft tissue sarcoma in adults: analysis of data from a prospective database of 1,772 sarcoma patients. Ann Surg 1993; 217:72–77.
- Potter BK, Adams SC, Pitcher JD Jr, Temple HT. Local recurrence of disease after unplanned excisions of high-grade soft tissue sarcomas. Clin Orthop Relat Res 2008; 466:3093–3100.
- Mariani L, Miceli R, Kattan MW, et al. Validation and adaptation of a nomogram for predicting the survival of patients with extremity soft tissue sarcoma using a three-grade system. Cancer 2005; 103:402–408.
Soft-tissue sarcomas are tumors of the mesenchymal system, and half develop in the extremities.1 Although patients with soft-tissue sarcomas have been treated with a combination of surgery, radiation therapy, and chemotherapy, it remains unclear whether either radiation or chemotherapy improves outcomes for these patients. Soft-tissue sarcomas are therefore currently treated with surgical resection when possible, with or without chemotherapy or radiation.
Even though multimodal therapy for patients with these tumors is controversial, a multidisciplinary conference among the many providers who may be involved in the management these patients—orthopedic, medical, and radiation oncologists, as well as the referring primary care physician, plastic and reconstructive surgeons, physical therapists, and radiologists and pathologists with expertise in these tumors—is helpful.2 This article presents an overview of the management of these patients, with a focus on the mainstay treatment, surgical resection. The roles of chemotherapy and radiation therapy for soft-tissue sarcomas, while touched upon here, are detailed in the final two articles in this supplement.
HISTOLOGIC GRADING AND THERAPY IMPLICATIONS
The prognosis of soft-tissue sarcomas correlates with histopathologic grade, and a three-grade system appears to be more accurate than a two-grade system.3 In general, low-grade lesions (grade 1) are unlikely to metastasize and are therefore less likely to need treatment with chemotherapy or radiation, as the risks of these therapies would most likely outweigh any benefit in terms of local control.
Specifically, the risk of radiation involves debilitation of local wound healing and the chance of dedifferentiation of low-grade lesions to higher-grade lesions with more metastatic potential. Grade 2 and 3 lesions are usually considered high-grade and are more likely to be treated with radiation and chemotherapy. Radiation is frequently used in patients with high-grade lesions when anticipated margins or actual margins are less than 1 cm.4–6
Chemotherapy’s lack of proven efficacy for soft-tissue sarcomas likely stems from poor understanding of the pathophysiology, molecular biology, and even some aspects of the natural history of these uncommon and heterogeneous tumors. There are more than 50 subtypes of soft-tissue sarcoma,7,8 and this heterogeneity has likely contributed to the difficulty of identifying chemotherapeutic agents that are highly active against these diseases.9
THE ROLE OF FAMILIAL GENETICS
Developing effective chemotherapeutic strategies may depend on grouping soft-tissue sarcomas more homogeneously. To compare like lesions with like lesions, molecular analysis and even molecular signatures may be of assistance. Along these lines, critical mutations and translocations have been described for several soft-tissue sarcoma subtypes.
Li-Fraumeni syndrome is an autosomal dominant cancer predisposition syndrome caused by germline mutations (ie, in every cell) in the p53 gene.10 Patients with Li-Fraumeni syndrome have an increased risk of developing soft-tissue sarcomas.1,11
Neurofibromatosis type 1 is caused by germline mutations in the NF1 gene, and malignant peripheral nerve sheath tumors occur within neurofibromas in neurofibromatosis patients and typically have additional mutations in CDKN2A or p53.9 INI1 loss is seen in all cases of extrarenal rhabdoid tumors and has been reported in a subset of epithelioid sarcomas (those occurring in proximal/axial regions).9,12 Delineation and greater understanding of these genetic abnormalities may lead to more effective medical therapy.
EVALUATION OF SUSPICIOUS SOFT-TISSUE MASSES
Figure 1 presents in flow chart form our general approach to the evaluation and management of patients with a soft-tissue mass suspicious for sarcoma—an approach detailed in the text below.
History and physical examination
Patients with soft-tissue sarcomas present with a mass that generally is increasing in size. The location and depth of the mass can be assessed on physical examination. In general, the deeper the mass, the more likely it is to be a sarcoma.13 Unlike bone sarcomas, soft-tissue sarcomas frequently are not associated with pain, so lack of pain does not make a mass more likely to be benign. In general, the only way to be sure that a mass is not malignant is to biopsy it. However, there are certain symptoms and signs that make a benign diagnosis much more likely. For example, very soft superficial masses that have not changed in size in years tend to be benign lipomas, and discolored lesions that go away with elevation of the affected body part tend to be hemangiomas.
Magnetic resonance imaging
Magnetic resonance imaging (MRI) is the primary imaging method for soft-tissue sarcomas. The benignity of a lesion such as a lipoma or hemangioma may be able to be determined with high certainty on MRI, in which case we call the imaging of the lesion “determinate.” Such lesions with determinate imaging (often referred to as “determinate lesions”) usually do not require a biopsy. However, the nature and identity of most lesions cannot be determined by MRI; although the MRI is still useful to help plan the biopsy in these cases, these lesions are termed “indeterminate” by MRI and should usually be biopsied.
Lesions that can be deemed determinate and usually be diagnosed as benign based on MRI findings include lipomas, hemangiomas, granuloma annulare, and ganglion cysts. However, most other soft-tissue lesions are indeterminate on MRI and, except in rare circumstances, require a biopsy to determine what they are and how they should be treated.
BIOPSY
The primary biopsy procedures for soft-tissue sarcomas are needle or open biopsy techniques and, in general, are similar to those for bone sarcomas, as reviewed in the previous article in this supplement. Regardless of the biopsy technique, hemostasis must be meticulous and patients are generally advised to not use the affected limb for at least several days after the biopsy to reduce the risk of a cancer cell–laden hematoma. It is preferable for the biopsy to be performed by or in consultation with the surgeon who will do the resection, if required.
Avoid transverse incisions
Lymph node biopsies
Lymph node biopsies are not generally indicated in patients with soft-tissue sarcoma. However, lymph node assessment and management should be considered in cases of clear cell sarcoma, epithelioid sarcoma, angiosarcoma, and embryonal/alveolar rhabdomyosarcoma, each of which has a greater than 10% incidence of lymph node metastasis.14 In this subset of soft-tissue sarcomas, a 5-year survival rate of 46% has been reported with therapeutic lymphadenectomy with curative intent versus nearly 0% with no lymphadenectomy or noncurative lymphadenectomy.14
Our approach in these sarcomas that go to the lymph nodes with increased relative frequency has been to first resect the sarcoma and then, after the margin is determined to be negative on the permanent pathology report, to schedule a nuclear medicine radiotracer study to analyze the drainage of the surgical bed. With this information we take the patient to the operating room and assess the location of the sentinel node (ie, node with the highest level of activity) through the skin using a radioactive counter with a sterile probe. We then make an incision in this area and find the lymph node. Upon removal of the “hot” lymph node, we reassess the radioactivity of the resected node and its node bed to be sure that we have the sentinel node. If this node or any node in the dissection has tumor in it, we do a therapeutic lymphadenectomy to remove all the lymph nodes in the area. For example, in the lower leg the lymphatic drainage is to the popliteal area, the inguinal area, or both. In the lower arm the lymphatic drainage is to the epitrochlear area and the axilla.
RESECTION
The resection surgery involves careful preoperative planning, almost always with an MRI and subsequent review by musculoskeletal tumor radiologists. In the operating room, general anesthesia is preferred to avoid ineffective blocks or overly effective blocks, which prevent neurologic examination immediately after the operation. If the functional loss is not too great, resection of the entire muscle or muscles involved is performed. If neurovascular structures are not encased (ie, not more than 50% surrounded in the case of arteries or motor nerves), then these structures are spared. If arteries are encased, the vessels are bypassed and the encased structure is left with the resection specimen. If the tumor is adjacent to but not encasing the neurovascular structures, the best course is to discuss with the radiation oncology team whether they prefer preoperative or postoperative radiation therapy. In general, for a high-grade lesion with adjacent neurovascular structues and no plane between the tumor and these structures, we ask our radiation oncologist colleagues to see the patient and discuss preoperative or perioperative (brachytherapy) radiation therapy. Postoperatively, where there is less than a 1-cm margin with no fascial boundary, we generally recommend radiation.
Margins
In our experience, margins of 1 cm or greater or resections with a fascial boundary are adequate and will leave patients with a much lower than 10% risk of recurrence. Others have postulated that margins that are smaller than this can have a very low rate of recurrence if perioperative (preoperative, intraoperative, or postoperative) radiation is given (personal communication from Drs. Jeffrey Eckardt and Dempsey Springfield). However, no well-controlled study has demonstrated how close the margin can be while still achieving an acceptable recurrence rate, and such a study would be very hard to perform given the rarity and heterogeneity of soft-tissue sarcomas and the variability in their assessment and surgical treatment.
Intralesional surgery leads to recurrence
Intralesional surgery will always lead to recurrence if the lesion is truly a soft-tissue sarcoma, even in spite of radiation therapy, chemotherapy, or both. Myomectomy and compartmental resections are frequently necessary to achieve a negative margin (normal tissue around the entire resection specimen). If intralesional surgery has been performed at an outside institution, we have generally recommended resection of the tumor bed, and in our experience this has reduced the recurrence rate after intralesional surgery to levels near those obtained when we perform the biopsy. In our experience, intralesional surgery without tumor bed resection will result in recurrence in nearly every case.
Reconstruction
Postoperative reconstruction of the defect involves closure of the fascia and skin with minimal tension, if possible. If there is tension, a vacuum-assisted closure dressing is placed on the wound and the patient returns for definitive closure, usually with a muscle flap. If the flap is a straightforward rotational flap, such as a medial gastrocnemius, or if only a split-thickness skin graft is required because there is healthy muscle in the floor of the open wound, this can be performed by experienced orthopedic surgeons. If these straightforward solutions are not possible, consultation with plastic surgeons is required, and they will cover the area with a complex rotational flap or, occasionally, with a free flap. For split-thickness skin grafts, it is prudent to make certain that the width of a #15 knife blade can pass between the blade and the housing of the Padgett dermatome and to take the skin from the extremity ipsilateral to the sarcoma (even with negative margins) to ensure that skin will not be contaminated with errant sarcoma cells.
Reconstruction following sarcoma resection is discussed in further detail in the next article in this supplement.
OUTCOMES AND FOLLOW-UP
The recurrence rate for soft-tissue sarcomas resected at Cleveland Clinic over the past 15 years has been less than 10%. This rate is comparable to the rates at other institutions that perform a high volume of sarcoma resections, but at institutions without a group dedicated to these procedures or without substantial experience in them, the recurrence rate is much higher, particularly with positive margins.15
Cure for soft-tissue sarcomas depends on being disease-free not only locally but also systemically. Most metastases from soft-tissue sarcomas are to the lung and, less commonly (as noted above), the lymph nodes. We assess local recurrence and metastatic disease at 3-month intervals for the first 2 years. Among patients who are disease-free at 2 years after the definitive surgery, the cure rate is 80% to 85%. After 2 years, we assess patients for presence of disease at 6-month intervals for the next 3 years and at yearly intervals thereafter.
Patients who have a recurrence are at increased risk for metastatic disease, and it is often very hard to achieve local control, as these patients frequently have had tumor contamination of the wound. At that point, unless the entire wound is excised or an amputation is performed, recurrences will continue. A nomogram has been validated for evaluating 10-year soft-tissue sarcoma–specific survival16 and is freely available at www.nomograms.org.
FUTURE DIRECTIONS
Future research challenges in this area include breaking down soft-tissue sarcoma subgroups more homogeneously, possibly with genetic markers, to better determine which lesions might benefit from chemotherapy. The goal of improved subtyping is to decrease the metastatic rate of soft-tissue sarcomas in much the same manner that directed chemotherapy has improved the metastasis and cure rates for patients with Ewing sarcoma and osteosarcoma.
Soft-tissue sarcomas are tumors of the mesenchymal system, and half develop in the extremities.1 Although patients with soft-tissue sarcomas have been treated with a combination of surgery, radiation therapy, and chemotherapy, it remains unclear whether either radiation or chemotherapy improves outcomes for these patients. Soft-tissue sarcomas are therefore currently treated with surgical resection when possible, with or without chemotherapy or radiation.
Even though multimodal therapy for patients with these tumors is controversial, a multidisciplinary conference among the many providers who may be involved in the management these patients—orthopedic, medical, and radiation oncologists, as well as the referring primary care physician, plastic and reconstructive surgeons, physical therapists, and radiologists and pathologists with expertise in these tumors—is helpful.2 This article presents an overview of the management of these patients, with a focus on the mainstay treatment, surgical resection. The roles of chemotherapy and radiation therapy for soft-tissue sarcomas, while touched upon here, are detailed in the final two articles in this supplement.
HISTOLOGIC GRADING AND THERAPY IMPLICATIONS
The prognosis of soft-tissue sarcomas correlates with histopathologic grade, and a three-grade system appears to be more accurate than a two-grade system.3 In general, low-grade lesions (grade 1) are unlikely to metastasize and are therefore less likely to need treatment with chemotherapy or radiation, as the risks of these therapies would most likely outweigh any benefit in terms of local control.
Specifically, the risk of radiation involves debilitation of local wound healing and the chance of dedifferentiation of low-grade lesions to higher-grade lesions with more metastatic potential. Grade 2 and 3 lesions are usually considered high-grade and are more likely to be treated with radiation and chemotherapy. Radiation is frequently used in patients with high-grade lesions when anticipated margins or actual margins are less than 1 cm.4–6
Chemotherapy’s lack of proven efficacy for soft-tissue sarcomas likely stems from poor understanding of the pathophysiology, molecular biology, and even some aspects of the natural history of these uncommon and heterogeneous tumors. There are more than 50 subtypes of soft-tissue sarcoma,7,8 and this heterogeneity has likely contributed to the difficulty of identifying chemotherapeutic agents that are highly active against these diseases.9
THE ROLE OF FAMILIAL GENETICS
Developing effective chemotherapeutic strategies may depend on grouping soft-tissue sarcomas more homogeneously. To compare like lesions with like lesions, molecular analysis and even molecular signatures may be of assistance. Along these lines, critical mutations and translocations have been described for several soft-tissue sarcoma subtypes.
Li-Fraumeni syndrome is an autosomal dominant cancer predisposition syndrome caused by germline mutations (ie, in every cell) in the p53 gene.10 Patients with Li-Fraumeni syndrome have an increased risk of developing soft-tissue sarcomas.1,11
Neurofibromatosis type 1 is caused by germline mutations in the NF1 gene, and malignant peripheral nerve sheath tumors occur within neurofibromas in neurofibromatosis patients and typically have additional mutations in CDKN2A or p53.9 INI1 loss is seen in all cases of extrarenal rhabdoid tumors and has been reported in a subset of epithelioid sarcomas (those occurring in proximal/axial regions).9,12 Delineation and greater understanding of these genetic abnormalities may lead to more effective medical therapy.
EVALUATION OF SUSPICIOUS SOFT-TISSUE MASSES
Figure 1 presents in flow chart form our general approach to the evaluation and management of patients with a soft-tissue mass suspicious for sarcoma—an approach detailed in the text below.
History and physical examination
Patients with soft-tissue sarcomas present with a mass that generally is increasing in size. The location and depth of the mass can be assessed on physical examination. In general, the deeper the mass, the more likely it is to be a sarcoma.13 Unlike bone sarcomas, soft-tissue sarcomas frequently are not associated with pain, so lack of pain does not make a mass more likely to be benign. In general, the only way to be sure that a mass is not malignant is to biopsy it. However, there are certain symptoms and signs that make a benign diagnosis much more likely. For example, very soft superficial masses that have not changed in size in years tend to be benign lipomas, and discolored lesions that go away with elevation of the affected body part tend to be hemangiomas.
Magnetic resonance imaging
Magnetic resonance imaging (MRI) is the primary imaging method for soft-tissue sarcomas. The benignity of a lesion such as a lipoma or hemangioma may be able to be determined with high certainty on MRI, in which case we call the imaging of the lesion “determinate.” Such lesions with determinate imaging (often referred to as “determinate lesions”) usually do not require a biopsy. However, the nature and identity of most lesions cannot be determined by MRI; although the MRI is still useful to help plan the biopsy in these cases, these lesions are termed “indeterminate” by MRI and should usually be biopsied.
Lesions that can be deemed determinate and usually be diagnosed as benign based on MRI findings include lipomas, hemangiomas, granuloma annulare, and ganglion cysts. However, most other soft-tissue lesions are indeterminate on MRI and, except in rare circumstances, require a biopsy to determine what they are and how they should be treated.
BIOPSY
The primary biopsy procedures for soft-tissue sarcomas are needle or open biopsy techniques and, in general, are similar to those for bone sarcomas, as reviewed in the previous article in this supplement. Regardless of the biopsy technique, hemostasis must be meticulous and patients are generally advised to not use the affected limb for at least several days after the biopsy to reduce the risk of a cancer cell–laden hematoma. It is preferable for the biopsy to be performed by or in consultation with the surgeon who will do the resection, if required.
Avoid transverse incisions
Lymph node biopsies
Lymph node biopsies are not generally indicated in patients with soft-tissue sarcoma. However, lymph node assessment and management should be considered in cases of clear cell sarcoma, epithelioid sarcoma, angiosarcoma, and embryonal/alveolar rhabdomyosarcoma, each of which has a greater than 10% incidence of lymph node metastasis.14 In this subset of soft-tissue sarcomas, a 5-year survival rate of 46% has been reported with therapeutic lymphadenectomy with curative intent versus nearly 0% with no lymphadenectomy or noncurative lymphadenectomy.14
Our approach in these sarcomas that go to the lymph nodes with increased relative frequency has been to first resect the sarcoma and then, after the margin is determined to be negative on the permanent pathology report, to schedule a nuclear medicine radiotracer study to analyze the drainage of the surgical bed. With this information we take the patient to the operating room and assess the location of the sentinel node (ie, node with the highest level of activity) through the skin using a radioactive counter with a sterile probe. We then make an incision in this area and find the lymph node. Upon removal of the “hot” lymph node, we reassess the radioactivity of the resected node and its node bed to be sure that we have the sentinel node. If this node or any node in the dissection has tumor in it, we do a therapeutic lymphadenectomy to remove all the lymph nodes in the area. For example, in the lower leg the lymphatic drainage is to the popliteal area, the inguinal area, or both. In the lower arm the lymphatic drainage is to the epitrochlear area and the axilla.
RESECTION
The resection surgery involves careful preoperative planning, almost always with an MRI and subsequent review by musculoskeletal tumor radiologists. In the operating room, general anesthesia is preferred to avoid ineffective blocks or overly effective blocks, which prevent neurologic examination immediately after the operation. If the functional loss is not too great, resection of the entire muscle or muscles involved is performed. If neurovascular structures are not encased (ie, not more than 50% surrounded in the case of arteries or motor nerves), then these structures are spared. If arteries are encased, the vessels are bypassed and the encased structure is left with the resection specimen. If the tumor is adjacent to but not encasing the neurovascular structures, the best course is to discuss with the radiation oncology team whether they prefer preoperative or postoperative radiation therapy. In general, for a high-grade lesion with adjacent neurovascular structues and no plane between the tumor and these structures, we ask our radiation oncologist colleagues to see the patient and discuss preoperative or perioperative (brachytherapy) radiation therapy. Postoperatively, where there is less than a 1-cm margin with no fascial boundary, we generally recommend radiation.
Margins
In our experience, margins of 1 cm or greater or resections with a fascial boundary are adequate and will leave patients with a much lower than 10% risk of recurrence. Others have postulated that margins that are smaller than this can have a very low rate of recurrence if perioperative (preoperative, intraoperative, or postoperative) radiation is given (personal communication from Drs. Jeffrey Eckardt and Dempsey Springfield). However, no well-controlled study has demonstrated how close the margin can be while still achieving an acceptable recurrence rate, and such a study would be very hard to perform given the rarity and heterogeneity of soft-tissue sarcomas and the variability in their assessment and surgical treatment.
Intralesional surgery leads to recurrence
Intralesional surgery will always lead to recurrence if the lesion is truly a soft-tissue sarcoma, even in spite of radiation therapy, chemotherapy, or both. Myomectomy and compartmental resections are frequently necessary to achieve a negative margin (normal tissue around the entire resection specimen). If intralesional surgery has been performed at an outside institution, we have generally recommended resection of the tumor bed, and in our experience this has reduced the recurrence rate after intralesional surgery to levels near those obtained when we perform the biopsy. In our experience, intralesional surgery without tumor bed resection will result in recurrence in nearly every case.
Reconstruction
Postoperative reconstruction of the defect involves closure of the fascia and skin with minimal tension, if possible. If there is tension, a vacuum-assisted closure dressing is placed on the wound and the patient returns for definitive closure, usually with a muscle flap. If the flap is a straightforward rotational flap, such as a medial gastrocnemius, or if only a split-thickness skin graft is required because there is healthy muscle in the floor of the open wound, this can be performed by experienced orthopedic surgeons. If these straightforward solutions are not possible, consultation with plastic surgeons is required, and they will cover the area with a complex rotational flap or, occasionally, with a free flap. For split-thickness skin grafts, it is prudent to make certain that the width of a #15 knife blade can pass between the blade and the housing of the Padgett dermatome and to take the skin from the extremity ipsilateral to the sarcoma (even with negative margins) to ensure that skin will not be contaminated with errant sarcoma cells.
Reconstruction following sarcoma resection is discussed in further detail in the next article in this supplement.
OUTCOMES AND FOLLOW-UP
The recurrence rate for soft-tissue sarcomas resected at Cleveland Clinic over the past 15 years has been less than 10%. This rate is comparable to the rates at other institutions that perform a high volume of sarcoma resections, but at institutions without a group dedicated to these procedures or without substantial experience in them, the recurrence rate is much higher, particularly with positive margins.15
Cure for soft-tissue sarcomas depends on being disease-free not only locally but also systemically. Most metastases from soft-tissue sarcomas are to the lung and, less commonly (as noted above), the lymph nodes. We assess local recurrence and metastatic disease at 3-month intervals for the first 2 years. Among patients who are disease-free at 2 years after the definitive surgery, the cure rate is 80% to 85%. After 2 years, we assess patients for presence of disease at 6-month intervals for the next 3 years and at yearly intervals thereafter.
Patients who have a recurrence are at increased risk for metastatic disease, and it is often very hard to achieve local control, as these patients frequently have had tumor contamination of the wound. At that point, unless the entire wound is excised or an amputation is performed, recurrences will continue. A nomogram has been validated for evaluating 10-year soft-tissue sarcoma–specific survival16 and is freely available at www.nomograms.org.
FUTURE DIRECTIONS
Future research challenges in this area include breaking down soft-tissue sarcoma subgroups more homogeneously, possibly with genetic markers, to better determine which lesions might benefit from chemotherapy. The goal of improved subtyping is to decrease the metastatic rate of soft-tissue sarcomas in much the same manner that directed chemotherapy has improved the metastasis and cure rates for patients with Ewing sarcoma and osteosarcoma.
- Simon MA, Springfield D, eds. Surgery for Bone and Soft-tissue Tumors. Philadelphia, PA: Lippincott-Raven; 1998.
- Glencross J, Balasubramanian SP, Bacon J, Robinson MH, Reed MW. An audit of the management of soft tissue sarcoma within a health region in the UK. Eur J Surg Oncol 2003; 29:670–675.
- Kandel RA, Bell RS, Wunder JS, et al. Comparison between a 2- and 3-grade system in predicting metastatic-free survival in extremity soft-tissue sarcoma. J Surg Oncol 1999; 72:77–82.
- Suit HD, Spiro I. Role of radiation in the management of adult patients with sarcoma of soft tissue. Semin Surg Oncol 1994; 10:347–356.
- Suit H, Spiro I. Preoperative radiation therapy for patients with sarcoma of the soft tissues. Cancer Treat Res 1993; 67:99–105.
- Suit HD, Mankin HJ, Wood WC, et al. Treatment of the patient with stage M0 soft tissue sarcoma. J Clin Oncol 1988; 6:854–862.
- Fletcher CDM, Unni KK, Mertens F. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002.
- Brennan MF, Singer S, Maki RG, O’Sullivan B. Sarcomas of the soft tissue and bone. In: DeVita Jr VT, Lawrence TS, Rosenbert SA, eds. Cancer: Principles & Practice of Oncology. 8th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008.
- Weiss SW, Goldblum JR. Enzinger and Weiss’s Soft Tissue Tumors. 5th ed. Philadelphia, PA: Mosby Elsevier; 2008.
- Gonzalez KD, Buzin CH, Noltner KA, et al. High frequency of de novo mutations in Li-Fraumeni syndrome. J Med Genet 2009; 46:689–693.
- Gonzalez KD, Noltner KA, Buzin CH, et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol 2009; 27:1250–1256.
- Modena P, Lualdi E, Facchinetti F, et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res 2005; 65:4012–4019.
- Peabody TD, Simon MA. Principles of staging of soft-tissue sarcomas. Clin Orthop Relat Res 1993; 289:19–31.
- Fong Y, Coit DG, Woodruff JM, Brennan MF. Lymph node metastasis from soft tissue sarcoma in adults: analysis of data from a prospective database of 1,772 sarcoma patients. Ann Surg 1993; 217:72–77.
- Potter BK, Adams SC, Pitcher JD Jr, Temple HT. Local recurrence of disease after unplanned excisions of high-grade soft tissue sarcomas. Clin Orthop Relat Res 2008; 466:3093–3100.
- Mariani L, Miceli R, Kattan MW, et al. Validation and adaptation of a nomogram for predicting the survival of patients with extremity soft tissue sarcoma using a three-grade system. Cancer 2005; 103:402–408.
- Simon MA, Springfield D, eds. Surgery for Bone and Soft-tissue Tumors. Philadelphia, PA: Lippincott-Raven; 1998.
- Glencross J, Balasubramanian SP, Bacon J, Robinson MH, Reed MW. An audit of the management of soft tissue sarcoma within a health region in the UK. Eur J Surg Oncol 2003; 29:670–675.
- Kandel RA, Bell RS, Wunder JS, et al. Comparison between a 2- and 3-grade system in predicting metastatic-free survival in extremity soft-tissue sarcoma. J Surg Oncol 1999; 72:77–82.
- Suit HD, Spiro I. Role of radiation in the management of adult patients with sarcoma of soft tissue. Semin Surg Oncol 1994; 10:347–356.
- Suit H, Spiro I. Preoperative radiation therapy for patients with sarcoma of the soft tissues. Cancer Treat Res 1993; 67:99–105.
- Suit HD, Mankin HJ, Wood WC, et al. Treatment of the patient with stage M0 soft tissue sarcoma. J Clin Oncol 1988; 6:854–862.
- Fletcher CDM, Unni KK, Mertens F. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002.
- Brennan MF, Singer S, Maki RG, O’Sullivan B. Sarcomas of the soft tissue and bone. In: DeVita Jr VT, Lawrence TS, Rosenbert SA, eds. Cancer: Principles & Practice of Oncology. 8th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2008.
- Weiss SW, Goldblum JR. Enzinger and Weiss’s Soft Tissue Tumors. 5th ed. Philadelphia, PA: Mosby Elsevier; 2008.
- Gonzalez KD, Buzin CH, Noltner KA, et al. High frequency of de novo mutations in Li-Fraumeni syndrome. J Med Genet 2009; 46:689–693.
- Gonzalez KD, Noltner KA, Buzin CH, et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol 2009; 27:1250–1256.
- Modena P, Lualdi E, Facchinetti F, et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res 2005; 65:4012–4019.
- Peabody TD, Simon MA. Principles of staging of soft-tissue sarcomas. Clin Orthop Relat Res 1993; 289:19–31.
- Fong Y, Coit DG, Woodruff JM, Brennan MF. Lymph node metastasis from soft tissue sarcoma in adults: analysis of data from a prospective database of 1,772 sarcoma patients. Ann Surg 1993; 217:72–77.
- Potter BK, Adams SC, Pitcher JD Jr, Temple HT. Local recurrence of disease after unplanned excisions of high-grade soft tissue sarcomas. Clin Orthop Relat Res 2008; 466:3093–3100.
- Mariani L, Miceli R, Kattan MW, et al. Validation and adaptation of a nomogram for predicting the survival of patients with extremity soft tissue sarcoma using a three-grade system. Cancer 2005; 103:402–408.