User login
The active ingredient of any pharmaceutical product is responsible for the agent’s efficacy and safety profile. This ingredient is extensively studied in clinical trials and evaluated by the US Food and Drug Administration before the product is commercially available. In dermatologic products, especially those for treating dermatoses, the vehicle in which the active ingredient is formulated also plays a role in drug delivery and indirectly impacts therapeutic outcomes, unlike excipients in oral medications. Topical vehicles must be stable, provide a suitable environment that will not degrade the active ingredient or affect its efficacy, and be cosmetically acceptable.1
Topical vehicles are formulated to maintain the stability of the active ingredient and allow it to readily penetrate the skin and reach its target area with minimal absorption into the bloodstream, thus avoiding systemic adverse events. A variety of vehicles can exist for a single active ingredient to accommodate different phases of disease and different anatomical sites where the disease may occur.2 For example, alcohol-based vehicles, sprays, and foams are preferred for the scalp where evaporation of the vehicle is beneficial to prevent greasiness of the hair, while ointments may be preferred due to their occlusive nature for areas with xerotic or thick skin from dermatoses.
Cosmetic acceptability of the vehicle may influence patient adherence to therapy. Housman et al3 assessed a variety of products formulated in different vehicles (ie, solutions, foams, emollients, gels, creams, ointments) for the treatment of psoriasis. Patients with psoriasis applied each test product to a quarter-sized area of normal skin on the forearm using a cotton swab and completed a preference questionnaire. By far, respondents significantly preferred solutions and foams over creams, gels, and ointments (P<.01). Side effects were rated to be the most important characteristics of topical therapy, followed by time needed for application, ease of application, and messiness.3 Presumably, if patients are frustrated with the topical product that they are using, adherence to the prescribed dosage and application instructions will diminish over time, leading to suboptimal steady-state levels of the product. If appropriate levels of the drug are not present at the target site, treatment will not be successful.
Steady-state levels of a topical drug at the site of action also are maintained via appropriate application frequency, most commonly once to 4 times daily for dermatologic products. Fluocinonide and halcinonide are class II (potent) corticosteroids indicated for the relief of inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses and usually are administered at least twice daily. In double-blind clinical studies comparing both products in the treatment of psoriasis, halcinonide resulted in more improved outcomes than fluocinonide.4-6 Sudilovsky and Clewe4 studied 140 patients with moderate to severe psoriasis. After 3 weeks of treatment, 44% showed superior results with halcinonide, 27% showed superior results with fluocinonide, 26% showed equal results with both products, and 3% showed no relief.4 Similarly, Close5 reported that 61% of patients showed superior results with halcinonide, 25% showed superior results with fluocinonide, 10% showed equal results with both products, and 4% showed no relief (N=50). Lynfield and Watsky6 reported that 56% of patients with severe psoriasis who were treated with halcinonide for 2 weeks showed improvement to normal or slight inflammation compared to 44% of patients treated with fluocinonide (N=59). All 3 studies used cream formulations of halcinonide and fluocinonide.
Recently, halcinonide cream was shown to have an immediate release into the stratum corneum that peaked within 1 hour of application and remained elevated for 6 hours before beginning to decline.7 These results support a biphasic release of halcinonide, which is in agreement with its formulation—that halcinonide exists in both a solution phase for immediate release into the skin and in a suspension phase that allows a sustained release after equilibrium is reached between the solution and suspension phases.8 Fluocinonide is not known to be formulated in a similar way. Its vehicle composition and penetration into the skin could explain the superior efficacy of halcinonide versus fluocinonide.
The current pilot study was conducted to compare the release pattern of fluocinonide cream versus halcinonide cream into the stratum corneum using an in vivo, noninvasive method. Results for halcin-onide have been previously published.7
Methods
Participants were sequestered in a controlled environment for the entire day to allow the skin to equilibrate prior to product application. The methodology for the application and quantification of halcinonide cream 0.1% into the stratum corneum of 5 participants using a tape-stripping protocol has been described elsewhere.7 Concordia Clinical Research institutional review board (Cedar Knolls, New Jersey) approved this study, which was conducted at Dermatology Consulting Services (High Point, North Carolina).
A 0.1-g dose of generic fluocinonide cream 0.05% was applied to four 2.5-cm circular sites on the forearm in 5 participants with normal skin until completely absorbed. Circular tape strips were subsequently placed on the application site at 1, 3, 6, and 9 hours posttreatment and were held for 10 seconds with a controlled pressure plunger to ensure adequate and consistent contact between the tape strip and the skin. The tape strip was removed with forceps, rolled with the skin scale inside, and placed in a glass vial. This procedure was repeated 6 times at 1 of 4 sites with a new tape strip at each time point to obtain samples from deeper skin layers. A total of 24 tape strips were collected from each participant.
All vials were frozen at -20°C and were shipped overnight to Robert Kellar, PhD, at the Center for Bioengineering Innovation at Northern Arizona University (Flagstaff, Arizona) for mass spectroscopy evaluation. Once received at the outside facility, the vials were stored at -20°C until analysis. Each sample was spiked with a known quantity of an appropriate reference standard and extracted with 1 mL acetonitrile at room temperature for 1 minute with agitation. New unused tape strips were spiked with a small amount of fluocinonide reference standard for extraction efficiency.
Extracts were evaporated to dryness under nitrogen gas, resuspended in 200 µL chromatography solvent, and quantified using liquid chromatography–mass spectrometry. To remove the skin scale from the tape strips, 10 mL of a solvent solution of 0.1 mg/mL fludrocortisone acetate in acetonitrile was dispensed into a 4 dram vial containing the tape strip. The vials were ultrasonicated and shaken for 10 to 15 minutes, and the samples were further diluted to 100-fold and were inverted several times to ensure complete dissolution of fluocinonide before liquid chromatography–mass spectrometry.
A standard curve ranging from the lower limit of quantification to the upper limit of quantification for the fluocinonide reference was used to determine the quantity of fluocinonide in each of the tape strips. Once the lower limit of quantification was reached in a given set of tape strip samples (1-, 3-, 6-, and 9-hour samples), the next 2 sequential tape strips in that set were analyzed to confirm fluocinonide was not detectable in deeper layers. Standard quality controls were analyzed to ensure run-to-run and sample-to-sample accuracy.
Each sample was analyzed in duplicate; 10 mg fluocinonide was used as a reference standard. The minimum detectable concentration of fluocinonide was 1 ng/mL.
Results
As expected, tape strip 1 from each participant contained the highest concentration of fluocinonide. This strip corresponded to the most superficial layer of skin. Concentrations decreased in deeper skin layers, as detected in strips 2 to 6.
In general, the average concentration of fluocin-onide in strip 1 for all 5 participants was highest at hour 1, with a subsequent decline at hours 3, 6, and 9; however, participant 1 showed a second peak in fluocinonide concentration at hour 6 (Figure 1). When the fluocinonide concentration in strips 1 to 6 was averaged for each participant at each time point, similar results were obtained: a general decline after hour 1, but a second prominent peak at hour 6 in participant 1 only. In participant 1, the average fluocinonide concentration for strips 1 to 6 was 393 ng/mL at hour 1 and declined to 208 ng/mL at hour 3; it increased to 451 ng/mL at hour 6 before declining again to 202 ng/mL at hour 9.
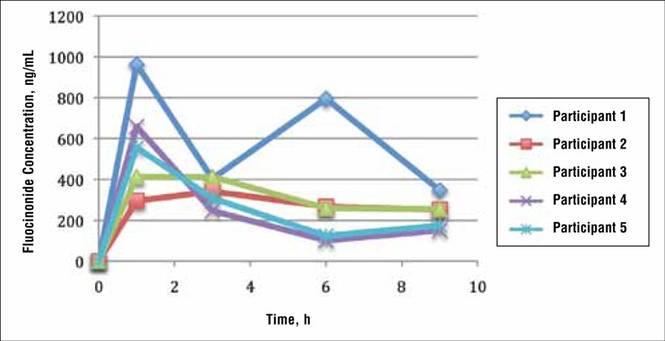
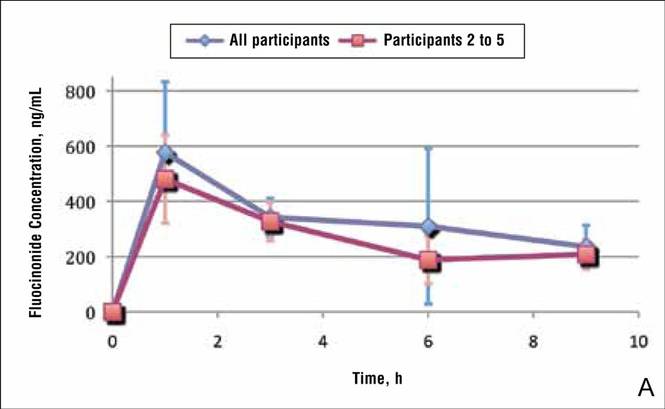
Because participant 1 was the only one to exhibit a second peak of fluocinonide concentration, it appears that measurements obtained from this participant may be outliers. When removing partici-pant 1 from the analysis of fluocinonide concentration in strip 1 at each time point, a clear decline is evident from hour 1 to hour 9 (Figure 2A, red line [partici-pants 2–5] vs blue line [participants 1–5]).
When the average concentration of fluocinonide was calculated in strips 1 to 6 from all participants, there was a general steady decline after hour 1 with a slight increase of 25 ng/mL at hour 6 (Figure 2B, blue line). This increase is due to the measurements obtained from participant 1; however, if partici-pant 1 is removed from the analysis, a constant decline is observed from hour 1 to hour 9 (Figure 2B, red line).
|
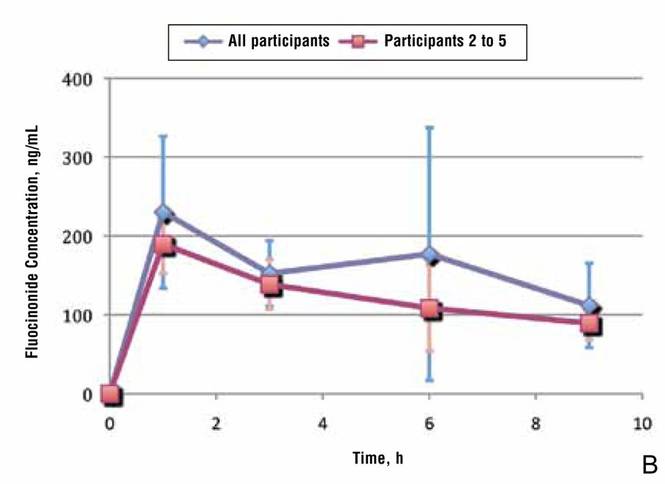
A prior study evaluated the penetration and absorption of halcinonide in the stratum corneum.7 In summary, halcinonide concentration peaked at hour 1 following application and remained elevated to hour 6, before beginning a slow decline. The average concentration of halcinonide from all participants in strips 1 to 6 reached 1350 ng/mL at hour 1, remained within 93% to 97% of this level (1253–1303 ng/mL) for the next 5 hours, and declined only 29% from the peak at hour 1 to hour 9 (958 ng/mL)(Figure 3, blue line).7 In contrast, the fluocinonide concentration in participants 2 to 5 from the current study reached 190 ng/mL at hour 1 and steadily declined 53% to 89 ng/mL by hour 9 (Figure 3, red line).
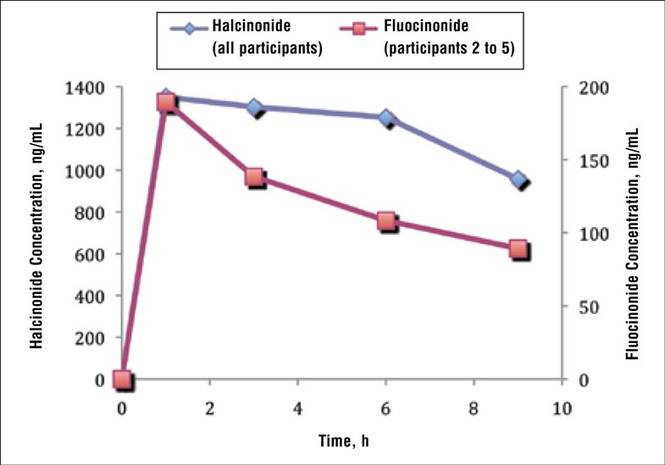
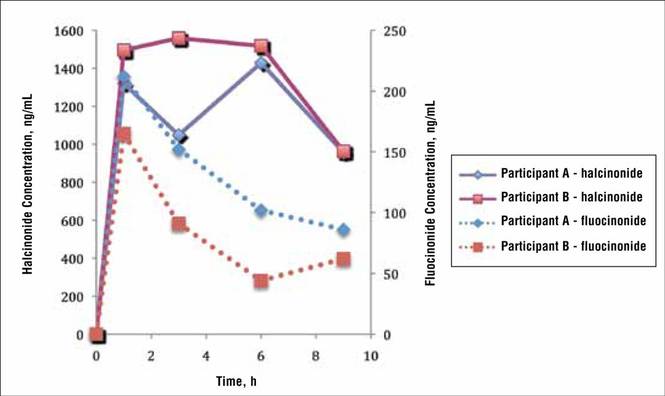
Two participants from the prior halcinonide study also were enrolled in the current fluocinonide study (referred to as participant A and B). In general, halcinonide levels in both participants remained elevated for 6 hours after application and declined 27.5% and 35.5%, respectively, by hour 9 (Figure 4). Participant A experienced a 20.5% dip in halcinonide concentration at hour 3 followed by an increase at hour 6; however, the halcinonide concentration at hour 9 was similar to hour 3.7 In contrast, fluocin-onide concentrations for these participants peaked at 1 hour and clearly declined approximately 60% over the next 8 hours.
Comment
The release of both fluocinonide and halcinonide into the skin was evaluated using dermal tape stripping on 4 sites on the forearms of healthy individuals. Cream formulations of each corticosteroid were evaluated in 5 participants, with 2 participants receiving both formulations during different study periods. In the prior study with halcinonide, the stratum corneum exhibited the highest concentration of the corticosteroid, with substantial declines beyond strip 6 (ie, strips 7–20).7 For this reason, only strips 1 to 6 were evaluated for corticosteroid penetration and absorption.
Results from strip 1 indicated immediate absorption of corticosteroid (fluocinonide and halcinonide) into the skin. Unlike the release of halcinonide, which demonstrated a clear sustained release over 6 hours before decreasing,7 fluocinonide concentrations began declining immediately after peaking at hour 1 and continued to decline up to hour 9. Only participant 1 exhibited a second peak of fluocinonide concentration at hour 6; the rest of the participants did not. This second peak is most likely an anomaly due to the small number of participants rather than a true elevation.
Given the rapid decline of fluocinonide concentration over the 9 hours compared with the more gradual decline of halcinonide concentration, there appears to be no evidence of a biphasic sustained release of fluocinonide from its vehicle. This difference in release pattern from each corticosteroid’s respective vehicle may explain in part the different clinical outcomes in comparative studies.4-6
It is known that vehicle composition affects corticosteroid diffusion from the vehicle to the skin surface and subsequent penetration into the skin.9 Either process can determine the overall effectiveness of the product. Ayres and Hooper10 evaluated the penetration of 4 topical preparations of cortisol. Product 1 delivered 16 times more cortisol to the skin than product 2, 8 times more than product 3, and 3 times more than product 4. Because all the preparations contained cortisol-free alcohol, these differences were attributed to the vehicle in which the cortisol was formulated. Products 1 and 4 both contained 10% urea, but the urea in product 1 was a powder in a cream base and the urea in product 4 was in a stabilizing emulsified base. Product 2 contained a propylene glycol/water base and product 3 was a water-miscible cream.10
Generic corticosteroid products have been observed in clinical practice and have been shown in vasoconstriction assays to be less and more potent than their brand-name equivalents.2,11 Vasoconstriction assays are the standard for assessing the potency of topical corticosteroids and predicting their clinical efficacy.2 One study reported significant differences in therapeutic effectiveness between generic formulations and their brand-name equivalents.12 Kenalog cream 0.1% (multiple manufacturers) was significantly more potent than any of the generic triamcinolone creams tested (P<.05); in fact, Kenalog cream 0.025% (multiple manufacturers) was statistically superior to all the generic triamcinolone creams 0.1%. Moreover, Artistocort A ointment 0.1% (Lederele Laboratories) and Valisone cream 0.1% (Schering Corporation) also were more potent than their generics at the same concentration in the same vehicle type.12 A second study also observed that 2 of 6 generic formulations had significantly less vasoconstriction than their respective brand-name formulations.11 A brand-name betamethasone valerate cream produced significantly greater vasoconstriction than its generic equivalent, and a brand-name betamethasone dipropionate cream produced greater vasoconstriction than one generic and equal vasoconstriction to another generic. Additionally, the vasoconstriction measured with Diprosone was greater than that measured with Diprolene, another brand-name product of betamethasone dipropionate.11 Diprosone and Diprolene differ in their vehicle content. The latter, a class I corticosteroid, contains a modified vehicle high in propylene glycol, whereas the former contains less propylene glycol and thus is classified as a class III corticosteroid. Propylene glycol allows hydrophobic molecules such as corticosteroids to dissolve more fully in the vehicle.12
Ostrenga et al1 studied the solubility of corticosteroids in different vehicles and, as expected, corticosteroids that fully solubilized in the vehicle exhibited better penetration into the skin on assessment with vasoconstriction assays. Corticosteroids in a suspension, on the other hand, showed slower penetration into the skin.1,13 A balance between the solution and suspension phase would allow a drug to rapidly penetrate the skin upon application, and when this pool of solubilized drug was depleted, additional drug could penetrate into the skin from the suspension phase. Based on the tape strip results from the current study it appears that halcinonide, which is manufactured in a biphasic formulation, follows this pattern of penetration and absorption into the stratum corneum. In contrast, fluocinonide appears to exist in a soluble state without much, if any, amount in a suspension phase because it had no sustained release during the 9 hours after application.
Common belief among dermatologists is that long-term use of corticosteroids leads to tachyphylaxis,14 which can be attributed to poor patient adherence. If patients skip doses, then the steady state of the product at the target site is not maintained. It is interesting to speculate that using agents with more sustained release beyond the time of application (such as halcinonide) may preserve steady-state levels even when patients are neglectful of the next medication application. Corticosteroids that work in 2 phases such as halcinonide may minimize tachyphylaxis experienced with prolonged use of corticosteroids.
Fluocinonide and halcinonide are both class II high-potency corticosteroids as shown on outcomes from vasoconstrictor assays, which assess the extent to which a corticosteroid causes cutaneous vasoconstriction or blanching in normal healthy individuals.15 The assay depends on the molecule diffusing from the vehicle, penetrating the skin, and causing a reaction (blanching) that is then evaluated. The assay cannot effectively evaluate the rate of continued diffusion and skin penetration beyond the appearance of blanching. In contrast, the tape-stripping method provides an inside look at the extent of penetration of the corticosteroid beyond the skin surface and the rate of its clearance from different skin layers. In the current study, the levels of fluocinonide declined after peaking at 1 hour after application, but the levels of halcinonide clearly remained elevated after peaking at the same time point. Most likely, vasoconstrictor studies would not be able to differentiate between the concentrations of the 2 products in the stratum corneum beyond the first hour after application.
Tape stripping, or dermatopharmacokinetics, has advantages over vasoconstriction assays in studying corticosteroid penetration and clearance from the stratum corneum. At one point, the US Food and Drug Administration had included tape stripping in its preliminary guidelines for generic topical bioequivalence studies until data from the same formulation generated from 2 different laboratories produced different results.16 Since that time, much work has been done with tape stripping to ensure its consistency. Weigmann et al17 demonstrated equivalent results with clobetasol using vasoconstriction and tape stripping, and Wiedersberg et al18 demonstrated the same with betamethasone. For the current study, the fluocinonide and halcinonide formulations were weighed prior to application so that the same dose was tested in all participants. A plunger was used to produce consistent pressure at all application sites to control for the amount of skin that was stripped off with the tape. Results for both corticosteroids were consistent between the participants. Variability in the data was detected; however, this observation is most likely due to the small number of participants in the studies.
Conclusion
In summary, this pilot study demonstrated that fluocinonide concentration in the stratum corneum peaks within the first hour of application before beginning a steady general decline. There was no evidence of sustained release. In contrast, halcin-onide demonstrated a sustained release for 6 hours after application. Halcinonide is formulated in a cream base in which the corticosteroid is present in a solution and suspension phase that allows for sustained delivery in skin over time. Fluocinonide does not appear to be formulated in the same way, and its concentrations in the stratum corneum begin to decline 1 hour after application.
Acknowledgement
Thank you to Robert Kellar, PhD, at the Center for Bioengineering Innovation at Northern Arizona University, Flagstaff, for conducting the liquid chromatography–mass spectrometry.
1. Ostrenga J, Haleblian J, Poulsen B, et al. Vehicle design for a new topical steroid, fluocinonide. J Invest Dermatol. 1971;56:392-399.
2. Rathi SK, D’Souza P. Rational and ethical use of topical corticosteroids based on safety and efficacy. Indian J Dermatol. 2012;57:251-259.
3. Housman TS, Mellen BG, Rapp SR, et al. Patients with psoriasis prefer solution and foam vehicles: a quantitative assessment of vehicle preference. Cutis. 2002;70:327-332.
4. Sudilovsky A, Clewe TH. Comparative efficacy of halcin-onide and fluocinonide creams in psoriasis and eczematous dermatoses. J Clin Pharmacol. 1975;15:779-784.
5. Close JE. Double-blind comparison of topical halcinonide and fluocinonide in the treatment of psoriasis. Int J Dermatol. 1976;15:534-537.
6. Lynfield Y, Watsky M. Psoriasis: topical corticosteroid therapy. Cutis. 1976;18:133, 136-137.
7. Draelos ZD. Demonstration of the biphasic release of 0.1% halcinonide cream. J Drugs Dermatol. 2015;14:89-90.
8. Bagatell FK. Halcinonide: a new potent topical anti-inflammatory drug. Cutis. 1974;14:459-462.
9. Ostrenga J, Steinmetz C, Poulsen B. Significance of vehicle composition. I. relationship between topical vehicle composition, skin penetrability, and clinical efficacy. J Pharm Sci. 1971;60:1175-1179.
10. Ayres PJ, Hooper G. Assessment of the skin penetration properties of different carrier vehicles for topically applied cortisol. Br J Dermatol. 1978;99:307-317.
11. Olsen EA. Double-blind controlled comparison of generic and trade-name topical steroids using the vasoconstriction assay. Arch Dermatol. 1991;127:197-201.
12. Stoughton RB. Are generic formulations equivalent to trade name topical glucocorticoids? Arch Dermatol. 1987;123:1312-1314.
13. Poulsen BJ, Young E, Coquilla V, et al. Effect of topical vehicle composition on the in vitro release of fluocinolone acetonide and its acetate ester. J Pharm Sci. 1968;57:928-933.
14. Taheri A, Cantrell J, Feldman SR. Tachyphylaxis to topical glucocorticoids: what is the evidence? Dermatol Online J. 2013;19:18954.
15. Ference JD, Last AR. Choosing topical corticosteroids. Am Fam Physician. 2009;79:135-140.
16. Pershing LK, Nelson JL, Corlett JL, et al. Assessment of dermatopharmacokinetic approach in the bioequivalence determination of topical tretinoin gel products. J Am Acad Dermatol. 2003;48:740-751.
17. Weigmann H, Lademann J, v Pelchrzim R, et al. Bioavailability of clobetasol propionate-quantification of drug concentrations in the stratum corneum by dermatopharmacokinetics using tape stripping. Skin Pharmacol Appl Skin Physiol. 1999;12:46-53.
18. Wiedersberg S, Naik A, Leopold CS, et al. Pharmacodynamics and dermatopharmacokinetics of betamethasone 17-valerate: assessment of topical bioavailability. Br J Dermatol. 2009;160:676-686.
The active ingredient of any pharmaceutical product is responsible for the agent’s efficacy and safety profile. This ingredient is extensively studied in clinical trials and evaluated by the US Food and Drug Administration before the product is commercially available. In dermatologic products, especially those for treating dermatoses, the vehicle in which the active ingredient is formulated also plays a role in drug delivery and indirectly impacts therapeutic outcomes, unlike excipients in oral medications. Topical vehicles must be stable, provide a suitable environment that will not degrade the active ingredient or affect its efficacy, and be cosmetically acceptable.1
Topical vehicles are formulated to maintain the stability of the active ingredient and allow it to readily penetrate the skin and reach its target area with minimal absorption into the bloodstream, thus avoiding systemic adverse events. A variety of vehicles can exist for a single active ingredient to accommodate different phases of disease and different anatomical sites where the disease may occur.2 For example, alcohol-based vehicles, sprays, and foams are preferred for the scalp where evaporation of the vehicle is beneficial to prevent greasiness of the hair, while ointments may be preferred due to their occlusive nature for areas with xerotic or thick skin from dermatoses.
Cosmetic acceptability of the vehicle may influence patient adherence to therapy. Housman et al3 assessed a variety of products formulated in different vehicles (ie, solutions, foams, emollients, gels, creams, ointments) for the treatment of psoriasis. Patients with psoriasis applied each test product to a quarter-sized area of normal skin on the forearm using a cotton swab and completed a preference questionnaire. By far, respondents significantly preferred solutions and foams over creams, gels, and ointments (P<.01). Side effects were rated to be the most important characteristics of topical therapy, followed by time needed for application, ease of application, and messiness.3 Presumably, if patients are frustrated with the topical product that they are using, adherence to the prescribed dosage and application instructions will diminish over time, leading to suboptimal steady-state levels of the product. If appropriate levels of the drug are not present at the target site, treatment will not be successful.
Steady-state levels of a topical drug at the site of action also are maintained via appropriate application frequency, most commonly once to 4 times daily for dermatologic products. Fluocinonide and halcinonide are class II (potent) corticosteroids indicated for the relief of inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses and usually are administered at least twice daily. In double-blind clinical studies comparing both products in the treatment of psoriasis, halcinonide resulted in more improved outcomes than fluocinonide.4-6 Sudilovsky and Clewe4 studied 140 patients with moderate to severe psoriasis. After 3 weeks of treatment, 44% showed superior results with halcinonide, 27% showed superior results with fluocinonide, 26% showed equal results with both products, and 3% showed no relief.4 Similarly, Close5 reported that 61% of patients showed superior results with halcinonide, 25% showed superior results with fluocinonide, 10% showed equal results with both products, and 4% showed no relief (N=50). Lynfield and Watsky6 reported that 56% of patients with severe psoriasis who were treated with halcinonide for 2 weeks showed improvement to normal or slight inflammation compared to 44% of patients treated with fluocinonide (N=59). All 3 studies used cream formulations of halcinonide and fluocinonide.
Recently, halcinonide cream was shown to have an immediate release into the stratum corneum that peaked within 1 hour of application and remained elevated for 6 hours before beginning to decline.7 These results support a biphasic release of halcinonide, which is in agreement with its formulation—that halcinonide exists in both a solution phase for immediate release into the skin and in a suspension phase that allows a sustained release after equilibrium is reached between the solution and suspension phases.8 Fluocinonide is not known to be formulated in a similar way. Its vehicle composition and penetration into the skin could explain the superior efficacy of halcinonide versus fluocinonide.
The current pilot study was conducted to compare the release pattern of fluocinonide cream versus halcinonide cream into the stratum corneum using an in vivo, noninvasive method. Results for halcin-onide have been previously published.7
Methods
Participants were sequestered in a controlled environment for the entire day to allow the skin to equilibrate prior to product application. The methodology for the application and quantification of halcinonide cream 0.1% into the stratum corneum of 5 participants using a tape-stripping protocol has been described elsewhere.7 Concordia Clinical Research institutional review board (Cedar Knolls, New Jersey) approved this study, which was conducted at Dermatology Consulting Services (High Point, North Carolina).
A 0.1-g dose of generic fluocinonide cream 0.05% was applied to four 2.5-cm circular sites on the forearm in 5 participants with normal skin until completely absorbed. Circular tape strips were subsequently placed on the application site at 1, 3, 6, and 9 hours posttreatment and were held for 10 seconds with a controlled pressure plunger to ensure adequate and consistent contact between the tape strip and the skin. The tape strip was removed with forceps, rolled with the skin scale inside, and placed in a glass vial. This procedure was repeated 6 times at 1 of 4 sites with a new tape strip at each time point to obtain samples from deeper skin layers. A total of 24 tape strips were collected from each participant.
All vials were frozen at -20°C and were shipped overnight to Robert Kellar, PhD, at the Center for Bioengineering Innovation at Northern Arizona University (Flagstaff, Arizona) for mass spectroscopy evaluation. Once received at the outside facility, the vials were stored at -20°C until analysis. Each sample was spiked with a known quantity of an appropriate reference standard and extracted with 1 mL acetonitrile at room temperature for 1 minute with agitation. New unused tape strips were spiked with a small amount of fluocinonide reference standard for extraction efficiency.
Extracts were evaporated to dryness under nitrogen gas, resuspended in 200 µL chromatography solvent, and quantified using liquid chromatography–mass spectrometry. To remove the skin scale from the tape strips, 10 mL of a solvent solution of 0.1 mg/mL fludrocortisone acetate in acetonitrile was dispensed into a 4 dram vial containing the tape strip. The vials were ultrasonicated and shaken for 10 to 15 minutes, and the samples were further diluted to 100-fold and were inverted several times to ensure complete dissolution of fluocinonide before liquid chromatography–mass spectrometry.
A standard curve ranging from the lower limit of quantification to the upper limit of quantification for the fluocinonide reference was used to determine the quantity of fluocinonide in each of the tape strips. Once the lower limit of quantification was reached in a given set of tape strip samples (1-, 3-, 6-, and 9-hour samples), the next 2 sequential tape strips in that set were analyzed to confirm fluocinonide was not detectable in deeper layers. Standard quality controls were analyzed to ensure run-to-run and sample-to-sample accuracy.
Each sample was analyzed in duplicate; 10 mg fluocinonide was used as a reference standard. The minimum detectable concentration of fluocinonide was 1 ng/mL.
Results
As expected, tape strip 1 from each participant contained the highest concentration of fluocinonide. This strip corresponded to the most superficial layer of skin. Concentrations decreased in deeper skin layers, as detected in strips 2 to 6.
In general, the average concentration of fluocin-onide in strip 1 for all 5 participants was highest at hour 1, with a subsequent decline at hours 3, 6, and 9; however, participant 1 showed a second peak in fluocinonide concentration at hour 6 (Figure 1). When the fluocinonide concentration in strips 1 to 6 was averaged for each participant at each time point, similar results were obtained: a general decline after hour 1, but a second prominent peak at hour 6 in participant 1 only. In participant 1, the average fluocinonide concentration for strips 1 to 6 was 393 ng/mL at hour 1 and declined to 208 ng/mL at hour 3; it increased to 451 ng/mL at hour 6 before declining again to 202 ng/mL at hour 9.
Because participant 1 was the only one to exhibit a second peak of fluocinonide concentration, it appears that measurements obtained from this participant may be outliers. When removing partici-pant 1 from the analysis of fluocinonide concentration in strip 1 at each time point, a clear decline is evident from hour 1 to hour 9 (Figure 2A, red line [partici-pants 2–5] vs blue line [participants 1–5]).
When the average concentration of fluocinonide was calculated in strips 1 to 6 from all participants, there was a general steady decline after hour 1 with a slight increase of 25 ng/mL at hour 6 (Figure 2B, blue line). This increase is due to the measurements obtained from participant 1; however, if partici-pant 1 is removed from the analysis, a constant decline is observed from hour 1 to hour 9 (Figure 2B, red line).
|
A prior study evaluated the penetration and absorption of halcinonide in the stratum corneum.7 In summary, halcinonide concentration peaked at hour 1 following application and remained elevated to hour 6, before beginning a slow decline. The average concentration of halcinonide from all participants in strips 1 to 6 reached 1350 ng/mL at hour 1, remained within 93% to 97% of this level (1253–1303 ng/mL) for the next 5 hours, and declined only 29% from the peak at hour 1 to hour 9 (958 ng/mL)(Figure 3, blue line).7 In contrast, the fluocinonide concentration in participants 2 to 5 from the current study reached 190 ng/mL at hour 1 and steadily declined 53% to 89 ng/mL by hour 9 (Figure 3, red line).
Two participants from the prior halcinonide study also were enrolled in the current fluocinonide study (referred to as participant A and B). In general, halcinonide levels in both participants remained elevated for 6 hours after application and declined 27.5% and 35.5%, respectively, by hour 9 (Figure 4). Participant A experienced a 20.5% dip in halcinonide concentration at hour 3 followed by an increase at hour 6; however, the halcinonide concentration at hour 9 was similar to hour 3.7 In contrast, fluocin-onide concentrations for these participants peaked at 1 hour and clearly declined approximately 60% over the next 8 hours.
Comment
The release of both fluocinonide and halcinonide into the skin was evaluated using dermal tape stripping on 4 sites on the forearms of healthy individuals. Cream formulations of each corticosteroid were evaluated in 5 participants, with 2 participants receiving both formulations during different study periods. In the prior study with halcinonide, the stratum corneum exhibited the highest concentration of the corticosteroid, with substantial declines beyond strip 6 (ie, strips 7–20).7 For this reason, only strips 1 to 6 were evaluated for corticosteroid penetration and absorption.
Results from strip 1 indicated immediate absorption of corticosteroid (fluocinonide and halcinonide) into the skin. Unlike the release of halcinonide, which demonstrated a clear sustained release over 6 hours before decreasing,7 fluocinonide concentrations began declining immediately after peaking at hour 1 and continued to decline up to hour 9. Only participant 1 exhibited a second peak of fluocinonide concentration at hour 6; the rest of the participants did not. This second peak is most likely an anomaly due to the small number of participants rather than a true elevation.
Given the rapid decline of fluocinonide concentration over the 9 hours compared with the more gradual decline of halcinonide concentration, there appears to be no evidence of a biphasic sustained release of fluocinonide from its vehicle. This difference in release pattern from each corticosteroid’s respective vehicle may explain in part the different clinical outcomes in comparative studies.4-6
It is known that vehicle composition affects corticosteroid diffusion from the vehicle to the skin surface and subsequent penetration into the skin.9 Either process can determine the overall effectiveness of the product. Ayres and Hooper10 evaluated the penetration of 4 topical preparations of cortisol. Product 1 delivered 16 times more cortisol to the skin than product 2, 8 times more than product 3, and 3 times more than product 4. Because all the preparations contained cortisol-free alcohol, these differences were attributed to the vehicle in which the cortisol was formulated. Products 1 and 4 both contained 10% urea, but the urea in product 1 was a powder in a cream base and the urea in product 4 was in a stabilizing emulsified base. Product 2 contained a propylene glycol/water base and product 3 was a water-miscible cream.10
Generic corticosteroid products have been observed in clinical practice and have been shown in vasoconstriction assays to be less and more potent than their brand-name equivalents.2,11 Vasoconstriction assays are the standard for assessing the potency of topical corticosteroids and predicting their clinical efficacy.2 One study reported significant differences in therapeutic effectiveness between generic formulations and their brand-name equivalents.12 Kenalog cream 0.1% (multiple manufacturers) was significantly more potent than any of the generic triamcinolone creams tested (P<.05); in fact, Kenalog cream 0.025% (multiple manufacturers) was statistically superior to all the generic triamcinolone creams 0.1%. Moreover, Artistocort A ointment 0.1% (Lederele Laboratories) and Valisone cream 0.1% (Schering Corporation) also were more potent than their generics at the same concentration in the same vehicle type.12 A second study also observed that 2 of 6 generic formulations had significantly less vasoconstriction than their respective brand-name formulations.11 A brand-name betamethasone valerate cream produced significantly greater vasoconstriction than its generic equivalent, and a brand-name betamethasone dipropionate cream produced greater vasoconstriction than one generic and equal vasoconstriction to another generic. Additionally, the vasoconstriction measured with Diprosone was greater than that measured with Diprolene, another brand-name product of betamethasone dipropionate.11 Diprosone and Diprolene differ in their vehicle content. The latter, a class I corticosteroid, contains a modified vehicle high in propylene glycol, whereas the former contains less propylene glycol and thus is classified as a class III corticosteroid. Propylene glycol allows hydrophobic molecules such as corticosteroids to dissolve more fully in the vehicle.12
Ostrenga et al1 studied the solubility of corticosteroids in different vehicles and, as expected, corticosteroids that fully solubilized in the vehicle exhibited better penetration into the skin on assessment with vasoconstriction assays. Corticosteroids in a suspension, on the other hand, showed slower penetration into the skin.1,13 A balance between the solution and suspension phase would allow a drug to rapidly penetrate the skin upon application, and when this pool of solubilized drug was depleted, additional drug could penetrate into the skin from the suspension phase. Based on the tape strip results from the current study it appears that halcinonide, which is manufactured in a biphasic formulation, follows this pattern of penetration and absorption into the stratum corneum. In contrast, fluocinonide appears to exist in a soluble state without much, if any, amount in a suspension phase because it had no sustained release during the 9 hours after application.
Common belief among dermatologists is that long-term use of corticosteroids leads to tachyphylaxis,14 which can be attributed to poor patient adherence. If patients skip doses, then the steady state of the product at the target site is not maintained. It is interesting to speculate that using agents with more sustained release beyond the time of application (such as halcinonide) may preserve steady-state levels even when patients are neglectful of the next medication application. Corticosteroids that work in 2 phases such as halcinonide may minimize tachyphylaxis experienced with prolonged use of corticosteroids.
Fluocinonide and halcinonide are both class II high-potency corticosteroids as shown on outcomes from vasoconstrictor assays, which assess the extent to which a corticosteroid causes cutaneous vasoconstriction or blanching in normal healthy individuals.15 The assay depends on the molecule diffusing from the vehicle, penetrating the skin, and causing a reaction (blanching) that is then evaluated. The assay cannot effectively evaluate the rate of continued diffusion and skin penetration beyond the appearance of blanching. In contrast, the tape-stripping method provides an inside look at the extent of penetration of the corticosteroid beyond the skin surface and the rate of its clearance from different skin layers. In the current study, the levels of fluocinonide declined after peaking at 1 hour after application, but the levels of halcinonide clearly remained elevated after peaking at the same time point. Most likely, vasoconstrictor studies would not be able to differentiate between the concentrations of the 2 products in the stratum corneum beyond the first hour after application.
Tape stripping, or dermatopharmacokinetics, has advantages over vasoconstriction assays in studying corticosteroid penetration and clearance from the stratum corneum. At one point, the US Food and Drug Administration had included tape stripping in its preliminary guidelines for generic topical bioequivalence studies until data from the same formulation generated from 2 different laboratories produced different results.16 Since that time, much work has been done with tape stripping to ensure its consistency. Weigmann et al17 demonstrated equivalent results with clobetasol using vasoconstriction and tape stripping, and Wiedersberg et al18 demonstrated the same with betamethasone. For the current study, the fluocinonide and halcinonide formulations were weighed prior to application so that the same dose was tested in all participants. A plunger was used to produce consistent pressure at all application sites to control for the amount of skin that was stripped off with the tape. Results for both corticosteroids were consistent between the participants. Variability in the data was detected; however, this observation is most likely due to the small number of participants in the studies.
Conclusion
In summary, this pilot study demonstrated that fluocinonide concentration in the stratum corneum peaks within the first hour of application before beginning a steady general decline. There was no evidence of sustained release. In contrast, halcin-onide demonstrated a sustained release for 6 hours after application. Halcinonide is formulated in a cream base in which the corticosteroid is present in a solution and suspension phase that allows for sustained delivery in skin over time. Fluocinonide does not appear to be formulated in the same way, and its concentrations in the stratum corneum begin to decline 1 hour after application.
Acknowledgement
Thank you to Robert Kellar, PhD, at the Center for Bioengineering Innovation at Northern Arizona University, Flagstaff, for conducting the liquid chromatography–mass spectrometry.
The active ingredient of any pharmaceutical product is responsible for the agent’s efficacy and safety profile. This ingredient is extensively studied in clinical trials and evaluated by the US Food and Drug Administration before the product is commercially available. In dermatologic products, especially those for treating dermatoses, the vehicle in which the active ingredient is formulated also plays a role in drug delivery and indirectly impacts therapeutic outcomes, unlike excipients in oral medications. Topical vehicles must be stable, provide a suitable environment that will not degrade the active ingredient or affect its efficacy, and be cosmetically acceptable.1
Topical vehicles are formulated to maintain the stability of the active ingredient and allow it to readily penetrate the skin and reach its target area with minimal absorption into the bloodstream, thus avoiding systemic adverse events. A variety of vehicles can exist for a single active ingredient to accommodate different phases of disease and different anatomical sites where the disease may occur.2 For example, alcohol-based vehicles, sprays, and foams are preferred for the scalp where evaporation of the vehicle is beneficial to prevent greasiness of the hair, while ointments may be preferred due to their occlusive nature for areas with xerotic or thick skin from dermatoses.
Cosmetic acceptability of the vehicle may influence patient adherence to therapy. Housman et al3 assessed a variety of products formulated in different vehicles (ie, solutions, foams, emollients, gels, creams, ointments) for the treatment of psoriasis. Patients with psoriasis applied each test product to a quarter-sized area of normal skin on the forearm using a cotton swab and completed a preference questionnaire. By far, respondents significantly preferred solutions and foams over creams, gels, and ointments (P<.01). Side effects were rated to be the most important characteristics of topical therapy, followed by time needed for application, ease of application, and messiness.3 Presumably, if patients are frustrated with the topical product that they are using, adherence to the prescribed dosage and application instructions will diminish over time, leading to suboptimal steady-state levels of the product. If appropriate levels of the drug are not present at the target site, treatment will not be successful.
Steady-state levels of a topical drug at the site of action also are maintained via appropriate application frequency, most commonly once to 4 times daily for dermatologic products. Fluocinonide and halcinonide are class II (potent) corticosteroids indicated for the relief of inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses and usually are administered at least twice daily. In double-blind clinical studies comparing both products in the treatment of psoriasis, halcinonide resulted in more improved outcomes than fluocinonide.4-6 Sudilovsky and Clewe4 studied 140 patients with moderate to severe psoriasis. After 3 weeks of treatment, 44% showed superior results with halcinonide, 27% showed superior results with fluocinonide, 26% showed equal results with both products, and 3% showed no relief.4 Similarly, Close5 reported that 61% of patients showed superior results with halcinonide, 25% showed superior results with fluocinonide, 10% showed equal results with both products, and 4% showed no relief (N=50). Lynfield and Watsky6 reported that 56% of patients with severe psoriasis who were treated with halcinonide for 2 weeks showed improvement to normal or slight inflammation compared to 44% of patients treated with fluocinonide (N=59). All 3 studies used cream formulations of halcinonide and fluocinonide.
Recently, halcinonide cream was shown to have an immediate release into the stratum corneum that peaked within 1 hour of application and remained elevated for 6 hours before beginning to decline.7 These results support a biphasic release of halcinonide, which is in agreement with its formulation—that halcinonide exists in both a solution phase for immediate release into the skin and in a suspension phase that allows a sustained release after equilibrium is reached between the solution and suspension phases.8 Fluocinonide is not known to be formulated in a similar way. Its vehicle composition and penetration into the skin could explain the superior efficacy of halcinonide versus fluocinonide.
The current pilot study was conducted to compare the release pattern of fluocinonide cream versus halcinonide cream into the stratum corneum using an in vivo, noninvasive method. Results for halcin-onide have been previously published.7
Methods
Participants were sequestered in a controlled environment for the entire day to allow the skin to equilibrate prior to product application. The methodology for the application and quantification of halcinonide cream 0.1% into the stratum corneum of 5 participants using a tape-stripping protocol has been described elsewhere.7 Concordia Clinical Research institutional review board (Cedar Knolls, New Jersey) approved this study, which was conducted at Dermatology Consulting Services (High Point, North Carolina).
A 0.1-g dose of generic fluocinonide cream 0.05% was applied to four 2.5-cm circular sites on the forearm in 5 participants with normal skin until completely absorbed. Circular tape strips were subsequently placed on the application site at 1, 3, 6, and 9 hours posttreatment and were held for 10 seconds with a controlled pressure plunger to ensure adequate and consistent contact between the tape strip and the skin. The tape strip was removed with forceps, rolled with the skin scale inside, and placed in a glass vial. This procedure was repeated 6 times at 1 of 4 sites with a new tape strip at each time point to obtain samples from deeper skin layers. A total of 24 tape strips were collected from each participant.
All vials were frozen at -20°C and were shipped overnight to Robert Kellar, PhD, at the Center for Bioengineering Innovation at Northern Arizona University (Flagstaff, Arizona) for mass spectroscopy evaluation. Once received at the outside facility, the vials were stored at -20°C until analysis. Each sample was spiked with a known quantity of an appropriate reference standard and extracted with 1 mL acetonitrile at room temperature for 1 minute with agitation. New unused tape strips were spiked with a small amount of fluocinonide reference standard for extraction efficiency.
Extracts were evaporated to dryness under nitrogen gas, resuspended in 200 µL chromatography solvent, and quantified using liquid chromatography–mass spectrometry. To remove the skin scale from the tape strips, 10 mL of a solvent solution of 0.1 mg/mL fludrocortisone acetate in acetonitrile was dispensed into a 4 dram vial containing the tape strip. The vials were ultrasonicated and shaken for 10 to 15 minutes, and the samples were further diluted to 100-fold and were inverted several times to ensure complete dissolution of fluocinonide before liquid chromatography–mass spectrometry.
A standard curve ranging from the lower limit of quantification to the upper limit of quantification for the fluocinonide reference was used to determine the quantity of fluocinonide in each of the tape strips. Once the lower limit of quantification was reached in a given set of tape strip samples (1-, 3-, 6-, and 9-hour samples), the next 2 sequential tape strips in that set were analyzed to confirm fluocinonide was not detectable in deeper layers. Standard quality controls were analyzed to ensure run-to-run and sample-to-sample accuracy.
Each sample was analyzed in duplicate; 10 mg fluocinonide was used as a reference standard. The minimum detectable concentration of fluocinonide was 1 ng/mL.
Results
As expected, tape strip 1 from each participant contained the highest concentration of fluocinonide. This strip corresponded to the most superficial layer of skin. Concentrations decreased in deeper skin layers, as detected in strips 2 to 6.
In general, the average concentration of fluocin-onide in strip 1 for all 5 participants was highest at hour 1, with a subsequent decline at hours 3, 6, and 9; however, participant 1 showed a second peak in fluocinonide concentration at hour 6 (Figure 1). When the fluocinonide concentration in strips 1 to 6 was averaged for each participant at each time point, similar results were obtained: a general decline after hour 1, but a second prominent peak at hour 6 in participant 1 only. In participant 1, the average fluocinonide concentration for strips 1 to 6 was 393 ng/mL at hour 1 and declined to 208 ng/mL at hour 3; it increased to 451 ng/mL at hour 6 before declining again to 202 ng/mL at hour 9.
Because participant 1 was the only one to exhibit a second peak of fluocinonide concentration, it appears that measurements obtained from this participant may be outliers. When removing partici-pant 1 from the analysis of fluocinonide concentration in strip 1 at each time point, a clear decline is evident from hour 1 to hour 9 (Figure 2A, red line [partici-pants 2–5] vs blue line [participants 1–5]).
When the average concentration of fluocinonide was calculated in strips 1 to 6 from all participants, there was a general steady decline after hour 1 with a slight increase of 25 ng/mL at hour 6 (Figure 2B, blue line). This increase is due to the measurements obtained from participant 1; however, if partici-pant 1 is removed from the analysis, a constant decline is observed from hour 1 to hour 9 (Figure 2B, red line).
|
A prior study evaluated the penetration and absorption of halcinonide in the stratum corneum.7 In summary, halcinonide concentration peaked at hour 1 following application and remained elevated to hour 6, before beginning a slow decline. The average concentration of halcinonide from all participants in strips 1 to 6 reached 1350 ng/mL at hour 1, remained within 93% to 97% of this level (1253–1303 ng/mL) for the next 5 hours, and declined only 29% from the peak at hour 1 to hour 9 (958 ng/mL)(Figure 3, blue line).7 In contrast, the fluocinonide concentration in participants 2 to 5 from the current study reached 190 ng/mL at hour 1 and steadily declined 53% to 89 ng/mL by hour 9 (Figure 3, red line).
Two participants from the prior halcinonide study also were enrolled in the current fluocinonide study (referred to as participant A and B). In general, halcinonide levels in both participants remained elevated for 6 hours after application and declined 27.5% and 35.5%, respectively, by hour 9 (Figure 4). Participant A experienced a 20.5% dip in halcinonide concentration at hour 3 followed by an increase at hour 6; however, the halcinonide concentration at hour 9 was similar to hour 3.7 In contrast, fluocin-onide concentrations for these participants peaked at 1 hour and clearly declined approximately 60% over the next 8 hours.
Comment
The release of both fluocinonide and halcinonide into the skin was evaluated using dermal tape stripping on 4 sites on the forearms of healthy individuals. Cream formulations of each corticosteroid were evaluated in 5 participants, with 2 participants receiving both formulations during different study periods. In the prior study with halcinonide, the stratum corneum exhibited the highest concentration of the corticosteroid, with substantial declines beyond strip 6 (ie, strips 7–20).7 For this reason, only strips 1 to 6 were evaluated for corticosteroid penetration and absorption.
Results from strip 1 indicated immediate absorption of corticosteroid (fluocinonide and halcinonide) into the skin. Unlike the release of halcinonide, which demonstrated a clear sustained release over 6 hours before decreasing,7 fluocinonide concentrations began declining immediately after peaking at hour 1 and continued to decline up to hour 9. Only participant 1 exhibited a second peak of fluocinonide concentration at hour 6; the rest of the participants did not. This second peak is most likely an anomaly due to the small number of participants rather than a true elevation.
Given the rapid decline of fluocinonide concentration over the 9 hours compared with the more gradual decline of halcinonide concentration, there appears to be no evidence of a biphasic sustained release of fluocinonide from its vehicle. This difference in release pattern from each corticosteroid’s respective vehicle may explain in part the different clinical outcomes in comparative studies.4-6
It is known that vehicle composition affects corticosteroid diffusion from the vehicle to the skin surface and subsequent penetration into the skin.9 Either process can determine the overall effectiveness of the product. Ayres and Hooper10 evaluated the penetration of 4 topical preparations of cortisol. Product 1 delivered 16 times more cortisol to the skin than product 2, 8 times more than product 3, and 3 times more than product 4. Because all the preparations contained cortisol-free alcohol, these differences were attributed to the vehicle in which the cortisol was formulated. Products 1 and 4 both contained 10% urea, but the urea in product 1 was a powder in a cream base and the urea in product 4 was in a stabilizing emulsified base. Product 2 contained a propylene glycol/water base and product 3 was a water-miscible cream.10
Generic corticosteroid products have been observed in clinical practice and have been shown in vasoconstriction assays to be less and more potent than their brand-name equivalents.2,11 Vasoconstriction assays are the standard for assessing the potency of topical corticosteroids and predicting their clinical efficacy.2 One study reported significant differences in therapeutic effectiveness between generic formulations and their brand-name equivalents.12 Kenalog cream 0.1% (multiple manufacturers) was significantly more potent than any of the generic triamcinolone creams tested (P<.05); in fact, Kenalog cream 0.025% (multiple manufacturers) was statistically superior to all the generic triamcinolone creams 0.1%. Moreover, Artistocort A ointment 0.1% (Lederele Laboratories) and Valisone cream 0.1% (Schering Corporation) also were more potent than their generics at the same concentration in the same vehicle type.12 A second study also observed that 2 of 6 generic formulations had significantly less vasoconstriction than their respective brand-name formulations.11 A brand-name betamethasone valerate cream produced significantly greater vasoconstriction than its generic equivalent, and a brand-name betamethasone dipropionate cream produced greater vasoconstriction than one generic and equal vasoconstriction to another generic. Additionally, the vasoconstriction measured with Diprosone was greater than that measured with Diprolene, another brand-name product of betamethasone dipropionate.11 Diprosone and Diprolene differ in their vehicle content. The latter, a class I corticosteroid, contains a modified vehicle high in propylene glycol, whereas the former contains less propylene glycol and thus is classified as a class III corticosteroid. Propylene glycol allows hydrophobic molecules such as corticosteroids to dissolve more fully in the vehicle.12
Ostrenga et al1 studied the solubility of corticosteroids in different vehicles and, as expected, corticosteroids that fully solubilized in the vehicle exhibited better penetration into the skin on assessment with vasoconstriction assays. Corticosteroids in a suspension, on the other hand, showed slower penetration into the skin.1,13 A balance between the solution and suspension phase would allow a drug to rapidly penetrate the skin upon application, and when this pool of solubilized drug was depleted, additional drug could penetrate into the skin from the suspension phase. Based on the tape strip results from the current study it appears that halcinonide, which is manufactured in a biphasic formulation, follows this pattern of penetration and absorption into the stratum corneum. In contrast, fluocinonide appears to exist in a soluble state without much, if any, amount in a suspension phase because it had no sustained release during the 9 hours after application.
Common belief among dermatologists is that long-term use of corticosteroids leads to tachyphylaxis,14 which can be attributed to poor patient adherence. If patients skip doses, then the steady state of the product at the target site is not maintained. It is interesting to speculate that using agents with more sustained release beyond the time of application (such as halcinonide) may preserve steady-state levels even when patients are neglectful of the next medication application. Corticosteroids that work in 2 phases such as halcinonide may minimize tachyphylaxis experienced with prolonged use of corticosteroids.
Fluocinonide and halcinonide are both class II high-potency corticosteroids as shown on outcomes from vasoconstrictor assays, which assess the extent to which a corticosteroid causes cutaneous vasoconstriction or blanching in normal healthy individuals.15 The assay depends on the molecule diffusing from the vehicle, penetrating the skin, and causing a reaction (blanching) that is then evaluated. The assay cannot effectively evaluate the rate of continued diffusion and skin penetration beyond the appearance of blanching. In contrast, the tape-stripping method provides an inside look at the extent of penetration of the corticosteroid beyond the skin surface and the rate of its clearance from different skin layers. In the current study, the levels of fluocinonide declined after peaking at 1 hour after application, but the levels of halcinonide clearly remained elevated after peaking at the same time point. Most likely, vasoconstrictor studies would not be able to differentiate between the concentrations of the 2 products in the stratum corneum beyond the first hour after application.
Tape stripping, or dermatopharmacokinetics, has advantages over vasoconstriction assays in studying corticosteroid penetration and clearance from the stratum corneum. At one point, the US Food and Drug Administration had included tape stripping in its preliminary guidelines for generic topical bioequivalence studies until data from the same formulation generated from 2 different laboratories produced different results.16 Since that time, much work has been done with tape stripping to ensure its consistency. Weigmann et al17 demonstrated equivalent results with clobetasol using vasoconstriction and tape stripping, and Wiedersberg et al18 demonstrated the same with betamethasone. For the current study, the fluocinonide and halcinonide formulations were weighed prior to application so that the same dose was tested in all participants. A plunger was used to produce consistent pressure at all application sites to control for the amount of skin that was stripped off with the tape. Results for both corticosteroids were consistent between the participants. Variability in the data was detected; however, this observation is most likely due to the small number of participants in the studies.
Conclusion
In summary, this pilot study demonstrated that fluocinonide concentration in the stratum corneum peaks within the first hour of application before beginning a steady general decline. There was no evidence of sustained release. In contrast, halcin-onide demonstrated a sustained release for 6 hours after application. Halcinonide is formulated in a cream base in which the corticosteroid is present in a solution and suspension phase that allows for sustained delivery in skin over time. Fluocinonide does not appear to be formulated in the same way, and its concentrations in the stratum corneum begin to decline 1 hour after application.
Acknowledgement
Thank you to Robert Kellar, PhD, at the Center for Bioengineering Innovation at Northern Arizona University, Flagstaff, for conducting the liquid chromatography–mass spectrometry.
1. Ostrenga J, Haleblian J, Poulsen B, et al. Vehicle design for a new topical steroid, fluocinonide. J Invest Dermatol. 1971;56:392-399.
2. Rathi SK, D’Souza P. Rational and ethical use of topical corticosteroids based on safety and efficacy. Indian J Dermatol. 2012;57:251-259.
3. Housman TS, Mellen BG, Rapp SR, et al. Patients with psoriasis prefer solution and foam vehicles: a quantitative assessment of vehicle preference. Cutis. 2002;70:327-332.
4. Sudilovsky A, Clewe TH. Comparative efficacy of halcin-onide and fluocinonide creams in psoriasis and eczematous dermatoses. J Clin Pharmacol. 1975;15:779-784.
5. Close JE. Double-blind comparison of topical halcinonide and fluocinonide in the treatment of psoriasis. Int J Dermatol. 1976;15:534-537.
6. Lynfield Y, Watsky M. Psoriasis: topical corticosteroid therapy. Cutis. 1976;18:133, 136-137.
7. Draelos ZD. Demonstration of the biphasic release of 0.1% halcinonide cream. J Drugs Dermatol. 2015;14:89-90.
8. Bagatell FK. Halcinonide: a new potent topical anti-inflammatory drug. Cutis. 1974;14:459-462.
9. Ostrenga J, Steinmetz C, Poulsen B. Significance of vehicle composition. I. relationship between topical vehicle composition, skin penetrability, and clinical efficacy. J Pharm Sci. 1971;60:1175-1179.
10. Ayres PJ, Hooper G. Assessment of the skin penetration properties of different carrier vehicles for topically applied cortisol. Br J Dermatol. 1978;99:307-317.
11. Olsen EA. Double-blind controlled comparison of generic and trade-name topical steroids using the vasoconstriction assay. Arch Dermatol. 1991;127:197-201.
12. Stoughton RB. Are generic formulations equivalent to trade name topical glucocorticoids? Arch Dermatol. 1987;123:1312-1314.
13. Poulsen BJ, Young E, Coquilla V, et al. Effect of topical vehicle composition on the in vitro release of fluocinolone acetonide and its acetate ester. J Pharm Sci. 1968;57:928-933.
14. Taheri A, Cantrell J, Feldman SR. Tachyphylaxis to topical glucocorticoids: what is the evidence? Dermatol Online J. 2013;19:18954.
15. Ference JD, Last AR. Choosing topical corticosteroids. Am Fam Physician. 2009;79:135-140.
16. Pershing LK, Nelson JL, Corlett JL, et al. Assessment of dermatopharmacokinetic approach in the bioequivalence determination of topical tretinoin gel products. J Am Acad Dermatol. 2003;48:740-751.
17. Weigmann H, Lademann J, v Pelchrzim R, et al. Bioavailability of clobetasol propionate-quantification of drug concentrations in the stratum corneum by dermatopharmacokinetics using tape stripping. Skin Pharmacol Appl Skin Physiol. 1999;12:46-53.
18. Wiedersberg S, Naik A, Leopold CS, et al. Pharmacodynamics and dermatopharmacokinetics of betamethasone 17-valerate: assessment of topical bioavailability. Br J Dermatol. 2009;160:676-686.
1. Ostrenga J, Haleblian J, Poulsen B, et al. Vehicle design for a new topical steroid, fluocinonide. J Invest Dermatol. 1971;56:392-399.
2. Rathi SK, D’Souza P. Rational and ethical use of topical corticosteroids based on safety and efficacy. Indian J Dermatol. 2012;57:251-259.
3. Housman TS, Mellen BG, Rapp SR, et al. Patients with psoriasis prefer solution and foam vehicles: a quantitative assessment of vehicle preference. Cutis. 2002;70:327-332.
4. Sudilovsky A, Clewe TH. Comparative efficacy of halcin-onide and fluocinonide creams in psoriasis and eczematous dermatoses. J Clin Pharmacol. 1975;15:779-784.
5. Close JE. Double-blind comparison of topical halcinonide and fluocinonide in the treatment of psoriasis. Int J Dermatol. 1976;15:534-537.
6. Lynfield Y, Watsky M. Psoriasis: topical corticosteroid therapy. Cutis. 1976;18:133, 136-137.
7. Draelos ZD. Demonstration of the biphasic release of 0.1% halcinonide cream. J Drugs Dermatol. 2015;14:89-90.
8. Bagatell FK. Halcinonide: a new potent topical anti-inflammatory drug. Cutis. 1974;14:459-462.
9. Ostrenga J, Steinmetz C, Poulsen B. Significance of vehicle composition. I. relationship between topical vehicle composition, skin penetrability, and clinical efficacy. J Pharm Sci. 1971;60:1175-1179.
10. Ayres PJ, Hooper G. Assessment of the skin penetration properties of different carrier vehicles for topically applied cortisol. Br J Dermatol. 1978;99:307-317.
11. Olsen EA. Double-blind controlled comparison of generic and trade-name topical steroids using the vasoconstriction assay. Arch Dermatol. 1991;127:197-201.
12. Stoughton RB. Are generic formulations equivalent to trade name topical glucocorticoids? Arch Dermatol. 1987;123:1312-1314.
13. Poulsen BJ, Young E, Coquilla V, et al. Effect of topical vehicle composition on the in vitro release of fluocinolone acetonide and its acetate ester. J Pharm Sci. 1968;57:928-933.
14. Taheri A, Cantrell J, Feldman SR. Tachyphylaxis to topical glucocorticoids: what is the evidence? Dermatol Online J. 2013;19:18954.
15. Ference JD, Last AR. Choosing topical corticosteroids. Am Fam Physician. 2009;79:135-140.
16. Pershing LK, Nelson JL, Corlett JL, et al. Assessment of dermatopharmacokinetic approach in the bioequivalence determination of topical tretinoin gel products. J Am Acad Dermatol. 2003;48:740-751.
17. Weigmann H, Lademann J, v Pelchrzim R, et al. Bioavailability of clobetasol propionate-quantification of drug concentrations in the stratum corneum by dermatopharmacokinetics using tape stripping. Skin Pharmacol Appl Skin Physiol. 1999;12:46-53.
18. Wiedersberg S, Naik A, Leopold CS, et al. Pharmacodynamics and dermatopharmacokinetics of betamethasone 17-valerate: assessment of topical bioavailability. Br J Dermatol. 2009;160:676-686.
Practice Points
- Fluocinonide concentration in the stratum corneum peaks within the first hour of application and then begins a steady decline.
- Halcinonide concentration also peaks within the first hour of application and remains elevated for 6 hours after application.
- Halcinonide, rather than fluocinonide, may provide clinical benefits in between doses because of its sustained release hours after application.
