User login
Heart Rhythm Society (HRS): Heart Rhythm 2013
Reablate, don't medicate, after failed AF ablation
DENVER – After a failed first ablation procedure for paroxysmal atrial fibrillation, redo ablation proved more effective than did antiarrhythmic drug therapy in a randomized trial, Dr. Jonathan Steinberg reported at the annual meeting of the Heart Rhythm Society.
The success rate of a first ablation procedure in patients with symptomatic paroxysmal AF is typically about 60%. What to do for the 40% who are nonresponders has been unclear, with no prior randomized clinical trial evidence available to guide decisions, noted Dr. Steinberg of Columbia University, New York.
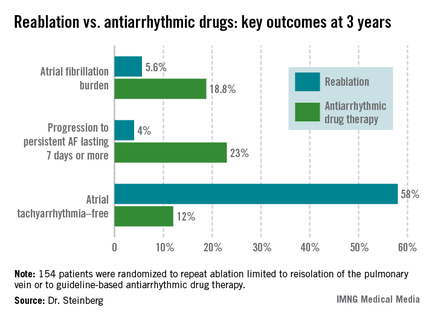
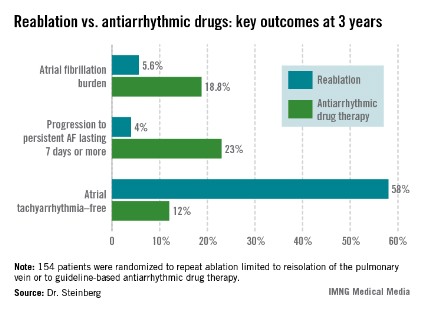
The study comprised 154 patients with recurrent symptomatic paroxysmal AF 3 months after an initial ablation procedure involving only pulmonary vein isolation. All participants received an implantable loop recorder to track atrial arrhythmic events.
They were then randomized to redo-ablation limited to reisolation of the pulmonary vein, which was successfully accomplished in all instances, or to guideline-based antiarrhythmic drug therapy. The choice of drug was left to individual investigator discretion. The three options were propafenone at 450-900 mg/day, sotalol at 160-320 mg/day, or flecainide at 200-400 mg/day. Propafenone was selected in the majority of cases, at an average dose of 579 mg/day.
The average AF burden as measured by implantable loop recorder at randomization was 15%. The primary study endpoint was AF burden at 36 months of follow-up, which was 5.6% in the redo-ablation group compared with 18.8% in the antiarrhythmic drug group.
Secondary endpoints uniformly favored redo-ablation as well.
Data from the implantable loop recorders was evaluated every 3 months during 3 years of follow-up. As early as 3 months into the study, the group given antiarrhythmic drugs had an AF burden of 3.3%, significantly higher than the 1.9% rate seen in the redo-ablation group. Thereafter, the drug therapy group experienced a gradual increase in AF burden throughout the first 12-15 months, followed by a much more substantial increase during the remainder of the study.
"The redo-ablation group had a different pattern" of AF burden, Dr. Steinberg observed. "It was low throughout the first 12-15 months, with just a slight increase, and it then rose only gradually over time until the 36-month end of the study."
Freedom from any atrial tachyarrhythmia at 1 year was 30% in the antiarrhythmic drug therapy group and 75% in the redo-ablation group. By 3 years, 12% of those in the drug therapy group were free of atrial tachyarrhythmias as were 58% in the redo-ablation group.
Complications in the redo-ablation group consisted of two cases of cardiac tamponade. In contrast, 49 patients, or 64%, in the antiarrhythmic drug therapy group discontinued medication due to intolerance or ineffectiveness.
Session cochair Dr. Gordon Tomaselli of Johns Hopkins University, Baltimore, said that in light of the potential proarrhythmic effects of virtually all antiarrhythmic drugs, it would have been useful to include a no-antiarrhythmic drug control group in the study.
Dr. Steinberg reported having no conflicts of interest.
DENVER – After a failed first ablation procedure for paroxysmal atrial fibrillation, redo ablation proved more effective than did antiarrhythmic drug therapy in a randomized trial, Dr. Jonathan Steinberg reported at the annual meeting of the Heart Rhythm Society.
The success rate of a first ablation procedure in patients with symptomatic paroxysmal AF is typically about 60%. What to do for the 40% who are nonresponders has been unclear, with no prior randomized clinical trial evidence available to guide decisions, noted Dr. Steinberg of Columbia University, New York.
The study comprised 154 patients with recurrent symptomatic paroxysmal AF 3 months after an initial ablation procedure involving only pulmonary vein isolation. All participants received an implantable loop recorder to track atrial arrhythmic events.
They were then randomized to redo-ablation limited to reisolation of the pulmonary vein, which was successfully accomplished in all instances, or to guideline-based antiarrhythmic drug therapy. The choice of drug was left to individual investigator discretion. The three options were propafenone at 450-900 mg/day, sotalol at 160-320 mg/day, or flecainide at 200-400 mg/day. Propafenone was selected in the majority of cases, at an average dose of 579 mg/day.
The average AF burden as measured by implantable loop recorder at randomization was 15%. The primary study endpoint was AF burden at 36 months of follow-up, which was 5.6% in the redo-ablation group compared with 18.8% in the antiarrhythmic drug group.
Secondary endpoints uniformly favored redo-ablation as well.
Data from the implantable loop recorders was evaluated every 3 months during 3 years of follow-up. As early as 3 months into the study, the group given antiarrhythmic drugs had an AF burden of 3.3%, significantly higher than the 1.9% rate seen in the redo-ablation group. Thereafter, the drug therapy group experienced a gradual increase in AF burden throughout the first 12-15 months, followed by a much more substantial increase during the remainder of the study.
"The redo-ablation group had a different pattern" of AF burden, Dr. Steinberg observed. "It was low throughout the first 12-15 months, with just a slight increase, and it then rose only gradually over time until the 36-month end of the study."
Freedom from any atrial tachyarrhythmia at 1 year was 30% in the antiarrhythmic drug therapy group and 75% in the redo-ablation group. By 3 years, 12% of those in the drug therapy group were free of atrial tachyarrhythmias as were 58% in the redo-ablation group.
Complications in the redo-ablation group consisted of two cases of cardiac tamponade. In contrast, 49 patients, or 64%, in the antiarrhythmic drug therapy group discontinued medication due to intolerance or ineffectiveness.
Session cochair Dr. Gordon Tomaselli of Johns Hopkins University, Baltimore, said that in light of the potential proarrhythmic effects of virtually all antiarrhythmic drugs, it would have been useful to include a no-antiarrhythmic drug control group in the study.
Dr. Steinberg reported having no conflicts of interest.
DENVER – After a failed first ablation procedure for paroxysmal atrial fibrillation, redo ablation proved more effective than did antiarrhythmic drug therapy in a randomized trial, Dr. Jonathan Steinberg reported at the annual meeting of the Heart Rhythm Society.
The success rate of a first ablation procedure in patients with symptomatic paroxysmal AF is typically about 60%. What to do for the 40% who are nonresponders has been unclear, with no prior randomized clinical trial evidence available to guide decisions, noted Dr. Steinberg of Columbia University, New York.
The study comprised 154 patients with recurrent symptomatic paroxysmal AF 3 months after an initial ablation procedure involving only pulmonary vein isolation. All participants received an implantable loop recorder to track atrial arrhythmic events.
They were then randomized to redo-ablation limited to reisolation of the pulmonary vein, which was successfully accomplished in all instances, or to guideline-based antiarrhythmic drug therapy. The choice of drug was left to individual investigator discretion. The three options were propafenone at 450-900 mg/day, sotalol at 160-320 mg/day, or flecainide at 200-400 mg/day. Propafenone was selected in the majority of cases, at an average dose of 579 mg/day.
The average AF burden as measured by implantable loop recorder at randomization was 15%. The primary study endpoint was AF burden at 36 months of follow-up, which was 5.6% in the redo-ablation group compared with 18.8% in the antiarrhythmic drug group.
Secondary endpoints uniformly favored redo-ablation as well.
Data from the implantable loop recorders was evaluated every 3 months during 3 years of follow-up. As early as 3 months into the study, the group given antiarrhythmic drugs had an AF burden of 3.3%, significantly higher than the 1.9% rate seen in the redo-ablation group. Thereafter, the drug therapy group experienced a gradual increase in AF burden throughout the first 12-15 months, followed by a much more substantial increase during the remainder of the study.
"The redo-ablation group had a different pattern" of AF burden, Dr. Steinberg observed. "It was low throughout the first 12-15 months, with just a slight increase, and it then rose only gradually over time until the 36-month end of the study."
Freedom from any atrial tachyarrhythmia at 1 year was 30% in the antiarrhythmic drug therapy group and 75% in the redo-ablation group. By 3 years, 12% of those in the drug therapy group were free of atrial tachyarrhythmias as were 58% in the redo-ablation group.
Complications in the redo-ablation group consisted of two cases of cardiac tamponade. In contrast, 49 patients, or 64%, in the antiarrhythmic drug therapy group discontinued medication due to intolerance or ineffectiveness.
Session cochair Dr. Gordon Tomaselli of Johns Hopkins University, Baltimore, said that in light of the potential proarrhythmic effects of virtually all antiarrhythmic drugs, it would have been useful to include a no-antiarrhythmic drug control group in the study.
Dr. Steinberg reported having no conflicts of interest.
AT HEART RHYTHM 2013
Major Finding: At 3 years after a first ablation procedure had failed for symptomatic paroxysmal atrial fibrillation, 12% of those randomized to drug therapy and 58% of those in the redo-ablation group were free of atrial tachyarrhythmias.
Data Source: A randomized, prospective, multicenter clinical trial involving 154 patients whose atrial arrhythmia status was monitored via implantable loop recorder.
Disclosures: The presenter reported having no conflicts of interest.
Shock-Less trial improves physicians' ICD programming
DENVER – The rate of adherence to evidence-based implantable cardioverter-defibrillator programming strategies known to reduce the rate of unnecessary shocks climbed significantly in a large, prospective study in which physicians received detailed reports on their own performance and how it stacked up to that of others.
The Shock-Less study was a real-world international study involving 4,131 ICD recipients and their physicians at 118 sites. The improved adherence to evidence-based programming achieved through the use of the individualized, multipage therapy programming reports (TPRs) translated into a highly significant 27% reduction in the risk of all-cause ICD shocks during follow-up, Dr. Marc T. Silver reported at the annual meeting of the Heart Rhythm Society.
This is welcome news for patients. It means less shock-related morbidity and potentially less mortality, said Dr. Silver of WakeMed in Raleigh, N.C.
The study included 2,693 patients who received their Medtronic primary or secondary prevention ICD before a center received its first Shock-Less report and 1,438 implanted after the report. A total of 265 all-cause shocks occurred in the "before" group, 116 in the "after" cohort.
In a multivariate logistic regression analysis adjusted for factors known to affect shock rates, including patient age, smoking status, New York Heart Association functional class, and atrial fibrillation, patients in the "after" group had a 27% reduction in the relative risk of both appropriate and inappropriate shocks (P = .002).
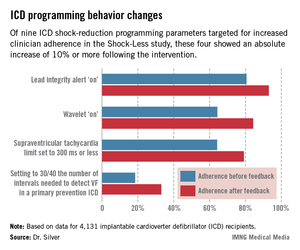
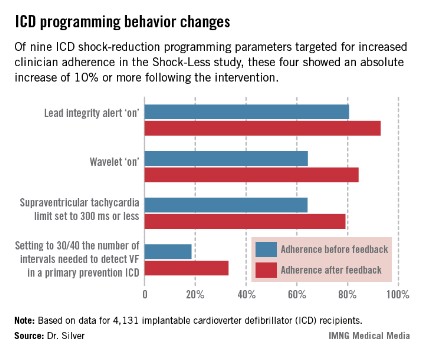
The TPRs provided ICD centers and their individual physicians with detailed feedback on rates of adherence to nine evidence-based programming settings that help reduce shocks. Most ICDs don’t arrive from the manufacturer with these settings in place. Some of the changes in programming were quite impressive (see graphic), including a near-doubling of the rate of primary prevention ICDs programmed to 30/40 as the number of intervals to detect ventricular fibrillation; this rate improved from 18.5% to 33.1%.
Dr. Silver offered personal testimony as to the power of the TPRs as a behavior-modification tool.

"Having received TPRs myself, it is a character-building experience. If you can get hold of information like this on your own practices, I guarantee you will leave a little smaller afterwards, like I did. Bigger in some way, smaller in others," he said.
Yet there remains a clear opportunity for further improvement in physician performance, Dr. Silver added.
"Achieving numbers in the 33% range for adherence to 30/40 [the number of intervals needed to detect ventricular fibrillation] is not what I think many of us would hope for one day," he observed.
Along those lines, audience member Dr. Thomas F. Deering of the Piedmont Heart Institute, Atlanta, commented that while the changes in physician behavior achieved through the Shock-Less project were significant, they were not sweeping in magnitude. Given the negative clinical consequences that result from lack of adherence to evidence-based programming, isn’t it time to request that the device industry change the default settings on their ICDs in accord with the evidence-based guidelines?, he asked.
"As someone who’s become very interested in physician behavior," Dr. Silver replied, "I regret to say that changing nominal settings on the devices may be the best way to move our profession forward. I say that with some degree of regret, but I think that’s the truth."
Dr. Silver reported serving as a consultant to Medtronic, which sponsored the Shock-Less study.
DENVER – The rate of adherence to evidence-based implantable cardioverter-defibrillator programming strategies known to reduce the rate of unnecessary shocks climbed significantly in a large, prospective study in which physicians received detailed reports on their own performance and how it stacked up to that of others.
The Shock-Less study was a real-world international study involving 4,131 ICD recipients and their physicians at 118 sites. The improved adherence to evidence-based programming achieved through the use of the individualized, multipage therapy programming reports (TPRs) translated into a highly significant 27% reduction in the risk of all-cause ICD shocks during follow-up, Dr. Marc T. Silver reported at the annual meeting of the Heart Rhythm Society.
This is welcome news for patients. It means less shock-related morbidity and potentially less mortality, said Dr. Silver of WakeMed in Raleigh, N.C.
The study included 2,693 patients who received their Medtronic primary or secondary prevention ICD before a center received its first Shock-Less report and 1,438 implanted after the report. A total of 265 all-cause shocks occurred in the "before" group, 116 in the "after" cohort.
In a multivariate logistic regression analysis adjusted for factors known to affect shock rates, including patient age, smoking status, New York Heart Association functional class, and atrial fibrillation, patients in the "after" group had a 27% reduction in the relative risk of both appropriate and inappropriate shocks (P = .002).
The TPRs provided ICD centers and their individual physicians with detailed feedback on rates of adherence to nine evidence-based programming settings that help reduce shocks. Most ICDs don’t arrive from the manufacturer with these settings in place. Some of the changes in programming were quite impressive (see graphic), including a near-doubling of the rate of primary prevention ICDs programmed to 30/40 as the number of intervals to detect ventricular fibrillation; this rate improved from 18.5% to 33.1%.
Dr. Silver offered personal testimony as to the power of the TPRs as a behavior-modification tool.
"Having received TPRs myself, it is a character-building experience. If you can get hold of information like this on your own practices, I guarantee you will leave a little smaller afterwards, like I did. Bigger in some way, smaller in others," he said.
Yet there remains a clear opportunity for further improvement in physician performance, Dr. Silver added.
"Achieving numbers in the 33% range for adherence to 30/40 [the number of intervals needed to detect ventricular fibrillation] is not what I think many of us would hope for one day," he observed.
Along those lines, audience member Dr. Thomas F. Deering of the Piedmont Heart Institute, Atlanta, commented that while the changes in physician behavior achieved through the Shock-Less project were significant, they were not sweeping in magnitude. Given the negative clinical consequences that result from lack of adherence to evidence-based programming, isn’t it time to request that the device industry change the default settings on their ICDs in accord with the evidence-based guidelines?, he asked.
"As someone who’s become very interested in physician behavior," Dr. Silver replied, "I regret to say that changing nominal settings on the devices may be the best way to move our profession forward. I say that with some degree of regret, but I think that’s the truth."
Dr. Silver reported serving as a consultant to Medtronic, which sponsored the Shock-Less study.
DENVER – The rate of adherence to evidence-based implantable cardioverter-defibrillator programming strategies known to reduce the rate of unnecessary shocks climbed significantly in a large, prospective study in which physicians received detailed reports on their own performance and how it stacked up to that of others.
The Shock-Less study was a real-world international study involving 4,131 ICD recipients and their physicians at 118 sites. The improved adherence to evidence-based programming achieved through the use of the individualized, multipage therapy programming reports (TPRs) translated into a highly significant 27% reduction in the risk of all-cause ICD shocks during follow-up, Dr. Marc T. Silver reported at the annual meeting of the Heart Rhythm Society.
This is welcome news for patients. It means less shock-related morbidity and potentially less mortality, said Dr. Silver of WakeMed in Raleigh, N.C.
The study included 2,693 patients who received their Medtronic primary or secondary prevention ICD before a center received its first Shock-Less report and 1,438 implanted after the report. A total of 265 all-cause shocks occurred in the "before" group, 116 in the "after" cohort.
In a multivariate logistic regression analysis adjusted for factors known to affect shock rates, including patient age, smoking status, New York Heart Association functional class, and atrial fibrillation, patients in the "after" group had a 27% reduction in the relative risk of both appropriate and inappropriate shocks (P = .002).
The TPRs provided ICD centers and their individual physicians with detailed feedback on rates of adherence to nine evidence-based programming settings that help reduce shocks. Most ICDs don’t arrive from the manufacturer with these settings in place. Some of the changes in programming were quite impressive (see graphic), including a near-doubling of the rate of primary prevention ICDs programmed to 30/40 as the number of intervals to detect ventricular fibrillation; this rate improved from 18.5% to 33.1%.
Dr. Silver offered personal testimony as to the power of the TPRs as a behavior-modification tool.
"Having received TPRs myself, it is a character-building experience. If you can get hold of information like this on your own practices, I guarantee you will leave a little smaller afterwards, like I did. Bigger in some way, smaller in others," he said.
Yet there remains a clear opportunity for further improvement in physician performance, Dr. Silver added.
"Achieving numbers in the 33% range for adherence to 30/40 [the number of intervals needed to detect ventricular fibrillation] is not what I think many of us would hope for one day," he observed.
Along those lines, audience member Dr. Thomas F. Deering of the Piedmont Heart Institute, Atlanta, commented that while the changes in physician behavior achieved through the Shock-Less project were significant, they were not sweeping in magnitude. Given the negative clinical consequences that result from lack of adherence to evidence-based programming, isn’t it time to request that the device industry change the default settings on their ICDs in accord with the evidence-based guidelines?, he asked.
"As someone who’s become very interested in physician behavior," Dr. Silver replied, "I regret to say that changing nominal settings on the devices may be the best way to move our profession forward. I say that with some degree of regret, but I think that’s the truth."
Dr. Silver reported serving as a consultant to Medtronic, which sponsored the Shock-Less study.
AT HEART RHYTHM 2013
Major Finding: A 27% reduction in shocks from ICDs ensued after physicians received detailed, structured reports on their rates of adherence to evidence-based shock-reduction programming strategies.
Data Source: Shock-Less is an international prospective cohort study involving 4,131 ICD recipients and their physicians at 118 centers.
Disclosures: Dr. Silver reported serving as a consultant to Medtronic, which sponsored the Shock-Less study.
Biomarkers predict response to cardiac resynchronization therapy
DENVER – Levels of troponin T and brain natriuretic peptide prior to implantation of a cardiac resynchronization device were strongly predictive of the risk of death or hospitalization for heart failure during the first year of device therapy in the BENEFIT study.
BENEFIT was a 1-year observational study undertaken to determine whether markers could predict which candidates for cardiac resynchronization therapy (CRT) were least likely to benefit from the costly device. At present, roughly 30% of patients who appropriately receive a CRT device do not respond to the treatment.
"We are all at this point frustrated with the persistent portion of CRT recipients who do not respond to therapy," BENEFIT principal investigator Dr. Alaa A. Shalaby said in presenting the study findings at the annual meeting of the Heart Rhythm Society. "We think our results suggest CRT should be offered earlier in the course of the disease"

BENEFIT included 267 CRT recipients at 33 centers. Patients were placed into one of three predefined risk categories based upon baseline levels of brain natriuretic peptide (BNP) and troponin T (TnT). On the basis os prior studies, high BNP was defined as 440 pg/mL or greater and a detectible TnT level was 0.01 ng/mL or greater. The low-risk group had an undetectable TnT and a BNP below 440 pg/mL. The intermediate-risk group had either an elevated BNP or TnT. The high-risk group had elevated BNP and TnT, reported Dr. Shalaby of the University of Pittsburgh.
The median baseline BNP in the study population was 198 pg/mL.
The three groups were similar in terms of baseline characteristics, including the proportion of patients with ischemic cardiomyopathy. Based on BNP and TnT results, 59% of patients were in the low-risk category, 33% had intermediate risk, and 8% were deemed high risk. The intermediate-risk group was evenly divided between patients who qualified on the basis of a high BNP and those with a detectable TnT.
During 12 months of follow-up there were 11 deaths and 19 heart failure hospitalizations. The incidence of either endpoint was 7% in the low-risk group, 15% in the intermediate-risk group, and 30% in the high-risk cohort.
After the researchers adjusted the results for age, left ventricular ejection fraction, QRS duration, and NYHA class, the risk of death or heart failure hospitalization was 2.5-fold greater in the group with intermediate-level baseline biomarkers as in the low-risk group. The high-biomarker group had a 7.3-fold increased risk, compared with the low-biomarker cohort. Both of these differences were statistically significant.
Changes in the biomarker levels after 6 and 12 months of CRT will be the subject of a future BENEFIT analysis, he noted.
The BENEFIT study was supported by St. Jude Medical. Dr. Shalaby reported having no conflicts of interest.
DENVER – Levels of troponin T and brain natriuretic peptide prior to implantation of a cardiac resynchronization device were strongly predictive of the risk of death or hospitalization for heart failure during the first year of device therapy in the BENEFIT study.
BENEFIT was a 1-year observational study undertaken to determine whether markers could predict which candidates for cardiac resynchronization therapy (CRT) were least likely to benefit from the costly device. At present, roughly 30% of patients who appropriately receive a CRT device do not respond to the treatment.
"We are all at this point frustrated with the persistent portion of CRT recipients who do not respond to therapy," BENEFIT principal investigator Dr. Alaa A. Shalaby said in presenting the study findings at the annual meeting of the Heart Rhythm Society. "We think our results suggest CRT should be offered earlier in the course of the disease"
BENEFIT included 267 CRT recipients at 33 centers. Patients were placed into one of three predefined risk categories based upon baseline levels of brain natriuretic peptide (BNP) and troponin T (TnT). On the basis os prior studies, high BNP was defined as 440 pg/mL or greater and a detectible TnT level was 0.01 ng/mL or greater. The low-risk group had an undetectable TnT and a BNP below 440 pg/mL. The intermediate-risk group had either an elevated BNP or TnT. The high-risk group had elevated BNP and TnT, reported Dr. Shalaby of the University of Pittsburgh.
The median baseline BNP in the study population was 198 pg/mL.
The three groups were similar in terms of baseline characteristics, including the proportion of patients with ischemic cardiomyopathy. Based on BNP and TnT results, 59% of patients were in the low-risk category, 33% had intermediate risk, and 8% were deemed high risk. The intermediate-risk group was evenly divided between patients who qualified on the basis of a high BNP and those with a detectable TnT.
During 12 months of follow-up there were 11 deaths and 19 heart failure hospitalizations. The incidence of either endpoint was 7% in the low-risk group, 15% in the intermediate-risk group, and 30% in the high-risk cohort.
After the researchers adjusted the results for age, left ventricular ejection fraction, QRS duration, and NYHA class, the risk of death or heart failure hospitalization was 2.5-fold greater in the group with intermediate-level baseline biomarkers as in the low-risk group. The high-biomarker group had a 7.3-fold increased risk, compared with the low-biomarker cohort. Both of these differences were statistically significant.
Changes in the biomarker levels after 6 and 12 months of CRT will be the subject of a future BENEFIT analysis, he noted.
The BENEFIT study was supported by St. Jude Medical. Dr. Shalaby reported having no conflicts of interest.
DENVER – Levels of troponin T and brain natriuretic peptide prior to implantation of a cardiac resynchronization device were strongly predictive of the risk of death or hospitalization for heart failure during the first year of device therapy in the BENEFIT study.
BENEFIT was a 1-year observational study undertaken to determine whether markers could predict which candidates for cardiac resynchronization therapy (CRT) were least likely to benefit from the costly device. At present, roughly 30% of patients who appropriately receive a CRT device do not respond to the treatment.
"We are all at this point frustrated with the persistent portion of CRT recipients who do not respond to therapy," BENEFIT principal investigator Dr. Alaa A. Shalaby said in presenting the study findings at the annual meeting of the Heart Rhythm Society. "We think our results suggest CRT should be offered earlier in the course of the disease"
BENEFIT included 267 CRT recipients at 33 centers. Patients were placed into one of three predefined risk categories based upon baseline levels of brain natriuretic peptide (BNP) and troponin T (TnT). On the basis os prior studies, high BNP was defined as 440 pg/mL or greater and a detectible TnT level was 0.01 ng/mL or greater. The low-risk group had an undetectable TnT and a BNP below 440 pg/mL. The intermediate-risk group had either an elevated BNP or TnT. The high-risk group had elevated BNP and TnT, reported Dr. Shalaby of the University of Pittsburgh.
The median baseline BNP in the study population was 198 pg/mL.
The three groups were similar in terms of baseline characteristics, including the proportion of patients with ischemic cardiomyopathy. Based on BNP and TnT results, 59% of patients were in the low-risk category, 33% had intermediate risk, and 8% were deemed high risk. The intermediate-risk group was evenly divided between patients who qualified on the basis of a high BNP and those with a detectable TnT.
During 12 months of follow-up there were 11 deaths and 19 heart failure hospitalizations. The incidence of either endpoint was 7% in the low-risk group, 15% in the intermediate-risk group, and 30% in the high-risk cohort.
After the researchers adjusted the results for age, left ventricular ejection fraction, QRS duration, and NYHA class, the risk of death or heart failure hospitalization was 2.5-fold greater in the group with intermediate-level baseline biomarkers as in the low-risk group. The high-biomarker group had a 7.3-fold increased risk, compared with the low-biomarker cohort. Both of these differences were statistically significant.
Changes in the biomarker levels after 6 and 12 months of CRT will be the subject of a future BENEFIT analysis, he noted.
The BENEFIT study was supported by St. Jude Medical. Dr. Shalaby reported having no conflicts of interest.
AT HEART RHYTHM 2013
Major finding: Cardiac resynchronization therapy device recipients with a preimplantation brain natriuretic peptide level of at least 440 pg/mL and a detectable troponin T level were 7.3-fold more likely to die or be hospitalized for heart failure during the first 12 months after device implantation than were those with an undetectable troponin T level and a lower brain natriuretic peptide level.
Data source: The BENEFIT study was a prospective, 33-center study in which 267 patients who received a CRT device were followed for 1 year.
Disclosures: BENEFIT was supported by St. Jude Medical. The presenter reported having no conflicts of interest.

Optim ICD leads show tiny failure rate - so far
DENVER – Failure rates for the St. Jude Medical Riata ST Optim and Durata implantable cardioverter-defibrillator leads were reassuringly low in an independent analysis of three large prospective registries.
The study was undertaken at company expense because St. Jude Medical’s earlier-generation, silicone-wrapped Riata and Riata ST ICD leads were so failure-prone, that they were recalled several years ago. Their replacements – the subject of the new study – are insulated with Optim, a proprietary silicone/polyurethane copolymer with exceptional mechanical strength, flexibility, and abrasion resistance, according to Dr. John A. Cairns, who presented the registry study analysis at the annual meeting of the Heart Rhythm Society.

The prospective, St. Jude Medical–sponsored OPTIMUM, SJ4, and SCORE registries totaled 11,005 Durata and Riata ST Optim leads implanted in 10,820 patients. Median follow-up to date in the study, conducted independently under the direction of the Population Health Research Institute at McMaster University in Hamilton, Ont., is 3.0 years. Sixty-four percent of the leads remain under active follow-up, according to Dr. Cairns, who chairs the study steering committee and is professor of medicine at the University of British Columbia, Vancouver, as well as president-elect of the Canadian Academy of Health Sciences.
Participation in the registries entails patient follow-up every 6 months, with device interrogation encouraged. Participating physicians are supposed to return inactivated leads to St. Jude Medical for detailed analysis.
To date, the all-cause mechanical failure rate in the combined analysis is 0.35%, with a 0.22% conductor fracture rate, an insulation abrasion rate of 0.07%, and no cases of externalized conductors.
A life-table analysis was used to derive the 99.4% estimated 5-year rate of freedom from all-cause mechanical failure. The 5-year freedom from conductor fracture was 99.6%, with a 99.9% freedom from insulation abrasion and 100% freedom from externalized conductors.

Dr. Cairns said the company registries have been rigorously conducted, with central reporting and formal definitions of adverse events. Those strengths, coupled with the independent outside analysis, make the resultant findings more "in the realm of clinical trials rather than so much of the anecdotal information that’s out there," he added.
Session cochair John D. Day said in an interview that he considered the data "great news for patients with those leads" and was "cautiously optimistic" regarding the long-term performance of the ICD leads. He noted, however, that as of this analysis only 551 leads had actually reached the 5-year follow-up point. Many thousands more will do so in the next several years, at which point definitive conclusions can be drawn, according to Dr. Day, director of heart rhythm specialists at Intermountain Healthcare, Salt Lake City.
Dr. Cairns reported serving as a consultant to St. Jude Medical.
DENVER – Failure rates for the St. Jude Medical Riata ST Optim and Durata implantable cardioverter-defibrillator leads were reassuringly low in an independent analysis of three large prospective registries.
The study was undertaken at company expense because St. Jude Medical’s earlier-generation, silicone-wrapped Riata and Riata ST ICD leads were so failure-prone, that they were recalled several years ago. Their replacements – the subject of the new study – are insulated with Optim, a proprietary silicone/polyurethane copolymer with exceptional mechanical strength, flexibility, and abrasion resistance, according to Dr. John A. Cairns, who presented the registry study analysis at the annual meeting of the Heart Rhythm Society.
The prospective, St. Jude Medical–sponsored OPTIMUM, SJ4, and SCORE registries totaled 11,005 Durata and Riata ST Optim leads implanted in 10,820 patients. Median follow-up to date in the study, conducted independently under the direction of the Population Health Research Institute at McMaster University in Hamilton, Ont., is 3.0 years. Sixty-four percent of the leads remain under active follow-up, according to Dr. Cairns, who chairs the study steering committee and is professor of medicine at the University of British Columbia, Vancouver, as well as president-elect of the Canadian Academy of Health Sciences.
Participation in the registries entails patient follow-up every 6 months, with device interrogation encouraged. Participating physicians are supposed to return inactivated leads to St. Jude Medical for detailed analysis.
To date, the all-cause mechanical failure rate in the combined analysis is 0.35%, with a 0.22% conductor fracture rate, an insulation abrasion rate of 0.07%, and no cases of externalized conductors.
A life-table analysis was used to derive the 99.4% estimated 5-year rate of freedom from all-cause mechanical failure. The 5-year freedom from conductor fracture was 99.6%, with a 99.9% freedom from insulation abrasion and 100% freedom from externalized conductors.
Dr. Cairns said the company registries have been rigorously conducted, with central reporting and formal definitions of adverse events. Those strengths, coupled with the independent outside analysis, make the resultant findings more "in the realm of clinical trials rather than so much of the anecdotal information that’s out there," he added.
Session cochair John D. Day said in an interview that he considered the data "great news for patients with those leads" and was "cautiously optimistic" regarding the long-term performance of the ICD leads. He noted, however, that as of this analysis only 551 leads had actually reached the 5-year follow-up point. Many thousands more will do so in the next several years, at which point definitive conclusions can be drawn, according to Dr. Day, director of heart rhythm specialists at Intermountain Healthcare, Salt Lake City.
Dr. Cairns reported serving as a consultant to St. Jude Medical.
DENVER – Failure rates for the St. Jude Medical Riata ST Optim and Durata implantable cardioverter-defibrillator leads were reassuringly low in an independent analysis of three large prospective registries.
The study was undertaken at company expense because St. Jude Medical’s earlier-generation, silicone-wrapped Riata and Riata ST ICD leads were so failure-prone, that they were recalled several years ago. Their replacements – the subject of the new study – are insulated with Optim, a proprietary silicone/polyurethane copolymer with exceptional mechanical strength, flexibility, and abrasion resistance, according to Dr. John A. Cairns, who presented the registry study analysis at the annual meeting of the Heart Rhythm Society.
The prospective, St. Jude Medical–sponsored OPTIMUM, SJ4, and SCORE registries totaled 11,005 Durata and Riata ST Optim leads implanted in 10,820 patients. Median follow-up to date in the study, conducted independently under the direction of the Population Health Research Institute at McMaster University in Hamilton, Ont., is 3.0 years. Sixty-four percent of the leads remain under active follow-up, according to Dr. Cairns, who chairs the study steering committee and is professor of medicine at the University of British Columbia, Vancouver, as well as president-elect of the Canadian Academy of Health Sciences.
Participation in the registries entails patient follow-up every 6 months, with device interrogation encouraged. Participating physicians are supposed to return inactivated leads to St. Jude Medical for detailed analysis.
To date, the all-cause mechanical failure rate in the combined analysis is 0.35%, with a 0.22% conductor fracture rate, an insulation abrasion rate of 0.07%, and no cases of externalized conductors.
A life-table analysis was used to derive the 99.4% estimated 5-year rate of freedom from all-cause mechanical failure. The 5-year freedom from conductor fracture was 99.6%, with a 99.9% freedom from insulation abrasion and 100% freedom from externalized conductors.
Dr. Cairns said the company registries have been rigorously conducted, with central reporting and formal definitions of adverse events. Those strengths, coupled with the independent outside analysis, make the resultant findings more "in the realm of clinical trials rather than so much of the anecdotal information that’s out there," he added.
Session cochair John D. Day said in an interview that he considered the data "great news for patients with those leads" and was "cautiously optimistic" regarding the long-term performance of the ICD leads. He noted, however, that as of this analysis only 551 leads had actually reached the 5-year follow-up point. Many thousands more will do so in the next several years, at which point definitive conclusions can be drawn, according to Dr. Day, director of heart rhythm specialists at Intermountain Healthcare, Salt Lake City.
Dr. Cairns reported serving as a consultant to St. Jude Medical.
AT HEART RHYTHM 2013
Major finding: The 5-year rate of freedom from mechanical failure for St. Jude Medical’s Durata and Riata ST Optim ICD leads was 99.4% in a large study featuring outside analysis.
Data source: The study included 11,005 Durata or Riata ST Optim leads implanted in more than 10,000 patients with a median 3 years’ follow-up in three prospective registries.
Disclosures: The study was sponsored by St. Jude Medical and independently conducted by the Population Health Research Institute at McMaster University. Dr. Cairns reported serving as a consultant to St. Jude Medical.

Novel contact force catheter advances AF ablation
DENVER – An investigational atrial fibrillation ablation catheter that provides the interventionalist with real-time measurement of contact force with the beating heart wall is safe and appears to improve 12-month success rates, according to the SMART-AF trial.
The principle underlying the development of this novel ablation catheter is that the durability of the therapeutic lesion created during AF ablation depends upon the amount of contact between the radiofrequency electrode and target tissue. Conventional catheters require the operator to assess this mainly visually, a hit-or-miss proposition. In contrast, the new catheter uses contact force sensing technology to provide the operator with quantitative feedback as to whether adequate tissue contact is occurring. The Biosense Webster ThermoCool SmartTouch contact force catheter is integrated with a standard 3-D navigation system, so the operator can see the data on the monitor during the procedure, Dr. Andrea Natale explained in presenting the SMART-AF results at the annual meeting of the Heart Rhythm Society.

The primary efficacy endpoint was freedom from documented AF, atrial flutter, and atrial tachycardia episodes through 12 months of follow-up, not including the standard 3-month blanking period immediately post ablation. Seventy-two percent of the 122 patients with symptomatic paroxysmal AF included in the efficacy analysis remained recurrence free at 12 months.
More impressively, the success rate climbed to 84% in cases where the interventionalist maintained contact force in the therapeutic range more than 82% of the time, while dropping to 61% when the contact force time was 82% or less, reported Dr. Natale, executive medical director of the Texas Cardiac Arrhythmia Institute, Austin.
While therapeutic efficacy proved to be a function of time spent in the therapeutic contact force range, adverse events were not, he noted.
The rate of serious adverse events occurring within 7 days of ablation was 9.9% among the 161 patients included in the SMART-AF safety analysis. The efficacy analysis included 39 fewer patients than the safety analysis. Those first 39 patients comprised investigators’ learning curve in the multicenter, prospective, single-arm study.
An independent safety committee deemed the single case of pericardial effusion that occurred in SMART-AF to be the only device-related adverse event. In addition, the committee judged the four cases of cardiac tamponade and the single case of pericarditis to be possibly device related. No strokes, MIs, or thromboembolisms occurred in the study.
The Food and Drug Administration allowed SMART-AF to use historical controls from earlier clinical trials utilizing a similar Biosense Webster ablation catheter which lacked contact force sensing. The required endpoints in SMART-AF were a 12-month freedom from recurrence greater than 50% and an early adverse event rate less than 16.6%, both of which were met.
SMART-AF investigators selected individual contact force target ranges based upon their early experience with the technology along with patient characteristics. To date, it hasn’t been possible to zero in on a fixed quantity of contact force that predicts treatment success regardless of who is doing the procedure.
All patients underwent circumferential pulmonary vein isolation, with additional radiofrequency lesions placed outside the pulmonary vein ostia as warranted.
The contact force sensing catheter utilizes a 7.5 F shaft with an 8 F electrode. Dr. Natale said it’s his anecdotal impression that the catheter results in reduced fluoroscopy and procedure times, but they weren’t measured in the study. What patients and physicians really care about, he added, is therapeutic success.
Asked if he thought ablation catheters using contact force sensing technology, if eventually approved by the FDA, would become the standard of care, he replied that he believes so, as long as the price is reasonable.
Dr. Michael R. Gold, who wasn’t involved in the SMART-AF study, commented that he found the results "very exciting."
"Our ability – using our fingers 3 feet away from a catheter – to know whether we’re making the right amount of contact is limited. Often, we worry, is it too little or too much? Experts like Dr. Natale do it wonderfully, but for the rest of us, it’s a challenge to try to do these ablations. So the concept of being able to know how much force you’re creating, and to optimize it while minimizing the risk of complications [such as] pushing too hard and poking a hole in the heart, is clearly the Holy Grail we’ve been trying to achieve. This is the first step in that direction," said Dr. Gold, professor of medicine, chief of cardiology, and medical director of the Heart and Vascular Center at the Medical University of South Carolina, Charleston.
Dr. Natale serves as a consultant to Biosense Webster, the sponsor of the SMART-AF study, as well as to other medical device companies.
DENVER – An investigational atrial fibrillation ablation catheter that provides the interventionalist with real-time measurement of contact force with the beating heart wall is safe and appears to improve 12-month success rates, according to the SMART-AF trial.
The principle underlying the development of this novel ablation catheter is that the durability of the therapeutic lesion created during AF ablation depends upon the amount of contact between the radiofrequency electrode and target tissue. Conventional catheters require the operator to assess this mainly visually, a hit-or-miss proposition. In contrast, the new catheter uses contact force sensing technology to provide the operator with quantitative feedback as to whether adequate tissue contact is occurring. The Biosense Webster ThermoCool SmartTouch contact force catheter is integrated with a standard 3-D navigation system, so the operator can see the data on the monitor during the procedure, Dr. Andrea Natale explained in presenting the SMART-AF results at the annual meeting of the Heart Rhythm Society.
The primary efficacy endpoint was freedom from documented AF, atrial flutter, and atrial tachycardia episodes through 12 months of follow-up, not including the standard 3-month blanking period immediately post ablation. Seventy-two percent of the 122 patients with symptomatic paroxysmal AF included in the efficacy analysis remained recurrence free at 12 months.
More impressively, the success rate climbed to 84% in cases where the interventionalist maintained contact force in the therapeutic range more than 82% of the time, while dropping to 61% when the contact force time was 82% or less, reported Dr. Natale, executive medical director of the Texas Cardiac Arrhythmia Institute, Austin.
While therapeutic efficacy proved to be a function of time spent in the therapeutic contact force range, adverse events were not, he noted.
The rate of serious adverse events occurring within 7 days of ablation was 9.9% among the 161 patients included in the SMART-AF safety analysis. The efficacy analysis included 39 fewer patients than the safety analysis. Those first 39 patients comprised investigators’ learning curve in the multicenter, prospective, single-arm study.
An independent safety committee deemed the single case of pericardial effusion that occurred in SMART-AF to be the only device-related adverse event. In addition, the committee judged the four cases of cardiac tamponade and the single case of pericarditis to be possibly device related. No strokes, MIs, or thromboembolisms occurred in the study.
The Food and Drug Administration allowed SMART-AF to use historical controls from earlier clinical trials utilizing a similar Biosense Webster ablation catheter which lacked contact force sensing. The required endpoints in SMART-AF were a 12-month freedom from recurrence greater than 50% and an early adverse event rate less than 16.6%, both of which were met.
SMART-AF investigators selected individual contact force target ranges based upon their early experience with the technology along with patient characteristics. To date, it hasn’t been possible to zero in on a fixed quantity of contact force that predicts treatment success regardless of who is doing the procedure.
All patients underwent circumferential pulmonary vein isolation, with additional radiofrequency lesions placed outside the pulmonary vein ostia as warranted.
The contact force sensing catheter utilizes a 7.5 F shaft with an 8 F electrode. Dr. Natale said it’s his anecdotal impression that the catheter results in reduced fluoroscopy and procedure times, but they weren’t measured in the study. What patients and physicians really care about, he added, is therapeutic success.
Asked if he thought ablation catheters using contact force sensing technology, if eventually approved by the FDA, would become the standard of care, he replied that he believes so, as long as the price is reasonable.
Dr. Michael R. Gold, who wasn’t involved in the SMART-AF study, commented that he found the results "very exciting."
"Our ability – using our fingers 3 feet away from a catheter – to know whether we’re making the right amount of contact is limited. Often, we worry, is it too little or too much? Experts like Dr. Natale do it wonderfully, but for the rest of us, it’s a challenge to try to do these ablations. So the concept of being able to know how much force you’re creating, and to optimize it while minimizing the risk of complications [such as] pushing too hard and poking a hole in the heart, is clearly the Holy Grail we’ve been trying to achieve. This is the first step in that direction," said Dr. Gold, professor of medicine, chief of cardiology, and medical director of the Heart and Vascular Center at the Medical University of South Carolina, Charleston.
Dr. Natale serves as a consultant to Biosense Webster, the sponsor of the SMART-AF study, as well as to other medical device companies.
DENVER – An investigational atrial fibrillation ablation catheter that provides the interventionalist with real-time measurement of contact force with the beating heart wall is safe and appears to improve 12-month success rates, according to the SMART-AF trial.
The principle underlying the development of this novel ablation catheter is that the durability of the therapeutic lesion created during AF ablation depends upon the amount of contact between the radiofrequency electrode and target tissue. Conventional catheters require the operator to assess this mainly visually, a hit-or-miss proposition. In contrast, the new catheter uses contact force sensing technology to provide the operator with quantitative feedback as to whether adequate tissue contact is occurring. The Biosense Webster ThermoCool SmartTouch contact force catheter is integrated with a standard 3-D navigation system, so the operator can see the data on the monitor during the procedure, Dr. Andrea Natale explained in presenting the SMART-AF results at the annual meeting of the Heart Rhythm Society.
The primary efficacy endpoint was freedom from documented AF, atrial flutter, and atrial tachycardia episodes through 12 months of follow-up, not including the standard 3-month blanking period immediately post ablation. Seventy-two percent of the 122 patients with symptomatic paroxysmal AF included in the efficacy analysis remained recurrence free at 12 months.
More impressively, the success rate climbed to 84% in cases where the interventionalist maintained contact force in the therapeutic range more than 82% of the time, while dropping to 61% when the contact force time was 82% or less, reported Dr. Natale, executive medical director of the Texas Cardiac Arrhythmia Institute, Austin.
While therapeutic efficacy proved to be a function of time spent in the therapeutic contact force range, adverse events were not, he noted.
The rate of serious adverse events occurring within 7 days of ablation was 9.9% among the 161 patients included in the SMART-AF safety analysis. The efficacy analysis included 39 fewer patients than the safety analysis. Those first 39 patients comprised investigators’ learning curve in the multicenter, prospective, single-arm study.
An independent safety committee deemed the single case of pericardial effusion that occurred in SMART-AF to be the only device-related adverse event. In addition, the committee judged the four cases of cardiac tamponade and the single case of pericarditis to be possibly device related. No strokes, MIs, or thromboembolisms occurred in the study.
The Food and Drug Administration allowed SMART-AF to use historical controls from earlier clinical trials utilizing a similar Biosense Webster ablation catheter which lacked contact force sensing. The required endpoints in SMART-AF were a 12-month freedom from recurrence greater than 50% and an early adverse event rate less than 16.6%, both of which were met.
SMART-AF investigators selected individual contact force target ranges based upon their early experience with the technology along with patient characteristics. To date, it hasn’t been possible to zero in on a fixed quantity of contact force that predicts treatment success regardless of who is doing the procedure.
All patients underwent circumferential pulmonary vein isolation, with additional radiofrequency lesions placed outside the pulmonary vein ostia as warranted.
The contact force sensing catheter utilizes a 7.5 F shaft with an 8 F electrode. Dr. Natale said it’s his anecdotal impression that the catheter results in reduced fluoroscopy and procedure times, but they weren’t measured in the study. What patients and physicians really care about, he added, is therapeutic success.
Asked if he thought ablation catheters using contact force sensing technology, if eventually approved by the FDA, would become the standard of care, he replied that he believes so, as long as the price is reasonable.
Dr. Michael R. Gold, who wasn’t involved in the SMART-AF study, commented that he found the results "very exciting."
"Our ability – using our fingers 3 feet away from a catheter – to know whether we’re making the right amount of contact is limited. Often, we worry, is it too little or too much? Experts like Dr. Natale do it wonderfully, but for the rest of us, it’s a challenge to try to do these ablations. So the concept of being able to know how much force you’re creating, and to optimize it while minimizing the risk of complications [such as] pushing too hard and poking a hole in the heart, is clearly the Holy Grail we’ve been trying to achieve. This is the first step in that direction," said Dr. Gold, professor of medicine, chief of cardiology, and medical director of the Heart and Vascular Center at the Medical University of South Carolina, Charleston.
Dr. Natale serves as a consultant to Biosense Webster, the sponsor of the SMART-AF study, as well as to other medical device companies.
AT HEART RHYTHM 2013
Major finding: Patients undergoing ablation of paroxysmal atrial fibrillation using an ablation catheter featuring contact force sensing technology had an impressive 84% rate of freedom from recurrence at 12 months if the operator kept contact force with the target tissue within the desired range more than 82% of the time.
Data source: SMART-AF, a multicenter, prospective, nonrandomized study with 161 evaluated patients.
Disclosures: SMART-AF was sponsored by Biosense Webster. The presenter serves as a consultant to the company.

Antibacterial envelope may limit cardiac device infections
DENVER – Encasing implantable cardioverter-defibrillators and cardiac resynchronization therapy devices in a proprietary antibiotic-impregnated envelope during implantation was associated with a low 90-day major infection rate in an interim analysis of a large prospective multicenter study.
The CITADEL/CENTURION study involves 1,000 patients who underwent ICD or CRT device replacement using the antibacterial envelope at 55 U.S. centers. A major device infection – defined as an infection involving the device pocket, deep tissue, or endocarditis – occurred in one patient through 3 months of prospective follow-up, for an infection incidence of 0.1%, Dr. Charles Henrikson reported at the annual meeting of the Heart Rhythm Society.

CITADEL/CENTURION is not a randomized trial. Instead, the control group consists of 533 Canadian patients in a published study, all of whom underwent ICD or CRT replacement because of device advisories or recalls. The major device infection rate in this retrospective study (JAMA 2006;295:1907-11) during a mean 2.7 months following device replacement was 1.9%, or roughly 19-fold greater than in CITADEL/CENTURION, noted Dr. Henrikson of Oregon Health and Science University, Portland.
Device replacement is a known risk factor for major device infections, which carry a substantial toll in terms of morbidity, mortality, and expense.
The antibacterial envelope, known as AIGISRx, has received Food and Drug Administration approval and is commercially available. The antibacterial envelope is designed to prevent device migration as well as infections. "It changes the device from something that’s slick and slides right in into something with an incredibly high coefficient of friction, so you need to make a larger pocket to put it in. That’s why we’re paying a lot of attention to the hematoma rate," according to Dr. Henrikson.
The device consists of a nonresorbable polypropylene mesh coated with a resorbable polymer layer, which releases rifampin and minocycline for 7-10 days. These two antibiotics are active against the pathogens that cause most device infections, including both methicillin-sensitive and -resistant Staphylococcus aureus as well as S. epidermis.
The indication for device replacement in CITADEL/CENTURION was end of battery life in about three-quarters of patients, with device upgrades and lead revisions accounting for nearly all of the rest. Explantation because of device infection was a contraindication to study participation. All subjects were on perioperative prophylactic antibiotics, as were the historical controls.
The chief device-related complication recorded during the study was major pocket hematoma requiring a change in patient management, typically open drainage or transfusion. The complication occurred in 1.5% of patients, as compared to 2.25% of the historical controls.
Two of the 1,000 patients developed generalized rashes thought by independent reviewers to be possibly related to AIGISRx.
The plan is to monitor CITADEL/CENTURION participants for device infections and mechanical complications through 12 months, Dr. Henrikson explained. Only the 3-month data were available for presentation at a late-breaking clinical trials session during Heart Rhythm 2013.
CITADEL/CENTURION is sponsored by TYRX, which markets AIGISRx. Dr. Henrikson reported having received research support from the company.
DENVER – Encasing implantable cardioverter-defibrillators and cardiac resynchronization therapy devices in a proprietary antibiotic-impregnated envelope during implantation was associated with a low 90-day major infection rate in an interim analysis of a large prospective multicenter study.
The CITADEL/CENTURION study involves 1,000 patients who underwent ICD or CRT device replacement using the antibacterial envelope at 55 U.S. centers. A major device infection – defined as an infection involving the device pocket, deep tissue, or endocarditis – occurred in one patient through 3 months of prospective follow-up, for an infection incidence of 0.1%, Dr. Charles Henrikson reported at the annual meeting of the Heart Rhythm Society.
CITADEL/CENTURION is not a randomized trial. Instead, the control group consists of 533 Canadian patients in a published study, all of whom underwent ICD or CRT replacement because of device advisories or recalls. The major device infection rate in this retrospective study (JAMA 2006;295:1907-11) during a mean 2.7 months following device replacement was 1.9%, or roughly 19-fold greater than in CITADEL/CENTURION, noted Dr. Henrikson of Oregon Health and Science University, Portland.
Device replacement is a known risk factor for major device infections, which carry a substantial toll in terms of morbidity, mortality, and expense.
The antibacterial envelope, known as AIGISRx, has received Food and Drug Administration approval and is commercially available. The antibacterial envelope is designed to prevent device migration as well as infections. "It changes the device from something that’s slick and slides right in into something with an incredibly high coefficient of friction, so you need to make a larger pocket to put it in. That’s why we’re paying a lot of attention to the hematoma rate," according to Dr. Henrikson.
The device consists of a nonresorbable polypropylene mesh coated with a resorbable polymer layer, which releases rifampin and minocycline for 7-10 days. These two antibiotics are active against the pathogens that cause most device infections, including both methicillin-sensitive and -resistant Staphylococcus aureus as well as S. epidermis.
The indication for device replacement in CITADEL/CENTURION was end of battery life in about three-quarters of patients, with device upgrades and lead revisions accounting for nearly all of the rest. Explantation because of device infection was a contraindication to study participation. All subjects were on perioperative prophylactic antibiotics, as were the historical controls.
The chief device-related complication recorded during the study was major pocket hematoma requiring a change in patient management, typically open drainage or transfusion. The complication occurred in 1.5% of patients, as compared to 2.25% of the historical controls.
Two of the 1,000 patients developed generalized rashes thought by independent reviewers to be possibly related to AIGISRx.
The plan is to monitor CITADEL/CENTURION participants for device infections and mechanical complications through 12 months, Dr. Henrikson explained. Only the 3-month data were available for presentation at a late-breaking clinical trials session during Heart Rhythm 2013.
CITADEL/CENTURION is sponsored by TYRX, which markets AIGISRx. Dr. Henrikson reported having received research support from the company.
DENVER – Encasing implantable cardioverter-defibrillators and cardiac resynchronization therapy devices in a proprietary antibiotic-impregnated envelope during implantation was associated with a low 90-day major infection rate in an interim analysis of a large prospective multicenter study.
The CITADEL/CENTURION study involves 1,000 patients who underwent ICD or CRT device replacement using the antibacterial envelope at 55 U.S. centers. A major device infection – defined as an infection involving the device pocket, deep tissue, or endocarditis – occurred in one patient through 3 months of prospective follow-up, for an infection incidence of 0.1%, Dr. Charles Henrikson reported at the annual meeting of the Heart Rhythm Society.
CITADEL/CENTURION is not a randomized trial. Instead, the control group consists of 533 Canadian patients in a published study, all of whom underwent ICD or CRT replacement because of device advisories or recalls. The major device infection rate in this retrospective study (JAMA 2006;295:1907-11) during a mean 2.7 months following device replacement was 1.9%, or roughly 19-fold greater than in CITADEL/CENTURION, noted Dr. Henrikson of Oregon Health and Science University, Portland.
Device replacement is a known risk factor for major device infections, which carry a substantial toll in terms of morbidity, mortality, and expense.
The antibacterial envelope, known as AIGISRx, has received Food and Drug Administration approval and is commercially available. The antibacterial envelope is designed to prevent device migration as well as infections. "It changes the device from something that’s slick and slides right in into something with an incredibly high coefficient of friction, so you need to make a larger pocket to put it in. That’s why we’re paying a lot of attention to the hematoma rate," according to Dr. Henrikson.
The device consists of a nonresorbable polypropylene mesh coated with a resorbable polymer layer, which releases rifampin and minocycline for 7-10 days. These two antibiotics are active against the pathogens that cause most device infections, including both methicillin-sensitive and -resistant Staphylococcus aureus as well as S. epidermis.
The indication for device replacement in CITADEL/CENTURION was end of battery life in about three-quarters of patients, with device upgrades and lead revisions accounting for nearly all of the rest. Explantation because of device infection was a contraindication to study participation. All subjects were on perioperative prophylactic antibiotics, as were the historical controls.
The chief device-related complication recorded during the study was major pocket hematoma requiring a change in patient management, typically open drainage or transfusion. The complication occurred in 1.5% of patients, as compared to 2.25% of the historical controls.
Two of the 1,000 patients developed generalized rashes thought by independent reviewers to be possibly related to AIGISRx.
The plan is to monitor CITADEL/CENTURION participants for device infections and mechanical complications through 12 months, Dr. Henrikson explained. Only the 3-month data were available for presentation at a late-breaking clinical trials session during Heart Rhythm 2013.
CITADEL/CENTURION is sponsored by TYRX, which markets AIGISRx. Dr. Henrikson reported having received research support from the company.
AT HEART RHYTHM 2013
Major finding: A major device infection – defined as an infection involving the device pocket, deep tissue, or endocarditis – occurred in one patient, for an infection incidence of 0.1%.
Data source: Three months of data from 1,000 patients in CITADEL/CENTURION study, an ongoing multicenter, prospective study.
Disclosures: The study is sponsored by TYRX, which markets the AIGISRx antibacterial envelope. The presenter reported receiving research support from the company.

Wearable defibrillator vest useful as bridge to ICD
DENVER – The LifeVest wearable automatic defibrillator provides a safe and highly effective bridging strategy while physicians decide whether a patient should get an implantable cardioverter defibrillator, according to findings from the WEARIT-II registry.
Experience gleaned from the first 882 of a planned 3,000 patients to be enrolled in this prospective, real-world registry indicates the defibrillator vest consistently recognizes and safely terminates life-threatening arrhythmias while avoiding unnecessary shocks for non–life-threatening arrhythmic events, Dr. Ilan Goldenberg reported at the annual meeting of the Heart Rhythm Society.

Indeed, the inappropriate shock rate in WEARIT-II was a mere 0.3%, far lower than with contemporary implantable cardioverter defibrillator (ICD) therapy, noted Dr. Goldenberg of the University of Rochester (N.Y.).
This bridging strategy deserves broad use in high-risk populations, he said. The WEARIT-II data show that bridging is particularly useful as a means of protecting patients with a transient arrhythmia risk as well as those whose long-term arrhythmia risk is undefined and requires further evaluation. Examples include patients who are post MI, or have new-onset heart failure with a depressed left ventricular ejection fraction (LVEF) of 35% or less, have recently undergone coronary revascularization, or are undergoing detailed evaluation of a possible inherited or congenital arrhythmic disorder.
Guidelines state that an ICD is indicated for primary prevention of sudden cardiac death in patients with an LVEF of 35% or less, but not within 40 days following an MI, or within 3 months after diagnosis of heart failure, or within 90 days following coronary revascularization. The reason for these mandatory delays is that many patients will experience improvement in LVEF in response to medical therapy such that they no longer qualify for ICD implantation. But if their physician is concerned about their arrhythmia risk during that waiting period, the wearable defibrillator is an excellent solution, Dr. Goldenberg continued.
There is a clear need for more selective prescription of ICDs for primary prevention of cardiac arrest. This was already evident more than a decade ago, when the MADIT-II (Multicenter Automatic Defibrillator Trial–II) demonstrated that only one-third of patients received appropriate ICD therapy during 4 years of follow-up (N. Engl. J. Med. 2002;346:877-83). More recently, the MADIT-Reduce Inappropriate Therapy trial reported that participating ICD recipients had a low appropriate shock rate of 3 shocks per 100 patient-years (N. Engl. J. Med. 2012;367:2275-83) . The wearable defibrillator bridging strategy offers a means of safely being more selective in ICD placement. Patients whose risk isn’t yet clearly defined can in effect have a nonpermanent trial run of up to 6 months’ duration using the LifeVest, the electrophysiologist explained.
The LifeVest is commercially available and routinely covered by insurers. It’s a thin, lightweight vest designed to be worn under clothes. It is attached to a waist battery pack. The LifeVest provides continuous heart rhythm monitoring and automatic defibrillation upon detection of a potentially fatal ventricular arrhythmia. The device has an override button that enables a patient experiencing a sustained ventricular tachyarrhythmia (VT) to allow the event to terminate spontaneously, thereby avoiding unnecessary shocks. Should the patient pass out while the VT continues, the LifeVest will deliver shock therapy.
The 882 patients in the WEAR-IT registry had a mean LVEF of 25%. A total of 771 patients had acquired heart conditions, most commonly nonischemic or ischemic cardiomyopathy. The other 111 patients had inherited or congenital conditions, such as long QT syndrome. Patients wore the LifeVest for an average of 81 days and for a mean of 21 hours daily.
Appropriate LifeVest shock therapy that terminated life-threatening fast VT or ventricular fibrillation occurred at a rate of 9 events per 100 patient-years. Sustained VT that was allowed to spontaneously terminate as a result of the patient’s use of the device’s override button occurred at a rate of 27 events per 100 patient-years.
The device also detected nonsustained VT at a rate of 47 events per 100 patient-years, atrial arrhythmias and other supraventricular tachycardias at 64 events per 100 patient-years, and asystole at 3 events per 100 patient-years.
Four deaths occurred. Three of those happened when the patient was not wearing the vest; the fourth was caused by asystole.
Upon ending their use of the LifeVest, 41% of patients did not receive an ICD because their LVEF improved. Moreover, the arrhythmias detected by the LifeVest affected patient disposition: 80% of patients who received an appropriate shock from the vest got an ICD, as did fewer than 40% of those with no arrhythmias detected during vest wear.
Patients within 40 days post MI or 90 days post revascularization had the highest arrhythmic event rates among those with acquired heart conditions; however, the event rate was even higher among those with inherited or congenital conditions.
Dr. Michael R. Gold commented that in his experience, another important group of candidates for the wearable defibrillator are arrhythmia-prone patients who develop a cardiac device infection requiring device removal.
"You’re worried about that patient yet you can’t implant another device because it may take weeks or months to clear the infection," noted Dr. Gold, professor of medicine, chief of cardiology, and medical director of the heart and vascular center at the Medical University of South Carolina, Charleston.
Dr. Goldenberg reported having received research grants from Zoll Medical, which sponsors the WEARIT-II registry and markets the LifeVest, as well as from Boston Scientific.
DENVER – The LifeVest wearable automatic defibrillator provides a safe and highly effective bridging strategy while physicians decide whether a patient should get an implantable cardioverter defibrillator, according to findings from the WEARIT-II registry.
Experience gleaned from the first 882 of a planned 3,000 patients to be enrolled in this prospective, real-world registry indicates the defibrillator vest consistently recognizes and safely terminates life-threatening arrhythmias while avoiding unnecessary shocks for non–life-threatening arrhythmic events, Dr. Ilan Goldenberg reported at the annual meeting of the Heart Rhythm Society.
Indeed, the inappropriate shock rate in WEARIT-II was a mere 0.3%, far lower than with contemporary implantable cardioverter defibrillator (ICD) therapy, noted Dr. Goldenberg of the University of Rochester (N.Y.).
This bridging strategy deserves broad use in high-risk populations, he said. The WEARIT-II data show that bridging is particularly useful as a means of protecting patients with a transient arrhythmia risk as well as those whose long-term arrhythmia risk is undefined and requires further evaluation. Examples include patients who are post MI, or have new-onset heart failure with a depressed left ventricular ejection fraction (LVEF) of 35% or less, have recently undergone coronary revascularization, or are undergoing detailed evaluation of a possible inherited or congenital arrhythmic disorder.
Guidelines state that an ICD is indicated for primary prevention of sudden cardiac death in patients with an LVEF of 35% or less, but not within 40 days following an MI, or within 3 months after diagnosis of heart failure, or within 90 days following coronary revascularization. The reason for these mandatory delays is that many patients will experience improvement in LVEF in response to medical therapy such that they no longer qualify for ICD implantation. But if their physician is concerned about their arrhythmia risk during that waiting period, the wearable defibrillator is an excellent solution, Dr. Goldenberg continued.
There is a clear need for more selective prescription of ICDs for primary prevention of cardiac arrest. This was already evident more than a decade ago, when the MADIT-II (Multicenter Automatic Defibrillator Trial–II) demonstrated that only one-third of patients received appropriate ICD therapy during 4 years of follow-up (N. Engl. J. Med. 2002;346:877-83). More recently, the MADIT-Reduce Inappropriate Therapy trial reported that participating ICD recipients had a low appropriate shock rate of 3 shocks per 100 patient-years (N. Engl. J. Med. 2012;367:2275-83) . The wearable defibrillator bridging strategy offers a means of safely being more selective in ICD placement. Patients whose risk isn’t yet clearly defined can in effect have a nonpermanent trial run of up to 6 months’ duration using the LifeVest, the electrophysiologist explained.
The LifeVest is commercially available and routinely covered by insurers. It’s a thin, lightweight vest designed to be worn under clothes. It is attached to a waist battery pack. The LifeVest provides continuous heart rhythm monitoring and automatic defibrillation upon detection of a potentially fatal ventricular arrhythmia. The device has an override button that enables a patient experiencing a sustained ventricular tachyarrhythmia (VT) to allow the event to terminate spontaneously, thereby avoiding unnecessary shocks. Should the patient pass out while the VT continues, the LifeVest will deliver shock therapy.
The 882 patients in the WEAR-IT registry had a mean LVEF of 25%. A total of 771 patients had acquired heart conditions, most commonly nonischemic or ischemic cardiomyopathy. The other 111 patients had inherited or congenital conditions, such as long QT syndrome. Patients wore the LifeVest for an average of 81 days and for a mean of 21 hours daily.
Appropriate LifeVest shock therapy that terminated life-threatening fast VT or ventricular fibrillation occurred at a rate of 9 events per 100 patient-years. Sustained VT that was allowed to spontaneously terminate as a result of the patient’s use of the device’s override button occurred at a rate of 27 events per 100 patient-years.
The device also detected nonsustained VT at a rate of 47 events per 100 patient-years, atrial arrhythmias and other supraventricular tachycardias at 64 events per 100 patient-years, and asystole at 3 events per 100 patient-years.
Four deaths occurred. Three of those happened when the patient was not wearing the vest; the fourth was caused by asystole.
Upon ending their use of the LifeVest, 41% of patients did not receive an ICD because their LVEF improved. Moreover, the arrhythmias detected by the LifeVest affected patient disposition: 80% of patients who received an appropriate shock from the vest got an ICD, as did fewer than 40% of those with no arrhythmias detected during vest wear.
Patients within 40 days post MI or 90 days post revascularization had the highest arrhythmic event rates among those with acquired heart conditions; however, the event rate was even higher among those with inherited or congenital conditions.
Dr. Michael R. Gold commented that in his experience, another important group of candidates for the wearable defibrillator are arrhythmia-prone patients who develop a cardiac device infection requiring device removal.
"You’re worried about that patient yet you can’t implant another device because it may take weeks or months to clear the infection," noted Dr. Gold, professor of medicine, chief of cardiology, and medical director of the heart and vascular center at the Medical University of South Carolina, Charleston.
Dr. Goldenberg reported having received research grants from Zoll Medical, which sponsors the WEARIT-II registry and markets the LifeVest, as well as from Boston Scientific.
DENVER – The LifeVest wearable automatic defibrillator provides a safe and highly effective bridging strategy while physicians decide whether a patient should get an implantable cardioverter defibrillator, according to findings from the WEARIT-II registry.
Experience gleaned from the first 882 of a planned 3,000 patients to be enrolled in this prospective, real-world registry indicates the defibrillator vest consistently recognizes and safely terminates life-threatening arrhythmias while avoiding unnecessary shocks for non–life-threatening arrhythmic events, Dr. Ilan Goldenberg reported at the annual meeting of the Heart Rhythm Society.
Indeed, the inappropriate shock rate in WEARIT-II was a mere 0.3%, far lower than with contemporary implantable cardioverter defibrillator (ICD) therapy, noted Dr. Goldenberg of the University of Rochester (N.Y.).
This bridging strategy deserves broad use in high-risk populations, he said. The WEARIT-II data show that bridging is particularly useful as a means of protecting patients with a transient arrhythmia risk as well as those whose long-term arrhythmia risk is undefined and requires further evaluation. Examples include patients who are post MI, or have new-onset heart failure with a depressed left ventricular ejection fraction (LVEF) of 35% or less, have recently undergone coronary revascularization, or are undergoing detailed evaluation of a possible inherited or congenital arrhythmic disorder.
Guidelines state that an ICD is indicated for primary prevention of sudden cardiac death in patients with an LVEF of 35% or less, but not within 40 days following an MI, or within 3 months after diagnosis of heart failure, or within 90 days following coronary revascularization. The reason for these mandatory delays is that many patients will experience improvement in LVEF in response to medical therapy such that they no longer qualify for ICD implantation. But if their physician is concerned about their arrhythmia risk during that waiting period, the wearable defibrillator is an excellent solution, Dr. Goldenberg continued.
There is a clear need for more selective prescription of ICDs for primary prevention of cardiac arrest. This was already evident more than a decade ago, when the MADIT-II (Multicenter Automatic Defibrillator Trial–II) demonstrated that only one-third of patients received appropriate ICD therapy during 4 years of follow-up (N. Engl. J. Med. 2002;346:877-83). More recently, the MADIT-Reduce Inappropriate Therapy trial reported that participating ICD recipients had a low appropriate shock rate of 3 shocks per 100 patient-years (N. Engl. J. Med. 2012;367:2275-83) . The wearable defibrillator bridging strategy offers a means of safely being more selective in ICD placement. Patients whose risk isn’t yet clearly defined can in effect have a nonpermanent trial run of up to 6 months’ duration using the LifeVest, the electrophysiologist explained.
The LifeVest is commercially available and routinely covered by insurers. It’s a thin, lightweight vest designed to be worn under clothes. It is attached to a waist battery pack. The LifeVest provides continuous heart rhythm monitoring and automatic defibrillation upon detection of a potentially fatal ventricular arrhythmia. The device has an override button that enables a patient experiencing a sustained ventricular tachyarrhythmia (VT) to allow the event to terminate spontaneously, thereby avoiding unnecessary shocks. Should the patient pass out while the VT continues, the LifeVest will deliver shock therapy.
The 882 patients in the WEAR-IT registry had a mean LVEF of 25%. A total of 771 patients had acquired heart conditions, most commonly nonischemic or ischemic cardiomyopathy. The other 111 patients had inherited or congenital conditions, such as long QT syndrome. Patients wore the LifeVest for an average of 81 days and for a mean of 21 hours daily.
Appropriate LifeVest shock therapy that terminated life-threatening fast VT or ventricular fibrillation occurred at a rate of 9 events per 100 patient-years. Sustained VT that was allowed to spontaneously terminate as a result of the patient’s use of the device’s override button occurred at a rate of 27 events per 100 patient-years.
The device also detected nonsustained VT at a rate of 47 events per 100 patient-years, atrial arrhythmias and other supraventricular tachycardias at 64 events per 100 patient-years, and asystole at 3 events per 100 patient-years.
Four deaths occurred. Three of those happened when the patient was not wearing the vest; the fourth was caused by asystole.
Upon ending their use of the LifeVest, 41% of patients did not receive an ICD because their LVEF improved. Moreover, the arrhythmias detected by the LifeVest affected patient disposition: 80% of patients who received an appropriate shock from the vest got an ICD, as did fewer than 40% of those with no arrhythmias detected during vest wear.
Patients within 40 days post MI or 90 days post revascularization had the highest arrhythmic event rates among those with acquired heart conditions; however, the event rate was even higher among those with inherited or congenital conditions.
Dr. Michael R. Gold commented that in his experience, another important group of candidates for the wearable defibrillator are arrhythmia-prone patients who develop a cardiac device infection requiring device removal.
"You’re worried about that patient yet you can’t implant another device because it may take weeks or months to clear the infection," noted Dr. Gold, professor of medicine, chief of cardiology, and medical director of the heart and vascular center at the Medical University of South Carolina, Charleston.
Dr. Goldenberg reported having received research grants from Zoll Medical, which sponsors the WEARIT-II registry and markets the LifeVest, as well as from Boston Scientific.
AT HEART RHYTHM 2013
Major finding: During an average of 81 days using the LifeVest wearable automatic defibrillator, patients experienced appropriate device shocks to terminate potentially fatal ventricular tachyarrhythmia/ventricular fibrillation at a rate of 9 events per 100 person-years while appropriately avoiding shocks for sustained VT with spontaneous termination at a rate of 27 events per 100 person-years. Only 0.3% of 882 vest users experienced an inappropriate shock.
Data source: The WEARIT-II registry, which to date includes 882 patients who have been prescribed the LifeVest wearable defibrillator.
Disclosures: The registry is sponsored by Zoll Medical, which markets the LifeVest. The presenter said he has received research grants from the company.

Dabigatran post-AF ablation may be riskier than warfarin
DENVER – Using dabigatran rather than warfarin for periprocedural anticoagulation in patients undergoing radiofrequency ablation for atrial fibrillation may entail a small but statistically increased risk of stroke or transient ischemic attack, according to a meta-analysis of 10 observational cohort studies.
Guidelines recommend periprocedural anticoagulation for at least 2 months post ablation. Warfarin continues to be the most widely used agent for this purpose, but new oral alternatives are attracting a great deal of interest from physicians and patients.

The meta-analysis, which included 1,501 patients on periprocedural dabigatran (Pradaxa) and 2,356 on warfarin, had dual primary end points. One was stroke or TIA, which occurred in 0.7% of the dabigatran group, compared with 0.2% of those on warfarin – a statistically significant difference (P = .0007), Dr. Benjamin A. Steinberg reported at the annual meeting of the Heart Rhythm Society.
The co-primary end point, major bleeding, was recorded in 1.6% of the dabigatran group, with a closely similar 1.7% incidence in the warfarin group, added Dr. Steinberg of Duke University in Durham, N.C.
Rates of cardiac tamponade, a secondary end point, were also similar: 1.1% with dabigatran and 0.9% with warfarin.
Although dabigatran is approved for the prevention of stroke or systemic embolism in patients with AF, a definitive determination of its safety and effectiveness in the setting of AF ablation therapy will require randomized trials, Dr. Steinberg observed.
Such trials would, of necessity, have to be quite large. The data from this meta-analysis suggest that, for every 200 patients undergoing AF ablation under the protection of dabigatran rather than warfarin, one additional case of stroke or TIA would occur.
The meta-analysis was supported in part by the Agency for Healthcare Research and Quality. Dr. Steinberg reported having no conflicts of interest.
DENVER – Using dabigatran rather than warfarin for periprocedural anticoagulation in patients undergoing radiofrequency ablation for atrial fibrillation may entail a small but statistically increased risk of stroke or transient ischemic attack, according to a meta-analysis of 10 observational cohort studies.
Guidelines recommend periprocedural anticoagulation for at least 2 months post ablation. Warfarin continues to be the most widely used agent for this purpose, but new oral alternatives are attracting a great deal of interest from physicians and patients.
The meta-analysis, which included 1,501 patients on periprocedural dabigatran (Pradaxa) and 2,356 on warfarin, had dual primary end points. One was stroke or TIA, which occurred in 0.7% of the dabigatran group, compared with 0.2% of those on warfarin – a statistically significant difference (P = .0007), Dr. Benjamin A. Steinberg reported at the annual meeting of the Heart Rhythm Society.
The co-primary end point, major bleeding, was recorded in 1.6% of the dabigatran group, with a closely similar 1.7% incidence in the warfarin group, added Dr. Steinberg of Duke University in Durham, N.C.
Rates of cardiac tamponade, a secondary end point, were also similar: 1.1% with dabigatran and 0.9% with warfarin.
Although dabigatran is approved for the prevention of stroke or systemic embolism in patients with AF, a definitive determination of its safety and effectiveness in the setting of AF ablation therapy will require randomized trials, Dr. Steinberg observed.
Such trials would, of necessity, have to be quite large. The data from this meta-analysis suggest that, for every 200 patients undergoing AF ablation under the protection of dabigatran rather than warfarin, one additional case of stroke or TIA would occur.
The meta-analysis was supported in part by the Agency for Healthcare Research and Quality. Dr. Steinberg reported having no conflicts of interest.
DENVER – Using dabigatran rather than warfarin for periprocedural anticoagulation in patients undergoing radiofrequency ablation for atrial fibrillation may entail a small but statistically increased risk of stroke or transient ischemic attack, according to a meta-analysis of 10 observational cohort studies.
Guidelines recommend periprocedural anticoagulation for at least 2 months post ablation. Warfarin continues to be the most widely used agent for this purpose, but new oral alternatives are attracting a great deal of interest from physicians and patients.
The meta-analysis, which included 1,501 patients on periprocedural dabigatran (Pradaxa) and 2,356 on warfarin, had dual primary end points. One was stroke or TIA, which occurred in 0.7% of the dabigatran group, compared with 0.2% of those on warfarin – a statistically significant difference (P = .0007), Dr. Benjamin A. Steinberg reported at the annual meeting of the Heart Rhythm Society.
The co-primary end point, major bleeding, was recorded in 1.6% of the dabigatran group, with a closely similar 1.7% incidence in the warfarin group, added Dr. Steinberg of Duke University in Durham, N.C.
Rates of cardiac tamponade, a secondary end point, were also similar: 1.1% with dabigatran and 0.9% with warfarin.
Although dabigatran is approved for the prevention of stroke or systemic embolism in patients with AF, a definitive determination of its safety and effectiveness in the setting of AF ablation therapy will require randomized trials, Dr. Steinberg observed.
Such trials would, of necessity, have to be quite large. The data from this meta-analysis suggest that, for every 200 patients undergoing AF ablation under the protection of dabigatran rather than warfarin, one additional case of stroke or TIA would occur.
The meta-analysis was supported in part by the Agency for Healthcare Research and Quality. Dr. Steinberg reported having no conflicts of interest.
AT HEART RHYTHM 2013
Major finding: The incidence of stroke or transient ischemic attack in patients on dabigatran for periprocedural anticoagulation in conjunction with radiofrequency ablation for atrial fibrillation was 0.7%, statistically greater than the 0.2% in patients on warfarin.
Data source: A meta-analysis of 10 observational cohort studies totaling 1,501 AF ablation patients on dabigatran and 2,356 on warfarin.
Disclosures: The meta-analysis was funded by the Agency for Healthcare Research and Quality. Dr. Steinberg reported having no conflicts of interest.

Cardiac implantable device infection rate is falling
DENVER – Whether cardiac electronic devices are implanted in an inpatient or outpatient setting doesn’t affect device infection rates, according to a national analysis of Medicare data for 1997-2010.
Outpatient implantations are on the upswing. From 1997 to 2010, the proportion of pacemaker implantations performed in Medicare patients on an outpatient basis climbed from 8.6% to 29%. In addition, the proportion of pacemaker revision procedures done in outpatient settings nearly doubled, from 37% to 72%, Dr. Arnold J. Greenspon reported at the annual meeting of the Heart Rhythm Society.

Similarly, outpatient installation of implantable cardioverter defibrillators (ICDs) accounted for just 6.2% of all ICD implantations in 1997, but 36% in 2010. The proportion of ICD revision procedures performed on an outpatient basis rose from 31% to 69% during this time period, added Dr. Greenspon, professor of medicine and director of the cardiac electrophysiology laboratory at Thomas Jefferson University, Philadelphia.
Device infection rates following primary implantation have been waning. The ICD infection rate dropped from 1.3% in 1997 to 0.8% in 2010. Pacemaker infections are less frequent: The rate was 0.9% in 1997, falling to just 0.2% in 2010.
However, the risk of deep infection warranting inpatient revision surgery has increased over time. The annual number of patients undergoing inpatient device removal on the basis of ICD diagnosis codes for deep infection or sepsis increased two- to -threefold during 1997-2010.
Risk factors for infection identified in this study included advanced age, renal failure and other medical comorbidities, and an increasing number of cardiac device procedures. Indeed, the pacemaker and ICD infection rates climbed to 14% and 9%, respectively, in patients who had undergone five device procedures.
This study was funded by Medtronic. Dr. Greenspon reported serving as a consultant to Medtronic, Boston Scientific, and St. Jude Medical.
DENVER – Whether cardiac electronic devices are implanted in an inpatient or outpatient setting doesn’t affect device infection rates, according to a national analysis of Medicare data for 1997-2010.
Outpatient implantations are on the upswing. From 1997 to 2010, the proportion of pacemaker implantations performed in Medicare patients on an outpatient basis climbed from 8.6% to 29%. In addition, the proportion of pacemaker revision procedures done in outpatient settings nearly doubled, from 37% to 72%, Dr. Arnold J. Greenspon reported at the annual meeting of the Heart Rhythm Society.
Similarly, outpatient installation of implantable cardioverter defibrillators (ICDs) accounted for just 6.2% of all ICD implantations in 1997, but 36% in 2010. The proportion of ICD revision procedures performed on an outpatient basis rose from 31% to 69% during this time period, added Dr. Greenspon, professor of medicine and director of the cardiac electrophysiology laboratory at Thomas Jefferson University, Philadelphia.
Device infection rates following primary implantation have been waning. The ICD infection rate dropped from 1.3% in 1997 to 0.8% in 2010. Pacemaker infections are less frequent: The rate was 0.9% in 1997, falling to just 0.2% in 2010.
However, the risk of deep infection warranting inpatient revision surgery has increased over time. The annual number of patients undergoing inpatient device removal on the basis of ICD diagnosis codes for deep infection or sepsis increased two- to -threefold during 1997-2010.
Risk factors for infection identified in this study included advanced age, renal failure and other medical comorbidities, and an increasing number of cardiac device procedures. Indeed, the pacemaker and ICD infection rates climbed to 14% and 9%, respectively, in patients who had undergone five device procedures.
This study was funded by Medtronic. Dr. Greenspon reported serving as a consultant to Medtronic, Boston Scientific, and St. Jude Medical.
DENVER – Whether cardiac electronic devices are implanted in an inpatient or outpatient setting doesn’t affect device infection rates, according to a national analysis of Medicare data for 1997-2010.
Outpatient implantations are on the upswing. From 1997 to 2010, the proportion of pacemaker implantations performed in Medicare patients on an outpatient basis climbed from 8.6% to 29%. In addition, the proportion of pacemaker revision procedures done in outpatient settings nearly doubled, from 37% to 72%, Dr. Arnold J. Greenspon reported at the annual meeting of the Heart Rhythm Society.
Similarly, outpatient installation of implantable cardioverter defibrillators (ICDs) accounted for just 6.2% of all ICD implantations in 1997, but 36% in 2010. The proportion of ICD revision procedures performed on an outpatient basis rose from 31% to 69% during this time period, added Dr. Greenspon, professor of medicine and director of the cardiac electrophysiology laboratory at Thomas Jefferson University, Philadelphia.
Device infection rates following primary implantation have been waning. The ICD infection rate dropped from 1.3% in 1997 to 0.8% in 2010. Pacemaker infections are less frequent: The rate was 0.9% in 1997, falling to just 0.2% in 2010.
However, the risk of deep infection warranting inpatient revision surgery has increased over time. The annual number of patients undergoing inpatient device removal on the basis of ICD diagnosis codes for deep infection or sepsis increased two- to -threefold during 1997-2010.
Risk factors for infection identified in this study included advanced age, renal failure and other medical comorbidities, and an increasing number of cardiac device procedures. Indeed, the pacemaker and ICD infection rates climbed to 14% and 9%, respectively, in patients who had undergone five device procedures.
This study was funded by Medtronic. Dr. Greenspon reported serving as a consultant to Medtronic, Boston Scientific, and St. Jude Medical.
AT HEART RHYTHM 2013
Major finding: Annual infection rates following primary pacemaker implantation declined nationally from 0.9% in 1997 to just 0.2% in 2010. The ICD infection rate dropped from 1.3% in 1997 to 0.8% in 2010.
Data source: An analysis of the Medicare database covering 1997-2010.
Disclosures: The study was sponsored by Medtronic. The presenter is a consultant to Medtronic and other medical device manufacturers.

Leadless cardiac pacemaker draws plaudits
DENVER – A first-in-man study has demonstrated that a novel leadless cardiac pacemaker is easy to implant percutaneously in the right ventricle, is readily retrievable when required, and performs like a conventional single-chamber permanent VVIR pacemaker.
"This is a relatively small feasibility study, but I think this new leadless pacemaker has the possibility of effecting a real paradigm shift in how we think about pacing. We’re going to be able to eliminate what has been the weak link in the pacemaker system: the lead," Dr. Vivek Reddy predicted in presenting the results of the LEADLESS study at the annual meeting of the Heart Rhythm Society.
More than 4.4 million people worldwide have a cardiac pacemaker. More than 65,000 of them develop chronic problems related to the pacemaker leads, including lead dislodgement, infection, lead failure, and hematoma. In one major study, the overall complication rate over the life of conventional pacemakers was close to 8%.

These complications come at considerable cost. A lead infection can easily add $50,000 to the cost of care, a lead revision $16,000, noted Dr. Reddy of Mount Sinai School of Medicine, New York.
The leadless pacemaker is about the size of a triple-A battery. Vascular access for its placement is gained in standard fashion through the femoral vein using a steerable 18F catheter. The device is typically placed in the right ventricular apex and fixed in place by a single turn of an attached helix. The single-chamber pacemaker uses low-power electronics and has an estimated battery life of 8-17 years, depending upon how much of the time it’s actively pacing. The pacemaker senses right ventricular blood temperature, using it as a guide to boost pacing rate in response to increased metabolic demand.
In the LEADLESS study, 32 of 33 patients at three European centers underwent successful pacemaker implantation. The procedure took roughly 30 minutes initially, but times came down with greater operator experience. The most common indication for a permanent single-chamber pacemaker among study participants was chronic atrial fibrillation with second- or third-degree atrioventricular heart block.
The pacemaker is designed so the interventionalist can quickly determine if the device has been placed in an optimal location; if not, it can be repositioned during the same procedure before being finally locked into place. Twenty-three of the 32 patients did not require any device repositioning.

During 3 months of study follow-up, two patients required device retrieval. In one, the leadless pacemaker had migrated through an open patent foramen ovale into the left ventricle. The other patient developed indications warranting an implantable cardioverter-defibrillator. Device retrieval was a straightforward matter and took 6 and 13 minutes, respectively, using a basket catheter.
The one serious adverse event in the study involved a patient who experienced cardiac perforation and tamponade during pacemaker implantation. Five days after an uncomplicated surgical repair, the patient had a large right-sided stroke and died.
Dr. Reddy said the leadless pacemaker will be commercialized in Europe later this year. Planning is underway for a large, multicenter U.S. trial that will probably start next year. And Nanostim, the company developing the device, is also working on a leadless atrial pacemaker that will be able to communicate with the right ventricular pacemaker in order to provide multichamber cardiac pacing.
Dr. Reddy’s presentation met with an enthusiastic response.
"This is fascinating, incredible technology," declared Dr. Andrea M. Russo of Cooper University Health Care in Camden, N.J.
"The idea of being able to put a pacemaker into someone without having leads and without having a device under the skin is probably the next phase of what we’d hope to do with cardiac pacing therapy," Dr. Michael R. Gold observed.
"I think most of us, if we were to make predictions about what’s in store 10 years from now, we’d be hoping that we’d no longer use leads, which are the component that fails most frequently in any device. If we can get rid of the leads, put these little pacemakers anywhere we want in the heart, and get them to cross-talk and communicate with each other, that’s very exciting," said Dr. Gold, professor of medicine, chief of cardiology, and medical director of the heart and vascular center at the Medical University of South Carolina, Charleston.
The LEADLESS study was funded by Nanostim. Dr. Reddy has received research grant support from and is a consultant to the company.
DENVER – A first-in-man study has demonstrated that a novel leadless cardiac pacemaker is easy to implant percutaneously in the right ventricle, is readily retrievable when required, and performs like a conventional single-chamber permanent VVIR pacemaker.
"This is a relatively small feasibility study, but I think this new leadless pacemaker has the possibility of effecting a real paradigm shift in how we think about pacing. We’re going to be able to eliminate what has been the weak link in the pacemaker system: the lead," Dr. Vivek Reddy predicted in presenting the results of the LEADLESS study at the annual meeting of the Heart Rhythm Society.
More than 4.4 million people worldwide have a cardiac pacemaker. More than 65,000 of them develop chronic problems related to the pacemaker leads, including lead dislodgement, infection, lead failure, and hematoma. In one major study, the overall complication rate over the life of conventional pacemakers was close to 8%.
These complications come at considerable cost. A lead infection can easily add $50,000 to the cost of care, a lead revision $16,000, noted Dr. Reddy of Mount Sinai School of Medicine, New York.
The leadless pacemaker is about the size of a triple-A battery. Vascular access for its placement is gained in standard fashion through the femoral vein using a steerable 18F catheter. The device is typically placed in the right ventricular apex and fixed in place by a single turn of an attached helix. The single-chamber pacemaker uses low-power electronics and has an estimated battery life of 8-17 years, depending upon how much of the time it’s actively pacing. The pacemaker senses right ventricular blood temperature, using it as a guide to boost pacing rate in response to increased metabolic demand.
In the LEADLESS study, 32 of 33 patients at three European centers underwent successful pacemaker implantation. The procedure took roughly 30 minutes initially, but times came down with greater operator experience. The most common indication for a permanent single-chamber pacemaker among study participants was chronic atrial fibrillation with second- or third-degree atrioventricular heart block.
The pacemaker is designed so the interventionalist can quickly determine if the device has been placed in an optimal location; if not, it can be repositioned during the same procedure before being finally locked into place. Twenty-three of the 32 patients did not require any device repositioning.
During 3 months of study follow-up, two patients required device retrieval. In one, the leadless pacemaker had migrated through an open patent foramen ovale into the left ventricle. The other patient developed indications warranting an implantable cardioverter-defibrillator. Device retrieval was a straightforward matter and took 6 and 13 minutes, respectively, using a basket catheter.
The one serious adverse event in the study involved a patient who experienced cardiac perforation and tamponade during pacemaker implantation. Five days after an uncomplicated surgical repair, the patient had a large right-sided stroke and died.
Dr. Reddy said the leadless pacemaker will be commercialized in Europe later this year. Planning is underway for a large, multicenter U.S. trial that will probably start next year. And Nanostim, the company developing the device, is also working on a leadless atrial pacemaker that will be able to communicate with the right ventricular pacemaker in order to provide multichamber cardiac pacing.
Dr. Reddy’s presentation met with an enthusiastic response.
"This is fascinating, incredible technology," declared Dr. Andrea M. Russo of Cooper University Health Care in Camden, N.J.
"The idea of being able to put a pacemaker into someone without having leads and without having a device under the skin is probably the next phase of what we’d hope to do with cardiac pacing therapy," Dr. Michael R. Gold observed.
"I think most of us, if we were to make predictions about what’s in store 10 years from now, we’d be hoping that we’d no longer use leads, which are the component that fails most frequently in any device. If we can get rid of the leads, put these little pacemakers anywhere we want in the heart, and get them to cross-talk and communicate with each other, that’s very exciting," said Dr. Gold, professor of medicine, chief of cardiology, and medical director of the heart and vascular center at the Medical University of South Carolina, Charleston.
The LEADLESS study was funded by Nanostim. Dr. Reddy has received research grant support from and is a consultant to the company.
DENVER – A first-in-man study has demonstrated that a novel leadless cardiac pacemaker is easy to implant percutaneously in the right ventricle, is readily retrievable when required, and performs like a conventional single-chamber permanent VVIR pacemaker.
"This is a relatively small feasibility study, but I think this new leadless pacemaker has the possibility of effecting a real paradigm shift in how we think about pacing. We’re going to be able to eliminate what has been the weak link in the pacemaker system: the lead," Dr. Vivek Reddy predicted in presenting the results of the LEADLESS study at the annual meeting of the Heart Rhythm Society.
More than 4.4 million people worldwide have a cardiac pacemaker. More than 65,000 of them develop chronic problems related to the pacemaker leads, including lead dislodgement, infection, lead failure, and hematoma. In one major study, the overall complication rate over the life of conventional pacemakers was close to 8%.
These complications come at considerable cost. A lead infection can easily add $50,000 to the cost of care, a lead revision $16,000, noted Dr. Reddy of Mount Sinai School of Medicine, New York.
The leadless pacemaker is about the size of a triple-A battery. Vascular access for its placement is gained in standard fashion through the femoral vein using a steerable 18F catheter. The device is typically placed in the right ventricular apex and fixed in place by a single turn of an attached helix. The single-chamber pacemaker uses low-power electronics and has an estimated battery life of 8-17 years, depending upon how much of the time it’s actively pacing. The pacemaker senses right ventricular blood temperature, using it as a guide to boost pacing rate in response to increased metabolic demand.
In the LEADLESS study, 32 of 33 patients at three European centers underwent successful pacemaker implantation. The procedure took roughly 30 minutes initially, but times came down with greater operator experience. The most common indication for a permanent single-chamber pacemaker among study participants was chronic atrial fibrillation with second- or third-degree atrioventricular heart block.
The pacemaker is designed so the interventionalist can quickly determine if the device has been placed in an optimal location; if not, it can be repositioned during the same procedure before being finally locked into place. Twenty-three of the 32 patients did not require any device repositioning.
During 3 months of study follow-up, two patients required device retrieval. In one, the leadless pacemaker had migrated through an open patent foramen ovale into the left ventricle. The other patient developed indications warranting an implantable cardioverter-defibrillator. Device retrieval was a straightforward matter and took 6 and 13 minutes, respectively, using a basket catheter.
The one serious adverse event in the study involved a patient who experienced cardiac perforation and tamponade during pacemaker implantation. Five days after an uncomplicated surgical repair, the patient had a large right-sided stroke and died.
Dr. Reddy said the leadless pacemaker will be commercialized in Europe later this year. Planning is underway for a large, multicenter U.S. trial that will probably start next year. And Nanostim, the company developing the device, is also working on a leadless atrial pacemaker that will be able to communicate with the right ventricular pacemaker in order to provide multichamber cardiac pacing.
Dr. Reddy’s presentation met with an enthusiastic response.
"This is fascinating, incredible technology," declared Dr. Andrea M. Russo of Cooper University Health Care in Camden, N.J.
"The idea of being able to put a pacemaker into someone without having leads and without having a device under the skin is probably the next phase of what we’d hope to do with cardiac pacing therapy," Dr. Michael R. Gold observed.
"I think most of us, if we were to make predictions about what’s in store 10 years from now, we’d be hoping that we’d no longer use leads, which are the component that fails most frequently in any device. If we can get rid of the leads, put these little pacemakers anywhere we want in the heart, and get them to cross-talk and communicate with each other, that’s very exciting," said Dr. Gold, professor of medicine, chief of cardiology, and medical director of the heart and vascular center at the Medical University of South Carolina, Charleston.
The LEADLESS study was funded by Nanostim. Dr. Reddy has received research grant support from and is a consultant to the company.
AT HEART RHYTHM 2013
Major finding: A leadless cardiac pacemaker the size of a triple-A battery was successfully implanted in 32 of 33 candidates for permanent single-chamber pacing. The device performed well during 3 months of follow-up.
Data source: The first-in-man, prospective, single-arm, nonrandomized study conducted at three European centers.
Disclosures: The LEADLESS study was funded by Nanostim. Dr. Reddy has received research grant support from and is a consultant to the company.
