User login
Make the Diagnosis - August 2017
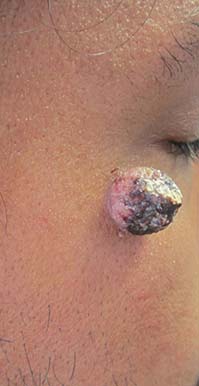
Trichofolliculoma
Trichofolliculoma is a rare, benign skin lesion that was first described in 1944. While the etiology is largely known, it is considered to be a hamartoma with follicular differentiation that results when the pluripotent skin cells cease to differentiate into the hair follicle. The lesion lacks specific predilection for gender or race but most commonly occurs on the face of adults. Clinically, trichofolliculoma presents as a solitary, skin-colored papule or nodule. It may contain a central sebum-producing pore or have a tuft of white hair protruding from the central pore, although neither of these manifestations are mandatory. Trichofolliculomas generally are asymptomatic and are not associated with any systemic or other cutaneous diseases. Therefore, no treatment is required. However, surgical excision or curettage and electrodesiccation may be performed for cosmetic reasons.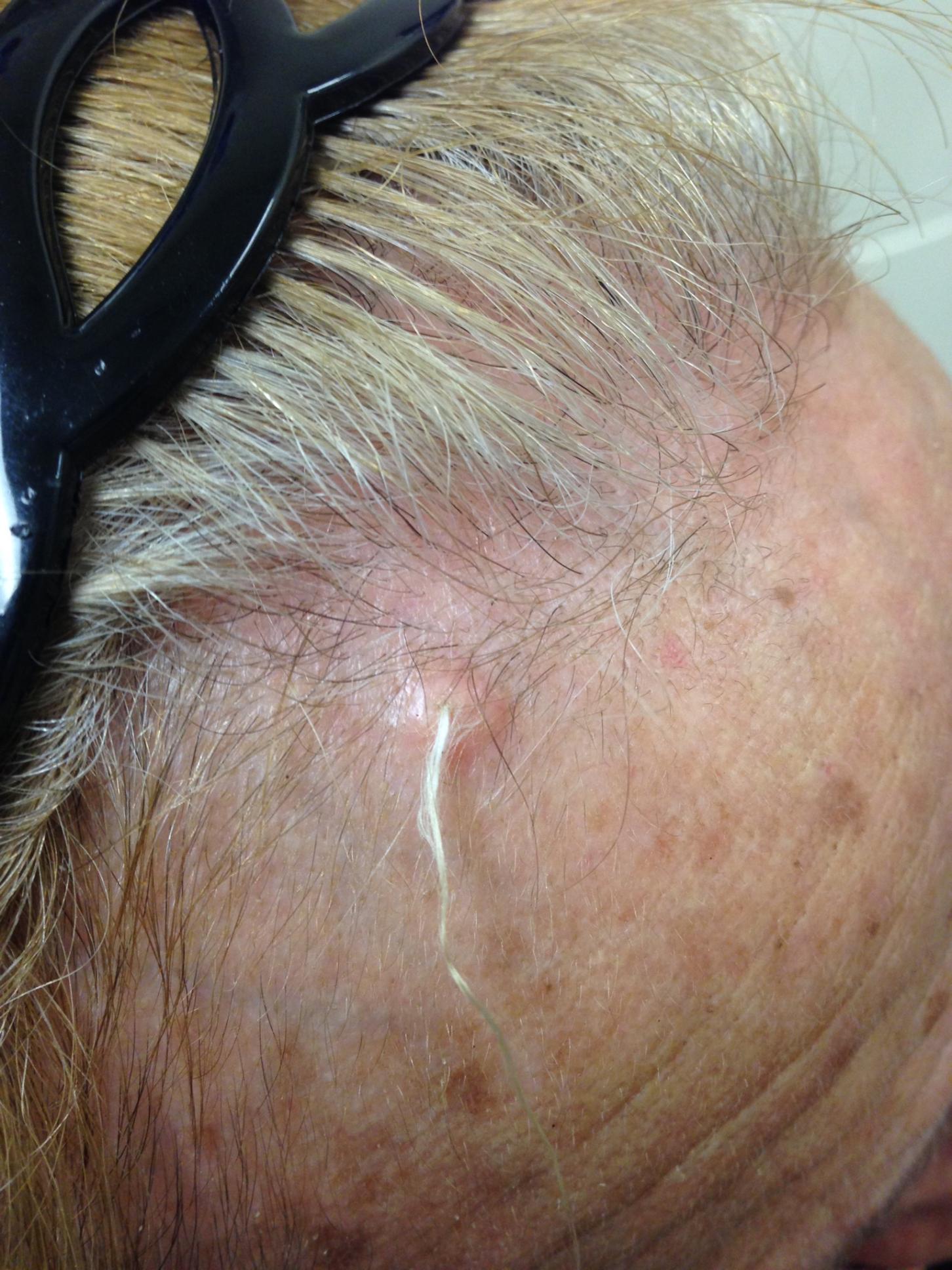
Definitive diagnosis of trichofolliculoma requires a biopsy. The histopathology shows a primary cystic structure within the dermis that may or may not be connected to the epidermis. There are multiple abnormal secondary follicles that radiate from the primary cystic structure, and the entire apparatus is surrounded by a well-circumscribed dense connective tissue. Immunohistochemical studies have revealed that trichofolliculoma is characterized by activated, aberrant cytokeratin-15 (CK15)–positive follicle stem cells that show outer root sheath differentiation while attempting to make hair. This results in disorder of the normal hair cycle.
The clinical differential diagnosis for trichofolliculoma includes dermal nevus, basal cell carcinoma, and pilar sheath acanthoma. These cutaneous entities may be ruled out by histopathology.
The histopathology of basal cell carcinoma consists of tumors of basaloid cells with little cytoplasm and hyperchromatic nuclei. There is palisading around the periphery of the tumor with stromal retraction that leads to a clefting appearance, as well as mucinous change in the stroma.
The histopathology of dermal nevus reveals small nests of melanocytes within the upper dermis that commonly localize around the pilosebaceous units. There is variability in pigmentation and cellularity of the melanocytes, and there is no junctional component.
The histopathology of pilar sheath acanthoma shows dermal proliferation of lobules comprising benign squamous epithelium that arrange themselves around small cystic spaces. The lobules themselves are surrounded by eosinophilic basement membrane.
The case and photo were submitted by Victoria Billero, MS; Adam Wulkan, MD; and Caroline Winslow, MD, all of the department of dermatology, University of Miami (Fla.).
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Trichofolliculoma
Trichofolliculoma is a rare, benign skin lesion that was first described in 1944. While the etiology is largely known, it is considered to be a hamartoma with follicular differentiation that results when the pluripotent skin cells cease to differentiate into the hair follicle. The lesion lacks specific predilection for gender or race but most commonly occurs on the face of adults. Clinically, trichofolliculoma presents as a solitary, skin-colored papule or nodule. It may contain a central sebum-producing pore or have a tuft of white hair protruding from the central pore, although neither of these manifestations are mandatory. Trichofolliculomas generally are asymptomatic and are not associated with any systemic or other cutaneous diseases. Therefore, no treatment is required. However, surgical excision or curettage and electrodesiccation may be performed for cosmetic reasons.
Definitive diagnosis of trichofolliculoma requires a biopsy. The histopathology shows a primary cystic structure within the dermis that may or may not be connected to the epidermis. There are multiple abnormal secondary follicles that radiate from the primary cystic structure, and the entire apparatus is surrounded by a well-circumscribed dense connective tissue. Immunohistochemical studies have revealed that trichofolliculoma is characterized by activated, aberrant cytokeratin-15 (CK15)–positive follicle stem cells that show outer root sheath differentiation while attempting to make hair. This results in disorder of the normal hair cycle.
The clinical differential diagnosis for trichofolliculoma includes dermal nevus, basal cell carcinoma, and pilar sheath acanthoma. These cutaneous entities may be ruled out by histopathology.
The histopathology of basal cell carcinoma consists of tumors of basaloid cells with little cytoplasm and hyperchromatic nuclei. There is palisading around the periphery of the tumor with stromal retraction that leads to a clefting appearance, as well as mucinous change in the stroma.
The histopathology of dermal nevus reveals small nests of melanocytes within the upper dermis that commonly localize around the pilosebaceous units. There is variability in pigmentation and cellularity of the melanocytes, and there is no junctional component.
The histopathology of pilar sheath acanthoma shows dermal proliferation of lobules comprising benign squamous epithelium that arrange themselves around small cystic spaces. The lobules themselves are surrounded by eosinophilic basement membrane.
The case and photo were submitted by Victoria Billero, MS; Adam Wulkan, MD; and Caroline Winslow, MD, all of the department of dermatology, University of Miami (Fla.).
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Trichofolliculoma
Trichofolliculoma is a rare, benign skin lesion that was first described in 1944. While the etiology is largely known, it is considered to be a hamartoma with follicular differentiation that results when the pluripotent skin cells cease to differentiate into the hair follicle. The lesion lacks specific predilection for gender or race but most commonly occurs on the face of adults. Clinically, trichofolliculoma presents as a solitary, skin-colored papule or nodule. It may contain a central sebum-producing pore or have a tuft of white hair protruding from the central pore, although neither of these manifestations are mandatory. Trichofolliculomas generally are asymptomatic and are not associated with any systemic or other cutaneous diseases. Therefore, no treatment is required. However, surgical excision or curettage and electrodesiccation may be performed for cosmetic reasons.
Definitive diagnosis of trichofolliculoma requires a biopsy. The histopathology shows a primary cystic structure within the dermis that may or may not be connected to the epidermis. There are multiple abnormal secondary follicles that radiate from the primary cystic structure, and the entire apparatus is surrounded by a well-circumscribed dense connective tissue. Immunohistochemical studies have revealed that trichofolliculoma is characterized by activated, aberrant cytokeratin-15 (CK15)–positive follicle stem cells that show outer root sheath differentiation while attempting to make hair. This results in disorder of the normal hair cycle.
The clinical differential diagnosis for trichofolliculoma includes dermal nevus, basal cell carcinoma, and pilar sheath acanthoma. These cutaneous entities may be ruled out by histopathology.
The histopathology of basal cell carcinoma consists of tumors of basaloid cells with little cytoplasm and hyperchromatic nuclei. There is palisading around the periphery of the tumor with stromal retraction that leads to a clefting appearance, as well as mucinous change in the stroma.
The histopathology of dermal nevus reveals small nests of melanocytes within the upper dermis that commonly localize around the pilosebaceous units. There is variability in pigmentation and cellularity of the melanocytes, and there is no junctional component.
The histopathology of pilar sheath acanthoma shows dermal proliferation of lobules comprising benign squamous epithelium that arrange themselves around small cystic spaces. The lobules themselves are surrounded by eosinophilic basement membrane.
The case and photo were submitted by Victoria Billero, MS; Adam Wulkan, MD; and Caroline Winslow, MD, all of the department of dermatology, University of Miami (Fla.).
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
A 67-year-old white female with no significant past medical history presented as a first-time patient to clinic for a full skin check. An incidental finding of a solitary, asymptomatic, non-tender 8 mm flesh-colored papule with a tuft of long white hair protruding through a central pore was observed on her right lateral forehead that had been present for longer than 10 years.
Make the Diagnosis - July 2017
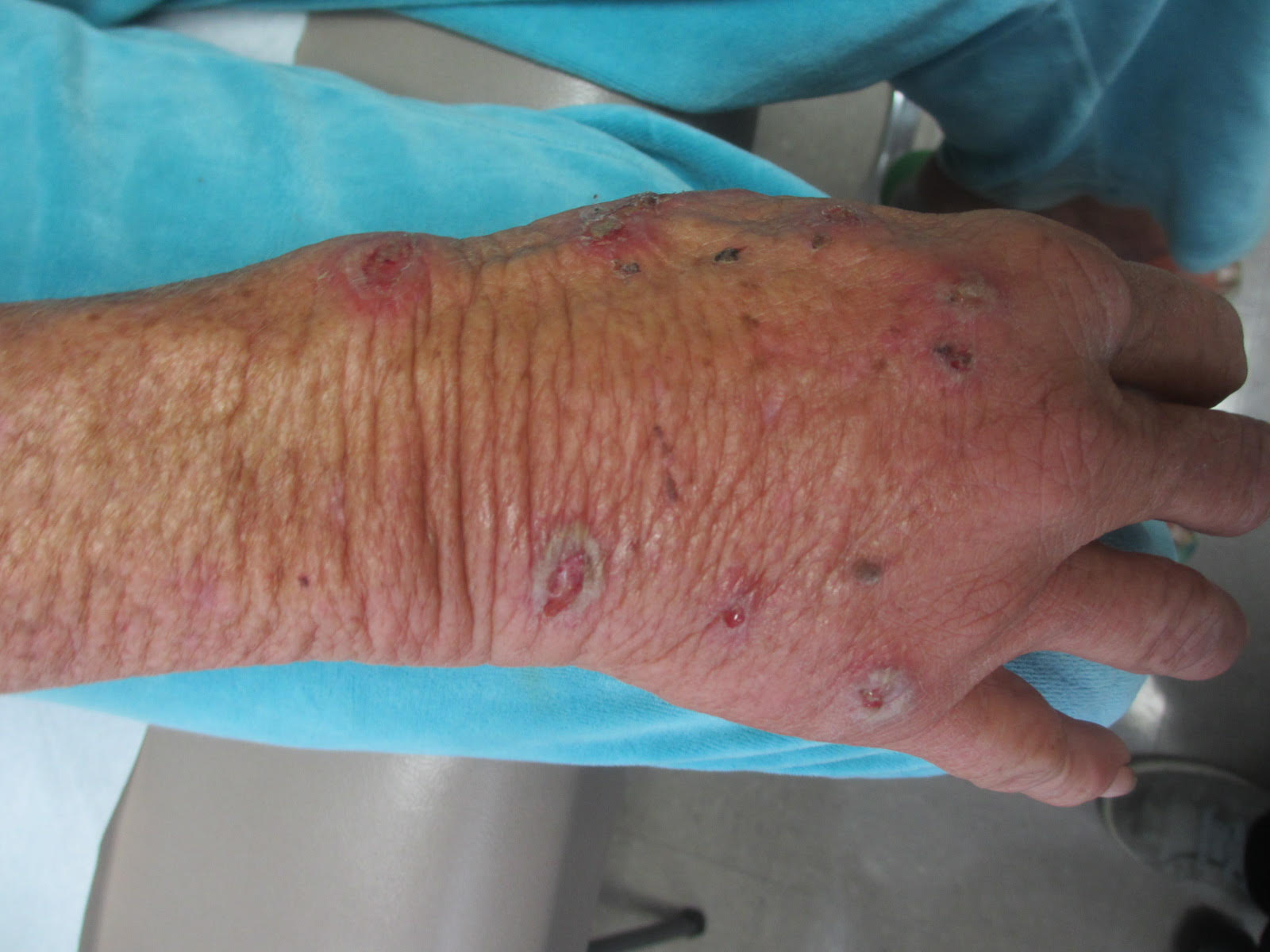
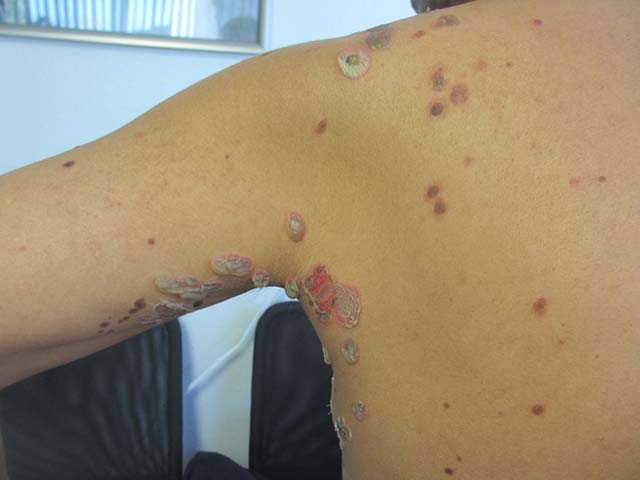
Sweet’s syndrome (acute febrile neutrophilic dermatosis)
A biopsy revealed an intact epidermis with intense infiltration of neutrophils in the superficial dermis, striking superficial edema, and a few lymphoid cells, consistent with Sweet’s syndrome. The patient was started on prednisone with a slow taper and had resolution of her symptoms.
Sweet’s syndrome (acute febrile neutrophilic dermatosis) is characterized by the sudden manifestation of painful edematous and erythematous papules, plaques, and/or nodules. As may be predicted by the descriptive name, the skin findings are often accompanied by fever, leukocytosis, and other extracutaneous manifestations. Sweet’s syndrome is generally divided into three categories: classical (idiopathic), drug associated, and malignancy associated.
Classical (idiopathic) Sweet’s syndrome
Drug-associated Sweet’s syndrome
Drug-associated Sweet’s syndrome must meet all the diagnostic criteria of the classical variant, as well as a temporal relationship for both the onset and resolution of symptoms associated with the initiation and cessation of a drug, respectively. Signs and symptoms of the disease process typically develop 2 weeks after the initial drug exposure. Granulocyte colony stimulating factor (G-CSF) is the most commonly reported inciting drug; however, an extensive list of potential offenders has been described in the literature. Although most cases will self-resolve within several weeks of drug cessation, treatment with corticosteroids can expedite recovery.
Malignancy-associated Sweet’s syndrome
Malignancy-associated Sweet’s syndrome may occur as the first revelation of an undiagnosed malignancy, as a complication of an already diagnosed malignancy, or as a warning sign of the recurrence of a previously diagnosed malignancy. Association with solid tumors is uncommon, with approximately 85% of reported cases occurring in patients with an underlying hematologic malignancy (most frequently, acute myeloblastic leukemia). The treatment of choice for malignancy-associated Sweet’s syndrome is targeted at eradicating the underlying malignancy; however, most patients will receive a course of corticosteroids to aid in a faster resolution of symptoms. Very few cases of Sweet’s syndrome associated with untreated melanoma have been reported in the literature, as patients with melanoma are much more likely to develop Sweet’s syndrome as a result of anti-neoplastic drug therapy than as a result of the tumor itself.
While it is not completely clear if this patient’s diagnosis was the classical type or malignancy associated, it is more likely the former as the patient improved with oral corticosteroids.
This case and photo are courtesy of Natasha Cowan, University of California, San Diego, and Nick Celano, MD, San Diego Family Dermatology.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Sweet’s syndrome (acute febrile neutrophilic dermatosis)
A biopsy revealed an intact epidermis with intense infiltration of neutrophils in the superficial dermis, striking superficial edema, and a few lymphoid cells, consistent with Sweet’s syndrome. The patient was started on prednisone with a slow taper and had resolution of her symptoms.
Sweet’s syndrome (acute febrile neutrophilic dermatosis) is characterized by the sudden manifestation of painful edematous and erythematous papules, plaques, and/or nodules. As may be predicted by the descriptive name, the skin findings are often accompanied by fever, leukocytosis, and other extracutaneous manifestations. Sweet’s syndrome is generally divided into three categories: classical (idiopathic), drug associated, and malignancy associated.
Classical (idiopathic) Sweet’s syndrome
Drug-associated Sweet’s syndrome
Drug-associated Sweet’s syndrome must meet all the diagnostic criteria of the classical variant, as well as a temporal relationship for both the onset and resolution of symptoms associated with the initiation and cessation of a drug, respectively. Signs and symptoms of the disease process typically develop 2 weeks after the initial drug exposure. Granulocyte colony stimulating factor (G-CSF) is the most commonly reported inciting drug; however, an extensive list of potential offenders has been described in the literature. Although most cases will self-resolve within several weeks of drug cessation, treatment with corticosteroids can expedite recovery.
Malignancy-associated Sweet’s syndrome
Malignancy-associated Sweet’s syndrome may occur as the first revelation of an undiagnosed malignancy, as a complication of an already diagnosed malignancy, or as a warning sign of the recurrence of a previously diagnosed malignancy. Association with solid tumors is uncommon, with approximately 85% of reported cases occurring in patients with an underlying hematologic malignancy (most frequently, acute myeloblastic leukemia). The treatment of choice for malignancy-associated Sweet’s syndrome is targeted at eradicating the underlying malignancy; however, most patients will receive a course of corticosteroids to aid in a faster resolution of symptoms. Very few cases of Sweet’s syndrome associated with untreated melanoma have been reported in the literature, as patients with melanoma are much more likely to develop Sweet’s syndrome as a result of anti-neoplastic drug therapy than as a result of the tumor itself.
While it is not completely clear if this patient’s diagnosis was the classical type or malignancy associated, it is more likely the former as the patient improved with oral corticosteroids.
This case and photo are courtesy of Natasha Cowan, University of California, San Diego, and Nick Celano, MD, San Diego Family Dermatology.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Sweet’s syndrome (acute febrile neutrophilic dermatosis)
A biopsy revealed an intact epidermis with intense infiltration of neutrophils in the superficial dermis, striking superficial edema, and a few lymphoid cells, consistent with Sweet’s syndrome. The patient was started on prednisone with a slow taper and had resolution of her symptoms.
Sweet’s syndrome (acute febrile neutrophilic dermatosis) is characterized by the sudden manifestation of painful edematous and erythematous papules, plaques, and/or nodules. As may be predicted by the descriptive name, the skin findings are often accompanied by fever, leukocytosis, and other extracutaneous manifestations. Sweet’s syndrome is generally divided into three categories: classical (idiopathic), drug associated, and malignancy associated.
Classical (idiopathic) Sweet’s syndrome
Drug-associated Sweet’s syndrome
Drug-associated Sweet’s syndrome must meet all the diagnostic criteria of the classical variant, as well as a temporal relationship for both the onset and resolution of symptoms associated with the initiation and cessation of a drug, respectively. Signs and symptoms of the disease process typically develop 2 weeks after the initial drug exposure. Granulocyte colony stimulating factor (G-CSF) is the most commonly reported inciting drug; however, an extensive list of potential offenders has been described in the literature. Although most cases will self-resolve within several weeks of drug cessation, treatment with corticosteroids can expedite recovery.
Malignancy-associated Sweet’s syndrome
Malignancy-associated Sweet’s syndrome may occur as the first revelation of an undiagnosed malignancy, as a complication of an already diagnosed malignancy, or as a warning sign of the recurrence of a previously diagnosed malignancy. Association with solid tumors is uncommon, with approximately 85% of reported cases occurring in patients with an underlying hematologic malignancy (most frequently, acute myeloblastic leukemia). The treatment of choice for malignancy-associated Sweet’s syndrome is targeted at eradicating the underlying malignancy; however, most patients will receive a course of corticosteroids to aid in a faster resolution of symptoms. Very few cases of Sweet’s syndrome associated with untreated melanoma have been reported in the literature, as patients with melanoma are much more likely to develop Sweet’s syndrome as a result of anti-neoplastic drug therapy than as a result of the tumor itself.
While it is not completely clear if this patient’s diagnosis was the classical type or malignancy associated, it is more likely the former as the patient improved with oral corticosteroids.
This case and photo are courtesy of Natasha Cowan, University of California, San Diego, and Nick Celano, MD, San Diego Family Dermatology.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
A 57-year-old homeless white female with history of untreated malignant melanoma presented with a one-week history of an itchy, painful rash on the right side of her body. Physical exam revealed scattered bullous and pustular edematous plaques on the right arm, hand, and lower leg. She had a fever of 101.5F and an elevated white blood cell count.
Make the Diagnosis - June 2017
Diagnosis: Porokeratotic eccrine ostial and dermal duct nevus (PEODDN)
Porokeratotic eccrine ostial and dermal duct nevus (PEODDN) is a rare, benign adnexal hamartoma first reported by Marsden et al. in 1979 as “comedo nevus of the palm” (Br J Dermatol. 1979 Dec;101[6]:717-22).
To date, 77 cases of PEODDN have been reported in the literature, with 53% having congenital onset. The average age of onset for the acquired lesions was 6 years. PEODDN affects males and females equally. Acral location is the most common (94%), but lesions have been reported less commonly on the trunk and face. A review of the literature found that most cases of PEODDN were independent, not associated with other conditions. Rarely, Bowen’s disease and squamous cell carcinoma have been reported to arise within PEODDN. Interestingly, two cases have been associated with keratitis-ichthyosis-deafness syndrome (KID).
Common differential diagnoses for PEODDN include porokeratosis of Mibelli, linear psoriasis, and linear epidermal nevus. Linear porokeratosis and porokeratosis of Mibelli are characterized by sharply demarcated hyperkeratotic annular lesions with distinct keratotic edges but do not have eccrine gland involvement on histopathology. Linear psoriasis, a rare form of psoriasis, is characterized by late onset linear psoriatic lesions along Blaschko lines. Linear epidermal nevus is a disease characterized by pruritic, erythematous scaly lesions following Blaschko lines that occurs in the first months of life and is slowly progressive. Histopathological features of PEODDN are diagnostic and distinguish it from these other clinical entities. A prominent parakeratotic column within an epidermal invagination that displays loss of the granular layer is found overlaying an eccrine duct with a dilated acrosyringium. Vacuolated and dyskeratotic keratinocytes are also typically present within the epidermal invagination.
The treatment for PEODDN remains elusive. Topical keratolytics, topical retinoids, topical steroids, topical calcipotriol, cryosurgery, phototherapy, and anthralin have been used to treat PEODDN unsuccessfully. Thus far, the most efficacious treatments include CO2 laser and surgical excision for small lesions. Given the young age of this patient, emollient therapy was chosen as treatment until the patient reaches an age at which the lesion is cosmetically more disturbing and other therapies may be safely attempted.
In conclusion, this patient represents a classic case of the rare entity, PEODDN, and draws attention to recent discoveries that a genetic mutation in GJB2 is causative. Because PEODDN shares its pathogenic mutation with KID syndrome, clinicians should be aware that, if the same mutation also affects germline cells, offspring have the potential to express manifestations of KID syndrome.
This case and photo are courtesy of Molly B. Hirt, a medical student at Indiana University, Indianapolis; Carrie L. Davis, MD, of the Dermatology Center of Southern Indiana and Indiana University, Bloomington; and Anita N. Haggstrom, MD, of the departments of dermatology and pediatrics, Indiana University, Indianapolis.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Diagnosis: Porokeratotic eccrine ostial and dermal duct nevus (PEODDN)
Porokeratotic eccrine ostial and dermal duct nevus (PEODDN) is a rare, benign adnexal hamartoma first reported by Marsden et al. in 1979 as “comedo nevus of the palm” (Br J Dermatol. 1979 Dec;101[6]:717-22).
To date, 77 cases of PEODDN have been reported in the literature, with 53% having congenital onset. The average age of onset for the acquired lesions was 6 years. PEODDN affects males and females equally. Acral location is the most common (94%), but lesions have been reported less commonly on the trunk and face. A review of the literature found that most cases of PEODDN were independent, not associated with other conditions. Rarely, Bowen’s disease and squamous cell carcinoma have been reported to arise within PEODDN. Interestingly, two cases have been associated with keratitis-ichthyosis-deafness syndrome (KID).
Common differential diagnoses for PEODDN include porokeratosis of Mibelli, linear psoriasis, and linear epidermal nevus. Linear porokeratosis and porokeratosis of Mibelli are characterized by sharply demarcated hyperkeratotic annular lesions with distinct keratotic edges but do not have eccrine gland involvement on histopathology. Linear psoriasis, a rare form of psoriasis, is characterized by late onset linear psoriatic lesions along Blaschko lines. Linear epidermal nevus is a disease characterized by pruritic, erythematous scaly lesions following Blaschko lines that occurs in the first months of life and is slowly progressive. Histopathological features of PEODDN are diagnostic and distinguish it from these other clinical entities. A prominent parakeratotic column within an epidermal invagination that displays loss of the granular layer is found overlaying an eccrine duct with a dilated acrosyringium. Vacuolated and dyskeratotic keratinocytes are also typically present within the epidermal invagination.
The treatment for PEODDN remains elusive. Topical keratolytics, topical retinoids, topical steroids, topical calcipotriol, cryosurgery, phototherapy, and anthralin have been used to treat PEODDN unsuccessfully. Thus far, the most efficacious treatments include CO2 laser and surgical excision for small lesions. Given the young age of this patient, emollient therapy was chosen as treatment until the patient reaches an age at which the lesion is cosmetically more disturbing and other therapies may be safely attempted.
In conclusion, this patient represents a classic case of the rare entity, PEODDN, and draws attention to recent discoveries that a genetic mutation in GJB2 is causative. Because PEODDN shares its pathogenic mutation with KID syndrome, clinicians should be aware that, if the same mutation also affects germline cells, offspring have the potential to express manifestations of KID syndrome.
This case and photo are courtesy of Molly B. Hirt, a medical student at Indiana University, Indianapolis; Carrie L. Davis, MD, of the Dermatology Center of Southern Indiana and Indiana University, Bloomington; and Anita N. Haggstrom, MD, of the departments of dermatology and pediatrics, Indiana University, Indianapolis.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Diagnosis: Porokeratotic eccrine ostial and dermal duct nevus (PEODDN)
Porokeratotic eccrine ostial and dermal duct nevus (PEODDN) is a rare, benign adnexal hamartoma first reported by Marsden et al. in 1979 as “comedo nevus of the palm” (Br J Dermatol. 1979 Dec;101[6]:717-22).
To date, 77 cases of PEODDN have been reported in the literature, with 53% having congenital onset. The average age of onset for the acquired lesions was 6 years. PEODDN affects males and females equally. Acral location is the most common (94%), but lesions have been reported less commonly on the trunk and face. A review of the literature found that most cases of PEODDN were independent, not associated with other conditions. Rarely, Bowen’s disease and squamous cell carcinoma have been reported to arise within PEODDN. Interestingly, two cases have been associated with keratitis-ichthyosis-deafness syndrome (KID).
Common differential diagnoses for PEODDN include porokeratosis of Mibelli, linear psoriasis, and linear epidermal nevus. Linear porokeratosis and porokeratosis of Mibelli are characterized by sharply demarcated hyperkeratotic annular lesions with distinct keratotic edges but do not have eccrine gland involvement on histopathology. Linear psoriasis, a rare form of psoriasis, is characterized by late onset linear psoriatic lesions along Blaschko lines. Linear epidermal nevus is a disease characterized by pruritic, erythematous scaly lesions following Blaschko lines that occurs in the first months of life and is slowly progressive. Histopathological features of PEODDN are diagnostic and distinguish it from these other clinical entities. A prominent parakeratotic column within an epidermal invagination that displays loss of the granular layer is found overlaying an eccrine duct with a dilated acrosyringium. Vacuolated and dyskeratotic keratinocytes are also typically present within the epidermal invagination.
The treatment for PEODDN remains elusive. Topical keratolytics, topical retinoids, topical steroids, topical calcipotriol, cryosurgery, phototherapy, and anthralin have been used to treat PEODDN unsuccessfully. Thus far, the most efficacious treatments include CO2 laser and surgical excision for small lesions. Given the young age of this patient, emollient therapy was chosen as treatment until the patient reaches an age at which the lesion is cosmetically more disturbing and other therapies may be safely attempted.
In conclusion, this patient represents a classic case of the rare entity, PEODDN, and draws attention to recent discoveries that a genetic mutation in GJB2 is causative. Because PEODDN shares its pathogenic mutation with KID syndrome, clinicians should be aware that, if the same mutation also affects germline cells, offspring have the potential to express manifestations of KID syndrome.
This case and photo are courtesy of Molly B. Hirt, a medical student at Indiana University, Indianapolis; Carrie L. Davis, MD, of the Dermatology Center of Southern Indiana and Indiana University, Bloomington; and Anita N. Haggstrom, MD, of the departments of dermatology and pediatrics, Indiana University, Indianapolis.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Make the Diagnosis - May 2017
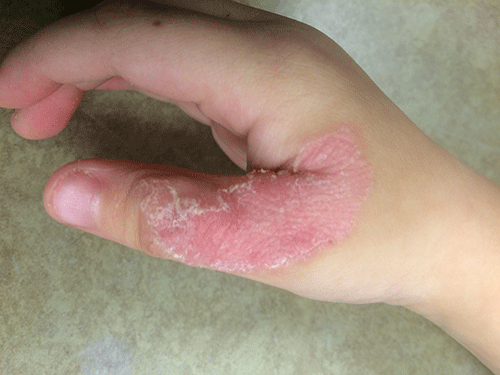
Dermatitis herpetiformis

The primary lesions of DH are vesicles and papules in a grouped or “herpetic” formation. However, as these lesions are extremely pruritic, the primary lesions may be absent in many cases and instead replaced by secondary excoriations and erosions. DH has a very classic distribution pattern, particularly involving the bilateral extensor surfaces, buttocks, and scalp. Although some cases of oral DH have been reported, mucosal involvement is generally considered to be very rare.
Despite its strong association with underlying celiac disease, most patients with DH do not report any associated gastrointestinal symptoms. Those with DH may present with any variety of other autoimmune conditions, with hypothyroidism being the most common. Interestingly, patients with DH have been shown to be at an increased development of non-Hodgkin lymphoma. It is not certain whether adherence to a strict gluten-free diet reduces this risk in this population.
Diagnosis can be made with a proper clinical history and examination, tissue pathology, direct immunofluorescence microscopy (DIF), and/or serology, with DIF being the most definitive. Perilesional skin is preferred for DIF, as lesional biopsies have been found to have higher rates of false negative results. The characteristic DIF finding diagnostic of DH is granular IgA deposits within dermal papillae, which was seen in this patient’s DIF.
Adequate treatment of DH can usually be accomplished with a combination of dapsone and a gluten-free diet. Initially, dapsone may be used for more immediate relief of associated pruritus and other bothersome symptoms. A strict gluten-free diet should be implemented as soon as possible, and dapsone can be tapered approximately 2-3 months after initiation as to avoid potential adverse effects with longterm treatment at higher doses.
The case and photo were submitted by Natasha Cowan, BS, University of California, San Diego, School of Medicine, and Nick Celano, MD, of San Diego Family Dermatology.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Dermatitis herpetiformis
The primary lesions of DH are vesicles and papules in a grouped or “herpetic” formation. However, as these lesions are extremely pruritic, the primary lesions may be absent in many cases and instead replaced by secondary excoriations and erosions. DH has a very classic distribution pattern, particularly involving the bilateral extensor surfaces, buttocks, and scalp. Although some cases of oral DH have been reported, mucosal involvement is generally considered to be very rare.
Despite its strong association with underlying celiac disease, most patients with DH do not report any associated gastrointestinal symptoms. Those with DH may present with any variety of other autoimmune conditions, with hypothyroidism being the most common. Interestingly, patients with DH have been shown to be at an increased development of non-Hodgkin lymphoma. It is not certain whether adherence to a strict gluten-free diet reduces this risk in this population.
Diagnosis can be made with a proper clinical history and examination, tissue pathology, direct immunofluorescence microscopy (DIF), and/or serology, with DIF being the most definitive. Perilesional skin is preferred for DIF, as lesional biopsies have been found to have higher rates of false negative results. The characteristic DIF finding diagnostic of DH is granular IgA deposits within dermal papillae, which was seen in this patient’s DIF.
Adequate treatment of DH can usually be accomplished with a combination of dapsone and a gluten-free diet. Initially, dapsone may be used for more immediate relief of associated pruritus and other bothersome symptoms. A strict gluten-free diet should be implemented as soon as possible, and dapsone can be tapered approximately 2-3 months after initiation as to avoid potential adverse effects with longterm treatment at higher doses.
The case and photo were submitted by Natasha Cowan, BS, University of California, San Diego, School of Medicine, and Nick Celano, MD, of San Diego Family Dermatology.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Dermatitis herpetiformis
The primary lesions of DH are vesicles and papules in a grouped or “herpetic” formation. However, as these lesions are extremely pruritic, the primary lesions may be absent in many cases and instead replaced by secondary excoriations and erosions. DH has a very classic distribution pattern, particularly involving the bilateral extensor surfaces, buttocks, and scalp. Although some cases of oral DH have been reported, mucosal involvement is generally considered to be very rare.
Despite its strong association with underlying celiac disease, most patients with DH do not report any associated gastrointestinal symptoms. Those with DH may present with any variety of other autoimmune conditions, with hypothyroidism being the most common. Interestingly, patients with DH have been shown to be at an increased development of non-Hodgkin lymphoma. It is not certain whether adherence to a strict gluten-free diet reduces this risk in this population.
Diagnosis can be made with a proper clinical history and examination, tissue pathology, direct immunofluorescence microscopy (DIF), and/or serology, with DIF being the most definitive. Perilesional skin is preferred for DIF, as lesional biopsies have been found to have higher rates of false negative results. The characteristic DIF finding diagnostic of DH is granular IgA deposits within dermal papillae, which was seen in this patient’s DIF.
Adequate treatment of DH can usually be accomplished with a combination of dapsone and a gluten-free diet. Initially, dapsone may be used for more immediate relief of associated pruritus and other bothersome symptoms. A strict gluten-free diet should be implemented as soon as possible, and dapsone can be tapered approximately 2-3 months after initiation as to avoid potential adverse effects with longterm treatment at higher doses.
The case and photo were submitted by Natasha Cowan, BS, University of California, San Diego, School of Medicine, and Nick Celano, MD, of San Diego Family Dermatology.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Make the Diagnosis - April 2017
Trichomycosis axillaris
Trichomycosis axillaris (TA) is a bacterial infection of the genus Corynebacterium that affects hairs in the axilla. Predisposing factors include poor local hygiene, hyperhidrosis, obesity, and warm, humid climates. Gram-positive diphtheroids, classically Corynebacterium tenuis, mix with sweat on hair shafts and subsequently produce a cementing material. In the majority of cases, bacterial infection causes keratin damage to the hair shaft with sparing of the cortex. However, recent cases have reported significant invasion of the hair cortex by bacterial structures.
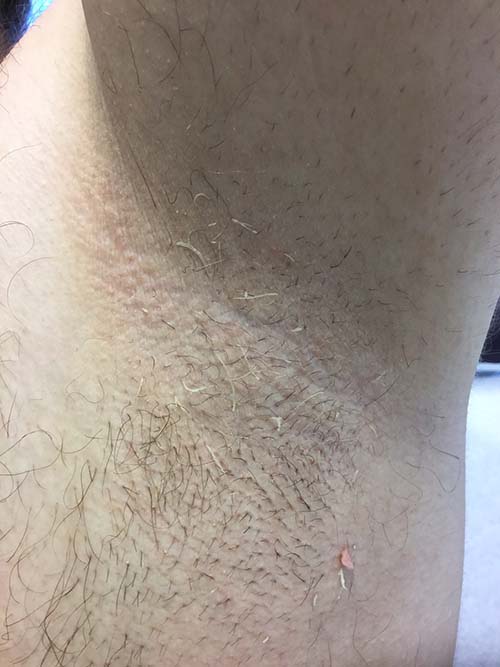
TA is often a clinical diagnosis. Histopathology reveals a discontinuous thick-layer coating of bacterial structures adhered to the hair shafts. Periodic acid–Schiff, gram, and Grocott silver stains are all positive. Examination of hairs with 20% hydrogen peroxide solution can provide a microscopic diagnosis. Wood lamp examination produces a weak, yellow fluorescence, and potassium hydroxide test demonstrates yellowish material of minimal translucency surrounding the hair cortex.
Complete resolution can be achieved by shaving off all the axillary hairs. It is imperative to maintain good hygiene and keep the area dry and clean. To prevent recurrence, topical benzoyl peroxide or topical antibiotics such as erythromycin and clindamycin can be applied. If axillary hyperhidrosis is concurrently present, aluminum chloride hexahydrate solution can decrease sweating and reduce the risk of bacterial regrowth. Injections of botulinum toxin can be used to treat the hyperhidrosis as well.
This patient responded well to topical erythromycin lotion.
This case and photo are courtesy of Jessica Cervantes, University of Miami Miller School of Medicine, and Dr. Bilu Martin.
Donna Bilu Martin, MD, is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/edermatologynews. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Trichomycosis axillaris
Trichomycosis axillaris (TA) is a bacterial infection of the genus Corynebacterium that affects hairs in the axilla. Predisposing factors include poor local hygiene, hyperhidrosis, obesity, and warm, humid climates. Gram-positive diphtheroids, classically Corynebacterium tenuis, mix with sweat on hair shafts and subsequently produce a cementing material. In the majority of cases, bacterial infection causes keratin damage to the hair shaft with sparing of the cortex. However, recent cases have reported significant invasion of the hair cortex by bacterial structures.
TA is often a clinical diagnosis. Histopathology reveals a discontinuous thick-layer coating of bacterial structures adhered to the hair shafts. Periodic acid–Schiff, gram, and Grocott silver stains are all positive. Examination of hairs with 20% hydrogen peroxide solution can provide a microscopic diagnosis. Wood lamp examination produces a weak, yellow fluorescence, and potassium hydroxide test demonstrates yellowish material of minimal translucency surrounding the hair cortex.
Complete resolution can be achieved by shaving off all the axillary hairs. It is imperative to maintain good hygiene and keep the area dry and clean. To prevent recurrence, topical benzoyl peroxide or topical antibiotics such as erythromycin and clindamycin can be applied. If axillary hyperhidrosis is concurrently present, aluminum chloride hexahydrate solution can decrease sweating and reduce the risk of bacterial regrowth. Injections of botulinum toxin can be used to treat the hyperhidrosis as well.
This patient responded well to topical erythromycin lotion.
This case and photo are courtesy of Jessica Cervantes, University of Miami Miller School of Medicine, and Dr. Bilu Martin.
Donna Bilu Martin, MD, is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/edermatologynews. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Trichomycosis axillaris
Trichomycosis axillaris (TA) is a bacterial infection of the genus Corynebacterium that affects hairs in the axilla. Predisposing factors include poor local hygiene, hyperhidrosis, obesity, and warm, humid climates. Gram-positive diphtheroids, classically Corynebacterium tenuis, mix with sweat on hair shafts and subsequently produce a cementing material. In the majority of cases, bacterial infection causes keratin damage to the hair shaft with sparing of the cortex. However, recent cases have reported significant invasion of the hair cortex by bacterial structures.
TA is often a clinical diagnosis. Histopathology reveals a discontinuous thick-layer coating of bacterial structures adhered to the hair shafts. Periodic acid–Schiff, gram, and Grocott silver stains are all positive. Examination of hairs with 20% hydrogen peroxide solution can provide a microscopic diagnosis. Wood lamp examination produces a weak, yellow fluorescence, and potassium hydroxide test demonstrates yellowish material of minimal translucency surrounding the hair cortex.
Complete resolution can be achieved by shaving off all the axillary hairs. It is imperative to maintain good hygiene and keep the area dry and clean. To prevent recurrence, topical benzoyl peroxide or topical antibiotics such as erythromycin and clindamycin can be applied. If axillary hyperhidrosis is concurrently present, aluminum chloride hexahydrate solution can decrease sweating and reduce the risk of bacterial regrowth. Injections of botulinum toxin can be used to treat the hyperhidrosis as well.
This patient responded well to topical erythromycin lotion.
This case and photo are courtesy of Jessica Cervantes, University of Miami Miller School of Medicine, and Dr. Bilu Martin.
Donna Bilu Martin, MD, is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/edermatologynews. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
A 35-year-old male with no significant past medical history presented with the chief complaint of occasional malodorous “crust in his underarms.” He used no prior treatments, and he did report axillary hyperhidrosis.
Make the Diagnosis - February 2017
Primary Cutaneous Cryptococcosis
The differential diagnosis upon initial examination may be wide. Patients generally present with a new skin lesion that proves to be refractory to conservative or traditional therapy. The upper extremities are the most common site of PCC. In almost all cases, a previous injury at the site of inoculation can be identified. Risk factors for exposure to Cryptococcus include residence in a rural area and contact with soil contaminated by avian droppings. The average time between skin injury and onset of symptoms has been reported as 2.5 days. Although rare, it is possible for a patient to present with multiple sites of infection in the absence of dissemination. This is almost exclusively seen in immunocompromised hosts.
Because of its widely variable clinical manifestations, PCC is a diagnosis made by culture and histology. Histopathology reveals numerous yeasts and a giant cell inflammatory process. In PCC, serology should not demonstrate Cryptococcus antigen, as the disease is localized to the skin. Patients diagnosed with PCC should undergo proper work-up to rule out the possibility of an underlying immunosuppressive condition, such as infection or malignancy.
Therapy for PCC can be solely medical or a combination of medical and surgical treatments. Surgical de-bulking in combination with 6-12 months of fluconazole is often used. Early treatment is essential, as cases of Cryptococcus dissemination secondary to PCC have been reported.
This patient's workup did not reveal any underlying immunodeficiencies. He worked with heating, ventilation and air condition (HVAC) in Spain. He was referred to infectious disease and admitted to the hospital for intravenous antifungal therapy.
Case and photo courtesy of: Natasha Cowan, BS, UCSD School of Medicine, and Brooke Resh Sateesh, MD, San Deigo Family Dermatology
Donna Bilu Martin, MD, is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Primary Cutaneous Cryptococcosis
The differential diagnosis upon initial examination may be wide. Patients generally present with a new skin lesion that proves to be refractory to conservative or traditional therapy. The upper extremities are the most common site of PCC. In almost all cases, a previous injury at the site of inoculation can be identified. Risk factors for exposure to Cryptococcus include residence in a rural area and contact with soil contaminated by avian droppings. The average time between skin injury and onset of symptoms has been reported as 2.5 days. Although rare, it is possible for a patient to present with multiple sites of infection in the absence of dissemination. This is almost exclusively seen in immunocompromised hosts.
Because of its widely variable clinical manifestations, PCC is a diagnosis made by culture and histology. Histopathology reveals numerous yeasts and a giant cell inflammatory process. In PCC, serology should not demonstrate Cryptococcus antigen, as the disease is localized to the skin. Patients diagnosed with PCC should undergo proper work-up to rule out the possibility of an underlying immunosuppressive condition, such as infection or malignancy.
Therapy for PCC can be solely medical or a combination of medical and surgical treatments. Surgical de-bulking in combination with 6-12 months of fluconazole is often used. Early treatment is essential, as cases of Cryptococcus dissemination secondary to PCC have been reported.
This patient's workup did not reveal any underlying immunodeficiencies. He worked with heating, ventilation and air condition (HVAC) in Spain. He was referred to infectious disease and admitted to the hospital for intravenous antifungal therapy.
Case and photo courtesy of: Natasha Cowan, BS, UCSD School of Medicine, and Brooke Resh Sateesh, MD, San Deigo Family Dermatology
Donna Bilu Martin, MD, is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Primary Cutaneous Cryptococcosis
The differential diagnosis upon initial examination may be wide. Patients generally present with a new skin lesion that proves to be refractory to conservative or traditional therapy. The upper extremities are the most common site of PCC. In almost all cases, a previous injury at the site of inoculation can be identified. Risk factors for exposure to Cryptococcus include residence in a rural area and contact with soil contaminated by avian droppings. The average time between skin injury and onset of symptoms has been reported as 2.5 days. Although rare, it is possible for a patient to present with multiple sites of infection in the absence of dissemination. This is almost exclusively seen in immunocompromised hosts.
Because of its widely variable clinical manifestations, PCC is a diagnosis made by culture and histology. Histopathology reveals numerous yeasts and a giant cell inflammatory process. In PCC, serology should not demonstrate Cryptococcus antigen, as the disease is localized to the skin. Patients diagnosed with PCC should undergo proper work-up to rule out the possibility of an underlying immunosuppressive condition, such as infection or malignancy.
Therapy for PCC can be solely medical or a combination of medical and surgical treatments. Surgical de-bulking in combination with 6-12 months of fluconazole is often used. Early treatment is essential, as cases of Cryptococcus dissemination secondary to PCC have been reported.
This patient's workup did not reveal any underlying immunodeficiencies. He worked with heating, ventilation and air condition (HVAC) in Spain. He was referred to infectious disease and admitted to the hospital for intravenous antifungal therapy.
Case and photo courtesy of: Natasha Cowan, BS, UCSD School of Medicine, and Brooke Resh Sateesh, MD, San Deigo Family Dermatology
Donna Bilu Martin, MD, is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
A 34-year-old healthy male with no significant past medical history presented with a rapidly enlarging lesion on the face for 2-3 weeks. He reported bleeding with mild contact/trauma. On physical examination, a 2.3cm pedunculated red-brown plaque with surrounding scattered hyperpigmented papules were present on the right cheek.
Make the Diagnosis - January 2017
Pemphigus vulgaris
Pemphigus vulgaris is the most common type of pemphigus. The average age of onset of 40-60 years. Clinically, patients may present with mucosal blisters and/or erosions. The most common site of mucosal lesions is the oral cavity, where the disease often manifests. Autoantibodies are produced against desmoglein 3 or both desmoglein 1 and desmoglein 3 in pemphigus vulgaris. Blistering is commonly induced with mechanical pressure at th edge of a blister or on normal skin, which is known as the Nikolsky sign. Pemphigus vulgaris has two uncommon clinical variants, pemphigus vegetans and pemphigus herpetiformis. Lack of prompt treatment of pemphigus vulgaris leads to epitope spreading and increased difficulty in management. Treatment with systemic glucocorticoids is the current standard of care to achieve control of the disease, and nonsteroidal immunomodulatory agents such as azathioprine, mycophenolate mofetil, and dapsone can be used in conjunction to help reduce adverse effects associated with long-term glucocorticoid therapy.
Pemphigus foliaceus results from autoantibodies against desmoglein 1. Patients usually present with small, scattered superficial cutaneous blisters that transition into scaly, crusted erosions. The scalp, neck, and trunk are the most common sites of presentation, with sparing of the mucous membranes. Like pemphigus vulgaris, the Nikolsky sign is frequently present in patients with pemphigus foliaceus. However, pemphigus foliaceus is readily distinguished from pemphigus vulgaris by its lack of mucous membrane involvement. The mainstays of treatment of pemphigus foliaceus are similar to those of pemphigus vulgaris, with systemic glucocorticoids and nonsteroidal adjuvant therapies playing a major role in controlling disease.
IgA pemphigus may occur at any age and is characterized by vesicles that progress into pustules commonly present on the trunk and proximal extremities. Erythematous plaques are frequently present alongside the vesicles and pustules. Like pemphigus foliaceus, IgA pemphigus usually spares the mucous membranes. The lesions may be pruritic but can also be asymptomatic. All other types of pemphigus are caused by IgG autoantibodies; however, IgA pemphigus results from IgA autoantibodies against target keratinocyte antigens. Several reports have shown dapsone to be a successful first-line adjuvant therapy for the treatment of IgA pemphigus.
Paraneoplastic pemphigus affects both genders and can occur at any age. It is most commonly the result of a malignancy. It is not clear which autoantibodies are actually responsible for the pathogenicity of most cases of paraneoplastic pemphigus. Clinically, it presents as a combination of severe erosive stomatitis, polymorphous cutaneous lesions, and possible pulmonary involvement. Severe, painful, and erosive mucositis is ubiquitous to the disease, and oral erosions are the most common initial presentation of paraneoplastic pemphigus, with a characteristic involvement of the tongue. Skin lesions commonly manifest after the onset of mucosal lesions. Cutaneous involvement is highly varied from patient to patient, with lesions resembling bullae, inflammatory violaceous papules, targetoid lesions, and desquamation. Management consists of treatment of the underlying neoplasm and control of the disease itself using a variety of agents including immunosuppressants and rituximab.
The patient’s biopsy came back consistent with pemphigus vulgaris. He was started on topical steroid gel for the mucosal lesions, topical steroid cream for the cutaneous lesions, and oral prednisone.
This case and photo were submitted by Natasha Cowan, University of California, San Diego, and Brooke Resh Sateesh, MD, San Diego Family Dermatology.
Donna Bilu Martin, MD, is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Pemphigus vulgaris
Pemphigus vulgaris is the most common type of pemphigus. The average age of onset of 40-60 years. Clinically, patients may present with mucosal blisters and/or erosions. The most common site of mucosal lesions is the oral cavity, where the disease often manifests. Autoantibodies are produced against desmoglein 3 or both desmoglein 1 and desmoglein 3 in pemphigus vulgaris. Blistering is commonly induced with mechanical pressure at th edge of a blister or on normal skin, which is known as the Nikolsky sign. Pemphigus vulgaris has two uncommon clinical variants, pemphigus vegetans and pemphigus herpetiformis. Lack of prompt treatment of pemphigus vulgaris leads to epitope spreading and increased difficulty in management. Treatment with systemic glucocorticoids is the current standard of care to achieve control of the disease, and nonsteroidal immunomodulatory agents such as azathioprine, mycophenolate mofetil, and dapsone can be used in conjunction to help reduce adverse effects associated with long-term glucocorticoid therapy.
Pemphigus foliaceus results from autoantibodies against desmoglein 1. Patients usually present with small, scattered superficial cutaneous blisters that transition into scaly, crusted erosions. The scalp, neck, and trunk are the most common sites of presentation, with sparing of the mucous membranes. Like pemphigus vulgaris, the Nikolsky sign is frequently present in patients with pemphigus foliaceus. However, pemphigus foliaceus is readily distinguished from pemphigus vulgaris by its lack of mucous membrane involvement. The mainstays of treatment of pemphigus foliaceus are similar to those of pemphigus vulgaris, with systemic glucocorticoids and nonsteroidal adjuvant therapies playing a major role in controlling disease.
IgA pemphigus may occur at any age and is characterized by vesicles that progress into pustules commonly present on the trunk and proximal extremities. Erythematous plaques are frequently present alongside the vesicles and pustules. Like pemphigus foliaceus, IgA pemphigus usually spares the mucous membranes. The lesions may be pruritic but can also be asymptomatic. All other types of pemphigus are caused by IgG autoantibodies; however, IgA pemphigus results from IgA autoantibodies against target keratinocyte antigens. Several reports have shown dapsone to be a successful first-line adjuvant therapy for the treatment of IgA pemphigus.
Paraneoplastic pemphigus affects both genders and can occur at any age. It is most commonly the result of a malignancy. It is not clear which autoantibodies are actually responsible for the pathogenicity of most cases of paraneoplastic pemphigus. Clinically, it presents as a combination of severe erosive stomatitis, polymorphous cutaneous lesions, and possible pulmonary involvement. Severe, painful, and erosive mucositis is ubiquitous to the disease, and oral erosions are the most common initial presentation of paraneoplastic pemphigus, with a characteristic involvement of the tongue. Skin lesions commonly manifest after the onset of mucosal lesions. Cutaneous involvement is highly varied from patient to patient, with lesions resembling bullae, inflammatory violaceous papules, targetoid lesions, and desquamation. Management consists of treatment of the underlying neoplasm and control of the disease itself using a variety of agents including immunosuppressants and rituximab.
The patient’s biopsy came back consistent with pemphigus vulgaris. He was started on topical steroid gel for the mucosal lesions, topical steroid cream for the cutaneous lesions, and oral prednisone.
This case and photo were submitted by Natasha Cowan, University of California, San Diego, and Brooke Resh Sateesh, MD, San Diego Family Dermatology.
Donna Bilu Martin, MD, is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Pemphigus vulgaris
Pemphigus vulgaris is the most common type of pemphigus. The average age of onset of 40-60 years. Clinically, patients may present with mucosal blisters and/or erosions. The most common site of mucosal lesions is the oral cavity, where the disease often manifests. Autoantibodies are produced against desmoglein 3 or both desmoglein 1 and desmoglein 3 in pemphigus vulgaris. Blistering is commonly induced with mechanical pressure at th edge of a blister or on normal skin, which is known as the Nikolsky sign. Pemphigus vulgaris has two uncommon clinical variants, pemphigus vegetans and pemphigus herpetiformis. Lack of prompt treatment of pemphigus vulgaris leads to epitope spreading and increased difficulty in management. Treatment with systemic glucocorticoids is the current standard of care to achieve control of the disease, and nonsteroidal immunomodulatory agents such as azathioprine, mycophenolate mofetil, and dapsone can be used in conjunction to help reduce adverse effects associated with long-term glucocorticoid therapy.
Pemphigus foliaceus results from autoantibodies against desmoglein 1. Patients usually present with small, scattered superficial cutaneous blisters that transition into scaly, crusted erosions. The scalp, neck, and trunk are the most common sites of presentation, with sparing of the mucous membranes. Like pemphigus vulgaris, the Nikolsky sign is frequently present in patients with pemphigus foliaceus. However, pemphigus foliaceus is readily distinguished from pemphigus vulgaris by its lack of mucous membrane involvement. The mainstays of treatment of pemphigus foliaceus are similar to those of pemphigus vulgaris, with systemic glucocorticoids and nonsteroidal adjuvant therapies playing a major role in controlling disease.
IgA pemphigus may occur at any age and is characterized by vesicles that progress into pustules commonly present on the trunk and proximal extremities. Erythematous plaques are frequently present alongside the vesicles and pustules. Like pemphigus foliaceus, IgA pemphigus usually spares the mucous membranes. The lesions may be pruritic but can also be asymptomatic. All other types of pemphigus are caused by IgG autoantibodies; however, IgA pemphigus results from IgA autoantibodies against target keratinocyte antigens. Several reports have shown dapsone to be a successful first-line adjuvant therapy for the treatment of IgA pemphigus.
Paraneoplastic pemphigus affects both genders and can occur at any age. It is most commonly the result of a malignancy. It is not clear which autoantibodies are actually responsible for the pathogenicity of most cases of paraneoplastic pemphigus. Clinically, it presents as a combination of severe erosive stomatitis, polymorphous cutaneous lesions, and possible pulmonary involvement. Severe, painful, and erosive mucositis is ubiquitous to the disease, and oral erosions are the most common initial presentation of paraneoplastic pemphigus, with a characteristic involvement of the tongue. Skin lesions commonly manifest after the onset of mucosal lesions. Cutaneous involvement is highly varied from patient to patient, with lesions resembling bullae, inflammatory violaceous papules, targetoid lesions, and desquamation. Management consists of treatment of the underlying neoplasm and control of the disease itself using a variety of agents including immunosuppressants and rituximab.
The patient’s biopsy came back consistent with pemphigus vulgaris. He was started on topical steroid gel for the mucosal lesions, topical steroid cream for the cutaneous lesions, and oral prednisone.
This case and photo were submitted by Natasha Cowan, University of California, San Diego, and Brooke Resh Sateesh, MD, San Diego Family Dermatology.
Donna Bilu Martin, MD, is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
A 50 year old Hispanic male presented with a two day history of blisters on his lips, extremities, and upper body. He complained of soreness on his lips. Bullae were flaccid and some had crusting.
Make the Diagnosis - December 2016
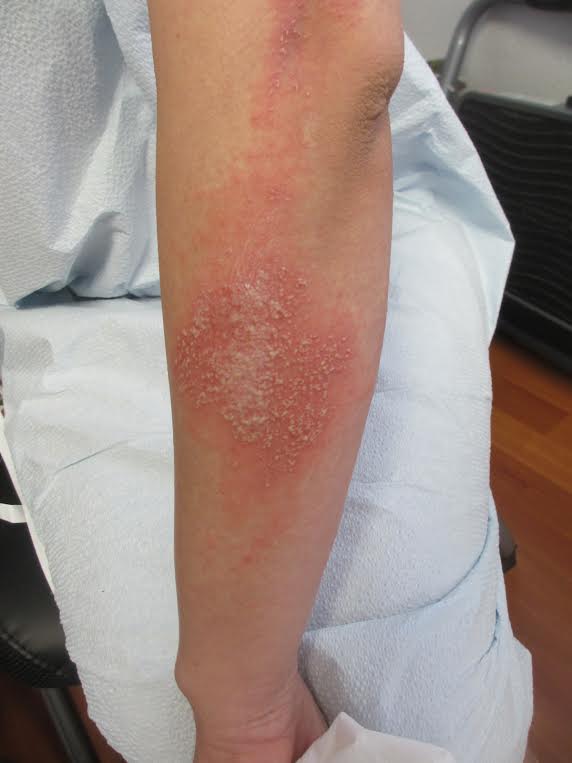
Acute generalized exanthematous pustulosis (AGEP)
Acute generalized exanthematous pustulosis (AGEP) is a fairly rare condition, with a reported incidence of one to five cases per million people per year. AGEP affects both sexes, but with a slight female predominance. Although the median age of occurrence is 56 years, people of all ages can develop AGEP. In approximately 90% of cases, AGEP is caused by a drug reaction, with the most common culprits being antibiotics, diltiazem, and antimalarials.
Fever, leukocytosis with an elevated neutrophil count ( greater than 7,000/microL), and mild eosinophilia are commonly observed during the acute phase of AGEP. Systemic involvement is fairly uncommon, with a study of 58 cases exhibiting organ involvement in 17% of patients. When systemic involvement was observed, hepatic, pulmonary, and renal dysfunction were most common. Hepatic dysfunction can result in elevated transaminase levels in some patients. Similarly, reduced creatinine clearance may be observed because of renal involvement.
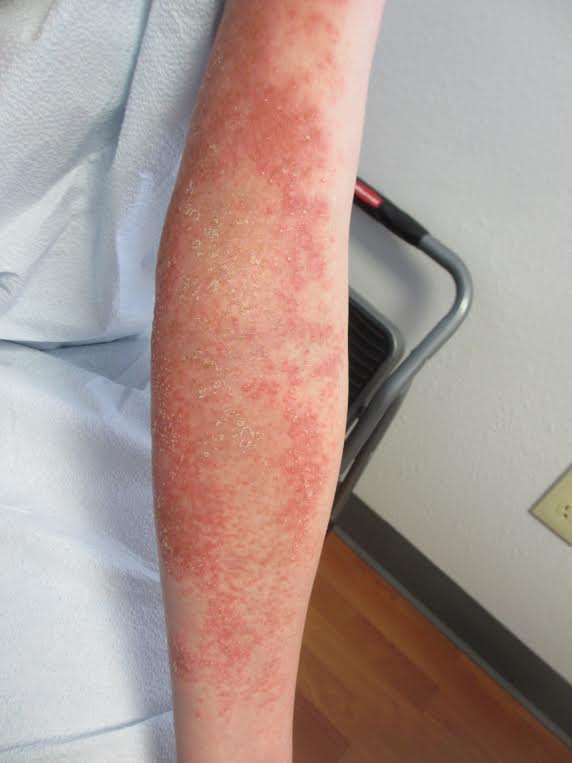
The management of AGEP involves removal of the offending agent. Antiseptic solutions and moist dressings may be used during the pustular phase to prevent infection. Emollients can aid in restoring skin barrier function during the desquamation phase. Antibiotics are not necessary prophylactically and should be avoided unless infection occurs. Pruritus and inflammation may be treated with topical corticosteroids in prolonged cases. Systemic corticosteroids are generally unwarranted as AGEP is a self-limited condition that is known to spontaneously resolve with the removal of the causative drug.
This case and photo were submitted by Natasha Cowan, BS, University of California, San Diego, and Brooke Resh Sateesh MD, San Diego Family Dermatology.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Acute generalized exanthematous pustulosis (AGEP)
Acute generalized exanthematous pustulosis (AGEP) is a fairly rare condition, with a reported incidence of one to five cases per million people per year. AGEP affects both sexes, but with a slight female predominance. Although the median age of occurrence is 56 years, people of all ages can develop AGEP. In approximately 90% of cases, AGEP is caused by a drug reaction, with the most common culprits being antibiotics, diltiazem, and antimalarials.
Fever, leukocytosis with an elevated neutrophil count ( greater than 7,000/microL), and mild eosinophilia are commonly observed during the acute phase of AGEP. Systemic involvement is fairly uncommon, with a study of 58 cases exhibiting organ involvement in 17% of patients. When systemic involvement was observed, hepatic, pulmonary, and renal dysfunction were most common. Hepatic dysfunction can result in elevated transaminase levels in some patients. Similarly, reduced creatinine clearance may be observed because of renal involvement.
The management of AGEP involves removal of the offending agent. Antiseptic solutions and moist dressings may be used during the pustular phase to prevent infection. Emollients can aid in restoring skin barrier function during the desquamation phase. Antibiotics are not necessary prophylactically and should be avoided unless infection occurs. Pruritus and inflammation may be treated with topical corticosteroids in prolonged cases. Systemic corticosteroids are generally unwarranted as AGEP is a self-limited condition that is known to spontaneously resolve with the removal of the causative drug.
This case and photo were submitted by Natasha Cowan, BS, University of California, San Diego, and Brooke Resh Sateesh MD, San Diego Family Dermatology.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
Acute generalized exanthematous pustulosis (AGEP)
Acute generalized exanthematous pustulosis (AGEP) is a fairly rare condition, with a reported incidence of one to five cases per million people per year. AGEP affects both sexes, but with a slight female predominance. Although the median age of occurrence is 56 years, people of all ages can develop AGEP. In approximately 90% of cases, AGEP is caused by a drug reaction, with the most common culprits being antibiotics, diltiazem, and antimalarials.
Fever, leukocytosis with an elevated neutrophil count ( greater than 7,000/microL), and mild eosinophilia are commonly observed during the acute phase of AGEP. Systemic involvement is fairly uncommon, with a study of 58 cases exhibiting organ involvement in 17% of patients. When systemic involvement was observed, hepatic, pulmonary, and renal dysfunction were most common. Hepatic dysfunction can result in elevated transaminase levels in some patients. Similarly, reduced creatinine clearance may be observed because of renal involvement.
The management of AGEP involves removal of the offending agent. Antiseptic solutions and moist dressings may be used during the pustular phase to prevent infection. Emollients can aid in restoring skin barrier function during the desquamation phase. Antibiotics are not necessary prophylactically and should be avoided unless infection occurs. Pruritus and inflammation may be treated with topical corticosteroids in prolonged cases. Systemic corticosteroids are generally unwarranted as AGEP is a self-limited condition that is known to spontaneously resolve with the removal of the causative drug.
This case and photo were submitted by Natasha Cowan, BS, University of California, San Diego, and Brooke Resh Sateesh MD, San Diego Family Dermatology.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@frontlinemedcom.com.
A 27 year old female presented with pustules on her upper extremities and bilateral antecubital fossa. She also had superficial desquamation with underlying erythema on her torso. Five days prior to presentation, she underwent rhinoplasty for a deviated nasal septum. She received cefazolin during the surgery. One day post surgery, she developed lesions on her arms that then spread to her chest and back. She was treated in the emergency room with prednisone, famotidine (Pepcid), and diphenhydramine (Benadryl). She returned to the emergency department 4 days later and was treated with vancomycin, piperacillin/tazobactam, and clindamycin for possible toxic shock syndrome. She was admitted to the hospital and dermatology was consulted. Her liver function tests and eosinophil count were normal.
Make the Diagnosis - November 2016
Cutaneous larva migrans
Cutaneous larva migrans (CLM) presents as a self-limited cutaneous eruption. It is also known as creeping eruption. Animal (dog and cat) hookworms such as Ancylostoma braziliense are responsible for most CLM infestations in the central and southeastern United States. The hookworms are often acquired while walking barefoot in contaminated sand. Carpenters, plumbers, and gardeners are also likely to be affected. Ova from the hookworms hatch into larvae that penetrate into skin.
On physical examination, lesions are erythematous, linear and serpiginous. Exposed sites, such as lower legs, hands, genitals, buttocks, and feet are most commonly involved. The migration begins about 4 days after onset. There is often intense pruritus and lesions advance 1-2 cm per day. Old lesions fade as the larvae migrate. They may have periods of rest, evidenced as papules. The differential diagnosis includes other parasites (uncinariasis, gnathostomiasis, strongyloidiasis, myiasis), dermatophytosis, phytophotodermatitis, erythema chronicum migrans, granuloma annulare, and bullous impetigo.
CLM is often a clinical diagnosis. Histopathology often reveals an inflammatory infiltrate or, if taken from an advancing lesion, part of the parasite. Serology may reveal an eosinophilia.
CLM is usually a limited infestation that typically resolves within 2-8 weeks, although longer timelines have been reported. Complications include cellulitis, abscess formation, and Loeffler syndrome (patchy lung infiltrate and eosinophilia). Antihistamines alone may be used to alleviate the accompanying pruritus, but curative treatment is often used. First-line curative therapies include oral albendazole and ivermectin but topical tiabendazole is also effective. Our patient responded well to oral ivermectin.
Case and photo courtesy of Mark Ash, MS; Brody School of Medicine,Greenville, North Carolina; and Donna Bilu Martin, MD; Premier Dermatology, MD, Aventura, Florida.
Cutaneous larva migrans
Cutaneous larva migrans (CLM) presents as a self-limited cutaneous eruption. It is also known as creeping eruption. Animal (dog and cat) hookworms such as Ancylostoma braziliense are responsible for most CLM infestations in the central and southeastern United States. The hookworms are often acquired while walking barefoot in contaminated sand. Carpenters, plumbers, and gardeners are also likely to be affected. Ova from the hookworms hatch into larvae that penetrate into skin.
On physical examination, lesions are erythematous, linear and serpiginous. Exposed sites, such as lower legs, hands, genitals, buttocks, and feet are most commonly involved. The migration begins about 4 days after onset. There is often intense pruritus and lesions advance 1-2 cm per day. Old lesions fade as the larvae migrate. They may have periods of rest, evidenced as papules. The differential diagnosis includes other parasites (uncinariasis, gnathostomiasis, strongyloidiasis, myiasis), dermatophytosis, phytophotodermatitis, erythema chronicum migrans, granuloma annulare, and bullous impetigo.
CLM is often a clinical diagnosis. Histopathology often reveals an inflammatory infiltrate or, if taken from an advancing lesion, part of the parasite. Serology may reveal an eosinophilia.
CLM is usually a limited infestation that typically resolves within 2-8 weeks, although longer timelines have been reported. Complications include cellulitis, abscess formation, and Loeffler syndrome (patchy lung infiltrate and eosinophilia). Antihistamines alone may be used to alleviate the accompanying pruritus, but curative treatment is often used. First-line curative therapies include oral albendazole and ivermectin but topical tiabendazole is also effective. Our patient responded well to oral ivermectin.
Case and photo courtesy of Mark Ash, MS; Brody School of Medicine,Greenville, North Carolina; and Donna Bilu Martin, MD; Premier Dermatology, MD, Aventura, Florida.
Cutaneous larva migrans
Cutaneous larva migrans (CLM) presents as a self-limited cutaneous eruption. It is also known as creeping eruption. Animal (dog and cat) hookworms such as Ancylostoma braziliense are responsible for most CLM infestations in the central and southeastern United States. The hookworms are often acquired while walking barefoot in contaminated sand. Carpenters, plumbers, and gardeners are also likely to be affected. Ova from the hookworms hatch into larvae that penetrate into skin.
On physical examination, lesions are erythematous, linear and serpiginous. Exposed sites, such as lower legs, hands, genitals, buttocks, and feet are most commonly involved. The migration begins about 4 days after onset. There is often intense pruritus and lesions advance 1-2 cm per day. Old lesions fade as the larvae migrate. They may have periods of rest, evidenced as papules. The differential diagnosis includes other parasites (uncinariasis, gnathostomiasis, strongyloidiasis, myiasis), dermatophytosis, phytophotodermatitis, erythema chronicum migrans, granuloma annulare, and bullous impetigo.
CLM is often a clinical diagnosis. Histopathology often reveals an inflammatory infiltrate or, if taken from an advancing lesion, part of the parasite. Serology may reveal an eosinophilia.
CLM is usually a limited infestation that typically resolves within 2-8 weeks, although longer timelines have been reported. Complications include cellulitis, abscess formation, and Loeffler syndrome (patchy lung infiltrate and eosinophilia). Antihistamines alone may be used to alleviate the accompanying pruritus, but curative treatment is often used. First-line curative therapies include oral albendazole and ivermectin but topical tiabendazole is also effective. Our patient responded well to oral ivermectin.
Case and photo courtesy of Mark Ash, MS; Brody School of Medicine,Greenville, North Carolina; and Donna Bilu Martin, MD; Premier Dermatology, MD, Aventura, Florida.
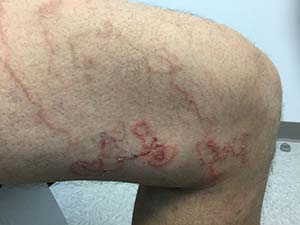
A 45 year old healthy male presented to the office with pruritic lesions on the leg for several weeks. He described a crawling sensation under the skin and states the lesions are slowly moving. On physical examination, erythematous, winding, linear tracts were present on his left thigh.
Make the Diagnosis - October 2016
Erythema multiforme
Erythema multiforme (EM) is a hypersensitivity reaction. Cutaneous lesions generally occur symmetrically on the extensor surfaces of the acral extremities and spread centripetally. Lesions are polymorphous and may be macular, papular, or bullous, with or without mucosal involvement. Classic targetoid lesions have a dusky central area, a paler edematous ring, and an erythematous outermost ring. EM can be triggered by multiple causes. 90% of cases are infectious, with herpes simplex virus infection most common and mycoplasma pneumoniae second. Medications (sulfonamides, anticonvulsants, etc), malignancy, autoimmune disease, immunizations, radiation, and sarcoidosis may also cause EM.
The differential diagnosis for EM includes urticaria, Stevens-Johnson syndrome, fixed drug eruption, bullous pemphigoid, paraneoplastic pemphigus, acute febrile neutophilic dermatosis (Sweet's syndrome), polymorphous light eruption (PMLE), and cutaneous small vessel vasculitis. The lesions of EM are fixed and all arise within 72 hours of onset, which can help distinguish between urticaria. The distribution of the lesions can help rule out Stevens-Johnson syndrome, which classically begins centrally and spreads distally and often will involve mucosal surfaces. Fixed drug eruption generally presents with fewer lesions and a preceding medication history. Bullous pemphigoid and paraneoplastic pemphigus are both chronic in their course and have distinguishing histopathologic characteristics that will not be present in erythema multiforme. Sweet's syndrome is also associated with infection; however, histology reveals a dense neutrophilic infiltrate with dermal edema on biopsy. History can be useful in distinguishing EM from PMLE: preceding infection strongly suggests EM, while ultraviolet radiation exposure is more likely to result in PMLE. Lastly, EM differs from cutaneous small vessel vasculitis through both histopathologic examination and direct immunofluorescence.
EM is usually a self-limited condition of about two weeks without significant sequelae, however, it may vary in both the length and severity of its course. A small subset of patients may experience episodic eruptions over a period of years, in what is known as recurrent EM. An even smaller subset of patients may continuously suffer from the lesions for months or even years, which is termed persistent EM. Treatment is aimed at the underlying condition or cause. Offending medications should be discontinued. Patients with mild cases of EM generally fare well with symptomatic treatment alone; however, more severe cases may require hospitalization for hydration, analgesia, and antiviral therapy. The use of systemic corticosteroids for EM is controversial and is generally reserved for severe cases with mucosal involvement. Prophylactic antiviral therapy is indicated for those with recurrent erythema multiforme of viral origin who suffer from six or more episodes per year. Very rarely, erythema multiforme may include ocular involvement leading to serious long-term sequelae, so patients with ocular symptoms should be immediately referred to an ophthalmologist for evaluation.
The patient's biopsy revealed necrotic keratinocytes and exocytosis of lymphocytes and was consistent with EM. She improved with oral antiviral therapy and topical steroid cream.
Erythema multiforme
Erythema multiforme (EM) is a hypersensitivity reaction. Cutaneous lesions generally occur symmetrically on the extensor surfaces of the acral extremities and spread centripetally. Lesions are polymorphous and may be macular, papular, or bullous, with or without mucosal involvement. Classic targetoid lesions have a dusky central area, a paler edematous ring, and an erythematous outermost ring. EM can be triggered by multiple causes. 90% of cases are infectious, with herpes simplex virus infection most common and mycoplasma pneumoniae second. Medications (sulfonamides, anticonvulsants, etc), malignancy, autoimmune disease, immunizations, radiation, and sarcoidosis may also cause EM.
The differential diagnosis for EM includes urticaria, Stevens-Johnson syndrome, fixed drug eruption, bullous pemphigoid, paraneoplastic pemphigus, acute febrile neutophilic dermatosis (Sweet's syndrome), polymorphous light eruption (PMLE), and cutaneous small vessel vasculitis. The lesions of EM are fixed and all arise within 72 hours of onset, which can help distinguish between urticaria. The distribution of the lesions can help rule out Stevens-Johnson syndrome, which classically begins centrally and spreads distally and often will involve mucosal surfaces. Fixed drug eruption generally presents with fewer lesions and a preceding medication history. Bullous pemphigoid and paraneoplastic pemphigus are both chronic in their course and have distinguishing histopathologic characteristics that will not be present in erythema multiforme. Sweet's syndrome is also associated with infection; however, histology reveals a dense neutrophilic infiltrate with dermal edema on biopsy. History can be useful in distinguishing EM from PMLE: preceding infection strongly suggests EM, while ultraviolet radiation exposure is more likely to result in PMLE. Lastly, EM differs from cutaneous small vessel vasculitis through both histopathologic examination and direct immunofluorescence.
EM is usually a self-limited condition of about two weeks without significant sequelae, however, it may vary in both the length and severity of its course. A small subset of patients may experience episodic eruptions over a period of years, in what is known as recurrent EM. An even smaller subset of patients may continuously suffer from the lesions for months or even years, which is termed persistent EM. Treatment is aimed at the underlying condition or cause. Offending medications should be discontinued. Patients with mild cases of EM generally fare well with symptomatic treatment alone; however, more severe cases may require hospitalization for hydration, analgesia, and antiviral therapy. The use of systemic corticosteroids for EM is controversial and is generally reserved for severe cases with mucosal involvement. Prophylactic antiviral therapy is indicated for those with recurrent erythema multiforme of viral origin who suffer from six or more episodes per year. Very rarely, erythema multiforme may include ocular involvement leading to serious long-term sequelae, so patients with ocular symptoms should be immediately referred to an ophthalmologist for evaluation.
The patient's biopsy revealed necrotic keratinocytes and exocytosis of lymphocytes and was consistent with EM. She improved with oral antiviral therapy and topical steroid cream.
Erythema multiforme
Erythema multiforme (EM) is a hypersensitivity reaction. Cutaneous lesions generally occur symmetrically on the extensor surfaces of the acral extremities and spread centripetally. Lesions are polymorphous and may be macular, papular, or bullous, with or without mucosal involvement. Classic targetoid lesions have a dusky central area, a paler edematous ring, and an erythematous outermost ring. EM can be triggered by multiple causes. 90% of cases are infectious, with herpes simplex virus infection most common and mycoplasma pneumoniae second. Medications (sulfonamides, anticonvulsants, etc), malignancy, autoimmune disease, immunizations, radiation, and sarcoidosis may also cause EM.
The differential diagnosis for EM includes urticaria, Stevens-Johnson syndrome, fixed drug eruption, bullous pemphigoid, paraneoplastic pemphigus, acute febrile neutophilic dermatosis (Sweet's syndrome), polymorphous light eruption (PMLE), and cutaneous small vessel vasculitis. The lesions of EM are fixed and all arise within 72 hours of onset, which can help distinguish between urticaria. The distribution of the lesions can help rule out Stevens-Johnson syndrome, which classically begins centrally and spreads distally and often will involve mucosal surfaces. Fixed drug eruption generally presents with fewer lesions and a preceding medication history. Bullous pemphigoid and paraneoplastic pemphigus are both chronic in their course and have distinguishing histopathologic characteristics that will not be present in erythema multiforme. Sweet's syndrome is also associated with infection; however, histology reveals a dense neutrophilic infiltrate with dermal edema on biopsy. History can be useful in distinguishing EM from PMLE: preceding infection strongly suggests EM, while ultraviolet radiation exposure is more likely to result in PMLE. Lastly, EM differs from cutaneous small vessel vasculitis through both histopathologic examination and direct immunofluorescence.
EM is usually a self-limited condition of about two weeks without significant sequelae, however, it may vary in both the length and severity of its course. A small subset of patients may experience episodic eruptions over a period of years, in what is known as recurrent EM. An even smaller subset of patients may continuously suffer from the lesions for months or even years, which is termed persistent EM. Treatment is aimed at the underlying condition or cause. Offending medications should be discontinued. Patients with mild cases of EM generally fare well with symptomatic treatment alone; however, more severe cases may require hospitalization for hydration, analgesia, and antiviral therapy. The use of systemic corticosteroids for EM is controversial and is generally reserved for severe cases with mucosal involvement. Prophylactic antiviral therapy is indicated for those with recurrent erythema multiforme of viral origin who suffer from six or more episodes per year. Very rarely, erythema multiforme may include ocular involvement leading to serious long-term sequelae, so patients with ocular symptoms should be immediately referred to an ophthalmologist for evaluation.
The patient's biopsy revealed necrotic keratinocytes and exocytosis of lymphocytes and was consistent with EM. She improved with oral antiviral therapy and topical steroid cream.
A healthy 30 year old female with a history of herpes simplex virus on the lips presented with three days of pruritic targetoid lesions on the arms and legs. She reports similar episodes in the past.