User login
Make the Diagnosis - July 2015
Diagnosis: Systemic sclerosis
Systemic sclerosis, or scleroderma, is a rare connective tissue disorder in which excessive collagen is deposited in the skin and internal organs. This disease predominantly affects women (3-6:1) between the ages of 20 and 60 years with no apparent racial predominance. Effective treatment is critical, as scleroderma carries a poor prognosis, with a mortality rate of up to 50% at 5 years in severe cases. The pathogenesis of systemic sclerosis is unknown, but three pathways are implicated, including immune deregulation, vascular abnormalities, and abnormal fibroblast activation.
Clinical presentation is variable because of the involvement of multiple organ systems. Common features include cutaneous pruritus, skin thickening, Raynaud's phenomenon, difficulty swallowing, shortness of breath, palpitations, nonproductive cough, and joint pain and swelling, as well as muscle pain and weakness. Laboratory findings may include elevated erythrocyte sedimentation rate, thrombocytopenia, hypergammaglobulinemia, increased urea and creatinine levels, and elevated C-reactive protein. Antinuclear antibodies are usually elevated, especially Scl-70, antimitochondrial, and anticentromere antibodies. Cardiac and pulmonary function should be assessed upon diagnosis. A Doppler echocardiogram may detect cardiac abnormalities, and chest x-ray or high-resolution CT is used to assess for pulmonary fibrosis.
Despite the severity of the disease, there are no Food and Drug Administration-approved disease-modifying agents for the treatment of scleroderma, and management often focuses on symptom relief. For example, patients with kidney involvement should be placed on an ACE inhibitor or angiotensin II inhibitor therapy, and patients with gastrointestinal tract involvement should use proton pump inhibitors and H2 blockers to control reflux. Bosentan and pentoxifylline, which target vascular abnormalities, also may help improve skin fibrosis. Steroids show benefits in the early stages of the disease, but carry a risk of scleroderma renal crisis with doses greater than 15 mg of prednisone daily. Mycophenolate mofetil and sirolimus have immunomodulatory and antifibrotic properties, which may be of benefit in this disease.
Cyclophosphamide is reserved for more severe cases. Other treatment modalities include rituximab, intravenous immunoglobulin, and autologous stem cell transplantation.
Diagnosis: Systemic sclerosis
Systemic sclerosis, or scleroderma, is a rare connective tissue disorder in which excessive collagen is deposited in the skin and internal organs. This disease predominantly affects women (3-6:1) between the ages of 20 and 60 years with no apparent racial predominance. Effective treatment is critical, as scleroderma carries a poor prognosis, with a mortality rate of up to 50% at 5 years in severe cases. The pathogenesis of systemic sclerosis is unknown, but three pathways are implicated, including immune deregulation, vascular abnormalities, and abnormal fibroblast activation.
Clinical presentation is variable because of the involvement of multiple organ systems. Common features include cutaneous pruritus, skin thickening, Raynaud's phenomenon, difficulty swallowing, shortness of breath, palpitations, nonproductive cough, and joint pain and swelling, as well as muscle pain and weakness. Laboratory findings may include elevated erythrocyte sedimentation rate, thrombocytopenia, hypergammaglobulinemia, increased urea and creatinine levels, and elevated C-reactive protein. Antinuclear antibodies are usually elevated, especially Scl-70, antimitochondrial, and anticentromere antibodies. Cardiac and pulmonary function should be assessed upon diagnosis. A Doppler echocardiogram may detect cardiac abnormalities, and chest x-ray or high-resolution CT is used to assess for pulmonary fibrosis.
Despite the severity of the disease, there are no Food and Drug Administration-approved disease-modifying agents for the treatment of scleroderma, and management often focuses on symptom relief. For example, patients with kidney involvement should be placed on an ACE inhibitor or angiotensin II inhibitor therapy, and patients with gastrointestinal tract involvement should use proton pump inhibitors and H2 blockers to control reflux. Bosentan and pentoxifylline, which target vascular abnormalities, also may help improve skin fibrosis. Steroids show benefits in the early stages of the disease, but carry a risk of scleroderma renal crisis with doses greater than 15 mg of prednisone daily. Mycophenolate mofetil and sirolimus have immunomodulatory and antifibrotic properties, which may be of benefit in this disease.
Cyclophosphamide is reserved for more severe cases. Other treatment modalities include rituximab, intravenous immunoglobulin, and autologous stem cell transplantation.
Diagnosis: Systemic sclerosis
Systemic sclerosis, or scleroderma, is a rare connective tissue disorder in which excessive collagen is deposited in the skin and internal organs. This disease predominantly affects women (3-6:1) between the ages of 20 and 60 years with no apparent racial predominance. Effective treatment is critical, as scleroderma carries a poor prognosis, with a mortality rate of up to 50% at 5 years in severe cases. The pathogenesis of systemic sclerosis is unknown, but three pathways are implicated, including immune deregulation, vascular abnormalities, and abnormal fibroblast activation.
Clinical presentation is variable because of the involvement of multiple organ systems. Common features include cutaneous pruritus, skin thickening, Raynaud's phenomenon, difficulty swallowing, shortness of breath, palpitations, nonproductive cough, and joint pain and swelling, as well as muscle pain and weakness. Laboratory findings may include elevated erythrocyte sedimentation rate, thrombocytopenia, hypergammaglobulinemia, increased urea and creatinine levels, and elevated C-reactive protein. Antinuclear antibodies are usually elevated, especially Scl-70, antimitochondrial, and anticentromere antibodies. Cardiac and pulmonary function should be assessed upon diagnosis. A Doppler echocardiogram may detect cardiac abnormalities, and chest x-ray or high-resolution CT is used to assess for pulmonary fibrosis.
Despite the severity of the disease, there are no Food and Drug Administration-approved disease-modifying agents for the treatment of scleroderma, and management often focuses on symptom relief. For example, patients with kidney involvement should be placed on an ACE inhibitor or angiotensin II inhibitor therapy, and patients with gastrointestinal tract involvement should use proton pump inhibitors and H2 blockers to control reflux. Bosentan and pentoxifylline, which target vascular abnormalities, also may help improve skin fibrosis. Steroids show benefits in the early stages of the disease, but carry a risk of scleroderma renal crisis with doses greater than 15 mg of prednisone daily. Mycophenolate mofetil and sirolimus have immunomodulatory and antifibrotic properties, which may be of benefit in this disease.
Cyclophosphamide is reserved for more severe cases. Other treatment modalities include rituximab, intravenous immunoglobulin, and autologous stem cell transplantation.

This case and photo were submitted by Charlotte E. LaSenna and Dr. Andrea Maderal of the University of Miami department of dermatology. Dr. Bilu Martin is in private practice at Premier Dermatology, MD in Aventura, Fla. To submit your case for possible publication, send an e-mail to dermnews@frontlinemedcom.com. A 55-year-old woman with an 8-year history of previously diagnosed vitiligo presented with worsening pruritus and swelling of the hands and feet for several months. Her medical history included liver disease. Upon physical examination, she was ill-appearing, with notable salt-and-pepper diffuse depigmentation, as well as pitting edema of the bilateral hands and face. Laboratory studies showed a positive ANA >1:2,560 with a centromere pattern, negative Scl-70, and positive antimitochondrial antibody at 158.5. Renal function and urinalysis were normal. Liver function tests were abnormal with elevated alkaline phosphatase and bilirubin.

Make the Diagnosis - June 2015
Diagnosis: Kaposi’s sarcoma
Kaposi's sarcoma is a vascular neoplasm with four principal clinical variants: HIV/AIDS-related Kaposi's sarcoma, classic Kaposi's sarcoma, African endemic Kaposi's sarcoma, and immunosuppression-associated Kaposi's sarcoma. The etiologic agent in all clinical variants is human herpes virus type 8 (HHV-8). HIV/AIDS-related Kaposi's sarcoma is primarily seen in men who have sex with men.
The four variants of Kaposi's sarcoma can have different clinical presentations. In HIV/AIDS-associated Kaposi's sarcoma, patients may present with a single lesion or with symmetric widespread lesions. Clinically, the lesions can range from faint erythematous macules, to small violaceous papules, to large plaques or ulcerated nodules. The lesions are generally asymptomatic.
Any mucocutaneous surface can be involved. Common body locations include the face (especially the nose), hard palate, trunk, penis, lower legs, and soles. The most common areas of internal involvement are the gastrointestinal system and lymphatics. Histologically, atypical, angular vessels with an associated inflammatory infiltrate containing plasma cells appear in the upper dermis in macular lesions. Nodules and tumors reveal a spindle cell neoplasm pattern. Lesions stain positive for human herpes virus 8 (HHV-8).
Since the introduction of highly active antiretroviral therapy (HAART), the incidence of Kaposi's sarcoma has greatly decreased. HAART is the most effective treatment method, and should be the initial therapy in most patients with mild to moderate disease.
However, some patients with HIV/AIDS-associated Kaposi's sarcoma require further treatment - those who have well-controlled HIV and undetectable viral loads. Other treatments include local destruction (cryotherapy), topical alitretinoin (9-cis-retinoic acid), intralesional interferon or vinblastine, superficial radiotherapy, liposomal doxorubicin, daunorubicin, or paclitaxel.
Diagnosis: Kaposi’s sarcoma
Kaposi's sarcoma is a vascular neoplasm with four principal clinical variants: HIV/AIDS-related Kaposi's sarcoma, classic Kaposi's sarcoma, African endemic Kaposi's sarcoma, and immunosuppression-associated Kaposi's sarcoma. The etiologic agent in all clinical variants is human herpes virus type 8 (HHV-8). HIV/AIDS-related Kaposi's sarcoma is primarily seen in men who have sex with men.
The four variants of Kaposi's sarcoma can have different clinical presentations. In HIV/AIDS-associated Kaposi's sarcoma, patients may present with a single lesion or with symmetric widespread lesions. Clinically, the lesions can range from faint erythematous macules, to small violaceous papules, to large plaques or ulcerated nodules. The lesions are generally asymptomatic.
Any mucocutaneous surface can be involved. Common body locations include the face (especially the nose), hard palate, trunk, penis, lower legs, and soles. The most common areas of internal involvement are the gastrointestinal system and lymphatics. Histologically, atypical, angular vessels with an associated inflammatory infiltrate containing plasma cells appear in the upper dermis in macular lesions. Nodules and tumors reveal a spindle cell neoplasm pattern. Lesions stain positive for human herpes virus 8 (HHV-8).
Since the introduction of highly active antiretroviral therapy (HAART), the incidence of Kaposi's sarcoma has greatly decreased. HAART is the most effective treatment method, and should be the initial therapy in most patients with mild to moderate disease.
However, some patients with HIV/AIDS-associated Kaposi's sarcoma require further treatment - those who have well-controlled HIV and undetectable viral loads. Other treatments include local destruction (cryotherapy), topical alitretinoin (9-cis-retinoic acid), intralesional interferon or vinblastine, superficial radiotherapy, liposomal doxorubicin, daunorubicin, or paclitaxel.
Diagnosis: Kaposi’s sarcoma
Kaposi's sarcoma is a vascular neoplasm with four principal clinical variants: HIV/AIDS-related Kaposi's sarcoma, classic Kaposi's sarcoma, African endemic Kaposi's sarcoma, and immunosuppression-associated Kaposi's sarcoma. The etiologic agent in all clinical variants is human herpes virus type 8 (HHV-8). HIV/AIDS-related Kaposi's sarcoma is primarily seen in men who have sex with men.
The four variants of Kaposi's sarcoma can have different clinical presentations. In HIV/AIDS-associated Kaposi's sarcoma, patients may present with a single lesion or with symmetric widespread lesions. Clinically, the lesions can range from faint erythematous macules, to small violaceous papules, to large plaques or ulcerated nodules. The lesions are generally asymptomatic.
Any mucocutaneous surface can be involved. Common body locations include the face (especially the nose), hard palate, trunk, penis, lower legs, and soles. The most common areas of internal involvement are the gastrointestinal system and lymphatics. Histologically, atypical, angular vessels with an associated inflammatory infiltrate containing plasma cells appear in the upper dermis in macular lesions. Nodules and tumors reveal a spindle cell neoplasm pattern. Lesions stain positive for human herpes virus 8 (HHV-8).
Since the introduction of highly active antiretroviral therapy (HAART), the incidence of Kaposi's sarcoma has greatly decreased. HAART is the most effective treatment method, and should be the initial therapy in most patients with mild to moderate disease.
However, some patients with HIV/AIDS-associated Kaposi's sarcoma require further treatment - those who have well-controlled HIV and undetectable viral loads. Other treatments include local destruction (cryotherapy), topical alitretinoin (9-cis-retinoic acid), intralesional interferon or vinblastine, superficial radiotherapy, liposomal doxorubicin, daunorubicin, or paclitaxel.
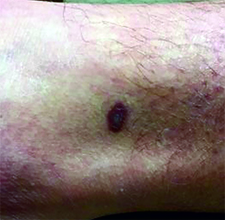
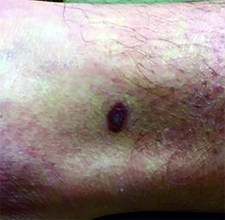
This case and photo were submitted by Dr. Ann Mazor Reed, Larkin Community Hospital, South Miami; and Dr. Donna Bilu Martin, Premier Dermatology, MD. Dr. Bilu Martin is in private practice at Premier Dermatology, MD in Aventura, Fla. To submit your case for possible publication, send an e-mail to dermnews@frontlinemedcom.com. A 39-year-old white male presented with a 2-month history involving asymptomatic violaceous plaques on his leg and posterior neck. He had no significant past medical history. He had no oral or mucosal involvement, no lymphadenopathy, and denied any systemic symptoms.
Make the Diagnosis - May 2015
Diagnosis: Granuloma annulare
Granuloma annulare (GA) is a self-limited cutaneous disorder predominantly seen in women that affects children and adults. The cause is unknown. Inciting factors can include herpes zoster infection, sun exposure, medications, and trauma.
Several clinical variants exist. Localized GA is the most common form, often presenting in the first three decades of life as an asymptomatic, erythematous, annular plaque with a firm border and central clearing localized to the wrists, ankles, and dorsal hands or feet. Generalized GA accounts for 15% of reported cases and presents in the fourth to seventh decades of life as multiple asymptomatic or pruritic skin-colored or erythematous papules and plaques on the trunk and extremities. Subcutaneous GA is more common in children and presents as multiple painless nodules on the scalp or extremities. Patch GA can be localized or generalized. Perforating GA presents as asymptomatic erythematous papules that evolve into yellow, umbilicated papules with a clear-to-white discharge.
Histopathologically, an interstitial or palisading pattern is seen with a dermal lymphohistiocytic infiltrate, degenerated collagen, and mucin deposition (visualized with alcian blue or colloidal iron stains). The interstitial pattern presents in the majority of cases.
Diagnosis of GA is predominantly clinically based. When the diagnosis is questionable or the presentation is atypical, biopsy is useful. Granuloma annulare is often self-limiting and resolves within 2 years, although recurrence is possible. First-line therapy for localized GA includes high-potency topical corticosteroids or intralesional corticosteroids. Other treatments include cryotherapy, phototherapy, and topical tacrolimus. For generalized GA, topical or intralesional corticosteroids may be used for select lesions. Topical calcineurin inhibitors, light therapy, hydroxychloroquine, isotretinion, and dapsone also have been reported as treatments in the literature.
Diagnosis: Granuloma annulare
Granuloma annulare (GA) is a self-limited cutaneous disorder predominantly seen in women that affects children and adults. The cause is unknown. Inciting factors can include herpes zoster infection, sun exposure, medications, and trauma.
Several clinical variants exist. Localized GA is the most common form, often presenting in the first three decades of life as an asymptomatic, erythematous, annular plaque with a firm border and central clearing localized to the wrists, ankles, and dorsal hands or feet. Generalized GA accounts for 15% of reported cases and presents in the fourth to seventh decades of life as multiple asymptomatic or pruritic skin-colored or erythematous papules and plaques on the trunk and extremities. Subcutaneous GA is more common in children and presents as multiple painless nodules on the scalp or extremities. Patch GA can be localized or generalized. Perforating GA presents as asymptomatic erythematous papules that evolve into yellow, umbilicated papules with a clear-to-white discharge.
Histopathologically, an interstitial or palisading pattern is seen with a dermal lymphohistiocytic infiltrate, degenerated collagen, and mucin deposition (visualized with alcian blue or colloidal iron stains). The interstitial pattern presents in the majority of cases.
Diagnosis of GA is predominantly clinically based. When the diagnosis is questionable or the presentation is atypical, biopsy is useful. Granuloma annulare is often self-limiting and resolves within 2 years, although recurrence is possible. First-line therapy for localized GA includes high-potency topical corticosteroids or intralesional corticosteroids. Other treatments include cryotherapy, phototherapy, and topical tacrolimus. For generalized GA, topical or intralesional corticosteroids may be used for select lesions. Topical calcineurin inhibitors, light therapy, hydroxychloroquine, isotretinion, and dapsone also have been reported as treatments in the literature.
Diagnosis: Granuloma annulare
Granuloma annulare (GA) is a self-limited cutaneous disorder predominantly seen in women that affects children and adults. The cause is unknown. Inciting factors can include herpes zoster infection, sun exposure, medications, and trauma.
Several clinical variants exist. Localized GA is the most common form, often presenting in the first three decades of life as an asymptomatic, erythematous, annular plaque with a firm border and central clearing localized to the wrists, ankles, and dorsal hands or feet. Generalized GA accounts for 15% of reported cases and presents in the fourth to seventh decades of life as multiple asymptomatic or pruritic skin-colored or erythematous papules and plaques on the trunk and extremities. Subcutaneous GA is more common in children and presents as multiple painless nodules on the scalp or extremities. Patch GA can be localized or generalized. Perforating GA presents as asymptomatic erythematous papules that evolve into yellow, umbilicated papules with a clear-to-white discharge.
Histopathologically, an interstitial or palisading pattern is seen with a dermal lymphohistiocytic infiltrate, degenerated collagen, and mucin deposition (visualized with alcian blue or colloidal iron stains). The interstitial pattern presents in the majority of cases.
Diagnosis of GA is predominantly clinically based. When the diagnosis is questionable or the presentation is atypical, biopsy is useful. Granuloma annulare is often self-limiting and resolves within 2 years, although recurrence is possible. First-line therapy for localized GA includes high-potency topical corticosteroids or intralesional corticosteroids. Other treatments include cryotherapy, phototherapy, and topical tacrolimus. For generalized GA, topical or intralesional corticosteroids may be used for select lesions. Topical calcineurin inhibitors, light therapy, hydroxychloroquine, isotretinion, and dapsone also have been reported as treatments in the literature.
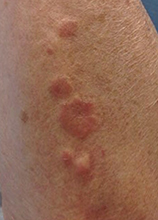
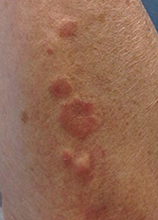
This case and photo were submitted by Orli Stern of Ross University and Dr. Donna Bilu Martin of Premier Dermatology, MD. A 60-year-old female with no significant past medical history presented with a 3-month history of asymptomatic, erythematous, firm, annular plaques on her bilateral proximal upper extremities and dorsal hands. The lesions have not been treated in the past.
Make the Diagnosis - April 2015
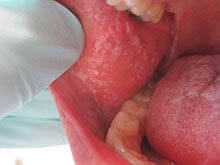
Diagnosis: Lichen Planus
Lichen planus is a common inflammatory condition involving the skin, nails, mucous membranes, and hair follicles. It has no racial predilection, and often affects men and women aged 20-60 years. It is less common in children, who account for only 4% of cases. The lesions are often atypical.
Clinically, patients often present with erythematous to violaceous small, flat-topped, polygonal papules that may coalesce into plaques. Lesions are generally pruritic, and may be tender or painful. Older lesions may be hyperpigmented. White streaks known as Wickham striae can cross the surface of lesions. These striae also can be present orally, such as in the patient described here. Oral lesions also may be atrophic or erosive.
Common body locations with involvement are the inner wrists, legs, torso, or genitals (glans penis). The face is rarely involved. Nail changes, such as longitudinal ridging and splitting, onycholysis, red lunula, yellow nail syndrome, and pterygium formation, can occur.
Lichen planus often spontaneously resolves on its own, with 2/3 of patients resolving in a year. Mucous membrane disease tends to be more chronic. The etiology of lichen planus is unknown. It may have an autoimmune mechanism in which T cells induce keratinocytes to undergo apoptosis. Between 4% and 60% of lichen planus patients also have hepatitis C infections. The differential diagnosis for cutaneous lesions include lichenoid drug eruption, guttate psoriasis, syphilis, and pityriasis lichenoides et varioliformis acuta. Oral lesions may resemble candidiasis, leukoplakia, malignancies, and bullous disease.
Topical and intralesional steroids are often effective for localized disease. Systemic corticosteroids can be useful when lesions are widespread. Phototherapy, isotretinoin, acitretin, hydroxychloroquine, and oral immunosuppressive agents (such as cyclosporine and mycophenolate mofetil) all have been described in the treatment of lichen planus.
Diagnosis: Lichen Planus
Lichen planus is a common inflammatory condition involving the skin, nails, mucous membranes, and hair follicles. It has no racial predilection, and often affects men and women aged 20-60 years. It is less common in children, who account for only 4% of cases. The lesions are often atypical.
Clinically, patients often present with erythematous to violaceous small, flat-topped, polygonal papules that may coalesce into plaques. Lesions are generally pruritic, and may be tender or painful. Older lesions may be hyperpigmented. White streaks known as Wickham striae can cross the surface of lesions. These striae also can be present orally, such as in the patient described here. Oral lesions also may be atrophic or erosive.
Common body locations with involvement are the inner wrists, legs, torso, or genitals (glans penis). The face is rarely involved. Nail changes, such as longitudinal ridging and splitting, onycholysis, red lunula, yellow nail syndrome, and pterygium formation, can occur.
Lichen planus often spontaneously resolves on its own, with 2/3 of patients resolving in a year. Mucous membrane disease tends to be more chronic. The etiology of lichen planus is unknown. It may have an autoimmune mechanism in which T cells induce keratinocytes to undergo apoptosis. Between 4% and 60% of lichen planus patients also have hepatitis C infections. The differential diagnosis for cutaneous lesions include lichenoid drug eruption, guttate psoriasis, syphilis, and pityriasis lichenoides et varioliformis acuta. Oral lesions may resemble candidiasis, leukoplakia, malignancies, and bullous disease.
Topical and intralesional steroids are often effective for localized disease. Systemic corticosteroids can be useful when lesions are widespread. Phototherapy, isotretinoin, acitretin, hydroxychloroquine, and oral immunosuppressive agents (such as cyclosporine and mycophenolate mofetil) all have been described in the treatment of lichen planus.
Diagnosis: Lichen Planus
Lichen planus is a common inflammatory condition involving the skin, nails, mucous membranes, and hair follicles. It has no racial predilection, and often affects men and women aged 20-60 years. It is less common in children, who account for only 4% of cases. The lesions are often atypical.
Clinically, patients often present with erythematous to violaceous small, flat-topped, polygonal papules that may coalesce into plaques. Lesions are generally pruritic, and may be tender or painful. Older lesions may be hyperpigmented. White streaks known as Wickham striae can cross the surface of lesions. These striae also can be present orally, such as in the patient described here. Oral lesions also may be atrophic or erosive.
Common body locations with involvement are the inner wrists, legs, torso, or genitals (glans penis). The face is rarely involved. Nail changes, such as longitudinal ridging and splitting, onycholysis, red lunula, yellow nail syndrome, and pterygium formation, can occur.
Lichen planus often spontaneously resolves on its own, with 2/3 of patients resolving in a year. Mucous membrane disease tends to be more chronic. The etiology of lichen planus is unknown. It may have an autoimmune mechanism in which T cells induce keratinocytes to undergo apoptosis. Between 4% and 60% of lichen planus patients also have hepatitis C infections. The differential diagnosis for cutaneous lesions include lichenoid drug eruption, guttate psoriasis, syphilis, and pityriasis lichenoides et varioliformis acuta. Oral lesions may resemble candidiasis, leukoplakia, malignancies, and bullous disease.
Topical and intralesional steroids are often effective for localized disease. Systemic corticosteroids can be useful when lesions are widespread. Phototherapy, isotretinoin, acitretin, hydroxychloroquine, and oral immunosuppressive agents (such as cyclosporine and mycophenolate mofetil) all have been described in the treatment of lichen planus.
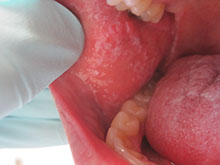
This case and photo were submitted by Dr. Damon McClain, a dermatologist in Camp Lejeune, N.C. A 34-year-old male presented with a 1-month history of an itchy rash on his penis and feet. Upon physical examination, these lesions were seen orally. Blood work, including hepatitis serologies, was negative. His skin lesions improved with topical steroids.
Make the Diagnosis - March 2015
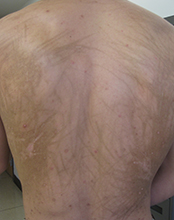
Diagnosis: Bleomycin-induced flagellate hyperpigmentation
Bleomycin is an antineoplastic agent. Most reported side effects involve the lung and skin since these organs have lower concentrations of the enzyme that detoxifies bleomycin. Other dermatologic side effects of bleomycin include Raynaud's phenomenon, hyperkeratosis, nailbed changes, and peeling of the skin on the palmar and plantar surfaces.
Flagellate erythema has been described in relation to bleomycin treatment along with other reported associations with peplomycin (a bleomycin derivative), docetaxel, dermatomyositis, adult-onset Still's disease, and shiitake mushroom dermatitis. The rash may appear following administration of bleomycin by any route and has been shown to be dose independent. Onset occurs anytime from 1 day to several months after exposure to the offending agent. Patients typically present with history of itching followed by the appearance of red linear streaks, which are most commonly found on the trunk. Over time, the erythema will resolve to postinflammatory hyperpigmentation.
The exact cause is unknown, but it is thought that scratching causes vasodilation with bleomycin accumulating in the skin. Diagnosis is made by physical examination of the characteristic appearance of the rash along with the history of chemotherapy. A skin biopsy may be performed. In most cases, the rash resolves spontaneously within 6-8 months. Severe rashes may warrant discontinuation of bleomycin. Using antihistamines and topical and oral corticosteroids in conjunction with bleomycin may reduce the incidence of flagellate erythema/hyperpigmentation.
Diagnosis: Bleomycin-induced flagellate hyperpigmentation
Bleomycin is an antineoplastic agent. Most reported side effects involve the lung and skin since these organs have lower concentrations of the enzyme that detoxifies bleomycin. Other dermatologic side effects of bleomycin include Raynaud's phenomenon, hyperkeratosis, nailbed changes, and peeling of the skin on the palmar and plantar surfaces.
Flagellate erythema has been described in relation to bleomycin treatment along with other reported associations with peplomycin (a bleomycin derivative), docetaxel, dermatomyositis, adult-onset Still's disease, and shiitake mushroom dermatitis. The rash may appear following administration of bleomycin by any route and has been shown to be dose independent. Onset occurs anytime from 1 day to several months after exposure to the offending agent. Patients typically present with history of itching followed by the appearance of red linear streaks, which are most commonly found on the trunk. Over time, the erythema will resolve to postinflammatory hyperpigmentation.
The exact cause is unknown, but it is thought that scratching causes vasodilation with bleomycin accumulating in the skin. Diagnosis is made by physical examination of the characteristic appearance of the rash along with the history of chemotherapy. A skin biopsy may be performed. In most cases, the rash resolves spontaneously within 6-8 months. Severe rashes may warrant discontinuation of bleomycin. Using antihistamines and topical and oral corticosteroids in conjunction with bleomycin may reduce the incidence of flagellate erythema/hyperpigmentation.
Diagnosis: Bleomycin-induced flagellate hyperpigmentation
Bleomycin is an antineoplastic agent. Most reported side effects involve the lung and skin since these organs have lower concentrations of the enzyme that detoxifies bleomycin. Other dermatologic side effects of bleomycin include Raynaud's phenomenon, hyperkeratosis, nailbed changes, and peeling of the skin on the palmar and plantar surfaces.
Flagellate erythema has been described in relation to bleomycin treatment along with other reported associations with peplomycin (a bleomycin derivative), docetaxel, dermatomyositis, adult-onset Still's disease, and shiitake mushroom dermatitis. The rash may appear following administration of bleomycin by any route and has been shown to be dose independent. Onset occurs anytime from 1 day to several months after exposure to the offending agent. Patients typically present with history of itching followed by the appearance of red linear streaks, which are most commonly found on the trunk. Over time, the erythema will resolve to postinflammatory hyperpigmentation.
The exact cause is unknown, but it is thought that scratching causes vasodilation with bleomycin accumulating in the skin. Diagnosis is made by physical examination of the characteristic appearance of the rash along with the history of chemotherapy. A skin biopsy may be performed. In most cases, the rash resolves spontaneously within 6-8 months. Severe rashes may warrant discontinuation of bleomycin. Using antihistamines and topical and oral corticosteroids in conjunction with bleomycin may reduce the incidence of flagellate erythema/hyperpigmentation.
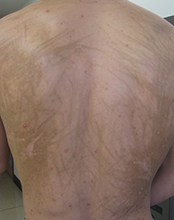
This case and photo were submitted by Dr. Damon McClain, a dermatologist in Camp Lejeune, N.C., and by Parteek Singla. A 26-year-old male presented with asymptomatic hyperpigmented streaks on his back. He was receiving chemotherapy for testicular cancer. He received dexamethasone with his first chemotherapy treatment and had no cutaneous eruption at that time. He was not given a steroid with his second dose of chemotherapy. The lesions began an hour after that dose. Initially, the lesions were red and pruritic, and then they turned brown. The patient had no nailfold changes on examination. He had no other medical problems and reported that he had not eaten any unusual foods.
Make the Diagnosis - February 2015
Diagnosis: Bullous impetigo
Bullous impetigo is a superficial skin infection that is most commonly seen in children (especially under the age of 2 years). It accounts for 10% of all pediatric skin disease and 25% of all impetigo.
Bullous impetigo is a variant of impetigo that produces exfoliative or epidermolytic toxins (ETA and ETB) in response to a staphylococcal infection. ETA and ETB are glutamate-specific serine proteases that bind and cleave desmoglein-1, a glycoprotein integral to intraepidermal keratinocyte adhesion. ETA and ETB produced at the site of a staphylococcal infection drive a cutaneous blistering response caused by intraepidermal cleavage below or within the stratum granulosum. While toxins typically act locally, they can spread systemically and produce generalized blistering. Therefore, lesions may be few and localized, or numerous and widespread.
A patient with bullous impetigo typically presents with a history of vesicular lesions that progress to flaccid bullae with little/no surrounding erythema. The color of the fluid contents can change from clear to cloudy/yellow. These bullae can easily rupture and leave a moist, erythematous base with surrounding honey-colored crust. Sometimes only the central portion of the bullae will drain, leaving a surrounding rim that retains fluid for days. Bullous impetigo most commonly affects moist, intertriginous areas. Most bullae heal without scarring, although hyperpigmentation has been reported in patients with darker skin types. Fever and other constitutional symptoms are uncommon.
The differential diagnosis of bullous impetigo includes contact dermatitis, bullous insect bites, thermal burns, pemphigus vulgaris, bullous pemphigoid, erythema multiforme, and dermatitis herpetiformis. It is especially important to rule out herpes simplex, varicella, bullous tinea, bullous fixed drug reaction, bullous drug eruption, and staphylococcal scalded skin syndrome.
The diagnosis of bullous impetigo is usually based upon clinical presentation. However, the diagnosis can easily be confirmed through bacterial culture of blister fluid that isolates Staphylococcus. Nikolsky’s sign is usually negative.
Bullous impetigo is self-limiting and, if left untreated, will typically resolve in weeks to months. The treatment of choice includes topical mupirocin ointment, along with local cleansing and crust removal. Oral antistaphylococcal drugs for 7-10 days are prescribed in the setting of widespread or complicated disease. Penicillinase-resistant penicillins and cephalosporins are effective against methicillin-susceptible Staphylococcus aureus. It is also important to consider methicillin-resistant S. aureus and treat with trimethoprim-sulfamethoxazole, clindamycin, or linezolid. Intravenous vancomycin should be reserved for patients with severe, widespread disease.
This patient was treated with cephalexin and mupirocin. Her lesions began drying up 2 days after the initiation of antibiotics. Bacterial culture results were positive for Staphylococcus.
Diagnosis: Bullous impetigo
Bullous impetigo is a superficial skin infection that is most commonly seen in children (especially under the age of 2 years). It accounts for 10% of all pediatric skin disease and 25% of all impetigo.
Bullous impetigo is a variant of impetigo that produces exfoliative or epidermolytic toxins (ETA and ETB) in response to a staphylococcal infection. ETA and ETB are glutamate-specific serine proteases that bind and cleave desmoglein-1, a glycoprotein integral to intraepidermal keratinocyte adhesion. ETA and ETB produced at the site of a staphylococcal infection drive a cutaneous blistering response caused by intraepidermal cleavage below or within the stratum granulosum. While toxins typically act locally, they can spread systemically and produce generalized blistering. Therefore, lesions may be few and localized, or numerous and widespread.
A patient with bullous impetigo typically presents with a history of vesicular lesions that progress to flaccid bullae with little/no surrounding erythema. The color of the fluid contents can change from clear to cloudy/yellow. These bullae can easily rupture and leave a moist, erythematous base with surrounding honey-colored crust. Sometimes only the central portion of the bullae will drain, leaving a surrounding rim that retains fluid for days. Bullous impetigo most commonly affects moist, intertriginous areas. Most bullae heal without scarring, although hyperpigmentation has been reported in patients with darker skin types. Fever and other constitutional symptoms are uncommon.
The differential diagnosis of bullous impetigo includes contact dermatitis, bullous insect bites, thermal burns, pemphigus vulgaris, bullous pemphigoid, erythema multiforme, and dermatitis herpetiformis. It is especially important to rule out herpes simplex, varicella, bullous tinea, bullous fixed drug reaction, bullous drug eruption, and staphylococcal scalded skin syndrome.
The diagnosis of bullous impetigo is usually based upon clinical presentation. However, the diagnosis can easily be confirmed through bacterial culture of blister fluid that isolates Staphylococcus. Nikolsky’s sign is usually negative.
Bullous impetigo is self-limiting and, if left untreated, will typically resolve in weeks to months. The treatment of choice includes topical mupirocin ointment, along with local cleansing and crust removal. Oral antistaphylococcal drugs for 7-10 days are prescribed in the setting of widespread or complicated disease. Penicillinase-resistant penicillins and cephalosporins are effective against methicillin-susceptible Staphylococcus aureus. It is also important to consider methicillin-resistant S. aureus and treat with trimethoprim-sulfamethoxazole, clindamycin, or linezolid. Intravenous vancomycin should be reserved for patients with severe, widespread disease.
This patient was treated with cephalexin and mupirocin. Her lesions began drying up 2 days after the initiation of antibiotics. Bacterial culture results were positive for Staphylococcus.
Diagnosis: Bullous impetigo
Bullous impetigo is a superficial skin infection that is most commonly seen in children (especially under the age of 2 years). It accounts for 10% of all pediatric skin disease and 25% of all impetigo.
Bullous impetigo is a variant of impetigo that produces exfoliative or epidermolytic toxins (ETA and ETB) in response to a staphylococcal infection. ETA and ETB are glutamate-specific serine proteases that bind and cleave desmoglein-1, a glycoprotein integral to intraepidermal keratinocyte adhesion. ETA and ETB produced at the site of a staphylococcal infection drive a cutaneous blistering response caused by intraepidermal cleavage below or within the stratum granulosum. While toxins typically act locally, they can spread systemically and produce generalized blistering. Therefore, lesions may be few and localized, or numerous and widespread.
A patient with bullous impetigo typically presents with a history of vesicular lesions that progress to flaccid bullae with little/no surrounding erythema. The color of the fluid contents can change from clear to cloudy/yellow. These bullae can easily rupture and leave a moist, erythematous base with surrounding honey-colored crust. Sometimes only the central portion of the bullae will drain, leaving a surrounding rim that retains fluid for days. Bullous impetigo most commonly affects moist, intertriginous areas. Most bullae heal without scarring, although hyperpigmentation has been reported in patients with darker skin types. Fever and other constitutional symptoms are uncommon.
The differential diagnosis of bullous impetigo includes contact dermatitis, bullous insect bites, thermal burns, pemphigus vulgaris, bullous pemphigoid, erythema multiforme, and dermatitis herpetiformis. It is especially important to rule out herpes simplex, varicella, bullous tinea, bullous fixed drug reaction, bullous drug eruption, and staphylococcal scalded skin syndrome.
The diagnosis of bullous impetigo is usually based upon clinical presentation. However, the diagnosis can easily be confirmed through bacterial culture of blister fluid that isolates Staphylococcus. Nikolsky’s sign is usually negative.
Bullous impetigo is self-limiting and, if left untreated, will typically resolve in weeks to months. The treatment of choice includes topical mupirocin ointment, along with local cleansing and crust removal. Oral antistaphylococcal drugs for 7-10 days are prescribed in the setting of widespread or complicated disease. Penicillinase-resistant penicillins and cephalosporins are effective against methicillin-susceptible Staphylococcus aureus. It is also important to consider methicillin-resistant S. aureus and treat with trimethoprim-sulfamethoxazole, clindamycin, or linezolid. Intravenous vancomycin should be reserved for patients with severe, widespread disease.
This patient was treated with cephalexin and mupirocin. Her lesions began drying up 2 days after the initiation of antibiotics. Bacterial culture results were positive for Staphylococcus.
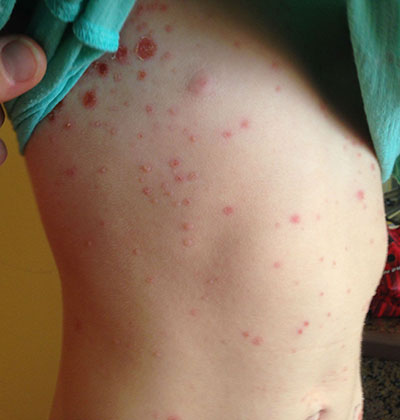
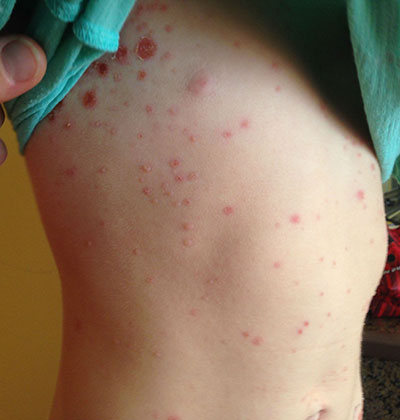
This case and photo were submitted by Tanya Greywal and Dr. Brooke Resh Sateesh. Dr. Resh Sateesh is in private practice in San Diego, Calif. A 2-year-old girl presented with a 1-week history of papules on her left abdomen. She had no systemic symptoms. However, the patient returned 2 days later with diffuse lesions on her trunk and legs.
Make the Diagnosis - January 2015
Diagnosis: Roseola
Roseola, also known as exanthema subitum or sixth disease, is a common viral infection that affects children by the age of 2 years, most commonly between ages 6 and 15 months. Two strains of viruses, human herpes virus 6 and 7, are the most common causes. Roseola spreads from person to person through respiratory secretions. Symptoms can vary in severity and can start with a sudden high fever, often greater than 103° F, that can last several days and may be associated with a febrile seizure. Associated symptoms may include sore throat, runny nose, cough, and lymphadenopathy.
After the fever resolves, erythematous macules and papules develop, starting on the chest, back, or abdomen and subsequently spreading to the neck and arms. The rash is not typically pruritic, is not contagious, and can last anywhere from several hours to several days. It is important to note that a person with roseola is contagious even if no rash is present.
Roseola can be difficult to diagnose given the nonspecific symptoms that are very similar to those of other childhood illnesses. Diagnosis is often clinical, based on history and physical, but can be confirmed by the characteristic rash or by detecting antibodies to roseola.
Treatment of roseola is supportive, with most children recovering within a week. Over-the-counter medications to treat fever, such as acetaminophen and ibuprofen, can be used, but aspirin should be avoided to treat fever in children younger than 2 years given the risk of Reye's syndrome. Antibodies developed against roseola are protective against future infections. Measures should be taken to avoid contact with those infected and hands should be washed frequently to avoid spreading the virus. Adults can contract roseola if they did not have it as a child; the disease is milder in adults but they can spread it to children.
Another common viral childhood illness is hand, foot, and mouth disease, caused by an enterovirus (coxsackievirus) that leads to sores and blisters on the hands, feet, and buttocks/legs that last for a week. The virus is spread through coughing and sneezing and through fecal-to-oral transmission. Symptoms include sore throat and fever with the rash. Diagnosis is clinical, based on history and physical exam, and the condition is treated supportively similar to roseola.
Another common illness is erythema infectiosum, also known as fifth disease, and is caused by parvovirus B19. The virus is common among school-aged children. The rash typically starts as erythematous patches on the cheeks giving the characteristic "slapped cheek" appearance. By the time the rash appears, the child is no longer contagious. The virus is spread through respiratory secretions.
Adults whose occupations put them in close contact with children can become infected with symptoms, including joint pain, rather than the rash. Infection in pregnant women and patients with sickle cell disease carries an increased risk of miscarriage and severe anemia, respectively. Similar to roseola and hand, foot, and mouth disease, diagnosis of fifth disease is clinical, and treatment is largely supportive.
Diagnosis: Roseola
Roseola, also known as exanthema subitum or sixth disease, is a common viral infection that affects children by the age of 2 years, most commonly between ages 6 and 15 months. Two strains of viruses, human herpes virus 6 and 7, are the most common causes. Roseola spreads from person to person through respiratory secretions. Symptoms can vary in severity and can start with a sudden high fever, often greater than 103° F, that can last several days and may be associated with a febrile seizure. Associated symptoms may include sore throat, runny nose, cough, and lymphadenopathy.
After the fever resolves, erythematous macules and papules develop, starting on the chest, back, or abdomen and subsequently spreading to the neck and arms. The rash is not typically pruritic, is not contagious, and can last anywhere from several hours to several days. It is important to note that a person with roseola is contagious even if no rash is present.
Roseola can be difficult to diagnose given the nonspecific symptoms that are very similar to those of other childhood illnesses. Diagnosis is often clinical, based on history and physical, but can be confirmed by the characteristic rash or by detecting antibodies to roseola.
Treatment of roseola is supportive, with most children recovering within a week. Over-the-counter medications to treat fever, such as acetaminophen and ibuprofen, can be used, but aspirin should be avoided to treat fever in children younger than 2 years given the risk of Reye's syndrome. Antibodies developed against roseola are protective against future infections. Measures should be taken to avoid contact with those infected and hands should be washed frequently to avoid spreading the virus. Adults can contract roseola if they did not have it as a child; the disease is milder in adults but they can spread it to children.
Another common viral childhood illness is hand, foot, and mouth disease, caused by an enterovirus (coxsackievirus) that leads to sores and blisters on the hands, feet, and buttocks/legs that last for a week. The virus is spread through coughing and sneezing and through fecal-to-oral transmission. Symptoms include sore throat and fever with the rash. Diagnosis is clinical, based on history and physical exam, and the condition is treated supportively similar to roseola.
Another common illness is erythema infectiosum, also known as fifth disease, and is caused by parvovirus B19. The virus is common among school-aged children. The rash typically starts as erythematous patches on the cheeks giving the characteristic "slapped cheek" appearance. By the time the rash appears, the child is no longer contagious. The virus is spread through respiratory secretions.
Adults whose occupations put them in close contact with children can become infected with symptoms, including joint pain, rather than the rash. Infection in pregnant women and patients with sickle cell disease carries an increased risk of miscarriage and severe anemia, respectively. Similar to roseola and hand, foot, and mouth disease, diagnosis of fifth disease is clinical, and treatment is largely supportive.
Diagnosis: Roseola
Roseola, also known as exanthema subitum or sixth disease, is a common viral infection that affects children by the age of 2 years, most commonly between ages 6 and 15 months. Two strains of viruses, human herpes virus 6 and 7, are the most common causes. Roseola spreads from person to person through respiratory secretions. Symptoms can vary in severity and can start with a sudden high fever, often greater than 103° F, that can last several days and may be associated with a febrile seizure. Associated symptoms may include sore throat, runny nose, cough, and lymphadenopathy.
After the fever resolves, erythematous macules and papules develop, starting on the chest, back, or abdomen and subsequently spreading to the neck and arms. The rash is not typically pruritic, is not contagious, and can last anywhere from several hours to several days. It is important to note that a person with roseola is contagious even if no rash is present.
Roseola can be difficult to diagnose given the nonspecific symptoms that are very similar to those of other childhood illnesses. Diagnosis is often clinical, based on history and physical, but can be confirmed by the characteristic rash or by detecting antibodies to roseola.
Treatment of roseola is supportive, with most children recovering within a week. Over-the-counter medications to treat fever, such as acetaminophen and ibuprofen, can be used, but aspirin should be avoided to treat fever in children younger than 2 years given the risk of Reye's syndrome. Antibodies developed against roseola are protective against future infections. Measures should be taken to avoid contact with those infected and hands should be washed frequently to avoid spreading the virus. Adults can contract roseola if they did not have it as a child; the disease is milder in adults but they can spread it to children.
Another common viral childhood illness is hand, foot, and mouth disease, caused by an enterovirus (coxsackievirus) that leads to sores and blisters on the hands, feet, and buttocks/legs that last for a week. The virus is spread through coughing and sneezing and through fecal-to-oral transmission. Symptoms include sore throat and fever with the rash. Diagnosis is clinical, based on history and physical exam, and the condition is treated supportively similar to roseola.
Another common illness is erythema infectiosum, also known as fifth disease, and is caused by parvovirus B19. The virus is common among school-aged children. The rash typically starts as erythematous patches on the cheeks giving the characteristic "slapped cheek" appearance. By the time the rash appears, the child is no longer contagious. The virus is spread through respiratory secretions.
Adults whose occupations put them in close contact with children can become infected with symptoms, including joint pain, rather than the rash. Infection in pregnant women and patients with sickle cell disease carries an increased risk of miscarriage and severe anemia, respectively. Similar to roseola and hand, foot, and mouth disease, diagnosis of fifth disease is clinical, and treatment is largely supportive.
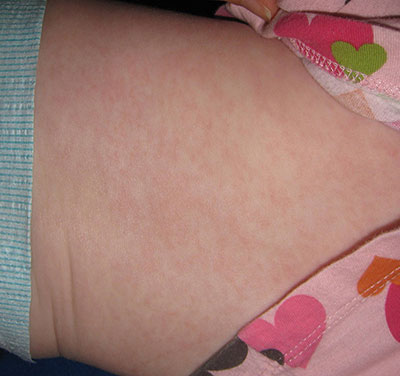
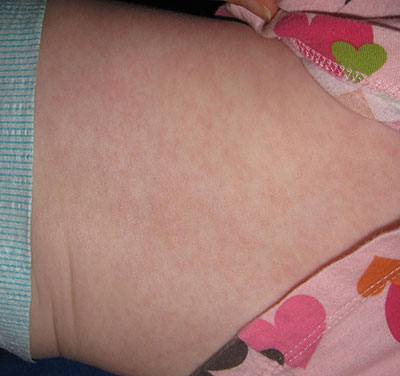
This case was submitted by Parteek Singla, B.S., and Dr. Donna Bilu Martin of Premier Dermatology, MD. An otherwise healthy 10-month-old female developed a high fever and runny nose that lasted for 4 days. On the fifth day, she developed a rash on her trunk that spread to her extremities and lasted for 1 day. Her illness resolved with no sequelae.
Case of the Month
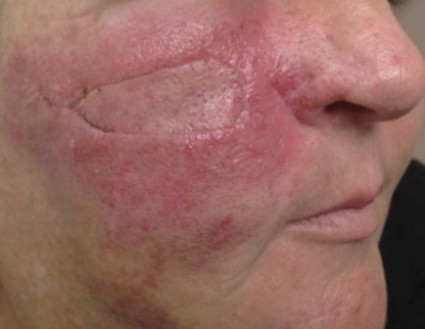
A 56-year-old Hispanic female with a past medical history significant for basal cell carcinoma presented with a history of itchy, erythematous papules on her right cheek. Four days prior, she presented for suture removal after reconstruction with an island pedicle flap following Mohs micrographic surgery. She experienced a similar rash on her forearm following another surgery in the past.
a) Cellulitis
b) Contact dermatitis
c) Herpes simplex virus
Diagnosis: Contact dermatitis secondary to Mastisol and Steri-Strips
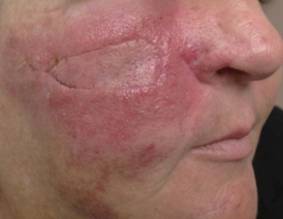
Contact dermatitis is a localized, pruritic, erythematous rash that occurs after contact with a certain allergen or irritant. The disorder is typically classified as either allergic contact dermatitis or irritant contact dermatitis.
Allergic contact dermatitis is a T cell–mediated, type-IV, delayed-type hypersensitivity reaction that requires prior sensitization with the causative agent before the patient becomes allergic to it. The typical rash of pruritus, erythema, edema, and vesicle formation occurs with further exposures.
The mechanism of immune response to a particular allergen requires that the antigen be of low molecular weight (less than 500 d) in order to penetrate the stratum corneum and gain access to the immunologic system. CD4, CD8, T regulatory cells, and natural killer T cells have all been implicated.
The process is composed of an afferent (sensitization) phase and an efferent (elicitation) phase.
Common haptens or immunogenic agents include nickel, urushiol from poison ivy resin, ultraviolet light, dyes, and fragrances.
This patient revealed a history of a similar reaction to the one presented in this case following wound dressing on her forearm with two products: Steri-Strips and Mastisol liquid adhesive. Unfortunately, she did not reveal this history until she had experienced the reaction a second time. The patient was instructed to apply hydrocortisone 1% cream twice daily to the red areas only and to follow up with a clinic visit in 4-5 days. Documentation of her allergy was included in her medical record.
This case was submitted by Dr. Keyvan Nouri; Dr. Katlein Franca; Jennifer Ledon; and Jessica Savas of the University of Miami.
–Donna Bilu Martin, M.D.
A 56-year-old Hispanic female with a past medical history significant for basal cell carcinoma presented with a history of itchy, erythematous papules on her right cheek. Four days prior, she presented for suture removal after reconstruction with an island pedicle flap following Mohs micrographic surgery. She experienced a similar rash on her forearm following another surgery in the past.
a) Cellulitis
b) Contact dermatitis
c) Herpes simplex virus
Diagnosis: Contact dermatitis secondary to Mastisol and Steri-Strips
Contact dermatitis is a localized, pruritic, erythematous rash that occurs after contact with a certain allergen or irritant. The disorder is typically classified as either allergic contact dermatitis or irritant contact dermatitis.
Allergic contact dermatitis is a T cell–mediated, type-IV, delayed-type hypersensitivity reaction that requires prior sensitization with the causative agent before the patient becomes allergic to it. The typical rash of pruritus, erythema, edema, and vesicle formation occurs with further exposures.
The mechanism of immune response to a particular allergen requires that the antigen be of low molecular weight (less than 500 d) in order to penetrate the stratum corneum and gain access to the immunologic system. CD4, CD8, T regulatory cells, and natural killer T cells have all been implicated.
The process is composed of an afferent (sensitization) phase and an efferent (elicitation) phase.
Common haptens or immunogenic agents include nickel, urushiol from poison ivy resin, ultraviolet light, dyes, and fragrances.
This patient revealed a history of a similar reaction to the one presented in this case following wound dressing on her forearm with two products: Steri-Strips and Mastisol liquid adhesive. Unfortunately, she did not reveal this history until she had experienced the reaction a second time. The patient was instructed to apply hydrocortisone 1% cream twice daily to the red areas only and to follow up with a clinic visit in 4-5 days. Documentation of her allergy was included in her medical record.
This case was submitted by Dr. Keyvan Nouri; Dr. Katlein Franca; Jennifer Ledon; and Jessica Savas of the University of Miami.
–Donna Bilu Martin, M.D.
A 56-year-old Hispanic female with a past medical history significant for basal cell carcinoma presented with a history of itchy, erythematous papules on her right cheek. Four days prior, she presented for suture removal after reconstruction with an island pedicle flap following Mohs micrographic surgery. She experienced a similar rash on her forearm following another surgery in the past.
a) Cellulitis
b) Contact dermatitis
c) Herpes simplex virus
Diagnosis: Contact dermatitis secondary to Mastisol and Steri-Strips
Contact dermatitis is a localized, pruritic, erythematous rash that occurs after contact with a certain allergen or irritant. The disorder is typically classified as either allergic contact dermatitis or irritant contact dermatitis.
Allergic contact dermatitis is a T cell–mediated, type-IV, delayed-type hypersensitivity reaction that requires prior sensitization with the causative agent before the patient becomes allergic to it. The typical rash of pruritus, erythema, edema, and vesicle formation occurs with further exposures.
The mechanism of immune response to a particular allergen requires that the antigen be of low molecular weight (less than 500 d) in order to penetrate the stratum corneum and gain access to the immunologic system. CD4, CD8, T regulatory cells, and natural killer T cells have all been implicated.
The process is composed of an afferent (sensitization) phase and an efferent (elicitation) phase.
Common haptens or immunogenic agents include nickel, urushiol from poison ivy resin, ultraviolet light, dyes, and fragrances.
This patient revealed a history of a similar reaction to the one presented in this case following wound dressing on her forearm with two products: Steri-Strips and Mastisol liquid adhesive. Unfortunately, she did not reveal this history until she had experienced the reaction a second time. The patient was instructed to apply hydrocortisone 1% cream twice daily to the red areas only and to follow up with a clinic visit in 4-5 days. Documentation of her allergy was included in her medical record.
This case was submitted by Dr. Keyvan Nouri; Dr. Katlein Franca; Jennifer Ledon; and Jessica Savas of the University of Miami.
–Donna Bilu Martin, M.D.
Make The Diagnosis
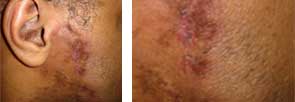
A 51 year-old-male presented with asymptomatic violaceous, indurated plaques on his left and right cheeks. He also had follicular plugging in the right ear. What’s your diagnosis?
Images courtesy Dr. Donna Bilu Martin
Diagnosis: Lupus Erythematosus Panniculitis
Lupus panniculitis, or lupus profundus, represents 2%-3% of all patients with lupus erythematosus. It most commonly occurs in adults aged 20-60. Patients present with tender subcutaneous nodules and plaques that tend to develop on the face, upper outer arms, shoulders, hips, and trunk. The distal extremities are usually spared. The overlying skin can show features of chronic cutaneous lupus including scaling, follicular plugging, atrophy, dyspigmentation, telangiectasias, and ulceration.
Histopathology reveals a primarily lobular panniculitis with a marked predominance of lymphocytes and scattered plasma cells. One characteristic feature is hyalin necrosis of fat lobules that can extend into the septa.
Treatment options include sunscreen, potent topical and intralesional corticosteroids, antimalarials, systemic steroids (in initial phases of disease), dapsone, cyclophosphamide, and thalidomide. This patient was treated with thalidomide, which resulted in improvement of his lesions.
This case was first presented at Maryland Derm, at the University of Maryland School of Medicine in Baltimore, by Dr. Bilu Martin and Dr. Anthony Gaspari.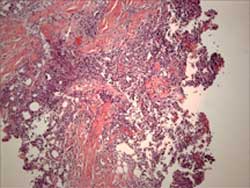
Image courtesy Dr. Donna Bilu Martin
Histology shows a lobular panniculitis with a marked predominance of lymphocytes and scattered plasma cells.
A 51 year-old-male presented with asymptomatic violaceous, indurated plaques on his left and right cheeks. He also had follicular plugging in the right ear. What’s your diagnosis?
Images courtesy Dr. Donna Bilu Martin
Diagnosis: Lupus Erythematosus Panniculitis
Lupus panniculitis, or lupus profundus, represents 2%-3% of all patients with lupus erythematosus. It most commonly occurs in adults aged 20-60. Patients present with tender subcutaneous nodules and plaques that tend to develop on the face, upper outer arms, shoulders, hips, and trunk. The distal extremities are usually spared. The overlying skin can show features of chronic cutaneous lupus including scaling, follicular plugging, atrophy, dyspigmentation, telangiectasias, and ulceration.
Histopathology reveals a primarily lobular panniculitis with a marked predominance of lymphocytes and scattered plasma cells. One characteristic feature is hyalin necrosis of fat lobules that can extend into the septa.
Treatment options include sunscreen, potent topical and intralesional corticosteroids, antimalarials, systemic steroids (in initial phases of disease), dapsone, cyclophosphamide, and thalidomide. This patient was treated with thalidomide, which resulted in improvement of his lesions.
This case was first presented at Maryland Derm, at the University of Maryland School of Medicine in Baltimore, by Dr. Bilu Martin and Dr. Anthony Gaspari.
Image courtesy Dr. Donna Bilu Martin
Histology shows a lobular panniculitis with a marked predominance of lymphocytes and scattered plasma cells.
A 51 year-old-male presented with asymptomatic violaceous, indurated plaques on his left and right cheeks. He also had follicular plugging in the right ear. What’s your diagnosis?
Images courtesy Dr. Donna Bilu Martin
Diagnosis: Lupus Erythematosus Panniculitis
Lupus panniculitis, or lupus profundus, represents 2%-3% of all patients with lupus erythematosus. It most commonly occurs in adults aged 20-60. Patients present with tender subcutaneous nodules and plaques that tend to develop on the face, upper outer arms, shoulders, hips, and trunk. The distal extremities are usually spared. The overlying skin can show features of chronic cutaneous lupus including scaling, follicular plugging, atrophy, dyspigmentation, telangiectasias, and ulceration.
Histopathology reveals a primarily lobular panniculitis with a marked predominance of lymphocytes and scattered plasma cells. One characteristic feature is hyalin necrosis of fat lobules that can extend into the septa.
Treatment options include sunscreen, potent topical and intralesional corticosteroids, antimalarials, systemic steroids (in initial phases of disease), dapsone, cyclophosphamide, and thalidomide. This patient was treated with thalidomide, which resulted in improvement of his lesions.
This case was first presented at Maryland Derm, at the University of Maryland School of Medicine in Baltimore, by Dr. Bilu Martin and Dr. Anthony Gaspari.
Image courtesy Dr. Donna Bilu Martin
Histology shows a lobular panniculitis with a marked predominance of lymphocytes and scattered plasma cells.
Make the Diagnosis
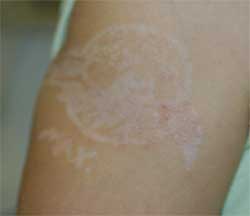
Diagnosis: Contact Dermatitis to Paraphenylenediamine
The patient’s mother reported blisters, erythema, and scabbing in the area of the tattoo. Six months later, the patient underwent paraphenylenediamine patch testing and exhibited a reaction.
The patient was treated with mild topical steroids and a 4-day prednisone course prior to presentation. A week of clobetasol ointment improved the pruritus and erythema.
Henna is a green powdered extract derived from the leaves of the Lawsonia alba plant. The active ingredient is lawsone. Middle Eastern and Indian cultures use the extract to dye the hair, skin, and nails. Contact with the skin for an extended period of time yields a brownish orange pigment. In Western countries, Henna tattoos have gained popularity as a temporary alternative to ink tattoos.
Henna may be used in its pure form, however, paraphenylenediamine (PPD) is often added to darken the pigment, expedite drying time, and improve design accuracy. PPD is an allergen found in hair dyes and photographic film processing. It is a potent T-cell stimulator, and its efficacy is directly related to concentration and duration of exposure. Patch tests among individuals with henna contact dermatitis are negative to pure henna powder but react strongly to PPD, which has lead to the assumption that PPD is the main allergen in henna paste.
Henna tattoo inks have been found to have PPD concentrations as high as 15%-30%, and, often, the inks are in contact with the skin for several days after application. The hypersensitivity can sensitize individuals to PPD-containing substances such as dark hair dyes and dark clothing. Cross reaction may cause hypersensitivity to natural rubber latex, azo dyes, thiurams, PABA sunscreen, para-aminosalicylic acid, and benzocaine.
The initial inflammatory response may present as erythematous, eczematous, pruritic, or papulovesicular eruption in the area or boundary of the original design. Edema, anaphylaxis, and collapse are less common manifestations. The inflammation can result in scarring, keloid formation, and permanent, post-inflammatory pigment changes.
As demonstrated in my patient, hypopigmentation occurs more frequently in children than adults. Therapy includes protection of the blistered area, antihistamines, treatment of infection, and aggressive topical corticosteroid therapy.
This case was first presented at Maryland Derm, at the University of Maryland School of Medicine in Baltimore, by Dr. Martin, Dr. Vera David, and Dr. Anthony Gaspari.
Diagnosis: Contact Dermatitis to Paraphenylenediamine
The patient’s mother reported blisters, erythema, and scabbing in the area of the tattoo. Six months later, the patient underwent paraphenylenediamine patch testing and exhibited a reaction.
The patient was treated with mild topical steroids and a 4-day prednisone course prior to presentation. A week of clobetasol ointment improved the pruritus and erythema.
Henna is a green powdered extract derived from the leaves of the Lawsonia alba plant. The active ingredient is lawsone. Middle Eastern and Indian cultures use the extract to dye the hair, skin, and nails. Contact with the skin for an extended period of time yields a brownish orange pigment. In Western countries, Henna tattoos have gained popularity as a temporary alternative to ink tattoos.
Henna may be used in its pure form, however, paraphenylenediamine (PPD) is often added to darken the pigment, expedite drying time, and improve design accuracy. PPD is an allergen found in hair dyes and photographic film processing. It is a potent T-cell stimulator, and its efficacy is directly related to concentration and duration of exposure. Patch tests among individuals with henna contact dermatitis are negative to pure henna powder but react strongly to PPD, which has lead to the assumption that PPD is the main allergen in henna paste.
Henna tattoo inks have been found to have PPD concentrations as high as 15%-30%, and, often, the inks are in contact with the skin for several days after application. The hypersensitivity can sensitize individuals to PPD-containing substances such as dark hair dyes and dark clothing. Cross reaction may cause hypersensitivity to natural rubber latex, azo dyes, thiurams, PABA sunscreen, para-aminosalicylic acid, and benzocaine.
The initial inflammatory response may present as erythematous, eczematous, pruritic, or papulovesicular eruption in the area or boundary of the original design. Edema, anaphylaxis, and collapse are less common manifestations. The inflammation can result in scarring, keloid formation, and permanent, post-inflammatory pigment changes.
As demonstrated in my patient, hypopigmentation occurs more frequently in children than adults. Therapy includes protection of the blistered area, antihistamines, treatment of infection, and aggressive topical corticosteroid therapy.
This case was first presented at Maryland Derm, at the University of Maryland School of Medicine in Baltimore, by Dr. Martin, Dr. Vera David, and Dr. Anthony Gaspari.
Diagnosis: Contact Dermatitis to Paraphenylenediamine
The patient’s mother reported blisters, erythema, and scabbing in the area of the tattoo. Six months later, the patient underwent paraphenylenediamine patch testing and exhibited a reaction.
The patient was treated with mild topical steroids and a 4-day prednisone course prior to presentation. A week of clobetasol ointment improved the pruritus and erythema.
Henna is a green powdered extract derived from the leaves of the Lawsonia alba plant. The active ingredient is lawsone. Middle Eastern and Indian cultures use the extract to dye the hair, skin, and nails. Contact with the skin for an extended period of time yields a brownish orange pigment. In Western countries, Henna tattoos have gained popularity as a temporary alternative to ink tattoos.
Henna may be used in its pure form, however, paraphenylenediamine (PPD) is often added to darken the pigment, expedite drying time, and improve design accuracy. PPD is an allergen found in hair dyes and photographic film processing. It is a potent T-cell stimulator, and its efficacy is directly related to concentration and duration of exposure. Patch tests among individuals with henna contact dermatitis are negative to pure henna powder but react strongly to PPD, which has lead to the assumption that PPD is the main allergen in henna paste.
Henna tattoo inks have been found to have PPD concentrations as high as 15%-30%, and, often, the inks are in contact with the skin for several days after application. The hypersensitivity can sensitize individuals to PPD-containing substances such as dark hair dyes and dark clothing. Cross reaction may cause hypersensitivity to natural rubber latex, azo dyes, thiurams, PABA sunscreen, para-aminosalicylic acid, and benzocaine.
The initial inflammatory response may present as erythematous, eczematous, pruritic, or papulovesicular eruption in the area or boundary of the original design. Edema, anaphylaxis, and collapse are less common manifestations. The inflammation can result in scarring, keloid formation, and permanent, post-inflammatory pigment changes.
As demonstrated in my patient, hypopigmentation occurs more frequently in children than adults. Therapy includes protection of the blistered area, antihistamines, treatment of infection, and aggressive topical corticosteroid therapy.
This case was first presented at Maryland Derm, at the University of Maryland School of Medicine in Baltimore, by Dr. Martin, Dr. Vera David, and Dr. Anthony Gaspari.