User login
PFO closure may reduce migraine days and prevent stroke
, according to a discussion at the 2023 Scottsdale Headache Symposium.
In two clinical trials evaluating whether PFO closure reduces migraine risk, the primary endpoints were not met, but a signal of benefit on secondary endpoints and the association between PFO, migraine, and stroke are among the reasons that PFO closure should be reevaluated, according to Andrew Charles MD, Director of the Goldberg Migraine Program, University of California, Los Angeles.

Other right-to-left shunt defects have also been associated with both migraine and stroke, leading Dr. Charles to suggest these defects are more a common denominator.
“Stroke during a migraine is, in fact, very uncommon,” Dr. Charles said. “This raises the possibility that it is not the migraine causing the stroke but rather there is a shared risk factor for stroke and migraine,” said Dr. Charles, referring to PFO as well as other right-to-left shunt defects, such as hereditary hemorrhaging telangiectasia in the lungs.
One Intervention, Two Potential Benefits
Fixing these defects is therefore at least theoretically attractive for preventing both migraine and stroke, but Dr. Charles said the opportunity for preventing both migraine and stroke is most attractive in migraine patients who have additional stroke risk factors.
Use of oral contraceptives, which produce a hypercoagulable state, is an example.
“Are these the people we should really be thinking about if they have PFO and migraine, particularly migraine with aura?” Dr. Charles asked.
The association between right-to-left shunts and migraine is strong. Although PFO is common, presenting in 20%-25% of the adult population, it has been found in up to 50% of individuals who have migraine with aura. In patients with migraine but no aura, the prevalence of PFO has been estimated to be approximately 35% or still somewhat elevated relative to the general population.
Primary Endpoint Missed in Clinical Trials
The question of whether risk of migraine can be reduced with repair of PFO or other right-to-left shunts remains unresolved. In two high-quality randomized trials undertaken in PFO repair, neither met its primary endpoint. In one of these, called PRIMA, which was terminated early for slow enrollment, the reduction in mean headache attacks was not significant relative to medical therapy.
In the second, called PREMIUM, device closure of PFO also failed to significantly reduce migraine attacks over sham procedure although it was associated with complete migraine remission (10% vs 1%).
A pooled analysis of these two studies that was conducted subsequently concluded that PFO closure reduces mean monthly migraine days (-3.1 vs. -1.9 days; P = -.02) and increases the likelihood of complete migraine cessation (9% vs. 0.7%; P < .001), but Dr. Charles pointed out the primary endpoint was migraine attacks not migraine days, so other analyses can only be considered hypothesis-generating.
There are several reasons to relook at the relationship between migraine and PFO but the potential to prevent both migraine and stroke with PFO closure could be one of the most important.
Several years ago, Dr. Charles and his coinvestigators from UCLA evaluated more than 700 ischemic strokes. Of these, 127 strokes were characterized as cryptogenic because of lack of another identifiable etiology. While 59% of these patients had PFO, which is several times higher than the general population, the prevalence of PFO in patients with a cryptogenic stroke and a history of migraine was 79% in this published study.
“So, in this group of patients who did not have any other clear cause for a stroke, a diagnosis of PFO was very much overrepresented,” Dr. Charles said.
Migraine Days Might Be a Better Endpoint
For patients with migraine who have risk factors for stroke, this makes PFO closure an attractive intervention, but a positive randomized trial is needed. Several are underway. Importantly, the trials now enrolling are using migraine days, which was significantly reduced in both PREMIUM and PRIMA, rather than migraine attacks as the primary endpoint.
“Migraine days is now accepted by the Food and Drug Administration as a criterion of benefit,” reported Jonathan Tobis, MD, Research Director, Interventional Cardiology, UCLA David Geffen School of Medicine, Los Angeles.
He explained that the FDA insisted on migraine attacks as the endpoint for the PREMIUM trial, but this was a far more challenging endpoint on which to show a statistical benefit. He emphasized that a new set of trials will now test efficacy on the basis of migraine days.
One of these trials, called RELIEF, which is randomizing patients to device closure of PFO or a sham procedure. Both groups are receiving clopidogrel or prasugrel based on a previous observation that patients who respond to these drugs are also more likely to respond to PFO closure.
Another trial, called COMPETE-2, is comparing PFO closure with a device to aspirin plus a sham closure. This trial is ongoing in China.
Stroke is not being evaluated as an endpoint in either trial, but Dr. Charles suggested that this does warrant attention.
“I would also just put it out there that, apart from simply migraine, this is a therapeutic approach that we might actually think about in terms of helping to prevent stroke in our migraine patients,” he said.
Senior author of a recent meta-analysis of trials evaluating PFO closure and control of migraine, Ling Liu, MD, Department of Neurology, University of Sichuan, Chengdu, China, agreed that PFO closure for the treatment of migraine deserves “a reevaluation.”
In his meta-analysis of three randomized trials, one pooled study, and eight retrospective case series with 1,165 patients, PFO closure was associated with a nearly 75% reduction (odds ratio [OR], 0.259; P = .0048) reduction in migraine days and 50% increase in resolution of migraine in patients with a history of migraine with aura (OR, 1.586; P = .227).
The incidence of stroke was not evaluated in this meta-analysis, but Dr. Liu believes that the evidence of reducing the burden of migraine with PFO closure is compelling. Given the evidence from this meta-analysis that PFO closure is safe, Dr. Liu maintained that a definitive trial is needed “especially for migraine with frequent aura.”
As an interventional cardiologist, Dr. Tobis said that when PFO closures is performed for prevention of stroke in patients with migraine, it often leads to reduced migraine activity and, in some cases, elimination of migraine. Like others, he believes new analyses should be conducted.
“Everyone involved in this field believes there is something there,” Dr. Tobis said. The missing link is a clinical trial to confirm it.
Dr. Charles and Dr. Liu report no potential conflicts of interest. Dr. Tobis reports a financial relationship with Holistick Medical.
, according to a discussion at the 2023 Scottsdale Headache Symposium.
In two clinical trials evaluating whether PFO closure reduces migraine risk, the primary endpoints were not met, but a signal of benefit on secondary endpoints and the association between PFO, migraine, and stroke are among the reasons that PFO closure should be reevaluated, according to Andrew Charles MD, Director of the Goldberg Migraine Program, University of California, Los Angeles.
Other right-to-left shunt defects have also been associated with both migraine and stroke, leading Dr. Charles to suggest these defects are more a common denominator.
“Stroke during a migraine is, in fact, very uncommon,” Dr. Charles said. “This raises the possibility that it is not the migraine causing the stroke but rather there is a shared risk factor for stroke and migraine,” said Dr. Charles, referring to PFO as well as other right-to-left shunt defects, such as hereditary hemorrhaging telangiectasia in the lungs.
One Intervention, Two Potential Benefits
Fixing these defects is therefore at least theoretically attractive for preventing both migraine and stroke, but Dr. Charles said the opportunity for preventing both migraine and stroke is most attractive in migraine patients who have additional stroke risk factors.
Use of oral contraceptives, which produce a hypercoagulable state, is an example.
“Are these the people we should really be thinking about if they have PFO and migraine, particularly migraine with aura?” Dr. Charles asked.
The association between right-to-left shunts and migraine is strong. Although PFO is common, presenting in 20%-25% of the adult population, it has been found in up to 50% of individuals who have migraine with aura. In patients with migraine but no aura, the prevalence of PFO has been estimated to be approximately 35% or still somewhat elevated relative to the general population.
Primary Endpoint Missed in Clinical Trials
The question of whether risk of migraine can be reduced with repair of PFO or other right-to-left shunts remains unresolved. In two high-quality randomized trials undertaken in PFO repair, neither met its primary endpoint. In one of these, called PRIMA, which was terminated early for slow enrollment, the reduction in mean headache attacks was not significant relative to medical therapy.
In the second, called PREMIUM, device closure of PFO also failed to significantly reduce migraine attacks over sham procedure although it was associated with complete migraine remission (10% vs 1%).
A pooled analysis of these two studies that was conducted subsequently concluded that PFO closure reduces mean monthly migraine days (-3.1 vs. -1.9 days; P = -.02) and increases the likelihood of complete migraine cessation (9% vs. 0.7%; P < .001), but Dr. Charles pointed out the primary endpoint was migraine attacks not migraine days, so other analyses can only be considered hypothesis-generating.
There are several reasons to relook at the relationship between migraine and PFO but the potential to prevent both migraine and stroke with PFO closure could be one of the most important.
Several years ago, Dr. Charles and his coinvestigators from UCLA evaluated more than 700 ischemic strokes. Of these, 127 strokes were characterized as cryptogenic because of lack of another identifiable etiology. While 59% of these patients had PFO, which is several times higher than the general population, the prevalence of PFO in patients with a cryptogenic stroke and a history of migraine was 79% in this published study.
“So, in this group of patients who did not have any other clear cause for a stroke, a diagnosis of PFO was very much overrepresented,” Dr. Charles said.
Migraine Days Might Be a Better Endpoint
For patients with migraine who have risk factors for stroke, this makes PFO closure an attractive intervention, but a positive randomized trial is needed. Several are underway. Importantly, the trials now enrolling are using migraine days, which was significantly reduced in both PREMIUM and PRIMA, rather than migraine attacks as the primary endpoint.
“Migraine days is now accepted by the Food and Drug Administration as a criterion of benefit,” reported Jonathan Tobis, MD, Research Director, Interventional Cardiology, UCLA David Geffen School of Medicine, Los Angeles.
He explained that the FDA insisted on migraine attacks as the endpoint for the PREMIUM trial, but this was a far more challenging endpoint on which to show a statistical benefit. He emphasized that a new set of trials will now test efficacy on the basis of migraine days.
One of these trials, called RELIEF, which is randomizing patients to device closure of PFO or a sham procedure. Both groups are receiving clopidogrel or prasugrel based on a previous observation that patients who respond to these drugs are also more likely to respond to PFO closure.
Another trial, called COMPETE-2, is comparing PFO closure with a device to aspirin plus a sham closure. This trial is ongoing in China.
Stroke is not being evaluated as an endpoint in either trial, but Dr. Charles suggested that this does warrant attention.
“I would also just put it out there that, apart from simply migraine, this is a therapeutic approach that we might actually think about in terms of helping to prevent stroke in our migraine patients,” he said.
Senior author of a recent meta-analysis of trials evaluating PFO closure and control of migraine, Ling Liu, MD, Department of Neurology, University of Sichuan, Chengdu, China, agreed that PFO closure for the treatment of migraine deserves “a reevaluation.”
In his meta-analysis of three randomized trials, one pooled study, and eight retrospective case series with 1,165 patients, PFO closure was associated with a nearly 75% reduction (odds ratio [OR], 0.259; P = .0048) reduction in migraine days and 50% increase in resolution of migraine in patients with a history of migraine with aura (OR, 1.586; P = .227).
The incidence of stroke was not evaluated in this meta-analysis, but Dr. Liu believes that the evidence of reducing the burden of migraine with PFO closure is compelling. Given the evidence from this meta-analysis that PFO closure is safe, Dr. Liu maintained that a definitive trial is needed “especially for migraine with frequent aura.”
As an interventional cardiologist, Dr. Tobis said that when PFO closures is performed for prevention of stroke in patients with migraine, it often leads to reduced migraine activity and, in some cases, elimination of migraine. Like others, he believes new analyses should be conducted.
“Everyone involved in this field believes there is something there,” Dr. Tobis said. The missing link is a clinical trial to confirm it.
Dr. Charles and Dr. Liu report no potential conflicts of interest. Dr. Tobis reports a financial relationship with Holistick Medical.
, according to a discussion at the 2023 Scottsdale Headache Symposium.
In two clinical trials evaluating whether PFO closure reduces migraine risk, the primary endpoints were not met, but a signal of benefit on secondary endpoints and the association between PFO, migraine, and stroke are among the reasons that PFO closure should be reevaluated, according to Andrew Charles MD, Director of the Goldberg Migraine Program, University of California, Los Angeles.
Other right-to-left shunt defects have also been associated with both migraine and stroke, leading Dr. Charles to suggest these defects are more a common denominator.
“Stroke during a migraine is, in fact, very uncommon,” Dr. Charles said. “This raises the possibility that it is not the migraine causing the stroke but rather there is a shared risk factor for stroke and migraine,” said Dr. Charles, referring to PFO as well as other right-to-left shunt defects, such as hereditary hemorrhaging telangiectasia in the lungs.
One Intervention, Two Potential Benefits
Fixing these defects is therefore at least theoretically attractive for preventing both migraine and stroke, but Dr. Charles said the opportunity for preventing both migraine and stroke is most attractive in migraine patients who have additional stroke risk factors.
Use of oral contraceptives, which produce a hypercoagulable state, is an example.
“Are these the people we should really be thinking about if they have PFO and migraine, particularly migraine with aura?” Dr. Charles asked.
The association between right-to-left shunts and migraine is strong. Although PFO is common, presenting in 20%-25% of the adult population, it has been found in up to 50% of individuals who have migraine with aura. In patients with migraine but no aura, the prevalence of PFO has been estimated to be approximately 35% or still somewhat elevated relative to the general population.
Primary Endpoint Missed in Clinical Trials
The question of whether risk of migraine can be reduced with repair of PFO or other right-to-left shunts remains unresolved. In two high-quality randomized trials undertaken in PFO repair, neither met its primary endpoint. In one of these, called PRIMA, which was terminated early for slow enrollment, the reduction in mean headache attacks was not significant relative to medical therapy.
In the second, called PREMIUM, device closure of PFO also failed to significantly reduce migraine attacks over sham procedure although it was associated with complete migraine remission (10% vs 1%).
A pooled analysis of these two studies that was conducted subsequently concluded that PFO closure reduces mean monthly migraine days (-3.1 vs. -1.9 days; P = -.02) and increases the likelihood of complete migraine cessation (9% vs. 0.7%; P < .001), but Dr. Charles pointed out the primary endpoint was migraine attacks not migraine days, so other analyses can only be considered hypothesis-generating.
There are several reasons to relook at the relationship between migraine and PFO but the potential to prevent both migraine and stroke with PFO closure could be one of the most important.
Several years ago, Dr. Charles and his coinvestigators from UCLA evaluated more than 700 ischemic strokes. Of these, 127 strokes were characterized as cryptogenic because of lack of another identifiable etiology. While 59% of these patients had PFO, which is several times higher than the general population, the prevalence of PFO in patients with a cryptogenic stroke and a history of migraine was 79% in this published study.
“So, in this group of patients who did not have any other clear cause for a stroke, a diagnosis of PFO was very much overrepresented,” Dr. Charles said.
Migraine Days Might Be a Better Endpoint
For patients with migraine who have risk factors for stroke, this makes PFO closure an attractive intervention, but a positive randomized trial is needed. Several are underway. Importantly, the trials now enrolling are using migraine days, which was significantly reduced in both PREMIUM and PRIMA, rather than migraine attacks as the primary endpoint.
“Migraine days is now accepted by the Food and Drug Administration as a criterion of benefit,” reported Jonathan Tobis, MD, Research Director, Interventional Cardiology, UCLA David Geffen School of Medicine, Los Angeles.
He explained that the FDA insisted on migraine attacks as the endpoint for the PREMIUM trial, but this was a far more challenging endpoint on which to show a statistical benefit. He emphasized that a new set of trials will now test efficacy on the basis of migraine days.
One of these trials, called RELIEF, which is randomizing patients to device closure of PFO or a sham procedure. Both groups are receiving clopidogrel or prasugrel based on a previous observation that patients who respond to these drugs are also more likely to respond to PFO closure.
Another trial, called COMPETE-2, is comparing PFO closure with a device to aspirin plus a sham closure. This trial is ongoing in China.
Stroke is not being evaluated as an endpoint in either trial, but Dr. Charles suggested that this does warrant attention.
“I would also just put it out there that, apart from simply migraine, this is a therapeutic approach that we might actually think about in terms of helping to prevent stroke in our migraine patients,” he said.
Senior author of a recent meta-analysis of trials evaluating PFO closure and control of migraine, Ling Liu, MD, Department of Neurology, University of Sichuan, Chengdu, China, agreed that PFO closure for the treatment of migraine deserves “a reevaluation.”
In his meta-analysis of three randomized trials, one pooled study, and eight retrospective case series with 1,165 patients, PFO closure was associated with a nearly 75% reduction (odds ratio [OR], 0.259; P = .0048) reduction in migraine days and 50% increase in resolution of migraine in patients with a history of migraine with aura (OR, 1.586; P = .227).
The incidence of stroke was not evaluated in this meta-analysis, but Dr. Liu believes that the evidence of reducing the burden of migraine with PFO closure is compelling. Given the evidence from this meta-analysis that PFO closure is safe, Dr. Liu maintained that a definitive trial is needed “especially for migraine with frequent aura.”
As an interventional cardiologist, Dr. Tobis said that when PFO closures is performed for prevention of stroke in patients with migraine, it often leads to reduced migraine activity and, in some cases, elimination of migraine. Like others, he believes new analyses should be conducted.
“Everyone involved in this field believes there is something there,” Dr. Tobis said. The missing link is a clinical trial to confirm it.
Dr. Charles and Dr. Liu report no potential conflicts of interest. Dr. Tobis reports a financial relationship with Holistick Medical.
FROM THE 2023 SCOTTSDALE HEADACHE SYMPOSIUM
Transapical valve replacement relieves mitral regurgitation
, relief of mitral regurgitation, and increases in cardiac hemodynamics and quality of life sustained at 1 year.
Further, patients with severe mitral annular calcification (MAC) showed improvements in hemodynamics, functional status, and quality of life after the procedure.
With 70 centers participating in the Tendyne SUMMIT trial, the first 100 trial roll-in patients accrued from the first one or two patients from each site without previous Tendyne TMVR experience.
“For this new procedure, with new operators, there was no intraprocedural mortality, and procedural survival was 100%,” co-primary investigator Jason Rogers, MD, of the University of California Davis Medical Center, Sacramento, told attendees at a Late-Breaking Clinical Science session at the Transcatheter Cardiovascular Therapeutics annual meeting.
“The survival was 74% at 12 months. The valve was very effective at eliminating much regurgitation, and 96.5% of patients had either zero or 1+ at a year, and 97% at 30 days had no mitral regurgitation,” he reported. As follow-up was during the COVID-19 pandemic, several of the deaths were attributed to COVID.
Device and trial designs
The Tendyne TMVR is placed through the cardiac apex. It has an outer frame contoured to comport with the shape of the native mitral valve. Inside is a circular, self-expanding, tri-leaflet bioprosthetic valve.
A unique aspect of the design is a tether attached to the outflow side of the valve to allow positioning and control of the valve. At the end of the tether is an apical pad that is placed over the apical access site to control bleeding. The device is currently limited to investigational use in the United States.
The trial enrolled patients with grade III/IV MR or severe MAC if valve anatomy was deemed amenable to transcatheter repair or met MitraClip indications and if these treatments were considered more appropriate than surgery.
Dr. Rogers reported on the first 100 roll-in (early experimental) patients who received Tendyne TMVR. There was a separate severe MAC cohort receiving Tendyne implantation (N = 103). A further 1:1 randomized study of 382 patients compared Tendyne investigational treatment with a MitraClip control group.
At baseline, the 100 roll-in patients had an average age of 75 years, 54% were men, 46% had a frailty score of 2 or greater, and 41% had been hospitalized in the prior 12 months for heart failure. Left ventricular ejection fraction (LVEF) was 48.6% ± 10.3%.
Improved cardiac function
Procedural survival was 100%, technical success 94%, and valve implantation occurred in 97%. Of the first 100 patients, 26 had died by 1 year, and two withdrew consent, leaving 72 for evaluation.
Immediate post-procedure survival was 98%, 87.9% at 3 months, 83.7% at 6 months, and 74.3% at 1 year. MR severity decreased from 29% 3+ and 69% 4+ at baseline to 96.5% 0/1+ and 3.5% 2+ at 1 year.
Cumulative adverse outcomes at 1 year were 27% all-cause mortality, 21.6% cardiovascular mortality, 5.4% all-cause stroke, 2.3% myocardial infarction (MI), 2.2% post-operative mitral reintervention, no major but 2.3% minor device thrombosis, and 32.4% major bleeding.
Most adverse events occurred peri-procedurally or within the first month, representing, “I think, a new procedure with new operators and a high real risk population,” Dr. Rogers said.
Echocardiography at 1 year compared with baseline showed significant changes with decreases in left ventricular end diastolic volume (LVEDV), increases in cardiac output (CO) and forward stroke volume, and no change in mitral valve gradient or left ventricular outflow tract (LVOT) gradient. New York Heart Association (NYHA) classification decreased from 69% class III/IV at baseline to 20% at 1 year, at which point 80% of patients were in class I/II.
“There was a consistent and steady improvement in KCCQ [Kansas City Cardiomyopathy Questionnaire] score, as expected, as patients recovered from this invasive procedure,” Dr. Rogers said. The 1-year score was 68.7, representing fair to good quality of life.
Outcomes with severe MAC
After screening for MR 3+ or greater, severe mitral stenosis, or moderate MR plus mitral stenosis, 103 eligible patients were treated with the Tendyne device. The median MAC volume of the cohort was 4000 mm3, with a maximum of 38,000 mm3.
Patients averaged 78 years old, 44.7% male, 55.3% had a frailty score of 2 or greater, 73.8% were in NYHA class III or greater, and 29.1% had been hospitalized within the prior 12 months for heart failure. Grade III or IV MR severity was present in 89%, with MR being primary in 90.3% of patients, and 10.7% had severe mitral stenosis.
Tendyne procedure survival was 98.1%, technical success was 94.2%, and valves were implanted in all patients. Emergency surgery or other intervention was required in 5.8%.
As co-presenter of the SUMMIT results, Vinod Thourani, MD, of the Piedmont Heart Institute in Atlanta, said at 30 days there was 6.8% all-cause mortality, all of it cardiovascular. There was one disabling stroke, one MI, no device thrombosis, and 21.4% major bleeding.
“At 1 month, there was less than grade 1 mitral regurgitation in all patients,” he reported, vs. 89% grade 3+/4+ at baseline. “At 1 month, it was an improvement in the NYHA classification to almost 70% in class I or II, which was improved from baseline of 26% in NYHA class I or II.”
Hemodynamic parameters all showed improvement, with a reduction in LVEF, LVEDV, and mitral valve gradient and increases in CO and forward stroke volume. There was no significant increase in LVOT gradient.
There was a small improvement in the KCCQ quality of life score from a baseline score of 49.2 to 52.3 at 30 days. “We’re expecting the KCCQ overall score to improve on 1 year follow up since the patients [are] still recovering from their thoracotomy incision,” Dr. Thourani predicted.
The primary endpoint will be evaluated at 1 year post procedure, he said at the meeting, sponsored by the Cardiovascular Research Foundation.
No good option
Designated discussant Joanna Chikwe, MD, chair of cardiac surgery at Cedars-Sinai Medical Center in Los Angeles, first thanked the presenters for their trial, saying, “What an absolute pleasure to be a mitral surgeon at a meeting where you’re presenting a solution for something that we find incredibly challenging. There’s no good transcatheter option for MAC. There’s no great surgical option for MAC.”
She noted the small size of the MAC cohort and asked what drove failure in patient screening, starting with 474 patients, identifying 120 who would be eligible, and enrolling 103 in the MAC cohort. The presenters identified neo-LVOT, the residual LVOT created after implanting the mitral valve prosthesis. Screening also eliminated patients with a too large or too small annulus.
Dr. Thourani said in Europe, surgeons have used anterior leaflet splitting before Tendyne, which may help to expand the population of eligible patients, but no leaflet modification was allowed in the SUMMIT trial.
Dr. Chikwe then pointed to the six deaths in the MAC arm and 11 deaths in the roll-in arm and asked about the mechanism of these deaths. “Was it [that] the 22% major bleeding is transapical? Really the Achilles heel of this procedure? Is this something that could become a transcatheter device?”
“We call it a transcatheter procedure, but it’s very much a surgical procedure,” Dr. Rogers answered. “And, you know, despite having great experienced sites...many surgeons don’t deal with the apex very much.” Furthermore, catheter insertion can lead to bleeding complications.
He noted that the roll-in patients were the first one or two cases at each site, and there have been improvements with site experience. The apical pads assist in hemostasis. He said the current design of the Tendyne catheter-delivered valve does not allow it to be adapted to a transfemoral transseptal approach.
Dr. Rogers is a consultant to and co-national principal investigator of the SUMMIT Pivotal Trial for Abbott. He is a consultant to Boston Scientific and a consultant/equity holder in Laminar. Dr. Thourani has received grant/research support from Abbott Vascular, Artivion, AtriCure, Boston Scientific, Croivalve, Edwards Lifesciences, JenaValve, Medtronic, and Trisol; consultant fees/honoraria from Abbott Vascular, Artivion, AtriCure, Boston Scientific, Croivalve, and Edwards Lifesciences; and has an executive role/ownership interest in DASI Simulations. Dr. Chikwe reports no relevant financial relationships. The SUMMIT trial was sponsored by Abbott.
A version of this article first appeared on Medscape.com.
, relief of mitral regurgitation, and increases in cardiac hemodynamics and quality of life sustained at 1 year.
Further, patients with severe mitral annular calcification (MAC) showed improvements in hemodynamics, functional status, and quality of life after the procedure.
With 70 centers participating in the Tendyne SUMMIT trial, the first 100 trial roll-in patients accrued from the first one or two patients from each site without previous Tendyne TMVR experience.
“For this new procedure, with new operators, there was no intraprocedural mortality, and procedural survival was 100%,” co-primary investigator Jason Rogers, MD, of the University of California Davis Medical Center, Sacramento, told attendees at a Late-Breaking Clinical Science session at the Transcatheter Cardiovascular Therapeutics annual meeting.
“The survival was 74% at 12 months. The valve was very effective at eliminating much regurgitation, and 96.5% of patients had either zero or 1+ at a year, and 97% at 30 days had no mitral regurgitation,” he reported. As follow-up was during the COVID-19 pandemic, several of the deaths were attributed to COVID.
Device and trial designs
The Tendyne TMVR is placed through the cardiac apex. It has an outer frame contoured to comport with the shape of the native mitral valve. Inside is a circular, self-expanding, tri-leaflet bioprosthetic valve.
A unique aspect of the design is a tether attached to the outflow side of the valve to allow positioning and control of the valve. At the end of the tether is an apical pad that is placed over the apical access site to control bleeding. The device is currently limited to investigational use in the United States.
The trial enrolled patients with grade III/IV MR or severe MAC if valve anatomy was deemed amenable to transcatheter repair or met MitraClip indications and if these treatments were considered more appropriate than surgery.
Dr. Rogers reported on the first 100 roll-in (early experimental) patients who received Tendyne TMVR. There was a separate severe MAC cohort receiving Tendyne implantation (N = 103). A further 1:1 randomized study of 382 patients compared Tendyne investigational treatment with a MitraClip control group.
At baseline, the 100 roll-in patients had an average age of 75 years, 54% were men, 46% had a frailty score of 2 or greater, and 41% had been hospitalized in the prior 12 months for heart failure. Left ventricular ejection fraction (LVEF) was 48.6% ± 10.3%.
Improved cardiac function
Procedural survival was 100%, technical success 94%, and valve implantation occurred in 97%. Of the first 100 patients, 26 had died by 1 year, and two withdrew consent, leaving 72 for evaluation.
Immediate post-procedure survival was 98%, 87.9% at 3 months, 83.7% at 6 months, and 74.3% at 1 year. MR severity decreased from 29% 3+ and 69% 4+ at baseline to 96.5% 0/1+ and 3.5% 2+ at 1 year.
Cumulative adverse outcomes at 1 year were 27% all-cause mortality, 21.6% cardiovascular mortality, 5.4% all-cause stroke, 2.3% myocardial infarction (MI), 2.2% post-operative mitral reintervention, no major but 2.3% minor device thrombosis, and 32.4% major bleeding.
Most adverse events occurred peri-procedurally or within the first month, representing, “I think, a new procedure with new operators and a high real risk population,” Dr. Rogers said.
Echocardiography at 1 year compared with baseline showed significant changes with decreases in left ventricular end diastolic volume (LVEDV), increases in cardiac output (CO) and forward stroke volume, and no change in mitral valve gradient or left ventricular outflow tract (LVOT) gradient. New York Heart Association (NYHA) classification decreased from 69% class III/IV at baseline to 20% at 1 year, at which point 80% of patients were in class I/II.
“There was a consistent and steady improvement in KCCQ [Kansas City Cardiomyopathy Questionnaire] score, as expected, as patients recovered from this invasive procedure,” Dr. Rogers said. The 1-year score was 68.7, representing fair to good quality of life.
Outcomes with severe MAC
After screening for MR 3+ or greater, severe mitral stenosis, or moderate MR plus mitral stenosis, 103 eligible patients were treated with the Tendyne device. The median MAC volume of the cohort was 4000 mm3, with a maximum of 38,000 mm3.
Patients averaged 78 years old, 44.7% male, 55.3% had a frailty score of 2 or greater, 73.8% were in NYHA class III or greater, and 29.1% had been hospitalized within the prior 12 months for heart failure. Grade III or IV MR severity was present in 89%, with MR being primary in 90.3% of patients, and 10.7% had severe mitral stenosis.
Tendyne procedure survival was 98.1%, technical success was 94.2%, and valves were implanted in all patients. Emergency surgery or other intervention was required in 5.8%.
As co-presenter of the SUMMIT results, Vinod Thourani, MD, of the Piedmont Heart Institute in Atlanta, said at 30 days there was 6.8% all-cause mortality, all of it cardiovascular. There was one disabling stroke, one MI, no device thrombosis, and 21.4% major bleeding.
“At 1 month, there was less than grade 1 mitral regurgitation in all patients,” he reported, vs. 89% grade 3+/4+ at baseline. “At 1 month, it was an improvement in the NYHA classification to almost 70% in class I or II, which was improved from baseline of 26% in NYHA class I or II.”
Hemodynamic parameters all showed improvement, with a reduction in LVEF, LVEDV, and mitral valve gradient and increases in CO and forward stroke volume. There was no significant increase in LVOT gradient.
There was a small improvement in the KCCQ quality of life score from a baseline score of 49.2 to 52.3 at 30 days. “We’re expecting the KCCQ overall score to improve on 1 year follow up since the patients [are] still recovering from their thoracotomy incision,” Dr. Thourani predicted.
The primary endpoint will be evaluated at 1 year post procedure, he said at the meeting, sponsored by the Cardiovascular Research Foundation.
No good option
Designated discussant Joanna Chikwe, MD, chair of cardiac surgery at Cedars-Sinai Medical Center in Los Angeles, first thanked the presenters for their trial, saying, “What an absolute pleasure to be a mitral surgeon at a meeting where you’re presenting a solution for something that we find incredibly challenging. There’s no good transcatheter option for MAC. There’s no great surgical option for MAC.”
She noted the small size of the MAC cohort and asked what drove failure in patient screening, starting with 474 patients, identifying 120 who would be eligible, and enrolling 103 in the MAC cohort. The presenters identified neo-LVOT, the residual LVOT created after implanting the mitral valve prosthesis. Screening also eliminated patients with a too large or too small annulus.
Dr. Thourani said in Europe, surgeons have used anterior leaflet splitting before Tendyne, which may help to expand the population of eligible patients, but no leaflet modification was allowed in the SUMMIT trial.
Dr. Chikwe then pointed to the six deaths in the MAC arm and 11 deaths in the roll-in arm and asked about the mechanism of these deaths. “Was it [that] the 22% major bleeding is transapical? Really the Achilles heel of this procedure? Is this something that could become a transcatheter device?”
“We call it a transcatheter procedure, but it’s very much a surgical procedure,” Dr. Rogers answered. “And, you know, despite having great experienced sites...many surgeons don’t deal with the apex very much.” Furthermore, catheter insertion can lead to bleeding complications.
He noted that the roll-in patients were the first one or two cases at each site, and there have been improvements with site experience. The apical pads assist in hemostasis. He said the current design of the Tendyne catheter-delivered valve does not allow it to be adapted to a transfemoral transseptal approach.
Dr. Rogers is a consultant to and co-national principal investigator of the SUMMIT Pivotal Trial for Abbott. He is a consultant to Boston Scientific and a consultant/equity holder in Laminar. Dr. Thourani has received grant/research support from Abbott Vascular, Artivion, AtriCure, Boston Scientific, Croivalve, Edwards Lifesciences, JenaValve, Medtronic, and Trisol; consultant fees/honoraria from Abbott Vascular, Artivion, AtriCure, Boston Scientific, Croivalve, and Edwards Lifesciences; and has an executive role/ownership interest in DASI Simulations. Dr. Chikwe reports no relevant financial relationships. The SUMMIT trial was sponsored by Abbott.
A version of this article first appeared on Medscape.com.
, relief of mitral regurgitation, and increases in cardiac hemodynamics and quality of life sustained at 1 year.
Further, patients with severe mitral annular calcification (MAC) showed improvements in hemodynamics, functional status, and quality of life after the procedure.
With 70 centers participating in the Tendyne SUMMIT trial, the first 100 trial roll-in patients accrued from the first one or two patients from each site without previous Tendyne TMVR experience.
“For this new procedure, with new operators, there was no intraprocedural mortality, and procedural survival was 100%,” co-primary investigator Jason Rogers, MD, of the University of California Davis Medical Center, Sacramento, told attendees at a Late-Breaking Clinical Science session at the Transcatheter Cardiovascular Therapeutics annual meeting.
“The survival was 74% at 12 months. The valve was very effective at eliminating much regurgitation, and 96.5% of patients had either zero or 1+ at a year, and 97% at 30 days had no mitral regurgitation,” he reported. As follow-up was during the COVID-19 pandemic, several of the deaths were attributed to COVID.
Device and trial designs
The Tendyne TMVR is placed through the cardiac apex. It has an outer frame contoured to comport with the shape of the native mitral valve. Inside is a circular, self-expanding, tri-leaflet bioprosthetic valve.
A unique aspect of the design is a tether attached to the outflow side of the valve to allow positioning and control of the valve. At the end of the tether is an apical pad that is placed over the apical access site to control bleeding. The device is currently limited to investigational use in the United States.
The trial enrolled patients with grade III/IV MR or severe MAC if valve anatomy was deemed amenable to transcatheter repair or met MitraClip indications and if these treatments were considered more appropriate than surgery.
Dr. Rogers reported on the first 100 roll-in (early experimental) patients who received Tendyne TMVR. There was a separate severe MAC cohort receiving Tendyne implantation (N = 103). A further 1:1 randomized study of 382 patients compared Tendyne investigational treatment with a MitraClip control group.
At baseline, the 100 roll-in patients had an average age of 75 years, 54% were men, 46% had a frailty score of 2 or greater, and 41% had been hospitalized in the prior 12 months for heart failure. Left ventricular ejection fraction (LVEF) was 48.6% ± 10.3%.
Improved cardiac function
Procedural survival was 100%, technical success 94%, and valve implantation occurred in 97%. Of the first 100 patients, 26 had died by 1 year, and two withdrew consent, leaving 72 for evaluation.
Immediate post-procedure survival was 98%, 87.9% at 3 months, 83.7% at 6 months, and 74.3% at 1 year. MR severity decreased from 29% 3+ and 69% 4+ at baseline to 96.5% 0/1+ and 3.5% 2+ at 1 year.
Cumulative adverse outcomes at 1 year were 27% all-cause mortality, 21.6% cardiovascular mortality, 5.4% all-cause stroke, 2.3% myocardial infarction (MI), 2.2% post-operative mitral reintervention, no major but 2.3% minor device thrombosis, and 32.4% major bleeding.
Most adverse events occurred peri-procedurally or within the first month, representing, “I think, a new procedure with new operators and a high real risk population,” Dr. Rogers said.
Echocardiography at 1 year compared with baseline showed significant changes with decreases in left ventricular end diastolic volume (LVEDV), increases in cardiac output (CO) and forward stroke volume, and no change in mitral valve gradient or left ventricular outflow tract (LVOT) gradient. New York Heart Association (NYHA) classification decreased from 69% class III/IV at baseline to 20% at 1 year, at which point 80% of patients were in class I/II.
“There was a consistent and steady improvement in KCCQ [Kansas City Cardiomyopathy Questionnaire] score, as expected, as patients recovered from this invasive procedure,” Dr. Rogers said. The 1-year score was 68.7, representing fair to good quality of life.
Outcomes with severe MAC
After screening for MR 3+ or greater, severe mitral stenosis, or moderate MR plus mitral stenosis, 103 eligible patients were treated with the Tendyne device. The median MAC volume of the cohort was 4000 mm3, with a maximum of 38,000 mm3.
Patients averaged 78 years old, 44.7% male, 55.3% had a frailty score of 2 or greater, 73.8% were in NYHA class III or greater, and 29.1% had been hospitalized within the prior 12 months for heart failure. Grade III or IV MR severity was present in 89%, with MR being primary in 90.3% of patients, and 10.7% had severe mitral stenosis.
Tendyne procedure survival was 98.1%, technical success was 94.2%, and valves were implanted in all patients. Emergency surgery or other intervention was required in 5.8%.
As co-presenter of the SUMMIT results, Vinod Thourani, MD, of the Piedmont Heart Institute in Atlanta, said at 30 days there was 6.8% all-cause mortality, all of it cardiovascular. There was one disabling stroke, one MI, no device thrombosis, and 21.4% major bleeding.
“At 1 month, there was less than grade 1 mitral regurgitation in all patients,” he reported, vs. 89% grade 3+/4+ at baseline. “At 1 month, it was an improvement in the NYHA classification to almost 70% in class I or II, which was improved from baseline of 26% in NYHA class I or II.”
Hemodynamic parameters all showed improvement, with a reduction in LVEF, LVEDV, and mitral valve gradient and increases in CO and forward stroke volume. There was no significant increase in LVOT gradient.
There was a small improvement in the KCCQ quality of life score from a baseline score of 49.2 to 52.3 at 30 days. “We’re expecting the KCCQ overall score to improve on 1 year follow up since the patients [are] still recovering from their thoracotomy incision,” Dr. Thourani predicted.
The primary endpoint will be evaluated at 1 year post procedure, he said at the meeting, sponsored by the Cardiovascular Research Foundation.
No good option
Designated discussant Joanna Chikwe, MD, chair of cardiac surgery at Cedars-Sinai Medical Center in Los Angeles, first thanked the presenters for their trial, saying, “What an absolute pleasure to be a mitral surgeon at a meeting where you’re presenting a solution for something that we find incredibly challenging. There’s no good transcatheter option for MAC. There’s no great surgical option for MAC.”
She noted the small size of the MAC cohort and asked what drove failure in patient screening, starting with 474 patients, identifying 120 who would be eligible, and enrolling 103 in the MAC cohort. The presenters identified neo-LVOT, the residual LVOT created after implanting the mitral valve prosthesis. Screening also eliminated patients with a too large or too small annulus.
Dr. Thourani said in Europe, surgeons have used anterior leaflet splitting before Tendyne, which may help to expand the population of eligible patients, but no leaflet modification was allowed in the SUMMIT trial.
Dr. Chikwe then pointed to the six deaths in the MAC arm and 11 deaths in the roll-in arm and asked about the mechanism of these deaths. “Was it [that] the 22% major bleeding is transapical? Really the Achilles heel of this procedure? Is this something that could become a transcatheter device?”
“We call it a transcatheter procedure, but it’s very much a surgical procedure,” Dr. Rogers answered. “And, you know, despite having great experienced sites...many surgeons don’t deal with the apex very much.” Furthermore, catheter insertion can lead to bleeding complications.
He noted that the roll-in patients were the first one or two cases at each site, and there have been improvements with site experience. The apical pads assist in hemostasis. He said the current design of the Tendyne catheter-delivered valve does not allow it to be adapted to a transfemoral transseptal approach.
Dr. Rogers is a consultant to and co-national principal investigator of the SUMMIT Pivotal Trial for Abbott. He is a consultant to Boston Scientific and a consultant/equity holder in Laminar. Dr. Thourani has received grant/research support from Abbott Vascular, Artivion, AtriCure, Boston Scientific, Croivalve, Edwards Lifesciences, JenaValve, Medtronic, and Trisol; consultant fees/honoraria from Abbott Vascular, Artivion, AtriCure, Boston Scientific, Croivalve, and Edwards Lifesciences; and has an executive role/ownership interest in DASI Simulations. Dr. Chikwe reports no relevant financial relationships. The SUMMIT trial was sponsored by Abbott.
A version of this article first appeared on Medscape.com.
FROM TCT 2023
Less severe strokes with LAA closure vs. DOAC in AFib?
TOPLINE:
Left atrial appendage closure was associated with about half as many disabling or fatal strokes and lower mortality after a stroke, compared with dual oral anticoagulant therapy in patients with atrial fibrillation (AFib), new observational research shows.
METHODOLOGY:
- The retrospective registry analysis included 447 adult patients with nonvalvular AFib, mean age 74 years, who were hospitalized with an ischemic stroke, 322 of whom were receiving direct oral anticoagulant (DOAC) therapy, mostly (84%) apixaban or rivaroxaban, and 125 were treated with left atrial appendage closure (LAAC), almost all (97%) with Watchman or Watchman-FLX devices.
- All patients received standard stroke care, monitoring, and treatment as well as physical therapy/rehabilitation.
- For the primary outcome, researchers used the modified Rankin Scale (mRS) to determine disabling (mRS score of 3-5) and fatal (mRS score of 6) strokes at discharge and at 3 months.
- The study adjusted for age, smoking, paroxysmal AFib, prior major bleeding, prior hemorrhagic stroke, medication adherence, and other risk factors.
TAKEAWAY:
- (38.3% vs. 70.3%; P < .001) and at 3 months (33.3% vs. 56.2%; P < .001), even though the LAAC group had more baseline comorbidity, for example, older age, more smokers, and more prior major bleeding.
- There was no significant difference in mortality between groups during hospitalization, but at 3 months, mortality was lower in the LAAC group (14.7% vs. 32.1%; P = .002).
- Multivariate linear regression analysis showed LAAC independently predicted more favorable mRS at discharge (2.8) and 3 months (1.4) (both P < .001) and was associated with less all-cause death at 3 months (odds ratio, 0.28; 95% confidence interval, 0.12-0.64; P = .002).
- Including those that excluded the 14.4% of LAAC patients who also received DOAC therapy, sensitivity analyses patients who got reduced dose DOACs and nonadherent patients yielded nearly identical outcomes to the full cohort analysis.
IN PRACTICE:
“Despite a higher baseline risk profile, patients treated with LAAC who developed IS had better outcomes than those receiving DOAC prophylaxis,” the authors conclude, adding that several ongoing prospective trials could, “shed light on the mechanism(s) responsible for differences in stroke severity.”
SOURCE:
The study was conducted by Mohit K. Turagam, MD, Icahn School of Medicine at Mount Sinai, New York, and colleagues. It was published online in JACC: Clinical Electrophysiology.
LIMITATIONS:
Despite sensitivity analyses and adjustment for risk factors, selection bias, missing data, and other confounding factors could have affected outcomes. The study didn’t evaluate recurrent IS or type and intensity of rehabilitation on outcomes. Lack of imaging data comparing stroke infarct size and volume limits understanding of exact mechanism driving higher stroke severity with DOACs. Because patients who died before reaching hospital weren’t captured in the registry, the actual mortality may be higher than reported.
DISCLOSURES:
Dr. Turagam has served as a consultant for Biosense Webster and Sanofi.
A version of this article first appeared on Medscape.com.
TOPLINE:
Left atrial appendage closure was associated with about half as many disabling or fatal strokes and lower mortality after a stroke, compared with dual oral anticoagulant therapy in patients with atrial fibrillation (AFib), new observational research shows.
METHODOLOGY:
- The retrospective registry analysis included 447 adult patients with nonvalvular AFib, mean age 74 years, who were hospitalized with an ischemic stroke, 322 of whom were receiving direct oral anticoagulant (DOAC) therapy, mostly (84%) apixaban or rivaroxaban, and 125 were treated with left atrial appendage closure (LAAC), almost all (97%) with Watchman or Watchman-FLX devices.
- All patients received standard stroke care, monitoring, and treatment as well as physical therapy/rehabilitation.
- For the primary outcome, researchers used the modified Rankin Scale (mRS) to determine disabling (mRS score of 3-5) and fatal (mRS score of 6) strokes at discharge and at 3 months.
- The study adjusted for age, smoking, paroxysmal AFib, prior major bleeding, prior hemorrhagic stroke, medication adherence, and other risk factors.
TAKEAWAY:
- (38.3% vs. 70.3%; P < .001) and at 3 months (33.3% vs. 56.2%; P < .001), even though the LAAC group had more baseline comorbidity, for example, older age, more smokers, and more prior major bleeding.
- There was no significant difference in mortality between groups during hospitalization, but at 3 months, mortality was lower in the LAAC group (14.7% vs. 32.1%; P = .002).
- Multivariate linear regression analysis showed LAAC independently predicted more favorable mRS at discharge (2.8) and 3 months (1.4) (both P < .001) and was associated with less all-cause death at 3 months (odds ratio, 0.28; 95% confidence interval, 0.12-0.64; P = .002).
- Including those that excluded the 14.4% of LAAC patients who also received DOAC therapy, sensitivity analyses patients who got reduced dose DOACs and nonadherent patients yielded nearly identical outcomes to the full cohort analysis.
IN PRACTICE:
“Despite a higher baseline risk profile, patients treated with LAAC who developed IS had better outcomes than those receiving DOAC prophylaxis,” the authors conclude, adding that several ongoing prospective trials could, “shed light on the mechanism(s) responsible for differences in stroke severity.”
SOURCE:
The study was conducted by Mohit K. Turagam, MD, Icahn School of Medicine at Mount Sinai, New York, and colleagues. It was published online in JACC: Clinical Electrophysiology.
LIMITATIONS:
Despite sensitivity analyses and adjustment for risk factors, selection bias, missing data, and other confounding factors could have affected outcomes. The study didn’t evaluate recurrent IS or type and intensity of rehabilitation on outcomes. Lack of imaging data comparing stroke infarct size and volume limits understanding of exact mechanism driving higher stroke severity with DOACs. Because patients who died before reaching hospital weren’t captured in the registry, the actual mortality may be higher than reported.
DISCLOSURES:
Dr. Turagam has served as a consultant for Biosense Webster and Sanofi.
A version of this article first appeared on Medscape.com.
TOPLINE:
Left atrial appendage closure was associated with about half as many disabling or fatal strokes and lower mortality after a stroke, compared with dual oral anticoagulant therapy in patients with atrial fibrillation (AFib), new observational research shows.
METHODOLOGY:
- The retrospective registry analysis included 447 adult patients with nonvalvular AFib, mean age 74 years, who were hospitalized with an ischemic stroke, 322 of whom were receiving direct oral anticoagulant (DOAC) therapy, mostly (84%) apixaban or rivaroxaban, and 125 were treated with left atrial appendage closure (LAAC), almost all (97%) with Watchman or Watchman-FLX devices.
- All patients received standard stroke care, monitoring, and treatment as well as physical therapy/rehabilitation.
- For the primary outcome, researchers used the modified Rankin Scale (mRS) to determine disabling (mRS score of 3-5) and fatal (mRS score of 6) strokes at discharge and at 3 months.
- The study adjusted for age, smoking, paroxysmal AFib, prior major bleeding, prior hemorrhagic stroke, medication adherence, and other risk factors.
TAKEAWAY:
- (38.3% vs. 70.3%; P < .001) and at 3 months (33.3% vs. 56.2%; P < .001), even though the LAAC group had more baseline comorbidity, for example, older age, more smokers, and more prior major bleeding.
- There was no significant difference in mortality between groups during hospitalization, but at 3 months, mortality was lower in the LAAC group (14.7% vs. 32.1%; P = .002).
- Multivariate linear regression analysis showed LAAC independently predicted more favorable mRS at discharge (2.8) and 3 months (1.4) (both P < .001) and was associated with less all-cause death at 3 months (odds ratio, 0.28; 95% confidence interval, 0.12-0.64; P = .002).
- Including those that excluded the 14.4% of LAAC patients who also received DOAC therapy, sensitivity analyses patients who got reduced dose DOACs and nonadherent patients yielded nearly identical outcomes to the full cohort analysis.
IN PRACTICE:
“Despite a higher baseline risk profile, patients treated with LAAC who developed IS had better outcomes than those receiving DOAC prophylaxis,” the authors conclude, adding that several ongoing prospective trials could, “shed light on the mechanism(s) responsible for differences in stroke severity.”
SOURCE:
The study was conducted by Mohit K. Turagam, MD, Icahn School of Medicine at Mount Sinai, New York, and colleagues. It was published online in JACC: Clinical Electrophysiology.
LIMITATIONS:
Despite sensitivity analyses and adjustment for risk factors, selection bias, missing data, and other confounding factors could have affected outcomes. The study didn’t evaluate recurrent IS or type and intensity of rehabilitation on outcomes. Lack of imaging data comparing stroke infarct size and volume limits understanding of exact mechanism driving higher stroke severity with DOACs. Because patients who died before reaching hospital weren’t captured in the registry, the actual mortality may be higher than reported.
DISCLOSURES:
Dr. Turagam has served as a consultant for Biosense Webster and Sanofi.
A version of this article first appeared on Medscape.com.
Alternative antirejection regimen is efficacious in pediatric heart transplant
Study challenges everolimus boxed warning
according to the first phase 3 trial to compare antirejection strategies in the pediatric setting.
Even though MMF and tacrolimus have never been evaluated for pediatric cardiac transplant in a controlled trial, this combination is widely considered a standard based on adult data, said Christopher Almond, MD, a professor of pediatric cardiology at Stanford (Calif.) Medicine.
Everolimus has not been widely used in an antirejection regimen in children following heart transplant in part because of a boxed warning. The warning was added to labeling when this agent was associated with increased infection and increased mortality in adults if given within 3 months of transplant.
In this non-inferiority trial, called TEAMMATE, patients were randomized to the MMF-based or everolimus-based regimen 6 months after transplant.
Everolimus- vs. MMF-based antirejection
The study enrolled 210 children and adolescents 21 years of age or younger. The control arm treatment consisted of MMF (660 mg/m2 every 12 hours) plus standard dose of tacrolimus (initially 7-10 ng/mL followed at 6 months by 5-8 ng/mL).
In the experimental arm, patients received everolimus (3-8 ng/mL) plus a low dose of tacrolimus (initially 3-5 ng/mL followed at 6 months by 2.5-4.5 ng/mL).
The primary endpoint was score on the major adverse transplant event (MATE-6) tool. Based on gradations of severity, this assigns values for cardiac allograft vasculopathy (CAV), chronic kidney disease (CKD), acute cellular rejection (ACR), antibody-mediated rejection, infection, and posttransplant lymphoproliferative disorder (PTLD).
Thirty months after randomization, the MATE-6 scores were 1.96 in the everolimus group and 2.18 in the MMF group, which conferred the everolimus-based regimen with a numerical but not a significant advantage over the MMF-based regimen. For the goal of noninferiority, the everolimus regimen “met the prespecified safety criterion for success,” Dr. Almond said.
Numerical advantage for everolimus on efficacy
The primary efficacy endpoint was the MATE-3 score, which is limited to CAV, CKD, and ACR. Again, the mean score on this metric (0.93 vs. 1.25) was lower on the everolimus-based regimen but not significantly different.
Looking at specific events in the MATE-6 score, the everolimus-based regimen was associated with lower numerical rates of CAV and CKD, but a higher rate of PTLD, Dr. Almond reported.
On the MATE-3 efficacy analysis, the everolimus-based regimen was again associated with lower numerical rates of CAV and CKD but higher rates of ACR.
In terms of adverse events, including those involving the gastrointestinal tract, blood cells, proteinuria, and interstitial lung disease, most did not differ markedly even if many were numerically more common in the MMF-based arm. The exception was aphthous stomatitis, which was more common on everolimus (32% vs. 7%; P < .001). There were more discontinuations for an adverse event in the MMF arm (21% vs. 12%; P < .001).
Other differences included a lower proportion of patients in the everolimus arm with anti-HLA antibodies (17% vs. 30%; P < .05). Total cholesterol levels at the end of the study were lower but not significantly different in the MMF group, while the higher median glomerular filtration rate was higher on everolimus, and this did reach statistical significance (P < .05).
Infection rates overall were similar, but cytomegalovirus (CMV) infection was more common on the MMF-based regimen. The 30% lower rate of CMV infection in the everolimus proved to be potentially clinically meaningful when it was considered in the context of MATE-3. When these two endpoints were combined (MATE-3 and CMV infection as a prespecified secondary endpoint, the difference was statistically significant (P = .03) in favor of the everolimus-based regimen,
Study supports safety of everolimus regimen
The take-home message is that the everolimus-based regimen, which “is safe in children and young adults when initiated at 6 months after transplant,” can be considered as an alternative to MFF, Dr. Almond concluded.
However, one of the coauthors of the study, Joseph Rossano, MD, chief of the division of cardiology, Children’s Hospital of Philadelphia, suggested a stronger message.
“These data provide compelling reasons to consider initiation of the combination of everolimus and tacrolimus at 6 months post transplant in pediatric heart transplant recipients,” Dr. Rossano said.
Even though the everolimus-based regimen met the terms of noninferiority overall, patients who received this combination rather than the MMF-based regimen “were less likely to have the combined endpoints of vasculopathy, CKD, rejection and CMV infection. Additionally, they were less likely to make donor specific antibodies,” he said.
He also said that this study challenges the current boxed warning for everolimus. He pointed out that the warning, based on early use of everolimus in adults, does not appear to be an issue for children treated at 6 months.
Early mortality based on infection “was not observed in our study,” he said.
The AHA-invited discussant, Antonio G. Cabrera, MD, division chief of pediatric cardiology, University of Utah, Salt Lake City, drew the same conclusions. Based on the study, the everolimus-based regimen can only be described as noninferior to the MMF-based regimen, but Dr. Cabrera listed the same relative advantages as Dr. Rossano, including better kidney function.
Overall, either regimen might be more appealing based on several variables, but Dr. Cabrera said these data suggest everolimus-based treatment “should be considered” as one of two evidence-based options,
Dr. Almond reported no potential financial conflicts of interest. Dr. Rossano reports financial relationships with Abiomed, Bayer, Cytokinetics, Merck, and Myokardia. Dr. Cabrera reported no potential financial conflicts of interest.
Study challenges everolimus boxed warning
Study challenges everolimus boxed warning
according to the first phase 3 trial to compare antirejection strategies in the pediatric setting.
Even though MMF and tacrolimus have never been evaluated for pediatric cardiac transplant in a controlled trial, this combination is widely considered a standard based on adult data, said Christopher Almond, MD, a professor of pediatric cardiology at Stanford (Calif.) Medicine.
Everolimus has not been widely used in an antirejection regimen in children following heart transplant in part because of a boxed warning. The warning was added to labeling when this agent was associated with increased infection and increased mortality in adults if given within 3 months of transplant.
In this non-inferiority trial, called TEAMMATE, patients were randomized to the MMF-based or everolimus-based regimen 6 months after transplant.
Everolimus- vs. MMF-based antirejection
The study enrolled 210 children and adolescents 21 years of age or younger. The control arm treatment consisted of MMF (660 mg/m2 every 12 hours) plus standard dose of tacrolimus (initially 7-10 ng/mL followed at 6 months by 5-8 ng/mL).
In the experimental arm, patients received everolimus (3-8 ng/mL) plus a low dose of tacrolimus (initially 3-5 ng/mL followed at 6 months by 2.5-4.5 ng/mL).
The primary endpoint was score on the major adverse transplant event (MATE-6) tool. Based on gradations of severity, this assigns values for cardiac allograft vasculopathy (CAV), chronic kidney disease (CKD), acute cellular rejection (ACR), antibody-mediated rejection, infection, and posttransplant lymphoproliferative disorder (PTLD).
Thirty months after randomization, the MATE-6 scores were 1.96 in the everolimus group and 2.18 in the MMF group, which conferred the everolimus-based regimen with a numerical but not a significant advantage over the MMF-based regimen. For the goal of noninferiority, the everolimus regimen “met the prespecified safety criterion for success,” Dr. Almond said.
Numerical advantage for everolimus on efficacy
The primary efficacy endpoint was the MATE-3 score, which is limited to CAV, CKD, and ACR. Again, the mean score on this metric (0.93 vs. 1.25) was lower on the everolimus-based regimen but not significantly different.
Looking at specific events in the MATE-6 score, the everolimus-based regimen was associated with lower numerical rates of CAV and CKD, but a higher rate of PTLD, Dr. Almond reported.
On the MATE-3 efficacy analysis, the everolimus-based regimen was again associated with lower numerical rates of CAV and CKD but higher rates of ACR.
In terms of adverse events, including those involving the gastrointestinal tract, blood cells, proteinuria, and interstitial lung disease, most did not differ markedly even if many were numerically more common in the MMF-based arm. The exception was aphthous stomatitis, which was more common on everolimus (32% vs. 7%; P < .001). There were more discontinuations for an adverse event in the MMF arm (21% vs. 12%; P < .001).
Other differences included a lower proportion of patients in the everolimus arm with anti-HLA antibodies (17% vs. 30%; P < .05). Total cholesterol levels at the end of the study were lower but not significantly different in the MMF group, while the higher median glomerular filtration rate was higher on everolimus, and this did reach statistical significance (P < .05).
Infection rates overall were similar, but cytomegalovirus (CMV) infection was more common on the MMF-based regimen. The 30% lower rate of CMV infection in the everolimus proved to be potentially clinically meaningful when it was considered in the context of MATE-3. When these two endpoints were combined (MATE-3 and CMV infection as a prespecified secondary endpoint, the difference was statistically significant (P = .03) in favor of the everolimus-based regimen,
Study supports safety of everolimus regimen
The take-home message is that the everolimus-based regimen, which “is safe in children and young adults when initiated at 6 months after transplant,” can be considered as an alternative to MFF, Dr. Almond concluded.
However, one of the coauthors of the study, Joseph Rossano, MD, chief of the division of cardiology, Children’s Hospital of Philadelphia, suggested a stronger message.
“These data provide compelling reasons to consider initiation of the combination of everolimus and tacrolimus at 6 months post transplant in pediatric heart transplant recipients,” Dr. Rossano said.
Even though the everolimus-based regimen met the terms of noninferiority overall, patients who received this combination rather than the MMF-based regimen “were less likely to have the combined endpoints of vasculopathy, CKD, rejection and CMV infection. Additionally, they were less likely to make donor specific antibodies,” he said.
He also said that this study challenges the current boxed warning for everolimus. He pointed out that the warning, based on early use of everolimus in adults, does not appear to be an issue for children treated at 6 months.
Early mortality based on infection “was not observed in our study,” he said.
The AHA-invited discussant, Antonio G. Cabrera, MD, division chief of pediatric cardiology, University of Utah, Salt Lake City, drew the same conclusions. Based on the study, the everolimus-based regimen can only be described as noninferior to the MMF-based regimen, but Dr. Cabrera listed the same relative advantages as Dr. Rossano, including better kidney function.
Overall, either regimen might be more appealing based on several variables, but Dr. Cabrera said these data suggest everolimus-based treatment “should be considered” as one of two evidence-based options,
Dr. Almond reported no potential financial conflicts of interest. Dr. Rossano reports financial relationships with Abiomed, Bayer, Cytokinetics, Merck, and Myokardia. Dr. Cabrera reported no potential financial conflicts of interest.
according to the first phase 3 trial to compare antirejection strategies in the pediatric setting.
Even though MMF and tacrolimus have never been evaluated for pediatric cardiac transplant in a controlled trial, this combination is widely considered a standard based on adult data, said Christopher Almond, MD, a professor of pediatric cardiology at Stanford (Calif.) Medicine.
Everolimus has not been widely used in an antirejection regimen in children following heart transplant in part because of a boxed warning. The warning was added to labeling when this agent was associated with increased infection and increased mortality in adults if given within 3 months of transplant.
In this non-inferiority trial, called TEAMMATE, patients were randomized to the MMF-based or everolimus-based regimen 6 months after transplant.
Everolimus- vs. MMF-based antirejection
The study enrolled 210 children and adolescents 21 years of age or younger. The control arm treatment consisted of MMF (660 mg/m2 every 12 hours) plus standard dose of tacrolimus (initially 7-10 ng/mL followed at 6 months by 5-8 ng/mL).
In the experimental arm, patients received everolimus (3-8 ng/mL) plus a low dose of tacrolimus (initially 3-5 ng/mL followed at 6 months by 2.5-4.5 ng/mL).
The primary endpoint was score on the major adverse transplant event (MATE-6) tool. Based on gradations of severity, this assigns values for cardiac allograft vasculopathy (CAV), chronic kidney disease (CKD), acute cellular rejection (ACR), antibody-mediated rejection, infection, and posttransplant lymphoproliferative disorder (PTLD).
Thirty months after randomization, the MATE-6 scores were 1.96 in the everolimus group and 2.18 in the MMF group, which conferred the everolimus-based regimen with a numerical but not a significant advantage over the MMF-based regimen. For the goal of noninferiority, the everolimus regimen “met the prespecified safety criterion for success,” Dr. Almond said.
Numerical advantage for everolimus on efficacy
The primary efficacy endpoint was the MATE-3 score, which is limited to CAV, CKD, and ACR. Again, the mean score on this metric (0.93 vs. 1.25) was lower on the everolimus-based regimen but not significantly different.
Looking at specific events in the MATE-6 score, the everolimus-based regimen was associated with lower numerical rates of CAV and CKD, but a higher rate of PTLD, Dr. Almond reported.
On the MATE-3 efficacy analysis, the everolimus-based regimen was again associated with lower numerical rates of CAV and CKD but higher rates of ACR.
In terms of adverse events, including those involving the gastrointestinal tract, blood cells, proteinuria, and interstitial lung disease, most did not differ markedly even if many were numerically more common in the MMF-based arm. The exception was aphthous stomatitis, which was more common on everolimus (32% vs. 7%; P < .001). There were more discontinuations for an adverse event in the MMF arm (21% vs. 12%; P < .001).
Other differences included a lower proportion of patients in the everolimus arm with anti-HLA antibodies (17% vs. 30%; P < .05). Total cholesterol levels at the end of the study were lower but not significantly different in the MMF group, while the higher median glomerular filtration rate was higher on everolimus, and this did reach statistical significance (P < .05).
Infection rates overall were similar, but cytomegalovirus (CMV) infection was more common on the MMF-based regimen. The 30% lower rate of CMV infection in the everolimus proved to be potentially clinically meaningful when it was considered in the context of MATE-3. When these two endpoints were combined (MATE-3 and CMV infection as a prespecified secondary endpoint, the difference was statistically significant (P = .03) in favor of the everolimus-based regimen,
Study supports safety of everolimus regimen
The take-home message is that the everolimus-based regimen, which “is safe in children and young adults when initiated at 6 months after transplant,” can be considered as an alternative to MFF, Dr. Almond concluded.
However, one of the coauthors of the study, Joseph Rossano, MD, chief of the division of cardiology, Children’s Hospital of Philadelphia, suggested a stronger message.
“These data provide compelling reasons to consider initiation of the combination of everolimus and tacrolimus at 6 months post transplant in pediatric heart transplant recipients,” Dr. Rossano said.
Even though the everolimus-based regimen met the terms of noninferiority overall, patients who received this combination rather than the MMF-based regimen “were less likely to have the combined endpoints of vasculopathy, CKD, rejection and CMV infection. Additionally, they were less likely to make donor specific antibodies,” he said.
He also said that this study challenges the current boxed warning for everolimus. He pointed out that the warning, based on early use of everolimus in adults, does not appear to be an issue for children treated at 6 months.
Early mortality based on infection “was not observed in our study,” he said.
The AHA-invited discussant, Antonio G. Cabrera, MD, division chief of pediatric cardiology, University of Utah, Salt Lake City, drew the same conclusions. Based on the study, the everolimus-based regimen can only be described as noninferior to the MMF-based regimen, but Dr. Cabrera listed the same relative advantages as Dr. Rossano, including better kidney function.
Overall, either regimen might be more appealing based on several variables, but Dr. Cabrera said these data suggest everolimus-based treatment “should be considered” as one of two evidence-based options,
Dr. Almond reported no potential financial conflicts of interest. Dr. Rossano reports financial relationships with Abiomed, Bayer, Cytokinetics, Merck, and Myokardia. Dr. Cabrera reported no potential financial conflicts of interest.
FROM AHA 2023
AI-ECG gets STEMI patients to cath lab sooner
PHILADELPHIA – An artificial intelligence platform that sends alerts based on electrocardiography results enabled cardiologists and emergency department physicians at a major hospital in Taiwan to move patients with ST-elevation myocardial infarction (STEMI) into the catheterization laboratory 9 minutes sooner than the conventional protocol that did not use AI.
“This is the first randomized clinical trial to demonstrate the reduction of electrocardiography to coronary cath lab activation time" from 52.3 to 43.3 minutes (P = .003), Chin Sheng Lin, MD, PhD, director of cardiology at the National Defense Medical Center Tri-Service General Hospital in Taipei City, said in presenting the results at the American Heart Association scientific sessions.
Dr. Lin reported results from the Artificial Intelligence Enabled Rapid Identify of ST-Elevation Myocardial Infarction Using Electrocardiogram (ARISE) trial. The trial included 43,994 patients who came to the hospital’s emergency and inpatient departments with at least one ECG but no history of coronary angiography (CAG) in the previous 3 days between May 2022 and April 2023.
They were randomly assigned by date to either AI-ECG for rapid identification and triage of STEMI or standard care. Overall, 145 patients were finally diagnosed with STEMI based on CAG, 77 in the intervention group and 68 in the control group. All patients were seen by one of 20 cardiologists who participated in the study.
Dr. Lin and his group developed an AI algorithm that captures the ECG readout in the emergency department, analyzes the data and then sends a high-risk alarm to the front-line physician and on-duty cardiologist to activate the primary percutaneous coronary intervention (PCI).
Trial results
The differentiation between groups was even more pronounced in ED patients during regular working hours, Dr. Lin said, at 61.6 minutes for the intervention group vs. 33.1 minutes for controls (P = .001).*
He noted that the AI group showed a trend towards fewer cases of clinically suspected STEMI but not getting CAG, 6.5% vs. 15.8%, for an odds ratio of 0.37 (95% confidence interval, 0.14-0.94).
The AI-ECG model also demonstrated a high diagnostic accuracy. “With this AI-ECG system, because it has a very high accuracy and a high positive predictive variable that reach 88%, we can send a message to the on-duty cardiologists and also the emergency room physician and they can send the patients to receive the operation or the PCI as soon as possible,” Dr. Lin said in an interview.
The time differential is critical, Dr. Lin said. “For the patient with acute myocardial infarction, 1 minute is critical, because the patients can die within minutes,” he said. “If we can save 9 minutes I think we can save more lives, but it needs a larger study to evaluate that.”
Dr. Lin acknowledged a few limitations with the trial, among them its single-center nature, relatively small sample size of STEMI patients and the short-term of follow-up. Future study should involve multiple centers along with a prehospital, emergency medical services AI-ECG model.
‘Novel’ for an AI trial
“This is an incredible application of an AI technology in a real-world problem,” said Brahmajee K. Nallamothu, MD, MPH, an interventional cardiologist at the University of Michigan, Ann Arbor, who did not participate in the study. “What I really love about this study is it’s actually a clinical problem that has large implications, particularly for under-resourced areas.”

Using a randomized clinical trial to evaluate the AI platform is “very, very novel,” he said, and called the time improvement “enormous.” Referencing Dr. Lin’s next steps for studying the AI-ECG platform, Dr. Nallamothu said, “if we could push this up even earlier to paramedics and EMTs and prehospital systems, there would be a lot of excitement there.”
He noted the sensitivity analysis resulted in a rate of 88.8% along with the positive predictive value of 88%. “Missing 1 out of 10 ST-elevation MIs in my eyes can still be considered a big deal, so we need to know if this is happening in particular types of patients, for example women versus men, or other groups.”
However, some investigations reported false activation rates as high as 33%, he said. “So, to say that, the positive predictive value is at 88% is really exciting and I think it can make a real inroads,” Dr. Nallamothu said.
Dr. Lin and Dr. Nallamothu have no relevant disclosures.
*Correction, 11/20/23: An earlier version of this article misstated in both trial arms the time to coronary catheterization lab activation.
PHILADELPHIA – An artificial intelligence platform that sends alerts based on electrocardiography results enabled cardiologists and emergency department physicians at a major hospital in Taiwan to move patients with ST-elevation myocardial infarction (STEMI) into the catheterization laboratory 9 minutes sooner than the conventional protocol that did not use AI.
“This is the first randomized clinical trial to demonstrate the reduction of electrocardiography to coronary cath lab activation time" from 52.3 to 43.3 minutes (P = .003), Chin Sheng Lin, MD, PhD, director of cardiology at the National Defense Medical Center Tri-Service General Hospital in Taipei City, said in presenting the results at the American Heart Association scientific sessions.
Dr. Lin reported results from the Artificial Intelligence Enabled Rapid Identify of ST-Elevation Myocardial Infarction Using Electrocardiogram (ARISE) trial. The trial included 43,994 patients who came to the hospital’s emergency and inpatient departments with at least one ECG but no history of coronary angiography (CAG) in the previous 3 days between May 2022 and April 2023.
They were randomly assigned by date to either AI-ECG for rapid identification and triage of STEMI or standard care. Overall, 145 patients were finally diagnosed with STEMI based on CAG, 77 in the intervention group and 68 in the control group. All patients were seen by one of 20 cardiologists who participated in the study.
Dr. Lin and his group developed an AI algorithm that captures the ECG readout in the emergency department, analyzes the data and then sends a high-risk alarm to the front-line physician and on-duty cardiologist to activate the primary percutaneous coronary intervention (PCI).
Trial results
The differentiation between groups was even more pronounced in ED patients during regular working hours, Dr. Lin said, at 61.6 minutes for the intervention group vs. 33.1 minutes for controls (P = .001).*
He noted that the AI group showed a trend towards fewer cases of clinically suspected STEMI but not getting CAG, 6.5% vs. 15.8%, for an odds ratio of 0.37 (95% confidence interval, 0.14-0.94).
The AI-ECG model also demonstrated a high diagnostic accuracy. “With this AI-ECG system, because it has a very high accuracy and a high positive predictive variable that reach 88%, we can send a message to the on-duty cardiologists and also the emergency room physician and they can send the patients to receive the operation or the PCI as soon as possible,” Dr. Lin said in an interview.
The time differential is critical, Dr. Lin said. “For the patient with acute myocardial infarction, 1 minute is critical, because the patients can die within minutes,” he said. “If we can save 9 minutes I think we can save more lives, but it needs a larger study to evaluate that.”
Dr. Lin acknowledged a few limitations with the trial, among them its single-center nature, relatively small sample size of STEMI patients and the short-term of follow-up. Future study should involve multiple centers along with a prehospital, emergency medical services AI-ECG model.
‘Novel’ for an AI trial
“This is an incredible application of an AI technology in a real-world problem,” said Brahmajee K. Nallamothu, MD, MPH, an interventional cardiologist at the University of Michigan, Ann Arbor, who did not participate in the study. “What I really love about this study is it’s actually a clinical problem that has large implications, particularly for under-resourced areas.”
Using a randomized clinical trial to evaluate the AI platform is “very, very novel,” he said, and called the time improvement “enormous.” Referencing Dr. Lin’s next steps for studying the AI-ECG platform, Dr. Nallamothu said, “if we could push this up even earlier to paramedics and EMTs and prehospital systems, there would be a lot of excitement there.”
He noted the sensitivity analysis resulted in a rate of 88.8% along with the positive predictive value of 88%. “Missing 1 out of 10 ST-elevation MIs in my eyes can still be considered a big deal, so we need to know if this is happening in particular types of patients, for example women versus men, or other groups.”
However, some investigations reported false activation rates as high as 33%, he said. “So, to say that, the positive predictive value is at 88% is really exciting and I think it can make a real inroads,” Dr. Nallamothu said.
Dr. Lin and Dr. Nallamothu have no relevant disclosures.
*Correction, 11/20/23: An earlier version of this article misstated in both trial arms the time to coronary catheterization lab activation.
PHILADELPHIA – An artificial intelligence platform that sends alerts based on electrocardiography results enabled cardiologists and emergency department physicians at a major hospital in Taiwan to move patients with ST-elevation myocardial infarction (STEMI) into the catheterization laboratory 9 minutes sooner than the conventional protocol that did not use AI.
“This is the first randomized clinical trial to demonstrate the reduction of electrocardiography to coronary cath lab activation time" from 52.3 to 43.3 minutes (P = .003), Chin Sheng Lin, MD, PhD, director of cardiology at the National Defense Medical Center Tri-Service General Hospital in Taipei City, said in presenting the results at the American Heart Association scientific sessions.
Dr. Lin reported results from the Artificial Intelligence Enabled Rapid Identify of ST-Elevation Myocardial Infarction Using Electrocardiogram (ARISE) trial. The trial included 43,994 patients who came to the hospital’s emergency and inpatient departments with at least one ECG but no history of coronary angiography (CAG) in the previous 3 days between May 2022 and April 2023.
They were randomly assigned by date to either AI-ECG for rapid identification and triage of STEMI or standard care. Overall, 145 patients were finally diagnosed with STEMI based on CAG, 77 in the intervention group and 68 in the control group. All patients were seen by one of 20 cardiologists who participated in the study.
Dr. Lin and his group developed an AI algorithm that captures the ECG readout in the emergency department, analyzes the data and then sends a high-risk alarm to the front-line physician and on-duty cardiologist to activate the primary percutaneous coronary intervention (PCI).
Trial results
The differentiation between groups was even more pronounced in ED patients during regular working hours, Dr. Lin said, at 61.6 minutes for the intervention group vs. 33.1 minutes for controls (P = .001).*
He noted that the AI group showed a trend towards fewer cases of clinically suspected STEMI but not getting CAG, 6.5% vs. 15.8%, for an odds ratio of 0.37 (95% confidence interval, 0.14-0.94).
The AI-ECG model also demonstrated a high diagnostic accuracy. “With this AI-ECG system, because it has a very high accuracy and a high positive predictive variable that reach 88%, we can send a message to the on-duty cardiologists and also the emergency room physician and they can send the patients to receive the operation or the PCI as soon as possible,” Dr. Lin said in an interview.
The time differential is critical, Dr. Lin said. “For the patient with acute myocardial infarction, 1 minute is critical, because the patients can die within minutes,” he said. “If we can save 9 minutes I think we can save more lives, but it needs a larger study to evaluate that.”
Dr. Lin acknowledged a few limitations with the trial, among them its single-center nature, relatively small sample size of STEMI patients and the short-term of follow-up. Future study should involve multiple centers along with a prehospital, emergency medical services AI-ECG model.
‘Novel’ for an AI trial
“This is an incredible application of an AI technology in a real-world problem,” said Brahmajee K. Nallamothu, MD, MPH, an interventional cardiologist at the University of Michigan, Ann Arbor, who did not participate in the study. “What I really love about this study is it’s actually a clinical problem that has large implications, particularly for under-resourced areas.”
Using a randomized clinical trial to evaluate the AI platform is “very, very novel,” he said, and called the time improvement “enormous.” Referencing Dr. Lin’s next steps for studying the AI-ECG platform, Dr. Nallamothu said, “if we could push this up even earlier to paramedics and EMTs and prehospital systems, there would be a lot of excitement there.”
He noted the sensitivity analysis resulted in a rate of 88.8% along with the positive predictive value of 88%. “Missing 1 out of 10 ST-elevation MIs in my eyes can still be considered a big deal, so we need to know if this is happening in particular types of patients, for example women versus men, or other groups.”
However, some investigations reported false activation rates as high as 33%, he said. “So, to say that, the positive predictive value is at 88% is really exciting and I think it can make a real inroads,” Dr. Nallamothu said.
Dr. Lin and Dr. Nallamothu have no relevant disclosures.
*Correction, 11/20/23: An earlier version of this article misstated in both trial arms the time to coronary catheterization lab activation.
AT AHA 2023
Angioplasty finally proven beneficial in stable angina: ORBITA-2
PHILADELPHIA – Percutaneous coronary intervention (PCI) in patients with stable coronary artery disease (CAD) reduces angina frequency, increases exercise capacity, and improves quality of life, results of a placebo-controlled, randomized trial show, confirming advantages that have never before been proven.
reported Christopher A. Rajkumar, MBBS, an interventional cardiology registrar at the Imperial College Healthcare Trust, London.
Results of the trial, ORBITA-2, were presented at the annual scientific sessions of the American Heart Association and simultaneously published online in the New England Journal of Medicine.
Symptom relief has long been a justification for PCI in patients with stable CAD, but the evidence has been derived from uncontrolled studies, Dr. Rajkumar said. However, the first ORBITA trial, which was also placebo controlled and randomized, failed to show benefit.
Dr. Rajkumar acknowledged that the benefit of PCI in ORBITA-2 was lower than previously reported in nonrandomized trials. He also noted that 59% of patients still had at least some angina symptoms following PCI.
Even though ORBITA-2 proves that PCI is better than no PCI, he agreed that well-informed patients, such as those who wish to avoid an invasive procedure, might still reasonably select antianginal medication over PCI. Current guidelines recommend PCI for patients with refractory angina despite medical therapy.
While Dr. Rajkumar was unwilling to speculate on how these data might change guidelines, he did say that patients with stable CAD and angina “now have a choice of two first-line evidence-based pathways.”
‘Remarkable’ trial
“ORBITA 2 is a rather remarkable trial because my surgical colleagues have been asking me for many decades whether PCI actually works,” said Martin B. Leon, MD, professor of medicine, Columbia University Irving Medical Center, New York. “Now I can say with confidence on the basis of a placebo-controlled trial that PCI certainly does have a favorable impact in patients with documented angina, severe coronary stenosis, and demonstrated ischemia.”
The key enrollment criteria for ORBITA-2 were angina, severe coronary stenosis in at least one vessel, and ischemia on stress imaging or invasive physiology. Unlike the previous ORBITA trial, which was limited to single-vessel disease and did not require objective evidence of ischemia, ORBITA 2 employed change in angina, rather than improved exercise capacity, as its primary endpoint.
Relative to sham PCI, patients randomly assigned to an interventional procedure had a more than twofold increase in the odds ratio of improved angina control (OR, 2.2; P < .001) based on a patient scoring system that captured angina symptoms as well as angina medication use on a smartphone application.
The advantage of PCI over sham PCI was also significant for all secondary outcomes. These included a nearly fourfold greater (OR, 3.76; P < .001) likelihood of improvement in the Canadian Cardiovascular Society angina grade and a 1-minute increase (from 10 min. 40 seconds to 11 min. 40 seconds) in treadmill exercise time (P = .008).
On quality of life measured with the self-assessment questionnaire and the EQ-5D-5L, almost all endpoints were highly statistically significant in favor of PCI (typically on the level of P < .001).
The study had a bold design: At enrollment patients stopped all antianginal medications to undergo dobutamine echocardiography and other baseline tests. They were stopped again 2 weeks later, when patients were randomized.
With a study protocol that enrolled patients off medication, “we intentionally diverged from the clinical guidelines,” Dr. Rajkumar said.
Of the 439 patients enrolled, 301 were randomly assigned at the end of the 2-week period, when patients were already sedated. Control patients remained sedated for at least 15 minutes. All 151 of those randomized to PCI and the 150 control patients were available for the intent-to-treat analysis at the end of 12 weeks.
The novel angina symptom burden score was created from daily angina episodes and units of daily antianginal medication captured on the smartphone app. On an ordinal scale, a score of 0 on any given day represented no anginal symptoms and no antianginal medication.
As angina severity or medication use increased, it raised the daily scores. If there was unacceptable angina (requiring the patient to be removed from the blind), acute coronary syndrome, or death, it produced the highest scores, which reached a maximum of 79.
The favorable OR for a lower symptom burden in the PCI group reflected a relative reduction in angina observed the first day after the procedure. Over the entire follow-up, more patients in the PCI group had an angina score of 0 and more of those who had angina did not take antianginal medications.
This objective evidence that PCI reduces symptoms and improves quality of life in patients with angina and stable CAD was met at the AHA late-breaking session with a sustained ovation.
ORBITA-2 addresses ORBITA criticisms
Connie N. Hess, MD, the AHA-invited discussant and an interventional cardiologist at the University of Colorado Medicine, Aurora, provided perspective on the differences between ORBITA 2 and ORBITA, which she said “addressed a fundamentally different hypothesis” by focusing on angina rather than exercise capacity.
Of the criticisms of the original ORBITA, which Dr. Hess noted was the first sham-controlled PCI trial ever conducted in stable CAD, one is that patients with multivessel disease were excluded, another was that objectively proven ischemia was not required, and a third was that the study of 6 weeks had a short duration.
“ORBITA 2 addressed many of these concerns,” Dr. Hess said, but, when noting that 80% of patients in the newer trial still had single vessel disease, she questioned whether the true effect of PCI for improving symptoms might still be underestimated.
ORBITA-2 was supported by the National Institute for Health and Care Research Imperial Biomedical Research Centre, the Medical Research Council, NIHR, the British Heart Foundation, Philips, and St. Mary’s Coronary Flow Trust. Dr. Rajkumar reported relevant financial relationships. Dr. Leon reported financial relationships with Abbott Vascular, Anteris, Boston Scientific, Edwards Lifesciences, Foldax, and Medtronic. Dr. Hess has financial relationships with more than 20 pharmaceutical companies, but none related specifically to this presentation.
PHILADELPHIA – Percutaneous coronary intervention (PCI) in patients with stable coronary artery disease (CAD) reduces angina frequency, increases exercise capacity, and improves quality of life, results of a placebo-controlled, randomized trial show, confirming advantages that have never before been proven.
reported Christopher A. Rajkumar, MBBS, an interventional cardiology registrar at the Imperial College Healthcare Trust, London.
Results of the trial, ORBITA-2, were presented at the annual scientific sessions of the American Heart Association and simultaneously published online in the New England Journal of Medicine.
Symptom relief has long been a justification for PCI in patients with stable CAD, but the evidence has been derived from uncontrolled studies, Dr. Rajkumar said. However, the first ORBITA trial, which was also placebo controlled and randomized, failed to show benefit.
Dr. Rajkumar acknowledged that the benefit of PCI in ORBITA-2 was lower than previously reported in nonrandomized trials. He also noted that 59% of patients still had at least some angina symptoms following PCI.
Even though ORBITA-2 proves that PCI is better than no PCI, he agreed that well-informed patients, such as those who wish to avoid an invasive procedure, might still reasonably select antianginal medication over PCI. Current guidelines recommend PCI for patients with refractory angina despite medical therapy.
While Dr. Rajkumar was unwilling to speculate on how these data might change guidelines, he did say that patients with stable CAD and angina “now have a choice of two first-line evidence-based pathways.”
‘Remarkable’ trial
“ORBITA 2 is a rather remarkable trial because my surgical colleagues have been asking me for many decades whether PCI actually works,” said Martin B. Leon, MD, professor of medicine, Columbia University Irving Medical Center, New York. “Now I can say with confidence on the basis of a placebo-controlled trial that PCI certainly does have a favorable impact in patients with documented angina, severe coronary stenosis, and demonstrated ischemia.”
The key enrollment criteria for ORBITA-2 were angina, severe coronary stenosis in at least one vessel, and ischemia on stress imaging or invasive physiology. Unlike the previous ORBITA trial, which was limited to single-vessel disease and did not require objective evidence of ischemia, ORBITA 2 employed change in angina, rather than improved exercise capacity, as its primary endpoint.
Relative to sham PCI, patients randomly assigned to an interventional procedure had a more than twofold increase in the odds ratio of improved angina control (OR, 2.2; P < .001) based on a patient scoring system that captured angina symptoms as well as angina medication use on a smartphone application.
The advantage of PCI over sham PCI was also significant for all secondary outcomes. These included a nearly fourfold greater (OR, 3.76; P < .001) likelihood of improvement in the Canadian Cardiovascular Society angina grade and a 1-minute increase (from 10 min. 40 seconds to 11 min. 40 seconds) in treadmill exercise time (P = .008).
On quality of life measured with the self-assessment questionnaire and the EQ-5D-5L, almost all endpoints were highly statistically significant in favor of PCI (typically on the level of P < .001).
The study had a bold design: At enrollment patients stopped all antianginal medications to undergo dobutamine echocardiography and other baseline tests. They were stopped again 2 weeks later, when patients were randomized.
With a study protocol that enrolled patients off medication, “we intentionally diverged from the clinical guidelines,” Dr. Rajkumar said.
Of the 439 patients enrolled, 301 were randomly assigned at the end of the 2-week period, when patients were already sedated. Control patients remained sedated for at least 15 minutes. All 151 of those randomized to PCI and the 150 control patients were available for the intent-to-treat analysis at the end of 12 weeks.
The novel angina symptom burden score was created from daily angina episodes and units of daily antianginal medication captured on the smartphone app. On an ordinal scale, a score of 0 on any given day represented no anginal symptoms and no antianginal medication.
As angina severity or medication use increased, it raised the daily scores. If there was unacceptable angina (requiring the patient to be removed from the blind), acute coronary syndrome, or death, it produced the highest scores, which reached a maximum of 79.
The favorable OR for a lower symptom burden in the PCI group reflected a relative reduction in angina observed the first day after the procedure. Over the entire follow-up, more patients in the PCI group had an angina score of 0 and more of those who had angina did not take antianginal medications.
This objective evidence that PCI reduces symptoms and improves quality of life in patients with angina and stable CAD was met at the AHA late-breaking session with a sustained ovation.
ORBITA-2 addresses ORBITA criticisms
Connie N. Hess, MD, the AHA-invited discussant and an interventional cardiologist at the University of Colorado Medicine, Aurora, provided perspective on the differences between ORBITA 2 and ORBITA, which she said “addressed a fundamentally different hypothesis” by focusing on angina rather than exercise capacity.
Of the criticisms of the original ORBITA, which Dr. Hess noted was the first sham-controlled PCI trial ever conducted in stable CAD, one is that patients with multivessel disease were excluded, another was that objectively proven ischemia was not required, and a third was that the study of 6 weeks had a short duration.
“ORBITA 2 addressed many of these concerns,” Dr. Hess said, but, when noting that 80% of patients in the newer trial still had single vessel disease, she questioned whether the true effect of PCI for improving symptoms might still be underestimated.
ORBITA-2 was supported by the National Institute for Health and Care Research Imperial Biomedical Research Centre, the Medical Research Council, NIHR, the British Heart Foundation, Philips, and St. Mary’s Coronary Flow Trust. Dr. Rajkumar reported relevant financial relationships. Dr. Leon reported financial relationships with Abbott Vascular, Anteris, Boston Scientific, Edwards Lifesciences, Foldax, and Medtronic. Dr. Hess has financial relationships with more than 20 pharmaceutical companies, but none related specifically to this presentation.
PHILADELPHIA – Percutaneous coronary intervention (PCI) in patients with stable coronary artery disease (CAD) reduces angina frequency, increases exercise capacity, and improves quality of life, results of a placebo-controlled, randomized trial show, confirming advantages that have never before been proven.
reported Christopher A. Rajkumar, MBBS, an interventional cardiology registrar at the Imperial College Healthcare Trust, London.
Results of the trial, ORBITA-2, were presented at the annual scientific sessions of the American Heart Association and simultaneously published online in the New England Journal of Medicine.
Symptom relief has long been a justification for PCI in patients with stable CAD, but the evidence has been derived from uncontrolled studies, Dr. Rajkumar said. However, the first ORBITA trial, which was also placebo controlled and randomized, failed to show benefit.
Dr. Rajkumar acknowledged that the benefit of PCI in ORBITA-2 was lower than previously reported in nonrandomized trials. He also noted that 59% of patients still had at least some angina symptoms following PCI.
Even though ORBITA-2 proves that PCI is better than no PCI, he agreed that well-informed patients, such as those who wish to avoid an invasive procedure, might still reasonably select antianginal medication over PCI. Current guidelines recommend PCI for patients with refractory angina despite medical therapy.
While Dr. Rajkumar was unwilling to speculate on how these data might change guidelines, he did say that patients with stable CAD and angina “now have a choice of two first-line evidence-based pathways.”
‘Remarkable’ trial
“ORBITA 2 is a rather remarkable trial because my surgical colleagues have been asking me for many decades whether PCI actually works,” said Martin B. Leon, MD, professor of medicine, Columbia University Irving Medical Center, New York. “Now I can say with confidence on the basis of a placebo-controlled trial that PCI certainly does have a favorable impact in patients with documented angina, severe coronary stenosis, and demonstrated ischemia.”
The key enrollment criteria for ORBITA-2 were angina, severe coronary stenosis in at least one vessel, and ischemia on stress imaging or invasive physiology. Unlike the previous ORBITA trial, which was limited to single-vessel disease and did not require objective evidence of ischemia, ORBITA 2 employed change in angina, rather than improved exercise capacity, as its primary endpoint.
Relative to sham PCI, patients randomly assigned to an interventional procedure had a more than twofold increase in the odds ratio of improved angina control (OR, 2.2; P < .001) based on a patient scoring system that captured angina symptoms as well as angina medication use on a smartphone application.
The advantage of PCI over sham PCI was also significant for all secondary outcomes. These included a nearly fourfold greater (OR, 3.76; P < .001) likelihood of improvement in the Canadian Cardiovascular Society angina grade and a 1-minute increase (from 10 min. 40 seconds to 11 min. 40 seconds) in treadmill exercise time (P = .008).
On quality of life measured with the self-assessment questionnaire and the EQ-5D-5L, almost all endpoints were highly statistically significant in favor of PCI (typically on the level of P < .001).
The study had a bold design: At enrollment patients stopped all antianginal medications to undergo dobutamine echocardiography and other baseline tests. They were stopped again 2 weeks later, when patients were randomized.
With a study protocol that enrolled patients off medication, “we intentionally diverged from the clinical guidelines,” Dr. Rajkumar said.
Of the 439 patients enrolled, 301 were randomly assigned at the end of the 2-week period, when patients were already sedated. Control patients remained sedated for at least 15 minutes. All 151 of those randomized to PCI and the 150 control patients were available for the intent-to-treat analysis at the end of 12 weeks.
The novel angina symptom burden score was created from daily angina episodes and units of daily antianginal medication captured on the smartphone app. On an ordinal scale, a score of 0 on any given day represented no anginal symptoms and no antianginal medication.
As angina severity or medication use increased, it raised the daily scores. If there was unacceptable angina (requiring the patient to be removed from the blind), acute coronary syndrome, or death, it produced the highest scores, which reached a maximum of 79.
The favorable OR for a lower symptom burden in the PCI group reflected a relative reduction in angina observed the first day after the procedure. Over the entire follow-up, more patients in the PCI group had an angina score of 0 and more of those who had angina did not take antianginal medications.
This objective evidence that PCI reduces symptoms and improves quality of life in patients with angina and stable CAD was met at the AHA late-breaking session with a sustained ovation.
ORBITA-2 addresses ORBITA criticisms
Connie N. Hess, MD, the AHA-invited discussant and an interventional cardiologist at the University of Colorado Medicine, Aurora, provided perspective on the differences between ORBITA 2 and ORBITA, which she said “addressed a fundamentally different hypothesis” by focusing on angina rather than exercise capacity.
Of the criticisms of the original ORBITA, which Dr. Hess noted was the first sham-controlled PCI trial ever conducted in stable CAD, one is that patients with multivessel disease were excluded, another was that objectively proven ischemia was not required, and a third was that the study of 6 weeks had a short duration.
“ORBITA 2 addressed many of these concerns,” Dr. Hess said, but, when noting that 80% of patients in the newer trial still had single vessel disease, she questioned whether the true effect of PCI for improving symptoms might still be underestimated.
ORBITA-2 was supported by the National Institute for Health and Care Research Imperial Biomedical Research Centre, the Medical Research Council, NIHR, the British Heart Foundation, Philips, and St. Mary’s Coronary Flow Trust. Dr. Rajkumar reported relevant financial relationships. Dr. Leon reported financial relationships with Abbott Vascular, Anteris, Boston Scientific, Edwards Lifesciences, Foldax, and Medtronic. Dr. Hess has financial relationships with more than 20 pharmaceutical companies, but none related specifically to this presentation.
AT AHA 2023
Excellent outcome of Ross procedure after 2 decades
TOPLINE:
a survival rate equivalent to that of the general population, results of a new study show. The need for reintervention in these patients is low.
METHODOLOGY:
- The study was a post hoc analysis of a randomized clinical trial that showed superior survival, freedom from reoperation, and quality of life at 10 years for patients who received the Ross procedure, compared with those who got homograft root replacement.
- This new analysis included 108 patients, median age 38 years and mostly male and of British origin, who underwent the Ross procedure. Of these, 45% had aortic regurgitation (AR) as the main hemodynamic lesion.
- The primary outcome was long-term survival, compared with an age-, sex-, and country of origin–matched general U.K. population using a novel, patient-level matching strategy. Secondary outcomes included freedom from any valve-related reintervention, autograft reintervention, or homograft reintervention.
TAKEAWAY:
- Survival at 25 years was 83.0% (95% confidence interval, 75.5%-91.2%), representing a relative survival of 99.1% (95% CI, 91.8%-100%), compared with the matched general population (survival in general population was 83.7%).
- At 25 years, freedom from any Ross-related reintervention was 71.1% (95% CI, 61.6%-82.0%); freedom from autograft reintervention was 80.3% (95% CI, 71.9%-89.6%); and freedom from homograft reintervention was 86.3% (95% CI, 79.0%-94.3%).
- There was no increased hazard for autograft deterioration in patients presenting with versus without preoperative AR, an important finding since it has been suggested Ross procedure benefits may not extend fully to patients with preoperative AR, said the authors.
- 86% of patients had New York Heart Association class I or II status at the latest clinical follow-up (approaching 25 years).
IN PRACTICE:
This study shows the Ross procedure “provided excellent survival into the third decade after surgery,” with the new data further supporting “the unique benefits” of the valve substitute in adults, the authors conclude.
Authors of an accompanying editorial, Tsuyoshi Kaneko, MD, Division of Cardiothoracic Surgery, Washington University School of Medicine, St. Louis, and Maral Ouzounian, MD, PhD, Peter Munk Cardiac Centre, Division of Cardiac Surgery, University Health Network, University of Toronto, write that the new evidence suggests the Ross procedure is “a truly attractive option in younger patients with long life expectancy.” However, they note that aortic regurgitation in the cohort worsened over time, potentially leading to late reinterventions; echocardiographic follow-up was available in only 71% of patients; and generalizing the Ross procedure to a broader group of surgeons is challenging.
SOURCE:
The study was conducted by Maximiliaan L. Notenboom, BSc, department of cardiothoracic surgery, Erasmus University Medical Center, Rotterdam, the Netherlands, and colleagues. It was published online in JAMA Cardiology.
LIMITATIONS:
The analysis reflects a single-surgeon experience, so it’s difficult to extrapolate the results, although the operative steps involved in the Ross procedure have now been clearly delineated, making the operation reproducible. The duration of echocardiographic follow-up was shorter and less complete than the clinical follow-up. Outcomes of the cohort that underwent homograft procedures in the randomized clinical trial were not reported, but since that procedure has nearly disappeared from practice, reporting on its long-term outcomes would be of limited clinical significance.
DISCLOSURES:
Mr. Notenboom has disclosed no relevant financial relationships. Co-author Fabio De Robertis, MD, department of cardiothoracic surgery and transplantation, Royal Brompton & Harefield Hospitals, London, received nonfinancial support from Edwards Lifescience for travel and personal fees from Bristol Myers Squibb for consulting outside the submitted work, and has a service agreement with Medtronic U.K., which paid a fee to the Royal Brompton & Harefield Hospitals Charity Fund.
Editorial co-author Kaneko received personal fees from Edwards Lifesciences, Medtronic, Abbott, and Johnson & Johnson outside the submitted work; Ouzounian received personal fees from Medtronic, Edwards Lifesciences, and Terumo Aortic outside the submitted work.
A version of this article appeared on Medscape.com.
TOPLINE:
a survival rate equivalent to that of the general population, results of a new study show. The need for reintervention in these patients is low.
METHODOLOGY:
- The study was a post hoc analysis of a randomized clinical trial that showed superior survival, freedom from reoperation, and quality of life at 10 years for patients who received the Ross procedure, compared with those who got homograft root replacement.
- This new analysis included 108 patients, median age 38 years and mostly male and of British origin, who underwent the Ross procedure. Of these, 45% had aortic regurgitation (AR) as the main hemodynamic lesion.
- The primary outcome was long-term survival, compared with an age-, sex-, and country of origin–matched general U.K. population using a novel, patient-level matching strategy. Secondary outcomes included freedom from any valve-related reintervention, autograft reintervention, or homograft reintervention.
TAKEAWAY:
- Survival at 25 years was 83.0% (95% confidence interval, 75.5%-91.2%), representing a relative survival of 99.1% (95% CI, 91.8%-100%), compared with the matched general population (survival in general population was 83.7%).
- At 25 years, freedom from any Ross-related reintervention was 71.1% (95% CI, 61.6%-82.0%); freedom from autograft reintervention was 80.3% (95% CI, 71.9%-89.6%); and freedom from homograft reintervention was 86.3% (95% CI, 79.0%-94.3%).
- There was no increased hazard for autograft deterioration in patients presenting with versus without preoperative AR, an important finding since it has been suggested Ross procedure benefits may not extend fully to patients with preoperative AR, said the authors.
- 86% of patients had New York Heart Association class I or II status at the latest clinical follow-up (approaching 25 years).
IN PRACTICE:
This study shows the Ross procedure “provided excellent survival into the third decade after surgery,” with the new data further supporting “the unique benefits” of the valve substitute in adults, the authors conclude.
Authors of an accompanying editorial, Tsuyoshi Kaneko, MD, Division of Cardiothoracic Surgery, Washington University School of Medicine, St. Louis, and Maral Ouzounian, MD, PhD, Peter Munk Cardiac Centre, Division of Cardiac Surgery, University Health Network, University of Toronto, write that the new evidence suggests the Ross procedure is “a truly attractive option in younger patients with long life expectancy.” However, they note that aortic regurgitation in the cohort worsened over time, potentially leading to late reinterventions; echocardiographic follow-up was available in only 71% of patients; and generalizing the Ross procedure to a broader group of surgeons is challenging.
SOURCE:
The study was conducted by Maximiliaan L. Notenboom, BSc, department of cardiothoracic surgery, Erasmus University Medical Center, Rotterdam, the Netherlands, and colleagues. It was published online in JAMA Cardiology.
LIMITATIONS:
The analysis reflects a single-surgeon experience, so it’s difficult to extrapolate the results, although the operative steps involved in the Ross procedure have now been clearly delineated, making the operation reproducible. The duration of echocardiographic follow-up was shorter and less complete than the clinical follow-up. Outcomes of the cohort that underwent homograft procedures in the randomized clinical trial were not reported, but since that procedure has nearly disappeared from practice, reporting on its long-term outcomes would be of limited clinical significance.
DISCLOSURES:
Mr. Notenboom has disclosed no relevant financial relationships. Co-author Fabio De Robertis, MD, department of cardiothoracic surgery and transplantation, Royal Brompton & Harefield Hospitals, London, received nonfinancial support from Edwards Lifescience for travel and personal fees from Bristol Myers Squibb for consulting outside the submitted work, and has a service agreement with Medtronic U.K., which paid a fee to the Royal Brompton & Harefield Hospitals Charity Fund.
Editorial co-author Kaneko received personal fees from Edwards Lifesciences, Medtronic, Abbott, and Johnson & Johnson outside the submitted work; Ouzounian received personal fees from Medtronic, Edwards Lifesciences, and Terumo Aortic outside the submitted work.
A version of this article appeared on Medscape.com.
TOPLINE:
a survival rate equivalent to that of the general population, results of a new study show. The need for reintervention in these patients is low.
METHODOLOGY:
- The study was a post hoc analysis of a randomized clinical trial that showed superior survival, freedom from reoperation, and quality of life at 10 years for patients who received the Ross procedure, compared with those who got homograft root replacement.
- This new analysis included 108 patients, median age 38 years and mostly male and of British origin, who underwent the Ross procedure. Of these, 45% had aortic regurgitation (AR) as the main hemodynamic lesion.
- The primary outcome was long-term survival, compared with an age-, sex-, and country of origin–matched general U.K. population using a novel, patient-level matching strategy. Secondary outcomes included freedom from any valve-related reintervention, autograft reintervention, or homograft reintervention.
TAKEAWAY:
- Survival at 25 years was 83.0% (95% confidence interval, 75.5%-91.2%), representing a relative survival of 99.1% (95% CI, 91.8%-100%), compared with the matched general population (survival in general population was 83.7%).
- At 25 years, freedom from any Ross-related reintervention was 71.1% (95% CI, 61.6%-82.0%); freedom from autograft reintervention was 80.3% (95% CI, 71.9%-89.6%); and freedom from homograft reintervention was 86.3% (95% CI, 79.0%-94.3%).
- There was no increased hazard for autograft deterioration in patients presenting with versus without preoperative AR, an important finding since it has been suggested Ross procedure benefits may not extend fully to patients with preoperative AR, said the authors.
- 86% of patients had New York Heart Association class I or II status at the latest clinical follow-up (approaching 25 years).
IN PRACTICE:
This study shows the Ross procedure “provided excellent survival into the third decade after surgery,” with the new data further supporting “the unique benefits” of the valve substitute in adults, the authors conclude.
Authors of an accompanying editorial, Tsuyoshi Kaneko, MD, Division of Cardiothoracic Surgery, Washington University School of Medicine, St. Louis, and Maral Ouzounian, MD, PhD, Peter Munk Cardiac Centre, Division of Cardiac Surgery, University Health Network, University of Toronto, write that the new evidence suggests the Ross procedure is “a truly attractive option in younger patients with long life expectancy.” However, they note that aortic regurgitation in the cohort worsened over time, potentially leading to late reinterventions; echocardiographic follow-up was available in only 71% of patients; and generalizing the Ross procedure to a broader group of surgeons is challenging.
SOURCE:
The study was conducted by Maximiliaan L. Notenboom, BSc, department of cardiothoracic surgery, Erasmus University Medical Center, Rotterdam, the Netherlands, and colleagues. It was published online in JAMA Cardiology.
LIMITATIONS:
The analysis reflects a single-surgeon experience, so it’s difficult to extrapolate the results, although the operative steps involved in the Ross procedure have now been clearly delineated, making the operation reproducible. The duration of echocardiographic follow-up was shorter and less complete than the clinical follow-up. Outcomes of the cohort that underwent homograft procedures in the randomized clinical trial were not reported, but since that procedure has nearly disappeared from practice, reporting on its long-term outcomes would be of limited clinical significance.
DISCLOSURES:
Mr. Notenboom has disclosed no relevant financial relationships. Co-author Fabio De Robertis, MD, department of cardiothoracic surgery and transplantation, Royal Brompton & Harefield Hospitals, London, received nonfinancial support from Edwards Lifescience for travel and personal fees from Bristol Myers Squibb for consulting outside the submitted work, and has a service agreement with Medtronic U.K., which paid a fee to the Royal Brompton & Harefield Hospitals Charity Fund.
Editorial co-author Kaneko received personal fees from Edwards Lifesciences, Medtronic, Abbott, and Johnson & Johnson outside the submitted work; Ouzounian received personal fees from Medtronic, Edwards Lifesciences, and Terumo Aortic outside the submitted work.
A version of this article appeared on Medscape.com.
Short aspirin therapy noninferior to DAPT for 1 year after PCI for ACS
SAN FRANCISCO – Stopping aspirin within 1 month of implanting a drug-eluting stent (DES) for acute coronary syndrome (ACS) followed by ticagrelor monotherapy was shown to be noninferior to 12 months of dual antiplatelet therapy (DAPT) in net adverse cardiovascular and bleeding events in the T-PASS trial.
of death, myocardial infarction, stent thrombosis, stroke, and major bleeding, primarily due to a significant reduction in bleeding events,” senior author Myeong-Ki Hong, MD, PhD, Yonsei University, Seoul, Korea, told attendees at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
“This study provides evidence that stopping aspirin within 1 month after implantation of drug-eluting stents for ticagrelor monotherapy is a reasonable alternative to 12-month DAPT as for adverse cardiovascular and bleeding events,” Dr. Hong concluded.
The study was published in Circulation ahead of print to coincide with the presentation.
Three months to 1 month
Previous trials (TICO and TWILIGHT) have shown that ticagrelor monotherapy after 3 months of DAPT can be safe and effectively prevent ischemic events after percutaneous coronary intervention (PCI) in ACS or high-risk PCI patients.
The current study aimed to investigate whether ticagrelor monotherapy after less than 1 month of DAPT was noninferior to 12 months of ticagrelor-based DAPT for preventing adverse cardiovascular and bleeding events in patients with ACS undergoing PCI with a DES implant.
T-PASS, carried out at 24 centers in Korea, enrolled ACS patients aged 19 years or older who received an ultrathin, bioresorbable polymer sirolimus-eluting stent (Orsiro, Biotronik). They were randomized 1:1 to ticagrelor monotherapy after less than 1 month of DAPT (n = 1,426) or to ticagrelor-based DAPT for 12 months (n = 1,424).
The primary outcome measure was net adverse clinical events (NACE) at 12 months, consisting of major bleeding plus major adverse cardiovascular events. All patients were included in the intention-to-treat analysis.
The study could enroll patients aged 19-80 years. It excluded anyone with active bleeding, at increased risk for bleeding, with anemia (hemoglobin ≤ 8 g/dL), platelets less than 100,000/mcL, need for oral anticoagulation therapy, current or potential pregnancy, or a life expectancy less than 1 year.
Baseline characteristics of the two groups were well balanced. The extended monotherapy and DAPT arms had an average age of 61 ± 10 years, were 84% and 83% male and had diabetes mellitus in 30% and 29%, respectively, with 74% of each group admitted via the emergency room. ST-elevation myocardial infarction occurred in 40% and 41% of patients in each group, respectively.
Results showed that stopping aspirin early was noninferior and possibly superior to 12 months of DAPT.
For the 12-month clinical outcome, fewer patients in the less than 1 month DAPT followed by ticagrelor monotherapy arm reached the primary clinical endpoint of NACE versus the ticagrelor-based 12-month DAPT arm, both in terms of noninferiority (P < .001) and superiority (P = .002). Similar results were found for the 1-month landmark analyses.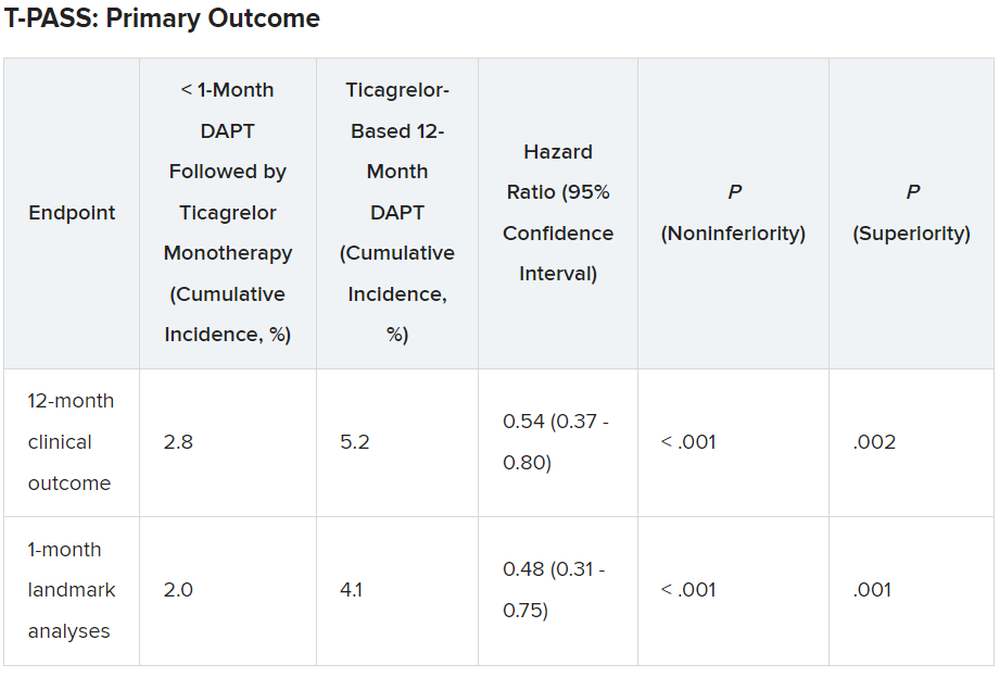
For both the 12-month clinical outcome and the 1-month landmark analyses, the curves for the two arms began to diverge at about 150 days, with the one for ticagrelor monotherapy essentially flattening out just after that and the one for the 12-month DAPT therapy continuing to rise out to the 1-year point.
In the less than 1 month DAPT arm, aspirin was stopped at a median of 16 days. Panelist Adnan Kastrati, MD, Deutsches Herzzentrum München, Technische Universität, Munich, Germany, asked Dr. Hong about the criteria for the point at which aspirin was stopped in the less than 1 month arm.
Dr. Hong replied: “Actually, we recommend less than 1 month, so therefore in some patients, it was the operator’s decision,” depending on risk factors for stopping or continuing aspirin. He said that in some patients it may be reasonable to stop aspirin even in 7-10 days. Fewer than 10% of patients in the less than 1 month arm continued on aspirin past 30 days, but a few continued on it to the 1-year point.
There was no difference between the less than 1 month DAPT followed by ticagrelor monotherapy arm and the 12-month DAPT arm in terms of major adverse cardiac and cerebrovascular events at 1 year (1.8% vs. 2.2%, respectively; hazard ratio, 0.84; 95% confidence interval, 0.50-1.41; log-rank, P = .51).
However, the 12-month DAPT arm showed a significantly greater incidence of major bleeding at 1 year: 3.4% versus 1.2% for less than 1 month aspirin arm (HR, 0.35; 95% CI, 0.20-0.61; log-rank, P < .001).
Dr. Hong said that a limitation of the study was that it was open label and not placebo controlled. However, an independent clinical event adjudication committee assessed all clinical outcomes.
Lead discussant Marco Valgimigli, MD, PhD, Cardiocentro Ticino Foundation, Lugano, Switzerland, noted that T-PASS is the fifth study to investigate ticagrelor monotherapy versus a DAPT, giving randomized data on almost 22,000 patients.
“T-PASS showed very consistently with the prior four studies that by dropping aspirin and continuation with ticagrelor therapy, compared with the standard DAPT regimen, is associated with no penalty ... and in fact leading to a very significant and clinically very convincing risk reduction, and I would like to underline major bleeding risk reduction,” he said, pointing out that this study comes from the same research group that carried out the TICO trial.
Dr. Hong has received institutional research grants from Samjin Pharmaceutical and Chong Kun Dang Pharmaceutical, and speaker’s fees from Medtronic and Edwards Lifesciences. Dr. Kastrati has disclosed no relevant financial relationships. Dr. Valgimigli has received grant support/research contracts from Terumo Medical and AstraZeneca; consultant fees/honoraria/speaker’s bureau for Terumo Medical Corporation, Bayer, Daiichi Sankyo/Eli Lilly, Amgen, Alvimedica, AstraZenca, Idorsia, Coreflow, Vifor, Bristol-Myers Squibb, and iVascular. The study was funded by Biotronik.
A version of this article first appeared on Medscape.com.
SAN FRANCISCO – Stopping aspirin within 1 month of implanting a drug-eluting stent (DES) for acute coronary syndrome (ACS) followed by ticagrelor monotherapy was shown to be noninferior to 12 months of dual antiplatelet therapy (DAPT) in net adverse cardiovascular and bleeding events in the T-PASS trial.
of death, myocardial infarction, stent thrombosis, stroke, and major bleeding, primarily due to a significant reduction in bleeding events,” senior author Myeong-Ki Hong, MD, PhD, Yonsei University, Seoul, Korea, told attendees at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
“This study provides evidence that stopping aspirin within 1 month after implantation of drug-eluting stents for ticagrelor monotherapy is a reasonable alternative to 12-month DAPT as for adverse cardiovascular and bleeding events,” Dr. Hong concluded.
The study was published in Circulation ahead of print to coincide with the presentation.
Three months to 1 month
Previous trials (TICO and TWILIGHT) have shown that ticagrelor monotherapy after 3 months of DAPT can be safe and effectively prevent ischemic events after percutaneous coronary intervention (PCI) in ACS or high-risk PCI patients.
The current study aimed to investigate whether ticagrelor monotherapy after less than 1 month of DAPT was noninferior to 12 months of ticagrelor-based DAPT for preventing adverse cardiovascular and bleeding events in patients with ACS undergoing PCI with a DES implant.
T-PASS, carried out at 24 centers in Korea, enrolled ACS patients aged 19 years or older who received an ultrathin, bioresorbable polymer sirolimus-eluting stent (Orsiro, Biotronik). They were randomized 1:1 to ticagrelor monotherapy after less than 1 month of DAPT (n = 1,426) or to ticagrelor-based DAPT for 12 months (n = 1,424).
The primary outcome measure was net adverse clinical events (NACE) at 12 months, consisting of major bleeding plus major adverse cardiovascular events. All patients were included in the intention-to-treat analysis.
The study could enroll patients aged 19-80 years. It excluded anyone with active bleeding, at increased risk for bleeding, with anemia (hemoglobin ≤ 8 g/dL), platelets less than 100,000/mcL, need for oral anticoagulation therapy, current or potential pregnancy, or a life expectancy less than 1 year.
Baseline characteristics of the two groups were well balanced. The extended monotherapy and DAPT arms had an average age of 61 ± 10 years, were 84% and 83% male and had diabetes mellitus in 30% and 29%, respectively, with 74% of each group admitted via the emergency room. ST-elevation myocardial infarction occurred in 40% and 41% of patients in each group, respectively.
Results showed that stopping aspirin early was noninferior and possibly superior to 12 months of DAPT.
For the 12-month clinical outcome, fewer patients in the less than 1 month DAPT followed by ticagrelor monotherapy arm reached the primary clinical endpoint of NACE versus the ticagrelor-based 12-month DAPT arm, both in terms of noninferiority (P < .001) and superiority (P = .002). Similar results were found for the 1-month landmark analyses.
For both the 12-month clinical outcome and the 1-month landmark analyses, the curves for the two arms began to diverge at about 150 days, with the one for ticagrelor monotherapy essentially flattening out just after that and the one for the 12-month DAPT therapy continuing to rise out to the 1-year point.
In the less than 1 month DAPT arm, aspirin was stopped at a median of 16 days. Panelist Adnan Kastrati, MD, Deutsches Herzzentrum München, Technische Universität, Munich, Germany, asked Dr. Hong about the criteria for the point at which aspirin was stopped in the less than 1 month arm.
Dr. Hong replied: “Actually, we recommend less than 1 month, so therefore in some patients, it was the operator’s decision,” depending on risk factors for stopping or continuing aspirin. He said that in some patients it may be reasonable to stop aspirin even in 7-10 days. Fewer than 10% of patients in the less than 1 month arm continued on aspirin past 30 days, but a few continued on it to the 1-year point.
There was no difference between the less than 1 month DAPT followed by ticagrelor monotherapy arm and the 12-month DAPT arm in terms of major adverse cardiac and cerebrovascular events at 1 year (1.8% vs. 2.2%, respectively; hazard ratio, 0.84; 95% confidence interval, 0.50-1.41; log-rank, P = .51).
However, the 12-month DAPT arm showed a significantly greater incidence of major bleeding at 1 year: 3.4% versus 1.2% for less than 1 month aspirin arm (HR, 0.35; 95% CI, 0.20-0.61; log-rank, P < .001).
Dr. Hong said that a limitation of the study was that it was open label and not placebo controlled. However, an independent clinical event adjudication committee assessed all clinical outcomes.
Lead discussant Marco Valgimigli, MD, PhD, Cardiocentro Ticino Foundation, Lugano, Switzerland, noted that T-PASS is the fifth study to investigate ticagrelor monotherapy versus a DAPT, giving randomized data on almost 22,000 patients.
“T-PASS showed very consistently with the prior four studies that by dropping aspirin and continuation with ticagrelor therapy, compared with the standard DAPT regimen, is associated with no penalty ... and in fact leading to a very significant and clinically very convincing risk reduction, and I would like to underline major bleeding risk reduction,” he said, pointing out that this study comes from the same research group that carried out the TICO trial.
Dr. Hong has received institutional research grants from Samjin Pharmaceutical and Chong Kun Dang Pharmaceutical, and speaker’s fees from Medtronic and Edwards Lifesciences. Dr. Kastrati has disclosed no relevant financial relationships. Dr. Valgimigli has received grant support/research contracts from Terumo Medical and AstraZeneca; consultant fees/honoraria/speaker’s bureau for Terumo Medical Corporation, Bayer, Daiichi Sankyo/Eli Lilly, Amgen, Alvimedica, AstraZenca, Idorsia, Coreflow, Vifor, Bristol-Myers Squibb, and iVascular. The study was funded by Biotronik.
A version of this article first appeared on Medscape.com.
SAN FRANCISCO – Stopping aspirin within 1 month of implanting a drug-eluting stent (DES) for acute coronary syndrome (ACS) followed by ticagrelor monotherapy was shown to be noninferior to 12 months of dual antiplatelet therapy (DAPT) in net adverse cardiovascular and bleeding events in the T-PASS trial.
of death, myocardial infarction, stent thrombosis, stroke, and major bleeding, primarily due to a significant reduction in bleeding events,” senior author Myeong-Ki Hong, MD, PhD, Yonsei University, Seoul, Korea, told attendees at the Transcatheter Cardiovascular Therapeutics annual meeting, sponsored by the Cardiovascular Research Foundation.
“This study provides evidence that stopping aspirin within 1 month after implantation of drug-eluting stents for ticagrelor monotherapy is a reasonable alternative to 12-month DAPT as for adverse cardiovascular and bleeding events,” Dr. Hong concluded.
The study was published in Circulation ahead of print to coincide with the presentation.
Three months to 1 month
Previous trials (TICO and TWILIGHT) have shown that ticagrelor monotherapy after 3 months of DAPT can be safe and effectively prevent ischemic events after percutaneous coronary intervention (PCI) in ACS or high-risk PCI patients.
The current study aimed to investigate whether ticagrelor monotherapy after less than 1 month of DAPT was noninferior to 12 months of ticagrelor-based DAPT for preventing adverse cardiovascular and bleeding events in patients with ACS undergoing PCI with a DES implant.
T-PASS, carried out at 24 centers in Korea, enrolled ACS patients aged 19 years or older who received an ultrathin, bioresorbable polymer sirolimus-eluting stent (Orsiro, Biotronik). They were randomized 1:1 to ticagrelor monotherapy after less than 1 month of DAPT (n = 1,426) or to ticagrelor-based DAPT for 12 months (n = 1,424).
The primary outcome measure was net adverse clinical events (NACE) at 12 months, consisting of major bleeding plus major adverse cardiovascular events. All patients were included in the intention-to-treat analysis.
The study could enroll patients aged 19-80 years. It excluded anyone with active bleeding, at increased risk for bleeding, with anemia (hemoglobin ≤ 8 g/dL), platelets less than 100,000/mcL, need for oral anticoagulation therapy, current or potential pregnancy, or a life expectancy less than 1 year.
Baseline characteristics of the two groups were well balanced. The extended monotherapy and DAPT arms had an average age of 61 ± 10 years, were 84% and 83% male and had diabetes mellitus in 30% and 29%, respectively, with 74% of each group admitted via the emergency room. ST-elevation myocardial infarction occurred in 40% and 41% of patients in each group, respectively.
Results showed that stopping aspirin early was noninferior and possibly superior to 12 months of DAPT.
For the 12-month clinical outcome, fewer patients in the less than 1 month DAPT followed by ticagrelor monotherapy arm reached the primary clinical endpoint of NACE versus the ticagrelor-based 12-month DAPT arm, both in terms of noninferiority (P < .001) and superiority (P = .002). Similar results were found for the 1-month landmark analyses.
For both the 12-month clinical outcome and the 1-month landmark analyses, the curves for the two arms began to diverge at about 150 days, with the one for ticagrelor monotherapy essentially flattening out just after that and the one for the 12-month DAPT therapy continuing to rise out to the 1-year point.
In the less than 1 month DAPT arm, aspirin was stopped at a median of 16 days. Panelist Adnan Kastrati, MD, Deutsches Herzzentrum München, Technische Universität, Munich, Germany, asked Dr. Hong about the criteria for the point at which aspirin was stopped in the less than 1 month arm.
Dr. Hong replied: “Actually, we recommend less than 1 month, so therefore in some patients, it was the operator’s decision,” depending on risk factors for stopping or continuing aspirin. He said that in some patients it may be reasonable to stop aspirin even in 7-10 days. Fewer than 10% of patients in the less than 1 month arm continued on aspirin past 30 days, but a few continued on it to the 1-year point.
There was no difference between the less than 1 month DAPT followed by ticagrelor monotherapy arm and the 12-month DAPT arm in terms of major adverse cardiac and cerebrovascular events at 1 year (1.8% vs. 2.2%, respectively; hazard ratio, 0.84; 95% confidence interval, 0.50-1.41; log-rank, P = .51).
However, the 12-month DAPT arm showed a significantly greater incidence of major bleeding at 1 year: 3.4% versus 1.2% for less than 1 month aspirin arm (HR, 0.35; 95% CI, 0.20-0.61; log-rank, P < .001).
Dr. Hong said that a limitation of the study was that it was open label and not placebo controlled. However, an independent clinical event adjudication committee assessed all clinical outcomes.
Lead discussant Marco Valgimigli, MD, PhD, Cardiocentro Ticino Foundation, Lugano, Switzerland, noted that T-PASS is the fifth study to investigate ticagrelor monotherapy versus a DAPT, giving randomized data on almost 22,000 patients.
“T-PASS showed very consistently with the prior four studies that by dropping aspirin and continuation with ticagrelor therapy, compared with the standard DAPT regimen, is associated with no penalty ... and in fact leading to a very significant and clinically very convincing risk reduction, and I would like to underline major bleeding risk reduction,” he said, pointing out that this study comes from the same research group that carried out the TICO trial.
Dr. Hong has received institutional research grants from Samjin Pharmaceutical and Chong Kun Dang Pharmaceutical, and speaker’s fees from Medtronic and Edwards Lifesciences. Dr. Kastrati has disclosed no relevant financial relationships. Dr. Valgimigli has received grant support/research contracts from Terumo Medical and AstraZeneca; consultant fees/honoraria/speaker’s bureau for Terumo Medical Corporation, Bayer, Daiichi Sankyo/Eli Lilly, Amgen, Alvimedica, AstraZenca, Idorsia, Coreflow, Vifor, Bristol-Myers Squibb, and iVascular. The study was funded by Biotronik.
A version of this article first appeared on Medscape.com.
AT TCT 2023
Drug-coated balloon beats conventional angioplasty for high-risk patients with in-stent restenosis
SAN FRANCISCO – For the treatment of coronary artery in-stent restenosis, angioplasty with a drug-coated balloon (AGENT DCB; Boston Scientific) was superior to conventional balloon angioplasty in preventing target lesion failure at 1 year in a high-risk patient population.
Approximate 50% reductions in the rates of target lesion restenosis and target vessel myocardial infarction (MI) accounted for the superior findings with the AGENT DCB over conventional balloon angioplasty.
Robert Yeh, MD, of Beth Israel Deaconess Medical Center in Boston reported at the annual Transcatheter Cardiovascular Therapeutics congress. “This represented a 38% relative risk reduction as well as a 10% absolute risk reduction in the endpoint. The P value for superiority was 0.0063, highly statistically significant.”
In-stent restenosis is clinically challenging and accounts for about 10% of all percutaneous coronary interventions. “Sometimes these patients have multiple layers, and that could be a third or fourth layer of stent, something that we try to avoid,” he said.
Drug-coated balloons, which are not currently approved in the United States, can deliver drugs that inhibit blockages from reforming, “without leaving additional layers of metal behind,” he added. Such devices are already available in Europe and Japan.
AGENT IDE was a prospective, multicenter, superiority trial that randomly assigned 480 patients 2:1 to the AGENT DCB (n = 321) or to conventional balloon angioplasty (n = 159). Randomization occurred after successful pre-dilation of the target vessel.
The trial included patients with in-stent restenosis previously treated with a bare metal or a drug-eluting stent with lesion lengths < 26 mm (reference vessel diameter: > 2 mm to ≤ 4), and percent diameter stenosis of more than 70% if they were asymptomatic or of more than 50% if they were symptomatic. Patients were excluded if they had a recent ST-elevation MI, bifurcation, saphenous vein or arterial graft, or thrombus in the target vessel.
All received dual antiplatelet therapy for at least 1 month and then antiplatelet monotherapy for the duration of the trial. The primary endpoint was target lesion failure at 1 year, a composite of target lesion restenosis, target vessel-related MI, or cardiac death. More than 93% of patients in each arm were available for evaluation of the primary endpoint.
The two groups were well balanced at baseline: Approximate age was 68 years, 27% were women, and three quarters were White. Approximately 28%-32% had had a prior coronary artery bypass graft, 20%-22% had previous heart failure, and about 22% had a history of left main coronary artery disease. Half had diabetes, and about half had stable angina.
Multiple stent layers were common in 43% of each group. Stenosis diameter was about 65% at baseline for the two groups and was reduced to 22% post procedure.
Outcomes all favored AGENT DCB
In the AGENT DCB group, the technical success rate was 92.9% vs 89.3% for balloon angioplasty. Intravascular imaging was used during the procedure in 72.3% of DCB cases and in 76.7% of balloon cases.
Besides demonstrating a nearly 38% reduction in the primary endpoint of target lesion failure at 1 year for the DCB over conventional balloon angioplasty, DCB nearly halved the rate of target lesion revascularization and target vessel MI and was superior on other measures of clinical outcome.
*Hazard ratio, 0.49; 95% CI, 0.31-0.79; ** HR, 0.51; 95% CI, 0.27-0.95
There was no stent rethrombosis with the DCB vs 3.9% with the conventional balloon angioplasty. Of note, there were no differences between the groups in terms of cardiac or noncardiac death.
Subgroup analyses of the primary outcome in terms of sex, age, diabetes, vessel size, or single or multiple stent layers all trended in favor of AGENT DCB but were not statistically significant for interaction.
The study is being expanded to include 600 patients. This device is a US Food and Drug Administration–designated breakthrough device, “and this pivotal trial will be the primary evidence used to support FDA approval,” Dr. Yeh said. “And given the marked superiority over conventional balloon angioplasty, I believe that the AGENT DCB is likely to become an important new treatment option for patients with coronary stenosis in the United States.”
Long overdue
Róisín Colleran, MBBCh, of the Cardiovascular Research Institute Dublin at Mater Private Hospital in Ireland, the designated discussant, first congratulated Dr. Yeh and his coinvestigators on the study’s conduct and findings.
“This study is long overdue,” she said. As Dr. Yeh noted, about 10% of PCI procedures are done for in-stent restenosis, Dr. Colleran said, but in 2023, there is still no coronary drug eluting balloon approved for this indication in the US, despite the class 1 recommendation in the 2014 European guidelines.
She pointed to the trial results, saying they are “clear...a significant reduction in target lesion failure driven by halving in rates of both target lesion revascularization and target vessel MI.”
Strengths of the study are it is the largest of its kind to date, with 480 patients, conducted at 40 US centers, using device-specific endpoints. There was a “very high” intravascular imaging rate of 75% in a cohort with a high risk for in-stent restenosis, consisting of 50% of patients with diabetes and more than 40% with multiple stents.
“The main limitation is the choice of comparator,” Dr. Colleran said. Balloon angioplasty is inferior to both stenting and drug coated balloon therapy for treatment of in-stent restenosis but is the standard of care in the United States, she noted. “I think...for regulatory reasons this was the comparator chosen,” she said.
“I think the implications are clear,” Dr. Colleran added. “This trial should provide a basis for regulatory approval of the drug coated balloon treatment of in-stent restenosis in the U.S. and finally provide this as an available treatment option for such patients.”
Dr. Yeh reported receiving grant/research support from Abbott Vascular, BD Bard, Boston Scientific, Cook Medical, Philips Medical, and Medtronic, and consulting for Abbott Vascular, Boston Scientific, CathWorks, Elixir Medical, Infraredx, Medtronic, Shockwave Medical, and Zol. Dr. Colleran had no disclosures. The trial was supported by Boston Scientific.
A version of this article first appeared on Medscape.com.
SAN FRANCISCO – For the treatment of coronary artery in-stent restenosis, angioplasty with a drug-coated balloon (AGENT DCB; Boston Scientific) was superior to conventional balloon angioplasty in preventing target lesion failure at 1 year in a high-risk patient population.
Approximate 50% reductions in the rates of target lesion restenosis and target vessel myocardial infarction (MI) accounted for the superior findings with the AGENT DCB over conventional balloon angioplasty.
Robert Yeh, MD, of Beth Israel Deaconess Medical Center in Boston reported at the annual Transcatheter Cardiovascular Therapeutics congress. “This represented a 38% relative risk reduction as well as a 10% absolute risk reduction in the endpoint. The P value for superiority was 0.0063, highly statistically significant.”
In-stent restenosis is clinically challenging and accounts for about 10% of all percutaneous coronary interventions. “Sometimes these patients have multiple layers, and that could be a third or fourth layer of stent, something that we try to avoid,” he said.
Drug-coated balloons, which are not currently approved in the United States, can deliver drugs that inhibit blockages from reforming, “without leaving additional layers of metal behind,” he added. Such devices are already available in Europe and Japan.
AGENT IDE was a prospective, multicenter, superiority trial that randomly assigned 480 patients 2:1 to the AGENT DCB (n = 321) or to conventional balloon angioplasty (n = 159). Randomization occurred after successful pre-dilation of the target vessel.
The trial included patients with in-stent restenosis previously treated with a bare metal or a drug-eluting stent with lesion lengths < 26 mm (reference vessel diameter: > 2 mm to ≤ 4), and percent diameter stenosis of more than 70% if they were asymptomatic or of more than 50% if they were symptomatic. Patients were excluded if they had a recent ST-elevation MI, bifurcation, saphenous vein or arterial graft, or thrombus in the target vessel.
All received dual antiplatelet therapy for at least 1 month and then antiplatelet monotherapy for the duration of the trial. The primary endpoint was target lesion failure at 1 year, a composite of target lesion restenosis, target vessel-related MI, or cardiac death. More than 93% of patients in each arm were available for evaluation of the primary endpoint.
The two groups were well balanced at baseline: Approximate age was 68 years, 27% were women, and three quarters were White. Approximately 28%-32% had had a prior coronary artery bypass graft, 20%-22% had previous heart failure, and about 22% had a history of left main coronary artery disease. Half had diabetes, and about half had stable angina.
Multiple stent layers were common in 43% of each group. Stenosis diameter was about 65% at baseline for the two groups and was reduced to 22% post procedure.
Outcomes all favored AGENT DCB
In the AGENT DCB group, the technical success rate was 92.9% vs 89.3% for balloon angioplasty. Intravascular imaging was used during the procedure in 72.3% of DCB cases and in 76.7% of balloon cases.
Besides demonstrating a nearly 38% reduction in the primary endpoint of target lesion failure at 1 year for the DCB over conventional balloon angioplasty, DCB nearly halved the rate of target lesion revascularization and target vessel MI and was superior on other measures of clinical outcome.
*Hazard ratio, 0.49; 95% CI, 0.31-0.79; ** HR, 0.51; 95% CI, 0.27-0.95
There was no stent rethrombosis with the DCB vs 3.9% with the conventional balloon angioplasty. Of note, there were no differences between the groups in terms of cardiac or noncardiac death.
Subgroup analyses of the primary outcome in terms of sex, age, diabetes, vessel size, or single or multiple stent layers all trended in favor of AGENT DCB but were not statistically significant for interaction.
The study is being expanded to include 600 patients. This device is a US Food and Drug Administration–designated breakthrough device, “and this pivotal trial will be the primary evidence used to support FDA approval,” Dr. Yeh said. “And given the marked superiority over conventional balloon angioplasty, I believe that the AGENT DCB is likely to become an important new treatment option for patients with coronary stenosis in the United States.”
Long overdue
Róisín Colleran, MBBCh, of the Cardiovascular Research Institute Dublin at Mater Private Hospital in Ireland, the designated discussant, first congratulated Dr. Yeh and his coinvestigators on the study’s conduct and findings.
“This study is long overdue,” she said. As Dr. Yeh noted, about 10% of PCI procedures are done for in-stent restenosis, Dr. Colleran said, but in 2023, there is still no coronary drug eluting balloon approved for this indication in the US, despite the class 1 recommendation in the 2014 European guidelines.
She pointed to the trial results, saying they are “clear...a significant reduction in target lesion failure driven by halving in rates of both target lesion revascularization and target vessel MI.”
Strengths of the study are it is the largest of its kind to date, with 480 patients, conducted at 40 US centers, using device-specific endpoints. There was a “very high” intravascular imaging rate of 75% in a cohort with a high risk for in-stent restenosis, consisting of 50% of patients with diabetes and more than 40% with multiple stents.
“The main limitation is the choice of comparator,” Dr. Colleran said. Balloon angioplasty is inferior to both stenting and drug coated balloon therapy for treatment of in-stent restenosis but is the standard of care in the United States, she noted. “I think...for regulatory reasons this was the comparator chosen,” she said.
“I think the implications are clear,” Dr. Colleran added. “This trial should provide a basis for regulatory approval of the drug coated balloon treatment of in-stent restenosis in the U.S. and finally provide this as an available treatment option for such patients.”
Dr. Yeh reported receiving grant/research support from Abbott Vascular, BD Bard, Boston Scientific, Cook Medical, Philips Medical, and Medtronic, and consulting for Abbott Vascular, Boston Scientific, CathWorks, Elixir Medical, Infraredx, Medtronic, Shockwave Medical, and Zol. Dr. Colleran had no disclosures. The trial was supported by Boston Scientific.
A version of this article first appeared on Medscape.com.
SAN FRANCISCO – For the treatment of coronary artery in-stent restenosis, angioplasty with a drug-coated balloon (AGENT DCB; Boston Scientific) was superior to conventional balloon angioplasty in preventing target lesion failure at 1 year in a high-risk patient population.
Approximate 50% reductions in the rates of target lesion restenosis and target vessel myocardial infarction (MI) accounted for the superior findings with the AGENT DCB over conventional balloon angioplasty.
Robert Yeh, MD, of Beth Israel Deaconess Medical Center in Boston reported at the annual Transcatheter Cardiovascular Therapeutics congress. “This represented a 38% relative risk reduction as well as a 10% absolute risk reduction in the endpoint. The P value for superiority was 0.0063, highly statistically significant.”
In-stent restenosis is clinically challenging and accounts for about 10% of all percutaneous coronary interventions. “Sometimes these patients have multiple layers, and that could be a third or fourth layer of stent, something that we try to avoid,” he said.
Drug-coated balloons, which are not currently approved in the United States, can deliver drugs that inhibit blockages from reforming, “without leaving additional layers of metal behind,” he added. Such devices are already available in Europe and Japan.
AGENT IDE was a prospective, multicenter, superiority trial that randomly assigned 480 patients 2:1 to the AGENT DCB (n = 321) or to conventional balloon angioplasty (n = 159). Randomization occurred after successful pre-dilation of the target vessel.
The trial included patients with in-stent restenosis previously treated with a bare metal or a drug-eluting stent with lesion lengths < 26 mm (reference vessel diameter: > 2 mm to ≤ 4), and percent diameter stenosis of more than 70% if they were asymptomatic or of more than 50% if they were symptomatic. Patients were excluded if they had a recent ST-elevation MI, bifurcation, saphenous vein or arterial graft, or thrombus in the target vessel.
All received dual antiplatelet therapy for at least 1 month and then antiplatelet monotherapy for the duration of the trial. The primary endpoint was target lesion failure at 1 year, a composite of target lesion restenosis, target vessel-related MI, or cardiac death. More than 93% of patients in each arm were available for evaluation of the primary endpoint.
The two groups were well balanced at baseline: Approximate age was 68 years, 27% were women, and three quarters were White. Approximately 28%-32% had had a prior coronary artery bypass graft, 20%-22% had previous heart failure, and about 22% had a history of left main coronary artery disease. Half had diabetes, and about half had stable angina.
Multiple stent layers were common in 43% of each group. Stenosis diameter was about 65% at baseline for the two groups and was reduced to 22% post procedure.
Outcomes all favored AGENT DCB
In the AGENT DCB group, the technical success rate was 92.9% vs 89.3% for balloon angioplasty. Intravascular imaging was used during the procedure in 72.3% of DCB cases and in 76.7% of balloon cases.
Besides demonstrating a nearly 38% reduction in the primary endpoint of target lesion failure at 1 year for the DCB over conventional balloon angioplasty, DCB nearly halved the rate of target lesion revascularization and target vessel MI and was superior on other measures of clinical outcome.
*Hazard ratio, 0.49; 95% CI, 0.31-0.79; ** HR, 0.51; 95% CI, 0.27-0.95
There was no stent rethrombosis with the DCB vs 3.9% with the conventional balloon angioplasty. Of note, there were no differences between the groups in terms of cardiac or noncardiac death.
Subgroup analyses of the primary outcome in terms of sex, age, diabetes, vessel size, or single or multiple stent layers all trended in favor of AGENT DCB but were not statistically significant for interaction.
The study is being expanded to include 600 patients. This device is a US Food and Drug Administration–designated breakthrough device, “and this pivotal trial will be the primary evidence used to support FDA approval,” Dr. Yeh said. “And given the marked superiority over conventional balloon angioplasty, I believe that the AGENT DCB is likely to become an important new treatment option for patients with coronary stenosis in the United States.”
Long overdue
Róisín Colleran, MBBCh, of the Cardiovascular Research Institute Dublin at Mater Private Hospital in Ireland, the designated discussant, first congratulated Dr. Yeh and his coinvestigators on the study’s conduct and findings.
“This study is long overdue,” she said. As Dr. Yeh noted, about 10% of PCI procedures are done for in-stent restenosis, Dr. Colleran said, but in 2023, there is still no coronary drug eluting balloon approved for this indication in the US, despite the class 1 recommendation in the 2014 European guidelines.
She pointed to the trial results, saying they are “clear...a significant reduction in target lesion failure driven by halving in rates of both target lesion revascularization and target vessel MI.”
Strengths of the study are it is the largest of its kind to date, with 480 patients, conducted at 40 US centers, using device-specific endpoints. There was a “very high” intravascular imaging rate of 75% in a cohort with a high risk for in-stent restenosis, consisting of 50% of patients with diabetes and more than 40% with multiple stents.
“The main limitation is the choice of comparator,” Dr. Colleran said. Balloon angioplasty is inferior to both stenting and drug coated balloon therapy for treatment of in-stent restenosis but is the standard of care in the United States, she noted. “I think...for regulatory reasons this was the comparator chosen,” she said.
“I think the implications are clear,” Dr. Colleran added. “This trial should provide a basis for regulatory approval of the drug coated balloon treatment of in-stent restenosis in the U.S. and finally provide this as an available treatment option for such patients.”
Dr. Yeh reported receiving grant/research support from Abbott Vascular, BD Bard, Boston Scientific, Cook Medical, Philips Medical, and Medtronic, and consulting for Abbott Vascular, Boston Scientific, CathWorks, Elixir Medical, Infraredx, Medtronic, Shockwave Medical, and Zol. Dr. Colleran had no disclosures. The trial was supported by Boston Scientific.
A version of this article first appeared on Medscape.com.
AT TCT 2023
Second pig heart recipient dies
the University of Maryland Medical Center (UMMC), Baltimore, reported in a statement.
Mr. Faucette, a former lab tech who was turned down repeatedly for a standard allograft transplantation because of his various medical conditions, received the pig heart transplant on Sept. 20, 2023.
He first came to UMMC as a patient on Sept. 14. When he was admitted, he was in end-stage heart failure. Shortly before the surgery, his heart stopped, and he required resuscitation.
On Sept. 15, the Food and Drug Administration granted an emergency authorization for the surgery through its single-patient investigational new drug compassionate use pathway.
“My only real hope left is to go with the pig heart, the xenotransplant,” Mr. Faucette said in an interview from his hospital room a few days before his surgery. “At least now I have hope, and I have a chance.” He made “significant progress” in the month after the surgery, participating in physical therapy and spending time with family, according to the university. But in the days before his death, the heart showed signs of rejection.
“Mr. Faucette’s last wish was for us to make the most of what we have learned from our experience, so others may be guaranteed a chance for a new heart when a human organ is unavailable,” said Bartley P. Griffith, MD, who transplanted the pig heart into Mr. Faucette at UMMC. “He then told the team of doctors and nurses who gathered around him that he loved us. We will miss him tremendously.”
Muhammad M. Mohiuddin, MD, professor of surgery and scientific/program director of the Cardiac Xenotransplantation Program at the University of Maryland School of Medicine, said that “Mr. Faucette was a scientist who not only read and interpreted his own biopsies, but who understood the important contribution he was making in advancing the field.
“As with the first patient, David Bennett Sr., we intend to conduct an extensive analysis to identify factors that can be prevented in future transplants; this will allow us to continue to move forward and educate our colleagues in the field on our experience,” Dr. Mohiuddin added.
The researchers don’t plan to make further comments until their investigation is complete, a university spokesperson said in an interview.
UMMC performed the first transplant of a genetically modified pig heart in January 2022. Mr. Bennett, the recipient of that heart, survived for 60 days. The researchers published their initial findings in The New England Journal of Medicine, and then the results of their follow-up investigation in The Lancet.
A version of this article first appeared on Medscape.com.
the University of Maryland Medical Center (UMMC), Baltimore, reported in a statement.
Mr. Faucette, a former lab tech who was turned down repeatedly for a standard allograft transplantation because of his various medical conditions, received the pig heart transplant on Sept. 20, 2023.
He first came to UMMC as a patient on Sept. 14. When he was admitted, he was in end-stage heart failure. Shortly before the surgery, his heart stopped, and he required resuscitation.
On Sept. 15, the Food and Drug Administration granted an emergency authorization for the surgery through its single-patient investigational new drug compassionate use pathway.
“My only real hope left is to go with the pig heart, the xenotransplant,” Mr. Faucette said in an interview from his hospital room a few days before his surgery. “At least now I have hope, and I have a chance.” He made “significant progress” in the month after the surgery, participating in physical therapy and spending time with family, according to the university. But in the days before his death, the heart showed signs of rejection.
“Mr. Faucette’s last wish was for us to make the most of what we have learned from our experience, so others may be guaranteed a chance for a new heart when a human organ is unavailable,” said Bartley P. Griffith, MD, who transplanted the pig heart into Mr. Faucette at UMMC. “He then told the team of doctors and nurses who gathered around him that he loved us. We will miss him tremendously.”
Muhammad M. Mohiuddin, MD, professor of surgery and scientific/program director of the Cardiac Xenotransplantation Program at the University of Maryland School of Medicine, said that “Mr. Faucette was a scientist who not only read and interpreted his own biopsies, but who understood the important contribution he was making in advancing the field.
“As with the first patient, David Bennett Sr., we intend to conduct an extensive analysis to identify factors that can be prevented in future transplants; this will allow us to continue to move forward and educate our colleagues in the field on our experience,” Dr. Mohiuddin added.
The researchers don’t plan to make further comments until their investigation is complete, a university spokesperson said in an interview.
UMMC performed the first transplant of a genetically modified pig heart in January 2022. Mr. Bennett, the recipient of that heart, survived for 60 days. The researchers published their initial findings in The New England Journal of Medicine, and then the results of their follow-up investigation in The Lancet.
A version of this article first appeared on Medscape.com.
the University of Maryland Medical Center (UMMC), Baltimore, reported in a statement.
Mr. Faucette, a former lab tech who was turned down repeatedly for a standard allograft transplantation because of his various medical conditions, received the pig heart transplant on Sept. 20, 2023.
He first came to UMMC as a patient on Sept. 14. When he was admitted, he was in end-stage heart failure. Shortly before the surgery, his heart stopped, and he required resuscitation.
On Sept. 15, the Food and Drug Administration granted an emergency authorization for the surgery through its single-patient investigational new drug compassionate use pathway.
“My only real hope left is to go with the pig heart, the xenotransplant,” Mr. Faucette said in an interview from his hospital room a few days before his surgery. “At least now I have hope, and I have a chance.” He made “significant progress” in the month after the surgery, participating in physical therapy and spending time with family, according to the university. But in the days before his death, the heart showed signs of rejection.
“Mr. Faucette’s last wish was for us to make the most of what we have learned from our experience, so others may be guaranteed a chance for a new heart when a human organ is unavailable,” said Bartley P. Griffith, MD, who transplanted the pig heart into Mr. Faucette at UMMC. “He then told the team of doctors and nurses who gathered around him that he loved us. We will miss him tremendously.”
Muhammad M. Mohiuddin, MD, professor of surgery and scientific/program director of the Cardiac Xenotransplantation Program at the University of Maryland School of Medicine, said that “Mr. Faucette was a scientist who not only read and interpreted his own biopsies, but who understood the important contribution he was making in advancing the field.
“As with the first patient, David Bennett Sr., we intend to conduct an extensive analysis to identify factors that can be prevented in future transplants; this will allow us to continue to move forward and educate our colleagues in the field on our experience,” Dr. Mohiuddin added.
The researchers don’t plan to make further comments until their investigation is complete, a university spokesperson said in an interview.
UMMC performed the first transplant of a genetically modified pig heart in January 2022. Mr. Bennett, the recipient of that heart, survived for 60 days. The researchers published their initial findings in The New England Journal of Medicine, and then the results of their follow-up investigation in The Lancet.
A version of this article first appeared on Medscape.com.