User login
Monitoring patients with vasculitis
Granulomatosis with polyangiitis (GPA), is one of the most common types of small-vessel vasculitis, with an estimated prevalence in the United States of 3 per 100,000 people. It is distinguished from other necrotizing vasculitides by its tendency to affect the upper and lower respiratory system and the kidneys. Despite the success of induction and maintenance treatments with cyclophosphamide (CYC), glucocorticoids, and less toxic immunosuppressive alternative therapies in improving the disease course, significant treatment-related toxicities and frequent disease relapses demand stringent patient-specific monitoring in order to provide early treatment of relapses and prevent or decrease morbidity.
SMALL-VESSEL VASCULITIS MANAGEMENT OVERVIEW
Granulomatosis with polyangiitis (formerly Wegener’s granulomatosis, or WG) is an antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis that often affects the respiratory system and kidneys across a broad spectrum of clinical presentations, from mild through life-threatening disease. Patients with severe disease present with significant multisystem manifestations, which, in addition to the respiratory system and kidneys, may involve the joints, eyes, and other organs.
Managing patients diagnosed with systemic small-vessel vasculitides such as GPA and microscopic polyangiitis (MPA) is an inexact science. The goals of treatment are to increase survival, induce and maintain remission, reduce relapses, and minimize treatment-related toxicity. Inducing and maintaining remission have become realistic goals because of the availability of medications that prolong life. On the other hand, extended periods of treatment associated with prolonged life increase the risk of treatment-related toxicity in patients who are inadequately monitored.
MONITORING CONSIDERATIONS
Achieving treatment goals requires long-term monitoring of both disease activity and treatment-related toxicities, with constant adjustments to meet the needs of the individual patient and address the often rapidly changing disease and treatment course. The monitoring protocol consists of regularly scheduled follow-up office visits, urine sediment analyses at every office visit whether or not the patient has relapse symptoms, laboratory tests at regular intervals as indicated by the patient’s medication plan and disease presentation, additional tests such as lung computed tomography (CT), and patient education regarding new symptoms and the frequency of office visits. A consistent monitoring strategy will help detect a relapse before it can produce more severe morbidity, identify treatment-related complications, and—equally important—identify the achievement of remission. An example of the consequences of inconsistent monitoring is presented in “Relapse in a nonadherent patient.”
Because there is no definitive cure for small-vessel vasculitis, relapse is always a possibility. The early diagnosis and treatment of relapse may prevent or decrease morbidity from disease, but strict monitoring is needed to identify relapse and initiate treatment before morbidity occurs (see “Relapse in a patient with new symptoms”). Repeat induction therapy following a relapse introduces risk of drug toxicity and requires careful monitoring, as does long-term maintenance therapy.
In addition to induction and maintenance therapy, several other situations, including prior therapeutic complications, serum creatinine levels, and risk of cardiovascular disease, require special monitoring attention.

Induction therapy: monitor response
Response to treatment during induction must be monitored to identify whether remission is achieved. Induction monitoring requires complete assessment of organ-system involvement at every visit with tools such as the Birmingham Vasculitis Activity Score (BVAS) and, when appropriate, the BVAS/WG. If new or worsening symptoms develop during induction therapy, then the patient needs assessment for continued disease activity as well as treatment complications such as infections related to immunosuppressive therapy.
During induction therapy with daily oral CYC, monitoring should include weekly complete blood cell counts to ensure early identification of leukopenia and other cytopenias. The risk of morbidities increases with the cumulative dose, so a stable blood count for 2 months does not obviate the risk of leukopenia. If persistent hematuria is present without cellular casts, cystoscopy is indicated to look for signs of hemorrhagic cystitis. Prophylaxis against Pneumocystis jirovecii is recommended in all patients who receive immunosuppressive therapy. Finally, bone density measurements should be done at baseline.
Maintenance therapy: frequency can be extended
Monitoring during maintenance therapy is similar to induction monitoring; however, when the dosage of methotrexate or azathioprine is stabilized, the frequency of some tests can be extended to monthly rather than weekly. For example, a complete blood cell count, comprehensive metabolic panel, sedimentation rate, C-reactive protein measurement, and urinalysis should be performed monthly. Follow-up visits should include urine sediment analyses and monitoring for cardiovascular disease risk factors. Medication monitoring should include cystoscopy for persistent hematuria without cellular casts, bone density measurements, and ophthalmologic examinations as frequently as indicated for each individual’s needs. P jirovecii prophylaxis should continue as long as the patient receives immunosuppressive medication.

Therapy-related complications
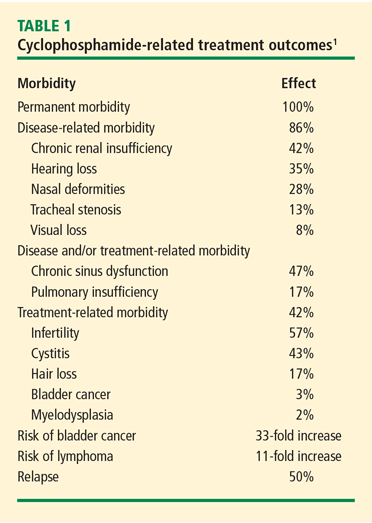
Bladder complications. In a retrospective analysis of 145 patients with GPA treated with CYC and followed for 0.5 to 27 years (median 8.5 years), nonglomerular hematuria developed in 50% of the patients and bladder carcinoma in 5%.2 The cumulative CYC dose (19 to 251 g) in this group was much higher than what is currently used. Cytologic examination of the urine showed 43% sensitivity for dysplasia (specificity 100%) and 29% sensitivity for atypia (specificity 89%). In contrast, in a retrospective outcomes analysis involving newly diagnosed patients with GPA treated with CYC or methotrexate, 82 patients followed for up to 12 years had no incidents of cystitis or bladder cancer.3 Patients in this study were treated with CYC for only 3 to 6 months and therefore received a lower cumulative dose.
To prevent cystitis during treatment with CYC, the patient should be well hydrated, especially in the morning when CYC should be taken. The bladder should be emptied frequently. The addition of mesna when administering intravenous CYC decreases the risk of cystitis. Serial cystoscopy and urine cytology should be used only in patients with nonglomerular hematuria.
Infertility. Preservation of ovarian function is a concern with CYC therapy in women of childbearing age. The cumulative dose threshold for gonadal failure is unknown, because data from cancer studies4 demonstrating gonadal failure involve higher cumulative CYC doses than are typical for vasculitis treatment. It is also unknown whether duration of amenorrhea predicts the recovery of menses or fertility. The primary option for preservation of ovarian function is the use of gonadotropin-releasing hormone agonists. Oral contraceptives also may be used, but the best prevention is to avoid CYC in these patients if possible.

Osteoporosis. At glucocorticoid dosages of 5 mg/day or greater, bone mineral density begins a rapid decline within the first 3 months and peaks at 6 months.5 The American College of Rheumatology has provided recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis.5 Table 2 presents recommendations for postmenopausal women and men aged 50 years and older who will use glucocorticoids for 3 months or more.5 Recommendations are also available for premenopausal women and men younger than 50 years of age who have a history of fragility fracture.
Leukopenia. Leukopenia should be avoided during CYC treatment. The target white blood cell count should be within the normal range. During treatment with daily oral CYC, the patient should be monitored with a weekly complete blood cell count and medication should be adjusted to maintain the target white blood cell count.
Upon completion of induction therapy, after 3 to 6 months, the patient is switched to maintenance therapy with an alternative immunosuppressive agent such as azathioprine or methotrexate, depending on the serum creatinine concentration and other factors. This transition, characterized by full-dose immunosuppressive therapy when the bone marrow has been previously suppressed by CYC treatment, may induce pancytopenia. Monitoring with weekly complete blood counts for at least 4 weeks after initiating maintenance therapy can help ensure stability during the transition period.

Monitor serum creatinine and adjust dosages
The serum creatinine concentration may increase as CYC treatment progresses; in some cases, the serum creatinine concentration increases before a response to treatment is seen. The CYC dosages should be adjusted as necessary in response to serum creatinine changes. Careful monitoring of serum creatinine is necessary during methotrexate therapy, as methotrexate treatment in the setting of renal insufficiency increases the risk of bone marrow suppression.
Cardiovascular disease in GPA and MPA
Premature atherosclerosis has been well described in patients with GPA.6 Within 5 years of diagnosis of GPA or MPA, a cardiovascular event will occur in 14% of patients.7 In the absence of specific guidelines for prevention of cardiovascular disease in patients with vasculitis, it is essential to monitor patients and treat modifiable traditional risk factors aggressively, especially in younger patients. Suppiah et al found that independent determinants of cardiovascular outcome included older age, diastolic hypertension, and positive proteinase-3–ANCA status in patients without prior cardiovascular disease.7
In the Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) study, Merkel et al showed an increased incidence of thrombosis in patients with active GPA8 (see “Relapse presenting as thrombosis,” left). As with cardiovascular disease, there are no specific guidelines for monitoring asymptomatic patients for thrombosis or for duration of anticoagulation in patients with GPA. It is recommended that patients be evaluated for active GPA or relapse in the setting of acute thrombosis whether or not symptoms of active GPA are present.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Talar-Williams C, Hijazi YM, Walther MM, et al. Cyclophosphamide-induced cystitis and bladder cancer in patients with Wegener granulomatosis. Ann Intern Med 1996; 124:477–484.
- Villa-Forte A, Clark TM, Gomes M, et al. Substitution of methotrexate for cyclophosphamide in Wegener granulomatosis: a 12-year single-practice experience. Medicine 2007; 86:269–277.
- Harel S, Fermé C, Poirot C. Management of fertility in patients treated for Hodgkin’s lymphoma [published online ahead of print August 9, 2011]. Haematologica 2011; 96:1692–1699. doi: 10.3324/haematol.2011.045856
- Grossman JM, Gordon R, Ranganath VK, et al. American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis [published online ahead of print July 26, 2010]. Arthritis Care Res (Hoboken) 2010; 62:1515–1526. doi: 10.1002/acr.20295
- Faurschou M, Mellemkjaer L, Sorensen IJ, Svalgaard Thomsen B, Dreyer L, Baslund B. Increased morbidity from ischemic heart disease in patients with Wegener’s granulomatosis. Arthritis Rheum 2009; 60:1187–1192.
- Suppiah R, Judge A, Batra R, et al. A model to predict cardiovascular events in patients with newly diagnosed Wegener’s granulomatosis and microscopic polyangiitis. Arthritis Care Res (Hoboken) 2011; 63:588–596.
- Merkel PA, Lo GH, Holbrook JT, et al; for Wegener’s Granulomatosis Etanercept Trial Research Group. Brief communication: high incidence of venous thrombotic events among patients with Wegener granulomatosis: the Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) study. Ann Intern Med 2005; 142:620–626.
Granulomatosis with polyangiitis (GPA), is one of the most common types of small-vessel vasculitis, with an estimated prevalence in the United States of 3 per 100,000 people. It is distinguished from other necrotizing vasculitides by its tendency to affect the upper and lower respiratory system and the kidneys. Despite the success of induction and maintenance treatments with cyclophosphamide (CYC), glucocorticoids, and less toxic immunosuppressive alternative therapies in improving the disease course, significant treatment-related toxicities and frequent disease relapses demand stringent patient-specific monitoring in order to provide early treatment of relapses and prevent or decrease morbidity.
SMALL-VESSEL VASCULITIS MANAGEMENT OVERVIEW
Granulomatosis with polyangiitis (formerly Wegener’s granulomatosis, or WG) is an antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis that often affects the respiratory system and kidneys across a broad spectrum of clinical presentations, from mild through life-threatening disease. Patients with severe disease present with significant multisystem manifestations, which, in addition to the respiratory system and kidneys, may involve the joints, eyes, and other organs.
Managing patients diagnosed with systemic small-vessel vasculitides such as GPA and microscopic polyangiitis (MPA) is an inexact science. The goals of treatment are to increase survival, induce and maintain remission, reduce relapses, and minimize treatment-related toxicity. Inducing and maintaining remission have become realistic goals because of the availability of medications that prolong life. On the other hand, extended periods of treatment associated with prolonged life increase the risk of treatment-related toxicity in patients who are inadequately monitored.
MONITORING CONSIDERATIONS
Achieving treatment goals requires long-term monitoring of both disease activity and treatment-related toxicities, with constant adjustments to meet the needs of the individual patient and address the often rapidly changing disease and treatment course. The monitoring protocol consists of regularly scheduled follow-up office visits, urine sediment analyses at every office visit whether or not the patient has relapse symptoms, laboratory tests at regular intervals as indicated by the patient’s medication plan and disease presentation, additional tests such as lung computed tomography (CT), and patient education regarding new symptoms and the frequency of office visits. A consistent monitoring strategy will help detect a relapse before it can produce more severe morbidity, identify treatment-related complications, and—equally important—identify the achievement of remission. An example of the consequences of inconsistent monitoring is presented in “Relapse in a nonadherent patient.”
Because there is no definitive cure for small-vessel vasculitis, relapse is always a possibility. The early diagnosis and treatment of relapse may prevent or decrease morbidity from disease, but strict monitoring is needed to identify relapse and initiate treatment before morbidity occurs (see “Relapse in a patient with new symptoms”). Repeat induction therapy following a relapse introduces risk of drug toxicity and requires careful monitoring, as does long-term maintenance therapy.
In addition to induction and maintenance therapy, several other situations, including prior therapeutic complications, serum creatinine levels, and risk of cardiovascular disease, require special monitoring attention.
Induction therapy: monitor response
Response to treatment during induction must be monitored to identify whether remission is achieved. Induction monitoring requires complete assessment of organ-system involvement at every visit with tools such as the Birmingham Vasculitis Activity Score (BVAS) and, when appropriate, the BVAS/WG. If new or worsening symptoms develop during induction therapy, then the patient needs assessment for continued disease activity as well as treatment complications such as infections related to immunosuppressive therapy.
During induction therapy with daily oral CYC, monitoring should include weekly complete blood cell counts to ensure early identification of leukopenia and other cytopenias. The risk of morbidities increases with the cumulative dose, so a stable blood count for 2 months does not obviate the risk of leukopenia. If persistent hematuria is present without cellular casts, cystoscopy is indicated to look for signs of hemorrhagic cystitis. Prophylaxis against Pneumocystis jirovecii is recommended in all patients who receive immunosuppressive therapy. Finally, bone density measurements should be done at baseline.
Maintenance therapy: frequency can be extended
Monitoring during maintenance therapy is similar to induction monitoring; however, when the dosage of methotrexate or azathioprine is stabilized, the frequency of some tests can be extended to monthly rather than weekly. For example, a complete blood cell count, comprehensive metabolic panel, sedimentation rate, C-reactive protein measurement, and urinalysis should be performed monthly. Follow-up visits should include urine sediment analyses and monitoring for cardiovascular disease risk factors. Medication monitoring should include cystoscopy for persistent hematuria without cellular casts, bone density measurements, and ophthalmologic examinations as frequently as indicated for each individual’s needs. P jirovecii prophylaxis should continue as long as the patient receives immunosuppressive medication.
Therapy-related complications
Bladder complications. In a retrospective analysis of 145 patients with GPA treated with CYC and followed for 0.5 to 27 years (median 8.5 years), nonglomerular hematuria developed in 50% of the patients and bladder carcinoma in 5%.2 The cumulative CYC dose (19 to 251 g) in this group was much higher than what is currently used. Cytologic examination of the urine showed 43% sensitivity for dysplasia (specificity 100%) and 29% sensitivity for atypia (specificity 89%). In contrast, in a retrospective outcomes analysis involving newly diagnosed patients with GPA treated with CYC or methotrexate, 82 patients followed for up to 12 years had no incidents of cystitis or bladder cancer.3 Patients in this study were treated with CYC for only 3 to 6 months and therefore received a lower cumulative dose.
To prevent cystitis during treatment with CYC, the patient should be well hydrated, especially in the morning when CYC should be taken. The bladder should be emptied frequently. The addition of mesna when administering intravenous CYC decreases the risk of cystitis. Serial cystoscopy and urine cytology should be used only in patients with nonglomerular hematuria.
Infertility. Preservation of ovarian function is a concern with CYC therapy in women of childbearing age. The cumulative dose threshold for gonadal failure is unknown, because data from cancer studies4 demonstrating gonadal failure involve higher cumulative CYC doses than are typical for vasculitis treatment. It is also unknown whether duration of amenorrhea predicts the recovery of menses or fertility. The primary option for preservation of ovarian function is the use of gonadotropin-releasing hormone agonists. Oral contraceptives also may be used, but the best prevention is to avoid CYC in these patients if possible.
Osteoporosis. At glucocorticoid dosages of 5 mg/day or greater, bone mineral density begins a rapid decline within the first 3 months and peaks at 6 months.5 The American College of Rheumatology has provided recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis.5 Table 2 presents recommendations for postmenopausal women and men aged 50 years and older who will use glucocorticoids for 3 months or more.5 Recommendations are also available for premenopausal women and men younger than 50 years of age who have a history of fragility fracture.
Leukopenia. Leukopenia should be avoided during CYC treatment. The target white blood cell count should be within the normal range. During treatment with daily oral CYC, the patient should be monitored with a weekly complete blood cell count and medication should be adjusted to maintain the target white blood cell count.
Upon completion of induction therapy, after 3 to 6 months, the patient is switched to maintenance therapy with an alternative immunosuppressive agent such as azathioprine or methotrexate, depending on the serum creatinine concentration and other factors. This transition, characterized by full-dose immunosuppressive therapy when the bone marrow has been previously suppressed by CYC treatment, may induce pancytopenia. Monitoring with weekly complete blood counts for at least 4 weeks after initiating maintenance therapy can help ensure stability during the transition period.
Monitor serum creatinine and adjust dosages
The serum creatinine concentration may increase as CYC treatment progresses; in some cases, the serum creatinine concentration increases before a response to treatment is seen. The CYC dosages should be adjusted as necessary in response to serum creatinine changes. Careful monitoring of serum creatinine is necessary during methotrexate therapy, as methotrexate treatment in the setting of renal insufficiency increases the risk of bone marrow suppression.
Cardiovascular disease in GPA and MPA
Premature atherosclerosis has been well described in patients with GPA.6 Within 5 years of diagnosis of GPA or MPA, a cardiovascular event will occur in 14% of patients.7 In the absence of specific guidelines for prevention of cardiovascular disease in patients with vasculitis, it is essential to monitor patients and treat modifiable traditional risk factors aggressively, especially in younger patients. Suppiah et al found that independent determinants of cardiovascular outcome included older age, diastolic hypertension, and positive proteinase-3–ANCA status in patients without prior cardiovascular disease.7
In the Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) study, Merkel et al showed an increased incidence of thrombosis in patients with active GPA8 (see “Relapse presenting as thrombosis,” left). As with cardiovascular disease, there are no specific guidelines for monitoring asymptomatic patients for thrombosis or for duration of anticoagulation in patients with GPA. It is recommended that patients be evaluated for active GPA or relapse in the setting of acute thrombosis whether or not symptoms of active GPA are present.
Granulomatosis with polyangiitis (GPA), is one of the most common types of small-vessel vasculitis, with an estimated prevalence in the United States of 3 per 100,000 people. It is distinguished from other necrotizing vasculitides by its tendency to affect the upper and lower respiratory system and the kidneys. Despite the success of induction and maintenance treatments with cyclophosphamide (CYC), glucocorticoids, and less toxic immunosuppressive alternative therapies in improving the disease course, significant treatment-related toxicities and frequent disease relapses demand stringent patient-specific monitoring in order to provide early treatment of relapses and prevent or decrease morbidity.
SMALL-VESSEL VASCULITIS MANAGEMENT OVERVIEW
Granulomatosis with polyangiitis (formerly Wegener’s granulomatosis, or WG) is an antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis that often affects the respiratory system and kidneys across a broad spectrum of clinical presentations, from mild through life-threatening disease. Patients with severe disease present with significant multisystem manifestations, which, in addition to the respiratory system and kidneys, may involve the joints, eyes, and other organs.
Managing patients diagnosed with systemic small-vessel vasculitides such as GPA and microscopic polyangiitis (MPA) is an inexact science. The goals of treatment are to increase survival, induce and maintain remission, reduce relapses, and minimize treatment-related toxicity. Inducing and maintaining remission have become realistic goals because of the availability of medications that prolong life. On the other hand, extended periods of treatment associated with prolonged life increase the risk of treatment-related toxicity in patients who are inadequately monitored.
MONITORING CONSIDERATIONS
Achieving treatment goals requires long-term monitoring of both disease activity and treatment-related toxicities, with constant adjustments to meet the needs of the individual patient and address the often rapidly changing disease and treatment course. The monitoring protocol consists of regularly scheduled follow-up office visits, urine sediment analyses at every office visit whether or not the patient has relapse symptoms, laboratory tests at regular intervals as indicated by the patient’s medication plan and disease presentation, additional tests such as lung computed tomography (CT), and patient education regarding new symptoms and the frequency of office visits. A consistent monitoring strategy will help detect a relapse before it can produce more severe morbidity, identify treatment-related complications, and—equally important—identify the achievement of remission. An example of the consequences of inconsistent monitoring is presented in “Relapse in a nonadherent patient.”
Because there is no definitive cure for small-vessel vasculitis, relapse is always a possibility. The early diagnosis and treatment of relapse may prevent or decrease morbidity from disease, but strict monitoring is needed to identify relapse and initiate treatment before morbidity occurs (see “Relapse in a patient with new symptoms”). Repeat induction therapy following a relapse introduces risk of drug toxicity and requires careful monitoring, as does long-term maintenance therapy.
In addition to induction and maintenance therapy, several other situations, including prior therapeutic complications, serum creatinine levels, and risk of cardiovascular disease, require special monitoring attention.
Induction therapy: monitor response
Response to treatment during induction must be monitored to identify whether remission is achieved. Induction monitoring requires complete assessment of organ-system involvement at every visit with tools such as the Birmingham Vasculitis Activity Score (BVAS) and, when appropriate, the BVAS/WG. If new or worsening symptoms develop during induction therapy, then the patient needs assessment for continued disease activity as well as treatment complications such as infections related to immunosuppressive therapy.
During induction therapy with daily oral CYC, monitoring should include weekly complete blood cell counts to ensure early identification of leukopenia and other cytopenias. The risk of morbidities increases with the cumulative dose, so a stable blood count for 2 months does not obviate the risk of leukopenia. If persistent hematuria is present without cellular casts, cystoscopy is indicated to look for signs of hemorrhagic cystitis. Prophylaxis against Pneumocystis jirovecii is recommended in all patients who receive immunosuppressive therapy. Finally, bone density measurements should be done at baseline.
Maintenance therapy: frequency can be extended
Monitoring during maintenance therapy is similar to induction monitoring; however, when the dosage of methotrexate or azathioprine is stabilized, the frequency of some tests can be extended to monthly rather than weekly. For example, a complete blood cell count, comprehensive metabolic panel, sedimentation rate, C-reactive protein measurement, and urinalysis should be performed monthly. Follow-up visits should include urine sediment analyses and monitoring for cardiovascular disease risk factors. Medication monitoring should include cystoscopy for persistent hematuria without cellular casts, bone density measurements, and ophthalmologic examinations as frequently as indicated for each individual’s needs. P jirovecii prophylaxis should continue as long as the patient receives immunosuppressive medication.
Therapy-related complications
Bladder complications. In a retrospective analysis of 145 patients with GPA treated with CYC and followed for 0.5 to 27 years (median 8.5 years), nonglomerular hematuria developed in 50% of the patients and bladder carcinoma in 5%.2 The cumulative CYC dose (19 to 251 g) in this group was much higher than what is currently used. Cytologic examination of the urine showed 43% sensitivity for dysplasia (specificity 100%) and 29% sensitivity for atypia (specificity 89%). In contrast, in a retrospective outcomes analysis involving newly diagnosed patients with GPA treated with CYC or methotrexate, 82 patients followed for up to 12 years had no incidents of cystitis or bladder cancer.3 Patients in this study were treated with CYC for only 3 to 6 months and therefore received a lower cumulative dose.
To prevent cystitis during treatment with CYC, the patient should be well hydrated, especially in the morning when CYC should be taken. The bladder should be emptied frequently. The addition of mesna when administering intravenous CYC decreases the risk of cystitis. Serial cystoscopy and urine cytology should be used only in patients with nonglomerular hematuria.
Infertility. Preservation of ovarian function is a concern with CYC therapy in women of childbearing age. The cumulative dose threshold for gonadal failure is unknown, because data from cancer studies4 demonstrating gonadal failure involve higher cumulative CYC doses than are typical for vasculitis treatment. It is also unknown whether duration of amenorrhea predicts the recovery of menses or fertility. The primary option for preservation of ovarian function is the use of gonadotropin-releasing hormone agonists. Oral contraceptives also may be used, but the best prevention is to avoid CYC in these patients if possible.
Osteoporosis. At glucocorticoid dosages of 5 mg/day or greater, bone mineral density begins a rapid decline within the first 3 months and peaks at 6 months.5 The American College of Rheumatology has provided recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis.5 Table 2 presents recommendations for postmenopausal women and men aged 50 years and older who will use glucocorticoids for 3 months or more.5 Recommendations are also available for premenopausal women and men younger than 50 years of age who have a history of fragility fracture.
Leukopenia. Leukopenia should be avoided during CYC treatment. The target white blood cell count should be within the normal range. During treatment with daily oral CYC, the patient should be monitored with a weekly complete blood cell count and medication should be adjusted to maintain the target white blood cell count.
Upon completion of induction therapy, after 3 to 6 months, the patient is switched to maintenance therapy with an alternative immunosuppressive agent such as azathioprine or methotrexate, depending on the serum creatinine concentration and other factors. This transition, characterized by full-dose immunosuppressive therapy when the bone marrow has been previously suppressed by CYC treatment, may induce pancytopenia. Monitoring with weekly complete blood counts for at least 4 weeks after initiating maintenance therapy can help ensure stability during the transition period.
Monitor serum creatinine and adjust dosages
The serum creatinine concentration may increase as CYC treatment progresses; in some cases, the serum creatinine concentration increases before a response to treatment is seen. The CYC dosages should be adjusted as necessary in response to serum creatinine changes. Careful monitoring of serum creatinine is necessary during methotrexate therapy, as methotrexate treatment in the setting of renal insufficiency increases the risk of bone marrow suppression.
Cardiovascular disease in GPA and MPA
Premature atherosclerosis has been well described in patients with GPA.6 Within 5 years of diagnosis of GPA or MPA, a cardiovascular event will occur in 14% of patients.7 In the absence of specific guidelines for prevention of cardiovascular disease in patients with vasculitis, it is essential to monitor patients and treat modifiable traditional risk factors aggressively, especially in younger patients. Suppiah et al found that independent determinants of cardiovascular outcome included older age, diastolic hypertension, and positive proteinase-3–ANCA status in patients without prior cardiovascular disease.7
In the Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) study, Merkel et al showed an increased incidence of thrombosis in patients with active GPA8 (see “Relapse presenting as thrombosis,” left). As with cardiovascular disease, there are no specific guidelines for monitoring asymptomatic patients for thrombosis or for duration of anticoagulation in patients with GPA. It is recommended that patients be evaluated for active GPA or relapse in the setting of acute thrombosis whether or not symptoms of active GPA are present.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Talar-Williams C, Hijazi YM, Walther MM, et al. Cyclophosphamide-induced cystitis and bladder cancer in patients with Wegener granulomatosis. Ann Intern Med 1996; 124:477–484.
- Villa-Forte A, Clark TM, Gomes M, et al. Substitution of methotrexate for cyclophosphamide in Wegener granulomatosis: a 12-year single-practice experience. Medicine 2007; 86:269–277.
- Harel S, Fermé C, Poirot C. Management of fertility in patients treated for Hodgkin’s lymphoma [published online ahead of print August 9, 2011]. Haematologica 2011; 96:1692–1699. doi: 10.3324/haematol.2011.045856
- Grossman JM, Gordon R, Ranganath VK, et al. American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis [published online ahead of print July 26, 2010]. Arthritis Care Res (Hoboken) 2010; 62:1515–1526. doi: 10.1002/acr.20295
- Faurschou M, Mellemkjaer L, Sorensen IJ, Svalgaard Thomsen B, Dreyer L, Baslund B. Increased morbidity from ischemic heart disease in patients with Wegener’s granulomatosis. Arthritis Rheum 2009; 60:1187–1192.
- Suppiah R, Judge A, Batra R, et al. A model to predict cardiovascular events in patients with newly diagnosed Wegener’s granulomatosis and microscopic polyangiitis. Arthritis Care Res (Hoboken) 2011; 63:588–596.
- Merkel PA, Lo GH, Holbrook JT, et al; for Wegener’s Granulomatosis Etanercept Trial Research Group. Brief communication: high incidence of venous thrombotic events among patients with Wegener granulomatosis: the Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) study. Ann Intern Med 2005; 142:620–626.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Talar-Williams C, Hijazi YM, Walther MM, et al. Cyclophosphamide-induced cystitis and bladder cancer in patients with Wegener granulomatosis. Ann Intern Med 1996; 124:477–484.
- Villa-Forte A, Clark TM, Gomes M, et al. Substitution of methotrexate for cyclophosphamide in Wegener granulomatosis: a 12-year single-practice experience. Medicine 2007; 86:269–277.
- Harel S, Fermé C, Poirot C. Management of fertility in patients treated for Hodgkin’s lymphoma [published online ahead of print August 9, 2011]. Haematologica 2011; 96:1692–1699. doi: 10.3324/haematol.2011.045856
- Grossman JM, Gordon R, Ranganath VK, et al. American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis [published online ahead of print July 26, 2010]. Arthritis Care Res (Hoboken) 2010; 62:1515–1526. doi: 10.1002/acr.20295
- Faurschou M, Mellemkjaer L, Sorensen IJ, Svalgaard Thomsen B, Dreyer L, Baslund B. Increased morbidity from ischemic heart disease in patients with Wegener’s granulomatosis. Arthritis Rheum 2009; 60:1187–1192.
- Suppiah R, Judge A, Batra R, et al. A model to predict cardiovascular events in patients with newly diagnosed Wegener’s granulomatosis and microscopic polyangiitis. Arthritis Care Res (Hoboken) 2011; 63:588–596.
- Merkel PA, Lo GH, Holbrook JT, et al; for Wegener’s Granulomatosis Etanercept Trial Research Group. Brief communication: high incidence of venous thrombotic events among patients with Wegener granulomatosis: the Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) study. Ann Intern Med 2005; 142:620–626.
In reply: Giant cell arteritis
In Reply: We know from autopsy studies that most patients with giant cell arteritis, if not all, develop aortitis at some point during the course of their disease, but we don’t know (and no study yet has completely addressed) the following questions:
- What is the most clinically appropriate and cost-effective method of screening?
- How often should we be screening these patients?
Given the high cost of the most accurate and detailed available test, ie, magnetic resonance angiography of the aorta, annual chest radiography has been recommended by some experts in the field.
Although the high frequency of thoracic aneurysm justifies high clinical vigilance, we don’t know the most adequate and cost-effective test for screening for aortic aneurysm. Until we have an answer to these questions it is difficult to formulate specific guidelines, and different experts will continue to have different practices that are based on their own experience.
At this time, I carefully listen for bruits and murmurs on physical examination and check the blood pressure in all four extremities during patient follow-up visits. If I detect any abnormalities suggesting pathology of the aorta or major branches, I order magnetic resonance angiography of the entire aorta and its main branches.
In Reply: We know from autopsy studies that most patients with giant cell arteritis, if not all, develop aortitis at some point during the course of their disease, but we don’t know (and no study yet has completely addressed) the following questions:
- What is the most clinically appropriate and cost-effective method of screening?
- How often should we be screening these patients?
Given the high cost of the most accurate and detailed available test, ie, magnetic resonance angiography of the aorta, annual chest radiography has been recommended by some experts in the field.
Although the high frequency of thoracic aneurysm justifies high clinical vigilance, we don’t know the most adequate and cost-effective test for screening for aortic aneurysm. Until we have an answer to these questions it is difficult to formulate specific guidelines, and different experts will continue to have different practices that are based on their own experience.
At this time, I carefully listen for bruits and murmurs on physical examination and check the blood pressure in all four extremities during patient follow-up visits. If I detect any abnormalities suggesting pathology of the aorta or major branches, I order magnetic resonance angiography of the entire aorta and its main branches.
In Reply: We know from autopsy studies that most patients with giant cell arteritis, if not all, develop aortitis at some point during the course of their disease, but we don’t know (and no study yet has completely addressed) the following questions:
- What is the most clinically appropriate and cost-effective method of screening?
- How often should we be screening these patients?
Given the high cost of the most accurate and detailed available test, ie, magnetic resonance angiography of the aorta, annual chest radiography has been recommended by some experts in the field.
Although the high frequency of thoracic aneurysm justifies high clinical vigilance, we don’t know the most adequate and cost-effective test for screening for aortic aneurysm. Until we have an answer to these questions it is difficult to formulate specific guidelines, and different experts will continue to have different practices that are based on their own experience.
At this time, I carefully listen for bruits and murmurs on physical examination and check the blood pressure in all four extremities during patient follow-up visits. If I detect any abnormalities suggesting pathology of the aorta or major branches, I order magnetic resonance angiography of the entire aorta and its main branches.
Giant cell arteritis: Suspect it, treat it promptly
Giant cell arteritis is the most common primary systemic vasculitis. The disease occurs almost exclusively in people over age 50, with an annual incidence of 15 to 25 per 100,000.1 Incidence rates vary significantly depending on ethnicity. The highest rates are in whites, particularly those of North European descent.2 Incidence rates progressively increase after age 50. The disease is more prevalent in women. Its cause is unknown; both genetic and environmental factors are thought to play a role.
INFLAMED ARTERIES
Giant cell arteritis is characterized by a granulomatous inflammatory infiltrate affecting large and medium-size arteries. Not all vessels are equally affected: the most susceptible are the cranial arteries, the aorta, and the aorta’s primary branches, particularly those in the upper extremities.
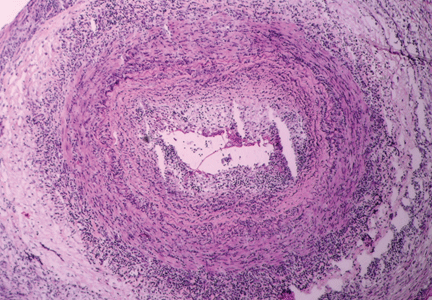
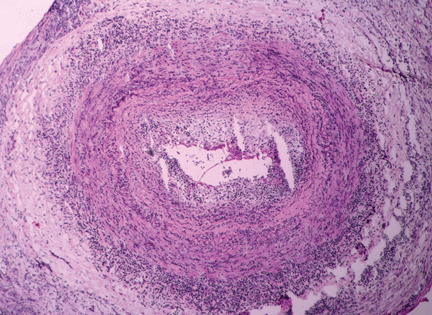
The disease is usually associated with an intense acute-phase response. Vessel wall inflammation results in intimal hyperplasia, luminal occlusion, and tissue ischemia. Typical histologic features include a mononuclear inflammatory infiltrate primarily composed of CD4+ T cells and activated macrophages. Multinucleated giant cells are seen in only about 50% of positive biopsies; therefore, their presence is not essential for the diagnosis.3
FOUR MAIN PHENOTYPES
Some of the possible symptoms of giant cell arteritis readily point to the correct diagnosis, eg, those due to cranial artery involvement, such as temporal headache, claudication of masticatory muscles, and visual changes. However, the clinical presentation can be quite varied.
There are four predominant clinical phenotypes, which may be present at the onset of disease or appear later as the disease progresses. Although they will be described separately in this review, these clinical presentations often overlap.
Cranial arteritis
Cranial arteritis is the clinical presentation most readily associated with giant cell arteritis. Clinical features result from involvement of branches of the external or internal carotid artery.
Headache, the most frequent symptom, is typically but not exclusively localized to the temporal areas.
Visual loss is due to involvement of the branches of the ophthalmic or posterior ciliary arteries, resulting in ischemia of the optic nerve or its tracts. It occurs in up to 20% of patients.4,5
Other symptoms and complications from cranial arteritis include tenderness of the scalp and temporal areas, claudication of the tongue or jaw muscles, stroke, and more rarely, tongue infarction.
Polymyalgia rheumatica
Polymyalgia rheumatica is a clinical syndrome that can occur by itself or in conjunction with giant cell arteritis. It may occur independently of giant cell arteritis, but also occurs in about 40% of patients with giant cell arteritis. It may precede, develop simultaneously with, or develop later during the course of the giant cell arteritis.6,7 It is a common clinical manifestation in relapses of giant cell arteritis, even in those who did not have symptoms of polymyalgia rheumatica at the time giant cell arteritis was diagnosed.
Polymyalgia rheumatica is characterized by aching of the shoulder and hip girdle, with morning stiffness. Fatigue and malaise are often present and may be severe. Some patients with polymyalgia rheumatica may also present with peripheral joint synovitis, which may be mistakenly diagnosed as rheumatoid arthritis.8 Muscle weakness and elevated muscle enzymes are not associated with polymyalgia rheumatica.
Polymyalgia rheumatica is a clinical diagnosis. Approximately 80% of patients with polymyalgia rheumatica have an elevated erythrocyte sedimentation rate or an elevated C-reactive protein level.9 When it occurs in the absence of giant cell arteritis, it is treated differently, with less intense doses of corticosteroids. All patients with polymyalgia rheumatica should be routinely questioned regarding symptoms of giant cell arteritis.
Nonspecific systemic inflammatory disease
Some patients with giant cell arteritis may present with a nonspecific systemic inflammatory disease characterized by some combination of fever, night sweats, fatigue, malaise, and weight loss. In these patients, the diagnosis may be delayed by the lack of localizing symptoms.
Laboratory tests typically show anemia, leukocytosis, and thrombocytosis. The erythrocyte sedimentation rate and the C-reactive protein level are usually very high.
Giant cell arteritis should be in the differential diagnosis when these signs and symptoms are found in patients over age 50.
Large-vessel vasculitis
Although thoracic aortic aneurysm and dissection have been described as late complications of giant cell arteritis, large-vessel vasculitis may precede or occur concomitantly with cranial arteritis early in the disease.10,11
Population-based surveys have shown that large-vessel vasculitis is extremely frequent in patients with giant cell arteritis. In a postmortem study of 11 patients with giant cell arteritis, all of them had evidence of arteritis involving the subclavian artery, the carotid artery, and the aorta.12
Patients may have no symptoms or may present with symptoms or signs of tissue ischemia such as claudication of the extremities, carotid artery tenderness, decreased or absent pulses, and large-vessel bruits on physical examination.
CONSIDER THE DIAGNOSIS IN OLDER PATIENTS
Giant cell arteritis should always be considered in patients over age 50 who have any of the clinical features described above. It is therefore very important to be familiar with its symptoms and signs.
A complete and detailed history and a detailed but focused physical examination that includes a comprehensive vascular examination are the first and most important steps in establishing the diagnosis. The vascular examination includes measuring the blood pressure in all four extremities, palpating the peripheral pulses, listening for bruits, and palpating the temporal arteries.
Temporal artery biopsy: The gold standard
Confirming the diagnosis of giant cell arteritis requires histologic findings of inflammation in the temporal artery. Superficial temporal artery biopsy is recommended for diagnostic confirmation in patients who have cranial symptoms and other signs suggesting the disease.
The biopsy should be performed on the same side as the symptoms or abnormal findings on examination. Performing a biopsy in both temporal arteries may increase the diagnostic yield but may not need to be done routinely.13
Although some experts recommend temporal artery biopsy in all patients in whom giant cell arteritis is suspected, biopsy has a lower diagnostic yield in patients who have no cranial symptoms. Interestingly, 5% to 15% of temporal artery biopsies performed in patients who had isolated polymyalgia rheumatica were found to be positive.14,15 Patients with polymyalgia rheumatica and no clinical symptoms to suggest giant cell arteritis generally are not biopsied.
The segmental nature of the inflammation involving the temporal artery in giant cell arteritis may result in negative biopsy results in patients with giant cell arteritis. A temporal artery biopsy length of 5 mm or less has a very low (8%) rate of positive results, whereas a length longer than 20 mm exceeds a 50% rate of positive results. Although the optimal length of a temporal artery specimen is still debated, a longer biopsy specimen should be obtained to increase the chance of arterial specimens showing inflammatory changes.16,17
Laboratory studies: Acute-phase reactants may be elevated
High levels of acute-phase reactants should increase one’s suspicion of giant cell arteritis. Elevations in the erythrocyte sedimentation rate and C-reactive protein and interleukin 6 levels reflect the inflammatory process in this disease.19 However, not all patients with giant cell arteritis have a high sedimentation rate, and as many as 20% of patients with biopsy-proven giant cell arteritis have a normal sedimentation rate before therapy.20 Therefore, a normal sedimentation rate does not exclude the diagnosis of giant cell arteritis and should not delay its diagnosis and treatment.
As a result of systemic inflammation, the patient may also present with normochromic normocytic anemia, leukocytosis, and thrombocytosis.
Imaging studies are controversial
Imaging studies are potentially useful diagnostic tools in large-vessel vasculitis but are still the subject of significant controversy.
Ultrasonography of the temporal artery has been a controversial subject in many studies.21,22 Color duplex ultrasonography of the temporal artery has been reported to be helpful in the diagnosis of giant cell arteritis (showing a “halo” around the arterial lumen), but further studies are needed to establish its clinical utility.
At this time, temporal artery biopsy remains the gold standard diagnostic test for giant cell arteritis, and ultrasonography is neither a substitute for biopsy nor a screening test for this disease.23 Some have suggested, however, that ultrasonography may help to identify the best site for biopsy of the temporal artery in some patients.
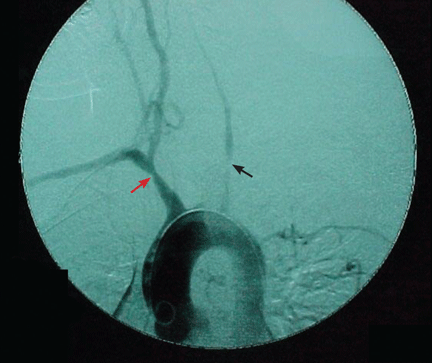
Arteriography is an accurate technique for evaluating the vessel lumen and allows for measuring central blood pressure and performing vascular interventions. However, because of potential complications, it has been largely replaced by noninvasive angiographic imaging to delineate vascular anatomy.
TREATMENT
Glucocorticoid therapy remains the standard of care
Once the diagnosis of giant cell arteritis is established, glucocorticoid treatment should be started. Glucocorticoids are the standard therapy, and they usually bring about a prompt clinical response. Although never evaluated in placebo-controlled trials, these drugs have been shown to prevent progression of visual loss in a retrospective study.26
In patients with visual symptoms or imminent visual loss, therapy should be started promptly once suspicion of giant cell arteritis is raised; ie, one should not wait until the diagnosis is confirmed by biopsy.
Ideally, a glucocorticoid should be started after a temporal artery biopsy is done, but treatment should not be delayed, as it rapidly suppresses the inflammatory response and may prevent complications from tissue ischemia, such as blindness. Visual loss is usually irreversible.
There is still a role for obtaining a temporal artery biopsy up to several weeks after glucocorticoid therapy is started, as the pathological abnormalities of arteritis do not rapidly resolve.27
Glucocorticoid therapy is highly effective in inducing disease remission in patients with giant cell arteritis. Nearly all patients respond to 1 mg/kg (40–60 mg) per day of prednisone or its equivalent.
The initial dosing is usually maintained for 4 weeks and then decreased slowly. The duration of therapy varies; most patients remain on therapy for at least 1 year, and some cannot stop it completely without recurrence of symptoms.
If a patient is about to lose his or her vision or has lost all or some vision in one eye, a higher initial dose of a glucocorticoid is usually used (ie, a pulse of 500 or 1,000 mg of intravenous methylprednisolone) and may be beneficial.28
Although a rapid clinical response to therapy is usually seen within 48 hours, some patients may have a more delayed clinical improvement.
Alternate-day therapy was compared with daily therapy and was found to be less effective, and as a result it is not recommended.29
Glucocorticoid therapy can cause significant toxicity in patients with giant cell arteritis, as they commonly must take these drugs for long periods. The rate of relapse in those who discontinue therapy is quite high—as high as 77% within 12 months.30
Given the concern about glucocorticoid toxicity, several studies have evaluated alternative strategies and other immunosuppressive drugs. However, no study has concluded that other medications are effective in the treatment of giant cell arteritis.
Mazlumzadeh et al31 evaluated the initial use of intravenous pulse methylprednisolone therapy (15 mg/kg ideal body weight on 3 consecutive days) in an attempt to decrease the glucocorticoid requirement. Although the group receiving this therapy had a lower relapse rate than in the placebo group, and their cumulative dose of glucocorticoid was lower (all patients also received oral prednisone), there was no reduction in the rate of glucocorticoid-associated toxicity.31 Care must be taken to prevent and monitor for corticosteroid complications such as osteoporosis, glaucoma, diabetes mellitus, and hypertension.
Methotrexate: Mixed results in clinical trials
Methotrexate has been evaluated in three prospective randomized trials,30,32,33 with mixed results.
Spiera et al32 enrolled 21 patients in a double-blind placebo-controlled trial: 12 patients received low-dose methotrexate (7.5 mg/week) and 9 received placebo. In addition, all 21 received a glucocorticoid. There was no significant difference between the methotrexate- and placebo-treated patients in the cumulative dose of glucocorticoid, duration of glucocorticoid therapy, time to taper off the glucocorticoid to less than 10 mg of prednisone per day, or glucocorticoidrelated adverse effects.
Jover et al,33 in another double-blind placebo-controlled trial, studied 42 patients with giant cell arteritis, half of whom were randomized to receive methotrexate 10 mg/week, while the other half received placebo. All patients received prednisone. Patients in the methotrexate group had fewer relapses and a 25% lower cumulative dose of prednisone during follow-up. However, the incidence of adverse events was similar in both groups. Methotrexate was discontinued in 3 patients who developed drug-related adverse events.
Hoffman et al30 randomized 98 patients to receive either methotrexate (up to 15 mg/week) or placebo in a double-blind fashion. All patients also received prednisone at an initial dose of 1 mg/kg/day (up to 60 mg/day). At completion of the study, no differences between the groups were noted in the rates of relapse or treatment-related morbidity or in the cumulative dose of glucocorticoid. However, treatment with methotrexate appeared to lower the rate of recurrence of isolated polymyalgia rheumatica in a small number of patients.30
Comment. Differences in the results of these trials may be attributed to several factors, including different definitions of relapses and different glucocorticoid doses and tapering regimens.
A meta-analysis of these three trials34 showed a reduction in the risk of relapse: 4 patients would have to be treated to prevent one first relapse, 5 would have to be treated to prevent one second relapse, and 11 would need to be treated to prevent one first relapse of cranial symptoms in the first 48 weeks. However, the main goal of methotrexate therapy is to decrease the frequency of adverse events from glucocorticoids, and this meta-analysis found no difference in rates of glucocorticoid-related adverse events in patients treated with methotrexate.
The study raises the question of whether methotrexate should be further evaluated in in different patient populations and at higher doses.34
Infliximab is not recommended
In a prospective study, patients with giant cell arteritis were randomly assigned to receive either infliximab (Remicade) 5 mg/kg every 8 weeks or placebo, in addition to standard glucocorticoid therapy. The study showed no significant difference in the relapse rate and a higher rate of infection in the infliximab group (71%) than in the placebo group (56%). Given the lack of any benefit observed in this study, infliximab is not recommended in the treatment of patients with giant cell arteritis.35
Aspirin is recommended
Daily low-dose aspirin therapy has been shown in several studies to be effective in preventing ischemic complications of giant cell arteritis, including stroke and visual loss. It is currently recommended that all patients with giant cell arteritis without a major contraindication take aspirin 81 mg daily.36–38
- Salvarani C, Gabriel SE, O’Fallon WM, Hunder GG. The incidence of giant cell arteritis in Olmsted County, Minnesota: apparent fluctuations in a cyclic pattern. Ann Intern Med 1995; 123:192–194.
- Baldursson O, Steinsson K, Björnsson J, Lie JT. Giant cell arteritis in Iceland. An epidemiologic and histopathologic analysis. Arthritis Rheum 1994; 37:1007–1012.
- Weyand CM, Goronzy JJ. Medium- and large-vessel vasculitis. N Engl J Med 2003; 349:160–169.
- Aiello PD, Trautmann JC, McPhee TJ, Kunselman AR, Hunder GG. Visual prognosis in giant cell arteritis. Ophthalmology 1993; 100:550–555.
- Salvarani C, Cimino L, Macchioni P, et al. Risk factors for visual loss in an Italian population-based cohort of patients with giant cell arteritis. Arthritis Rheum 2005; 53:293–297.
- Bahlas S, Ramos-Remus C, Davis P. Clinical outcome of 149 patients with polymyalgia rheumatica and giant cell arteritis. J Rheumatol 1998; 25:99–104.
- Gonzalez-Gay MA, Barros S, Lopez-Diaz MJ, Garcia-Porrua C, Sanchez-Andrade A, Llorca J. Giant cell arteritis: disease patterns of clinical presentation in a series of 240 patients. Medicine (Baltimore) 2005; 84:269–276.
- Salvarani C, Cantini F, Macchioni P, et al. Distal musculoskeletal manifestations in polymyalgia rheumatica: a prospective followup study. Arthritis Rheum 1998; 41:1221–1226.
- Salvarani C, Cantini F, Boiardi L, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med 2002; 347:261–271.
- Lie JT. Aortic and extracranial large vessel giant cell arteritis: a review of 72 cases with histopathologic documentation. Semin Arthritis Rheum 1995; 24:422–431.
- Evans JM, O’Fallon WM, Hunder GG. Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis. A population-based study. Ann Intern Med 1995; 122:502–507.
- Ostberg G. An arteritis with special reference to polymyalgia arteritica. Acta Pathol Microbiol Scand Suppl 1973; 237(suppl 237):1–59.
- Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol 1999; 128:211–215.
- González-Gay MA, Garcia-Porrua C, Rivas MJ, Rodriguez-Ledo P, Llorca J. Epidemiology of biopsy proven giant cell arteritis in northwestern Spain: trend over an 18 year period. Ann Rheum Dis 2001; 60:367–371.
- Rodriguez-Valverde V, Sarabia JM, González-Gay MA, et al. Risk factors and predictive models of giant cell arteritis in polymyalgia rheumatica. Am J Med 1997; 102:331–336.
- Mahr A, Saba M, Kambouchner M, et al. Temporal artery biopsy for diagnosing giant cell arteritis: the longer, the better? Ann Rheum Dis 2006; 65:826–828.
- Breuer GS, Nesher R, Nesher G. Effect of biopsy length on the rate of positive temporal artery biopsies. Clin Exp Rheumatol 2009; 27(1 suppl 52):S10–S13.
- Weyand CM, Goronzy JJ. Giant-cell arteritis and polymyalgia rheumatica. Ann Intern Med 2003; 139:505–515.
- Salvarani C, Cantini F, Boiardi L, Hunder GG. Laboratory investigations useful in giant cell arteritis and Takayasu’s arteritis. Clin Exp Rheumatol 2003; 21(6 suppl 32):S23–S28.
- Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurence in a population-based study. Arthritis Rheum 2001; 45:140–145.
- Schmidt WA, Kraft HE, Vorpahl K, Völker L, Gromnica-Ihle EJ. Color duplex ultrasonography in the diagnosis of temporal arteritis. N Engl J Med 1997; 337:1336–1342.
- Karassa FB, Matsagas MI, Schmidt WA, Ioannidis JP. Meta-analysis: test performance of ultrasonography for giant-cell arteritis. Ann Intern Med 2005; 142:359–369.
- Maldini C, Dépinay-Dhellemmes C, Tra TT, et al. Limited value of temporal artery ultrasonography examinations for diagnosis of giant cell arteritis: analysis of 77 subjects. J Rheumatol 2010; Epub ahead of print.
- Both M, Ahmadi-Simab K, Reuter M, et al. MRI and FDG-PET in the assessment of inflammatory aortic arch syndrome in complicated courses of giant cell arteritis. Ann Rheum Dis 2008; 67:1030–1033.
- Tso E, Flamm SD, White RD, Schvartzman PR, Mascha E, Hoffman GS. Takayasu arteritis: utility and limitations of magnetic resonance imaging in diagnosis and treatment. Arthritis Rheum 2002; 46:1634–1642.
- Birkhead NC, Wagener HP, Shick RM. Treatment of temporal arteritis with adrenal corticosteroids; results in fifty-five cases in which lesion was proved at biopsy. J Am Med Assoc 1957; 163:821–827.
- Ray-Chaudhuri N, Kiné DA, Tijani SO, et al. Effect of prior steroid treatment on temporal artery biopsy findings in giant cell arteritis. Br J Ophthalmol 2002; 86:530–532.
- Chan CC, Paine M, O’Day J. Steroid management in giant cell arteritis. Br J Ophthalmol 2001; 85:1061–1064.
- Hunder GG, Sheps SG, Allen GL, Joyce JW. Daily and alternate-day corticosteroid regimens in treatment of giant cell arteritis: comparison in a prospective study. Ann Intern Med 1975; 82:613–618.
- Hoffman GS, Cid MC, Hellmann DB, et al; International Network for the Study of Systemic Vasculitides. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum 2002; 46:1309–1318.
- Mazlumzadeh M, Hunder GG, Easley KA, et al. Treatment of giant cell arteritis using induction therapy with high-dose glucocorticoids: a double-blind, placebo-controlled, randomized prospective clinical trial. Arthritis Rheum 2006; 54:3310–3318.
- Spiera RF, Mitnick HJ, Kupersmith M, et al. A prospective, doubleblind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA). Clin Exp Rheumatol 2001; 19:495–501.
- Jover JA, Hernández-García C, Morado IC, Vargas E, Bañares A, Fernández-Gutiérrez B. Combined treatment of giant-cell arteritis with methotrexate and prednisone. a randomized, double-blind, placebo-controlled trial. Ann Intern Med 2001; 134:106–114.
- Mahr AD, Jover JA, Spiera RF, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum 2007; 56:2789–2797.
- Hoffman GS, Cid MC, Rendt-Zagar KE, et al; Infliximab-GCA Study Group. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med 2007; 146:621–630.
- Weyand CM, Kaiser M, Yang H, Younge B, Goronzy JJ. Therapeutic effects of acetylsalicylic acid in giant cell arteritis. Arthritis Rheum 2002; 46:457–466.
- Nesher G, Berkun Y, Mates M, Baras M, Rubinow A, Sonnenblick M. Low-dose aspirin and prevention of cranial ischemic complications in giant cell arteritis. Arthritis Rheum 2004; 50:1332–1337.
- Lee MS, Smith SD, Galor A, Hoffman GS. Antiplatelet and anticoagulant therapy in patients with giant cell arteritis. Arthritis Rheum 2006; 54:3306–3309.
Giant cell arteritis is the most common primary systemic vasculitis. The disease occurs almost exclusively in people over age 50, with an annual incidence of 15 to 25 per 100,000.1 Incidence rates vary significantly depending on ethnicity. The highest rates are in whites, particularly those of North European descent.2 Incidence rates progressively increase after age 50. The disease is more prevalent in women. Its cause is unknown; both genetic and environmental factors are thought to play a role.
INFLAMED ARTERIES
Giant cell arteritis is characterized by a granulomatous inflammatory infiltrate affecting large and medium-size arteries. Not all vessels are equally affected: the most susceptible are the cranial arteries, the aorta, and the aorta’s primary branches, particularly those in the upper extremities.
The disease is usually associated with an intense acute-phase response. Vessel wall inflammation results in intimal hyperplasia, luminal occlusion, and tissue ischemia. Typical histologic features include a mononuclear inflammatory infiltrate primarily composed of CD4+ T cells and activated macrophages. Multinucleated giant cells are seen in only about 50% of positive biopsies; therefore, their presence is not essential for the diagnosis.3
FOUR MAIN PHENOTYPES
Some of the possible symptoms of giant cell arteritis readily point to the correct diagnosis, eg, those due to cranial artery involvement, such as temporal headache, claudication of masticatory muscles, and visual changes. However, the clinical presentation can be quite varied.
There are four predominant clinical phenotypes, which may be present at the onset of disease or appear later as the disease progresses. Although they will be described separately in this review, these clinical presentations often overlap.
Cranial arteritis
Cranial arteritis is the clinical presentation most readily associated with giant cell arteritis. Clinical features result from involvement of branches of the external or internal carotid artery.
Headache, the most frequent symptom, is typically but not exclusively localized to the temporal areas.
Visual loss is due to involvement of the branches of the ophthalmic or posterior ciliary arteries, resulting in ischemia of the optic nerve or its tracts. It occurs in up to 20% of patients.4,5
Other symptoms and complications from cranial arteritis include tenderness of the scalp and temporal areas, claudication of the tongue or jaw muscles, stroke, and more rarely, tongue infarction.
Polymyalgia rheumatica
Polymyalgia rheumatica is a clinical syndrome that can occur by itself or in conjunction with giant cell arteritis. It may occur independently of giant cell arteritis, but also occurs in about 40% of patients with giant cell arteritis. It may precede, develop simultaneously with, or develop later during the course of the giant cell arteritis.6,7 It is a common clinical manifestation in relapses of giant cell arteritis, even in those who did not have symptoms of polymyalgia rheumatica at the time giant cell arteritis was diagnosed.
Polymyalgia rheumatica is characterized by aching of the shoulder and hip girdle, with morning stiffness. Fatigue and malaise are often present and may be severe. Some patients with polymyalgia rheumatica may also present with peripheral joint synovitis, which may be mistakenly diagnosed as rheumatoid arthritis.8 Muscle weakness and elevated muscle enzymes are not associated with polymyalgia rheumatica.
Polymyalgia rheumatica is a clinical diagnosis. Approximately 80% of patients with polymyalgia rheumatica have an elevated erythrocyte sedimentation rate or an elevated C-reactive protein level.9 When it occurs in the absence of giant cell arteritis, it is treated differently, with less intense doses of corticosteroids. All patients with polymyalgia rheumatica should be routinely questioned regarding symptoms of giant cell arteritis.
Nonspecific systemic inflammatory disease
Some patients with giant cell arteritis may present with a nonspecific systemic inflammatory disease characterized by some combination of fever, night sweats, fatigue, malaise, and weight loss. In these patients, the diagnosis may be delayed by the lack of localizing symptoms.
Laboratory tests typically show anemia, leukocytosis, and thrombocytosis. The erythrocyte sedimentation rate and the C-reactive protein level are usually very high.
Giant cell arteritis should be in the differential diagnosis when these signs and symptoms are found in patients over age 50.
Large-vessel vasculitis
Although thoracic aortic aneurysm and dissection have been described as late complications of giant cell arteritis, large-vessel vasculitis may precede or occur concomitantly with cranial arteritis early in the disease.10,11
Population-based surveys have shown that large-vessel vasculitis is extremely frequent in patients with giant cell arteritis. In a postmortem study of 11 patients with giant cell arteritis, all of them had evidence of arteritis involving the subclavian artery, the carotid artery, and the aorta.12
Patients may have no symptoms or may present with symptoms or signs of tissue ischemia such as claudication of the extremities, carotid artery tenderness, decreased or absent pulses, and large-vessel bruits on physical examination.
CONSIDER THE DIAGNOSIS IN OLDER PATIENTS
Giant cell arteritis should always be considered in patients over age 50 who have any of the clinical features described above. It is therefore very important to be familiar with its symptoms and signs.
A complete and detailed history and a detailed but focused physical examination that includes a comprehensive vascular examination are the first and most important steps in establishing the diagnosis. The vascular examination includes measuring the blood pressure in all four extremities, palpating the peripheral pulses, listening for bruits, and palpating the temporal arteries.
Temporal artery biopsy: The gold standard
Confirming the diagnosis of giant cell arteritis requires histologic findings of inflammation in the temporal artery. Superficial temporal artery biopsy is recommended for diagnostic confirmation in patients who have cranial symptoms and other signs suggesting the disease.
The biopsy should be performed on the same side as the symptoms or abnormal findings on examination. Performing a biopsy in both temporal arteries may increase the diagnostic yield but may not need to be done routinely.13
Although some experts recommend temporal artery biopsy in all patients in whom giant cell arteritis is suspected, biopsy has a lower diagnostic yield in patients who have no cranial symptoms. Interestingly, 5% to 15% of temporal artery biopsies performed in patients who had isolated polymyalgia rheumatica were found to be positive.14,15 Patients with polymyalgia rheumatica and no clinical symptoms to suggest giant cell arteritis generally are not biopsied.
The segmental nature of the inflammation involving the temporal artery in giant cell arteritis may result in negative biopsy results in patients with giant cell arteritis. A temporal artery biopsy length of 5 mm or less has a very low (8%) rate of positive results, whereas a length longer than 20 mm exceeds a 50% rate of positive results. Although the optimal length of a temporal artery specimen is still debated, a longer biopsy specimen should be obtained to increase the chance of arterial specimens showing inflammatory changes.16,17
Laboratory studies: Acute-phase reactants may be elevated
High levels of acute-phase reactants should increase one’s suspicion of giant cell arteritis. Elevations in the erythrocyte sedimentation rate and C-reactive protein and interleukin 6 levels reflect the inflammatory process in this disease.19 However, not all patients with giant cell arteritis have a high sedimentation rate, and as many as 20% of patients with biopsy-proven giant cell arteritis have a normal sedimentation rate before therapy.20 Therefore, a normal sedimentation rate does not exclude the diagnosis of giant cell arteritis and should not delay its diagnosis and treatment.
As a result of systemic inflammation, the patient may also present with normochromic normocytic anemia, leukocytosis, and thrombocytosis.
Imaging studies are controversial
Imaging studies are potentially useful diagnostic tools in large-vessel vasculitis but are still the subject of significant controversy.
Ultrasonography of the temporal artery has been a controversial subject in many studies.21,22 Color duplex ultrasonography of the temporal artery has been reported to be helpful in the diagnosis of giant cell arteritis (showing a “halo” around the arterial lumen), but further studies are needed to establish its clinical utility.
At this time, temporal artery biopsy remains the gold standard diagnostic test for giant cell arteritis, and ultrasonography is neither a substitute for biopsy nor a screening test for this disease.23 Some have suggested, however, that ultrasonography may help to identify the best site for biopsy of the temporal artery in some patients.
Arteriography is an accurate technique for evaluating the vessel lumen and allows for measuring central blood pressure and performing vascular interventions. However, because of potential complications, it has been largely replaced by noninvasive angiographic imaging to delineate vascular anatomy.
TREATMENT
Glucocorticoid therapy remains the standard of care
Once the diagnosis of giant cell arteritis is established, glucocorticoid treatment should be started. Glucocorticoids are the standard therapy, and they usually bring about a prompt clinical response. Although never evaluated in placebo-controlled trials, these drugs have been shown to prevent progression of visual loss in a retrospective study.26
In patients with visual symptoms or imminent visual loss, therapy should be started promptly once suspicion of giant cell arteritis is raised; ie, one should not wait until the diagnosis is confirmed by biopsy.
Ideally, a glucocorticoid should be started after a temporal artery biopsy is done, but treatment should not be delayed, as it rapidly suppresses the inflammatory response and may prevent complications from tissue ischemia, such as blindness. Visual loss is usually irreversible.
There is still a role for obtaining a temporal artery biopsy up to several weeks after glucocorticoid therapy is started, as the pathological abnormalities of arteritis do not rapidly resolve.27
Glucocorticoid therapy is highly effective in inducing disease remission in patients with giant cell arteritis. Nearly all patients respond to 1 mg/kg (40–60 mg) per day of prednisone or its equivalent.
The initial dosing is usually maintained for 4 weeks and then decreased slowly. The duration of therapy varies; most patients remain on therapy for at least 1 year, and some cannot stop it completely without recurrence of symptoms.
If a patient is about to lose his or her vision or has lost all or some vision in one eye, a higher initial dose of a glucocorticoid is usually used (ie, a pulse of 500 or 1,000 mg of intravenous methylprednisolone) and may be beneficial.28
Although a rapid clinical response to therapy is usually seen within 48 hours, some patients may have a more delayed clinical improvement.
Alternate-day therapy was compared with daily therapy and was found to be less effective, and as a result it is not recommended.29
Glucocorticoid therapy can cause significant toxicity in patients with giant cell arteritis, as they commonly must take these drugs for long periods. The rate of relapse in those who discontinue therapy is quite high—as high as 77% within 12 months.30
Given the concern about glucocorticoid toxicity, several studies have evaluated alternative strategies and other immunosuppressive drugs. However, no study has concluded that other medications are effective in the treatment of giant cell arteritis.
Mazlumzadeh et al31 evaluated the initial use of intravenous pulse methylprednisolone therapy (15 mg/kg ideal body weight on 3 consecutive days) in an attempt to decrease the glucocorticoid requirement. Although the group receiving this therapy had a lower relapse rate than in the placebo group, and their cumulative dose of glucocorticoid was lower (all patients also received oral prednisone), there was no reduction in the rate of glucocorticoid-associated toxicity.31 Care must be taken to prevent and monitor for corticosteroid complications such as osteoporosis, glaucoma, diabetes mellitus, and hypertension.
Methotrexate: Mixed results in clinical trials
Methotrexate has been evaluated in three prospective randomized trials,30,32,33 with mixed results.
Spiera et al32 enrolled 21 patients in a double-blind placebo-controlled trial: 12 patients received low-dose methotrexate (7.5 mg/week) and 9 received placebo. In addition, all 21 received a glucocorticoid. There was no significant difference between the methotrexate- and placebo-treated patients in the cumulative dose of glucocorticoid, duration of glucocorticoid therapy, time to taper off the glucocorticoid to less than 10 mg of prednisone per day, or glucocorticoidrelated adverse effects.
Jover et al,33 in another double-blind placebo-controlled trial, studied 42 patients with giant cell arteritis, half of whom were randomized to receive methotrexate 10 mg/week, while the other half received placebo. All patients received prednisone. Patients in the methotrexate group had fewer relapses and a 25% lower cumulative dose of prednisone during follow-up. However, the incidence of adverse events was similar in both groups. Methotrexate was discontinued in 3 patients who developed drug-related adverse events.
Hoffman et al30 randomized 98 patients to receive either methotrexate (up to 15 mg/week) or placebo in a double-blind fashion. All patients also received prednisone at an initial dose of 1 mg/kg/day (up to 60 mg/day). At completion of the study, no differences between the groups were noted in the rates of relapse or treatment-related morbidity or in the cumulative dose of glucocorticoid. However, treatment with methotrexate appeared to lower the rate of recurrence of isolated polymyalgia rheumatica in a small number of patients.30
Comment. Differences in the results of these trials may be attributed to several factors, including different definitions of relapses and different glucocorticoid doses and tapering regimens.
A meta-analysis of these three trials34 showed a reduction in the risk of relapse: 4 patients would have to be treated to prevent one first relapse, 5 would have to be treated to prevent one second relapse, and 11 would need to be treated to prevent one first relapse of cranial symptoms in the first 48 weeks. However, the main goal of methotrexate therapy is to decrease the frequency of adverse events from glucocorticoids, and this meta-analysis found no difference in rates of glucocorticoid-related adverse events in patients treated with methotrexate.
The study raises the question of whether methotrexate should be further evaluated in in different patient populations and at higher doses.34
Infliximab is not recommended
In a prospective study, patients with giant cell arteritis were randomly assigned to receive either infliximab (Remicade) 5 mg/kg every 8 weeks or placebo, in addition to standard glucocorticoid therapy. The study showed no significant difference in the relapse rate and a higher rate of infection in the infliximab group (71%) than in the placebo group (56%). Given the lack of any benefit observed in this study, infliximab is not recommended in the treatment of patients with giant cell arteritis.35
Aspirin is recommended
Daily low-dose aspirin therapy has been shown in several studies to be effective in preventing ischemic complications of giant cell arteritis, including stroke and visual loss. It is currently recommended that all patients with giant cell arteritis without a major contraindication take aspirin 81 mg daily.36–38
Giant cell arteritis is the most common primary systemic vasculitis. The disease occurs almost exclusively in people over age 50, with an annual incidence of 15 to 25 per 100,000.1 Incidence rates vary significantly depending on ethnicity. The highest rates are in whites, particularly those of North European descent.2 Incidence rates progressively increase after age 50. The disease is more prevalent in women. Its cause is unknown; both genetic and environmental factors are thought to play a role.
INFLAMED ARTERIES
Giant cell arteritis is characterized by a granulomatous inflammatory infiltrate affecting large and medium-size arteries. Not all vessels are equally affected: the most susceptible are the cranial arteries, the aorta, and the aorta’s primary branches, particularly those in the upper extremities.
The disease is usually associated with an intense acute-phase response. Vessel wall inflammation results in intimal hyperplasia, luminal occlusion, and tissue ischemia. Typical histologic features include a mononuclear inflammatory infiltrate primarily composed of CD4+ T cells and activated macrophages. Multinucleated giant cells are seen in only about 50% of positive biopsies; therefore, their presence is not essential for the diagnosis.3
FOUR MAIN PHENOTYPES
Some of the possible symptoms of giant cell arteritis readily point to the correct diagnosis, eg, those due to cranial artery involvement, such as temporal headache, claudication of masticatory muscles, and visual changes. However, the clinical presentation can be quite varied.
There are four predominant clinical phenotypes, which may be present at the onset of disease or appear later as the disease progresses. Although they will be described separately in this review, these clinical presentations often overlap.
Cranial arteritis
Cranial arteritis is the clinical presentation most readily associated with giant cell arteritis. Clinical features result from involvement of branches of the external or internal carotid artery.
Headache, the most frequent symptom, is typically but not exclusively localized to the temporal areas.
Visual loss is due to involvement of the branches of the ophthalmic or posterior ciliary arteries, resulting in ischemia of the optic nerve or its tracts. It occurs in up to 20% of patients.4,5
Other symptoms and complications from cranial arteritis include tenderness of the scalp and temporal areas, claudication of the tongue or jaw muscles, stroke, and more rarely, tongue infarction.
Polymyalgia rheumatica
Polymyalgia rheumatica is a clinical syndrome that can occur by itself or in conjunction with giant cell arteritis. It may occur independently of giant cell arteritis, but also occurs in about 40% of patients with giant cell arteritis. It may precede, develop simultaneously with, or develop later during the course of the giant cell arteritis.6,7 It is a common clinical manifestation in relapses of giant cell arteritis, even in those who did not have symptoms of polymyalgia rheumatica at the time giant cell arteritis was diagnosed.
Polymyalgia rheumatica is characterized by aching of the shoulder and hip girdle, with morning stiffness. Fatigue and malaise are often present and may be severe. Some patients with polymyalgia rheumatica may also present with peripheral joint synovitis, which may be mistakenly diagnosed as rheumatoid arthritis.8 Muscle weakness and elevated muscle enzymes are not associated with polymyalgia rheumatica.
Polymyalgia rheumatica is a clinical diagnosis. Approximately 80% of patients with polymyalgia rheumatica have an elevated erythrocyte sedimentation rate or an elevated C-reactive protein level.9 When it occurs in the absence of giant cell arteritis, it is treated differently, with less intense doses of corticosteroids. All patients with polymyalgia rheumatica should be routinely questioned regarding symptoms of giant cell arteritis.
Nonspecific systemic inflammatory disease
Some patients with giant cell arteritis may present with a nonspecific systemic inflammatory disease characterized by some combination of fever, night sweats, fatigue, malaise, and weight loss. In these patients, the diagnosis may be delayed by the lack of localizing symptoms.
Laboratory tests typically show anemia, leukocytosis, and thrombocytosis. The erythrocyte sedimentation rate and the C-reactive protein level are usually very high.
Giant cell arteritis should be in the differential diagnosis when these signs and symptoms are found in patients over age 50.
Large-vessel vasculitis
Although thoracic aortic aneurysm and dissection have been described as late complications of giant cell arteritis, large-vessel vasculitis may precede or occur concomitantly with cranial arteritis early in the disease.10,11
Population-based surveys have shown that large-vessel vasculitis is extremely frequent in patients with giant cell arteritis. In a postmortem study of 11 patients with giant cell arteritis, all of them had evidence of arteritis involving the subclavian artery, the carotid artery, and the aorta.12
Patients may have no symptoms or may present with symptoms or signs of tissue ischemia such as claudication of the extremities, carotid artery tenderness, decreased or absent pulses, and large-vessel bruits on physical examination.
CONSIDER THE DIAGNOSIS IN OLDER PATIENTS
Giant cell arteritis should always be considered in patients over age 50 who have any of the clinical features described above. It is therefore very important to be familiar with its symptoms and signs.
A complete and detailed history and a detailed but focused physical examination that includes a comprehensive vascular examination are the first and most important steps in establishing the diagnosis. The vascular examination includes measuring the blood pressure in all four extremities, palpating the peripheral pulses, listening for bruits, and palpating the temporal arteries.
Temporal artery biopsy: The gold standard
Confirming the diagnosis of giant cell arteritis requires histologic findings of inflammation in the temporal artery. Superficial temporal artery biopsy is recommended for diagnostic confirmation in patients who have cranial symptoms and other signs suggesting the disease.
The biopsy should be performed on the same side as the symptoms or abnormal findings on examination. Performing a biopsy in both temporal arteries may increase the diagnostic yield but may not need to be done routinely.13
Although some experts recommend temporal artery biopsy in all patients in whom giant cell arteritis is suspected, biopsy has a lower diagnostic yield in patients who have no cranial symptoms. Interestingly, 5% to 15% of temporal artery biopsies performed in patients who had isolated polymyalgia rheumatica were found to be positive.14,15 Patients with polymyalgia rheumatica and no clinical symptoms to suggest giant cell arteritis generally are not biopsied.
The segmental nature of the inflammation involving the temporal artery in giant cell arteritis may result in negative biopsy results in patients with giant cell arteritis. A temporal artery biopsy length of 5 mm or less has a very low (8%) rate of positive results, whereas a length longer than 20 mm exceeds a 50% rate of positive results. Although the optimal length of a temporal artery specimen is still debated, a longer biopsy specimen should be obtained to increase the chance of arterial specimens showing inflammatory changes.16,17
Laboratory studies: Acute-phase reactants may be elevated
High levels of acute-phase reactants should increase one’s suspicion of giant cell arteritis. Elevations in the erythrocyte sedimentation rate and C-reactive protein and interleukin 6 levels reflect the inflammatory process in this disease.19 However, not all patients with giant cell arteritis have a high sedimentation rate, and as many as 20% of patients with biopsy-proven giant cell arteritis have a normal sedimentation rate before therapy.20 Therefore, a normal sedimentation rate does not exclude the diagnosis of giant cell arteritis and should not delay its diagnosis and treatment.
As a result of systemic inflammation, the patient may also present with normochromic normocytic anemia, leukocytosis, and thrombocytosis.
Imaging studies are controversial
Imaging studies are potentially useful diagnostic tools in large-vessel vasculitis but are still the subject of significant controversy.
Ultrasonography of the temporal artery has been a controversial subject in many studies.21,22 Color duplex ultrasonography of the temporal artery has been reported to be helpful in the diagnosis of giant cell arteritis (showing a “halo” around the arterial lumen), but further studies are needed to establish its clinical utility.
At this time, temporal artery biopsy remains the gold standard diagnostic test for giant cell arteritis, and ultrasonography is neither a substitute for biopsy nor a screening test for this disease.23 Some have suggested, however, that ultrasonography may help to identify the best site for biopsy of the temporal artery in some patients.
Arteriography is an accurate technique for evaluating the vessel lumen and allows for measuring central blood pressure and performing vascular interventions. However, because of potential complications, it has been largely replaced by noninvasive angiographic imaging to delineate vascular anatomy.
TREATMENT
Glucocorticoid therapy remains the standard of care
Once the diagnosis of giant cell arteritis is established, glucocorticoid treatment should be started. Glucocorticoids are the standard therapy, and they usually bring about a prompt clinical response. Although never evaluated in placebo-controlled trials, these drugs have been shown to prevent progression of visual loss in a retrospective study.26
In patients with visual symptoms or imminent visual loss, therapy should be started promptly once suspicion of giant cell arteritis is raised; ie, one should not wait until the diagnosis is confirmed by biopsy.
Ideally, a glucocorticoid should be started after a temporal artery biopsy is done, but treatment should not be delayed, as it rapidly suppresses the inflammatory response and may prevent complications from tissue ischemia, such as blindness. Visual loss is usually irreversible.
There is still a role for obtaining a temporal artery biopsy up to several weeks after glucocorticoid therapy is started, as the pathological abnormalities of arteritis do not rapidly resolve.27
Glucocorticoid therapy is highly effective in inducing disease remission in patients with giant cell arteritis. Nearly all patients respond to 1 mg/kg (40–60 mg) per day of prednisone or its equivalent.
The initial dosing is usually maintained for 4 weeks and then decreased slowly. The duration of therapy varies; most patients remain on therapy for at least 1 year, and some cannot stop it completely without recurrence of symptoms.
If a patient is about to lose his or her vision or has lost all or some vision in one eye, a higher initial dose of a glucocorticoid is usually used (ie, a pulse of 500 or 1,000 mg of intravenous methylprednisolone) and may be beneficial.28
Although a rapid clinical response to therapy is usually seen within 48 hours, some patients may have a more delayed clinical improvement.
Alternate-day therapy was compared with daily therapy and was found to be less effective, and as a result it is not recommended.29
Glucocorticoid therapy can cause significant toxicity in patients with giant cell arteritis, as they commonly must take these drugs for long periods. The rate of relapse in those who discontinue therapy is quite high—as high as 77% within 12 months.30
Given the concern about glucocorticoid toxicity, several studies have evaluated alternative strategies and other immunosuppressive drugs. However, no study has concluded that other medications are effective in the treatment of giant cell arteritis.
Mazlumzadeh et al31 evaluated the initial use of intravenous pulse methylprednisolone therapy (15 mg/kg ideal body weight on 3 consecutive days) in an attempt to decrease the glucocorticoid requirement. Although the group receiving this therapy had a lower relapse rate than in the placebo group, and their cumulative dose of glucocorticoid was lower (all patients also received oral prednisone), there was no reduction in the rate of glucocorticoid-associated toxicity.31 Care must be taken to prevent and monitor for corticosteroid complications such as osteoporosis, glaucoma, diabetes mellitus, and hypertension.
Methotrexate: Mixed results in clinical trials
Methotrexate has been evaluated in three prospective randomized trials,30,32,33 with mixed results.
Spiera et al32 enrolled 21 patients in a double-blind placebo-controlled trial: 12 patients received low-dose methotrexate (7.5 mg/week) and 9 received placebo. In addition, all 21 received a glucocorticoid. There was no significant difference between the methotrexate- and placebo-treated patients in the cumulative dose of glucocorticoid, duration of glucocorticoid therapy, time to taper off the glucocorticoid to less than 10 mg of prednisone per day, or glucocorticoidrelated adverse effects.
Jover et al,33 in another double-blind placebo-controlled trial, studied 42 patients with giant cell arteritis, half of whom were randomized to receive methotrexate 10 mg/week, while the other half received placebo. All patients received prednisone. Patients in the methotrexate group had fewer relapses and a 25% lower cumulative dose of prednisone during follow-up. However, the incidence of adverse events was similar in both groups. Methotrexate was discontinued in 3 patients who developed drug-related adverse events.
Hoffman et al30 randomized 98 patients to receive either methotrexate (up to 15 mg/week) or placebo in a double-blind fashion. All patients also received prednisone at an initial dose of 1 mg/kg/day (up to 60 mg/day). At completion of the study, no differences between the groups were noted in the rates of relapse or treatment-related morbidity or in the cumulative dose of glucocorticoid. However, treatment with methotrexate appeared to lower the rate of recurrence of isolated polymyalgia rheumatica in a small number of patients.30
Comment. Differences in the results of these trials may be attributed to several factors, including different definitions of relapses and different glucocorticoid doses and tapering regimens.
A meta-analysis of these three trials34 showed a reduction in the risk of relapse: 4 patients would have to be treated to prevent one first relapse, 5 would have to be treated to prevent one second relapse, and 11 would need to be treated to prevent one first relapse of cranial symptoms in the first 48 weeks. However, the main goal of methotrexate therapy is to decrease the frequency of adverse events from glucocorticoids, and this meta-analysis found no difference in rates of glucocorticoid-related adverse events in patients treated with methotrexate.
The study raises the question of whether methotrexate should be further evaluated in in different patient populations and at higher doses.34
Infliximab is not recommended
In a prospective study, patients with giant cell arteritis were randomly assigned to receive either infliximab (Remicade) 5 mg/kg every 8 weeks or placebo, in addition to standard glucocorticoid therapy. The study showed no significant difference in the relapse rate and a higher rate of infection in the infliximab group (71%) than in the placebo group (56%). Given the lack of any benefit observed in this study, infliximab is not recommended in the treatment of patients with giant cell arteritis.35
Aspirin is recommended
Daily low-dose aspirin therapy has been shown in several studies to be effective in preventing ischemic complications of giant cell arteritis, including stroke and visual loss. It is currently recommended that all patients with giant cell arteritis without a major contraindication take aspirin 81 mg daily.36–38
- Salvarani C, Gabriel SE, O’Fallon WM, Hunder GG. The incidence of giant cell arteritis in Olmsted County, Minnesota: apparent fluctuations in a cyclic pattern. Ann Intern Med 1995; 123:192–194.
- Baldursson O, Steinsson K, Björnsson J, Lie JT. Giant cell arteritis in Iceland. An epidemiologic and histopathologic analysis. Arthritis Rheum 1994; 37:1007–1012.
- Weyand CM, Goronzy JJ. Medium- and large-vessel vasculitis. N Engl J Med 2003; 349:160–169.
- Aiello PD, Trautmann JC, McPhee TJ, Kunselman AR, Hunder GG. Visual prognosis in giant cell arteritis. Ophthalmology 1993; 100:550–555.
- Salvarani C, Cimino L, Macchioni P, et al. Risk factors for visual loss in an Italian population-based cohort of patients with giant cell arteritis. Arthritis Rheum 2005; 53:293–297.
- Bahlas S, Ramos-Remus C, Davis P. Clinical outcome of 149 patients with polymyalgia rheumatica and giant cell arteritis. J Rheumatol 1998; 25:99–104.
- Gonzalez-Gay MA, Barros S, Lopez-Diaz MJ, Garcia-Porrua C, Sanchez-Andrade A, Llorca J. Giant cell arteritis: disease patterns of clinical presentation in a series of 240 patients. Medicine (Baltimore) 2005; 84:269–276.
- Salvarani C, Cantini F, Macchioni P, et al. Distal musculoskeletal manifestations in polymyalgia rheumatica: a prospective followup study. Arthritis Rheum 1998; 41:1221–1226.
- Salvarani C, Cantini F, Boiardi L, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med 2002; 347:261–271.
- Lie JT. Aortic and extracranial large vessel giant cell arteritis: a review of 72 cases with histopathologic documentation. Semin Arthritis Rheum 1995; 24:422–431.
- Evans JM, O’Fallon WM, Hunder GG. Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis. A population-based study. Ann Intern Med 1995; 122:502–507.
- Ostberg G. An arteritis with special reference to polymyalgia arteritica. Acta Pathol Microbiol Scand Suppl 1973; 237(suppl 237):1–59.
- Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol 1999; 128:211–215.
- González-Gay MA, Garcia-Porrua C, Rivas MJ, Rodriguez-Ledo P, Llorca J. Epidemiology of biopsy proven giant cell arteritis in northwestern Spain: trend over an 18 year period. Ann Rheum Dis 2001; 60:367–371.
- Rodriguez-Valverde V, Sarabia JM, González-Gay MA, et al. Risk factors and predictive models of giant cell arteritis in polymyalgia rheumatica. Am J Med 1997; 102:331–336.
- Mahr A, Saba M, Kambouchner M, et al. Temporal artery biopsy for diagnosing giant cell arteritis: the longer, the better? Ann Rheum Dis 2006; 65:826–828.
- Breuer GS, Nesher R, Nesher G. Effect of biopsy length on the rate of positive temporal artery biopsies. Clin Exp Rheumatol 2009; 27(1 suppl 52):S10–S13.
- Weyand CM, Goronzy JJ. Giant-cell arteritis and polymyalgia rheumatica. Ann Intern Med 2003; 139:505–515.
- Salvarani C, Cantini F, Boiardi L, Hunder GG. Laboratory investigations useful in giant cell arteritis and Takayasu’s arteritis. Clin Exp Rheumatol 2003; 21(6 suppl 32):S23–S28.
- Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurence in a population-based study. Arthritis Rheum 2001; 45:140–145.
- Schmidt WA, Kraft HE, Vorpahl K, Völker L, Gromnica-Ihle EJ. Color duplex ultrasonography in the diagnosis of temporal arteritis. N Engl J Med 1997; 337:1336–1342.
- Karassa FB, Matsagas MI, Schmidt WA, Ioannidis JP. Meta-analysis: test performance of ultrasonography for giant-cell arteritis. Ann Intern Med 2005; 142:359–369.
- Maldini C, Dépinay-Dhellemmes C, Tra TT, et al. Limited value of temporal artery ultrasonography examinations for diagnosis of giant cell arteritis: analysis of 77 subjects. J Rheumatol 2010; Epub ahead of print.
- Both M, Ahmadi-Simab K, Reuter M, et al. MRI and FDG-PET in the assessment of inflammatory aortic arch syndrome in complicated courses of giant cell arteritis. Ann Rheum Dis 2008; 67:1030–1033.
- Tso E, Flamm SD, White RD, Schvartzman PR, Mascha E, Hoffman GS. Takayasu arteritis: utility and limitations of magnetic resonance imaging in diagnosis and treatment. Arthritis Rheum 2002; 46:1634–1642.
- Birkhead NC, Wagener HP, Shick RM. Treatment of temporal arteritis with adrenal corticosteroids; results in fifty-five cases in which lesion was proved at biopsy. J Am Med Assoc 1957; 163:821–827.
- Ray-Chaudhuri N, Kiné DA, Tijani SO, et al. Effect of prior steroid treatment on temporal artery biopsy findings in giant cell arteritis. Br J Ophthalmol 2002; 86:530–532.
- Chan CC, Paine M, O’Day J. Steroid management in giant cell arteritis. Br J Ophthalmol 2001; 85:1061–1064.
- Hunder GG, Sheps SG, Allen GL, Joyce JW. Daily and alternate-day corticosteroid regimens in treatment of giant cell arteritis: comparison in a prospective study. Ann Intern Med 1975; 82:613–618.
- Hoffman GS, Cid MC, Hellmann DB, et al; International Network for the Study of Systemic Vasculitides. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum 2002; 46:1309–1318.
- Mazlumzadeh M, Hunder GG, Easley KA, et al. Treatment of giant cell arteritis using induction therapy with high-dose glucocorticoids: a double-blind, placebo-controlled, randomized prospective clinical trial. Arthritis Rheum 2006; 54:3310–3318.
- Spiera RF, Mitnick HJ, Kupersmith M, et al. A prospective, doubleblind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA). Clin Exp Rheumatol 2001; 19:495–501.
- Jover JA, Hernández-García C, Morado IC, Vargas E, Bañares A, Fernández-Gutiérrez B. Combined treatment of giant-cell arteritis with methotrexate and prednisone. a randomized, double-blind, placebo-controlled trial. Ann Intern Med 2001; 134:106–114.
- Mahr AD, Jover JA, Spiera RF, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum 2007; 56:2789–2797.
- Hoffman GS, Cid MC, Rendt-Zagar KE, et al; Infliximab-GCA Study Group. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med 2007; 146:621–630.
- Weyand CM, Kaiser M, Yang H, Younge B, Goronzy JJ. Therapeutic effects of acetylsalicylic acid in giant cell arteritis. Arthritis Rheum 2002; 46:457–466.
- Nesher G, Berkun Y, Mates M, Baras M, Rubinow A, Sonnenblick M. Low-dose aspirin and prevention of cranial ischemic complications in giant cell arteritis. Arthritis Rheum 2004; 50:1332–1337.
- Lee MS, Smith SD, Galor A, Hoffman GS. Antiplatelet and anticoagulant therapy in patients with giant cell arteritis. Arthritis Rheum 2006; 54:3306–3309.
- Salvarani C, Gabriel SE, O’Fallon WM, Hunder GG. The incidence of giant cell arteritis in Olmsted County, Minnesota: apparent fluctuations in a cyclic pattern. Ann Intern Med 1995; 123:192–194.
- Baldursson O, Steinsson K, Björnsson J, Lie JT. Giant cell arteritis in Iceland. An epidemiologic and histopathologic analysis. Arthritis Rheum 1994; 37:1007–1012.
- Weyand CM, Goronzy JJ. Medium- and large-vessel vasculitis. N Engl J Med 2003; 349:160–169.
- Aiello PD, Trautmann JC, McPhee TJ, Kunselman AR, Hunder GG. Visual prognosis in giant cell arteritis. Ophthalmology 1993; 100:550–555.
- Salvarani C, Cimino L, Macchioni P, et al. Risk factors for visual loss in an Italian population-based cohort of patients with giant cell arteritis. Arthritis Rheum 2005; 53:293–297.
- Bahlas S, Ramos-Remus C, Davis P. Clinical outcome of 149 patients with polymyalgia rheumatica and giant cell arteritis. J Rheumatol 1998; 25:99–104.
- Gonzalez-Gay MA, Barros S, Lopez-Diaz MJ, Garcia-Porrua C, Sanchez-Andrade A, Llorca J. Giant cell arteritis: disease patterns of clinical presentation in a series of 240 patients. Medicine (Baltimore) 2005; 84:269–276.
- Salvarani C, Cantini F, Macchioni P, et al. Distal musculoskeletal manifestations in polymyalgia rheumatica: a prospective followup study. Arthritis Rheum 1998; 41:1221–1226.
- Salvarani C, Cantini F, Boiardi L, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. N Engl J Med 2002; 347:261–271.
- Lie JT. Aortic and extracranial large vessel giant cell arteritis: a review of 72 cases with histopathologic documentation. Semin Arthritis Rheum 1995; 24:422–431.
- Evans JM, O’Fallon WM, Hunder GG. Increased incidence of aortic aneurysm and dissection in giant cell (temporal) arteritis. A population-based study. Ann Intern Med 1995; 122:502–507.
- Ostberg G. An arteritis with special reference to polymyalgia arteritica. Acta Pathol Microbiol Scand Suppl 1973; 237(suppl 237):1–59.
- Boyev LR, Miller NR, Green WR. Efficacy of unilateral versus bilateral temporal artery biopsies for the diagnosis of giant cell arteritis. Am J Ophthalmol 1999; 128:211–215.
- González-Gay MA, Garcia-Porrua C, Rivas MJ, Rodriguez-Ledo P, Llorca J. Epidemiology of biopsy proven giant cell arteritis in northwestern Spain: trend over an 18 year period. Ann Rheum Dis 2001; 60:367–371.
- Rodriguez-Valverde V, Sarabia JM, González-Gay MA, et al. Risk factors and predictive models of giant cell arteritis in polymyalgia rheumatica. Am J Med 1997; 102:331–336.
- Mahr A, Saba M, Kambouchner M, et al. Temporal artery biopsy for diagnosing giant cell arteritis: the longer, the better? Ann Rheum Dis 2006; 65:826–828.
- Breuer GS, Nesher R, Nesher G. Effect of biopsy length on the rate of positive temporal artery biopsies. Clin Exp Rheumatol 2009; 27(1 suppl 52):S10–S13.
- Weyand CM, Goronzy JJ. Giant-cell arteritis and polymyalgia rheumatica. Ann Intern Med 2003; 139:505–515.
- Salvarani C, Cantini F, Boiardi L, Hunder GG. Laboratory investigations useful in giant cell arteritis and Takayasu’s arteritis. Clin Exp Rheumatol 2003; 21(6 suppl 32):S23–S28.
- Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurence in a population-based study. Arthritis Rheum 2001; 45:140–145.
- Schmidt WA, Kraft HE, Vorpahl K, Völker L, Gromnica-Ihle EJ. Color duplex ultrasonography in the diagnosis of temporal arteritis. N Engl J Med 1997; 337:1336–1342.
- Karassa FB, Matsagas MI, Schmidt WA, Ioannidis JP. Meta-analysis: test performance of ultrasonography for giant-cell arteritis. Ann Intern Med 2005; 142:359–369.
- Maldini C, Dépinay-Dhellemmes C, Tra TT, et al. Limited value of temporal artery ultrasonography examinations for diagnosis of giant cell arteritis: analysis of 77 subjects. J Rheumatol 2010; Epub ahead of print.
- Both M, Ahmadi-Simab K, Reuter M, et al. MRI and FDG-PET in the assessment of inflammatory aortic arch syndrome in complicated courses of giant cell arteritis. Ann Rheum Dis 2008; 67:1030–1033.
- Tso E, Flamm SD, White RD, Schvartzman PR, Mascha E, Hoffman GS. Takayasu arteritis: utility and limitations of magnetic resonance imaging in diagnosis and treatment. Arthritis Rheum 2002; 46:1634–1642.
- Birkhead NC, Wagener HP, Shick RM. Treatment of temporal arteritis with adrenal corticosteroids; results in fifty-five cases in which lesion was proved at biopsy. J Am Med Assoc 1957; 163:821–827.
- Ray-Chaudhuri N, Kiné DA, Tijani SO, et al. Effect of prior steroid treatment on temporal artery biopsy findings in giant cell arteritis. Br J Ophthalmol 2002; 86:530–532.
- Chan CC, Paine M, O’Day J. Steroid management in giant cell arteritis. Br J Ophthalmol 2001; 85:1061–1064.
- Hunder GG, Sheps SG, Allen GL, Joyce JW. Daily and alternate-day corticosteroid regimens in treatment of giant cell arteritis: comparison in a prospective study. Ann Intern Med 1975; 82:613–618.
- Hoffman GS, Cid MC, Hellmann DB, et al; International Network for the Study of Systemic Vasculitides. A multicenter, randomized, double-blind, placebo-controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum 2002; 46:1309–1318.
- Mazlumzadeh M, Hunder GG, Easley KA, et al. Treatment of giant cell arteritis using induction therapy with high-dose glucocorticoids: a double-blind, placebo-controlled, randomized prospective clinical trial. Arthritis Rheum 2006; 54:3310–3318.
- Spiera RF, Mitnick HJ, Kupersmith M, et al. A prospective, doubleblind, randomized, placebo controlled trial of methotrexate in the treatment of giant cell arteritis (GCA). Clin Exp Rheumatol 2001; 19:495–501.
- Jover JA, Hernández-García C, Morado IC, Vargas E, Bañares A, Fernández-Gutiérrez B. Combined treatment of giant-cell arteritis with methotrexate and prednisone. a randomized, double-blind, placebo-controlled trial. Ann Intern Med 2001; 134:106–114.
- Mahr AD, Jover JA, Spiera RF, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum 2007; 56:2789–2797.
- Hoffman GS, Cid MC, Rendt-Zagar KE, et al; Infliximab-GCA Study Group. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med 2007; 146:621–630.
- Weyand CM, Kaiser M, Yang H, Younge B, Goronzy JJ. Therapeutic effects of acetylsalicylic acid in giant cell arteritis. Arthritis Rheum 2002; 46:457–466.
- Nesher G, Berkun Y, Mates M, Baras M, Rubinow A, Sonnenblick M. Low-dose aspirin and prevention of cranial ischemic complications in giant cell arteritis. Arthritis Rheum 2004; 50:1332–1337.
- Lee MS, Smith SD, Galor A, Hoffman GS. Antiplatelet and anticoagulant therapy in patients with giant cell arteritis. Arthritis Rheum 2006; 54:3306–3309.
KEY POINTS
- Giant cell arteritis is often associated with an intense acute-phase response and cranial symptoms.
- Large-vessel involvement is commonly present and may result in serious complications such as visual loss, stroke, limb claudication, and aortic aneurysm.
- The diagnosis is usually confirmed by an abnormal temporal artery biopsy.
- Symptoms of giant cell arteritis usually respond rapidly and completely to glucocorticoid therapy, still the mainstay of treatment. Most patients need prolonged therapy.
- Several studies have evaluated alternative drugs in an attempt to avoid toxicity from long-term use of glucocorticoids. Results have been mixed, and further study is needed.