User login
A 40-year-old man with spells of generalized weakness and paresthesias
A 40-year-old man who works as a roofer began, 1 week ago, to experience episodes of generalized weakness, perioral numbness, and diffuse paresthesias. In the past he has had recurring nosebleeds but no history of other medical conditions.
His recent “spells” come on abruptly and spontaneously, without warning, and last about 15 minutes. He never loses consciousness, but he reports a feeling of derealization or an out-of-body experience—he can hear the people around him talking during the spells, but he feels that everything is far away. He has been having about three episodes per day. They typically occur after mild exertion or heavy lifting, and each episode resolves with complete rest. He has had no nausea, vomiting, loss of bowel or bladder control, fever, chills, or traumatic brain injury.
The patient first reported to the emergency department of a local hospital for evaluation. There, he underwent computed tomography (CT) of the head without contrast, which showed nothing abnormal. However, he had an episode while in the emergency department, which prompted his physician to admit him to the hospital.
In the hospital, he underwent an extensive medical evaluation. CT angiography revealed no evidence of vasculitis or occlusive disease. Results of electroencephalography during these spells were normal. Results of magnetic resonance imaging of the cervical and lumbar spine were also normal.
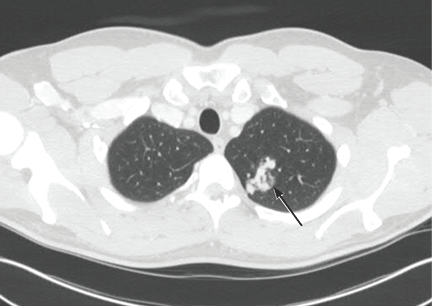
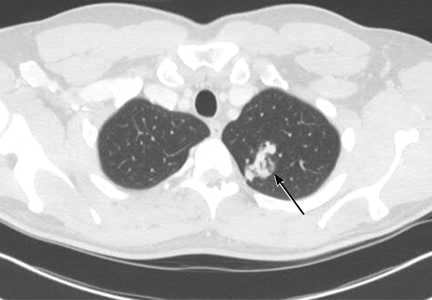
A neurologist was consulted. Concerned that the spells were due to paradoxical emboli coming through a patent foramen ovale, the neurologist recommended transthoracic echocardiography with agitated saline. This study showed a normal ejection fraction and a right-to-left shunt through a left pulmonary arteriovenous malformation (AVM). Unfortunately, the shunt fraction could not be estimated because the patient had another episode during the procedure, and so the procedure was cut short. CT of the chest confirmed a large AVM in the upper lobe of the left lung (Figure 1).
The patient is transferred
The patient’s physician requested that he be transferred to Mayo Clinic for further evaluation.
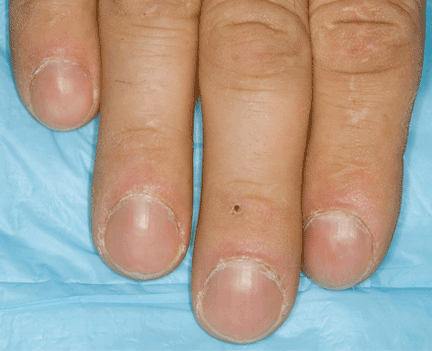
When he arrived, we performed a complete physical examination, in which we noted scattered erythematous maculopapular telangiectases in the lower lips and significant digital clubbing (Figure 2). He could not recall any family members having rheumatologic or cardiovascular diseases, but he recalled that his father has oral telangiectases and recurrent epistaxis.
His examination was interrupted by yet another spell, during which his oxygen saturation fell to 85%. We immediately started giving him oxygen by nasal cannula, which raised his oxygen saturation to 96%, and the spell promptly ended.
After his physical examination was completed and his records from the other hospital were reviewed, a diagnosis was made. No further diagnostic studies were pursued.
WHICH IS THE MOST LIKELY DIAGNOSIS?
1. Based on the information available, which of the following is the most likely diagnosis?
- Generalized tonic-clonic seizures
- Osler-Weber-Rendu disease
- Subarachnoid hemorrhage
- Conversion disorder
- Atrial septal defect
Generalized tonic-clonic seizures begin with abrupt loss of consciousness, followed by stiffening of the body and extremities. This is the tonic phase, which may last for 1 minute. The clonic phase follows, characterized by abnormal jerking and teeth-clenching (raising the concern that the patient will bite his or her tongue). The clonic phase lasts 1 to 2 minutes. After a seizure, confusion and headache are common. On electroencephalography, epileptiform abnormalities are documented in about 23% of patients with a first documented seizure.1
Our patient’s history of remaining fully conscious and of having normal electroencephalographic findings during his spells does not suggest generalized tonic-clonic seizures.
Osler-Weber-Rendu disease is also known as hereditary hemorrhagic telangiectasia (HHT). Its pathophysiology is complex, and it is believed to be related to mutations in an endothelial protein2 that lead to abnormal vascular structures. The estimated prevalence in European studies is 1 in 5,000; in Japanese studies it is 1 in 8,000.3–4
The diagnosis of HHT is based on four clinical criteria:
- Spontaneous and recurrent epistaxis
- Multiple mucocutaneous telangiectases
- Pulmonary, cerebral, or gastrointestinal AVMs
- A first-degree relative with the disease.
The presence of three or four of these criteria establishes a “definite” diagnosis, while fewer than two makes it “unlikely.”5 Since the spectrum of this disease is wide, varying from mild epistaxis to iron-deficiency anemia, its diagnosis is often missed.6
Our patient meets at least three of the criteria—recurrent epistaxis, oral telangiectases, and a CT-documented pulmonary AVM. His father has a history of oral telangiectases and epistaxis but was never formally diagnosed with HHT. The patient presented with spells of weakness and paresthesias from worsening hypoxemia due to an enlarged pulmonary AVM. Thus, based on these features, HHT is the most likely diagnosis.
Subarachnoid hemorrhage is commonly from a ruptured cerebral aneurysm. Common symptoms include sudden, severe headaches with focal neurologic deficits, a stiff neck, brief loss of consciousness, nausea, and vomiting.7
Our patient’s CT scan showed no intracranial bleeding, and CT angiography showed no evidence of aneurysm. Thus, he has neither clinical nor radiographic features of subarachnoid hemorrhage.
Conversion disorder is typically associated with psychological stressors.8 It is characterized by the sudden onset of neurologic deficits such as blindness, paralysis, and numbness that cannot be explained by a general medical condition.
Our patient has a known pulmonary AVM with clinical and laboratory findings of hypoxemia that explain his spells. Therefore, the diagnosis of conversion disorder cannot be made.
A right-to-left intracardiac shunt can be present in patients with patent foramen ovale, atrial septal defects with shunt reversal, Eisenmenger syndrome, or tetralogy of Fallot (even in adults). It can present with hypoxemia and neurologic weakness.
Our patient’s echocardiogram ruled out these conditions.
MANIFESTATIONS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
2. Which is the most common clinical manifestation of HHT?
- Epistaxis
- Mucocutaneous telangiectases
- Hematochezia
- Dyspnea
Epistaxis is the most common presentation, occurring in more than 90% of patients.9 Many patients experience only mild occasional nosebleeds that are not frequent or severe enough to cause anemia or to lead to medical treatment or consultation. Others, however, have heavy, frequent bleeding that requires invasive interventions.10
Mucocutaneous telangiectases are the second most common clinical manifestation, documented in about 75% of patients. They are cosmetically unpleasant but rarely bleed. They occur most commonly on the face, lips, tongue, and fingertips, and they increase in size and number with age.11
Gastrointestinal bleeding, sometimes manifesting as hematochezia, occurs in one-third of people with HHT. It most commonly presents with iron-deficiency anemia in patients over age 40.12
Dyspnea. Pulmonary AVMs occur in 30% to 50% of affected people, but interestingly, most patients with pulmonary AVMs have no respiratory symptoms, including dyspnea.
In pulmonary AVMs, abnormal vessels replace normal capillary beds, creating a capillary-free communication between the pulmonary and systemic circulations. This abnormal connection prevents blood from the pulmonary arterial system from being oxygenated, resulting in hypoxemia and secondary polycythemia, as in our patient. One-third of patients have evidence of right-to-left shunting, such as the clubbing in our patient.9,13
Other, less common complications of HHT include seizures or hemorrhage from cerebral AVMs and stroke and brain abscesses from paradoxical embolization due to the loss of the capillary filter in the pulmonary vascular bed. Hepatic involvement may result in portal hypertension and hepatic encephalopathy.14
Back to our patient
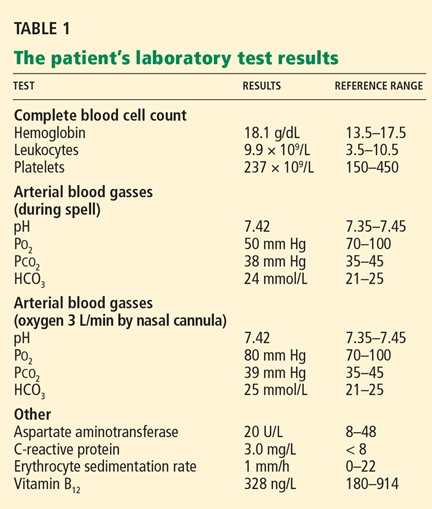
As mentioned above, during one of the patient’s spells of paresthesia and weakness, we noted his oxygen saturation by oximetry was 85%. At that time, his arterial Po2 was also low at 50 mm Hg (normal 70–100). With oxygen supplementation, his spell completely resolved and his Po2 improved to 80 mm Hg. Though the shunt fraction of his pulmonary AVM was never measured, it was likely less than 30% of the cardiac output, as his hypoxemia improved with oxygen supplementation alone.15 When he was taken off oxygen supplementation, his spells recurred, but with oxygen support he remained clinically stable.
MANAGEMENT
3. Which is the next logical step in our patient’s management?
- Consult a surgeon for lobectomy
- Consult an interventional radiologist for embolization therapy
- Transfer to the intensive care unit for elective intubation
- Observe with close follow-up
Untreated pulmonary AVMs enlarge at an estimated rate of 0.3 mm/year. The estimated death rate is up to 15.8% per year, with most deaths resulting from stroke, cerebral abscess, hemoptysis, and hemothorax.16–18 Common indications for treatment are progressively enlarging lesions, symptomatic hypoxemia, and paradoxical embolization.19 Pulmonary AVMs in which the feeding artery is 3 mm or more in diameter require treatment.
Embolization therapy, in which the AVM is occluded angiographically, is considered a first-line treatment for pulmonary AVM, with a procedural success rate (defined as involution of the AVM) of 97%.20 Embolization therapy allows patients to avoid major surgery, with its potential complications, and it has a shorter recovery time.
Surgical procedures such as excision, vascular ligation, or lobectomy can be considered if the lesion cannot be treated by embolization or if the patient has an anaphylactic allergy to contrast dyes.
This patient had no clinical signs of impending respiratory failure requiring elective intubation.
Since he was experiencing symptoms, there is no role for observation in this case.
Back to our patient
An interventional radiologist was consulted, and the patient underwent bilateral pulmonary artery angiography with successful coil embolization of his large left-upper-lobe AVM. He was weaned off oxygen and had no further spells of generalized weakness and paresthesias.
Given his father’s history of recurrent epistaxis and oral telangiectases, the patient asks about the risk of his children acquiring this disease.
GENETICS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
4. Which of the following is the inheritance pattern for HHT?
- Autosomal dominant
- Autosomal recessive
- Maternal inheritance
- X-linked recessive
The inheritance pattern is autosomal dominant with variable expression and penetrance. At least four different mutations have been identified in genes on chromosomes 9 and 12 that result in abnormal vascular malformations.21–24 The other modes of inheritance have not been described in HHT.
RECOMMENDATIONS FOR OUR PATIENT
5. Which of the following is not recommended for our patient?
- Consideration of genetic testing
- Consideration of screening of first-degree relatives
- Dental prophylaxis
- Scuba diving
Genetic testing. The molecular diagnosis of HHT is primarily based on sequencing of the entire coding regions of the ENG and ALK1 genes on chromosomes 9 and 12, respectively. The interpretation of these results is quite complex. The clinical genetics laboratories in North America that currently offer molecular diagnostic testing for HHT recommend that testing be coordinated and ordered through a center that specializes in this disease or by a genetics professional. Testing of the index case is performed to confirm the clinical diagnosis and also to determine if genetic testing will be possible in at-risk relatives. Further genetic testing should be pursued in at-risk family members only if the index case has a positive result.25
Screening of relatives. Given that HHT is an autosomal dominant disease, the current practice is to offer molecular genetic screening early in life for first-degree relatives.25,26 The external signs such as telangiectases and nosebleeds may not manifest until the second or third decade of life. However, AVMs in the brain, spinal cord, lungs, and liver are usually congenital and may present suddenly and with serious complications, even in childhood.
Dental prophylaxis. People with HHT and a pulmonary AVM are at risk of bacteremia and consequent brain abscesses after dental procedures. Antibiotic prophylaxis is therefore highly recommended.27
One sport to avoid. There have been several case reports of paradoxical air emboli occurring in patients with HHT complicated by a pulmonary AVM. Hsu et al28 reported a 31-year-old man with an undiagnosed large pulmonary AVM and HHT who became comatose with diffuse bilateral hemispheric brain swelling on head CT after scuba diving, due to air embolism.
The HHT Foundation International recommends that people with this disease avoid scuba diving (the only sport to be avoided) owing to the risk of air emboli from small lung AVMs. It also recommends that patients alert health care providers about their risk of air embolism whenever intravenous access is being established.
Back to our patient
The patient met with a geneticist, and blood was collected for genetic testing before he was sent home. Additionally, the need to screen his first-degree relatives was thoroughly discussed. Four days after discharge he returned to work, and his spells have not recurred. He has a follow-up appointment scheduled with a pulmonologist specializing in this disease for the results of genetic testing and for continued management.
TAKE-HOME POINTS
- The diagnosis of HHT is based on the following four clinical criteria: spontaneous or recurrent epistaxis, multiple mucocutaneous telangiectases, visceral involvement (eg, cerebral, pulmonary, or gastrointestinal AVM), and a first-degree relative with this disease.
- The diagnosis may be confirmed with genetic testing.
- The diagnosis may be underreported, given the wide spectrum of disease presentation, from inconsequential epistaxis to massive gastrointestinal bleeding.
- HHT is autosomal dominant, and therefore all first-degree relatives should be screened.
- Krumholz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2007; 69:1996–2007.
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest 1999; 104:1343–1351.
- Dakeishi M, Shioya T, Wada Y, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat 2002; 19:140–148.
- Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995; 333:918–924.
- Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91:66–67.
- Gallitelli M, Pasculli G, Fiore T, Carella A, Sabbà C. Emergencies in hereditary haemorrhagic telangiectasia. QJM 2006; 99:15–22.
- Gorelick PB, Hier DB, Caplan LR, Langenberg P. Headache in acute cerebrovascular disease. Neurology 1986; 36:1445–1450.
- Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry 2006; 163:1510–1517.
- Shovlin CL, Letarte M. Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54:714–729.
- AAssar OS, Friedman CM, White RI. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 1991; 101:977–980.
- McAllister KA, Lennon F, Bowles-Biesecker B, et al. Genetic heterogenicity in hereditary haemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet 1994; 31:927–932.
- Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32:291–297.
- Shovlin CL, Jaskson JE, Bamford KB, et al. Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax 2008; 63:259–266.
- Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343:931–936.
- Kolleft MH, Micek ST. Critical care. In:Cooper DH, Krainik AJ, Lubner SJ, Reno HEL, editors. Washington Manual of Medical Therapeutics. 32nd ed. Philadelphia: Lippincott Williams & Wilkins, 2007:224–230.
- Swanson KL, Prakash UB, Stanson AW. Pulmonary arteriovenous fistulas: Mayo Clinic experience: 1872–1997. Mayo Clin Proc 1999; 74:671–680.
- Dines DE, Arms RA, Bernatz PE, Gomes MR. Pulmonary arteriovenous fistulas. Mayo Clinic Proc 1974; 49:460–465.
- Sluiter-Eringa H, Orie NG, Sluiter HJ. Pulmonary arteriovenous fistula: diagnosis and prognosis in noncompliant patients. Am Rev Respir Dis 1969; 100:177–188.
- Dines DE, Seward JB, Bernatz PE. Pulmonary arteriovenous fistula. Mayo Clin Proc 1983; 58:176–181.
- Pollak JS, Saluja S, Thabet A, Henderson KJ, Denbow N, White RI. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radio 2006; 17:35–44.
- Berg JN, Gallion CJ, Stenzel TT, et al. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997; 61:60–67.
- McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary hemorrhagic telangiectasias type 1. Nat Genet 1994; 8:345–351.
- Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase gene in hereditary haemorrhagic telangeictasia type 2. Nat Genet 1996; 13:189–195.
- Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet 2006; 43:97–110.
- Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med 2004; 6:175–191.
- Cohen JH, Faughnan ME, Letarte M, Vandezande K, Kennedy SJ, Krahn MD. Cost comparison of genetic and clinical screening in families with hereditary hemorrhagic telangiectasia. Am J Med Genet A 2005; 137:153–160.
- Shovlin C, Bamfort K, Wray D. Post-NICE 2008: Antibiotic prophylaxis prior to dental procedures for patients with pulmonary arteriovenous malformations (PAVMs) and hereditary haemorrhagic telangiectasia. Br Dent J 2008; 205:531–533.
- Hsu YL, Wang HC, Yang PC. Desbaric air embolism during diving: an unusual complication of Osler-Weber-Rendu disease. Br J Sports Med 2004; 38:E6.
A 40-year-old man who works as a roofer began, 1 week ago, to experience episodes of generalized weakness, perioral numbness, and diffuse paresthesias. In the past he has had recurring nosebleeds but no history of other medical conditions.
His recent “spells” come on abruptly and spontaneously, without warning, and last about 15 minutes. He never loses consciousness, but he reports a feeling of derealization or an out-of-body experience—he can hear the people around him talking during the spells, but he feels that everything is far away. He has been having about three episodes per day. They typically occur after mild exertion or heavy lifting, and each episode resolves with complete rest. He has had no nausea, vomiting, loss of bowel or bladder control, fever, chills, or traumatic brain injury.
The patient first reported to the emergency department of a local hospital for evaluation. There, he underwent computed tomography (CT) of the head without contrast, which showed nothing abnormal. However, he had an episode while in the emergency department, which prompted his physician to admit him to the hospital.
In the hospital, he underwent an extensive medical evaluation. CT angiography revealed no evidence of vasculitis or occlusive disease. Results of electroencephalography during these spells were normal. Results of magnetic resonance imaging of the cervical and lumbar spine were also normal.
A neurologist was consulted. Concerned that the spells were due to paradoxical emboli coming through a patent foramen ovale, the neurologist recommended transthoracic echocardiography with agitated saline. This study showed a normal ejection fraction and a right-to-left shunt through a left pulmonary arteriovenous malformation (AVM). Unfortunately, the shunt fraction could not be estimated because the patient had another episode during the procedure, and so the procedure was cut short. CT of the chest confirmed a large AVM in the upper lobe of the left lung (Figure 1).
The patient is transferred
The patient’s physician requested that he be transferred to Mayo Clinic for further evaluation.
When he arrived, we performed a complete physical examination, in which we noted scattered erythematous maculopapular telangiectases in the lower lips and significant digital clubbing (Figure 2). He could not recall any family members having rheumatologic or cardiovascular diseases, but he recalled that his father has oral telangiectases and recurrent epistaxis.
His examination was interrupted by yet another spell, during which his oxygen saturation fell to 85%. We immediately started giving him oxygen by nasal cannula, which raised his oxygen saturation to 96%, and the spell promptly ended.
After his physical examination was completed and his records from the other hospital were reviewed, a diagnosis was made. No further diagnostic studies were pursued.
WHICH IS THE MOST LIKELY DIAGNOSIS?
1. Based on the information available, which of the following is the most likely diagnosis?
- Generalized tonic-clonic seizures
- Osler-Weber-Rendu disease
- Subarachnoid hemorrhage
- Conversion disorder
- Atrial septal defect
Generalized tonic-clonic seizures begin with abrupt loss of consciousness, followed by stiffening of the body and extremities. This is the tonic phase, which may last for 1 minute. The clonic phase follows, characterized by abnormal jerking and teeth-clenching (raising the concern that the patient will bite his or her tongue). The clonic phase lasts 1 to 2 minutes. After a seizure, confusion and headache are common. On electroencephalography, epileptiform abnormalities are documented in about 23% of patients with a first documented seizure.1
Our patient’s history of remaining fully conscious and of having normal electroencephalographic findings during his spells does not suggest generalized tonic-clonic seizures.
Osler-Weber-Rendu disease is also known as hereditary hemorrhagic telangiectasia (HHT). Its pathophysiology is complex, and it is believed to be related to mutations in an endothelial protein2 that lead to abnormal vascular structures. The estimated prevalence in European studies is 1 in 5,000; in Japanese studies it is 1 in 8,000.3–4
The diagnosis of HHT is based on four clinical criteria:
- Spontaneous and recurrent epistaxis
- Multiple mucocutaneous telangiectases
- Pulmonary, cerebral, or gastrointestinal AVMs
- A first-degree relative with the disease.
The presence of three or four of these criteria establishes a “definite” diagnosis, while fewer than two makes it “unlikely.”5 Since the spectrum of this disease is wide, varying from mild epistaxis to iron-deficiency anemia, its diagnosis is often missed.6
Our patient meets at least three of the criteria—recurrent epistaxis, oral telangiectases, and a CT-documented pulmonary AVM. His father has a history of oral telangiectases and epistaxis but was never formally diagnosed with HHT. The patient presented with spells of weakness and paresthesias from worsening hypoxemia due to an enlarged pulmonary AVM. Thus, based on these features, HHT is the most likely diagnosis.
Subarachnoid hemorrhage is commonly from a ruptured cerebral aneurysm. Common symptoms include sudden, severe headaches with focal neurologic deficits, a stiff neck, brief loss of consciousness, nausea, and vomiting.7
Our patient’s CT scan showed no intracranial bleeding, and CT angiography showed no evidence of aneurysm. Thus, he has neither clinical nor radiographic features of subarachnoid hemorrhage.
Conversion disorder is typically associated with psychological stressors.8 It is characterized by the sudden onset of neurologic deficits such as blindness, paralysis, and numbness that cannot be explained by a general medical condition.
Our patient has a known pulmonary AVM with clinical and laboratory findings of hypoxemia that explain his spells. Therefore, the diagnosis of conversion disorder cannot be made.
A right-to-left intracardiac shunt can be present in patients with patent foramen ovale, atrial septal defects with shunt reversal, Eisenmenger syndrome, or tetralogy of Fallot (even in adults). It can present with hypoxemia and neurologic weakness.
Our patient’s echocardiogram ruled out these conditions.
MANIFESTATIONS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
2. Which is the most common clinical manifestation of HHT?
- Epistaxis
- Mucocutaneous telangiectases
- Hematochezia
- Dyspnea
Epistaxis is the most common presentation, occurring in more than 90% of patients.9 Many patients experience only mild occasional nosebleeds that are not frequent or severe enough to cause anemia or to lead to medical treatment or consultation. Others, however, have heavy, frequent bleeding that requires invasive interventions.10
Mucocutaneous telangiectases are the second most common clinical manifestation, documented in about 75% of patients. They are cosmetically unpleasant but rarely bleed. They occur most commonly on the face, lips, tongue, and fingertips, and they increase in size and number with age.11
Gastrointestinal bleeding, sometimes manifesting as hematochezia, occurs in one-third of people with HHT. It most commonly presents with iron-deficiency anemia in patients over age 40.12
Dyspnea. Pulmonary AVMs occur in 30% to 50% of affected people, but interestingly, most patients with pulmonary AVMs have no respiratory symptoms, including dyspnea.
In pulmonary AVMs, abnormal vessels replace normal capillary beds, creating a capillary-free communication between the pulmonary and systemic circulations. This abnormal connection prevents blood from the pulmonary arterial system from being oxygenated, resulting in hypoxemia and secondary polycythemia, as in our patient. One-third of patients have evidence of right-to-left shunting, such as the clubbing in our patient.9,13
Other, less common complications of HHT include seizures or hemorrhage from cerebral AVMs and stroke and brain abscesses from paradoxical embolization due to the loss of the capillary filter in the pulmonary vascular bed. Hepatic involvement may result in portal hypertension and hepatic encephalopathy.14
Back to our patient
As mentioned above, during one of the patient’s spells of paresthesia and weakness, we noted his oxygen saturation by oximetry was 85%. At that time, his arterial Po2 was also low at 50 mm Hg (normal 70–100). With oxygen supplementation, his spell completely resolved and his Po2 improved to 80 mm Hg. Though the shunt fraction of his pulmonary AVM was never measured, it was likely less than 30% of the cardiac output, as his hypoxemia improved with oxygen supplementation alone.15 When he was taken off oxygen supplementation, his spells recurred, but with oxygen support he remained clinically stable.
MANAGEMENT
3. Which is the next logical step in our patient’s management?
- Consult a surgeon for lobectomy
- Consult an interventional radiologist for embolization therapy
- Transfer to the intensive care unit for elective intubation
- Observe with close follow-up
Untreated pulmonary AVMs enlarge at an estimated rate of 0.3 mm/year. The estimated death rate is up to 15.8% per year, with most deaths resulting from stroke, cerebral abscess, hemoptysis, and hemothorax.16–18 Common indications for treatment are progressively enlarging lesions, symptomatic hypoxemia, and paradoxical embolization.19 Pulmonary AVMs in which the feeding artery is 3 mm or more in diameter require treatment.
Embolization therapy, in which the AVM is occluded angiographically, is considered a first-line treatment for pulmonary AVM, with a procedural success rate (defined as involution of the AVM) of 97%.20 Embolization therapy allows patients to avoid major surgery, with its potential complications, and it has a shorter recovery time.
Surgical procedures such as excision, vascular ligation, or lobectomy can be considered if the lesion cannot be treated by embolization or if the patient has an anaphylactic allergy to contrast dyes.
This patient had no clinical signs of impending respiratory failure requiring elective intubation.
Since he was experiencing symptoms, there is no role for observation in this case.
Back to our patient
An interventional radiologist was consulted, and the patient underwent bilateral pulmonary artery angiography with successful coil embolization of his large left-upper-lobe AVM. He was weaned off oxygen and had no further spells of generalized weakness and paresthesias.
Given his father’s history of recurrent epistaxis and oral telangiectases, the patient asks about the risk of his children acquiring this disease.
GENETICS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
4. Which of the following is the inheritance pattern for HHT?
- Autosomal dominant
- Autosomal recessive
- Maternal inheritance
- X-linked recessive
The inheritance pattern is autosomal dominant with variable expression and penetrance. At least four different mutations have been identified in genes on chromosomes 9 and 12 that result in abnormal vascular malformations.21–24 The other modes of inheritance have not been described in HHT.
RECOMMENDATIONS FOR OUR PATIENT
5. Which of the following is not recommended for our patient?
- Consideration of genetic testing
- Consideration of screening of first-degree relatives
- Dental prophylaxis
- Scuba diving
Genetic testing. The molecular diagnosis of HHT is primarily based on sequencing of the entire coding regions of the ENG and ALK1 genes on chromosomes 9 and 12, respectively. The interpretation of these results is quite complex. The clinical genetics laboratories in North America that currently offer molecular diagnostic testing for HHT recommend that testing be coordinated and ordered through a center that specializes in this disease or by a genetics professional. Testing of the index case is performed to confirm the clinical diagnosis and also to determine if genetic testing will be possible in at-risk relatives. Further genetic testing should be pursued in at-risk family members only if the index case has a positive result.25
Screening of relatives. Given that HHT is an autosomal dominant disease, the current practice is to offer molecular genetic screening early in life for first-degree relatives.25,26 The external signs such as telangiectases and nosebleeds may not manifest until the second or third decade of life. However, AVMs in the brain, spinal cord, lungs, and liver are usually congenital and may present suddenly and with serious complications, even in childhood.
Dental prophylaxis. People with HHT and a pulmonary AVM are at risk of bacteremia and consequent brain abscesses after dental procedures. Antibiotic prophylaxis is therefore highly recommended.27
One sport to avoid. There have been several case reports of paradoxical air emboli occurring in patients with HHT complicated by a pulmonary AVM. Hsu et al28 reported a 31-year-old man with an undiagnosed large pulmonary AVM and HHT who became comatose with diffuse bilateral hemispheric brain swelling on head CT after scuba diving, due to air embolism.
The HHT Foundation International recommends that people with this disease avoid scuba diving (the only sport to be avoided) owing to the risk of air emboli from small lung AVMs. It also recommends that patients alert health care providers about their risk of air embolism whenever intravenous access is being established.
Back to our patient
The patient met with a geneticist, and blood was collected for genetic testing before he was sent home. Additionally, the need to screen his first-degree relatives was thoroughly discussed. Four days after discharge he returned to work, and his spells have not recurred. He has a follow-up appointment scheduled with a pulmonologist specializing in this disease for the results of genetic testing and for continued management.
TAKE-HOME POINTS
- The diagnosis of HHT is based on the following four clinical criteria: spontaneous or recurrent epistaxis, multiple mucocutaneous telangiectases, visceral involvement (eg, cerebral, pulmonary, or gastrointestinal AVM), and a first-degree relative with this disease.
- The diagnosis may be confirmed with genetic testing.
- The diagnosis may be underreported, given the wide spectrum of disease presentation, from inconsequential epistaxis to massive gastrointestinal bleeding.
- HHT is autosomal dominant, and therefore all first-degree relatives should be screened.
A 40-year-old man who works as a roofer began, 1 week ago, to experience episodes of generalized weakness, perioral numbness, and diffuse paresthesias. In the past he has had recurring nosebleeds but no history of other medical conditions.
His recent “spells” come on abruptly and spontaneously, without warning, and last about 15 minutes. He never loses consciousness, but he reports a feeling of derealization or an out-of-body experience—he can hear the people around him talking during the spells, but he feels that everything is far away. He has been having about three episodes per day. They typically occur after mild exertion or heavy lifting, and each episode resolves with complete rest. He has had no nausea, vomiting, loss of bowel or bladder control, fever, chills, or traumatic brain injury.
The patient first reported to the emergency department of a local hospital for evaluation. There, he underwent computed tomography (CT) of the head without contrast, which showed nothing abnormal. However, he had an episode while in the emergency department, which prompted his physician to admit him to the hospital.
In the hospital, he underwent an extensive medical evaluation. CT angiography revealed no evidence of vasculitis or occlusive disease. Results of electroencephalography during these spells were normal. Results of magnetic resonance imaging of the cervical and lumbar spine were also normal.
A neurologist was consulted. Concerned that the spells were due to paradoxical emboli coming through a patent foramen ovale, the neurologist recommended transthoracic echocardiography with agitated saline. This study showed a normal ejection fraction and a right-to-left shunt through a left pulmonary arteriovenous malformation (AVM). Unfortunately, the shunt fraction could not be estimated because the patient had another episode during the procedure, and so the procedure was cut short. CT of the chest confirmed a large AVM in the upper lobe of the left lung (Figure 1).
The patient is transferred
The patient’s physician requested that he be transferred to Mayo Clinic for further evaluation.
When he arrived, we performed a complete physical examination, in which we noted scattered erythematous maculopapular telangiectases in the lower lips and significant digital clubbing (Figure 2). He could not recall any family members having rheumatologic or cardiovascular diseases, but he recalled that his father has oral telangiectases and recurrent epistaxis.
His examination was interrupted by yet another spell, during which his oxygen saturation fell to 85%. We immediately started giving him oxygen by nasal cannula, which raised his oxygen saturation to 96%, and the spell promptly ended.
After his physical examination was completed and his records from the other hospital were reviewed, a diagnosis was made. No further diagnostic studies were pursued.
WHICH IS THE MOST LIKELY DIAGNOSIS?
1. Based on the information available, which of the following is the most likely diagnosis?
- Generalized tonic-clonic seizures
- Osler-Weber-Rendu disease
- Subarachnoid hemorrhage
- Conversion disorder
- Atrial septal defect
Generalized tonic-clonic seizures begin with abrupt loss of consciousness, followed by stiffening of the body and extremities. This is the tonic phase, which may last for 1 minute. The clonic phase follows, characterized by abnormal jerking and teeth-clenching (raising the concern that the patient will bite his or her tongue). The clonic phase lasts 1 to 2 minutes. After a seizure, confusion and headache are common. On electroencephalography, epileptiform abnormalities are documented in about 23% of patients with a first documented seizure.1
Our patient’s history of remaining fully conscious and of having normal electroencephalographic findings during his spells does not suggest generalized tonic-clonic seizures.
Osler-Weber-Rendu disease is also known as hereditary hemorrhagic telangiectasia (HHT). Its pathophysiology is complex, and it is believed to be related to mutations in an endothelial protein2 that lead to abnormal vascular structures. The estimated prevalence in European studies is 1 in 5,000; in Japanese studies it is 1 in 8,000.3–4
The diagnosis of HHT is based on four clinical criteria:
- Spontaneous and recurrent epistaxis
- Multiple mucocutaneous telangiectases
- Pulmonary, cerebral, or gastrointestinal AVMs
- A first-degree relative with the disease.
The presence of three or four of these criteria establishes a “definite” diagnosis, while fewer than two makes it “unlikely.”5 Since the spectrum of this disease is wide, varying from mild epistaxis to iron-deficiency anemia, its diagnosis is often missed.6
Our patient meets at least three of the criteria—recurrent epistaxis, oral telangiectases, and a CT-documented pulmonary AVM. His father has a history of oral telangiectases and epistaxis but was never formally diagnosed with HHT. The patient presented with spells of weakness and paresthesias from worsening hypoxemia due to an enlarged pulmonary AVM. Thus, based on these features, HHT is the most likely diagnosis.
Subarachnoid hemorrhage is commonly from a ruptured cerebral aneurysm. Common symptoms include sudden, severe headaches with focal neurologic deficits, a stiff neck, brief loss of consciousness, nausea, and vomiting.7
Our patient’s CT scan showed no intracranial bleeding, and CT angiography showed no evidence of aneurysm. Thus, he has neither clinical nor radiographic features of subarachnoid hemorrhage.
Conversion disorder is typically associated with psychological stressors.8 It is characterized by the sudden onset of neurologic deficits such as blindness, paralysis, and numbness that cannot be explained by a general medical condition.
Our patient has a known pulmonary AVM with clinical and laboratory findings of hypoxemia that explain his spells. Therefore, the diagnosis of conversion disorder cannot be made.
A right-to-left intracardiac shunt can be present in patients with patent foramen ovale, atrial septal defects with shunt reversal, Eisenmenger syndrome, or tetralogy of Fallot (even in adults). It can present with hypoxemia and neurologic weakness.
Our patient’s echocardiogram ruled out these conditions.
MANIFESTATIONS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
2. Which is the most common clinical manifestation of HHT?
- Epistaxis
- Mucocutaneous telangiectases
- Hematochezia
- Dyspnea
Epistaxis is the most common presentation, occurring in more than 90% of patients.9 Many patients experience only mild occasional nosebleeds that are not frequent or severe enough to cause anemia or to lead to medical treatment or consultation. Others, however, have heavy, frequent bleeding that requires invasive interventions.10
Mucocutaneous telangiectases are the second most common clinical manifestation, documented in about 75% of patients. They are cosmetically unpleasant but rarely bleed. They occur most commonly on the face, lips, tongue, and fingertips, and they increase in size and number with age.11
Gastrointestinal bleeding, sometimes manifesting as hematochezia, occurs in one-third of people with HHT. It most commonly presents with iron-deficiency anemia in patients over age 40.12
Dyspnea. Pulmonary AVMs occur in 30% to 50% of affected people, but interestingly, most patients with pulmonary AVMs have no respiratory symptoms, including dyspnea.
In pulmonary AVMs, abnormal vessels replace normal capillary beds, creating a capillary-free communication between the pulmonary and systemic circulations. This abnormal connection prevents blood from the pulmonary arterial system from being oxygenated, resulting in hypoxemia and secondary polycythemia, as in our patient. One-third of patients have evidence of right-to-left shunting, such as the clubbing in our patient.9,13
Other, less common complications of HHT include seizures or hemorrhage from cerebral AVMs and stroke and brain abscesses from paradoxical embolization due to the loss of the capillary filter in the pulmonary vascular bed. Hepatic involvement may result in portal hypertension and hepatic encephalopathy.14
Back to our patient
As mentioned above, during one of the patient’s spells of paresthesia and weakness, we noted his oxygen saturation by oximetry was 85%. At that time, his arterial Po2 was also low at 50 mm Hg (normal 70–100). With oxygen supplementation, his spell completely resolved and his Po2 improved to 80 mm Hg. Though the shunt fraction of his pulmonary AVM was never measured, it was likely less than 30% of the cardiac output, as his hypoxemia improved with oxygen supplementation alone.15 When he was taken off oxygen supplementation, his spells recurred, but with oxygen support he remained clinically stable.
MANAGEMENT
3. Which is the next logical step in our patient’s management?
- Consult a surgeon for lobectomy
- Consult an interventional radiologist for embolization therapy
- Transfer to the intensive care unit for elective intubation
- Observe with close follow-up
Untreated pulmonary AVMs enlarge at an estimated rate of 0.3 mm/year. The estimated death rate is up to 15.8% per year, with most deaths resulting from stroke, cerebral abscess, hemoptysis, and hemothorax.16–18 Common indications for treatment are progressively enlarging lesions, symptomatic hypoxemia, and paradoxical embolization.19 Pulmonary AVMs in which the feeding artery is 3 mm or more in diameter require treatment.
Embolization therapy, in which the AVM is occluded angiographically, is considered a first-line treatment for pulmonary AVM, with a procedural success rate (defined as involution of the AVM) of 97%.20 Embolization therapy allows patients to avoid major surgery, with its potential complications, and it has a shorter recovery time.
Surgical procedures such as excision, vascular ligation, or lobectomy can be considered if the lesion cannot be treated by embolization or if the patient has an anaphylactic allergy to contrast dyes.
This patient had no clinical signs of impending respiratory failure requiring elective intubation.
Since he was experiencing symptoms, there is no role for observation in this case.
Back to our patient
An interventional radiologist was consulted, and the patient underwent bilateral pulmonary artery angiography with successful coil embolization of his large left-upper-lobe AVM. He was weaned off oxygen and had no further spells of generalized weakness and paresthesias.
Given his father’s history of recurrent epistaxis and oral telangiectases, the patient asks about the risk of his children acquiring this disease.
GENETICS OF HEREDITARY HEMORRHAGIC TELANGIECTASIA
4. Which of the following is the inheritance pattern for HHT?
- Autosomal dominant
- Autosomal recessive
- Maternal inheritance
- X-linked recessive
The inheritance pattern is autosomal dominant with variable expression and penetrance. At least four different mutations have been identified in genes on chromosomes 9 and 12 that result in abnormal vascular malformations.21–24 The other modes of inheritance have not been described in HHT.
RECOMMENDATIONS FOR OUR PATIENT
5. Which of the following is not recommended for our patient?
- Consideration of genetic testing
- Consideration of screening of first-degree relatives
- Dental prophylaxis
- Scuba diving
Genetic testing. The molecular diagnosis of HHT is primarily based on sequencing of the entire coding regions of the ENG and ALK1 genes on chromosomes 9 and 12, respectively. The interpretation of these results is quite complex. The clinical genetics laboratories in North America that currently offer molecular diagnostic testing for HHT recommend that testing be coordinated and ordered through a center that specializes in this disease or by a genetics professional. Testing of the index case is performed to confirm the clinical diagnosis and also to determine if genetic testing will be possible in at-risk relatives. Further genetic testing should be pursued in at-risk family members only if the index case has a positive result.25
Screening of relatives. Given that HHT is an autosomal dominant disease, the current practice is to offer molecular genetic screening early in life for first-degree relatives.25,26 The external signs such as telangiectases and nosebleeds may not manifest until the second or third decade of life. However, AVMs in the brain, spinal cord, lungs, and liver are usually congenital and may present suddenly and with serious complications, even in childhood.
Dental prophylaxis. People with HHT and a pulmonary AVM are at risk of bacteremia and consequent brain abscesses after dental procedures. Antibiotic prophylaxis is therefore highly recommended.27
One sport to avoid. There have been several case reports of paradoxical air emboli occurring in patients with HHT complicated by a pulmonary AVM. Hsu et al28 reported a 31-year-old man with an undiagnosed large pulmonary AVM and HHT who became comatose with diffuse bilateral hemispheric brain swelling on head CT after scuba diving, due to air embolism.
The HHT Foundation International recommends that people with this disease avoid scuba diving (the only sport to be avoided) owing to the risk of air emboli from small lung AVMs. It also recommends that patients alert health care providers about their risk of air embolism whenever intravenous access is being established.
Back to our patient
The patient met with a geneticist, and blood was collected for genetic testing before he was sent home. Additionally, the need to screen his first-degree relatives was thoroughly discussed. Four days after discharge he returned to work, and his spells have not recurred. He has a follow-up appointment scheduled with a pulmonologist specializing in this disease for the results of genetic testing and for continued management.
TAKE-HOME POINTS
- The diagnosis of HHT is based on the following four clinical criteria: spontaneous or recurrent epistaxis, multiple mucocutaneous telangiectases, visceral involvement (eg, cerebral, pulmonary, or gastrointestinal AVM), and a first-degree relative with this disease.
- The diagnosis may be confirmed with genetic testing.
- The diagnosis may be underreported, given the wide spectrum of disease presentation, from inconsequential epistaxis to massive gastrointestinal bleeding.
- HHT is autosomal dominant, and therefore all first-degree relatives should be screened.
- Krumholz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2007; 69:1996–2007.
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest 1999; 104:1343–1351.
- Dakeishi M, Shioya T, Wada Y, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat 2002; 19:140–148.
- Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995; 333:918–924.
- Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91:66–67.
- Gallitelli M, Pasculli G, Fiore T, Carella A, Sabbà C. Emergencies in hereditary haemorrhagic telangiectasia. QJM 2006; 99:15–22.
- Gorelick PB, Hier DB, Caplan LR, Langenberg P. Headache in acute cerebrovascular disease. Neurology 1986; 36:1445–1450.
- Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry 2006; 163:1510–1517.
- Shovlin CL, Letarte M. Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54:714–729.
- AAssar OS, Friedman CM, White RI. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 1991; 101:977–980.
- McAllister KA, Lennon F, Bowles-Biesecker B, et al. Genetic heterogenicity in hereditary haemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet 1994; 31:927–932.
- Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32:291–297.
- Shovlin CL, Jaskson JE, Bamford KB, et al. Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax 2008; 63:259–266.
- Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343:931–936.
- Kolleft MH, Micek ST. Critical care. In:Cooper DH, Krainik AJ, Lubner SJ, Reno HEL, editors. Washington Manual of Medical Therapeutics. 32nd ed. Philadelphia: Lippincott Williams & Wilkins, 2007:224–230.
- Swanson KL, Prakash UB, Stanson AW. Pulmonary arteriovenous fistulas: Mayo Clinic experience: 1872–1997. Mayo Clin Proc 1999; 74:671–680.
- Dines DE, Arms RA, Bernatz PE, Gomes MR. Pulmonary arteriovenous fistulas. Mayo Clinic Proc 1974; 49:460–465.
- Sluiter-Eringa H, Orie NG, Sluiter HJ. Pulmonary arteriovenous fistula: diagnosis and prognosis in noncompliant patients. Am Rev Respir Dis 1969; 100:177–188.
- Dines DE, Seward JB, Bernatz PE. Pulmonary arteriovenous fistula. Mayo Clin Proc 1983; 58:176–181.
- Pollak JS, Saluja S, Thabet A, Henderson KJ, Denbow N, White RI. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radio 2006; 17:35–44.
- Berg JN, Gallion CJ, Stenzel TT, et al. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997; 61:60–67.
- McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary hemorrhagic telangiectasias type 1. Nat Genet 1994; 8:345–351.
- Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase gene in hereditary haemorrhagic telangeictasia type 2. Nat Genet 1996; 13:189–195.
- Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet 2006; 43:97–110.
- Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med 2004; 6:175–191.
- Cohen JH, Faughnan ME, Letarte M, Vandezande K, Kennedy SJ, Krahn MD. Cost comparison of genetic and clinical screening in families with hereditary hemorrhagic telangiectasia. Am J Med Genet A 2005; 137:153–160.
- Shovlin C, Bamfort K, Wray D. Post-NICE 2008: Antibiotic prophylaxis prior to dental procedures for patients with pulmonary arteriovenous malformations (PAVMs) and hereditary haemorrhagic telangiectasia. Br Dent J 2008; 205:531–533.
- Hsu YL, Wang HC, Yang PC. Desbaric air embolism during diving: an unusual complication of Osler-Weber-Rendu disease. Br J Sports Med 2004; 38:E6.
- Krumholz A, Wiebe S, Gronseth G, et al. Practice parameter: evaluating an apparent unprovoked first seizure in adults (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2007; 69:1996–2007.
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest 1999; 104:1343–1351.
- Dakeishi M, Shioya T, Wada Y, et al. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat 2002; 19:140–148.
- Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med 1995; 333:918–924.
- Shovlin CL, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet 2000; 91:66–67.
- Gallitelli M, Pasculli G, Fiore T, Carella A, Sabbà C. Emergencies in hereditary haemorrhagic telangiectasia. QJM 2006; 99:15–22.
- Gorelick PB, Hier DB, Caplan LR, Langenberg P. Headache in acute cerebrovascular disease. Neurology 1986; 36:1445–1450.
- Stonnington CM, Barry JJ, Fisher RS. Conversion disorder. Am J Psychiatry 2006; 163:1510–1517.
- Shovlin CL, Letarte M. Hereditary hemorrhagic telangiectasia and pulmonary arteriovenous malformations: issues in clinical management and review of pathogenic mechanisms. Thorax 1999; 54:714–729.
- AAssar OS, Friedman CM, White RI. The natural history of epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope 1991; 101:977–980.
- McAllister KA, Lennon F, Bowles-Biesecker B, et al. Genetic heterogenicity in hereditary haemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet 1994; 31:927–932.
- Plauchu H, de Chadarevian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet 1989; 32:291–297.
- Shovlin CL, Jaskson JE, Bamford KB, et al. Primary determinants of ischaemic stroke/brain abscess risks are independent of severity of pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia. Thorax 2008; 63:259–266.
- Garcia-Tsao G, Korzenik JR, Young L, et al. Liver disease in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2000; 343:931–936.
- Kolleft MH, Micek ST. Critical care. In:Cooper DH, Krainik AJ, Lubner SJ, Reno HEL, editors. Washington Manual of Medical Therapeutics. 32nd ed. Philadelphia: Lippincott Williams & Wilkins, 2007:224–230.
- Swanson KL, Prakash UB, Stanson AW. Pulmonary arteriovenous fistulas: Mayo Clinic experience: 1872–1997. Mayo Clin Proc 1999; 74:671–680.
- Dines DE, Arms RA, Bernatz PE, Gomes MR. Pulmonary arteriovenous fistulas. Mayo Clinic Proc 1974; 49:460–465.
- Sluiter-Eringa H, Orie NG, Sluiter HJ. Pulmonary arteriovenous fistula: diagnosis and prognosis in noncompliant patients. Am Rev Respir Dis 1969; 100:177–188.
- Dines DE, Seward JB, Bernatz PE. Pulmonary arteriovenous fistula. Mayo Clin Proc 1983; 58:176–181.
- Pollak JS, Saluja S, Thabet A, Henderson KJ, Denbow N, White RI. Clinical and anatomic outcomes after embolotherapy of pulmonary arteriovenous malformations. J Vasc Interv Radio 2006; 17:35–44.
- Berg JN, Gallion CJ, Stenzel TT, et al. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet 1997; 61:60–67.
- McAllister KA, Grogg KM, Johnson DW, et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary hemorrhagic telangiectasias type 1. Nat Genet 1994; 8:345–351.
- Johnson DW, Berg JN, Baldwin MA, et al. Mutations in the activin receptor-like kinase gene in hereditary haemorrhagic telangeictasia type 2. Nat Genet 1996; 13:189–195.
- Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet 2006; 43:97–110.
- Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med 2004; 6:175–191.
- Cohen JH, Faughnan ME, Letarte M, Vandezande K, Kennedy SJ, Krahn MD. Cost comparison of genetic and clinical screening in families with hereditary hemorrhagic telangiectasia. Am J Med Genet A 2005; 137:153–160.
- Shovlin C, Bamfort K, Wray D. Post-NICE 2008: Antibiotic prophylaxis prior to dental procedures for patients with pulmonary arteriovenous malformations (PAVMs) and hereditary haemorrhagic telangiectasia. Br Dent J 2008; 205:531–533.
- Hsu YL, Wang HC, Yang PC. Desbaric air embolism during diving: an unusual complication of Osler-Weber-Rendu disease. Br J Sports Med 2004; 38:E6.