User login
Progressive Papular Eruption on the Face and Groin
The Diagnosis: Xanthoma Disseminatum
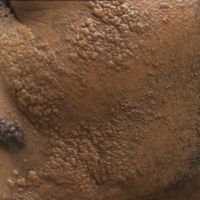
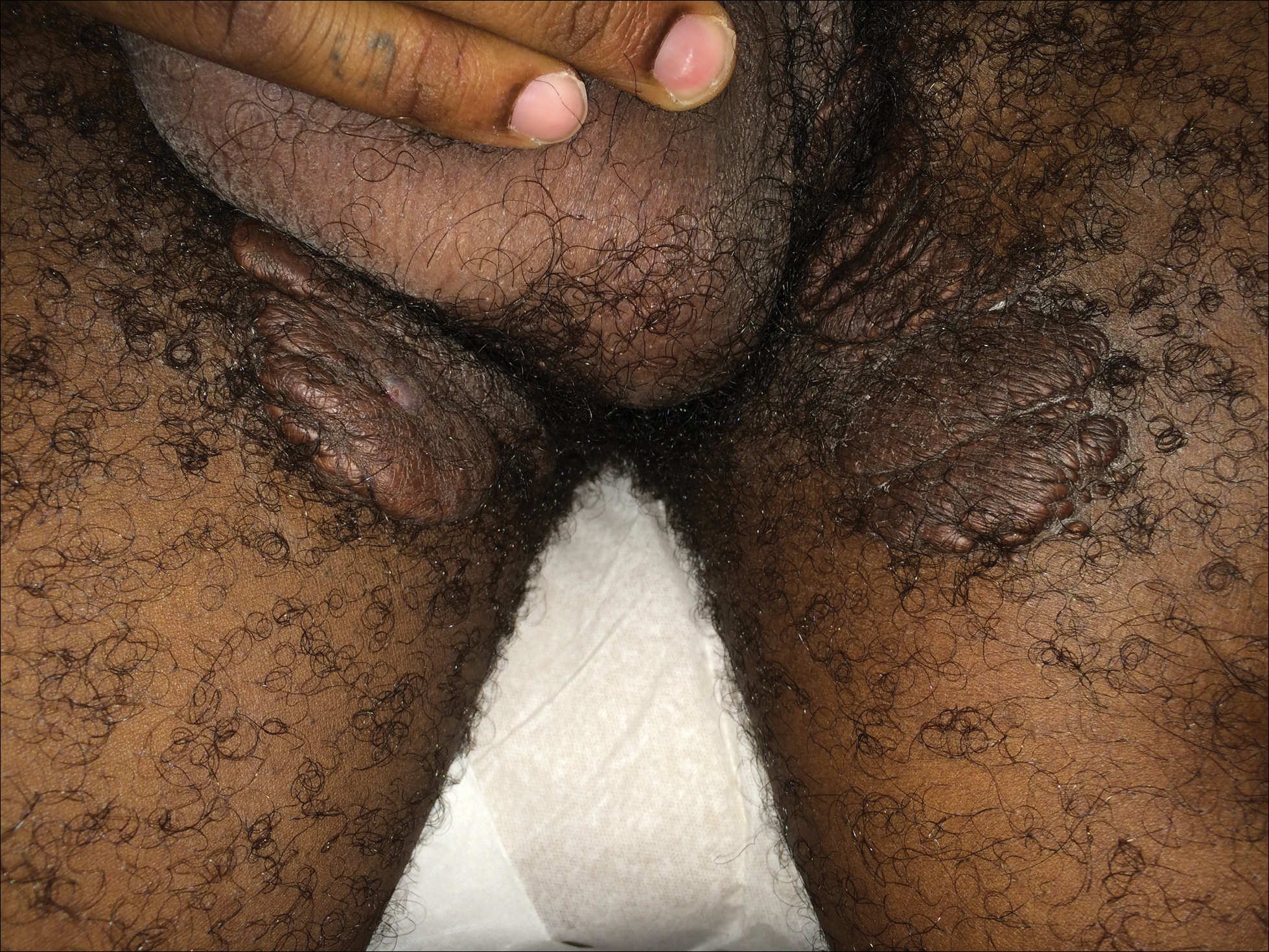
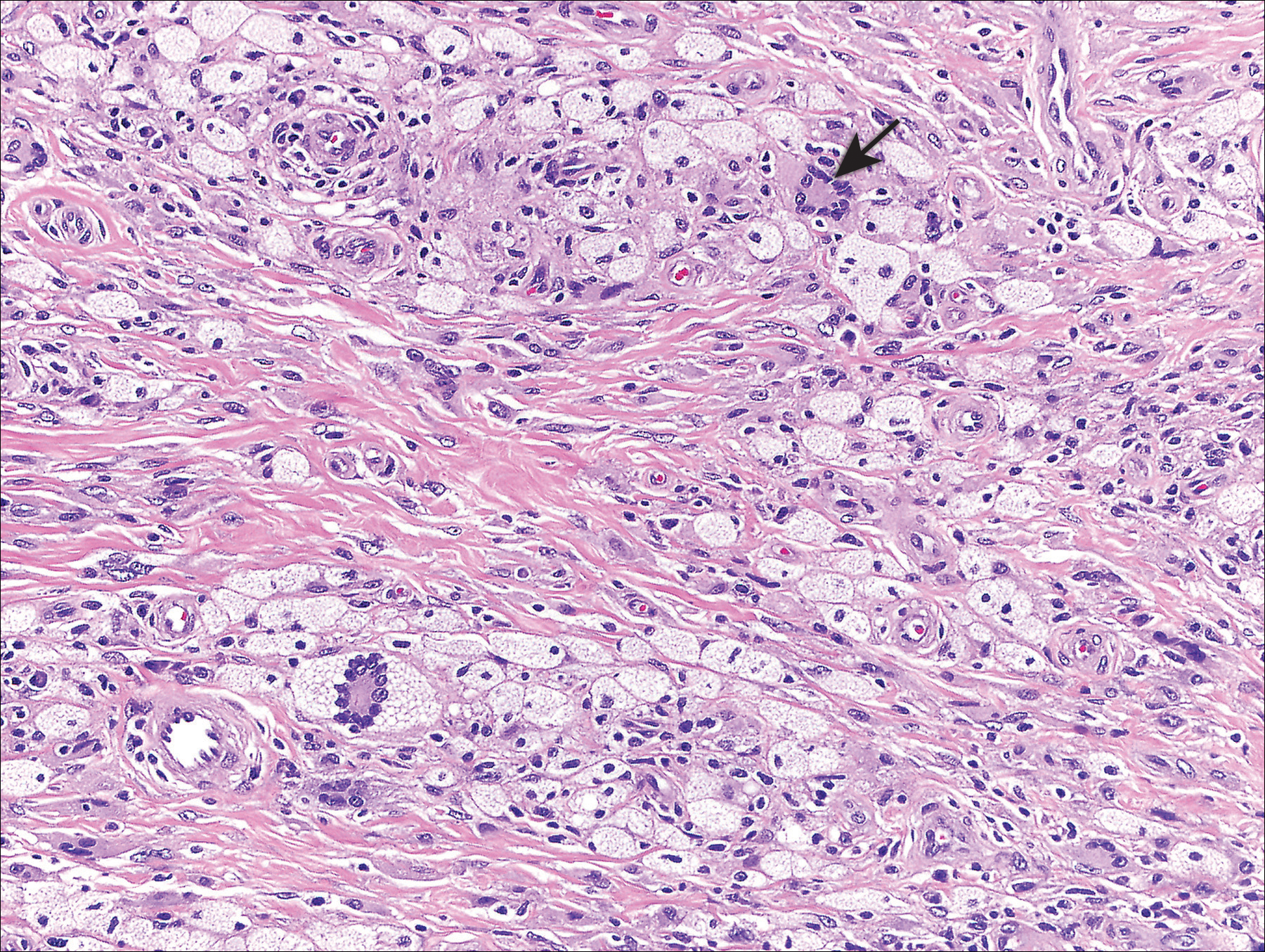
Genital examination revealed approximately 1.5×3-cm soft, yellow-pink plaques extending from the bilateral inguinal folds to the proximal medial thighs (Figure 1). There was no mucosal, axillary, extensor extremity, or palmoplantar involvement. Histopathologic examination of a biopsy from a plaque on the left side of the lower abdomen revealed sheets of foamy histiocytes distributed throughout a fibrotic dermis. Both mononucleated and multinucleated histiocytes were present, including many Touton giant cells (Figure 2). A patchy infiltrate of lymphocytes and rare eosinophils also was noted. The histiocytes labeled with factor XIIIa but not with S-100. Laboratory tests were performed with the following pertinent findings: low-density lipoprotein, 150 mg/dL (reference range, <130 mg/dL); high-density lipoprotein, 30 mg/dL (reference range, >40 mg/dL). Total cholesterol and triglyceride levels were within reference range, and complete blood cell count and basic metabolic panel were normal.
Xanthoma disseminatum (XD)(also known as Montgomery syndrome) is a rare, nonfamilial, normolipemic non-Langerhans cell histiocytosis characterized by extensive lipid deposition in the skin, mucous membranes, and internal organs. The pathogenesis of XD is poorly understood, but it may represent a macrophage-mediated reactive process triggered by superantigens.1
Xanthoma disseminatum most commonly affects males aged 5 to 25 years.2 Clinically, it is characterized by red-brown to yellowish papules and plaques symmetrically distributed over the eyelids, trunk, face, and proximal extremities. There is a predilection for involvement of flexural and intertriginous surfaces and tendency for extension along Langer lines. Extracutaneous involvement can be a notable cause of morbidity and mortality, underscoring the importance of distinguishing XD from other clinically similar xanthomatoses. Mucous membrane involvement occurs in 40% to 60% of patients.3 The oropharynx, larynx, and corneal and conjunctival membranes are most commonly affected, resulting in dysphagia, dysphonia or dyspnea, and visual impairment, respectively. Symptoms of internal organ involvement can be manifold, including pain or limited mobility secondary to osteolytic bone lesions or muscle or synovial membrane involvement, as well as seizures, strabismus, and cerebellar ataxia due to central nervous system lesions.2-4 Approximately 40% of patients develop diabetes insipidus secondary to involvement of the pituitary meninges.3
The differential diagnosis of XD includes juvenile xanthogranuloma, papular xanthomas, eruptive xanthomas, generalized eruptive histiocytosis, progressive nodular histiocytosis, multicentric reticulohistiocytosis, eruptive syringomas, sarcoidosis, and Langerhans cell histiocytosis; the latter should be considered, especially when there is concomitant diabetes insipidus.5 Laboratory studies typically are unremarkable. Although the majority of patients are normolipemic, rates of hyperlipemia within this group are comparable to the general population, occasionally rendering it difficult for the clinician to distinguish XD from hyperlipemic xanthomatoses. As such, diagnosis and differentiation from other xanthomatous processes rests on clinicopathological correlation. Histopathology reveals dermal collections of histiocytes, some with foamy cytoplasm, that range in appearance from spindled to scalloped to Touton-like. Early histopathology demonstrates scalloped macrophages with few foamy cells; a mixture of foamy cells, scalloped cells, inflammatory cells, and Touton and foreign body giant cells is characteristic of late lesions. Immunohistochemistry stains positive for non-Langerhans cell surface markers CD68 and factor XIIIa. Electron microscopy demonstrates dense and myeloid bodies, cholesterol crystals, and lipid vacuoles.5
Three subtypes of XD have been described based on the distinct clinical courses that have been observed in patients: a common, persistent, cutaneous form; a self-limited form with spontaneous resolution; and a progressive subtype with internal organ involvement. No consistently efficacious therapies have been identified, but isolated case reports attest to the efficacy of various agents, including azathioprine, clofibrate, cyclophosphamide, glucocorticoids, chlorambucil, and combination or monotherapy with lipid-lowering agents.3,5,6 Surgical resection, cryotherapy, radiotherapy, and CO2 laser therapy may offer some temporary benefit but do not alter the typically relapsing course of the disease.7,8 Remission and long-term control of lesions was reported with use of 2-chlorodeoxyadenosine, a purine nucleoside analogue, for 5 of 8 patients in a case series.3
- Zelger B, Cerio R, Orchard G, et al. Histologic and immunohistochemical study comparing xanthoma disseminatum and histiocytosis X. Arch Dermatol. 1992;128:1207-1212.
- Mahajan V, Sharma A, Chauhan P, et al. Xanthoma disseminatum: a red herring xanthomatosis. Indian J Dermatol Venereol Leprol. 2013;79:253-254.
- Khezri F, Gibson LE, Tefferi A. Xanthoma disseminatum: effective therapy with 2-chlorodeoxyadenosine in a case series. Arch Dermatol. 2011;147:459-464.
- Weiss N, Keller C. Xanthoma disseminatum: a rare normolipemic xanthomatosis. Clin Investig. 1993;71:233-238.
- Park HY, Cho DH, Joe DH, et al. A case of xanthoma disseminatum with spontaneous resolution over 10 years: review of the literature on long-term follow-up [published online May 26, 2011]. Dermatology. 2011;222:236-243.
- Kim SM, Waters P, Vincent A, et al. Sjogren's syndrome myelopathy: spinal cord involvement in Sjogren's syndrome might be a manifestation of neuromyelitis optica. Mult Scler. 2009;15:1062-1068.
- Eisendle K, Linder D, Ratzinger G, et al. Inflammation and lipid accumulation in xanthoma disseminatum: therapeutic considerations. J Am Acad Dermatol. 2008;58(2 suppl):S47-S49.
- Kim JY, Jung HD, Choe YS, et al. A case of xanthoma disseminatum accentuating over the eyelids. Ann Dermatol. 2010;22:353-357.
The Diagnosis: Xanthoma Disseminatum
Genital examination revealed approximately 1.5×3-cm soft, yellow-pink plaques extending from the bilateral inguinal folds to the proximal medial thighs (Figure 1). There was no mucosal, axillary, extensor extremity, or palmoplantar involvement. Histopathologic examination of a biopsy from a plaque on the left side of the lower abdomen revealed sheets of foamy histiocytes distributed throughout a fibrotic dermis. Both mononucleated and multinucleated histiocytes were present, including many Touton giant cells (Figure 2). A patchy infiltrate of lymphocytes and rare eosinophils also was noted. The histiocytes labeled with factor XIIIa but not with S-100. Laboratory tests were performed with the following pertinent findings: low-density lipoprotein, 150 mg/dL (reference range, <130 mg/dL); high-density lipoprotein, 30 mg/dL (reference range, >40 mg/dL). Total cholesterol and triglyceride levels were within reference range, and complete blood cell count and basic metabolic panel were normal.
Xanthoma disseminatum (XD)(also known as Montgomery syndrome) is a rare, nonfamilial, normolipemic non-Langerhans cell histiocytosis characterized by extensive lipid deposition in the skin, mucous membranes, and internal organs. The pathogenesis of XD is poorly understood, but it may represent a macrophage-mediated reactive process triggered by superantigens.1
Xanthoma disseminatum most commonly affects males aged 5 to 25 years.2 Clinically, it is characterized by red-brown to yellowish papules and plaques symmetrically distributed over the eyelids, trunk, face, and proximal extremities. There is a predilection for involvement of flexural and intertriginous surfaces and tendency for extension along Langer lines. Extracutaneous involvement can be a notable cause of morbidity and mortality, underscoring the importance of distinguishing XD from other clinically similar xanthomatoses. Mucous membrane involvement occurs in 40% to 60% of patients.3 The oropharynx, larynx, and corneal and conjunctival membranes are most commonly affected, resulting in dysphagia, dysphonia or dyspnea, and visual impairment, respectively. Symptoms of internal organ involvement can be manifold, including pain or limited mobility secondary to osteolytic bone lesions or muscle or synovial membrane involvement, as well as seizures, strabismus, and cerebellar ataxia due to central nervous system lesions.2-4 Approximately 40% of patients develop diabetes insipidus secondary to involvement of the pituitary meninges.3
The differential diagnosis of XD includes juvenile xanthogranuloma, papular xanthomas, eruptive xanthomas, generalized eruptive histiocytosis, progressive nodular histiocytosis, multicentric reticulohistiocytosis, eruptive syringomas, sarcoidosis, and Langerhans cell histiocytosis; the latter should be considered, especially when there is concomitant diabetes insipidus.5 Laboratory studies typically are unremarkable. Although the majority of patients are normolipemic, rates of hyperlipemia within this group are comparable to the general population, occasionally rendering it difficult for the clinician to distinguish XD from hyperlipemic xanthomatoses. As such, diagnosis and differentiation from other xanthomatous processes rests on clinicopathological correlation. Histopathology reveals dermal collections of histiocytes, some with foamy cytoplasm, that range in appearance from spindled to scalloped to Touton-like. Early histopathology demonstrates scalloped macrophages with few foamy cells; a mixture of foamy cells, scalloped cells, inflammatory cells, and Touton and foreign body giant cells is characteristic of late lesions. Immunohistochemistry stains positive for non-Langerhans cell surface markers CD68 and factor XIIIa. Electron microscopy demonstrates dense and myeloid bodies, cholesterol crystals, and lipid vacuoles.5
Three subtypes of XD have been described based on the distinct clinical courses that have been observed in patients: a common, persistent, cutaneous form; a self-limited form with spontaneous resolution; and a progressive subtype with internal organ involvement. No consistently efficacious therapies have been identified, but isolated case reports attest to the efficacy of various agents, including azathioprine, clofibrate, cyclophosphamide, glucocorticoids, chlorambucil, and combination or monotherapy with lipid-lowering agents.3,5,6 Surgical resection, cryotherapy, radiotherapy, and CO2 laser therapy may offer some temporary benefit but do not alter the typically relapsing course of the disease.7,8 Remission and long-term control of lesions was reported with use of 2-chlorodeoxyadenosine, a purine nucleoside analogue, for 5 of 8 patients in a case series.3
The Diagnosis: Xanthoma Disseminatum
Genital examination revealed approximately 1.5×3-cm soft, yellow-pink plaques extending from the bilateral inguinal folds to the proximal medial thighs (Figure 1). There was no mucosal, axillary, extensor extremity, or palmoplantar involvement. Histopathologic examination of a biopsy from a plaque on the left side of the lower abdomen revealed sheets of foamy histiocytes distributed throughout a fibrotic dermis. Both mononucleated and multinucleated histiocytes were present, including many Touton giant cells (Figure 2). A patchy infiltrate of lymphocytes and rare eosinophils also was noted. The histiocytes labeled with factor XIIIa but not with S-100. Laboratory tests were performed with the following pertinent findings: low-density lipoprotein, 150 mg/dL (reference range, <130 mg/dL); high-density lipoprotein, 30 mg/dL (reference range, >40 mg/dL). Total cholesterol and triglyceride levels were within reference range, and complete blood cell count and basic metabolic panel were normal.
Xanthoma disseminatum (XD)(also known as Montgomery syndrome) is a rare, nonfamilial, normolipemic non-Langerhans cell histiocytosis characterized by extensive lipid deposition in the skin, mucous membranes, and internal organs. The pathogenesis of XD is poorly understood, but it may represent a macrophage-mediated reactive process triggered by superantigens.1
Xanthoma disseminatum most commonly affects males aged 5 to 25 years.2 Clinically, it is characterized by red-brown to yellowish papules and plaques symmetrically distributed over the eyelids, trunk, face, and proximal extremities. There is a predilection for involvement of flexural and intertriginous surfaces and tendency for extension along Langer lines. Extracutaneous involvement can be a notable cause of morbidity and mortality, underscoring the importance of distinguishing XD from other clinically similar xanthomatoses. Mucous membrane involvement occurs in 40% to 60% of patients.3 The oropharynx, larynx, and corneal and conjunctival membranes are most commonly affected, resulting in dysphagia, dysphonia or dyspnea, and visual impairment, respectively. Symptoms of internal organ involvement can be manifold, including pain or limited mobility secondary to osteolytic bone lesions or muscle or synovial membrane involvement, as well as seizures, strabismus, and cerebellar ataxia due to central nervous system lesions.2-4 Approximately 40% of patients develop diabetes insipidus secondary to involvement of the pituitary meninges.3
The differential diagnosis of XD includes juvenile xanthogranuloma, papular xanthomas, eruptive xanthomas, generalized eruptive histiocytosis, progressive nodular histiocytosis, multicentric reticulohistiocytosis, eruptive syringomas, sarcoidosis, and Langerhans cell histiocytosis; the latter should be considered, especially when there is concomitant diabetes insipidus.5 Laboratory studies typically are unremarkable. Although the majority of patients are normolipemic, rates of hyperlipemia within this group are comparable to the general population, occasionally rendering it difficult for the clinician to distinguish XD from hyperlipemic xanthomatoses. As such, diagnosis and differentiation from other xanthomatous processes rests on clinicopathological correlation. Histopathology reveals dermal collections of histiocytes, some with foamy cytoplasm, that range in appearance from spindled to scalloped to Touton-like. Early histopathology demonstrates scalloped macrophages with few foamy cells; a mixture of foamy cells, scalloped cells, inflammatory cells, and Touton and foreign body giant cells is characteristic of late lesions. Immunohistochemistry stains positive for non-Langerhans cell surface markers CD68 and factor XIIIa. Electron microscopy demonstrates dense and myeloid bodies, cholesterol crystals, and lipid vacuoles.5
Three subtypes of XD have been described based on the distinct clinical courses that have been observed in patients: a common, persistent, cutaneous form; a self-limited form with spontaneous resolution; and a progressive subtype with internal organ involvement. No consistently efficacious therapies have been identified, but isolated case reports attest to the efficacy of various agents, including azathioprine, clofibrate, cyclophosphamide, glucocorticoids, chlorambucil, and combination or monotherapy with lipid-lowering agents.3,5,6 Surgical resection, cryotherapy, radiotherapy, and CO2 laser therapy may offer some temporary benefit but do not alter the typically relapsing course of the disease.7,8 Remission and long-term control of lesions was reported with use of 2-chlorodeoxyadenosine, a purine nucleoside analogue, for 5 of 8 patients in a case series.3
- Zelger B, Cerio R, Orchard G, et al. Histologic and immunohistochemical study comparing xanthoma disseminatum and histiocytosis X. Arch Dermatol. 1992;128:1207-1212.
- Mahajan V, Sharma A, Chauhan P, et al. Xanthoma disseminatum: a red herring xanthomatosis. Indian J Dermatol Venereol Leprol. 2013;79:253-254.
- Khezri F, Gibson LE, Tefferi A. Xanthoma disseminatum: effective therapy with 2-chlorodeoxyadenosine in a case series. Arch Dermatol. 2011;147:459-464.
- Weiss N, Keller C. Xanthoma disseminatum: a rare normolipemic xanthomatosis. Clin Investig. 1993;71:233-238.
- Park HY, Cho DH, Joe DH, et al. A case of xanthoma disseminatum with spontaneous resolution over 10 years: review of the literature on long-term follow-up [published online May 26, 2011]. Dermatology. 2011;222:236-243.
- Kim SM, Waters P, Vincent A, et al. Sjogren's syndrome myelopathy: spinal cord involvement in Sjogren's syndrome might be a manifestation of neuromyelitis optica. Mult Scler. 2009;15:1062-1068.
- Eisendle K, Linder D, Ratzinger G, et al. Inflammation and lipid accumulation in xanthoma disseminatum: therapeutic considerations. J Am Acad Dermatol. 2008;58(2 suppl):S47-S49.
- Kim JY, Jung HD, Choe YS, et al. A case of xanthoma disseminatum accentuating over the eyelids. Ann Dermatol. 2010;22:353-357.
- Zelger B, Cerio R, Orchard G, et al. Histologic and immunohistochemical study comparing xanthoma disseminatum and histiocytosis X. Arch Dermatol. 1992;128:1207-1212.
- Mahajan V, Sharma A, Chauhan P, et al. Xanthoma disseminatum: a red herring xanthomatosis. Indian J Dermatol Venereol Leprol. 2013;79:253-254.
- Khezri F, Gibson LE, Tefferi A. Xanthoma disseminatum: effective therapy with 2-chlorodeoxyadenosine in a case series. Arch Dermatol. 2011;147:459-464.
- Weiss N, Keller C. Xanthoma disseminatum: a rare normolipemic xanthomatosis. Clin Investig. 1993;71:233-238.
- Park HY, Cho DH, Joe DH, et al. A case of xanthoma disseminatum with spontaneous resolution over 10 years: review of the literature on long-term follow-up [published online May 26, 2011]. Dermatology. 2011;222:236-243.
- Kim SM, Waters P, Vincent A, et al. Sjogren's syndrome myelopathy: spinal cord involvement in Sjogren's syndrome might be a manifestation of neuromyelitis optica. Mult Scler. 2009;15:1062-1068.
- Eisendle K, Linder D, Ratzinger G, et al. Inflammation and lipid accumulation in xanthoma disseminatum: therapeutic considerations. J Am Acad Dermatol. 2008;58(2 suppl):S47-S49.
- Kim JY, Jung HD, Choe YS, et al. A case of xanthoma disseminatum accentuating over the eyelids. Ann Dermatol. 2010;22:353-357.

A 28-year-old man presented for evaluation of numerous papules on the face and groin that first appeared in adolescence and had been increasing in size and number over the last several years. The lesions occasionally were pruritic. Review of systems was noncontributory. His medical history was notable for asthma, and there were no affected family members. Physical examination revealed numerous symmetrically distributed, soft, yellow-pink, 1- to 5-mm papules coalescing into plaques on the bilateral malar cheeks extending to the medial canthi and the maxillary, mandibular, zygomatic, and submental regions, as well as the bilateral external auditory meatus.