User login
IV bevacizumab improves severe bleeding in HHT
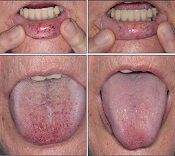
Intravenous (IV) bevacizumab “dramatically” improves severe bleeding associated with hereditary hemorrhagic telangiectasia (HHT), according to researchers.
In a retrospective study, HHT patients with severe bleeding had a substantial reduction in nose bleeds and gastrointestinal (GI) bleeding after treatment with IV bevacizumab.
In addition, patients were able to stop or considerably reduce red blood cell (RBC) transfusions.
The researchers therefore believe that IV bevacizumab should be considered as a first-line therapy for the treatment of refractory bleeding in patients with severe HHT.
The team detailed this research and in Mayo Clinic Proceedings alongside a related editorial.
“Some HHT patients suffer from severe epistaxis and gastrointestinal bleeding, which can result in severe anemia and years of blood transfusions,” said study author Vivek N. Iyer, MD, of the Mayo Clinic in Rochester, Minnesota.
“Both problems also appear to sometimes worsen with age. In some patients, both epistaxis and GI bleeding can become refractory/resistant to existing treatment options, leaving patients severely anemic and dependent on iron infusions or blood transfusions. Quality of life is very poor in these cases.”
With this in mind, Dr Iyer and his colleagues analyzed the records of 34 patients who were treated with IV bevacizumab for severe HHT-related bleeding from June 2013 through January 2017.
Patient characteristics
Patients had a median age of 63 (range, 57 to 72). Sixty-two percent of patients were female.
The primary source of bleeding was epistaxis in 15 patients, GI bleeding in 4 patients, and combined epistaxis and GI bleeding in 15 patients.
Prior epistaxis treatments included potassium-titanyl-phosphate/other laser procedures (62%), sclerotherapy (26%), endovascular angiographic embolization (21%), septodermoplasty (24%), subcutaneous bevacizumab injections (21%), and bevacizumab nasal spray (29%).
Seventy-one percent of patients underwent upper endoscopy (100% of these with telangiectasias), and 53% underwent colonoscopy (56% of these with telangiectasias).
Forty-one percent of patients had IV iron supplementation in the past 6 months.
Twenty-eight patients had received RBC transfusions. Sixteen patients were transfusion-dependent and had received a median of 75 transfusions before starting treatment with bevacizumab. The median duration of transfusion dependence was 6 years.
Treatment
The typical initial dosing cycle of bevacizumab consisted of 8 doses (4 doses each administered 2 weeks apart, followed by 4 doses each administered 1 month apart) over a period of around 22 weeks.
Further maintenance doses after the initial dosing cycle were individualized in each patient.
At last follow-up, 3 patients were still receiving bevacizumab from the initial dosing protocol. For the 31 patients who had completed the first dosing cycle, the median duration of follow-up was 13.6 months.
Eighteen patients required at least 1 top-up dose of IV bevacizumab because of worsening bleeding and/or anemia.
Efficacy
The median follow-up was 17.6 months (range, 3 to 42.5 months).
An Epistaxis Severity Score (ESS) questionnaire was used to assess the severity of nose bleeds both at the beginning of the study and after starting bevacizumab.
One month after starting treatment, there was a significant reduction in ESS scores (P<0.001). This improvement was maintained after patients completed the initial treatment cycle.
The median ESS score was 6.5 at baseline, 3.3 at 1 month, 4.0 at 3 months, 2.3 at the end of the first cycle, 2.0 at 1 to 3 months after the first cycle, 3.2 at 4 to 6 months, and 2.8 at 7 to 12 months.
GI bleeding also improved, with resolution or improvement of anemia in all 19 patients with this condition.
There was a reduction in RBC transfusions as well. The proportion of patients receiving transfusions was 53% in the 6 months before IV bevacizumab, 15% in the 1 to 3 months after starting IV bevacizumab, 14% at 4 to 6 months, 8% at 7 to 9 months, and 9% at 9 to 12 months.
Eighty-eight percent (n=14) of the transfusion-dependent patients had received a transfusion in the 6 months prior to starting IV bevacizumab. This compares to 31% (n=5) of patients in the 1 to 3 months after the start of treatment, 29% (n=4) at 4 to 6 months, 14% (n=2) at 7 to 9 months, and 8% (n=1) at 9 to 12 months.
Safety
Four patients had hypertension (HTN). One had pre-existing HTN and had to double the daily dose of lisinopril from 10 mg to 20 mg.
Two HTN patients had to start antihypertensive medications. The fourth patient experienced hypertensive urgency with a temporary decline in renal function. However, the patient was able to resume bevacizumab.
Two patients had infusion-related chills and fever, but premedication with acetaminophen and diphenhydramine prevented these events from recurring.
Three patients died during follow-up. Causes of death were stroke, infective endocarditis (methicillin-sensitive Staphylococcus aureus) with multiple cerebral infarcts, and postoperative respiratory failure (after left atrial appendectomy for paroxysmal atrial fibrillation).
None of these deaths were directly linked to bevacizumab.
“This study provides good-quality evidence for the excellent efficacy and safety of intravenous bevacizumab in the treatment of these patients,” Dr Iyer said. “Intravenous bevacizumab should be considered as a standard, first-line treatment option for HHT patients with severe bleeding and transfusion-dependent anemia.” ![]()
Intravenous (IV) bevacizumab “dramatically” improves severe bleeding associated with hereditary hemorrhagic telangiectasia (HHT), according to researchers.
In a retrospective study, HHT patients with severe bleeding had a substantial reduction in nose bleeds and gastrointestinal (GI) bleeding after treatment with IV bevacizumab.
In addition, patients were able to stop or considerably reduce red blood cell (RBC) transfusions.
The researchers therefore believe that IV bevacizumab should be considered as a first-line therapy for the treatment of refractory bleeding in patients with severe HHT.
The team detailed this research and in Mayo Clinic Proceedings alongside a related editorial.
“Some HHT patients suffer from severe epistaxis and gastrointestinal bleeding, which can result in severe anemia and years of blood transfusions,” said study author Vivek N. Iyer, MD, of the Mayo Clinic in Rochester, Minnesota.
“Both problems also appear to sometimes worsen with age. In some patients, both epistaxis and GI bleeding can become refractory/resistant to existing treatment options, leaving patients severely anemic and dependent on iron infusions or blood transfusions. Quality of life is very poor in these cases.”
With this in mind, Dr Iyer and his colleagues analyzed the records of 34 patients who were treated with IV bevacizumab for severe HHT-related bleeding from June 2013 through January 2017.
Patient characteristics
Patients had a median age of 63 (range, 57 to 72). Sixty-two percent of patients were female.
The primary source of bleeding was epistaxis in 15 patients, GI bleeding in 4 patients, and combined epistaxis and GI bleeding in 15 patients.
Prior epistaxis treatments included potassium-titanyl-phosphate/other laser procedures (62%), sclerotherapy (26%), endovascular angiographic embolization (21%), septodermoplasty (24%), subcutaneous bevacizumab injections (21%), and bevacizumab nasal spray (29%).
Seventy-one percent of patients underwent upper endoscopy (100% of these with telangiectasias), and 53% underwent colonoscopy (56% of these with telangiectasias).
Forty-one percent of patients had IV iron supplementation in the past 6 months.
Twenty-eight patients had received RBC transfusions. Sixteen patients were transfusion-dependent and had received a median of 75 transfusions before starting treatment with bevacizumab. The median duration of transfusion dependence was 6 years.
Treatment
The typical initial dosing cycle of bevacizumab consisted of 8 doses (4 doses each administered 2 weeks apart, followed by 4 doses each administered 1 month apart) over a period of around 22 weeks.
Further maintenance doses after the initial dosing cycle were individualized in each patient.
At last follow-up, 3 patients were still receiving bevacizumab from the initial dosing protocol. For the 31 patients who had completed the first dosing cycle, the median duration of follow-up was 13.6 months.
Eighteen patients required at least 1 top-up dose of IV bevacizumab because of worsening bleeding and/or anemia.
Efficacy
The median follow-up was 17.6 months (range, 3 to 42.5 months).
An Epistaxis Severity Score (ESS) questionnaire was used to assess the severity of nose bleeds both at the beginning of the study and after starting bevacizumab.
One month after starting treatment, there was a significant reduction in ESS scores (P<0.001). This improvement was maintained after patients completed the initial treatment cycle.
The median ESS score was 6.5 at baseline, 3.3 at 1 month, 4.0 at 3 months, 2.3 at the end of the first cycle, 2.0 at 1 to 3 months after the first cycle, 3.2 at 4 to 6 months, and 2.8 at 7 to 12 months.
GI bleeding also improved, with resolution or improvement of anemia in all 19 patients with this condition.
There was a reduction in RBC transfusions as well. The proportion of patients receiving transfusions was 53% in the 6 months before IV bevacizumab, 15% in the 1 to 3 months after starting IV bevacizumab, 14% at 4 to 6 months, 8% at 7 to 9 months, and 9% at 9 to 12 months.
Eighty-eight percent (n=14) of the transfusion-dependent patients had received a transfusion in the 6 months prior to starting IV bevacizumab. This compares to 31% (n=5) of patients in the 1 to 3 months after the start of treatment, 29% (n=4) at 4 to 6 months, 14% (n=2) at 7 to 9 months, and 8% (n=1) at 9 to 12 months.
Safety
Four patients had hypertension (HTN). One had pre-existing HTN and had to double the daily dose of lisinopril from 10 mg to 20 mg.
Two HTN patients had to start antihypertensive medications. The fourth patient experienced hypertensive urgency with a temporary decline in renal function. However, the patient was able to resume bevacizumab.
Two patients had infusion-related chills and fever, but premedication with acetaminophen and diphenhydramine prevented these events from recurring.
Three patients died during follow-up. Causes of death were stroke, infective endocarditis (methicillin-sensitive Staphylococcus aureus) with multiple cerebral infarcts, and postoperative respiratory failure (after left atrial appendectomy for paroxysmal atrial fibrillation).
None of these deaths were directly linked to bevacizumab.
“This study provides good-quality evidence for the excellent efficacy and safety of intravenous bevacizumab in the treatment of these patients,” Dr Iyer said. “Intravenous bevacizumab should be considered as a standard, first-line treatment option for HHT patients with severe bleeding and transfusion-dependent anemia.” ![]()
Intravenous (IV) bevacizumab “dramatically” improves severe bleeding associated with hereditary hemorrhagic telangiectasia (HHT), according to researchers.
In a retrospective study, HHT patients with severe bleeding had a substantial reduction in nose bleeds and gastrointestinal (GI) bleeding after treatment with IV bevacizumab.
In addition, patients were able to stop or considerably reduce red blood cell (RBC) transfusions.
The researchers therefore believe that IV bevacizumab should be considered as a first-line therapy for the treatment of refractory bleeding in patients with severe HHT.
The team detailed this research and in Mayo Clinic Proceedings alongside a related editorial.
“Some HHT patients suffer from severe epistaxis and gastrointestinal bleeding, which can result in severe anemia and years of blood transfusions,” said study author Vivek N. Iyer, MD, of the Mayo Clinic in Rochester, Minnesota.
“Both problems also appear to sometimes worsen with age. In some patients, both epistaxis and GI bleeding can become refractory/resistant to existing treatment options, leaving patients severely anemic and dependent on iron infusions or blood transfusions. Quality of life is very poor in these cases.”
With this in mind, Dr Iyer and his colleagues analyzed the records of 34 patients who were treated with IV bevacizumab for severe HHT-related bleeding from June 2013 through January 2017.
Patient characteristics
Patients had a median age of 63 (range, 57 to 72). Sixty-two percent of patients were female.
The primary source of bleeding was epistaxis in 15 patients, GI bleeding in 4 patients, and combined epistaxis and GI bleeding in 15 patients.
Prior epistaxis treatments included potassium-titanyl-phosphate/other laser procedures (62%), sclerotherapy (26%), endovascular angiographic embolization (21%), septodermoplasty (24%), subcutaneous bevacizumab injections (21%), and bevacizumab nasal spray (29%).
Seventy-one percent of patients underwent upper endoscopy (100% of these with telangiectasias), and 53% underwent colonoscopy (56% of these with telangiectasias).
Forty-one percent of patients had IV iron supplementation in the past 6 months.
Twenty-eight patients had received RBC transfusions. Sixteen patients were transfusion-dependent and had received a median of 75 transfusions before starting treatment with bevacizumab. The median duration of transfusion dependence was 6 years.
Treatment
The typical initial dosing cycle of bevacizumab consisted of 8 doses (4 doses each administered 2 weeks apart, followed by 4 doses each administered 1 month apart) over a period of around 22 weeks.
Further maintenance doses after the initial dosing cycle were individualized in each patient.
At last follow-up, 3 patients were still receiving bevacizumab from the initial dosing protocol. For the 31 patients who had completed the first dosing cycle, the median duration of follow-up was 13.6 months.
Eighteen patients required at least 1 top-up dose of IV bevacizumab because of worsening bleeding and/or anemia.
Efficacy
The median follow-up was 17.6 months (range, 3 to 42.5 months).
An Epistaxis Severity Score (ESS) questionnaire was used to assess the severity of nose bleeds both at the beginning of the study and after starting bevacizumab.
One month after starting treatment, there was a significant reduction in ESS scores (P<0.001). This improvement was maintained after patients completed the initial treatment cycle.
The median ESS score was 6.5 at baseline, 3.3 at 1 month, 4.0 at 3 months, 2.3 at the end of the first cycle, 2.0 at 1 to 3 months after the first cycle, 3.2 at 4 to 6 months, and 2.8 at 7 to 12 months.
GI bleeding also improved, with resolution or improvement of anemia in all 19 patients with this condition.
There was a reduction in RBC transfusions as well. The proportion of patients receiving transfusions was 53% in the 6 months before IV bevacizumab, 15% in the 1 to 3 months after starting IV bevacizumab, 14% at 4 to 6 months, 8% at 7 to 9 months, and 9% at 9 to 12 months.
Eighty-eight percent (n=14) of the transfusion-dependent patients had received a transfusion in the 6 months prior to starting IV bevacizumab. This compares to 31% (n=5) of patients in the 1 to 3 months after the start of treatment, 29% (n=4) at 4 to 6 months, 14% (n=2) at 7 to 9 months, and 8% (n=1) at 9 to 12 months.
Safety
Four patients had hypertension (HTN). One had pre-existing HTN and had to double the daily dose of lisinopril from 10 mg to 20 mg.
Two HTN patients had to start antihypertensive medications. The fourth patient experienced hypertensive urgency with a temporary decline in renal function. However, the patient was able to resume bevacizumab.
Two patients had infusion-related chills and fever, but premedication with acetaminophen and diphenhydramine prevented these events from recurring.
Three patients died during follow-up. Causes of death were stroke, infective endocarditis (methicillin-sensitive Staphylococcus aureus) with multiple cerebral infarcts, and postoperative respiratory failure (after left atrial appendectomy for paroxysmal atrial fibrillation).
None of these deaths were directly linked to bevacizumab.
“This study provides good-quality evidence for the excellent efficacy and safety of intravenous bevacizumab in the treatment of these patients,” Dr Iyer said. “Intravenous bevacizumab should be considered as a standard, first-line treatment option for HHT patients with severe bleeding and transfusion-dependent anemia.” ![]()
Trial protocols redacted by industry sponsors

New research has revealed a lack of public information regarding protocols for industry-sponsored, randomized drug trials in Denmark.
First, researchers found it difficult to obtain protocols for commercially sponsored trials, with some sponsors taking legal action in an attempt to keep protocols private.
When the researchers did receive protocols, many had widespread redactions.
The researchers reported these findings in the Journal of the Royal Society of Medicine.
“We wished to compare the information in the protocols with the information provided to the patients in order to evaluate whether the trials were ethical and necessary and whether essential information about the benefits and the harms of the drugs had been hidden from the patients,” said study author Peter Gøtzsche, MD, director of the Nordic Cochrane Centre in Copenhagen, Denmark.
To that end, Dr Gøtzsche and his colleagues used the Danish Freedom of Information Act to request access to 78 protocols for randomized trials that were approved by a research ethics committee from October 2012 to March 2013.
The researchers said several companies refused to provide protocols and involved their lawyers. In fact, Sanofi-Aventis sued the National Committee on Health Research Ethics but lost.
Three years after this project was started, the researchers were able to obtain all the protocols they had requested. Eight protocols were excluded from analysis because they did not meet the research inclusion criteria.
Seventeen of 34 protocols for commercially sponsored trials were unredacted, compared to 34 of 36 non-commercially sponsored trials.
The researchers said the redactions “were most widespread in those sections of the protocol where there is empirical evidence of substantial problems with the trustworthiness of published drug trials.”
This includes data analysis, the handling of missing data, the detection/analysis of adverse events, the definition of patient outcomes, interim analyses and premature study termination, the sponsor’s access to incoming data while the study is ongoing, ownership of the data, and investigators’ publication rights.
“The amount of redactions in the protocols we received was so vast that it made them rather useless for assessing the ethical justification for the studies and to identify discrepancies with subsequent publications,” Dr Gøtzsche said.
“We could not identify any legitimate rationale for the redactions. The current mistrust in industry-sponsored drug trials can only change if the industry offers unconditional access to its trial protocols and other relevant documents and data.” ![]()
New research has revealed a lack of public information regarding protocols for industry-sponsored, randomized drug trials in Denmark.
First, researchers found it difficult to obtain protocols for commercially sponsored trials, with some sponsors taking legal action in an attempt to keep protocols private.
When the researchers did receive protocols, many had widespread redactions.
The researchers reported these findings in the Journal of the Royal Society of Medicine.
“We wished to compare the information in the protocols with the information provided to the patients in order to evaluate whether the trials were ethical and necessary and whether essential information about the benefits and the harms of the drugs had been hidden from the patients,” said study author Peter Gøtzsche, MD, director of the Nordic Cochrane Centre in Copenhagen, Denmark.
To that end, Dr Gøtzsche and his colleagues used the Danish Freedom of Information Act to request access to 78 protocols for randomized trials that were approved by a research ethics committee from October 2012 to March 2013.
The researchers said several companies refused to provide protocols and involved their lawyers. In fact, Sanofi-Aventis sued the National Committee on Health Research Ethics but lost.
Three years after this project was started, the researchers were able to obtain all the protocols they had requested. Eight protocols were excluded from analysis because they did not meet the research inclusion criteria.
Seventeen of 34 protocols for commercially sponsored trials were unredacted, compared to 34 of 36 non-commercially sponsored trials.
The researchers said the redactions “were most widespread in those sections of the protocol where there is empirical evidence of substantial problems with the trustworthiness of published drug trials.”
This includes data analysis, the handling of missing data, the detection/analysis of adverse events, the definition of patient outcomes, interim analyses and premature study termination, the sponsor’s access to incoming data while the study is ongoing, ownership of the data, and investigators’ publication rights.
“The amount of redactions in the protocols we received was so vast that it made them rather useless for assessing the ethical justification for the studies and to identify discrepancies with subsequent publications,” Dr Gøtzsche said.
“We could not identify any legitimate rationale for the redactions. The current mistrust in industry-sponsored drug trials can only change if the industry offers unconditional access to its trial protocols and other relevant documents and data.” ![]()
New research has revealed a lack of public information regarding protocols for industry-sponsored, randomized drug trials in Denmark.
First, researchers found it difficult to obtain protocols for commercially sponsored trials, with some sponsors taking legal action in an attempt to keep protocols private.
When the researchers did receive protocols, many had widespread redactions.
The researchers reported these findings in the Journal of the Royal Society of Medicine.
“We wished to compare the information in the protocols with the information provided to the patients in order to evaluate whether the trials were ethical and necessary and whether essential information about the benefits and the harms of the drugs had been hidden from the patients,” said study author Peter Gøtzsche, MD, director of the Nordic Cochrane Centre in Copenhagen, Denmark.
To that end, Dr Gøtzsche and his colleagues used the Danish Freedom of Information Act to request access to 78 protocols for randomized trials that were approved by a research ethics committee from October 2012 to March 2013.
The researchers said several companies refused to provide protocols and involved their lawyers. In fact, Sanofi-Aventis sued the National Committee on Health Research Ethics but lost.
Three years after this project was started, the researchers were able to obtain all the protocols they had requested. Eight protocols were excluded from analysis because they did not meet the research inclusion criteria.
Seventeen of 34 protocols for commercially sponsored trials were unredacted, compared to 34 of 36 non-commercially sponsored trials.
The researchers said the redactions “were most widespread in those sections of the protocol where there is empirical evidence of substantial problems with the trustworthiness of published drug trials.”
This includes data analysis, the handling of missing data, the detection/analysis of adverse events, the definition of patient outcomes, interim analyses and premature study termination, the sponsor’s access to incoming data while the study is ongoing, ownership of the data, and investigators’ publication rights.
“The amount of redactions in the protocols we received was so vast that it made them rather useless for assessing the ethical justification for the studies and to identify discrepancies with subsequent publications,” Dr Gøtzsche said.
“We could not identify any legitimate rationale for the redactions. The current mistrust in industry-sponsored drug trials can only change if the industry offers unconditional access to its trial protocols and other relevant documents and data.” ![]()
No need for structured PE assessment, team says

There is no need for a structured algorithm to rule out pulmonary embolism (PE) in patients who visit the emergency department (ED) for syncope, according to researchers.
The team analyzed data on nearly 1.7 million patients who presented to the ED for syncope in 4 different countries.
PE occurred in less than 1% of these patients and in less than 3% of patients who were hospitalized.
The researchers therefore concluded that PE should be considered in such patients, but a systematic protocol for ruling out PE is not necessary.
“Our results are saying that there is no need, so far, for using a structured clinical algorithm to assess pulmonary emboli in all the patients presenting [to the ED with syncope],” said Nicola Montano, MD, PhD, of the University of Milan in Italy.
Dr Montano and his colleagues described these results in JAMA Internal Medicine.
The team noted that, in the PESIT trial, researchers used a standardized algorithm to evaluate PE prevalence in patients hospitalized after a first syncope episode.
The algorithm was based on pretest clinical probability and results of the D-dimer assay. Any patient with positive D-dimer results or high pretest PE probability underwent computed tomography or ventilation perfusion lung scanning.
In this study, the prevalence of PE was 17.3%.
A subsequent study showed a much lower prevalence of PE and deep vein thrombosis in patients hospitalized for syncope. The prevalence of venous thromboembolism (VTE) in this study was 1.4%.
The differences between these studies and the fact that both studies included only hospitalized patients prompted Dr Montano and his colleagues to conduct the current study.
The team set out to determine PE/VTE prevalence in unselected patients with syncope presenting to the ED.
Results
The researchers analyzed administrative data from 5 databases in 4 countries—Canada, Denmark, Italy, and the US—collected from January 1, 2000, through September 30, 2016.
The data included 1,671,944 adults who presented to the ED for syncope. The prevalence of PE at ED or hospital discharge ranged from 0.06% to 0.55%. Among the hospitalized patients only, the prevalence of PE ranged from 0.15% to 2.10%.
At 90 days of follow-up, the prevalence of PE ranged from 0.14% to 0.83% for all patients and from 0.35% to 2.63% for hospitalized patients.
The prevalence of VTE at 90 days ranged from 0.30% to 1.37% for all patients and from 0.75% to 3.86% for hospitalized patients.
The researchers said the main limitation of this study is the use of administrative data because some patients with syncope, PE, or VTE may have been missed. ![]()
There is no need for a structured algorithm to rule out pulmonary embolism (PE) in patients who visit the emergency department (ED) for syncope, according to researchers.
The team analyzed data on nearly 1.7 million patients who presented to the ED for syncope in 4 different countries.
PE occurred in less than 1% of these patients and in less than 3% of patients who were hospitalized.
The researchers therefore concluded that PE should be considered in such patients, but a systematic protocol for ruling out PE is not necessary.
“Our results are saying that there is no need, so far, for using a structured clinical algorithm to assess pulmonary emboli in all the patients presenting [to the ED with syncope],” said Nicola Montano, MD, PhD, of the University of Milan in Italy.
Dr Montano and his colleagues described these results in JAMA Internal Medicine.
The team noted that, in the PESIT trial, researchers used a standardized algorithm to evaluate PE prevalence in patients hospitalized after a first syncope episode.
The algorithm was based on pretest clinical probability and results of the D-dimer assay. Any patient with positive D-dimer results or high pretest PE probability underwent computed tomography or ventilation perfusion lung scanning.
In this study, the prevalence of PE was 17.3%.
A subsequent study showed a much lower prevalence of PE and deep vein thrombosis in patients hospitalized for syncope. The prevalence of venous thromboembolism (VTE) in this study was 1.4%.
The differences between these studies and the fact that both studies included only hospitalized patients prompted Dr Montano and his colleagues to conduct the current study.
The team set out to determine PE/VTE prevalence in unselected patients with syncope presenting to the ED.
Results
The researchers analyzed administrative data from 5 databases in 4 countries—Canada, Denmark, Italy, and the US—collected from January 1, 2000, through September 30, 2016.
The data included 1,671,944 adults who presented to the ED for syncope. The prevalence of PE at ED or hospital discharge ranged from 0.06% to 0.55%. Among the hospitalized patients only, the prevalence of PE ranged from 0.15% to 2.10%.
At 90 days of follow-up, the prevalence of PE ranged from 0.14% to 0.83% for all patients and from 0.35% to 2.63% for hospitalized patients.
The prevalence of VTE at 90 days ranged from 0.30% to 1.37% for all patients and from 0.75% to 3.86% for hospitalized patients.
The researchers said the main limitation of this study is the use of administrative data because some patients with syncope, PE, or VTE may have been missed. ![]()
There is no need for a structured algorithm to rule out pulmonary embolism (PE) in patients who visit the emergency department (ED) for syncope, according to researchers.
The team analyzed data on nearly 1.7 million patients who presented to the ED for syncope in 4 different countries.
PE occurred in less than 1% of these patients and in less than 3% of patients who were hospitalized.
The researchers therefore concluded that PE should be considered in such patients, but a systematic protocol for ruling out PE is not necessary.
“Our results are saying that there is no need, so far, for using a structured clinical algorithm to assess pulmonary emboli in all the patients presenting [to the ED with syncope],” said Nicola Montano, MD, PhD, of the University of Milan in Italy.
Dr Montano and his colleagues described these results in JAMA Internal Medicine.
The team noted that, in the PESIT trial, researchers used a standardized algorithm to evaluate PE prevalence in patients hospitalized after a first syncope episode.
The algorithm was based on pretest clinical probability and results of the D-dimer assay. Any patient with positive D-dimer results or high pretest PE probability underwent computed tomography or ventilation perfusion lung scanning.
In this study, the prevalence of PE was 17.3%.
A subsequent study showed a much lower prevalence of PE and deep vein thrombosis in patients hospitalized for syncope. The prevalence of venous thromboembolism (VTE) in this study was 1.4%.
The differences between these studies and the fact that both studies included only hospitalized patients prompted Dr Montano and his colleagues to conduct the current study.
The team set out to determine PE/VTE prevalence in unselected patients with syncope presenting to the ED.
Results
The researchers analyzed administrative data from 5 databases in 4 countries—Canada, Denmark, Italy, and the US—collected from January 1, 2000, through September 30, 2016.
The data included 1,671,944 adults who presented to the ED for syncope. The prevalence of PE at ED or hospital discharge ranged from 0.06% to 0.55%. Among the hospitalized patients only, the prevalence of PE ranged from 0.15% to 2.10%.
At 90 days of follow-up, the prevalence of PE ranged from 0.14% to 0.83% for all patients and from 0.35% to 2.63% for hospitalized patients.
The prevalence of VTE at 90 days ranged from 0.30% to 1.37% for all patients and from 0.75% to 3.86% for hospitalized patients.
The researchers said the main limitation of this study is the use of administrative data because some patients with syncope, PE, or VTE may have been missed. ![]()
Phototherapeutic technology could fight MM, other cancers

Preclinical research suggests that light-triggered, chemotherapy-loaded nanoparticles could treat multiple myeloma (MM) and other malignancies.
Researchers showed that light emitted as part of traditional cancer-imaging techniques could also trigger a light-sensitive chemotherapy drug.
When this drug was packaged into nanoparticles that target lit-up cancer cells, the drug produced toxic free radicals that killed the cancer cells.
Researchers found this technique to be effective in mice with MM and aggressive, metastatic breast cancer.
“Our study shows that this phototherapeutic technology is particularly suited to attacking small tumors that spread to different parts of the body, including deep in the bone marrow,” said Samuel Achilefu, PhD, of Washington University in St. Louis, Missouri.
Dr Achilefu and his colleagues described the technology in Nature Communications.
The technology harnesses the chemotherapy drug titanocene. When used alone, titanocene did not work well in clinical trials, even at relatively high doses. However, when it is exposed to the radiation emitted by visible light, titanocene produces reactive particles that are toxic to cells, even at low doses.
Dr Achilefu and his colleagues packaged low doses of titanocene inside nanoparticles targeted to proteins on the surface of cancer cells. Specifically, the team used nanomicelles targeting VLA-4, “an attractive target for precision imaging and therapy” in MM, according to the researchers.
When these nanomicelles made contact with MM cells, their membranes fused together, releasing titanocene into the cells.
The researchers then delivered the imaging agent fluorodeoxyglucose (FDG). MM cells took up the FDG at high rates, causing the cells to glow in a positron emission tomography scan. This glow also triggered the titanocene, releasing free radicals and killing the MM cells.
This treatment strategy was used on mice with MM once a week for 4 weeks. In the weeks following, the treated mice had significantly smaller tumors and survived longer than control mice. Fifty percent of treated mice survived at least 90 days, and 50% of control mice survived 62 days.
This strategy also produced an anti-tumor effect in mice with breast cancer, although, in these experiments, the researchers used human serum albumin nanoparticles.
The effect in breast cancer was less pronounced than in MM. The researchers said this was likely due to the extreme aggressiveness of the breast cancer cell line used.
The team also found that certain MM cells were resistant to this treatment technique.
“This is an opportunity to learn because it’s similar to what is seen in patients—some of the cells become dormant but don’t die after treatment,” Dr Achilefu said. “When we looked closer at the cells that were resistant to our phototherapy, we saw that the surface protein we are targeting was not there.”
Specifically, the resistant cells had downregulated expression of CD49d, and the researchers believe this may have impaired the binding of nanomicelles to the MM cells.
“So next, we want to find out if we can pinpoint another surface protein to target and kill these resistant cells along with the myeloma cells that did respond to the original therapy, which could lead to complete remission,” Dr Achilefu said.
Furthermore, Dr Achilefu envisions that, one day, doctors might be able to use this technology to prevent cancer from recurring.
“We are interested in exploring whether this is something a patient in remission could take once a year for prevention,” Dr Achilefu said. “The toxicity appears to be low, so we imagine an outpatient procedure that could involve zapping any cancerous cells, making cancer a chronic condition that could be controlled long-term.”
Dr Achilefu and his colleagues believe this phototherapeutic technology is less toxic than standard radiation and chemotherapy because the titanocene and FDG are targeted to the same place at the same time only in cancer cells.
The body rids itself of titanocene through the liver, while FDG is cleared through the kidneys. The fact that these components are disposed of separately minimizes damage to other organs. When separated, the components are not toxic, according to the researchers. ![]()
Preclinical research suggests that light-triggered, chemotherapy-loaded nanoparticles could treat multiple myeloma (MM) and other malignancies.
Researchers showed that light emitted as part of traditional cancer-imaging techniques could also trigger a light-sensitive chemotherapy drug.
When this drug was packaged into nanoparticles that target lit-up cancer cells, the drug produced toxic free radicals that killed the cancer cells.
Researchers found this technique to be effective in mice with MM and aggressive, metastatic breast cancer.
“Our study shows that this phototherapeutic technology is particularly suited to attacking small tumors that spread to different parts of the body, including deep in the bone marrow,” said Samuel Achilefu, PhD, of Washington University in St. Louis, Missouri.
Dr Achilefu and his colleagues described the technology in Nature Communications.
The technology harnesses the chemotherapy drug titanocene. When used alone, titanocene did not work well in clinical trials, even at relatively high doses. However, when it is exposed to the radiation emitted by visible light, titanocene produces reactive particles that are toxic to cells, even at low doses.
Dr Achilefu and his colleagues packaged low doses of titanocene inside nanoparticles targeted to proteins on the surface of cancer cells. Specifically, the team used nanomicelles targeting VLA-4, “an attractive target for precision imaging and therapy” in MM, according to the researchers.
When these nanomicelles made contact with MM cells, their membranes fused together, releasing titanocene into the cells.
The researchers then delivered the imaging agent fluorodeoxyglucose (FDG). MM cells took up the FDG at high rates, causing the cells to glow in a positron emission tomography scan. This glow also triggered the titanocene, releasing free radicals and killing the MM cells.
This treatment strategy was used on mice with MM once a week for 4 weeks. In the weeks following, the treated mice had significantly smaller tumors and survived longer than control mice. Fifty percent of treated mice survived at least 90 days, and 50% of control mice survived 62 days.
This strategy also produced an anti-tumor effect in mice with breast cancer, although, in these experiments, the researchers used human serum albumin nanoparticles.
The effect in breast cancer was less pronounced than in MM. The researchers said this was likely due to the extreme aggressiveness of the breast cancer cell line used.
The team also found that certain MM cells were resistant to this treatment technique.
“This is an opportunity to learn because it’s similar to what is seen in patients—some of the cells become dormant but don’t die after treatment,” Dr Achilefu said. “When we looked closer at the cells that were resistant to our phototherapy, we saw that the surface protein we are targeting was not there.”
Specifically, the resistant cells had downregulated expression of CD49d, and the researchers believe this may have impaired the binding of nanomicelles to the MM cells.
“So next, we want to find out if we can pinpoint another surface protein to target and kill these resistant cells along with the myeloma cells that did respond to the original therapy, which could lead to complete remission,” Dr Achilefu said.
Furthermore, Dr Achilefu envisions that, one day, doctors might be able to use this technology to prevent cancer from recurring.
“We are interested in exploring whether this is something a patient in remission could take once a year for prevention,” Dr Achilefu said. “The toxicity appears to be low, so we imagine an outpatient procedure that could involve zapping any cancerous cells, making cancer a chronic condition that could be controlled long-term.”
Dr Achilefu and his colleagues believe this phototherapeutic technology is less toxic than standard radiation and chemotherapy because the titanocene and FDG are targeted to the same place at the same time only in cancer cells.
The body rids itself of titanocene through the liver, while FDG is cleared through the kidneys. The fact that these components are disposed of separately minimizes damage to other organs. When separated, the components are not toxic, according to the researchers. ![]()
Preclinical research suggests that light-triggered, chemotherapy-loaded nanoparticles could treat multiple myeloma (MM) and other malignancies.
Researchers showed that light emitted as part of traditional cancer-imaging techniques could also trigger a light-sensitive chemotherapy drug.
When this drug was packaged into nanoparticles that target lit-up cancer cells, the drug produced toxic free radicals that killed the cancer cells.
Researchers found this technique to be effective in mice with MM and aggressive, metastatic breast cancer.
“Our study shows that this phototherapeutic technology is particularly suited to attacking small tumors that spread to different parts of the body, including deep in the bone marrow,” said Samuel Achilefu, PhD, of Washington University in St. Louis, Missouri.
Dr Achilefu and his colleagues described the technology in Nature Communications.
The technology harnesses the chemotherapy drug titanocene. When used alone, titanocene did not work well in clinical trials, even at relatively high doses. However, when it is exposed to the radiation emitted by visible light, titanocene produces reactive particles that are toxic to cells, even at low doses.
Dr Achilefu and his colleagues packaged low doses of titanocene inside nanoparticles targeted to proteins on the surface of cancer cells. Specifically, the team used nanomicelles targeting VLA-4, “an attractive target for precision imaging and therapy” in MM, according to the researchers.
When these nanomicelles made contact with MM cells, their membranes fused together, releasing titanocene into the cells.
The researchers then delivered the imaging agent fluorodeoxyglucose (FDG). MM cells took up the FDG at high rates, causing the cells to glow in a positron emission tomography scan. This glow also triggered the titanocene, releasing free radicals and killing the MM cells.
This treatment strategy was used on mice with MM once a week for 4 weeks. In the weeks following, the treated mice had significantly smaller tumors and survived longer than control mice. Fifty percent of treated mice survived at least 90 days, and 50% of control mice survived 62 days.
This strategy also produced an anti-tumor effect in mice with breast cancer, although, in these experiments, the researchers used human serum albumin nanoparticles.
The effect in breast cancer was less pronounced than in MM. The researchers said this was likely due to the extreme aggressiveness of the breast cancer cell line used.
The team also found that certain MM cells were resistant to this treatment technique.
“This is an opportunity to learn because it’s similar to what is seen in patients—some of the cells become dormant but don’t die after treatment,” Dr Achilefu said. “When we looked closer at the cells that were resistant to our phototherapy, we saw that the surface protein we are targeting was not there.”
Specifically, the resistant cells had downregulated expression of CD49d, and the researchers believe this may have impaired the binding of nanomicelles to the MM cells.
“So next, we want to find out if we can pinpoint another surface protein to target and kill these resistant cells along with the myeloma cells that did respond to the original therapy, which could lead to complete remission,” Dr Achilefu said.
Furthermore, Dr Achilefu envisions that, one day, doctors might be able to use this technology to prevent cancer from recurring.
“We are interested in exploring whether this is something a patient in remission could take once a year for prevention,” Dr Achilefu said. “The toxicity appears to be low, so we imagine an outpatient procedure that could involve zapping any cancerous cells, making cancer a chronic condition that could be controlled long-term.”
Dr Achilefu and his colleagues believe this phototherapeutic technology is less toxic than standard radiation and chemotherapy because the titanocene and FDG are targeted to the same place at the same time only in cancer cells.
The body rids itself of titanocene through the liver, while FDG is cleared through the kidneys. The fact that these components are disposed of separately minimizes damage to other organs. When separated, the components are not toxic, according to the researchers. ![]()
Zika vaccine candidate receives fast track designation
The US Food and Drug Administration (FDA) has granted fast track designation to TAK-426, a candidate vaccine for Zika virus.
TAK-426 is a purified, inactivated, alum-adjuvanted, whole Zika virus vaccine candidate being developed by Takeda Pharmaceutical Company Limited with the aid of federal funding.
TAK-426 is currently being studied in a phase 1 trial, ZIK-101 (NCT03343626).
This randomized, placebo-controlled, double-blind trial was designed to evaluate the safety and immunogenicity of TAK-426 in 240 male and female subjects between the ages of 18 and 49.
The trial is taking place in the continental US and US territories. It is being conducted under a US Investigational New Drug application.
Takeda says that, if initial data from ZIK-101 are supportive, the company will work to progress into phase 2 development as soon as possible.
Takeda’s Zika program is supported by federal funds from the Biomedical Advanced Research and Development Authority (BARDA) within the Office of the Assistant Secretary for Preparedness and Response in the US Department of Health and Human Services (contract No. HHSO100201600015C).
“We recognize the public health threat posed by the Zika virus,” said Laurence De Moerlooze, PhD, lead of the global Zika program at Takeda.
“As soon as Takeda received funding from BARDA, we mobilized a team and prioritized development of this vaccine candidate, initiating a phase 1 trial within 15 months of contract signature. With fast track designation, the ongoing support of BARDA, and the abilities of our organization, we are confident that we will continue to make expedient progress.”
About fast track designation
The FDA’s fast track designation is a process designed to facilitate the development and expedite the review of drugs and vaccines with the potential to treat serious or life-threatening conditions and address unmet medical needs.
The fast track process allows for more frequent interactions with the FDA, rolling reviews of a product application, and eligibility for priority review if relevant criteria are met. The FDA’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months. ![]()
The US Food and Drug Administration (FDA) has granted fast track designation to TAK-426, a candidate vaccine for Zika virus.
TAK-426 is a purified, inactivated, alum-adjuvanted, whole Zika virus vaccine candidate being developed by Takeda Pharmaceutical Company Limited with the aid of federal funding.
TAK-426 is currently being studied in a phase 1 trial, ZIK-101 (NCT03343626).
This randomized, placebo-controlled, double-blind trial was designed to evaluate the safety and immunogenicity of TAK-426 in 240 male and female subjects between the ages of 18 and 49.
The trial is taking place in the continental US and US territories. It is being conducted under a US Investigational New Drug application.
Takeda says that, if initial data from ZIK-101 are supportive, the company will work to progress into phase 2 development as soon as possible.
Takeda’s Zika program is supported by federal funds from the Biomedical Advanced Research and Development Authority (BARDA) within the Office of the Assistant Secretary for Preparedness and Response in the US Department of Health and Human Services (contract No. HHSO100201600015C).
“We recognize the public health threat posed by the Zika virus,” said Laurence De Moerlooze, PhD, lead of the global Zika program at Takeda.
“As soon as Takeda received funding from BARDA, we mobilized a team and prioritized development of this vaccine candidate, initiating a phase 1 trial within 15 months of contract signature. With fast track designation, the ongoing support of BARDA, and the abilities of our organization, we are confident that we will continue to make expedient progress.”
About fast track designation
The FDA’s fast track designation is a process designed to facilitate the development and expedite the review of drugs and vaccines with the potential to treat serious or life-threatening conditions and address unmet medical needs.
The fast track process allows for more frequent interactions with the FDA, rolling reviews of a product application, and eligibility for priority review if relevant criteria are met. The FDA’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months. ![]()
The US Food and Drug Administration (FDA) has granted fast track designation to TAK-426, a candidate vaccine for Zika virus.
TAK-426 is a purified, inactivated, alum-adjuvanted, whole Zika virus vaccine candidate being developed by Takeda Pharmaceutical Company Limited with the aid of federal funding.
TAK-426 is currently being studied in a phase 1 trial, ZIK-101 (NCT03343626).
This randomized, placebo-controlled, double-blind trial was designed to evaluate the safety and immunogenicity of TAK-426 in 240 male and female subjects between the ages of 18 and 49.
The trial is taking place in the continental US and US territories. It is being conducted under a US Investigational New Drug application.
Takeda says that, if initial data from ZIK-101 are supportive, the company will work to progress into phase 2 development as soon as possible.
Takeda’s Zika program is supported by federal funds from the Biomedical Advanced Research and Development Authority (BARDA) within the Office of the Assistant Secretary for Preparedness and Response in the US Department of Health and Human Services (contract No. HHSO100201600015C).
“We recognize the public health threat posed by the Zika virus,” said Laurence De Moerlooze, PhD, lead of the global Zika program at Takeda.
“As soon as Takeda received funding from BARDA, we mobilized a team and prioritized development of this vaccine candidate, initiating a phase 1 trial within 15 months of contract signature. With fast track designation, the ongoing support of BARDA, and the abilities of our organization, we are confident that we will continue to make expedient progress.”
About fast track designation
The FDA’s fast track designation is a process designed to facilitate the development and expedite the review of drugs and vaccines with the potential to treat serious or life-threatening conditions and address unmet medical needs.
The fast track process allows for more frequent interactions with the FDA, rolling reviews of a product application, and eligibility for priority review if relevant criteria are met. The FDA’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months. ![]()
IMPDH inhibitors could treat ALL

A mutation that leads to relapse in patients with acute lymphoblastic leukemia (ALL) also causes a weakness that could be exploited to kill leukemia cells, according to research published in Nature.
Investigators found evidence to suggest that mutations in the NT5C2 gene make leukemic cells resistant to a common chemotherapy drug but vulnerable to a class of drugs called IMPDH inhibitors.
“Increased sensitivity to IMPDH inhibition shows proof of principle that the pathway for resistance provides a new therapeutic target,” said Adolfo Ferrando, MD, PhD, of Columbia University’s Herbert Irving Comprehensive Cancer Center in New York, New York.
Dr Ferrando’s lab had previously found that cancer cells from relapsed ALL patients frequently have mutations in NT5C2, which drives resistance to 6-mercaptopurine.
However, the investigators didn’t know how these mutations emerge as cancer recurs after remission.
In analyzing samples from ALL patients, the team found they could detect the NT5C2 mutation R367Q in cancer cells before patients were clinically diagnosed as relapsed. However, the mutation was not detectable in most cases at the time of diagnosis.
These findings suggest that cells with the R367Q mutation only multiply in response to treatment, and the mutation may help predict relapse.
“This seems to be a late mutation involved in disease progression,” Dr Ferrando said. “Our data support that it may not be present at diagnosis in many cases, and that, in cases where it may be present, it represents a very minor population.”
The investigators also found the R367Q mutation impaired leukemia cell growth and leukemia-initiating cell activity. This was “associated with excess export of purines to the extracellular space and depletion of the intracellular purine-nucleotide pool.”
These findings led the investigators to test mizoribine, an IMPDH inhibitor, in Nt5c2+/R367Q mutant and Nt5c2+/co-R367Q wild-type ALL lymphoblasts. The team found the mutant cells were significantly more sensitive to mizoribine.
In Nt5c2+/R367Q leukemia-bearing mice, treatment with mizoribine produced a “marked” anti-leukemic response and significantly prolonged survival.
In immunodeficient mice transplanted with an NT5C2(R367Q) xenograft, mizoribine decreased tumor burden and tumor infiltration.
“IMPDH inhibitors could eventually emerge as relevant antileukemic drugs, but this would require additional preclinical work before clinical testing,” Dr Ferrando said. ![]()
A mutation that leads to relapse in patients with acute lymphoblastic leukemia (ALL) also causes a weakness that could be exploited to kill leukemia cells, according to research published in Nature.
Investigators found evidence to suggest that mutations in the NT5C2 gene make leukemic cells resistant to a common chemotherapy drug but vulnerable to a class of drugs called IMPDH inhibitors.
“Increased sensitivity to IMPDH inhibition shows proof of principle that the pathway for resistance provides a new therapeutic target,” said Adolfo Ferrando, MD, PhD, of Columbia University’s Herbert Irving Comprehensive Cancer Center in New York, New York.
Dr Ferrando’s lab had previously found that cancer cells from relapsed ALL patients frequently have mutations in NT5C2, which drives resistance to 6-mercaptopurine.
However, the investigators didn’t know how these mutations emerge as cancer recurs after remission.
In analyzing samples from ALL patients, the team found they could detect the NT5C2 mutation R367Q in cancer cells before patients were clinically diagnosed as relapsed. However, the mutation was not detectable in most cases at the time of diagnosis.
These findings suggest that cells with the R367Q mutation only multiply in response to treatment, and the mutation may help predict relapse.
“This seems to be a late mutation involved in disease progression,” Dr Ferrando said. “Our data support that it may not be present at diagnosis in many cases, and that, in cases where it may be present, it represents a very minor population.”
The investigators also found the R367Q mutation impaired leukemia cell growth and leukemia-initiating cell activity. This was “associated with excess export of purines to the extracellular space and depletion of the intracellular purine-nucleotide pool.”
These findings led the investigators to test mizoribine, an IMPDH inhibitor, in Nt5c2+/R367Q mutant and Nt5c2+/co-R367Q wild-type ALL lymphoblasts. The team found the mutant cells were significantly more sensitive to mizoribine.
In Nt5c2+/R367Q leukemia-bearing mice, treatment with mizoribine produced a “marked” anti-leukemic response and significantly prolonged survival.
In immunodeficient mice transplanted with an NT5C2(R367Q) xenograft, mizoribine decreased tumor burden and tumor infiltration.
“IMPDH inhibitors could eventually emerge as relevant antileukemic drugs, but this would require additional preclinical work before clinical testing,” Dr Ferrando said. ![]()
A mutation that leads to relapse in patients with acute lymphoblastic leukemia (ALL) also causes a weakness that could be exploited to kill leukemia cells, according to research published in Nature.
Investigators found evidence to suggest that mutations in the NT5C2 gene make leukemic cells resistant to a common chemotherapy drug but vulnerable to a class of drugs called IMPDH inhibitors.
“Increased sensitivity to IMPDH inhibition shows proof of principle that the pathway for resistance provides a new therapeutic target,” said Adolfo Ferrando, MD, PhD, of Columbia University’s Herbert Irving Comprehensive Cancer Center in New York, New York.
Dr Ferrando’s lab had previously found that cancer cells from relapsed ALL patients frequently have mutations in NT5C2, which drives resistance to 6-mercaptopurine.
However, the investigators didn’t know how these mutations emerge as cancer recurs after remission.
In analyzing samples from ALL patients, the team found they could detect the NT5C2 mutation R367Q in cancer cells before patients were clinically diagnosed as relapsed. However, the mutation was not detectable in most cases at the time of diagnosis.
These findings suggest that cells with the R367Q mutation only multiply in response to treatment, and the mutation may help predict relapse.
“This seems to be a late mutation involved in disease progression,” Dr Ferrando said. “Our data support that it may not be present at diagnosis in many cases, and that, in cases where it may be present, it represents a very minor population.”
The investigators also found the R367Q mutation impaired leukemia cell growth and leukemia-initiating cell activity. This was “associated with excess export of purines to the extracellular space and depletion of the intracellular purine-nucleotide pool.”
These findings led the investigators to test mizoribine, an IMPDH inhibitor, in Nt5c2+/R367Q mutant and Nt5c2+/co-R367Q wild-type ALL lymphoblasts. The team found the mutant cells were significantly more sensitive to mizoribine.
In Nt5c2+/R367Q leukemia-bearing mice, treatment with mizoribine produced a “marked” anti-leukemic response and significantly prolonged survival.
In immunodeficient mice transplanted with an NT5C2(R367Q) xenograft, mizoribine decreased tumor burden and tumor infiltration.
“IMPDH inhibitors could eventually emerge as relevant antileukemic drugs, but this would require additional preclinical work before clinical testing,” Dr Ferrando said. ![]()
FDA clears assay for myeloma patients

The US Food and Drug Administration (FDA) has granted 510(k) clearance for Sebia’s Hydrashift 2/4 daratumumab immunofixation assay.
This in vitro diagnostic test allows for assessment of response in patients with multiple myeloma by mitigating potential interference caused by the anti-CD38 antibody daratumumab (Darzalex®).
Daratumumab can interfere with the visualization of M-proteins in immunofixation electrophoresis.
The Hydrashift 2/4 daratumumab assay is intended to be used with Sebia’s Hydragel IF kit to provide qualitative detection of monoclonal proteins in human serum by immunofixation electrophoresis.
The assay is performed on Sebia’s Hydrasys 2 agarose gel platform.
The Hydrashift 2/4 daratumumab assay is the result of a collaboration between Sebia and Janssen Biotech, Inc. Sebia received development rights from Janssen and is the worldwide supplier of this assay.
The Hydrashift 2/4 daratumumab assay received the CE mark in November 2016. ![]()
The US Food and Drug Administration (FDA) has granted 510(k) clearance for Sebia’s Hydrashift 2/4 daratumumab immunofixation assay.
This in vitro diagnostic test allows for assessment of response in patients with multiple myeloma by mitigating potential interference caused by the anti-CD38 antibody daratumumab (Darzalex®).
Daratumumab can interfere with the visualization of M-proteins in immunofixation electrophoresis.
The Hydrashift 2/4 daratumumab assay is intended to be used with Sebia’s Hydragel IF kit to provide qualitative detection of monoclonal proteins in human serum by immunofixation electrophoresis.
The assay is performed on Sebia’s Hydrasys 2 agarose gel platform.
The Hydrashift 2/4 daratumumab assay is the result of a collaboration between Sebia and Janssen Biotech, Inc. Sebia received development rights from Janssen and is the worldwide supplier of this assay.
The Hydrashift 2/4 daratumumab assay received the CE mark in November 2016. ![]()
The US Food and Drug Administration (FDA) has granted 510(k) clearance for Sebia’s Hydrashift 2/4 daratumumab immunofixation assay.
This in vitro diagnostic test allows for assessment of response in patients with multiple myeloma by mitigating potential interference caused by the anti-CD38 antibody daratumumab (Darzalex®).
Daratumumab can interfere with the visualization of M-proteins in immunofixation electrophoresis.
The Hydrashift 2/4 daratumumab assay is intended to be used with Sebia’s Hydragel IF kit to provide qualitative detection of monoclonal proteins in human serum by immunofixation electrophoresis.
The assay is performed on Sebia’s Hydrasys 2 agarose gel platform.
The Hydrashift 2/4 daratumumab assay is the result of a collaboration between Sebia and Janssen Biotech, Inc. Sebia received development rights from Janssen and is the worldwide supplier of this assay.
The Hydrashift 2/4 daratumumab assay received the CE mark in November 2016.
CHMP recommends approval of emicizumab

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval of emicizumab (Hemlibra®), a bispecific factor IXa- and factor X-directed antibody.
The recommendation is for emicizumab to be used as routine prophylaxis in patients of all ages who have hemophilia A with factor VIII inhibitors.
The marketing authorization application for emicizumab is being reviewed under accelerated assessment, a procedure granted to medicines the CHMP believes are of major interest for public health and therapeutic innovation.
The CHMP’s opinion on emicizumab will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The CHMP’s recommendation for emicizumab is based on results from 2 phase 3 trials—HAVEN 1 and HAVEN 2.
Results from HAVEN 1 were published in NEJM and presented at the 26th ISTH Congress in July 2017. Updated results from HAVEN 2 were presented at the 2017 ASH Annual Meeting.
HAVEN 1
The study enrolled 109 patients (age 12 and older) with hemophilia A and FVIII inhibitors who were previously treated with bypassing agents (BPAs) on-demand or as prophylaxis.
The patients were randomized to receive emicizumab prophylaxis or no prophylaxis. On-demand treatment of breakthrough bleeds with BPAs was allowed.
There was a significant reduction in treated bleeds of 87% with emicizumab prophylaxis compared to no prophylaxis (95% CI: 72.3; 94.3, P<0.0001). And there was an 80% reduction in all bleeds with emicizumab (95% CI: 62.5; 89.8, P<0.0001).
Adverse events (AEs) occurring in at least 5% of patients treated with emicizumab were local injection site reactions, headache, fatigue, upper respiratory tract infection, and arthralgia.
Two patients experienced thromboembolic events (TEs), and 3 had thrombotic microangiopathy (TMA) while receiving emicizumab prophylaxis and more than 100 u/kg/day of activated prothrombin complex concentrate, on average, for 24 hours or more before the event. Two of these patients had also received recombinant factor VIIa.
Neither TE required anticoagulation therapy, and 1 patient restarted emicizumab. The cases of TMA observed were transient, and 1 patient restarted emicizumab.
HAVEN 2
In this single-arm trial, researchers evaluated emicizumab prophylaxis in 60 patients, ages 1 to 17, who had hemophilia A with FVIII inhibitors.
The efficacy analysis included 57 patients who were younger than 12. The 3 older patients were only included in the safety analysis.
Of the 57 patients, 64.9% had 0 bleeds, 94.7% had 0 treated bleeds, and 98.2% had 0 treated spontaneous bleeds and 0 treated joint bleeds. None of the patients had treated target joint bleeds.
Forty patients had a total of 201 AEs. The most common of these were viral upper respiratory tract infections (16.7%) and injection site reactions (16.7%).
There were no TEs or TMA events, and none of the patients tested positive for anti-drug antibodies. None of the 7 serious AEs in this trial were considered treatment-related.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval of emicizumab (Hemlibra®), a bispecific factor IXa- and factor X-directed antibody.
The recommendation is for emicizumab to be used as routine prophylaxis in patients of all ages who have hemophilia A with factor VIII inhibitors.
The marketing authorization application for emicizumab is being reviewed under accelerated assessment, a procedure granted to medicines the CHMP believes are of major interest for public health and therapeutic innovation.
The CHMP’s opinion on emicizumab will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The CHMP’s recommendation for emicizumab is based on results from 2 phase 3 trials—HAVEN 1 and HAVEN 2.
Results from HAVEN 1 were published in NEJM and presented at the 26th ISTH Congress in July 2017. Updated results from HAVEN 2 were presented at the 2017 ASH Annual Meeting.
HAVEN 1
The study enrolled 109 patients (age 12 and older) with hemophilia A and FVIII inhibitors who were previously treated with bypassing agents (BPAs) on-demand or as prophylaxis.
The patients were randomized to receive emicizumab prophylaxis or no prophylaxis. On-demand treatment of breakthrough bleeds with BPAs was allowed.
There was a significant reduction in treated bleeds of 87% with emicizumab prophylaxis compared to no prophylaxis (95% CI: 72.3; 94.3, P<0.0001). And there was an 80% reduction in all bleeds with emicizumab (95% CI: 62.5; 89.8, P<0.0001).
Adverse events (AEs) occurring in at least 5% of patients treated with emicizumab were local injection site reactions, headache, fatigue, upper respiratory tract infection, and arthralgia.
Two patients experienced thromboembolic events (TEs), and 3 had thrombotic microangiopathy (TMA) while receiving emicizumab prophylaxis and more than 100 u/kg/day of activated prothrombin complex concentrate, on average, for 24 hours or more before the event. Two of these patients had also received recombinant factor VIIa.
Neither TE required anticoagulation therapy, and 1 patient restarted emicizumab. The cases of TMA observed were transient, and 1 patient restarted emicizumab.
HAVEN 2
In this single-arm trial, researchers evaluated emicizumab prophylaxis in 60 patients, ages 1 to 17, who had hemophilia A with FVIII inhibitors.
The efficacy analysis included 57 patients who were younger than 12. The 3 older patients were only included in the safety analysis.
Of the 57 patients, 64.9% had 0 bleeds, 94.7% had 0 treated bleeds, and 98.2% had 0 treated spontaneous bleeds and 0 treated joint bleeds. None of the patients had treated target joint bleeds.
Forty patients had a total of 201 AEs. The most common of these were viral upper respiratory tract infections (16.7%) and injection site reactions (16.7%).
There were no TEs or TMA events, and none of the patients tested positive for anti-drug antibodies. None of the 7 serious AEs in this trial were considered treatment-related.
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended approval of emicizumab (Hemlibra®), a bispecific factor IXa- and factor X-directed antibody.
The recommendation is for emicizumab to be used as routine prophylaxis in patients of all ages who have hemophilia A with factor VIII inhibitors.
The marketing authorization application for emicizumab is being reviewed under accelerated assessment, a procedure granted to medicines the CHMP believes are of major interest for public health and therapeutic innovation.
The CHMP’s opinion on emicizumab will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The CHMP’s recommendation for emicizumab is based on results from 2 phase 3 trials—HAVEN 1 and HAVEN 2.
Results from HAVEN 1 were published in NEJM and presented at the 26th ISTH Congress in July 2017. Updated results from HAVEN 2 were presented at the 2017 ASH Annual Meeting.
HAVEN 1
The study enrolled 109 patients (age 12 and older) with hemophilia A and FVIII inhibitors who were previously treated with bypassing agents (BPAs) on-demand or as prophylaxis.
The patients were randomized to receive emicizumab prophylaxis or no prophylaxis. On-demand treatment of breakthrough bleeds with BPAs was allowed.
There was a significant reduction in treated bleeds of 87% with emicizumab prophylaxis compared to no prophylaxis (95% CI: 72.3; 94.3, P<0.0001). And there was an 80% reduction in all bleeds with emicizumab (95% CI: 62.5; 89.8, P<0.0001).
Adverse events (AEs) occurring in at least 5% of patients treated with emicizumab were local injection site reactions, headache, fatigue, upper respiratory tract infection, and arthralgia.
Two patients experienced thromboembolic events (TEs), and 3 had thrombotic microangiopathy (TMA) while receiving emicizumab prophylaxis and more than 100 u/kg/day of activated prothrombin complex concentrate, on average, for 24 hours or more before the event. Two of these patients had also received recombinant factor VIIa.
Neither TE required anticoagulation therapy, and 1 patient restarted emicizumab. The cases of TMA observed were transient, and 1 patient restarted emicizumab.
HAVEN 2
In this single-arm trial, researchers evaluated emicizumab prophylaxis in 60 patients, ages 1 to 17, who had hemophilia A with FVIII inhibitors.
The efficacy analysis included 57 patients who were younger than 12. The 3 older patients were only included in the safety analysis.
Of the 57 patients, 64.9% had 0 bleeds, 94.7% had 0 treated bleeds, and 98.2% had 0 treated spontaneous bleeds and 0 treated joint bleeds. None of the patients had treated target joint bleeds.
Forty patients had a total of 201 AEs. The most common of these were viral upper respiratory tract infections (16.7%) and injection site reactions (16.7%).
There were no TEs or TMA events, and none of the patients tested positive for anti-drug antibodies. None of the 7 serious AEs in this trial were considered treatment-related.
Real-world data show risk of major bleeding, stroke with NOACs
LOS ANGELES—A real-world analysis has quantified the risks of stroke and major bleeding in patients with non-valvular atrial fibrillation (NVAF) starting treatment with novel oral anticoagulants (NOACs).
The data showed that patients receiving dabigatran had a significantly lower rate of major bleeding but a similar rate of stroke as patients receiving rivaroxaban.
Rates of stroke and major bleeding were not significantly different in patients receiving dabigatran and those receiving apixaban.
These findings were presented at the International Stroke Conference 2018 (abstract 100). This research was supported by Boehringer Ingelheim, makers of dabigatran. The researchers are employed by, or have received payments from, the company.
“With an increasing number of the 2.7 million Americans living with atrial fibrillation being treated with NOACs, real-world analyses like this that compare their effectiveness and safety are important,” said lead investigator Todd C. Villines, MD, of Walter Reed National Military Medical Center in Bethesda, Maryland.
“As a researcher and treating physician, I hope that this large-scale, US practice-based comparison will provide additional insight on available NOAC therapies . . . .”
In this retrospective, observational study, Dr Villines and his colleagues assessed the safety and effectiveness of NOACs in NVAF patients treated through the US Department of Defense Military Health System.
The researchers analyzed data from patients newly initiating treatment with apixaban, dabigatran, or rivaroxaban.
The team examined 2 cohorts. One resulted in 12,763 propensity-score-matched patients receiving dabigatran (150 mg bid) or rivaroxaban (20 mg daily). The other resulted in 4802 propensity-score- matched patients receiving dabigatran (150 mg bid) or apixaban (5 mg bid).
The primary outcomes in this study were the risk of major bleeding and stroke.
Dabigatran-treated patients had a significantly lower rate of major bleeding than rivaroxaban-treated patients—2.08% (266/12,763) and 2.53% (323/12,763), respectively (hazard ratio [HR]=0.82; 95% confidence interval [CI], 0.70-0.97; P=0.0182).
However, rates of stroke were not significantly different in the dabigatran and rivaroxaban groups—0.60% (77/12,763) and 0.78% (100/12,763), respectively (HR=0.77; 95% CI 0.57-1.04; P=0.0844).
Likewise, there was no significant difference in stroke rates for patients receiving dabigatran and apixaban—0.44% (21/4802) and 0.35% (17/4802), respectively (HR=1.26; 95% CI, 0.66-2.39; P=0.4892].
And there was no significant difference in major bleeding—1.60% (77/4802) and 1.21% (58/4802), respectively (HR=1.37; 95% CI, 0.97-1.94; P=0.0702).
The researchers said limitations of this study include the potential for residual confounding as an observational, on-treatment study. In addition, the study included data from electronic health records, which may not have been optimal to identify baseline risk and outcomes. Finally, there were limited dabigatran users available for matching with apixaban users.
LOS ANGELES—A real-world analysis has quantified the risks of stroke and major bleeding in patients with non-valvular atrial fibrillation (NVAF) starting treatment with novel oral anticoagulants (NOACs).
The data showed that patients receiving dabigatran had a significantly lower rate of major bleeding but a similar rate of stroke as patients receiving rivaroxaban.
Rates of stroke and major bleeding were not significantly different in patients receiving dabigatran and those receiving apixaban.
These findings were presented at the International Stroke Conference 2018 (abstract 100). This research was supported by Boehringer Ingelheim, makers of dabigatran. The researchers are employed by, or have received payments from, the company.
“With an increasing number of the 2.7 million Americans living with atrial fibrillation being treated with NOACs, real-world analyses like this that compare their effectiveness and safety are important,” said lead investigator Todd C. Villines, MD, of Walter Reed National Military Medical Center in Bethesda, Maryland.
“As a researcher and treating physician, I hope that this large-scale, US practice-based comparison will provide additional insight on available NOAC therapies . . . .”
In this retrospective, observational study, Dr Villines and his colleagues assessed the safety and effectiveness of NOACs in NVAF patients treated through the US Department of Defense Military Health System.
The researchers analyzed data from patients newly initiating treatment with apixaban, dabigatran, or rivaroxaban.
The team examined 2 cohorts. One resulted in 12,763 propensity-score-matched patients receiving dabigatran (150 mg bid) or rivaroxaban (20 mg daily). The other resulted in 4802 propensity-score- matched patients receiving dabigatran (150 mg bid) or apixaban (5 mg bid).
The primary outcomes in this study were the risk of major bleeding and stroke.
Dabigatran-treated patients had a significantly lower rate of major bleeding than rivaroxaban-treated patients—2.08% (266/12,763) and 2.53% (323/12,763), respectively (hazard ratio [HR]=0.82; 95% confidence interval [CI], 0.70-0.97; P=0.0182).
However, rates of stroke were not significantly different in the dabigatran and rivaroxaban groups—0.60% (77/12,763) and 0.78% (100/12,763), respectively (HR=0.77; 95% CI 0.57-1.04; P=0.0844).
Likewise, there was no significant difference in stroke rates for patients receiving dabigatran and apixaban—0.44% (21/4802) and 0.35% (17/4802), respectively (HR=1.26; 95% CI, 0.66-2.39; P=0.4892].
And there was no significant difference in major bleeding—1.60% (77/4802) and 1.21% (58/4802), respectively (HR=1.37; 95% CI, 0.97-1.94; P=0.0702).
The researchers said limitations of this study include the potential for residual confounding as an observational, on-treatment study. In addition, the study included data from electronic health records, which may not have been optimal to identify baseline risk and outcomes. Finally, there were limited dabigatran users available for matching with apixaban users.
LOS ANGELES—A real-world analysis has quantified the risks of stroke and major bleeding in patients with non-valvular atrial fibrillation (NVAF) starting treatment with novel oral anticoagulants (NOACs).
The data showed that patients receiving dabigatran had a significantly lower rate of major bleeding but a similar rate of stroke as patients receiving rivaroxaban.
Rates of stroke and major bleeding were not significantly different in patients receiving dabigatran and those receiving apixaban.
These findings were presented at the International Stroke Conference 2018 (abstract 100). This research was supported by Boehringer Ingelheim, makers of dabigatran. The researchers are employed by, or have received payments from, the company.
“With an increasing number of the 2.7 million Americans living with atrial fibrillation being treated with NOACs, real-world analyses like this that compare their effectiveness and safety are important,” said lead investigator Todd C. Villines, MD, of Walter Reed National Military Medical Center in Bethesda, Maryland.
“As a researcher and treating physician, I hope that this large-scale, US practice-based comparison will provide additional insight on available NOAC therapies . . . .”
In this retrospective, observational study, Dr Villines and his colleagues assessed the safety and effectiveness of NOACs in NVAF patients treated through the US Department of Defense Military Health System.
The researchers analyzed data from patients newly initiating treatment with apixaban, dabigatran, or rivaroxaban.
The team examined 2 cohorts. One resulted in 12,763 propensity-score-matched patients receiving dabigatran (150 mg bid) or rivaroxaban (20 mg daily). The other resulted in 4802 propensity-score- matched patients receiving dabigatran (150 mg bid) or apixaban (5 mg bid).
The primary outcomes in this study were the risk of major bleeding and stroke.
Dabigatran-treated patients had a significantly lower rate of major bleeding than rivaroxaban-treated patients—2.08% (266/12,763) and 2.53% (323/12,763), respectively (hazard ratio [HR]=0.82; 95% confidence interval [CI], 0.70-0.97; P=0.0182).
However, rates of stroke were not significantly different in the dabigatran and rivaroxaban groups—0.60% (77/12,763) and 0.78% (100/12,763), respectively (HR=0.77; 95% CI 0.57-1.04; P=0.0844).
Likewise, there was no significant difference in stroke rates for patients receiving dabigatran and apixaban—0.44% (21/4802) and 0.35% (17/4802), respectively (HR=1.26; 95% CI, 0.66-2.39; P=0.4892].
And there was no significant difference in major bleeding—1.60% (77/4802) and 1.21% (58/4802), respectively (HR=1.37; 95% CI, 0.97-1.94; P=0.0702).
The researchers said limitations of this study include the potential for residual confounding as an observational, on-treatment study. In addition, the study included data from electronic health records, which may not have been optimal to identify baseline risk and outcomes. Finally, there were limited dabigatran users available for matching with apixaban users.
T-cell therapy produces durable responses in rel/ref HL

Engineered T cells can produce durable responses in patients with Epstein Barr virus–positive (EBV+), relapsed/refractory Hodgkin lymphoma (HL), according to research published in the Journal of Clinical Oncology.
These T cells, known as DNRII-LSTs, produced responses in 4 of the 8 patients studied.
This included 3 complete responses (CRs), the longest of which has exceeded 7 years.
What’s more, these responses were achieved without the use of lymphodepleting chemotherapy.
“While the study is small, its findings are incredibly encouraging for our [patients’] families and for the cancer field,” said study author Catherine M. Bollard, MD, MBChB, of Children’s National Health System in Washington, DC.
To engineer the DNRII-LSTs, Dr Bollard and her colleagues forced expression of a dominant-negative TGF-beta receptor type 2 (DNRII) on LMP-specific T cells (LSTs), which are T cells directed to the EBV latency-associated antigens LMP-1 and LMP-2.
The goal of forcing DNRII expression was to enable the LSTs to resist the hostile tumor environment so they could seek out and kill the tumor cells.
Dr Bollard and her colleagues administered DNRII-LSTs to 8 patients with EBV+ HL. The patients ranged in age from 27 to 47.
Seven of the 8 patients had active disease at the time of DNRII-LST infusion. Two patients had stage IVB HL, 1 had stage IIIB, and 2 had stage IIB. Four patients had nodular-sclerosing HL.
Six patients had relapsed twice. The remaining 2 patients had relapsed 3 and 4 times, respectively. All patients had previously received an autologous stem cell transplant and a range of multi-agent chemotherapy regimens (eg, ABVD, R-ICE, and MOPP).
For this study, the patients received 2 to 12 infusions of DNRII-LSTs, at doses ranging from 2 × 107 to 1.5 × 108 cells/m2.
Results
The researchers found that autologous DNRII-LSTs (given to 7 patients) did not cause autoimmunity, and donor-derived DNRII-LSTs (n=1) did not induce graft-vs-host disease.
The team also noted there were no toxicities resulting from cytokine release syndrome.
Four patients achieved a response to treatment—3 CRs and a partial response.
All complete responders are still in CR, but the partial responder progressed at 19 months and ultimately died of sepsis (2 years after the first dose of DNRII-LSTs).
The other 4 patients had stable disease (SD) for 4 months to 13 months after treatment with DNRII-LSTs.
One patient with SD died of disease progression 2 years after receiving DNRII-LSTs, and another died of transplant complications less than 2 years after the last dose of DNRII-LSTs.
One patient with SD went on to receive additional therapy and is still alive more than 6 years after receiving DNRII-LSTs (currently receiving nivolumab). Another SD patient went on to receive additional therapy, achieved a CR, and is still alive.
One of the patients who achieved a CR to DNRII-LSTs remains in CR more than 7 years after the last dose. Another patient’s CR has exceeded 2 years, and another’s has exceeded 5 years.
All 3 of these patients received doses of 2 × 107 cells/m2. The patients with the longest and shortest CRs each received 2 infusions of DNRII-LSTs. The patient with the CR exceeding 5 years received 12 infusions.
“These results come 18 years after this revolutionary approach was first conceptualized,” Dr Bollard said. “I started work in this area in 2000. At that time, the oncology community had little enthusiasm for the use of T-cell therapies to treat cancer.”
“Even then, when T-cell therapy was in its relative infancy, some research institutions began to see more than 90% complete responses and cure rates in some settings. This most recent study points to the potential of specialized T cells to fight even more types of immune-evading tumors.”
Engineered T cells can produce durable responses in patients with Epstein Barr virus–positive (EBV+), relapsed/refractory Hodgkin lymphoma (HL), according to research published in the Journal of Clinical Oncology.
These T cells, known as DNRII-LSTs, produced responses in 4 of the 8 patients studied.
This included 3 complete responses (CRs), the longest of which has exceeded 7 years.
What’s more, these responses were achieved without the use of lymphodepleting chemotherapy.
“While the study is small, its findings are incredibly encouraging for our [patients’] families and for the cancer field,” said study author Catherine M. Bollard, MD, MBChB, of Children’s National Health System in Washington, DC.
To engineer the DNRII-LSTs, Dr Bollard and her colleagues forced expression of a dominant-negative TGF-beta receptor type 2 (DNRII) on LMP-specific T cells (LSTs), which are T cells directed to the EBV latency-associated antigens LMP-1 and LMP-2.
The goal of forcing DNRII expression was to enable the LSTs to resist the hostile tumor environment so they could seek out and kill the tumor cells.
Dr Bollard and her colleagues administered DNRII-LSTs to 8 patients with EBV+ HL. The patients ranged in age from 27 to 47.
Seven of the 8 patients had active disease at the time of DNRII-LST infusion. Two patients had stage IVB HL, 1 had stage IIIB, and 2 had stage IIB. Four patients had nodular-sclerosing HL.
Six patients had relapsed twice. The remaining 2 patients had relapsed 3 and 4 times, respectively. All patients had previously received an autologous stem cell transplant and a range of multi-agent chemotherapy regimens (eg, ABVD, R-ICE, and MOPP).
For this study, the patients received 2 to 12 infusions of DNRII-LSTs, at doses ranging from 2 × 107 to 1.5 × 108 cells/m2.
Results
The researchers found that autologous DNRII-LSTs (given to 7 patients) did not cause autoimmunity, and donor-derived DNRII-LSTs (n=1) did not induce graft-vs-host disease.
The team also noted there were no toxicities resulting from cytokine release syndrome.
Four patients achieved a response to treatment—3 CRs and a partial response.
All complete responders are still in CR, but the partial responder progressed at 19 months and ultimately died of sepsis (2 years after the first dose of DNRII-LSTs).
The other 4 patients had stable disease (SD) for 4 months to 13 months after treatment with DNRII-LSTs.
One patient with SD died of disease progression 2 years after receiving DNRII-LSTs, and another died of transplant complications less than 2 years after the last dose of DNRII-LSTs.
One patient with SD went on to receive additional therapy and is still alive more than 6 years after receiving DNRII-LSTs (currently receiving nivolumab). Another SD patient went on to receive additional therapy, achieved a CR, and is still alive.
One of the patients who achieved a CR to DNRII-LSTs remains in CR more than 7 years after the last dose. Another patient’s CR has exceeded 2 years, and another’s has exceeded 5 years.
All 3 of these patients received doses of 2 × 107 cells/m2. The patients with the longest and shortest CRs each received 2 infusions of DNRII-LSTs. The patient with the CR exceeding 5 years received 12 infusions.
“These results come 18 years after this revolutionary approach was first conceptualized,” Dr Bollard said. “I started work in this area in 2000. At that time, the oncology community had little enthusiasm for the use of T-cell therapies to treat cancer.”
“Even then, when T-cell therapy was in its relative infancy, some research institutions began to see more than 90% complete responses and cure rates in some settings. This most recent study points to the potential of specialized T cells to fight even more types of immune-evading tumors.”
Engineered T cells can produce durable responses in patients with Epstein Barr virus–positive (EBV+), relapsed/refractory Hodgkin lymphoma (HL), according to research published in the Journal of Clinical Oncology.
These T cells, known as DNRII-LSTs, produced responses in 4 of the 8 patients studied.
This included 3 complete responses (CRs), the longest of which has exceeded 7 years.
What’s more, these responses were achieved without the use of lymphodepleting chemotherapy.
“While the study is small, its findings are incredibly encouraging for our [patients’] families and for the cancer field,” said study author Catherine M. Bollard, MD, MBChB, of Children’s National Health System in Washington, DC.
To engineer the DNRII-LSTs, Dr Bollard and her colleagues forced expression of a dominant-negative TGF-beta receptor type 2 (DNRII) on LMP-specific T cells (LSTs), which are T cells directed to the EBV latency-associated antigens LMP-1 and LMP-2.
The goal of forcing DNRII expression was to enable the LSTs to resist the hostile tumor environment so they could seek out and kill the tumor cells.
Dr Bollard and her colleagues administered DNRII-LSTs to 8 patients with EBV+ HL. The patients ranged in age from 27 to 47.
Seven of the 8 patients had active disease at the time of DNRII-LST infusion. Two patients had stage IVB HL, 1 had stage IIIB, and 2 had stage IIB. Four patients had nodular-sclerosing HL.
Six patients had relapsed twice. The remaining 2 patients had relapsed 3 and 4 times, respectively. All patients had previously received an autologous stem cell transplant and a range of multi-agent chemotherapy regimens (eg, ABVD, R-ICE, and MOPP).
For this study, the patients received 2 to 12 infusions of DNRII-LSTs, at doses ranging from 2 × 107 to 1.5 × 108 cells/m2.
Results
The researchers found that autologous DNRII-LSTs (given to 7 patients) did not cause autoimmunity, and donor-derived DNRII-LSTs (n=1) did not induce graft-vs-host disease.
The team also noted there were no toxicities resulting from cytokine release syndrome.
Four patients achieved a response to treatment—3 CRs and a partial response.
All complete responders are still in CR, but the partial responder progressed at 19 months and ultimately died of sepsis (2 years after the first dose of DNRII-LSTs).
The other 4 patients had stable disease (SD) for 4 months to 13 months after treatment with DNRII-LSTs.
One patient with SD died of disease progression 2 years after receiving DNRII-LSTs, and another died of transplant complications less than 2 years after the last dose of DNRII-LSTs.
One patient with SD went on to receive additional therapy and is still alive more than 6 years after receiving DNRII-LSTs (currently receiving nivolumab). Another SD patient went on to receive additional therapy, achieved a CR, and is still alive.
One of the patients who achieved a CR to DNRII-LSTs remains in CR more than 7 years after the last dose. Another patient’s CR has exceeded 2 years, and another’s has exceeded 5 years.
All 3 of these patients received doses of 2 × 107 cells/m2. The patients with the longest and shortest CRs each received 2 infusions of DNRII-LSTs. The patient with the CR exceeding 5 years received 12 infusions.
“These results come 18 years after this revolutionary approach was first conceptualized,” Dr Bollard said. “I started work in this area in 2000. At that time, the oncology community had little enthusiasm for the use of T-cell therapies to treat cancer.”
“Even then, when T-cell therapy was in its relative infancy, some research institutions began to see more than 90% complete responses and cure rates in some settings. This most recent study points to the potential of specialized T cells to fight even more types of immune-evading tumors.”