User login
NOACs may do less harm to kidneys than warfarin

New research suggests warfarin may produce worse renal outcomes than non-vitamin K antagonist oral anticoagulants (NOACs) in patients with atrial fibrillation.
In a study of close to 10,000 patients, dabigatran and rivaroxaban were associated with lower risks of adverse renal outcomes than warfarin.
However, risks with warfarin and apixaban were not significantly different.
This research was published in the Journal of the American College of Cardiology.*
“Kidney function decline in patients taking oral anticoagulant drugs is an important topic that has been overlooked in previous clinical trials,” said study author Xiaoxi Yao, PhD, of Mayo Clinic in Rochester, Minnesota.
“Even our past work at Mayo Clinic has been primarily focused on risks for stroke or bleeding.”
For the current study, Dr Yao and her colleagues examined the de-identified records of 9769 patients from the OptumLabs Data Warehouse.
The patients had atrial fibrillation and started taking oral anticoagulants—apixaban, dabigatran, rivaroxaban, or warfarin—between Oct. 1, 2010, and April 30, 2016.
The researchers looked at 4 indicators of kidney function in these patients:
- A 30% or greater decline in estimated glomerular filtration rate (eGFR)
- Doubled serum creatinine level
- Acute kidney injury (AKI)
- Kidney failure.
For the entire study population, the cumulative risk of each event occurring within 2 years of beginning anticoagulation was as follows:
- 24.4% for ≥30% eGFR
- 4.0% for doubled creatinine level
- 14.8% for AKI
- 1.7% for kidney failure.
“Our study demonstrated that renal function decline is very common among atrial fibrillation patients on blood thinners,” Dr Yao said. “About 1 in 4 patients had significantly reduced kidney function within 2 years of being on any of these medications, and 1 in 7 patients had acute kidney injury.”
“In general, patients with atrial fibrillation taking blood-thinning medications tend to have declining kidney function over time,” added study author Peter Noseworthy, MD, of Mayo Clinic in Rochester, Minnesota.
“However, our findings indicate that the non-vitamin K antagonist oral anticoagulants, as a group, are associated with less injury to kidneys than warfarin.”
When the researchers compared all 3 NOACs to warfarin, they found NOAC use was associated with a reduced risk of:
- ≥30% decline in eGFR—hazard ratio (HR)=0.77 (P<0.001)
- Doubling of creatinine—HR=0.62 (P=0.03)
- AKI—HR=0.68 (P<0.001).
However, results differed in one-to-one comparisons.
There was no significant difference between warfarin and apixaban for any of the renal endpoints measured. The HRs (for apixaban vs warfarin) were:
- 0.88 for ≥30% decline in eGFR (P=0.25)
- 0.80 for doubling of creatinine (P=0.51)
- 0.84 for AKI (P=0.16)
- 1.02 for kidney failure (P=0.95).
Dabigatran, on the other hand, was associated with lower risks of ≥30% decline in eGFR and AKI than warfarin. The HRs were as follows:
- 0.72 for ≥30% decline in eGFR (P=0.01)
- 0.64 for doubling of creatinine (P=0.24)
- 0.55 for AKI (P<0.001)
- 0.45 for kidney failure (P=0.21).
Rivaroxaban was associated with lower risks of ≥30% decline in eGFR, doubling of creatinine, and AKI. The HRs were as follows:
- 0.73 for ≥30% decline in eGFR (P<0.001)
- 0.46 for doubling of creatinine (P<0.01)
- 0.69 for AKI (P<0.001)
- 0.63 for kidney failure (P=0.13).
“Patients with atrial fibrillation already face a high risk of kidney disease, perhaps because many such patients have risk factors, such as advanced age, diabetes, and hypertension,” Dr Yao said. “Many drugs these patients are taking rely on kidney function for drug elimination. Therefore, it is particularly important for these patients to choose a drug that minimizes the impact on kidneys.”
“Since non-vitamin K antagonist oral anticoagulants have a different drug mechanism than warfarin, researchers have hypothesized that non-vitamin K antagonist oral anticoagulants may be related to better renal outcomes. Our study is among the first few studies confirming this hypothesis.” ![]()
*This study was funded by the Mayo Clinic Robert D. and Patricia E. Kern Center for the Science of Health Care Delivery, which receives no industry funding. However, study authors did declare industry relationships.
New research suggests warfarin may produce worse renal outcomes than non-vitamin K antagonist oral anticoagulants (NOACs) in patients with atrial fibrillation.
In a study of close to 10,000 patients, dabigatran and rivaroxaban were associated with lower risks of adverse renal outcomes than warfarin.
However, risks with warfarin and apixaban were not significantly different.
This research was published in the Journal of the American College of Cardiology.*
“Kidney function decline in patients taking oral anticoagulant drugs is an important topic that has been overlooked in previous clinical trials,” said study author Xiaoxi Yao, PhD, of Mayo Clinic in Rochester, Minnesota.
“Even our past work at Mayo Clinic has been primarily focused on risks for stroke or bleeding.”
For the current study, Dr Yao and her colleagues examined the de-identified records of 9769 patients from the OptumLabs Data Warehouse.
The patients had atrial fibrillation and started taking oral anticoagulants—apixaban, dabigatran, rivaroxaban, or warfarin—between Oct. 1, 2010, and April 30, 2016.
The researchers looked at 4 indicators of kidney function in these patients:
- A 30% or greater decline in estimated glomerular filtration rate (eGFR)
- Doubled serum creatinine level
- Acute kidney injury (AKI)
- Kidney failure.
For the entire study population, the cumulative risk of each event occurring within 2 years of beginning anticoagulation was as follows:
- 24.4% for ≥30% eGFR
- 4.0% for doubled creatinine level
- 14.8% for AKI
- 1.7% for kidney failure.
“Our study demonstrated that renal function decline is very common among atrial fibrillation patients on blood thinners,” Dr Yao said. “About 1 in 4 patients had significantly reduced kidney function within 2 years of being on any of these medications, and 1 in 7 patients had acute kidney injury.”
“In general, patients with atrial fibrillation taking blood-thinning medications tend to have declining kidney function over time,” added study author Peter Noseworthy, MD, of Mayo Clinic in Rochester, Minnesota.
“However, our findings indicate that the non-vitamin K antagonist oral anticoagulants, as a group, are associated with less injury to kidneys than warfarin.”
When the researchers compared all 3 NOACs to warfarin, they found NOAC use was associated with a reduced risk of:
- ≥30% decline in eGFR—hazard ratio (HR)=0.77 (P<0.001)
- Doubling of creatinine—HR=0.62 (P=0.03)
- AKI—HR=0.68 (P<0.001).
However, results differed in one-to-one comparisons.
There was no significant difference between warfarin and apixaban for any of the renal endpoints measured. The HRs (for apixaban vs warfarin) were:
- 0.88 for ≥30% decline in eGFR (P=0.25)
- 0.80 for doubling of creatinine (P=0.51)
- 0.84 for AKI (P=0.16)
- 1.02 for kidney failure (P=0.95).
Dabigatran, on the other hand, was associated with lower risks of ≥30% decline in eGFR and AKI than warfarin. The HRs were as follows:
- 0.72 for ≥30% decline in eGFR (P=0.01)
- 0.64 for doubling of creatinine (P=0.24)
- 0.55 for AKI (P<0.001)
- 0.45 for kidney failure (P=0.21).
Rivaroxaban was associated with lower risks of ≥30% decline in eGFR, doubling of creatinine, and AKI. The HRs were as follows:
- 0.73 for ≥30% decline in eGFR (P<0.001)
- 0.46 for doubling of creatinine (P<0.01)
- 0.69 for AKI (P<0.001)
- 0.63 for kidney failure (P=0.13).
“Patients with atrial fibrillation already face a high risk of kidney disease, perhaps because many such patients have risk factors, such as advanced age, diabetes, and hypertension,” Dr Yao said. “Many drugs these patients are taking rely on kidney function for drug elimination. Therefore, it is particularly important for these patients to choose a drug that minimizes the impact on kidneys.”
“Since non-vitamin K antagonist oral anticoagulants have a different drug mechanism than warfarin, researchers have hypothesized that non-vitamin K antagonist oral anticoagulants may be related to better renal outcomes. Our study is among the first few studies confirming this hypothesis.” ![]()
*This study was funded by the Mayo Clinic Robert D. and Patricia E. Kern Center for the Science of Health Care Delivery, which receives no industry funding. However, study authors did declare industry relationships.
New research suggests warfarin may produce worse renal outcomes than non-vitamin K antagonist oral anticoagulants (NOACs) in patients with atrial fibrillation.
In a study of close to 10,000 patients, dabigatran and rivaroxaban were associated with lower risks of adverse renal outcomes than warfarin.
However, risks with warfarin and apixaban were not significantly different.
This research was published in the Journal of the American College of Cardiology.*
“Kidney function decline in patients taking oral anticoagulant drugs is an important topic that has been overlooked in previous clinical trials,” said study author Xiaoxi Yao, PhD, of Mayo Clinic in Rochester, Minnesota.
“Even our past work at Mayo Clinic has been primarily focused on risks for stroke or bleeding.”
For the current study, Dr Yao and her colleagues examined the de-identified records of 9769 patients from the OptumLabs Data Warehouse.
The patients had atrial fibrillation and started taking oral anticoagulants—apixaban, dabigatran, rivaroxaban, or warfarin—between Oct. 1, 2010, and April 30, 2016.
The researchers looked at 4 indicators of kidney function in these patients:
- A 30% or greater decline in estimated glomerular filtration rate (eGFR)
- Doubled serum creatinine level
- Acute kidney injury (AKI)
- Kidney failure.
For the entire study population, the cumulative risk of each event occurring within 2 years of beginning anticoagulation was as follows:
- 24.4% for ≥30% eGFR
- 4.0% for doubled creatinine level
- 14.8% for AKI
- 1.7% for kidney failure.
“Our study demonstrated that renal function decline is very common among atrial fibrillation patients on blood thinners,” Dr Yao said. “About 1 in 4 patients had significantly reduced kidney function within 2 years of being on any of these medications, and 1 in 7 patients had acute kidney injury.”
“In general, patients with atrial fibrillation taking blood-thinning medications tend to have declining kidney function over time,” added study author Peter Noseworthy, MD, of Mayo Clinic in Rochester, Minnesota.
“However, our findings indicate that the non-vitamin K antagonist oral anticoagulants, as a group, are associated with less injury to kidneys than warfarin.”
When the researchers compared all 3 NOACs to warfarin, they found NOAC use was associated with a reduced risk of:
- ≥30% decline in eGFR—hazard ratio (HR)=0.77 (P<0.001)
- Doubling of creatinine—HR=0.62 (P=0.03)
- AKI—HR=0.68 (P<0.001).
However, results differed in one-to-one comparisons.
There was no significant difference between warfarin and apixaban for any of the renal endpoints measured. The HRs (for apixaban vs warfarin) were:
- 0.88 for ≥30% decline in eGFR (P=0.25)
- 0.80 for doubling of creatinine (P=0.51)
- 0.84 for AKI (P=0.16)
- 1.02 for kidney failure (P=0.95).
Dabigatran, on the other hand, was associated with lower risks of ≥30% decline in eGFR and AKI than warfarin. The HRs were as follows:
- 0.72 for ≥30% decline in eGFR (P=0.01)
- 0.64 for doubling of creatinine (P=0.24)
- 0.55 for AKI (P<0.001)
- 0.45 for kidney failure (P=0.21).
Rivaroxaban was associated with lower risks of ≥30% decline in eGFR, doubling of creatinine, and AKI. The HRs were as follows:
- 0.73 for ≥30% decline in eGFR (P<0.001)
- 0.46 for doubling of creatinine (P<0.01)
- 0.69 for AKI (P<0.001)
- 0.63 for kidney failure (P=0.13).
“Patients with atrial fibrillation already face a high risk of kidney disease, perhaps because many such patients have risk factors, such as advanced age, diabetes, and hypertension,” Dr Yao said. “Many drugs these patients are taking rely on kidney function for drug elimination. Therefore, it is particularly important for these patients to choose a drug that minimizes the impact on kidneys.”
“Since non-vitamin K antagonist oral anticoagulants have a different drug mechanism than warfarin, researchers have hypothesized that non-vitamin K antagonist oral anticoagulants may be related to better renal outcomes. Our study is among the first few studies confirming this hypothesis.” ![]()
*This study was funded by the Mayo Clinic Robert D. and Patricia E. Kern Center for the Science of Health Care Delivery, which receives no industry funding. However, study authors did declare industry relationships.
Technique may predict treatment outcomes in MM
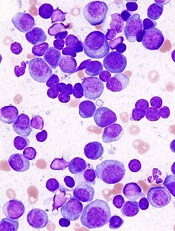
A cell-measuring technique can help predict how multiple myeloma (MM) patients will respond to different therapies, according to research published in Nature Communications.
The technique involves a microfluidic device known as a serial suspended microchannel resonator (sSMR).
The sSMR measures cell mass and allows researchers to calculate growth rates of single cells over short periods of time.
The device consists of a series of sensors that weigh cells as they flow through tiny channels.
The sSMR can measure 50 to 100 cells per hour.
Over a 20-minute period, each cell is weighed 10 times. This is enough to get an accurate mass accumulation rate (MAR), which is a measurement of the rate at which the cells gain mass.
With previous work, researchers showed that MAR can reveal drug susceptibility. A decrease in MAR following drug treatment means cells are sensitive to the drug. If cells are resistant, there is no change in MAR.
For the current study, Scott Manalis, PhD, of the Massachusetts Institute of Technology in Cambridge, and his colleagues tested drugs on cells from MM patients.
The team compared MAR results obtained via sSMR to outcomes of treatment in 9 patients.
For each patient, the researchers tracked the cells’ response to 3 different drugs—bortezomib, lenalidomide, and dexamethasone—plus combinations of those drugs.
In all 9 cases, the sSMR results matched the outcomes seen in patients, as measured by clinical biomarkers in the bloodstream.
“When the clinical biomarkers showed that the patients should be sensitive to a drug, we also saw sensitivity by our measurement,” said study author Mark Stevens, PhD, of the Massachusetts Institute of Technology and Dana-Farber Cancer Institute.
“[I]n cases where the patients were resistant, we saw that in the clinical biomarkers as well as our measurement.”
The researchers believe their device could potentially be used in MM patients at the time of relapse when the disease may have developed resistance to specific therapies.
“At this time of relapse, we would take a bone marrow biopsy from a patient, and we would test each therapy individually or in combinations that are typically used in the clinic,” Dr Stevens said. “At that point, we’d be able to inform the clinician as to which therapy or combinations of therapies this patient seems to be most sensitive or most resistant to.”
Now, the researchers are planning to conduct a larger clinical study to validate this approach. They also aim to investigate the possibility of using this technology for other cancers.
Some of the researchers involved in this study are cofounders of Travera and Affinity Biosensors, 2 companies that develop techniques relevant to this research. ![]()
A cell-measuring technique can help predict how multiple myeloma (MM) patients will respond to different therapies, according to research published in Nature Communications.
The technique involves a microfluidic device known as a serial suspended microchannel resonator (sSMR).
The sSMR measures cell mass and allows researchers to calculate growth rates of single cells over short periods of time.
The device consists of a series of sensors that weigh cells as they flow through tiny channels.
The sSMR can measure 50 to 100 cells per hour.
Over a 20-minute period, each cell is weighed 10 times. This is enough to get an accurate mass accumulation rate (MAR), which is a measurement of the rate at which the cells gain mass.
With previous work, researchers showed that MAR can reveal drug susceptibility. A decrease in MAR following drug treatment means cells are sensitive to the drug. If cells are resistant, there is no change in MAR.
For the current study, Scott Manalis, PhD, of the Massachusetts Institute of Technology in Cambridge, and his colleagues tested drugs on cells from MM patients.
The team compared MAR results obtained via sSMR to outcomes of treatment in 9 patients.
For each patient, the researchers tracked the cells’ response to 3 different drugs—bortezomib, lenalidomide, and dexamethasone—plus combinations of those drugs.
In all 9 cases, the sSMR results matched the outcomes seen in patients, as measured by clinical biomarkers in the bloodstream.
“When the clinical biomarkers showed that the patients should be sensitive to a drug, we also saw sensitivity by our measurement,” said study author Mark Stevens, PhD, of the Massachusetts Institute of Technology and Dana-Farber Cancer Institute.
“[I]n cases where the patients were resistant, we saw that in the clinical biomarkers as well as our measurement.”
The researchers believe their device could potentially be used in MM patients at the time of relapse when the disease may have developed resistance to specific therapies.
“At this time of relapse, we would take a bone marrow biopsy from a patient, and we would test each therapy individually or in combinations that are typically used in the clinic,” Dr Stevens said. “At that point, we’d be able to inform the clinician as to which therapy or combinations of therapies this patient seems to be most sensitive or most resistant to.”
Now, the researchers are planning to conduct a larger clinical study to validate this approach. They also aim to investigate the possibility of using this technology for other cancers.
Some of the researchers involved in this study are cofounders of Travera and Affinity Biosensors, 2 companies that develop techniques relevant to this research. ![]()
A cell-measuring technique can help predict how multiple myeloma (MM) patients will respond to different therapies, according to research published in Nature Communications.
The technique involves a microfluidic device known as a serial suspended microchannel resonator (sSMR).
The sSMR measures cell mass and allows researchers to calculate growth rates of single cells over short periods of time.
The device consists of a series of sensors that weigh cells as they flow through tiny channels.
The sSMR can measure 50 to 100 cells per hour.
Over a 20-minute period, each cell is weighed 10 times. This is enough to get an accurate mass accumulation rate (MAR), which is a measurement of the rate at which the cells gain mass.
With previous work, researchers showed that MAR can reveal drug susceptibility. A decrease in MAR following drug treatment means cells are sensitive to the drug. If cells are resistant, there is no change in MAR.
For the current study, Scott Manalis, PhD, of the Massachusetts Institute of Technology in Cambridge, and his colleagues tested drugs on cells from MM patients.
The team compared MAR results obtained via sSMR to outcomes of treatment in 9 patients.
For each patient, the researchers tracked the cells’ response to 3 different drugs—bortezomib, lenalidomide, and dexamethasone—plus combinations of those drugs.
In all 9 cases, the sSMR results matched the outcomes seen in patients, as measured by clinical biomarkers in the bloodstream.
“When the clinical biomarkers showed that the patients should be sensitive to a drug, we also saw sensitivity by our measurement,” said study author Mark Stevens, PhD, of the Massachusetts Institute of Technology and Dana-Farber Cancer Institute.
“[I]n cases where the patients were resistant, we saw that in the clinical biomarkers as well as our measurement.”
The researchers believe their device could potentially be used in MM patients at the time of relapse when the disease may have developed resistance to specific therapies.
“At this time of relapse, we would take a bone marrow biopsy from a patient, and we would test each therapy individually or in combinations that are typically used in the clinic,” Dr Stevens said. “At that point, we’d be able to inform the clinician as to which therapy or combinations of therapies this patient seems to be most sensitive or most resistant to.”
Now, the researchers are planning to conduct a larger clinical study to validate this approach. They also aim to investigate the possibility of using this technology for other cancers.
Some of the researchers involved in this study are cofounders of Travera and Affinity Biosensors, 2 companies that develop techniques relevant to this research. ![]()
Antimalarial might fight Zika virus

Preclinical research suggests a drug used to prevent and treat malaria may also be effective against Zika virus.
Investigators found that chloroquine can protect human neural progenitors from infection with Zika virus.
The drug also decreased Zika-induced mortality in one mouse model and hindered transmission of Zika virus from mother to fetus in another mouse model.
Alexey Terskikh, PhD, of Sanford Burnham Prebys Medical Discovery Institute in La Jolla, California, and his colleagues conducted this research and disclosed the results in Scientific Reports.
“There is still an urgent need to bolster our preparedness and capacity to respond to the next Zika outbreak,” Dr Terskikh said. “Our latest research suggests the antimalaria drug chloroquine may be an effective drug to treat and prevent Zika infections.”
The investigators first found that chloroquine reduced Zika infection in primary human fetal neural precursor cells.
The team also discovered that chloroquine reduced the percentage of Zika-positive cells and the level of apoptosis in neurospheres derived from human induced pluripotent stem cells.
The investigators then tested chloroquine in AG129 mice, which lack receptors for type I and II interferons and have been used to model Zika virus infection.
Some of these mice received chloroquine (50 mg/kg/day) in their drinking water for 2 days and were then infected with Zika virus. The mice continued to receive chloroquine at the same dose for 5 days and then received 5 mg/kg/day until the end of the experiment.
Control AG129 mice were infected with Zika virus and received regular drinking water.
Compared to controls, chloroquine-treated mice had significantly prolonged survival and a significant reduction in Zika-induced weight loss (P<0.01 for both).
Next, the investigators used SJL mice to study horizontal and vertical transmission of Zika. They observed successful transmission of the virus from males to females and from mothers to pups.
The team then analyzed the effects of chloroquine on fetal transmission of Zika. Pregnant SJL mice were infected with Zika and given drinking water containing chloroquine (30 mg/kg/day) starting on day E13.5. The mice were euthanized on E18.5, and maternal blood and fetal brain samples were collected.
Chloroquine treatment resulted in a roughly 20-fold reduction in Zika virus titers in the maternal blood and fetal brain.
“Although chloroquine didn’t completely clear Zika from infected mice, it did reduce the viral load, suggesting it could limit the neurological damage found in newborns infected by the virus,” Dr Terskikh said.
“Chloroquine has a long history of successfully treating malaria, and there are no reports of it causing birth defects. Additional studies are certainly needed to determine the precise details of how it works. But given its low cost, availability, and safety history, further study in a clinical trial to test its effectiveness against Zika virus in humans is merited.” ![]()
Preclinical research suggests a drug used to prevent and treat malaria may also be effective against Zika virus.
Investigators found that chloroquine can protect human neural progenitors from infection with Zika virus.
The drug also decreased Zika-induced mortality in one mouse model and hindered transmission of Zika virus from mother to fetus in another mouse model.
Alexey Terskikh, PhD, of Sanford Burnham Prebys Medical Discovery Institute in La Jolla, California, and his colleagues conducted this research and disclosed the results in Scientific Reports.
“There is still an urgent need to bolster our preparedness and capacity to respond to the next Zika outbreak,” Dr Terskikh said. “Our latest research suggests the antimalaria drug chloroquine may be an effective drug to treat and prevent Zika infections.”
The investigators first found that chloroquine reduced Zika infection in primary human fetal neural precursor cells.
The team also discovered that chloroquine reduced the percentage of Zika-positive cells and the level of apoptosis in neurospheres derived from human induced pluripotent stem cells.
The investigators then tested chloroquine in AG129 mice, which lack receptors for type I and II interferons and have been used to model Zika virus infection.
Some of these mice received chloroquine (50 mg/kg/day) in their drinking water for 2 days and were then infected with Zika virus. The mice continued to receive chloroquine at the same dose for 5 days and then received 5 mg/kg/day until the end of the experiment.
Control AG129 mice were infected with Zika virus and received regular drinking water.
Compared to controls, chloroquine-treated mice had significantly prolonged survival and a significant reduction in Zika-induced weight loss (P<0.01 for both).
Next, the investigators used SJL mice to study horizontal and vertical transmission of Zika. They observed successful transmission of the virus from males to females and from mothers to pups.
The team then analyzed the effects of chloroquine on fetal transmission of Zika. Pregnant SJL mice were infected with Zika and given drinking water containing chloroquine (30 mg/kg/day) starting on day E13.5. The mice were euthanized on E18.5, and maternal blood and fetal brain samples were collected.
Chloroquine treatment resulted in a roughly 20-fold reduction in Zika virus titers in the maternal blood and fetal brain.
“Although chloroquine didn’t completely clear Zika from infected mice, it did reduce the viral load, suggesting it could limit the neurological damage found in newborns infected by the virus,” Dr Terskikh said.
“Chloroquine has a long history of successfully treating malaria, and there are no reports of it causing birth defects. Additional studies are certainly needed to determine the precise details of how it works. But given its low cost, availability, and safety history, further study in a clinical trial to test its effectiveness against Zika virus in humans is merited.” ![]()
Preclinical research suggests a drug used to prevent and treat malaria may also be effective against Zika virus.
Investigators found that chloroquine can protect human neural progenitors from infection with Zika virus.
The drug also decreased Zika-induced mortality in one mouse model and hindered transmission of Zika virus from mother to fetus in another mouse model.
Alexey Terskikh, PhD, of Sanford Burnham Prebys Medical Discovery Institute in La Jolla, California, and his colleagues conducted this research and disclosed the results in Scientific Reports.
“There is still an urgent need to bolster our preparedness and capacity to respond to the next Zika outbreak,” Dr Terskikh said. “Our latest research suggests the antimalaria drug chloroquine may be an effective drug to treat and prevent Zika infections.”
The investigators first found that chloroquine reduced Zika infection in primary human fetal neural precursor cells.
The team also discovered that chloroquine reduced the percentage of Zika-positive cells and the level of apoptosis in neurospheres derived from human induced pluripotent stem cells.
The investigators then tested chloroquine in AG129 mice, which lack receptors for type I and II interferons and have been used to model Zika virus infection.
Some of these mice received chloroquine (50 mg/kg/day) in their drinking water for 2 days and were then infected with Zika virus. The mice continued to receive chloroquine at the same dose for 5 days and then received 5 mg/kg/day until the end of the experiment.
Control AG129 mice were infected with Zika virus and received regular drinking water.
Compared to controls, chloroquine-treated mice had significantly prolonged survival and a significant reduction in Zika-induced weight loss (P<0.01 for both).
Next, the investigators used SJL mice to study horizontal and vertical transmission of Zika. They observed successful transmission of the virus from males to females and from mothers to pups.
The team then analyzed the effects of chloroquine on fetal transmission of Zika. Pregnant SJL mice were infected with Zika and given drinking water containing chloroquine (30 mg/kg/day) starting on day E13.5. The mice were euthanized on E18.5, and maternal blood and fetal brain samples were collected.
Chloroquine treatment resulted in a roughly 20-fold reduction in Zika virus titers in the maternal blood and fetal brain.
“Although chloroquine didn’t completely clear Zika from infected mice, it did reduce the viral load, suggesting it could limit the neurological damage found in newborns infected by the virus,” Dr Terskikh said.
“Chloroquine has a long history of successfully treating malaria, and there are no reports of it causing birth defects. Additional studies are certainly needed to determine the precise details of how it works. But given its low cost, availability, and safety history, further study in a clinical trial to test its effectiveness against Zika virus in humans is merited.” ![]()
Team discovers mechanism of resistance in AML

Researchers say they have uncovered a target to overcome drug resistance in acute myeloid leukemia (AML).
The team discovered how a linkage between 2 proteins enables AML cells to resist chemotherapy and showed that disrupting the linkage could render the cells vulnerable to treatment.
The researchers believe their discovery could lead to drugs to enhance chemotherapy in patients with AML and other cancers.
John Schuetz, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues described this research in Nature Communications.
The team launched their experiments based on previous findings that a protein called ABCC4 was greatly elevated in aggressive cases of AML.
Dr Schuetz and his colleagues searched for other proteins that might interact with ABCC4 and enable its function. The team’s screening of candidate proteins yielded one, MPP1, which was also greatly increased in AML.
The researchers found the 2 proteins are connected, and the connection enables cells to assume the characteristics of highly proliferative leukemia cells.
These experiments involved genetically altering hematopoietic progenitor cells to have high MPP1 and ABCC4 levels. The cells were grown in culture and then replated to see if they would continue to grow, as such self-renewal is a hallmark of leukemia cells.
The researchers found that serial regrowth depended on the cells having high levels of both ABCC4 and MPP1.
“Typically, if you take normal progenitors and you replate, you could do that one time, maybe twice,” Dr Schuetz said. “But our big surprise was that overexpressing MPP1—analogous to what you would see in leukemia—allows those progenitors to self-renew, to be replated over and over, to form new colonies.”
The experiments also revealed that MPP1 and ABCC4 functioned at the cell membrane, where they could play a role in the machinery that would rid the leukemia cells of chemotherapy drugs.
“When we disrupted their interaction, ABCC4 moved off the membrane and the cells became more sensitive to drugs used in AML—drugs that are pumped out of the cell by ABCC4,” Dr Schuetz said.
By screening thousands of compounds, the researchers identified some that could disrupt the ABCC4-MPP1 connection. One, called Antimycin-A, reversed drug resistance in AML cell lines and in cells from AML patients.
Antimycin-A is too toxic to be used in chemotherapy, but the researchers believe identification of the compound should aid the search for other, less-toxic drugs to disrupt the ABCC4-MPP1 interaction.
The team’s findings could also enable clinicians to identify AML patients with high levels of ABCC4 and MPP1. In such patients, drugs that disrupt ABCC4-MPP1 might enhance the effectiveness of standard chemotherapy, Dr Schuetz said.
He also noted that other cancers, including breast and colon cancer and medulloblastoma, show high levels of both ABCC4 and MPP1. Chemotherapy for those cancers might also be enhanced by drugs that disrupt ABCC4-MPP1. ![]()
Researchers say they have uncovered a target to overcome drug resistance in acute myeloid leukemia (AML).
The team discovered how a linkage between 2 proteins enables AML cells to resist chemotherapy and showed that disrupting the linkage could render the cells vulnerable to treatment.
The researchers believe their discovery could lead to drugs to enhance chemotherapy in patients with AML and other cancers.
John Schuetz, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues described this research in Nature Communications.
The team launched their experiments based on previous findings that a protein called ABCC4 was greatly elevated in aggressive cases of AML.
Dr Schuetz and his colleagues searched for other proteins that might interact with ABCC4 and enable its function. The team’s screening of candidate proteins yielded one, MPP1, which was also greatly increased in AML.
The researchers found the 2 proteins are connected, and the connection enables cells to assume the characteristics of highly proliferative leukemia cells.
These experiments involved genetically altering hematopoietic progenitor cells to have high MPP1 and ABCC4 levels. The cells were grown in culture and then replated to see if they would continue to grow, as such self-renewal is a hallmark of leukemia cells.
The researchers found that serial regrowth depended on the cells having high levels of both ABCC4 and MPP1.
“Typically, if you take normal progenitors and you replate, you could do that one time, maybe twice,” Dr Schuetz said. “But our big surprise was that overexpressing MPP1—analogous to what you would see in leukemia—allows those progenitors to self-renew, to be replated over and over, to form new colonies.”
The experiments also revealed that MPP1 and ABCC4 functioned at the cell membrane, where they could play a role in the machinery that would rid the leukemia cells of chemotherapy drugs.
“When we disrupted their interaction, ABCC4 moved off the membrane and the cells became more sensitive to drugs used in AML—drugs that are pumped out of the cell by ABCC4,” Dr Schuetz said.
By screening thousands of compounds, the researchers identified some that could disrupt the ABCC4-MPP1 connection. One, called Antimycin-A, reversed drug resistance in AML cell lines and in cells from AML patients.
Antimycin-A is too toxic to be used in chemotherapy, but the researchers believe identification of the compound should aid the search for other, less-toxic drugs to disrupt the ABCC4-MPP1 interaction.
The team’s findings could also enable clinicians to identify AML patients with high levels of ABCC4 and MPP1. In such patients, drugs that disrupt ABCC4-MPP1 might enhance the effectiveness of standard chemotherapy, Dr Schuetz said.
He also noted that other cancers, including breast and colon cancer and medulloblastoma, show high levels of both ABCC4 and MPP1. Chemotherapy for those cancers might also be enhanced by drugs that disrupt ABCC4-MPP1. ![]()
Researchers say they have uncovered a target to overcome drug resistance in acute myeloid leukemia (AML).
The team discovered how a linkage between 2 proteins enables AML cells to resist chemotherapy and showed that disrupting the linkage could render the cells vulnerable to treatment.
The researchers believe their discovery could lead to drugs to enhance chemotherapy in patients with AML and other cancers.
John Schuetz, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues described this research in Nature Communications.
The team launched their experiments based on previous findings that a protein called ABCC4 was greatly elevated in aggressive cases of AML.
Dr Schuetz and his colleagues searched for other proteins that might interact with ABCC4 and enable its function. The team’s screening of candidate proteins yielded one, MPP1, which was also greatly increased in AML.
The researchers found the 2 proteins are connected, and the connection enables cells to assume the characteristics of highly proliferative leukemia cells.
These experiments involved genetically altering hematopoietic progenitor cells to have high MPP1 and ABCC4 levels. The cells were grown in culture and then replated to see if they would continue to grow, as such self-renewal is a hallmark of leukemia cells.
The researchers found that serial regrowth depended on the cells having high levels of both ABCC4 and MPP1.
“Typically, if you take normal progenitors and you replate, you could do that one time, maybe twice,” Dr Schuetz said. “But our big surprise was that overexpressing MPP1—analogous to what you would see in leukemia—allows those progenitors to self-renew, to be replated over and over, to form new colonies.”
The experiments also revealed that MPP1 and ABCC4 functioned at the cell membrane, where they could play a role in the machinery that would rid the leukemia cells of chemotherapy drugs.
“When we disrupted their interaction, ABCC4 moved off the membrane and the cells became more sensitive to drugs used in AML—drugs that are pumped out of the cell by ABCC4,” Dr Schuetz said.
By screening thousands of compounds, the researchers identified some that could disrupt the ABCC4-MPP1 connection. One, called Antimycin-A, reversed drug resistance in AML cell lines and in cells from AML patients.
Antimycin-A is too toxic to be used in chemotherapy, but the researchers believe identification of the compound should aid the search for other, less-toxic drugs to disrupt the ABCC4-MPP1 interaction.
The team’s findings could also enable clinicians to identify AML patients with high levels of ABCC4 and MPP1. In such patients, drugs that disrupt ABCC4-MPP1 might enhance the effectiveness of standard chemotherapy, Dr Schuetz said.
He also noted that other cancers, including breast and colon cancer and medulloblastoma, show high levels of both ABCC4 and MPP1. Chemotherapy for those cancers might also be enhanced by drugs that disrupt ABCC4-MPP1. ![]()
Product approved to treat patients with hemophilia A and inhibitors

The US Food and Drug Administration (FDA) has approved use of emicizumab-kxwh (Hemlibra®), a bispecific factor IXa- and factor X-directed antibody.
Emicizumab is approved as routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children who have hemophilia A and factor VIII (FVIII) inhibitors.
Emicizumab can be self-administered once-weekly via subcutaneous injection.
The labeling for emicizumab contains a boxed warning noting that patients who received emicizumab in conjunction with activated prothrombin complex concentrate developed thrombotic microangiopathy (TMA) and thromboembolic events (TEs).
Therefore, patients should discontinue prophylactic use of bypassing agents (BPAs) the day before starting prophylaxis with emicizumab.
The FDA granted the approval of emicizumab to Genentech, Inc. The agency granted the application for emicizumab priority review, and the product received breakthrough therapy and orphan drug designations.
Access to emicizumab
According to Genentech, emicizumab will be available shortly.
The company said it will be offering comprehensive services to help minimize barriers to access and reimbursement. Patients can call 866-436-5427 (866-HEMLIBRA) for more information.
For people who qualify, Genentech plans to offer patient assistance programs through Genentech Access Solutions. More information is available at 866-422-2377 (866-4ACCESS) or http://www.Genentech-Access.com.
Emicizumab trials
The biologics license application for emicizumab was supported by results from a pair of phase 3 studies—HAVEN 1 and HAVEN 2.
Results from HAVEN 1 were published in NEJM and presented at the 26th ISTH Congress in July. Interim results from HAVEN 2 were presented at ISTH as well.
HAVEN 1
The study enrolled 109 patients (age 12 and older) with hemophilia A and FVIII inhibitors who were previously treated with BPAs on-demand or as prophylaxis.
The patients were randomized to receive emicizumab prophylaxis or no prophylaxis. On-demand treatment of breakthrough bleeds with BPAs was allowed.
There was a significant reduction in treated bleeds of 87% with emicizumab prophylaxis compared to no prophylaxis (95% CI: 72.3; 94.3, P<0.0001). And there was an 80% reduction in all bleeds with emicizumab (95% CI: 62.5; 89.8, P<0.0001).
Adverse events occurring in at least 5% of patients treated with emicizumab were local injection site reactions, headache, fatigue, upper respiratory tract infection, and arthralgia.
Two patients experienced TEs, and 3 had TMA while receiving emicizumab prophylaxis and more than 100 u/kg/day of activated prothrombin complex concentrate, on average, for 24 hours or more before the event. Two of these patients had also received recombinant factor VIIa.
Neither TE required anticoagulation therapy, and 1 patient restarted emicizumab. The cases of TMA observed were transient, and 1 patient restarted emicizumab.
HAVEN 2
In this single-arm trial, researchers evaluated emicizumab prophylaxis in children younger than 12 years of age who had hemophilia A with FVIII inhibitors.
The interim efficacy analysis, after at least 12 weeks of treatment, included 23 children.
After a median observation time of 38.1 weeks, 87% (95% CI: 66.4; 97.2) of children who received emicizumab experienced 0 treated bleeds. The percentage with 0 treated or non-treated bleeds was lower, at 34.8% (95% CI: 16.4; 57.3).
The most common adverse events (observed in at least 10% of patients) were mild injection site reactions and nasopharyngitis. No TEs or TMAs were observed.
HAVEN 3 and 4
Emicizumab is now being studied in 2 additional phase 3 trials.
In HAVEN 3, researchers are evaluating emicizumab prophylaxis dosed once weekly or once every other week in patients age 12 and older with hemophilia A without FVIII inhibitors.
In HAVEN 4, researchers are evaluating emicizumab prophylaxis dosed every 4 weeks in patients age 12 and older with hemophilia A, with or without inhibitors. ![]()
The US Food and Drug Administration (FDA) has approved use of emicizumab-kxwh (Hemlibra®), a bispecific factor IXa- and factor X-directed antibody.
Emicizumab is approved as routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children who have hemophilia A and factor VIII (FVIII) inhibitors.
Emicizumab can be self-administered once-weekly via subcutaneous injection.
The labeling for emicizumab contains a boxed warning noting that patients who received emicizumab in conjunction with activated prothrombin complex concentrate developed thrombotic microangiopathy (TMA) and thromboembolic events (TEs).
Therefore, patients should discontinue prophylactic use of bypassing agents (BPAs) the day before starting prophylaxis with emicizumab.
The FDA granted the approval of emicizumab to Genentech, Inc. The agency granted the application for emicizumab priority review, and the product received breakthrough therapy and orphan drug designations.
Access to emicizumab
According to Genentech, emicizumab will be available shortly.
The company said it will be offering comprehensive services to help minimize barriers to access and reimbursement. Patients can call 866-436-5427 (866-HEMLIBRA) for more information.
For people who qualify, Genentech plans to offer patient assistance programs through Genentech Access Solutions. More information is available at 866-422-2377 (866-4ACCESS) or http://www.Genentech-Access.com.
Emicizumab trials
The biologics license application for emicizumab was supported by results from a pair of phase 3 studies—HAVEN 1 and HAVEN 2.
Results from HAVEN 1 were published in NEJM and presented at the 26th ISTH Congress in July. Interim results from HAVEN 2 were presented at ISTH as well.
HAVEN 1
The study enrolled 109 patients (age 12 and older) with hemophilia A and FVIII inhibitors who were previously treated with BPAs on-demand or as prophylaxis.
The patients were randomized to receive emicizumab prophylaxis or no prophylaxis. On-demand treatment of breakthrough bleeds with BPAs was allowed.
There was a significant reduction in treated bleeds of 87% with emicizumab prophylaxis compared to no prophylaxis (95% CI: 72.3; 94.3, P<0.0001). And there was an 80% reduction in all bleeds with emicizumab (95% CI: 62.5; 89.8, P<0.0001).
Adverse events occurring in at least 5% of patients treated with emicizumab were local injection site reactions, headache, fatigue, upper respiratory tract infection, and arthralgia.
Two patients experienced TEs, and 3 had TMA while receiving emicizumab prophylaxis and more than 100 u/kg/day of activated prothrombin complex concentrate, on average, for 24 hours or more before the event. Two of these patients had also received recombinant factor VIIa.
Neither TE required anticoagulation therapy, and 1 patient restarted emicizumab. The cases of TMA observed were transient, and 1 patient restarted emicizumab.
HAVEN 2
In this single-arm trial, researchers evaluated emicizumab prophylaxis in children younger than 12 years of age who had hemophilia A with FVIII inhibitors.
The interim efficacy analysis, after at least 12 weeks of treatment, included 23 children.
After a median observation time of 38.1 weeks, 87% (95% CI: 66.4; 97.2) of children who received emicizumab experienced 0 treated bleeds. The percentage with 0 treated or non-treated bleeds was lower, at 34.8% (95% CI: 16.4; 57.3).
The most common adverse events (observed in at least 10% of patients) were mild injection site reactions and nasopharyngitis. No TEs or TMAs were observed.
HAVEN 3 and 4
Emicizumab is now being studied in 2 additional phase 3 trials.
In HAVEN 3, researchers are evaluating emicizumab prophylaxis dosed once weekly or once every other week in patients age 12 and older with hemophilia A without FVIII inhibitors.
In HAVEN 4, researchers are evaluating emicizumab prophylaxis dosed every 4 weeks in patients age 12 and older with hemophilia A, with or without inhibitors. ![]()
The US Food and Drug Administration (FDA) has approved use of emicizumab-kxwh (Hemlibra®), a bispecific factor IXa- and factor X-directed antibody.
Emicizumab is approved as routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children who have hemophilia A and factor VIII (FVIII) inhibitors.
Emicizumab can be self-administered once-weekly via subcutaneous injection.
The labeling for emicizumab contains a boxed warning noting that patients who received emicizumab in conjunction with activated prothrombin complex concentrate developed thrombotic microangiopathy (TMA) and thromboembolic events (TEs).
Therefore, patients should discontinue prophylactic use of bypassing agents (BPAs) the day before starting prophylaxis with emicizumab.
The FDA granted the approval of emicizumab to Genentech, Inc. The agency granted the application for emicizumab priority review, and the product received breakthrough therapy and orphan drug designations.
Access to emicizumab
According to Genentech, emicizumab will be available shortly.
The company said it will be offering comprehensive services to help minimize barriers to access and reimbursement. Patients can call 866-436-5427 (866-HEMLIBRA) for more information.
For people who qualify, Genentech plans to offer patient assistance programs through Genentech Access Solutions. More information is available at 866-422-2377 (866-4ACCESS) or http://www.Genentech-Access.com.
Emicizumab trials
The biologics license application for emicizumab was supported by results from a pair of phase 3 studies—HAVEN 1 and HAVEN 2.
Results from HAVEN 1 were published in NEJM and presented at the 26th ISTH Congress in July. Interim results from HAVEN 2 were presented at ISTH as well.
HAVEN 1
The study enrolled 109 patients (age 12 and older) with hemophilia A and FVIII inhibitors who were previously treated with BPAs on-demand or as prophylaxis.
The patients were randomized to receive emicizumab prophylaxis or no prophylaxis. On-demand treatment of breakthrough bleeds with BPAs was allowed.
There was a significant reduction in treated bleeds of 87% with emicizumab prophylaxis compared to no prophylaxis (95% CI: 72.3; 94.3, P<0.0001). And there was an 80% reduction in all bleeds with emicizumab (95% CI: 62.5; 89.8, P<0.0001).
Adverse events occurring in at least 5% of patients treated with emicizumab were local injection site reactions, headache, fatigue, upper respiratory tract infection, and arthralgia.
Two patients experienced TEs, and 3 had TMA while receiving emicizumab prophylaxis and more than 100 u/kg/day of activated prothrombin complex concentrate, on average, for 24 hours or more before the event. Two of these patients had also received recombinant factor VIIa.
Neither TE required anticoagulation therapy, and 1 patient restarted emicizumab. The cases of TMA observed were transient, and 1 patient restarted emicizumab.
HAVEN 2
In this single-arm trial, researchers evaluated emicizumab prophylaxis in children younger than 12 years of age who had hemophilia A with FVIII inhibitors.
The interim efficacy analysis, after at least 12 weeks of treatment, included 23 children.
After a median observation time of 38.1 weeks, 87% (95% CI: 66.4; 97.2) of children who received emicizumab experienced 0 treated bleeds. The percentage with 0 treated or non-treated bleeds was lower, at 34.8% (95% CI: 16.4; 57.3).
The most common adverse events (observed in at least 10% of patients) were mild injection site reactions and nasopharyngitis. No TEs or TMAs were observed.
HAVEN 3 and 4
Emicizumab is now being studied in 2 additional phase 3 trials.
In HAVEN 3, researchers are evaluating emicizumab prophylaxis dosed once weekly or once every other week in patients age 12 and older with hemophilia A without FVIII inhibitors.
In HAVEN 4, researchers are evaluating emicizumab prophylaxis dosed every 4 weeks in patients age 12 and older with hemophilia A, with or without inhibitors. ![]()
FDA expands approval of obinutuzumab

The US Food and Drug Administration (FDA) has expanded the approved use of obinutuzumab (Gazyva®).
The drug is now approved for use in combination with chemotherapy to treat patients with previously untreated follicular lymphoma (FL) that is advanced (stage II bulky, stage III, or stage IV) .
In patients who respond to this treatment, obinutuzumab monotherapy can be given as maintenance.
The FDA granted this new approval of obinutuzumab to Genentech, Inc. The application for obinutuzumab in this indication received priority review.
The latest FDA approval means obinutuzumab is available in the US for the following indications:
- In combination with chlorambucil to treat chronic lymphocytic leukemia (CLL) in adults who have not had previous CLL treatment
- In combination with bendamustine, followed by obinutuzumab alone, to treat FL in adults who did not respond to a rituximab-containing regimen or whose FL returned after such treatment
- In combination with chemotherapy, followed by obinutuzumab alone in responders, to treat stage II bulky, stage III, or stage IV FL in adults who have not had previous FL treatment.
Phase 3 results
The latest approval of obinutuzumab is based on results from the phase 3 GALLIUM study, which were presented at the 2016 ASH Annual Meeting and published in NEJM in October.
The following are updated data from the obinutuzumab prescribing information.
GALLIUM included 1385 patients with previously untreated non-Hodgkin lymphoma, and 1202 of these patients had advanced FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone), CVP (cyclophosphamide, vincristine, and prednisolone), and bendamustine.
At a median observation time of 38 months, the overall response rate was 91% in the obinutuzumab arm and 88% in the rituximab arm. The complete response rates were 28% and 27%, respectively.
The median progression-free survival was not reached in either arm. The hazard ratio, for obinutuzumab compared to rituximab, was 0.72 (95% CI, 0.56-0.93, P=0.0118).
Safety was evaluated based on all 1385 patients in the study, 86% of whom had previously untreated FL and 14% of whom had marginal zone lymphoma.
Serious adverse events (AEs) occurred in 50% of patients in the obinutuzumab arm and 43% in the rituximab arm. Fatal AEs occurred in 5% and 4%, respectively. Infections and second malignancies were the leading causes of these deaths.
The most common AEs (incidence ≥ 20%) observed at least 2% more patients in the obinutuzumab arm were infusion-related reactions, neutropenia, upper respiratory tract infection, cough, constipation, and diarrhea.
The most common grade 3 to 5 AEs (incidence ≥ 5%) observed more frequently in the obinutuzumab arm were neutropenia, infusion-related reactions, febrile neutropenia, and thrombocytopenia. ![]()
The US Food and Drug Administration (FDA) has expanded the approved use of obinutuzumab (Gazyva®).
The drug is now approved for use in combination with chemotherapy to treat patients with previously untreated follicular lymphoma (FL) that is advanced (stage II bulky, stage III, or stage IV) .
In patients who respond to this treatment, obinutuzumab monotherapy can be given as maintenance.
The FDA granted this new approval of obinutuzumab to Genentech, Inc. The application for obinutuzumab in this indication received priority review.
The latest FDA approval means obinutuzumab is available in the US for the following indications:
- In combination with chlorambucil to treat chronic lymphocytic leukemia (CLL) in adults who have not had previous CLL treatment
- In combination with bendamustine, followed by obinutuzumab alone, to treat FL in adults who did not respond to a rituximab-containing regimen or whose FL returned after such treatment
- In combination with chemotherapy, followed by obinutuzumab alone in responders, to treat stage II bulky, stage III, or stage IV FL in adults who have not had previous FL treatment.
Phase 3 results
The latest approval of obinutuzumab is based on results from the phase 3 GALLIUM study, which were presented at the 2016 ASH Annual Meeting and published in NEJM in October.
The following are updated data from the obinutuzumab prescribing information.
GALLIUM included 1385 patients with previously untreated non-Hodgkin lymphoma, and 1202 of these patients had advanced FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone), CVP (cyclophosphamide, vincristine, and prednisolone), and bendamustine.
At a median observation time of 38 months, the overall response rate was 91% in the obinutuzumab arm and 88% in the rituximab arm. The complete response rates were 28% and 27%, respectively.
The median progression-free survival was not reached in either arm. The hazard ratio, for obinutuzumab compared to rituximab, was 0.72 (95% CI, 0.56-0.93, P=0.0118).
Safety was evaluated based on all 1385 patients in the study, 86% of whom had previously untreated FL and 14% of whom had marginal zone lymphoma.
Serious adverse events (AEs) occurred in 50% of patients in the obinutuzumab arm and 43% in the rituximab arm. Fatal AEs occurred in 5% and 4%, respectively. Infections and second malignancies were the leading causes of these deaths.
The most common AEs (incidence ≥ 20%) observed at least 2% more patients in the obinutuzumab arm were infusion-related reactions, neutropenia, upper respiratory tract infection, cough, constipation, and diarrhea.
The most common grade 3 to 5 AEs (incidence ≥ 5%) observed more frequently in the obinutuzumab arm were neutropenia, infusion-related reactions, febrile neutropenia, and thrombocytopenia. ![]()
The US Food and Drug Administration (FDA) has expanded the approved use of obinutuzumab (Gazyva®).
The drug is now approved for use in combination with chemotherapy to treat patients with previously untreated follicular lymphoma (FL) that is advanced (stage II bulky, stage III, or stage IV) .
In patients who respond to this treatment, obinutuzumab monotherapy can be given as maintenance.
The FDA granted this new approval of obinutuzumab to Genentech, Inc. The application for obinutuzumab in this indication received priority review.
The latest FDA approval means obinutuzumab is available in the US for the following indications:
- In combination with chlorambucil to treat chronic lymphocytic leukemia (CLL) in adults who have not had previous CLL treatment
- In combination with bendamustine, followed by obinutuzumab alone, to treat FL in adults who did not respond to a rituximab-containing regimen or whose FL returned after such treatment
- In combination with chemotherapy, followed by obinutuzumab alone in responders, to treat stage II bulky, stage III, or stage IV FL in adults who have not had previous FL treatment.
Phase 3 results
The latest approval of obinutuzumab is based on results from the phase 3 GALLIUM study, which were presented at the 2016 ASH Annual Meeting and published in NEJM in October.
The following are updated data from the obinutuzumab prescribing information.
GALLIUM included 1385 patients with previously untreated non-Hodgkin lymphoma, and 1202 of these patients had advanced FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone), CVP (cyclophosphamide, vincristine, and prednisolone), and bendamustine.
At a median observation time of 38 months, the overall response rate was 91% in the obinutuzumab arm and 88% in the rituximab arm. The complete response rates were 28% and 27%, respectively.
The median progression-free survival was not reached in either arm. The hazard ratio, for obinutuzumab compared to rituximab, was 0.72 (95% CI, 0.56-0.93, P=0.0118).
Safety was evaluated based on all 1385 patients in the study, 86% of whom had previously untreated FL and 14% of whom had marginal zone lymphoma.
Serious adverse events (AEs) occurred in 50% of patients in the obinutuzumab arm and 43% in the rituximab arm. Fatal AEs occurred in 5% and 4%, respectively. Infections and second malignancies were the leading causes of these deaths.
The most common AEs (incidence ≥ 20%) observed at least 2% more patients in the obinutuzumab arm were infusion-related reactions, neutropenia, upper respiratory tract infection, cough, constipation, and diarrhea.
The most common grade 3 to 5 AEs (incidence ≥ 5%) observed more frequently in the obinutuzumab arm were neutropenia, infusion-related reactions, febrile neutropenia, and thrombocytopenia. ![]()
CAR T-cell therapy on fast track with FDA, EMA

A chimeric antigen receptor (CAR) T-cell therapy, bb2121, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) and was granted access to the European Medicines Agency’s (EMA’s) PRIority MEdicines (PRIME) program.
The CAR T-cell therapy is designed to target B-cell maturation antigen in previously treated patients with multiple myeloma.
bb2121 is being developed by bluebird bio, Inc., and Celgene Corporation.
The EMA’s and FDA’s decisions on bb2121 were based on preliminary data from an ongoing phase 1 study, CRB-401 (NCT02658929).
Results from this study were presented at the 2017 ASCO Annual Meeting (abstract 3010).
The study enrolled patients with relapsed and/or refractory multiple myeloma. As of the May 4, 2017 data cut-off, 21 patients had been enrolled.
Patients had a median of 7 prior lines of therapy (range, 3-14). Their previous treatments included lenalidomide and bortezomib (100%), pomalidomide and carfilzomib (91%), daratumumab (71%), and autologous stem cell transplant (100% at least once).
Twenty-nine percent of patients were refractory to bortezomib, lenalidomide, carfilzomib, pomalidomide, and daratumumab.
Patients received a conditioning regimen of cyclophosphamide and fludarabine, followed by an infusion of bb2121 at 1 of 4 doses: 50 x 106, 150 x 106, 450 x 106 and 800 x 106 CAR+ T cells.
All 21 patients were evaluable for safety.
The most common treatment-emergent grade 3-4 adverse events were cytopenias commonly associated with the lymphodepletion regimen, as well as grade 3 events of hyponatremia (n=4), upper respiratory infection (n=2), syncope (n=2), and cytokine release syndrome (CRS, n=2).
In all, 71% of patients (15/21) had CRS, mostly grade 1 and 2. For the 2 patients with grade 3 CRS, it resolved within 24 hours. To manage CRS, 4 patients received tocilizumab, and 1 (with grade 2) received steroids as well.
Eighteen patients were evaluable for efficacy, and the overall response rate was 89% (16/18).
There were 4 complete responses—2 in the 150 x 106 cohort, 1 in the 450 x 106 cohort, and 1 in the 800 x 106 cohort. The complete responder in the 450 x 106 cohort ultimately died of cardiopulmonary arrest that was deemed unrelated to treatment.
Five patients had a partial response—2 in the 450 x 106 cohort and 1 in each of the other cohorts. Seven patients had a very good partial response—5 in the 450 x 106 cohort and 1 each in the 150 x 106 cohort and 800 x 106 cohort.
Updated data from this study are scheduled to be presented at the 2017 ASH Annual Meeting (abstract 740).
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
About PRIME
The EMA launched its PRIME program to enhance support for the development of medicines that target an unmet medical need.
The program involves enhanced interaction and early dialogue with developers of promising medicines to optimize development plans and speed up evaluation so these medicines can reach patients earlier.
PRIME focuses on medicines that may offer a major therapeutic advantage over existing treatments or benefit patients without treatment options. To be accepted for PRIME, a medicine must have demonstrated the potential to benefit patients with unmet medical needs based on early clinical data. ![]()
A chimeric antigen receptor (CAR) T-cell therapy, bb2121, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) and was granted access to the European Medicines Agency’s (EMA’s) PRIority MEdicines (PRIME) program.
The CAR T-cell therapy is designed to target B-cell maturation antigen in previously treated patients with multiple myeloma.
bb2121 is being developed by bluebird bio, Inc., and Celgene Corporation.
The EMA’s and FDA’s decisions on bb2121 were based on preliminary data from an ongoing phase 1 study, CRB-401 (NCT02658929).
Results from this study were presented at the 2017 ASCO Annual Meeting (abstract 3010).
The study enrolled patients with relapsed and/or refractory multiple myeloma. As of the May 4, 2017 data cut-off, 21 patients had been enrolled.
Patients had a median of 7 prior lines of therapy (range, 3-14). Their previous treatments included lenalidomide and bortezomib (100%), pomalidomide and carfilzomib (91%), daratumumab (71%), and autologous stem cell transplant (100% at least once).
Twenty-nine percent of patients were refractory to bortezomib, lenalidomide, carfilzomib, pomalidomide, and daratumumab.
Patients received a conditioning regimen of cyclophosphamide and fludarabine, followed by an infusion of bb2121 at 1 of 4 doses: 50 x 106, 150 x 106, 450 x 106 and 800 x 106 CAR+ T cells.
All 21 patients were evaluable for safety.
The most common treatment-emergent grade 3-4 adverse events were cytopenias commonly associated with the lymphodepletion regimen, as well as grade 3 events of hyponatremia (n=4), upper respiratory infection (n=2), syncope (n=2), and cytokine release syndrome (CRS, n=2).
In all, 71% of patients (15/21) had CRS, mostly grade 1 and 2. For the 2 patients with grade 3 CRS, it resolved within 24 hours. To manage CRS, 4 patients received tocilizumab, and 1 (with grade 2) received steroids as well.
Eighteen patients were evaluable for efficacy, and the overall response rate was 89% (16/18).
There were 4 complete responses—2 in the 150 x 106 cohort, 1 in the 450 x 106 cohort, and 1 in the 800 x 106 cohort. The complete responder in the 450 x 106 cohort ultimately died of cardiopulmonary arrest that was deemed unrelated to treatment.
Five patients had a partial response—2 in the 450 x 106 cohort and 1 in each of the other cohorts. Seven patients had a very good partial response—5 in the 450 x 106 cohort and 1 each in the 150 x 106 cohort and 800 x 106 cohort.
Updated data from this study are scheduled to be presented at the 2017 ASH Annual Meeting (abstract 740).
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
About PRIME
The EMA launched its PRIME program to enhance support for the development of medicines that target an unmet medical need.
The program involves enhanced interaction and early dialogue with developers of promising medicines to optimize development plans and speed up evaluation so these medicines can reach patients earlier.
PRIME focuses on medicines that may offer a major therapeutic advantage over existing treatments or benefit patients without treatment options. To be accepted for PRIME, a medicine must have demonstrated the potential to benefit patients with unmet medical needs based on early clinical data. ![]()
A chimeric antigen receptor (CAR) T-cell therapy, bb2121, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) and was granted access to the European Medicines Agency’s (EMA’s) PRIority MEdicines (PRIME) program.
The CAR T-cell therapy is designed to target B-cell maturation antigen in previously treated patients with multiple myeloma.
bb2121 is being developed by bluebird bio, Inc., and Celgene Corporation.
The EMA’s and FDA’s decisions on bb2121 were based on preliminary data from an ongoing phase 1 study, CRB-401 (NCT02658929).
Results from this study were presented at the 2017 ASCO Annual Meeting (abstract 3010).
The study enrolled patients with relapsed and/or refractory multiple myeloma. As of the May 4, 2017 data cut-off, 21 patients had been enrolled.
Patients had a median of 7 prior lines of therapy (range, 3-14). Their previous treatments included lenalidomide and bortezomib (100%), pomalidomide and carfilzomib (91%), daratumumab (71%), and autologous stem cell transplant (100% at least once).
Twenty-nine percent of patients were refractory to bortezomib, lenalidomide, carfilzomib, pomalidomide, and daratumumab.
Patients received a conditioning regimen of cyclophosphamide and fludarabine, followed by an infusion of bb2121 at 1 of 4 doses: 50 x 106, 150 x 106, 450 x 106 and 800 x 106 CAR+ T cells.
All 21 patients were evaluable for safety.
The most common treatment-emergent grade 3-4 adverse events were cytopenias commonly associated with the lymphodepletion regimen, as well as grade 3 events of hyponatremia (n=4), upper respiratory infection (n=2), syncope (n=2), and cytokine release syndrome (CRS, n=2).
In all, 71% of patients (15/21) had CRS, mostly grade 1 and 2. For the 2 patients with grade 3 CRS, it resolved within 24 hours. To manage CRS, 4 patients received tocilizumab, and 1 (with grade 2) received steroids as well.
Eighteen patients were evaluable for efficacy, and the overall response rate was 89% (16/18).
There were 4 complete responses—2 in the 150 x 106 cohort, 1 in the 450 x 106 cohort, and 1 in the 800 x 106 cohort. The complete responder in the 450 x 106 cohort ultimately died of cardiopulmonary arrest that was deemed unrelated to treatment.
Five patients had a partial response—2 in the 450 x 106 cohort and 1 in each of the other cohorts. Seven patients had a very good partial response—5 in the 450 x 106 cohort and 1 each in the 150 x 106 cohort and 800 x 106 cohort.
Updated data from this study are scheduled to be presented at the 2017 ASH Annual Meeting (abstract 740).
About breakthrough designation
The FDA’s breakthrough designation is intended to expedite the development and review of new treatments for serious or life-threatening conditions.
The designation entitles the company developing a therapy to more intensive FDA guidance on an efficient and accelerated development program, as well as eligibility for other actions to expedite FDA review, such as rolling submission and priority review.
To earn breakthrough designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
About PRIME
The EMA launched its PRIME program to enhance support for the development of medicines that target an unmet medical need.
The program involves enhanced interaction and early dialogue with developers of promising medicines to optimize development plans and speed up evaluation so these medicines can reach patients earlier.
PRIME focuses on medicines that may offer a major therapeutic advantage over existing treatments or benefit patients without treatment options. To be accepted for PRIME, a medicine must have demonstrated the potential to benefit patients with unmet medical needs based on early clinical data.
Rigosertib produces better OS in MDS than tAML

Rigosertib has demonstrated activity and tolerability in patients with myelodysplastic syndromes (MDS) and acute myeloid leukemia transformed from MDS (tAML), according to researchers.
In a phase 1/2 study, rigosertib produced responses in a quarter of MDS/tAML patients and enabled stable disease in another quarter.
Overall survival (OS) was about a year longer for responders than for non-responders.
MDS patients were more likely to respond to rigosertib and therefore enjoyed longer OS than tAML patients.
Overall, rigosertib was considered well-tolerated. There were no treatment-related deaths, though 18% of patients experienced treatment-related serious adverse events (AEs).
Lewis Silverman, MD, of Icahn School of Medicine at Mount Sinai in New York, New York, and his colleagues described these results in Leukemia Research.
The study was sponsored by Onconova Therapeutics, Inc., the company developing rigosertib.
Rigosertib is an inhibitor of Ras-effector pathways that interacts with the Ras binding domains common to several signaling proteins, including Raf and PI3 kinase.
Dr Silverman and his colleagues tested intravenous rigosertib in a dose-escalation, phase 1/2 study of 22 patients. Patients had tAML (n=13), high-risk MDS (n=6), intermediate-2-risk MDS (n=2), or chronic myelomonocytic leukemia (n=1).
All patients had relapsed or were refractory to standard therapy and had no approved options for second-line therapies. The patients’ median age was 78 (range, 59-84), and 90% were male.
Patients received 3- to 7-day continuous infusions of rigosertib at doses ranging from 650 mg/m2/day to 1700 mg/m2/day in 14-day cycles.
The mean number of treatment cycles was 5.6 ± 5.8 (range, 1-23). The maximum tolerated dose of rigosertib was 1700 mg/m2/day, and the recommended phase 2 dose was 1375 mg/m2/day.
Safety
All patients had at least 1 AE. The most common AEs of any grade were fatigue (n=16, 73%), diarrhea (n=12, 55%), pyrexia (n=12, 55%), dyspnea (n=11, 50%), insomnia (n=11, 50%), anemia (n=10, 46%), constipation (n=9, 41%), nausea (n=9, 41%), cough (n=9, 41%), and decreased appetite (n=9, 41%).
The most common grade 3 or higher AEs were anemia (n=9, 41%), thrombocytopenia (n=5, 23%), pneumonia (n=5, 23%), hypoglycemia (n=4, 18%), hyponatremia (n=4, 18%), and hypophosphatemia (n=4, 18%).
Four patients (18%) had treatment-related serious AEs. This included hematuria and pollakiuria (n=1), dysuria and pollakiuria (n=1), asthenia (n=1), and dyspnea (n=1). Thirteen patients (59%) stopped treatment due to AEs.
Ten patients, who remained on study from 1 to 19 months, died within 30 days of stopping rigosertib. There were no treatment-related deaths.
Efficacy
Nineteen patients were evaluable for efficacy.
Five patients responded to treatment. Four patients with MDS had a marrow complete response, and 1 with tAML had a marrow partial response. Two of the patients with marrow complete response also had hematologic improvements.
Five patients had stable disease, 3 with MDS and 2 with tAML.
The median OS was 15.7 months for responders and 2.0 months for non-responders (P=0.0070). The median OS was 12.0 months for MDS patients and 2.0 months for tAML patients (P<0.0001).
“The publication of results from this historical study provides support of the relationship between bone marrow blast response and improvement in overall survival in this group of patients with MDS and acute myeloid leukemia for whom no FDA-approved treatments are currently available,” said Ramesh Kumar, president and chief executive officer of Onconova Therapeutics, Inc.
He added that these data are “fundamental to the rationale” of ongoing studies of rigosertib in high-risk MDS patients.
Rigosertib has demonstrated activity and tolerability in patients with myelodysplastic syndromes (MDS) and acute myeloid leukemia transformed from MDS (tAML), according to researchers.
In a phase 1/2 study, rigosertib produced responses in a quarter of MDS/tAML patients and enabled stable disease in another quarter.
Overall survival (OS) was about a year longer for responders than for non-responders.
MDS patients were more likely to respond to rigosertib and therefore enjoyed longer OS than tAML patients.
Overall, rigosertib was considered well-tolerated. There were no treatment-related deaths, though 18% of patients experienced treatment-related serious adverse events (AEs).
Lewis Silverman, MD, of Icahn School of Medicine at Mount Sinai in New York, New York, and his colleagues described these results in Leukemia Research.
The study was sponsored by Onconova Therapeutics, Inc., the company developing rigosertib.
Rigosertib is an inhibitor of Ras-effector pathways that interacts with the Ras binding domains common to several signaling proteins, including Raf and PI3 kinase.
Dr Silverman and his colleagues tested intravenous rigosertib in a dose-escalation, phase 1/2 study of 22 patients. Patients had tAML (n=13), high-risk MDS (n=6), intermediate-2-risk MDS (n=2), or chronic myelomonocytic leukemia (n=1).
All patients had relapsed or were refractory to standard therapy and had no approved options for second-line therapies. The patients’ median age was 78 (range, 59-84), and 90% were male.
Patients received 3- to 7-day continuous infusions of rigosertib at doses ranging from 650 mg/m2/day to 1700 mg/m2/day in 14-day cycles.
The mean number of treatment cycles was 5.6 ± 5.8 (range, 1-23). The maximum tolerated dose of rigosertib was 1700 mg/m2/day, and the recommended phase 2 dose was 1375 mg/m2/day.
Safety
All patients had at least 1 AE. The most common AEs of any grade were fatigue (n=16, 73%), diarrhea (n=12, 55%), pyrexia (n=12, 55%), dyspnea (n=11, 50%), insomnia (n=11, 50%), anemia (n=10, 46%), constipation (n=9, 41%), nausea (n=9, 41%), cough (n=9, 41%), and decreased appetite (n=9, 41%).
The most common grade 3 or higher AEs were anemia (n=9, 41%), thrombocytopenia (n=5, 23%), pneumonia (n=5, 23%), hypoglycemia (n=4, 18%), hyponatremia (n=4, 18%), and hypophosphatemia (n=4, 18%).
Four patients (18%) had treatment-related serious AEs. This included hematuria and pollakiuria (n=1), dysuria and pollakiuria (n=1), asthenia (n=1), and dyspnea (n=1). Thirteen patients (59%) stopped treatment due to AEs.
Ten patients, who remained on study from 1 to 19 months, died within 30 days of stopping rigosertib. There were no treatment-related deaths.
Efficacy
Nineteen patients were evaluable for efficacy.
Five patients responded to treatment. Four patients with MDS had a marrow complete response, and 1 with tAML had a marrow partial response. Two of the patients with marrow complete response also had hematologic improvements.
Five patients had stable disease, 3 with MDS and 2 with tAML.
The median OS was 15.7 months for responders and 2.0 months for non-responders (P=0.0070). The median OS was 12.0 months for MDS patients and 2.0 months for tAML patients (P<0.0001).
“The publication of results from this historical study provides support of the relationship between bone marrow blast response and improvement in overall survival in this group of patients with MDS and acute myeloid leukemia for whom no FDA-approved treatments are currently available,” said Ramesh Kumar, president and chief executive officer of Onconova Therapeutics, Inc.
He added that these data are “fundamental to the rationale” of ongoing studies of rigosertib in high-risk MDS patients.
Rigosertib has demonstrated activity and tolerability in patients with myelodysplastic syndromes (MDS) and acute myeloid leukemia transformed from MDS (tAML), according to researchers.
In a phase 1/2 study, rigosertib produced responses in a quarter of MDS/tAML patients and enabled stable disease in another quarter.
Overall survival (OS) was about a year longer for responders than for non-responders.
MDS patients were more likely to respond to rigosertib and therefore enjoyed longer OS than tAML patients.
Overall, rigosertib was considered well-tolerated. There were no treatment-related deaths, though 18% of patients experienced treatment-related serious adverse events (AEs).
Lewis Silverman, MD, of Icahn School of Medicine at Mount Sinai in New York, New York, and his colleagues described these results in Leukemia Research.
The study was sponsored by Onconova Therapeutics, Inc., the company developing rigosertib.
Rigosertib is an inhibitor of Ras-effector pathways that interacts with the Ras binding domains common to several signaling proteins, including Raf and PI3 kinase.
Dr Silverman and his colleagues tested intravenous rigosertib in a dose-escalation, phase 1/2 study of 22 patients. Patients had tAML (n=13), high-risk MDS (n=6), intermediate-2-risk MDS (n=2), or chronic myelomonocytic leukemia (n=1).
All patients had relapsed or were refractory to standard therapy and had no approved options for second-line therapies. The patients’ median age was 78 (range, 59-84), and 90% were male.
Patients received 3- to 7-day continuous infusions of rigosertib at doses ranging from 650 mg/m2/day to 1700 mg/m2/day in 14-day cycles.
The mean number of treatment cycles was 5.6 ± 5.8 (range, 1-23). The maximum tolerated dose of rigosertib was 1700 mg/m2/day, and the recommended phase 2 dose was 1375 mg/m2/day.
Safety
All patients had at least 1 AE. The most common AEs of any grade were fatigue (n=16, 73%), diarrhea (n=12, 55%), pyrexia (n=12, 55%), dyspnea (n=11, 50%), insomnia (n=11, 50%), anemia (n=10, 46%), constipation (n=9, 41%), nausea (n=9, 41%), cough (n=9, 41%), and decreased appetite (n=9, 41%).
The most common grade 3 or higher AEs were anemia (n=9, 41%), thrombocytopenia (n=5, 23%), pneumonia (n=5, 23%), hypoglycemia (n=4, 18%), hyponatremia (n=4, 18%), and hypophosphatemia (n=4, 18%).
Four patients (18%) had treatment-related serious AEs. This included hematuria and pollakiuria (n=1), dysuria and pollakiuria (n=1), asthenia (n=1), and dyspnea (n=1). Thirteen patients (59%) stopped treatment due to AEs.
Ten patients, who remained on study from 1 to 19 months, died within 30 days of stopping rigosertib. There were no treatment-related deaths.
Efficacy
Nineteen patients were evaluable for efficacy.
Five patients responded to treatment. Four patients with MDS had a marrow complete response, and 1 with tAML had a marrow partial response. Two of the patients with marrow complete response also had hematologic improvements.
Five patients had stable disease, 3 with MDS and 2 with tAML.
The median OS was 15.7 months for responders and 2.0 months for non-responders (P=0.0070). The median OS was 12.0 months for MDS patients and 2.0 months for tAML patients (P<0.0001).
“The publication of results from this historical study provides support of the relationship between bone marrow blast response and improvement in overall survival in this group of patients with MDS and acute myeloid leukemia for whom no FDA-approved treatments are currently available,” said Ramesh Kumar, president and chief executive officer of Onconova Therapeutics, Inc.
He added that these data are “fundamental to the rationale” of ongoing studies of rigosertib in high-risk MDS patients.
Drug can treat severely ill SCD patients, case suggests

ATLANTA—Results of a case study suggest voxelotor (previously GBT440) can be effective in severely ill patients with sickle cell disease (SCD).
Voxelotor is currently under investigation in the phase 3 HOPE study, which includes SCD patients age 12 and older.
A 67-year-old male SCD patient could not participate in the study due to severe, transfusion-refractory anemia, so he received voxelotor via compassionate access.
This patient’s results were presented at the Sickle Cell Disease Association of America (SCDAA) 45th Annual National Convention.
The patient had the HbSS genotype with severe anemia that was refractory to transfusion. The patient had developed red cell antibodies after receiving multiple transfusions, and these antibodies prevented further transfusions to correct his anemia.
The patient also had moderate chronic obstructive pulmonary disease requiring supplemental oxygen therapy, recurrent and frequent pain exacerbations, extreme fatigue, and clinical depression.
The patient received voxelotor at 900 mg orally once daily. He responded to the treatment within 1 to 2 weeks, experiencing improvements in pain, fatigue, and overall mental health (as measured by the Patient Health Quality-9 score).
The patient’s hemoglobin levels rose quickly, to approximately 1.5 g/dL above baseline, with a sustained increase over 66 weeks in the range of 1 to 1.5 g/dL.
There were reductions in reticulocyte count and bilirubin as well, both consistent with diminished hemolysis.
The patient’s blood oxygen saturation improved on the standard walk test, from 86 mmHg at baseline to 96 mmHg at 65 weeks, and he discontinued continuous oxygen supplementation.
The patient has not been hospitalized due to sickle cell pain since he started taking voxelotor.
He has experienced a treatment-related side effect—grade 2 diarrhea. This occurred 9 weeks after he started voxelotor treatment, when the dose was increased to 1500 mg daily, but it resolved upon return to 900 mg. The patient has experienced no other treatment-related side effects.
Clinical and laboratory improvements have continued for more than 17 months, and the patient remains on treatment today under compassionate use access.
“This severely ill SCD patient’s clinical response, assessed by both objective and subjective measures, illustrates why we are encouraged by the voxelotor program,” said Ted W. Love, MD, president and chief executive officer of Global Blood Therapeutics, the company developing voxelotor.
“We plan to present additional data from other severely ill sickle cell patients who have received voxelotor via single-patient compassionate access treatment at FSCDR [the Foundation for Sickle Cell Disease Research] at an upcoming medical meeting. Of course, controlled clinical trials are needed to assess the efficacy and safety of voxelotor in SCD patients, including those with severe anemia.”
ATLANTA—Results of a case study suggest voxelotor (previously GBT440) can be effective in severely ill patients with sickle cell disease (SCD).
Voxelotor is currently under investigation in the phase 3 HOPE study, which includes SCD patients age 12 and older.
A 67-year-old male SCD patient could not participate in the study due to severe, transfusion-refractory anemia, so he received voxelotor via compassionate access.
This patient’s results were presented at the Sickle Cell Disease Association of America (SCDAA) 45th Annual National Convention.
The patient had the HbSS genotype with severe anemia that was refractory to transfusion. The patient had developed red cell antibodies after receiving multiple transfusions, and these antibodies prevented further transfusions to correct his anemia.
The patient also had moderate chronic obstructive pulmonary disease requiring supplemental oxygen therapy, recurrent and frequent pain exacerbations, extreme fatigue, and clinical depression.
The patient received voxelotor at 900 mg orally once daily. He responded to the treatment within 1 to 2 weeks, experiencing improvements in pain, fatigue, and overall mental health (as measured by the Patient Health Quality-9 score).
The patient’s hemoglobin levels rose quickly, to approximately 1.5 g/dL above baseline, with a sustained increase over 66 weeks in the range of 1 to 1.5 g/dL.
There were reductions in reticulocyte count and bilirubin as well, both consistent with diminished hemolysis.
The patient’s blood oxygen saturation improved on the standard walk test, from 86 mmHg at baseline to 96 mmHg at 65 weeks, and he discontinued continuous oxygen supplementation.
The patient has not been hospitalized due to sickle cell pain since he started taking voxelotor.
He has experienced a treatment-related side effect—grade 2 diarrhea. This occurred 9 weeks after he started voxelotor treatment, when the dose was increased to 1500 mg daily, but it resolved upon return to 900 mg. The patient has experienced no other treatment-related side effects.
Clinical and laboratory improvements have continued for more than 17 months, and the patient remains on treatment today under compassionate use access.
“This severely ill SCD patient’s clinical response, assessed by both objective and subjective measures, illustrates why we are encouraged by the voxelotor program,” said Ted W. Love, MD, president and chief executive officer of Global Blood Therapeutics, the company developing voxelotor.
“We plan to present additional data from other severely ill sickle cell patients who have received voxelotor via single-patient compassionate access treatment at FSCDR [the Foundation for Sickle Cell Disease Research] at an upcoming medical meeting. Of course, controlled clinical trials are needed to assess the efficacy and safety of voxelotor in SCD patients, including those with severe anemia.”
ATLANTA—Results of a case study suggest voxelotor (previously GBT440) can be effective in severely ill patients with sickle cell disease (SCD).
Voxelotor is currently under investigation in the phase 3 HOPE study, which includes SCD patients age 12 and older.
A 67-year-old male SCD patient could not participate in the study due to severe, transfusion-refractory anemia, so he received voxelotor via compassionate access.
This patient’s results were presented at the Sickle Cell Disease Association of America (SCDAA) 45th Annual National Convention.
The patient had the HbSS genotype with severe anemia that was refractory to transfusion. The patient had developed red cell antibodies after receiving multiple transfusions, and these antibodies prevented further transfusions to correct his anemia.
The patient also had moderate chronic obstructive pulmonary disease requiring supplemental oxygen therapy, recurrent and frequent pain exacerbations, extreme fatigue, and clinical depression.
The patient received voxelotor at 900 mg orally once daily. He responded to the treatment within 1 to 2 weeks, experiencing improvements in pain, fatigue, and overall mental health (as measured by the Patient Health Quality-9 score).
The patient’s hemoglobin levels rose quickly, to approximately 1.5 g/dL above baseline, with a sustained increase over 66 weeks in the range of 1 to 1.5 g/dL.
There were reductions in reticulocyte count and bilirubin as well, both consistent with diminished hemolysis.
The patient’s blood oxygen saturation improved on the standard walk test, from 86 mmHg at baseline to 96 mmHg at 65 weeks, and he discontinued continuous oxygen supplementation.
The patient has not been hospitalized due to sickle cell pain since he started taking voxelotor.
He has experienced a treatment-related side effect—grade 2 diarrhea. This occurred 9 weeks after he started voxelotor treatment, when the dose was increased to 1500 mg daily, but it resolved upon return to 900 mg. The patient has experienced no other treatment-related side effects.
Clinical and laboratory improvements have continued for more than 17 months, and the patient remains on treatment today under compassionate use access.
“This severely ill SCD patient’s clinical response, assessed by both objective and subjective measures, illustrates why we are encouraged by the voxelotor program,” said Ted W. Love, MD, president and chief executive officer of Global Blood Therapeutics, the company developing voxelotor.
“We plan to present additional data from other severely ill sickle cell patients who have received voxelotor via single-patient compassionate access treatment at FSCDR [the Foundation for Sickle Cell Disease Research] at an upcoming medical meeting. Of course, controlled clinical trials are needed to assess the efficacy and safety of voxelotor in SCD patients, including those with severe anemia.”
FDA approves generic clofarabine

The US Food and Drug Administration (FDA) has approved Dr. Reddy’s Laboratories Ltd.’s Clofarabine Injection, a therapeutic equivalent generic version of Clolar® (clofarabine) Injection.
The generic drug is now approved to treat patients age 1 to 21 who have relapsed or refractory acute lymphoblastic leukemia and have received at least 2 prior treatment regimens.
Dr. Reddy’s Clofarabine Injection is available in single-dose, 20 mL flint vials containing 20 mg of clofarabine in 20 mL of solution (1 mg/mL).
The US Food and Drug Administration (FDA) has approved Dr. Reddy’s Laboratories Ltd.’s Clofarabine Injection, a therapeutic equivalent generic version of Clolar® (clofarabine) Injection.
The generic drug is now approved to treat patients age 1 to 21 who have relapsed or refractory acute lymphoblastic leukemia and have received at least 2 prior treatment regimens.
Dr. Reddy’s Clofarabine Injection is available in single-dose, 20 mL flint vials containing 20 mg of clofarabine in 20 mL of solution (1 mg/mL).
The US Food and Drug Administration (FDA) has approved Dr. Reddy’s Laboratories Ltd.’s Clofarabine Injection, a therapeutic equivalent generic version of Clolar® (clofarabine) Injection.
The generic drug is now approved to treat patients age 1 to 21 who have relapsed or refractory acute lymphoblastic leukemia and have received at least 2 prior treatment regimens.
Dr. Reddy’s Clofarabine Injection is available in single-dose, 20 mL flint vials containing 20 mg of clofarabine in 20 mL of solution (1 mg/mL).