User login
Pyogenic Hepatic Abscess in an Immunocompetent Patient With Poor Oral Health and COVID-19 Infection
Pyogenic hepatic abscess (PHA) is a collection of pus in the liver caused by bacterial infection of the liver parenchyma. This potentially life-threatening condition has a mortality rate reported to be as high as 47%.1 The incidence of PHA is reported to be 2.3 per 100,000 individuals and is more common in immunosuppressed individuals and those with diabetes mellitus, cancer, and liver transplant.2,3 PHA infections are usually polymicrobial and most commonly include enteric organisms like Escherichia coli and Klebsiella pneumoniae.4
We present a rare cause of PHA with Fusobacterium nucleatum (F nucleatum) in an immunocompetent patient with poor oral health, history of diverticulitis, and recent COVID-19 infection whose only symptoms were chest pain and a 4-week history of fever and malaise.
Case Presentation
A 52-year-old man initially presented to the C.W. Bill Young Veterans Affairs Medical Center (CWBYVAMC) emergency department in Bay Pines, Florida, for fever, malaise, and right-sided chest pain on inspiration. The fever and malaise began while he was on vacation 4 weeks prior. He originally presented to an outside hospital where he tested positive for COVID-19 and was recommended ibuprofen and rest. His symptoms did not improve, and he returned a second time to the outside hospital 2 weeks later and was diagnosed with pneumonia and placed on outpatient antibiotics. The patient subsequently returned to CWBYVAMC 2 weeks after starting antibiotics when he began to develop right-sided inspiratory chest pain. He reported no other recent travel and no abdominal pain. The patient’s history was significant for diverticulitis 2 years before. A colonoscopy was performed during that time and showed no masses.
On presentation, the patient was febrile with a temperature of 100.8 °F; otherwise, his vital signs were stable. Physical examinations, including abdominal, respiratory, and cardiovascular, were unremarkable. The initial laboratory workup revealed a white blood cell (WBC) count of 18.7 K/μL (reference range, 5-10 K/μL) and microcytic anemia with a hemoglobin level of 8.8 g/dL. The comprehensive metabolic panel revealed normal aspartate transaminase, alanine transaminase, and total bilirubin levels and elevated alkaline phosphatase of 215 U/L (reference range, 44-147 U/L), revealing possible mild intrahepatic cholestasis. Urinalysis showed trace proteinuria and urobilinogen. Coagulation studies showed elevated D-dimer and procalcitonin levels at 1.9 ng/mL (reference range, < 0.1 ng/mL) and 1.21 ng/mL (reference range, < 0.5 ng/mL), respectively, with normal prothrombin and partial thromboplastin times. The patient had a normal troponin, fecal, and blood culture; entamoeba serology was negative.
A computed tomograph (CT) angiography of the chest was performed to rule out pulmonary embolism, revealing liver lesions suspicious for abscess or metastatic disease. Minimal pleural effusion was detected bilaterally. A subsequent CT 
Following the procedure, the patient developed shaking chills, hypertension, fever, and acute hypoxic respiratory failure. He improved with oxygen and was transferred to the intensive care unit (ICU) where he had an increase in temperature and became septic without shock. A repeat blood culture was negative. An echocardiogram revealed no vegetation. Vancomycin was added for empiric coverage of potentially resistant organisms. The patient clinically improved and was able to leave the ICU 2 days later on hospital day 4.
The patient’s renal function worsened on day 5, and piperacillin-tazobactam and vancomycin were discontinued due to possible acute interstitial nephritis and renal toxicity. He started cefepime and continued metronidazole, and his renal function returned to normal 2 days later. Vancomycin was then re-administered. The results of the culture taken from the abscess came back positive for monomicrobial growth of F nucleatum on hospital day 9. 
Due to the patient’s persisting fever and WBC count, a repeat CT of the abdomen on hospital day 10 revealed a partial decrease in the abscess with a persistent collection superior to the location of the initial pigtail catheter placement. A second pigtail catheter was then placed near the dome of the liver 1 day later on hospital day 11. Following the procedure, the patient improved significantly. The repeat CT after 1 week showed marked overall resolution of the abscess, and the repeat culture of the abscess did not reveal any organism growth. Vancomycin was discontinued on day 19, and the drains were removed on hospital day 20. He was discharged home in stable condition on metronidazole and cefdinir for 21 days with follow-up appointments for CT of the abdomen and with primary care, infectious disease, and a dental specialist.
Discussion
F nucleatum is a gram-negative, nonmotile, spindle-shaped rod found in dental plaques.5 The incidence of F nucleatum bacteremia is 0.34 per 100,000 people and increases with age, with the median age being 53.5 years.6 Although our patient did not present with F nucleatum bacteremia, it is possible that bacteremia was present before hospitalization but resolved by the time the sample was drawn for culture. F nucleatum bacteremia can lead to a variety of presentations. The most common primary diagnoses are intra-abdominal infections (eg, PHA, respiratory tract infections, and hematological disorders).1,6
PHA Presentation
The most common presenting symptoms of PHA are fever (88%), abdominal pain (79%), and vomiting (50%).4 The patient’s presentation of inspiratory right-sided chest pain is likely due to irritation of the diaphragmatic pleura of the right lung secondary to the abscess formation. The patient did not experience abdominal pain throughout the course of this disease or on palpation of his right upper quadrant. To our knowledge, this is the only case of PHA in the literature of a patient with inspiratory chest pain without respiratory infection, abdominal pain, and cardiac abnormalities. There was no radiologic evidence or signs of hypoxia on admission to CWBYVAMC, which makes respiratory infection an unlikely cause of the chest pain. Moreover, the patient presented with new-onset chest pain 2 weeks after the diagnosis of pneumonia.
Common laboratory findings of PHA include transaminitis, leukocytosis, and bilirubinemia.4 Of note, increased procalcitonin has also been associated with PHA and extreme elevation (> 200 μg/L) may be a useful biomarker to identify F nucleatum infections before the presence of leukocytosis.3 CT of PHA usually reveals right lobe involvement, and F nucleatum infection usually demonstrates multiple abscesses.4,7
Contributing Factors in F nucleatum PHA
F nucleatum is associated with several oral diseases, such as periodontitis and gingivitis.8 It is important to do an oral inspection on patients with F nucleatum infections because it can spread from oral cavities to different body parts.
F nucleatum is also found in the gut.9 Any disease that can cause a break in the gastrointestinal mucosa may result in F nucleatum bacteremia and PHA. This may be why F nucleatum has been associated with a variety of different diseases, such as diverticulitis, inflammatory bowel disease, appendicitis, and colorectal cancer.10,11 Our patient had a history of diverticulosis with diverticulitis. Bawa and colleagues described a patient with recurrent diverticulitis who developed F nucleatum bacteremia and PHA.11 Our patient did not have any signs of diverticulitis.
Our patient’s COVID-19 infection also had a role in delaying the appropriate treatment of PHA. Without any symptoms of PHA, a diagnosis is difficult in a patient with a positive COVID-19 test, and treatment was delayed 1 month. Moreover, COVID-19 has been reported to delay the diagnosis of PHA even in the absence of a positive COVID-19 test. Collins and Diamond presented a patient during the COVID-19 pandemic who developed a periodontal abscess, which resulted in F nucleatum bacteremia and PHA due to delayed hospital presentation after the patient’s practitioners recommended self-isolation, despite a negative COVID-19 test.12 This highlights the impact that COVID-19 may have on the timely diagnosis and treatment of patients with PHA.
Malignancy has been associated with F nucleatum bacteremia.1,13 Possibly the association is due to gastrointestinal mucosa malignancy’s ability to cause micro-abrasions, resulting in F nucleatum bacteremia.10 Additionally, F nucleatum may promote the development of colorectal neoplasms.8 Due to this association, screening for colorectal cancer in patients with F nucleatum infection is important. In our patient, a colonoscopy was performed during the patient’s hospitalization for diverticulitis 2 years prior. No signs of colorectal neoplasm were noted
Conclusions
PHA due to F nucleatum is a rare but potentially life-threatening condition that must be diagnosed and treated promptly. It usually presents with fever, abdominal pain, and vomiting but can present with chest pain in the absence of a respiratory infection, cardiac abnormalities, and abdominal pain, as in our patient. A wide spectrum of infections can occur with F nucleatum, including PHA.
Suspicion for infection with this organism should be kept high in middle-aged and older individuals who present with an indolent disease course and have risk factors, such as poor oral health and comorbidities. Suspicion should be kept high even in the event of COVID-19 infection, especially in individuals with prolonged fever without other signs indicating respiratory infection. We believe that the most likely causes of this patient’s infection were his dental caries and periodontal disease. The timing of his symptoms is not consistent with his previous episode of diverticulitis. Due to the mortality of PHA, diagnosis and treatment must be prompt. Initial treatment with drainage and empiric anaerobic coverage is recommended, followed by a tailored antibiotic regiment if indicated by culture, and further drainage if suggested by imaging.
1. Yang CC, Ye JJ, Hsu PC, et al. Characteristics and outcomes of Fusobacterium nucleatum bacteremia—a 6-year experience at a tertiary care hospital in northern Taiwan. Diagn Microbiol Infect Dis. 2011;70(2):167-174. doi:10.1016/j.diagmicrobio.2010.12.017
2. Kaplan GG, Gregson DB, Laupland KB. Population-based study of the epidemiology of and the risk factors for pyogenic liver abscess. Clin Gastroenterol Hepatol. 2004;2(11):1032-1038. doi:10.1016/s1542-3565(04)00459-8
3. Cao SA, Hinchey S. Identification and management of fusobacterium nucleatum liver abscess and bacteremia in a young healthy man. Cureus. 2020;12(12):e12303. doi:10.7759/cureus.12303
4. Abbas MT, Khan FY, Muhsin SA, Al-Dehwe B, Abukamar M, Elzouki AN. Epidemiology, clinical features and outcome of liver abscess: a single reference center experience in Qatar. Oman Med J. 2014;29(4):260-263. doi:10.5001/omj.2014.69
5. Bolstad AI, Jensen HB, Bakken V. Taxonomy, biology, and periodontal aspects of Fusobacterium nucleatum. Clin Microbiol Rev. 1996;9(1):55-71. doi:10.1128/CMR.9.1.55
6. Afra K, Laupland K, Leal J, Lloyd T, Gregson D. Incidence, risk factors, and outcomes of Fusobacterium species bacteremia. BMC Infect Dis. 2013;13:264. doi:10.1186/1471-2334-13-264
7. Crippin JS, Wang KK. An unrecognized etiology for pyogenic hepatic abscesses in normal hosts: dental disease. Am J Gastroenterol. 1992;87(12):1740-1743.
8. Shang FM, Liu HL. Fusobacterium nucleatum and colorectal cancer: a review. World J Gastrointest Oncol. 2018;10(3):71-81. doi:10.4251/wjgo.v10.i3.71
9. Allen-Vercoe E, Strauss J, Chadee K. Fusobacterium nucleatum: an emerging gut pathogen? Gut Microbes. 2011;2(5):294-298. doi:10.4161/gmic.2.5.18603
10. Han YW. Fusobacterium nucleatum: a commensal-turned pathogen. Curr Opin Microbiol. 2015;23:141-147. doi:10.1016/j.mib.2014.11.013
11. Bawa A, Kainat A, Raza H, George TB, Omer H, Pillai AC. Fusobacterium bacteremia causing hepatic abscess in a patient with diverticulitis. Cureus. 2022;14(7):e26938. doi:10.7759/cureus.26938
12. Collins L, Diamond T. Fusobacterium nucleatum causing a pyogenic liver abscess: a rare complication of periodontal disease that occurred during the COVID-19 pandemic. BMJ Case Rep. 2021;14(1):e240080. doi:10.1136/bcr-2020-240080
13. Nohrstrom E, Mattila T, Pettila V, et al. Clinical spectrum of bacteraemic Fusobacterium infections: from septic shock to nosocomial bacteraemia. Scand J Infect Dis. 2011;43(6-7):463-470. doi:10.3109/00365548.2011.565071
Pyogenic hepatic abscess (PHA) is a collection of pus in the liver caused by bacterial infection of the liver parenchyma. This potentially life-threatening condition has a mortality rate reported to be as high as 47%.1 The incidence of PHA is reported to be 2.3 per 100,000 individuals and is more common in immunosuppressed individuals and those with diabetes mellitus, cancer, and liver transplant.2,3 PHA infections are usually polymicrobial and most commonly include enteric organisms like Escherichia coli and Klebsiella pneumoniae.4
We present a rare cause of PHA with Fusobacterium nucleatum (F nucleatum) in an immunocompetent patient with poor oral health, history of diverticulitis, and recent COVID-19 infection whose only symptoms were chest pain and a 4-week history of fever and malaise.
Case Presentation
A 52-year-old man initially presented to the C.W. Bill Young Veterans Affairs Medical Center (CWBYVAMC) emergency department in Bay Pines, Florida, for fever, malaise, and right-sided chest pain on inspiration. The fever and malaise began while he was on vacation 4 weeks prior. He originally presented to an outside hospital where he tested positive for COVID-19 and was recommended ibuprofen and rest. His symptoms did not improve, and he returned a second time to the outside hospital 2 weeks later and was diagnosed with pneumonia and placed on outpatient antibiotics. The patient subsequently returned to CWBYVAMC 2 weeks after starting antibiotics when he began to develop right-sided inspiratory chest pain. He reported no other recent travel and no abdominal pain. The patient’s history was significant for diverticulitis 2 years before. A colonoscopy was performed during that time and showed no masses.
On presentation, the patient was febrile with a temperature of 100.8 °F; otherwise, his vital signs were stable. Physical examinations, including abdominal, respiratory, and cardiovascular, were unremarkable. The initial laboratory workup revealed a white blood cell (WBC) count of 18.7 K/μL (reference range, 5-10 K/μL) and microcytic anemia with a hemoglobin level of 8.8 g/dL. The comprehensive metabolic panel revealed normal aspartate transaminase, alanine transaminase, and total bilirubin levels and elevated alkaline phosphatase of 215 U/L (reference range, 44-147 U/L), revealing possible mild intrahepatic cholestasis. Urinalysis showed trace proteinuria and urobilinogen. Coagulation studies showed elevated D-dimer and procalcitonin levels at 1.9 ng/mL (reference range, < 0.1 ng/mL) and 1.21 ng/mL (reference range, < 0.5 ng/mL), respectively, with normal prothrombin and partial thromboplastin times. The patient had a normal troponin, fecal, and blood culture; entamoeba serology was negative.
A computed tomograph (CT) angiography of the chest was performed to rule out pulmonary embolism, revealing liver lesions suspicious for abscess or metastatic disease. Minimal pleural effusion was detected bilaterally. A subsequent CT
Following the procedure, the patient developed shaking chills, hypertension, fever, and acute hypoxic respiratory failure. He improved with oxygen and was transferred to the intensive care unit (ICU) where he had an increase in temperature and became septic without shock. A repeat blood culture was negative. An echocardiogram revealed no vegetation. Vancomycin was added for empiric coverage of potentially resistant organisms. The patient clinically improved and was able to leave the ICU 2 days later on hospital day 4.
The patient’s renal function worsened on day 5, and piperacillin-tazobactam and vancomycin were discontinued due to possible acute interstitial nephritis and renal toxicity. He started cefepime and continued metronidazole, and his renal function returned to normal 2 days later. Vancomycin was then re-administered. The results of the culture taken from the abscess came back positive for monomicrobial growth of F nucleatum on hospital day 9.
Due to the patient’s persisting fever and WBC count, a repeat CT of the abdomen on hospital day 10 revealed a partial decrease in the abscess with a persistent collection superior to the location of the initial pigtail catheter placement. A second pigtail catheter was then placed near the dome of the liver 1 day later on hospital day 11. Following the procedure, the patient improved significantly. The repeat CT after 1 week showed marked overall resolution of the abscess, and the repeat culture of the abscess did not reveal any organism growth. Vancomycin was discontinued on day 19, and the drains were removed on hospital day 20. He was discharged home in stable condition on metronidazole and cefdinir for 21 days with follow-up appointments for CT of the abdomen and with primary care, infectious disease, and a dental specialist.
Discussion
F nucleatum is a gram-negative, nonmotile, spindle-shaped rod found in dental plaques.5 The incidence of F nucleatum bacteremia is 0.34 per 100,000 people and increases with age, with the median age being 53.5 years.6 Although our patient did not present with F nucleatum bacteremia, it is possible that bacteremia was present before hospitalization but resolved by the time the sample was drawn for culture. F nucleatum bacteremia can lead to a variety of presentations. The most common primary diagnoses are intra-abdominal infections (eg, PHA, respiratory tract infections, and hematological disorders).1,6
PHA Presentation
The most common presenting symptoms of PHA are fever (88%), abdominal pain (79%), and vomiting (50%).4 The patient’s presentation of inspiratory right-sided chest pain is likely due to irritation of the diaphragmatic pleura of the right lung secondary to the abscess formation. The patient did not experience abdominal pain throughout the course of this disease or on palpation of his right upper quadrant. To our knowledge, this is the only case of PHA in the literature of a patient with inspiratory chest pain without respiratory infection, abdominal pain, and cardiac abnormalities. There was no radiologic evidence or signs of hypoxia on admission to CWBYVAMC, which makes respiratory infection an unlikely cause of the chest pain. Moreover, the patient presented with new-onset chest pain 2 weeks after the diagnosis of pneumonia.
Common laboratory findings of PHA include transaminitis, leukocytosis, and bilirubinemia.4 Of note, increased procalcitonin has also been associated with PHA and extreme elevation (> 200 μg/L) may be a useful biomarker to identify F nucleatum infections before the presence of leukocytosis.3 CT of PHA usually reveals right lobe involvement, and F nucleatum infection usually demonstrates multiple abscesses.4,7
Contributing Factors in F nucleatum PHA
F nucleatum is associated with several oral diseases, such as periodontitis and gingivitis.8 It is important to do an oral inspection on patients with F nucleatum infections because it can spread from oral cavities to different body parts.
F nucleatum is also found in the gut.9 Any disease that can cause a break in the gastrointestinal mucosa may result in F nucleatum bacteremia and PHA. This may be why F nucleatum has been associated with a variety of different diseases, such as diverticulitis, inflammatory bowel disease, appendicitis, and colorectal cancer.10,11 Our patient had a history of diverticulosis with diverticulitis. Bawa and colleagues described a patient with recurrent diverticulitis who developed F nucleatum bacteremia and PHA.11 Our patient did not have any signs of diverticulitis.
Our patient’s COVID-19 infection also had a role in delaying the appropriate treatment of PHA. Without any symptoms of PHA, a diagnosis is difficult in a patient with a positive COVID-19 test, and treatment was delayed 1 month. Moreover, COVID-19 has been reported to delay the diagnosis of PHA even in the absence of a positive COVID-19 test. Collins and Diamond presented a patient during the COVID-19 pandemic who developed a periodontal abscess, which resulted in F nucleatum bacteremia and PHA due to delayed hospital presentation after the patient’s practitioners recommended self-isolation, despite a negative COVID-19 test.12 This highlights the impact that COVID-19 may have on the timely diagnosis and treatment of patients with PHA.
Malignancy has been associated with F nucleatum bacteremia.1,13 Possibly the association is due to gastrointestinal mucosa malignancy’s ability to cause micro-abrasions, resulting in F nucleatum bacteremia.10 Additionally, F nucleatum may promote the development of colorectal neoplasms.8 Due to this association, screening for colorectal cancer in patients with F nucleatum infection is important. In our patient, a colonoscopy was performed during the patient’s hospitalization for diverticulitis 2 years prior. No signs of colorectal neoplasm were noted
Conclusions
PHA due to F nucleatum is a rare but potentially life-threatening condition that must be diagnosed and treated promptly. It usually presents with fever, abdominal pain, and vomiting but can present with chest pain in the absence of a respiratory infection, cardiac abnormalities, and abdominal pain, as in our patient. A wide spectrum of infections can occur with F nucleatum, including PHA.
Suspicion for infection with this organism should be kept high in middle-aged and older individuals who present with an indolent disease course and have risk factors, such as poor oral health and comorbidities. Suspicion should be kept high even in the event of COVID-19 infection, especially in individuals with prolonged fever without other signs indicating respiratory infection. We believe that the most likely causes of this patient’s infection were his dental caries and periodontal disease. The timing of his symptoms is not consistent with his previous episode of diverticulitis. Due to the mortality of PHA, diagnosis and treatment must be prompt. Initial treatment with drainage and empiric anaerobic coverage is recommended, followed by a tailored antibiotic regiment if indicated by culture, and further drainage if suggested by imaging.
Pyogenic hepatic abscess (PHA) is a collection of pus in the liver caused by bacterial infection of the liver parenchyma. This potentially life-threatening condition has a mortality rate reported to be as high as 47%.1 The incidence of PHA is reported to be 2.3 per 100,000 individuals and is more common in immunosuppressed individuals and those with diabetes mellitus, cancer, and liver transplant.2,3 PHA infections are usually polymicrobial and most commonly include enteric organisms like Escherichia coli and Klebsiella pneumoniae.4
We present a rare cause of PHA with Fusobacterium nucleatum (F nucleatum) in an immunocompetent patient with poor oral health, history of diverticulitis, and recent COVID-19 infection whose only symptoms were chest pain and a 4-week history of fever and malaise.
Case Presentation
A 52-year-old man initially presented to the C.W. Bill Young Veterans Affairs Medical Center (CWBYVAMC) emergency department in Bay Pines, Florida, for fever, malaise, and right-sided chest pain on inspiration. The fever and malaise began while he was on vacation 4 weeks prior. He originally presented to an outside hospital where he tested positive for COVID-19 and was recommended ibuprofen and rest. His symptoms did not improve, and he returned a second time to the outside hospital 2 weeks later and was diagnosed with pneumonia and placed on outpatient antibiotics. The patient subsequently returned to CWBYVAMC 2 weeks after starting antibiotics when he began to develop right-sided inspiratory chest pain. He reported no other recent travel and no abdominal pain. The patient’s history was significant for diverticulitis 2 years before. A colonoscopy was performed during that time and showed no masses.
On presentation, the patient was febrile with a temperature of 100.8 °F; otherwise, his vital signs were stable. Physical examinations, including abdominal, respiratory, and cardiovascular, were unremarkable. The initial laboratory workup revealed a white blood cell (WBC) count of 18.7 K/μL (reference range, 5-10 K/μL) and microcytic anemia with a hemoglobin level of 8.8 g/dL. The comprehensive metabolic panel revealed normal aspartate transaminase, alanine transaminase, and total bilirubin levels and elevated alkaline phosphatase of 215 U/L (reference range, 44-147 U/L), revealing possible mild intrahepatic cholestasis. Urinalysis showed trace proteinuria and urobilinogen. Coagulation studies showed elevated D-dimer and procalcitonin levels at 1.9 ng/mL (reference range, < 0.1 ng/mL) and 1.21 ng/mL (reference range, < 0.5 ng/mL), respectively, with normal prothrombin and partial thromboplastin times. The patient had a normal troponin, fecal, and blood culture; entamoeba serology was negative.
A computed tomograph (CT) angiography of the chest was performed to rule out pulmonary embolism, revealing liver lesions suspicious for abscess or metastatic disease. Minimal pleural effusion was detected bilaterally. A subsequent CT
Following the procedure, the patient developed shaking chills, hypertension, fever, and acute hypoxic respiratory failure. He improved with oxygen and was transferred to the intensive care unit (ICU) where he had an increase in temperature and became septic without shock. A repeat blood culture was negative. An echocardiogram revealed no vegetation. Vancomycin was added for empiric coverage of potentially resistant organisms. The patient clinically improved and was able to leave the ICU 2 days later on hospital day 4.
The patient’s renal function worsened on day 5, and piperacillin-tazobactam and vancomycin were discontinued due to possible acute interstitial nephritis and renal toxicity. He started cefepime and continued metronidazole, and his renal function returned to normal 2 days later. Vancomycin was then re-administered. The results of the culture taken from the abscess came back positive for monomicrobial growth of F nucleatum on hospital day 9.
Due to the patient’s persisting fever and WBC count, a repeat CT of the abdomen on hospital day 10 revealed a partial decrease in the abscess with a persistent collection superior to the location of the initial pigtail catheter placement. A second pigtail catheter was then placed near the dome of the liver 1 day later on hospital day 11. Following the procedure, the patient improved significantly. The repeat CT after 1 week showed marked overall resolution of the abscess, and the repeat culture of the abscess did not reveal any organism growth. Vancomycin was discontinued on day 19, and the drains were removed on hospital day 20. He was discharged home in stable condition on metronidazole and cefdinir for 21 days with follow-up appointments for CT of the abdomen and with primary care, infectious disease, and a dental specialist.
Discussion
F nucleatum is a gram-negative, nonmotile, spindle-shaped rod found in dental plaques.5 The incidence of F nucleatum bacteremia is 0.34 per 100,000 people and increases with age, with the median age being 53.5 years.6 Although our patient did not present with F nucleatum bacteremia, it is possible that bacteremia was present before hospitalization but resolved by the time the sample was drawn for culture. F nucleatum bacteremia can lead to a variety of presentations. The most common primary diagnoses are intra-abdominal infections (eg, PHA, respiratory tract infections, and hematological disorders).1,6
PHA Presentation
The most common presenting symptoms of PHA are fever (88%), abdominal pain (79%), and vomiting (50%).4 The patient’s presentation of inspiratory right-sided chest pain is likely due to irritation of the diaphragmatic pleura of the right lung secondary to the abscess formation. The patient did not experience abdominal pain throughout the course of this disease or on palpation of his right upper quadrant. To our knowledge, this is the only case of PHA in the literature of a patient with inspiratory chest pain without respiratory infection, abdominal pain, and cardiac abnormalities. There was no radiologic evidence or signs of hypoxia on admission to CWBYVAMC, which makes respiratory infection an unlikely cause of the chest pain. Moreover, the patient presented with new-onset chest pain 2 weeks after the diagnosis of pneumonia.
Common laboratory findings of PHA include transaminitis, leukocytosis, and bilirubinemia.4 Of note, increased procalcitonin has also been associated with PHA and extreme elevation (> 200 μg/L) may be a useful biomarker to identify F nucleatum infections before the presence of leukocytosis.3 CT of PHA usually reveals right lobe involvement, and F nucleatum infection usually demonstrates multiple abscesses.4,7
Contributing Factors in F nucleatum PHA
F nucleatum is associated with several oral diseases, such as periodontitis and gingivitis.8 It is important to do an oral inspection on patients with F nucleatum infections because it can spread from oral cavities to different body parts.
F nucleatum is also found in the gut.9 Any disease that can cause a break in the gastrointestinal mucosa may result in F nucleatum bacteremia and PHA. This may be why F nucleatum has been associated with a variety of different diseases, such as diverticulitis, inflammatory bowel disease, appendicitis, and colorectal cancer.10,11 Our patient had a history of diverticulosis with diverticulitis. Bawa and colleagues described a patient with recurrent diverticulitis who developed F nucleatum bacteremia and PHA.11 Our patient did not have any signs of diverticulitis.
Our patient’s COVID-19 infection also had a role in delaying the appropriate treatment of PHA. Without any symptoms of PHA, a diagnosis is difficult in a patient with a positive COVID-19 test, and treatment was delayed 1 month. Moreover, COVID-19 has been reported to delay the diagnosis of PHA even in the absence of a positive COVID-19 test. Collins and Diamond presented a patient during the COVID-19 pandemic who developed a periodontal abscess, which resulted in F nucleatum bacteremia and PHA due to delayed hospital presentation after the patient’s practitioners recommended self-isolation, despite a negative COVID-19 test.12 This highlights the impact that COVID-19 may have on the timely diagnosis and treatment of patients with PHA.
Malignancy has been associated with F nucleatum bacteremia.1,13 Possibly the association is due to gastrointestinal mucosa malignancy’s ability to cause micro-abrasions, resulting in F nucleatum bacteremia.10 Additionally, F nucleatum may promote the development of colorectal neoplasms.8 Due to this association, screening for colorectal cancer in patients with F nucleatum infection is important. In our patient, a colonoscopy was performed during the patient’s hospitalization for diverticulitis 2 years prior. No signs of colorectal neoplasm were noted
Conclusions
PHA due to F nucleatum is a rare but potentially life-threatening condition that must be diagnosed and treated promptly. It usually presents with fever, abdominal pain, and vomiting but can present with chest pain in the absence of a respiratory infection, cardiac abnormalities, and abdominal pain, as in our patient. A wide spectrum of infections can occur with F nucleatum, including PHA.
Suspicion for infection with this organism should be kept high in middle-aged and older individuals who present with an indolent disease course and have risk factors, such as poor oral health and comorbidities. Suspicion should be kept high even in the event of COVID-19 infection, especially in individuals with prolonged fever without other signs indicating respiratory infection. We believe that the most likely causes of this patient’s infection were his dental caries and periodontal disease. The timing of his symptoms is not consistent with his previous episode of diverticulitis. Due to the mortality of PHA, diagnosis and treatment must be prompt. Initial treatment with drainage and empiric anaerobic coverage is recommended, followed by a tailored antibiotic regiment if indicated by culture, and further drainage if suggested by imaging.
1. Yang CC, Ye JJ, Hsu PC, et al. Characteristics and outcomes of Fusobacterium nucleatum bacteremia—a 6-year experience at a tertiary care hospital in northern Taiwan. Diagn Microbiol Infect Dis. 2011;70(2):167-174. doi:10.1016/j.diagmicrobio.2010.12.017
2. Kaplan GG, Gregson DB, Laupland KB. Population-based study of the epidemiology of and the risk factors for pyogenic liver abscess. Clin Gastroenterol Hepatol. 2004;2(11):1032-1038. doi:10.1016/s1542-3565(04)00459-8
3. Cao SA, Hinchey S. Identification and management of fusobacterium nucleatum liver abscess and bacteremia in a young healthy man. Cureus. 2020;12(12):e12303. doi:10.7759/cureus.12303
4. Abbas MT, Khan FY, Muhsin SA, Al-Dehwe B, Abukamar M, Elzouki AN. Epidemiology, clinical features and outcome of liver abscess: a single reference center experience in Qatar. Oman Med J. 2014;29(4):260-263. doi:10.5001/omj.2014.69
5. Bolstad AI, Jensen HB, Bakken V. Taxonomy, biology, and periodontal aspects of Fusobacterium nucleatum. Clin Microbiol Rev. 1996;9(1):55-71. doi:10.1128/CMR.9.1.55
6. Afra K, Laupland K, Leal J, Lloyd T, Gregson D. Incidence, risk factors, and outcomes of Fusobacterium species bacteremia. BMC Infect Dis. 2013;13:264. doi:10.1186/1471-2334-13-264
7. Crippin JS, Wang KK. An unrecognized etiology for pyogenic hepatic abscesses in normal hosts: dental disease. Am J Gastroenterol. 1992;87(12):1740-1743.
8. Shang FM, Liu HL. Fusobacterium nucleatum and colorectal cancer: a review. World J Gastrointest Oncol. 2018;10(3):71-81. doi:10.4251/wjgo.v10.i3.71
9. Allen-Vercoe E, Strauss J, Chadee K. Fusobacterium nucleatum: an emerging gut pathogen? Gut Microbes. 2011;2(5):294-298. doi:10.4161/gmic.2.5.18603
10. Han YW. Fusobacterium nucleatum: a commensal-turned pathogen. Curr Opin Microbiol. 2015;23:141-147. doi:10.1016/j.mib.2014.11.013
11. Bawa A, Kainat A, Raza H, George TB, Omer H, Pillai AC. Fusobacterium bacteremia causing hepatic abscess in a patient with diverticulitis. Cureus. 2022;14(7):e26938. doi:10.7759/cureus.26938
12. Collins L, Diamond T. Fusobacterium nucleatum causing a pyogenic liver abscess: a rare complication of periodontal disease that occurred during the COVID-19 pandemic. BMJ Case Rep. 2021;14(1):e240080. doi:10.1136/bcr-2020-240080
13. Nohrstrom E, Mattila T, Pettila V, et al. Clinical spectrum of bacteraemic Fusobacterium infections: from septic shock to nosocomial bacteraemia. Scand J Infect Dis. 2011;43(6-7):463-470. doi:10.3109/00365548.2011.565071
1. Yang CC, Ye JJ, Hsu PC, et al. Characteristics and outcomes of Fusobacterium nucleatum bacteremia—a 6-year experience at a tertiary care hospital in northern Taiwan. Diagn Microbiol Infect Dis. 2011;70(2):167-174. doi:10.1016/j.diagmicrobio.2010.12.017
2. Kaplan GG, Gregson DB, Laupland KB. Population-based study of the epidemiology of and the risk factors for pyogenic liver abscess. Clin Gastroenterol Hepatol. 2004;2(11):1032-1038. doi:10.1016/s1542-3565(04)00459-8
3. Cao SA, Hinchey S. Identification and management of fusobacterium nucleatum liver abscess and bacteremia in a young healthy man. Cureus. 2020;12(12):e12303. doi:10.7759/cureus.12303
4. Abbas MT, Khan FY, Muhsin SA, Al-Dehwe B, Abukamar M, Elzouki AN. Epidemiology, clinical features and outcome of liver abscess: a single reference center experience in Qatar. Oman Med J. 2014;29(4):260-263. doi:10.5001/omj.2014.69
5. Bolstad AI, Jensen HB, Bakken V. Taxonomy, biology, and periodontal aspects of Fusobacterium nucleatum. Clin Microbiol Rev. 1996;9(1):55-71. doi:10.1128/CMR.9.1.55
6. Afra K, Laupland K, Leal J, Lloyd T, Gregson D. Incidence, risk factors, and outcomes of Fusobacterium species bacteremia. BMC Infect Dis. 2013;13:264. doi:10.1186/1471-2334-13-264
7. Crippin JS, Wang KK. An unrecognized etiology for pyogenic hepatic abscesses in normal hosts: dental disease. Am J Gastroenterol. 1992;87(12):1740-1743.
8. Shang FM, Liu HL. Fusobacterium nucleatum and colorectal cancer: a review. World J Gastrointest Oncol. 2018;10(3):71-81. doi:10.4251/wjgo.v10.i3.71
9. Allen-Vercoe E, Strauss J, Chadee K. Fusobacterium nucleatum: an emerging gut pathogen? Gut Microbes. 2011;2(5):294-298. doi:10.4161/gmic.2.5.18603
10. Han YW. Fusobacterium nucleatum: a commensal-turned pathogen. Curr Opin Microbiol. 2015;23:141-147. doi:10.1016/j.mib.2014.11.013
11. Bawa A, Kainat A, Raza H, George TB, Omer H, Pillai AC. Fusobacterium bacteremia causing hepatic abscess in a patient with diverticulitis. Cureus. 2022;14(7):e26938. doi:10.7759/cureus.26938
12. Collins L, Diamond T. Fusobacterium nucleatum causing a pyogenic liver abscess: a rare complication of periodontal disease that occurred during the COVID-19 pandemic. BMJ Case Rep. 2021;14(1):e240080. doi:10.1136/bcr-2020-240080
13. Nohrstrom E, Mattila T, Pettila V, et al. Clinical spectrum of bacteraemic Fusobacterium infections: from septic shock to nosocomial bacteraemia. Scand J Infect Dis. 2011;43(6-7):463-470. doi:10.3109/00365548.2011.565071
Postprandial Right Upper Quadrant Abdominal Pain
A 53-year-old male patient presented to the emergency department following a primary care office visit with sudden onset right upper quadrant abdominal pain that persisted for 3 weeks, worsening over the last 2 days. The abdominal pain worsened after eating or drinking and mildly improved with omeprazole. Associated symptoms included intermittent fever, night sweats, fatigue, and bloating since onset without vomiting or diarrhea. He reported a “complicated” cholecystectomy at an outside facility 6 months prior and that his “gallbladder was adhered to his duodenum,” though outside records were not available. Additional medical history included diverticulosis with prior flares of diverticulitis but no recent flares or treatments. His home medications included acetaminophen, naproxen, intranasal fluticasone, omeprazole, gabapentin, baclofen, trazodone, and antihistamines. He reported no tobacco or illicit drug use and stated he consumed a 6 pack of beer every 6 weeks.
Initial vital signs in the emergency department demonstrated an afebrile oral temperature with unremarkable blood pressure and pulse. He was alert and oriented and did not appear in significant acute distress. Physical examination of the abdomen demonstrated a nondistended abdomen, normal active bowel sounds in all 4 quadrants, and mild right upper and lower quadrant tenderness to soft and deep palpation with release.
Significant laboratory values included elevated C-reactive protein of 44.1 mg/L and mild leukocytosis of 11.1 K/µL (reference range, 4.00-10.60 K/µL). The basic metabolic panel, liver-associated enzymes, and lipase levels were within normal limits.
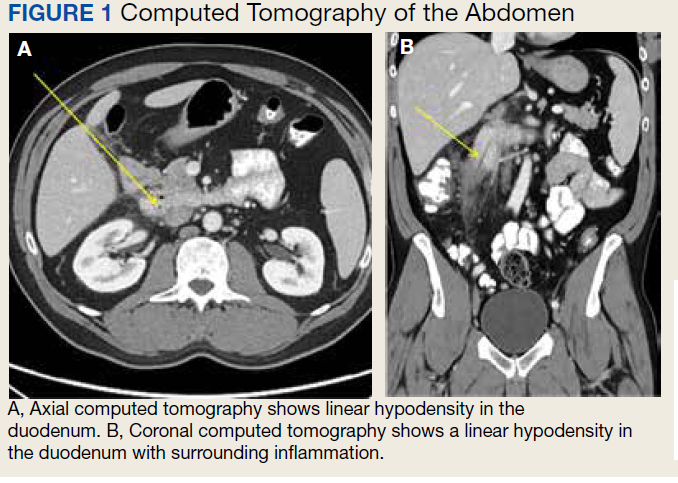
The initial imaging study was a computed tomography (CT) of the abdomen and pelvis with oral and IV contrast. The radiology report depicted a thin, needle-like hypodense foreign body approximately 8 cm in length in the proximal duodenum, slightly protruding extraluminally, and at least a moderate amount of surrounding inflammation without abscess or free air (Figure 1).
- What is your diagnosis?
- How would you treat this patient?
Our Diagnosis
Based on the clinical history of postprandial abdominal pain with prior cholecystectomy and leukocytosis, the initial differential diagnosis included peptic ulcer disease, gastroesophageal reflux, or delayed sequela of the cholecystectomy 6 months prior. Although suspicion remained for possible delayed postoperative complications from the cholecystectomy, ultrasound and hepatobiliary iminodiacetic acid (HIDA) scan were not pursued based on CT imaging findings. The needle-like hypodensity in the duodenum with surrounding inflammation visualized on CT was concerning for an unidentified penetrating foreign body with a possible retroperitoneal microperforation.
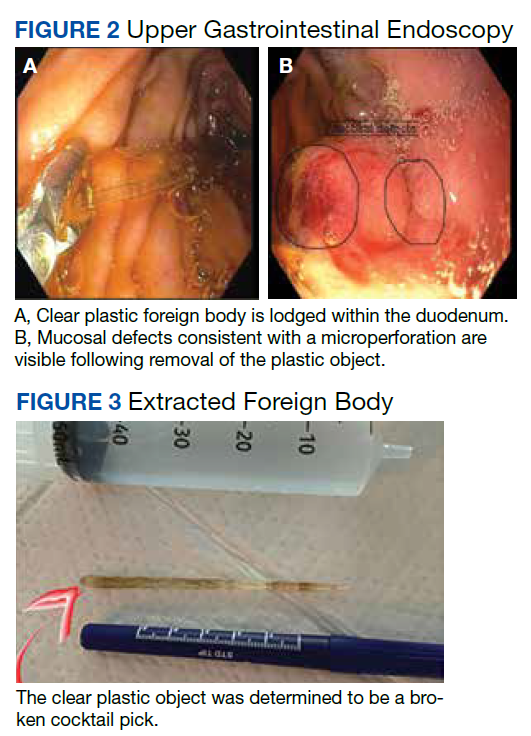
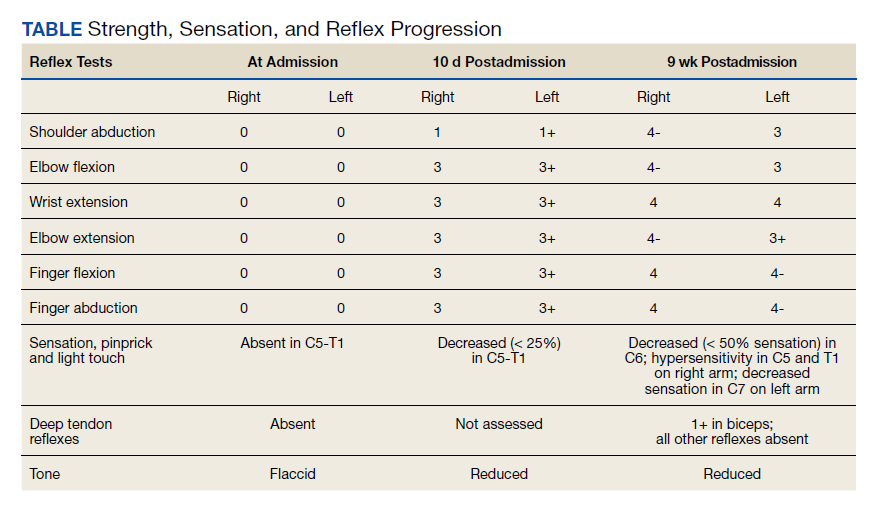
After these imaging findings were relayed from Radiology to the Gastroenterology Service, the patient underwent an upper gastrointestinal (GI) endoscopy to further evaluate the duodenum. Inspection revealed mild gastritis and a linear, clear piece of plastic with both ends firmly lodged within the mucosa from the distal duodenal bulb to the second portion of the duodenum; a significant mucosal defect of the bowel wall was visualized after careful extraction of the foreign body (Figure 2). The patient was diagnosed with a small duodenal perforation, which was sealed endoscopically with 2 endoclips. The extracted piece of plastic was examined and determined to be a broken cocktail pick (Figure 3). During discussion with the patient postprocedure, he stated that he ingested several olive martinis (which were served with cocktail picks) approximately 3 weeks prior to presentation and did not recall ingesting the cocktail pick. A repeat abdominal CT following the endoscopy demonstrated no leak or free air from the site of the repaired duodenal perforation (Figure 4). The patient avoided surgery and was permitted to resume a liquid diet prior to discharge.
Discussion
Foreign body ingestion in adults is most commonly unintentional with fish bones being the most common culprit.1 In unintentional instances of foreign body ingestion, many patients are not aware of the event, with dentures posing a significant well-known risk factor due to lack of palatal sensory feedback.2 Most ingested foreign bodies pass uninhibited through the GI tract without complications. However, less than 1% of ingested foreign bodies cause potentially life-threatening GI perforations.3
The risk of GI perforation due to foreign body ingestion is greatest with elongated, sharp objects, such as needles, bones, toothpicks, and cocktail picks. These objects tend to lodge at areas of narrowing or angulation, such as the appendix, ileocecal region, or as in this case, the duodenum.3 Passage of a foreign body through the duodenum is more likely to be inhibited if the object is longer than 6 cm and with a diameter > 2.5 cm.4 Signs of duodenal perforation are often subtle compared with jejunal or ileal perforations. Patients are commonly afebrile with normal white blood cell counts and are more likely to have chronic symptoms for > 3 days before the appropriate diagnosis of foreign body ingestion is made.1 Duodenal perforations may be more stable clinically compared with distal GI perforations in part due to the retroperitoneal location with relatively fewer bacteria present intraluminally. GI perforations may not occur acutely during passage of the foreign body but can present weeks, months, or even years later.5 Delayed onset of symptoms may happen when the foreign body becomes lodged and only partially perforates the bowel wall, resulting in a chronic inflammatory process. Other possible complications include fistulization and abscess formation from migrating linear sharp objects through the bowel wall, which is most observed with toothpicks and cocktail picks, specifically.5
Foreign bodies identified on plain radiographs commonly include radiopaque objects, such as glass, metallic objects, most animal bones and some fish bones, and some medications. However, radiolucent objects, such as toothpicks and cocktail picks, wood, plastic, most fish bones, and most medicines, often will not appear on radiographs. The diagnosis of ingested foreign body can therefore easily be delayed or overlooked on plain radiographs due to ingestion of radiolucent objects or lack of adequate patient history. A high index of suspicion is needed in such instances. The modality of choice for identifying GI perforation due to ingested foreign objects is CT.5 All of these commonly missed materials on radiographs will be visible on CT with variable densities. As an added benefit, CT also may reveal ingested objects not visualized on radiographs and show ancillary signs of perforation, such as extraluminal free air, localized inflammation, and fluid collections or abscess surrounding a segment of thickened bowel.5
Most ingested foreign bodies will pass through the GI system and can be managed with careful observation alone. However, upper endoscopy is emergently indicated in 3 scenarios of foreign body ingestion: (1) complete occlusion of the esophagus with salivary pooling due to risk of aspiration; (2) ingestion of batteries due to toxic substances; and (3) ingestion of sharp or pointed foreign bodies due to risk of perforation.4 Overall, endoscopic intervention is required in 20% of cases and surgical intervention remains rare at 1%.4 In the case of this patient, an emergent upper endoscopy was needed due to suspected duodenal perforation.
Treatment of duodenal perforations due to foreign bodies may involve conservative, surgical, or endoscopic management. Contained, small perforations in a stable patient may be treated conservatively with IV fluids, antibiotics, and proton pump inhibitors as they self-seal with omentum if the foreign body has passed.6 Retained duodenal foreign bodies pose a risk of persistent perforation or fistulization and must be removed. Anterior duodenal perforations pose a risk of peritonitis, whereas posterior duodenal perforations, although retroperitoneal and sparing the peritoneal cavity, may result in localized abscess formation necessitating foreign body removal. Endoscopic clipping is a modernized, less invasive way to close GI perforations. Through-the-scope clips (TTSCs) can close luminal defects < 2 cm in size.7 Defects > 1 cm may be repaired with combined TTSCs and endoloop or omental patching. Over-the-scope clips can close full thickness defects up to 2 to 3 cm with the advantage of being able to close leaks and fistulas involving inflamed or indurated tissue.7
Conclusions
Intestinal perforations related to foreign body ingestion are a rare complication occurring in < 1% of patients. Although most ingested foreign objects will pass through the GI tract, elongated or sharp objects pose a risk for perforation. In many cases, a history of foreign body ingestion is not obtained, and a high index of suspicion is required. Duodenal perforations due to foreign body ingestion should be included in the differential among the more common diagnoses of peptic ulcers, pancreatitis, and gallbladder disease in the setting of postprandial right upper quadrant abdominal pain. CT is the best modality for identifying foreign bodies, including objects that may be missed on plain radiographs.
1. Goh BK, Chow PK, Quah HM, et al. Perforation of the gastrointestinal tract secondary to ingestion of foreign bodies. World J Surg. 2006;(30)372-377. doi:10.1007/s00268-005-0490-2
2. Bunker PG. The role of dentistry in problems of foreign body in the air and food passage. J Am Dent Assoc. 1962;(64):782-787. doi:10.14219/jada.archive.1962.0160
3. Hunter TB, Taljanovic MS. Foreign bodies. Radiographics. 2003;23(3):731-757. doi:10.1148/rg.233025137
4. Ambe P, Weber SA, Schauer M, Knoefel WT. Swallowed foreign bodies in adults. Dtsch Arztebl Int. 2012;109(50):869-875. doi:10.3238/arztebl.2012.0869
5. Kuzmich S, Burke CJ, Harvey CJ, et al. Perforation of gastrointestinal tract by poorly conspicuous ingested foreign bodies: radiological diagnosis. Br J Radiol. 2015;88(1050):20150086. doi:10.1259/bjr.20150086
6. Hill AG. Management of perforated duodenal ulcer. In: Holzheimer RG, Mannick JA, eds. Surgical Treatment: Evidence-Based and Problem-Oriented. Zuckschwerdt; 2001.
7. Rogalski P, Daniluk J, Baniukiewicz A, Wroblewski E, Dabrowski A. Endoscopic management of gastrointestinal perforations, leaks and fistulas. World J Gastroenterol. 2015;21(37):10542-10552. doi:10.3748/wjg.v21.i37.10542
A 53-year-old male patient presented to the emergency department following a primary care office visit with sudden onset right upper quadrant abdominal pain that persisted for 3 weeks, worsening over the last 2 days. The abdominal pain worsened after eating or drinking and mildly improved with omeprazole. Associated symptoms included intermittent fever, night sweats, fatigue, and bloating since onset without vomiting or diarrhea. He reported a “complicated” cholecystectomy at an outside facility 6 months prior and that his “gallbladder was adhered to his duodenum,” though outside records were not available. Additional medical history included diverticulosis with prior flares of diverticulitis but no recent flares or treatments. His home medications included acetaminophen, naproxen, intranasal fluticasone, omeprazole, gabapentin, baclofen, trazodone, and antihistamines. He reported no tobacco or illicit drug use and stated he consumed a 6 pack of beer every 6 weeks.
Initial vital signs in the emergency department demonstrated an afebrile oral temperature with unremarkable blood pressure and pulse. He was alert and oriented and did not appear in significant acute distress. Physical examination of the abdomen demonstrated a nondistended abdomen, normal active bowel sounds in all 4 quadrants, and mild right upper and lower quadrant tenderness to soft and deep palpation with release.
Significant laboratory values included elevated C-reactive protein of 44.1 mg/L and mild leukocytosis of 11.1 K/µL (reference range, 4.00-10.60 K/µL). The basic metabolic panel, liver-associated enzymes, and lipase levels were within normal limits.
The initial imaging study was a computed tomography (CT) of the abdomen and pelvis with oral and IV contrast. The radiology report depicted a thin, needle-like hypodense foreign body approximately 8 cm in length in the proximal duodenum, slightly protruding extraluminally, and at least a moderate amount of surrounding inflammation without abscess or free air (Figure 1).
- What is your diagnosis?
- How would you treat this patient?
Our Diagnosis
Based on the clinical history of postprandial abdominal pain with prior cholecystectomy and leukocytosis, the initial differential diagnosis included peptic ulcer disease, gastroesophageal reflux, or delayed sequela of the cholecystectomy 6 months prior. Although suspicion remained for possible delayed postoperative complications from the cholecystectomy, ultrasound and hepatobiliary iminodiacetic acid (HIDA) scan were not pursued based on CT imaging findings. The needle-like hypodensity in the duodenum with surrounding inflammation visualized on CT was concerning for an unidentified penetrating foreign body with a possible retroperitoneal microperforation.
After these imaging findings were relayed from Radiology to the Gastroenterology Service, the patient underwent an upper gastrointestinal (GI) endoscopy to further evaluate the duodenum. Inspection revealed mild gastritis and a linear, clear piece of plastic with both ends firmly lodged within the mucosa from the distal duodenal bulb to the second portion of the duodenum; a significant mucosal defect of the bowel wall was visualized after careful extraction of the foreign body (Figure 2). The patient was diagnosed with a small duodenal perforation, which was sealed endoscopically with 2 endoclips. The extracted piece of plastic was examined and determined to be a broken cocktail pick (Figure 3). During discussion with the patient postprocedure, he stated that he ingested several olive martinis (which were served with cocktail picks) approximately 3 weeks prior to presentation and did not recall ingesting the cocktail pick. A repeat abdominal CT following the endoscopy demonstrated no leak or free air from the site of the repaired duodenal perforation (Figure 4). The patient avoided surgery and was permitted to resume a liquid diet prior to discharge.
Discussion
Foreign body ingestion in adults is most commonly unintentional with fish bones being the most common culprit.1 In unintentional instances of foreign body ingestion, many patients are not aware of the event, with dentures posing a significant well-known risk factor due to lack of palatal sensory feedback.2 Most ingested foreign bodies pass uninhibited through the GI tract without complications. However, less than 1% of ingested foreign bodies cause potentially life-threatening GI perforations.3
The risk of GI perforation due to foreign body ingestion is greatest with elongated, sharp objects, such as needles, bones, toothpicks, and cocktail picks. These objects tend to lodge at areas of narrowing or angulation, such as the appendix, ileocecal region, or as in this case, the duodenum.3 Passage of a foreign body through the duodenum is more likely to be inhibited if the object is longer than 6 cm and with a diameter > 2.5 cm.4 Signs of duodenal perforation are often subtle compared with jejunal or ileal perforations. Patients are commonly afebrile with normal white blood cell counts and are more likely to have chronic symptoms for > 3 days before the appropriate diagnosis of foreign body ingestion is made.1 Duodenal perforations may be more stable clinically compared with distal GI perforations in part due to the retroperitoneal location with relatively fewer bacteria present intraluminally. GI perforations may not occur acutely during passage of the foreign body but can present weeks, months, or even years later.5 Delayed onset of symptoms may happen when the foreign body becomes lodged and only partially perforates the bowel wall, resulting in a chronic inflammatory process. Other possible complications include fistulization and abscess formation from migrating linear sharp objects through the bowel wall, which is most observed with toothpicks and cocktail picks, specifically.5
Foreign bodies identified on plain radiographs commonly include radiopaque objects, such as glass, metallic objects, most animal bones and some fish bones, and some medications. However, radiolucent objects, such as toothpicks and cocktail picks, wood, plastic, most fish bones, and most medicines, often will not appear on radiographs. The diagnosis of ingested foreign body can therefore easily be delayed or overlooked on plain radiographs due to ingestion of radiolucent objects or lack of adequate patient history. A high index of suspicion is needed in such instances. The modality of choice for identifying GI perforation due to ingested foreign objects is CT.5 All of these commonly missed materials on radiographs will be visible on CT with variable densities. As an added benefit, CT also may reveal ingested objects not visualized on radiographs and show ancillary signs of perforation, such as extraluminal free air, localized inflammation, and fluid collections or abscess surrounding a segment of thickened bowel.5
Most ingested foreign bodies will pass through the GI system and can be managed with careful observation alone. However, upper endoscopy is emergently indicated in 3 scenarios of foreign body ingestion: (1) complete occlusion of the esophagus with salivary pooling due to risk of aspiration; (2) ingestion of batteries due to toxic substances; and (3) ingestion of sharp or pointed foreign bodies due to risk of perforation.4 Overall, endoscopic intervention is required in 20% of cases and surgical intervention remains rare at 1%.4 In the case of this patient, an emergent upper endoscopy was needed due to suspected duodenal perforation.
Treatment of duodenal perforations due to foreign bodies may involve conservative, surgical, or endoscopic management. Contained, small perforations in a stable patient may be treated conservatively with IV fluids, antibiotics, and proton pump inhibitors as they self-seal with omentum if the foreign body has passed.6 Retained duodenal foreign bodies pose a risk of persistent perforation or fistulization and must be removed. Anterior duodenal perforations pose a risk of peritonitis, whereas posterior duodenal perforations, although retroperitoneal and sparing the peritoneal cavity, may result in localized abscess formation necessitating foreign body removal. Endoscopic clipping is a modernized, less invasive way to close GI perforations. Through-the-scope clips (TTSCs) can close luminal defects < 2 cm in size.7 Defects > 1 cm may be repaired with combined TTSCs and endoloop or omental patching. Over-the-scope clips can close full thickness defects up to 2 to 3 cm with the advantage of being able to close leaks and fistulas involving inflamed or indurated tissue.7
Conclusions
Intestinal perforations related to foreign body ingestion are a rare complication occurring in < 1% of patients. Although most ingested foreign objects will pass through the GI tract, elongated or sharp objects pose a risk for perforation. In many cases, a history of foreign body ingestion is not obtained, and a high index of suspicion is required. Duodenal perforations due to foreign body ingestion should be included in the differential among the more common diagnoses of peptic ulcers, pancreatitis, and gallbladder disease in the setting of postprandial right upper quadrant abdominal pain. CT is the best modality for identifying foreign bodies, including objects that may be missed on plain radiographs.
A 53-year-old male patient presented to the emergency department following a primary care office visit with sudden onset right upper quadrant abdominal pain that persisted for 3 weeks, worsening over the last 2 days. The abdominal pain worsened after eating or drinking and mildly improved with omeprazole. Associated symptoms included intermittent fever, night sweats, fatigue, and bloating since onset without vomiting or diarrhea. He reported a “complicated” cholecystectomy at an outside facility 6 months prior and that his “gallbladder was adhered to his duodenum,” though outside records were not available. Additional medical history included diverticulosis with prior flares of diverticulitis but no recent flares or treatments. His home medications included acetaminophen, naproxen, intranasal fluticasone, omeprazole, gabapentin, baclofen, trazodone, and antihistamines. He reported no tobacco or illicit drug use and stated he consumed a 6 pack of beer every 6 weeks.
Initial vital signs in the emergency department demonstrated an afebrile oral temperature with unremarkable blood pressure and pulse. He was alert and oriented and did not appear in significant acute distress. Physical examination of the abdomen demonstrated a nondistended abdomen, normal active bowel sounds in all 4 quadrants, and mild right upper and lower quadrant tenderness to soft and deep palpation with release.
Significant laboratory values included elevated C-reactive protein of 44.1 mg/L and mild leukocytosis of 11.1 K/µL (reference range, 4.00-10.60 K/µL). The basic metabolic panel, liver-associated enzymes, and lipase levels were within normal limits.
The initial imaging study was a computed tomography (CT) of the abdomen and pelvis with oral and IV contrast. The radiology report depicted a thin, needle-like hypodense foreign body approximately 8 cm in length in the proximal duodenum, slightly protruding extraluminally, and at least a moderate amount of surrounding inflammation without abscess or free air (Figure 1).
- What is your diagnosis?
- How would you treat this patient?
Our Diagnosis
Based on the clinical history of postprandial abdominal pain with prior cholecystectomy and leukocytosis, the initial differential diagnosis included peptic ulcer disease, gastroesophageal reflux, or delayed sequela of the cholecystectomy 6 months prior. Although suspicion remained for possible delayed postoperative complications from the cholecystectomy, ultrasound and hepatobiliary iminodiacetic acid (HIDA) scan were not pursued based on CT imaging findings. The needle-like hypodensity in the duodenum with surrounding inflammation visualized on CT was concerning for an unidentified penetrating foreign body with a possible retroperitoneal microperforation.
After these imaging findings were relayed from Radiology to the Gastroenterology Service, the patient underwent an upper gastrointestinal (GI) endoscopy to further evaluate the duodenum. Inspection revealed mild gastritis and a linear, clear piece of plastic with both ends firmly lodged within the mucosa from the distal duodenal bulb to the second portion of the duodenum; a significant mucosal defect of the bowel wall was visualized after careful extraction of the foreign body (Figure 2). The patient was diagnosed with a small duodenal perforation, which was sealed endoscopically with 2 endoclips. The extracted piece of plastic was examined and determined to be a broken cocktail pick (Figure 3). During discussion with the patient postprocedure, he stated that he ingested several olive martinis (which were served with cocktail picks) approximately 3 weeks prior to presentation and did not recall ingesting the cocktail pick. A repeat abdominal CT following the endoscopy demonstrated no leak or free air from the site of the repaired duodenal perforation (Figure 4). The patient avoided surgery and was permitted to resume a liquid diet prior to discharge.
Discussion
Foreign body ingestion in adults is most commonly unintentional with fish bones being the most common culprit.1 In unintentional instances of foreign body ingestion, many patients are not aware of the event, with dentures posing a significant well-known risk factor due to lack of palatal sensory feedback.2 Most ingested foreign bodies pass uninhibited through the GI tract without complications. However, less than 1% of ingested foreign bodies cause potentially life-threatening GI perforations.3
The risk of GI perforation due to foreign body ingestion is greatest with elongated, sharp objects, such as needles, bones, toothpicks, and cocktail picks. These objects tend to lodge at areas of narrowing or angulation, such as the appendix, ileocecal region, or as in this case, the duodenum.3 Passage of a foreign body through the duodenum is more likely to be inhibited if the object is longer than 6 cm and with a diameter > 2.5 cm.4 Signs of duodenal perforation are often subtle compared with jejunal or ileal perforations. Patients are commonly afebrile with normal white blood cell counts and are more likely to have chronic symptoms for > 3 days before the appropriate diagnosis of foreign body ingestion is made.1 Duodenal perforations may be more stable clinically compared with distal GI perforations in part due to the retroperitoneal location with relatively fewer bacteria present intraluminally. GI perforations may not occur acutely during passage of the foreign body but can present weeks, months, or even years later.5 Delayed onset of symptoms may happen when the foreign body becomes lodged and only partially perforates the bowel wall, resulting in a chronic inflammatory process. Other possible complications include fistulization and abscess formation from migrating linear sharp objects through the bowel wall, which is most observed with toothpicks and cocktail picks, specifically.5
Foreign bodies identified on plain radiographs commonly include radiopaque objects, such as glass, metallic objects, most animal bones and some fish bones, and some medications. However, radiolucent objects, such as toothpicks and cocktail picks, wood, plastic, most fish bones, and most medicines, often will not appear on radiographs. The diagnosis of ingested foreign body can therefore easily be delayed or overlooked on plain radiographs due to ingestion of radiolucent objects or lack of adequate patient history. A high index of suspicion is needed in such instances. The modality of choice for identifying GI perforation due to ingested foreign objects is CT.5 All of these commonly missed materials on radiographs will be visible on CT with variable densities. As an added benefit, CT also may reveal ingested objects not visualized on radiographs and show ancillary signs of perforation, such as extraluminal free air, localized inflammation, and fluid collections or abscess surrounding a segment of thickened bowel.5
Most ingested foreign bodies will pass through the GI system and can be managed with careful observation alone. However, upper endoscopy is emergently indicated in 3 scenarios of foreign body ingestion: (1) complete occlusion of the esophagus with salivary pooling due to risk of aspiration; (2) ingestion of batteries due to toxic substances; and (3) ingestion of sharp or pointed foreign bodies due to risk of perforation.4 Overall, endoscopic intervention is required in 20% of cases and surgical intervention remains rare at 1%.4 In the case of this patient, an emergent upper endoscopy was needed due to suspected duodenal perforation.
Treatment of duodenal perforations due to foreign bodies may involve conservative, surgical, or endoscopic management. Contained, small perforations in a stable patient may be treated conservatively with IV fluids, antibiotics, and proton pump inhibitors as they self-seal with omentum if the foreign body has passed.6 Retained duodenal foreign bodies pose a risk of persistent perforation or fistulization and must be removed. Anterior duodenal perforations pose a risk of peritonitis, whereas posterior duodenal perforations, although retroperitoneal and sparing the peritoneal cavity, may result in localized abscess formation necessitating foreign body removal. Endoscopic clipping is a modernized, less invasive way to close GI perforations. Through-the-scope clips (TTSCs) can close luminal defects < 2 cm in size.7 Defects > 1 cm may be repaired with combined TTSCs and endoloop or omental patching. Over-the-scope clips can close full thickness defects up to 2 to 3 cm with the advantage of being able to close leaks and fistulas involving inflamed or indurated tissue.7
Conclusions
Intestinal perforations related to foreign body ingestion are a rare complication occurring in < 1% of patients. Although most ingested foreign objects will pass through the GI tract, elongated or sharp objects pose a risk for perforation. In many cases, a history of foreign body ingestion is not obtained, and a high index of suspicion is required. Duodenal perforations due to foreign body ingestion should be included in the differential among the more common diagnoses of peptic ulcers, pancreatitis, and gallbladder disease in the setting of postprandial right upper quadrant abdominal pain. CT is the best modality for identifying foreign bodies, including objects that may be missed on plain radiographs.
1. Goh BK, Chow PK, Quah HM, et al. Perforation of the gastrointestinal tract secondary to ingestion of foreign bodies. World J Surg. 2006;(30)372-377. doi:10.1007/s00268-005-0490-2
2. Bunker PG. The role of dentistry in problems of foreign body in the air and food passage. J Am Dent Assoc. 1962;(64):782-787. doi:10.14219/jada.archive.1962.0160
3. Hunter TB, Taljanovic MS. Foreign bodies. Radiographics. 2003;23(3):731-757. doi:10.1148/rg.233025137
4. Ambe P, Weber SA, Schauer M, Knoefel WT. Swallowed foreign bodies in adults. Dtsch Arztebl Int. 2012;109(50):869-875. doi:10.3238/arztebl.2012.0869
5. Kuzmich S, Burke CJ, Harvey CJ, et al. Perforation of gastrointestinal tract by poorly conspicuous ingested foreign bodies: radiological diagnosis. Br J Radiol. 2015;88(1050):20150086. doi:10.1259/bjr.20150086
6. Hill AG. Management of perforated duodenal ulcer. In: Holzheimer RG, Mannick JA, eds. Surgical Treatment: Evidence-Based and Problem-Oriented. Zuckschwerdt; 2001.
7. Rogalski P, Daniluk J, Baniukiewicz A, Wroblewski E, Dabrowski A. Endoscopic management of gastrointestinal perforations, leaks and fistulas. World J Gastroenterol. 2015;21(37):10542-10552. doi:10.3748/wjg.v21.i37.10542
1. Goh BK, Chow PK, Quah HM, et al. Perforation of the gastrointestinal tract secondary to ingestion of foreign bodies. World J Surg. 2006;(30)372-377. doi:10.1007/s00268-005-0490-2
2. Bunker PG. The role of dentistry in problems of foreign body in the air and food passage. J Am Dent Assoc. 1962;(64):782-787. doi:10.14219/jada.archive.1962.0160
3. Hunter TB, Taljanovic MS. Foreign bodies. Radiographics. 2003;23(3):731-757. doi:10.1148/rg.233025137
4. Ambe P, Weber SA, Schauer M, Knoefel WT. Swallowed foreign bodies in adults. Dtsch Arztebl Int. 2012;109(50):869-875. doi:10.3238/arztebl.2012.0869
5. Kuzmich S, Burke CJ, Harvey CJ, et al. Perforation of gastrointestinal tract by poorly conspicuous ingested foreign bodies: radiological diagnosis. Br J Radiol. 2015;88(1050):20150086. doi:10.1259/bjr.20150086
6. Hill AG. Management of perforated duodenal ulcer. In: Holzheimer RG, Mannick JA, eds. Surgical Treatment: Evidence-Based and Problem-Oriented. Zuckschwerdt; 2001.
7. Rogalski P, Daniluk J, Baniukiewicz A, Wroblewski E, Dabrowski A. Endoscopic management of gastrointestinal perforations, leaks and fistulas. World J Gastroenterol. 2015;21(37):10542-10552. doi:10.3748/wjg.v21.i37.10542
Rhabdomyolysis Occurring After Use of Cocaine Contaminated With Fentanyl Causing Bilateral Brachial Plexopathy
The brachial plexus is a group of interwoven nerves arising from the cervical spinal cord and coursing through the neck, shoulder, and axilla with terminal branches extending to the distal arm.1 Disorders of the brachial plexus are more rare than other isolated peripheral nerve disorders, trauma being the most common etiology.1 Traction, neoplasms, radiation exposure, external compression, and inflammatory processes, such as Parsonage-Turner syndrome, have also been described as less common etiologies.2
Rhabdomyolysis, a condition in which muscle breakdown occurs, is an uncommon and perhaps underrecognized cause of brachial plexopathy. Rhabdomyolysis is often caused by muscle overuse, trauma, prolonged immobilization, drugs, or toxins. Substances indicated as precipitating factors include alcohol, opioids, cocaine, and amphetamines.3,4 As rhabdomyolysis progresses, swelling and edema can compress surrounding structures. Therefore, in cases of rhabdomyolysis involving the muscles of the neck and shoulder girdle, external compression of the brachial plexus can potentially cause brachial plexopathy. Rare cases of this phenomenon occurring as a sequela of substance use have been described.1,5-9 Few cases have been reported in the literature.
The following case report describes a patient who
Case Presentation
A 68-year-old male patient with a history of polysubstance use disorder presented to the emergency department with complete loss of sensory and motor function of both arms. He had fallen asleep on his couch the previous evening with his arms crossed over his chest in the prone position.
On admission, the patient presented with an agitated mental status. The patient presented with 0/5 strength bilaterally in the upper extremities (UEs) accompanied by numbness and tingling. Radial pulses were palpable in both arms. All UE reflexes were absent, but patellar reflex was intact bilaterally. On hospital day 2, the patient was awake, alert, and oriented to person, place, and time and could provide a full history. The patient’s cranial nerves were intact with shoulder shrug testing mildly weak at 4/5 strength.
Serum electrolytes and glucose levels were normal. The creatine phosphokinase (CPK) level was elevated at 21,292 IU/L. Creatinine and blood urea nitrogen levels were elevated at 1.7 mg/dL and 32 mg/dL, respectively. Serum B12, thyroid-stimulating hormone, and hemoglobin A1c levels were normal.

Due to the absence of evidence of spinal cord injury, presence of normal motor and sensory function of the lower extremities, an elevated CPK level, signal hyperintensities of the muscles of the shoulder girdle, and the patient’s history, the leading diagnosis at this time was brachial plexopathy secondary to focal rhabdomyolysis.
Over the next week, the patient regained some motor function of the left hand and some sensory function bilaterally. At 8 weeks postadmission, a nerve conduction study showed prolonged latencies in the median and ulnar nerves bilaterally. The following week, the patient reported pain in both shoulders (left greater than the right) as well as weakness of shoulder movement on the left greater than the right. There was pain in the right arm throughout. On examination, there was improved function of the arms distal to the elbow, which was better on the right side despite the associated pain (Table). There was atrophy of the left scapular muscles, hypothenar eminence, and deltoid muscle. There was weakness of the left triceps, with slight fourth and fifth finger flexion. The patient was unable to elevate or abduct the left shoulder but could elevate the right shoulder up to 45°. Sensation was decreased over the right outer arm and left posterior upper arm, with hypersensitivity in the right medial upper and lower arm. Deep tendon reflexes were absent in the upper arm aside from the biceps reflex (1+). All reflexes of the lower extremities were normal. It is interesting to note the relative greater improvement on the right despite the edema found on initial imaging being more prominent on the right.
Discussion
Rhabdomyolysis is a condition defined by myocyte necrosis that results in release of cellular contents and local edema. Inciting events may be traumatic, metabolic, ischemic, or substance induced. Common substances indicated include cocaine, amphetamines, acetaminophen, opioids, and alcohol.10 It classically presents with muscle pain and a marked elevation in serum CPK level, but other metabolic disturbances, acute kidney injury, or toxic hepatitis may also occur. A more uncommon sequela of rhabdomyolysis is plexopathy caused by edematous swelling and compression of the surrounding structures.
Rare cases of brachial plexopathy caused by rhabdomyolysis following substance use have been described. In many of these cases, rhabdomyolysis occurred after alcohol use with or without concurrent use of prescription opioids or heroin.7-9 One case following use of 3,4-methylenedioxy-N-methylamptamine (MDMA) and marijuana use was reported.1 Another case of concurrent brachial plexopathy and Horner syndrome in a 29-year-old male patient following ingestion of alcohol and opioids has also been described.5 The rate of occurrence of this phenomenon in the general population is unknown.
The pathophysiology of rhabdomyolysis caused by substance use has not been definitively identified, but it is hypothesized that the cause is 2-fold. The first insult is the direct toxicity of the substances to myocytes.8,9 The second factor is prolonged immobilization in a position that compresses the affected musculature and blood supply, causing both mechanical stress and ischemia to the muscles and brachial plexus. This prolonged immobilization can frequently follow use of substances, such as alcohol or opioids.9 Cases have been reported wherein rhabdomyolysis causing brachial plexopathy occurred despite relatively normal positioning of the arms and shoulders during sleep.9 In our case, the patient had fallen asleep with his arms crossed over his chest in the prone position with his head turned, though he could not recall to which side. Although he stated that he had slept in this position regularly, the effects of fentanyl may have prevented the patient from waking to adjust his posture. This position had potential to compress the musculature of the neck and shoulders and restrict blood flow, resulting in the focal rhabdomyolysis seen in this patient. In theory, the position could also cause a stretch injury of the brachial plexus, although a pure stretch injury would more likely present unilaterally and without evidence of rhabdomyolysis.
Chronic ethanol use may have been a major contributor by both sensitizing the muscles to toxicity of other substances and induction of CYP450 enzymes that are normally responsible for metabolizing other drugs.8 Alcohol also inhibits gluconeogenesis and leads to hyperpolarization of myocytes, further contributing to their susceptibility to damage.9 Our patient had a prior history of alcohol use years before this event, but not at the time of this event.
Our patient had other known risk factors for rhabdomyolysis, including his long-term statin therapy, but it is unclear whether these were contributing factors in his case.10 Of the medications that are known to cause rhabdomyolysis, statins are among the most commonly described, although the mechanism through which this process occurs is not clear. A case of rhabdomyolysis following use of cocaine and heroin in a patient on long-standing statin therapy has been described.13 Our review of the literature found no cases of statin-induced rhabdomyolysis associated with brachial plexopathy. It is possible that concurrent statin therapy has an additive effect to other substances in inducing rhabdomyolysis.
Parsonage-Turner syndrome, also known as neuralgic amyotrophy, should also be included in the differential diagnosis. While there have been multiple etiologies proposed for Parsonage-Turner syndrome, it is generally thought to begin as a primary inflammatory process targeting the brachial plexus. One case report describes Parsonage-Turner syndrome progressing to secondary rhabdomyolysis.6 In this case, no primary etiology was identified, so the Parsonage-Turner syndrome diagnosis was made with secondary rhabdomyolysis.6 We believe it is possible that this case and others may have been misdiagnosed as Parsonage-Turner syndrome.
Aside from physical rehabilitation programs, cases of plexopathy secondary to rhabdomyolysis similar to our patient have largely been treated with supportive therapy and symptom management. Pain management was the primary goal in this patient, which was achieved with moderate success using a combination of muscle relaxants, antiepileptics, tramadol, and serotonin-norepinephrine reuptake inhibitors. Some surgical approaches have been reported in the literature. One case of rhabdomyolysis of the shoulder girdle causing a similar process benefitted from fasciotomy and surgical decompression.7 This patient had a complete recovery of all motor functions aside from shoulder abduction at 8 weeks postoperation, but neuropathic pain persisted in both arms. It is possible our patient may have benefitted from a similar treatment. Further research is necessary to determine the utility of this type of procedure when treating such cases.
Conclusions
This case report adds to the literature describing focal rhabdomyolysis causing secondary bilateral brachial plexopathy after substance use. Further research is needed to establish a definitive pathophysiology as well as treatment guidelines. Evidence-based treatment could mean better outcomes and quicker recoveries for future patients with this condition.
1. Eker Büyüks¸ireci D, Polat M, Zinnurog˘lu M, Cengiz B, Kaymak Karatas¸ GK. Bilateral pan-plexus lesion after substance use: A case report. Turk J Phys Med Rehabil. 2019;65(4):411-414. doi:10.5606/tftrd.2019.3157
2. Rubin DI. Brachial and lumbosacral plexopathies: a review. Clin Neurophysiol Pract. 2020;5:173-193. doi:10.1016/j.cnp.2020.07.005
3. Oshima Y. Characteristics of drug-associated rhabdomyolysis: analysis of 8,610 cases reported to the US Food and Drug Administration. Intern Med. 2011;50(8):845-853. doi:10.2169/internalmedicine.50.4484
4. Waldman W, Kabata PM, Dines AM, et al. Rhabdomyolysis related to acute recreational drug toxicity-a euro-den study. PLoS One. 2021;16(3):e0246297. doi:10.1371/journal.pone.0246297
5. Lee SC, Geannette C, Wolfe SW, Feinberg JH, Sneag DB. Rhabdomyolysis resulting in concurrent Horner’s syndrome and brachial plexopathy: a case report. Skeletal Radiology. 2017;46(8):1131-1136. doi:10.1007/s00256-017-2634-5
6. Goetsch MR, Shen J, Jones JA, Memon A, Chatham W. Neuralgic amyotrophy presenting with multifocal myonecrosis and rhabdomyolysis. Cureus. 2020;12(3):e7382. doi:10.7759/cureus.7382
7. Tonetti DA, Tarkin IS, Bandi K, Moossy JJ. Complete bilateral brachial plexus injury from rhabdomyolysis and compartment syndrome: surgical case report. Oper Neurosurg (Hagerstown). 2019;17(2):E68-e72. doi:10.1093/ons/opy289
8. Riggs JE, Schochet SS Jr, Hogg JP. Focal rhabdomyolysis and brachial plexopathy: an association with heroin and chronic ethanol use. Mil Med. 1999;164(3):228-229.
9. Maddison P. Acute rhabdomyolysis and brachial plexopathy following alcohol ingestion. Muscle Nerve. 2002;25(2):283-285. doi:10.1002/mus.10021.abs
10. Giannoglou GD, Chatzizisis YS, Misirli G. The syndrome of rhabdomyolysis: pathophysiology and diagnosis. Eur J Intern Med. 2007;18(2):90-100. doi:10.1016/j.ejim.2006.09.020
11. Meacham MC, Lynch KL, Coffin PO, Wade A, Wheeler E, Riley ED. Addressing overdose risk among unstably housed women in San Francisco, California: an examination of potential fentanyl contamination of multiple substances. Harm Reduct J. 2020;17(1). doi:10.1186/s12954-020-00361-8
12. Klar SA, Brodkin E, Gibson E, et al. Notes from the field: furanyl-fentanyl overdose events caused by smoking contaminated crack cocaine - British Columbia, Canada, July 15-18, 2016. MMWR Morb Mortal Wkly Rep. 2016;65(37):1015-1016. doi:10.15585/mmwr.mm6537a6
13. Mitaritonno M, Lupo M, Greco I, Mazza A, Cervellin G. Severe rhabdomyolysis induced by co-administration of cocaine and heroin in a 45 years old man treated with rosuvastatin: a case report. Acta Biomed. 2021;92(S1):e2021089. doi:10.23750/abm.v92iS1.8858
The brachial plexus is a group of interwoven nerves arising from the cervical spinal cord and coursing through the neck, shoulder, and axilla with terminal branches extending to the distal arm.1 Disorders of the brachial plexus are more rare than other isolated peripheral nerve disorders, trauma being the most common etiology.1 Traction, neoplasms, radiation exposure, external compression, and inflammatory processes, such as Parsonage-Turner syndrome, have also been described as less common etiologies.2
Rhabdomyolysis, a condition in which muscle breakdown occurs, is an uncommon and perhaps underrecognized cause of brachial plexopathy. Rhabdomyolysis is often caused by muscle overuse, trauma, prolonged immobilization, drugs, or toxins. Substances indicated as precipitating factors include alcohol, opioids, cocaine, and amphetamines.3,4 As rhabdomyolysis progresses, swelling and edema can compress surrounding structures. Therefore, in cases of rhabdomyolysis involving the muscles of the neck and shoulder girdle, external compression of the brachial plexus can potentially cause brachial plexopathy. Rare cases of this phenomenon occurring as a sequela of substance use have been described.1,5-9 Few cases have been reported in the literature.
The following case report describes a patient who
Case Presentation
A 68-year-old male patient with a history of polysubstance use disorder presented to the emergency department with complete loss of sensory and motor function of both arms. He had fallen asleep on his couch the previous evening with his arms crossed over his chest in the prone position.
On admission, the patient presented with an agitated mental status. The patient presented with 0/5 strength bilaterally in the upper extremities (UEs) accompanied by numbness and tingling. Radial pulses were palpable in both arms. All UE reflexes were absent, but patellar reflex was intact bilaterally. On hospital day 2, the patient was awake, alert, and oriented to person, place, and time and could provide a full history. The patient’s cranial nerves were intact with shoulder shrug testing mildly weak at 4/5 strength.
Serum electrolytes and glucose levels were normal. The creatine phosphokinase (CPK) level was elevated at 21,292 IU/L. Creatinine and blood urea nitrogen levels were elevated at 1.7 mg/dL and 32 mg/dL, respectively. Serum B12, thyroid-stimulating hormone, and hemoglobin A1c levels were normal.
Due to the absence of evidence of spinal cord injury, presence of normal motor and sensory function of the lower extremities, an elevated CPK level, signal hyperintensities of the muscles of the shoulder girdle, and the patient’s history, the leading diagnosis at this time was brachial plexopathy secondary to focal rhabdomyolysis.
Over the next week, the patient regained some motor function of the left hand and some sensory function bilaterally. At 8 weeks postadmission, a nerve conduction study showed prolonged latencies in the median and ulnar nerves bilaterally. The following week, the patient reported pain in both shoulders (left greater than the right) as well as weakness of shoulder movement on the left greater than the right. There was pain in the right arm throughout. On examination, there was improved function of the arms distal to the elbow, which was better on the right side despite the associated pain (Table). There was atrophy of the left scapular muscles, hypothenar eminence, and deltoid muscle. There was weakness of the left triceps, with slight fourth and fifth finger flexion. The patient was unable to elevate or abduct the left shoulder but could elevate the right shoulder up to 45°. Sensation was decreased over the right outer arm and left posterior upper arm, with hypersensitivity in the right medial upper and lower arm. Deep tendon reflexes were absent in the upper arm aside from the biceps reflex (1+). All reflexes of the lower extremities were normal. It is interesting to note the relative greater improvement on the right despite the edema found on initial imaging being more prominent on the right.
Discussion
Rhabdomyolysis is a condition defined by myocyte necrosis that results in release of cellular contents and local edema. Inciting events may be traumatic, metabolic, ischemic, or substance induced. Common substances indicated include cocaine, amphetamines, acetaminophen, opioids, and alcohol.10 It classically presents with muscle pain and a marked elevation in serum CPK level, but other metabolic disturbances, acute kidney injury, or toxic hepatitis may also occur. A more uncommon sequela of rhabdomyolysis is plexopathy caused by edematous swelling and compression of the surrounding structures.
Rare cases of brachial plexopathy caused by rhabdomyolysis following substance use have been described. In many of these cases, rhabdomyolysis occurred after alcohol use with or without concurrent use of prescription opioids or heroin.7-9 One case following use of 3,4-methylenedioxy-N-methylamptamine (MDMA) and marijuana use was reported.1 Another case of concurrent brachial plexopathy and Horner syndrome in a 29-year-old male patient following ingestion of alcohol and opioids has also been described.5 The rate of occurrence of this phenomenon in the general population is unknown.
The pathophysiology of rhabdomyolysis caused by substance use has not been definitively identified, but it is hypothesized that the cause is 2-fold. The first insult is the direct toxicity of the substances to myocytes.8,9 The second factor is prolonged immobilization in a position that compresses the affected musculature and blood supply, causing both mechanical stress and ischemia to the muscles and brachial plexus. This prolonged immobilization can frequently follow use of substances, such as alcohol or opioids.9 Cases have been reported wherein rhabdomyolysis causing brachial plexopathy occurred despite relatively normal positioning of the arms and shoulders during sleep.9 In our case, the patient had fallen asleep with his arms crossed over his chest in the prone position with his head turned, though he could not recall to which side. Although he stated that he had slept in this position regularly, the effects of fentanyl may have prevented the patient from waking to adjust his posture. This position had potential to compress the musculature of the neck and shoulders and restrict blood flow, resulting in the focal rhabdomyolysis seen in this patient. In theory, the position could also cause a stretch injury of the brachial plexus, although a pure stretch injury would more likely present unilaterally and without evidence of rhabdomyolysis.
Chronic ethanol use may have been a major contributor by both sensitizing the muscles to toxicity of other substances and induction of CYP450 enzymes that are normally responsible for metabolizing other drugs.8 Alcohol also inhibits gluconeogenesis and leads to hyperpolarization of myocytes, further contributing to their susceptibility to damage.9 Our patient had a prior history of alcohol use years before this event, but not at the time of this event.
Our patient had other known risk factors for rhabdomyolysis, including his long-term statin therapy, but it is unclear whether these were contributing factors in his case.10 Of the medications that are known to cause rhabdomyolysis, statins are among the most commonly described, although the mechanism through which this process occurs is not clear. A case of rhabdomyolysis following use of cocaine and heroin in a patient on long-standing statin therapy has been described.13 Our review of the literature found no cases of statin-induced rhabdomyolysis associated with brachial plexopathy. It is possible that concurrent statin therapy has an additive effect to other substances in inducing rhabdomyolysis.
Parsonage-Turner syndrome, also known as neuralgic amyotrophy, should also be included in the differential diagnosis. While there have been multiple etiologies proposed for Parsonage-Turner syndrome, it is generally thought to begin as a primary inflammatory process targeting the brachial plexus. One case report describes Parsonage-Turner syndrome progressing to secondary rhabdomyolysis.6 In this case, no primary etiology was identified, so the Parsonage-Turner syndrome diagnosis was made with secondary rhabdomyolysis.6 We believe it is possible that this case and others may have been misdiagnosed as Parsonage-Turner syndrome.
Aside from physical rehabilitation programs, cases of plexopathy secondary to rhabdomyolysis similar to our patient have largely been treated with supportive therapy and symptom management. Pain management was the primary goal in this patient, which was achieved with moderate success using a combination of muscle relaxants, antiepileptics, tramadol, and serotonin-norepinephrine reuptake inhibitors. Some surgical approaches have been reported in the literature. One case of rhabdomyolysis of the shoulder girdle causing a similar process benefitted from fasciotomy and surgical decompression.7 This patient had a complete recovery of all motor functions aside from shoulder abduction at 8 weeks postoperation, but neuropathic pain persisted in both arms. It is possible our patient may have benefitted from a similar treatment. Further research is necessary to determine the utility of this type of procedure when treating such cases.
Conclusions
This case report adds to the literature describing focal rhabdomyolysis causing secondary bilateral brachial plexopathy after substance use. Further research is needed to establish a definitive pathophysiology as well as treatment guidelines. Evidence-based treatment could mean better outcomes and quicker recoveries for future patients with this condition.
The brachial plexus is a group of interwoven nerves arising from the cervical spinal cord and coursing through the neck, shoulder, and axilla with terminal branches extending to the distal arm.1 Disorders of the brachial plexus are more rare than other isolated peripheral nerve disorders, trauma being the most common etiology.1 Traction, neoplasms, radiation exposure, external compression, and inflammatory processes, such as Parsonage-Turner syndrome, have also been described as less common etiologies.2
Rhabdomyolysis, a condition in which muscle breakdown occurs, is an uncommon and perhaps underrecognized cause of brachial plexopathy. Rhabdomyolysis is often caused by muscle overuse, trauma, prolonged immobilization, drugs, or toxins. Substances indicated as precipitating factors include alcohol, opioids, cocaine, and amphetamines.3,4 As rhabdomyolysis progresses, swelling and edema can compress surrounding structures. Therefore, in cases of rhabdomyolysis involving the muscles of the neck and shoulder girdle, external compression of the brachial plexus can potentially cause brachial plexopathy. Rare cases of this phenomenon occurring as a sequela of substance use have been described.1,5-9 Few cases have been reported in the literature.
The following case report describes a patient who
Case Presentation
A 68-year-old male patient with a history of polysubstance use disorder presented to the emergency department with complete loss of sensory and motor function of both arms. He had fallen asleep on his couch the previous evening with his arms crossed over his chest in the prone position.
On admission, the patient presented with an agitated mental status. The patient presented with 0/5 strength bilaterally in the upper extremities (UEs) accompanied by numbness and tingling. Radial pulses were palpable in both arms. All UE reflexes were absent, but patellar reflex was intact bilaterally. On hospital day 2, the patient was awake, alert, and oriented to person, place, and time and could provide a full history. The patient’s cranial nerves were intact with shoulder shrug testing mildly weak at 4/5 strength.
Serum electrolytes and glucose levels were normal. The creatine phosphokinase (CPK) level was elevated at 21,292 IU/L. Creatinine and blood urea nitrogen levels were elevated at 1.7 mg/dL and 32 mg/dL, respectively. Serum B12, thyroid-stimulating hormone, and hemoglobin A1c levels were normal.
Due to the absence of evidence of spinal cord injury, presence of normal motor and sensory function of the lower extremities, an elevated CPK level, signal hyperintensities of the muscles of the shoulder girdle, and the patient’s history, the leading diagnosis at this time was brachial plexopathy secondary to focal rhabdomyolysis.
Over the next week, the patient regained some motor function of the left hand and some sensory function bilaterally. At 8 weeks postadmission, a nerve conduction study showed prolonged latencies in the median and ulnar nerves bilaterally. The following week, the patient reported pain in both shoulders (left greater than the right) as well as weakness of shoulder movement on the left greater than the right. There was pain in the right arm throughout. On examination, there was improved function of the arms distal to the elbow, which was better on the right side despite the associated pain (Table). There was atrophy of the left scapular muscles, hypothenar eminence, and deltoid muscle. There was weakness of the left triceps, with slight fourth and fifth finger flexion. The patient was unable to elevate or abduct the left shoulder but could elevate the right shoulder up to 45°. Sensation was decreased over the right outer arm and left posterior upper arm, with hypersensitivity in the right medial upper and lower arm. Deep tendon reflexes were absent in the upper arm aside from the biceps reflex (1+). All reflexes of the lower extremities were normal. It is interesting to note the relative greater improvement on the right despite the edema found on initial imaging being more prominent on the right.
Discussion
Rhabdomyolysis is a condition defined by myocyte necrosis that results in release of cellular contents and local edema. Inciting events may be traumatic, metabolic, ischemic, or substance induced. Common substances indicated include cocaine, amphetamines, acetaminophen, opioids, and alcohol.10 It classically presents with muscle pain and a marked elevation in serum CPK level, but other metabolic disturbances, acute kidney injury, or toxic hepatitis may also occur. A more uncommon sequela of rhabdomyolysis is plexopathy caused by edematous swelling and compression of the surrounding structures.
Rare cases of brachial plexopathy caused by rhabdomyolysis following substance use have been described. In many of these cases, rhabdomyolysis occurred after alcohol use with or without concurrent use of prescription opioids or heroin.7-9 One case following use of 3,4-methylenedioxy-N-methylamptamine (MDMA) and marijuana use was reported.1 Another case of concurrent brachial plexopathy and Horner syndrome in a 29-year-old male patient following ingestion of alcohol and opioids has also been described.5 The rate of occurrence of this phenomenon in the general population is unknown.
The pathophysiology of rhabdomyolysis caused by substance use has not been definitively identified, but it is hypothesized that the cause is 2-fold. The first insult is the direct toxicity of the substances to myocytes.8,9 The second factor is prolonged immobilization in a position that compresses the affected musculature and blood supply, causing both mechanical stress and ischemia to the muscles and brachial plexus. This prolonged immobilization can frequently follow use of substances, such as alcohol or opioids.9 Cases have been reported wherein rhabdomyolysis causing brachial plexopathy occurred despite relatively normal positioning of the arms and shoulders during sleep.9 In our case, the patient had fallen asleep with his arms crossed over his chest in the prone position with his head turned, though he could not recall to which side. Although he stated that he had slept in this position regularly, the effects of fentanyl may have prevented the patient from waking to adjust his posture. This position had potential to compress the musculature of the neck and shoulders and restrict blood flow, resulting in the focal rhabdomyolysis seen in this patient. In theory, the position could also cause a stretch injury of the brachial plexus, although a pure stretch injury would more likely present unilaterally and without evidence of rhabdomyolysis.
Chronic ethanol use may have been a major contributor by both sensitizing the muscles to toxicity of other substances and induction of CYP450 enzymes that are normally responsible for metabolizing other drugs.8 Alcohol also inhibits gluconeogenesis and leads to hyperpolarization of myocytes, further contributing to their susceptibility to damage.9 Our patient had a prior history of alcohol use years before this event, but not at the time of this event.
Our patient had other known risk factors for rhabdomyolysis, including his long-term statin therapy, but it is unclear whether these were contributing factors in his case.10 Of the medications that are known to cause rhabdomyolysis, statins are among the most commonly described, although the mechanism through which this process occurs is not clear. A case of rhabdomyolysis following use of cocaine and heroin in a patient on long-standing statin therapy has been described.13 Our review of the literature found no cases of statin-induced rhabdomyolysis associated with brachial plexopathy. It is possible that concurrent statin therapy has an additive effect to other substances in inducing rhabdomyolysis.
Parsonage-Turner syndrome, also known as neuralgic amyotrophy, should also be included in the differential diagnosis. While there have been multiple etiologies proposed for Parsonage-Turner syndrome, it is generally thought to begin as a primary inflammatory process targeting the brachial plexus. One case report describes Parsonage-Turner syndrome progressing to secondary rhabdomyolysis.6 In this case, no primary etiology was identified, so the Parsonage-Turner syndrome diagnosis was made with secondary rhabdomyolysis.6 We believe it is possible that this case and others may have been misdiagnosed as Parsonage-Turner syndrome.
Aside from physical rehabilitation programs, cases of plexopathy secondary to rhabdomyolysis similar to our patient have largely been treated with supportive therapy and symptom management. Pain management was the primary goal in this patient, which was achieved with moderate success using a combination of muscle relaxants, antiepileptics, tramadol, and serotonin-norepinephrine reuptake inhibitors. Some surgical approaches have been reported in the literature. One case of rhabdomyolysis of the shoulder girdle causing a similar process benefitted from fasciotomy and surgical decompression.7 This patient had a complete recovery of all motor functions aside from shoulder abduction at 8 weeks postoperation, but neuropathic pain persisted in both arms. It is possible our patient may have benefitted from a similar treatment. Further research is necessary to determine the utility of this type of procedure when treating such cases.
Conclusions
This case report adds to the literature describing focal rhabdomyolysis causing secondary bilateral brachial plexopathy after substance use. Further research is needed to establish a definitive pathophysiology as well as treatment guidelines. Evidence-based treatment could mean better outcomes and quicker recoveries for future patients with this condition.
1. Eker Büyüks¸ireci D, Polat M, Zinnurog˘lu M, Cengiz B, Kaymak Karatas¸ GK. Bilateral pan-plexus lesion after substance use: A case report. Turk J Phys Med Rehabil. 2019;65(4):411-414. doi:10.5606/tftrd.2019.3157
2. Rubin DI. Brachial and lumbosacral plexopathies: a review. Clin Neurophysiol Pract. 2020;5:173-193. doi:10.1016/j.cnp.2020.07.005
3. Oshima Y. Characteristics of drug-associated rhabdomyolysis: analysis of 8,610 cases reported to the US Food and Drug Administration. Intern Med. 2011;50(8):845-853. doi:10.2169/internalmedicine.50.4484
4. Waldman W, Kabata PM, Dines AM, et al. Rhabdomyolysis related to acute recreational drug toxicity-a euro-den study. PLoS One. 2021;16(3):e0246297. doi:10.1371/journal.pone.0246297
5. Lee SC, Geannette C, Wolfe SW, Feinberg JH, Sneag DB. Rhabdomyolysis resulting in concurrent Horner’s syndrome and brachial plexopathy: a case report. Skeletal Radiology. 2017;46(8):1131-1136. doi:10.1007/s00256-017-2634-5
6. Goetsch MR, Shen J, Jones JA, Memon A, Chatham W. Neuralgic amyotrophy presenting with multifocal myonecrosis and rhabdomyolysis. Cureus. 2020;12(3):e7382. doi:10.7759/cureus.7382
7. Tonetti DA, Tarkin IS, Bandi K, Moossy JJ. Complete bilateral brachial plexus injury from rhabdomyolysis and compartment syndrome: surgical case report. Oper Neurosurg (Hagerstown). 2019;17(2):E68-e72. doi:10.1093/ons/opy289
8. Riggs JE, Schochet SS Jr, Hogg JP. Focal rhabdomyolysis and brachial plexopathy: an association with heroin and chronic ethanol use. Mil Med. 1999;164(3):228-229.
9. Maddison P. Acute rhabdomyolysis and brachial plexopathy following alcohol ingestion. Muscle Nerve. 2002;25(2):283-285. doi:10.1002/mus.10021.abs
10. Giannoglou GD, Chatzizisis YS, Misirli G. The syndrome of rhabdomyolysis: pathophysiology and diagnosis. Eur J Intern Med. 2007;18(2):90-100. doi:10.1016/j.ejim.2006.09.020
11. Meacham MC, Lynch KL, Coffin PO, Wade A, Wheeler E, Riley ED. Addressing overdose risk among unstably housed women in San Francisco, California: an examination of potential fentanyl contamination of multiple substances. Harm Reduct J. 2020;17(1). doi:10.1186/s12954-020-00361-8
12. Klar SA, Brodkin E, Gibson E, et al. Notes from the field: furanyl-fentanyl overdose events caused by smoking contaminated crack cocaine - British Columbia, Canada, July 15-18, 2016. MMWR Morb Mortal Wkly Rep. 2016;65(37):1015-1016. doi:10.15585/mmwr.mm6537a6
13. Mitaritonno M, Lupo M, Greco I, Mazza A, Cervellin G. Severe rhabdomyolysis induced by co-administration of cocaine and heroin in a 45 years old man treated with rosuvastatin: a case report. Acta Biomed. 2021;92(S1):e2021089. doi:10.23750/abm.v92iS1.8858
1. Eker Büyüks¸ireci D, Polat M, Zinnurog˘lu M, Cengiz B, Kaymak Karatas¸ GK. Bilateral pan-plexus lesion after substance use: A case report. Turk J Phys Med Rehabil. 2019;65(4):411-414. doi:10.5606/tftrd.2019.3157
2. Rubin DI. Brachial and lumbosacral plexopathies: a review. Clin Neurophysiol Pract. 2020;5:173-193. doi:10.1016/j.cnp.2020.07.005
3. Oshima Y. Characteristics of drug-associated rhabdomyolysis: analysis of 8,610 cases reported to the US Food and Drug Administration. Intern Med. 2011;50(8):845-853. doi:10.2169/internalmedicine.50.4484
4. Waldman W, Kabata PM, Dines AM, et al. Rhabdomyolysis related to acute recreational drug toxicity-a euro-den study. PLoS One. 2021;16(3):e0246297. doi:10.1371/journal.pone.0246297
5. Lee SC, Geannette C, Wolfe SW, Feinberg JH, Sneag DB. Rhabdomyolysis resulting in concurrent Horner’s syndrome and brachial plexopathy: a case report. Skeletal Radiology. 2017;46(8):1131-1136. doi:10.1007/s00256-017-2634-5
6. Goetsch MR, Shen J, Jones JA, Memon A, Chatham W. Neuralgic amyotrophy presenting with multifocal myonecrosis and rhabdomyolysis. Cureus. 2020;12(3):e7382. doi:10.7759/cureus.7382
7. Tonetti DA, Tarkin IS, Bandi K, Moossy JJ. Complete bilateral brachial plexus injury from rhabdomyolysis and compartment syndrome: surgical case report. Oper Neurosurg (Hagerstown). 2019;17(2):E68-e72. doi:10.1093/ons/opy289
8. Riggs JE, Schochet SS Jr, Hogg JP. Focal rhabdomyolysis and brachial plexopathy: an association with heroin and chronic ethanol use. Mil Med. 1999;164(3):228-229.
9. Maddison P. Acute rhabdomyolysis and brachial plexopathy following alcohol ingestion. Muscle Nerve. 2002;25(2):283-285. doi:10.1002/mus.10021.abs
10. Giannoglou GD, Chatzizisis YS, Misirli G. The syndrome of rhabdomyolysis: pathophysiology and diagnosis. Eur J Intern Med. 2007;18(2):90-100. doi:10.1016/j.ejim.2006.09.020
11. Meacham MC, Lynch KL, Coffin PO, Wade A, Wheeler E, Riley ED. Addressing overdose risk among unstably housed women in San Francisco, California: an examination of potential fentanyl contamination of multiple substances. Harm Reduct J. 2020;17(1). doi:10.1186/s12954-020-00361-8
12. Klar SA, Brodkin E, Gibson E, et al. Notes from the field: furanyl-fentanyl overdose events caused by smoking contaminated crack cocaine - British Columbia, Canada, July 15-18, 2016. MMWR Morb Mortal Wkly Rep. 2016;65(37):1015-1016. doi:10.15585/mmwr.mm6537a6
13. Mitaritonno M, Lupo M, Greco I, Mazza A, Cervellin G. Severe rhabdomyolysis induced by co-administration of cocaine and heroin in a 45 years old man treated with rosuvastatin: a case report. Acta Biomed. 2021;92(S1):e2021089. doi:10.23750/abm.v92iS1.8858
Autonomic Dysfunction in the Setting of CADASIL Syndrome
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) syndrome is the most common monogenic inherited cause of stroke. CADASIL syndrome is a nonsclerotic angiopathy resulting from a mutation of the NOTCH3 gene on chromosome 19p13, encoding a receptor expressed by vascular smooth muscle cells.1 This mutation results in migraine, recurrent ischemic stroke, affective disorders, and dementia, with migraine often manifesting earliest.2,3
The onset of stroke symptoms occurs typically in ages ≥ 60 years with some patients experiencing stroke as early as in their 30s.1,4 Presentation varies among patients even within the same family.5 CADASIL syndrome is frequently mistaken for other more common neurologic conditions due to the low prevalence of CADASIL syndrome, reported to be between 2 and 5 per 100,000.3,6 The cumulative nature of multiple ischemic episodes seen in 85% of symptomatic individuals leads to disability. Dementia is often hallmarked as one of the features of end-stage CADASIL syndrome.7 Extent and severity of brain tissue damage are shown to be the most critical factors of clinical symptoms.8 There is no specific treatment for CADASIL syndrome other than addressing risk factors.9
Symptoms are traditionally described to be limited to the central nervous system (CNS); however, reports of other organ system effects exist. Twenty-six percent of premature mortality relating to CADASIL syndrome is sudden unexpected death, which several authors have postulated could be attributed to cardiac events.10,11
The NOTCH3 gene encodes a protein expressed during gastrulation and in the CNS during embryological development. The expression of this protein decreases with time and has limited expression in adulthood.12 The pathophysiology of CADASIL syndrome includes myriad changes, including cerebral vessels narrowed by intimal thickening due to expansion of the extracellular matrix, degeneration of smooth muscle cells of the cerebral vessel walls, and osmiophilic material deposition in patients with CADASIL syndrome.13 Granular osmiophilic material in the vascular basal lamina can be observed on electron microscopy of patients with CADASIL syndrome and are used for diagnostic purposes.14
CADASIL syndrome often presents a diagnostic dilemma for physicians and is easy to misdiagnose in the early stages. The diagnostic dilemma arises given the subacute onset of CADASIL syndrome with vague early presenting symptoms, such as headache, prior to more specific findings (ie, multiple early strokes or transient ischemic attacks [TIA]). Patients presenting with CADASIL syndrome may be misdiagnosed with other neurologic conditions, including migraine or multiple sclerosis (MS).15 Especially in the case of MS, lesions visible on magnetic resonance imaging (MRI) may be differentiated by the higher rates of temporo polar lesions seen in CADASIL syndrome in comparison with those in MS.3
It is important to consider CADASIL syndrome in patients presenting at a young age with stroke due to the compounding effects of multiple ischemic episodes and subsequent motor/sensory and neuropsychologic deficits. This necessitates increasing awareness of CADASIL syndrome in the neurologic and radiologic community and the importance of educating families of patients on the importance of being evaluated. This diagnostic dilemma can lead to delay in appropriate therapy and control of related modifiable risk factors, including hypertension, hyperlipidemia, etc. Delays in initiation of anti-stroke pharmacotherapy can lead to additional morbidity and mortality in these patients.
The radiology of CADASIL syndrome is unique and particularly important due to the possible confusion with MS. MRI is an important tool in the evaluation of the cerebral pathology of CADASIL syndrome, revealing white matter and microangiopathic signal abnormalities, indicative of ischemic infarcts, lacunar strokes, and diffuse leukoencephalopathy.13,16 MRI lesions are often seen in the basal ganglia, thalamus, external capsule, and pons.7 The lesions also are seen in the periventricular region, explaining its misperception as MS.17 In addition, cerebral microhemorrhages have been seen. To further differentiate these lesions, the anterior temporal lobe should be observed for gliosis or hyperintensities, which correlates with CADASIL syndrome.18 Location of hyperintensity in the temporal lobes, relative sparing of the occipital/orbitofrontal white matter, corpus callosum, subcortical u-fibers, and cortex is helpful in differentiating from other etiologies, such as microvascular white matter ischemic disease, MS, and mitochondrial encephalopathy with lactic acidosis and strokelike symptoms (MELAS).
Case Presentation
A patient aged > 50 years presented to the emergency department (ED) due to numbness of the right perioral area, gait difficulties, difficulty speaking, and increasing right lower extremity weakness with no numbness or paresthesia. The patient’s medical history is relevant for CADASIL syndrome, hypertension, prior cerebrovascular accident, recurrent TIAs, multinodular goiter with a history of radioactive iodine treatment, and neurogenic bladder controlled with oxybutynin since age 30 years. The patient had a significant stroke history: the first stroke occurred at age 36 years and 3 more strokes at ages 38, 44, and 53 years and 4 TIAs over that period. This patient reported no recent headache or memory changes and had no history of smoking, alcohol, or recreational drug use. Family history was pertinent for the mother’s death secondary to stroke, with a history of multiple strokes beginning at a young, undetermined age and no major motor, sensory, or neuropsychologic deficits prior to her death. A sister and first cousin had been diagnosed with MS.
On triage in the ED, stroke alert was called but tissue plasminogen activator was not given due to time eligibility. The patient’s numbness and weakness were improved within 7 hours, but she continued to have difficulty with dysarthric speech and unsteady gait following this incident. Antihypertensive medications were discontinued on admission to allow for permissive hypertension to improve cerebral blood flow. A brain MRI revealed bilateral increased T2 fluid-attenuated inversion recovery (FLAIR) signal in the anterior temporal lobes, confluent increased T2 FLAIR signal in the periventricular/deep white matter, bilateral basal ganglia chronic lacunar infarcts, and several chronic microbleeds (Figure 1). There was no evidence for an acute infarct on the MRI. Recrudescence of prior stroke symptoms secondary to CADASIL syndrome was suspected as a primary diagnosis with a differential of TIA.
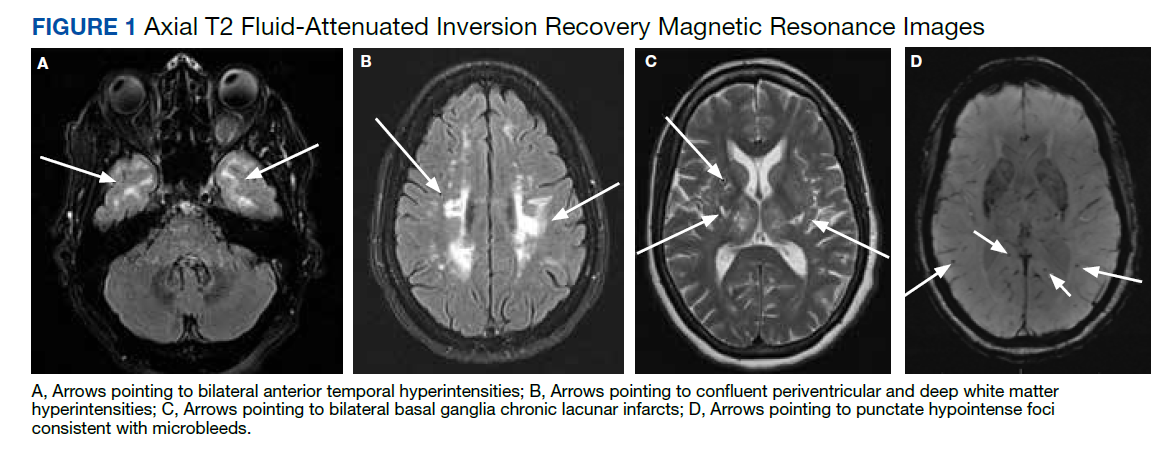
Starting the second day of admission, the patient had intermittent sinus bradycardia with the lowest heart rate (HR) in the range of 40 beats per minute (bpm) while awake with an unchanged neurologic examination. Each episode was transient, lasting less than an hour per staff documentation. The electrocardiogram (ECG) on admission demonstrated normal sinus rhythm in the range of 70 to 80 bpm.
The patient was asymptomatic and normotensive during the episodes of bradycardia. The patient had not yet resumed any antihypertensives. An echocardiogram was unremarkable with a left ventricular ejection fraction of 55 to 60%, normal anatomy, and no significant pericardial effusion. Carotid artery duplex examination demonstrated patent vessels with anterograde vertebral flow bilaterally. Due to the unknown cause of the bradycardia, the patient was discharged with a 14-day ambulatory cardiac monitor, advised to continue statin, aspirin, and lisinopril, and given a referral to continue with outpatient physical therapy and occupational therapy.
The patient’s ambulatory cardiac monitoring showed dominant sinus rhythm, with the HR in the range of 40 to 170 bpm with an overall average 70 to 80 bpm. The patient’s HR spent 5% of the recording time under 50 bpm and 14% of the time > 100. There was no evidence of heart block. No symptoms were recorded per the patient’s symptom diary during the entire 2 weeks of monitoring. Further follow-up showed that the patient presented to a primary care practitioner 1 month later with similar symptoms and was sent to the ED of an outside hospital without admission. The ECG was again unremarkable, demonstrating only sinus bradycardia with normal T waves, QT interval, without ST elevations or depressions. About 3 weeks later, the patient presented to the ED again with chest pain and was discharged with a diagnosis of atypical chest pain possibly related to anxiety without findings consistent with acute coronary syndrome (ACS).
Discussion
This patient with CADASIL syndrome and significant stroke history with cardiac symptoms demonstrates 3 important discussion points: the difficulty of early diagnosis, high rates of morbidity/mortality, and the need for further research into the cardiac effects of CADASIL syndrome. Due to this patient’s bradycardic episodes while being monitored on telemetry, it is possible that the cause of the strokelike symptoms was a TIA, secondary to decreased perfusion pressure, explaining the lack of acute ischemia on imaging. With regards to the history of thyroid dysfunction, this particular episode of bradycardia was unlikely to be related as the thyroid-stimulating hormone was reflective of subclinical hyperthyroidism with T4 levels within normal limits.
This case demonstrates a potential link between CADASIL syndrome and autonomic dysfunction. Similar to general stroke patients, patients with CADASIL syndrome are at an increased risk of hypoperfusion injury secondary to cardiovascular and autonomic dysfunction. This raises a question of initial and surveillance screening tests on diagnosis of CADASIL syndrome. It may be appropriate to obtain routine echocardiogram and ECG and other arrhythmia screening tests in these patients, especially during or following an ischemic episode. However, more evidence is required to support creation of a formal recommendation.
In a study of cardiac rhythm abnormalities in a half-million adults, 1.57% of women aged 55 to 64 years were found to have rhythm abnormality with 0.27% having a bradyarrhythmia.19 In the setting of neurologic disease, ECG changes such as arrhythmias and repolarization changes are regularly noted.20 However, it is unlikely that the bradycardia would be causing the brain lesions. In CADASIL syndrome, there is relative sparing of the occipital, orbitofrontal subcortical white matter, subcortical fibers, and cortex. Specifically, within CADASIL syndrome, a study of 23 patients showed no ECG changes regarding infarction/ischemia, conduction disturbances, or arrhythmias compared with that of controls.21
Further research into the cardiac effects of CADASIL syndrome is needed. As CADASIL syndrome is primarily a disorder of the vasculature, the disease has potential to affect the heart in addition to the brain.1 This theory is well supported by the embryologic effects of the NOTCH3 receptor pathways, which are responsible for the development of the cardiovascular system.22 Anecdotal evidence supports this theory as few case reports have been published that describe various cardiac abnormalities in patients with CADASIL syndrome, including myocardial infarction (MI), conduction abnormalities, and arrhythmias.2, 23-25
There have only been 2 published studies regarding investigations into CADASIL syndrome and cardiac disease. The first paper was a case-control study that investigated ECG changes in the setting of CADASIL syndrome. The study found no evidence for MI, ischemia, conduction disorder, or arrhythmias in patients with CADASIL syndrome.21 Unfortunately, this study was underpowered and limited in scope, only investigating a single ECG recording from 23 patients with CADASIL syndrome in a single clinic.21 Other cardiac markers, such as echocardiogram, stress test, and contractility, and longitudinal cardiac outcomes were not investigated in this study.21 The second paper was also a case-control study by Rufa and colleagues that investigated HR variability and other ECG changes during a 10-minute rest recording on 23 patients with CADASIL syndrome and compared the results to 22 age- and gender-matched patients in good health.11
This study found reduced HR variability and an increased ratio of low-frequency to high-frequency variability, which the authors claimed demonstrates autonomic dysfunction in patients with CADASIL syndrome.11 Rufa and colleagues concluded that patients with CADASIL syndrome are at higher risk for cardiac arrhythmias.11 This study also found no evidence for MI, ischemia, conduction disorder, or arrhythmias in the patients with CADASIL syndrome compared with that of age-matched controls.11 Similar to the first paper, this study is underpowered, only looks at a single timepoint recording, and uses incomplete and indirect measurements of cardiac function.
There is a need for a longitudinal review of cardiac outcomes in the CADASIL syndrome population to determine whether these patients require additional surveillance or prophylaxis. While the variability in HR of our patient cannot be definitively attributed solely to CADASIL syndrome, the subsequent admissions demonstrate that long-term monitoring may be warranted.
Conclusions
CADASIL syndrome is an autosomal dominant NOTCH3 signaling disease that affects the small vessel vasculature and leads to early ischemic events, headache, dementia, and death. CADASIL syndrome is frequently misdiagnosed due to insidious onset and vague presenting symptoms. Delay in diagnosis often results in nonoptimized medical management. Current guidelines recommend following poststroke protocol and minimizing individual risk factors by using antiplatelet, antihypertensive, and dyslipidemia medications. This case demonstrates a classic presentation of CADASIL syndrome with lesser described cardiac symptoms. Few cases of unusual cardiac symptoms in the setting of CADASIL syndrome have been reported. The relationship between cardiovascular disease and CADASIL syndrome is not well described. Further research is needed to elucidate any links between CADASIL syndrome and cardiovascular disease and to optimize management for these patients.
1. Moreton FC, Razvi SS, Davidson R, Muir KW. Changing clinical patterns and increasing prevalence in CADASIL. Acta Neurol Scand. 2014;130(3):197-203. doi:10.1111/ane.12266
2. Lesnik Oberstein SA, Jukema JW, Van Duinen SG, Macfarlane PW, van Houwelingen HC, Breuning MH, et al. Myocardial infarction in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Medicine (Baltimore). 2003;82(4):251-256. doi:10.1097/01.md.0000085054.63483.40
3. Di Donato I, Bianchi S, De Stefano N, Dichgans M, Dotti MT, Duering M, et al. Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) as a model of small vessel disease: update on clinical, diagnostic, and management aspects. BMC Med. 2017;15(1):41. doi:10.1186/s12916-017-0778-8
4. Dunphy L, Rani A, Duodu Y, Behnam Y. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL) presenting with stroke in a young man. BMJ Case Rep. 2019 ;12(7):e229609. doi:10.1136/bcr-2019-229609
5. Bianchi S, Zicari E, Carluccio A, Di Donato I, Pescini F, Nannucci S, et al. CADASIL in central Italy: a retrospective clinical and genetic study in 229 patients. J Neurol. 2015;262(1):134-141. doi:10.1007/s00415-014-7533-2
6. Phillips CD, Zuckerman SJ, Medical Education Commission. CADASIL can mimic multiple sclerosis. J La State Med Soc. 2010 May-Jun;162(3):174.
7. Hervé D, Chabriat H. CADASIL. J Geriatr Psychiatry Neurol. 2010;23(4):269-276. doi:10.1177/0891988710383570
8. Yamamoto Y, Hase Y, Ihara M, Khundakar A, Roeber S, Duering M, et al. Neuronal densities and vascular pathology in the hippocampal formation in CADASIL. Neurobiol Aging. 2021;97:33-40. doi:10.1016/j.neurobiolaging.2020.09.016
9. Ferrante EA, Cudrici CD, Boehm M. CADASIL: new advances in basic science and clinical perspectives. Curr Opin Hematol. 2019;26(3):193-198. doi:10.1097/MOH.0000000000000497
10. Opherk C, Peters N, Herzog J, Luedtke R, Dichgans M. Long-term prognosis and causes of death in CADASIL: a retrospective study in 411 patients. Brain. 2004;127(pt 11):2533-2539.
11. Rufa A, Guideri F, Acampa M, Cevenini G, Bianchi S, De Stefano N, et al. Cardiac autonomic nervous system and risk of arrhythmias in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Stroke. 2007 Feb;38(2):276-280. doi:10.1093/brain/awh282
12. Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383(6602):707-710. doi:10.1038/383707a0
13. Kalaria RN, Viitanen M, Kalimo H, Dichgans M, Tabira T, CASASIL Group of Vas-Cog. The pathogenesis of CADASIL: an update. J Neurol Sci. 2004;226(1-2):35-39. doi:10.1016/j.jns.2004.09.008
14. Reddy SPK, Vishnu VY, Goyal V, Singh MB, Arora S, Garg A, et al. CADASIL syndrome and stroke in young people. QJM. 2020 Feb 1;113(2):118-119. doi:10.1093/qjmed/hcz243
15. Carone DA. CADASIL and multiple sclerosis: A case report of prolonged misdiagnosis. Applied neuropsychology Adult. 2017;24(3):294-297. doi:10.1080/23279095.2016.1214132
16. Zhu S, Nahas SJ. CADASIL: Imaging characteristics and clinical correlation. Curr Pain Headache Rep. 2016;20(10):57. doi:10.1007/s11916-016-0584-6
17. Kalaria RN, Low WC, Oakley AE, Slade JY, Ince PG, Morris CM, et al. CADASIL and genetics of cerebral ischaemia. J Neural Transm Suppl. 2002;(63):75-90. doi:10.1007/978-3-7091-6137-1_5
18. O’Sullivan M, Jarosz JM, Martin RJ, Deasy N, Powell JF, Markus HS. MRI hyperintensities of the temporal lobe and external capsule in patients with CADASIL. Neurology. 2001;56(5):628-634. doi:10.1212/wnl.56.5.628
19. Khurshid S, Choi SH, Weng L-C, Wang EY, Trinquart L, Benjamin EJ, et al. Frequency of cardiac rhythm abnormalities in a half million adults. Circ ArrhythmElectrophysiol. 2018;11(7):e006273. doi:10.1161/CIRCEP.118.006273
20. Samuels MA. The brain–heart connection. Circulation. 2007;116(1):77-84. doi:10.1161/CIRCULATIONAHA. 106.678995
21. Cumurciuc R, Henry P, Gobron C, Vicaut E, Bousser MG, Chabriat H, et al. Electrocardiogram in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy patients without any clinical evidence of coronary artery disease: a case-control study. Stroke. 2006;37(4):1100-1102. doi:10.1161/01.STR.0000209242.68844.20
22. Luxán G, D’Amato G, MacGrogan D, de la Pompa JL. Endocardial notch signaling in cardiac development and disease. Circ Res. 2016;118(1):e1-e18. doi:10.1161/CIRCRESAHA.115.305350
23. Rubin CB, Hahn V, Kobayashi T, Litwack A. A report of accelerated coronary artery disease associated with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Case Rep Cardiol. 2015;2015:167513. doi:10.1155/2015/167513
24. Langer C, Adukauskaite A, Plank F, Feuchtner G, Cartes-Zumelzu F. Cerebral autosomal dominant arteriopathy (CADASIL) with cardiac involvement (ANOCA) and subcortical leukencephalopathy. J Cardiovasc Comput Tomogr. 2020;14(5):e1-e6. doi:10.1016/j.jcct.2018.08.005
25. Pettersen JA, Keith J, Gao F, Spence JD, Black SE. CADASIL accelerated by acute hypotension: Arterial and venous contribution to leukoaraiosis. Neurology. 2017;88(11):1077-1080. doi:10.1212/WNL.0000000000003717
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) syndrome is the most common monogenic inherited cause of stroke. CADASIL syndrome is a nonsclerotic angiopathy resulting from a mutation of the NOTCH3 gene on chromosome 19p13, encoding a receptor expressed by vascular smooth muscle cells.1 This mutation results in migraine, recurrent ischemic stroke, affective disorders, and dementia, with migraine often manifesting earliest.2,3
The onset of stroke symptoms occurs typically in ages ≥ 60 years with some patients experiencing stroke as early as in their 30s.1,4 Presentation varies among patients even within the same family.5 CADASIL syndrome is frequently mistaken for other more common neurologic conditions due to the low prevalence of CADASIL syndrome, reported to be between 2 and 5 per 100,000.3,6 The cumulative nature of multiple ischemic episodes seen in 85% of symptomatic individuals leads to disability. Dementia is often hallmarked as one of the features of end-stage CADASIL syndrome.7 Extent and severity of brain tissue damage are shown to be the most critical factors of clinical symptoms.8 There is no specific treatment for CADASIL syndrome other than addressing risk factors.9
Symptoms are traditionally described to be limited to the central nervous system (CNS); however, reports of other organ system effects exist. Twenty-six percent of premature mortality relating to CADASIL syndrome is sudden unexpected death, which several authors have postulated could be attributed to cardiac events.10,11
The NOTCH3 gene encodes a protein expressed during gastrulation and in the CNS during embryological development. The expression of this protein decreases with time and has limited expression in adulthood.12 The pathophysiology of CADASIL syndrome includes myriad changes, including cerebral vessels narrowed by intimal thickening due to expansion of the extracellular matrix, degeneration of smooth muscle cells of the cerebral vessel walls, and osmiophilic material deposition in patients with CADASIL syndrome.13 Granular osmiophilic material in the vascular basal lamina can be observed on electron microscopy of patients with CADASIL syndrome and are used for diagnostic purposes.14
CADASIL syndrome often presents a diagnostic dilemma for physicians and is easy to misdiagnose in the early stages. The diagnostic dilemma arises given the subacute onset of CADASIL syndrome with vague early presenting symptoms, such as headache, prior to more specific findings (ie, multiple early strokes or transient ischemic attacks [TIA]). Patients presenting with CADASIL syndrome may be misdiagnosed with other neurologic conditions, including migraine or multiple sclerosis (MS).15 Especially in the case of MS, lesions visible on magnetic resonance imaging (MRI) may be differentiated by the higher rates of temporo polar lesions seen in CADASIL syndrome in comparison with those in MS.3
It is important to consider CADASIL syndrome in patients presenting at a young age with stroke due to the compounding effects of multiple ischemic episodes and subsequent motor/sensory and neuropsychologic deficits. This necessitates increasing awareness of CADASIL syndrome in the neurologic and radiologic community and the importance of educating families of patients on the importance of being evaluated. This diagnostic dilemma can lead to delay in appropriate therapy and control of related modifiable risk factors, including hypertension, hyperlipidemia, etc. Delays in initiation of anti-stroke pharmacotherapy can lead to additional morbidity and mortality in these patients.
The radiology of CADASIL syndrome is unique and particularly important due to the possible confusion with MS. MRI is an important tool in the evaluation of the cerebral pathology of CADASIL syndrome, revealing white matter and microangiopathic signal abnormalities, indicative of ischemic infarcts, lacunar strokes, and diffuse leukoencephalopathy.13,16 MRI lesions are often seen in the basal ganglia, thalamus, external capsule, and pons.7 The lesions also are seen in the periventricular region, explaining its misperception as MS.17 In addition, cerebral microhemorrhages have been seen. To further differentiate these lesions, the anterior temporal lobe should be observed for gliosis or hyperintensities, which correlates with CADASIL syndrome.18 Location of hyperintensity in the temporal lobes, relative sparing of the occipital/orbitofrontal white matter, corpus callosum, subcortical u-fibers, and cortex is helpful in differentiating from other etiologies, such as microvascular white matter ischemic disease, MS, and mitochondrial encephalopathy with lactic acidosis and strokelike symptoms (MELAS).
Case Presentation
A patient aged > 50 years presented to the emergency department (ED) due to numbness of the right perioral area, gait difficulties, difficulty speaking, and increasing right lower extremity weakness with no numbness or paresthesia. The patient’s medical history is relevant for CADASIL syndrome, hypertension, prior cerebrovascular accident, recurrent TIAs, multinodular goiter with a history of radioactive iodine treatment, and neurogenic bladder controlled with oxybutynin since age 30 years. The patient had a significant stroke history: the first stroke occurred at age 36 years and 3 more strokes at ages 38, 44, and 53 years and 4 TIAs over that period. This patient reported no recent headache or memory changes and had no history of smoking, alcohol, or recreational drug use. Family history was pertinent for the mother’s death secondary to stroke, with a history of multiple strokes beginning at a young, undetermined age and no major motor, sensory, or neuropsychologic deficits prior to her death. A sister and first cousin had been diagnosed with MS.
On triage in the ED, stroke alert was called but tissue plasminogen activator was not given due to time eligibility. The patient’s numbness and weakness were improved within 7 hours, but she continued to have difficulty with dysarthric speech and unsteady gait following this incident. Antihypertensive medications were discontinued on admission to allow for permissive hypertension to improve cerebral blood flow. A brain MRI revealed bilateral increased T2 fluid-attenuated inversion recovery (FLAIR) signal in the anterior temporal lobes, confluent increased T2 FLAIR signal in the periventricular/deep white matter, bilateral basal ganglia chronic lacunar infarcts, and several chronic microbleeds (Figure 1). There was no evidence for an acute infarct on the MRI. Recrudescence of prior stroke symptoms secondary to CADASIL syndrome was suspected as a primary diagnosis with a differential of TIA.
Starting the second day of admission, the patient had intermittent sinus bradycardia with the lowest heart rate (HR) in the range of 40 beats per minute (bpm) while awake with an unchanged neurologic examination. Each episode was transient, lasting less than an hour per staff documentation. The electrocardiogram (ECG) on admission demonstrated normal sinus rhythm in the range of 70 to 80 bpm.
The patient was asymptomatic and normotensive during the episodes of bradycardia. The patient had not yet resumed any antihypertensives. An echocardiogram was unremarkable with a left ventricular ejection fraction of 55 to 60%, normal anatomy, and no significant pericardial effusion. Carotid artery duplex examination demonstrated patent vessels with anterograde vertebral flow bilaterally. Due to the unknown cause of the bradycardia, the patient was discharged with a 14-day ambulatory cardiac monitor, advised to continue statin, aspirin, and lisinopril, and given a referral to continue with outpatient physical therapy and occupational therapy.
The patient’s ambulatory cardiac monitoring showed dominant sinus rhythm, with the HR in the range of 40 to 170 bpm with an overall average 70 to 80 bpm. The patient’s HR spent 5% of the recording time under 50 bpm and 14% of the time > 100. There was no evidence of heart block. No symptoms were recorded per the patient’s symptom diary during the entire 2 weeks of monitoring. Further follow-up showed that the patient presented to a primary care practitioner 1 month later with similar symptoms and was sent to the ED of an outside hospital without admission. The ECG was again unremarkable, demonstrating only sinus bradycardia with normal T waves, QT interval, without ST elevations or depressions. About 3 weeks later, the patient presented to the ED again with chest pain and was discharged with a diagnosis of atypical chest pain possibly related to anxiety without findings consistent with acute coronary syndrome (ACS).
Discussion
This patient with CADASIL syndrome and significant stroke history with cardiac symptoms demonstrates 3 important discussion points: the difficulty of early diagnosis, high rates of morbidity/mortality, and the need for further research into the cardiac effects of CADASIL syndrome. Due to this patient’s bradycardic episodes while being monitored on telemetry, it is possible that the cause of the strokelike symptoms was a TIA, secondary to decreased perfusion pressure, explaining the lack of acute ischemia on imaging. With regards to the history of thyroid dysfunction, this particular episode of bradycardia was unlikely to be related as the thyroid-stimulating hormone was reflective of subclinical hyperthyroidism with T4 levels within normal limits.
This case demonstrates a potential link between CADASIL syndrome and autonomic dysfunction. Similar to general stroke patients, patients with CADASIL syndrome are at an increased risk of hypoperfusion injury secondary to cardiovascular and autonomic dysfunction. This raises a question of initial and surveillance screening tests on diagnosis of CADASIL syndrome. It may be appropriate to obtain routine echocardiogram and ECG and other arrhythmia screening tests in these patients, especially during or following an ischemic episode. However, more evidence is required to support creation of a formal recommendation.
In a study of cardiac rhythm abnormalities in a half-million adults, 1.57% of women aged 55 to 64 years were found to have rhythm abnormality with 0.27% having a bradyarrhythmia.19 In the setting of neurologic disease, ECG changes such as arrhythmias and repolarization changes are regularly noted.20 However, it is unlikely that the bradycardia would be causing the brain lesions. In CADASIL syndrome, there is relative sparing of the occipital, orbitofrontal subcortical white matter, subcortical fibers, and cortex. Specifically, within CADASIL syndrome, a study of 23 patients showed no ECG changes regarding infarction/ischemia, conduction disturbances, or arrhythmias compared with that of controls.21
Further research into the cardiac effects of CADASIL syndrome is needed. As CADASIL syndrome is primarily a disorder of the vasculature, the disease has potential to affect the heart in addition to the brain.1 This theory is well supported by the embryologic effects of the NOTCH3 receptor pathways, which are responsible for the development of the cardiovascular system.22 Anecdotal evidence supports this theory as few case reports have been published that describe various cardiac abnormalities in patients with CADASIL syndrome, including myocardial infarction (MI), conduction abnormalities, and arrhythmias.2, 23-25
There have only been 2 published studies regarding investigations into CADASIL syndrome and cardiac disease. The first paper was a case-control study that investigated ECG changes in the setting of CADASIL syndrome. The study found no evidence for MI, ischemia, conduction disorder, or arrhythmias in patients with CADASIL syndrome.21 Unfortunately, this study was underpowered and limited in scope, only investigating a single ECG recording from 23 patients with CADASIL syndrome in a single clinic.21 Other cardiac markers, such as echocardiogram, stress test, and contractility, and longitudinal cardiac outcomes were not investigated in this study.21 The second paper was also a case-control study by Rufa and colleagues that investigated HR variability and other ECG changes during a 10-minute rest recording on 23 patients with CADASIL syndrome and compared the results to 22 age- and gender-matched patients in good health.11
This study found reduced HR variability and an increased ratio of low-frequency to high-frequency variability, which the authors claimed demonstrates autonomic dysfunction in patients with CADASIL syndrome.11 Rufa and colleagues concluded that patients with CADASIL syndrome are at higher risk for cardiac arrhythmias.11 This study also found no evidence for MI, ischemia, conduction disorder, or arrhythmias in the patients with CADASIL syndrome compared with that of age-matched controls.11 Similar to the first paper, this study is underpowered, only looks at a single timepoint recording, and uses incomplete and indirect measurements of cardiac function.
There is a need for a longitudinal review of cardiac outcomes in the CADASIL syndrome population to determine whether these patients require additional surveillance or prophylaxis. While the variability in HR of our patient cannot be definitively attributed solely to CADASIL syndrome, the subsequent admissions demonstrate that long-term monitoring may be warranted.
Conclusions
CADASIL syndrome is an autosomal dominant NOTCH3 signaling disease that affects the small vessel vasculature and leads to early ischemic events, headache, dementia, and death. CADASIL syndrome is frequently misdiagnosed due to insidious onset and vague presenting symptoms. Delay in diagnosis often results in nonoptimized medical management. Current guidelines recommend following poststroke protocol and minimizing individual risk factors by using antiplatelet, antihypertensive, and dyslipidemia medications. This case demonstrates a classic presentation of CADASIL syndrome with lesser described cardiac symptoms. Few cases of unusual cardiac symptoms in the setting of CADASIL syndrome have been reported. The relationship between cardiovascular disease and CADASIL syndrome is not well described. Further research is needed to elucidate any links between CADASIL syndrome and cardiovascular disease and to optimize management for these patients.
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) syndrome is the most common monogenic inherited cause of stroke. CADASIL syndrome is a nonsclerotic angiopathy resulting from a mutation of the NOTCH3 gene on chromosome 19p13, encoding a receptor expressed by vascular smooth muscle cells.1 This mutation results in migraine, recurrent ischemic stroke, affective disorders, and dementia, with migraine often manifesting earliest.2,3
The onset of stroke symptoms occurs typically in ages ≥ 60 years with some patients experiencing stroke as early as in their 30s.1,4 Presentation varies among patients even within the same family.5 CADASIL syndrome is frequently mistaken for other more common neurologic conditions due to the low prevalence of CADASIL syndrome, reported to be between 2 and 5 per 100,000.3,6 The cumulative nature of multiple ischemic episodes seen in 85% of symptomatic individuals leads to disability. Dementia is often hallmarked as one of the features of end-stage CADASIL syndrome.7 Extent and severity of brain tissue damage are shown to be the most critical factors of clinical symptoms.8 There is no specific treatment for CADASIL syndrome other than addressing risk factors.9
Symptoms are traditionally described to be limited to the central nervous system (CNS); however, reports of other organ system effects exist. Twenty-six percent of premature mortality relating to CADASIL syndrome is sudden unexpected death, which several authors have postulated could be attributed to cardiac events.10,11
The NOTCH3 gene encodes a protein expressed during gastrulation and in the CNS during embryological development. The expression of this protein decreases with time and has limited expression in adulthood.12 The pathophysiology of CADASIL syndrome includes myriad changes, including cerebral vessels narrowed by intimal thickening due to expansion of the extracellular matrix, degeneration of smooth muscle cells of the cerebral vessel walls, and osmiophilic material deposition in patients with CADASIL syndrome.13 Granular osmiophilic material in the vascular basal lamina can be observed on electron microscopy of patients with CADASIL syndrome and are used for diagnostic purposes.14
CADASIL syndrome often presents a diagnostic dilemma for physicians and is easy to misdiagnose in the early stages. The diagnostic dilemma arises given the subacute onset of CADASIL syndrome with vague early presenting symptoms, such as headache, prior to more specific findings (ie, multiple early strokes or transient ischemic attacks [TIA]). Patients presenting with CADASIL syndrome may be misdiagnosed with other neurologic conditions, including migraine or multiple sclerosis (MS).15 Especially in the case of MS, lesions visible on magnetic resonance imaging (MRI) may be differentiated by the higher rates of temporo polar lesions seen in CADASIL syndrome in comparison with those in MS.3
It is important to consider CADASIL syndrome in patients presenting at a young age with stroke due to the compounding effects of multiple ischemic episodes and subsequent motor/sensory and neuropsychologic deficits. This necessitates increasing awareness of CADASIL syndrome in the neurologic and radiologic community and the importance of educating families of patients on the importance of being evaluated. This diagnostic dilemma can lead to delay in appropriate therapy and control of related modifiable risk factors, including hypertension, hyperlipidemia, etc. Delays in initiation of anti-stroke pharmacotherapy can lead to additional morbidity and mortality in these patients.
The radiology of CADASIL syndrome is unique and particularly important due to the possible confusion with MS. MRI is an important tool in the evaluation of the cerebral pathology of CADASIL syndrome, revealing white matter and microangiopathic signal abnormalities, indicative of ischemic infarcts, lacunar strokes, and diffuse leukoencephalopathy.13,16 MRI lesions are often seen in the basal ganglia, thalamus, external capsule, and pons.7 The lesions also are seen in the periventricular region, explaining its misperception as MS.17 In addition, cerebral microhemorrhages have been seen. To further differentiate these lesions, the anterior temporal lobe should be observed for gliosis or hyperintensities, which correlates with CADASIL syndrome.18 Location of hyperintensity in the temporal lobes, relative sparing of the occipital/orbitofrontal white matter, corpus callosum, subcortical u-fibers, and cortex is helpful in differentiating from other etiologies, such as microvascular white matter ischemic disease, MS, and mitochondrial encephalopathy with lactic acidosis and strokelike symptoms (MELAS).
Case Presentation
A patient aged > 50 years presented to the emergency department (ED) due to numbness of the right perioral area, gait difficulties, difficulty speaking, and increasing right lower extremity weakness with no numbness or paresthesia. The patient’s medical history is relevant for CADASIL syndrome, hypertension, prior cerebrovascular accident, recurrent TIAs, multinodular goiter with a history of radioactive iodine treatment, and neurogenic bladder controlled with oxybutynin since age 30 years. The patient had a significant stroke history: the first stroke occurred at age 36 years and 3 more strokes at ages 38, 44, and 53 years and 4 TIAs over that period. This patient reported no recent headache or memory changes and had no history of smoking, alcohol, or recreational drug use. Family history was pertinent for the mother’s death secondary to stroke, with a history of multiple strokes beginning at a young, undetermined age and no major motor, sensory, or neuropsychologic deficits prior to her death. A sister and first cousin had been diagnosed with MS.
On triage in the ED, stroke alert was called but tissue plasminogen activator was not given due to time eligibility. The patient’s numbness and weakness were improved within 7 hours, but she continued to have difficulty with dysarthric speech and unsteady gait following this incident. Antihypertensive medications were discontinued on admission to allow for permissive hypertension to improve cerebral blood flow. A brain MRI revealed bilateral increased T2 fluid-attenuated inversion recovery (FLAIR) signal in the anterior temporal lobes, confluent increased T2 FLAIR signal in the periventricular/deep white matter, bilateral basal ganglia chronic lacunar infarcts, and several chronic microbleeds (Figure 1). There was no evidence for an acute infarct on the MRI. Recrudescence of prior stroke symptoms secondary to CADASIL syndrome was suspected as a primary diagnosis with a differential of TIA.
Starting the second day of admission, the patient had intermittent sinus bradycardia with the lowest heart rate (HR) in the range of 40 beats per minute (bpm) while awake with an unchanged neurologic examination. Each episode was transient, lasting less than an hour per staff documentation. The electrocardiogram (ECG) on admission demonstrated normal sinus rhythm in the range of 70 to 80 bpm.
The patient was asymptomatic and normotensive during the episodes of bradycardia. The patient had not yet resumed any antihypertensives. An echocardiogram was unremarkable with a left ventricular ejection fraction of 55 to 60%, normal anatomy, and no significant pericardial effusion. Carotid artery duplex examination demonstrated patent vessels with anterograde vertebral flow bilaterally. Due to the unknown cause of the bradycardia, the patient was discharged with a 14-day ambulatory cardiac monitor, advised to continue statin, aspirin, and lisinopril, and given a referral to continue with outpatient physical therapy and occupational therapy.
The patient’s ambulatory cardiac monitoring showed dominant sinus rhythm, with the HR in the range of 40 to 170 bpm with an overall average 70 to 80 bpm. The patient’s HR spent 5% of the recording time under 50 bpm and 14% of the time > 100. There was no evidence of heart block. No symptoms were recorded per the patient’s symptom diary during the entire 2 weeks of monitoring. Further follow-up showed that the patient presented to a primary care practitioner 1 month later with similar symptoms and was sent to the ED of an outside hospital without admission. The ECG was again unremarkable, demonstrating only sinus bradycardia with normal T waves, QT interval, without ST elevations or depressions. About 3 weeks later, the patient presented to the ED again with chest pain and was discharged with a diagnosis of atypical chest pain possibly related to anxiety without findings consistent with acute coronary syndrome (ACS).
Discussion
This patient with CADASIL syndrome and significant stroke history with cardiac symptoms demonstrates 3 important discussion points: the difficulty of early diagnosis, high rates of morbidity/mortality, and the need for further research into the cardiac effects of CADASIL syndrome. Due to this patient’s bradycardic episodes while being monitored on telemetry, it is possible that the cause of the strokelike symptoms was a TIA, secondary to decreased perfusion pressure, explaining the lack of acute ischemia on imaging. With regards to the history of thyroid dysfunction, this particular episode of bradycardia was unlikely to be related as the thyroid-stimulating hormone was reflective of subclinical hyperthyroidism with T4 levels within normal limits.
This case demonstrates a potential link between CADASIL syndrome and autonomic dysfunction. Similar to general stroke patients, patients with CADASIL syndrome are at an increased risk of hypoperfusion injury secondary to cardiovascular and autonomic dysfunction. This raises a question of initial and surveillance screening tests on diagnosis of CADASIL syndrome. It may be appropriate to obtain routine echocardiogram and ECG and other arrhythmia screening tests in these patients, especially during or following an ischemic episode. However, more evidence is required to support creation of a formal recommendation.
In a study of cardiac rhythm abnormalities in a half-million adults, 1.57% of women aged 55 to 64 years were found to have rhythm abnormality with 0.27% having a bradyarrhythmia.19 In the setting of neurologic disease, ECG changes such as arrhythmias and repolarization changes are regularly noted.20 However, it is unlikely that the bradycardia would be causing the brain lesions. In CADASIL syndrome, there is relative sparing of the occipital, orbitofrontal subcortical white matter, subcortical fibers, and cortex. Specifically, within CADASIL syndrome, a study of 23 patients showed no ECG changes regarding infarction/ischemia, conduction disturbances, or arrhythmias compared with that of controls.21
Further research into the cardiac effects of CADASIL syndrome is needed. As CADASIL syndrome is primarily a disorder of the vasculature, the disease has potential to affect the heart in addition to the brain.1 This theory is well supported by the embryologic effects of the NOTCH3 receptor pathways, which are responsible for the development of the cardiovascular system.22 Anecdotal evidence supports this theory as few case reports have been published that describe various cardiac abnormalities in patients with CADASIL syndrome, including myocardial infarction (MI), conduction abnormalities, and arrhythmias.2, 23-25
There have only been 2 published studies regarding investigations into CADASIL syndrome and cardiac disease. The first paper was a case-control study that investigated ECG changes in the setting of CADASIL syndrome. The study found no evidence for MI, ischemia, conduction disorder, or arrhythmias in patients with CADASIL syndrome.21 Unfortunately, this study was underpowered and limited in scope, only investigating a single ECG recording from 23 patients with CADASIL syndrome in a single clinic.21 Other cardiac markers, such as echocardiogram, stress test, and contractility, and longitudinal cardiac outcomes were not investigated in this study.21 The second paper was also a case-control study by Rufa and colleagues that investigated HR variability and other ECG changes during a 10-minute rest recording on 23 patients with CADASIL syndrome and compared the results to 22 age- and gender-matched patients in good health.11
This study found reduced HR variability and an increased ratio of low-frequency to high-frequency variability, which the authors claimed demonstrates autonomic dysfunction in patients with CADASIL syndrome.11 Rufa and colleagues concluded that patients with CADASIL syndrome are at higher risk for cardiac arrhythmias.11 This study also found no evidence for MI, ischemia, conduction disorder, or arrhythmias in the patients with CADASIL syndrome compared with that of age-matched controls.11 Similar to the first paper, this study is underpowered, only looks at a single timepoint recording, and uses incomplete and indirect measurements of cardiac function.
There is a need for a longitudinal review of cardiac outcomes in the CADASIL syndrome population to determine whether these patients require additional surveillance or prophylaxis. While the variability in HR of our patient cannot be definitively attributed solely to CADASIL syndrome, the subsequent admissions demonstrate that long-term monitoring may be warranted.
Conclusions
CADASIL syndrome is an autosomal dominant NOTCH3 signaling disease that affects the small vessel vasculature and leads to early ischemic events, headache, dementia, and death. CADASIL syndrome is frequently misdiagnosed due to insidious onset and vague presenting symptoms. Delay in diagnosis often results in nonoptimized medical management. Current guidelines recommend following poststroke protocol and minimizing individual risk factors by using antiplatelet, antihypertensive, and dyslipidemia medications. This case demonstrates a classic presentation of CADASIL syndrome with lesser described cardiac symptoms. Few cases of unusual cardiac symptoms in the setting of CADASIL syndrome have been reported. The relationship between cardiovascular disease and CADASIL syndrome is not well described. Further research is needed to elucidate any links between CADASIL syndrome and cardiovascular disease and to optimize management for these patients.
1. Moreton FC, Razvi SS, Davidson R, Muir KW. Changing clinical patterns and increasing prevalence in CADASIL. Acta Neurol Scand. 2014;130(3):197-203. doi:10.1111/ane.12266
2. Lesnik Oberstein SA, Jukema JW, Van Duinen SG, Macfarlane PW, van Houwelingen HC, Breuning MH, et al. Myocardial infarction in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Medicine (Baltimore). 2003;82(4):251-256. doi:10.1097/01.md.0000085054.63483.40
3. Di Donato I, Bianchi S, De Stefano N, Dichgans M, Dotti MT, Duering M, et al. Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) as a model of small vessel disease: update on clinical, diagnostic, and management aspects. BMC Med. 2017;15(1):41. doi:10.1186/s12916-017-0778-8
4. Dunphy L, Rani A, Duodu Y, Behnam Y. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL) presenting with stroke in a young man. BMJ Case Rep. 2019 ;12(7):e229609. doi:10.1136/bcr-2019-229609
5. Bianchi S, Zicari E, Carluccio A, Di Donato I, Pescini F, Nannucci S, et al. CADASIL in central Italy: a retrospective clinical and genetic study in 229 patients. J Neurol. 2015;262(1):134-141. doi:10.1007/s00415-014-7533-2
6. Phillips CD, Zuckerman SJ, Medical Education Commission. CADASIL can mimic multiple sclerosis. J La State Med Soc. 2010 May-Jun;162(3):174.
7. Hervé D, Chabriat H. CADASIL. J Geriatr Psychiatry Neurol. 2010;23(4):269-276. doi:10.1177/0891988710383570
8. Yamamoto Y, Hase Y, Ihara M, Khundakar A, Roeber S, Duering M, et al. Neuronal densities and vascular pathology in the hippocampal formation in CADASIL. Neurobiol Aging. 2021;97:33-40. doi:10.1016/j.neurobiolaging.2020.09.016
9. Ferrante EA, Cudrici CD, Boehm M. CADASIL: new advances in basic science and clinical perspectives. Curr Opin Hematol. 2019;26(3):193-198. doi:10.1097/MOH.0000000000000497
10. Opherk C, Peters N, Herzog J, Luedtke R, Dichgans M. Long-term prognosis and causes of death in CADASIL: a retrospective study in 411 patients. Brain. 2004;127(pt 11):2533-2539.
11. Rufa A, Guideri F, Acampa M, Cevenini G, Bianchi S, De Stefano N, et al. Cardiac autonomic nervous system and risk of arrhythmias in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Stroke. 2007 Feb;38(2):276-280. doi:10.1093/brain/awh282
12. Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383(6602):707-710. doi:10.1038/383707a0
13. Kalaria RN, Viitanen M, Kalimo H, Dichgans M, Tabira T, CASASIL Group of Vas-Cog. The pathogenesis of CADASIL: an update. J Neurol Sci. 2004;226(1-2):35-39. doi:10.1016/j.jns.2004.09.008
14. Reddy SPK, Vishnu VY, Goyal V, Singh MB, Arora S, Garg A, et al. CADASIL syndrome and stroke in young people. QJM. 2020 Feb 1;113(2):118-119. doi:10.1093/qjmed/hcz243
15. Carone DA. CADASIL and multiple sclerosis: A case report of prolonged misdiagnosis. Applied neuropsychology Adult. 2017;24(3):294-297. doi:10.1080/23279095.2016.1214132
16. Zhu S, Nahas SJ. CADASIL: Imaging characteristics and clinical correlation. Curr Pain Headache Rep. 2016;20(10):57. doi:10.1007/s11916-016-0584-6
17. Kalaria RN, Low WC, Oakley AE, Slade JY, Ince PG, Morris CM, et al. CADASIL and genetics of cerebral ischaemia. J Neural Transm Suppl. 2002;(63):75-90. doi:10.1007/978-3-7091-6137-1_5
18. O’Sullivan M, Jarosz JM, Martin RJ, Deasy N, Powell JF, Markus HS. MRI hyperintensities of the temporal lobe and external capsule in patients with CADASIL. Neurology. 2001;56(5):628-634. doi:10.1212/wnl.56.5.628
19. Khurshid S, Choi SH, Weng L-C, Wang EY, Trinquart L, Benjamin EJ, et al. Frequency of cardiac rhythm abnormalities in a half million adults. Circ ArrhythmElectrophysiol. 2018;11(7):e006273. doi:10.1161/CIRCEP.118.006273
20. Samuels MA. The brain–heart connection. Circulation. 2007;116(1):77-84. doi:10.1161/CIRCULATIONAHA. 106.678995
21. Cumurciuc R, Henry P, Gobron C, Vicaut E, Bousser MG, Chabriat H, et al. Electrocardiogram in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy patients without any clinical evidence of coronary artery disease: a case-control study. Stroke. 2006;37(4):1100-1102. doi:10.1161/01.STR.0000209242.68844.20
22. Luxán G, D’Amato G, MacGrogan D, de la Pompa JL. Endocardial notch signaling in cardiac development and disease. Circ Res. 2016;118(1):e1-e18. doi:10.1161/CIRCRESAHA.115.305350
23. Rubin CB, Hahn V, Kobayashi T, Litwack A. A report of accelerated coronary artery disease associated with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Case Rep Cardiol. 2015;2015:167513. doi:10.1155/2015/167513
24. Langer C, Adukauskaite A, Plank F, Feuchtner G, Cartes-Zumelzu F. Cerebral autosomal dominant arteriopathy (CADASIL) with cardiac involvement (ANOCA) and subcortical leukencephalopathy. J Cardiovasc Comput Tomogr. 2020;14(5):e1-e6. doi:10.1016/j.jcct.2018.08.005
25. Pettersen JA, Keith J, Gao F, Spence JD, Black SE. CADASIL accelerated by acute hypotension: Arterial and venous contribution to leukoaraiosis. Neurology. 2017;88(11):1077-1080. doi:10.1212/WNL.0000000000003717
1. Moreton FC, Razvi SS, Davidson R, Muir KW. Changing clinical patterns and increasing prevalence in CADASIL. Acta Neurol Scand. 2014;130(3):197-203. doi:10.1111/ane.12266
2. Lesnik Oberstein SA, Jukema JW, Van Duinen SG, Macfarlane PW, van Houwelingen HC, Breuning MH, et al. Myocardial infarction in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Medicine (Baltimore). 2003;82(4):251-256. doi:10.1097/01.md.0000085054.63483.40
3. Di Donato I, Bianchi S, De Stefano N, Dichgans M, Dotti MT, Duering M, et al. Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) as a model of small vessel disease: update on clinical, diagnostic, and management aspects. BMC Med. 2017;15(1):41. doi:10.1186/s12916-017-0778-8
4. Dunphy L, Rani A, Duodu Y, Behnam Y. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL) presenting with stroke in a young man. BMJ Case Rep. 2019 ;12(7):e229609. doi:10.1136/bcr-2019-229609
5. Bianchi S, Zicari E, Carluccio A, Di Donato I, Pescini F, Nannucci S, et al. CADASIL in central Italy: a retrospective clinical and genetic study in 229 patients. J Neurol. 2015;262(1):134-141. doi:10.1007/s00415-014-7533-2
6. Phillips CD, Zuckerman SJ, Medical Education Commission. CADASIL can mimic multiple sclerosis. J La State Med Soc. 2010 May-Jun;162(3):174.
7. Hervé D, Chabriat H. CADASIL. J Geriatr Psychiatry Neurol. 2010;23(4):269-276. doi:10.1177/0891988710383570
8. Yamamoto Y, Hase Y, Ihara M, Khundakar A, Roeber S, Duering M, et al. Neuronal densities and vascular pathology in the hippocampal formation in CADASIL. Neurobiol Aging. 2021;97:33-40. doi:10.1016/j.neurobiolaging.2020.09.016
9. Ferrante EA, Cudrici CD, Boehm M. CADASIL: new advances in basic science and clinical perspectives. Curr Opin Hematol. 2019;26(3):193-198. doi:10.1097/MOH.0000000000000497
10. Opherk C, Peters N, Herzog J, Luedtke R, Dichgans M. Long-term prognosis and causes of death in CADASIL: a retrospective study in 411 patients. Brain. 2004;127(pt 11):2533-2539.
11. Rufa A, Guideri F, Acampa M, Cevenini G, Bianchi S, De Stefano N, et al. Cardiac autonomic nervous system and risk of arrhythmias in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Stroke. 2007 Feb;38(2):276-280. doi:10.1093/brain/awh282
12. Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383(6602):707-710. doi:10.1038/383707a0
13. Kalaria RN, Viitanen M, Kalimo H, Dichgans M, Tabira T, CASASIL Group of Vas-Cog. The pathogenesis of CADASIL: an update. J Neurol Sci. 2004;226(1-2):35-39. doi:10.1016/j.jns.2004.09.008
14. Reddy SPK, Vishnu VY, Goyal V, Singh MB, Arora S, Garg A, et al. CADASIL syndrome and stroke in young people. QJM. 2020 Feb 1;113(2):118-119. doi:10.1093/qjmed/hcz243
15. Carone DA. CADASIL and multiple sclerosis: A case report of prolonged misdiagnosis. Applied neuropsychology Adult. 2017;24(3):294-297. doi:10.1080/23279095.2016.1214132
16. Zhu S, Nahas SJ. CADASIL: Imaging characteristics and clinical correlation. Curr Pain Headache Rep. 2016;20(10):57. doi:10.1007/s11916-016-0584-6
17. Kalaria RN, Low WC, Oakley AE, Slade JY, Ince PG, Morris CM, et al. CADASIL and genetics of cerebral ischaemia. J Neural Transm Suppl. 2002;(63):75-90. doi:10.1007/978-3-7091-6137-1_5
18. O’Sullivan M, Jarosz JM, Martin RJ, Deasy N, Powell JF, Markus HS. MRI hyperintensities of the temporal lobe and external capsule in patients with CADASIL. Neurology. 2001;56(5):628-634. doi:10.1212/wnl.56.5.628
19. Khurshid S, Choi SH, Weng L-C, Wang EY, Trinquart L, Benjamin EJ, et al. Frequency of cardiac rhythm abnormalities in a half million adults. Circ ArrhythmElectrophysiol. 2018;11(7):e006273. doi:10.1161/CIRCEP.118.006273
20. Samuels MA. The brain–heart connection. Circulation. 2007;116(1):77-84. doi:10.1161/CIRCULATIONAHA. 106.678995
21. Cumurciuc R, Henry P, Gobron C, Vicaut E, Bousser MG, Chabriat H, et al. Electrocardiogram in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy patients without any clinical evidence of coronary artery disease: a case-control study. Stroke. 2006;37(4):1100-1102. doi:10.1161/01.STR.0000209242.68844.20
22. Luxán G, D’Amato G, MacGrogan D, de la Pompa JL. Endocardial notch signaling in cardiac development and disease. Circ Res. 2016;118(1):e1-e18. doi:10.1161/CIRCRESAHA.115.305350
23. Rubin CB, Hahn V, Kobayashi T, Litwack A. A report of accelerated coronary artery disease associated with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Case Rep Cardiol. 2015;2015:167513. doi:10.1155/2015/167513
24. Langer C, Adukauskaite A, Plank F, Feuchtner G, Cartes-Zumelzu F. Cerebral autosomal dominant arteriopathy (CADASIL) with cardiac involvement (ANOCA) and subcortical leukencephalopathy. J Cardiovasc Comput Tomogr. 2020;14(5):e1-e6. doi:10.1016/j.jcct.2018.08.005
25. Pettersen JA, Keith J, Gao F, Spence JD, Black SE. CADASIL accelerated by acute hypotension: Arterial and venous contribution to leukoaraiosis. Neurology. 2017;88(11):1077-1080. doi:10.1212/WNL.0000000000003717
Clinical Presentation of Subacute Combined Degeneration in a Patient With Chronic B12 Deficiency
Subacute combined degeneration (SCD) is an acquired neurologic complication of vitamin B12 (cobalamin) or, rarely, vitamin B9 (folate) deficiency. SCD is characterized by progressive demyelination of the dorsal and lateral spinal cord, resulting in peripheral neuropathy; gait ataxia; impaired proprioception, vibration, and fine touch; optic neuropathy; and cognitive impairment.1 In addition to SCD, other neurologic manifestations of B12 deficiency include dementia, depression, visual symptoms due to optic atrophy, and behavioral changes.2 The prevalence of SCD in the US has not been well documented, but B12 deficiency is reported at 6% in those aged < 60 years and 20% in those > 60 years.3
Causes of B12 and B9 deficiency include advanced age, low nutritional intake (eg, vegan diet), impaired absorption (eg, inflammatory bowel disease, autoimmune pernicious anemia, gastrectomy, pancreatic disease), alcohol use, tapeworm infection, medications, and high metabolic states.2,4 Impaired B12 absorption is common in patients taking medications, such as metformin and proton pump inhibitors (PPI), due to suppression of ileal membrane transport and intrinsic factor activity.5-7 B-vitamin deficiency can be exacerbated by states of increased cellular turnover, such as polycythemia vera, due to elevated DNA synthesis.
Patients may experience permanent neurologic damage when the diagnosis and treatment of SCD are missed or delayed. Early diagnosis of SCD can be challenging due to lack of specific hematologic markers. In addition, many other conditions such as diabetic neuropathy, malnutrition, toxic neuropathy, sarcoidosis, HIV, multiple sclerosis, polycythemia vera, and iron deficiency anemia have similar presentations and clinical findings.8 Anemia and/or macrocytosis are not specific to B12 deficiency.4 In addition, patients with B12 deficiency may have a normal complete blood count (CBC); those with concomitant iron deficiency may have minimal or no mean corpuscular volume (MCV) elevation.4 In patients suspected to have B12 deficiency based on clinical presentation or laboratory findings of macrocytosis, serum methylmalonic acid (MMA) can serve as a direct measure of B12 activity, with levels > 0.75 μmol/L almost always indicating cobalamin deficiency. 9 On the other hand, plasma total homocysteine (tHcy) is a sensitive marker for B12 deficiency. The active form of B12, holotranscobalamin, has also emerged as a specific measure of B12 deficiency.9 However, in patients with SCD, measurement of these markers may be unnecessary due to the severity of their clinical symptoms.
The diagnosis of SCD is further complicated because not all individuals who develop B12 or B9 deficiency will develop SCD. It is difficult to determine which patients will develop SCD because the minimum level of serum B12 required for normal function is unknown, and recent studies indicate that SCD may occur even at low-normal B12 and B9 levels.2,4,10 Commonly, a serum B12 level of < 200 pg/mL is considered deficient, while a level between 200 and 300 pg/mL is considered borderline.4 The goal level of serum B12 is > 300 pg/mL, which is considered normal.4 While serologic findings of B-vitamin deficiency are only moderately specific, radiographic findings are highly sensitive and specific for SCD. According to Briani and colleagues, the most consistent finding in SCD on magnetic resonance imaging (MRI) is a “symmetrical, abnormally increased T2 signal intensity, commonly confined to posterior or posterior and lateral columns in the cervical and thoracic spinal cord.”2
We present a case of SCD in a patient with low-normal vitamin B12 levels who presented with progressive sensorimotor deficits and vision loss. The patient was subsequently diagnosed with SCD by radiologic workup. His course was complicated by worsening neurologic deficits despite B12 replacement. The progression of his clinical symptoms demonstrates the need for prompt, aggressive B12 replacement in patients diagnosed with SCD.
Case Presentation
A 63-year-old man presented for neurologic evaluation of progressive gait disturbance, paresthesia, blurred vision, and increasing falls despite use of a walker. Pertinent medical history included polycythemia vera requiring phlebotomy for approximately 9 years, alcohol use disorder (18 servings weekly), type 2 diabetes mellitus, and a remote episode of transient ischemic attack (TIA). The patient reported a 5-year history of burning pain in all extremities. A prior physician diagnosis attributed the symptoms to polyneuropathy secondary to iron deficiency anemia in the setting of chronic phlebotomy for polycythemia vera and high erythrogenesis. He was prescribed gabapentin 600 mg 3 times daily for pain control. B12 deficiency was considered an unlikely etiology due to a low-normal serum level of 305 pg/mL (reference range, 190-950 pg/mL) and normocytosis, with MCV of 88 fL (reference range, 80-100 fL). The patient also reported a 3-year history of blurred vision, which was initially attributed to be secondary to diabetic retinopathy. One week prior to presenting to our clinic, he was evaluated by ophthalmology for new-onset, bilateral central visual field defects, and he was diagnosed with nutritional optic neuropathy.
Ophthalmology suspected B12 deficiency. Notable findings included reduced deep tendon reflexes (DTRs) in the upper extremities and absent DTRs in the lower extremities, reduced sensation to light touch in all extremities, absent sensation to pinprick, vibration, and temperature in the lower extremities, positive Romberg sign, and a wide-based antalgic gait with the ankles externally rotated bilaterally (Table 1)
Previous cardiac evaluation failed to provide a diagnosis for syncopal episodes. MRI of the brain revealed nonspecific white matter changes consistent with chronic microvascular ischemic disease. Electromyography was limited due to pain but showed severe peripheral neuropathy. Laboratory results showed megalocytosis, low-normal serum B12 levels, and low serum folate levels (Table 2). The patient was diagnosed with polyneuropathy and was given intramuscular (IM) vitamin B12 1000 mcg once and a daily multivitamin (containing 25 mcg of B12). He was counseled on alcohol abstinence and medication adherence and was scheduled for follow-up in 3 months. He continued outpatient phlebotomy every 6 weeks for polycythemia.
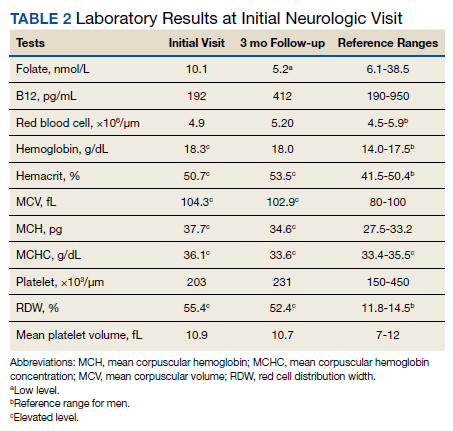
At 3-month follow-up, the patient reported medication adherence, continued alcohol use, and worsening of symptoms. Falls, which now occurred 2 to 3 times weekly despite proper use of a walker, were described as sudden loss of bilateral lower extremity strength without loss of consciousness, palpitations, or other prodrome. Laboratory results showed minimal changes. Physical examination of the patient demonstrated similar deficits as on initial presentation. The patient received one additional B12 1000 mcg IM. Gabapentin was replaced with pregabalin 75 mg twice daily due to persistent uncontrolled pain and paresthesia. The patient was scheduled for a 3-month followup (6 months from initial visit) and repeat serology.
At 6-month follow-up, the patient showed continued progression of disease with significant difficulty using the walker, worsening falls, and wheelchair use required. Physical examination showed decreased sensation bilaterally up to the knees, absent bilateral patellar and Achilles reflexes, and unsteady gait. Laboratory results showed persistent subclinical B12 deficiency. MRI of the brain and spine showed high T2 signaling in a pattern highly specific for SCD. A formal diagnosis of SCD was made. The patient received an additional B12 1000 mcg IM once. Follow-up phone call with the patient 1 month later revealed no progression or improvement of symptoms.
Radiographic Findings
MRI of the cervical and thoracic spine demonstrated abnormal high T2 signal starting from C2 and extending along the course of the cervical and thoracic spinal cord (Figure). MRI in SCD classically shows symmetric, bilateral high T2 signal within the dorsal columns; on axial images, there is typically an inverted “V” sign.2,4 There can also be abnormal cerebral white matter change; however, MRI of the brain in this patient did not show any abnormalities.2 The imaging differential for this appearance includes other metabolic deficiencies/toxicities: copper deficiency; vitamin E deficiency; methotrexateinduced myelopathy, and infectious causes: HIV vacuolar myelopathy; and neurosyphilis (tabes dorsalis).4
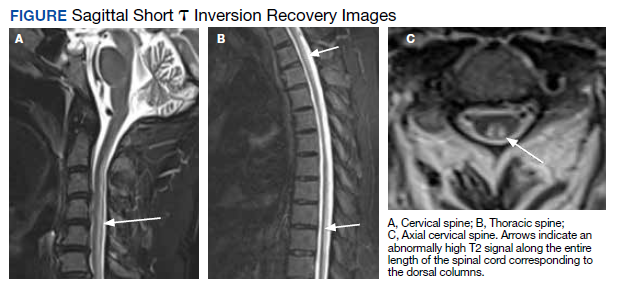
Discussion
This case demonstrates the clinical and radiographic findings of SCD and underscores the need for high-intensity dosing of B12 replacement in patients with SCD to prevent progression of the disease and development of morbidities.
Symptoms of SCD may manifest even when the vitamin levels are in low-normal levels. Its presentation is often nonspecific, thus radiologic workup is beneficial to elucidate the clinical picture. We support the use of spinal MRI in patients with clinical suspicion of SCD to help rule out other causes of myelopathy. However, an MRI is not indicated in all patients with B12 deficiency, especially those without myelopathic symptoms. Additionally, follow-up spinal MRIs are useful in monitoring the progression or improvement of SCD after B12 replacement.2 It is important to note that the MRI findings in SCD are not specific to B12 deficiency; other causes may present with similar radiographic findings.4 Therefore, radiologic findings must be correlated with a patient’s clinical presentation.
B12 replacement improves and may resolve clinical symptoms and abnormal radiographic findings of SCD. The treatment duration of B12 deficiency depends on the underlying etiology. Reversible causes, such as metformin use > 4 months, PPI use > 12 months, and dietary deficiency, require treatment until appropriate levels are reached and symptoms are resolved.4,11 The need for chronic metformin and PPI use should also be reassessed regularly. In patients who require long-term metformin use, IM administration of B12 1000 mcg annually should be considered, which will ensure adequate storage for more than 1 year.12,13 In patients who require long-term PPI use, the risk and benefits of continued use should be measured, and if needed, the lowest possible effective PPI dose is recommended.14 Irreversible causes of B12 deficiency, such as advanced age, prior gastrectomy, chronic pancreatitis, or autoimmune pernicious anemia, require lifelong supplementation of B12.4,11
In general, oral vitamin B12 replacement at 1000 to 2000 mcg daily may be as effective as parenteral replacement in patients with mild to moderate deficiency or neurologic symptoms.11 On the other hand, patients with SCD often require parenteral replacement of B12 due to the severity of their deficiency or neurologic symptoms, need for more rapid improvement in symptoms, and prevention of irreversible neurological deficits. 4,11 Appropriate B12 replacement in SCD requires intensive initial therapy which may involve IM B12 1000 mcg every other day for 2 weeks and additional IM supplementation every 2 to 3 months afterward until resolution of deficiency.4,14 IM replacement may also be considered in patients who are nonadherent to oral replacement or have an underlying gastrointestinal condition that impairs enteral absorption.4,11
B12 deficiency is frequently undertreated and can lead to progression of disease with significant morbidity. The need for highintensity dosing of B12 replacement is crucial in patients with SCD. Failure to respond to treatment, as shown from the lack of improvement of serum markers or symptoms, likely suggests undertreatment, treatment nonadherence, iron deficiency anemia, an unidentified malabsorption syndrome, or other diagnoses. In our case, significant undertreatment, compounded by his suspected iron deficiency anemia secondary to his polycythemia vera and chronic phlebotomies, are the most likel etiologies for his lack of clinical improvement.
Multiple factors may affect the prognosis of SCD. Males aged < 50 years with absence of anemia, spinal cord atrophy, Romberg sign, Babinski sign, or sensory deficits on examination have increased likelihood of eventual recovery of signs and symptoms of SCD; those with less spinal cord involvement (< 7 cord segments), contrast enhancement, and spinal cord edema also have improved outcomes.4,15
Conclusion
SCD is a rare but serious complication of chronic vitamin B12 deficiency that presents with a variety of neurological findings and may be easily confused with other illnesses. The condition is easily overlooked or misdiagnosed; thus, it is crucial to differentiate B12 deficiency from other common causes of neurologic symptoms. Specific findings on MRI are useful to support the clinical diagnosis of SCD and guide clinical decisions. Given the prevalence of B12 deficiency in the older adult population, clinicians should remain alert to the possibility of these conditions in patients who present with progressive neuropathy. Once a patient is diagnosed with SCD secondary to a B12 deficiency, appropriate B12 replacement is critical. Appropriate B12 replacement is aggressive and involves IM B12 1000 mcg every other day for 2 to 3 weeks, followed by additional IM administration every 2 months before transitioning to oral therapy. As seen in this case, failure to adequately replenish B12 can lead to progression or lack of resolution of SCD symptoms.
1. Gürsoy AE, Kolukısa M, Babacan-Yıldız G, Celebi A. Subacute Combined Degeneration of the Spinal Cord due to Different Etiologies and Improvement of MRI Findings. Case Rep Neurol Med. 2013;2013:159649. doi:10.1155/2013/159649
2. Briani C, Dalla Torre C, Citton V, et al. Cobalamin deficiency: clinical picture and radiological findings. Nutrients. 2013;5(11):4521-4539. Published 2013 Nov 15. doi:10.3390/nu5114521
3. Hunt A, Harrington D, Robinson S. Vitamin B12 deficiency. BMJ. 2014;349:g5226. Published 2014 Sep 4. doi:10.1136/bmj.g5226
4. Qudsiya Z, De Jesus O. Subacute combined degeneration of the spinal cord. [Updated 2021 Feb 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Updated August 30, 2021. Accessed January 5, 2022. https://www.ncbi.nlm.nih.gov/books /NBK559316/
5. de Jager J, Kooy A, Lehert P, et al. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. BMJ. 2010;340:c2181. Published 2010 May 20. doi:10.1136/bmj.c2181
6. Aroda VR, Edelstein SL, Goldberg RB, et al. Longterm Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J Clin Endocrinol Metab. 2016;101(4):1754-1761. doi:10.1210/jc.2015-3754
7. Lam JR, Schneider JL, Zhao W, Corley DA. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency. JAMA. 2013;310(22):2435-2442. doi:10.1001/jama.2013.280490
8. Mihalj M, Titlic´ M, Bonacin D, Dogaš Z. Sensomotor axonal peripheral neuropathy as a first complication of polycythemia rubra vera: A report of 3 cases. Am J Case Rep. 2013;14:385-387. Published 2013 Sep 25. doi:10.12659/AJCR.884016
9. Devalia V, Hamilton MS, Molloy AM; British Committee for Standards in Haematology. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496-513. doi:10.1111/bjh.12959
10. Cao J, Xu S, Liu C. Is serum vitamin B12 decrease a necessity for the diagnosis of subacute combined degeneration?: A meta-analysis. Medicine (Baltimore). 2020;99(14):e19700.doi:10.1097/MD.0000000000019700
11. Langan RC, Goodbred AJ. Vitamin B12 Deficiency: Recognition and Management. Am Fam Physician. 2017;96(6):384-389.
12. Mazokopakis EE, Starakis IK. Recommendations for diagnosis and management of metformin-induced vitamin B12 (Cbl) deficiency. Diabetes Res Clin Pract. 2012;97(3):359-367. doi:10.1016/j.diabres.2012.06.001
13. Mahajan R, Gupta K. Revisiting Metformin: Annual Vitamin B12 Supplementation may become Mandatory with Long-Term Metformin Use. J Young Pharm. 2010;2(4):428-429. doi:10.4103/0975-1483.71621
14. Parks NE. Metabolic and Toxic Myelopathies. Continuum (Minneap Minn). 2021;27(1):143-162. doi:10.1212/CON.0000000000000963
15. Vasconcelos OM, Poehm EH, McCarter RJ, Campbell WW, Quezado ZM. Potential outcome factors in subacute combined degeneration: review of observational studies. J Gen Intern Med. 2006;21(10):1063-1068. doi:10.1111/j.1525-1497.2006.00525.x
Subacute combined degeneration (SCD) is an acquired neurologic complication of vitamin B12 (cobalamin) or, rarely, vitamin B9 (folate) deficiency. SCD is characterized by progressive demyelination of the dorsal and lateral spinal cord, resulting in peripheral neuropathy; gait ataxia; impaired proprioception, vibration, and fine touch; optic neuropathy; and cognitive impairment.1 In addition to SCD, other neurologic manifestations of B12 deficiency include dementia, depression, visual symptoms due to optic atrophy, and behavioral changes.2 The prevalence of SCD in the US has not been well documented, but B12 deficiency is reported at 6% in those aged < 60 years and 20% in those > 60 years.3
Causes of B12 and B9 deficiency include advanced age, low nutritional intake (eg, vegan diet), impaired absorption (eg, inflammatory bowel disease, autoimmune pernicious anemia, gastrectomy, pancreatic disease), alcohol use, tapeworm infection, medications, and high metabolic states.2,4 Impaired B12 absorption is common in patients taking medications, such as metformin and proton pump inhibitors (PPI), due to suppression of ileal membrane transport and intrinsic factor activity.5-7 B-vitamin deficiency can be exacerbated by states of increased cellular turnover, such as polycythemia vera, due to elevated DNA synthesis.
Patients may experience permanent neurologic damage when the diagnosis and treatment of SCD are missed or delayed. Early diagnosis of SCD can be challenging due to lack of specific hematologic markers. In addition, many other conditions such as diabetic neuropathy, malnutrition, toxic neuropathy, sarcoidosis, HIV, multiple sclerosis, polycythemia vera, and iron deficiency anemia have similar presentations and clinical findings.8 Anemia and/or macrocytosis are not specific to B12 deficiency.4 In addition, patients with B12 deficiency may have a normal complete blood count (CBC); those with concomitant iron deficiency may have minimal or no mean corpuscular volume (MCV) elevation.4 In patients suspected to have B12 deficiency based on clinical presentation or laboratory findings of macrocytosis, serum methylmalonic acid (MMA) can serve as a direct measure of B12 activity, with levels > 0.75 μmol/L almost always indicating cobalamin deficiency. 9 On the other hand, plasma total homocysteine (tHcy) is a sensitive marker for B12 deficiency. The active form of B12, holotranscobalamin, has also emerged as a specific measure of B12 deficiency.9 However, in patients with SCD, measurement of these markers may be unnecessary due to the severity of their clinical symptoms.
The diagnosis of SCD is further complicated because not all individuals who develop B12 or B9 deficiency will develop SCD. It is difficult to determine which patients will develop SCD because the minimum level of serum B12 required for normal function is unknown, and recent studies indicate that SCD may occur even at low-normal B12 and B9 levels.2,4,10 Commonly, a serum B12 level of < 200 pg/mL is considered deficient, while a level between 200 and 300 pg/mL is considered borderline.4 The goal level of serum B12 is > 300 pg/mL, which is considered normal.4 While serologic findings of B-vitamin deficiency are only moderately specific, radiographic findings are highly sensitive and specific for SCD. According to Briani and colleagues, the most consistent finding in SCD on magnetic resonance imaging (MRI) is a “symmetrical, abnormally increased T2 signal intensity, commonly confined to posterior or posterior and lateral columns in the cervical and thoracic spinal cord.”2
We present a case of SCD in a patient with low-normal vitamin B12 levels who presented with progressive sensorimotor deficits and vision loss. The patient was subsequently diagnosed with SCD by radiologic workup. His course was complicated by worsening neurologic deficits despite B12 replacement. The progression of his clinical symptoms demonstrates the need for prompt, aggressive B12 replacement in patients diagnosed with SCD.
Case Presentation
A 63-year-old man presented for neurologic evaluation of progressive gait disturbance, paresthesia, blurred vision, and increasing falls despite use of a walker. Pertinent medical history included polycythemia vera requiring phlebotomy for approximately 9 years, alcohol use disorder (18 servings weekly), type 2 diabetes mellitus, and a remote episode of transient ischemic attack (TIA). The patient reported a 5-year history of burning pain in all extremities. A prior physician diagnosis attributed the symptoms to polyneuropathy secondary to iron deficiency anemia in the setting of chronic phlebotomy for polycythemia vera and high erythrogenesis. He was prescribed gabapentin 600 mg 3 times daily for pain control. B12 deficiency was considered an unlikely etiology due to a low-normal serum level of 305 pg/mL (reference range, 190-950 pg/mL) and normocytosis, with MCV of 88 fL (reference range, 80-100 fL). The patient also reported a 3-year history of blurred vision, which was initially attributed to be secondary to diabetic retinopathy. One week prior to presenting to our clinic, he was evaluated by ophthalmology for new-onset, bilateral central visual field defects, and he was diagnosed with nutritional optic neuropathy.
Ophthalmology suspected B12 deficiency. Notable findings included reduced deep tendon reflexes (DTRs) in the upper extremities and absent DTRs in the lower extremities, reduced sensation to light touch in all extremities, absent sensation to pinprick, vibration, and temperature in the lower extremities, positive Romberg sign, and a wide-based antalgic gait with the ankles externally rotated bilaterally (Table 1)
Previous cardiac evaluation failed to provide a diagnosis for syncopal episodes. MRI of the brain revealed nonspecific white matter changes consistent with chronic microvascular ischemic disease. Electromyography was limited due to pain but showed severe peripheral neuropathy. Laboratory results showed megalocytosis, low-normal serum B12 levels, and low serum folate levels (Table 2). The patient was diagnosed with polyneuropathy and was given intramuscular (IM) vitamin B12 1000 mcg once and a daily multivitamin (containing 25 mcg of B12). He was counseled on alcohol abstinence and medication adherence and was scheduled for follow-up in 3 months. He continued outpatient phlebotomy every 6 weeks for polycythemia.
At 3-month follow-up, the patient reported medication adherence, continued alcohol use, and worsening of symptoms. Falls, which now occurred 2 to 3 times weekly despite proper use of a walker, were described as sudden loss of bilateral lower extremity strength without loss of consciousness, palpitations, or other prodrome. Laboratory results showed minimal changes. Physical examination of the patient demonstrated similar deficits as on initial presentation. The patient received one additional B12 1000 mcg IM. Gabapentin was replaced with pregabalin 75 mg twice daily due to persistent uncontrolled pain and paresthesia. The patient was scheduled for a 3-month followup (6 months from initial visit) and repeat serology.
At 6-month follow-up, the patient showed continued progression of disease with significant difficulty using the walker, worsening falls, and wheelchair use required. Physical examination showed decreased sensation bilaterally up to the knees, absent bilateral patellar and Achilles reflexes, and unsteady gait. Laboratory results showed persistent subclinical B12 deficiency. MRI of the brain and spine showed high T2 signaling in a pattern highly specific for SCD. A formal diagnosis of SCD was made. The patient received an additional B12 1000 mcg IM once. Follow-up phone call with the patient 1 month later revealed no progression or improvement of symptoms.
Radiographic Findings
MRI of the cervical and thoracic spine demonstrated abnormal high T2 signal starting from C2 and extending along the course of the cervical and thoracic spinal cord (Figure). MRI in SCD classically shows symmetric, bilateral high T2 signal within the dorsal columns; on axial images, there is typically an inverted “V” sign.2,4 There can also be abnormal cerebral white matter change; however, MRI of the brain in this patient did not show any abnormalities.2 The imaging differential for this appearance includes other metabolic deficiencies/toxicities: copper deficiency; vitamin E deficiency; methotrexateinduced myelopathy, and infectious causes: HIV vacuolar myelopathy; and neurosyphilis (tabes dorsalis).4
Discussion
This case demonstrates the clinical and radiographic findings of SCD and underscores the need for high-intensity dosing of B12 replacement in patients with SCD to prevent progression of the disease and development of morbidities.
Symptoms of SCD may manifest even when the vitamin levels are in low-normal levels. Its presentation is often nonspecific, thus radiologic workup is beneficial to elucidate the clinical picture. We support the use of spinal MRI in patients with clinical suspicion of SCD to help rule out other causes of myelopathy. However, an MRI is not indicated in all patients with B12 deficiency, especially those without myelopathic symptoms. Additionally, follow-up spinal MRIs are useful in monitoring the progression or improvement of SCD after B12 replacement.2 It is important to note that the MRI findings in SCD are not specific to B12 deficiency; other causes may present with similar radiographic findings.4 Therefore, radiologic findings must be correlated with a patient’s clinical presentation.
B12 replacement improves and may resolve clinical symptoms and abnormal radiographic findings of SCD. The treatment duration of B12 deficiency depends on the underlying etiology. Reversible causes, such as metformin use > 4 months, PPI use > 12 months, and dietary deficiency, require treatment until appropriate levels are reached and symptoms are resolved.4,11 The need for chronic metformin and PPI use should also be reassessed regularly. In patients who require long-term metformin use, IM administration of B12 1000 mcg annually should be considered, which will ensure adequate storage for more than 1 year.12,13 In patients who require long-term PPI use, the risk and benefits of continued use should be measured, and if needed, the lowest possible effective PPI dose is recommended.14 Irreversible causes of B12 deficiency, such as advanced age, prior gastrectomy, chronic pancreatitis, or autoimmune pernicious anemia, require lifelong supplementation of B12.4,11
In general, oral vitamin B12 replacement at 1000 to 2000 mcg daily may be as effective as parenteral replacement in patients with mild to moderate deficiency or neurologic symptoms.11 On the other hand, patients with SCD often require parenteral replacement of B12 due to the severity of their deficiency or neurologic symptoms, need for more rapid improvement in symptoms, and prevention of irreversible neurological deficits. 4,11 Appropriate B12 replacement in SCD requires intensive initial therapy which may involve IM B12 1000 mcg every other day for 2 weeks and additional IM supplementation every 2 to 3 months afterward until resolution of deficiency.4,14 IM replacement may also be considered in patients who are nonadherent to oral replacement or have an underlying gastrointestinal condition that impairs enteral absorption.4,11
B12 deficiency is frequently undertreated and can lead to progression of disease with significant morbidity. The need for highintensity dosing of B12 replacement is crucial in patients with SCD. Failure to respond to treatment, as shown from the lack of improvement of serum markers or symptoms, likely suggests undertreatment, treatment nonadherence, iron deficiency anemia, an unidentified malabsorption syndrome, or other diagnoses. In our case, significant undertreatment, compounded by his suspected iron deficiency anemia secondary to his polycythemia vera and chronic phlebotomies, are the most likel etiologies for his lack of clinical improvement.
Multiple factors may affect the prognosis of SCD. Males aged < 50 years with absence of anemia, spinal cord atrophy, Romberg sign, Babinski sign, or sensory deficits on examination have increased likelihood of eventual recovery of signs and symptoms of SCD; those with less spinal cord involvement (< 7 cord segments), contrast enhancement, and spinal cord edema also have improved outcomes.4,15
Conclusion
SCD is a rare but serious complication of chronic vitamin B12 deficiency that presents with a variety of neurological findings and may be easily confused with other illnesses. The condition is easily overlooked or misdiagnosed; thus, it is crucial to differentiate B12 deficiency from other common causes of neurologic symptoms. Specific findings on MRI are useful to support the clinical diagnosis of SCD and guide clinical decisions. Given the prevalence of B12 deficiency in the older adult population, clinicians should remain alert to the possibility of these conditions in patients who present with progressive neuropathy. Once a patient is diagnosed with SCD secondary to a B12 deficiency, appropriate B12 replacement is critical. Appropriate B12 replacement is aggressive and involves IM B12 1000 mcg every other day for 2 to 3 weeks, followed by additional IM administration every 2 months before transitioning to oral therapy. As seen in this case, failure to adequately replenish B12 can lead to progression or lack of resolution of SCD symptoms.
Subacute combined degeneration (SCD) is an acquired neurologic complication of vitamin B12 (cobalamin) or, rarely, vitamin B9 (folate) deficiency. SCD is characterized by progressive demyelination of the dorsal and lateral spinal cord, resulting in peripheral neuropathy; gait ataxia; impaired proprioception, vibration, and fine touch; optic neuropathy; and cognitive impairment.1 In addition to SCD, other neurologic manifestations of B12 deficiency include dementia, depression, visual symptoms due to optic atrophy, and behavioral changes.2 The prevalence of SCD in the US has not been well documented, but B12 deficiency is reported at 6% in those aged < 60 years and 20% in those > 60 years.3
Causes of B12 and B9 deficiency include advanced age, low nutritional intake (eg, vegan diet), impaired absorption (eg, inflammatory bowel disease, autoimmune pernicious anemia, gastrectomy, pancreatic disease), alcohol use, tapeworm infection, medications, and high metabolic states.2,4 Impaired B12 absorption is common in patients taking medications, such as metformin and proton pump inhibitors (PPI), due to suppression of ileal membrane transport and intrinsic factor activity.5-7 B-vitamin deficiency can be exacerbated by states of increased cellular turnover, such as polycythemia vera, due to elevated DNA synthesis.
Patients may experience permanent neurologic damage when the diagnosis and treatment of SCD are missed or delayed. Early diagnosis of SCD can be challenging due to lack of specific hematologic markers. In addition, many other conditions such as diabetic neuropathy, malnutrition, toxic neuropathy, sarcoidosis, HIV, multiple sclerosis, polycythemia vera, and iron deficiency anemia have similar presentations and clinical findings.8 Anemia and/or macrocytosis are not specific to B12 deficiency.4 In addition, patients with B12 deficiency may have a normal complete blood count (CBC); those with concomitant iron deficiency may have minimal or no mean corpuscular volume (MCV) elevation.4 In patients suspected to have B12 deficiency based on clinical presentation or laboratory findings of macrocytosis, serum methylmalonic acid (MMA) can serve as a direct measure of B12 activity, with levels > 0.75 μmol/L almost always indicating cobalamin deficiency. 9 On the other hand, plasma total homocysteine (tHcy) is a sensitive marker for B12 deficiency. The active form of B12, holotranscobalamin, has also emerged as a specific measure of B12 deficiency.9 However, in patients with SCD, measurement of these markers may be unnecessary due to the severity of their clinical symptoms.
The diagnosis of SCD is further complicated because not all individuals who develop B12 or B9 deficiency will develop SCD. It is difficult to determine which patients will develop SCD because the minimum level of serum B12 required for normal function is unknown, and recent studies indicate that SCD may occur even at low-normal B12 and B9 levels.2,4,10 Commonly, a serum B12 level of < 200 pg/mL is considered deficient, while a level between 200 and 300 pg/mL is considered borderline.4 The goal level of serum B12 is > 300 pg/mL, which is considered normal.4 While serologic findings of B-vitamin deficiency are only moderately specific, radiographic findings are highly sensitive and specific for SCD. According to Briani and colleagues, the most consistent finding in SCD on magnetic resonance imaging (MRI) is a “symmetrical, abnormally increased T2 signal intensity, commonly confined to posterior or posterior and lateral columns in the cervical and thoracic spinal cord.”2
We present a case of SCD in a patient with low-normal vitamin B12 levels who presented with progressive sensorimotor deficits and vision loss. The patient was subsequently diagnosed with SCD by radiologic workup. His course was complicated by worsening neurologic deficits despite B12 replacement. The progression of his clinical symptoms demonstrates the need for prompt, aggressive B12 replacement in patients diagnosed with SCD.
Case Presentation
A 63-year-old man presented for neurologic evaluation of progressive gait disturbance, paresthesia, blurred vision, and increasing falls despite use of a walker. Pertinent medical history included polycythemia vera requiring phlebotomy for approximately 9 years, alcohol use disorder (18 servings weekly), type 2 diabetes mellitus, and a remote episode of transient ischemic attack (TIA). The patient reported a 5-year history of burning pain in all extremities. A prior physician diagnosis attributed the symptoms to polyneuropathy secondary to iron deficiency anemia in the setting of chronic phlebotomy for polycythemia vera and high erythrogenesis. He was prescribed gabapentin 600 mg 3 times daily for pain control. B12 deficiency was considered an unlikely etiology due to a low-normal serum level of 305 pg/mL (reference range, 190-950 pg/mL) and normocytosis, with MCV of 88 fL (reference range, 80-100 fL). The patient also reported a 3-year history of blurred vision, which was initially attributed to be secondary to diabetic retinopathy. One week prior to presenting to our clinic, he was evaluated by ophthalmology for new-onset, bilateral central visual field defects, and he was diagnosed with nutritional optic neuropathy.
Ophthalmology suspected B12 deficiency. Notable findings included reduced deep tendon reflexes (DTRs) in the upper extremities and absent DTRs in the lower extremities, reduced sensation to light touch in all extremities, absent sensation to pinprick, vibration, and temperature in the lower extremities, positive Romberg sign, and a wide-based antalgic gait with the ankles externally rotated bilaterally (Table 1)
Previous cardiac evaluation failed to provide a diagnosis for syncopal episodes. MRI of the brain revealed nonspecific white matter changes consistent with chronic microvascular ischemic disease. Electromyography was limited due to pain but showed severe peripheral neuropathy. Laboratory results showed megalocytosis, low-normal serum B12 levels, and low serum folate levels (Table 2). The patient was diagnosed with polyneuropathy and was given intramuscular (IM) vitamin B12 1000 mcg once and a daily multivitamin (containing 25 mcg of B12). He was counseled on alcohol abstinence and medication adherence and was scheduled for follow-up in 3 months. He continued outpatient phlebotomy every 6 weeks for polycythemia.
At 3-month follow-up, the patient reported medication adherence, continued alcohol use, and worsening of symptoms. Falls, which now occurred 2 to 3 times weekly despite proper use of a walker, were described as sudden loss of bilateral lower extremity strength without loss of consciousness, palpitations, or other prodrome. Laboratory results showed minimal changes. Physical examination of the patient demonstrated similar deficits as on initial presentation. The patient received one additional B12 1000 mcg IM. Gabapentin was replaced with pregabalin 75 mg twice daily due to persistent uncontrolled pain and paresthesia. The patient was scheduled for a 3-month followup (6 months from initial visit) and repeat serology.
At 6-month follow-up, the patient showed continued progression of disease with significant difficulty using the walker, worsening falls, and wheelchair use required. Physical examination showed decreased sensation bilaterally up to the knees, absent bilateral patellar and Achilles reflexes, and unsteady gait. Laboratory results showed persistent subclinical B12 deficiency. MRI of the brain and spine showed high T2 signaling in a pattern highly specific for SCD. A formal diagnosis of SCD was made. The patient received an additional B12 1000 mcg IM once. Follow-up phone call with the patient 1 month later revealed no progression or improvement of symptoms.
Radiographic Findings
MRI of the cervical and thoracic spine demonstrated abnormal high T2 signal starting from C2 and extending along the course of the cervical and thoracic spinal cord (Figure). MRI in SCD classically shows symmetric, bilateral high T2 signal within the dorsal columns; on axial images, there is typically an inverted “V” sign.2,4 There can also be abnormal cerebral white matter change; however, MRI of the brain in this patient did not show any abnormalities.2 The imaging differential for this appearance includes other metabolic deficiencies/toxicities: copper deficiency; vitamin E deficiency; methotrexateinduced myelopathy, and infectious causes: HIV vacuolar myelopathy; and neurosyphilis (tabes dorsalis).4
Discussion
This case demonstrates the clinical and radiographic findings of SCD and underscores the need for high-intensity dosing of B12 replacement in patients with SCD to prevent progression of the disease and development of morbidities.
Symptoms of SCD may manifest even when the vitamin levels are in low-normal levels. Its presentation is often nonspecific, thus radiologic workup is beneficial to elucidate the clinical picture. We support the use of spinal MRI in patients with clinical suspicion of SCD to help rule out other causes of myelopathy. However, an MRI is not indicated in all patients with B12 deficiency, especially those without myelopathic symptoms. Additionally, follow-up spinal MRIs are useful in monitoring the progression or improvement of SCD after B12 replacement.2 It is important to note that the MRI findings in SCD are not specific to B12 deficiency; other causes may present with similar radiographic findings.4 Therefore, radiologic findings must be correlated with a patient’s clinical presentation.
B12 replacement improves and may resolve clinical symptoms and abnormal radiographic findings of SCD. The treatment duration of B12 deficiency depends on the underlying etiology. Reversible causes, such as metformin use > 4 months, PPI use > 12 months, and dietary deficiency, require treatment until appropriate levels are reached and symptoms are resolved.4,11 The need for chronic metformin and PPI use should also be reassessed regularly. In patients who require long-term metformin use, IM administration of B12 1000 mcg annually should be considered, which will ensure adequate storage for more than 1 year.12,13 In patients who require long-term PPI use, the risk and benefits of continued use should be measured, and if needed, the lowest possible effective PPI dose is recommended.14 Irreversible causes of B12 deficiency, such as advanced age, prior gastrectomy, chronic pancreatitis, or autoimmune pernicious anemia, require lifelong supplementation of B12.4,11
In general, oral vitamin B12 replacement at 1000 to 2000 mcg daily may be as effective as parenteral replacement in patients with mild to moderate deficiency or neurologic symptoms.11 On the other hand, patients with SCD often require parenteral replacement of B12 due to the severity of their deficiency or neurologic symptoms, need for more rapid improvement in symptoms, and prevention of irreversible neurological deficits. 4,11 Appropriate B12 replacement in SCD requires intensive initial therapy which may involve IM B12 1000 mcg every other day for 2 weeks and additional IM supplementation every 2 to 3 months afterward until resolution of deficiency.4,14 IM replacement may also be considered in patients who are nonadherent to oral replacement or have an underlying gastrointestinal condition that impairs enteral absorption.4,11
B12 deficiency is frequently undertreated and can lead to progression of disease with significant morbidity. The need for highintensity dosing of B12 replacement is crucial in patients with SCD. Failure to respond to treatment, as shown from the lack of improvement of serum markers or symptoms, likely suggests undertreatment, treatment nonadherence, iron deficiency anemia, an unidentified malabsorption syndrome, or other diagnoses. In our case, significant undertreatment, compounded by his suspected iron deficiency anemia secondary to his polycythemia vera and chronic phlebotomies, are the most likel etiologies for his lack of clinical improvement.
Multiple factors may affect the prognosis of SCD. Males aged < 50 years with absence of anemia, spinal cord atrophy, Romberg sign, Babinski sign, or sensory deficits on examination have increased likelihood of eventual recovery of signs and symptoms of SCD; those with less spinal cord involvement (< 7 cord segments), contrast enhancement, and spinal cord edema also have improved outcomes.4,15
Conclusion
SCD is a rare but serious complication of chronic vitamin B12 deficiency that presents with a variety of neurological findings and may be easily confused with other illnesses. The condition is easily overlooked or misdiagnosed; thus, it is crucial to differentiate B12 deficiency from other common causes of neurologic symptoms. Specific findings on MRI are useful to support the clinical diagnosis of SCD and guide clinical decisions. Given the prevalence of B12 deficiency in the older adult population, clinicians should remain alert to the possibility of these conditions in patients who present with progressive neuropathy. Once a patient is diagnosed with SCD secondary to a B12 deficiency, appropriate B12 replacement is critical. Appropriate B12 replacement is aggressive and involves IM B12 1000 mcg every other day for 2 to 3 weeks, followed by additional IM administration every 2 months before transitioning to oral therapy. As seen in this case, failure to adequately replenish B12 can lead to progression or lack of resolution of SCD symptoms.
1. Gürsoy AE, Kolukısa M, Babacan-Yıldız G, Celebi A. Subacute Combined Degeneration of the Spinal Cord due to Different Etiologies and Improvement of MRI Findings. Case Rep Neurol Med. 2013;2013:159649. doi:10.1155/2013/159649
2. Briani C, Dalla Torre C, Citton V, et al. Cobalamin deficiency: clinical picture and radiological findings. Nutrients. 2013;5(11):4521-4539. Published 2013 Nov 15. doi:10.3390/nu5114521
3. Hunt A, Harrington D, Robinson S. Vitamin B12 deficiency. BMJ. 2014;349:g5226. Published 2014 Sep 4. doi:10.1136/bmj.g5226
4. Qudsiya Z, De Jesus O. Subacute combined degeneration of the spinal cord. [Updated 2021 Feb 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Updated August 30, 2021. Accessed January 5, 2022. https://www.ncbi.nlm.nih.gov/books /NBK559316/
5. de Jager J, Kooy A, Lehert P, et al. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. BMJ. 2010;340:c2181. Published 2010 May 20. doi:10.1136/bmj.c2181
6. Aroda VR, Edelstein SL, Goldberg RB, et al. Longterm Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J Clin Endocrinol Metab. 2016;101(4):1754-1761. doi:10.1210/jc.2015-3754
7. Lam JR, Schneider JL, Zhao W, Corley DA. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency. JAMA. 2013;310(22):2435-2442. doi:10.1001/jama.2013.280490
8. Mihalj M, Titlic´ M, Bonacin D, Dogaš Z. Sensomotor axonal peripheral neuropathy as a first complication of polycythemia rubra vera: A report of 3 cases. Am J Case Rep. 2013;14:385-387. Published 2013 Sep 25. doi:10.12659/AJCR.884016
9. Devalia V, Hamilton MS, Molloy AM; British Committee for Standards in Haematology. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496-513. doi:10.1111/bjh.12959
10. Cao J, Xu S, Liu C. Is serum vitamin B12 decrease a necessity for the diagnosis of subacute combined degeneration?: A meta-analysis. Medicine (Baltimore). 2020;99(14):e19700.doi:10.1097/MD.0000000000019700
11. Langan RC, Goodbred AJ. Vitamin B12 Deficiency: Recognition and Management. Am Fam Physician. 2017;96(6):384-389.
12. Mazokopakis EE, Starakis IK. Recommendations for diagnosis and management of metformin-induced vitamin B12 (Cbl) deficiency. Diabetes Res Clin Pract. 2012;97(3):359-367. doi:10.1016/j.diabres.2012.06.001
13. Mahajan R, Gupta K. Revisiting Metformin: Annual Vitamin B12 Supplementation may become Mandatory with Long-Term Metformin Use. J Young Pharm. 2010;2(4):428-429. doi:10.4103/0975-1483.71621
14. Parks NE. Metabolic and Toxic Myelopathies. Continuum (Minneap Minn). 2021;27(1):143-162. doi:10.1212/CON.0000000000000963
15. Vasconcelos OM, Poehm EH, McCarter RJ, Campbell WW, Quezado ZM. Potential outcome factors in subacute combined degeneration: review of observational studies. J Gen Intern Med. 2006;21(10):1063-1068. doi:10.1111/j.1525-1497.2006.00525.x
1. Gürsoy AE, Kolukısa M, Babacan-Yıldız G, Celebi A. Subacute Combined Degeneration of the Spinal Cord due to Different Etiologies and Improvement of MRI Findings. Case Rep Neurol Med. 2013;2013:159649. doi:10.1155/2013/159649
2. Briani C, Dalla Torre C, Citton V, et al. Cobalamin deficiency: clinical picture and radiological findings. Nutrients. 2013;5(11):4521-4539. Published 2013 Nov 15. doi:10.3390/nu5114521
3. Hunt A, Harrington D, Robinson S. Vitamin B12 deficiency. BMJ. 2014;349:g5226. Published 2014 Sep 4. doi:10.1136/bmj.g5226
4. Qudsiya Z, De Jesus O. Subacute combined degeneration of the spinal cord. [Updated 2021 Feb 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. Updated August 30, 2021. Accessed January 5, 2022. https://www.ncbi.nlm.nih.gov/books /NBK559316/
5. de Jager J, Kooy A, Lehert P, et al. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. BMJ. 2010;340:c2181. Published 2010 May 20. doi:10.1136/bmj.c2181
6. Aroda VR, Edelstein SL, Goldberg RB, et al. Longterm Metformin Use and Vitamin B12 Deficiency in the Diabetes Prevention Program Outcomes Study. J Clin Endocrinol Metab. 2016;101(4):1754-1761. doi:10.1210/jc.2015-3754
7. Lam JR, Schneider JL, Zhao W, Corley DA. Proton pump inhibitor and histamine 2 receptor antagonist use and vitamin B12 deficiency. JAMA. 2013;310(22):2435-2442. doi:10.1001/jama.2013.280490
8. Mihalj M, Titlic´ M, Bonacin D, Dogaš Z. Sensomotor axonal peripheral neuropathy as a first complication of polycythemia rubra vera: A report of 3 cases. Am J Case Rep. 2013;14:385-387. Published 2013 Sep 25. doi:10.12659/AJCR.884016
9. Devalia V, Hamilton MS, Molloy AM; British Committee for Standards in Haematology. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol. 2014;166(4):496-513. doi:10.1111/bjh.12959
10. Cao J, Xu S, Liu C. Is serum vitamin B12 decrease a necessity for the diagnosis of subacute combined degeneration?: A meta-analysis. Medicine (Baltimore). 2020;99(14):e19700.doi:10.1097/MD.0000000000019700
11. Langan RC, Goodbred AJ. Vitamin B12 Deficiency: Recognition and Management. Am Fam Physician. 2017;96(6):384-389.
12. Mazokopakis EE, Starakis IK. Recommendations for diagnosis and management of metformin-induced vitamin B12 (Cbl) deficiency. Diabetes Res Clin Pract. 2012;97(3):359-367. doi:10.1016/j.diabres.2012.06.001
13. Mahajan R, Gupta K. Revisiting Metformin: Annual Vitamin B12 Supplementation may become Mandatory with Long-Term Metformin Use. J Young Pharm. 2010;2(4):428-429. doi:10.4103/0975-1483.71621
14. Parks NE. Metabolic and Toxic Myelopathies. Continuum (Minneap Minn). 2021;27(1):143-162. doi:10.1212/CON.0000000000000963
15. Vasconcelos OM, Poehm EH, McCarter RJ, Campbell WW, Quezado ZM. Potential outcome factors in subacute combined degeneration: review of observational studies. J Gen Intern Med. 2006;21(10):1063-1068. doi:10.1111/j.1525-1497.2006.00525.x
Emphysematous Aortitis due to Klebsiella Pneumoniae in a Patient With Poorly Controlled Diabetes Mellitus
Patients with poorly controlled diabetes mellitus and an infectious source can be predisposed to infectious aortitis.
Aortitis is the all-encompassing term ascribed to the inflammatory process in the aortic wall that can be either infective or noninfective in origin, commonly autoimmune or inflammatory large-vessel vasculitis.1 Infectious aortitis, also known as bacterial, microbial, or cryptogenic aortitis, as well as mycotic or infected aneurysm, is a rare entity in the current antibiotic era but potentially a life-threatening disorder.2 The potential complications of infectious aortitis include emphysematous aortitis (EA), pseudoaneurysm, aortic rupture, septic emboli, and fistula formation (eg, aorto-enteric fistula).2,3
EA is a rare but serious inflammatory condition of the aorta with a nonspecific clinical presentation associated with high morbidity and mortality.2-6 The condition is characterized by a localized collection of gas and purulent exudate at the aortic wall.1,3 A few cases of EA have previously been reported; however, no known cases have been reported in the literature due to Klebsiella pneumoniae (K pneumoniae).
The pathophysiology of EA is the presence of underlying damage to the arterial wall caused by a hematogenously inoculated gas-producing organism.2,3 Most reported cases of EA are due to endovascular graft complications. Under normal circumstances, the aortic intima is highly resistant to infectious pathogens; however, certain risk factors, such as diabetes mellitus (DM), atherosclerotic disease, preexisting aneurysm, cystic medial necrosis, vascular malformation, presence of medical devices, surgery, or impaired immunity can alter the integrity of the aortic intimal layer and predispose the aortic intima to infection.1,4-7 Bacteria are the most common causative organisms that can infect the aorta, especially Staphylococcus, Enterococcus, Streptococcus, Salmonella, and spirochete Treponema pallidum (syphilis).1,2,4,8 The site of the primary infection remains unclear in some patients.2,3,5,6 Infection of the aorta can arise by several mechanisms: direct extension of a local infection to an existing intimal injury or atherosclerotic plaque (the most common mechanism), septic embolism from endocarditis, direct bacterial inoculation from traumatic contamination, contiguous infection extending to the aorta wall, or a distant source of bacteremia.2,3
Clinical manifestations of EA depend on the site and the extent of infection. The diagnosis should be considered in patients with atherosclerosis, fever, abdominal pain, and leukocytosis.2,4-8 The differential diagnosis for EA includes (1) noninfective causes of aortitis, including rheumatoid arthritis and systemic lupus erythematosus; (2) tuberculous aortitis; (3) syphilitic aortitis; and (4) idiopathic isolated aortitis. Establishing an early diagnosis of infectious aortitis is extremely important because this condition is associated with a high rate of morbidity and mortality secondary to aortic rupture.2-7
Imaging is critical for a reliable and quick diagnosis of acute aortic pathology. Noninvasive cross-sectional imaging modalities, such as contrast-enhanced computed tomography (CT), magnetic resonance imaging, nuclear medicine, or positron emission tomography, are used for both the initial diagnosis and follow-up of aortitis.1 CT is the primary imaging method in most medical centers because it is widely available with short acquisition time in critically ill patients.3 CT allows rapid detection of abnormalities in wall thickness, diameter, and density, and enhancement of periaortic structures, enabling reliable exclusion of other aortic pathologies that may resemble acute aortitis. Also, CT aids in planning the optimal therapeutic approach.1,3,5-8
This case illustrates EA associated with infection by K pneumoniae in a patient with poorly controlled type 2 DM (T2DM). In this single case, our patient presented to the Bay Pines Veterans Affairs Healthcare System (BPVAHS) in Florida with recent superficial soft tissue injury, severe hyperglycemia, worsening abdominal pain, and leukocytosis without fever or chills. The correct diagnosis of EA was confirmed by characteristic CT findings.
Case Presentation
A 72-year-old male with a history of T2DM, hypertension, atherosclerotic vascular disease, obstructive lung disease, and smoking 1.5 packs per day for 40 years presented with diabetic ketoacidosis, a urinary tract infection, and abdominal pain of 1-week duration that started to worsen the morning he arrived at the BPVAHS emergency department. He reported no nausea, vomiting, diarrhea, constipation, chest pain, shortness of breath, fever, chills, fatigue, or dysuria. He had a nonhealing laceration on his left medial foot that occurred 18 days before admission and was treated at an outside hospital.
The patient’s surgical history included a left common femoral endarterectomy and a left femoral popliteal above-knee reverse saphenous vein bypass 4 years ago for severe critical limb ischemia due to occlusion of his left superficial femoral artery with distal embolization to the first and fifth toes. About 6 months later, he developed disabling claudication in his left lower extremity due to distal popliteal artery occlusion and had another bypass surgery to the below-knee popliteal artery with a reverse saphenous vein graft harvested from the right thigh.
On initial examination, his vital signs were within normal limits except for a blood pressure of 177/87 mm Hg. His physical examination demonstrated a nondistended abdomen with normal bowel sounds, mild lower quadrant tenderness on the left more than on the right, intermittent abdominal pain located around umbilicus with radiation to the back, and a negative psoas sign. His left medial foot had a nonhealing laceration with black sutures in place, with minimal erythema in the surrounding tissue and scab formation. He also had mild costovertebral tenderness on the left.
Initial laboratory investigation results were notable for a glucose level of 609 mg/dL and a white blood cell count of 14.6 × 103 cells/mcL with 86.5% of neutrophils. A CT scan of his abdomen revealed extensive atherosclerosis of the abdominal aorta and periaortic aneurysmal fluid collection with multiple foci of gas (Figure 1). Additionally, the aneurysmal fluid collection involved the proximal segment of the left common femoral artery, suspicious for left femoral arteritis (Figure 2). The patient was started on broad-spectrum antibiotics, morphine, and an insulin drip. Both urine and blood cultures were positive for K pneumoniae susceptible to multiple antibiotics. He was transferred to a tertiary medical center and was referred for a vascular surgery consultation.
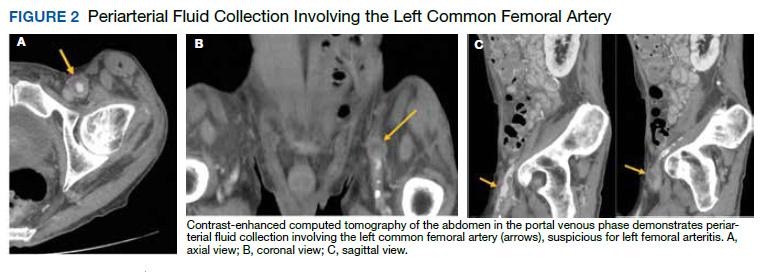
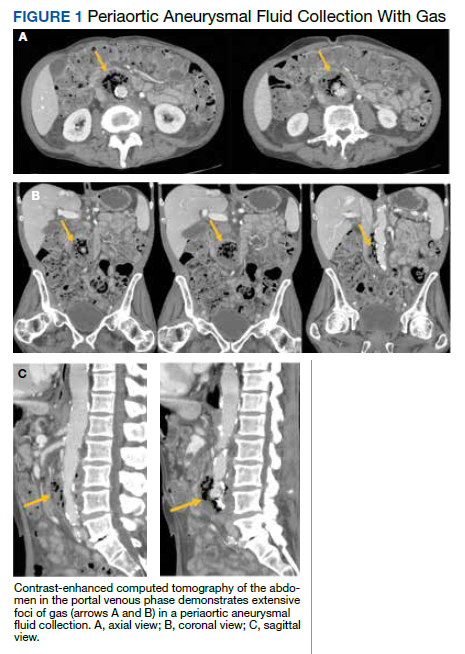
The patient underwent surgical resection of the infected infrarenal EA and infected left common femoral artery with right axillary-bifemoral bypass with an 8-mm PTFE (polytetrafluoroethylene) graft. During the surgery, excision of the wall of the left common femoral artery and infrarenal aorta revealed frank pus with purulent fluid, which was sent to cytology for analysis and culture. His intraoperative cultures grew K pneumoniae sensitive to multiple antibiotics, including ceftriaxone, sulfamethoxazole/trimethoprim, and ampicillin/sulbactam. The vascular surgery team recommended inpatient admission and administration of 6 weeks of IV antibiotics postoperatively with ceftriaxone, followed by outpatient oral suppression therapy after discharge. The patient tolerated the surgery well and was discharged after 6 weeks of IV ceftriaxone followed by outpatient oral suppression therapy. However, the patient was transferred back to BPVAHS for continued care and rehabilitation placement.
The patient’s subsequent course was complicated by multiple hospital admissions, including aspiration pneumonia, hypoglycemia, diarrhea, and anemia. On one of his CT abdomen/pelvic examinations, a cysticlike mass was noted in the pancreatic head with a possible pancreatic duodenal fistula (this mass was not mentioned on the initial presurgical CT, although it can be seen in retrospect (Figure 3). Gastroenterology was consulted.
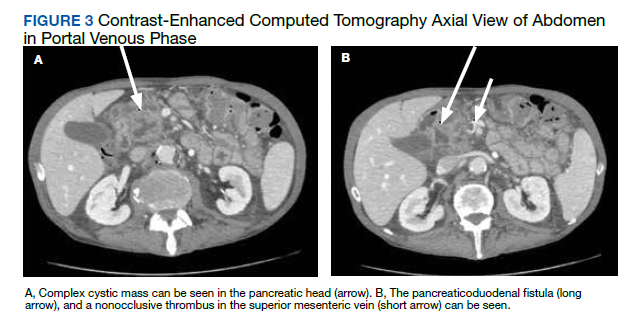
An upper endoscopy was performed that confirmed the fistula at the second portion of the duodenum. Findings from an endoscopic ultrasonography performed at an outside institution were concerning for a main duct intraductal papillary mucinous neoplasm (IPMN) with fistula, with biopsy results pending.
Discussion
This case contributes to the evidence that poorly controlled T2DM can be a predisposing factor for multiple vascular complications, including the infection of the aortic wall with progression to EA. Klebsiella species are considered opportunistic, Gram-negative pathogens that may disseminate to other tissues, causing life-threatening infections, including pneumonia, UTIs, bacteremia, and sepsis.9K pneumoniae infections are particularly challenging in neonates, the elderly, and immunocompromised individuals.9 CT is sensitive and specific in the detection of this pathologic entity.1,3 In patients with a suspected infectious etiology, the presence of foci of gas on CT in solid organ tissues is usually associated with an anaerobic infection. Gas can also be produced by Gram-negative facultative anaerobes that can ferment glucose in necrotic tissues.9
Although any microorganism can infect the aorta, K pneumoniae cultured from the blood specimen, urine culture, and intraoperative specimens in our patient was responsible for the formed gas in the aortic wall. Occurrence of spontaneous gas by this microorganism is usually associated with conditions leading to either increased vulnerability to infections and/or enhanced bacterial virulence.9 Although a relationship between EA and T2DM has not been proved, it is well known that patients with T2DM have a defect in their host-defense mechanisms, making them more susceptible to infections such as EA. Furthermore, because patients with T2DM are prone to the development of Gram-negative sepsis, organisms such as K pneumoniae would tend to emerge. Patients with poorly controlled T2DM and the presence of an infectious source can be predisposed to infectious aortitis, eventually leading to a gas-forming infection of the aorta.5,7
We postulate that the hematogenous spread of bacteria from a laceration in the leg as well as the presence of the pancreaticoduodenal fistula was likely the cause of the infectious EA in this case, considering the patient’s underlying uncontrolled T2DM. The patient’s prior left lower extremity vascular graft also may have provided a nidus for spreading to the aorta. Other reported underlying diseases of EA include aortic atherosclerosis, T2DM, diverticulitis, colon cancer, underlying aneurysm, immune-compromised status, and the presence of a medical device or open surgery.4-7,9
To our knowledge, this is the first case of EA associated with a pancreaticoduodenal fistula related to intraductal papillary mucinous neoplasm (IPMN). Fistulation of a main duct IPMN is rare, occurring in just 6.6% of cases.10 It can occur both before and after malignant degeneration.
EA requires rapid diagnosis, antibiotic therapy, and consultation with a vascular surgeon for immediate resection of the infected artery and graft bypass. The initial treatment of suspected infectious aortitis is IV antibiotics with broad antimicrobial coverage of the most likely pathologic organisms, particularly staphylococcal species and Gram-negative rods. Surgical debridement and revascularization should be completed early because of the high mortality rate of this condition. The intent of surgery is to control sepsis and reconstruct the arterial vasculature. Patients should remain on parenteral or oral antibiotics for at least 6 weeks to ensure full clearance of the infection.8 They should be followed up closely with serial blood cultures and CT scans.8 The rarity of the disorder, low level of awareness, varying presentations, and lack of evidence delineating pathogenesis and causality contribute to the challenge of recognizing, diagnosing, and treating EA in patients with T2DM and inflammation.
Conclusions
This case report can help bring awareness of this rare and potentially life-threatening condition in patients with T2DM. Clinicians should be aware of the risk of AE, particularly in patients with several additional risk factors: recent skin/soft tissue trauma, prior vascular graft surgery, and an underlying pancreatic mass. CT is the imaging method of choice that helps to rapidly choose a necessary emergent treatment approach.
1. Litmanovich DE, Yıldırım A, Bankier AA. Insights into imaging of aortitis. Insights Imaging. 2012;3(6):545-560. doi:10.1007/s13244-012-0192-x
2. Lopes RJ, Almeida J, Dias PJ, Pinho P, Maciel MJ. Infectious thoracic aortitis: a literature review. Clin Cardiol. 2009;32(9):488-490. doi:10.1002/clc.20578
3. Murphy DJ, Keraliya AR, Agrawal MD, Aghayev A, Steigner ML. Cross-sectional imaging of aortic infections. Insights Imaging. 2016;7(6):801-818. doi:10.1007/s13244-016-0522-5
4. Md Noh MSF, Abdul Rashid AM, Ar A, B N, Mohammed Y, A RE. Emphysematous aortitis: report of two cases and CT imaging findings. BJR Case Rep. 2017;3(3):20170006. doi:10.1259/bjrcr.20170006
5. Harris C, Geffen J, Rizg K, et al. A rare report of infectious emphysematous aortitis secondary to Clostridium septicum without prior vascular intervention. Case Rep Vasc Med. 2017;2017:4984325. doi:10.1155/2017/4984325
6. Ito F, Inokuchi R, Matsumoto A, et al. Presence of periaortic gas in Clostridium septicum-infected aortic aneurysm aids in early diagnosis: a case report and systematic review of the literature. J Med Case Rep. 2017;11(1):268. doi:10.1186/s13256-017-1422-0
7. Urgiles S, Matos-Casano H, Win KZ, Berardo J, Bhatt U, Shah J. Emphysematous aortitis due to Clostridium septicum in an 89-year-old female with ileus. Case Rep Infect Dis. 2019;2019:1094837. doi:10.1155/2019/1094837
8. Foote EA, Postier RG, Greenfield RA, Bronze MS. Infectious aortitis. Curr Treat Options Cardiovasc Med. 2005;7(2):89-97. doi:10.1007/s11936-005-0010-6
9. Paczosa MK, Mecsas J. Klebsiella pneumoniae: going on the offense with a strong defense. Microbiol Mol Biol Rev. 2016;80(3):629-661. doi:10.1128/mmbr.00078-15
10. Kobayashi G, Fujita N, Noda Y, et al. Intraductal papillary mucinous neoplasms of the pancreas showing fistula formation into other organs. J Gastroenterol. 2010;45(10):1080-1089. doi:10.1007/s00535-010-0263-z
Patients with poorly controlled diabetes mellitus and an infectious source can be predisposed to infectious aortitis.
Patients with poorly controlled diabetes mellitus and an infectious source can be predisposed to infectious aortitis.
Aortitis is the all-encompassing term ascribed to the inflammatory process in the aortic wall that can be either infective or noninfective in origin, commonly autoimmune or inflammatory large-vessel vasculitis.1 Infectious aortitis, also known as bacterial, microbial, or cryptogenic aortitis, as well as mycotic or infected aneurysm, is a rare entity in the current antibiotic era but potentially a life-threatening disorder.2 The potential complications of infectious aortitis include emphysematous aortitis (EA), pseudoaneurysm, aortic rupture, septic emboli, and fistula formation (eg, aorto-enteric fistula).2,3
EA is a rare but serious inflammatory condition of the aorta with a nonspecific clinical presentation associated with high morbidity and mortality.2-6 The condition is characterized by a localized collection of gas and purulent exudate at the aortic wall.1,3 A few cases of EA have previously been reported; however, no known cases have been reported in the literature due to Klebsiella pneumoniae (K pneumoniae).
The pathophysiology of EA is the presence of underlying damage to the arterial wall caused by a hematogenously inoculated gas-producing organism.2,3 Most reported cases of EA are due to endovascular graft complications. Under normal circumstances, the aortic intima is highly resistant to infectious pathogens; however, certain risk factors, such as diabetes mellitus (DM), atherosclerotic disease, preexisting aneurysm, cystic medial necrosis, vascular malformation, presence of medical devices, surgery, or impaired immunity can alter the integrity of the aortic intimal layer and predispose the aortic intima to infection.1,4-7 Bacteria are the most common causative organisms that can infect the aorta, especially Staphylococcus, Enterococcus, Streptococcus, Salmonella, and spirochete Treponema pallidum (syphilis).1,2,4,8 The site of the primary infection remains unclear in some patients.2,3,5,6 Infection of the aorta can arise by several mechanisms: direct extension of a local infection to an existing intimal injury or atherosclerotic plaque (the most common mechanism), septic embolism from endocarditis, direct bacterial inoculation from traumatic contamination, contiguous infection extending to the aorta wall, or a distant source of bacteremia.2,3
Clinical manifestations of EA depend on the site and the extent of infection. The diagnosis should be considered in patients with atherosclerosis, fever, abdominal pain, and leukocytosis.2,4-8 The differential diagnosis for EA includes (1) noninfective causes of aortitis, including rheumatoid arthritis and systemic lupus erythematosus; (2) tuberculous aortitis; (3) syphilitic aortitis; and (4) idiopathic isolated aortitis. Establishing an early diagnosis of infectious aortitis is extremely important because this condition is associated with a high rate of morbidity and mortality secondary to aortic rupture.2-7
Imaging is critical for a reliable and quick diagnosis of acute aortic pathology. Noninvasive cross-sectional imaging modalities, such as contrast-enhanced computed tomography (CT), magnetic resonance imaging, nuclear medicine, or positron emission tomography, are used for both the initial diagnosis and follow-up of aortitis.1 CT is the primary imaging method in most medical centers because it is widely available with short acquisition time in critically ill patients.3 CT allows rapid detection of abnormalities in wall thickness, diameter, and density, and enhancement of periaortic structures, enabling reliable exclusion of other aortic pathologies that may resemble acute aortitis. Also, CT aids in planning the optimal therapeutic approach.1,3,5-8
This case illustrates EA associated with infection by K pneumoniae in a patient with poorly controlled type 2 DM (T2DM). In this single case, our patient presented to the Bay Pines Veterans Affairs Healthcare System (BPVAHS) in Florida with recent superficial soft tissue injury, severe hyperglycemia, worsening abdominal pain, and leukocytosis without fever or chills. The correct diagnosis of EA was confirmed by characteristic CT findings.
Case Presentation
A 72-year-old male with a history of T2DM, hypertension, atherosclerotic vascular disease, obstructive lung disease, and smoking 1.5 packs per day for 40 years presented with diabetic ketoacidosis, a urinary tract infection, and abdominal pain of 1-week duration that started to worsen the morning he arrived at the BPVAHS emergency department. He reported no nausea, vomiting, diarrhea, constipation, chest pain, shortness of breath, fever, chills, fatigue, or dysuria. He had a nonhealing laceration on his left medial foot that occurred 18 days before admission and was treated at an outside hospital.
The patient’s surgical history included a left common femoral endarterectomy and a left femoral popliteal above-knee reverse saphenous vein bypass 4 years ago for severe critical limb ischemia due to occlusion of his left superficial femoral artery with distal embolization to the first and fifth toes. About 6 months later, he developed disabling claudication in his left lower extremity due to distal popliteal artery occlusion and had another bypass surgery to the below-knee popliteal artery with a reverse saphenous vein graft harvested from the right thigh.
On initial examination, his vital signs were within normal limits except for a blood pressure of 177/87 mm Hg. His physical examination demonstrated a nondistended abdomen with normal bowel sounds, mild lower quadrant tenderness on the left more than on the right, intermittent abdominal pain located around umbilicus with radiation to the back, and a negative psoas sign. His left medial foot had a nonhealing laceration with black sutures in place, with minimal erythema in the surrounding tissue and scab formation. He also had mild costovertebral tenderness on the left.
Initial laboratory investigation results were notable for a glucose level of 609 mg/dL and a white blood cell count of 14.6 × 103 cells/mcL with 86.5% of neutrophils. A CT scan of his abdomen revealed extensive atherosclerosis of the abdominal aorta and periaortic aneurysmal fluid collection with multiple foci of gas (Figure 1). Additionally, the aneurysmal fluid collection involved the proximal segment of the left common femoral artery, suspicious for left femoral arteritis (Figure 2). The patient was started on broad-spectrum antibiotics, morphine, and an insulin drip. Both urine and blood cultures were positive for K pneumoniae susceptible to multiple antibiotics. He was transferred to a tertiary medical center and was referred for a vascular surgery consultation.
The patient underwent surgical resection of the infected infrarenal EA and infected left common femoral artery with right axillary-bifemoral bypass with an 8-mm PTFE (polytetrafluoroethylene) graft. During the surgery, excision of the wall of the left common femoral artery and infrarenal aorta revealed frank pus with purulent fluid, which was sent to cytology for analysis and culture. His intraoperative cultures grew K pneumoniae sensitive to multiple antibiotics, including ceftriaxone, sulfamethoxazole/trimethoprim, and ampicillin/sulbactam. The vascular surgery team recommended inpatient admission and administration of 6 weeks of IV antibiotics postoperatively with ceftriaxone, followed by outpatient oral suppression therapy after discharge. The patient tolerated the surgery well and was discharged after 6 weeks of IV ceftriaxone followed by outpatient oral suppression therapy. However, the patient was transferred back to BPVAHS for continued care and rehabilitation placement.
The patient’s subsequent course was complicated by multiple hospital admissions, including aspiration pneumonia, hypoglycemia, diarrhea, and anemia. On one of his CT abdomen/pelvic examinations, a cysticlike mass was noted in the pancreatic head with a possible pancreatic duodenal fistula (this mass was not mentioned on the initial presurgical CT, although it can be seen in retrospect (Figure 3). Gastroenterology was consulted.
An upper endoscopy was performed that confirmed the fistula at the second portion of the duodenum. Findings from an endoscopic ultrasonography performed at an outside institution were concerning for a main duct intraductal papillary mucinous neoplasm (IPMN) with fistula, with biopsy results pending.
Discussion
This case contributes to the evidence that poorly controlled T2DM can be a predisposing factor for multiple vascular complications, including the infection of the aortic wall with progression to EA. Klebsiella species are considered opportunistic, Gram-negative pathogens that may disseminate to other tissues, causing life-threatening infections, including pneumonia, UTIs, bacteremia, and sepsis.9K pneumoniae infections are particularly challenging in neonates, the elderly, and immunocompromised individuals.9 CT is sensitive and specific in the detection of this pathologic entity.1,3 In patients with a suspected infectious etiology, the presence of foci of gas on CT in solid organ tissues is usually associated with an anaerobic infection. Gas can also be produced by Gram-negative facultative anaerobes that can ferment glucose in necrotic tissues.9
Although any microorganism can infect the aorta, K pneumoniae cultured from the blood specimen, urine culture, and intraoperative specimens in our patient was responsible for the formed gas in the aortic wall. Occurrence of spontaneous gas by this microorganism is usually associated with conditions leading to either increased vulnerability to infections and/or enhanced bacterial virulence.9 Although a relationship between EA and T2DM has not been proved, it is well known that patients with T2DM have a defect in their host-defense mechanisms, making them more susceptible to infections such as EA. Furthermore, because patients with T2DM are prone to the development of Gram-negative sepsis, organisms such as K pneumoniae would tend to emerge. Patients with poorly controlled T2DM and the presence of an infectious source can be predisposed to infectious aortitis, eventually leading to a gas-forming infection of the aorta.5,7
We postulate that the hematogenous spread of bacteria from a laceration in the leg as well as the presence of the pancreaticoduodenal fistula was likely the cause of the infectious EA in this case, considering the patient’s underlying uncontrolled T2DM. The patient’s prior left lower extremity vascular graft also may have provided a nidus for spreading to the aorta. Other reported underlying diseases of EA include aortic atherosclerosis, T2DM, diverticulitis, colon cancer, underlying aneurysm, immune-compromised status, and the presence of a medical device or open surgery.4-7,9
To our knowledge, this is the first case of EA associated with a pancreaticoduodenal fistula related to intraductal papillary mucinous neoplasm (IPMN). Fistulation of a main duct IPMN is rare, occurring in just 6.6% of cases.10 It can occur both before and after malignant degeneration.
EA requires rapid diagnosis, antibiotic therapy, and consultation with a vascular surgeon for immediate resection of the infected artery and graft bypass. The initial treatment of suspected infectious aortitis is IV antibiotics with broad antimicrobial coverage of the most likely pathologic organisms, particularly staphylococcal species and Gram-negative rods. Surgical debridement and revascularization should be completed early because of the high mortality rate of this condition. The intent of surgery is to control sepsis and reconstruct the arterial vasculature. Patients should remain on parenteral or oral antibiotics for at least 6 weeks to ensure full clearance of the infection.8 They should be followed up closely with serial blood cultures and CT scans.8 The rarity of the disorder, low level of awareness, varying presentations, and lack of evidence delineating pathogenesis and causality contribute to the challenge of recognizing, diagnosing, and treating EA in patients with T2DM and inflammation.
Conclusions
This case report can help bring awareness of this rare and potentially life-threatening condition in patients with T2DM. Clinicians should be aware of the risk of AE, particularly in patients with several additional risk factors: recent skin/soft tissue trauma, prior vascular graft surgery, and an underlying pancreatic mass. CT is the imaging method of choice that helps to rapidly choose a necessary emergent treatment approach.
Aortitis is the all-encompassing term ascribed to the inflammatory process in the aortic wall that can be either infective or noninfective in origin, commonly autoimmune or inflammatory large-vessel vasculitis.1 Infectious aortitis, also known as bacterial, microbial, or cryptogenic aortitis, as well as mycotic or infected aneurysm, is a rare entity in the current antibiotic era but potentially a life-threatening disorder.2 The potential complications of infectious aortitis include emphysematous aortitis (EA), pseudoaneurysm, aortic rupture, septic emboli, and fistula formation (eg, aorto-enteric fistula).2,3
EA is a rare but serious inflammatory condition of the aorta with a nonspecific clinical presentation associated with high morbidity and mortality.2-6 The condition is characterized by a localized collection of gas and purulent exudate at the aortic wall.1,3 A few cases of EA have previously been reported; however, no known cases have been reported in the literature due to Klebsiella pneumoniae (K pneumoniae).
The pathophysiology of EA is the presence of underlying damage to the arterial wall caused by a hematogenously inoculated gas-producing organism.2,3 Most reported cases of EA are due to endovascular graft complications. Under normal circumstances, the aortic intima is highly resistant to infectious pathogens; however, certain risk factors, such as diabetes mellitus (DM), atherosclerotic disease, preexisting aneurysm, cystic medial necrosis, vascular malformation, presence of medical devices, surgery, or impaired immunity can alter the integrity of the aortic intimal layer and predispose the aortic intima to infection.1,4-7 Bacteria are the most common causative organisms that can infect the aorta, especially Staphylococcus, Enterococcus, Streptococcus, Salmonella, and spirochete Treponema pallidum (syphilis).1,2,4,8 The site of the primary infection remains unclear in some patients.2,3,5,6 Infection of the aorta can arise by several mechanisms: direct extension of a local infection to an existing intimal injury or atherosclerotic plaque (the most common mechanism), septic embolism from endocarditis, direct bacterial inoculation from traumatic contamination, contiguous infection extending to the aorta wall, or a distant source of bacteremia.2,3
Clinical manifestations of EA depend on the site and the extent of infection. The diagnosis should be considered in patients with atherosclerosis, fever, abdominal pain, and leukocytosis.2,4-8 The differential diagnosis for EA includes (1) noninfective causes of aortitis, including rheumatoid arthritis and systemic lupus erythematosus; (2) tuberculous aortitis; (3) syphilitic aortitis; and (4) idiopathic isolated aortitis. Establishing an early diagnosis of infectious aortitis is extremely important because this condition is associated with a high rate of morbidity and mortality secondary to aortic rupture.2-7
Imaging is critical for a reliable and quick diagnosis of acute aortic pathology. Noninvasive cross-sectional imaging modalities, such as contrast-enhanced computed tomography (CT), magnetic resonance imaging, nuclear medicine, or positron emission tomography, are used for both the initial diagnosis and follow-up of aortitis.1 CT is the primary imaging method in most medical centers because it is widely available with short acquisition time in critically ill patients.3 CT allows rapid detection of abnormalities in wall thickness, diameter, and density, and enhancement of periaortic structures, enabling reliable exclusion of other aortic pathologies that may resemble acute aortitis. Also, CT aids in planning the optimal therapeutic approach.1,3,5-8
This case illustrates EA associated with infection by K pneumoniae in a patient with poorly controlled type 2 DM (T2DM). In this single case, our patient presented to the Bay Pines Veterans Affairs Healthcare System (BPVAHS) in Florida with recent superficial soft tissue injury, severe hyperglycemia, worsening abdominal pain, and leukocytosis without fever or chills. The correct diagnosis of EA was confirmed by characteristic CT findings.
Case Presentation
A 72-year-old male with a history of T2DM, hypertension, atherosclerotic vascular disease, obstructive lung disease, and smoking 1.5 packs per day for 40 years presented with diabetic ketoacidosis, a urinary tract infection, and abdominal pain of 1-week duration that started to worsen the morning he arrived at the BPVAHS emergency department. He reported no nausea, vomiting, diarrhea, constipation, chest pain, shortness of breath, fever, chills, fatigue, or dysuria. He had a nonhealing laceration on his left medial foot that occurred 18 days before admission and was treated at an outside hospital.
The patient’s surgical history included a left common femoral endarterectomy and a left femoral popliteal above-knee reverse saphenous vein bypass 4 years ago for severe critical limb ischemia due to occlusion of his left superficial femoral artery with distal embolization to the first and fifth toes. About 6 months later, he developed disabling claudication in his left lower extremity due to distal popliteal artery occlusion and had another bypass surgery to the below-knee popliteal artery with a reverse saphenous vein graft harvested from the right thigh.
On initial examination, his vital signs were within normal limits except for a blood pressure of 177/87 mm Hg. His physical examination demonstrated a nondistended abdomen with normal bowel sounds, mild lower quadrant tenderness on the left more than on the right, intermittent abdominal pain located around umbilicus with radiation to the back, and a negative psoas sign. His left medial foot had a nonhealing laceration with black sutures in place, with minimal erythema in the surrounding tissue and scab formation. He also had mild costovertebral tenderness on the left.
Initial laboratory investigation results were notable for a glucose level of 609 mg/dL and a white blood cell count of 14.6 × 103 cells/mcL with 86.5% of neutrophils. A CT scan of his abdomen revealed extensive atherosclerosis of the abdominal aorta and periaortic aneurysmal fluid collection with multiple foci of gas (Figure 1). Additionally, the aneurysmal fluid collection involved the proximal segment of the left common femoral artery, suspicious for left femoral arteritis (Figure 2). The patient was started on broad-spectrum antibiotics, morphine, and an insulin drip. Both urine and blood cultures were positive for K pneumoniae susceptible to multiple antibiotics. He was transferred to a tertiary medical center and was referred for a vascular surgery consultation.
The patient underwent surgical resection of the infected infrarenal EA and infected left common femoral artery with right axillary-bifemoral bypass with an 8-mm PTFE (polytetrafluoroethylene) graft. During the surgery, excision of the wall of the left common femoral artery and infrarenal aorta revealed frank pus with purulent fluid, which was sent to cytology for analysis and culture. His intraoperative cultures grew K pneumoniae sensitive to multiple antibiotics, including ceftriaxone, sulfamethoxazole/trimethoprim, and ampicillin/sulbactam. The vascular surgery team recommended inpatient admission and administration of 6 weeks of IV antibiotics postoperatively with ceftriaxone, followed by outpatient oral suppression therapy after discharge. The patient tolerated the surgery well and was discharged after 6 weeks of IV ceftriaxone followed by outpatient oral suppression therapy. However, the patient was transferred back to BPVAHS for continued care and rehabilitation placement.
The patient’s subsequent course was complicated by multiple hospital admissions, including aspiration pneumonia, hypoglycemia, diarrhea, and anemia. On one of his CT abdomen/pelvic examinations, a cysticlike mass was noted in the pancreatic head with a possible pancreatic duodenal fistula (this mass was not mentioned on the initial presurgical CT, although it can be seen in retrospect (Figure 3). Gastroenterology was consulted.
An upper endoscopy was performed that confirmed the fistula at the second portion of the duodenum. Findings from an endoscopic ultrasonography performed at an outside institution were concerning for a main duct intraductal papillary mucinous neoplasm (IPMN) with fistula, with biopsy results pending.
Discussion
This case contributes to the evidence that poorly controlled T2DM can be a predisposing factor for multiple vascular complications, including the infection of the aortic wall with progression to EA. Klebsiella species are considered opportunistic, Gram-negative pathogens that may disseminate to other tissues, causing life-threatening infections, including pneumonia, UTIs, bacteremia, and sepsis.9K pneumoniae infections are particularly challenging in neonates, the elderly, and immunocompromised individuals.9 CT is sensitive and specific in the detection of this pathologic entity.1,3 In patients with a suspected infectious etiology, the presence of foci of gas on CT in solid organ tissues is usually associated with an anaerobic infection. Gas can also be produced by Gram-negative facultative anaerobes that can ferment glucose in necrotic tissues.9
Although any microorganism can infect the aorta, K pneumoniae cultured from the blood specimen, urine culture, and intraoperative specimens in our patient was responsible for the formed gas in the aortic wall. Occurrence of spontaneous gas by this microorganism is usually associated with conditions leading to either increased vulnerability to infections and/or enhanced bacterial virulence.9 Although a relationship between EA and T2DM has not been proved, it is well known that patients with T2DM have a defect in their host-defense mechanisms, making them more susceptible to infections such as EA. Furthermore, because patients with T2DM are prone to the development of Gram-negative sepsis, organisms such as K pneumoniae would tend to emerge. Patients with poorly controlled T2DM and the presence of an infectious source can be predisposed to infectious aortitis, eventually leading to a gas-forming infection of the aorta.5,7
We postulate that the hematogenous spread of bacteria from a laceration in the leg as well as the presence of the pancreaticoduodenal fistula was likely the cause of the infectious EA in this case, considering the patient’s underlying uncontrolled T2DM. The patient’s prior left lower extremity vascular graft also may have provided a nidus for spreading to the aorta. Other reported underlying diseases of EA include aortic atherosclerosis, T2DM, diverticulitis, colon cancer, underlying aneurysm, immune-compromised status, and the presence of a medical device or open surgery.4-7,9
To our knowledge, this is the first case of EA associated with a pancreaticoduodenal fistula related to intraductal papillary mucinous neoplasm (IPMN). Fistulation of a main duct IPMN is rare, occurring in just 6.6% of cases.10 It can occur both before and after malignant degeneration.
EA requires rapid diagnosis, antibiotic therapy, and consultation with a vascular surgeon for immediate resection of the infected artery and graft bypass. The initial treatment of suspected infectious aortitis is IV antibiotics with broad antimicrobial coverage of the most likely pathologic organisms, particularly staphylococcal species and Gram-negative rods. Surgical debridement and revascularization should be completed early because of the high mortality rate of this condition. The intent of surgery is to control sepsis and reconstruct the arterial vasculature. Patients should remain on parenteral or oral antibiotics for at least 6 weeks to ensure full clearance of the infection.8 They should be followed up closely with serial blood cultures and CT scans.8 The rarity of the disorder, low level of awareness, varying presentations, and lack of evidence delineating pathogenesis and causality contribute to the challenge of recognizing, diagnosing, and treating EA in patients with T2DM and inflammation.
Conclusions
This case report can help bring awareness of this rare and potentially life-threatening condition in patients with T2DM. Clinicians should be aware of the risk of AE, particularly in patients with several additional risk factors: recent skin/soft tissue trauma, prior vascular graft surgery, and an underlying pancreatic mass. CT is the imaging method of choice that helps to rapidly choose a necessary emergent treatment approach.
1. Litmanovich DE, Yıldırım A, Bankier AA. Insights into imaging of aortitis. Insights Imaging. 2012;3(6):545-560. doi:10.1007/s13244-012-0192-x
2. Lopes RJ, Almeida J, Dias PJ, Pinho P, Maciel MJ. Infectious thoracic aortitis: a literature review. Clin Cardiol. 2009;32(9):488-490. doi:10.1002/clc.20578
3. Murphy DJ, Keraliya AR, Agrawal MD, Aghayev A, Steigner ML. Cross-sectional imaging of aortic infections. Insights Imaging. 2016;7(6):801-818. doi:10.1007/s13244-016-0522-5
4. Md Noh MSF, Abdul Rashid AM, Ar A, B N, Mohammed Y, A RE. Emphysematous aortitis: report of two cases and CT imaging findings. BJR Case Rep. 2017;3(3):20170006. doi:10.1259/bjrcr.20170006
5. Harris C, Geffen J, Rizg K, et al. A rare report of infectious emphysematous aortitis secondary to Clostridium septicum without prior vascular intervention. Case Rep Vasc Med. 2017;2017:4984325. doi:10.1155/2017/4984325
6. Ito F, Inokuchi R, Matsumoto A, et al. Presence of periaortic gas in Clostridium septicum-infected aortic aneurysm aids in early diagnosis: a case report and systematic review of the literature. J Med Case Rep. 2017;11(1):268. doi:10.1186/s13256-017-1422-0
7. Urgiles S, Matos-Casano H, Win KZ, Berardo J, Bhatt U, Shah J. Emphysematous aortitis due to Clostridium septicum in an 89-year-old female with ileus. Case Rep Infect Dis. 2019;2019:1094837. doi:10.1155/2019/1094837
8. Foote EA, Postier RG, Greenfield RA, Bronze MS. Infectious aortitis. Curr Treat Options Cardiovasc Med. 2005;7(2):89-97. doi:10.1007/s11936-005-0010-6
9. Paczosa MK, Mecsas J. Klebsiella pneumoniae: going on the offense with a strong defense. Microbiol Mol Biol Rev. 2016;80(3):629-661. doi:10.1128/mmbr.00078-15
10. Kobayashi G, Fujita N, Noda Y, et al. Intraductal papillary mucinous neoplasms of the pancreas showing fistula formation into other organs. J Gastroenterol. 2010;45(10):1080-1089. doi:10.1007/s00535-010-0263-z
1. Litmanovich DE, Yıldırım A, Bankier AA. Insights into imaging of aortitis. Insights Imaging. 2012;3(6):545-560. doi:10.1007/s13244-012-0192-x
2. Lopes RJ, Almeida J, Dias PJ, Pinho P, Maciel MJ. Infectious thoracic aortitis: a literature review. Clin Cardiol. 2009;32(9):488-490. doi:10.1002/clc.20578
3. Murphy DJ, Keraliya AR, Agrawal MD, Aghayev A, Steigner ML. Cross-sectional imaging of aortic infections. Insights Imaging. 2016;7(6):801-818. doi:10.1007/s13244-016-0522-5
4. Md Noh MSF, Abdul Rashid AM, Ar A, B N, Mohammed Y, A RE. Emphysematous aortitis: report of two cases and CT imaging findings. BJR Case Rep. 2017;3(3):20170006. doi:10.1259/bjrcr.20170006
5. Harris C, Geffen J, Rizg K, et al. A rare report of infectious emphysematous aortitis secondary to Clostridium septicum without prior vascular intervention. Case Rep Vasc Med. 2017;2017:4984325. doi:10.1155/2017/4984325
6. Ito F, Inokuchi R, Matsumoto A, et al. Presence of periaortic gas in Clostridium septicum-infected aortic aneurysm aids in early diagnosis: a case report and systematic review of the literature. J Med Case Rep. 2017;11(1):268. doi:10.1186/s13256-017-1422-0
7. Urgiles S, Matos-Casano H, Win KZ, Berardo J, Bhatt U, Shah J. Emphysematous aortitis due to Clostridium septicum in an 89-year-old female with ileus. Case Rep Infect Dis. 2019;2019:1094837. doi:10.1155/2019/1094837
8. Foote EA, Postier RG, Greenfield RA, Bronze MS. Infectious aortitis. Curr Treat Options Cardiovasc Med. 2005;7(2):89-97. doi:10.1007/s11936-005-0010-6
9. Paczosa MK, Mecsas J. Klebsiella pneumoniae: going on the offense with a strong defense. Microbiol Mol Biol Rev. 2016;80(3):629-661. doi:10.1128/mmbr.00078-15
10. Kobayashi G, Fujita N, Noda Y, et al. Intraductal papillary mucinous neoplasms of the pancreas showing fistula formation into other organs. J Gastroenterol. 2010;45(10):1080-1089. doi:10.1007/s00535-010-0263-z
Orbital Varix Masquerading as an Intraorbital Lymphoma
Clinical context was paramount to the diagnosis and management of a patient with periorbital pain and a history of systemic lymphoma.
We present a case of an orbital varix masquerading as an orbital lymphoma. Our case underscores the importance of clinical correlation and thorough study of the ordered films by the ordering health care provider.
Case Presentation
An 84-year-old female veteran presented to the Bay Pines Veterans Affairs Healthcare System emergency department. She had a past ocular history of nonproliferative diabetic retinopathy in both eyes (OU) and senile cataracts OU. She had a complicated medical history most notable for congestive heart failure and Stage IV B cell follicular lymphoma, having received 6 rounds of chemotherapy, and has since been on rituximab maintenance therapy for the past few years.
The patient reported dyspnea on exertion, 30-pound weight gain, and ocular pain in her right eye (OD), more so than her left eye (OS) that was severe enough to wake her from sleep. She endorsed an associated headache but reported no visual loss or any other ocular symptoms other than conjunctival injection. On examination, the patient demonstrated jugular venous distension. X-ray imaging obtained in the emergency department demonstrated bilateral pleural effusions. Our patient was admitted subsequently for an exacerbation of congestive heart failure. She was monitored for euvolemia and discharged 4 days later.
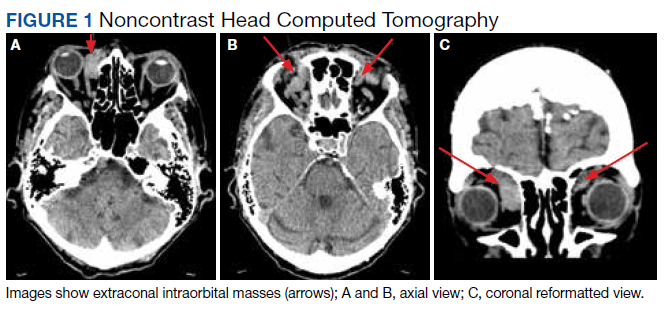
During admission, imaging of the orbits was obtained. Computed tomography (CT) of the head without contrast demonstrated at least 4 intraorbital masses in the right orbit, measuring up to 22 mm in maximum diameter and at least 3 intraorbital masses in the left orbit, measuring up to 16 mm in diameter (Figure 1). Magnetic resonance imaging (MRI) with contrast of the brain and orbits was ordered, which demonstrated multiple bilateral uniformly enhancing, primarily extraconal masses present in both orbits, the largest of which occupied the superomedial aspect of the right orbit and measured 12 x 18 x 20 mm. Further, the ophthalmic veins were noted to be engorged. The cavernous did not demonstrate any thrombosis. No other ocular structures were compromised, although there was compression of the extraocular muscles in both orbits (Figures 2, 3, 4, 5, and 6). At that time, the reading radiologist suggested the most likely diagnosis was metastatic orbital lymphoma given the clinical history, which became the working diagnosis.
A few days after admission, the patient received an ophthalmic evaluation at the eye clinic. Visual acuity (VA) at this time was 20/200 that pinholed (PH) 20/70 OD and 20/30 without pinhole improvement OS. Refraction was -2.50 + 1.50 × 120 OD and -0.25 + 0.50 × 065 OS, which yielded visual acuities of 20/60 and 20/30, respectively. There was no afferent pupillary defect and pupils were symmetric. Goldmann tonometry demonstrated pressures of 11 mm of mercury OU at 1630. Slit-lamp and dilated fundus examinations were within normal limits except for 2+ nuclear sclerotic cataracts, large cups of 0.6 OD and 0.7 OS, and a mild epiretinal membrane OD. The decision was made to refer the patient to oculoplastic service for biopsy of the lesion to rule out a metastatic lymphoid solid tumor. At this juncture, the working diagnosis continued to be metastatic orbital lymphoma.
The patient underwent right anterior orbitotomy. Intraoperatively, after dissection to the lesion was accomplished, it was noted that the mass displayed a blue to purple hue consistent with a vascular malformation. It was decided to continue careful dissection instead of obtaining a biopsy. Continued dissection further corroborated a vascular lesion. Meticulous hemostasis was maintained during the dissection; however, dissection was halted after about 35-mm depth into the orbit, given concern for damaging the optic nerve. The feeding vessel to the lesion was tied off with two 5-0 vicryl sutures, and the specimen was cut distal to the ligation. During the procedure, pupillary function was continually checked. The rest of the surgery proceeded without any difficulty, and the specimen was sent off to pathology.
Pathology returned as an orbital varix with no thrombosis or malignant tissue. Surgery to remove lesions of the left orbit was deferred given radiologic findings consistent with vascular lesions, similar to the removed lesion from the right orbit. The patient is currently without residual periorbital pain after diuresis, and the patient’s oncological management continues to be maintenance rituximab. The remaining lesions will be monitored with yearly serial imaging.
Discussion
In a study of 242 patients, Bacorn and colleagues found that a clinician’s preoperative assessment correlated with histopathologic diagnosis in 75.7% of cases, whereas the radiology report was correct in only 52.4% of cases.1 Retrospective analysis identified clues that could have been used to more rapidly elucidate the true diagnosis for our patient.
In regard to symptomatology, orbital varices present with intermittent proptosis, vision loss, and rarely, periorbital pain unless thrombosed.2,3 The severity of periorbital pain experienced by our patient is atypical of an orbital varix especially in the absence of a phlebolith. A specific feature of orbital varix is enlargement with the Valsalva maneuver.3 Although the patient did not report the notedsymptoms, more pointed questioning may have helped elucidate our patient’s true diagnosis sooner.
Radiologically, the presence of a partial flow void (decreased signal on T2) is useful for confirming the vascular nature of a lesion as was present in our case. Specific to the radiologic evaluation of orbital varices, it is recommended to obtain imaging with and without the Valsalva maneuver.4 Ultrasound is a superb tool in our armamentarium to image orbital lesions. B-scan ultrasound with and without Valsalva should be able to demonstrate variation in size when standing (minimal distension) vs lying flat with Valsalva (maximal distension).4 Further, Doppler ultrasound would be able to demonstrate changes in flow within the lesion when comparing previously mentioned maneuvers.4 Orbital lymphoma would not demonstrate this variation.
The size change of an orbital varix lesion may be further demonstrated on head CT with contrast. On CT, an orbital varix will demonstrate isodensity to other venous structures, whereas orbital lymphomas will be hyperdense when compared to extraocular muscles.4,5 Further, a head CT without contrast may demonstrate phleboliths within an orbital varix.4 MRI should be performed with the Valsalva maneuver. On T1 and T2 studies, orbital varices demonstrate hypointensity when compared to extraocular muscles (EOMs).4 Lymphomas demonstrate a very specific radiologic pattern on MRI. On T1, they demonstrate isointensity to hypointensity when compared to EOMS, and on T2, they demonstrate iso- to hyperintensity when compared to EOMs.5 With respect to fluorodeoxyglucose (FDG) positron emission tomography (PET), our patient’s orbital lesion did not demonstrate FDG uptake. In patients where lymphoma previously demonstrated FDG PET uptake, the absence of such uptake strongly argues against malignant nature of the lesion (Figure 7).
Prominently enhancing lesions are more likely to represent varices, aneurysms, or other highly or completely vascular lesions. Any intraorbital intervention should be conducted as though a vascular lesion is within the differential, and appropriate care should be taken even if not specifically enunciated in the radiologic report.
Management of orbital varices is not standardized; however, these lesions tend to be observed if no significant proptosis, pain, thrombosis, diplopia, or compression of the optic nerve is present. In such cases, surgical intervention is performed; however, the lesions may recur. Our patient’s presentation coincided with her heart failure exacerbation most likely secondary to flow disruption and fluid overload in the venous system, thereby exacerbating her orbital varices. The resolution of our patient’s orbital pain in the left orbit was likely due to improved distension after achieving euvolemia after diuresis. In cases where varices are secondary to a correctable etiologies, treatment of these etiologies are in order. Chen and colleagues reported a case of pulsatile proptosis associated with fluid overload in a newly diagnosed case of heart failure secondary to mitral regurgitation.6 Thus, orbital pain due to worsened orbital varices may represent a symptom of fluid overload and the provider may look for etiologies of this disease process.
Conclusions
We present a case of an orbital varix masquerading as an orbital lymphoma. While the ruling out of a diagnosis that might portend a poor prognosis is always of paramount importance, proper use of investigative studies and a thorough history could have helped elucidate the true diagnosis sooner: In this case an orbital varix masquerading as an orbital lymphoma. Mainly, the use of the Valsalva maneuver during the physical examination (resulting in proptosis) and during radiologic studies might have obviated the need for formal biopsy. Furthermore, orbital pain may be a presenting symptom of fluid overload in patients with a history of orbital varices.
1. Bacorn C, Gokoffski KK, Lin LK. Clinical correlation recommended: accuracy of clinician versus radiologic interpretation of the imaging of orbital lesions. Orbit. 2021;40(2):133-137. doi:10.1080/01676830.2020.1752742
2. Shams PN, Cugati S, Wells T, Huilgol S, Selva D. Orbital varix thrombosis and review of orbital vascular anomalies in blue rubber bleb nevus syndrome. Ophthalmic Plast Reconstr Surg. 2015;31(4):e82-e86. doi:10.1097/IOP.0000000000000107
3. Islam N, Mireskandari K, Rose GE. Orbital varices and orbital wall defects. Br J Ophthalmol. 2004;88(8):1092-1093.
4. Smoker WR, Gentry LR, Yee NK, Reede DL, Nerad JA. Vascular lesions of the orbit: more than meets the eye. Radiographics. 2008;28(1):185-325. doi:10.1148/rg.281075040
5. Karcioglu ZA, ed. Orbital Tumors. New York; 2005. Chap 13:133-140.
6. Chen Z, Jones H. A case of tricuspid regurgitation and congestive cardiac failure presenting with orbital pulsation. JRSM Cardiovasc Dis. 2012;1(1):cvd.2012.012005. Published 2012 Apr 5. doi:10.1258/cvd.2012.012005
Clinical context was paramount to the diagnosis and management of a patient with periorbital pain and a history of systemic lymphoma.
Clinical context was paramount to the diagnosis and management of a patient with periorbital pain and a history of systemic lymphoma.
We present a case of an orbital varix masquerading as an orbital lymphoma. Our case underscores the importance of clinical correlation and thorough study of the ordered films by the ordering health care provider.
Case Presentation
An 84-year-old female veteran presented to the Bay Pines Veterans Affairs Healthcare System emergency department. She had a past ocular history of nonproliferative diabetic retinopathy in both eyes (OU) and senile cataracts OU. She had a complicated medical history most notable for congestive heart failure and Stage IV B cell follicular lymphoma, having received 6 rounds of chemotherapy, and has since been on rituximab maintenance therapy for the past few years.
The patient reported dyspnea on exertion, 30-pound weight gain, and ocular pain in her right eye (OD), more so than her left eye (OS) that was severe enough to wake her from sleep. She endorsed an associated headache but reported no visual loss or any other ocular symptoms other than conjunctival injection. On examination, the patient demonstrated jugular venous distension. X-ray imaging obtained in the emergency department demonstrated bilateral pleural effusions. Our patient was admitted subsequently for an exacerbation of congestive heart failure. She was monitored for euvolemia and discharged 4 days later.
During admission, imaging of the orbits was obtained. Computed tomography (CT) of the head without contrast demonstrated at least 4 intraorbital masses in the right orbit, measuring up to 22 mm in maximum diameter and at least 3 intraorbital masses in the left orbit, measuring up to 16 mm in diameter (Figure 1). Magnetic resonance imaging (MRI) with contrast of the brain and orbits was ordered, which demonstrated multiple bilateral uniformly enhancing, primarily extraconal masses present in both orbits, the largest of which occupied the superomedial aspect of the right orbit and measured 12 x 18 x 20 mm. Further, the ophthalmic veins were noted to be engorged. The cavernous did not demonstrate any thrombosis. No other ocular structures were compromised, although there was compression of the extraocular muscles in both orbits (Figures 2, 3, 4, 5, and 6). At that time, the reading radiologist suggested the most likely diagnosis was metastatic orbital lymphoma given the clinical history, which became the working diagnosis.
A few days after admission, the patient received an ophthalmic evaluation at the eye clinic. Visual acuity (VA) at this time was 20/200 that pinholed (PH) 20/70 OD and 20/30 without pinhole improvement OS. Refraction was -2.50 + 1.50 × 120 OD and -0.25 + 0.50 × 065 OS, which yielded visual acuities of 20/60 and 20/30, respectively. There was no afferent pupillary defect and pupils were symmetric. Goldmann tonometry demonstrated pressures of 11 mm of mercury OU at 1630. Slit-lamp and dilated fundus examinations were within normal limits except for 2+ nuclear sclerotic cataracts, large cups of 0.6 OD and 0.7 OS, and a mild epiretinal membrane OD. The decision was made to refer the patient to oculoplastic service for biopsy of the lesion to rule out a metastatic lymphoid solid tumor. At this juncture, the working diagnosis continued to be metastatic orbital lymphoma.
The patient underwent right anterior orbitotomy. Intraoperatively, after dissection to the lesion was accomplished, it was noted that the mass displayed a blue to purple hue consistent with a vascular malformation. It was decided to continue careful dissection instead of obtaining a biopsy. Continued dissection further corroborated a vascular lesion. Meticulous hemostasis was maintained during the dissection; however, dissection was halted after about 35-mm depth into the orbit, given concern for damaging the optic nerve. The feeding vessel to the lesion was tied off with two 5-0 vicryl sutures, and the specimen was cut distal to the ligation. During the procedure, pupillary function was continually checked. The rest of the surgery proceeded without any difficulty, and the specimen was sent off to pathology.
Pathology returned as an orbital varix with no thrombosis or malignant tissue. Surgery to remove lesions of the left orbit was deferred given radiologic findings consistent with vascular lesions, similar to the removed lesion from the right orbit. The patient is currently without residual periorbital pain after diuresis, and the patient’s oncological management continues to be maintenance rituximab. The remaining lesions will be monitored with yearly serial imaging.
Discussion
In a study of 242 patients, Bacorn and colleagues found that a clinician’s preoperative assessment correlated with histopathologic diagnosis in 75.7% of cases, whereas the radiology report was correct in only 52.4% of cases.1 Retrospective analysis identified clues that could have been used to more rapidly elucidate the true diagnosis for our patient.
In regard to symptomatology, orbital varices present with intermittent proptosis, vision loss, and rarely, periorbital pain unless thrombosed.2,3 The severity of periorbital pain experienced by our patient is atypical of an orbital varix especially in the absence of a phlebolith. A specific feature of orbital varix is enlargement with the Valsalva maneuver.3 Although the patient did not report the notedsymptoms, more pointed questioning may have helped elucidate our patient’s true diagnosis sooner.
Radiologically, the presence of a partial flow void (decreased signal on T2) is useful for confirming the vascular nature of a lesion as was present in our case. Specific to the radiologic evaluation of orbital varices, it is recommended to obtain imaging with and without the Valsalva maneuver.4 Ultrasound is a superb tool in our armamentarium to image orbital lesions. B-scan ultrasound with and without Valsalva should be able to demonstrate variation in size when standing (minimal distension) vs lying flat with Valsalva (maximal distension).4 Further, Doppler ultrasound would be able to demonstrate changes in flow within the lesion when comparing previously mentioned maneuvers.4 Orbital lymphoma would not demonstrate this variation.
The size change of an orbital varix lesion may be further demonstrated on head CT with contrast. On CT, an orbital varix will demonstrate isodensity to other venous structures, whereas orbital lymphomas will be hyperdense when compared to extraocular muscles.4,5 Further, a head CT without contrast may demonstrate phleboliths within an orbital varix.4 MRI should be performed with the Valsalva maneuver. On T1 and T2 studies, orbital varices demonstrate hypointensity when compared to extraocular muscles (EOMs).4 Lymphomas demonstrate a very specific radiologic pattern on MRI. On T1, they demonstrate isointensity to hypointensity when compared to EOMS, and on T2, they demonstrate iso- to hyperintensity when compared to EOMs.5 With respect to fluorodeoxyglucose (FDG) positron emission tomography (PET), our patient’s orbital lesion did not demonstrate FDG uptake. In patients where lymphoma previously demonstrated FDG PET uptake, the absence of such uptake strongly argues against malignant nature of the lesion (Figure 7).
Prominently enhancing lesions are more likely to represent varices, aneurysms, or other highly or completely vascular lesions. Any intraorbital intervention should be conducted as though a vascular lesion is within the differential, and appropriate care should be taken even if not specifically enunciated in the radiologic report.
Management of orbital varices is not standardized; however, these lesions tend to be observed if no significant proptosis, pain, thrombosis, diplopia, or compression of the optic nerve is present. In such cases, surgical intervention is performed; however, the lesions may recur. Our patient’s presentation coincided with her heart failure exacerbation most likely secondary to flow disruption and fluid overload in the venous system, thereby exacerbating her orbital varices. The resolution of our patient’s orbital pain in the left orbit was likely due to improved distension after achieving euvolemia after diuresis. In cases where varices are secondary to a correctable etiologies, treatment of these etiologies are in order. Chen and colleagues reported a case of pulsatile proptosis associated with fluid overload in a newly diagnosed case of heart failure secondary to mitral regurgitation.6 Thus, orbital pain due to worsened orbital varices may represent a symptom of fluid overload and the provider may look for etiologies of this disease process.
Conclusions
We present a case of an orbital varix masquerading as an orbital lymphoma. While the ruling out of a diagnosis that might portend a poor prognosis is always of paramount importance, proper use of investigative studies and a thorough history could have helped elucidate the true diagnosis sooner: In this case an orbital varix masquerading as an orbital lymphoma. Mainly, the use of the Valsalva maneuver during the physical examination (resulting in proptosis) and during radiologic studies might have obviated the need for formal biopsy. Furthermore, orbital pain may be a presenting symptom of fluid overload in patients with a history of orbital varices.
We present a case of an orbital varix masquerading as an orbital lymphoma. Our case underscores the importance of clinical correlation and thorough study of the ordered films by the ordering health care provider.
Case Presentation
An 84-year-old female veteran presented to the Bay Pines Veterans Affairs Healthcare System emergency department. She had a past ocular history of nonproliferative diabetic retinopathy in both eyes (OU) and senile cataracts OU. She had a complicated medical history most notable for congestive heart failure and Stage IV B cell follicular lymphoma, having received 6 rounds of chemotherapy, and has since been on rituximab maintenance therapy for the past few years.
The patient reported dyspnea on exertion, 30-pound weight gain, and ocular pain in her right eye (OD), more so than her left eye (OS) that was severe enough to wake her from sleep. She endorsed an associated headache but reported no visual loss or any other ocular symptoms other than conjunctival injection. On examination, the patient demonstrated jugular venous distension. X-ray imaging obtained in the emergency department demonstrated bilateral pleural effusions. Our patient was admitted subsequently for an exacerbation of congestive heart failure. She was monitored for euvolemia and discharged 4 days later.
During admission, imaging of the orbits was obtained. Computed tomography (CT) of the head without contrast demonstrated at least 4 intraorbital masses in the right orbit, measuring up to 22 mm in maximum diameter and at least 3 intraorbital masses in the left orbit, measuring up to 16 mm in diameter (Figure 1). Magnetic resonance imaging (MRI) with contrast of the brain and orbits was ordered, which demonstrated multiple bilateral uniformly enhancing, primarily extraconal masses present in both orbits, the largest of which occupied the superomedial aspect of the right orbit and measured 12 x 18 x 20 mm. Further, the ophthalmic veins were noted to be engorged. The cavernous did not demonstrate any thrombosis. No other ocular structures were compromised, although there was compression of the extraocular muscles in both orbits (Figures 2, 3, 4, 5, and 6). At that time, the reading radiologist suggested the most likely diagnosis was metastatic orbital lymphoma given the clinical history, which became the working diagnosis.
A few days after admission, the patient received an ophthalmic evaluation at the eye clinic. Visual acuity (VA) at this time was 20/200 that pinholed (PH) 20/70 OD and 20/30 without pinhole improvement OS. Refraction was -2.50 + 1.50 × 120 OD and -0.25 + 0.50 × 065 OS, which yielded visual acuities of 20/60 and 20/30, respectively. There was no afferent pupillary defect and pupils were symmetric. Goldmann tonometry demonstrated pressures of 11 mm of mercury OU at 1630. Slit-lamp and dilated fundus examinations were within normal limits except for 2+ nuclear sclerotic cataracts, large cups of 0.6 OD and 0.7 OS, and a mild epiretinal membrane OD. The decision was made to refer the patient to oculoplastic service for biopsy of the lesion to rule out a metastatic lymphoid solid tumor. At this juncture, the working diagnosis continued to be metastatic orbital lymphoma.
The patient underwent right anterior orbitotomy. Intraoperatively, after dissection to the lesion was accomplished, it was noted that the mass displayed a blue to purple hue consistent with a vascular malformation. It was decided to continue careful dissection instead of obtaining a biopsy. Continued dissection further corroborated a vascular lesion. Meticulous hemostasis was maintained during the dissection; however, dissection was halted after about 35-mm depth into the orbit, given concern for damaging the optic nerve. The feeding vessel to the lesion was tied off with two 5-0 vicryl sutures, and the specimen was cut distal to the ligation. During the procedure, pupillary function was continually checked. The rest of the surgery proceeded without any difficulty, and the specimen was sent off to pathology.
Pathology returned as an orbital varix with no thrombosis or malignant tissue. Surgery to remove lesions of the left orbit was deferred given radiologic findings consistent with vascular lesions, similar to the removed lesion from the right orbit. The patient is currently without residual periorbital pain after diuresis, and the patient’s oncological management continues to be maintenance rituximab. The remaining lesions will be monitored with yearly serial imaging.
Discussion
In a study of 242 patients, Bacorn and colleagues found that a clinician’s preoperative assessment correlated with histopathologic diagnosis in 75.7% of cases, whereas the radiology report was correct in only 52.4% of cases.1 Retrospective analysis identified clues that could have been used to more rapidly elucidate the true diagnosis for our patient.
In regard to symptomatology, orbital varices present with intermittent proptosis, vision loss, and rarely, periorbital pain unless thrombosed.2,3 The severity of periorbital pain experienced by our patient is atypical of an orbital varix especially in the absence of a phlebolith. A specific feature of orbital varix is enlargement with the Valsalva maneuver.3 Although the patient did not report the notedsymptoms, more pointed questioning may have helped elucidate our patient’s true diagnosis sooner.
Radiologically, the presence of a partial flow void (decreased signal on T2) is useful for confirming the vascular nature of a lesion as was present in our case. Specific to the radiologic evaluation of orbital varices, it is recommended to obtain imaging with and without the Valsalva maneuver.4 Ultrasound is a superb tool in our armamentarium to image orbital lesions. B-scan ultrasound with and without Valsalva should be able to demonstrate variation in size when standing (minimal distension) vs lying flat with Valsalva (maximal distension).4 Further, Doppler ultrasound would be able to demonstrate changes in flow within the lesion when comparing previously mentioned maneuvers.4 Orbital lymphoma would not demonstrate this variation.
The size change of an orbital varix lesion may be further demonstrated on head CT with contrast. On CT, an orbital varix will demonstrate isodensity to other venous structures, whereas orbital lymphomas will be hyperdense when compared to extraocular muscles.4,5 Further, a head CT without contrast may demonstrate phleboliths within an orbital varix.4 MRI should be performed with the Valsalva maneuver. On T1 and T2 studies, orbital varices demonstrate hypointensity when compared to extraocular muscles (EOMs).4 Lymphomas demonstrate a very specific radiologic pattern on MRI. On T1, they demonstrate isointensity to hypointensity when compared to EOMS, and on T2, they demonstrate iso- to hyperintensity when compared to EOMs.5 With respect to fluorodeoxyglucose (FDG) positron emission tomography (PET), our patient’s orbital lesion did not demonstrate FDG uptake. In patients where lymphoma previously demonstrated FDG PET uptake, the absence of such uptake strongly argues against malignant nature of the lesion (Figure 7).
Prominently enhancing lesions are more likely to represent varices, aneurysms, or other highly or completely vascular lesions. Any intraorbital intervention should be conducted as though a vascular lesion is within the differential, and appropriate care should be taken even if not specifically enunciated in the radiologic report.
Management of orbital varices is not standardized; however, these lesions tend to be observed if no significant proptosis, pain, thrombosis, diplopia, or compression of the optic nerve is present. In such cases, surgical intervention is performed; however, the lesions may recur. Our patient’s presentation coincided with her heart failure exacerbation most likely secondary to flow disruption and fluid overload in the venous system, thereby exacerbating her orbital varices. The resolution of our patient’s orbital pain in the left orbit was likely due to improved distension after achieving euvolemia after diuresis. In cases where varices are secondary to a correctable etiologies, treatment of these etiologies are in order. Chen and colleagues reported a case of pulsatile proptosis associated with fluid overload in a newly diagnosed case of heart failure secondary to mitral regurgitation.6 Thus, orbital pain due to worsened orbital varices may represent a symptom of fluid overload and the provider may look for etiologies of this disease process.
Conclusions
We present a case of an orbital varix masquerading as an orbital lymphoma. While the ruling out of a diagnosis that might portend a poor prognosis is always of paramount importance, proper use of investigative studies and a thorough history could have helped elucidate the true diagnosis sooner: In this case an orbital varix masquerading as an orbital lymphoma. Mainly, the use of the Valsalva maneuver during the physical examination (resulting in proptosis) and during radiologic studies might have obviated the need for formal biopsy. Furthermore, orbital pain may be a presenting symptom of fluid overload in patients with a history of orbital varices.
1. Bacorn C, Gokoffski KK, Lin LK. Clinical correlation recommended: accuracy of clinician versus radiologic interpretation of the imaging of orbital lesions. Orbit. 2021;40(2):133-137. doi:10.1080/01676830.2020.1752742
2. Shams PN, Cugati S, Wells T, Huilgol S, Selva D. Orbital varix thrombosis and review of orbital vascular anomalies in blue rubber bleb nevus syndrome. Ophthalmic Plast Reconstr Surg. 2015;31(4):e82-e86. doi:10.1097/IOP.0000000000000107
3. Islam N, Mireskandari K, Rose GE. Orbital varices and orbital wall defects. Br J Ophthalmol. 2004;88(8):1092-1093.
4. Smoker WR, Gentry LR, Yee NK, Reede DL, Nerad JA. Vascular lesions of the orbit: more than meets the eye. Radiographics. 2008;28(1):185-325. doi:10.1148/rg.281075040
5. Karcioglu ZA, ed. Orbital Tumors. New York; 2005. Chap 13:133-140.
6. Chen Z, Jones H. A case of tricuspid regurgitation and congestive cardiac failure presenting with orbital pulsation. JRSM Cardiovasc Dis. 2012;1(1):cvd.2012.012005. Published 2012 Apr 5. doi:10.1258/cvd.2012.012005
1. Bacorn C, Gokoffski KK, Lin LK. Clinical correlation recommended: accuracy of clinician versus radiologic interpretation of the imaging of orbital lesions. Orbit. 2021;40(2):133-137. doi:10.1080/01676830.2020.1752742
2. Shams PN, Cugati S, Wells T, Huilgol S, Selva D. Orbital varix thrombosis and review of orbital vascular anomalies in blue rubber bleb nevus syndrome. Ophthalmic Plast Reconstr Surg. 2015;31(4):e82-e86. doi:10.1097/IOP.0000000000000107
3. Islam N, Mireskandari K, Rose GE. Orbital varices and orbital wall defects. Br J Ophthalmol. 2004;88(8):1092-1093.
4. Smoker WR, Gentry LR, Yee NK, Reede DL, Nerad JA. Vascular lesions of the orbit: more than meets the eye. Radiographics. 2008;28(1):185-325. doi:10.1148/rg.281075040
5. Karcioglu ZA, ed. Orbital Tumors. New York; 2005. Chap 13:133-140.
6. Chen Z, Jones H. A case of tricuspid regurgitation and congestive cardiac failure presenting with orbital pulsation. JRSM Cardiovasc Dis. 2012;1(1):cvd.2012.012005. Published 2012 Apr 5. doi:10.1258/cvd.2012.012005
Hemiballismus in Patients With Poorly Controlled Type 2 Diabetes Mellitus
Hemiballismus is an acquired hyperkinetic movement disorder characterized by unilateral, involuntary, often large-amplitude limb movements. Ballistic movements are now considered to be on the choreiform spectrum.1 Movements usually involve both the arm and leg, and in half of cases, facial movements such as tongue clucking and grimacing are seen.2,3 Presentations of hemiballismus vary in severity from intermittent to nearly continuous movements, which, in some cases, may lead to exhaustion, injury, or disability. Some patients are unable to ambulate or feed themselves with the affected limb.
Background
The 2 most common causes of hemichorea-hemiballismus are stroke and hyperglycemia, with an incidence of 4% and unknown incidence, respectively.1,3,4 Other causes include HIV, traumatic brain injury, encephalitis, vasculitis, mass effect, multiple sclerosis, and adverse drug reactions. 4-7 Acute or subacute hemiballismus is classically attributed to a lesion in subthalamic nucleus (STN), but this is true only in a minority of cases. Hemiballismus can be caused by any abnormality in various subnuclei of the basal ganglia, including the classic location in the STN, striatum, and globus pallidus.4 Evidence shows the lesions typically involve a functional network connected to the posterolateral putamen.8
Although not commonly recognized, hyperglycemia in patients with type 2 diabetes mellitus (T2DM) is the second most common cause of hemichoreahemiballismus. 3 Over the past 90 years, numerous case reports have described patients with DM with acute and subacute onset of hemiballistic and hemichoreiform movements while in a hyperglycemic state or after its resolution. Reported cases have been limited to small numbers of patients with only a few larger-scale reviews of more than 20 patients.7,9 Most reported cases involve geriatric patients and more commonly, females of Eastern Asian descent with an average age of onset of 71 years.4,10 Patients typically present with glucose levels from 500 to 1,000 mg/dL and hemoglobin A1c (HbA1c) levels almost double the normal values. Interestingly, neuroimaging findings in these patients have consistently shown hyperintense signal in the contralateral basal ganglia on T1-weighted magnetic resonance images (MRIs). Noncontrast computed tomography (CT) shows well-defined unilateral increased density in the contralateral basal ganglia without mass effect.1,9,11
This report aims to illustrate and enhance the understanding of hemiballismus associated with hyperglycemia. One patient presented to the US Department of Veterans Affairs (VA) Bay Pines VA Healthcare System (BPVAHCS) in Florida, which motivated us to search for other similar cases. We reviewed the charts of 2 other patients who presented to BPVAHCS over the past 10 years. The first case presented with severe hyperglycemia and abnormal movements that were not clearly diagnosed as hemiballismus. MRI findings were characteristic and assisted in making the diagnosis. The second case was misdiagnosed as hemiballismus secondary to ischemic stroke. The third case was initially diagnosed as conversion disorder until movements worsened and the correct diagnosis of hyperglycemia-induced hemichorea hemiballismus was confirmed by the pathognomonic neuroimaging findings.
Case Presentations
Case 1
A 65-year-old male with a history of uncontrolled T2DM presented with repetitive twitching and kicking movements that involved his left upper and lower extremities for 3 weeks. The patient reported that he did not take his medications or follow the recommended diabetes diet. His HbA1c on admission was 12.2% with a serum glucose of 254 mg/dL. The MRI showed a hyperintense T1 signal within the right basal ganglia including the right caudate with sparing of the internal capsule (Figure 1). There was no associated mass effect or restricted diffusion. It was compatible with a diagnosis of hyperglycemia- induced hemichorea-hemiballismus. The patient was advised to resume taking glipizide 10 mg daily, metformin 1,000 mg by mouth twice daily, and to begin 10 units of 70/30 insulin aspart 15 minutes before meals twice daily, and to follow a low carbohydrate diet, with reduce dietary intake of sugar. At his 1-month follow-up visit, the patient reported an improvement in his involuntary movements. At the 5-month follow-up, the patient’s HbA1c level was 10.4% and his hyperkinetic movements had completely resolved.
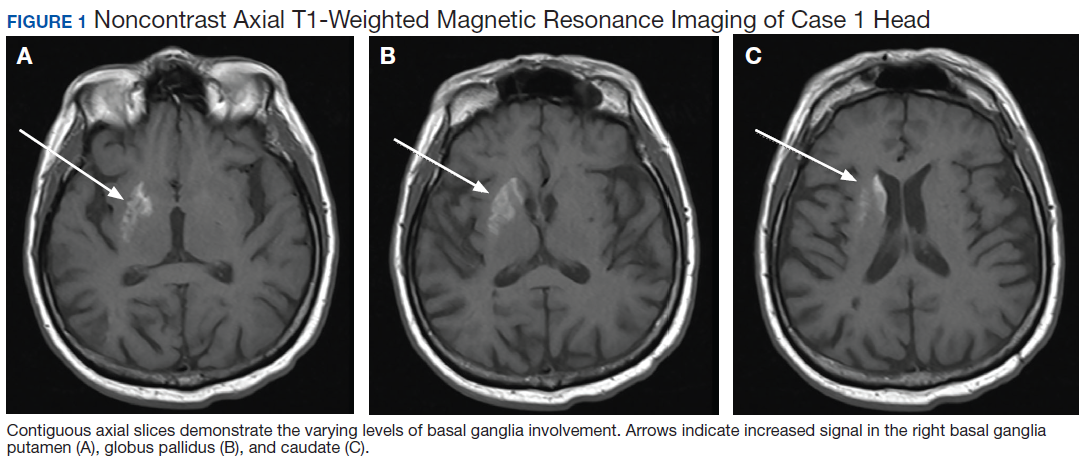
Case 2
of T2DM, hypertension, and hyperlipidemia was admitted due to increased jerky movements in the left upper extremity. On admission, his vital signs were within normal limits and his physical examination demonstrated choreoathetoid movements with ballistic components of his left upper extremity. His laboratory results showed a glucose level of 528 mg/dL with a HbA1c of 16.3%. An initial CT obtained in the emergency department (ED) demonstrated a well-defined hyperdensity in the striatal (caudate and lentiform nucleus) region (Figure 2). There was no associated edema/mass effect that would be typical for an intracranial hemorrhage.
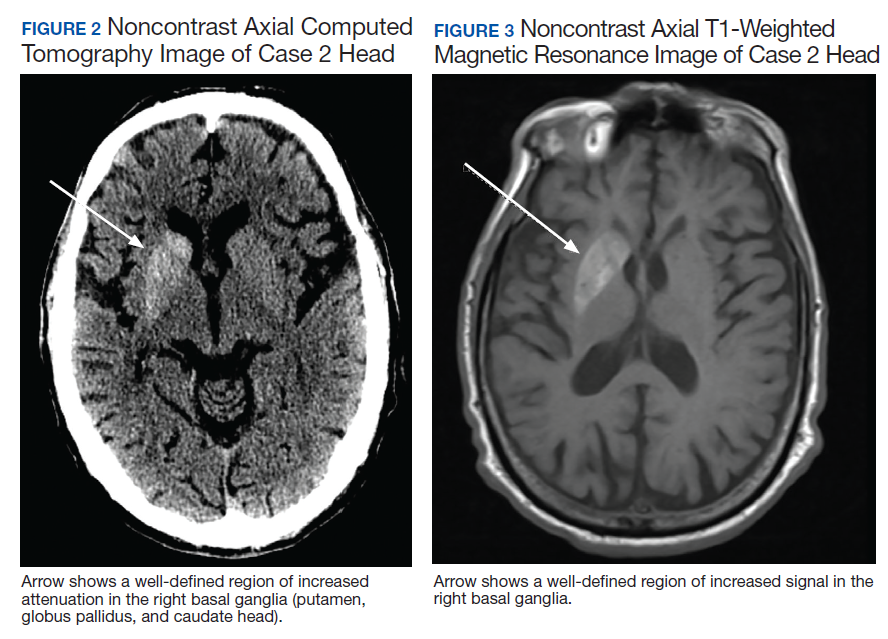
An MRI obtained 1 week later showed hyperintense TI signal corresponding to the basal ganglia (Figure 3). In addition, there was a questionable lacunar infarct in the right internal capsule. Due to lack of awareness regarding hyperglycemic associated basal ganglia changes, the patient’s movement disorder was presumed to be ischemic in etiology. The patient was prescribed oral amantadine 100 mg 3 times daily for the hemiballismus in conjunction with treatment of his T2DM. The only follow-up occurred 5 weeks later, which showed no improvement of uncontrollable movements. Imaging at that time (not available) indicated the persistence of the abnormal signal in the right basal ganglia. This patient died later that year without further follow-up.
Case 3
A 78-year-old white male with a history of syncope, transient ischemic attacks (TIAs), and poorly controlled T2DM presented with a 1-month history of progressively worsening involuntary, left-sided movements that began in his left shoulder and advanced to involve his arm, hand, and leg, and the left side of his face with grimacing and clucking of his tongue. Three weeks earlier, the patient had been discharged from the ED with a diagnosis of conversion disorder particularly because he experienced decreased movements when given a dose of Vitamin D. It was overlooked that administration of haloperidol had occurred a few hours before, and because the sounds made by his tongue were not felt to be consistent with a known movement disorder. A MRI of the brain was read as normal.
The patient returned 3 weeks later (the original presentation) due to his inability to perform activities of daily living because of his worsening involuntary movements. On admission, his HbA1c was 11.1% and his glucose was 167 mg/dL. On chart review, it was revealed that the patient’s HbA1c had been > 9% for the past 3 years with an increase from 10.1% to 11.1% in the 3 months preceding the onset of his symptoms.
On admission a MRI showed a unilateral right-sided T1 hyperintensity in the basal ganglia, no acute ischemia (Figure 4). In retrospect, subtle increased T1 signal can be seen on the earlier MRI (Figure 5). In view of the patient’s left-sided symptoms, DM, and MRI findings, a diagnosis of hyperglycemia-induced hemichorea- hemiballismus was made as the etiology of the patient’s symptoms.
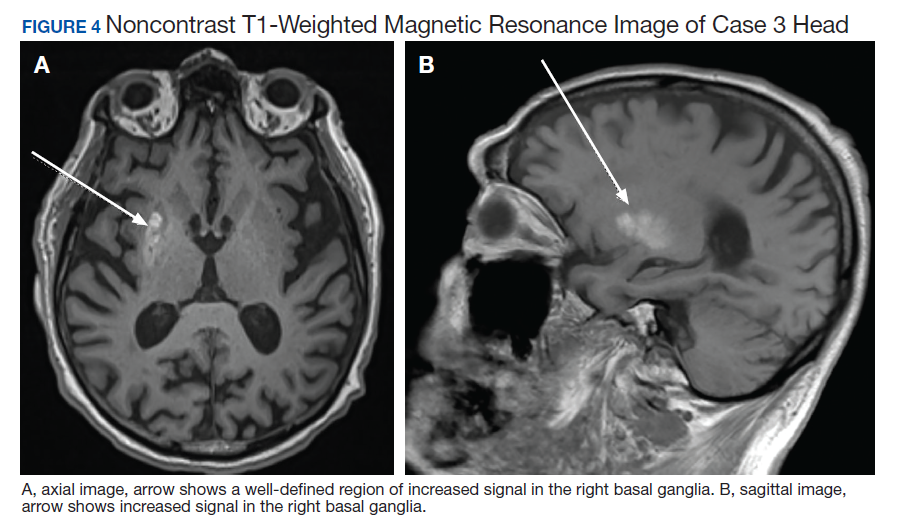
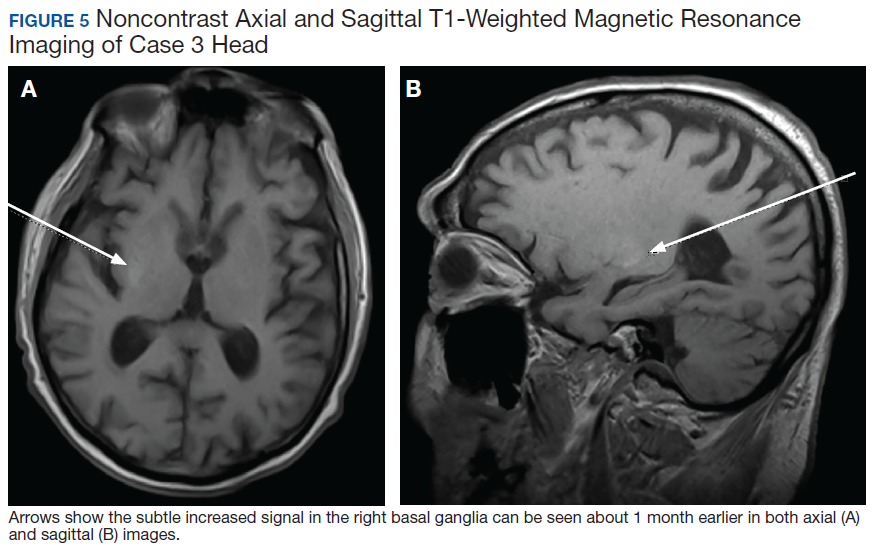
The patient was prescribed numerous medications to control his hyperkinesia including (and in combination): benztropine, gabapentin, baclofen, diphenhydramine, benzodiazepines, risperidone, olanzapine, and valproic acid, which did not control his movements. Ultimately, his hyperglycemic hemiballismus improved with tight glycemic control and oral tetrabenazine 12.5 mg twice daily. This patient underwent a protracted course of treatment with 17 days of inpatient medical admission, 3 weeks inpatient rehabilitation, and subsequent transfer to an assisted living facility.
Discussion
The 3 cases presented in this report contribute to the evidence that severe persistent hyperglycemia can result in movement disorders that mimic those seen after basal ganglia strokes. As with Case 2, past literature describes many cases of acute hyperglycemic episodes with glucose ranging from 500 to 1,000 mg/mL presenting with hemiballismus.1,3 However, there are many cases that describe hemiballismus occurring after glycemic correction, persisting despite glycemic correction, and presenting without an acute hyperglycemic episode, but in the setting of elevated HbA1c, as in Case 3.12,13 Notably, all 3 cases in this series had marked elevation in their HbA1c levels, which suggests that a more chronic hyperglycemic state or multiple shorter periods of hyperglycemia may be necessary to produce the described hyperkinetic movements.
Case reports describe the pathognomonic T1 hyperintensity of the basal ganglia that is identified in all 3 cases presented here. While the exact etiology remains unclear, the to metabolic derangements caused by hyperviscosity of the blood in the small end arteries feeding the basal ganglia.3,11 These abnormalities in turn interrupt the signaling cascade with abnormal firing rates or firing patterns, leading to reduced inhibition of the motor thalamus and ultimately present as hemiballismus.1,3,7 While most cases presented with unilateral hyperkinesis and associated contralateral basal ganglia abnormalities, there are reports of both unilateral and bilateral movements associated with bilateral basal ganglia hyperintensities on imaging. 9 The predilection for unilateral brain lesions may be explained by the varying degree of small vessel disease in different areas of the brain leading to perfusion deficits worsened by hyper viscosity. Further research into this is required to elucidate the exact pathophysiologic mechanism.
The course of disease for patients ranges from resolution within hours of tight glycemic control to persistent movements for > 3 months with a gradual improvement in severity.12,13 Treatments center on the importance of tight glycemic control to protect against the protracted course described in Case 3. Swift recognition of this rare condition is critical because improved glycemic control decreases the severity and duration of this disease. The significant disability associated with Case 3 highlights the need for prompt recognition and early, aggressive glycemic management to prevent the progression of hemiballismus. In addition to glycemic control, various CNS medications such as typical and atypical antipsychotics and tetrabenazine are firstline therapy with chemodenervation and surgical lesioning in cases unresponsive to medication therapy.
When unrecognized, hyperglycemic hemiballismus is associated with significant morbidity and mortality. The patients presented in this report were subject to either delayed diagnosis or misdiagnosis as stroke or psychiatric disorder. The rarity of the disorder, lack of evidence delineating pathogenesis and causality, low level of awareness, and varying presentations of patients all contribute to the challenge of recognizing, diagnosing, and treating hemiballismus due to hyperglycemia. This challenge can subsequently result in deteriorating symptoms, prolonged hospital stays, and unnecessary health care costs.
Conclusion
While hemiballismus due to severe persistent hyperglycemia is rare, the goal of this report is to highlight its occurrence in patients with T2DM. Further research can help develop a standardized, effective treatment strategy for these patients. Currently, lowering and maintaining appropriate glucose and HbA1c levels is the most effective treatment approach. Potential areas of research include alternative medical and surgical treatment interventions for patients while glycemic control is being achieved or for those who fail to benefit from glycemic control alone. Some success has been demonstrated with the use of antidopaminergic medications such as atypical antipsychotics and tetrabenazine and these medications should be considered when tight, sustained glycemic control alone is not successful in treating this disorder in the acute stages. Hopefully, with increasing awareness and recognition of hemiballismus related to hyperglycemia, more large-scale clinical trials can be conducted that will result in an effective treatment strategy for this devastating disorder.
1. Hawley JS, Weiner WJ. Hemiballismus: current concepts and review. Parkinsonism Relat Disord. 2012;18(2):125‐129. doi:10.1016/j.parkreldis.2011.08.015
2. Gasca-Salas C, Lang AE. Paroxysmal Hemiballism/ Hemichorea Resulting from Transient Ischemic Attacks. Mov Disord Clin Pract. 2015;3(3):303‐305. doi:10.1002/mdc3.12268
3. Garcia-Grimshaw MA, Jimenez-Ruiz A, Ornelas-Velazquez A, Luna-Armenta A, Gutierrez-Manjarrez FA. New-onset diabetes presenting as monoballism secondary to a mixed hyperglycemic crisis. Cureus. 2018;10(6):e2882. doi:10.7759/cureus.2882
4. Postuma RB, Lang AE. Hemiballism: revisiting a classic disorder. Lancet Neurol. 2003;2(11):661‐668. doi:10.1016/s1474-4422(03)00554-4
5. Gallo BV, Shulman LM, Weiner WJ, Petito CK, Berger JR. HIV encephalitis presenting with severe generalized chorea. Neurology. 1996;46(4):1163‐1165. doi:10.1212/wnl.46.4.1163
6. Provenzale JM, Glass JP. Hemiballismus: CT and MR findings. J Comput Assist Tomogr. 1995;19(4):537‐540.
7. Hodde M, Rowe KE, Surapaneni K, Terrigno P, Brighenti A, Altschuler EL. Management of severe hemiballismus: treatment challenges in the acute inpatient rehabilitation setting: a case presentation. PMR. 2017;9(7):732‐735. doi:10.1016/j.pmrj.2016.10.023
8. Laganiere S, Boes AD, Fox MD. Network localization of hemichorea-hemiballismus. Neurology. 2016;86(23):2187‐2195. doi:10.1212/WNL.0000000000002741
9. Cosentino C, Torres L, Nuñez Y, Suarez R, Velez M, Flores M. Hemichorea/hemiballism associated with hyperglycemia: report of 20 cases. Tremor Other Hyperkinet Mov (NY). 2016;6:402. doi:10.7916/D8DN454P
10. Oh SH, Lee KY, Im JH, Lee MS. Chorea associated with non-ketotic hyperglycemia and hyperintensity basal ganglia lesion on T1-weighted brain MRI study: a metaanalysis of 53 cases including four present cases. J Neurol Sci. 2002;200(1-2):57‐62. doi:10.1016/s0022-510x(02)00133-8
11. Carrion DM, Carrion AF. Non-ketotic hyperglycaemia hemichorea-hemiballismus and acute ischaemic stroke. BMJ Case Rep. 2013;2013:bcr2012008359. doi:10.1136/bcr-2012-008359
12. Cho HS, Hong CT, Chan L. Hemichorea after hyperglycemia correction: a case report and a short review of hyperglycemia-related hemichorea at the euglycemic state. Medicine (Baltimore). 2018;97(10):e0076. doi:10.1097/MD.0000000000010076
13. Lin YC, Lin YC. Prolonged hemiballism after the remission of non-ketotic hyperosmolar syndrome. BMJ Case Rep. 2012;2012:bcr0120125627. doi:10.1136/bcr.01.2012.5627
Hemiballismus is an acquired hyperkinetic movement disorder characterized by unilateral, involuntary, often large-amplitude limb movements. Ballistic movements are now considered to be on the choreiform spectrum.1 Movements usually involve both the arm and leg, and in half of cases, facial movements such as tongue clucking and grimacing are seen.2,3 Presentations of hemiballismus vary in severity from intermittent to nearly continuous movements, which, in some cases, may lead to exhaustion, injury, or disability. Some patients are unable to ambulate or feed themselves with the affected limb.
Background
The 2 most common causes of hemichorea-hemiballismus are stroke and hyperglycemia, with an incidence of 4% and unknown incidence, respectively.1,3,4 Other causes include HIV, traumatic brain injury, encephalitis, vasculitis, mass effect, multiple sclerosis, and adverse drug reactions. 4-7 Acute or subacute hemiballismus is classically attributed to a lesion in subthalamic nucleus (STN), but this is true only in a minority of cases. Hemiballismus can be caused by any abnormality in various subnuclei of the basal ganglia, including the classic location in the STN, striatum, and globus pallidus.4 Evidence shows the lesions typically involve a functional network connected to the posterolateral putamen.8
Although not commonly recognized, hyperglycemia in patients with type 2 diabetes mellitus (T2DM) is the second most common cause of hemichoreahemiballismus. 3 Over the past 90 years, numerous case reports have described patients with DM with acute and subacute onset of hemiballistic and hemichoreiform movements while in a hyperglycemic state or after its resolution. Reported cases have been limited to small numbers of patients with only a few larger-scale reviews of more than 20 patients.7,9 Most reported cases involve geriatric patients and more commonly, females of Eastern Asian descent with an average age of onset of 71 years.4,10 Patients typically present with glucose levels from 500 to 1,000 mg/dL and hemoglobin A1c (HbA1c) levels almost double the normal values. Interestingly, neuroimaging findings in these patients have consistently shown hyperintense signal in the contralateral basal ganglia on T1-weighted magnetic resonance images (MRIs). Noncontrast computed tomography (CT) shows well-defined unilateral increased density in the contralateral basal ganglia without mass effect.1,9,11
This report aims to illustrate and enhance the understanding of hemiballismus associated with hyperglycemia. One patient presented to the US Department of Veterans Affairs (VA) Bay Pines VA Healthcare System (BPVAHCS) in Florida, which motivated us to search for other similar cases. We reviewed the charts of 2 other patients who presented to BPVAHCS over the past 10 years. The first case presented with severe hyperglycemia and abnormal movements that were not clearly diagnosed as hemiballismus. MRI findings were characteristic and assisted in making the diagnosis. The second case was misdiagnosed as hemiballismus secondary to ischemic stroke. The third case was initially diagnosed as conversion disorder until movements worsened and the correct diagnosis of hyperglycemia-induced hemichorea hemiballismus was confirmed by the pathognomonic neuroimaging findings.
Case Presentations
Case 1
A 65-year-old male with a history of uncontrolled T2DM presented with repetitive twitching and kicking movements that involved his left upper and lower extremities for 3 weeks. The patient reported that he did not take his medications or follow the recommended diabetes diet. His HbA1c on admission was 12.2% with a serum glucose of 254 mg/dL. The MRI showed a hyperintense T1 signal within the right basal ganglia including the right caudate with sparing of the internal capsule (Figure 1). There was no associated mass effect or restricted diffusion. It was compatible with a diagnosis of hyperglycemia- induced hemichorea-hemiballismus. The patient was advised to resume taking glipizide 10 mg daily, metformin 1,000 mg by mouth twice daily, and to begin 10 units of 70/30 insulin aspart 15 minutes before meals twice daily, and to follow a low carbohydrate diet, with reduce dietary intake of sugar. At his 1-month follow-up visit, the patient reported an improvement in his involuntary movements. At the 5-month follow-up, the patient’s HbA1c level was 10.4% and his hyperkinetic movements had completely resolved.
Case 2
of T2DM, hypertension, and hyperlipidemia was admitted due to increased jerky movements in the left upper extremity. On admission, his vital signs were within normal limits and his physical examination demonstrated choreoathetoid movements with ballistic components of his left upper extremity. His laboratory results showed a glucose level of 528 mg/dL with a HbA1c of 16.3%. An initial CT obtained in the emergency department (ED) demonstrated a well-defined hyperdensity in the striatal (caudate and lentiform nucleus) region (Figure 2). There was no associated edema/mass effect that would be typical for an intracranial hemorrhage.
An MRI obtained 1 week later showed hyperintense TI signal corresponding to the basal ganglia (Figure 3). In addition, there was a questionable lacunar infarct in the right internal capsule. Due to lack of awareness regarding hyperglycemic associated basal ganglia changes, the patient’s movement disorder was presumed to be ischemic in etiology. The patient was prescribed oral amantadine 100 mg 3 times daily for the hemiballismus in conjunction with treatment of his T2DM. The only follow-up occurred 5 weeks later, which showed no improvement of uncontrollable movements. Imaging at that time (not available) indicated the persistence of the abnormal signal in the right basal ganglia. This patient died later that year without further follow-up.
Case 3
A 78-year-old white male with a history of syncope, transient ischemic attacks (TIAs), and poorly controlled T2DM presented with a 1-month history of progressively worsening involuntary, left-sided movements that began in his left shoulder and advanced to involve his arm, hand, and leg, and the left side of his face with grimacing and clucking of his tongue. Three weeks earlier, the patient had been discharged from the ED with a diagnosis of conversion disorder particularly because he experienced decreased movements when given a dose of Vitamin D. It was overlooked that administration of haloperidol had occurred a few hours before, and because the sounds made by his tongue were not felt to be consistent with a known movement disorder. A MRI of the brain was read as normal.
The patient returned 3 weeks later (the original presentation) due to his inability to perform activities of daily living because of his worsening involuntary movements. On admission, his HbA1c was 11.1% and his glucose was 167 mg/dL. On chart review, it was revealed that the patient’s HbA1c had been > 9% for the past 3 years with an increase from 10.1% to 11.1% in the 3 months preceding the onset of his symptoms.
On admission a MRI showed a unilateral right-sided T1 hyperintensity in the basal ganglia, no acute ischemia (Figure 4). In retrospect, subtle increased T1 signal can be seen on the earlier MRI (Figure 5). In view of the patient’s left-sided symptoms, DM, and MRI findings, a diagnosis of hyperglycemia-induced hemichorea- hemiballismus was made as the etiology of the patient’s symptoms.
The patient was prescribed numerous medications to control his hyperkinesia including (and in combination): benztropine, gabapentin, baclofen, diphenhydramine, benzodiazepines, risperidone, olanzapine, and valproic acid, which did not control his movements. Ultimately, his hyperglycemic hemiballismus improved with tight glycemic control and oral tetrabenazine 12.5 mg twice daily. This patient underwent a protracted course of treatment with 17 days of inpatient medical admission, 3 weeks inpatient rehabilitation, and subsequent transfer to an assisted living facility.
Discussion
The 3 cases presented in this report contribute to the evidence that severe persistent hyperglycemia can result in movement disorders that mimic those seen after basal ganglia strokes. As with Case 2, past literature describes many cases of acute hyperglycemic episodes with glucose ranging from 500 to 1,000 mg/mL presenting with hemiballismus.1,3 However, there are many cases that describe hemiballismus occurring after glycemic correction, persisting despite glycemic correction, and presenting without an acute hyperglycemic episode, but in the setting of elevated HbA1c, as in Case 3.12,13 Notably, all 3 cases in this series had marked elevation in their HbA1c levels, which suggests that a more chronic hyperglycemic state or multiple shorter periods of hyperglycemia may be necessary to produce the described hyperkinetic movements.
Case reports describe the pathognomonic T1 hyperintensity of the basal ganglia that is identified in all 3 cases presented here. While the exact etiology remains unclear, the to metabolic derangements caused by hyperviscosity of the blood in the small end arteries feeding the basal ganglia.3,11 These abnormalities in turn interrupt the signaling cascade with abnormal firing rates or firing patterns, leading to reduced inhibition of the motor thalamus and ultimately present as hemiballismus.1,3,7 While most cases presented with unilateral hyperkinesis and associated contralateral basal ganglia abnormalities, there are reports of both unilateral and bilateral movements associated with bilateral basal ganglia hyperintensities on imaging. 9 The predilection for unilateral brain lesions may be explained by the varying degree of small vessel disease in different areas of the brain leading to perfusion deficits worsened by hyper viscosity. Further research into this is required to elucidate the exact pathophysiologic mechanism.
The course of disease for patients ranges from resolution within hours of tight glycemic control to persistent movements for > 3 months with a gradual improvement in severity.12,13 Treatments center on the importance of tight glycemic control to protect against the protracted course described in Case 3. Swift recognition of this rare condition is critical because improved glycemic control decreases the severity and duration of this disease. The significant disability associated with Case 3 highlights the need for prompt recognition and early, aggressive glycemic management to prevent the progression of hemiballismus. In addition to glycemic control, various CNS medications such as typical and atypical antipsychotics and tetrabenazine are firstline therapy with chemodenervation and surgical lesioning in cases unresponsive to medication therapy.
When unrecognized, hyperglycemic hemiballismus is associated with significant morbidity and mortality. The patients presented in this report were subject to either delayed diagnosis or misdiagnosis as stroke or psychiatric disorder. The rarity of the disorder, lack of evidence delineating pathogenesis and causality, low level of awareness, and varying presentations of patients all contribute to the challenge of recognizing, diagnosing, and treating hemiballismus due to hyperglycemia. This challenge can subsequently result in deteriorating symptoms, prolonged hospital stays, and unnecessary health care costs.
Conclusion
While hemiballismus due to severe persistent hyperglycemia is rare, the goal of this report is to highlight its occurrence in patients with T2DM. Further research can help develop a standardized, effective treatment strategy for these patients. Currently, lowering and maintaining appropriate glucose and HbA1c levels is the most effective treatment approach. Potential areas of research include alternative medical and surgical treatment interventions for patients while glycemic control is being achieved or for those who fail to benefit from glycemic control alone. Some success has been demonstrated with the use of antidopaminergic medications such as atypical antipsychotics and tetrabenazine and these medications should be considered when tight, sustained glycemic control alone is not successful in treating this disorder in the acute stages. Hopefully, with increasing awareness and recognition of hemiballismus related to hyperglycemia, more large-scale clinical trials can be conducted that will result in an effective treatment strategy for this devastating disorder.
Hemiballismus is an acquired hyperkinetic movement disorder characterized by unilateral, involuntary, often large-amplitude limb movements. Ballistic movements are now considered to be on the choreiform spectrum.1 Movements usually involve both the arm and leg, and in half of cases, facial movements such as tongue clucking and grimacing are seen.2,3 Presentations of hemiballismus vary in severity from intermittent to nearly continuous movements, which, in some cases, may lead to exhaustion, injury, or disability. Some patients are unable to ambulate or feed themselves with the affected limb.
Background
The 2 most common causes of hemichorea-hemiballismus are stroke and hyperglycemia, with an incidence of 4% and unknown incidence, respectively.1,3,4 Other causes include HIV, traumatic brain injury, encephalitis, vasculitis, mass effect, multiple sclerosis, and adverse drug reactions. 4-7 Acute or subacute hemiballismus is classically attributed to a lesion in subthalamic nucleus (STN), but this is true only in a minority of cases. Hemiballismus can be caused by any abnormality in various subnuclei of the basal ganglia, including the classic location in the STN, striatum, and globus pallidus.4 Evidence shows the lesions typically involve a functional network connected to the posterolateral putamen.8
Although not commonly recognized, hyperglycemia in patients with type 2 diabetes mellitus (T2DM) is the second most common cause of hemichoreahemiballismus. 3 Over the past 90 years, numerous case reports have described patients with DM with acute and subacute onset of hemiballistic and hemichoreiform movements while in a hyperglycemic state or after its resolution. Reported cases have been limited to small numbers of patients with only a few larger-scale reviews of more than 20 patients.7,9 Most reported cases involve geriatric patients and more commonly, females of Eastern Asian descent with an average age of onset of 71 years.4,10 Patients typically present with glucose levels from 500 to 1,000 mg/dL and hemoglobin A1c (HbA1c) levels almost double the normal values. Interestingly, neuroimaging findings in these patients have consistently shown hyperintense signal in the contralateral basal ganglia on T1-weighted magnetic resonance images (MRIs). Noncontrast computed tomography (CT) shows well-defined unilateral increased density in the contralateral basal ganglia without mass effect.1,9,11
This report aims to illustrate and enhance the understanding of hemiballismus associated with hyperglycemia. One patient presented to the US Department of Veterans Affairs (VA) Bay Pines VA Healthcare System (BPVAHCS) in Florida, which motivated us to search for other similar cases. We reviewed the charts of 2 other patients who presented to BPVAHCS over the past 10 years. The first case presented with severe hyperglycemia and abnormal movements that were not clearly diagnosed as hemiballismus. MRI findings were characteristic and assisted in making the diagnosis. The second case was misdiagnosed as hemiballismus secondary to ischemic stroke. The third case was initially diagnosed as conversion disorder until movements worsened and the correct diagnosis of hyperglycemia-induced hemichorea hemiballismus was confirmed by the pathognomonic neuroimaging findings.
Case Presentations
Case 1
A 65-year-old male with a history of uncontrolled T2DM presented with repetitive twitching and kicking movements that involved his left upper and lower extremities for 3 weeks. The patient reported that he did not take his medications or follow the recommended diabetes diet. His HbA1c on admission was 12.2% with a serum glucose of 254 mg/dL. The MRI showed a hyperintense T1 signal within the right basal ganglia including the right caudate with sparing of the internal capsule (Figure 1). There was no associated mass effect or restricted diffusion. It was compatible with a diagnosis of hyperglycemia- induced hemichorea-hemiballismus. The patient was advised to resume taking glipizide 10 mg daily, metformin 1,000 mg by mouth twice daily, and to begin 10 units of 70/30 insulin aspart 15 minutes before meals twice daily, and to follow a low carbohydrate diet, with reduce dietary intake of sugar. At his 1-month follow-up visit, the patient reported an improvement in his involuntary movements. At the 5-month follow-up, the patient’s HbA1c level was 10.4% and his hyperkinetic movements had completely resolved.
Case 2
of T2DM, hypertension, and hyperlipidemia was admitted due to increased jerky movements in the left upper extremity. On admission, his vital signs were within normal limits and his physical examination demonstrated choreoathetoid movements with ballistic components of his left upper extremity. His laboratory results showed a glucose level of 528 mg/dL with a HbA1c of 16.3%. An initial CT obtained in the emergency department (ED) demonstrated a well-defined hyperdensity in the striatal (caudate and lentiform nucleus) region (Figure 2). There was no associated edema/mass effect that would be typical for an intracranial hemorrhage.
An MRI obtained 1 week later showed hyperintense TI signal corresponding to the basal ganglia (Figure 3). In addition, there was a questionable lacunar infarct in the right internal capsule. Due to lack of awareness regarding hyperglycemic associated basal ganglia changes, the patient’s movement disorder was presumed to be ischemic in etiology. The patient was prescribed oral amantadine 100 mg 3 times daily for the hemiballismus in conjunction with treatment of his T2DM. The only follow-up occurred 5 weeks later, which showed no improvement of uncontrollable movements. Imaging at that time (not available) indicated the persistence of the abnormal signal in the right basal ganglia. This patient died later that year without further follow-up.
Case 3
A 78-year-old white male with a history of syncope, transient ischemic attacks (TIAs), and poorly controlled T2DM presented with a 1-month history of progressively worsening involuntary, left-sided movements that began in his left shoulder and advanced to involve his arm, hand, and leg, and the left side of his face with grimacing and clucking of his tongue. Three weeks earlier, the patient had been discharged from the ED with a diagnosis of conversion disorder particularly because he experienced decreased movements when given a dose of Vitamin D. It was overlooked that administration of haloperidol had occurred a few hours before, and because the sounds made by his tongue were not felt to be consistent with a known movement disorder. A MRI of the brain was read as normal.
The patient returned 3 weeks later (the original presentation) due to his inability to perform activities of daily living because of his worsening involuntary movements. On admission, his HbA1c was 11.1% and his glucose was 167 mg/dL. On chart review, it was revealed that the patient’s HbA1c had been > 9% for the past 3 years with an increase from 10.1% to 11.1% in the 3 months preceding the onset of his symptoms.
On admission a MRI showed a unilateral right-sided T1 hyperintensity in the basal ganglia, no acute ischemia (Figure 4). In retrospect, subtle increased T1 signal can be seen on the earlier MRI (Figure 5). In view of the patient’s left-sided symptoms, DM, and MRI findings, a diagnosis of hyperglycemia-induced hemichorea- hemiballismus was made as the etiology of the patient’s symptoms.
The patient was prescribed numerous medications to control his hyperkinesia including (and in combination): benztropine, gabapentin, baclofen, diphenhydramine, benzodiazepines, risperidone, olanzapine, and valproic acid, which did not control his movements. Ultimately, his hyperglycemic hemiballismus improved with tight glycemic control and oral tetrabenazine 12.5 mg twice daily. This patient underwent a protracted course of treatment with 17 days of inpatient medical admission, 3 weeks inpatient rehabilitation, and subsequent transfer to an assisted living facility.
Discussion
The 3 cases presented in this report contribute to the evidence that severe persistent hyperglycemia can result in movement disorders that mimic those seen after basal ganglia strokes. As with Case 2, past literature describes many cases of acute hyperglycemic episodes with glucose ranging from 500 to 1,000 mg/mL presenting with hemiballismus.1,3 However, there are many cases that describe hemiballismus occurring after glycemic correction, persisting despite glycemic correction, and presenting without an acute hyperglycemic episode, but in the setting of elevated HbA1c, as in Case 3.12,13 Notably, all 3 cases in this series had marked elevation in their HbA1c levels, which suggests that a more chronic hyperglycemic state or multiple shorter periods of hyperglycemia may be necessary to produce the described hyperkinetic movements.
Case reports describe the pathognomonic T1 hyperintensity of the basal ganglia that is identified in all 3 cases presented here. While the exact etiology remains unclear, the to metabolic derangements caused by hyperviscosity of the blood in the small end arteries feeding the basal ganglia.3,11 These abnormalities in turn interrupt the signaling cascade with abnormal firing rates or firing patterns, leading to reduced inhibition of the motor thalamus and ultimately present as hemiballismus.1,3,7 While most cases presented with unilateral hyperkinesis and associated contralateral basal ganglia abnormalities, there are reports of both unilateral and bilateral movements associated with bilateral basal ganglia hyperintensities on imaging. 9 The predilection for unilateral brain lesions may be explained by the varying degree of small vessel disease in different areas of the brain leading to perfusion deficits worsened by hyper viscosity. Further research into this is required to elucidate the exact pathophysiologic mechanism.
The course of disease for patients ranges from resolution within hours of tight glycemic control to persistent movements for > 3 months with a gradual improvement in severity.12,13 Treatments center on the importance of tight glycemic control to protect against the protracted course described in Case 3. Swift recognition of this rare condition is critical because improved glycemic control decreases the severity and duration of this disease. The significant disability associated with Case 3 highlights the need for prompt recognition and early, aggressive glycemic management to prevent the progression of hemiballismus. In addition to glycemic control, various CNS medications such as typical and atypical antipsychotics and tetrabenazine are firstline therapy with chemodenervation and surgical lesioning in cases unresponsive to medication therapy.
When unrecognized, hyperglycemic hemiballismus is associated with significant morbidity and mortality. The patients presented in this report were subject to either delayed diagnosis or misdiagnosis as stroke or psychiatric disorder. The rarity of the disorder, lack of evidence delineating pathogenesis and causality, low level of awareness, and varying presentations of patients all contribute to the challenge of recognizing, diagnosing, and treating hemiballismus due to hyperglycemia. This challenge can subsequently result in deteriorating symptoms, prolonged hospital stays, and unnecessary health care costs.
Conclusion
While hemiballismus due to severe persistent hyperglycemia is rare, the goal of this report is to highlight its occurrence in patients with T2DM. Further research can help develop a standardized, effective treatment strategy for these patients. Currently, lowering and maintaining appropriate glucose and HbA1c levels is the most effective treatment approach. Potential areas of research include alternative medical and surgical treatment interventions for patients while glycemic control is being achieved or for those who fail to benefit from glycemic control alone. Some success has been demonstrated with the use of antidopaminergic medications such as atypical antipsychotics and tetrabenazine and these medications should be considered when tight, sustained glycemic control alone is not successful in treating this disorder in the acute stages. Hopefully, with increasing awareness and recognition of hemiballismus related to hyperglycemia, more large-scale clinical trials can be conducted that will result in an effective treatment strategy for this devastating disorder.
1. Hawley JS, Weiner WJ. Hemiballismus: current concepts and review. Parkinsonism Relat Disord. 2012;18(2):125‐129. doi:10.1016/j.parkreldis.2011.08.015
2. Gasca-Salas C, Lang AE. Paroxysmal Hemiballism/ Hemichorea Resulting from Transient Ischemic Attacks. Mov Disord Clin Pract. 2015;3(3):303‐305. doi:10.1002/mdc3.12268
3. Garcia-Grimshaw MA, Jimenez-Ruiz A, Ornelas-Velazquez A, Luna-Armenta A, Gutierrez-Manjarrez FA. New-onset diabetes presenting as monoballism secondary to a mixed hyperglycemic crisis. Cureus. 2018;10(6):e2882. doi:10.7759/cureus.2882
4. Postuma RB, Lang AE. Hemiballism: revisiting a classic disorder. Lancet Neurol. 2003;2(11):661‐668. doi:10.1016/s1474-4422(03)00554-4
5. Gallo BV, Shulman LM, Weiner WJ, Petito CK, Berger JR. HIV encephalitis presenting with severe generalized chorea. Neurology. 1996;46(4):1163‐1165. doi:10.1212/wnl.46.4.1163
6. Provenzale JM, Glass JP. Hemiballismus: CT and MR findings. J Comput Assist Tomogr. 1995;19(4):537‐540.
7. Hodde M, Rowe KE, Surapaneni K, Terrigno P, Brighenti A, Altschuler EL. Management of severe hemiballismus: treatment challenges in the acute inpatient rehabilitation setting: a case presentation. PMR. 2017;9(7):732‐735. doi:10.1016/j.pmrj.2016.10.023
8. Laganiere S, Boes AD, Fox MD. Network localization of hemichorea-hemiballismus. Neurology. 2016;86(23):2187‐2195. doi:10.1212/WNL.0000000000002741
9. Cosentino C, Torres L, Nuñez Y, Suarez R, Velez M, Flores M. Hemichorea/hemiballism associated with hyperglycemia: report of 20 cases. Tremor Other Hyperkinet Mov (NY). 2016;6:402. doi:10.7916/D8DN454P
10. Oh SH, Lee KY, Im JH, Lee MS. Chorea associated with non-ketotic hyperglycemia and hyperintensity basal ganglia lesion on T1-weighted brain MRI study: a metaanalysis of 53 cases including four present cases. J Neurol Sci. 2002;200(1-2):57‐62. doi:10.1016/s0022-510x(02)00133-8
11. Carrion DM, Carrion AF. Non-ketotic hyperglycaemia hemichorea-hemiballismus and acute ischaemic stroke. BMJ Case Rep. 2013;2013:bcr2012008359. doi:10.1136/bcr-2012-008359
12. Cho HS, Hong CT, Chan L. Hemichorea after hyperglycemia correction: a case report and a short review of hyperglycemia-related hemichorea at the euglycemic state. Medicine (Baltimore). 2018;97(10):e0076. doi:10.1097/MD.0000000000010076
13. Lin YC, Lin YC. Prolonged hemiballism after the remission of non-ketotic hyperosmolar syndrome. BMJ Case Rep. 2012;2012:bcr0120125627. doi:10.1136/bcr.01.2012.5627
1. Hawley JS, Weiner WJ. Hemiballismus: current concepts and review. Parkinsonism Relat Disord. 2012;18(2):125‐129. doi:10.1016/j.parkreldis.2011.08.015
2. Gasca-Salas C, Lang AE. Paroxysmal Hemiballism/ Hemichorea Resulting from Transient Ischemic Attacks. Mov Disord Clin Pract. 2015;3(3):303‐305. doi:10.1002/mdc3.12268
3. Garcia-Grimshaw MA, Jimenez-Ruiz A, Ornelas-Velazquez A, Luna-Armenta A, Gutierrez-Manjarrez FA. New-onset diabetes presenting as monoballism secondary to a mixed hyperglycemic crisis. Cureus. 2018;10(6):e2882. doi:10.7759/cureus.2882
4. Postuma RB, Lang AE. Hemiballism: revisiting a classic disorder. Lancet Neurol. 2003;2(11):661‐668. doi:10.1016/s1474-4422(03)00554-4
5. Gallo BV, Shulman LM, Weiner WJ, Petito CK, Berger JR. HIV encephalitis presenting with severe generalized chorea. Neurology. 1996;46(4):1163‐1165. doi:10.1212/wnl.46.4.1163
6. Provenzale JM, Glass JP. Hemiballismus: CT and MR findings. J Comput Assist Tomogr. 1995;19(4):537‐540.
7. Hodde M, Rowe KE, Surapaneni K, Terrigno P, Brighenti A, Altschuler EL. Management of severe hemiballismus: treatment challenges in the acute inpatient rehabilitation setting: a case presentation. PMR. 2017;9(7):732‐735. doi:10.1016/j.pmrj.2016.10.023
8. Laganiere S, Boes AD, Fox MD. Network localization of hemichorea-hemiballismus. Neurology. 2016;86(23):2187‐2195. doi:10.1212/WNL.0000000000002741
9. Cosentino C, Torres L, Nuñez Y, Suarez R, Velez M, Flores M. Hemichorea/hemiballism associated with hyperglycemia: report of 20 cases. Tremor Other Hyperkinet Mov (NY). 2016;6:402. doi:10.7916/D8DN454P
10. Oh SH, Lee KY, Im JH, Lee MS. Chorea associated with non-ketotic hyperglycemia and hyperintensity basal ganglia lesion on T1-weighted brain MRI study: a metaanalysis of 53 cases including four present cases. J Neurol Sci. 2002;200(1-2):57‐62. doi:10.1016/s0022-510x(02)00133-8
11. Carrion DM, Carrion AF. Non-ketotic hyperglycaemia hemichorea-hemiballismus and acute ischaemic stroke. BMJ Case Rep. 2013;2013:bcr2012008359. doi:10.1136/bcr-2012-008359
12. Cho HS, Hong CT, Chan L. Hemichorea after hyperglycemia correction: a case report and a short review of hyperglycemia-related hemichorea at the euglycemic state. Medicine (Baltimore). 2018;97(10):e0076. doi:10.1097/MD.0000000000010076
13. Lin YC, Lin YC. Prolonged hemiballism after the remission of non-ketotic hyperosmolar syndrome. BMJ Case Rep. 2012;2012:bcr0120125627. doi:10.1136/bcr.01.2012.5627
Review of Radiologic Considerations in an Immunocompetent Patient With Primary Central Nervous System Lymphoma (FULL)
Central nervous system (CNS) lymphoma can be classified into 2 categories: primary CNS lymphoma (PCNSL), which includes disease limited to brain, eyes, spinal cord; and leptomeninges without coexisting or previous systemic lymphoma. Secondary CNS lymphoma (SCNSL) is essentially metastatic disease from a systemic primary site.1 The focus of this case presentation is PCNSL, with an emphasis on imaging characteristics and differential diagnosis.
The median age at diagnosis for PCNSL is 65 years, and the overall incidence has been decreasing since the mid-1990s, likely related to the increased use of highly-active antiretroviral therapy (HAART) in patients with AIDS.2,3 Although overall incidence has decreased, incidence in the elderly population has increased.4 Historically, PCNSL has been considered an AIDS-defining illness.5 These patients, among other immunocompromised patients, such as those on chronic immunosuppressive therapy, are at a higher risk for developing the malignancy.6
Clinical presentation varies because of the location of CNS involvement and may present with headache, mood or personality disturbances, or focal neurologic deficits. Seizures are less likely due to the tendency of PCNSL to spare gray matter. Initial workup generally includes a head computed tomography (CT) scan, as well as a contrast-enhanced magnetic resonance image (MRI), which may help direct clinicians to the appropriate diagnosis. However, there is significant overlap between the imaging characteristics of PCNSL and numerous other disease processes, including glioblastoma and demyelination. The imaging characteristics of PCNSL are considerably different depending on the patient’s immune status.7
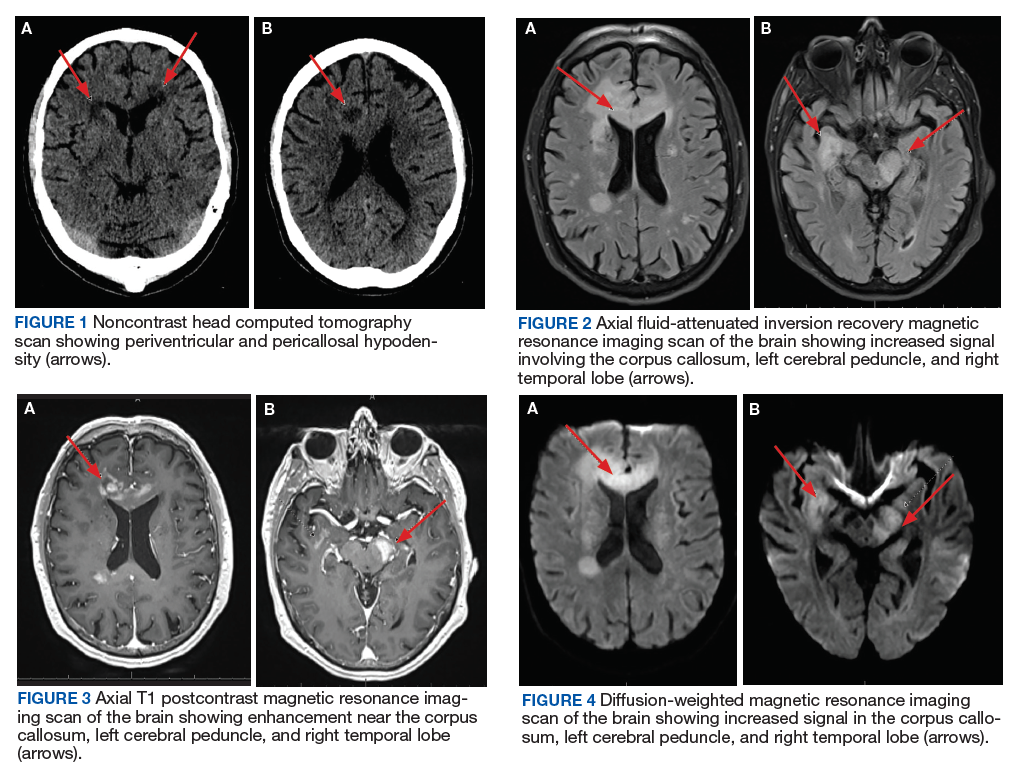
This case illustrates a rare presentation of PCNSL in an immunocompetent patient whose MRI characteristics were seemingly more consistent with those seen in patients with immunodeficiency. The main differential diagnoses and key imaging characteristics, which may help obtain accurate diagnosis, will be discussed.
Case Presentation
A 72-year-old male veteran presented with a 2-month history of subjective weakness in his upper and lower extremities progressing to multiple falls at home. He had no significant medical history other than a thymectomy at age 15 for an enlarged thymus, which per patient report, was benign. An initial laboratory test that included vitamin B12, folate, thyroid-stimulating hormone, complete blood cell count, and comprehensive metabolic panel, were unremarkable, with a white blood cell count of 8.5 K/uL. The initial neurologic evaluation did not show any focal neurologic deficits; however, during the initial hospital stay, the patient developed increasing lower extremity weakness on examination. A noncontrast CT head scan showed extensive nonspecific hypodensities within the periventricular white matter (Figure 1). A contrast-enhanced MRI showed enhancing lesions involving the corpus callosum, left cerebral peduncle, and right temporal lobe (Figures 2, 3, and 4). These lesions also exhibited significant restricted diffusion and a mild amount of surrounding vasogenic edema. The working diagnosis after the MRI included primary CNS lymphoma, multifocal glioblastoma, and tumefactive demyelinating disease. The patient was started on IV steroids and transferred for neurosurgical evaluation and biopsy at an outside hospital. The frontal lesion was biopsied, and the initial frozen section was consistent with lymphoma; a bone marrow biopsy was negative. The workup for immunodeficiency was unremarkable. Pathology revealed high-grade B-cell lymphoma, and the patient began a chemotherapy regimen.
Discussion
The workup of altered mental status, focal neurologic deficits, headaches, or other neurologic conditions often begins with a noncontrast CT scan. On CT, PCNSL generally appears isodense to hyperdense to gray matter, but appearance is variable. The often hyperdense appearance is attributable to the hypercellular nature of lymphoma. Many times, as in this case, CT may show only vague hypodensities, some of which may be associated with surrounding edema. This presentation is nonspecific and may be seen with advancing age due to changes of chronic microvascular ischemia as well as demyelination, other malignancies, and several other disease processes, both benign and malignant. After the initial CT scan, further workup requires evaluation with MRI. PCNSL exhibits restricted diffusion and variable signal intensity on T2-weighted imaging.
PCNSL is frequently centrally located within the periventricular white matter, often within the frontal lobe but can involve other lobes, the basal ganglia, brainstem, cerebellum, or less likely, the spinal canal.7 Contrary to primary CNS disease, secondary lymphoma within the CNS has been described classically as affecting a leptomeningeal (pia and arachnoid mater) distribution two-thirds of the time, with parenchymal involvement occurring in the other one-third of patients. A recent study by Malikova and colleagues found parenchymal involvement may be much more common than previously thought.1 Leptomeningeal spread of disease often involves the cranial nerves, subependymal regions, spinal cord, or spinal nerve roots. Dural involvement in primary or secondary lymphoma is rare.
PCNSL nearly always shows enhancement. Linear enhancement along perivascular spaces is highly characteristic of PCNSL. The typical appearance of PCNSL associated with immunodeficiency varies from that seen in an otherwise immunocompetent patient. Patients with immunodeficiency usually have multifocal involvement, central necrosis leading to a ring enhancement appearance, and have more propensity for spontaneous hemorrhage.7 Immunocompetent patients are less likely to present with multifocal disease and rarely show ring enhancement. Also, spontaneous hemorrhage is rare in immunocompetent patients. In our case, extensive multifocal involvement was present, whereas typically immunocompetent patients will present with a solitary homogeneously enhancing parenchymal mass.
The primary differential for PCNSL includes malignant glioma, tumefactive multiple sclerosis, metastatic disease, and in an immunocompromised patient, toxoplasmosis. The degree of associated vasogenic edema and mass effect is generally lower in PCNSL than that of malignant gliomas and metastasis. Also, PCNSL tends to spare the cerebral cortex.8
Classically, PCNSL, malignant gliomas, and demyelinating disease have been considered the main differential for lesions that cross midline and involve both cerebral hemispheres. Lymphoma generally exhibits more restricted diffusion than malignant gliomas and metastasis, attributable to the highly cellular nature of lymphoma.7 Tumefactive multiple sclerosis is associated with relatively minimal mass effect for lesion size and exhibits less restricted diffusion values when compared to high grade gliomas and PCNSL. One fairly specific finding for tumefactive demyelinating lesions is incomplete rim enhancement.9 Unfortunately, an MRI is not reliable in differentiating these entities, and biopsy is required for definitive diagnosis. Many advancing imaging modalities may help provide the correct diagnosis of PCNSL, including diffusion-weighted and apparent diffusion coefficient imaging, diffusion tensor imaging, MR spectroscopy and PET imaging.7
Conclusion
With the increasing use of HAART, the paradigm of PCNSL is shifting toward one predominantly affecting immunocompetent patients. PCNSL should be considered in any patient with multiple enhancing CNS lesions, regardless of immune status. Several key imaging characteristics may help differentiate PCNSL and other disease processes; however, at this time, biopsy is recommended for definitive diagnosis.
1. Malikova H, Burghardtova M, Koubska E, Mandys V, Kozak T, Weichet J. Secondary central nervous system lymphoma: spectrum of morphological MRI appearances. Neuropsychiatr Dis Treat. 2018;4:733-740.
2. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-Oncol. 2012;14(suppl 5):v1-v49.
3. Diamond C, Taylor TH, Aboumrad T, Anton-Culver H. Changes in acquired immunodeficiency syndrome-related non-Hodgkin lymphoma in the era of highly active antiretroviral therapy: incidence, presentation, treatment, and survival. Cancer. 2006;106(1):128-135.
4. O’Neill BP, Decker PA, Tieu C, Cerhan JR. The changing incidence of primary central nervous system lymphoma is driven primarily by the changing incidence in young and middle-aged men and differs from time trends in systemic diffuse large B-cell non-Hodgkins lymphoma. Am J Hematol. 2013;88(12):997-1000.
5. [no authors listed]. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm Rep. 1992;41(rr-17):1-19.
6. Maiuri F. Central nervous system lymphomas and immunodeficiency. Neurological Research. 1989;11(1):2-5.
7. Haldorsen IS, Espeland A, Larsson EM. Central nervous system lymphoma: characteristic findings on traditional and advanced imaging. AJNR Am J Neuroradiol. 2010;32(6):984-992.
8. Gómez Roselló E, Quiles Granado AM, Laguillo Sala G, Gutiérrez S. Primary central nervous system lymphoma in immunocompetent patients: spectrum of findings and differential characteristics. Radiología. 2018;60(4):280-289.
9. Mabray MC, Cohen BA, Villanueva-Meyer JE, et al. Performance of Apparent Diffusion Coefficient Values and Conventional MRI Features in Differentiating Tumefactive Demyelinating Lesions From Primary Brain Neoplasms. American Journal of Roentgenology. 2015;205(5):1075-1085.
Central nervous system (CNS) lymphoma can be classified into 2 categories: primary CNS lymphoma (PCNSL), which includes disease limited to brain, eyes, spinal cord; and leptomeninges without coexisting or previous systemic lymphoma. Secondary CNS lymphoma (SCNSL) is essentially metastatic disease from a systemic primary site.1 The focus of this case presentation is PCNSL, with an emphasis on imaging characteristics and differential diagnosis.
The median age at diagnosis for PCNSL is 65 years, and the overall incidence has been decreasing since the mid-1990s, likely related to the increased use of highly-active antiretroviral therapy (HAART) in patients with AIDS.2,3 Although overall incidence has decreased, incidence in the elderly population has increased.4 Historically, PCNSL has been considered an AIDS-defining illness.5 These patients, among other immunocompromised patients, such as those on chronic immunosuppressive therapy, are at a higher risk for developing the malignancy.6
Clinical presentation varies because of the location of CNS involvement and may present with headache, mood or personality disturbances, or focal neurologic deficits. Seizures are less likely due to the tendency of PCNSL to spare gray matter. Initial workup generally includes a head computed tomography (CT) scan, as well as a contrast-enhanced magnetic resonance image (MRI), which may help direct clinicians to the appropriate diagnosis. However, there is significant overlap between the imaging characteristics of PCNSL and numerous other disease processes, including glioblastoma and demyelination. The imaging characteristics of PCNSL are considerably different depending on the patient’s immune status.7
This case illustrates a rare presentation of PCNSL in an immunocompetent patient whose MRI characteristics were seemingly more consistent with those seen in patients with immunodeficiency. The main differential diagnoses and key imaging characteristics, which may help obtain accurate diagnosis, will be discussed.
Case Presentation
A 72-year-old male veteran presented with a 2-month history of subjective weakness in his upper and lower extremities progressing to multiple falls at home. He had no significant medical history other than a thymectomy at age 15 for an enlarged thymus, which per patient report, was benign. An initial laboratory test that included vitamin B12, folate, thyroid-stimulating hormone, complete blood cell count, and comprehensive metabolic panel, were unremarkable, with a white blood cell count of 8.5 K/uL. The initial neurologic evaluation did not show any focal neurologic deficits; however, during the initial hospital stay, the patient developed increasing lower extremity weakness on examination. A noncontrast CT head scan showed extensive nonspecific hypodensities within the periventricular white matter (Figure 1). A contrast-enhanced MRI showed enhancing lesions involving the corpus callosum, left cerebral peduncle, and right temporal lobe (Figures 2, 3, and 4). These lesions also exhibited significant restricted diffusion and a mild amount of surrounding vasogenic edema. The working diagnosis after the MRI included primary CNS lymphoma, multifocal glioblastoma, and tumefactive demyelinating disease. The patient was started on IV steroids and transferred for neurosurgical evaluation and biopsy at an outside hospital. The frontal lesion was biopsied, and the initial frozen section was consistent with lymphoma; a bone marrow biopsy was negative. The workup for immunodeficiency was unremarkable. Pathology revealed high-grade B-cell lymphoma, and the patient began a chemotherapy regimen.
Discussion
The workup of altered mental status, focal neurologic deficits, headaches, or other neurologic conditions often begins with a noncontrast CT scan. On CT, PCNSL generally appears isodense to hyperdense to gray matter, but appearance is variable. The often hyperdense appearance is attributable to the hypercellular nature of lymphoma. Many times, as in this case, CT may show only vague hypodensities, some of which may be associated with surrounding edema. This presentation is nonspecific and may be seen with advancing age due to changes of chronic microvascular ischemia as well as demyelination, other malignancies, and several other disease processes, both benign and malignant. After the initial CT scan, further workup requires evaluation with MRI. PCNSL exhibits restricted diffusion and variable signal intensity on T2-weighted imaging.
PCNSL is frequently centrally located within the periventricular white matter, often within the frontal lobe but can involve other lobes, the basal ganglia, brainstem, cerebellum, or less likely, the spinal canal.7 Contrary to primary CNS disease, secondary lymphoma within the CNS has been described classically as affecting a leptomeningeal (pia and arachnoid mater) distribution two-thirds of the time, with parenchymal involvement occurring in the other one-third of patients. A recent study by Malikova and colleagues found parenchymal involvement may be much more common than previously thought.1 Leptomeningeal spread of disease often involves the cranial nerves, subependymal regions, spinal cord, or spinal nerve roots. Dural involvement in primary or secondary lymphoma is rare.
PCNSL nearly always shows enhancement. Linear enhancement along perivascular spaces is highly characteristic of PCNSL. The typical appearance of PCNSL associated with immunodeficiency varies from that seen in an otherwise immunocompetent patient. Patients with immunodeficiency usually have multifocal involvement, central necrosis leading to a ring enhancement appearance, and have more propensity for spontaneous hemorrhage.7 Immunocompetent patients are less likely to present with multifocal disease and rarely show ring enhancement. Also, spontaneous hemorrhage is rare in immunocompetent patients. In our case, extensive multifocal involvement was present, whereas typically immunocompetent patients will present with a solitary homogeneously enhancing parenchymal mass.
The primary differential for PCNSL includes malignant glioma, tumefactive multiple sclerosis, metastatic disease, and in an immunocompromised patient, toxoplasmosis. The degree of associated vasogenic edema and mass effect is generally lower in PCNSL than that of malignant gliomas and metastasis. Also, PCNSL tends to spare the cerebral cortex.8
Classically, PCNSL, malignant gliomas, and demyelinating disease have been considered the main differential for lesions that cross midline and involve both cerebral hemispheres. Lymphoma generally exhibits more restricted diffusion than malignant gliomas and metastasis, attributable to the highly cellular nature of lymphoma.7 Tumefactive multiple sclerosis is associated with relatively minimal mass effect for lesion size and exhibits less restricted diffusion values when compared to high grade gliomas and PCNSL. One fairly specific finding for tumefactive demyelinating lesions is incomplete rim enhancement.9 Unfortunately, an MRI is not reliable in differentiating these entities, and biopsy is required for definitive diagnosis. Many advancing imaging modalities may help provide the correct diagnosis of PCNSL, including diffusion-weighted and apparent diffusion coefficient imaging, diffusion tensor imaging, MR spectroscopy and PET imaging.7
Conclusion
With the increasing use of HAART, the paradigm of PCNSL is shifting toward one predominantly affecting immunocompetent patients. PCNSL should be considered in any patient with multiple enhancing CNS lesions, regardless of immune status. Several key imaging characteristics may help differentiate PCNSL and other disease processes; however, at this time, biopsy is recommended for definitive diagnosis.
Central nervous system (CNS) lymphoma can be classified into 2 categories: primary CNS lymphoma (PCNSL), which includes disease limited to brain, eyes, spinal cord; and leptomeninges without coexisting or previous systemic lymphoma. Secondary CNS lymphoma (SCNSL) is essentially metastatic disease from a systemic primary site.1 The focus of this case presentation is PCNSL, with an emphasis on imaging characteristics and differential diagnosis.
The median age at diagnosis for PCNSL is 65 years, and the overall incidence has been decreasing since the mid-1990s, likely related to the increased use of highly-active antiretroviral therapy (HAART) in patients with AIDS.2,3 Although overall incidence has decreased, incidence in the elderly population has increased.4 Historically, PCNSL has been considered an AIDS-defining illness.5 These patients, among other immunocompromised patients, such as those on chronic immunosuppressive therapy, are at a higher risk for developing the malignancy.6
Clinical presentation varies because of the location of CNS involvement and may present with headache, mood or personality disturbances, or focal neurologic deficits. Seizures are less likely due to the tendency of PCNSL to spare gray matter. Initial workup generally includes a head computed tomography (CT) scan, as well as a contrast-enhanced magnetic resonance image (MRI), which may help direct clinicians to the appropriate diagnosis. However, there is significant overlap between the imaging characteristics of PCNSL and numerous other disease processes, including glioblastoma and demyelination. The imaging characteristics of PCNSL are considerably different depending on the patient’s immune status.7
This case illustrates a rare presentation of PCNSL in an immunocompetent patient whose MRI characteristics were seemingly more consistent with those seen in patients with immunodeficiency. The main differential diagnoses and key imaging characteristics, which may help obtain accurate diagnosis, will be discussed.
Case Presentation
A 72-year-old male veteran presented with a 2-month history of subjective weakness in his upper and lower extremities progressing to multiple falls at home. He had no significant medical history other than a thymectomy at age 15 for an enlarged thymus, which per patient report, was benign. An initial laboratory test that included vitamin B12, folate, thyroid-stimulating hormone, complete blood cell count, and comprehensive metabolic panel, were unremarkable, with a white blood cell count of 8.5 K/uL. The initial neurologic evaluation did not show any focal neurologic deficits; however, during the initial hospital stay, the patient developed increasing lower extremity weakness on examination. A noncontrast CT head scan showed extensive nonspecific hypodensities within the periventricular white matter (Figure 1). A contrast-enhanced MRI showed enhancing lesions involving the corpus callosum, left cerebral peduncle, and right temporal lobe (Figures 2, 3, and 4). These lesions also exhibited significant restricted diffusion and a mild amount of surrounding vasogenic edema. The working diagnosis after the MRI included primary CNS lymphoma, multifocal glioblastoma, and tumefactive demyelinating disease. The patient was started on IV steroids and transferred for neurosurgical evaluation and biopsy at an outside hospital. The frontal lesion was biopsied, and the initial frozen section was consistent with lymphoma; a bone marrow biopsy was negative. The workup for immunodeficiency was unremarkable. Pathology revealed high-grade B-cell lymphoma, and the patient began a chemotherapy regimen.
Discussion
The workup of altered mental status, focal neurologic deficits, headaches, or other neurologic conditions often begins with a noncontrast CT scan. On CT, PCNSL generally appears isodense to hyperdense to gray matter, but appearance is variable. The often hyperdense appearance is attributable to the hypercellular nature of lymphoma. Many times, as in this case, CT may show only vague hypodensities, some of which may be associated with surrounding edema. This presentation is nonspecific and may be seen with advancing age due to changes of chronic microvascular ischemia as well as demyelination, other malignancies, and several other disease processes, both benign and malignant. After the initial CT scan, further workup requires evaluation with MRI. PCNSL exhibits restricted diffusion and variable signal intensity on T2-weighted imaging.
PCNSL is frequently centrally located within the periventricular white matter, often within the frontal lobe but can involve other lobes, the basal ganglia, brainstem, cerebellum, or less likely, the spinal canal.7 Contrary to primary CNS disease, secondary lymphoma within the CNS has been described classically as affecting a leptomeningeal (pia and arachnoid mater) distribution two-thirds of the time, with parenchymal involvement occurring in the other one-third of patients. A recent study by Malikova and colleagues found parenchymal involvement may be much more common than previously thought.1 Leptomeningeal spread of disease often involves the cranial nerves, subependymal regions, spinal cord, or spinal nerve roots. Dural involvement in primary or secondary lymphoma is rare.
PCNSL nearly always shows enhancement. Linear enhancement along perivascular spaces is highly characteristic of PCNSL. The typical appearance of PCNSL associated with immunodeficiency varies from that seen in an otherwise immunocompetent patient. Patients with immunodeficiency usually have multifocal involvement, central necrosis leading to a ring enhancement appearance, and have more propensity for spontaneous hemorrhage.7 Immunocompetent patients are less likely to present with multifocal disease and rarely show ring enhancement. Also, spontaneous hemorrhage is rare in immunocompetent patients. In our case, extensive multifocal involvement was present, whereas typically immunocompetent patients will present with a solitary homogeneously enhancing parenchymal mass.
The primary differential for PCNSL includes malignant glioma, tumefactive multiple sclerosis, metastatic disease, and in an immunocompromised patient, toxoplasmosis. The degree of associated vasogenic edema and mass effect is generally lower in PCNSL than that of malignant gliomas and metastasis. Also, PCNSL tends to spare the cerebral cortex.8
Classically, PCNSL, malignant gliomas, and demyelinating disease have been considered the main differential for lesions that cross midline and involve both cerebral hemispheres. Lymphoma generally exhibits more restricted diffusion than malignant gliomas and metastasis, attributable to the highly cellular nature of lymphoma.7 Tumefactive multiple sclerosis is associated with relatively minimal mass effect for lesion size and exhibits less restricted diffusion values when compared to high grade gliomas and PCNSL. One fairly specific finding for tumefactive demyelinating lesions is incomplete rim enhancement.9 Unfortunately, an MRI is not reliable in differentiating these entities, and biopsy is required for definitive diagnosis. Many advancing imaging modalities may help provide the correct diagnosis of PCNSL, including diffusion-weighted and apparent diffusion coefficient imaging, diffusion tensor imaging, MR spectroscopy and PET imaging.7
Conclusion
With the increasing use of HAART, the paradigm of PCNSL is shifting toward one predominantly affecting immunocompetent patients. PCNSL should be considered in any patient with multiple enhancing CNS lesions, regardless of immune status. Several key imaging characteristics may help differentiate PCNSL and other disease processes; however, at this time, biopsy is recommended for definitive diagnosis.
1. Malikova H, Burghardtova M, Koubska E, Mandys V, Kozak T, Weichet J. Secondary central nervous system lymphoma: spectrum of morphological MRI appearances. Neuropsychiatr Dis Treat. 2018;4:733-740.
2. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-Oncol. 2012;14(suppl 5):v1-v49.
3. Diamond C, Taylor TH, Aboumrad T, Anton-Culver H. Changes in acquired immunodeficiency syndrome-related non-Hodgkin lymphoma in the era of highly active antiretroviral therapy: incidence, presentation, treatment, and survival. Cancer. 2006;106(1):128-135.
4. O’Neill BP, Decker PA, Tieu C, Cerhan JR. The changing incidence of primary central nervous system lymphoma is driven primarily by the changing incidence in young and middle-aged men and differs from time trends in systemic diffuse large B-cell non-Hodgkins lymphoma. Am J Hematol. 2013;88(12):997-1000.
5. [no authors listed]. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm Rep. 1992;41(rr-17):1-19.
6. Maiuri F. Central nervous system lymphomas and immunodeficiency. Neurological Research. 1989;11(1):2-5.
7. Haldorsen IS, Espeland A, Larsson EM. Central nervous system lymphoma: characteristic findings on traditional and advanced imaging. AJNR Am J Neuroradiol. 2010;32(6):984-992.
8. Gómez Roselló E, Quiles Granado AM, Laguillo Sala G, Gutiérrez S. Primary central nervous system lymphoma in immunocompetent patients: spectrum of findings and differential characteristics. Radiología. 2018;60(4):280-289.
9. Mabray MC, Cohen BA, Villanueva-Meyer JE, et al. Performance of Apparent Diffusion Coefficient Values and Conventional MRI Features in Differentiating Tumefactive Demyelinating Lesions From Primary Brain Neoplasms. American Journal of Roentgenology. 2015;205(5):1075-1085.
1. Malikova H, Burghardtova M, Koubska E, Mandys V, Kozak T, Weichet J. Secondary central nervous system lymphoma: spectrum of morphological MRI appearances. Neuropsychiatr Dis Treat. 2018;4:733-740.
2. Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-Oncol. 2012;14(suppl 5):v1-v49.
3. Diamond C, Taylor TH, Aboumrad T, Anton-Culver H. Changes in acquired immunodeficiency syndrome-related non-Hodgkin lymphoma in the era of highly active antiretroviral therapy: incidence, presentation, treatment, and survival. Cancer. 2006;106(1):128-135.
4. O’Neill BP, Decker PA, Tieu C, Cerhan JR. The changing incidence of primary central nervous system lymphoma is driven primarily by the changing incidence in young and middle-aged men and differs from time trends in systemic diffuse large B-cell non-Hodgkins lymphoma. Am J Hematol. 2013;88(12):997-1000.
5. [no authors listed]. 1993 revised classification system for HIV infection and expanded surveillance case definition for AIDS among adolescents and adults. MMWR Recomm Rep. 1992;41(rr-17):1-19.
6. Maiuri F. Central nervous system lymphomas and immunodeficiency. Neurological Research. 1989;11(1):2-5.
7. Haldorsen IS, Espeland A, Larsson EM. Central nervous system lymphoma: characteristic findings on traditional and advanced imaging. AJNR Am J Neuroradiol. 2010;32(6):984-992.
8. Gómez Roselló E, Quiles Granado AM, Laguillo Sala G, Gutiérrez S. Primary central nervous system lymphoma in immunocompetent patients: spectrum of findings and differential characteristics. Radiología. 2018;60(4):280-289.
9. Mabray MC, Cohen BA, Villanueva-Meyer JE, et al. Performance of Apparent Diffusion Coefficient Values and Conventional MRI Features in Differentiating Tumefactive Demyelinating Lesions From Primary Brain Neoplasms. American Journal of Roentgenology. 2015;205(5):1075-1085.
Acute Encephalopathy Following Hyperbaric Oxygen Therapy in a Patient on Metronidazole
Altered mental status (AMS) is a common presentation to the emergency department (ED) for older patients and is often due to underlying drug-associated adverse effects (AEs), medical or psychiatric illness, or neurologic disease. EDs often have protocols for diagnosing and managing AMS to assess the underlying etiology. A formal assessment with a full history and physical examination is paramount to diagnosing the cause of AMS.
Oral metronidazole is a commonly used antibiotic for anaerobic bacterial infections and Clostridium difficile-associated diarrhea and colitis.1Metronidazole produces cytotoxic intermediates that cause DNA strand breakage and destabilization, resulting in bactericidal activity in host cells.2Common AEs include gastrointestinal symptoms such as nausea, vomiting, and diarrhea; less common AEs can involve the nervous system and include seizures, peripheral neuropathy, dizziness, ataxia, and encephalopathy.3,4A pattern of magnetic resonance image (MRI) abnormalities typically located at the cerebellar dentate nucleus midbrain, dorsal pons, medulla, and splenium of the corpus callosum have been associated with metronidazole usage.5
Hyperbaric oxygen therapy (HBOT) is a treatment modality used as the primary therapy for decompression sickness, arterial gas embolism, and carbon monoxide poisoning. HBOT is used as adjuvant therapy for osteonecrosis caused by radiation or bisphosphonate use.6,7 HBOT increases the partial pressure of oxygen in plasma and increases the amount of oxygen delivered to tissues throughout the body.8Hyperoxia, defined as an elevated partial pressure of oxygen leading to excess oxygenation to tissues and organs, increases production of reactive oxygen and nitrogen species, which are signaling factors in a variety of pathways that stimulate angiogenesis.8 AEs of HBOT include barotrauma-related injuries and oxygen toxicity, such as respiratory distress or central nervous system (CNS) symptoms.9 Severe CNS AEs occur in 1% to 2% of patients undergoing therapy and manifest as generalized tonic-clonic seizures, typically in patients with preexisting neurologic disorders, brain injury, or lowered seizure threshold.7,8,10 There have been no documented incidences of HBOT inducing acute encephalopathy.
Case Presentation
A 63-year-old male smoker with no history of alcohol use presented to the ED with an acute onset of lightheadedness, confusion, and poor coordination following his second HBOT for radiation-induced osteonecrosis of the mandible. The patient reported chronic, slowly progressive pain and numbness of the feet that began 4 years earlier. He noted marked worsening of pain and difficulty standing and walking 3 to 4 months prior to presentation.
Ten years prior, the patient was diagnosed with cancer of the right tonsil. A tonsillectomy with wide margins was performed, followed by 35 rounds of radiation treatment and 2 rounds of chemotherapy with cisplatin.
In May 2017, the patient presented with a lump in the right cheek that was diagnosed as osteonecrosis of the mandible. An oral surgeon prescribed metronidazole 500 mg qid and amoxicillin 500 mg tid. The patient was adherent until presentation in November 2017. Following lack of improvement of the osteonecrosis from antibiotic therapy, oral surgery was planned, and the patient was referred for HBOT with a planned 20 HBOT preoperative treatments and 10 postoperative treatments.
Following his first 2-hour HBOT treatment on November 13, 2017, the patient complained of light-headedness, confusion, and incoordination. While driving on a familiar route to his home, he collided with a tree that was 6 feet from the curb. The patient attempted to drive another vehicle later that day, resulting in a second motor vehicle accident. There was no significant injury reported in either accident.
His partner described the patient’s episode of disorientation lasting 6 to 8 hours, during which he “looked drunk” and was unable to sit in a chair without falling. The following morning, the patient had improved mental status but had not returned to baseline. His second HBOT treatment took place that day, and again, the patient acutely experienced light-headedness and confusion following completion. Therapy was suspended, and the patient was referred to the ED for further evaluation. Mild facial asymmetry without weakness, decreased sensation from toes to knees bilaterally, and absent Achilles reflexes bilaterally were found on neurologic examination. He exhibited past-pointing on finger-to-nose testing bilaterally. He was able to ambulate independently, but he could not perform tandem gait.
An MRI of the brain showed abnormal T2 hyperintensity found bilaterally at the dentate nuclei and inferior colliculi. The splenium of the corpus callosum also showed mild involvement with hyperintense lesions. Laboratory tests of the patient’s complete blood count; comprehensive metabolic panel; vitamins B1, B6, B12; and folic acid levels had no notable abnormalities and were within normal limits.
Metronidazole and HBOT therapy were discontinued, and all of the patient’s symptoms resolved within 2 weeks. A repeat examination and MRI performed 1 month later showed resolution of all the patient’s clinical findings and MRI abnormalities. HBOT was resumed without the recurrence of previously described symptoms.
Discussion
This patient’s encephalopathic symptoms correlate temporally with the onset of HBOT. There is no medical literature suggesting a relationship between HBOT and encephalopathic symptoms with MRI abnormalities, and in fact, some studies suggest HBOT as a treatment for hypoxic-ischemic encephalopathy in neonates.11 This led us to believe that the HBOT may have exacerbated some underlying condition, evidenced by the specific MRI findings of T2 fluid-attenuated inversion recovery (FLAIR) hyperintensities in the dentate nuclei and inferior colliculi (Figures 1 and 2). 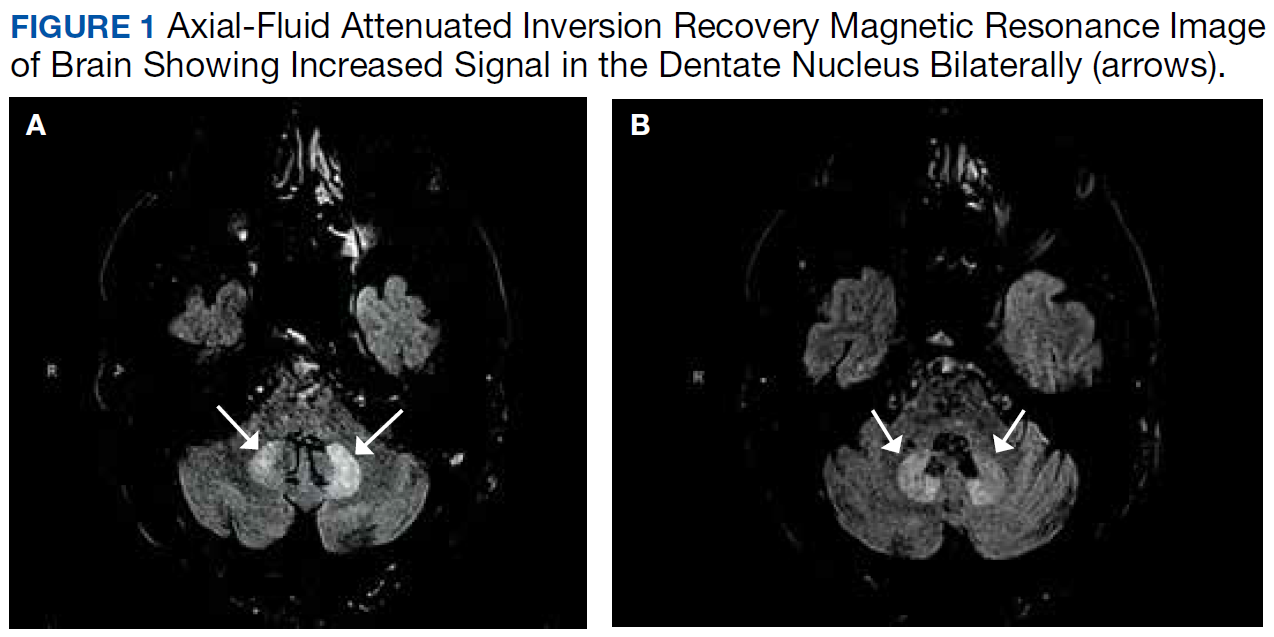
Differential diagnoses for T2 hyperintense lesions in the dentate nuclei include metronidazole toxicity, acute Wernicke encephalopathy (WE), and methyl bromide intoxication. Diseases that would have presented in infancy with similar MRI findings (Canavan disease, maple-syrup urine disease, and glutaric aciduria type 1) were not considered plausible.12-14 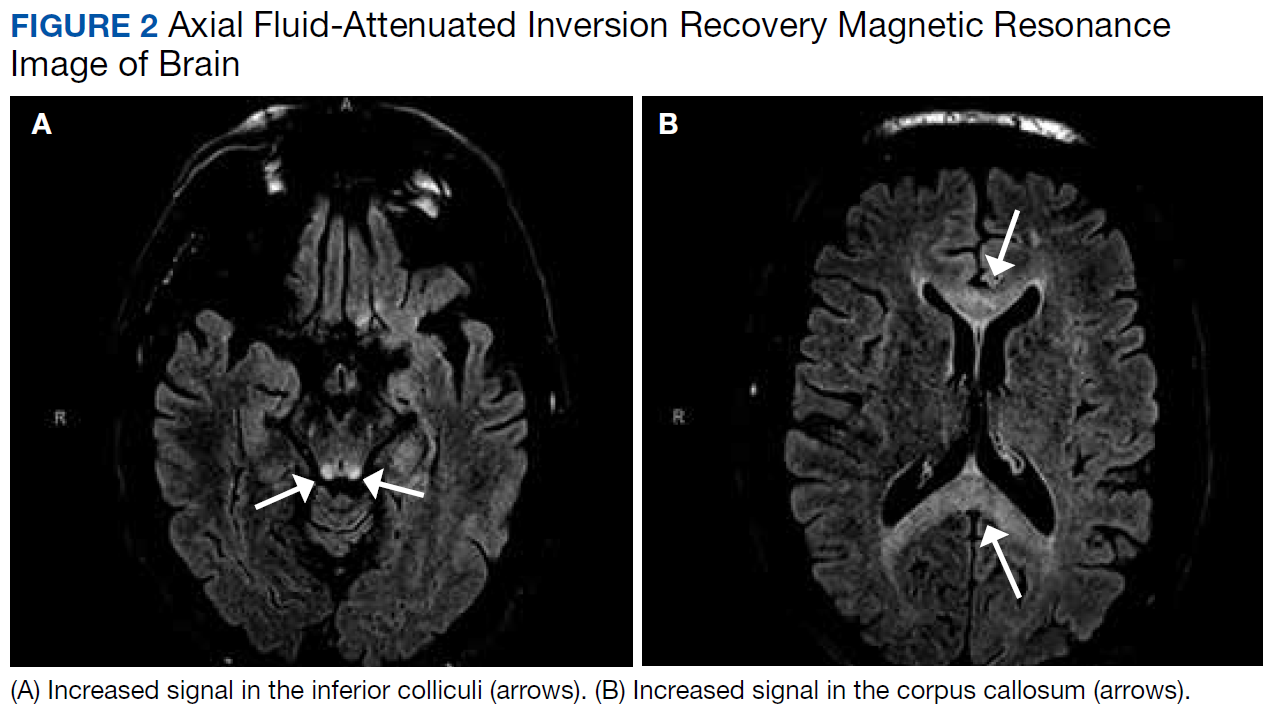
Despite his denial of alcohol use, the patient was at risk for malnutrition secondary to his mandibular lesion and difficulty eating. Clinically, he presented with episodes of confusion and ataxia, consistent with 2 of the classic triad of symptoms of WE (no ocular abnormalities noted on exam). Typical MRI findings in WE include signal intensity alterations (including T2 hyperintensities) in the medial thalami, mammillary bodies, collicular bodies, and periaqueductal and periventricular regions.14,15 Atypical MRI findings in WE include symmetric signal intensity changes in the cerebellum, dentate nuclei, caudate nuclei, red nuclei, cranial nerve nuclei, and splenium.14 Of note, atypical MRI findings were more common in patients without alcohol use disorders and WE, and typical MRI findings were more common in patients with alcohol use disorders.14 However, this patient’s report of no alcohol use and the serum thiamine level being within normal limits (173 nmol/L; range 78-185 nmol/L) made acute WE less likely than metronidonazale-induced encephalopathy (MIE).
The most common neurologic AE of metronidazole is distal symmetric sensory polyneuropathy, which also can have motor or autonomic features.16,17 While our patient had a history of peripheral neuropathy, he noted marked worsening of foot pain 3 months after initiating metronidazole therapy. A potential mechanism involves metronidazole or its cytotoxic intermediates binding neuronal ribonucleic acids, thus inhibiting protein synthesis and resulting in degenerative neuronal changes and reversible axonal swelling (as opposed to the DNA interference attributed to the drug’s mechanism of bactericidal action).18 Neuropathies may result from prolonged high-dose metronidazole therapy (cumulative dose > 42 g),3 but they also have been seen in short-term use of high dosages.17
CNS AEs are much rarer and are thought to be associated with metronidazole’s ability to cross the blood-brain barrier. These patients present as a toxic encephalopathy with cerebellar dysfunction (dysarthria, ataxia) as the most common presentation, followed by AMS and seizures.4 Our patient presented with acute confusion and ataxia. Animal studies suggest that γ-aminobutyric acid (GABA) receptor modulation in the cerebellar and vestibular systems may contribute to this neurotoxicity, but no definitive mechanism of injury has been found.19
On MRI, MIE most commonly presents with hyperintense lesions in the bilateral cerebellar dentate nucleus on T2-weighted and FLAIR images.5,20 The midbrain, dorsal pons, medulla, and corpus callosum also can show increased signal intensity.5 This AE does not seem to be dose- or duration-dependent, and most cases report complete or partial resolution of symptoms following discontinuation of the drug, though this is not absolute.4,13,21 The patient’s MRI findings were highly consistent with MIE (Figure 2).
Conclusion
This patient’s highly specific MRI findings, neurologic examination consistent with confusion, ataxia, length-dependent sensory neuropathy, and 360-g cumulative dose of metronidazole over the previous 6 months suggest he experienced MIE. The mechanism of how HBOT precipitated the patient’s altered mental status, incoordination, and worsening of peripheral neuropathy is unknown. Although encephalopathy with MRI abnormalities as described is not a reported AE of HBOT, it may be unrecognized. It is possible that without HBOT the patient would have remained asymptomatic apart from his peripheral neuropathy.
We propose HBOT may exacerbate or increase the risk of a patient developing MIE. Our patient was able to safely resume HBOT after metronidazole was discontinued, suggesting that the combination was the causation for the development of encephalopathy. We do not believe any similar cases have been reported.
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541.
2. Edwards DI. The action of metronidazole on DNA. J Antimicrob Chemother. 1977;3(1):43-48.
3. Goolsby TA, Jakeman B, Gaynes RP. Clinical relevance of metronidazole and peripheral neuropathy: a systematic review of the literature. Int J Antimicrob Agents. 2018;51(3):319-325.
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systematic review. Clin Neuropharmacol. 2011;34(6):241-247.
5. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658.
6. Ceponis P, Keilman C, Guerry C, Freiberger JJ. Hyperbaric oxygen therapy and osteonecrosis. Oral Dis. 2017;23(2):141-151.
7. Leach R, Rees P, Wilmshurst P. Hyperbaric oxygen therapy. BMJ. 1998;317(7166):1140-1143.
8. Thom SR. Hyperbaric oxygen–its mechanisms and efficacy. Plastic Reconstr Surg. 2011;127(suppl 1):131S-141S.
9. Plafki C, Peters P, Almeling M, Welslau W, Busch R. Complications and side effects of hyperbaric oxygen therapy. Aviation Space Environ Med. 2000;71(2):119-124.
10. Hadanny A, Meir O, Bechor Y, Fishlev G, Bergan J, Efrati S. Seizures during hyperbaric oxygen therapy: retrospective analysis of 62,614 treatment sessions. Undersea Hyperb Med. 2016;43(1):21-28.
11. Liu Z, Xiong T, Meads C. Clinical effectiveness of treatment with hyperbaric oxygen for neonatal hypoxic-ischaemic encephalopathy: systematic review of Chinese literature. BMJ. 2006;333(7564):374.
12. Bond KM, Brinjikji W, Eckel LJ, Kallmes DF, McDonald RJ, Carr CM. Dentate update: imaging features of entities that affect the dentate nucleus. AJNR Am J Neuroradiol. 2017;38(8):1467-1474.
13. Agarwal A, Kanekar S, Sabat S, Thamburaj K. Metronidazole-induced cerebellar toxicity. Neurol Int. 2016;8(1):6365.
14. Zuccoli G, Pipitone N. Neuroimaging findings in acute Wernicke’s encephalopathy: review of the literature. AJR Am J Roentgenol. 2009;192(2):501-508.
15. Jung YC, Chanraud S, Sullivan EV. Neuroimaging of Wernicke’s encephalopathy and Korsakoff’s syndrome. Neuropsychol Rev. 2012;22(2):170-180.
16. Hobson-Webb LD, Roach ES, Donofrio PD. Metronidazole: newly recognized cause of autonomic neuropathy. J Child Neurol. 2006;21(5):429-431.
17. Nath Chaurasia R. Rapid onset metronidazole induced sensory neuropathy: case series and review of literature. Int J Neurorehabilitation. 2015;02:152.
18. Bradley WG, Karlsson IJ, Rassol CG. Metronidazole neuropathy. Br Med J. 1977;2(6087):610-611.
19. Evans J, Levesque D, Knowles K, Longshore R, Plummer S. Diazepam as a treatment for metronidazole toxicosis in dogs: a retrospective study of 21 cases. J Vet Intern Med. 2003;17(3):304-310.
20. Farmakiotis D, Zeluff B. Images in clinical medicine. Metronidazole-associated encephalopathy. N Engl J Med. 2016;374(15):1465.
21. Hobbs K, Stern-Nezer S, Buckwalter MS, Fischbein N, Finley Caulfield A. Metronidazole-induced encephalopathy: not always a reversible situation. Neurocrit Care. 2015;22(3):429-436.
Altered mental status (AMS) is a common presentation to the emergency department (ED) for older patients and is often due to underlying drug-associated adverse effects (AEs), medical or psychiatric illness, or neurologic disease. EDs often have protocols for diagnosing and managing AMS to assess the underlying etiology. A formal assessment with a full history and physical examination is paramount to diagnosing the cause of AMS.
Oral metronidazole is a commonly used antibiotic for anaerobic bacterial infections and Clostridium difficile-associated diarrhea and colitis.1Metronidazole produces cytotoxic intermediates that cause DNA strand breakage and destabilization, resulting in bactericidal activity in host cells.2Common AEs include gastrointestinal symptoms such as nausea, vomiting, and diarrhea; less common AEs can involve the nervous system and include seizures, peripheral neuropathy, dizziness, ataxia, and encephalopathy.3,4A pattern of magnetic resonance image (MRI) abnormalities typically located at the cerebellar dentate nucleus midbrain, dorsal pons, medulla, and splenium of the corpus callosum have been associated with metronidazole usage.5
Hyperbaric oxygen therapy (HBOT) is a treatment modality used as the primary therapy for decompression sickness, arterial gas embolism, and carbon monoxide poisoning. HBOT is used as adjuvant therapy for osteonecrosis caused by radiation or bisphosphonate use.6,7 HBOT increases the partial pressure of oxygen in plasma and increases the amount of oxygen delivered to tissues throughout the body.8Hyperoxia, defined as an elevated partial pressure of oxygen leading to excess oxygenation to tissues and organs, increases production of reactive oxygen and nitrogen species, which are signaling factors in a variety of pathways that stimulate angiogenesis.8 AEs of HBOT include barotrauma-related injuries and oxygen toxicity, such as respiratory distress or central nervous system (CNS) symptoms.9 Severe CNS AEs occur in 1% to 2% of patients undergoing therapy and manifest as generalized tonic-clonic seizures, typically in patients with preexisting neurologic disorders, brain injury, or lowered seizure threshold.7,8,10 There have been no documented incidences of HBOT inducing acute encephalopathy.
Case Presentation
A 63-year-old male smoker with no history of alcohol use presented to the ED with an acute onset of lightheadedness, confusion, and poor coordination following his second HBOT for radiation-induced osteonecrosis of the mandible. The patient reported chronic, slowly progressive pain and numbness of the feet that began 4 years earlier. He noted marked worsening of pain and difficulty standing and walking 3 to 4 months prior to presentation.
Ten years prior, the patient was diagnosed with cancer of the right tonsil. A tonsillectomy with wide margins was performed, followed by 35 rounds of radiation treatment and 2 rounds of chemotherapy with cisplatin.
In May 2017, the patient presented with a lump in the right cheek that was diagnosed as osteonecrosis of the mandible. An oral surgeon prescribed metronidazole 500 mg qid and amoxicillin 500 mg tid. The patient was adherent until presentation in November 2017. Following lack of improvement of the osteonecrosis from antibiotic therapy, oral surgery was planned, and the patient was referred for HBOT with a planned 20 HBOT preoperative treatments and 10 postoperative treatments.
Following his first 2-hour HBOT treatment on November 13, 2017, the patient complained of light-headedness, confusion, and incoordination. While driving on a familiar route to his home, he collided with a tree that was 6 feet from the curb. The patient attempted to drive another vehicle later that day, resulting in a second motor vehicle accident. There was no significant injury reported in either accident.
His partner described the patient’s episode of disorientation lasting 6 to 8 hours, during which he “looked drunk” and was unable to sit in a chair without falling. The following morning, the patient had improved mental status but had not returned to baseline. His second HBOT treatment took place that day, and again, the patient acutely experienced light-headedness and confusion following completion. Therapy was suspended, and the patient was referred to the ED for further evaluation. Mild facial asymmetry without weakness, decreased sensation from toes to knees bilaterally, and absent Achilles reflexes bilaterally were found on neurologic examination. He exhibited past-pointing on finger-to-nose testing bilaterally. He was able to ambulate independently, but he could not perform tandem gait.
An MRI of the brain showed abnormal T2 hyperintensity found bilaterally at the dentate nuclei and inferior colliculi. The splenium of the corpus callosum also showed mild involvement with hyperintense lesions. Laboratory tests of the patient’s complete blood count; comprehensive metabolic panel; vitamins B1, B6, B12; and folic acid levels had no notable abnormalities and were within normal limits.
Metronidazole and HBOT therapy were discontinued, and all of the patient’s symptoms resolved within 2 weeks. A repeat examination and MRI performed 1 month later showed resolution of all the patient’s clinical findings and MRI abnormalities. HBOT was resumed without the recurrence of previously described symptoms.
Discussion
This patient’s encephalopathic symptoms correlate temporally with the onset of HBOT. There is no medical literature suggesting a relationship between HBOT and encephalopathic symptoms with MRI abnormalities, and in fact, some studies suggest HBOT as a treatment for hypoxic-ischemic encephalopathy in neonates.11 This led us to believe that the HBOT may have exacerbated some underlying condition, evidenced by the specific MRI findings of T2 fluid-attenuated inversion recovery (FLAIR) hyperintensities in the dentate nuclei and inferior colliculi (Figures 1 and 2).
Differential diagnoses for T2 hyperintense lesions in the dentate nuclei include metronidazole toxicity, acute Wernicke encephalopathy (WE), and methyl bromide intoxication. Diseases that would have presented in infancy with similar MRI findings (Canavan disease, maple-syrup urine disease, and glutaric aciduria type 1) were not considered plausible.12-14
Despite his denial of alcohol use, the patient was at risk for malnutrition secondary to his mandibular lesion and difficulty eating. Clinically, he presented with episodes of confusion and ataxia, consistent with 2 of the classic triad of symptoms of WE (no ocular abnormalities noted on exam). Typical MRI findings in WE include signal intensity alterations (including T2 hyperintensities) in the medial thalami, mammillary bodies, collicular bodies, and periaqueductal and periventricular regions.14,15 Atypical MRI findings in WE include symmetric signal intensity changes in the cerebellum, dentate nuclei, caudate nuclei, red nuclei, cranial nerve nuclei, and splenium.14 Of note, atypical MRI findings were more common in patients without alcohol use disorders and WE, and typical MRI findings were more common in patients with alcohol use disorders.14 However, this patient’s report of no alcohol use and the serum thiamine level being within normal limits (173 nmol/L; range 78-185 nmol/L) made acute WE less likely than metronidonazale-induced encephalopathy (MIE).
The most common neurologic AE of metronidazole is distal symmetric sensory polyneuropathy, which also can have motor or autonomic features.16,17 While our patient had a history of peripheral neuropathy, he noted marked worsening of foot pain 3 months after initiating metronidazole therapy. A potential mechanism involves metronidazole or its cytotoxic intermediates binding neuronal ribonucleic acids, thus inhibiting protein synthesis and resulting in degenerative neuronal changes and reversible axonal swelling (as opposed to the DNA interference attributed to the drug’s mechanism of bactericidal action).18 Neuropathies may result from prolonged high-dose metronidazole therapy (cumulative dose > 42 g),3 but they also have been seen in short-term use of high dosages.17
CNS AEs are much rarer and are thought to be associated with metronidazole’s ability to cross the blood-brain barrier. These patients present as a toxic encephalopathy with cerebellar dysfunction (dysarthria, ataxia) as the most common presentation, followed by AMS and seizures.4 Our patient presented with acute confusion and ataxia. Animal studies suggest that γ-aminobutyric acid (GABA) receptor modulation in the cerebellar and vestibular systems may contribute to this neurotoxicity, but no definitive mechanism of injury has been found.19
On MRI, MIE most commonly presents with hyperintense lesions in the bilateral cerebellar dentate nucleus on T2-weighted and FLAIR images.5,20 The midbrain, dorsal pons, medulla, and corpus callosum also can show increased signal intensity.5 This AE does not seem to be dose- or duration-dependent, and most cases report complete or partial resolution of symptoms following discontinuation of the drug, though this is not absolute.4,13,21 The patient’s MRI findings were highly consistent with MIE (Figure 2).
Conclusion
This patient’s highly specific MRI findings, neurologic examination consistent with confusion, ataxia, length-dependent sensory neuropathy, and 360-g cumulative dose of metronidazole over the previous 6 months suggest he experienced MIE. The mechanism of how HBOT precipitated the patient’s altered mental status, incoordination, and worsening of peripheral neuropathy is unknown. Although encephalopathy with MRI abnormalities as described is not a reported AE of HBOT, it may be unrecognized. It is possible that without HBOT the patient would have remained asymptomatic apart from his peripheral neuropathy.
We propose HBOT may exacerbate or increase the risk of a patient developing MIE. Our patient was able to safely resume HBOT after metronidazole was discontinued, suggesting that the combination was the causation for the development of encephalopathy. We do not believe any similar cases have been reported.
Altered mental status (AMS) is a common presentation to the emergency department (ED) for older patients and is often due to underlying drug-associated adverse effects (AEs), medical or psychiatric illness, or neurologic disease. EDs often have protocols for diagnosing and managing AMS to assess the underlying etiology. A formal assessment with a full history and physical examination is paramount to diagnosing the cause of AMS.
Oral metronidazole is a commonly used antibiotic for anaerobic bacterial infections and Clostridium difficile-associated diarrhea and colitis.1Metronidazole produces cytotoxic intermediates that cause DNA strand breakage and destabilization, resulting in bactericidal activity in host cells.2Common AEs include gastrointestinal symptoms such as nausea, vomiting, and diarrhea; less common AEs can involve the nervous system and include seizures, peripheral neuropathy, dizziness, ataxia, and encephalopathy.3,4A pattern of magnetic resonance image (MRI) abnormalities typically located at the cerebellar dentate nucleus midbrain, dorsal pons, medulla, and splenium of the corpus callosum have been associated with metronidazole usage.5
Hyperbaric oxygen therapy (HBOT) is a treatment modality used as the primary therapy for decompression sickness, arterial gas embolism, and carbon monoxide poisoning. HBOT is used as adjuvant therapy for osteonecrosis caused by radiation or bisphosphonate use.6,7 HBOT increases the partial pressure of oxygen in plasma and increases the amount of oxygen delivered to tissues throughout the body.8Hyperoxia, defined as an elevated partial pressure of oxygen leading to excess oxygenation to tissues and organs, increases production of reactive oxygen and nitrogen species, which are signaling factors in a variety of pathways that stimulate angiogenesis.8 AEs of HBOT include barotrauma-related injuries and oxygen toxicity, such as respiratory distress or central nervous system (CNS) symptoms.9 Severe CNS AEs occur in 1% to 2% of patients undergoing therapy and manifest as generalized tonic-clonic seizures, typically in patients with preexisting neurologic disorders, brain injury, or lowered seizure threshold.7,8,10 There have been no documented incidences of HBOT inducing acute encephalopathy.
Case Presentation
A 63-year-old male smoker with no history of alcohol use presented to the ED with an acute onset of lightheadedness, confusion, and poor coordination following his second HBOT for radiation-induced osteonecrosis of the mandible. The patient reported chronic, slowly progressive pain and numbness of the feet that began 4 years earlier. He noted marked worsening of pain and difficulty standing and walking 3 to 4 months prior to presentation.
Ten years prior, the patient was diagnosed with cancer of the right tonsil. A tonsillectomy with wide margins was performed, followed by 35 rounds of radiation treatment and 2 rounds of chemotherapy with cisplatin.
In May 2017, the patient presented with a lump in the right cheek that was diagnosed as osteonecrosis of the mandible. An oral surgeon prescribed metronidazole 500 mg qid and amoxicillin 500 mg tid. The patient was adherent until presentation in November 2017. Following lack of improvement of the osteonecrosis from antibiotic therapy, oral surgery was planned, and the patient was referred for HBOT with a planned 20 HBOT preoperative treatments and 10 postoperative treatments.
Following his first 2-hour HBOT treatment on November 13, 2017, the patient complained of light-headedness, confusion, and incoordination. While driving on a familiar route to his home, he collided with a tree that was 6 feet from the curb. The patient attempted to drive another vehicle later that day, resulting in a second motor vehicle accident. There was no significant injury reported in either accident.
His partner described the patient’s episode of disorientation lasting 6 to 8 hours, during which he “looked drunk” and was unable to sit in a chair without falling. The following morning, the patient had improved mental status but had not returned to baseline. His second HBOT treatment took place that day, and again, the patient acutely experienced light-headedness and confusion following completion. Therapy was suspended, and the patient was referred to the ED for further evaluation. Mild facial asymmetry without weakness, decreased sensation from toes to knees bilaterally, and absent Achilles reflexes bilaterally were found on neurologic examination. He exhibited past-pointing on finger-to-nose testing bilaterally. He was able to ambulate independently, but he could not perform tandem gait.
An MRI of the brain showed abnormal T2 hyperintensity found bilaterally at the dentate nuclei and inferior colliculi. The splenium of the corpus callosum also showed mild involvement with hyperintense lesions. Laboratory tests of the patient’s complete blood count; comprehensive metabolic panel; vitamins B1, B6, B12; and folic acid levels had no notable abnormalities and were within normal limits.
Metronidazole and HBOT therapy were discontinued, and all of the patient’s symptoms resolved within 2 weeks. A repeat examination and MRI performed 1 month later showed resolution of all the patient’s clinical findings and MRI abnormalities. HBOT was resumed without the recurrence of previously described symptoms.
Discussion
This patient’s encephalopathic symptoms correlate temporally with the onset of HBOT. There is no medical literature suggesting a relationship between HBOT and encephalopathic symptoms with MRI abnormalities, and in fact, some studies suggest HBOT as a treatment for hypoxic-ischemic encephalopathy in neonates.11 This led us to believe that the HBOT may have exacerbated some underlying condition, evidenced by the specific MRI findings of T2 fluid-attenuated inversion recovery (FLAIR) hyperintensities in the dentate nuclei and inferior colliculi (Figures 1 and 2).
Differential diagnoses for T2 hyperintense lesions in the dentate nuclei include metronidazole toxicity, acute Wernicke encephalopathy (WE), and methyl bromide intoxication. Diseases that would have presented in infancy with similar MRI findings (Canavan disease, maple-syrup urine disease, and glutaric aciduria type 1) were not considered plausible.12-14
Despite his denial of alcohol use, the patient was at risk for malnutrition secondary to his mandibular lesion and difficulty eating. Clinically, he presented with episodes of confusion and ataxia, consistent with 2 of the classic triad of symptoms of WE (no ocular abnormalities noted on exam). Typical MRI findings in WE include signal intensity alterations (including T2 hyperintensities) in the medial thalami, mammillary bodies, collicular bodies, and periaqueductal and periventricular regions.14,15 Atypical MRI findings in WE include symmetric signal intensity changes in the cerebellum, dentate nuclei, caudate nuclei, red nuclei, cranial nerve nuclei, and splenium.14 Of note, atypical MRI findings were more common in patients without alcohol use disorders and WE, and typical MRI findings were more common in patients with alcohol use disorders.14 However, this patient’s report of no alcohol use and the serum thiamine level being within normal limits (173 nmol/L; range 78-185 nmol/L) made acute WE less likely than metronidonazale-induced encephalopathy (MIE).
The most common neurologic AE of metronidazole is distal symmetric sensory polyneuropathy, which also can have motor or autonomic features.16,17 While our patient had a history of peripheral neuropathy, he noted marked worsening of foot pain 3 months after initiating metronidazole therapy. A potential mechanism involves metronidazole or its cytotoxic intermediates binding neuronal ribonucleic acids, thus inhibiting protein synthesis and resulting in degenerative neuronal changes and reversible axonal swelling (as opposed to the DNA interference attributed to the drug’s mechanism of bactericidal action).18 Neuropathies may result from prolonged high-dose metronidazole therapy (cumulative dose > 42 g),3 but they also have been seen in short-term use of high dosages.17
CNS AEs are much rarer and are thought to be associated with metronidazole’s ability to cross the blood-brain barrier. These patients present as a toxic encephalopathy with cerebellar dysfunction (dysarthria, ataxia) as the most common presentation, followed by AMS and seizures.4 Our patient presented with acute confusion and ataxia. Animal studies suggest that γ-aminobutyric acid (GABA) receptor modulation in the cerebellar and vestibular systems may contribute to this neurotoxicity, but no definitive mechanism of injury has been found.19
On MRI, MIE most commonly presents with hyperintense lesions in the bilateral cerebellar dentate nucleus on T2-weighted and FLAIR images.5,20 The midbrain, dorsal pons, medulla, and corpus callosum also can show increased signal intensity.5 This AE does not seem to be dose- or duration-dependent, and most cases report complete or partial resolution of symptoms following discontinuation of the drug, though this is not absolute.4,13,21 The patient’s MRI findings were highly consistent with MIE (Figure 2).
Conclusion
This patient’s highly specific MRI findings, neurologic examination consistent with confusion, ataxia, length-dependent sensory neuropathy, and 360-g cumulative dose of metronidazole over the previous 6 months suggest he experienced MIE. The mechanism of how HBOT precipitated the patient’s altered mental status, incoordination, and worsening of peripheral neuropathy is unknown. Although encephalopathy with MRI abnormalities as described is not a reported AE of HBOT, it may be unrecognized. It is possible that without HBOT the patient would have remained asymptomatic apart from his peripheral neuropathy.
We propose HBOT may exacerbate or increase the risk of a patient developing MIE. Our patient was able to safely resume HBOT after metronidazole was discontinued, suggesting that the combination was the causation for the development of encephalopathy. We do not believe any similar cases have been reported.
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541.
2. Edwards DI. The action of metronidazole on DNA. J Antimicrob Chemother. 1977;3(1):43-48.
3. Goolsby TA, Jakeman B, Gaynes RP. Clinical relevance of metronidazole and peripheral neuropathy: a systematic review of the literature. Int J Antimicrob Agents. 2018;51(3):319-325.
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systematic review. Clin Neuropharmacol. 2011;34(6):241-247.
5. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658.
6. Ceponis P, Keilman C, Guerry C, Freiberger JJ. Hyperbaric oxygen therapy and osteonecrosis. Oral Dis. 2017;23(2):141-151.
7. Leach R, Rees P, Wilmshurst P. Hyperbaric oxygen therapy. BMJ. 1998;317(7166):1140-1143.
8. Thom SR. Hyperbaric oxygen–its mechanisms and efficacy. Plastic Reconstr Surg. 2011;127(suppl 1):131S-141S.
9. Plafki C, Peters P, Almeling M, Welslau W, Busch R. Complications and side effects of hyperbaric oxygen therapy. Aviation Space Environ Med. 2000;71(2):119-124.
10. Hadanny A, Meir O, Bechor Y, Fishlev G, Bergan J, Efrati S. Seizures during hyperbaric oxygen therapy: retrospective analysis of 62,614 treatment sessions. Undersea Hyperb Med. 2016;43(1):21-28.
11. Liu Z, Xiong T, Meads C. Clinical effectiveness of treatment with hyperbaric oxygen for neonatal hypoxic-ischaemic encephalopathy: systematic review of Chinese literature. BMJ. 2006;333(7564):374.
12. Bond KM, Brinjikji W, Eckel LJ, Kallmes DF, McDonald RJ, Carr CM. Dentate update: imaging features of entities that affect the dentate nucleus. AJNR Am J Neuroradiol. 2017;38(8):1467-1474.
13. Agarwal A, Kanekar S, Sabat S, Thamburaj K. Metronidazole-induced cerebellar toxicity. Neurol Int. 2016;8(1):6365.
14. Zuccoli G, Pipitone N. Neuroimaging findings in acute Wernicke’s encephalopathy: review of the literature. AJR Am J Roentgenol. 2009;192(2):501-508.
15. Jung YC, Chanraud S, Sullivan EV. Neuroimaging of Wernicke’s encephalopathy and Korsakoff’s syndrome. Neuropsychol Rev. 2012;22(2):170-180.
16. Hobson-Webb LD, Roach ES, Donofrio PD. Metronidazole: newly recognized cause of autonomic neuropathy. J Child Neurol. 2006;21(5):429-431.
17. Nath Chaurasia R. Rapid onset metronidazole induced sensory neuropathy: case series and review of literature. Int J Neurorehabilitation. 2015;02:152.
18. Bradley WG, Karlsson IJ, Rassol CG. Metronidazole neuropathy. Br Med J. 1977;2(6087):610-611.
19. Evans J, Levesque D, Knowles K, Longshore R, Plummer S. Diazepam as a treatment for metronidazole toxicosis in dogs: a retrospective study of 21 cases. J Vet Intern Med. 2003;17(3):304-310.
20. Farmakiotis D, Zeluff B. Images in clinical medicine. Metronidazole-associated encephalopathy. N Engl J Med. 2016;374(15):1465.
21. Hobbs K, Stern-Nezer S, Buckwalter MS, Fischbein N, Finley Caulfield A. Metronidazole-induced encephalopathy: not always a reversible situation. Neurocrit Care. 2015;22(3):429-436.
1. Samuelson J. Why metronidazole is active against both bacteria and parasites. Antimicrob Agents Chemother. 1999;43(7):1533-1541.
2. Edwards DI. The action of metronidazole on DNA. J Antimicrob Chemother. 1977;3(1):43-48.
3. Goolsby TA, Jakeman B, Gaynes RP. Clinical relevance of metronidazole and peripheral neuropathy: a systematic review of the literature. Int J Antimicrob Agents. 2018;51(3):319-325.
4. Kuriyama A, Jackson JL, Doi A, Kamiya T. Metronidazole-induced central nervous system toxicity: a systematic review. Clin Neuropharmacol. 2011;34(6):241-247.
5. Kim E, Na DG, Kim EY, Kim JH, Son KR, Chang KH. MR imaging of metronidazole-induced encephalopathy: lesion distribution and diffusion-weighted imaging findings. AJNR Am J Neuroradiol. 2007;28(9):1652-1658.
6. Ceponis P, Keilman C, Guerry C, Freiberger JJ. Hyperbaric oxygen therapy and osteonecrosis. Oral Dis. 2017;23(2):141-151.
7. Leach R, Rees P, Wilmshurst P. Hyperbaric oxygen therapy. BMJ. 1998;317(7166):1140-1143.
8. Thom SR. Hyperbaric oxygen–its mechanisms and efficacy. Plastic Reconstr Surg. 2011;127(suppl 1):131S-141S.
9. Plafki C, Peters P, Almeling M, Welslau W, Busch R. Complications and side effects of hyperbaric oxygen therapy. Aviation Space Environ Med. 2000;71(2):119-124.
10. Hadanny A, Meir O, Bechor Y, Fishlev G, Bergan J, Efrati S. Seizures during hyperbaric oxygen therapy: retrospective analysis of 62,614 treatment sessions. Undersea Hyperb Med. 2016;43(1):21-28.
11. Liu Z, Xiong T, Meads C. Clinical effectiveness of treatment with hyperbaric oxygen for neonatal hypoxic-ischaemic encephalopathy: systematic review of Chinese literature. BMJ. 2006;333(7564):374.
12. Bond KM, Brinjikji W, Eckel LJ, Kallmes DF, McDonald RJ, Carr CM. Dentate update: imaging features of entities that affect the dentate nucleus. AJNR Am J Neuroradiol. 2017;38(8):1467-1474.
13. Agarwal A, Kanekar S, Sabat S, Thamburaj K. Metronidazole-induced cerebellar toxicity. Neurol Int. 2016;8(1):6365.
14. Zuccoli G, Pipitone N. Neuroimaging findings in acute Wernicke’s encephalopathy: review of the literature. AJR Am J Roentgenol. 2009;192(2):501-508.
15. Jung YC, Chanraud S, Sullivan EV. Neuroimaging of Wernicke’s encephalopathy and Korsakoff’s syndrome. Neuropsychol Rev. 2012;22(2):170-180.
16. Hobson-Webb LD, Roach ES, Donofrio PD. Metronidazole: newly recognized cause of autonomic neuropathy. J Child Neurol. 2006;21(5):429-431.
17. Nath Chaurasia R. Rapid onset metronidazole induced sensory neuropathy: case series and review of literature. Int J Neurorehabilitation. 2015;02:152.
18. Bradley WG, Karlsson IJ, Rassol CG. Metronidazole neuropathy. Br Med J. 1977;2(6087):610-611.
19. Evans J, Levesque D, Knowles K, Longshore R, Plummer S. Diazepam as a treatment for metronidazole toxicosis in dogs: a retrospective study of 21 cases. J Vet Intern Med. 2003;17(3):304-310.
20. Farmakiotis D, Zeluff B. Images in clinical medicine. Metronidazole-associated encephalopathy. N Engl J Med. 2016;374(15):1465.
21. Hobbs K, Stern-Nezer S, Buckwalter MS, Fischbein N, Finley Caulfield A. Metronidazole-induced encephalopathy: not always a reversible situation. Neurocrit Care. 2015;22(3):429-436.