User login
An evidence-based approach to treating pediatric anxiety disorders
Anxiety disorders are remarkably common among pediatric patients1,2 and are associated with significant morbidity3 and increased risk of suicidality in adolescents.4,5 Effective diagnosis and treatment of pediatric anxiety disorders are critical for reducing psychosocial morbidity,3,6 suicidality, and the risk of secondary mood disorders.7
This article summarizes open-label studies and randomized controlled trials (RCTs) of selective serotonin reuptake inhibitors (SSRIs), selective serotonin-norepinephrine reuptake inhibitors, atypical anxiolytics, and benzodiazepines in children and adolescents with generalized anxiety disorder (GAD), social phobia, separation anxiety disorder, and panic disorder. Although we focus on psychopharmacologic treatments, the best outcomes generally are observed with multimodal treatments that combine psychotherapy and pharmacotherapy.
Generalized anxiety disorder
Researchers have evaluated SSRIs, benzodiazepines, and buspirone in pediatric patients with GAD. In a double-blind, placebo-controlled trial of 22 patients age 5 to 17, sertraline, 50 mg/d, was associated with improvement in Hamilton Anxiety Rating Scale (HAM-A), Clinical Global Impression-Severity (CGI-S), and Clinical Global Impression-Improvement (CGI-I) scores over 9 weeks.8 The Child-Adolescent Anxiety Multimodal Study compared cognitive-behavioral therapy (CBT) to sertraline or sertraline plus CBT in 488 patients age 7 to 17, 78% of whom had GAD.9 Sertraline monotherapy was superior to placebo and not statistically different from CBT, while combination treatment was superior to both monotherapy conditions in improving CGI score. In both trials, sertraline was well tolerated.
One study evaluated fluoxetine, 5 to 40 mg/d, or CBT in 14 youths with GAD; both treatments improved symptoms.10 In a study of 320 GAD patients age 6 to 17, venlafaxine extended-release (XR) initiated at 37.5 mg/d was associated with improved HAM-A scores.11 In general, venlafaxine was well tolerated; adverse effects included increased blood pressure, asthenia, pain, anorexia, somnolence, weight loss, and possibly treatment-emergent suicidal ideation.
Two RCTs of buspirone, 15 to 60 mg/d, that evaluated 559 children and adolescents age 6 to 17 with GAD did not observe significant differences between buspirone and placebo.12 By contrast, 2 open-label studies of youths with anxiety suggested improvement associated with buspirone.12 Treatment-emergent adverse events included nausea, stomachache, and headache.
Clinical trials of benzodiazepines in anxious children and adolescents have yielded mixed results. A 4-week, open-label trial of alprazolam, 0.5 mg to 1.5 mg/d, in 12 adolescents with overanxious disorder—the DSM-III forerunner of GAD—found improvements in anxiety, depression, psychomotor excitation, and hyperactivity, but patients experienced sedation, activation, headache, and nausea.13 However, a double-blind RCT in 30 youths age 8 to 16 found no statistically significant difference between alprazolam and placebo.14 Alprazolam generally was well tolerated; fatigue and dry mouth were reported, but no withdrawal symptoms. Additionally, benzodiazepine use may be associated with tolerance and—in young children—disinhibition.
Social phobia
Researchers have evaluated paroxetine, citalopram, fluoxetine, and venlafaxine for treating social phobia in pediatric patients. In an RCT, 78% of paroxetine-treated patients with social phobia responded compared with 38% for placebo over 16 weeks. Adverse events—including withdrawal symptoms—were twice as likely in patients who received paroxetine. Additionally, 4 paroxetine patients exhibited suicidal ideation vs 0 patients who received placebo.15
In an RCT of 293 children and adolescents age 8 to 17 with social phobia, venlafaxine XR was initiated at 37.5 mg/d and titrated to 112.5 mg/d, 150 mg/d, or 225 mg/d, depending on body weight.16 The venlafaxine group experienced significantly improved anxiety symptoms and the medication generally was well tolerated, although 3 venlafaxine-treated patients developed suicidal ideation compared with 0 in the placebo group.
An RCT compared Social Effectiveness Therapy for Children (SET-C) and fluoxetine, 10 to 40 mg/d, for 139 patients age 7 to 17 with social phobia.17 SET-C is a CBT for children and adolescents that focuses on increasing interpersonal skills and becoming more comfortable in social situations; it involves psychoeducation, social skills training, and exposure exercises. At endpoint, 53% of patients in the SET-C group no longer met diagnostic criteria for social phobia. Fluoxetine was well tolerated; no severe adverse events were reported.
In an open-label study of sertraline (mean dose = 123 mg/d) for 14 young persons with social phobia, 36% of patients responded and 29% partially responded at 8 weeks.18 Adverse events generally were mild and included nausea, diarrhea, and headache. In a 12-week study, 12 pediatric patients with social phobia received citalopram, 10 to 40 mg/d, and eight 15-minute counseling sessions. At endpoint, clinicians rated 83% of patients as much improved or very much improved. The medication generally was well tolerated.19
Separation anxiety disorder
In a 4-week, double-blind crossover pilot study, researchers randomly assigned 15 children age 7 to 13 with separation anxiety disorder to clonazepam, up to 2 mg/d, or placebo.20 There was no significant difference in CGI-I score between clonazepam and placebo. Side effects—including drowsiness, irritability and “oppositional behavior”—were more frequent in patients treated with clonazepam.
Panic disorder
Only 2 open-label studies of SSRIs have been conducted in pediatric patients with panic disorder. The first evaluated the effectiveness and tolerability of fluoxetine, sertraline, or paroxetine over 6 months in 12 patients; 67% no longer met criteria for panic disorder at endpoint.21 In this study, benzodiazepines—including clonazepam and lorazepam—were used in 67% of patients at the start of SSRI treatment. The authors suggested this strategy may be clinically useful for patients with panic disorder.
In the second study, Fairbanks et al22 examined the use of fluoxetine for 6 to 9 weeks in 16 outpatients with mixed anxiety disorders who did not respond to psychotherapy. Patients age ≤12 were given 5 to 40 mg/d and those age ≥13 received 5 to 80 mg/d. Fluoxetine was associated with clinically significant improvement in 3 of the 5 patients who had panic disorder. Although overall fluoxetine was well tolerated, drowsiness, dyssomnia, decreased appetite, nausea, and abdominal pain were the most common side effects. Fluoxetine was not associated with suicidal ideation.
Mixed anxiety disorders
Most trials of pediatric anxiety have evaluated patients with “mixed anxiety disorders” because GAD, social phobia, and separation anxiety disorder are highly comorbid and share diagnostic features (Figure 1).9 An RCT of fluvoxamine, up to 300 mg/d, in 128 pediatric patients with ≥1 anxiety disorders found significant differences in CGI-I and endpoint Pediatric Anxiety Rating Scale (PARS) scores.23 Fluvoxamine was well tolerated but associated with increased motor activity and abdominal discomfort compared with placebo.
Two open-label trials of pediatric patients with mixed anxiety disorders suggested fluoxetine may be beneficial. Fairbanks et al22 documented clinical improvement in 10 of 10 patients with separation anxiety disorder, 8 of 10 with social phobia, 4 of 6 with specific phobia, 3 of 5 with panic disorder, and 1 of 7 with GAD. Birmaher et al24 evaluated 21 pediatric patients with overanxious disorder, social phobia, or separation anxiety who had not responded to psychotherapy and were not depressed; all patients received flexibly-dosed fluoxetine for up to 10 months. Fluoxetine was well tolerated and 81% of patients improved.
Finally, in a 12-week RCT of 74 patients age 7 to 17 with GAD, separation anxiety disorder, and/or social phobia, fluoxetine, 10 to 20 mg/d, was associated with improved scores on the Screen for Anxiety Related Emotional Disorders, PARS, CGI-I, CGI-S, and Children’s Global Assessment Scale.25 A follow-up open-label trial suggested that maintenance treatment is associated with sustained improvement.26
Figure 1: The pediatric anxiety disorders triad: Comorbidity is common
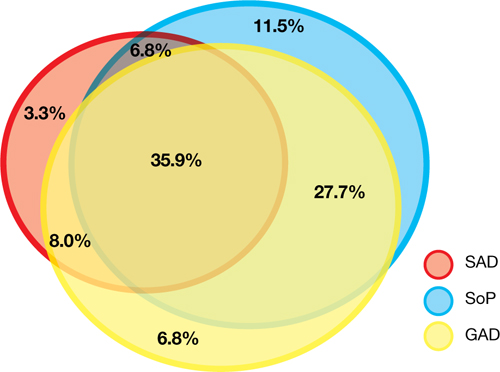
In the Child-Adolescent Multimodal Treatment Study, GAD was the most common disorder; however, GAD, SAD, and SoP were highly comorbid
GAD: generalized anxiety disorder; SAD: separation anxiety disorder; SoP: social phobia
Source: Reference 9
Anxiety disorders with ADHD
Anxiety disorders often are comorbid with attention-deficit/hyperactivity disorder (ADHD). An RCT of patients age 8 to 17 with ADHD and comorbid anxiety found that atomoxetine was associated with improved PARS scores and ADHD symptoms.27 The target dose was 1.2 mg/kg/d. Atomoxetine was well-tolerated; decreased appetite was the only significant adverse event in the treatment group vs placebo.
Multimodal treatment
Although this article reviews evidence for psychopharmacologic treatments, psychotherapeutic treatment of young patients with anxiety disorders has seen significant advances.28 Most psychotherapy studies have evaluated the efficacy of CBT,29-31 although there is evidence for psychodynamic therapy and interpersonal therapy.32 The American Academy of Child & Adolescent Psychiatry recommends a multimodal treatment approach because combination treatment appears to be more effective than monotherapy.8,28,33 Also, clinicians who treat pediatric patients who have an anxiety disorder should evaluate the family’s role on anxiety symptoms and may consider family therapy.
Treatment considerations
Evidence supports the efficacy of sertraline, citalopram, paroxetine, fluvoxamine, fluoxetine, and venlafaxine for treating children and adolescents with anxiety disorders (Figure 2).8,9,11,15,16,23,25 Some practitioners suggest using differing dosing strategies for pediatric anxiety disorders compared with those used to treat adults (Table).34 When considering SSRIs for children and adolescents, keep in mind the “black-box” warning regarding suicidality in these patients. Carefully monitor patients for treatment-emergent suicidality and routinely reassess for the presence and severity of suicidal ideation and suicide risk.
Figure 2: Number needed to treat for SSRIs and SNRIs in pediatric anxiety disorders

GAD: generalized anxiety disorder; RUPP: Research Unit on Pediatric Psychopharmacology; SAD: separation anxiety disorder; SNRI: serotonin-norepinephrine reuptake inhibitor; SoP: social phobia; SSRI: selective serotonin reuptake inhibitorTable
Practical dosing of SSRIs and SNRIs in pediatric patients with anxietya
| Medication | Initial child dose (age <12; mg/d) | Initial adolescent dose (age 12 to 17; mg/d) | Target dose (mg/d) |
|---|---|---|---|
| Citalopram | 5 to 10 | 10 | 20 to 40 |
| Escitalopram | 2.5 to 5 | 5 to 10 | 10 to 20 |
| Fluoxetineb | 10 | 20 | 20 to 40 (children), 40 to 60 (adolescents) |
| Paroxetineb | 5 to 10 | 10 | 20 |
| Sertralinec | 10 to 12.5 | 25 | 150 |
| Venlafaxine | 37.5 | 37.5 | 150 |
| aGeneralized anxiety disorder, social phobia, and separation anxiety disorder bMay consider cytochrome P450 genotyping for 2D6, which may suggest an alternate dosing strategy cSertraline is available in a liquid formulation (20 mg/mL) SNRI: serotonin-norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor Source: Adapted from reference 34 | |||
Related Resources
- Connolly SD, Bernstein GA; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
- Anxiety and Depression Association of America. www.adaa.org.
- American Academy of Child & Adolescent Psychiatry. www.aacap.org.
Drug Brand Names
- Alprazolam • Xanax
- Atomoxetine • Strattera
- Buspirone • BuSpar
- Citalopram • Celexa
- Clonazepam • Klonopin
- Fluoxetine • Prozac
- Fluvoxamine • Luvox, Luvox CR
- Lorazepam • Ativan
- Paroxetine • Paxil, Paxil CR
- Sertraline • Zoloft
- Venlafaxine • Effexor, Effexor XR
Disclosures
Dr. Strawn has received research support from the American Academy of Child & Adolescent Psychiatry, Eli Lilly and Company, and Shire, and is an employee of the University of Cincinnati, Cincinnati, OH.
Dr. McReynolds was employed by Eli Lilly and Company from 1997 to 2005.
1. Beesdo K, Knappe S, Pine DS. Anxiety and anxiety disorders in children and adolescents: developmental issues and implications for DSM-V. Psychiatr Clin North Am. 2009;32(3):483-524.
2. Beesdo K, Pine DS, Lieb R, et al. Incidence and risk patterns of anxiety and depressive disorders and categorization of generalized anxiety disorder. Arch Gen Psychiatry. 2010;67(1):47-57.
3. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first grade children: prediction to anxious symptoms and adaptive functioning in fifth grade. J Child Psychol Psychiatry. 1995;36(3):427-437.
4. Foley DL, Goldston DB, Costello EJ, et al. Proximal psychiatric risk factors for suicidality in youth: the Great Smoky Mountains Study. Arch Gen Psychiatry. 2006;63(9):1017-1024.
5. Jacobson CM, Muehlenkamp JJ, Miller AL, et al. Psychiatric impairment among adolescents engaging in different types of deliberate self-harm. J Clin Child Adolesc Psychol. 2008;37(2):363-375.
6. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first-grade children. J Abnorm Child Psychol. 1994;22(4):441-455.
7. Pine DS, Cohen P, Gurley D, et al. The risk for early-adulthood anxiety and depressive disorders in adolescents with anxiety and depressive disorders. Arch Gen Psychiatry. 1998;55(1):56-64.
8. Rynn MA, Siqueland L, Rickels K. Placebo-controlled trial of sertraline in the treatment of children with generalized anxiety disorders. Am J Psychiatry. 2001;158(12):2008-2014.
9. Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359(26):2753-2766.
10. Maslowsky J, Mogg K, Bradley BP, et al. A preliminary investigation of neural correlates of treatment in adolescents with generalized anxiety disorder. J Child Adolesc Psychopharmacol. 2010;20(2):105-111.
11. Rynn MA, Riddle MA, Yeung PP, et al. Efficacy and safety of extended-release venlafaxine in the treatment of generalized anxiety disorder in children and adolescents: two placebo-controlled trials. Am J Psychiatry. 2007;164(2):290-300.
12. BuSpar [package insert] Princeton NJ: Bristol-Myers Squibb; 2010.
13. Simeon JG, Ferguson HB. Alprazolam effects in children with anxiety disorders. Can J Psychiatry. 1987;32(7):570-574.
14. Simeon JG, Ferguson HB, Knott V, et al. Clinical, cognitive, and neurophysiological effects of alprazolam in children and adolescents with overanxious and avoidant disorders. J Am Acad Child Adolesc Psychiatry. 1992;31(1):29-33.
15. Wagner KD, Berard R, Stein MB, et al. A multicenter, randomized, double-blind, placebo-controlled trial of paroxetine in children and adolescents with social anxiety disorder. Arch Gen Psychiatry. 2004;61(11):1153-1162.
16. March JS, Entusah AR, Rynn M, et al. A randomized controlled trial of venlafaxine ER versus placebo in pediatric social anxiety disorder. Biol Psychiatry. 2007;62(10):1149-1154.
17. Beidel DC, Turner SM, Sallee FR, et al. SET-C versus fluoxetine in the treatment of childhood social phobia. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1622-1632.
18. Compton SN, Grant PJ, Chrisman AK, et al. Sertraline in children and adolescents with social anxiety disorder: an open trial. J Am Acad Child Adolesc Psychiatry. 2001;40(5):564-571.
19. Chavira DA, Stein MB. Combined psychoeducation and treatment with selective serotonin reuptake inhibitors for youth with generalized social anxiety disorder. J Child Adolesc Psychopharmacol. 2002;12(1):47-54.
20. Graae F, Milner J, Rizzotto L, et al. Clonazepam in childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(3):372-376.
21. Renaud J, Birmaher B, Wassick SC, et al. Use of selective serotonin reuptake inhibitors for the treatment of childhood panic disorder: a pilot study. J Child Adolesc Psychopharmacol. 1999;9(2):73-83.
22. Fairbanks JM, Pine DS, Tancer NK, et al. Open fluoxetine treatment of mixed anxiety disorders in children and adolescents. J Child Adolesc Psychopharmacol. 1997;7(1):17-29.
23. The Research Unit on Pediatric Psychopharmacology Anxiety Study Group. Fluvoxamine for the treatment of anxiety disorders in children and adolescents. N Engl J Med. 2001;344(17):1279-1285.
24. Birmaher B, Waterman GS, Ryan N, et al. Fluoxetine for childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(7):993-999.
25. Birmaher B, Axelson DA, Monk K, et al. Fluoxetine for the treatment of childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2003;42(4):415-423.
26. Clark DB, Birmaher B, Axelson D, et al. Fluoxetine for the treatment of childhood anxiety disorders: open-label, long-term extension to a controlled trial. J Am Acad Child Adolesc Psychiatry. 2005;44(12):1263-1270.
27. Geller D, Donnelly C, Lopez F, et al. Atomoxetine treatment for pediatric patients with attention-deficit/hyperactivity disorder with comorbid anxiety disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(9):1119-1127.
28. Connolly SD, Bernstein GA. Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
29. Kendall PC. Treating anxiety disorders in children: results of a randomized clinical trial. J Consult Clin Psychol. 1994;62(1):100-110.
30. Kendall PC, Flannery-Schroeder E, Panichelli-Mindel SM, et al. Therapy for youths with anxiety disorders: a second randomized clinical trial. J Consult Clin Psychol. 1997;65(3):366-380.
31. Reynolds S, Wilson C, Austin J, et al. Effects of psychotherapy for anxiety in children and adolescents: a meta-analytic review. Clin Psychol Rev. 2012;32(4):251-262.
32. Strawn JR, Wehry AM, DelBello MP, et al. Establishing the neurobiologic basis of treatment in children and adolescents with generalized anxiety disorder. Depress Anxiety. 2012;29(4):328-339.
33. Ginsburg GS, Kendall PC, Sakolsky D, et al. Remission after acute treatment in children and adolescents with anxiety disorders: findings from the CAMS. J Consult Clin Psychol. 2011;79(6):806-813.
34. Findling RL, Kowatch RA. How (not) to dose antidepressants and antipsychotics for children. Current Psychiatry. 2007;6(6):79-83.
Anxiety disorders are remarkably common among pediatric patients1,2 and are associated with significant morbidity3 and increased risk of suicidality in adolescents.4,5 Effective diagnosis and treatment of pediatric anxiety disorders are critical for reducing psychosocial morbidity,3,6 suicidality, and the risk of secondary mood disorders.7
This article summarizes open-label studies and randomized controlled trials (RCTs) of selective serotonin reuptake inhibitors (SSRIs), selective serotonin-norepinephrine reuptake inhibitors, atypical anxiolytics, and benzodiazepines in children and adolescents with generalized anxiety disorder (GAD), social phobia, separation anxiety disorder, and panic disorder. Although we focus on psychopharmacologic treatments, the best outcomes generally are observed with multimodal treatments that combine psychotherapy and pharmacotherapy.
Generalized anxiety disorder
Researchers have evaluated SSRIs, benzodiazepines, and buspirone in pediatric patients with GAD. In a double-blind, placebo-controlled trial of 22 patients age 5 to 17, sertraline, 50 mg/d, was associated with improvement in Hamilton Anxiety Rating Scale (HAM-A), Clinical Global Impression-Severity (CGI-S), and Clinical Global Impression-Improvement (CGI-I) scores over 9 weeks.8 The Child-Adolescent Anxiety Multimodal Study compared cognitive-behavioral therapy (CBT) to sertraline or sertraline plus CBT in 488 patients age 7 to 17, 78% of whom had GAD.9 Sertraline monotherapy was superior to placebo and not statistically different from CBT, while combination treatment was superior to both monotherapy conditions in improving CGI score. In both trials, sertraline was well tolerated.
One study evaluated fluoxetine, 5 to 40 mg/d, or CBT in 14 youths with GAD; both treatments improved symptoms.10 In a study of 320 GAD patients age 6 to 17, venlafaxine extended-release (XR) initiated at 37.5 mg/d was associated with improved HAM-A scores.11 In general, venlafaxine was well tolerated; adverse effects included increased blood pressure, asthenia, pain, anorexia, somnolence, weight loss, and possibly treatment-emergent suicidal ideation.
Two RCTs of buspirone, 15 to 60 mg/d, that evaluated 559 children and adolescents age 6 to 17 with GAD did not observe significant differences between buspirone and placebo.12 By contrast, 2 open-label studies of youths with anxiety suggested improvement associated with buspirone.12 Treatment-emergent adverse events included nausea, stomachache, and headache.
Clinical trials of benzodiazepines in anxious children and adolescents have yielded mixed results. A 4-week, open-label trial of alprazolam, 0.5 mg to 1.5 mg/d, in 12 adolescents with overanxious disorder—the DSM-III forerunner of GAD—found improvements in anxiety, depression, psychomotor excitation, and hyperactivity, but patients experienced sedation, activation, headache, and nausea.13 However, a double-blind RCT in 30 youths age 8 to 16 found no statistically significant difference between alprazolam and placebo.14 Alprazolam generally was well tolerated; fatigue and dry mouth were reported, but no withdrawal symptoms. Additionally, benzodiazepine use may be associated with tolerance and—in young children—disinhibition.
Social phobia
Researchers have evaluated paroxetine, citalopram, fluoxetine, and venlafaxine for treating social phobia in pediatric patients. In an RCT, 78% of paroxetine-treated patients with social phobia responded compared with 38% for placebo over 16 weeks. Adverse events—including withdrawal symptoms—were twice as likely in patients who received paroxetine. Additionally, 4 paroxetine patients exhibited suicidal ideation vs 0 patients who received placebo.15
In an RCT of 293 children and adolescents age 8 to 17 with social phobia, venlafaxine XR was initiated at 37.5 mg/d and titrated to 112.5 mg/d, 150 mg/d, or 225 mg/d, depending on body weight.16 The venlafaxine group experienced significantly improved anxiety symptoms and the medication generally was well tolerated, although 3 venlafaxine-treated patients developed suicidal ideation compared with 0 in the placebo group.
An RCT compared Social Effectiveness Therapy for Children (SET-C) and fluoxetine, 10 to 40 mg/d, for 139 patients age 7 to 17 with social phobia.17 SET-C is a CBT for children and adolescents that focuses on increasing interpersonal skills and becoming more comfortable in social situations; it involves psychoeducation, social skills training, and exposure exercises. At endpoint, 53% of patients in the SET-C group no longer met diagnostic criteria for social phobia. Fluoxetine was well tolerated; no severe adverse events were reported.
In an open-label study of sertraline (mean dose = 123 mg/d) for 14 young persons with social phobia, 36% of patients responded and 29% partially responded at 8 weeks.18 Adverse events generally were mild and included nausea, diarrhea, and headache. In a 12-week study, 12 pediatric patients with social phobia received citalopram, 10 to 40 mg/d, and eight 15-minute counseling sessions. At endpoint, clinicians rated 83% of patients as much improved or very much improved. The medication generally was well tolerated.19
Separation anxiety disorder
In a 4-week, double-blind crossover pilot study, researchers randomly assigned 15 children age 7 to 13 with separation anxiety disorder to clonazepam, up to 2 mg/d, or placebo.20 There was no significant difference in CGI-I score between clonazepam and placebo. Side effects—including drowsiness, irritability and “oppositional behavior”—were more frequent in patients treated with clonazepam.
Panic disorder
Only 2 open-label studies of SSRIs have been conducted in pediatric patients with panic disorder. The first evaluated the effectiveness and tolerability of fluoxetine, sertraline, or paroxetine over 6 months in 12 patients; 67% no longer met criteria for panic disorder at endpoint.21 In this study, benzodiazepines—including clonazepam and lorazepam—were used in 67% of patients at the start of SSRI treatment. The authors suggested this strategy may be clinically useful for patients with panic disorder.
In the second study, Fairbanks et al22 examined the use of fluoxetine for 6 to 9 weeks in 16 outpatients with mixed anxiety disorders who did not respond to psychotherapy. Patients age ≤12 were given 5 to 40 mg/d and those age ≥13 received 5 to 80 mg/d. Fluoxetine was associated with clinically significant improvement in 3 of the 5 patients who had panic disorder. Although overall fluoxetine was well tolerated, drowsiness, dyssomnia, decreased appetite, nausea, and abdominal pain were the most common side effects. Fluoxetine was not associated with suicidal ideation.
Mixed anxiety disorders
Most trials of pediatric anxiety have evaluated patients with “mixed anxiety disorders” because GAD, social phobia, and separation anxiety disorder are highly comorbid and share diagnostic features (Figure 1).9 An RCT of fluvoxamine, up to 300 mg/d, in 128 pediatric patients with ≥1 anxiety disorders found significant differences in CGI-I and endpoint Pediatric Anxiety Rating Scale (PARS) scores.23 Fluvoxamine was well tolerated but associated with increased motor activity and abdominal discomfort compared with placebo.
Two open-label trials of pediatric patients with mixed anxiety disorders suggested fluoxetine may be beneficial. Fairbanks et al22 documented clinical improvement in 10 of 10 patients with separation anxiety disorder, 8 of 10 with social phobia, 4 of 6 with specific phobia, 3 of 5 with panic disorder, and 1 of 7 with GAD. Birmaher et al24 evaluated 21 pediatric patients with overanxious disorder, social phobia, or separation anxiety who had not responded to psychotherapy and were not depressed; all patients received flexibly-dosed fluoxetine for up to 10 months. Fluoxetine was well tolerated and 81% of patients improved.
Finally, in a 12-week RCT of 74 patients age 7 to 17 with GAD, separation anxiety disorder, and/or social phobia, fluoxetine, 10 to 20 mg/d, was associated with improved scores on the Screen for Anxiety Related Emotional Disorders, PARS, CGI-I, CGI-S, and Children’s Global Assessment Scale.25 A follow-up open-label trial suggested that maintenance treatment is associated with sustained improvement.26
Figure 1: The pediatric anxiety disorders triad: Comorbidity is common
In the Child-Adolescent Multimodal Treatment Study, GAD was the most common disorder; however, GAD, SAD, and SoP were highly comorbid
GAD: generalized anxiety disorder; SAD: separation anxiety disorder; SoP: social phobia
Source: Reference 9
Anxiety disorders with ADHD
Anxiety disorders often are comorbid with attention-deficit/hyperactivity disorder (ADHD). An RCT of patients age 8 to 17 with ADHD and comorbid anxiety found that atomoxetine was associated with improved PARS scores and ADHD symptoms.27 The target dose was 1.2 mg/kg/d. Atomoxetine was well-tolerated; decreased appetite was the only significant adverse event in the treatment group vs placebo.
Multimodal treatment
Although this article reviews evidence for psychopharmacologic treatments, psychotherapeutic treatment of young patients with anxiety disorders has seen significant advances.28 Most psychotherapy studies have evaluated the efficacy of CBT,29-31 although there is evidence for psychodynamic therapy and interpersonal therapy.32 The American Academy of Child & Adolescent Psychiatry recommends a multimodal treatment approach because combination treatment appears to be more effective than monotherapy.8,28,33 Also, clinicians who treat pediatric patients who have an anxiety disorder should evaluate the family’s role on anxiety symptoms and may consider family therapy.
Treatment considerations
Evidence supports the efficacy of sertraline, citalopram, paroxetine, fluvoxamine, fluoxetine, and venlafaxine for treating children and adolescents with anxiety disorders (Figure 2).8,9,11,15,16,23,25 Some practitioners suggest using differing dosing strategies for pediatric anxiety disorders compared with those used to treat adults (Table).34 When considering SSRIs for children and adolescents, keep in mind the “black-box” warning regarding suicidality in these patients. Carefully monitor patients for treatment-emergent suicidality and routinely reassess for the presence and severity of suicidal ideation and suicide risk.
Figure 2: Number needed to treat for SSRIs and SNRIs in pediatric anxiety disorders
GAD: generalized anxiety disorder; RUPP: Research Unit on Pediatric Psychopharmacology; SAD: separation anxiety disorder; SNRI: serotonin-norepinephrine reuptake inhibitor; SoP: social phobia; SSRI: selective serotonin reuptake inhibitorTable
Practical dosing of SSRIs and SNRIs in pediatric patients with anxietya
| Medication | Initial child dose (age <12; mg/d) | Initial adolescent dose (age 12 to 17; mg/d) | Target dose (mg/d) |
|---|---|---|---|
| Citalopram | 5 to 10 | 10 | 20 to 40 |
| Escitalopram | 2.5 to 5 | 5 to 10 | 10 to 20 |
| Fluoxetineb | 10 | 20 | 20 to 40 (children), 40 to 60 (adolescents) |
| Paroxetineb | 5 to 10 | 10 | 20 |
| Sertralinec | 10 to 12.5 | 25 | 150 |
| Venlafaxine | 37.5 | 37.5 | 150 |
| aGeneralized anxiety disorder, social phobia, and separation anxiety disorder bMay consider cytochrome P450 genotyping for 2D6, which may suggest an alternate dosing strategy cSertraline is available in a liquid formulation (20 mg/mL) SNRI: serotonin-norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor Source: Adapted from reference 34 | |||
Related Resources
- Connolly SD, Bernstein GA; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
- Anxiety and Depression Association of America. www.adaa.org.
- American Academy of Child & Adolescent Psychiatry. www.aacap.org.
Drug Brand Names
- Alprazolam • Xanax
- Atomoxetine • Strattera
- Buspirone • BuSpar
- Citalopram • Celexa
- Clonazepam • Klonopin
- Fluoxetine • Prozac
- Fluvoxamine • Luvox, Luvox CR
- Lorazepam • Ativan
- Paroxetine • Paxil, Paxil CR
- Sertraline • Zoloft
- Venlafaxine • Effexor, Effexor XR
Disclosures
Dr. Strawn has received research support from the American Academy of Child & Adolescent Psychiatry, Eli Lilly and Company, and Shire, and is an employee of the University of Cincinnati, Cincinnati, OH.
Dr. McReynolds was employed by Eli Lilly and Company from 1997 to 2005.
Anxiety disorders are remarkably common among pediatric patients1,2 and are associated with significant morbidity3 and increased risk of suicidality in adolescents.4,5 Effective diagnosis and treatment of pediatric anxiety disorders are critical for reducing psychosocial morbidity,3,6 suicidality, and the risk of secondary mood disorders.7
This article summarizes open-label studies and randomized controlled trials (RCTs) of selective serotonin reuptake inhibitors (SSRIs), selective serotonin-norepinephrine reuptake inhibitors, atypical anxiolytics, and benzodiazepines in children and adolescents with generalized anxiety disorder (GAD), social phobia, separation anxiety disorder, and panic disorder. Although we focus on psychopharmacologic treatments, the best outcomes generally are observed with multimodal treatments that combine psychotherapy and pharmacotherapy.
Generalized anxiety disorder
Researchers have evaluated SSRIs, benzodiazepines, and buspirone in pediatric patients with GAD. In a double-blind, placebo-controlled trial of 22 patients age 5 to 17, sertraline, 50 mg/d, was associated with improvement in Hamilton Anxiety Rating Scale (HAM-A), Clinical Global Impression-Severity (CGI-S), and Clinical Global Impression-Improvement (CGI-I) scores over 9 weeks.8 The Child-Adolescent Anxiety Multimodal Study compared cognitive-behavioral therapy (CBT) to sertraline or sertraline plus CBT in 488 patients age 7 to 17, 78% of whom had GAD.9 Sertraline monotherapy was superior to placebo and not statistically different from CBT, while combination treatment was superior to both monotherapy conditions in improving CGI score. In both trials, sertraline was well tolerated.
One study evaluated fluoxetine, 5 to 40 mg/d, or CBT in 14 youths with GAD; both treatments improved symptoms.10 In a study of 320 GAD patients age 6 to 17, venlafaxine extended-release (XR) initiated at 37.5 mg/d was associated with improved HAM-A scores.11 In general, venlafaxine was well tolerated; adverse effects included increased blood pressure, asthenia, pain, anorexia, somnolence, weight loss, and possibly treatment-emergent suicidal ideation.
Two RCTs of buspirone, 15 to 60 mg/d, that evaluated 559 children and adolescents age 6 to 17 with GAD did not observe significant differences between buspirone and placebo.12 By contrast, 2 open-label studies of youths with anxiety suggested improvement associated with buspirone.12 Treatment-emergent adverse events included nausea, stomachache, and headache.
Clinical trials of benzodiazepines in anxious children and adolescents have yielded mixed results. A 4-week, open-label trial of alprazolam, 0.5 mg to 1.5 mg/d, in 12 adolescents with overanxious disorder—the DSM-III forerunner of GAD—found improvements in anxiety, depression, psychomotor excitation, and hyperactivity, but patients experienced sedation, activation, headache, and nausea.13 However, a double-blind RCT in 30 youths age 8 to 16 found no statistically significant difference between alprazolam and placebo.14 Alprazolam generally was well tolerated; fatigue and dry mouth were reported, but no withdrawal symptoms. Additionally, benzodiazepine use may be associated with tolerance and—in young children—disinhibition.
Social phobia
Researchers have evaluated paroxetine, citalopram, fluoxetine, and venlafaxine for treating social phobia in pediatric patients. In an RCT, 78% of paroxetine-treated patients with social phobia responded compared with 38% for placebo over 16 weeks. Adverse events—including withdrawal symptoms—were twice as likely in patients who received paroxetine. Additionally, 4 paroxetine patients exhibited suicidal ideation vs 0 patients who received placebo.15
In an RCT of 293 children and adolescents age 8 to 17 with social phobia, venlafaxine XR was initiated at 37.5 mg/d and titrated to 112.5 mg/d, 150 mg/d, or 225 mg/d, depending on body weight.16 The venlafaxine group experienced significantly improved anxiety symptoms and the medication generally was well tolerated, although 3 venlafaxine-treated patients developed suicidal ideation compared with 0 in the placebo group.
An RCT compared Social Effectiveness Therapy for Children (SET-C) and fluoxetine, 10 to 40 mg/d, for 139 patients age 7 to 17 with social phobia.17 SET-C is a CBT for children and adolescents that focuses on increasing interpersonal skills and becoming more comfortable in social situations; it involves psychoeducation, social skills training, and exposure exercises. At endpoint, 53% of patients in the SET-C group no longer met diagnostic criteria for social phobia. Fluoxetine was well tolerated; no severe adverse events were reported.
In an open-label study of sertraline (mean dose = 123 mg/d) for 14 young persons with social phobia, 36% of patients responded and 29% partially responded at 8 weeks.18 Adverse events generally were mild and included nausea, diarrhea, and headache. In a 12-week study, 12 pediatric patients with social phobia received citalopram, 10 to 40 mg/d, and eight 15-minute counseling sessions. At endpoint, clinicians rated 83% of patients as much improved or very much improved. The medication generally was well tolerated.19
Separation anxiety disorder
In a 4-week, double-blind crossover pilot study, researchers randomly assigned 15 children age 7 to 13 with separation anxiety disorder to clonazepam, up to 2 mg/d, or placebo.20 There was no significant difference in CGI-I score between clonazepam and placebo. Side effects—including drowsiness, irritability and “oppositional behavior”—were more frequent in patients treated with clonazepam.
Panic disorder
Only 2 open-label studies of SSRIs have been conducted in pediatric patients with panic disorder. The first evaluated the effectiveness and tolerability of fluoxetine, sertraline, or paroxetine over 6 months in 12 patients; 67% no longer met criteria for panic disorder at endpoint.21 In this study, benzodiazepines—including clonazepam and lorazepam—were used in 67% of patients at the start of SSRI treatment. The authors suggested this strategy may be clinically useful for patients with panic disorder.
In the second study, Fairbanks et al22 examined the use of fluoxetine for 6 to 9 weeks in 16 outpatients with mixed anxiety disorders who did not respond to psychotherapy. Patients age ≤12 were given 5 to 40 mg/d and those age ≥13 received 5 to 80 mg/d. Fluoxetine was associated with clinically significant improvement in 3 of the 5 patients who had panic disorder. Although overall fluoxetine was well tolerated, drowsiness, dyssomnia, decreased appetite, nausea, and abdominal pain were the most common side effects. Fluoxetine was not associated with suicidal ideation.
Mixed anxiety disorders
Most trials of pediatric anxiety have evaluated patients with “mixed anxiety disorders” because GAD, social phobia, and separation anxiety disorder are highly comorbid and share diagnostic features (Figure 1).9 An RCT of fluvoxamine, up to 300 mg/d, in 128 pediatric patients with ≥1 anxiety disorders found significant differences in CGI-I and endpoint Pediatric Anxiety Rating Scale (PARS) scores.23 Fluvoxamine was well tolerated but associated with increased motor activity and abdominal discomfort compared with placebo.
Two open-label trials of pediatric patients with mixed anxiety disorders suggested fluoxetine may be beneficial. Fairbanks et al22 documented clinical improvement in 10 of 10 patients with separation anxiety disorder, 8 of 10 with social phobia, 4 of 6 with specific phobia, 3 of 5 with panic disorder, and 1 of 7 with GAD. Birmaher et al24 evaluated 21 pediatric patients with overanxious disorder, social phobia, or separation anxiety who had not responded to psychotherapy and were not depressed; all patients received flexibly-dosed fluoxetine for up to 10 months. Fluoxetine was well tolerated and 81% of patients improved.
Finally, in a 12-week RCT of 74 patients age 7 to 17 with GAD, separation anxiety disorder, and/or social phobia, fluoxetine, 10 to 20 mg/d, was associated with improved scores on the Screen for Anxiety Related Emotional Disorders, PARS, CGI-I, CGI-S, and Children’s Global Assessment Scale.25 A follow-up open-label trial suggested that maintenance treatment is associated with sustained improvement.26
Figure 1: The pediatric anxiety disorders triad: Comorbidity is common
In the Child-Adolescent Multimodal Treatment Study, GAD was the most common disorder; however, GAD, SAD, and SoP were highly comorbid
GAD: generalized anxiety disorder; SAD: separation anxiety disorder; SoP: social phobia
Source: Reference 9
Anxiety disorders with ADHD
Anxiety disorders often are comorbid with attention-deficit/hyperactivity disorder (ADHD). An RCT of patients age 8 to 17 with ADHD and comorbid anxiety found that atomoxetine was associated with improved PARS scores and ADHD symptoms.27 The target dose was 1.2 mg/kg/d. Atomoxetine was well-tolerated; decreased appetite was the only significant adverse event in the treatment group vs placebo.
Multimodal treatment
Although this article reviews evidence for psychopharmacologic treatments, psychotherapeutic treatment of young patients with anxiety disorders has seen significant advances.28 Most psychotherapy studies have evaluated the efficacy of CBT,29-31 although there is evidence for psychodynamic therapy and interpersonal therapy.32 The American Academy of Child & Adolescent Psychiatry recommends a multimodal treatment approach because combination treatment appears to be more effective than monotherapy.8,28,33 Also, clinicians who treat pediatric patients who have an anxiety disorder should evaluate the family’s role on anxiety symptoms and may consider family therapy.
Treatment considerations
Evidence supports the efficacy of sertraline, citalopram, paroxetine, fluvoxamine, fluoxetine, and venlafaxine for treating children and adolescents with anxiety disorders (Figure 2).8,9,11,15,16,23,25 Some practitioners suggest using differing dosing strategies for pediatric anxiety disorders compared with those used to treat adults (Table).34 When considering SSRIs for children and adolescents, keep in mind the “black-box” warning regarding suicidality in these patients. Carefully monitor patients for treatment-emergent suicidality and routinely reassess for the presence and severity of suicidal ideation and suicide risk.
Figure 2: Number needed to treat for SSRIs and SNRIs in pediatric anxiety disorders
GAD: generalized anxiety disorder; RUPP: Research Unit on Pediatric Psychopharmacology; SAD: separation anxiety disorder; SNRI: serotonin-norepinephrine reuptake inhibitor; SoP: social phobia; SSRI: selective serotonin reuptake inhibitorTable
Practical dosing of SSRIs and SNRIs in pediatric patients with anxietya
| Medication | Initial child dose (age <12; mg/d) | Initial adolescent dose (age 12 to 17; mg/d) | Target dose (mg/d) |
|---|---|---|---|
| Citalopram | 5 to 10 | 10 | 20 to 40 |
| Escitalopram | 2.5 to 5 | 5 to 10 | 10 to 20 |
| Fluoxetineb | 10 | 20 | 20 to 40 (children), 40 to 60 (adolescents) |
| Paroxetineb | 5 to 10 | 10 | 20 |
| Sertralinec | 10 to 12.5 | 25 | 150 |
| Venlafaxine | 37.5 | 37.5 | 150 |
| aGeneralized anxiety disorder, social phobia, and separation anxiety disorder bMay consider cytochrome P450 genotyping for 2D6, which may suggest an alternate dosing strategy cSertraline is available in a liquid formulation (20 mg/mL) SNRI: serotonin-norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor Source: Adapted from reference 34 | |||
Related Resources
- Connolly SD, Bernstein GA; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
- Anxiety and Depression Association of America. www.adaa.org.
- American Academy of Child & Adolescent Psychiatry. www.aacap.org.
Drug Brand Names
- Alprazolam • Xanax
- Atomoxetine • Strattera
- Buspirone • BuSpar
- Citalopram • Celexa
- Clonazepam • Klonopin
- Fluoxetine • Prozac
- Fluvoxamine • Luvox, Luvox CR
- Lorazepam • Ativan
- Paroxetine • Paxil, Paxil CR
- Sertraline • Zoloft
- Venlafaxine • Effexor, Effexor XR
Disclosures
Dr. Strawn has received research support from the American Academy of Child & Adolescent Psychiatry, Eli Lilly and Company, and Shire, and is an employee of the University of Cincinnati, Cincinnati, OH.
Dr. McReynolds was employed by Eli Lilly and Company from 1997 to 2005.
1. Beesdo K, Knappe S, Pine DS. Anxiety and anxiety disorders in children and adolescents: developmental issues and implications for DSM-V. Psychiatr Clin North Am. 2009;32(3):483-524.
2. Beesdo K, Pine DS, Lieb R, et al. Incidence and risk patterns of anxiety and depressive disorders and categorization of generalized anxiety disorder. Arch Gen Psychiatry. 2010;67(1):47-57.
3. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first grade children: prediction to anxious symptoms and adaptive functioning in fifth grade. J Child Psychol Psychiatry. 1995;36(3):427-437.
4. Foley DL, Goldston DB, Costello EJ, et al. Proximal psychiatric risk factors for suicidality in youth: the Great Smoky Mountains Study. Arch Gen Psychiatry. 2006;63(9):1017-1024.
5. Jacobson CM, Muehlenkamp JJ, Miller AL, et al. Psychiatric impairment among adolescents engaging in different types of deliberate self-harm. J Clin Child Adolesc Psychol. 2008;37(2):363-375.
6. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first-grade children. J Abnorm Child Psychol. 1994;22(4):441-455.
7. Pine DS, Cohen P, Gurley D, et al. The risk for early-adulthood anxiety and depressive disorders in adolescents with anxiety and depressive disorders. Arch Gen Psychiatry. 1998;55(1):56-64.
8. Rynn MA, Siqueland L, Rickels K. Placebo-controlled trial of sertraline in the treatment of children with generalized anxiety disorders. Am J Psychiatry. 2001;158(12):2008-2014.
9. Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359(26):2753-2766.
10. Maslowsky J, Mogg K, Bradley BP, et al. A preliminary investigation of neural correlates of treatment in adolescents with generalized anxiety disorder. J Child Adolesc Psychopharmacol. 2010;20(2):105-111.
11. Rynn MA, Riddle MA, Yeung PP, et al. Efficacy and safety of extended-release venlafaxine in the treatment of generalized anxiety disorder in children and adolescents: two placebo-controlled trials. Am J Psychiatry. 2007;164(2):290-300.
12. BuSpar [package insert] Princeton NJ: Bristol-Myers Squibb; 2010.
13. Simeon JG, Ferguson HB. Alprazolam effects in children with anxiety disorders. Can J Psychiatry. 1987;32(7):570-574.
14. Simeon JG, Ferguson HB, Knott V, et al. Clinical, cognitive, and neurophysiological effects of alprazolam in children and adolescents with overanxious and avoidant disorders. J Am Acad Child Adolesc Psychiatry. 1992;31(1):29-33.
15. Wagner KD, Berard R, Stein MB, et al. A multicenter, randomized, double-blind, placebo-controlled trial of paroxetine in children and adolescents with social anxiety disorder. Arch Gen Psychiatry. 2004;61(11):1153-1162.
16. March JS, Entusah AR, Rynn M, et al. A randomized controlled trial of venlafaxine ER versus placebo in pediatric social anxiety disorder. Biol Psychiatry. 2007;62(10):1149-1154.
17. Beidel DC, Turner SM, Sallee FR, et al. SET-C versus fluoxetine in the treatment of childhood social phobia. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1622-1632.
18. Compton SN, Grant PJ, Chrisman AK, et al. Sertraline in children and adolescents with social anxiety disorder: an open trial. J Am Acad Child Adolesc Psychiatry. 2001;40(5):564-571.
19. Chavira DA, Stein MB. Combined psychoeducation and treatment with selective serotonin reuptake inhibitors for youth with generalized social anxiety disorder. J Child Adolesc Psychopharmacol. 2002;12(1):47-54.
20. Graae F, Milner J, Rizzotto L, et al. Clonazepam in childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(3):372-376.
21. Renaud J, Birmaher B, Wassick SC, et al. Use of selective serotonin reuptake inhibitors for the treatment of childhood panic disorder: a pilot study. J Child Adolesc Psychopharmacol. 1999;9(2):73-83.
22. Fairbanks JM, Pine DS, Tancer NK, et al. Open fluoxetine treatment of mixed anxiety disorders in children and adolescents. J Child Adolesc Psychopharmacol. 1997;7(1):17-29.
23. The Research Unit on Pediatric Psychopharmacology Anxiety Study Group. Fluvoxamine for the treatment of anxiety disorders in children and adolescents. N Engl J Med. 2001;344(17):1279-1285.
24. Birmaher B, Waterman GS, Ryan N, et al. Fluoxetine for childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(7):993-999.
25. Birmaher B, Axelson DA, Monk K, et al. Fluoxetine for the treatment of childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2003;42(4):415-423.
26. Clark DB, Birmaher B, Axelson D, et al. Fluoxetine for the treatment of childhood anxiety disorders: open-label, long-term extension to a controlled trial. J Am Acad Child Adolesc Psychiatry. 2005;44(12):1263-1270.
27. Geller D, Donnelly C, Lopez F, et al. Atomoxetine treatment for pediatric patients with attention-deficit/hyperactivity disorder with comorbid anxiety disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(9):1119-1127.
28. Connolly SD, Bernstein GA. Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
29. Kendall PC. Treating anxiety disorders in children: results of a randomized clinical trial. J Consult Clin Psychol. 1994;62(1):100-110.
30. Kendall PC, Flannery-Schroeder E, Panichelli-Mindel SM, et al. Therapy for youths with anxiety disorders: a second randomized clinical trial. J Consult Clin Psychol. 1997;65(3):366-380.
31. Reynolds S, Wilson C, Austin J, et al. Effects of psychotherapy for anxiety in children and adolescents: a meta-analytic review. Clin Psychol Rev. 2012;32(4):251-262.
32. Strawn JR, Wehry AM, DelBello MP, et al. Establishing the neurobiologic basis of treatment in children and adolescents with generalized anxiety disorder. Depress Anxiety. 2012;29(4):328-339.
33. Ginsburg GS, Kendall PC, Sakolsky D, et al. Remission after acute treatment in children and adolescents with anxiety disorders: findings from the CAMS. J Consult Clin Psychol. 2011;79(6):806-813.
34. Findling RL, Kowatch RA. How (not) to dose antidepressants and antipsychotics for children. Current Psychiatry. 2007;6(6):79-83.
1. Beesdo K, Knappe S, Pine DS. Anxiety and anxiety disorders in children and adolescents: developmental issues and implications for DSM-V. Psychiatr Clin North Am. 2009;32(3):483-524.
2. Beesdo K, Pine DS, Lieb R, et al. Incidence and risk patterns of anxiety and depressive disorders and categorization of generalized anxiety disorder. Arch Gen Psychiatry. 2010;67(1):47-57.
3. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first grade children: prediction to anxious symptoms and adaptive functioning in fifth grade. J Child Psychol Psychiatry. 1995;36(3):427-437.
4. Foley DL, Goldston DB, Costello EJ, et al. Proximal psychiatric risk factors for suicidality in youth: the Great Smoky Mountains Study. Arch Gen Psychiatry. 2006;63(9):1017-1024.
5. Jacobson CM, Muehlenkamp JJ, Miller AL, et al. Psychiatric impairment among adolescents engaging in different types of deliberate self-harm. J Clin Child Adolesc Psychol. 2008;37(2):363-375.
6. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first-grade children. J Abnorm Child Psychol. 1994;22(4):441-455.
7. Pine DS, Cohen P, Gurley D, et al. The risk for early-adulthood anxiety and depressive disorders in adolescents with anxiety and depressive disorders. Arch Gen Psychiatry. 1998;55(1):56-64.
8. Rynn MA, Siqueland L, Rickels K. Placebo-controlled trial of sertraline in the treatment of children with generalized anxiety disorders. Am J Psychiatry. 2001;158(12):2008-2014.
9. Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359(26):2753-2766.
10. Maslowsky J, Mogg K, Bradley BP, et al. A preliminary investigation of neural correlates of treatment in adolescents with generalized anxiety disorder. J Child Adolesc Psychopharmacol. 2010;20(2):105-111.
11. Rynn MA, Riddle MA, Yeung PP, et al. Efficacy and safety of extended-release venlafaxine in the treatment of generalized anxiety disorder in children and adolescents: two placebo-controlled trials. Am J Psychiatry. 2007;164(2):290-300.
12. BuSpar [package insert] Princeton NJ: Bristol-Myers Squibb; 2010.
13. Simeon JG, Ferguson HB. Alprazolam effects in children with anxiety disorders. Can J Psychiatry. 1987;32(7):570-574.
14. Simeon JG, Ferguson HB, Knott V, et al. Clinical, cognitive, and neurophysiological effects of alprazolam in children and adolescents with overanxious and avoidant disorders. J Am Acad Child Adolesc Psychiatry. 1992;31(1):29-33.
15. Wagner KD, Berard R, Stein MB, et al. A multicenter, randomized, double-blind, placebo-controlled trial of paroxetine in children and adolescents with social anxiety disorder. Arch Gen Psychiatry. 2004;61(11):1153-1162.
16. March JS, Entusah AR, Rynn M, et al. A randomized controlled trial of venlafaxine ER versus placebo in pediatric social anxiety disorder. Biol Psychiatry. 2007;62(10):1149-1154.
17. Beidel DC, Turner SM, Sallee FR, et al. SET-C versus fluoxetine in the treatment of childhood social phobia. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1622-1632.
18. Compton SN, Grant PJ, Chrisman AK, et al. Sertraline in children and adolescents with social anxiety disorder: an open trial. J Am Acad Child Adolesc Psychiatry. 2001;40(5):564-571.
19. Chavira DA, Stein MB. Combined psychoeducation and treatment with selective serotonin reuptake inhibitors for youth with generalized social anxiety disorder. J Child Adolesc Psychopharmacol. 2002;12(1):47-54.
20. Graae F, Milner J, Rizzotto L, et al. Clonazepam in childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(3):372-376.
21. Renaud J, Birmaher B, Wassick SC, et al. Use of selective serotonin reuptake inhibitors for the treatment of childhood panic disorder: a pilot study. J Child Adolesc Psychopharmacol. 1999;9(2):73-83.
22. Fairbanks JM, Pine DS, Tancer NK, et al. Open fluoxetine treatment of mixed anxiety disorders in children and adolescents. J Child Adolesc Psychopharmacol. 1997;7(1):17-29.
23. The Research Unit on Pediatric Psychopharmacology Anxiety Study Group. Fluvoxamine for the treatment of anxiety disorders in children and adolescents. N Engl J Med. 2001;344(17):1279-1285.
24. Birmaher B, Waterman GS, Ryan N, et al. Fluoxetine for childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(7):993-999.
25. Birmaher B, Axelson DA, Monk K, et al. Fluoxetine for the treatment of childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2003;42(4):415-423.
26. Clark DB, Birmaher B, Axelson D, et al. Fluoxetine for the treatment of childhood anxiety disorders: open-label, long-term extension to a controlled trial. J Am Acad Child Adolesc Psychiatry. 2005;44(12):1263-1270.
27. Geller D, Donnelly C, Lopez F, et al. Atomoxetine treatment for pediatric patients with attention-deficit/hyperactivity disorder with comorbid anxiety disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(9):1119-1127.
28. Connolly SD, Bernstein GA. Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
29. Kendall PC. Treating anxiety disorders in children: results of a randomized clinical trial. J Consult Clin Psychol. 1994;62(1):100-110.
30. Kendall PC, Flannery-Schroeder E, Panichelli-Mindel SM, et al. Therapy for youths with anxiety disorders: a second randomized clinical trial. J Consult Clin Psychol. 1997;65(3):366-380.
31. Reynolds S, Wilson C, Austin J, et al. Effects of psychotherapy for anxiety in children and adolescents: a meta-analytic review. Clin Psychol Rev. 2012;32(4):251-262.
32. Strawn JR, Wehry AM, DelBello MP, et al. Establishing the neurobiologic basis of treatment in children and adolescents with generalized anxiety disorder. Depress Anxiety. 2012;29(4):328-339.
33. Ginsburg GS, Kendall PC, Sakolsky D, et al. Remission after acute treatment in children and adolescents with anxiety disorders: findings from the CAMS. J Consult Clin Psychol. 2011;79(6):806-813.
34. Findling RL, Kowatch RA. How (not) to dose antidepressants and antipsychotics for children. Current Psychiatry. 2007;6(6):79-83.
Neuroleptic malignant syndrome: Answers to 6 tough questions
Diagnosis and treatment of neuroleptic malignant syndrome (NMS) are controversial because this potentially life-threatening syndrome is rare and its presentation varies. These factors make it difficult to evaluate treatments in controlled clinical trials, and data about the relative efficacy of specific interventions are scarce. It may be possible, however, to develop rational treatment guidelines using empiric clinical data.1,2
This article examines the evidence related to 6 controversial aspects of NMS diagnosis and treatment:
- most-reliable risk factors
- NMS as a spectrum disorder
- what causes NMS
- NMS triggered by first-generation vs second-generation antipsychotics
- first-line interventions
- restarting antipsychotics after an NMS episode.
1. Are there reliable risk factors for NMS?
In small case-controlled studies, agitation, dehydration, and exhaustion were the most consistently found systemic factors believed to predispose patients taking antipsychotics to NMS (Table 1).3-5 Catatonia and organic brain syndromes may be separate risk factors.1,6
Preliminary studies also have implicated dopamine receptor abnormalities caused by genetic polymorphisms or effects of low serum iron.1,7,8 Pharmacologic studies have suggested that higher doses, rapid titration, and IM injections of antipsychotics are associated with increased NMS risk.3,5 Some studies suggest that 15% to 20% of NMS patients have a history of NMS episodes.1,2 In addition, high-potency first-generation antipsychotics (FGAs)—especially haloperidol—are assumed to carry higher risk than low-potency drugs and second-generation antipsychotics (SGAs), although this hypothesis remains difficult to prove.9-11
These risk factors, however, are not practical for estimating NMS risk in a given patient because they are relatively common compared with the low risk of NMS occurrence. For the vast majority of patients with psychotic symptoms, the benefits of properly indicated antipsychotic pharmacotherapy will outweigh the risks.
Table 1
| Systemic |
| Agitation |
| Dehydration |
| Exhaustion |
| Low serum iron concentrations (normal: 60 to 170 mcg/dL) |
| Diagnoses |
| History of NMS |
| Catatonia |
| Organic brain syndromes |
| Central nervous system |
| Dopamine receptor dysfunction |
| Basal ganglia dysfunction |
| Sympathetic nervous system dysfunction |
| Pharmacologic treatment* |
| Intramuscular or intravenous injections |
| High-potency dopamine antagonists |
| Rapid dose titration |
| High doses |
| FGAs compared with SGAs (?) |
*For individual patients, these common risk factors must be weighted again the benefits of antipsychotic therapy FGAs: first-generation antipsychotics; SGAs:second-generation antipsychotics; NMS: neuroleptic malignant syndromeSource: References 1-5
2. Is NMS related to parkinsonism, catatonia, or malignant hyperthermia?
Parkinsonsim. Some researchers have described NMS as an extreme parkinsonian crisis resulting from overwhelming blockade of dopamine pathways in the brain.1,2,12 In this view, NMS resembles the parkinsonian-hyperthermia syndrome that can occur in Parkinson's disease patients following abrupt discontinuation or loss of efficacy of dopaminergic therapy, which can be treated by reinstituting dopaminergic agents.13 Evidence to support this view includes:
- Parkinsonian signs are a cardinal feature of NMS.
- Withdrawal of dopamine agonists precipitates the syndrome.
- All triggering drugs are dopamine receptor antagonists.
- Risks of NMS correlates with drugs' dopamine receptor affinity.
- Dopaminergic agonists may be an effective treatment.
- Lesions in dopaminergic pathways produce a similar syndrome.
- Patients with NMS have demonstrated low cerebrospinal fluid concentrations of the dopamine metabolite homovanillic acid.14
Catatonia. Fink et al15 and others16-18 have persuasively argued that NMS represents a form of drug-induced malignant catatonia. Evidence supporting this includes:
- The 2 disorders share neuropsychiatric symptoms.
- Catalonic signs are common in NMS.19
- Malignant catatonia and NMS share physiologic and labratory signs.20
- Reintroduction of antipsychotics can acutely worsen both conditions.
- Benzodiazepines and electroconvulsive therapy (ECT) are effective treatments for both disorders.15-18
Lee21 examined the relationship between catatonic features and treatment response of 14 NMS patients. Most patients with catatonic symptoms responded to benzodiazepines, whereas none of those did who had an extrapyramidal-hyperthermic presentation without catatonia. Lee concluded that NMS is heterogeneous and may occur in catatonic and noncatatonic forms that differ in treatment response.
- similar clinical signs of rigidity, hyperthermia, and hypermetabolism
- similar psychologic and labratory signs, such as rhabdomyolysis
- hyperthermia in both responding to dantrolene.
Although the 2 are similar in presentation, malignant hyperthermia occurs intraoperatively and reflects a pharmacogenetic disorder of calcium regulation in skeletal muscle. Additionally, rigidity in malignant hyperthermia does not respond to peripheral-acting muscle relaxants.1,22 Evidence suggests that patients who have previously experienced an NMS episodes are not at risk for malignant hyperthermia.22
3. What is the pathophysiology of NMS?
NMS pathophysiology is complex and likely involves interplay between multiple central and systemic pathways and neurotransmitters. As described above, compelling evidence suggests that dopamine blockade plays a central role.12
Dopamine blockade in the hypothalamus is believed to contribute to thermoregulatory failure, and blockade in the nigrostriatal system likely contributes to muscle rigidity and hypermetabolism. The loss of dopaminergic input to the anterior cingulate-medial orbitofrontal circuit and the lateral orbitofrontal circuit likely con-tributes to the mental status changes and catatonic features seen in NMS.12
Some researchers have proposed competing or complementary hypotheses, however. For example, Gurrera23 proposed that patients who are prone to developing NMS have a vulnerability to a hyperactive and dysregulated sympathetic nervous system, and this trait—together with dopamine system disruption induced by dopamine-blocking agents—produces NMS. Other investigators have implicated serotonin, norepinephrine, gamma-aminobutyric acid and glutaminergic mechanisms.1,12,24,25
4. Are FGAs or SGAs more likely to cause NMS?
NMS is assumed to occur less frequently in patients treated with SGAs than in those receiving FGAs, although this hypothesisis unproven. Isolated reports of NMS have been associated with nearly every SGA.9-11 It is difficult to prove FGA vs SGA liabilities because:
- NMS is rare.
- Dosing practices may be more conser-vative now than in the past.
- Most clinicians are aware of the earlysigns of NMS.
In an epidemiological study of a large database, Stubner et al26 found that patients receiving SGAs had a lower risk of NMS than those treated with haloperidol.26 In this study, the overall rate of NMS was 0.02%.
NMS hotline data. We recently examined which medication classes were implicated in 111 NMS cases reported to the Neuroleptic Malignant Syndrome Information Service hotline (1-888-NMS-TEMP) between 1997 and 2006 (Figure). We included only cases of definite or probable NMS (as diagnosed by hotline consultants) in which a single antipsychotic was administered. Slightly more cases were attributed to FGAs (51%) than SGAs (45%). The remaining cases were attributed to neuroleptics used in medical settings (such as promethazineor prochlorperazine). Because they are now prescribed less often, FGAs accounted for a disproportionate number of NMS cases reported to the hotline. Haloperidol accounted for the majority of FGA cases and 44% of all cases. If we had excluded haloperidol and compared the NMS risk of SGAs to only intermediate- or low-potency FGAs, the relative advantage of SGAs would have been lost. On the other hand, it is clear that SGAs still carry a risk for NMS. Analyses suggest that the SGA-associated classic features of NMS—fever, muscle rigidity, and autonomic and mental status changes—are retained in patients receiving SGAs, although some may not develop the severe rigidity and extreme temperatures common in patients receiving FGAs.9-11 The milder clinical characteristics associated with SGAs may reflect more conservative prescribing patterns or increased awareness and earlier recognition of NMS, which would prevent fulminant presentations.
5. What is the evidence for specific NMS treatments?
NMS is rare, its presentation varies, and its progression is unpredictable. These factors make it difficult to evaluate treatments in controlled clinical trials, and data about the relative efficacy of specific interventions are scarce.
Even so, the notion that NMS represents an extreme variant of drug-induced parkinsonism or catatonia suggests that specific NMS treatments could be based on symptom severity or stage of presentation. We propose a treatment guideline basedon theoretical mechanisms and anecdotal data (Algorithm).2,27-29
Support. After immediate withdrawal of the offending medication, supportive therapy is the cornerstone of NMS treatment.1,2,27
For patients presenting with mild signs and symptoms, supportive care and careful clinical monitoring may be sufficient. Extreme hyperthermia demands volume resuscitation and cooling measures, intensive medical care, and careful monitoring for complications.
Treatment. Despite a lack of consensus on drug treatments for uncomplicated NMS, approximately 40% of patients with acute NMS receive pharmacologic treatments.2
Lorazepam, 1 to 2 mg parenterally, is a reasonable first-line therapy for NMS, especially in individuals with catatonic features.4,15-18,21,30,31 Some investigators recommend higher doses.15 Benzodiazepines are preferred if sedation is required in agitated NMS patients.4,15-18
Dopaminergic agents such as bromocriptine and amantadine enhance dopaminergic transmission to reverse parkinsonian symptoms and have been reported to reduce time to recovery and halve mortality rates when used alone or in conjunction with other treatments.13,27,32,33 Rapid discontinuation of these agents can result in rebound symptoms, although this may be true for any specific drug treatment of NMS.1,31,32
Dantrolene uncouples excitation-contraction coupling by enhancing calcium sequestration in sarcoplasmic reticulumin skeletal muscle and has been used to treat NMS hypermetabolic symptoms. Some reviews found improvement in up to 80% of NMS patients treated with dantrolene monotherapy.27,32-35 Compared with supportive care, time to recovery may be reduced—and mortality decreased by almost one-half—when dantrolene is used alone or in combination with other medications.
Not all case reports have shown that dantrolene, benzodiazepines, ordopaminergic agonists are effective in treating NMS.31,36 In our opinion, only advanced NMS cases—with extreme temperature elevations, severe rigidity, and evidence of systemic hypermetabolism—benefit from dantrolene treatment.1,2
ECT has been used successfully to reduce mortality from NMS and other catatonic-spectrum disorders. It is usually employed after supportive therapy and psychopharmacologic interventions fail.2,15,16,27,37 ECT for acute NMS typically consists of a series of 6 to 10 treatments with bilateral electrode placement. Daily ECT may be needed initially.15
6. Are antipsychotics contraindicated following an NMS episode?
The rate of NMS recurrence on retreatment with an antipsychotic has varied.38 We estimate that up to 30% of patients may be at risk of NMS recurrence when rechallenged with an antipsychotic.1 By following proper precautions (Table 2), however, you can safely treat most patients who require continued antipsychotic therapy.1,2 When you restart treatment, a lower-potency antipsychotic from a different chemical class may be a safer option than retrying the triggering agent, according to retrospective analyses of limited available data. A patient who develops NMS on a FGA might benefit from an SGA trial, although some risk of recurrence remains.1,10
Current Psychiatry 2007;6(8):89-95.
Drug brand names
- Amantadine • Symmetrel
- Bromocriptine • Parlodel
- Chlorpromazine • Thorazine
- Dantrolene • Dantrium
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Lorazepam • Ativan
- Loxapine • Loxitane
- Perphenazine • Trilafon
- Prochlorperazine • Compazine, Compro
- Promethazine • Phenergan
- Thioridazine • Mellaril
Disclosure
Dr. Strawn is an American Psychiatric Institute for Research and Education (APIRE)/Janssen Scholar.
Dr. Keck has received research support from or served as a consultant to Abbott Laboratories, American Diabetes Association, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb, GlaxoSmithKline, Eli Lilly and Company, Janssen Pharmaceutica, National Institute of Mental Health, National Institute of Drug Abuse, Pfizer, Stanley Medical Research Institute, and UCB Pharma.
Dr. Caroff has received research support from Bristol-Myers Squibb, Ortho-McNeil Neurologics, and Pfizer.
1. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A, eds. Neuroleptic malignant syndrome and related conditions 2nd ed. Washington, DC: American Psychiatric Publishing Inc; 2003; 1-44.
2. Strawn JR, Keck PE, Jr, Caroff SN. Neuroleptic malignant syndrome Am J Psychiatry 2007;164:870-6.
3. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome Arch Gen Psychiatry 1989;46:914-18.
4. Rosebush PI, Stewart TD. A prospective analysis of 24 episodes of neuroleptic malignant syndrome Am J Psychiatry 1989;146:717-25.
5. Berardi D, Amore M, Keck PE, Jr, et al. Clinical and pharmacologic risk factors for neuroleptic malignant syndrome: a case-control study. Biol Psychiatry 1998;44:748-54.
6. White DA, Robins AH. Catatonia: harbinger of the neuroleptic malignant syndrome Br J Psychiatry 1991;158:419-21.
7. Rosebush PI, Mazurek MF. Serum iron and neuroleptic malignant syndrome. Lancet 1991;338:149-51.
8. Lee JW. Serum iron in catatonia and neuroleptic malignant syndrome Biol Psychiatry 1998;44:499-507.
9. Ananth J, Parameswaran S, Gunatilake S, et al. Neuroleptic malignant syndrome and atypical antipsychotic drugs J Clin Psychiatry 2004;65:464-70.
10. Caroff SN, Mann SC, Campbell EC. Atypical antipsychotics and neuroleptic malignant syndrome Psychiatr Ann 2000;30:314-21.
11. Hasan S, Buckley P. Novel antipsychotics and the neuroleptic malignant syndrome Am J Psychiatry 1998;155:1113-16.
12. Mann SC, Caroff SN, Fricchione G, Campbell EC. Central dopamine hypoactivity and the pathogenesis of neuroleptic malignant syndrome Psychiatr Ann 2000;30:363-74.
13. Factor SA, Santiago A. Parkinsonism-hyperpyrexia syndrome in Parkinson’s disease. In: Frucht SJ, Fahn S, eds. Movement disorder emergencies: diagnosis and treatment. Totowa, NJ: Humana Press; 2005; 29-40.
14. Nisijima K, Ishiguro T. Cerebrospinal fluid levels of monoamine metabolites and gamma-aminobutyric acid in neuroleptic malignant syndrome. J Psychiatr Res 1995;27:233-44.
15. Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Prog Neuropsychopharmacol Biol Psychiatry 2006;30:1182-3.
16. Fricchione G, Bush G, Fozdar M, et al. Recognition and treatment of the catatonic syndrome. J Intensive Care Med 1997;12:135-47.
17. Philbrick KL, Rummans TA. Malignant catatonia. J Neuropsychiatry Clin Neurosci 1994;6:1-13.
18. Mann SC, Caroff SN, Bleier HR, et al. Lethal catatonia. Am J Psychiatry 1986;143:1374-81.
19. Koch M, Chandragiri S, Rizvi S, et al. Catatonic signs in neuroleptic malignant syndrome. Compr Psychiatry 2000;41:73-5.
20. Lee JW. Laboratory findings. In: Caroff SN, Mann SC, Francis A, Fricchoine GL, eds. Catatonia: from psychopathology to neurobiology Washington, DC: American Psychiatric Press, Inc; 2004; 65-75.
21. Lee JW. Catatonic variants, hyperthermic extrapyramidal reactions, and subtypes of neuroleptic malignant syndrome. Ann Clin Psychiatry 2007;19:9-16.
22. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the perioperative setting. Am J Anesthesiol 2001;28:387-93.
23. Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry 1999;156:169-80.
24. Carroll BT. The universal field hypothesis of catatonia and neuroleptic malignant syndrome. CNS Spectr 2000;5:26-33.
25. Weller M, Kornhuber J. A rationale for NMDA receptor antagonist therapy of the neuroleptic malignant syndrome. Med Hypotheses 1992;38:329-33.
26. Stubner S, Rustenbeck E, Grohmann R, et al. Severe and uncommon involuntary movement disorders due to psychotropic drugs. Pharmacopsychiatry 2004;37(suppl 1):S54-S64.
27. Davis JM, Caroff SN, Mann SC. Treatment of neuroleptic malignant syndrome. Psychiatr Ann 2000;30:325-31.
28. Adityanjee PA, Singh S, Singh G, Ong S. Spectrum concept of neuroleptic malignant syndrome. Br J Psychiatry 1988;153:107-11.
29. Woodbury MM, Woodbury MA. Neuroleptic-induced catatonia as a stage in the progression toward neuroleptic malignant syndrome. J Am Acad Child Adolesc Psychiatry 1992;31:1161-4.
30. Francis A, Chondragivi S, Rizvi S, et al. Is lorazepam a treatment for neuroleptic malignant syndrome? CNS Spectr 2000;5:54-7.
31. Rosebush PI, Stewart T, Mazurek MF. The treatment of neuroleptic malignant syndrome. Are dantrolene and bromocriptine useful adjuncts to supportive care? Br J Psychiatry 1991;159:709-12.
32. Sakkas P, Davis JM, Janicak PG, Wang ZY. Drug treatment of the neuroleptic malignant syndrome. Psychopharmacol Bull 1991;27:381-4.
33. Rosenberg MR, Green M. Neuroleptic malignant syndrome: review of response to therapy. Arch Intern Med 1989;149:1927-31.
34. Yamawaki S, Morio M, Kazamutsuri G, et al. Clinical evaluation and effective usage of dantrolene sodium in neuroleptic malignant syndrome. Kiso to Rinsyou (Clinical Reports) 1993;27:1045-66.
35. Tsutsumi Y, Yamamoto K, Matsuura S, et al. The treatment of neuroleptic malignant syndrome using dantrolene sodium. Psychiatry Clin Neurosci 1998;52:433-8.
36. Reulbach U, Dutsch C, Biermann T, et al. Managing an effective treatment for neuroleptic malignant syndrome. Crit Care 2007;11:R4.-
37. Troller JN, Sachdev PS. Electroconvulsive treatment of neuroleptic malignant syndrome: a review and report of cases. Aust N Z J Psychiatry 1999;33:650-9.
38. Pope HG, Aizley HG, Keck PE, Jr, McElroy SL. Neuroleptic malignant syndrome: long term follow-up of 20 cases. J Clin Psychiatry 1991;52:208-12.
Diagnosis and treatment of neuroleptic malignant syndrome (NMS) are controversial because this potentially life-threatening syndrome is rare and its presentation varies. These factors make it difficult to evaluate treatments in controlled clinical trials, and data about the relative efficacy of specific interventions are scarce. It may be possible, however, to develop rational treatment guidelines using empiric clinical data.1,2
This article examines the evidence related to 6 controversial aspects of NMS diagnosis and treatment:
- most-reliable risk factors
- NMS as a spectrum disorder
- what causes NMS
- NMS triggered by first-generation vs second-generation antipsychotics
- first-line interventions
- restarting antipsychotics after an NMS episode.
1. Are there reliable risk factors for NMS?
In small case-controlled studies, agitation, dehydration, and exhaustion were the most consistently found systemic factors believed to predispose patients taking antipsychotics to NMS (Table 1).3-5 Catatonia and organic brain syndromes may be separate risk factors.1,6
Preliminary studies also have implicated dopamine receptor abnormalities caused by genetic polymorphisms or effects of low serum iron.1,7,8 Pharmacologic studies have suggested that higher doses, rapid titration, and IM injections of antipsychotics are associated with increased NMS risk.3,5 Some studies suggest that 15% to 20% of NMS patients have a history of NMS episodes.1,2 In addition, high-potency first-generation antipsychotics (FGAs)—especially haloperidol—are assumed to carry higher risk than low-potency drugs and second-generation antipsychotics (SGAs), although this hypothesis remains difficult to prove.9-11
These risk factors, however, are not practical for estimating NMS risk in a given patient because they are relatively common compared with the low risk of NMS occurrence. For the vast majority of patients with psychotic symptoms, the benefits of properly indicated antipsychotic pharmacotherapy will outweigh the risks.
Table 1
| Systemic |
| Agitation |
| Dehydration |
| Exhaustion |
| Low serum iron concentrations (normal: 60 to 170 mcg/dL) |
| Diagnoses |
| History of NMS |
| Catatonia |
| Organic brain syndromes |
| Central nervous system |
| Dopamine receptor dysfunction |
| Basal ganglia dysfunction |
| Sympathetic nervous system dysfunction |
| Pharmacologic treatment* |
| Intramuscular or intravenous injections |
| High-potency dopamine antagonists |
| Rapid dose titration |
| High doses |
| FGAs compared with SGAs (?) |
*For individual patients, these common risk factors must be weighted again the benefits of antipsychotic therapy FGAs: first-generation antipsychotics; SGAs:second-generation antipsychotics; NMS: neuroleptic malignant syndromeSource: References 1-5
2. Is NMS related to parkinsonism, catatonia, or malignant hyperthermia?
Parkinsonsim. Some researchers have described NMS as an extreme parkinsonian crisis resulting from overwhelming blockade of dopamine pathways in the brain.1,2,12 In this view, NMS resembles the parkinsonian-hyperthermia syndrome that can occur in Parkinson's disease patients following abrupt discontinuation or loss of efficacy of dopaminergic therapy, which can be treated by reinstituting dopaminergic agents.13 Evidence to support this view includes:
- Parkinsonian signs are a cardinal feature of NMS.
- Withdrawal of dopamine agonists precipitates the syndrome.
- All triggering drugs are dopamine receptor antagonists.
- Risks of NMS correlates with drugs' dopamine receptor affinity.
- Dopaminergic agonists may be an effective treatment.
- Lesions in dopaminergic pathways produce a similar syndrome.
- Patients with NMS have demonstrated low cerebrospinal fluid concentrations of the dopamine metabolite homovanillic acid.14
Catatonia. Fink et al15 and others16-18 have persuasively argued that NMS represents a form of drug-induced malignant catatonia. Evidence supporting this includes:
- The 2 disorders share neuropsychiatric symptoms.
- Catalonic signs are common in NMS.19
- Malignant catatonia and NMS share physiologic and labratory signs.20
- Reintroduction of antipsychotics can acutely worsen both conditions.
- Benzodiazepines and electroconvulsive therapy (ECT) are effective treatments for both disorders.15-18
Lee21 examined the relationship between catatonic features and treatment response of 14 NMS patients. Most patients with catatonic symptoms responded to benzodiazepines, whereas none of those did who had an extrapyramidal-hyperthermic presentation without catatonia. Lee concluded that NMS is heterogeneous and may occur in catatonic and noncatatonic forms that differ in treatment response.
- similar clinical signs of rigidity, hyperthermia, and hypermetabolism
- similar psychologic and labratory signs, such as rhabdomyolysis
- hyperthermia in both responding to dantrolene.
Although the 2 are similar in presentation, malignant hyperthermia occurs intraoperatively and reflects a pharmacogenetic disorder of calcium regulation in skeletal muscle. Additionally, rigidity in malignant hyperthermia does not respond to peripheral-acting muscle relaxants.1,22 Evidence suggests that patients who have previously experienced an NMS episodes are not at risk for malignant hyperthermia.22
3. What is the pathophysiology of NMS?
NMS pathophysiology is complex and likely involves interplay between multiple central and systemic pathways and neurotransmitters. As described above, compelling evidence suggests that dopamine blockade plays a central role.12
Dopamine blockade in the hypothalamus is believed to contribute to thermoregulatory failure, and blockade in the nigrostriatal system likely contributes to muscle rigidity and hypermetabolism. The loss of dopaminergic input to the anterior cingulate-medial orbitofrontal circuit and the lateral orbitofrontal circuit likely con-tributes to the mental status changes and catatonic features seen in NMS.12
Some researchers have proposed competing or complementary hypotheses, however. For example, Gurrera23 proposed that patients who are prone to developing NMS have a vulnerability to a hyperactive and dysregulated sympathetic nervous system, and this trait—together with dopamine system disruption induced by dopamine-blocking agents—produces NMS. Other investigators have implicated serotonin, norepinephrine, gamma-aminobutyric acid and glutaminergic mechanisms.1,12,24,25
4. Are FGAs or SGAs more likely to cause NMS?
NMS is assumed to occur less frequently in patients treated with SGAs than in those receiving FGAs, although this hypothesisis unproven. Isolated reports of NMS have been associated with nearly every SGA.9-11 It is difficult to prove FGA vs SGA liabilities because:
- NMS is rare.
- Dosing practices may be more conser-vative now than in the past.
- Most clinicians are aware of the earlysigns of NMS.
In an epidemiological study of a large database, Stubner et al26 found that patients receiving SGAs had a lower risk of NMS than those treated with haloperidol.26 In this study, the overall rate of NMS was 0.02%.
NMS hotline data. We recently examined which medication classes were implicated in 111 NMS cases reported to the Neuroleptic Malignant Syndrome Information Service hotline (1-888-NMS-TEMP) between 1997 and 2006 (Figure). We included only cases of definite or probable NMS (as diagnosed by hotline consultants) in which a single antipsychotic was administered. Slightly more cases were attributed to FGAs (51%) than SGAs (45%). The remaining cases were attributed to neuroleptics used in medical settings (such as promethazineor prochlorperazine). Because they are now prescribed less often, FGAs accounted for a disproportionate number of NMS cases reported to the hotline. Haloperidol accounted for the majority of FGA cases and 44% of all cases. If we had excluded haloperidol and compared the NMS risk of SGAs to only intermediate- or low-potency FGAs, the relative advantage of SGAs would have been lost. On the other hand, it is clear that SGAs still carry a risk for NMS. Analyses suggest that the SGA-associated classic features of NMS—fever, muscle rigidity, and autonomic and mental status changes—are retained in patients receiving SGAs, although some may not develop the severe rigidity and extreme temperatures common in patients receiving FGAs.9-11 The milder clinical characteristics associated with SGAs may reflect more conservative prescribing patterns or increased awareness and earlier recognition of NMS, which would prevent fulminant presentations.
5. What is the evidence for specific NMS treatments?
NMS is rare, its presentation varies, and its progression is unpredictable. These factors make it difficult to evaluate treatments in controlled clinical trials, and data about the relative efficacy of specific interventions are scarce.
Even so, the notion that NMS represents an extreme variant of drug-induced parkinsonism or catatonia suggests that specific NMS treatments could be based on symptom severity or stage of presentation. We propose a treatment guideline basedon theoretical mechanisms and anecdotal data (Algorithm).2,27-29
Support. After immediate withdrawal of the offending medication, supportive therapy is the cornerstone of NMS treatment.1,2,27
For patients presenting with mild signs and symptoms, supportive care and careful clinical monitoring may be sufficient. Extreme hyperthermia demands volume resuscitation and cooling measures, intensive medical care, and careful monitoring for complications.
Treatment. Despite a lack of consensus on drug treatments for uncomplicated NMS, approximately 40% of patients with acute NMS receive pharmacologic treatments.2
Lorazepam, 1 to 2 mg parenterally, is a reasonable first-line therapy for NMS, especially in individuals with catatonic features.4,15-18,21,30,31 Some investigators recommend higher doses.15 Benzodiazepines are preferred if sedation is required in agitated NMS patients.4,15-18
Dopaminergic agents such as bromocriptine and amantadine enhance dopaminergic transmission to reverse parkinsonian symptoms and have been reported to reduce time to recovery and halve mortality rates when used alone or in conjunction with other treatments.13,27,32,33 Rapid discontinuation of these agents can result in rebound symptoms, although this may be true for any specific drug treatment of NMS.1,31,32
Dantrolene uncouples excitation-contraction coupling by enhancing calcium sequestration in sarcoplasmic reticulumin skeletal muscle and has been used to treat NMS hypermetabolic symptoms. Some reviews found improvement in up to 80% of NMS patients treated with dantrolene monotherapy.27,32-35 Compared with supportive care, time to recovery may be reduced—and mortality decreased by almost one-half—when dantrolene is used alone or in combination with other medications.
Not all case reports have shown that dantrolene, benzodiazepines, ordopaminergic agonists are effective in treating NMS.31,36 In our opinion, only advanced NMS cases—with extreme temperature elevations, severe rigidity, and evidence of systemic hypermetabolism—benefit from dantrolene treatment.1,2
ECT has been used successfully to reduce mortality from NMS and other catatonic-spectrum disorders. It is usually employed after supportive therapy and psychopharmacologic interventions fail.2,15,16,27,37 ECT for acute NMS typically consists of a series of 6 to 10 treatments with bilateral electrode placement. Daily ECT may be needed initially.15
6. Are antipsychotics contraindicated following an NMS episode?
The rate of NMS recurrence on retreatment with an antipsychotic has varied.38 We estimate that up to 30% of patients may be at risk of NMS recurrence when rechallenged with an antipsychotic.1 By following proper precautions (Table 2), however, you can safely treat most patients who require continued antipsychotic therapy.1,2 When you restart treatment, a lower-potency antipsychotic from a different chemical class may be a safer option than retrying the triggering agent, according to retrospective analyses of limited available data. A patient who develops NMS on a FGA might benefit from an SGA trial, although some risk of recurrence remains.1,10
Current Psychiatry 2007;6(8):89-95.
Drug brand names
- Amantadine • Symmetrel
- Bromocriptine • Parlodel
- Chlorpromazine • Thorazine
- Dantrolene • Dantrium
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Lorazepam • Ativan
- Loxapine • Loxitane
- Perphenazine • Trilafon
- Prochlorperazine • Compazine, Compro
- Promethazine • Phenergan
- Thioridazine • Mellaril
Disclosure
Dr. Strawn is an American Psychiatric Institute for Research and Education (APIRE)/Janssen Scholar.
Dr. Keck has received research support from or served as a consultant to Abbott Laboratories, American Diabetes Association, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb, GlaxoSmithKline, Eli Lilly and Company, Janssen Pharmaceutica, National Institute of Mental Health, National Institute of Drug Abuse, Pfizer, Stanley Medical Research Institute, and UCB Pharma.
Dr. Caroff has received research support from Bristol-Myers Squibb, Ortho-McNeil Neurologics, and Pfizer.
Diagnosis and treatment of neuroleptic malignant syndrome (NMS) are controversial because this potentially life-threatening syndrome is rare and its presentation varies. These factors make it difficult to evaluate treatments in controlled clinical trials, and data about the relative efficacy of specific interventions are scarce. It may be possible, however, to develop rational treatment guidelines using empiric clinical data.1,2
This article examines the evidence related to 6 controversial aspects of NMS diagnosis and treatment:
- most-reliable risk factors
- NMS as a spectrum disorder
- what causes NMS
- NMS triggered by first-generation vs second-generation antipsychotics
- first-line interventions
- restarting antipsychotics after an NMS episode.
1. Are there reliable risk factors for NMS?
In small case-controlled studies, agitation, dehydration, and exhaustion were the most consistently found systemic factors believed to predispose patients taking antipsychotics to NMS (Table 1).3-5 Catatonia and organic brain syndromes may be separate risk factors.1,6
Preliminary studies also have implicated dopamine receptor abnormalities caused by genetic polymorphisms or effects of low serum iron.1,7,8 Pharmacologic studies have suggested that higher doses, rapid titration, and IM injections of antipsychotics are associated with increased NMS risk.3,5 Some studies suggest that 15% to 20% of NMS patients have a history of NMS episodes.1,2 In addition, high-potency first-generation antipsychotics (FGAs)—especially haloperidol—are assumed to carry higher risk than low-potency drugs and second-generation antipsychotics (SGAs), although this hypothesis remains difficult to prove.9-11
These risk factors, however, are not practical for estimating NMS risk in a given patient because they are relatively common compared with the low risk of NMS occurrence. For the vast majority of patients with psychotic symptoms, the benefits of properly indicated antipsychotic pharmacotherapy will outweigh the risks.
Table 1
| Systemic |
| Agitation |
| Dehydration |
| Exhaustion |
| Low serum iron concentrations (normal: 60 to 170 mcg/dL) |
| Diagnoses |
| History of NMS |
| Catatonia |
| Organic brain syndromes |
| Central nervous system |
| Dopamine receptor dysfunction |
| Basal ganglia dysfunction |
| Sympathetic nervous system dysfunction |
| Pharmacologic treatment* |
| Intramuscular or intravenous injections |
| High-potency dopamine antagonists |
| Rapid dose titration |
| High doses |
| FGAs compared with SGAs (?) |
*For individual patients, these common risk factors must be weighted again the benefits of antipsychotic therapy FGAs: first-generation antipsychotics; SGAs:second-generation antipsychotics; NMS: neuroleptic malignant syndromeSource: References 1-5
2. Is NMS related to parkinsonism, catatonia, or malignant hyperthermia?
Parkinsonsim. Some researchers have described NMS as an extreme parkinsonian crisis resulting from overwhelming blockade of dopamine pathways in the brain.1,2,12 In this view, NMS resembles the parkinsonian-hyperthermia syndrome that can occur in Parkinson's disease patients following abrupt discontinuation or loss of efficacy of dopaminergic therapy, which can be treated by reinstituting dopaminergic agents.13 Evidence to support this view includes:
- Parkinsonian signs are a cardinal feature of NMS.
- Withdrawal of dopamine agonists precipitates the syndrome.
- All triggering drugs are dopamine receptor antagonists.
- Risks of NMS correlates with drugs' dopamine receptor affinity.
- Dopaminergic agonists may be an effective treatment.
- Lesions in dopaminergic pathways produce a similar syndrome.
- Patients with NMS have demonstrated low cerebrospinal fluid concentrations of the dopamine metabolite homovanillic acid.14
Catatonia. Fink et al15 and others16-18 have persuasively argued that NMS represents a form of drug-induced malignant catatonia. Evidence supporting this includes:
- The 2 disorders share neuropsychiatric symptoms.
- Catalonic signs are common in NMS.19
- Malignant catatonia and NMS share physiologic and labratory signs.20
- Reintroduction of antipsychotics can acutely worsen both conditions.
- Benzodiazepines and electroconvulsive therapy (ECT) are effective treatments for both disorders.15-18
Lee21 examined the relationship between catatonic features and treatment response of 14 NMS patients. Most patients with catatonic symptoms responded to benzodiazepines, whereas none of those did who had an extrapyramidal-hyperthermic presentation without catatonia. Lee concluded that NMS is heterogeneous and may occur in catatonic and noncatatonic forms that differ in treatment response.
- similar clinical signs of rigidity, hyperthermia, and hypermetabolism
- similar psychologic and labratory signs, such as rhabdomyolysis
- hyperthermia in both responding to dantrolene.
Although the 2 are similar in presentation, malignant hyperthermia occurs intraoperatively and reflects a pharmacogenetic disorder of calcium regulation in skeletal muscle. Additionally, rigidity in malignant hyperthermia does not respond to peripheral-acting muscle relaxants.1,22 Evidence suggests that patients who have previously experienced an NMS episodes are not at risk for malignant hyperthermia.22
3. What is the pathophysiology of NMS?
NMS pathophysiology is complex and likely involves interplay between multiple central and systemic pathways and neurotransmitters. As described above, compelling evidence suggests that dopamine blockade plays a central role.12
Dopamine blockade in the hypothalamus is believed to contribute to thermoregulatory failure, and blockade in the nigrostriatal system likely contributes to muscle rigidity and hypermetabolism. The loss of dopaminergic input to the anterior cingulate-medial orbitofrontal circuit and the lateral orbitofrontal circuit likely con-tributes to the mental status changes and catatonic features seen in NMS.12
Some researchers have proposed competing or complementary hypotheses, however. For example, Gurrera23 proposed that patients who are prone to developing NMS have a vulnerability to a hyperactive and dysregulated sympathetic nervous system, and this trait—together with dopamine system disruption induced by dopamine-blocking agents—produces NMS. Other investigators have implicated serotonin, norepinephrine, gamma-aminobutyric acid and glutaminergic mechanisms.1,12,24,25
4. Are FGAs or SGAs more likely to cause NMS?
NMS is assumed to occur less frequently in patients treated with SGAs than in those receiving FGAs, although this hypothesisis unproven. Isolated reports of NMS have been associated with nearly every SGA.9-11 It is difficult to prove FGA vs SGA liabilities because:
- NMS is rare.
- Dosing practices may be more conser-vative now than in the past.
- Most clinicians are aware of the earlysigns of NMS.
In an epidemiological study of a large database, Stubner et al26 found that patients receiving SGAs had a lower risk of NMS than those treated with haloperidol.26 In this study, the overall rate of NMS was 0.02%.
NMS hotline data. We recently examined which medication classes were implicated in 111 NMS cases reported to the Neuroleptic Malignant Syndrome Information Service hotline (1-888-NMS-TEMP) between 1997 and 2006 (Figure). We included only cases of definite or probable NMS (as diagnosed by hotline consultants) in which a single antipsychotic was administered. Slightly more cases were attributed to FGAs (51%) than SGAs (45%). The remaining cases were attributed to neuroleptics used in medical settings (such as promethazineor prochlorperazine). Because they are now prescribed less often, FGAs accounted for a disproportionate number of NMS cases reported to the hotline. Haloperidol accounted for the majority of FGA cases and 44% of all cases. If we had excluded haloperidol and compared the NMS risk of SGAs to only intermediate- or low-potency FGAs, the relative advantage of SGAs would have been lost. On the other hand, it is clear that SGAs still carry a risk for NMS. Analyses suggest that the SGA-associated classic features of NMS—fever, muscle rigidity, and autonomic and mental status changes—are retained in patients receiving SGAs, although some may not develop the severe rigidity and extreme temperatures common in patients receiving FGAs.9-11 The milder clinical characteristics associated with SGAs may reflect more conservative prescribing patterns or increased awareness and earlier recognition of NMS, which would prevent fulminant presentations.
5. What is the evidence for specific NMS treatments?
NMS is rare, its presentation varies, and its progression is unpredictable. These factors make it difficult to evaluate treatments in controlled clinical trials, and data about the relative efficacy of specific interventions are scarce.
Even so, the notion that NMS represents an extreme variant of drug-induced parkinsonism or catatonia suggests that specific NMS treatments could be based on symptom severity or stage of presentation. We propose a treatment guideline basedon theoretical mechanisms and anecdotal data (Algorithm).2,27-29
Support. After immediate withdrawal of the offending medication, supportive therapy is the cornerstone of NMS treatment.1,2,27
For patients presenting with mild signs and symptoms, supportive care and careful clinical monitoring may be sufficient. Extreme hyperthermia demands volume resuscitation and cooling measures, intensive medical care, and careful monitoring for complications.
Treatment. Despite a lack of consensus on drug treatments for uncomplicated NMS, approximately 40% of patients with acute NMS receive pharmacologic treatments.2
Lorazepam, 1 to 2 mg parenterally, is a reasonable first-line therapy for NMS, especially in individuals with catatonic features.4,15-18,21,30,31 Some investigators recommend higher doses.15 Benzodiazepines are preferred if sedation is required in agitated NMS patients.4,15-18
Dopaminergic agents such as bromocriptine and amantadine enhance dopaminergic transmission to reverse parkinsonian symptoms and have been reported to reduce time to recovery and halve mortality rates when used alone or in conjunction with other treatments.13,27,32,33 Rapid discontinuation of these agents can result in rebound symptoms, although this may be true for any specific drug treatment of NMS.1,31,32
Dantrolene uncouples excitation-contraction coupling by enhancing calcium sequestration in sarcoplasmic reticulumin skeletal muscle and has been used to treat NMS hypermetabolic symptoms. Some reviews found improvement in up to 80% of NMS patients treated with dantrolene monotherapy.27,32-35 Compared with supportive care, time to recovery may be reduced—and mortality decreased by almost one-half—when dantrolene is used alone or in combination with other medications.
Not all case reports have shown that dantrolene, benzodiazepines, ordopaminergic agonists are effective in treating NMS.31,36 In our opinion, only advanced NMS cases—with extreme temperature elevations, severe rigidity, and evidence of systemic hypermetabolism—benefit from dantrolene treatment.1,2
ECT has been used successfully to reduce mortality from NMS and other catatonic-spectrum disorders. It is usually employed after supportive therapy and psychopharmacologic interventions fail.2,15,16,27,37 ECT for acute NMS typically consists of a series of 6 to 10 treatments with bilateral electrode placement. Daily ECT may be needed initially.15
6. Are antipsychotics contraindicated following an NMS episode?
The rate of NMS recurrence on retreatment with an antipsychotic has varied.38 We estimate that up to 30% of patients may be at risk of NMS recurrence when rechallenged with an antipsychotic.1 By following proper precautions (Table 2), however, you can safely treat most patients who require continued antipsychotic therapy.1,2 When you restart treatment, a lower-potency antipsychotic from a different chemical class may be a safer option than retrying the triggering agent, according to retrospective analyses of limited available data. A patient who develops NMS on a FGA might benefit from an SGA trial, although some risk of recurrence remains.1,10
Current Psychiatry 2007;6(8):89-95.
Drug brand names
- Amantadine • Symmetrel
- Bromocriptine • Parlodel
- Chlorpromazine • Thorazine
- Dantrolene • Dantrium
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Lorazepam • Ativan
- Loxapine • Loxitane
- Perphenazine • Trilafon
- Prochlorperazine • Compazine, Compro
- Promethazine • Phenergan
- Thioridazine • Mellaril
Disclosure
Dr. Strawn is an American Psychiatric Institute for Research and Education (APIRE)/Janssen Scholar.
Dr. Keck has received research support from or served as a consultant to Abbott Laboratories, American Diabetes Association, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb, GlaxoSmithKline, Eli Lilly and Company, Janssen Pharmaceutica, National Institute of Mental Health, National Institute of Drug Abuse, Pfizer, Stanley Medical Research Institute, and UCB Pharma.
Dr. Caroff has received research support from Bristol-Myers Squibb, Ortho-McNeil Neurologics, and Pfizer.
1. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A, eds. Neuroleptic malignant syndrome and related conditions 2nd ed. Washington, DC: American Psychiatric Publishing Inc; 2003; 1-44.
2. Strawn JR, Keck PE, Jr, Caroff SN. Neuroleptic malignant syndrome Am J Psychiatry 2007;164:870-6.
3. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome Arch Gen Psychiatry 1989;46:914-18.
4. Rosebush PI, Stewart TD. A prospective analysis of 24 episodes of neuroleptic malignant syndrome Am J Psychiatry 1989;146:717-25.
5. Berardi D, Amore M, Keck PE, Jr, et al. Clinical and pharmacologic risk factors for neuroleptic malignant syndrome: a case-control study. Biol Psychiatry 1998;44:748-54.
6. White DA, Robins AH. Catatonia: harbinger of the neuroleptic malignant syndrome Br J Psychiatry 1991;158:419-21.
7. Rosebush PI, Mazurek MF. Serum iron and neuroleptic malignant syndrome. Lancet 1991;338:149-51.
8. Lee JW. Serum iron in catatonia and neuroleptic malignant syndrome Biol Psychiatry 1998;44:499-507.
9. Ananth J, Parameswaran S, Gunatilake S, et al. Neuroleptic malignant syndrome and atypical antipsychotic drugs J Clin Psychiatry 2004;65:464-70.
10. Caroff SN, Mann SC, Campbell EC. Atypical antipsychotics and neuroleptic malignant syndrome Psychiatr Ann 2000;30:314-21.
11. Hasan S, Buckley P. Novel antipsychotics and the neuroleptic malignant syndrome Am J Psychiatry 1998;155:1113-16.
12. Mann SC, Caroff SN, Fricchione G, Campbell EC. Central dopamine hypoactivity and the pathogenesis of neuroleptic malignant syndrome Psychiatr Ann 2000;30:363-74.
13. Factor SA, Santiago A. Parkinsonism-hyperpyrexia syndrome in Parkinson’s disease. In: Frucht SJ, Fahn S, eds. Movement disorder emergencies: diagnosis and treatment. Totowa, NJ: Humana Press; 2005; 29-40.
14. Nisijima K, Ishiguro T. Cerebrospinal fluid levels of monoamine metabolites and gamma-aminobutyric acid in neuroleptic malignant syndrome. J Psychiatr Res 1995;27:233-44.
15. Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Prog Neuropsychopharmacol Biol Psychiatry 2006;30:1182-3.
16. Fricchione G, Bush G, Fozdar M, et al. Recognition and treatment of the catatonic syndrome. J Intensive Care Med 1997;12:135-47.
17. Philbrick KL, Rummans TA. Malignant catatonia. J Neuropsychiatry Clin Neurosci 1994;6:1-13.
18. Mann SC, Caroff SN, Bleier HR, et al. Lethal catatonia. Am J Psychiatry 1986;143:1374-81.
19. Koch M, Chandragiri S, Rizvi S, et al. Catatonic signs in neuroleptic malignant syndrome. Compr Psychiatry 2000;41:73-5.
20. Lee JW. Laboratory findings. In: Caroff SN, Mann SC, Francis A, Fricchoine GL, eds. Catatonia: from psychopathology to neurobiology Washington, DC: American Psychiatric Press, Inc; 2004; 65-75.
21. Lee JW. Catatonic variants, hyperthermic extrapyramidal reactions, and subtypes of neuroleptic malignant syndrome. Ann Clin Psychiatry 2007;19:9-16.
22. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the perioperative setting. Am J Anesthesiol 2001;28:387-93.
23. Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry 1999;156:169-80.
24. Carroll BT. The universal field hypothesis of catatonia and neuroleptic malignant syndrome. CNS Spectr 2000;5:26-33.
25. Weller M, Kornhuber J. A rationale for NMDA receptor antagonist therapy of the neuroleptic malignant syndrome. Med Hypotheses 1992;38:329-33.
26. Stubner S, Rustenbeck E, Grohmann R, et al. Severe and uncommon involuntary movement disorders due to psychotropic drugs. Pharmacopsychiatry 2004;37(suppl 1):S54-S64.
27. Davis JM, Caroff SN, Mann SC. Treatment of neuroleptic malignant syndrome. Psychiatr Ann 2000;30:325-31.
28. Adityanjee PA, Singh S, Singh G, Ong S. Spectrum concept of neuroleptic malignant syndrome. Br J Psychiatry 1988;153:107-11.
29. Woodbury MM, Woodbury MA. Neuroleptic-induced catatonia as a stage in the progression toward neuroleptic malignant syndrome. J Am Acad Child Adolesc Psychiatry 1992;31:1161-4.
30. Francis A, Chondragivi S, Rizvi S, et al. Is lorazepam a treatment for neuroleptic malignant syndrome? CNS Spectr 2000;5:54-7.
31. Rosebush PI, Stewart T, Mazurek MF. The treatment of neuroleptic malignant syndrome. Are dantrolene and bromocriptine useful adjuncts to supportive care? Br J Psychiatry 1991;159:709-12.
32. Sakkas P, Davis JM, Janicak PG, Wang ZY. Drug treatment of the neuroleptic malignant syndrome. Psychopharmacol Bull 1991;27:381-4.
33. Rosenberg MR, Green M. Neuroleptic malignant syndrome: review of response to therapy. Arch Intern Med 1989;149:1927-31.
34. Yamawaki S, Morio M, Kazamutsuri G, et al. Clinical evaluation and effective usage of dantrolene sodium in neuroleptic malignant syndrome. Kiso to Rinsyou (Clinical Reports) 1993;27:1045-66.
35. Tsutsumi Y, Yamamoto K, Matsuura S, et al. The treatment of neuroleptic malignant syndrome using dantrolene sodium. Psychiatry Clin Neurosci 1998;52:433-8.
36. Reulbach U, Dutsch C, Biermann T, et al. Managing an effective treatment for neuroleptic malignant syndrome. Crit Care 2007;11:R4.-
37. Troller JN, Sachdev PS. Electroconvulsive treatment of neuroleptic malignant syndrome: a review and report of cases. Aust N Z J Psychiatry 1999;33:650-9.
38. Pope HG, Aizley HG, Keck PE, Jr, McElroy SL. Neuroleptic malignant syndrome: long term follow-up of 20 cases. J Clin Psychiatry 1991;52:208-12.
1. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A, eds. Neuroleptic malignant syndrome and related conditions 2nd ed. Washington, DC: American Psychiatric Publishing Inc; 2003; 1-44.
2. Strawn JR, Keck PE, Jr, Caroff SN. Neuroleptic malignant syndrome Am J Psychiatry 2007;164:870-6.
3. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome Arch Gen Psychiatry 1989;46:914-18.
4. Rosebush PI, Stewart TD. A prospective analysis of 24 episodes of neuroleptic malignant syndrome Am J Psychiatry 1989;146:717-25.
5. Berardi D, Amore M, Keck PE, Jr, et al. Clinical and pharmacologic risk factors for neuroleptic malignant syndrome: a case-control study. Biol Psychiatry 1998;44:748-54.
6. White DA, Robins AH. Catatonia: harbinger of the neuroleptic malignant syndrome Br J Psychiatry 1991;158:419-21.
7. Rosebush PI, Mazurek MF. Serum iron and neuroleptic malignant syndrome. Lancet 1991;338:149-51.
8. Lee JW. Serum iron in catatonia and neuroleptic malignant syndrome Biol Psychiatry 1998;44:499-507.
9. Ananth J, Parameswaran S, Gunatilake S, et al. Neuroleptic malignant syndrome and atypical antipsychotic drugs J Clin Psychiatry 2004;65:464-70.
10. Caroff SN, Mann SC, Campbell EC. Atypical antipsychotics and neuroleptic malignant syndrome Psychiatr Ann 2000;30:314-21.
11. Hasan S, Buckley P. Novel antipsychotics and the neuroleptic malignant syndrome Am J Psychiatry 1998;155:1113-16.
12. Mann SC, Caroff SN, Fricchione G, Campbell EC. Central dopamine hypoactivity and the pathogenesis of neuroleptic malignant syndrome Psychiatr Ann 2000;30:363-74.
13. Factor SA, Santiago A. Parkinsonism-hyperpyrexia syndrome in Parkinson’s disease. In: Frucht SJ, Fahn S, eds. Movement disorder emergencies: diagnosis and treatment. Totowa, NJ: Humana Press; 2005; 29-40.
14. Nisijima K, Ishiguro T. Cerebrospinal fluid levels of monoamine metabolites and gamma-aminobutyric acid in neuroleptic malignant syndrome. J Psychiatr Res 1995;27:233-44.
15. Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Prog Neuropsychopharmacol Biol Psychiatry 2006;30:1182-3.
16. Fricchione G, Bush G, Fozdar M, et al. Recognition and treatment of the catatonic syndrome. J Intensive Care Med 1997;12:135-47.
17. Philbrick KL, Rummans TA. Malignant catatonia. J Neuropsychiatry Clin Neurosci 1994;6:1-13.
18. Mann SC, Caroff SN, Bleier HR, et al. Lethal catatonia. Am J Psychiatry 1986;143:1374-81.
19. Koch M, Chandragiri S, Rizvi S, et al. Catatonic signs in neuroleptic malignant syndrome. Compr Psychiatry 2000;41:73-5.
20. Lee JW. Laboratory findings. In: Caroff SN, Mann SC, Francis A, Fricchoine GL, eds. Catatonia: from psychopathology to neurobiology Washington, DC: American Psychiatric Press, Inc; 2004; 65-75.
21. Lee JW. Catatonic variants, hyperthermic extrapyramidal reactions, and subtypes of neuroleptic malignant syndrome. Ann Clin Psychiatry 2007;19:9-16.
22. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the perioperative setting. Am J Anesthesiol 2001;28:387-93.
23. Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry 1999;156:169-80.
24. Carroll BT. The universal field hypothesis of catatonia and neuroleptic malignant syndrome. CNS Spectr 2000;5:26-33.
25. Weller M, Kornhuber J. A rationale for NMDA receptor antagonist therapy of the neuroleptic malignant syndrome. Med Hypotheses 1992;38:329-33.
26. Stubner S, Rustenbeck E, Grohmann R, et al. Severe and uncommon involuntary movement disorders due to psychotropic drugs. Pharmacopsychiatry 2004;37(suppl 1):S54-S64.
27. Davis JM, Caroff SN, Mann SC. Treatment of neuroleptic malignant syndrome. Psychiatr Ann 2000;30:325-31.
28. Adityanjee PA, Singh S, Singh G, Ong S. Spectrum concept of neuroleptic malignant syndrome. Br J Psychiatry 1988;153:107-11.
29. Woodbury MM, Woodbury MA. Neuroleptic-induced catatonia as a stage in the progression toward neuroleptic malignant syndrome. J Am Acad Child Adolesc Psychiatry 1992;31:1161-4.
30. Francis A, Chondragivi S, Rizvi S, et al. Is lorazepam a treatment for neuroleptic malignant syndrome? CNS Spectr 2000;5:54-7.
31. Rosebush PI, Stewart T, Mazurek MF. The treatment of neuroleptic malignant syndrome. Are dantrolene and bromocriptine useful adjuncts to supportive care? Br J Psychiatry 1991;159:709-12.
32. Sakkas P, Davis JM, Janicak PG, Wang ZY. Drug treatment of the neuroleptic malignant syndrome. Psychopharmacol Bull 1991;27:381-4.
33. Rosenberg MR, Green M. Neuroleptic malignant syndrome: review of response to therapy. Arch Intern Med 1989;149:1927-31.
34. Yamawaki S, Morio M, Kazamutsuri G, et al. Clinical evaluation and effective usage of dantrolene sodium in neuroleptic malignant syndrome. Kiso to Rinsyou (Clinical Reports) 1993;27:1045-66.
35. Tsutsumi Y, Yamamoto K, Matsuura S, et al. The treatment of neuroleptic malignant syndrome using dantrolene sodium. Psychiatry Clin Neurosci 1998;52:433-8.
36. Reulbach U, Dutsch C, Biermann T, et al. Managing an effective treatment for neuroleptic malignant syndrome. Crit Care 2007;11:R4.-
37. Troller JN, Sachdev PS. Electroconvulsive treatment of neuroleptic malignant syndrome: a review and report of cases. Aust N Z J Psychiatry 1999;33:650-9.
38. Pope HG, Aizley HG, Keck PE, Jr, McElroy SL. Neuroleptic malignant syndrome: long term follow-up of 20 cases. J Clin Psychiatry 1991;52:208-12.