User login
Optimizing benzodiazepine treatment of anxiety disorders
Though once the main treatment for anxiety disorders—often as monotherapy1—benzodiazepines are now primarily used as adjunctive agents.2-4 Their ability to produce rapid anxiolysis represents a significant therapeutic advantage, but in recent decades their tolerability, class-specific risks, and lack of antidepressant properties contributed to benzodiazepines being largely replaced by selective serotonin reuptake inhibitors (SSRIs) for the pharmacologic treatment of anxiety. This shift within the pharmacologic armamentarium has decreased many clinicians’ familiarity with benzodiazepines.
While benzodiazepines continue to have an important role in managing anxiety disorders, particularly treatment-resistant anxiety,4 clinicians must consider the limitations of these agents. Benzodiazepines can be associated with abuse and dependence, and overdose risk when combined with opiates.5,6 They may cause memory impairment7,8 and conflicting data suggest they may contribute to the risk of developing cognitive disorders.9-11 Benzodiazepines also have been associated with falls and fractures,12 and worse outcomes in patients with posttraumatic stress disorder.13 Some studies of patients with chronic obstructive pulmonary disease (COPD) found benzodiazepines may increase the risk of COPD exacerbations and accidental overdose,14 though others found that was not always the case.15 Benzodiazepines may be associated with an increased risk of spontaneous abortion when used early in pregnancy.16 Prospective research in women who were breastfeeding found benzodiazepines may cause sedation in up to 2% of infants.17
Despite the potential for adverse effects, benzodiazepine use remains common.18 These medications have a rapid onset of action, are useful for breakthrough symptoms, may enhance treatment adherence, and alleviate activating symptoms of SSRIs. Like other commonly used medications, benzodiazepines have the potential for both harm and benefit.19 Similar to other medications with tolerability concerns but established efficacy, particularly in treatment-resistant anxiety disorders, it is important to balance “overprescribing … to patients at risk and underusing these effective medications when indicated.”19 Though the use of benzodiazepines has been discouraged and perceptions have shifted, knowledge of benzodiazepines and benzodiazepine pharmacology also has been degraded contemporaneously.
This article provides a synthesis of the clinically relevant pharmacology of benzodiazepines, with a focus on orally administered benzodiazepines, which are more common in outpatient clinical practice. Specifically, this review describes the pharmacology of benzodiazepines, benzodiazepine medication interactions, the relationship between pharmacologic characteristics and treatment response/tolerability, and selection and dosing of oral benzodiazepines (Table20).

Benzodiazepine pharmacodynamics
Benzodiazepines act at the gamma-aminobutyric acid (GABA)-A receptor complex and bind allosterically.21-23 Comprised of 5 glycoprotein subunits (2 alpha subunits, 2 beta subunits, and 1 gamma subunit), the receptor has 2 distinct sites at which the endogenous inhibitory transmitter GABA binds and 1 benzodiazepine binding site. Benzodiazepines bind within a socket created by the alpha and gamma subunits22 and after binding induce a conformational change in the receptor, which enhances GABA binding. There are 2 types of benzodiazepine receptors: BZ1 and BZ2. The subunits play a critical role in driving the pharmacologic characteristics of the receptor.24 BZ1 and BZ2 receptors bind benzodiazepines, although they are differentially distributed within the brain. Binding at BZ1 receptors—which are distributed in cortical, thalamic, and cerebellar regions—contributes to sedation and deleterious effects of benzodiazepines on memory (eg, anterograde amnesia). BZ2 receptors (which contain gamma-2 subunits) are responsible for anxiolytic and muscle-relaxing effects. They are distributed throughout limbic regions and motor tracts, including motor neurons and neurons in the dorsal horn of the spinal cord.24
Benzodiazepines—positive GABA-A receptor allosteric modulators—produce phasic inhibition, largely through the alpha and gamma subunits discussed above. In contrast, newer positive allosteric modulators (eg, zuranolone) bind at the alpha/beta subunits.25 Mechanistically, endogenous neuroactive steroids and nonbenzodiazepine GABA-A–positive allosteric modulators such as zuranolone and ganaxolone also differ in their regulation of GABA-A (downregulated with benzodiazepines and hypothetically upregulated with zuranolone)26 and their synaptic effects (benzodiazepines synaptically vs endogenous neurosteroids and nonbenzodiazpine positive allosteric modulators extrasynaptically).27
From a developmental perspective, benzodiazepines may have less efficacy for anxiolysis and worse tolerability in some pediatric patients,28 although they generally appear effective for immediate use to treat anxiety in acute settings.29 The differences in efficacy and tolerability may be related to pharmacodynamic differences between pediatric populations and adults. GABA receptor expression and function do not reach adult levels until age 14 to 17½ for subcortical regions and age 18 to 22 for cortical regions, although girls reach adult expression of GABA receptors slightly earlier than boys.30 D
Continue to: Pharmacology and clinical effects
Pharmacology and clinical effects
Benzodiazepine pharmacokinetics are intimately linked with the onset of action and duration of clinical effect and vary based on the route of administration, absorption, and distribution/redistribution.31 In this review, we focus on oral administration as opposed to IV, IM, sublingual, or intranasal administration.
Absorption
Benzodiazepines are rapidly absorbed after oral administration and quickly enter the systemic circulation. However, absorption rates vary depending on specific aspects of the gastrointestinal milieu and intrinsic properties of the benzodiazepine. For example, alprazolam is more rapidly absorbed than most other benzodiazepines, with a Tmax of 1.8 hours compared to lorazepam, which has a Tmax of approximately 2 hours. These pharmacokinetic effects instantiate differences in tolerability and efficacy. Thus, following single doses of alprazolam and diazepam, self-rated sedating effects and impairment on a task of working memory suggest that effects have a more rapid onset for alprazolam relative to lorazepam.32 Food and concomitant medications can significantly affect benzodiazepine absorption. A single-dose, 3-way crossover study demonstrated that taking diazepam concomitantly with an antacid (eg, aluminum hydroxide) decreased peak concentrations and prolonged absorption by approximately 30 minutes. However, total absorption of the medication was unaffected.33 Additionally, administration of diazepam with food significantly slows absorption from 1 hour 15 minutes to approximately 2 hours 30 minutes and increases benzodiazepine absorption by 25% (Figure 134); the fat content of the meal appears important in moderating this effect.35 The impact of food on alprazolam varies by formulation. For example, when administered in an extended-release (XR) formulation with a high-fat meal, alprazolam absorption increases by one-third, while absorption for administration of the orally disintegrating tablet with a high-fat meal increases from 1 hour 30 minutes to 2 hours. Similarly, for lorazepam, administration with a meal delays absorption by approximately 2 hours; however, this effect does not appear present with the XR formulation. Administering benzodiazepines with food can be clinically leveraged to either accelerate the onset of action or decrease peak-associated adverse effects. Thus, when a highly lipophilic benzodiazepine is needed to treat acute anxiety or prior to an expected anxiogenic stimuli, administering the medication without food may produce a faster onset of action.

CNS penetration
Benzodiazepines enter the CNS by passive diffusion. Because of this, lipophilicity at physiologic pH influences the rate at which a benzodiazepine crosses the blood-brain barrier. The rate at which benzodiazepines enter the CNS influences their clinical effects and the speed at which both efficacy (ie, anxiolysis) and adverse effects (ie, sedation, slowed cognition) are observed. In general, more lipophilic medications initiate their anxiolytic effect more quickly. However, by quickly leaving the CNS (through the same mechanism that allowed them to enter the CNS at such speed), their effects rapidly cease as they redistribute into fat. Thus, highly lipophilic benzodiazepines produce more intense effects compared to less lipophilic benzodiazepines. For these reasons, lipophilicity is more important than half-life for determining the duration of effect in most patients.
Lipophilicity and duration of effect
Benzodiazepines and their metabolites tend to be highly protein-bound and distributed in fat- and lipid-enriched areas such as the CNS. As a result, the more lipophilic agents generally have the highest rates of absorption and the fastest onset of clinical effects. The duration of action for many benzodiazepines is determined by the rate and extent of distribution (a function of lipophilicity) rather than by the rate of elimination. For example, diazepam has a longer half-life than lorazepam, but its duration of action following a single dose is shorter. This is because diazepam is more lipophilic and therefore more extensively distributed (particularly to adipose tissue). This results in it leaving the brain and blood and distributing to other tissues. In turn, its CNS effect (ie, anxiolytic effects) are more quickly terminated.
By contrast, less lipophilic benzodiazepines maintain their CNS concentrations longer; they have a longer duration of action because of their slower redistribution, which culminates in a shorter half-life, and are less extensively distributed to peripheral tissues. In essence, this means that (other things being equal) a less lipophilic benzodiazepine produces a more sustained anxiolytic effect compared to a highly lipophilic benzodiazepine.36 Lipophilicity is also important in predicting some cognitive adverse effects, including amnesia. Benzodiazepines with high lipophilicity have greater absorption and faster onset of action as well as more rapid amnestic effects.37,38 These effects may relate to overall efficacy differences for oral benzodiazepines. A recent meta-analysis by Stimpfl et al36 found that less lipophilic benzodiazepines produced a greater response compared to more lipophilic benzodiazepines.
Continue to: Metabolism
Metabolism
Regarding cytochrome P450 (CYP) metabolism, polymorphic CYP2C19 and CYP3A4/5 are involved in the metabolism of several benzodiazepines39 and CYP2B6 has been recognized as a contributor to diazepam metabolism. CYP3A5 gene polymorphisms may produce variation in alprazolam metabolism; however, the predominant cytochrome involved in the metabolism of oxidatively metabolized benzodiazepines (ie, benzodiazepines other than lorazepam, oxazepam, and temazepam) is primarily CYP3A4, and most effects on CYP3A4 activity are related to concomitant medications and other nongenetic factors.
Drug-drug interactions
Apart from lorazepam,40,41 oxazepam,42,43 and temazepam, most benzodiazepines are metabolized through oxidative mechanisms that involve CYP3A4 (Figure 220).39 As such, their metabolism is influenced by medications that impact CYP3A4, including antifungals (eg, ketoconazole), calcium channel blockers (eg, verapamil, diltiazem), nefazodone, some protease inhibitors, and macrolide antibiotics. Research has examined the impact of low-dose estrogen oral contraceptives (OCPs) on exposure (eg, plasma concentrations) of several benzodiazepines. The mechanism for this interaction is likely complex and putatively involves multiple pathways, including inhibition of CYP3A4 by OCPs. The effects of OCPs on benzodiazepine pharmacokinetics vary based on the metabolism of the benzodiazepine. In general, medications oxidized and nitroreduced (eg, chlordiazepoxide, alprazolam, diazepam, and nitrazepam) have decreased clearance in patients treated with OCPs. Regarding nonoxidatively metabolized benzodiazepines, data are mixed. Research found no OCP-related effects on the pharmacokinetics of nonoxidatively metabolized benzodiazepines44; another study suggested that clearance of these medications—through increased glucuronidation—may be increased.31 The effect of smoking on benzodiazepine concentration has been well documented. Smoking increases the clearance of orally administered diazepam,45 but not IV diazepam, midazolam, or lorazepam, suggesting that this represents a first-pass effect.46 For alprazolam, plasma concentrations are reduced by 15% to 30% in smokers and total body clearance is 24% greater compared to nonsmokers, which results in an approximately 50% increase in half-life in nonsmokers compared to smokers.47 The most notable interaction between benzodiazepines and SSRIs is seen with fluvoxamine. Because fluvoxamine moderately inhibits CYP2C19 and CYP3A4 and potently inhibits CYP1A2,48 the clearance of oxidatively metabolized benzodiazepines is reduced.49 Additionally, the effects of grapefruit juice—a potent inhibitor of CYP3A4—has been evaluated for several benzodiazepines. Yasui et al50 found grapefruit juice did not alter alprazolam plasma concentrations. However, in separate research, grapefruit juice tripled diazepam exposure, increased peak concentrations 1.5-fold, and prolonged absorption.51

Hepatic disease
Exposure to benzodiazepines—other than lorazepam, oxazepam, and temazepam—is influenced by intrinsic hepatic disease and requires dose adjustment in individuals with significant hepatic impairment. The impact of hepatic disease on the clinical pharmacology of benzodiazepines may relate to 2 factors: protein binding and metabolism. In a study of individuals with cirrhosis, lorazepam binding was decreased, although its metabolism and clearance were largely unaffected.40
Aging and benzodiazepine metabolism/clearance
Aging is associated with myriad physiologic changes (eg, decrease in renal clearance after age 40, changes in body fat distribution, changes in activity of cytochromes) that are relevant to benzodiazepine pharmacology. They may underlie differences in the tolerability of benzodiazepines and other clinically relevant characteristics (eg, duration of action, accumulation).
Several studies have evaluated the impact of aging on the clearance and disposition of selected benzodiazepines. The respective half-lives of chlordiazepoxide and diazepam increase from 4- to 6-fold from age 20 to 80. Further, with chronic dosing, highly lipophilic benzodiazepines may require additional attention in geriatric patients. In a study that included individuals up to age 78, steady-state plasma concentrations of diazepam and its metabolite, desmethyldiazepam (DMDZ), were 30% to 35% higher in older patients compared to younger individuals.52 In this study, the half-lives for the young and older patients were 31 hours and 86 hours, respectively, for diazepam, and 40 hours and 80 hours, respectively, for the active metabolite. The half-life of diazepam is increased by “1 hour for each year of age beginning with a half-life of 20 hours at 20 years of age, as the volume of distribution is increased, and clearance is decreased.”52 Clinically, this implies that in older adults, clinicians should expect lower peak concentrations (Cmax), higher trough concentrations (Cmin), and that diazepam will take longer to reach steady-state concentrations. Taken together, these findings raised concern that “slow accumulation and delayed washout of diazepam and DMDZ is probable.”52 These findings—which may have more clinical relevance than those of single-dose studies—suggest that the effects related to diazepam would also take longer to resolve in older patients. Finally, lorazepam clearance or distribution does not appear to be affected by aging, at least in patients age 15 to 73.40 Alprazolam is more slowly cleared in geriatric patients and its effects may be potentiated by reduced protein binding.
Continue to: Obesity
Obesity
The distribution of medications, including benzodiazepines, is altered in patients who are obese because of increased adipose tissue.53,54 This increase in the volume of distribution can attenuate the onset of action, increase medication accumulation in fat, and potentiate the duration of action.55,56
Obesity may also affect hepatic metabolism by induction of CYP1A2, CYP2C9, and CYP2C19, and inhibition of CYP3A4.57 Triazolam, which is metabolized by CYP3A4, is associated with a greater exposure (ie, plasma concentrations) in individuals who are obese.58 However, when considering differences in benzodiazepine pharmacokinetics in patients who are obese, clinicians must remember that elimination half-life depends on both volume of distribution and clearance. In
How quickly do benzodiazepines work?
Benzodiazepines act quickly. Meta-analyses36 suggest that improvement in anxiety symptoms compared to placebo is greatest initially and then the rate of improvement slows over successive weeks. Research on benzodiazepines reveals statistically significant differences between benzodiazepines and placebo within the first week of treatment, with >80% of the expected improvement by Week 8 of treatment emerging by Week 4 (Figure 336). The rapid reduction in anxiety symptoms seen with benzodiazepines has important treatment implications, given that traditional psychotherapeutic and antidepressant treatments are slow to produce improvements. Consistent data suggesting that benzodiazepines work faster than other treatments support that they may have a role during the initiation of other treatments.

What is the ‘best’ dose?
As seen with other classes of psychotropic medications,4 the relationship between benzodiazepine dose and response is complex. In a recent meta-analysis of 65 placebo-controlled trials of benzodiazepines in adults with anxiety disorders, there was a superior response over time for low-dose benzodiazepines (<3 mg/d in lorazepam equivalents) compared to a medium dose (3 to 6 mg/d; P = .042); high-dose benzodiazepines (>6 mg/d) yielded less improvement compared to medium doses (P = .001).36 A study of adults with panic disorder similarly found the greatest responses with alprazolam plasma concentrations of 20 to 40 ng/mL, with no additional benefit at <20 ng/mL or >40 ng/mL.49 As plasma concentrations increase, adverse effects such as sedation also increase, which may confound the observed loss of a dose-response relationship at higher doses and plasma concentrations.62 This may, in part, account for the observation that higher doses of benzodiazepines are associated with greater depressive symptoms and disrupted sleep.63 As such, low doses may represent a delicate equipoise between efficacy and tolerability, yielding the most optimal clinical response.
Which benzodiazepine should I prescribe?
Comparing benzodiazepines is difficult, given the differences in dosing and disorders studied and differences in how each individual clinical trial was conducted. A meta-analysis by Stimpfl et al36 that used Bayesian hierarchical modeling, which allowed some of this heterogeneity to be addressed, found that relative to the reference benzodiazepine (lorazepam), clonazepam had the greatest trajectory/magnitude of response (other specific benzodiazepines did not statistically differ from lorazepam) (Figure 436).

Continue to: Another aspect of the superiority...
Another aspect of the superiority of clonazepam in some research relates to its pharmacokinetic properties, particularly when compared with benzodiazepines that have very short half-lives. Short half-life benzodiazepines have been associated with rebound anxiety, which is defined as “the relative worsening of symptoms on discontinuation of treatment as compared to baseline symptoms” and is distinct from withdrawal.64 While it is difficult to assess this in clinical trials, Herman et al65 provided insight into the contribution of rebound anxiety in a study of patients with panic disorder treated with alprazolam who experienced “interdose anxiety symptoms.” Of the 48 patients in this study, 41 switched to clonazepam, and most who switched (82%) experienced improvement. The improvement was attributed to the decreased frequency of clonazepam (vs alprazolam) administration and lack of interdose anxiety. When selecting an oral benzodiazepine, consider the duration, onset of action, and differences in metabolism that produce varying levels of effectiveness for individual patients. In situations where rapid onset is desired, a short-acting benzodiazepine may be preferable, while a longer-acting benzodiazepine would be preferable in situations where the patient needs sustained effects.
Regarding lipophilicity, differences among benzodiazepines could contribute to differences in psychological dependence and differential utility in some situations. For example, alprazolam rapidly enters the CNS, producing an immediate anxiolytic effect. However, its egress from the CNS is equally rapid, and its anxiolytic effects disappear quickly. This may be desirable for addressing acute, predictable anxiety, but could have unintended consequences in treating chronic anxiety, where it could facilitate psychological dependence.
Practical considerations
When prescribing benzodiazepines, consider a myriad of patient- and medication-specific factors, as these have clinically relevant implications on treatment response. This information, taken together, supports the importance of an individualized approach to benzodiazepine use. Before selecting a benzodiazepine and during treatment, important elements of the patient’s history must be considered, including age, body weight, concomitant medication use (eg, antacids, CYP3A4 inhibitors, OCPs), smoking status, and history of hepatic or renal disease.
Patients age <18 are unlikely to have full expression of GABA receptors in the brain30 and therefore benzodiazepines may not be as efficacious for anxiolysis in this population. Moreover, compared to younger patients, older patients may experience higher steady-state concentrations of benzodiazepines, especially lipophilic agents, due to an increased volume of distribution and decreased clearance. In patients treated with OCPs, some benzodiazepines may take longer to reach steady-state, and dose adjustments may need to be considered. In patients who smoke, clearance of some oral benzodiazepines is also accelerated, potentially decreasing half-life by up to 50%.
When dosing and titrating benzodiazepines, consider the patient’s body weight, particularly if they are obese. The effects of obesity on benzodiazepine pharmacokinetics are complex. For glucuronidated benzodiazepines, clearance is increased in patients who are obese; however, the volume of distribution is also increased in such patients, meaning it will take longer for benzodiazepines to achieve steady-state in these individuals compared to patients who are not obese. These effects suggest it may take longer to achieve a response at a given dose in patients who are obese compared to individuals who are not obese.
Continue to: The properties of individual benzodiazepines...
The properties of individual benzodiazepines should also be considered when selecting a benzodiazepine treatment. If circumstances necessitate rapid symptom relief, a lipophilic benzodiazepine, such as diazepam, may be preferred for quick onset and offset of action. Onset of action may also be hastened by taking the benzodiazepine without food; conversely, if peak adverse effects are problematic, concurrent consumption of a high-fat meal may help decrease peak concentration and prolonging absorption. In other circumstances, such as if sustained anxiolysis is desired, a clinician may opt for a less lipophilic benzodiazepine, such as clonazepam. Finally, in terms of general treatment response, benzodiazepines separate from placebo in the first week of treatment, which supports the idea they may be useful during the introduction of other medications (eg, SSRIs) that take a longer time to achieve clinical effect.
Bottom Line
The pharmacokinetics of benzodiazepines are intimately linked with the onset of action and duration of clinical effect and vary based on individual absorption and distribution/redistribution. Benzodiazepines’ clinical profile derives from their pharmacokinetic differences and is influenced by many factors, including age, body weight, concomitant medication use, smoking status, and hepatic or renal disease. Consider these factors to individualize the approach to using benzodiazepines and optimize tolerability and efficacy.
Related Resources
- Weber SR, Duchemin AM. Benzodiazepines: sensible prescribing in light of the risks. Current Psychiatry. 2018;17(2):22-27.
- Balon R. Benzodiazepines for anxious depression. Current Psychiatry. 2018;17(8):9-12.
Drug Brand Names
Alprazolam • Xanax
Chlordiazepoxide • Librium
Clobazam • Onfi
Clonazepam • Klonopin
Clorazepate • Gen-Xene
Diazepam • Valium
Diltiazem • Cardizem
Fluvoxamine • Luvox
Ganaxolone • Ztalmy
Ketoconazole • Nizoral
Lorazepam • Ativan
Midazolam • Versed
Temazepam • Restoril
Triazolam • Halcion
Verapamil • Calan
1. Rickels K, Moeller HJ. Benzodiazepines in anxiety disorders: reassessment of usefulness and safety. World J Biol Psychiatry. 2019;20(7):514-518. doi:10.1080/15622975.2018.1500031
2. Stevens JC, Pollack MH. Benzodiazepines in clinical practice: consideration of their long-term use and alternative agents. J Clin Psychiatry. 2005;66(Suppl 2):21-27.
3. Pollack MH, van Ameringen M, Simon NM, et al. A double-blind randomized controlled trial of augmentation and switch strategies for refractory social anxiety disorder. Am J Psychiatry. 2014;171(1):44-53. doi:10.1176/appi.ajp.2013.12101353
4. Strawn JR, Geracioti L, Rajdev N, et al. Pharmacotherapy for generalized anxiety disorder in adult and pediatric patients: an evidence-based treatment review. Expert Opin Pharmacother. 2018;19(10):1057-1070. doi:10.1080/14656566.2018.1491966
5. Karaca-Mandic P, Meara E, Morden NE. The growing problem of co-treatment with opioids and benzodiazepines. BMJ. 2017;356:j1224. doi:10.1136/bmj.j1224
6. Bachhuber MA, Hennessy S, Cunningham CO, et al. Increasing benzodiazepine prescriptions and overdose mortality in the United States, 1996-2013. Am J Public Health. 2016;106(4):686-688. doi:10.2105/AJPH.2016.303061
7. Bentué-Ferrer D, Akwa Y. Benzodiazepines: Effects on memory functioning. In: Pandi-Perumal SR, Verster J, Monti J, et al, eds. Sleep Disorders: Diagnosis and Therapeutics. CRC Press; 2008:104-114. doi:10.3109/9780203091715-15
8. Pomara N, Facelle TM, Roth AE, et al. Dose-dependent retrograde facilitation of verbal memory in healthy elderly after acute oral lorazepam administration.Psychopharmacology (Berl). 2006;185(4):487-494. doi:10.1007/s00213-006-0336-0
9. Gray SL, Dublin S, Yu O, et al. Benzodiazepine use and risk of incident dementia or cognitive decline: prospective population based study. BMJ. 2016;352:i90. doi:10.1136/bmj.i90
10. Biétry FA, Pfeil AM, Reich O, et al. Benzodiazepine use and risk of developing Alzheimer’s disease: a case-control study based on Swiss claims data. CNS Drugs. 2017;31(3):245-251. doi:10.1007/s40263-016-0404-x
11. de Gage SB, Moride Y, Ducruet T, et al. Benzodiazepine use and risk of Alzheimer’s disease: case-control study. BMJ. 2014;349g5205. doi:10.1136/bmj.g5205
12. Shah R, Raji MA, Westra J, et al. Association of co-prescribing of opioid and benzodiazepine substitutes with incident falls and fractures among older adults: a cohort study. BMJ Open. 2021;11(12):e052057. doi:10.1136/bmjopen-2021-052057
13. Guina J, Rossetter SR, DeRhodes BJ, et al. Benzodiazepines for PTSD: a systematic review and meta-analysis. J Psychiatr Pract. 2015;21(4):281-303.
14. Ekström MP, Bornefalk-Hermansson A, Abernethy AP, et al. Safety of benzodiazepines and opioids in very severe respiratory disease: national prospective study. BMJ. 2014;348:g445. doi:10.1136/bmj.g445
15. Donovan LM, Malte CA, Spece LJ, et al. Center predictors of long-term benzodiazepine use in chronic obstructive pulmonary disease and post-traumatic stress disorder. Ann Am Thorac Soc. 2019;16(9):1151-1157. doi:10.1513/AnnalsATS.201901-048OC
16. Sheehy O, Zhao JP, Bérard A. Association between incident exposure to benzodiazepines in early pregnancy and risk of spontaneous abortion. JAMA Psychiatry. 2019;76(9):948-957. doi:10.1001/jamapsychiatry.2019.0963
17. Kelly LE, Poon S, Madadi P, et al. Neonatal benzodiazepines exposure during breastfeeding. J Pediatr. 2012;161(3):448-451. doi:10.1016/j.jpeds.2012.03.003
18. Agarwal SD, Landon BE. Patterns in outpatient benzodiazepine prescribing in the United States. JAMA Netw Open. 2019;2(1):e187399. doi:10.1001/jamanetworkopen.2018.7399
19. Hirschtritt ME, Olfson M, Kroenke K. Balancing the risks and benefits of benzodiazepines. JAMA. 2021;325(4):347-348. doi:10.1001/jama.2020.22106
20. Brunton LL, Hilal-Dandan R, Knollman BC, eds. Goodman & Gilman’s: The Pharmacological Basis of Therapeutics. McGraw-Hill Education; 2018.
21. Nutt DJ, Malizia AL. New insights into the role of the GABA(A)-benzodiazepine receptor in psychiatric disorder. British J Psychiatry. 2001;179:390-396. doi:10.1192/bjp.179.5.390
22. Sigel E. Mapping of the benzodiazepine recognition site on GABA(A) receptors. Curr Top Med Chem. 2002;2(8):833-839. doi:10.2174/1568026023393444
23. Savic
24. Smith TA. Type A gamma-aminobutyric acid (GABAA) receptor subunits and benzodiazepine binding: significance to clinical syndromes and their treatment. Br J Biomed Sci. 2001;58(2):111-121.
25. Althaus AL, Ackley MA, Belfort GM, et al. Preclinical characterization of zuranolone (SAGE-217), a selective neuroactive steroid GABAA receptor positive allosteric modulator. Neuropharmacology. 2020;181:108333. doi:10.1016/j.neuropharm.2020.108333
26. Jacob TC, Michels G, Silayeva L, et al. Benzodiazepine treatment induces subtype-specific changes in GABA(A) receptor trafficking and decreases synaptic inhibition. Proc Natl Acad Sci U S A. 2012;109(45):18595-18600. doi:10.1073/pnas.1204994109
27. Nicholson MW, Sweeney A, Pekle E, et al. Diazepam-induced loss of inhibitory synapses mediated by PLCδ/ Ca2+/calcineurin signalling downstream of GABAA receptors. Mol Psychiatry. 2018;23(9):1851-1867. doi:10.1038/s41380-018-0100-y
28. Dobson ET, Bloch MH, Strawn JR. Efficacy and tolerability of pharmacotherapy for pediatric anxiety disorders: a network meta-analysis. J Clin Psychiatry. 2019;80(1):17r12064. doi:10.4088/JCP.17r12064
29. Kuang H, Johnson JA, Mulqueen JM, et al. The efficacy of benzodiazepines as acute anxiolytics in children: a meta-analysis. Depress Anxiety. 2017;34(10):888-896. doi:10.1002/da.22643
30. Chugani DC, Muzik O, Juhász C, et al. Postnatal maturation of human GABAA receptors measured with positron emission tomography. Ann Neurol. 2001;49(5):618-626. doi:10.1002/ana.1003
31. Jochemsen R, Breimer DD. Pharmacokinetics of benzodiazepines: metabolic pathways and plasma level profiles. Curr Med Res Opin. 1984;8(Suppl 4):60-79. doi:10.1185/03007998409109545
32. Greenblatt DJ, Harmatz JS, Dorsey C, et al. Comparative single-dose kinetics and dynamics of lorazepam, alprazolam, prazepam, and placebo. Clin Pharmacol Ther. 1988;44(3)326-334. doi:10.1038/clpt.1988.158
33. Shader RI, Georgotas A, Greenblatt DJ, et al. Impaired absorption of desmethydiazepam from clorazepate by magnesium aluminum hydroxide. Clin Pharmacol Ther. 1978;24(3):308-315. doi:10.1002/cpt1978243308
34. Greenblatt DJ, Allen MD, MacLaughlin DS, et al. Diazepam absorption: effect of antacids and food. Clin Pharmacol Ther. 1978;24(5):600-609. doi:10.1002/cpt1978245600
35. Yamazaki A, Kumagai Y, Fujita T, et al. Different effects of light food on pharmacokinetics and pharmacodynamics of three benzodiazepines, quazepam, nitrazepam and diazepam. J Clin Pharm Ther. 2007;32(1):31-39. doi:10.1111/j.1365-2710.2007.00795.x
36. Stimpfl J, Mills JA, Strawn JR. Pharmacologic predictors of benzodiazepine response trajectory in anxiety disorders: a Bayesian hierarchical modeling meta-analysis. CNS Spectr. 2023;28(1):53-60. doi:10.1017/S1092852921000870
37. Griffin CE 3rd, Kaye AM, Bueno FR, et al. Benzodiazepine pharmacology and central nervous system-mediated effects. Ochsner J. 2013;13(2):214-223.
38. Buffett-Jerrott SE, Stewart SH. Cognitive and sedative effects of benzodiazepine use. Curr Pharm Des. 2005;8(1):45-58. doi:10.2174/1381612023396654
39. Fukasawa T, Suzuki A, Otani K. Effects of genetic polymorphism of cytochrome P450 enzymes on the pharmacokinetics of benzodiazepines. J Clin Pharm Ther. 2007;32(4):333-341. doi:10.1111/j.1365-2710.2007.00829.x
40. Kraus JW, Desmond PV, Marshall JP, et al. Effects of aging and liver disease on disposition of lorazepam. Clin Pharmacol Ther. 1978;24(4):411-419. doi:10.1002/cpt1978244411
41. Greenblatt DJ. Clinical pharmacokinetics of oxazepam and lorazepam. Clin Pharmacokinet. 1981;6(2):89-105. doi:10.2165/00003088-198106020-00001
42. Walkenstein SS, Wiser R, Gudmundsen CH, et al. Absorption, metabolism, and excretion of oxazepam and its succinate half‐ester. J Pharm Sci. 1964;53(10):1181-1186. doi:10.1002/jps.2600531010
43. Shull HJ, Wilkinson GR, Johnson R, et al. Normal disposition of oxazepam in acute viral hepatitis and cirrhosis. Ann Intern Med. 1976;84(4):420-425. doi:10.7326/0003-4819-84-4-420
44. Abernethy DR, Greenblatt DJ, Ochs HR, et al. Lorazepam and oxazepam kinetics in women on low-dose oral contraceptives. Clin Pharmacol Ther. 1983;33(5):628-632. doi:10.1038/clpt.1983.85
45. Greenblatt DJ, Allen MD, Harmatz JS, et al. Diazepam disposition determinants. Clin Pharmacol Ther. 1980;27(3):301-312. doi:10.1038/clpt.1980.40
46. Ochs HR, Greenblatt DJ, Knüchel M. Kinetics of diazepam, midazolam, and lorazepam, in cigarette smokers. Chest. 1985;87(2):223-226. doi:10.1378/chest.87.2.223
47. Smith RB, Gwilt PR, Wright CE 3rd. Single- and multiple-dose pharmacokinetics of oral alprazolam in healthy smoking and nonsmoking men. Clin Pharm. 1983;2(2):139-143.
48. Figgitt DP, McClellan KJ. Fluvoxamine. An updated review of its use in the management of adults with anxiety disorders. Drugs. 2000;60(4):925-954. doi:10.2165/00003495-200060040-00006
49. Greenblatt DJ, Wright CE. Clinical pharmacokinetics of alprazolam. Therapeutic implications. Clin Pharmacokinet. 1993;24(6):453-471. doi:10.2165/00003088-199324060-00003
50. Yasui N, Kondo T, Furukori H, et al. Effects of repeated ingestion of grapefruit juice on the single and multiple oral-dose pharmacokinetics and pharmacodynamics of alprazolam. Psychopharmacology (Berl). 2000;150(2):185-190. doi:10.1007/s002130000438
51. Özdemir M, Aktan Y, Boydagˇ BS, et al. Interaction between grapefruit juice and diazepam in humans. Eur J Drug Metab Pharmacokinet. 1998;23(1):55-59. doi:10.1007/BF03189827
52. Greenblatt DJ, Harmatz JS, Zhang Q, et al. Slow accumulation and elimination of diazepam and its active metabolite with extended treatment in the elderly. J Clin Pharmacol. 2021;61(2):193-203. doi:10.1002/jcph.1726
53. Abernethy DR, Greenblatt DJ. Drug disposition in obese humans: an update. Clin Pharmacokinet. 1986;11(3):199-213. doi:10.2165/00003088-198611030-00002
54. Hanley MJ, Abernethy DR, Greenblatt DJ. Effect of obesity on the pharmacokinetics of drugs in humans. Clin Pharmacokinet. 2010;49(2):71-87. doi:10.2165/11318100-000000000-00000
55. Bauer LA. Drug Dosing in special populations: renal and hepatic disease, dialysis, heart failure, obesity, and drug interactions. In: Weitz M, Thomas, CM, eds. Applied Clinical Pharmacokinetics. 3rd ed. McGraw-Hill Education; 2014. https://accesspharmacy.mhmedical.com/book.aspx?bookid=1374
56. Kendrick JG, Carr RR, Ensom MHH. Pharmacokinetics and drug dosing in obese children. J Pediatr Pharmacol Ther. 2010;15(2):94-109. doi:10.5863/1551-6776-15.2.94
57. Brill MJE, Diepstraten J, van Rongen A, et al. Impact of obesity on drug metabolism and elimination in adults and children. Clin Pharmacokinet. 2012;51(5):277-304. doi:10.2165/11599410-000000000-00000
58. Derry CL, Kroboth PD, Pittenger AL, et al. Pharmacokinetics and pharmacodynamics of triazolam after two intermittent doses in obese and normal-weight men. J Clin Psychopharmacol. 1995;15(3):197-205. doi:10.1097/00004714-199506000-00008
59. Abernethy DR, Greenblatt DJ, Divoll M, et al. The influence of obesity on the pharmacokinetics of oral alprazolam and triazolam. Clin Pharmacokinet. 1984;9(2):177-183. doi:10.2165/00003088-198409020-00005
60. Abernethy DR, Greenblatt DJ, Divoll M, et al. Prolonged accumulation of diazepam in obesity. J Clin Pharmacol. 1983;23(8-9):369-376. doi:10.1002/j.1552-4604.1983.tb02750.x
61. Abernethy DR, Greenblatt DJ, Divoll M, et al. Enhanced glucuronide conjugation of drugs in obesity: studies of lorazepam, oxazepam, and acetaminophen. J Lab Clin Med. 1983;101(6):873-880.
62. Greenblatt DJ, von Moltke LL, Harmatz JS, et al. Alprazolam pharmacokinetics, metabolism, and plasma levels: clinical implications. J Clin Psychiatry. 1993;54 Suppl:4-11.
63. Chen YT, Liu CY, Chang CM, et al. Perceptions, clinical characteristics, and other factors associated with prolonged and high daily dose of benzodiazepine use among patients with anxiety or depressive disorders. J Affect Disord. 2020;271:215-223. doi:10.1016/j.jad.2020.03.077
64. Herman JB, Brotman AW, Rosenbaum JF. Rebound anxiety in panic disorder patients treated with shorter-acting benzodiazepines. J Clin Psychiatry. 1987;48(Suppl):22-28.
65. Herman JB, Rosenbaum JF, Brotman AW. The alprazolam to clonazepam switch for the treatment of panic disorder. J Clin Psychopharmacol. 1987;7(3):175-178.
Though once the main treatment for anxiety disorders—often as monotherapy1—benzodiazepines are now primarily used as adjunctive agents.2-4 Their ability to produce rapid anxiolysis represents a significant therapeutic advantage, but in recent decades their tolerability, class-specific risks, and lack of antidepressant properties contributed to benzodiazepines being largely replaced by selective serotonin reuptake inhibitors (SSRIs) for the pharmacologic treatment of anxiety. This shift within the pharmacologic armamentarium has decreased many clinicians’ familiarity with benzodiazepines.
While benzodiazepines continue to have an important role in managing anxiety disorders, particularly treatment-resistant anxiety,4 clinicians must consider the limitations of these agents. Benzodiazepines can be associated with abuse and dependence, and overdose risk when combined with opiates.5,6 They may cause memory impairment7,8 and conflicting data suggest they may contribute to the risk of developing cognitive disorders.9-11 Benzodiazepines also have been associated with falls and fractures,12 and worse outcomes in patients with posttraumatic stress disorder.13 Some studies of patients with chronic obstructive pulmonary disease (COPD) found benzodiazepines may increase the risk of COPD exacerbations and accidental overdose,14 though others found that was not always the case.15 Benzodiazepines may be associated with an increased risk of spontaneous abortion when used early in pregnancy.16 Prospective research in women who were breastfeeding found benzodiazepines may cause sedation in up to 2% of infants.17
Despite the potential for adverse effects, benzodiazepine use remains common.18 These medications have a rapid onset of action, are useful for breakthrough symptoms, may enhance treatment adherence, and alleviate activating symptoms of SSRIs. Like other commonly used medications, benzodiazepines have the potential for both harm and benefit.19 Similar to other medications with tolerability concerns but established efficacy, particularly in treatment-resistant anxiety disorders, it is important to balance “overprescribing … to patients at risk and underusing these effective medications when indicated.”19 Though the use of benzodiazepines has been discouraged and perceptions have shifted, knowledge of benzodiazepines and benzodiazepine pharmacology also has been degraded contemporaneously.
This article provides a synthesis of the clinically relevant pharmacology of benzodiazepines, with a focus on orally administered benzodiazepines, which are more common in outpatient clinical practice. Specifically, this review describes the pharmacology of benzodiazepines, benzodiazepine medication interactions, the relationship between pharmacologic characteristics and treatment response/tolerability, and selection and dosing of oral benzodiazepines (Table20).
Benzodiazepine pharmacodynamics
Benzodiazepines act at the gamma-aminobutyric acid (GABA)-A receptor complex and bind allosterically.21-23 Comprised of 5 glycoprotein subunits (2 alpha subunits, 2 beta subunits, and 1 gamma subunit), the receptor has 2 distinct sites at which the endogenous inhibitory transmitter GABA binds and 1 benzodiazepine binding site. Benzodiazepines bind within a socket created by the alpha and gamma subunits22 and after binding induce a conformational change in the receptor, which enhances GABA binding. There are 2 types of benzodiazepine receptors: BZ1 and BZ2. The subunits play a critical role in driving the pharmacologic characteristics of the receptor.24 BZ1 and BZ2 receptors bind benzodiazepines, although they are differentially distributed within the brain. Binding at BZ1 receptors—which are distributed in cortical, thalamic, and cerebellar regions—contributes to sedation and deleterious effects of benzodiazepines on memory (eg, anterograde amnesia). BZ2 receptors (which contain gamma-2 subunits) are responsible for anxiolytic and muscle-relaxing effects. They are distributed throughout limbic regions and motor tracts, including motor neurons and neurons in the dorsal horn of the spinal cord.24
Benzodiazepines—positive GABA-A receptor allosteric modulators—produce phasic inhibition, largely through the alpha and gamma subunits discussed above. In contrast, newer positive allosteric modulators (eg, zuranolone) bind at the alpha/beta subunits.25 Mechanistically, endogenous neuroactive steroids and nonbenzodiazepine GABA-A–positive allosteric modulators such as zuranolone and ganaxolone also differ in their regulation of GABA-A (downregulated with benzodiazepines and hypothetically upregulated with zuranolone)26 and their synaptic effects (benzodiazepines synaptically vs endogenous neurosteroids and nonbenzodiazpine positive allosteric modulators extrasynaptically).27
From a developmental perspective, benzodiazepines may have less efficacy for anxiolysis and worse tolerability in some pediatric patients,28 although they generally appear effective for immediate use to treat anxiety in acute settings.29 The differences in efficacy and tolerability may be related to pharmacodynamic differences between pediatric populations and adults. GABA receptor expression and function do not reach adult levels until age 14 to 17½ for subcortical regions and age 18 to 22 for cortical regions, although girls reach adult expression of GABA receptors slightly earlier than boys.30 D
Continue to: Pharmacology and clinical effects
Pharmacology and clinical effects
Benzodiazepine pharmacokinetics are intimately linked with the onset of action and duration of clinical effect and vary based on the route of administration, absorption, and distribution/redistribution.31 In this review, we focus on oral administration as opposed to IV, IM, sublingual, or intranasal administration.
Absorption
Benzodiazepines are rapidly absorbed after oral administration and quickly enter the systemic circulation. However, absorption rates vary depending on specific aspects of the gastrointestinal milieu and intrinsic properties of the benzodiazepine. For example, alprazolam is more rapidly absorbed than most other benzodiazepines, with a Tmax of 1.8 hours compared to lorazepam, which has a Tmax of approximately 2 hours. These pharmacokinetic effects instantiate differences in tolerability and efficacy. Thus, following single doses of alprazolam and diazepam, self-rated sedating effects and impairment on a task of working memory suggest that effects have a more rapid onset for alprazolam relative to lorazepam.32 Food and concomitant medications can significantly affect benzodiazepine absorption. A single-dose, 3-way crossover study demonstrated that taking diazepam concomitantly with an antacid (eg, aluminum hydroxide) decreased peak concentrations and prolonged absorption by approximately 30 minutes. However, total absorption of the medication was unaffected.33 Additionally, administration of diazepam with food significantly slows absorption from 1 hour 15 minutes to approximately 2 hours 30 minutes and increases benzodiazepine absorption by 25% (Figure 134); the fat content of the meal appears important in moderating this effect.35 The impact of food on alprazolam varies by formulation. For example, when administered in an extended-release (XR) formulation with a high-fat meal, alprazolam absorption increases by one-third, while absorption for administration of the orally disintegrating tablet with a high-fat meal increases from 1 hour 30 minutes to 2 hours. Similarly, for lorazepam, administration with a meal delays absorption by approximately 2 hours; however, this effect does not appear present with the XR formulation. Administering benzodiazepines with food can be clinically leveraged to either accelerate the onset of action or decrease peak-associated adverse effects. Thus, when a highly lipophilic benzodiazepine is needed to treat acute anxiety or prior to an expected anxiogenic stimuli, administering the medication without food may produce a faster onset of action.
CNS penetration
Benzodiazepines enter the CNS by passive diffusion. Because of this, lipophilicity at physiologic pH influences the rate at which a benzodiazepine crosses the blood-brain barrier. The rate at which benzodiazepines enter the CNS influences their clinical effects and the speed at which both efficacy (ie, anxiolysis) and adverse effects (ie, sedation, slowed cognition) are observed. In general, more lipophilic medications initiate their anxiolytic effect more quickly. However, by quickly leaving the CNS (through the same mechanism that allowed them to enter the CNS at such speed), their effects rapidly cease as they redistribute into fat. Thus, highly lipophilic benzodiazepines produce more intense effects compared to less lipophilic benzodiazepines. For these reasons, lipophilicity is more important than half-life for determining the duration of effect in most patients.
Lipophilicity and duration of effect
Benzodiazepines and their metabolites tend to be highly protein-bound and distributed in fat- and lipid-enriched areas such as the CNS. As a result, the more lipophilic agents generally have the highest rates of absorption and the fastest onset of clinical effects. The duration of action for many benzodiazepines is determined by the rate and extent of distribution (a function of lipophilicity) rather than by the rate of elimination. For example, diazepam has a longer half-life than lorazepam, but its duration of action following a single dose is shorter. This is because diazepam is more lipophilic and therefore more extensively distributed (particularly to adipose tissue). This results in it leaving the brain and blood and distributing to other tissues. In turn, its CNS effect (ie, anxiolytic effects) are more quickly terminated.
By contrast, less lipophilic benzodiazepines maintain their CNS concentrations longer; they have a longer duration of action because of their slower redistribution, which culminates in a shorter half-life, and are less extensively distributed to peripheral tissues. In essence, this means that (other things being equal) a less lipophilic benzodiazepine produces a more sustained anxiolytic effect compared to a highly lipophilic benzodiazepine.36 Lipophilicity is also important in predicting some cognitive adverse effects, including amnesia. Benzodiazepines with high lipophilicity have greater absorption and faster onset of action as well as more rapid amnestic effects.37,38 These effects may relate to overall efficacy differences for oral benzodiazepines. A recent meta-analysis by Stimpfl et al36 found that less lipophilic benzodiazepines produced a greater response compared to more lipophilic benzodiazepines.
Continue to: Metabolism
Metabolism
Regarding cytochrome P450 (CYP) metabolism, polymorphic CYP2C19 and CYP3A4/5 are involved in the metabolism of several benzodiazepines39 and CYP2B6 has been recognized as a contributor to diazepam metabolism. CYP3A5 gene polymorphisms may produce variation in alprazolam metabolism; however, the predominant cytochrome involved in the metabolism of oxidatively metabolized benzodiazepines (ie, benzodiazepines other than lorazepam, oxazepam, and temazepam) is primarily CYP3A4, and most effects on CYP3A4 activity are related to concomitant medications and other nongenetic factors.
Drug-drug interactions
Apart from lorazepam,40,41 oxazepam,42,43 and temazepam, most benzodiazepines are metabolized through oxidative mechanisms that involve CYP3A4 (Figure 220).39 As such, their metabolism is influenced by medications that impact CYP3A4, including antifungals (eg, ketoconazole), calcium channel blockers (eg, verapamil, diltiazem), nefazodone, some protease inhibitors, and macrolide antibiotics. Research has examined the impact of low-dose estrogen oral contraceptives (OCPs) on exposure (eg, plasma concentrations) of several benzodiazepines. The mechanism for this interaction is likely complex and putatively involves multiple pathways, including inhibition of CYP3A4 by OCPs. The effects of OCPs on benzodiazepine pharmacokinetics vary based on the metabolism of the benzodiazepine. In general, medications oxidized and nitroreduced (eg, chlordiazepoxide, alprazolam, diazepam, and nitrazepam) have decreased clearance in patients treated with OCPs. Regarding nonoxidatively metabolized benzodiazepines, data are mixed. Research found no OCP-related effects on the pharmacokinetics of nonoxidatively metabolized benzodiazepines44; another study suggested that clearance of these medications—through increased glucuronidation—may be increased.31 The effect of smoking on benzodiazepine concentration has been well documented. Smoking increases the clearance of orally administered diazepam,45 but not IV diazepam, midazolam, or lorazepam, suggesting that this represents a first-pass effect.46 For alprazolam, plasma concentrations are reduced by 15% to 30% in smokers and total body clearance is 24% greater compared to nonsmokers, which results in an approximately 50% increase in half-life in nonsmokers compared to smokers.47 The most notable interaction between benzodiazepines and SSRIs is seen with fluvoxamine. Because fluvoxamine moderately inhibits CYP2C19 and CYP3A4 and potently inhibits CYP1A2,48 the clearance of oxidatively metabolized benzodiazepines is reduced.49 Additionally, the effects of grapefruit juice—a potent inhibitor of CYP3A4—has been evaluated for several benzodiazepines. Yasui et al50 found grapefruit juice did not alter alprazolam plasma concentrations. However, in separate research, grapefruit juice tripled diazepam exposure, increased peak concentrations 1.5-fold, and prolonged absorption.51
Hepatic disease
Exposure to benzodiazepines—other than lorazepam, oxazepam, and temazepam—is influenced by intrinsic hepatic disease and requires dose adjustment in individuals with significant hepatic impairment. The impact of hepatic disease on the clinical pharmacology of benzodiazepines may relate to 2 factors: protein binding and metabolism. In a study of individuals with cirrhosis, lorazepam binding was decreased, although its metabolism and clearance were largely unaffected.40
Aging and benzodiazepine metabolism/clearance
Aging is associated with myriad physiologic changes (eg, decrease in renal clearance after age 40, changes in body fat distribution, changes in activity of cytochromes) that are relevant to benzodiazepine pharmacology. They may underlie differences in the tolerability of benzodiazepines and other clinically relevant characteristics (eg, duration of action, accumulation).
Several studies have evaluated the impact of aging on the clearance and disposition of selected benzodiazepines. The respective half-lives of chlordiazepoxide and diazepam increase from 4- to 6-fold from age 20 to 80. Further, with chronic dosing, highly lipophilic benzodiazepines may require additional attention in geriatric patients. In a study that included individuals up to age 78, steady-state plasma concentrations of diazepam and its metabolite, desmethyldiazepam (DMDZ), were 30% to 35% higher in older patients compared to younger individuals.52 In this study, the half-lives for the young and older patients were 31 hours and 86 hours, respectively, for diazepam, and 40 hours and 80 hours, respectively, for the active metabolite. The half-life of diazepam is increased by “1 hour for each year of age beginning with a half-life of 20 hours at 20 years of age, as the volume of distribution is increased, and clearance is decreased.”52 Clinically, this implies that in older adults, clinicians should expect lower peak concentrations (Cmax), higher trough concentrations (Cmin), and that diazepam will take longer to reach steady-state concentrations. Taken together, these findings raised concern that “slow accumulation and delayed washout of diazepam and DMDZ is probable.”52 These findings—which may have more clinical relevance than those of single-dose studies—suggest that the effects related to diazepam would also take longer to resolve in older patients. Finally, lorazepam clearance or distribution does not appear to be affected by aging, at least in patients age 15 to 73.40 Alprazolam is more slowly cleared in geriatric patients and its effects may be potentiated by reduced protein binding.
Continue to: Obesity
Obesity
The distribution of medications, including benzodiazepines, is altered in patients who are obese because of increased adipose tissue.53,54 This increase in the volume of distribution can attenuate the onset of action, increase medication accumulation in fat, and potentiate the duration of action.55,56
Obesity may also affect hepatic metabolism by induction of CYP1A2, CYP2C9, and CYP2C19, and inhibition of CYP3A4.57 Triazolam, which is metabolized by CYP3A4, is associated with a greater exposure (ie, plasma concentrations) in individuals who are obese.58 However, when considering differences in benzodiazepine pharmacokinetics in patients who are obese, clinicians must remember that elimination half-life depends on both volume of distribution and clearance. In
How quickly do benzodiazepines work?
Benzodiazepines act quickly. Meta-analyses36 suggest that improvement in anxiety symptoms compared to placebo is greatest initially and then the rate of improvement slows over successive weeks. Research on benzodiazepines reveals statistically significant differences between benzodiazepines and placebo within the first week of treatment, with >80% of the expected improvement by Week 8 of treatment emerging by Week 4 (Figure 336). The rapid reduction in anxiety symptoms seen with benzodiazepines has important treatment implications, given that traditional psychotherapeutic and antidepressant treatments are slow to produce improvements. Consistent data suggesting that benzodiazepines work faster than other treatments support that they may have a role during the initiation of other treatments.
What is the ‘best’ dose?
As seen with other classes of psychotropic medications,4 the relationship between benzodiazepine dose and response is complex. In a recent meta-analysis of 65 placebo-controlled trials of benzodiazepines in adults with anxiety disorders, there was a superior response over time for low-dose benzodiazepines (<3 mg/d in lorazepam equivalents) compared to a medium dose (3 to 6 mg/d; P = .042); high-dose benzodiazepines (>6 mg/d) yielded less improvement compared to medium doses (P = .001).36 A study of adults with panic disorder similarly found the greatest responses with alprazolam plasma concentrations of 20 to 40 ng/mL, with no additional benefit at <20 ng/mL or >40 ng/mL.49 As plasma concentrations increase, adverse effects such as sedation also increase, which may confound the observed loss of a dose-response relationship at higher doses and plasma concentrations.62 This may, in part, account for the observation that higher doses of benzodiazepines are associated with greater depressive symptoms and disrupted sleep.63 As such, low doses may represent a delicate equipoise between efficacy and tolerability, yielding the most optimal clinical response.
Which benzodiazepine should I prescribe?
Comparing benzodiazepines is difficult, given the differences in dosing and disorders studied and differences in how each individual clinical trial was conducted. A meta-analysis by Stimpfl et al36 that used Bayesian hierarchical modeling, which allowed some of this heterogeneity to be addressed, found that relative to the reference benzodiazepine (lorazepam), clonazepam had the greatest trajectory/magnitude of response (other specific benzodiazepines did not statistically differ from lorazepam) (Figure 436).
Continue to: Another aspect of the superiority...
Another aspect of the superiority of clonazepam in some research relates to its pharmacokinetic properties, particularly when compared with benzodiazepines that have very short half-lives. Short half-life benzodiazepines have been associated with rebound anxiety, which is defined as “the relative worsening of symptoms on discontinuation of treatment as compared to baseline symptoms” and is distinct from withdrawal.64 While it is difficult to assess this in clinical trials, Herman et al65 provided insight into the contribution of rebound anxiety in a study of patients with panic disorder treated with alprazolam who experienced “interdose anxiety symptoms.” Of the 48 patients in this study, 41 switched to clonazepam, and most who switched (82%) experienced improvement. The improvement was attributed to the decreased frequency of clonazepam (vs alprazolam) administration and lack of interdose anxiety. When selecting an oral benzodiazepine, consider the duration, onset of action, and differences in metabolism that produce varying levels of effectiveness for individual patients. In situations where rapid onset is desired, a short-acting benzodiazepine may be preferable, while a longer-acting benzodiazepine would be preferable in situations where the patient needs sustained effects.
Regarding lipophilicity, differences among benzodiazepines could contribute to differences in psychological dependence and differential utility in some situations. For example, alprazolam rapidly enters the CNS, producing an immediate anxiolytic effect. However, its egress from the CNS is equally rapid, and its anxiolytic effects disappear quickly. This may be desirable for addressing acute, predictable anxiety, but could have unintended consequences in treating chronic anxiety, where it could facilitate psychological dependence.
Practical considerations
When prescribing benzodiazepines, consider a myriad of patient- and medication-specific factors, as these have clinically relevant implications on treatment response. This information, taken together, supports the importance of an individualized approach to benzodiazepine use. Before selecting a benzodiazepine and during treatment, important elements of the patient’s history must be considered, including age, body weight, concomitant medication use (eg, antacids, CYP3A4 inhibitors, OCPs), smoking status, and history of hepatic or renal disease.
Patients age <18 are unlikely to have full expression of GABA receptors in the brain30 and therefore benzodiazepines may not be as efficacious for anxiolysis in this population. Moreover, compared to younger patients, older patients may experience higher steady-state concentrations of benzodiazepines, especially lipophilic agents, due to an increased volume of distribution and decreased clearance. In patients treated with OCPs, some benzodiazepines may take longer to reach steady-state, and dose adjustments may need to be considered. In patients who smoke, clearance of some oral benzodiazepines is also accelerated, potentially decreasing half-life by up to 50%.
When dosing and titrating benzodiazepines, consider the patient’s body weight, particularly if they are obese. The effects of obesity on benzodiazepine pharmacokinetics are complex. For glucuronidated benzodiazepines, clearance is increased in patients who are obese; however, the volume of distribution is also increased in such patients, meaning it will take longer for benzodiazepines to achieve steady-state in these individuals compared to patients who are not obese. These effects suggest it may take longer to achieve a response at a given dose in patients who are obese compared to individuals who are not obese.
Continue to: The properties of individual benzodiazepines...
The properties of individual benzodiazepines should also be considered when selecting a benzodiazepine treatment. If circumstances necessitate rapid symptom relief, a lipophilic benzodiazepine, such as diazepam, may be preferred for quick onset and offset of action. Onset of action may also be hastened by taking the benzodiazepine without food; conversely, if peak adverse effects are problematic, concurrent consumption of a high-fat meal may help decrease peak concentration and prolonging absorption. In other circumstances, such as if sustained anxiolysis is desired, a clinician may opt for a less lipophilic benzodiazepine, such as clonazepam. Finally, in terms of general treatment response, benzodiazepines separate from placebo in the first week of treatment, which supports the idea they may be useful during the introduction of other medications (eg, SSRIs) that take a longer time to achieve clinical effect.
Bottom Line
The pharmacokinetics of benzodiazepines are intimately linked with the onset of action and duration of clinical effect and vary based on individual absorption and distribution/redistribution. Benzodiazepines’ clinical profile derives from their pharmacokinetic differences and is influenced by many factors, including age, body weight, concomitant medication use, smoking status, and hepatic or renal disease. Consider these factors to individualize the approach to using benzodiazepines and optimize tolerability and efficacy.
Related Resources
- Weber SR, Duchemin AM. Benzodiazepines: sensible prescribing in light of the risks. Current Psychiatry. 2018;17(2):22-27.
- Balon R. Benzodiazepines for anxious depression. Current Psychiatry. 2018;17(8):9-12.
Drug Brand Names
Alprazolam • Xanax
Chlordiazepoxide • Librium
Clobazam • Onfi
Clonazepam • Klonopin
Clorazepate • Gen-Xene
Diazepam • Valium
Diltiazem • Cardizem
Fluvoxamine • Luvox
Ganaxolone • Ztalmy
Ketoconazole • Nizoral
Lorazepam • Ativan
Midazolam • Versed
Temazepam • Restoril
Triazolam • Halcion
Verapamil • Calan
Though once the main treatment for anxiety disorders—often as monotherapy1—benzodiazepines are now primarily used as adjunctive agents.2-4 Their ability to produce rapid anxiolysis represents a significant therapeutic advantage, but in recent decades their tolerability, class-specific risks, and lack of antidepressant properties contributed to benzodiazepines being largely replaced by selective serotonin reuptake inhibitors (SSRIs) for the pharmacologic treatment of anxiety. This shift within the pharmacologic armamentarium has decreased many clinicians’ familiarity with benzodiazepines.
While benzodiazepines continue to have an important role in managing anxiety disorders, particularly treatment-resistant anxiety,4 clinicians must consider the limitations of these agents. Benzodiazepines can be associated with abuse and dependence, and overdose risk when combined with opiates.5,6 They may cause memory impairment7,8 and conflicting data suggest they may contribute to the risk of developing cognitive disorders.9-11 Benzodiazepines also have been associated with falls and fractures,12 and worse outcomes in patients with posttraumatic stress disorder.13 Some studies of patients with chronic obstructive pulmonary disease (COPD) found benzodiazepines may increase the risk of COPD exacerbations and accidental overdose,14 though others found that was not always the case.15 Benzodiazepines may be associated with an increased risk of spontaneous abortion when used early in pregnancy.16 Prospective research in women who were breastfeeding found benzodiazepines may cause sedation in up to 2% of infants.17
Despite the potential for adverse effects, benzodiazepine use remains common.18 These medications have a rapid onset of action, are useful for breakthrough symptoms, may enhance treatment adherence, and alleviate activating symptoms of SSRIs. Like other commonly used medications, benzodiazepines have the potential for both harm and benefit.19 Similar to other medications with tolerability concerns but established efficacy, particularly in treatment-resistant anxiety disorders, it is important to balance “overprescribing … to patients at risk and underusing these effective medications when indicated.”19 Though the use of benzodiazepines has been discouraged and perceptions have shifted, knowledge of benzodiazepines and benzodiazepine pharmacology also has been degraded contemporaneously.
This article provides a synthesis of the clinically relevant pharmacology of benzodiazepines, with a focus on orally administered benzodiazepines, which are more common in outpatient clinical practice. Specifically, this review describes the pharmacology of benzodiazepines, benzodiazepine medication interactions, the relationship between pharmacologic characteristics and treatment response/tolerability, and selection and dosing of oral benzodiazepines (Table20).
Benzodiazepine pharmacodynamics
Benzodiazepines act at the gamma-aminobutyric acid (GABA)-A receptor complex and bind allosterically.21-23 Comprised of 5 glycoprotein subunits (2 alpha subunits, 2 beta subunits, and 1 gamma subunit), the receptor has 2 distinct sites at which the endogenous inhibitory transmitter GABA binds and 1 benzodiazepine binding site. Benzodiazepines bind within a socket created by the alpha and gamma subunits22 and after binding induce a conformational change in the receptor, which enhances GABA binding. There are 2 types of benzodiazepine receptors: BZ1 and BZ2. The subunits play a critical role in driving the pharmacologic characteristics of the receptor.24 BZ1 and BZ2 receptors bind benzodiazepines, although they are differentially distributed within the brain. Binding at BZ1 receptors—which are distributed in cortical, thalamic, and cerebellar regions—contributes to sedation and deleterious effects of benzodiazepines on memory (eg, anterograde amnesia). BZ2 receptors (which contain gamma-2 subunits) are responsible for anxiolytic and muscle-relaxing effects. They are distributed throughout limbic regions and motor tracts, including motor neurons and neurons in the dorsal horn of the spinal cord.24
Benzodiazepines—positive GABA-A receptor allosteric modulators—produce phasic inhibition, largely through the alpha and gamma subunits discussed above. In contrast, newer positive allosteric modulators (eg, zuranolone) bind at the alpha/beta subunits.25 Mechanistically, endogenous neuroactive steroids and nonbenzodiazepine GABA-A–positive allosteric modulators such as zuranolone and ganaxolone also differ in their regulation of GABA-A (downregulated with benzodiazepines and hypothetically upregulated with zuranolone)26 and their synaptic effects (benzodiazepines synaptically vs endogenous neurosteroids and nonbenzodiazpine positive allosteric modulators extrasynaptically).27
From a developmental perspective, benzodiazepines may have less efficacy for anxiolysis and worse tolerability in some pediatric patients,28 although they generally appear effective for immediate use to treat anxiety in acute settings.29 The differences in efficacy and tolerability may be related to pharmacodynamic differences between pediatric populations and adults. GABA receptor expression and function do not reach adult levels until age 14 to 17½ for subcortical regions and age 18 to 22 for cortical regions, although girls reach adult expression of GABA receptors slightly earlier than boys.30 D
Continue to: Pharmacology and clinical effects
Pharmacology and clinical effects
Benzodiazepine pharmacokinetics are intimately linked with the onset of action and duration of clinical effect and vary based on the route of administration, absorption, and distribution/redistribution.31 In this review, we focus on oral administration as opposed to IV, IM, sublingual, or intranasal administration.
Absorption
Benzodiazepines are rapidly absorbed after oral administration and quickly enter the systemic circulation. However, absorption rates vary depending on specific aspects of the gastrointestinal milieu and intrinsic properties of the benzodiazepine. For example, alprazolam is more rapidly absorbed than most other benzodiazepines, with a Tmax of 1.8 hours compared to lorazepam, which has a Tmax of approximately 2 hours. These pharmacokinetic effects instantiate differences in tolerability and efficacy. Thus, following single doses of alprazolam and diazepam, self-rated sedating effects and impairment on a task of working memory suggest that effects have a more rapid onset for alprazolam relative to lorazepam.32 Food and concomitant medications can significantly affect benzodiazepine absorption. A single-dose, 3-way crossover study demonstrated that taking diazepam concomitantly with an antacid (eg, aluminum hydroxide) decreased peak concentrations and prolonged absorption by approximately 30 minutes. However, total absorption of the medication was unaffected.33 Additionally, administration of diazepam with food significantly slows absorption from 1 hour 15 minutes to approximately 2 hours 30 minutes and increases benzodiazepine absorption by 25% (Figure 134); the fat content of the meal appears important in moderating this effect.35 The impact of food on alprazolam varies by formulation. For example, when administered in an extended-release (XR) formulation with a high-fat meal, alprazolam absorption increases by one-third, while absorption for administration of the orally disintegrating tablet with a high-fat meal increases from 1 hour 30 minutes to 2 hours. Similarly, for lorazepam, administration with a meal delays absorption by approximately 2 hours; however, this effect does not appear present with the XR formulation. Administering benzodiazepines with food can be clinically leveraged to either accelerate the onset of action or decrease peak-associated adverse effects. Thus, when a highly lipophilic benzodiazepine is needed to treat acute anxiety or prior to an expected anxiogenic stimuli, administering the medication without food may produce a faster onset of action.
CNS penetration
Benzodiazepines enter the CNS by passive diffusion. Because of this, lipophilicity at physiologic pH influences the rate at which a benzodiazepine crosses the blood-brain barrier. The rate at which benzodiazepines enter the CNS influences their clinical effects and the speed at which both efficacy (ie, anxiolysis) and adverse effects (ie, sedation, slowed cognition) are observed. In general, more lipophilic medications initiate their anxiolytic effect more quickly. However, by quickly leaving the CNS (through the same mechanism that allowed them to enter the CNS at such speed), their effects rapidly cease as they redistribute into fat. Thus, highly lipophilic benzodiazepines produce more intense effects compared to less lipophilic benzodiazepines. For these reasons, lipophilicity is more important than half-life for determining the duration of effect in most patients.
Lipophilicity and duration of effect
Benzodiazepines and their metabolites tend to be highly protein-bound and distributed in fat- and lipid-enriched areas such as the CNS. As a result, the more lipophilic agents generally have the highest rates of absorption and the fastest onset of clinical effects. The duration of action for many benzodiazepines is determined by the rate and extent of distribution (a function of lipophilicity) rather than by the rate of elimination. For example, diazepam has a longer half-life than lorazepam, but its duration of action following a single dose is shorter. This is because diazepam is more lipophilic and therefore more extensively distributed (particularly to adipose tissue). This results in it leaving the brain and blood and distributing to other tissues. In turn, its CNS effect (ie, anxiolytic effects) are more quickly terminated.
By contrast, less lipophilic benzodiazepines maintain their CNS concentrations longer; they have a longer duration of action because of their slower redistribution, which culminates in a shorter half-life, and are less extensively distributed to peripheral tissues. In essence, this means that (other things being equal) a less lipophilic benzodiazepine produces a more sustained anxiolytic effect compared to a highly lipophilic benzodiazepine.36 Lipophilicity is also important in predicting some cognitive adverse effects, including amnesia. Benzodiazepines with high lipophilicity have greater absorption and faster onset of action as well as more rapid amnestic effects.37,38 These effects may relate to overall efficacy differences for oral benzodiazepines. A recent meta-analysis by Stimpfl et al36 found that less lipophilic benzodiazepines produced a greater response compared to more lipophilic benzodiazepines.
Continue to: Metabolism
Metabolism
Regarding cytochrome P450 (CYP) metabolism, polymorphic CYP2C19 and CYP3A4/5 are involved in the metabolism of several benzodiazepines39 and CYP2B6 has been recognized as a contributor to diazepam metabolism. CYP3A5 gene polymorphisms may produce variation in alprazolam metabolism; however, the predominant cytochrome involved in the metabolism of oxidatively metabolized benzodiazepines (ie, benzodiazepines other than lorazepam, oxazepam, and temazepam) is primarily CYP3A4, and most effects on CYP3A4 activity are related to concomitant medications and other nongenetic factors.
Drug-drug interactions
Apart from lorazepam,40,41 oxazepam,42,43 and temazepam, most benzodiazepines are metabolized through oxidative mechanisms that involve CYP3A4 (Figure 220).39 As such, their metabolism is influenced by medications that impact CYP3A4, including antifungals (eg, ketoconazole), calcium channel blockers (eg, verapamil, diltiazem), nefazodone, some protease inhibitors, and macrolide antibiotics. Research has examined the impact of low-dose estrogen oral contraceptives (OCPs) on exposure (eg, plasma concentrations) of several benzodiazepines. The mechanism for this interaction is likely complex and putatively involves multiple pathways, including inhibition of CYP3A4 by OCPs. The effects of OCPs on benzodiazepine pharmacokinetics vary based on the metabolism of the benzodiazepine. In general, medications oxidized and nitroreduced (eg, chlordiazepoxide, alprazolam, diazepam, and nitrazepam) have decreased clearance in patients treated with OCPs. Regarding nonoxidatively metabolized benzodiazepines, data are mixed. Research found no OCP-related effects on the pharmacokinetics of nonoxidatively metabolized benzodiazepines44; another study suggested that clearance of these medications—through increased glucuronidation—may be increased.31 The effect of smoking on benzodiazepine concentration has been well documented. Smoking increases the clearance of orally administered diazepam,45 but not IV diazepam, midazolam, or lorazepam, suggesting that this represents a first-pass effect.46 For alprazolam, plasma concentrations are reduced by 15% to 30% in smokers and total body clearance is 24% greater compared to nonsmokers, which results in an approximately 50% increase in half-life in nonsmokers compared to smokers.47 The most notable interaction between benzodiazepines and SSRIs is seen with fluvoxamine. Because fluvoxamine moderately inhibits CYP2C19 and CYP3A4 and potently inhibits CYP1A2,48 the clearance of oxidatively metabolized benzodiazepines is reduced.49 Additionally, the effects of grapefruit juice—a potent inhibitor of CYP3A4—has been evaluated for several benzodiazepines. Yasui et al50 found grapefruit juice did not alter alprazolam plasma concentrations. However, in separate research, grapefruit juice tripled diazepam exposure, increased peak concentrations 1.5-fold, and prolonged absorption.51
Hepatic disease
Exposure to benzodiazepines—other than lorazepam, oxazepam, and temazepam—is influenced by intrinsic hepatic disease and requires dose adjustment in individuals with significant hepatic impairment. The impact of hepatic disease on the clinical pharmacology of benzodiazepines may relate to 2 factors: protein binding and metabolism. In a study of individuals with cirrhosis, lorazepam binding was decreased, although its metabolism and clearance were largely unaffected.40
Aging and benzodiazepine metabolism/clearance
Aging is associated with myriad physiologic changes (eg, decrease in renal clearance after age 40, changes in body fat distribution, changes in activity of cytochromes) that are relevant to benzodiazepine pharmacology. They may underlie differences in the tolerability of benzodiazepines and other clinically relevant characteristics (eg, duration of action, accumulation).
Several studies have evaluated the impact of aging on the clearance and disposition of selected benzodiazepines. The respective half-lives of chlordiazepoxide and diazepam increase from 4- to 6-fold from age 20 to 80. Further, with chronic dosing, highly lipophilic benzodiazepines may require additional attention in geriatric patients. In a study that included individuals up to age 78, steady-state plasma concentrations of diazepam and its metabolite, desmethyldiazepam (DMDZ), were 30% to 35% higher in older patients compared to younger individuals.52 In this study, the half-lives for the young and older patients were 31 hours and 86 hours, respectively, for diazepam, and 40 hours and 80 hours, respectively, for the active metabolite. The half-life of diazepam is increased by “1 hour for each year of age beginning with a half-life of 20 hours at 20 years of age, as the volume of distribution is increased, and clearance is decreased.”52 Clinically, this implies that in older adults, clinicians should expect lower peak concentrations (Cmax), higher trough concentrations (Cmin), and that diazepam will take longer to reach steady-state concentrations. Taken together, these findings raised concern that “slow accumulation and delayed washout of diazepam and DMDZ is probable.”52 These findings—which may have more clinical relevance than those of single-dose studies—suggest that the effects related to diazepam would also take longer to resolve in older patients. Finally, lorazepam clearance or distribution does not appear to be affected by aging, at least in patients age 15 to 73.40 Alprazolam is more slowly cleared in geriatric patients and its effects may be potentiated by reduced protein binding.
Continue to: Obesity
Obesity
The distribution of medications, including benzodiazepines, is altered in patients who are obese because of increased adipose tissue.53,54 This increase in the volume of distribution can attenuate the onset of action, increase medication accumulation in fat, and potentiate the duration of action.55,56
Obesity may also affect hepatic metabolism by induction of CYP1A2, CYP2C9, and CYP2C19, and inhibition of CYP3A4.57 Triazolam, which is metabolized by CYP3A4, is associated with a greater exposure (ie, plasma concentrations) in individuals who are obese.58 However, when considering differences in benzodiazepine pharmacokinetics in patients who are obese, clinicians must remember that elimination half-life depends on both volume of distribution and clearance. In
How quickly do benzodiazepines work?
Benzodiazepines act quickly. Meta-analyses36 suggest that improvement in anxiety symptoms compared to placebo is greatest initially and then the rate of improvement slows over successive weeks. Research on benzodiazepines reveals statistically significant differences between benzodiazepines and placebo within the first week of treatment, with >80% of the expected improvement by Week 8 of treatment emerging by Week 4 (Figure 336). The rapid reduction in anxiety symptoms seen with benzodiazepines has important treatment implications, given that traditional psychotherapeutic and antidepressant treatments are slow to produce improvements. Consistent data suggesting that benzodiazepines work faster than other treatments support that they may have a role during the initiation of other treatments.
What is the ‘best’ dose?
As seen with other classes of psychotropic medications,4 the relationship between benzodiazepine dose and response is complex. In a recent meta-analysis of 65 placebo-controlled trials of benzodiazepines in adults with anxiety disorders, there was a superior response over time for low-dose benzodiazepines (<3 mg/d in lorazepam equivalents) compared to a medium dose (3 to 6 mg/d; P = .042); high-dose benzodiazepines (>6 mg/d) yielded less improvement compared to medium doses (P = .001).36 A study of adults with panic disorder similarly found the greatest responses with alprazolam plasma concentrations of 20 to 40 ng/mL, with no additional benefit at <20 ng/mL or >40 ng/mL.49 As plasma concentrations increase, adverse effects such as sedation also increase, which may confound the observed loss of a dose-response relationship at higher doses and plasma concentrations.62 This may, in part, account for the observation that higher doses of benzodiazepines are associated with greater depressive symptoms and disrupted sleep.63 As such, low doses may represent a delicate equipoise between efficacy and tolerability, yielding the most optimal clinical response.
Which benzodiazepine should I prescribe?
Comparing benzodiazepines is difficult, given the differences in dosing and disorders studied and differences in how each individual clinical trial was conducted. A meta-analysis by Stimpfl et al36 that used Bayesian hierarchical modeling, which allowed some of this heterogeneity to be addressed, found that relative to the reference benzodiazepine (lorazepam), clonazepam had the greatest trajectory/magnitude of response (other specific benzodiazepines did not statistically differ from lorazepam) (Figure 436).
Continue to: Another aspect of the superiority...
Another aspect of the superiority of clonazepam in some research relates to its pharmacokinetic properties, particularly when compared with benzodiazepines that have very short half-lives. Short half-life benzodiazepines have been associated with rebound anxiety, which is defined as “the relative worsening of symptoms on discontinuation of treatment as compared to baseline symptoms” and is distinct from withdrawal.64 While it is difficult to assess this in clinical trials, Herman et al65 provided insight into the contribution of rebound anxiety in a study of patients with panic disorder treated with alprazolam who experienced “interdose anxiety symptoms.” Of the 48 patients in this study, 41 switched to clonazepam, and most who switched (82%) experienced improvement. The improvement was attributed to the decreased frequency of clonazepam (vs alprazolam) administration and lack of interdose anxiety. When selecting an oral benzodiazepine, consider the duration, onset of action, and differences in metabolism that produce varying levels of effectiveness for individual patients. In situations where rapid onset is desired, a short-acting benzodiazepine may be preferable, while a longer-acting benzodiazepine would be preferable in situations where the patient needs sustained effects.
Regarding lipophilicity, differences among benzodiazepines could contribute to differences in psychological dependence and differential utility in some situations. For example, alprazolam rapidly enters the CNS, producing an immediate anxiolytic effect. However, its egress from the CNS is equally rapid, and its anxiolytic effects disappear quickly. This may be desirable for addressing acute, predictable anxiety, but could have unintended consequences in treating chronic anxiety, where it could facilitate psychological dependence.
Practical considerations
When prescribing benzodiazepines, consider a myriad of patient- and medication-specific factors, as these have clinically relevant implications on treatment response. This information, taken together, supports the importance of an individualized approach to benzodiazepine use. Before selecting a benzodiazepine and during treatment, important elements of the patient’s history must be considered, including age, body weight, concomitant medication use (eg, antacids, CYP3A4 inhibitors, OCPs), smoking status, and history of hepatic or renal disease.
Patients age <18 are unlikely to have full expression of GABA receptors in the brain30 and therefore benzodiazepines may not be as efficacious for anxiolysis in this population. Moreover, compared to younger patients, older patients may experience higher steady-state concentrations of benzodiazepines, especially lipophilic agents, due to an increased volume of distribution and decreased clearance. In patients treated with OCPs, some benzodiazepines may take longer to reach steady-state, and dose adjustments may need to be considered. In patients who smoke, clearance of some oral benzodiazepines is also accelerated, potentially decreasing half-life by up to 50%.
When dosing and titrating benzodiazepines, consider the patient’s body weight, particularly if they are obese. The effects of obesity on benzodiazepine pharmacokinetics are complex. For glucuronidated benzodiazepines, clearance is increased in patients who are obese; however, the volume of distribution is also increased in such patients, meaning it will take longer for benzodiazepines to achieve steady-state in these individuals compared to patients who are not obese. These effects suggest it may take longer to achieve a response at a given dose in patients who are obese compared to individuals who are not obese.
Continue to: The properties of individual benzodiazepines...
The properties of individual benzodiazepines should also be considered when selecting a benzodiazepine treatment. If circumstances necessitate rapid symptom relief, a lipophilic benzodiazepine, such as diazepam, may be preferred for quick onset and offset of action. Onset of action may also be hastened by taking the benzodiazepine without food; conversely, if peak adverse effects are problematic, concurrent consumption of a high-fat meal may help decrease peak concentration and prolonging absorption. In other circumstances, such as if sustained anxiolysis is desired, a clinician may opt for a less lipophilic benzodiazepine, such as clonazepam. Finally, in terms of general treatment response, benzodiazepines separate from placebo in the first week of treatment, which supports the idea they may be useful during the introduction of other medications (eg, SSRIs) that take a longer time to achieve clinical effect.
Bottom Line
The pharmacokinetics of benzodiazepines are intimately linked with the onset of action and duration of clinical effect and vary based on individual absorption and distribution/redistribution. Benzodiazepines’ clinical profile derives from their pharmacokinetic differences and is influenced by many factors, including age, body weight, concomitant medication use, smoking status, and hepatic or renal disease. Consider these factors to individualize the approach to using benzodiazepines and optimize tolerability and efficacy.
Related Resources
- Weber SR, Duchemin AM. Benzodiazepines: sensible prescribing in light of the risks. Current Psychiatry. 2018;17(2):22-27.
- Balon R. Benzodiazepines for anxious depression. Current Psychiatry. 2018;17(8):9-12.
Drug Brand Names
Alprazolam • Xanax
Chlordiazepoxide • Librium
Clobazam • Onfi
Clonazepam • Klonopin
Clorazepate • Gen-Xene
Diazepam • Valium
Diltiazem • Cardizem
Fluvoxamine • Luvox
Ganaxolone • Ztalmy
Ketoconazole • Nizoral
Lorazepam • Ativan
Midazolam • Versed
Temazepam • Restoril
Triazolam • Halcion
Verapamil • Calan
1. Rickels K, Moeller HJ. Benzodiazepines in anxiety disorders: reassessment of usefulness and safety. World J Biol Psychiatry. 2019;20(7):514-518. doi:10.1080/15622975.2018.1500031
2. Stevens JC, Pollack MH. Benzodiazepines in clinical practice: consideration of their long-term use and alternative agents. J Clin Psychiatry. 2005;66(Suppl 2):21-27.
3. Pollack MH, van Ameringen M, Simon NM, et al. A double-blind randomized controlled trial of augmentation and switch strategies for refractory social anxiety disorder. Am J Psychiatry. 2014;171(1):44-53. doi:10.1176/appi.ajp.2013.12101353
4. Strawn JR, Geracioti L, Rajdev N, et al. Pharmacotherapy for generalized anxiety disorder in adult and pediatric patients: an evidence-based treatment review. Expert Opin Pharmacother. 2018;19(10):1057-1070. doi:10.1080/14656566.2018.1491966
5. Karaca-Mandic P, Meara E, Morden NE. The growing problem of co-treatment with opioids and benzodiazepines. BMJ. 2017;356:j1224. doi:10.1136/bmj.j1224
6. Bachhuber MA, Hennessy S, Cunningham CO, et al. Increasing benzodiazepine prescriptions and overdose mortality in the United States, 1996-2013. Am J Public Health. 2016;106(4):686-688. doi:10.2105/AJPH.2016.303061
7. Bentué-Ferrer D, Akwa Y. Benzodiazepines: Effects on memory functioning. In: Pandi-Perumal SR, Verster J, Monti J, et al, eds. Sleep Disorders: Diagnosis and Therapeutics. CRC Press; 2008:104-114. doi:10.3109/9780203091715-15
8. Pomara N, Facelle TM, Roth AE, et al. Dose-dependent retrograde facilitation of verbal memory in healthy elderly after acute oral lorazepam administration.Psychopharmacology (Berl). 2006;185(4):487-494. doi:10.1007/s00213-006-0336-0
9. Gray SL, Dublin S, Yu O, et al. Benzodiazepine use and risk of incident dementia or cognitive decline: prospective population based study. BMJ. 2016;352:i90. doi:10.1136/bmj.i90
10. Biétry FA, Pfeil AM, Reich O, et al. Benzodiazepine use and risk of developing Alzheimer’s disease: a case-control study based on Swiss claims data. CNS Drugs. 2017;31(3):245-251. doi:10.1007/s40263-016-0404-x
11. de Gage SB, Moride Y, Ducruet T, et al. Benzodiazepine use and risk of Alzheimer’s disease: case-control study. BMJ. 2014;349g5205. doi:10.1136/bmj.g5205
12. Shah R, Raji MA, Westra J, et al. Association of co-prescribing of opioid and benzodiazepine substitutes with incident falls and fractures among older adults: a cohort study. BMJ Open. 2021;11(12):e052057. doi:10.1136/bmjopen-2021-052057
13. Guina J, Rossetter SR, DeRhodes BJ, et al. Benzodiazepines for PTSD: a systematic review and meta-analysis. J Psychiatr Pract. 2015;21(4):281-303.
14. Ekström MP, Bornefalk-Hermansson A, Abernethy AP, et al. Safety of benzodiazepines and opioids in very severe respiratory disease: national prospective study. BMJ. 2014;348:g445. doi:10.1136/bmj.g445
15. Donovan LM, Malte CA, Spece LJ, et al. Center predictors of long-term benzodiazepine use in chronic obstructive pulmonary disease and post-traumatic stress disorder. Ann Am Thorac Soc. 2019;16(9):1151-1157. doi:10.1513/AnnalsATS.201901-048OC
16. Sheehy O, Zhao JP, Bérard A. Association between incident exposure to benzodiazepines in early pregnancy and risk of spontaneous abortion. JAMA Psychiatry. 2019;76(9):948-957. doi:10.1001/jamapsychiatry.2019.0963
17. Kelly LE, Poon S, Madadi P, et al. Neonatal benzodiazepines exposure during breastfeeding. J Pediatr. 2012;161(3):448-451. doi:10.1016/j.jpeds.2012.03.003
18. Agarwal SD, Landon BE. Patterns in outpatient benzodiazepine prescribing in the United States. JAMA Netw Open. 2019;2(1):e187399. doi:10.1001/jamanetworkopen.2018.7399
19. Hirschtritt ME, Olfson M, Kroenke K. Balancing the risks and benefits of benzodiazepines. JAMA. 2021;325(4):347-348. doi:10.1001/jama.2020.22106
20. Brunton LL, Hilal-Dandan R, Knollman BC, eds. Goodman & Gilman’s: The Pharmacological Basis of Therapeutics. McGraw-Hill Education; 2018.
21. Nutt DJ, Malizia AL. New insights into the role of the GABA(A)-benzodiazepine receptor in psychiatric disorder. British J Psychiatry. 2001;179:390-396. doi:10.1192/bjp.179.5.390
22. Sigel E. Mapping of the benzodiazepine recognition site on GABA(A) receptors. Curr Top Med Chem. 2002;2(8):833-839. doi:10.2174/1568026023393444
23. Savic
24. Smith TA. Type A gamma-aminobutyric acid (GABAA) receptor subunits and benzodiazepine binding: significance to clinical syndromes and their treatment. Br J Biomed Sci. 2001;58(2):111-121.
25. Althaus AL, Ackley MA, Belfort GM, et al. Preclinical characterization of zuranolone (SAGE-217), a selective neuroactive steroid GABAA receptor positive allosteric modulator. Neuropharmacology. 2020;181:108333. doi:10.1016/j.neuropharm.2020.108333
26. Jacob TC, Michels G, Silayeva L, et al. Benzodiazepine treatment induces subtype-specific changes in GABA(A) receptor trafficking and decreases synaptic inhibition. Proc Natl Acad Sci U S A. 2012;109(45):18595-18600. doi:10.1073/pnas.1204994109
27. Nicholson MW, Sweeney A, Pekle E, et al. Diazepam-induced loss of inhibitory synapses mediated by PLCδ/ Ca2+/calcineurin signalling downstream of GABAA receptors. Mol Psychiatry. 2018;23(9):1851-1867. doi:10.1038/s41380-018-0100-y
28. Dobson ET, Bloch MH, Strawn JR. Efficacy and tolerability of pharmacotherapy for pediatric anxiety disorders: a network meta-analysis. J Clin Psychiatry. 2019;80(1):17r12064. doi:10.4088/JCP.17r12064
29. Kuang H, Johnson JA, Mulqueen JM, et al. The efficacy of benzodiazepines as acute anxiolytics in children: a meta-analysis. Depress Anxiety. 2017;34(10):888-896. doi:10.1002/da.22643
30. Chugani DC, Muzik O, Juhász C, et al. Postnatal maturation of human GABAA receptors measured with positron emission tomography. Ann Neurol. 2001;49(5):618-626. doi:10.1002/ana.1003
31. Jochemsen R, Breimer DD. Pharmacokinetics of benzodiazepines: metabolic pathways and plasma level profiles. Curr Med Res Opin. 1984;8(Suppl 4):60-79. doi:10.1185/03007998409109545
32. Greenblatt DJ, Harmatz JS, Dorsey C, et al. Comparative single-dose kinetics and dynamics of lorazepam, alprazolam, prazepam, and placebo. Clin Pharmacol Ther. 1988;44(3)326-334. doi:10.1038/clpt.1988.158
33. Shader RI, Georgotas A, Greenblatt DJ, et al. Impaired absorption of desmethydiazepam from clorazepate by magnesium aluminum hydroxide. Clin Pharmacol Ther. 1978;24(3):308-315. doi:10.1002/cpt1978243308
34. Greenblatt DJ, Allen MD, MacLaughlin DS, et al. Diazepam absorption: effect of antacids and food. Clin Pharmacol Ther. 1978;24(5):600-609. doi:10.1002/cpt1978245600
35. Yamazaki A, Kumagai Y, Fujita T, et al. Different effects of light food on pharmacokinetics and pharmacodynamics of three benzodiazepines, quazepam, nitrazepam and diazepam. J Clin Pharm Ther. 2007;32(1):31-39. doi:10.1111/j.1365-2710.2007.00795.x
36. Stimpfl J, Mills JA, Strawn JR. Pharmacologic predictors of benzodiazepine response trajectory in anxiety disorders: a Bayesian hierarchical modeling meta-analysis. CNS Spectr. 2023;28(1):53-60. doi:10.1017/S1092852921000870
37. Griffin CE 3rd, Kaye AM, Bueno FR, et al. Benzodiazepine pharmacology and central nervous system-mediated effects. Ochsner J. 2013;13(2):214-223.
38. Buffett-Jerrott SE, Stewart SH. Cognitive and sedative effects of benzodiazepine use. Curr Pharm Des. 2005;8(1):45-58. doi:10.2174/1381612023396654
39. Fukasawa T, Suzuki A, Otani K. Effects of genetic polymorphism of cytochrome P450 enzymes on the pharmacokinetics of benzodiazepines. J Clin Pharm Ther. 2007;32(4):333-341. doi:10.1111/j.1365-2710.2007.00829.x
40. Kraus JW, Desmond PV, Marshall JP, et al. Effects of aging and liver disease on disposition of lorazepam. Clin Pharmacol Ther. 1978;24(4):411-419. doi:10.1002/cpt1978244411
41. Greenblatt DJ. Clinical pharmacokinetics of oxazepam and lorazepam. Clin Pharmacokinet. 1981;6(2):89-105. doi:10.2165/00003088-198106020-00001
42. Walkenstein SS, Wiser R, Gudmundsen CH, et al. Absorption, metabolism, and excretion of oxazepam and its succinate half‐ester. J Pharm Sci. 1964;53(10):1181-1186. doi:10.1002/jps.2600531010
43. Shull HJ, Wilkinson GR, Johnson R, et al. Normal disposition of oxazepam in acute viral hepatitis and cirrhosis. Ann Intern Med. 1976;84(4):420-425. doi:10.7326/0003-4819-84-4-420
44. Abernethy DR, Greenblatt DJ, Ochs HR, et al. Lorazepam and oxazepam kinetics in women on low-dose oral contraceptives. Clin Pharmacol Ther. 1983;33(5):628-632. doi:10.1038/clpt.1983.85
45. Greenblatt DJ, Allen MD, Harmatz JS, et al. Diazepam disposition determinants. Clin Pharmacol Ther. 1980;27(3):301-312. doi:10.1038/clpt.1980.40
46. Ochs HR, Greenblatt DJ, Knüchel M. Kinetics of diazepam, midazolam, and lorazepam, in cigarette smokers. Chest. 1985;87(2):223-226. doi:10.1378/chest.87.2.223
47. Smith RB, Gwilt PR, Wright CE 3rd. Single- and multiple-dose pharmacokinetics of oral alprazolam in healthy smoking and nonsmoking men. Clin Pharm. 1983;2(2):139-143.
48. Figgitt DP, McClellan KJ. Fluvoxamine. An updated review of its use in the management of adults with anxiety disorders. Drugs. 2000;60(4):925-954. doi:10.2165/00003495-200060040-00006
49. Greenblatt DJ, Wright CE. Clinical pharmacokinetics of alprazolam. Therapeutic implications. Clin Pharmacokinet. 1993;24(6):453-471. doi:10.2165/00003088-199324060-00003
50. Yasui N, Kondo T, Furukori H, et al. Effects of repeated ingestion of grapefruit juice on the single and multiple oral-dose pharmacokinetics and pharmacodynamics of alprazolam. Psychopharmacology (Berl). 2000;150(2):185-190. doi:10.1007/s002130000438
51. Özdemir M, Aktan Y, Boydagˇ BS, et al. Interaction between grapefruit juice and diazepam in humans. Eur J Drug Metab Pharmacokinet. 1998;23(1):55-59. doi:10.1007/BF03189827
52. Greenblatt DJ, Harmatz JS, Zhang Q, et al. Slow accumulation and elimination of diazepam and its active metabolite with extended treatment in the elderly. J Clin Pharmacol. 2021;61(2):193-203. doi:10.1002/jcph.1726
53. Abernethy DR, Greenblatt DJ. Drug disposition in obese humans: an update. Clin Pharmacokinet. 1986;11(3):199-213. doi:10.2165/00003088-198611030-00002
54. Hanley MJ, Abernethy DR, Greenblatt DJ. Effect of obesity on the pharmacokinetics of drugs in humans. Clin Pharmacokinet. 2010;49(2):71-87. doi:10.2165/11318100-000000000-00000
55. Bauer LA. Drug Dosing in special populations: renal and hepatic disease, dialysis, heart failure, obesity, and drug interactions. In: Weitz M, Thomas, CM, eds. Applied Clinical Pharmacokinetics. 3rd ed. McGraw-Hill Education; 2014. https://accesspharmacy.mhmedical.com/book.aspx?bookid=1374
56. Kendrick JG, Carr RR, Ensom MHH. Pharmacokinetics and drug dosing in obese children. J Pediatr Pharmacol Ther. 2010;15(2):94-109. doi:10.5863/1551-6776-15.2.94
57. Brill MJE, Diepstraten J, van Rongen A, et al. Impact of obesity on drug metabolism and elimination in adults and children. Clin Pharmacokinet. 2012;51(5):277-304. doi:10.2165/11599410-000000000-00000
58. Derry CL, Kroboth PD, Pittenger AL, et al. Pharmacokinetics and pharmacodynamics of triazolam after two intermittent doses in obese and normal-weight men. J Clin Psychopharmacol. 1995;15(3):197-205. doi:10.1097/00004714-199506000-00008
59. Abernethy DR, Greenblatt DJ, Divoll M, et al. The influence of obesity on the pharmacokinetics of oral alprazolam and triazolam. Clin Pharmacokinet. 1984;9(2):177-183. doi:10.2165/00003088-198409020-00005
60. Abernethy DR, Greenblatt DJ, Divoll M, et al. Prolonged accumulation of diazepam in obesity. J Clin Pharmacol. 1983;23(8-9):369-376. doi:10.1002/j.1552-4604.1983.tb02750.x
61. Abernethy DR, Greenblatt DJ, Divoll M, et al. Enhanced glucuronide conjugation of drugs in obesity: studies of lorazepam, oxazepam, and acetaminophen. J Lab Clin Med. 1983;101(6):873-880.
62. Greenblatt DJ, von Moltke LL, Harmatz JS, et al. Alprazolam pharmacokinetics, metabolism, and plasma levels: clinical implications. J Clin Psychiatry. 1993;54 Suppl:4-11.
63. Chen YT, Liu CY, Chang CM, et al. Perceptions, clinical characteristics, and other factors associated with prolonged and high daily dose of benzodiazepine use among patients with anxiety or depressive disorders. J Affect Disord. 2020;271:215-223. doi:10.1016/j.jad.2020.03.077
64. Herman JB, Brotman AW, Rosenbaum JF. Rebound anxiety in panic disorder patients treated with shorter-acting benzodiazepines. J Clin Psychiatry. 1987;48(Suppl):22-28.
65. Herman JB, Rosenbaum JF, Brotman AW. The alprazolam to clonazepam switch for the treatment of panic disorder. J Clin Psychopharmacol. 1987;7(3):175-178.
1. Rickels K, Moeller HJ. Benzodiazepines in anxiety disorders: reassessment of usefulness and safety. World J Biol Psychiatry. 2019;20(7):514-518. doi:10.1080/15622975.2018.1500031
2. Stevens JC, Pollack MH. Benzodiazepines in clinical practice: consideration of their long-term use and alternative agents. J Clin Psychiatry. 2005;66(Suppl 2):21-27.
3. Pollack MH, van Ameringen M, Simon NM, et al. A double-blind randomized controlled trial of augmentation and switch strategies for refractory social anxiety disorder. Am J Psychiatry. 2014;171(1):44-53. doi:10.1176/appi.ajp.2013.12101353
4. Strawn JR, Geracioti L, Rajdev N, et al. Pharmacotherapy for generalized anxiety disorder in adult and pediatric patients: an evidence-based treatment review. Expert Opin Pharmacother. 2018;19(10):1057-1070. doi:10.1080/14656566.2018.1491966
5. Karaca-Mandic P, Meara E, Morden NE. The growing problem of co-treatment with opioids and benzodiazepines. BMJ. 2017;356:j1224. doi:10.1136/bmj.j1224
6. Bachhuber MA, Hennessy S, Cunningham CO, et al. Increasing benzodiazepine prescriptions and overdose mortality in the United States, 1996-2013. Am J Public Health. 2016;106(4):686-688. doi:10.2105/AJPH.2016.303061
7. Bentué-Ferrer D, Akwa Y. Benzodiazepines: Effects on memory functioning. In: Pandi-Perumal SR, Verster J, Monti J, et al, eds. Sleep Disorders: Diagnosis and Therapeutics. CRC Press; 2008:104-114. doi:10.3109/9780203091715-15
8. Pomara N, Facelle TM, Roth AE, et al. Dose-dependent retrograde facilitation of verbal memory in healthy elderly after acute oral lorazepam administration.Psychopharmacology (Berl). 2006;185(4):487-494. doi:10.1007/s00213-006-0336-0
9. Gray SL, Dublin S, Yu O, et al. Benzodiazepine use and risk of incident dementia or cognitive decline: prospective population based study. BMJ. 2016;352:i90. doi:10.1136/bmj.i90
10. Biétry FA, Pfeil AM, Reich O, et al. Benzodiazepine use and risk of developing Alzheimer’s disease: a case-control study based on Swiss claims data. CNS Drugs. 2017;31(3):245-251. doi:10.1007/s40263-016-0404-x
11. de Gage SB, Moride Y, Ducruet T, et al. Benzodiazepine use and risk of Alzheimer’s disease: case-control study. BMJ. 2014;349g5205. doi:10.1136/bmj.g5205
12. Shah R, Raji MA, Westra J, et al. Association of co-prescribing of opioid and benzodiazepine substitutes with incident falls and fractures among older adults: a cohort study. BMJ Open. 2021;11(12):e052057. doi:10.1136/bmjopen-2021-052057
13. Guina J, Rossetter SR, DeRhodes BJ, et al. Benzodiazepines for PTSD: a systematic review and meta-analysis. J Psychiatr Pract. 2015;21(4):281-303.
14. Ekström MP, Bornefalk-Hermansson A, Abernethy AP, et al. Safety of benzodiazepines and opioids in very severe respiratory disease: national prospective study. BMJ. 2014;348:g445. doi:10.1136/bmj.g445
15. Donovan LM, Malte CA, Spece LJ, et al. Center predictors of long-term benzodiazepine use in chronic obstructive pulmonary disease and post-traumatic stress disorder. Ann Am Thorac Soc. 2019;16(9):1151-1157. doi:10.1513/AnnalsATS.201901-048OC
16. Sheehy O, Zhao JP, Bérard A. Association between incident exposure to benzodiazepines in early pregnancy and risk of spontaneous abortion. JAMA Psychiatry. 2019;76(9):948-957. doi:10.1001/jamapsychiatry.2019.0963
17. Kelly LE, Poon S, Madadi P, et al. Neonatal benzodiazepines exposure during breastfeeding. J Pediatr. 2012;161(3):448-451. doi:10.1016/j.jpeds.2012.03.003
18. Agarwal SD, Landon BE. Patterns in outpatient benzodiazepine prescribing in the United States. JAMA Netw Open. 2019;2(1):e187399. doi:10.1001/jamanetworkopen.2018.7399
19. Hirschtritt ME, Olfson M, Kroenke K. Balancing the risks and benefits of benzodiazepines. JAMA. 2021;325(4):347-348. doi:10.1001/jama.2020.22106
20. Brunton LL, Hilal-Dandan R, Knollman BC, eds. Goodman & Gilman’s: The Pharmacological Basis of Therapeutics. McGraw-Hill Education; 2018.
21. Nutt DJ, Malizia AL. New insights into the role of the GABA(A)-benzodiazepine receptor in psychiatric disorder. British J Psychiatry. 2001;179:390-396. doi:10.1192/bjp.179.5.390
22. Sigel E. Mapping of the benzodiazepine recognition site on GABA(A) receptors. Curr Top Med Chem. 2002;2(8):833-839. doi:10.2174/1568026023393444
23. Savic
24. Smith TA. Type A gamma-aminobutyric acid (GABAA) receptor subunits and benzodiazepine binding: significance to clinical syndromes and their treatment. Br J Biomed Sci. 2001;58(2):111-121.
25. Althaus AL, Ackley MA, Belfort GM, et al. Preclinical characterization of zuranolone (SAGE-217), a selective neuroactive steroid GABAA receptor positive allosteric modulator. Neuropharmacology. 2020;181:108333. doi:10.1016/j.neuropharm.2020.108333
26. Jacob TC, Michels G, Silayeva L, et al. Benzodiazepine treatment induces subtype-specific changes in GABA(A) receptor trafficking and decreases synaptic inhibition. Proc Natl Acad Sci U S A. 2012;109(45):18595-18600. doi:10.1073/pnas.1204994109
27. Nicholson MW, Sweeney A, Pekle E, et al. Diazepam-induced loss of inhibitory synapses mediated by PLCδ/ Ca2+/calcineurin signalling downstream of GABAA receptors. Mol Psychiatry. 2018;23(9):1851-1867. doi:10.1038/s41380-018-0100-y
28. Dobson ET, Bloch MH, Strawn JR. Efficacy and tolerability of pharmacotherapy for pediatric anxiety disorders: a network meta-analysis. J Clin Psychiatry. 2019;80(1):17r12064. doi:10.4088/JCP.17r12064
29. Kuang H, Johnson JA, Mulqueen JM, et al. The efficacy of benzodiazepines as acute anxiolytics in children: a meta-analysis. Depress Anxiety. 2017;34(10):888-896. doi:10.1002/da.22643
30. Chugani DC, Muzik O, Juhász C, et al. Postnatal maturation of human GABAA receptors measured with positron emission tomography. Ann Neurol. 2001;49(5):618-626. doi:10.1002/ana.1003
31. Jochemsen R, Breimer DD. Pharmacokinetics of benzodiazepines: metabolic pathways and plasma level profiles. Curr Med Res Opin. 1984;8(Suppl 4):60-79. doi:10.1185/03007998409109545
32. Greenblatt DJ, Harmatz JS, Dorsey C, et al. Comparative single-dose kinetics and dynamics of lorazepam, alprazolam, prazepam, and placebo. Clin Pharmacol Ther. 1988;44(3)326-334. doi:10.1038/clpt.1988.158
33. Shader RI, Georgotas A, Greenblatt DJ, et al. Impaired absorption of desmethydiazepam from clorazepate by magnesium aluminum hydroxide. Clin Pharmacol Ther. 1978;24(3):308-315. doi:10.1002/cpt1978243308
34. Greenblatt DJ, Allen MD, MacLaughlin DS, et al. Diazepam absorption: effect of antacids and food. Clin Pharmacol Ther. 1978;24(5):600-609. doi:10.1002/cpt1978245600
35. Yamazaki A, Kumagai Y, Fujita T, et al. Different effects of light food on pharmacokinetics and pharmacodynamics of three benzodiazepines, quazepam, nitrazepam and diazepam. J Clin Pharm Ther. 2007;32(1):31-39. doi:10.1111/j.1365-2710.2007.00795.x
36. Stimpfl J, Mills JA, Strawn JR. Pharmacologic predictors of benzodiazepine response trajectory in anxiety disorders: a Bayesian hierarchical modeling meta-analysis. CNS Spectr. 2023;28(1):53-60. doi:10.1017/S1092852921000870
37. Griffin CE 3rd, Kaye AM, Bueno FR, et al. Benzodiazepine pharmacology and central nervous system-mediated effects. Ochsner J. 2013;13(2):214-223.
38. Buffett-Jerrott SE, Stewart SH. Cognitive and sedative effects of benzodiazepine use. Curr Pharm Des. 2005;8(1):45-58. doi:10.2174/1381612023396654
39. Fukasawa T, Suzuki A, Otani K. Effects of genetic polymorphism of cytochrome P450 enzymes on the pharmacokinetics of benzodiazepines. J Clin Pharm Ther. 2007;32(4):333-341. doi:10.1111/j.1365-2710.2007.00829.x
40. Kraus JW, Desmond PV, Marshall JP, et al. Effects of aging and liver disease on disposition of lorazepam. Clin Pharmacol Ther. 1978;24(4):411-419. doi:10.1002/cpt1978244411
41. Greenblatt DJ. Clinical pharmacokinetics of oxazepam and lorazepam. Clin Pharmacokinet. 1981;6(2):89-105. doi:10.2165/00003088-198106020-00001
42. Walkenstein SS, Wiser R, Gudmundsen CH, et al. Absorption, metabolism, and excretion of oxazepam and its succinate half‐ester. J Pharm Sci. 1964;53(10):1181-1186. doi:10.1002/jps.2600531010
43. Shull HJ, Wilkinson GR, Johnson R, et al. Normal disposition of oxazepam in acute viral hepatitis and cirrhosis. Ann Intern Med. 1976;84(4):420-425. doi:10.7326/0003-4819-84-4-420
44. Abernethy DR, Greenblatt DJ, Ochs HR, et al. Lorazepam and oxazepam kinetics in women on low-dose oral contraceptives. Clin Pharmacol Ther. 1983;33(5):628-632. doi:10.1038/clpt.1983.85
45. Greenblatt DJ, Allen MD, Harmatz JS, et al. Diazepam disposition determinants. Clin Pharmacol Ther. 1980;27(3):301-312. doi:10.1038/clpt.1980.40
46. Ochs HR, Greenblatt DJ, Knüchel M. Kinetics of diazepam, midazolam, and lorazepam, in cigarette smokers. Chest. 1985;87(2):223-226. doi:10.1378/chest.87.2.223
47. Smith RB, Gwilt PR, Wright CE 3rd. Single- and multiple-dose pharmacokinetics of oral alprazolam in healthy smoking and nonsmoking men. Clin Pharm. 1983;2(2):139-143.
48. Figgitt DP, McClellan KJ. Fluvoxamine. An updated review of its use in the management of adults with anxiety disorders. Drugs. 2000;60(4):925-954. doi:10.2165/00003495-200060040-00006
49. Greenblatt DJ, Wright CE. Clinical pharmacokinetics of alprazolam. Therapeutic implications. Clin Pharmacokinet. 1993;24(6):453-471. doi:10.2165/00003088-199324060-00003
50. Yasui N, Kondo T, Furukori H, et al. Effects of repeated ingestion of grapefruit juice on the single and multiple oral-dose pharmacokinetics and pharmacodynamics of alprazolam. Psychopharmacology (Berl). 2000;150(2):185-190. doi:10.1007/s002130000438
51. Özdemir M, Aktan Y, Boydagˇ BS, et al. Interaction between grapefruit juice and diazepam in humans. Eur J Drug Metab Pharmacokinet. 1998;23(1):55-59. doi:10.1007/BF03189827
52. Greenblatt DJ, Harmatz JS, Zhang Q, et al. Slow accumulation and elimination of diazepam and its active metabolite with extended treatment in the elderly. J Clin Pharmacol. 2021;61(2):193-203. doi:10.1002/jcph.1726
53. Abernethy DR, Greenblatt DJ. Drug disposition in obese humans: an update. Clin Pharmacokinet. 1986;11(3):199-213. doi:10.2165/00003088-198611030-00002
54. Hanley MJ, Abernethy DR, Greenblatt DJ. Effect of obesity on the pharmacokinetics of drugs in humans. Clin Pharmacokinet. 2010;49(2):71-87. doi:10.2165/11318100-000000000-00000
55. Bauer LA. Drug Dosing in special populations: renal and hepatic disease, dialysis, heart failure, obesity, and drug interactions. In: Weitz M, Thomas, CM, eds. Applied Clinical Pharmacokinetics. 3rd ed. McGraw-Hill Education; 2014. https://accesspharmacy.mhmedical.com/book.aspx?bookid=1374
56. Kendrick JG, Carr RR, Ensom MHH. Pharmacokinetics and drug dosing in obese children. J Pediatr Pharmacol Ther. 2010;15(2):94-109. doi:10.5863/1551-6776-15.2.94
57. Brill MJE, Diepstraten J, van Rongen A, et al. Impact of obesity on drug metabolism and elimination in adults and children. Clin Pharmacokinet. 2012;51(5):277-304. doi:10.2165/11599410-000000000-00000
58. Derry CL, Kroboth PD, Pittenger AL, et al. Pharmacokinetics and pharmacodynamics of triazolam after two intermittent doses in obese and normal-weight men. J Clin Psychopharmacol. 1995;15(3):197-205. doi:10.1097/00004714-199506000-00008
59. Abernethy DR, Greenblatt DJ, Divoll M, et al. The influence of obesity on the pharmacokinetics of oral alprazolam and triazolam. Clin Pharmacokinet. 1984;9(2):177-183. doi:10.2165/00003088-198409020-00005
60. Abernethy DR, Greenblatt DJ, Divoll M, et al. Prolonged accumulation of diazepam in obesity. J Clin Pharmacol. 1983;23(8-9):369-376. doi:10.1002/j.1552-4604.1983.tb02750.x
61. Abernethy DR, Greenblatt DJ, Divoll M, et al. Enhanced glucuronide conjugation of drugs in obesity: studies of lorazepam, oxazepam, and acetaminophen. J Lab Clin Med. 1983;101(6):873-880.
62. Greenblatt DJ, von Moltke LL, Harmatz JS, et al. Alprazolam pharmacokinetics, metabolism, and plasma levels: clinical implications. J Clin Psychiatry. 1993;54 Suppl:4-11.
63. Chen YT, Liu CY, Chang CM, et al. Perceptions, clinical characteristics, and other factors associated with prolonged and high daily dose of benzodiazepine use among patients with anxiety or depressive disorders. J Affect Disord. 2020;271:215-223. doi:10.1016/j.jad.2020.03.077
64. Herman JB, Brotman AW, Rosenbaum JF. Rebound anxiety in panic disorder patients treated with shorter-acting benzodiazepines. J Clin Psychiatry. 1987;48(Suppl):22-28.
65. Herman JB, Rosenbaum JF, Brotman AW. The alprazolam to clonazepam switch for the treatment of panic disorder. J Clin Psychopharmacol. 1987;7(3):175-178.

Pediatric insomnia: Treatment
Children and adolescents who do not receive sufficient sleep can experience worsening inattention, daytime fatigue, and cognitive and behavioral difficulties. Assessment and treatment of insomnia and other sleep difficulties in young patients is critical as poor sleep increases their risk for depression, self-harm, and suicide.
In Part 1 of this article (Pediatric insomnia: Assessment and diagnosis,
Psychotherapeutic interventions
Regardless of the source of a child’s insomnia or co-occurring disorders, healthy sleep practices are the first line behavioral treatment, including for youth with attention-deficit/hyperactivity disorder (ADHD), anxiety disorders, obsessive-compulsive disorder, and depressive disorders.
Healthy sleep practices/sleep hygiene
Developmentally appropriate bedtimes and routines (Table). Helping children establish a regular, consistent bedtime is key in promoting healthy sleep. Ideally, the bedtime routine involves 3 to 4 activities each night in the same order, and these activities should be relaxing and soothing (eg, taking a bath, putting on pajamas, reading books). Setting age-appropriate bedtimes also is important. If an older child is asked to go to bed at 8 pm but cannot fall asleep for an hour, they may not have insomnia but instead a developmentally inappropriate bedtime. Several studies found that children younger than age 10 should go to bed no later than 9 pm. Bedtimes later than 9 pm for young children are correlated with shorter sleep duration.1

Consistent sleep schedules. Another important aspect of healthy sleep is working with parents to enforce a consistent bedtime and wake-up time, including weekdays and weekends. Ideally, bedtime on weekdays and weekends should not vary by more than 1 hour. Helping children wake up at the same time each day helps to set and regulate their circadian rhythm. Keeping these schedules consistent on vacations and school holidays also is helpful. For adolescents, the weekday/weekend bedtimes can vary by up to 2 hours because adolescents have a delayed circadian rhythm and wake-up times for high school can be early.
Environmental factors. An important piece of parental education is stimulus control and the ingredients of healthy sleep. Healthy sleep ingredients include a dark, quiet, consistent, and cool bedroom; a comfortable bed, the child feeling safe, and limited environmental stimuli.
Continue to: Cognitive-behavioral therapy for insomnia...
Cognitive-behavioral therapy for insomnia
Relaxation. Pediatric patients can be taught relaxation, mindfulness, meditation, and progressive muscle relaxation techniques to help lower overall stress. This can be especially helpful for youth with sleep disorders or anxiety. Guided relaxation apps are popular among children and teens, and various apps offer soothing sounds, deep breathing, progressive muscle relaxation, and guided imagery. This can be taught in psychotherapy sessions and used at home to promote gains in between sessions.
Stimulus control. Stimulus control involves using the bed exclusively for sleep and avoiding nonsleep activities in bed (eg, reading, watching television, using a computer, worrying). These activities promote wakefulness and insomnia. This may mean the child does not get into bed until they cannot keep their eyes open, even if that delays bedtime. If the child is still awake within 15 to 20 minutes, they should be encouraged to get out of bed and engage in a nonstimulating activity such as meditation, reading, or sitting quietly in the dark or low light. This recommendation can run counter to parents’ intuition that children with sleep problems should go to bed earlier. Using the bed only for sleep conditions the child to falling asleep or being asleep when in bed.
Sleep restriction. Sleep restriction involves restricting sleep to a set number of hours in order to increase their sleep efficiency (time slept in bed divided by total time spent in bed x 100). Restricting sleep to 6 to 7 hours increases sleep efficiency, consolidates sleep, and extinguishes the association of being awake in bed. For older adolescents, sleep restriction may help to limit their time in bed to either falling asleep or being asleep. This is intended to be used as a short-term strategy and only after other sleep hygiene measures (bedtime routine, environmental factors, etc) have been put into place for several weeks. While this strategy sounds unappealing to most individuals with insomnia, it can lead to lasting change due to the use of behavioral conditioning in bed. Because sleep restriction can lead to significant daytime sleepiness and impairment during the day, sleep should not be restricted to <6 hours a day for children and adolescents. Once the adolescent is sleeping more consistently and sleep efficiency reaches 85% or higher, time in bed for sleep is increased.2
Cognitive restructuring. Some children and adolescents develop maladaptive thoughts about sleep that further promote insomnia. These thoughts might include “I will never get to sleep,” “I am going to have a terrible day if I cannot fall asleep,” or “I will fail my test tomorrow if I am unable to sleep.” Such maladaptive thoughts are often untrue but promote wakefulness in the early or middle part of the night. Cognitive restructuring involves helping the child identify each problematic thought, challenge how accurate each thought is with evidence, and replace the problematic thought with a more helpful thought. For instance, an adolescent can recognize that even if they have a sleepless night, their catastrophic outcome (eg, “I will not be able to function”) is likely untrue. A psychologist can help review evidence for this, including previous times when the adolescent has not slept well and managed to get through the next day.
When is pharmacologic treatment needed?
Pharmacologic treatment may be indicated if a child does not show significant improvement following behavioral intervention (Figure). However, it is critical to exclude other primary causes of dyssomnia (eg, obstructive sleep apnea, iron deficiency anemia) before pursuing pharmacotherapy, because pharmacotherapy could mask an underlying disorder. Moreover, while there is relatively limited evidence for psychopharmacologic interventions for sleep difficulties in children and adolescents, a large survey of child and adolescent psychiatrists (N = 1,273) suggested that medications were considered for one-quarter of pediatric patients with insomnia.3 Further, patients with specific comorbidities such as neurodevelopmental disorders may be more likely to be prescribed soporifics.4

Continue to: What is the evidence for pharmacotherapy?...
What is the evidence for pharmacotherapy?
Antihistamines. Histamine antagonists—which promote sleep by blocking the wakefulness-promoting and circadian-related effects of histamine—are the most commonly used medications to treat pediatric insomnia, despite a dearth of data from prospective trials.5,6 In 1 small study, Russo et al7 found diphenhydramine, 1 mg/kg at bedtime, reduced sleep latency and nighttime awakenings, and increased sleep duration in patients ages 2 to 12; similar effects have been observed in pediatric burn patients.8 There are some limited data for other H1 antagonists (eg, hydroxyzine) in pediatric insomnia.9-11
Alpha-2 agonists increase rapid eye movement sleep via dose-dependent downregulation of noradrenergic signaling12 and thus have been commonly prescribed for insomnia in children and adolescents. In fact, the nonselective alpha-2 agonist clonidine is among the most prescribed medications for youth with insomnia, and may be efficacious in youth with neurodevelopmental disorders and ADHD.13 In small retrospective studies, clonidine decreased sleep latency and nighttime awakenings in addition to increasing sleep duration.14 Also, clonidine was well tolerated but associated with daytime somnolence. Guanfacine—a selective alpha-2 agonist—is also commonly prescribed for insomnia in youth, although results of trials have been equivocal.15 Given the more rapid absorption and shorter Tmax of clonidine relative to guanfacine, the former may be preferred as a soporific.
Melatonin and melatonin agonists. The primary regulator of the sleep-wake cycle is melatonin, an endogenous hormone produced by the pineal gland in response to changes in retinal light perception. Exogenous melatonin supplementation may be the preferred initial pharmacotherapy for sleep-onset insomnia due to its chronobiotic properties.16 In clinical studies, both immediate-release17,18 and extended-release19 melatonin reduced sleep-onset latency and increased total sleep duration in pediatric patients, although the increase in total duration of sleep was greater with extended-release preparations. Additionally, tolerability data for melatonin in pediatric patients are encouraging. A 2-year randomized trial of prolonged-release melatonin for insomnia in pediatric patients found no adverse effects with regard to growth, body mass index, or pubertal development.20 Additionally, significant improvements in sleep quality, sleep patterns, and caregiver satisfaction were maintained throughout the trial, and no withdrawal symptoms were observed upon discontinuation.
Melatonin may have a particularly important role in circadian rhythm sleep disorders. In this regard, low-dose melatonin (0.5 mg), when timed relative to the endogenous dim light melatonin onset (DLMO), is more effective in shifting sleep phase than higher doses, which suggests that timing may have greater impact than dosage.21 Data regarding melatonin administration with respect to DLMO suggest that the optimal administration time is 4 to 6 hours before a child’s preferred bedtime, and doses of 0.5 to 1 mg have been effective when given in this window.22 Variation across studies has contributed to a lack of consensus regarding pediatric melatonin dosing. For example, .05 mg/kg may be a minimal effective dose when given 1 to 2 hours before bedtime18; however, in surveys doses vary considerably, with typical doses of 2.5 to 3 mg for prepubertal children and 5 mg for adolescents.5 Of note, in patients with decreased cytochrome P450 (CYP) 1A2 activity, lack of diurnal variation in melatonin serum concentration may decrease the effectiveness of melatonin.16Ramelteon is a potent agonist of the melatonin MT1 and MT2 receptors, with a significantly higher binding affinity than melatonin in vitro. In case reports, ramelteon was well-tolerated, improved delayed sleep onset, and decreased nighttime awakenings.23
Zolpidem, eszopiclone and zaleplon. Studies of selective GABAergic modulators and benzodiazepines have not produced positive results in prospective trials of youth with insomnia. Zolpidem was studied in children and adolescents (N = 201) with ADHD; although sleep latency did not differ between zolpidem and placebo, some significant improvements were observed in adolescents.24 Zolpidem was generally well tolerated, with approximately 10% of youth discontinuing due to adverse effects. Additionally, eszopiclone—which has a longer duration of action compared with zolpidem—has been studied in children and adolescents with ADHD (N = 486) who were also evaluated with a sleep study. No differences were observed between placebo and eszopiclone in terms of sleep latency and approximately 10% of patients discontinued treatment as a result of adverse events.25 We were unable to locate any prospective trials of zaleplon or benzodiazepine receptor agonists for insomnia in youth, although some reports suggest that clonazepam may have a possible role for specific parasomnias.26,27Dual orexin receptor antagonists. Suvorexant, an antagonist of the wakefulness-promoting neuropeptide orexin, improved subjective sleep quality in a prospective trial of adolescents with insomnia (N = 30), although dropout was high (44%).28 Of those patients, reasons for discontinuation included loss to follow-up, lack of effectiveness, and abnormal dreams. We were unable to locate any trials of lemborexant in pediatric patients.
Atypical antidepressants. Trazodone is commonly prescribed for insomnia in pediatric patients with comorbid mood or anxiety disorders. In open-label studies of children and toddlers, trazodone may be well-tolerated and improve sleep.29 Additionally, development of a physiologically based pharmacokinetic model to inform trazodone dosing for youth with insomnia is underway.30 Some studies in adolescents with depression suggest that caution should be used when combining trazodone with medications that inhibit CYP2D6. In the Treatment of SSRI-Resistant Depression in Adolescents study, none of the patients who were treated with trazodone (vs other soporifics) improved.31 This may relate to CYP2D6 interactions and accumulation of methyl-chloro-piperazine (mCPP), a trazodone metabolite that is associated with dysphoria, irritability, and depression.31 This finding has been replicated in a separate cohort of depressed adolescents.32
Because of its antihistaminergic effects, mirtazapine has been used to treat insomnia in adults. One open-label study of mirtazapine in children and young adults ages 3 to 23 with neurodevelopmental disorders suggested that mirtazapine improved behavioral symptoms and insomnia, and was associated with few treatment-limiting adverse effects.33
Tricyclic antidepressants. In a retrospective study of youth with insomnia who failed behavioral interventions and melatonin (N = 29), doxepin, a potent H1 antagonist, improved subjective sleep in one-half of patients.34
Continue to: Consultation with pediatric sleep medicine specialists...
Consultation with pediatric sleep medicine specialists
It often will behoove the psychiatric clinician to review their concerns with a behavioral sleep medicine specialist or a psychologist with specific expertise in the psychotherapeutic treatment of sleep who can provide important guidance regarding the key aspects of treatment. When discussing a particular patient’s presentation with the pediatric behavioral sleep psychologist/specialist, consider the following questions:
- Is the child’s sleep disorder the primary problem, or is the child’s insomnia secondary to another diagnosis (psychiatric or nonpsychiatric)?
- What are the primary sleep-related problems the child/family presents with? How long have the symptoms been present?
- Is the sleep disorder interfering with the child’s functioning, either academically or socially? Does the child’s sleep problem interfere with other family members’ sleep?
- Does the child have a comorbid psychological conditions such as ADHD, depression, or anxiety, and/or is undergoing treatment for these disorders, which may play a role in the sleep problem?
- Is a sleep study warranted?
A sleep study, also known as polysomnography (PSG), is a diagnostic test in which physiologic parameters are continuously recorded during a period of sleep via electroencephalography, electromyography, electrooculogram, electrocardiogram, airflow sensors, pulse oximeter, body position monitors, and video. In 2012, the American Academy of Sleep Medicine published evidenced-based practice parameters for respiratory and nonrespiratory indications for PSG.35 It is most commonly indicated to rule out obstructive sleep apnea in pediatric patients who exhibit snoring, gasping, irregular breathing, witnessed apneic events, unusual head positioning, or other signs of obstructive breathing during sleep. Nonrespiratory indications for PSG include children suspected of having periodic limb movement disorder and suspected narcolepsy. Children with frequent parasomnias, epilepsy, or nocturnal enuresis should be clinically screened for presence of comorbid sleep disorders, and PSG would be indicated if there are concerns about a possible sleep-disordered breathing disorder. PSG is also recommended for children with hypersomnia, to differentiate a parasomnia from sleep-related epilepsy, and for children suspected of having restless leg syndrome.36 PSG is not typically indicated in the initial evaluation of insomnia (unless there is evidence of a comorbid sleep disorder), circadian rhythm disorders (ie, delayed sleep phase syndrome), or for evaluation of children with sleep-related bruxism.3 Special considerations for PSG in children include ensuring a parent or guardian is always with the child, providing developmentally appropriate sleeping arrangements, arranging family tours of the sleep lab prior to the study, and accommodating for earlier bedtimes.37
Bottom Line
Techniques to promote healthy sleep in pediatric patients include behavioral interventions such as setting a developmentally appropriate bedtime and a consistent wake time, establishing bedtime routines, and encouraging relaxation/ wind-down period before bed. Cognitive-behavioral therapy for insomnia (CBT-I) may include cognitive restructuring of anxious thoughts, relaxation training, stimulus control, and sleep restriction. Use of medications may be indicated for children and teens who have not responded to CBT-I or soporific dosing of melatonin.
1. Mindell JA, Li AM, Sadeh A, et al. Bedtime routines for young children: a dose-dependent association with sleep outcomes. Sleep. 2015;38(5):717-722.
2. Kansagra S. Sleep disorders in adolescents. Pediatrics. 2020;145(Suppl 2):S204-S209.
3. Owens JA, Mindell JA. Pediatric insomnia. Pediatr Clin North Am. 2011;58(3):555-569.
4. Bruni O, Angriman M, Melegari MG, et al. Pharmacotherapeutic management of sleep disorders in children with neurodevelopmental disorders. Expert Opin Pharmacother. 2019;20(18):2257-2271.
5. Owens JA, Rosen CL, Mindell JA, et al. Use of pharmacotherapy for insomnia in child psychiatry practice: a national survey. Sleep Med. 2010;11(7):692-700.
6. Schnoes CJ, Kuhn BR, Workman EF, et al. Pediatric prescribing practices for clonidine and other pharmacologic agents for children with sleep disturbance. Clin Pediatr (Phila). 2006;45(3):229-238.
7. Russo RM, Gururaj VJ, Allen JE. The effectiveness of diphenhydramine HCI in pediatric sleep disorders. J Clin Pharmacol. 1976;16(5-6):284-288.
8. Yangzom N, Gottschlich MM, Ossege J, et al. The effect of diphenhydramine on sleep in pediatric burn patients: a secondary analysis. J Burn Care Res. 2015;36(2):266-271.
9. Ghanizadeh A, Zare S. A preliminary randomised double-blind placebo-controlled clinical trial of hydroxyzine for treating sleep bruxism in children. J Oral Rehabil. 2013;40(6):413-417.
10. Bektas O, Arıca B, Teber S, et al. Chloral hydrate and/or hydroxyzine for sedation in pediatric EEG recording. Brain Dev. 2014;36(2):130-136.
11. Ottaviano S, Giannotti F, Cortesi F. The effect of niaprazine on some common sleep disorders in children. A double-blind clinical trial by means of continuous home-videorecorded sleep. Childs Nerv Syst. 1991;7(6):332-335.
12. Nguyen M, Tharani S, Rahmani M, et al. A review of the use of clonidine as a sleep aid in the child and adolescent population. Clin Pediatr (Phila). 2014;53(3):211-216.
13. Prince JB, Wilens TE, Biederman J, et al. Clonidine for sleep disturbances associated with attention-deficit hyperactivity disorder: a systematic chart review of 62 cases. J Am Acad Child Adolesc Psychiatry. 1996;35(5):599-605.

14. Ingrassia A, Turk J. The use of clonidine for severe and intractable sleep problems in children with neurodevelopmental disorders--a case series. Eur Child Adolesc Psychiatry. 2005;14(1):34-40.
15. Politte LC, Scahill L, Figueroa J, et al. A randomized, placebo-controlled trial of extended-release guanfacine in children with autism spectrum disorder and ADHD symptoms: an analysis of secondary outcome measures. Neuropsychopharmacology. 2018;43(8):1772-1778.
16. Bruni O, Alonso-Alconada D, Besag F, et al. Current role of melatonin in pediatric neurology: clinical recommendations. Eur J Paediatr Neurol. 2015;19(2):122-1233.
17. Jain SV, Horn PS, Simakajornboon N, et al. Melatonin improves sleep in children with epilepsy: a randomized, double-blind, crossover study. Sleep Med. 2015;16(5):637-644.
18. van Geijlswijk IM, van der Heijden KB, Egberts AC, et al. Dose finding of melatonin for chronic idiopathic childhood sleep onset insomnia: an RCT. Psychopharmacology (Berl). 2010;212(3):379-391.
19. Gringras P, Nir T, Breddy J, et al. Efficacy and safety of pediatric prolonged-release melatonin for insomnia in children with autism spectrum disorder. J Am Acad Child Adolesc Psychiatry. 2017;56(11):948-957.e4.
20. Malow BA, Findling RL, Schroder CM, et al. Sleep, growth, and puberty after two years of prolonged-release melatonin in children with autism spectrum disorder. J Am Acad Child Adolesc Psychiatry. 2021;60(2):252-261.e3.
21. Burgess HJ, Emens JS. Drugs used in circadian sleep-wake rhythm disturbances. Sleep Med Clin. 2020;15(2):301-310.
22. Arns M, Kooij JJS, Coogan AN. Review: identification and management of circadian rhythm sleep disorders as a transdiagnostic feature in child and adolescent psychiatry. J Am Acad Child Adolesc Psychiatry. 2021;60(9):1085-1095.
23. Kawabe K, Horiuchi F, Oka Y, et al. The melatonin receptor agonist ramelteon effectively treats insomnia and behavioral symptoms in autistic disorder. Case Rep Psychiatry. 2014;2014:561071.
24. Blumer JL, Findling RL, Shih WJ, et al. Controlled clinical trial of zolpidem for the treatment of insomnia associated with attention-deficit/hyperactivity disorder in children 6 to 17 years of age. Pediatrics. 2009;123(5):e770-e776.
25. Sangal RB, Blumer JL, Lankford DA, et al. Eszopiclone for insomnia associated with attention-deficit/hyperactivity disorder. Pediatrics. 2014;134(4):e1095-e1103.
26. Arens R, Wright B, Elliott J, et al. Periodic limb movement in sleep in children with Williams syndrome. J Pediatr. 1998;133(5):670-674.
27. Thirumalai SS, Shubin RA, Robinson R. Rapid eye movement sleep behavior disorder in children with autism. J Child Neurol. 2002;17(3):173-178.
28. Kawabe K, Horiuchi F, Ochi M, et al. Suvorexant for the treatment of insomnia in adolescents. J Child Adolesc Psychopharmacol. 2017;27(9):792-795.
29. Pranzatelli MR, Tate ED, Dukart WS, et al. Sleep disturbance and rage attacks in opsoclonus-myoclonus syndrome: Response to trazodone. J Pediatr. 2005;147(3):372-378.
30. Oggianu L, Ke AB, Chetty M, et al. Estimation of an appropriate dose of trazodone for pediatric insomnia and the potential for a trazodone-atomoxetine interaction. CPT Pharmacometrics Syst Pharmacol. 2020;9(2):77-86.
31. Shamseddeen W, Clarke G, Keller MB, et al. Adjunctive sleep medications and depression outcome in the treatment of serotonin-selective reuptake inhibitor resistant depression in adolescents study. J Child Adolesc Psychopharmacol. 2012;22(1):29-36.
32. Sultan MA, Courtney DB. Adjunctive trazodone and depression outcome in adolescents treated with serotonin re-uptake inhibitors. J Can Acad Child Adolesc Psychiatry. 2017;26(3):233-240.
33. Posey DJ, Guenin KD, Kohn AE, et al. A naturalistic open-label study of mirtazapine in autistic and other pervasive developmental disorders. J Child Adolesc Psychopharmacol. 2001;11(3):267-277.
34. Shah YD, Stringel V, Pavkovic I, et al. Doxepin in children and adolescents with symptoms of insomnia: a single-center experience. J Clin Sleep Med. 2020;16(5):743-747.
35. Aurora RN, Lamm CI, Zak RS, et al. Practice parameters for the non-respiratory indications for polysomnography and multiple sleep latency testing for children. Sleep. 2012;35(11):1467-1473.
36. de Zambotti M, Goldstone A, Colrain IM, et al. Insomnia disorder in adolescence: diagnosis, impact, and treatment. Sleep Med Rev. 2018;39:12-24.
37. Beck SE, Marcus CL. Pediatric polysomnography. Sleep Med Clin. 2009;4(3):393-406.
Children and adolescents who do not receive sufficient sleep can experience worsening inattention, daytime fatigue, and cognitive and behavioral difficulties. Assessment and treatment of insomnia and other sleep difficulties in young patients is critical as poor sleep increases their risk for depression, self-harm, and suicide.
In Part 1 of this article (Pediatric insomnia: Assessment and diagnosis,
Psychotherapeutic interventions
Regardless of the source of a child’s insomnia or co-occurring disorders, healthy sleep practices are the first line behavioral treatment, including for youth with attention-deficit/hyperactivity disorder (ADHD), anxiety disorders, obsessive-compulsive disorder, and depressive disorders.
Healthy sleep practices/sleep hygiene
Developmentally appropriate bedtimes and routines (Table). Helping children establish a regular, consistent bedtime is key in promoting healthy sleep. Ideally, the bedtime routine involves 3 to 4 activities each night in the same order, and these activities should be relaxing and soothing (eg, taking a bath, putting on pajamas, reading books). Setting age-appropriate bedtimes also is important. If an older child is asked to go to bed at 8 pm but cannot fall asleep for an hour, they may not have insomnia but instead a developmentally inappropriate bedtime. Several studies found that children younger than age 10 should go to bed no later than 9 pm. Bedtimes later than 9 pm for young children are correlated with shorter sleep duration.1
Consistent sleep schedules. Another important aspect of healthy sleep is working with parents to enforce a consistent bedtime and wake-up time, including weekdays and weekends. Ideally, bedtime on weekdays and weekends should not vary by more than 1 hour. Helping children wake up at the same time each day helps to set and regulate their circadian rhythm. Keeping these schedules consistent on vacations and school holidays also is helpful. For adolescents, the weekday/weekend bedtimes can vary by up to 2 hours because adolescents have a delayed circadian rhythm and wake-up times for high school can be early.
Environmental factors. An important piece of parental education is stimulus control and the ingredients of healthy sleep. Healthy sleep ingredients include a dark, quiet, consistent, and cool bedroom; a comfortable bed, the child feeling safe, and limited environmental stimuli.
Continue to: Cognitive-behavioral therapy for insomnia...
Cognitive-behavioral therapy for insomnia
Relaxation. Pediatric patients can be taught relaxation, mindfulness, meditation, and progressive muscle relaxation techniques to help lower overall stress. This can be especially helpful for youth with sleep disorders or anxiety. Guided relaxation apps are popular among children and teens, and various apps offer soothing sounds, deep breathing, progressive muscle relaxation, and guided imagery. This can be taught in psychotherapy sessions and used at home to promote gains in between sessions.
Stimulus control. Stimulus control involves using the bed exclusively for sleep and avoiding nonsleep activities in bed (eg, reading, watching television, using a computer, worrying). These activities promote wakefulness and insomnia. This may mean the child does not get into bed until they cannot keep their eyes open, even if that delays bedtime. If the child is still awake within 15 to 20 minutes, they should be encouraged to get out of bed and engage in a nonstimulating activity such as meditation, reading, or sitting quietly in the dark or low light. This recommendation can run counter to parents’ intuition that children with sleep problems should go to bed earlier. Using the bed only for sleep conditions the child to falling asleep or being asleep when in bed.
Sleep restriction. Sleep restriction involves restricting sleep to a set number of hours in order to increase their sleep efficiency (time slept in bed divided by total time spent in bed x 100). Restricting sleep to 6 to 7 hours increases sleep efficiency, consolidates sleep, and extinguishes the association of being awake in bed. For older adolescents, sleep restriction may help to limit their time in bed to either falling asleep or being asleep. This is intended to be used as a short-term strategy and only after other sleep hygiene measures (bedtime routine, environmental factors, etc) have been put into place for several weeks. While this strategy sounds unappealing to most individuals with insomnia, it can lead to lasting change due to the use of behavioral conditioning in bed. Because sleep restriction can lead to significant daytime sleepiness and impairment during the day, sleep should not be restricted to <6 hours a day for children and adolescents. Once the adolescent is sleeping more consistently and sleep efficiency reaches 85% or higher, time in bed for sleep is increased.2
Cognitive restructuring. Some children and adolescents develop maladaptive thoughts about sleep that further promote insomnia. These thoughts might include “I will never get to sleep,” “I am going to have a terrible day if I cannot fall asleep,” or “I will fail my test tomorrow if I am unable to sleep.” Such maladaptive thoughts are often untrue but promote wakefulness in the early or middle part of the night. Cognitive restructuring involves helping the child identify each problematic thought, challenge how accurate each thought is with evidence, and replace the problematic thought with a more helpful thought. For instance, an adolescent can recognize that even if they have a sleepless night, their catastrophic outcome (eg, “I will not be able to function”) is likely untrue. A psychologist can help review evidence for this, including previous times when the adolescent has not slept well and managed to get through the next day.
When is pharmacologic treatment needed?
Pharmacologic treatment may be indicated if a child does not show significant improvement following behavioral intervention (Figure). However, it is critical to exclude other primary causes of dyssomnia (eg, obstructive sleep apnea, iron deficiency anemia) before pursuing pharmacotherapy, because pharmacotherapy could mask an underlying disorder. Moreover, while there is relatively limited evidence for psychopharmacologic interventions for sleep difficulties in children and adolescents, a large survey of child and adolescent psychiatrists (N = 1,273) suggested that medications were considered for one-quarter of pediatric patients with insomnia.3 Further, patients with specific comorbidities such as neurodevelopmental disorders may be more likely to be prescribed soporifics.4
Continue to: What is the evidence for pharmacotherapy?...
What is the evidence for pharmacotherapy?
Antihistamines. Histamine antagonists—which promote sleep by blocking the wakefulness-promoting and circadian-related effects of histamine—are the most commonly used medications to treat pediatric insomnia, despite a dearth of data from prospective trials.5,6 In 1 small study, Russo et al7 found diphenhydramine, 1 mg/kg at bedtime, reduced sleep latency and nighttime awakenings, and increased sleep duration in patients ages 2 to 12; similar effects have been observed in pediatric burn patients.8 There are some limited data for other H1 antagonists (eg, hydroxyzine) in pediatric insomnia.9-11
Alpha-2 agonists increase rapid eye movement sleep via dose-dependent downregulation of noradrenergic signaling12 and thus have been commonly prescribed for insomnia in children and adolescents. In fact, the nonselective alpha-2 agonist clonidine is among the most prescribed medications for youth with insomnia, and may be efficacious in youth with neurodevelopmental disorders and ADHD.13 In small retrospective studies, clonidine decreased sleep latency and nighttime awakenings in addition to increasing sleep duration.14 Also, clonidine was well tolerated but associated with daytime somnolence. Guanfacine—a selective alpha-2 agonist—is also commonly prescribed for insomnia in youth, although results of trials have been equivocal.15 Given the more rapid absorption and shorter Tmax of clonidine relative to guanfacine, the former may be preferred as a soporific.
Melatonin and melatonin agonists. The primary regulator of the sleep-wake cycle is melatonin, an endogenous hormone produced by the pineal gland in response to changes in retinal light perception. Exogenous melatonin supplementation may be the preferred initial pharmacotherapy for sleep-onset insomnia due to its chronobiotic properties.16 In clinical studies, both immediate-release17,18 and extended-release19 melatonin reduced sleep-onset latency and increased total sleep duration in pediatric patients, although the increase in total duration of sleep was greater with extended-release preparations. Additionally, tolerability data for melatonin in pediatric patients are encouraging. A 2-year randomized trial of prolonged-release melatonin for insomnia in pediatric patients found no adverse effects with regard to growth, body mass index, or pubertal development.20 Additionally, significant improvements in sleep quality, sleep patterns, and caregiver satisfaction were maintained throughout the trial, and no withdrawal symptoms were observed upon discontinuation.
Melatonin may have a particularly important role in circadian rhythm sleep disorders. In this regard, low-dose melatonin (0.5 mg), when timed relative to the endogenous dim light melatonin onset (DLMO), is more effective in shifting sleep phase than higher doses, which suggests that timing may have greater impact than dosage.21 Data regarding melatonin administration with respect to DLMO suggest that the optimal administration time is 4 to 6 hours before a child’s preferred bedtime, and doses of 0.5 to 1 mg have been effective when given in this window.22 Variation across studies has contributed to a lack of consensus regarding pediatric melatonin dosing. For example, .05 mg/kg may be a minimal effective dose when given 1 to 2 hours before bedtime18; however, in surveys doses vary considerably, with typical doses of 2.5 to 3 mg for prepubertal children and 5 mg for adolescents.5 Of note, in patients with decreased cytochrome P450 (CYP) 1A2 activity, lack of diurnal variation in melatonin serum concentration may decrease the effectiveness of melatonin.16Ramelteon is a potent agonist of the melatonin MT1 and MT2 receptors, with a significantly higher binding affinity than melatonin in vitro. In case reports, ramelteon was well-tolerated, improved delayed sleep onset, and decreased nighttime awakenings.23
Zolpidem, eszopiclone and zaleplon. Studies of selective GABAergic modulators and benzodiazepines have not produced positive results in prospective trials of youth with insomnia. Zolpidem was studied in children and adolescents (N = 201) with ADHD; although sleep latency did not differ between zolpidem and placebo, some significant improvements were observed in adolescents.24 Zolpidem was generally well tolerated, with approximately 10% of youth discontinuing due to adverse effects. Additionally, eszopiclone—which has a longer duration of action compared with zolpidem—has been studied in children and adolescents with ADHD (N = 486) who were also evaluated with a sleep study. No differences were observed between placebo and eszopiclone in terms of sleep latency and approximately 10% of patients discontinued treatment as a result of adverse events.25 We were unable to locate any prospective trials of zaleplon or benzodiazepine receptor agonists for insomnia in youth, although some reports suggest that clonazepam may have a possible role for specific parasomnias.26,27Dual orexin receptor antagonists. Suvorexant, an antagonist of the wakefulness-promoting neuropeptide orexin, improved subjective sleep quality in a prospective trial of adolescents with insomnia (N = 30), although dropout was high (44%).28 Of those patients, reasons for discontinuation included loss to follow-up, lack of effectiveness, and abnormal dreams. We were unable to locate any trials of lemborexant in pediatric patients.
Atypical antidepressants. Trazodone is commonly prescribed for insomnia in pediatric patients with comorbid mood or anxiety disorders. In open-label studies of children and toddlers, trazodone may be well-tolerated and improve sleep.29 Additionally, development of a physiologically based pharmacokinetic model to inform trazodone dosing for youth with insomnia is underway.30 Some studies in adolescents with depression suggest that caution should be used when combining trazodone with medications that inhibit CYP2D6. In the Treatment of SSRI-Resistant Depression in Adolescents study, none of the patients who were treated with trazodone (vs other soporifics) improved.31 This may relate to CYP2D6 interactions and accumulation of methyl-chloro-piperazine (mCPP), a trazodone metabolite that is associated with dysphoria, irritability, and depression.31 This finding has been replicated in a separate cohort of depressed adolescents.32
Because of its antihistaminergic effects, mirtazapine has been used to treat insomnia in adults. One open-label study of mirtazapine in children and young adults ages 3 to 23 with neurodevelopmental disorders suggested that mirtazapine improved behavioral symptoms and insomnia, and was associated with few treatment-limiting adverse effects.33
Tricyclic antidepressants. In a retrospective study of youth with insomnia who failed behavioral interventions and melatonin (N = 29), doxepin, a potent H1 antagonist, improved subjective sleep in one-half of patients.34
Continue to: Consultation with pediatric sleep medicine specialists...
Consultation with pediatric sleep medicine specialists
It often will behoove the psychiatric clinician to review their concerns with a behavioral sleep medicine specialist or a psychologist with specific expertise in the psychotherapeutic treatment of sleep who can provide important guidance regarding the key aspects of treatment. When discussing a particular patient’s presentation with the pediatric behavioral sleep psychologist/specialist, consider the following questions:
- Is the child’s sleep disorder the primary problem, or is the child’s insomnia secondary to another diagnosis (psychiatric or nonpsychiatric)?
- What are the primary sleep-related problems the child/family presents with? How long have the symptoms been present?
- Is the sleep disorder interfering with the child’s functioning, either academically or socially? Does the child’s sleep problem interfere with other family members’ sleep?
- Does the child have a comorbid psychological conditions such as ADHD, depression, or anxiety, and/or is undergoing treatment for these disorders, which may play a role in the sleep problem?
- Is a sleep study warranted?
A sleep study, also known as polysomnography (PSG), is a diagnostic test in which physiologic parameters are continuously recorded during a period of sleep via electroencephalography, electromyography, electrooculogram, electrocardiogram, airflow sensors, pulse oximeter, body position monitors, and video. In 2012, the American Academy of Sleep Medicine published evidenced-based practice parameters for respiratory and nonrespiratory indications for PSG.35 It is most commonly indicated to rule out obstructive sleep apnea in pediatric patients who exhibit snoring, gasping, irregular breathing, witnessed apneic events, unusual head positioning, or other signs of obstructive breathing during sleep. Nonrespiratory indications for PSG include children suspected of having periodic limb movement disorder and suspected narcolepsy. Children with frequent parasomnias, epilepsy, or nocturnal enuresis should be clinically screened for presence of comorbid sleep disorders, and PSG would be indicated if there are concerns about a possible sleep-disordered breathing disorder. PSG is also recommended for children with hypersomnia, to differentiate a parasomnia from sleep-related epilepsy, and for children suspected of having restless leg syndrome.36 PSG is not typically indicated in the initial evaluation of insomnia (unless there is evidence of a comorbid sleep disorder), circadian rhythm disorders (ie, delayed sleep phase syndrome), or for evaluation of children with sleep-related bruxism.3 Special considerations for PSG in children include ensuring a parent or guardian is always with the child, providing developmentally appropriate sleeping arrangements, arranging family tours of the sleep lab prior to the study, and accommodating for earlier bedtimes.37
Bottom Line
Techniques to promote healthy sleep in pediatric patients include behavioral interventions such as setting a developmentally appropriate bedtime and a consistent wake time, establishing bedtime routines, and encouraging relaxation/ wind-down period before bed. Cognitive-behavioral therapy for insomnia (CBT-I) may include cognitive restructuring of anxious thoughts, relaxation training, stimulus control, and sleep restriction. Use of medications may be indicated for children and teens who have not responded to CBT-I or soporific dosing of melatonin.
Children and adolescents who do not receive sufficient sleep can experience worsening inattention, daytime fatigue, and cognitive and behavioral difficulties. Assessment and treatment of insomnia and other sleep difficulties in young patients is critical as poor sleep increases their risk for depression, self-harm, and suicide.
In Part 1 of this article (Pediatric insomnia: Assessment and diagnosis,
Psychotherapeutic interventions
Regardless of the source of a child’s insomnia or co-occurring disorders, healthy sleep practices are the first line behavioral treatment, including for youth with attention-deficit/hyperactivity disorder (ADHD), anxiety disorders, obsessive-compulsive disorder, and depressive disorders.
Healthy sleep practices/sleep hygiene
Developmentally appropriate bedtimes and routines (Table). Helping children establish a regular, consistent bedtime is key in promoting healthy sleep. Ideally, the bedtime routine involves 3 to 4 activities each night in the same order, and these activities should be relaxing and soothing (eg, taking a bath, putting on pajamas, reading books). Setting age-appropriate bedtimes also is important. If an older child is asked to go to bed at 8 pm but cannot fall asleep for an hour, they may not have insomnia but instead a developmentally inappropriate bedtime. Several studies found that children younger than age 10 should go to bed no later than 9 pm. Bedtimes later than 9 pm for young children are correlated with shorter sleep duration.1
Consistent sleep schedules. Another important aspect of healthy sleep is working with parents to enforce a consistent bedtime and wake-up time, including weekdays and weekends. Ideally, bedtime on weekdays and weekends should not vary by more than 1 hour. Helping children wake up at the same time each day helps to set and regulate their circadian rhythm. Keeping these schedules consistent on vacations and school holidays also is helpful. For adolescents, the weekday/weekend bedtimes can vary by up to 2 hours because adolescents have a delayed circadian rhythm and wake-up times for high school can be early.
Environmental factors. An important piece of parental education is stimulus control and the ingredients of healthy sleep. Healthy sleep ingredients include a dark, quiet, consistent, and cool bedroom; a comfortable bed, the child feeling safe, and limited environmental stimuli.
Continue to: Cognitive-behavioral therapy for insomnia...
Cognitive-behavioral therapy for insomnia
Relaxation. Pediatric patients can be taught relaxation, mindfulness, meditation, and progressive muscle relaxation techniques to help lower overall stress. This can be especially helpful for youth with sleep disorders or anxiety. Guided relaxation apps are popular among children and teens, and various apps offer soothing sounds, deep breathing, progressive muscle relaxation, and guided imagery. This can be taught in psychotherapy sessions and used at home to promote gains in between sessions.
Stimulus control. Stimulus control involves using the bed exclusively for sleep and avoiding nonsleep activities in bed (eg, reading, watching television, using a computer, worrying). These activities promote wakefulness and insomnia. This may mean the child does not get into bed until they cannot keep their eyes open, even if that delays bedtime. If the child is still awake within 15 to 20 minutes, they should be encouraged to get out of bed and engage in a nonstimulating activity such as meditation, reading, or sitting quietly in the dark or low light. This recommendation can run counter to parents’ intuition that children with sleep problems should go to bed earlier. Using the bed only for sleep conditions the child to falling asleep or being asleep when in bed.
Sleep restriction. Sleep restriction involves restricting sleep to a set number of hours in order to increase their sleep efficiency (time slept in bed divided by total time spent in bed x 100). Restricting sleep to 6 to 7 hours increases sleep efficiency, consolidates sleep, and extinguishes the association of being awake in bed. For older adolescents, sleep restriction may help to limit their time in bed to either falling asleep or being asleep. This is intended to be used as a short-term strategy and only after other sleep hygiene measures (bedtime routine, environmental factors, etc) have been put into place for several weeks. While this strategy sounds unappealing to most individuals with insomnia, it can lead to lasting change due to the use of behavioral conditioning in bed. Because sleep restriction can lead to significant daytime sleepiness and impairment during the day, sleep should not be restricted to <6 hours a day for children and adolescents. Once the adolescent is sleeping more consistently and sleep efficiency reaches 85% or higher, time in bed for sleep is increased.2
Cognitive restructuring. Some children and adolescents develop maladaptive thoughts about sleep that further promote insomnia. These thoughts might include “I will never get to sleep,” “I am going to have a terrible day if I cannot fall asleep,” or “I will fail my test tomorrow if I am unable to sleep.” Such maladaptive thoughts are often untrue but promote wakefulness in the early or middle part of the night. Cognitive restructuring involves helping the child identify each problematic thought, challenge how accurate each thought is with evidence, and replace the problematic thought with a more helpful thought. For instance, an adolescent can recognize that even if they have a sleepless night, their catastrophic outcome (eg, “I will not be able to function”) is likely untrue. A psychologist can help review evidence for this, including previous times when the adolescent has not slept well and managed to get through the next day.
When is pharmacologic treatment needed?
Pharmacologic treatment may be indicated if a child does not show significant improvement following behavioral intervention (Figure). However, it is critical to exclude other primary causes of dyssomnia (eg, obstructive sleep apnea, iron deficiency anemia) before pursuing pharmacotherapy, because pharmacotherapy could mask an underlying disorder. Moreover, while there is relatively limited evidence for psychopharmacologic interventions for sleep difficulties in children and adolescents, a large survey of child and adolescent psychiatrists (N = 1,273) suggested that medications were considered for one-quarter of pediatric patients with insomnia.3 Further, patients with specific comorbidities such as neurodevelopmental disorders may be more likely to be prescribed soporifics.4
Continue to: What is the evidence for pharmacotherapy?...
What is the evidence for pharmacotherapy?
Antihistamines. Histamine antagonists—which promote sleep by blocking the wakefulness-promoting and circadian-related effects of histamine—are the most commonly used medications to treat pediatric insomnia, despite a dearth of data from prospective trials.5,6 In 1 small study, Russo et al7 found diphenhydramine, 1 mg/kg at bedtime, reduced sleep latency and nighttime awakenings, and increased sleep duration in patients ages 2 to 12; similar effects have been observed in pediatric burn patients.8 There are some limited data for other H1 antagonists (eg, hydroxyzine) in pediatric insomnia.9-11
Alpha-2 agonists increase rapid eye movement sleep via dose-dependent downregulation of noradrenergic signaling12 and thus have been commonly prescribed for insomnia in children and adolescents. In fact, the nonselective alpha-2 agonist clonidine is among the most prescribed medications for youth with insomnia, and may be efficacious in youth with neurodevelopmental disorders and ADHD.13 In small retrospective studies, clonidine decreased sleep latency and nighttime awakenings in addition to increasing sleep duration.14 Also, clonidine was well tolerated but associated with daytime somnolence. Guanfacine—a selective alpha-2 agonist—is also commonly prescribed for insomnia in youth, although results of trials have been equivocal.15 Given the more rapid absorption and shorter Tmax of clonidine relative to guanfacine, the former may be preferred as a soporific.
Melatonin and melatonin agonists. The primary regulator of the sleep-wake cycle is melatonin, an endogenous hormone produced by the pineal gland in response to changes in retinal light perception. Exogenous melatonin supplementation may be the preferred initial pharmacotherapy for sleep-onset insomnia due to its chronobiotic properties.16 In clinical studies, both immediate-release17,18 and extended-release19 melatonin reduced sleep-onset latency and increased total sleep duration in pediatric patients, although the increase in total duration of sleep was greater with extended-release preparations. Additionally, tolerability data for melatonin in pediatric patients are encouraging. A 2-year randomized trial of prolonged-release melatonin for insomnia in pediatric patients found no adverse effects with regard to growth, body mass index, or pubertal development.20 Additionally, significant improvements in sleep quality, sleep patterns, and caregiver satisfaction were maintained throughout the trial, and no withdrawal symptoms were observed upon discontinuation.
Melatonin may have a particularly important role in circadian rhythm sleep disorders. In this regard, low-dose melatonin (0.5 mg), when timed relative to the endogenous dim light melatonin onset (DLMO), is more effective in shifting sleep phase than higher doses, which suggests that timing may have greater impact than dosage.21 Data regarding melatonin administration with respect to DLMO suggest that the optimal administration time is 4 to 6 hours before a child’s preferred bedtime, and doses of 0.5 to 1 mg have been effective when given in this window.22 Variation across studies has contributed to a lack of consensus regarding pediatric melatonin dosing. For example, .05 mg/kg may be a minimal effective dose when given 1 to 2 hours before bedtime18; however, in surveys doses vary considerably, with typical doses of 2.5 to 3 mg for prepubertal children and 5 mg for adolescents.5 Of note, in patients with decreased cytochrome P450 (CYP) 1A2 activity, lack of diurnal variation in melatonin serum concentration may decrease the effectiveness of melatonin.16Ramelteon is a potent agonist of the melatonin MT1 and MT2 receptors, with a significantly higher binding affinity than melatonin in vitro. In case reports, ramelteon was well-tolerated, improved delayed sleep onset, and decreased nighttime awakenings.23
Zolpidem, eszopiclone and zaleplon. Studies of selective GABAergic modulators and benzodiazepines have not produced positive results in prospective trials of youth with insomnia. Zolpidem was studied in children and adolescents (N = 201) with ADHD; although sleep latency did not differ between zolpidem and placebo, some significant improvements were observed in adolescents.24 Zolpidem was generally well tolerated, with approximately 10% of youth discontinuing due to adverse effects. Additionally, eszopiclone—which has a longer duration of action compared with zolpidem—has been studied in children and adolescents with ADHD (N = 486) who were also evaluated with a sleep study. No differences were observed between placebo and eszopiclone in terms of sleep latency and approximately 10% of patients discontinued treatment as a result of adverse events.25 We were unable to locate any prospective trials of zaleplon or benzodiazepine receptor agonists for insomnia in youth, although some reports suggest that clonazepam may have a possible role for specific parasomnias.26,27Dual orexin receptor antagonists. Suvorexant, an antagonist of the wakefulness-promoting neuropeptide orexin, improved subjective sleep quality in a prospective trial of adolescents with insomnia (N = 30), although dropout was high (44%).28 Of those patients, reasons for discontinuation included loss to follow-up, lack of effectiveness, and abnormal dreams. We were unable to locate any trials of lemborexant in pediatric patients.
Atypical antidepressants. Trazodone is commonly prescribed for insomnia in pediatric patients with comorbid mood or anxiety disorders. In open-label studies of children and toddlers, trazodone may be well-tolerated and improve sleep.29 Additionally, development of a physiologically based pharmacokinetic model to inform trazodone dosing for youth with insomnia is underway.30 Some studies in adolescents with depression suggest that caution should be used when combining trazodone with medications that inhibit CYP2D6. In the Treatment of SSRI-Resistant Depression in Adolescents study, none of the patients who were treated with trazodone (vs other soporifics) improved.31 This may relate to CYP2D6 interactions and accumulation of methyl-chloro-piperazine (mCPP), a trazodone metabolite that is associated with dysphoria, irritability, and depression.31 This finding has been replicated in a separate cohort of depressed adolescents.32
Because of its antihistaminergic effects, mirtazapine has been used to treat insomnia in adults. One open-label study of mirtazapine in children and young adults ages 3 to 23 with neurodevelopmental disorders suggested that mirtazapine improved behavioral symptoms and insomnia, and was associated with few treatment-limiting adverse effects.33
Tricyclic antidepressants. In a retrospective study of youth with insomnia who failed behavioral interventions and melatonin (N = 29), doxepin, a potent H1 antagonist, improved subjective sleep in one-half of patients.34
Continue to: Consultation with pediatric sleep medicine specialists...
Consultation with pediatric sleep medicine specialists
It often will behoove the psychiatric clinician to review their concerns with a behavioral sleep medicine specialist or a psychologist with specific expertise in the psychotherapeutic treatment of sleep who can provide important guidance regarding the key aspects of treatment. When discussing a particular patient’s presentation with the pediatric behavioral sleep psychologist/specialist, consider the following questions:
- Is the child’s sleep disorder the primary problem, or is the child’s insomnia secondary to another diagnosis (psychiatric or nonpsychiatric)?
- What are the primary sleep-related problems the child/family presents with? How long have the symptoms been present?
- Is the sleep disorder interfering with the child’s functioning, either academically or socially? Does the child’s sleep problem interfere with other family members’ sleep?
- Does the child have a comorbid psychological conditions such as ADHD, depression, or anxiety, and/or is undergoing treatment for these disorders, which may play a role in the sleep problem?
- Is a sleep study warranted?
A sleep study, also known as polysomnography (PSG), is a diagnostic test in which physiologic parameters are continuously recorded during a period of sleep via electroencephalography, electromyography, electrooculogram, electrocardiogram, airflow sensors, pulse oximeter, body position monitors, and video. In 2012, the American Academy of Sleep Medicine published evidenced-based practice parameters for respiratory and nonrespiratory indications for PSG.35 It is most commonly indicated to rule out obstructive sleep apnea in pediatric patients who exhibit snoring, gasping, irregular breathing, witnessed apneic events, unusual head positioning, or other signs of obstructive breathing during sleep. Nonrespiratory indications for PSG include children suspected of having periodic limb movement disorder and suspected narcolepsy. Children with frequent parasomnias, epilepsy, or nocturnal enuresis should be clinically screened for presence of comorbid sleep disorders, and PSG would be indicated if there are concerns about a possible sleep-disordered breathing disorder. PSG is also recommended for children with hypersomnia, to differentiate a parasomnia from sleep-related epilepsy, and for children suspected of having restless leg syndrome.36 PSG is not typically indicated in the initial evaluation of insomnia (unless there is evidence of a comorbid sleep disorder), circadian rhythm disorders (ie, delayed sleep phase syndrome), or for evaluation of children with sleep-related bruxism.3 Special considerations for PSG in children include ensuring a parent or guardian is always with the child, providing developmentally appropriate sleeping arrangements, arranging family tours of the sleep lab prior to the study, and accommodating for earlier bedtimes.37
Bottom Line
Techniques to promote healthy sleep in pediatric patients include behavioral interventions such as setting a developmentally appropriate bedtime and a consistent wake time, establishing bedtime routines, and encouraging relaxation/ wind-down period before bed. Cognitive-behavioral therapy for insomnia (CBT-I) may include cognitive restructuring of anxious thoughts, relaxation training, stimulus control, and sleep restriction. Use of medications may be indicated for children and teens who have not responded to CBT-I or soporific dosing of melatonin.
1. Mindell JA, Li AM, Sadeh A, et al. Bedtime routines for young children: a dose-dependent association with sleep outcomes. Sleep. 2015;38(5):717-722.
2. Kansagra S. Sleep disorders in adolescents. Pediatrics. 2020;145(Suppl 2):S204-S209.
3. Owens JA, Mindell JA. Pediatric insomnia. Pediatr Clin North Am. 2011;58(3):555-569.
4. Bruni O, Angriman M, Melegari MG, et al. Pharmacotherapeutic management of sleep disorders in children with neurodevelopmental disorders. Expert Opin Pharmacother. 2019;20(18):2257-2271.
5. Owens JA, Rosen CL, Mindell JA, et al. Use of pharmacotherapy for insomnia in child psychiatry practice: a national survey. Sleep Med. 2010;11(7):692-700.
6. Schnoes CJ, Kuhn BR, Workman EF, et al. Pediatric prescribing practices for clonidine and other pharmacologic agents for children with sleep disturbance. Clin Pediatr (Phila). 2006;45(3):229-238.
7. Russo RM, Gururaj VJ, Allen JE. The effectiveness of diphenhydramine HCI in pediatric sleep disorders. J Clin Pharmacol. 1976;16(5-6):284-288.
8. Yangzom N, Gottschlich MM, Ossege J, et al. The effect of diphenhydramine on sleep in pediatric burn patients: a secondary analysis. J Burn Care Res. 2015;36(2):266-271.
9. Ghanizadeh A, Zare S. A preliminary randomised double-blind placebo-controlled clinical trial of hydroxyzine for treating sleep bruxism in children. J Oral Rehabil. 2013;40(6):413-417.
10. Bektas O, Arıca B, Teber S, et al. Chloral hydrate and/or hydroxyzine for sedation in pediatric EEG recording. Brain Dev. 2014;36(2):130-136.
11. Ottaviano S, Giannotti F, Cortesi F. The effect of niaprazine on some common sleep disorders in children. A double-blind clinical trial by means of continuous home-videorecorded sleep. Childs Nerv Syst. 1991;7(6):332-335.
12. Nguyen M, Tharani S, Rahmani M, et al. A review of the use of clonidine as a sleep aid in the child and adolescent population. Clin Pediatr (Phila). 2014;53(3):211-216.
13. Prince JB, Wilens TE, Biederman J, et al. Clonidine for sleep disturbances associated with attention-deficit hyperactivity disorder: a systematic chart review of 62 cases. J Am Acad Child Adolesc Psychiatry. 1996;35(5):599-605.
14. Ingrassia A, Turk J. The use of clonidine for severe and intractable sleep problems in children with neurodevelopmental disorders--a case series. Eur Child Adolesc Psychiatry. 2005;14(1):34-40.
15. Politte LC, Scahill L, Figueroa J, et al. A randomized, placebo-controlled trial of extended-release guanfacine in children with autism spectrum disorder and ADHD symptoms: an analysis of secondary outcome measures. Neuropsychopharmacology. 2018;43(8):1772-1778.
16. Bruni O, Alonso-Alconada D, Besag F, et al. Current role of melatonin in pediatric neurology: clinical recommendations. Eur J Paediatr Neurol. 2015;19(2):122-1233.
17. Jain SV, Horn PS, Simakajornboon N, et al. Melatonin improves sleep in children with epilepsy: a randomized, double-blind, crossover study. Sleep Med. 2015;16(5):637-644.
18. van Geijlswijk IM, van der Heijden KB, Egberts AC, et al. Dose finding of melatonin for chronic idiopathic childhood sleep onset insomnia: an RCT. Psychopharmacology (Berl). 2010;212(3):379-391.
19. Gringras P, Nir T, Breddy J, et al. Efficacy and safety of pediatric prolonged-release melatonin for insomnia in children with autism spectrum disorder. J Am Acad Child Adolesc Psychiatry. 2017;56(11):948-957.e4.
20. Malow BA, Findling RL, Schroder CM, et al. Sleep, growth, and puberty after two years of prolonged-release melatonin in children with autism spectrum disorder. J Am Acad Child Adolesc Psychiatry. 2021;60(2):252-261.e3.
21. Burgess HJ, Emens JS. Drugs used in circadian sleep-wake rhythm disturbances. Sleep Med Clin. 2020;15(2):301-310.
22. Arns M, Kooij JJS, Coogan AN. Review: identification and management of circadian rhythm sleep disorders as a transdiagnostic feature in child and adolescent psychiatry. J Am Acad Child Adolesc Psychiatry. 2021;60(9):1085-1095.
23. Kawabe K, Horiuchi F, Oka Y, et al. The melatonin receptor agonist ramelteon effectively treats insomnia and behavioral symptoms in autistic disorder. Case Rep Psychiatry. 2014;2014:561071.
24. Blumer JL, Findling RL, Shih WJ, et al. Controlled clinical trial of zolpidem for the treatment of insomnia associated with attention-deficit/hyperactivity disorder in children 6 to 17 years of age. Pediatrics. 2009;123(5):e770-e776.
25. Sangal RB, Blumer JL, Lankford DA, et al. Eszopiclone for insomnia associated with attention-deficit/hyperactivity disorder. Pediatrics. 2014;134(4):e1095-e1103.
26. Arens R, Wright B, Elliott J, et al. Periodic limb movement in sleep in children with Williams syndrome. J Pediatr. 1998;133(5):670-674.
27. Thirumalai SS, Shubin RA, Robinson R. Rapid eye movement sleep behavior disorder in children with autism. J Child Neurol. 2002;17(3):173-178.
28. Kawabe K, Horiuchi F, Ochi M, et al. Suvorexant for the treatment of insomnia in adolescents. J Child Adolesc Psychopharmacol. 2017;27(9):792-795.
29. Pranzatelli MR, Tate ED, Dukart WS, et al. Sleep disturbance and rage attacks in opsoclonus-myoclonus syndrome: Response to trazodone. J Pediatr. 2005;147(3):372-378.
30. Oggianu L, Ke AB, Chetty M, et al. Estimation of an appropriate dose of trazodone for pediatric insomnia and the potential for a trazodone-atomoxetine interaction. CPT Pharmacometrics Syst Pharmacol. 2020;9(2):77-86.
31. Shamseddeen W, Clarke G, Keller MB, et al. Adjunctive sleep medications and depression outcome in the treatment of serotonin-selective reuptake inhibitor resistant depression in adolescents study. J Child Adolesc Psychopharmacol. 2012;22(1):29-36.
32. Sultan MA, Courtney DB. Adjunctive trazodone and depression outcome in adolescents treated with serotonin re-uptake inhibitors. J Can Acad Child Adolesc Psychiatry. 2017;26(3):233-240.
33. Posey DJ, Guenin KD, Kohn AE, et al. A naturalistic open-label study of mirtazapine in autistic and other pervasive developmental disorders. J Child Adolesc Psychopharmacol. 2001;11(3):267-277.
34. Shah YD, Stringel V, Pavkovic I, et al. Doxepin in children and adolescents with symptoms of insomnia: a single-center experience. J Clin Sleep Med. 2020;16(5):743-747.
35. Aurora RN, Lamm CI, Zak RS, et al. Practice parameters for the non-respiratory indications for polysomnography and multiple sleep latency testing for children. Sleep. 2012;35(11):1467-1473.
36. de Zambotti M, Goldstone A, Colrain IM, et al. Insomnia disorder in adolescence: diagnosis, impact, and treatment. Sleep Med Rev. 2018;39:12-24.
37. Beck SE, Marcus CL. Pediatric polysomnography. Sleep Med Clin. 2009;4(3):393-406.
1. Mindell JA, Li AM, Sadeh A, et al. Bedtime routines for young children: a dose-dependent association with sleep outcomes. Sleep. 2015;38(5):717-722.
2. Kansagra S. Sleep disorders in adolescents. Pediatrics. 2020;145(Suppl 2):S204-S209.
3. Owens JA, Mindell JA. Pediatric insomnia. Pediatr Clin North Am. 2011;58(3):555-569.
4. Bruni O, Angriman M, Melegari MG, et al. Pharmacotherapeutic management of sleep disorders in children with neurodevelopmental disorders. Expert Opin Pharmacother. 2019;20(18):2257-2271.
5. Owens JA, Rosen CL, Mindell JA, et al. Use of pharmacotherapy for insomnia in child psychiatry practice: a national survey. Sleep Med. 2010;11(7):692-700.
6. Schnoes CJ, Kuhn BR, Workman EF, et al. Pediatric prescribing practices for clonidine and other pharmacologic agents for children with sleep disturbance. Clin Pediatr (Phila). 2006;45(3):229-238.
7. Russo RM, Gururaj VJ, Allen JE. The effectiveness of diphenhydramine HCI in pediatric sleep disorders. J Clin Pharmacol. 1976;16(5-6):284-288.
8. Yangzom N, Gottschlich MM, Ossege J, et al. The effect of diphenhydramine on sleep in pediatric burn patients: a secondary analysis. J Burn Care Res. 2015;36(2):266-271.
9. Ghanizadeh A, Zare S. A preliminary randomised double-blind placebo-controlled clinical trial of hydroxyzine for treating sleep bruxism in children. J Oral Rehabil. 2013;40(6):413-417.
10. Bektas O, Arıca B, Teber S, et al. Chloral hydrate and/or hydroxyzine for sedation in pediatric EEG recording. Brain Dev. 2014;36(2):130-136.
11. Ottaviano S, Giannotti F, Cortesi F. The effect of niaprazine on some common sleep disorders in children. A double-blind clinical trial by means of continuous home-videorecorded sleep. Childs Nerv Syst. 1991;7(6):332-335.
12. Nguyen M, Tharani S, Rahmani M, et al. A review of the use of clonidine as a sleep aid in the child and adolescent population. Clin Pediatr (Phila). 2014;53(3):211-216.
13. Prince JB, Wilens TE, Biederman J, et al. Clonidine for sleep disturbances associated with attention-deficit hyperactivity disorder: a systematic chart review of 62 cases. J Am Acad Child Adolesc Psychiatry. 1996;35(5):599-605.
14. Ingrassia A, Turk J. The use of clonidine for severe and intractable sleep problems in children with neurodevelopmental disorders--a case series. Eur Child Adolesc Psychiatry. 2005;14(1):34-40.
15. Politte LC, Scahill L, Figueroa J, et al. A randomized, placebo-controlled trial of extended-release guanfacine in children with autism spectrum disorder and ADHD symptoms: an analysis of secondary outcome measures. Neuropsychopharmacology. 2018;43(8):1772-1778.
16. Bruni O, Alonso-Alconada D, Besag F, et al. Current role of melatonin in pediatric neurology: clinical recommendations. Eur J Paediatr Neurol. 2015;19(2):122-1233.
17. Jain SV, Horn PS, Simakajornboon N, et al. Melatonin improves sleep in children with epilepsy: a randomized, double-blind, crossover study. Sleep Med. 2015;16(5):637-644.
18. van Geijlswijk IM, van der Heijden KB, Egberts AC, et al. Dose finding of melatonin for chronic idiopathic childhood sleep onset insomnia: an RCT. Psychopharmacology (Berl). 2010;212(3):379-391.
19. Gringras P, Nir T, Breddy J, et al. Efficacy and safety of pediatric prolonged-release melatonin for insomnia in children with autism spectrum disorder. J Am Acad Child Adolesc Psychiatry. 2017;56(11):948-957.e4.
20. Malow BA, Findling RL, Schroder CM, et al. Sleep, growth, and puberty after two years of prolonged-release melatonin in children with autism spectrum disorder. J Am Acad Child Adolesc Psychiatry. 2021;60(2):252-261.e3.
21. Burgess HJ, Emens JS. Drugs used in circadian sleep-wake rhythm disturbances. Sleep Med Clin. 2020;15(2):301-310.
22. Arns M, Kooij JJS, Coogan AN. Review: identification and management of circadian rhythm sleep disorders as a transdiagnostic feature in child and adolescent psychiatry. J Am Acad Child Adolesc Psychiatry. 2021;60(9):1085-1095.
23. Kawabe K, Horiuchi F, Oka Y, et al. The melatonin receptor agonist ramelteon effectively treats insomnia and behavioral symptoms in autistic disorder. Case Rep Psychiatry. 2014;2014:561071.
24. Blumer JL, Findling RL, Shih WJ, et al. Controlled clinical trial of zolpidem for the treatment of insomnia associated with attention-deficit/hyperactivity disorder in children 6 to 17 years of age. Pediatrics. 2009;123(5):e770-e776.
25. Sangal RB, Blumer JL, Lankford DA, et al. Eszopiclone for insomnia associated with attention-deficit/hyperactivity disorder. Pediatrics. 2014;134(4):e1095-e1103.
26. Arens R, Wright B, Elliott J, et al. Periodic limb movement in sleep in children with Williams syndrome. J Pediatr. 1998;133(5):670-674.
27. Thirumalai SS, Shubin RA, Robinson R. Rapid eye movement sleep behavior disorder in children with autism. J Child Neurol. 2002;17(3):173-178.
28. Kawabe K, Horiuchi F, Ochi M, et al. Suvorexant for the treatment of insomnia in adolescents. J Child Adolesc Psychopharmacol. 2017;27(9):792-795.
29. Pranzatelli MR, Tate ED, Dukart WS, et al. Sleep disturbance and rage attacks in opsoclonus-myoclonus syndrome: Response to trazodone. J Pediatr. 2005;147(3):372-378.
30. Oggianu L, Ke AB, Chetty M, et al. Estimation of an appropriate dose of trazodone for pediatric insomnia and the potential for a trazodone-atomoxetine interaction. CPT Pharmacometrics Syst Pharmacol. 2020;9(2):77-86.
31. Shamseddeen W, Clarke G, Keller MB, et al. Adjunctive sleep medications and depression outcome in the treatment of serotonin-selective reuptake inhibitor resistant depression in adolescents study. J Child Adolesc Psychopharmacol. 2012;22(1):29-36.
32. Sultan MA, Courtney DB. Adjunctive trazodone and depression outcome in adolescents treated with serotonin re-uptake inhibitors. J Can Acad Child Adolesc Psychiatry. 2017;26(3):233-240.
33. Posey DJ, Guenin KD, Kohn AE, et al. A naturalistic open-label study of mirtazapine in autistic and other pervasive developmental disorders. J Child Adolesc Psychopharmacol. 2001;11(3):267-277.
34. Shah YD, Stringel V, Pavkovic I, et al. Doxepin in children and adolescents with symptoms of insomnia: a single-center experience. J Clin Sleep Med. 2020;16(5):743-747.
35. Aurora RN, Lamm CI, Zak RS, et al. Practice parameters for the non-respiratory indications for polysomnography and multiple sleep latency testing for children. Sleep. 2012;35(11):1467-1473.
36. de Zambotti M, Goldstone A, Colrain IM, et al. Insomnia disorder in adolescence: diagnosis, impact, and treatment. Sleep Med Rev. 2018;39:12-24.
37. Beck SE, Marcus CL. Pediatric polysomnography. Sleep Med Clin. 2009;4(3):393-406.

Pediatric insomnia: Assessment and diagnosis
FIRST OF 2 PARTS
A thorough evaluation can identify modifiable factors and guide treatment
Sleep problems are common among children and adolescents,1 with prevalence rates of 25% to 40%.2-4 Young children most commonly exhibit what is referred to as bedtime problems and night wakenings, whereas children in middle childhood (age 4 to 12) through adolescence (age 13 to 17) report insomnia. For many children, these problems persist.3 Insufficient sleep in children and adolescents worsens inattention, daytime fatigue, and cognitive and behavioral deficits.5 Assessment and treatment of sleep problems in children and adolescents is critical because poor sleep among youth increases the risk for depression, self-harm, and suicide,6,7 increases family stress, and decreases parental well-being.1
This 2-part article describes the assessment, diagnosis, and treatment of sleep problems among children and adolescents. In part 1, we focus on:
- sleep architecture (circadian rhythms, stages of sleep)
- sleep in healthy youth (age 6 to 17) and those with attention-deficit/hyperactivity disorder (ADHD), depressive disorders, and anxiety
- how to assess sleep, and the differential diagnosis of behavioral sleep problems in pediatric patients.
In Part 2, we will cover psychotherapeutic and psychopharmacologic interventions for youth with insomnia, and describe an effective approach to consultation with pediatric sleep medicine specialists.
How much sleep do children and adolescents need?
Throughout their development, children spend 40% to 50% of their time asleep. Sleep schedules are based on circadian rhythms, which are physical, mental, and behavioral changes that follow an approximately 24-hour cycle. Human circadian rhythm varies between 24 and 25 hours and is vital in determining our sleep patterns. Exposure to sunlight drives our circadian rhythm, sending signals to our bodies to “turn on” melatonin production at night (ie, 9
Box
Sleep architecture consists of 3 states: wake; non-rapid eye movement (NREM) sleep; and rapid eye movement (REM) sleep (“dreaming” sleep).2 These stages have distinct polysomnographic features of electroencephalographic EEG patterns, eye movements, and muscle tone.2 NREM sleep can be further divided into 3 stages: stage 1 (N1), stage 2 (N2), and stage 3 (N3). Stage 1 is the lightest stage and lasts for 30 seconds to 5 minutes; it is easy to wake up from stage 1 sleep. During stage 2 sleep, the body moves into a deeper sleep stage that is considered “true” sleep. This sleep stage is characterized by bursts of rhythmic rapid EEG activity known as spindles, as well as high-amplitude slow-wave spikes called K complexes.2 Stage 2 sleep lasts for 10 to 45 minutes. Stage 3, better known as “deep sleep,” slow-wave sleep, or delta sleep, is the most restorative sleep.2 Respiration is low and parasympathetic activity is high.2 It is difficult to be awakened during deep sleep, and if aroused, the person likely will feel confused or groggy. Deep sleep is followed by a return to lighter stage of sleep before the first REM sleep period begins.
REM sleep is the active stage of sleep. Breathing and heart rate become irregular, and the body experiences muscle atonia, or temporary paralysis, of arms and legs. When in REM sleep, individuals have the highest brain metabolic rates, and periodic bursts of eye movements.2 Most individuals move through stages of NREM and REM sleep in predicable ways, meaning they experience NREM sleep, return to a lighter stage of sleep after deep sleep, then move into REM sleep before the cycle repeats. It takes approximately 90 minutes for most adults to complete the NREM sleep cycle, and then REM sleep occurs before returning to NREM sleep.
In children, especially in infants and babies, sleep cycles are closer to 50 to 60 minutes. Newborns spend approximately 50% of their sleep in REM sleep, whereas adults spend 20% to 25% of their sleep in REM sleep. Children will spend more time in REM sleep until the third and fourth years of life, at which point REM gradually decreases to 20% to 25% by adulthood.
Sleep needs also change predictably throughout the lifespan. The National Sleep Foundation guidelines for sleep duration provide clinicians and parents with a range of recommended sleep for each stage of development. Infants require 14 to 17 hours of sleep, whereas adolescents need 8 to 10 hours by age 14 to 17.8 The key for clinicians is to determine if the child is within the recommended range, and how they are functioning on the number of hours of sleep they report. This allows for variation in how much sleep an individual child might need while acknowledging that some children within a specific age group might need more or less sleep than other children of the same age.
Sleep in healthy youth: Middle childhood
School-age children (age 6 to 12) typically need 9 to 10 hours of sleep over a 24-hour period.2 This developmental period is especially important for children to develop healthy sleep habits; however, developmentally appropriate cognitive and social/emotional factors might interfere with the quality and quantity of sleep. Middle childhood is a time when children can understand the dangers of the outside world (ie, violence, health problems) and resulting anxiety can disrupt sleep. Parents usually are less involved in bedtime as children approach adolescence, which leads to later bedtimes. At this stage, many children begin to take on more serious roles in their academics and extracurricular activities, peer relationships become more important, and use of electronics (eg, television, video games, internet, and handheld devices) increases—all of which compete with sleep.9 Frequent sleep issues during middle childhood include:
- irregular sleep-wake schedules
- later bedtimes
- decreased nighttime sleep
- increased caffeine intake
- reduced parental presence at bedtime
- daytime sleepiness.3
In school-age children, regular napping, falling asleep during short car rides, and daytime fatigue at school or home are cause for concern. When these symptoms are present, an evaluation is warranted.
Sleep in healthy youth: Adolescence
The National Sleep Foundation recommends adolescents obtain 8 to 10 hours of sleep per night; for some adolescents, as much as 11 hours of sleep per night might be appropriate.8 However, this contrasts with findings from the National Sleep Foundation’s Sleep in America Poll, which revealed that 75% of 12th graders report <8 hours of sleep nightly.10 Many adolescents experience delayed sleep phase syndrome or delayed sleep-wake phase disorder, which involves a persistent phase shift of >2 hours in the sleep-wake schedule that conflicts with the adolescent’s school, work, or lifestyle demands.11 Such circadian rhythm disorders typically result from a poor match between the sleep-wake schedule and the demands of the adolescent’s life, or a failure to synchronize their internal clock with a 24-hour circadian clock.12 Children typically become tired after sunset, but puberty is associated with reduced slow-wave sleep and changes in circadian rhythms. As a result, a 3-hour delay (delayed phase preference) is common among adolescents. At approximately age 20, people start to become tired after sunset and awaken earlier in the morning—a pattern driven by sunlight and the timing of melatonin release that will remain stable until the sixth decade of life.
Continue to: Effects of chronic sleep deprivation...
Effects of chronic sleep deprivation
Most older studies of sleep loss examined the impact of total sleep loss (sleep deprivation) rather than the effect of partial sleep loss or sleep restriction, a more commonly experienced phenomenon. More recent research shows that a cumulative sleep deficit could cause the body to override voluntary wakefulness and a sleep-deprived individual can experience brief “microsleeps” where they are unaware and lose attention/wakefulness for several seconds.2 This can be deadly if a sleep-deprived adolescent experiences microsleeps while driving.13
There is a well-studied correlation between chronic sleep deprivation and increased body mass index in children.14 This might be caused by reduction in physical activity as well as alterations in the “hunger hormones”—ghrelin and leptin—that have been observed with sleep deprivation.15-17 Other studies have noted decreased glucose tolerance, reduced insulin sensitivity, and catecholamine and cortisol secretion abnormalities, which place children at higher risk for metabolic syndrome and hypertension.13,18 Sleep deprivation also is associated with mood and anxiety disorders and is an independent risk factor for substance use and suicidal ideation among adolescents.19 Sleep deprivation increases impairments in impulse control, concentration, and attention, which could be especially problematic in school-age children.
How sleep is assessed
The sleep history is the first step in evaluating a child or adolescent for a sleep disorder. The sleep history includes exploring the chief complaint, sleep patterns and schedules, bedtime routines, and nocturnal and daytime behaviors (Table).

Chief complaint
Behavioral sleep specialists will assess the primary problem with everyone involved in the child’s bedtime.20 This might include parents (custodial and noncustodial), grandparents, or stepparents as well as the child/adolescent. This important step can reveal a sleep disorder or an inappropriately early bedtime relative to the child’s development. During this assessment, ask detailed questions about how long the sleep problem has persisted, the frequency of sleep problems, and any precipitating stressors. Parents and caregivers can review strategies they have tried, and for how long and to what extent interventions were implemented consistently to result in change.
Sleep patterns and schedules
Review the child/adolescent’s typical sleep patterns and behaviors. Ask parents and caregivers, as well as the patient, about general sleep schedules for the past few weeks or a typical 2-week time period.2 A behavioral assessment of sleep should include asking families about how the child/adolescent sleeps during the week and over the weekend, and if school-year sleep differs from summer or holiday sleep schedules. These questions can illuminate how long a sleep problem has been occurring and what sleep habits might be contributing to the problem. Bedtime
Determine if there is a set bedtime or if the child goes to bed when they wish. It is important to ascertain if the bedtime is age-appropriate, if weekday and weekend bedtimes differ, and to what extent extracurricular activities or school demands impact bedtime. Assess the consistency of the bedtime, the nature of bedtime routines (eg, is the child engaging in stimulating activities before bed), where the bedtime routine occurs (eg, sibling’s room, parents’ room, child’s room), and what role (if any) electronic devices play.2
Nocturnal behaviors
Assessment should include a series of questions and age-specific questionnaires to focus on what behaviors occur at night, including awakenings. Parents should be asked how frequent night awakenings occur, how long arousals last, and how the child signals for the parent (eg, calling out, climbing into parents’ bed).2 Additionally, ask how parents respond and what is required to help the child fall back asleep (eg, rocking, soothing, feeding). The presence of nightmares, night terrors, parasomnias, and sleep-related breathing disorders also must be assessed.20
Daytime behaviors
A sleep history should include assessment of daytime functioning, including daytime sleepiness, fatigue, morning waking, and functioning during school, extracurriculars, and homework. For children and teens, falling asleep in the car, while in school, or during passive activities (meals, conversation) suggests insufficient sleep, sleep disruption, or excessive daytime sleepiness.2
Continue to: Sleep disruption in youth with psychiatric disorders...
Sleep disruption in youth with psychiatric disorders
Disordered sleep is common across psychiatric disorders. The National Comorbidity Survey Adolescent Supplement—a nationally representative cross-sectional survey of adolescents (N = 10,123)—found that a later weeknight bedtime, shorter weeknight sleep duration, and greater weekend bedtime delay increased the risk of developing a mood, anxiety, or substance use (including nicotine) disorder, and suicidality. These risk factors also were associated with lower “perceived mental and physical health.”21 Clinicians should routinely obtain a sleep history in children and adolescents with these disorders. Consider using the sleep screening tool BEARS:
- Bedtime issues
- Excessive daytime sleepiness
- Awakenings
- Regularity and duration of sleep
- Snoring.
ADHD
Up to one-half of children and adolescents with ADHD experience sleep problems,22,23 including delayed sleep onset, bedtime resistance, daytime fatigue, and feeling groggy in the morning beyond what is typical (>20 minutes). Pharmacotherapy for ADHD contributes to sleep disturbances24,25 while sleep deprivation exacerbates inattention and hyperactivity. In youth with ADHD, restless leg syndrome, periodic limb movement disorder, and sleep-disordered breathing disorder are more common than in the general population.
Depressive disorders
Up to three-quarters of depressed children and 90% of depressed adolescents report sleep disturbances, including initial, middle, and terminal insomnia as well as hypersomnia.26 Disrupted sleep in pediatric patients with major depressive disorder could be moderated by the patient’s age, with depressive symptoms more common among adolescents (age 12 to 17) than among younger children (age 6 to 11).27 Successful treatment of depression fails to relieve dyssomnia in 10% of children. Sleep problems that persist after successfully treating a depressive episode could increase the risk of another depressive episode.28
Anxiety disorders
Sleep problems are common among children and adolescents with anxiety disorders.29 Longitudinal data from >900 children found that symptoms of sleep disturbance in early childhood were correlated with experiencing an anxiety disorder 20 years later.30 Fears related to the dark or monsters under the bed that are developmentally appropriate for younger children may interfere with sleep. However, in anxious children, fears might also be related to separation, sleeping alone, worry about the loss of a loved one, concerns about personal safety, fear of frightening dreams, or concerns about academics and social relationships. Anxious individuals ruminate about their worries, and this might be especially true for children at bedtime, when there are limited distractions from ruminative fears.31 Bedtime resistance, parental involvement in bedtime rituals, and cultural factors related to sleep also could play a role for children with anxiety symptoms and sleep problems.
Having an anxiety disorder is significantly associated with an increased risk of insomnia; however, 73% of the time anxiety symptoms precede an insomnia diagnosis.29 Sleep problems and anxiety symptoms might have a reciprocal influence on one another; tiredness that results from sleep problems could exacerbate anxiety, which further worsens sleep problems.
A bridge to treatment
A thorough assessment can help identify modifiable factors and guide treatment selections. In Part 2 of this article, we will describe healthy sleep practices, cognitive-behavioral therapy for insomnia, when pharmacotherapy might be indicated, and the evidence supporting several medications commonly used to treat pediatric insomnia. We also will discuss factors to consider when seeking consultation with a pediatric behavioral sleep specialist.
1. Meltzer LJ, Mindell JA. Systematic review and meta-analysis of behavioral interventions for pediatric insomnia. J Pediatr Psychol. 2014;39(8):932-948. doi:10.1093/jpepsy/jsu041
2. Owens JA, Mindell JA. Pediatric insomnia. Pediatr Clin North Am. 2011;58(3):555-569. doi:10.1016/j.pcl.2011.03.011
3. Meltzer LJ, Plaufcan MR, Thomas JH, et al. Sleep problems and sleep disorders in pediatric primary care: treatment recommendations, persistence, and health care utilization. J Clin Sleep Med. 2014;10(4):421-426. doi:10.5664/jcsm.3620
4. Moore M, Meltzer LJ, Mindell JA. Bedtime problems and night wakings in children. Prim Care. 2008;35(3):569-581, viii. doi:10.1016/j.pop.2008.06.002
5. Williamson AA, Mindell JA, Hiscock H, et al. Longitudinal sleep problem trajectories are associated with multiple impairments in child well-being. J Child Psychol Psychiatry. 2020;61(10):1092-1103. doi:10.1111/jcpp.13303
6. Roberts RE, Roberts CR, Chen IG. Impact of insomnia on future functioning of adolescents. J Psychosom Res. 2002; 53(1):561-569. doi:10.1016/s0022-3999(02)00446-4
7. Singareddy R, Krishnamurthy VB, Vgontzas AN, et al. Subjective and objective sleep and self-harm behaviors in young children: a general population study. Psychiatry Res. 2013;209(3):549-553. doi:10.1016/j.psychres.2013.03.036
8. Hirshkowitz M, Whiton K, Albert SM, et al. National Sleep Foundation’s updated sleep duration recommendations: final report. Sleep Health. 2015;1(4):233-243. doi:10.1016/j.sleh.2015.10.004
9. Calamaro CJ, Mason TBA, Ratcliffe SJ. Adolescents living the 24/7 lifestyle: Effects of caffeine and technology on sleep duration and daytime functioning. Pediatrics. 2009;123(6):e1005-1010. doi:10.1542/peds.2008-3641
10. Mindell JA, Owens JA, Carskadon MA. Developmental features of sleep. Child Adolesc Psychiatr Clin N Am. 1999;8(4):695-725.
11. Moore M, Meltzer LJ. The sleepy adolescent: causes and consequences of sleepiness in teens. Paediatr Respir Rev. 2008;9(2):114-120. doi:10.1016/j.prrv.2008.01.001
12. Crowley SJ, Acebo C, Carskadon MA. Sleep, circadian rhythms, and delayed phase in adolescence. Sleep Med. 2007;8(6):602-612. doi:10.1016/j.sleep.2006.12.002
13. Millman RP; Working Group on Sleepiness in Adolescents/Young Adults; AAP Committee on Adolescence. Excessive sleepiness in adolescents and young adults: causes, consequences, and treatment strategies. Pediatrics. 2005;115(6):1774-1786. doi:10.1542/peds.2005-0772
14. Kaczor M, Skalski M. Prevalence and consequences of insomnia in pediatric population. Psychiatr Pol. 2016;50(3):555-569. doi:10.12740/PP/61226
15. Gomes TN, Dos Santos FK, Santos D, et al. Correlates of sedentary time in children: a multilevel modelling approach. BMC Public Health. 2014;14:890. doi:10.1186/1471-2458-14-890
16. Stone MR, Stevens D, Faulkner GEJ. Maintaining recommended sleep throughout the week is associated with increased physical activity in children. Prev Med. 2013;56(2):112-117. doi:10.1016/j.ypmed.2012.11.015
17. Hart CN, Fava JL, Subak LL, et al. Time in bed is associated with decreased physical activity and higher BMI in women seeking weight loss treatment. ISRN Obes. 2012;2012:320157. doi:10.5402/2012/320157
18. Tasali E, Leproult R, Ehrmann DA, et al. Slow-wave sleep and the risk of type 2 diabetes in humans. Proc Natl Acad Sci U S A. 2008;105(3):1044-1049. doi:10.1073/pnas.0706446105
19. de Zambotti M, Goldstone A, Colrain IM, et al. Insomnia disorder in adolescence: diagnosis, impact, and treatment. Sleep Med Rev. 2018;39:12-24. doi:10.1016/j.smrv.2017.06.009
20. Mindell JA, Owens JA. A clinical guide to pediatric sleep: diagnosis and management of sleep problems. 3rd ed. Lippincott Williams & Wilkins; 2015.
21. Zhang J, Paksarian D, Lamers F, et al. Sleep patterns and mental health correlates in US adolescents. J Pediatr. 2017;182:137-143. doi:10.1016/j.jpeds.2016.11.007
22. Gregory AM, Agnew-Blais JC, Matthews T, et al. ADHD and sleep quality: longitudinal analyses from childhood to early adulthood in a twin cohort. J Clin Child Adolesc Psychol. 2017;46(2):284-294. doi:10.1080/15374416.2016.1183499
23. Weiss MD, Salpekar J. Sleep problems in the child with attention-deficit hyperactivity disorder: Defining aetiology and appropriate treatments. CNS Drugs. 2010;24(10):811-828. doi:10.2165/11538990-000000000-00000
24. Galland BC, Tripp EG, Taylor BJ. The sleep of children with attention deficit hyperactivity disorder on and off methylphenidate: a matched case-control study. J Sleep Res. 2010;19(2):366-373. doi:10.1111/j.1365-2869.2009.00795.x
25. Becker SP, Froehlich TE, Epstein JN. Effects of methylphenidate on sleep functioning in children with attention-deficit/hyperactivity disorder. J Dev Behav Pediatr. 2016;37(5):395-404. doi:10.1097/DBP.0000000000000285
26. Roberts RE, Duong HT. Depression and insomnia among adolescents: a prospective perspective. J Affect Disord. 2013;148(1):66-71. doi:10.1016/j.jad.2012.11.049
27. Emslie GJ, Rush AJ, Weinberg WA, et al. Sleep EEG features of adolescents with major depression. Biol Psychiatry. 1994;36(9):573-581. doi:10.1016/0006-3223(94)90067-1
28. Alfano CA, Zakem AH, Costa NM, et al. Sleep problems and their relation to cognitive factors, anxiety, and depressive symptoms in children and adolescents. Depress Anxiety. 2009;26(6):503-512. doi:10.1002/da.20443
29. Alfano CA, Ginsburg GS, Kingery JN. Sleep-related problems among children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):224-232. doi:10.1097/01.chi.0000242233.06011.8e
30. Gregory AM, Caspi A, Eley TC, et al. Prospective longitudinal associations between persistent sleep problems in childhood and anxiety and depression disorders in adulthood. J Abnorm Child Psychol. 2005;33(2):157-163. doi: 10.1007/s10802-005-1824-0
31. Chorney DB, Detweiler MF, Morris TL, et al. The interplay of sleep disturbance, anxiety, and depression in children. J Pediatr Psychol. 2008;33(4):339-348. doi:10.1093/jpepsy/jsm105
32. Sadeh A. Stress, trauma, and sleep in children. Child Adolesc Psychiatr Clin N Am. 1996;5(3):685-700. doi:10.1016/S1056-4993(18)30356-0
33. Glod CA, Teicher MH, Hartman CR, et al. Increased nocturnal activity and impaired sleep maintenance in abused children. J Am Acad Child Adolesc Psychiatry. 1997;36(9):1236-1243. doi:10.1097/00004583-199709000-00016
34. Strawn JR, Lu L, Peris TS, et al. Research review: pediatric anxiety disorders: what have we learnt in the last 10 years? J Child Psychol Psychiatry. 2021;62(2):114-139. doi:10.1111/jcpp.13262
35. Wehry AM, Beesdo-Baum K, Hennelly MM, et al. Assessment and treatment of anxiety disorders in children and adolescents. Curr Psychiatry Rep. 2015;17(7):52. doi:10.1007/s11920-015-0591-z
36. Hamill Skoch S, Mills JA, Ramsey L, et al. Letter to editor: sleep disturbances in selective serotonin reuptake inhibitor-treated youth with anxiety disorders and obsessive compulsive disorder— a bayesian hierarchical modeling meta-analysis. J Child Adolesc Psychopharmacol. 2021;31(5):387-388. doi:10.1089/cap.2020.0169
FIRST OF 2 PARTS
A thorough evaluation can identify modifiable factors and guide treatment
Sleep problems are common among children and adolescents,1 with prevalence rates of 25% to 40%.2-4 Young children most commonly exhibit what is referred to as bedtime problems and night wakenings, whereas children in middle childhood (age 4 to 12) through adolescence (age 13 to 17) report insomnia. For many children, these problems persist.3 Insufficient sleep in children and adolescents worsens inattention, daytime fatigue, and cognitive and behavioral deficits.5 Assessment and treatment of sleep problems in children and adolescents is critical because poor sleep among youth increases the risk for depression, self-harm, and suicide,6,7 increases family stress, and decreases parental well-being.1
This 2-part article describes the assessment, diagnosis, and treatment of sleep problems among children and adolescents. In part 1, we focus on:
- sleep architecture (circadian rhythms, stages of sleep)
- sleep in healthy youth (age 6 to 17) and those with attention-deficit/hyperactivity disorder (ADHD), depressive disorders, and anxiety
- how to assess sleep, and the differential diagnosis of behavioral sleep problems in pediatric patients.
In Part 2, we will cover psychotherapeutic and psychopharmacologic interventions for youth with insomnia, and describe an effective approach to consultation with pediatric sleep medicine specialists.
How much sleep do children and adolescents need?
Throughout their development, children spend 40% to 50% of their time asleep. Sleep schedules are based on circadian rhythms, which are physical, mental, and behavioral changes that follow an approximately 24-hour cycle. Human circadian rhythm varies between 24 and 25 hours and is vital in determining our sleep patterns. Exposure to sunlight drives our circadian rhythm, sending signals to our bodies to “turn on” melatonin production at night (ie, 9
Box
Sleep architecture consists of 3 states: wake; non-rapid eye movement (NREM) sleep; and rapid eye movement (REM) sleep (“dreaming” sleep).2 These stages have distinct polysomnographic features of electroencephalographic EEG patterns, eye movements, and muscle tone.2 NREM sleep can be further divided into 3 stages: stage 1 (N1), stage 2 (N2), and stage 3 (N3). Stage 1 is the lightest stage and lasts for 30 seconds to 5 minutes; it is easy to wake up from stage 1 sleep. During stage 2 sleep, the body moves into a deeper sleep stage that is considered “true” sleep. This sleep stage is characterized by bursts of rhythmic rapid EEG activity known as spindles, as well as high-amplitude slow-wave spikes called K complexes.2 Stage 2 sleep lasts for 10 to 45 minutes. Stage 3, better known as “deep sleep,” slow-wave sleep, or delta sleep, is the most restorative sleep.2 Respiration is low and parasympathetic activity is high.2 It is difficult to be awakened during deep sleep, and if aroused, the person likely will feel confused or groggy. Deep sleep is followed by a return to lighter stage of sleep before the first REM sleep period begins.
REM sleep is the active stage of sleep. Breathing and heart rate become irregular, and the body experiences muscle atonia, or temporary paralysis, of arms and legs. When in REM sleep, individuals have the highest brain metabolic rates, and periodic bursts of eye movements.2 Most individuals move through stages of NREM and REM sleep in predicable ways, meaning they experience NREM sleep, return to a lighter stage of sleep after deep sleep, then move into REM sleep before the cycle repeats. It takes approximately 90 minutes for most adults to complete the NREM sleep cycle, and then REM sleep occurs before returning to NREM sleep.
In children, especially in infants and babies, sleep cycles are closer to 50 to 60 minutes. Newborns spend approximately 50% of their sleep in REM sleep, whereas adults spend 20% to 25% of their sleep in REM sleep. Children will spend more time in REM sleep until the third and fourth years of life, at which point REM gradually decreases to 20% to 25% by adulthood.
Sleep needs also change predictably throughout the lifespan. The National Sleep Foundation guidelines for sleep duration provide clinicians and parents with a range of recommended sleep for each stage of development. Infants require 14 to 17 hours of sleep, whereas adolescents need 8 to 10 hours by age 14 to 17.8 The key for clinicians is to determine if the child is within the recommended range, and how they are functioning on the number of hours of sleep they report. This allows for variation in how much sleep an individual child might need while acknowledging that some children within a specific age group might need more or less sleep than other children of the same age.
Sleep in healthy youth: Middle childhood
School-age children (age 6 to 12) typically need 9 to 10 hours of sleep over a 24-hour period.2 This developmental period is especially important for children to develop healthy sleep habits; however, developmentally appropriate cognitive and social/emotional factors might interfere with the quality and quantity of sleep. Middle childhood is a time when children can understand the dangers of the outside world (ie, violence, health problems) and resulting anxiety can disrupt sleep. Parents usually are less involved in bedtime as children approach adolescence, which leads to later bedtimes. At this stage, many children begin to take on more serious roles in their academics and extracurricular activities, peer relationships become more important, and use of electronics (eg, television, video games, internet, and handheld devices) increases—all of which compete with sleep.9 Frequent sleep issues during middle childhood include:
- irregular sleep-wake schedules
- later bedtimes
- decreased nighttime sleep
- increased caffeine intake
- reduced parental presence at bedtime
- daytime sleepiness.3
In school-age children, regular napping, falling asleep during short car rides, and daytime fatigue at school or home are cause for concern. When these symptoms are present, an evaluation is warranted.
Sleep in healthy youth: Adolescence
The National Sleep Foundation recommends adolescents obtain 8 to 10 hours of sleep per night; for some adolescents, as much as 11 hours of sleep per night might be appropriate.8 However, this contrasts with findings from the National Sleep Foundation’s Sleep in America Poll, which revealed that 75% of 12th graders report <8 hours of sleep nightly.10 Many adolescents experience delayed sleep phase syndrome or delayed sleep-wake phase disorder, which involves a persistent phase shift of >2 hours in the sleep-wake schedule that conflicts with the adolescent’s school, work, or lifestyle demands.11 Such circadian rhythm disorders typically result from a poor match between the sleep-wake schedule and the demands of the adolescent’s life, or a failure to synchronize their internal clock with a 24-hour circadian clock.12 Children typically become tired after sunset, but puberty is associated with reduced slow-wave sleep and changes in circadian rhythms. As a result, a 3-hour delay (delayed phase preference) is common among adolescents. At approximately age 20, people start to become tired after sunset and awaken earlier in the morning—a pattern driven by sunlight and the timing of melatonin release that will remain stable until the sixth decade of life.
Continue to: Effects of chronic sleep deprivation...
Effects of chronic sleep deprivation
Most older studies of sleep loss examined the impact of total sleep loss (sleep deprivation) rather than the effect of partial sleep loss or sleep restriction, a more commonly experienced phenomenon. More recent research shows that a cumulative sleep deficit could cause the body to override voluntary wakefulness and a sleep-deprived individual can experience brief “microsleeps” where they are unaware and lose attention/wakefulness for several seconds.2 This can be deadly if a sleep-deprived adolescent experiences microsleeps while driving.13
There is a well-studied correlation between chronic sleep deprivation and increased body mass index in children.14 This might be caused by reduction in physical activity as well as alterations in the “hunger hormones”—ghrelin and leptin—that have been observed with sleep deprivation.15-17 Other studies have noted decreased glucose tolerance, reduced insulin sensitivity, and catecholamine and cortisol secretion abnormalities, which place children at higher risk for metabolic syndrome and hypertension.13,18 Sleep deprivation also is associated with mood and anxiety disorders and is an independent risk factor for substance use and suicidal ideation among adolescents.19 Sleep deprivation increases impairments in impulse control, concentration, and attention, which could be especially problematic in school-age children.
How sleep is assessed
The sleep history is the first step in evaluating a child or adolescent for a sleep disorder. The sleep history includes exploring the chief complaint, sleep patterns and schedules, bedtime routines, and nocturnal and daytime behaviors (Table).
Chief complaint
Behavioral sleep specialists will assess the primary problem with everyone involved in the child’s bedtime.20 This might include parents (custodial and noncustodial), grandparents, or stepparents as well as the child/adolescent. This important step can reveal a sleep disorder or an inappropriately early bedtime relative to the child’s development. During this assessment, ask detailed questions about how long the sleep problem has persisted, the frequency of sleep problems, and any precipitating stressors. Parents and caregivers can review strategies they have tried, and for how long and to what extent interventions were implemented consistently to result in change.
Sleep patterns and schedules
Review the child/adolescent’s typical sleep patterns and behaviors. Ask parents and caregivers, as well as the patient, about general sleep schedules for the past few weeks or a typical 2-week time period.2 A behavioral assessment of sleep should include asking families about how the child/adolescent sleeps during the week and over the weekend, and if school-year sleep differs from summer or holiday sleep schedules. These questions can illuminate how long a sleep problem has been occurring and what sleep habits might be contributing to the problem. Bedtime
Determine if there is a set bedtime or if the child goes to bed when they wish. It is important to ascertain if the bedtime is age-appropriate, if weekday and weekend bedtimes differ, and to what extent extracurricular activities or school demands impact bedtime. Assess the consistency of the bedtime, the nature of bedtime routines (eg, is the child engaging in stimulating activities before bed), where the bedtime routine occurs (eg, sibling’s room, parents’ room, child’s room), and what role (if any) electronic devices play.2
Nocturnal behaviors
Assessment should include a series of questions and age-specific questionnaires to focus on what behaviors occur at night, including awakenings. Parents should be asked how frequent night awakenings occur, how long arousals last, and how the child signals for the parent (eg, calling out, climbing into parents’ bed).2 Additionally, ask how parents respond and what is required to help the child fall back asleep (eg, rocking, soothing, feeding). The presence of nightmares, night terrors, parasomnias, and sleep-related breathing disorders also must be assessed.20
Daytime behaviors
A sleep history should include assessment of daytime functioning, including daytime sleepiness, fatigue, morning waking, and functioning during school, extracurriculars, and homework. For children and teens, falling asleep in the car, while in school, or during passive activities (meals, conversation) suggests insufficient sleep, sleep disruption, or excessive daytime sleepiness.2
Continue to: Sleep disruption in youth with psychiatric disorders...
Sleep disruption in youth with psychiatric disorders
Disordered sleep is common across psychiatric disorders. The National Comorbidity Survey Adolescent Supplement—a nationally representative cross-sectional survey of adolescents (N = 10,123)—found that a later weeknight bedtime, shorter weeknight sleep duration, and greater weekend bedtime delay increased the risk of developing a mood, anxiety, or substance use (including nicotine) disorder, and suicidality. These risk factors also were associated with lower “perceived mental and physical health.”21 Clinicians should routinely obtain a sleep history in children and adolescents with these disorders. Consider using the sleep screening tool BEARS:
- Bedtime issues
- Excessive daytime sleepiness
- Awakenings
- Regularity and duration of sleep
- Snoring.
ADHD
Up to one-half of children and adolescents with ADHD experience sleep problems,22,23 including delayed sleep onset, bedtime resistance, daytime fatigue, and feeling groggy in the morning beyond what is typical (>20 minutes). Pharmacotherapy for ADHD contributes to sleep disturbances24,25 while sleep deprivation exacerbates inattention and hyperactivity. In youth with ADHD, restless leg syndrome, periodic limb movement disorder, and sleep-disordered breathing disorder are more common than in the general population.
Depressive disorders
Up to three-quarters of depressed children and 90% of depressed adolescents report sleep disturbances, including initial, middle, and terminal insomnia as well as hypersomnia.26 Disrupted sleep in pediatric patients with major depressive disorder could be moderated by the patient’s age, with depressive symptoms more common among adolescents (age 12 to 17) than among younger children (age 6 to 11).27 Successful treatment of depression fails to relieve dyssomnia in 10% of children. Sleep problems that persist after successfully treating a depressive episode could increase the risk of another depressive episode.28
Anxiety disorders
Sleep problems are common among children and adolescents with anxiety disorders.29 Longitudinal data from >900 children found that symptoms of sleep disturbance in early childhood were correlated with experiencing an anxiety disorder 20 years later.30 Fears related to the dark or monsters under the bed that are developmentally appropriate for younger children may interfere with sleep. However, in anxious children, fears might also be related to separation, sleeping alone, worry about the loss of a loved one, concerns about personal safety, fear of frightening dreams, or concerns about academics and social relationships. Anxious individuals ruminate about their worries, and this might be especially true for children at bedtime, when there are limited distractions from ruminative fears.31 Bedtime resistance, parental involvement in bedtime rituals, and cultural factors related to sleep also could play a role for children with anxiety symptoms and sleep problems.
Having an anxiety disorder is significantly associated with an increased risk of insomnia; however, 73% of the time anxiety symptoms precede an insomnia diagnosis.29 Sleep problems and anxiety symptoms might have a reciprocal influence on one another; tiredness that results from sleep problems could exacerbate anxiety, which further worsens sleep problems.
A bridge to treatment
A thorough assessment can help identify modifiable factors and guide treatment selections. In Part 2 of this article, we will describe healthy sleep practices, cognitive-behavioral therapy for insomnia, when pharmacotherapy might be indicated, and the evidence supporting several medications commonly used to treat pediatric insomnia. We also will discuss factors to consider when seeking consultation with a pediatric behavioral sleep specialist.
FIRST OF 2 PARTS
A thorough evaluation can identify modifiable factors and guide treatment
Sleep problems are common among children and adolescents,1 with prevalence rates of 25% to 40%.2-4 Young children most commonly exhibit what is referred to as bedtime problems and night wakenings, whereas children in middle childhood (age 4 to 12) through adolescence (age 13 to 17) report insomnia. For many children, these problems persist.3 Insufficient sleep in children and adolescents worsens inattention, daytime fatigue, and cognitive and behavioral deficits.5 Assessment and treatment of sleep problems in children and adolescents is critical because poor sleep among youth increases the risk for depression, self-harm, and suicide,6,7 increases family stress, and decreases parental well-being.1
This 2-part article describes the assessment, diagnosis, and treatment of sleep problems among children and adolescents. In part 1, we focus on:
- sleep architecture (circadian rhythms, stages of sleep)
- sleep in healthy youth (age 6 to 17) and those with attention-deficit/hyperactivity disorder (ADHD), depressive disorders, and anxiety
- how to assess sleep, and the differential diagnosis of behavioral sleep problems in pediatric patients.
In Part 2, we will cover psychotherapeutic and psychopharmacologic interventions for youth with insomnia, and describe an effective approach to consultation with pediatric sleep medicine specialists.
How much sleep do children and adolescents need?
Throughout their development, children spend 40% to 50% of their time asleep. Sleep schedules are based on circadian rhythms, which are physical, mental, and behavioral changes that follow an approximately 24-hour cycle. Human circadian rhythm varies between 24 and 25 hours and is vital in determining our sleep patterns. Exposure to sunlight drives our circadian rhythm, sending signals to our bodies to “turn on” melatonin production at night (ie, 9
Box
Sleep architecture consists of 3 states: wake; non-rapid eye movement (NREM) sleep; and rapid eye movement (REM) sleep (“dreaming” sleep).2 These stages have distinct polysomnographic features of electroencephalographic EEG patterns, eye movements, and muscle tone.2 NREM sleep can be further divided into 3 stages: stage 1 (N1), stage 2 (N2), and stage 3 (N3). Stage 1 is the lightest stage and lasts for 30 seconds to 5 minutes; it is easy to wake up from stage 1 sleep. During stage 2 sleep, the body moves into a deeper sleep stage that is considered “true” sleep. This sleep stage is characterized by bursts of rhythmic rapid EEG activity known as spindles, as well as high-amplitude slow-wave spikes called K complexes.2 Stage 2 sleep lasts for 10 to 45 minutes. Stage 3, better known as “deep sleep,” slow-wave sleep, or delta sleep, is the most restorative sleep.2 Respiration is low and parasympathetic activity is high.2 It is difficult to be awakened during deep sleep, and if aroused, the person likely will feel confused or groggy. Deep sleep is followed by a return to lighter stage of sleep before the first REM sleep period begins.
REM sleep is the active stage of sleep. Breathing and heart rate become irregular, and the body experiences muscle atonia, or temporary paralysis, of arms and legs. When in REM sleep, individuals have the highest brain metabolic rates, and periodic bursts of eye movements.2 Most individuals move through stages of NREM and REM sleep in predicable ways, meaning they experience NREM sleep, return to a lighter stage of sleep after deep sleep, then move into REM sleep before the cycle repeats. It takes approximately 90 minutes for most adults to complete the NREM sleep cycle, and then REM sleep occurs before returning to NREM sleep.
In children, especially in infants and babies, sleep cycles are closer to 50 to 60 minutes. Newborns spend approximately 50% of their sleep in REM sleep, whereas adults spend 20% to 25% of their sleep in REM sleep. Children will spend more time in REM sleep until the third and fourth years of life, at which point REM gradually decreases to 20% to 25% by adulthood.
Sleep needs also change predictably throughout the lifespan. The National Sleep Foundation guidelines for sleep duration provide clinicians and parents with a range of recommended sleep for each stage of development. Infants require 14 to 17 hours of sleep, whereas adolescents need 8 to 10 hours by age 14 to 17.8 The key for clinicians is to determine if the child is within the recommended range, and how they are functioning on the number of hours of sleep they report. This allows for variation in how much sleep an individual child might need while acknowledging that some children within a specific age group might need more or less sleep than other children of the same age.
Sleep in healthy youth: Middle childhood
School-age children (age 6 to 12) typically need 9 to 10 hours of sleep over a 24-hour period.2 This developmental period is especially important for children to develop healthy sleep habits; however, developmentally appropriate cognitive and social/emotional factors might interfere with the quality and quantity of sleep. Middle childhood is a time when children can understand the dangers of the outside world (ie, violence, health problems) and resulting anxiety can disrupt sleep. Parents usually are less involved in bedtime as children approach adolescence, which leads to later bedtimes. At this stage, many children begin to take on more serious roles in their academics and extracurricular activities, peer relationships become more important, and use of electronics (eg, television, video games, internet, and handheld devices) increases—all of which compete with sleep.9 Frequent sleep issues during middle childhood include:
- irregular sleep-wake schedules
- later bedtimes
- decreased nighttime sleep
- increased caffeine intake
- reduced parental presence at bedtime
- daytime sleepiness.3
In school-age children, regular napping, falling asleep during short car rides, and daytime fatigue at school or home are cause for concern. When these symptoms are present, an evaluation is warranted.
Sleep in healthy youth: Adolescence
The National Sleep Foundation recommends adolescents obtain 8 to 10 hours of sleep per night; for some adolescents, as much as 11 hours of sleep per night might be appropriate.8 However, this contrasts with findings from the National Sleep Foundation’s Sleep in America Poll, which revealed that 75% of 12th graders report <8 hours of sleep nightly.10 Many adolescents experience delayed sleep phase syndrome or delayed sleep-wake phase disorder, which involves a persistent phase shift of >2 hours in the sleep-wake schedule that conflicts with the adolescent’s school, work, or lifestyle demands.11 Such circadian rhythm disorders typically result from a poor match between the sleep-wake schedule and the demands of the adolescent’s life, or a failure to synchronize their internal clock with a 24-hour circadian clock.12 Children typically become tired after sunset, but puberty is associated with reduced slow-wave sleep and changes in circadian rhythms. As a result, a 3-hour delay (delayed phase preference) is common among adolescents. At approximately age 20, people start to become tired after sunset and awaken earlier in the morning—a pattern driven by sunlight and the timing of melatonin release that will remain stable until the sixth decade of life.
Continue to: Effects of chronic sleep deprivation...
Effects of chronic sleep deprivation
Most older studies of sleep loss examined the impact of total sleep loss (sleep deprivation) rather than the effect of partial sleep loss or sleep restriction, a more commonly experienced phenomenon. More recent research shows that a cumulative sleep deficit could cause the body to override voluntary wakefulness and a sleep-deprived individual can experience brief “microsleeps” where they are unaware and lose attention/wakefulness for several seconds.2 This can be deadly if a sleep-deprived adolescent experiences microsleeps while driving.13
There is a well-studied correlation between chronic sleep deprivation and increased body mass index in children.14 This might be caused by reduction in physical activity as well as alterations in the “hunger hormones”—ghrelin and leptin—that have been observed with sleep deprivation.15-17 Other studies have noted decreased glucose tolerance, reduced insulin sensitivity, and catecholamine and cortisol secretion abnormalities, which place children at higher risk for metabolic syndrome and hypertension.13,18 Sleep deprivation also is associated with mood and anxiety disorders and is an independent risk factor for substance use and suicidal ideation among adolescents.19 Sleep deprivation increases impairments in impulse control, concentration, and attention, which could be especially problematic in school-age children.
How sleep is assessed
The sleep history is the first step in evaluating a child or adolescent for a sleep disorder. The sleep history includes exploring the chief complaint, sleep patterns and schedules, bedtime routines, and nocturnal and daytime behaviors (Table).
Chief complaint
Behavioral sleep specialists will assess the primary problem with everyone involved in the child’s bedtime.20 This might include parents (custodial and noncustodial), grandparents, or stepparents as well as the child/adolescent. This important step can reveal a sleep disorder or an inappropriately early bedtime relative to the child’s development. During this assessment, ask detailed questions about how long the sleep problem has persisted, the frequency of sleep problems, and any precipitating stressors. Parents and caregivers can review strategies they have tried, and for how long and to what extent interventions were implemented consistently to result in change.
Sleep patterns and schedules
Review the child/adolescent’s typical sleep patterns and behaviors. Ask parents and caregivers, as well as the patient, about general sleep schedules for the past few weeks or a typical 2-week time period.2 A behavioral assessment of sleep should include asking families about how the child/adolescent sleeps during the week and over the weekend, and if school-year sleep differs from summer or holiday sleep schedules. These questions can illuminate how long a sleep problem has been occurring and what sleep habits might be contributing to the problem. Bedtime
Determine if there is a set bedtime or if the child goes to bed when they wish. It is important to ascertain if the bedtime is age-appropriate, if weekday and weekend bedtimes differ, and to what extent extracurricular activities or school demands impact bedtime. Assess the consistency of the bedtime, the nature of bedtime routines (eg, is the child engaging in stimulating activities before bed), where the bedtime routine occurs (eg, sibling’s room, parents’ room, child’s room), and what role (if any) electronic devices play.2
Nocturnal behaviors
Assessment should include a series of questions and age-specific questionnaires to focus on what behaviors occur at night, including awakenings. Parents should be asked how frequent night awakenings occur, how long arousals last, and how the child signals for the parent (eg, calling out, climbing into parents’ bed).2 Additionally, ask how parents respond and what is required to help the child fall back asleep (eg, rocking, soothing, feeding). The presence of nightmares, night terrors, parasomnias, and sleep-related breathing disorders also must be assessed.20
Daytime behaviors
A sleep history should include assessment of daytime functioning, including daytime sleepiness, fatigue, morning waking, and functioning during school, extracurriculars, and homework. For children and teens, falling asleep in the car, while in school, or during passive activities (meals, conversation) suggests insufficient sleep, sleep disruption, or excessive daytime sleepiness.2
Continue to: Sleep disruption in youth with psychiatric disorders...
Sleep disruption in youth with psychiatric disorders
Disordered sleep is common across psychiatric disorders. The National Comorbidity Survey Adolescent Supplement—a nationally representative cross-sectional survey of adolescents (N = 10,123)—found that a later weeknight bedtime, shorter weeknight sleep duration, and greater weekend bedtime delay increased the risk of developing a mood, anxiety, or substance use (including nicotine) disorder, and suicidality. These risk factors also were associated with lower “perceived mental and physical health.”21 Clinicians should routinely obtain a sleep history in children and adolescents with these disorders. Consider using the sleep screening tool BEARS:
- Bedtime issues
- Excessive daytime sleepiness
- Awakenings
- Regularity and duration of sleep
- Snoring.
ADHD
Up to one-half of children and adolescents with ADHD experience sleep problems,22,23 including delayed sleep onset, bedtime resistance, daytime fatigue, and feeling groggy in the morning beyond what is typical (>20 minutes). Pharmacotherapy for ADHD contributes to sleep disturbances24,25 while sleep deprivation exacerbates inattention and hyperactivity. In youth with ADHD, restless leg syndrome, periodic limb movement disorder, and sleep-disordered breathing disorder are more common than in the general population.
Depressive disorders
Up to three-quarters of depressed children and 90% of depressed adolescents report sleep disturbances, including initial, middle, and terminal insomnia as well as hypersomnia.26 Disrupted sleep in pediatric patients with major depressive disorder could be moderated by the patient’s age, with depressive symptoms more common among adolescents (age 12 to 17) than among younger children (age 6 to 11).27 Successful treatment of depression fails to relieve dyssomnia in 10% of children. Sleep problems that persist after successfully treating a depressive episode could increase the risk of another depressive episode.28
Anxiety disorders
Sleep problems are common among children and adolescents with anxiety disorders.29 Longitudinal data from >900 children found that symptoms of sleep disturbance in early childhood were correlated with experiencing an anxiety disorder 20 years later.30 Fears related to the dark or monsters under the bed that are developmentally appropriate for younger children may interfere with sleep. However, in anxious children, fears might also be related to separation, sleeping alone, worry about the loss of a loved one, concerns about personal safety, fear of frightening dreams, or concerns about academics and social relationships. Anxious individuals ruminate about their worries, and this might be especially true for children at bedtime, when there are limited distractions from ruminative fears.31 Bedtime resistance, parental involvement in bedtime rituals, and cultural factors related to sleep also could play a role for children with anxiety symptoms and sleep problems.
Having an anxiety disorder is significantly associated with an increased risk of insomnia; however, 73% of the time anxiety symptoms precede an insomnia diagnosis.29 Sleep problems and anxiety symptoms might have a reciprocal influence on one another; tiredness that results from sleep problems could exacerbate anxiety, which further worsens sleep problems.
A bridge to treatment
A thorough assessment can help identify modifiable factors and guide treatment selections. In Part 2 of this article, we will describe healthy sleep practices, cognitive-behavioral therapy for insomnia, when pharmacotherapy might be indicated, and the evidence supporting several medications commonly used to treat pediatric insomnia. We also will discuss factors to consider when seeking consultation with a pediatric behavioral sleep specialist.
1. Meltzer LJ, Mindell JA. Systematic review and meta-analysis of behavioral interventions for pediatric insomnia. J Pediatr Psychol. 2014;39(8):932-948. doi:10.1093/jpepsy/jsu041
2. Owens JA, Mindell JA. Pediatric insomnia. Pediatr Clin North Am. 2011;58(3):555-569. doi:10.1016/j.pcl.2011.03.011
3. Meltzer LJ, Plaufcan MR, Thomas JH, et al. Sleep problems and sleep disorders in pediatric primary care: treatment recommendations, persistence, and health care utilization. J Clin Sleep Med. 2014;10(4):421-426. doi:10.5664/jcsm.3620
4. Moore M, Meltzer LJ, Mindell JA. Bedtime problems and night wakings in children. Prim Care. 2008;35(3):569-581, viii. doi:10.1016/j.pop.2008.06.002
5. Williamson AA, Mindell JA, Hiscock H, et al. Longitudinal sleep problem trajectories are associated with multiple impairments in child well-being. J Child Psychol Psychiatry. 2020;61(10):1092-1103. doi:10.1111/jcpp.13303
6. Roberts RE, Roberts CR, Chen IG. Impact of insomnia on future functioning of adolescents. J Psychosom Res. 2002; 53(1):561-569. doi:10.1016/s0022-3999(02)00446-4
7. Singareddy R, Krishnamurthy VB, Vgontzas AN, et al. Subjective and objective sleep and self-harm behaviors in young children: a general population study. Psychiatry Res. 2013;209(3):549-553. doi:10.1016/j.psychres.2013.03.036
8. Hirshkowitz M, Whiton K, Albert SM, et al. National Sleep Foundation’s updated sleep duration recommendations: final report. Sleep Health. 2015;1(4):233-243. doi:10.1016/j.sleh.2015.10.004
9. Calamaro CJ, Mason TBA, Ratcliffe SJ. Adolescents living the 24/7 lifestyle: Effects of caffeine and technology on sleep duration and daytime functioning. Pediatrics. 2009;123(6):e1005-1010. doi:10.1542/peds.2008-3641
10. Mindell JA, Owens JA, Carskadon MA. Developmental features of sleep. Child Adolesc Psychiatr Clin N Am. 1999;8(4):695-725.
11. Moore M, Meltzer LJ. The sleepy adolescent: causes and consequences of sleepiness in teens. Paediatr Respir Rev. 2008;9(2):114-120. doi:10.1016/j.prrv.2008.01.001
12. Crowley SJ, Acebo C, Carskadon MA. Sleep, circadian rhythms, and delayed phase in adolescence. Sleep Med. 2007;8(6):602-612. doi:10.1016/j.sleep.2006.12.002
13. Millman RP; Working Group on Sleepiness in Adolescents/Young Adults; AAP Committee on Adolescence. Excessive sleepiness in adolescents and young adults: causes, consequences, and treatment strategies. Pediatrics. 2005;115(6):1774-1786. doi:10.1542/peds.2005-0772
14. Kaczor M, Skalski M. Prevalence and consequences of insomnia in pediatric population. Psychiatr Pol. 2016;50(3):555-569. doi:10.12740/PP/61226
15. Gomes TN, Dos Santos FK, Santos D, et al. Correlates of sedentary time in children: a multilevel modelling approach. BMC Public Health. 2014;14:890. doi:10.1186/1471-2458-14-890
16. Stone MR, Stevens D, Faulkner GEJ. Maintaining recommended sleep throughout the week is associated with increased physical activity in children. Prev Med. 2013;56(2):112-117. doi:10.1016/j.ypmed.2012.11.015
17. Hart CN, Fava JL, Subak LL, et al. Time in bed is associated with decreased physical activity and higher BMI in women seeking weight loss treatment. ISRN Obes. 2012;2012:320157. doi:10.5402/2012/320157
18. Tasali E, Leproult R, Ehrmann DA, et al. Slow-wave sleep and the risk of type 2 diabetes in humans. Proc Natl Acad Sci U S A. 2008;105(3):1044-1049. doi:10.1073/pnas.0706446105
19. de Zambotti M, Goldstone A, Colrain IM, et al. Insomnia disorder in adolescence: diagnosis, impact, and treatment. Sleep Med Rev. 2018;39:12-24. doi:10.1016/j.smrv.2017.06.009
20. Mindell JA, Owens JA. A clinical guide to pediatric sleep: diagnosis and management of sleep problems. 3rd ed. Lippincott Williams & Wilkins; 2015.
21. Zhang J, Paksarian D, Lamers F, et al. Sleep patterns and mental health correlates in US adolescents. J Pediatr. 2017;182:137-143. doi:10.1016/j.jpeds.2016.11.007
22. Gregory AM, Agnew-Blais JC, Matthews T, et al. ADHD and sleep quality: longitudinal analyses from childhood to early adulthood in a twin cohort. J Clin Child Adolesc Psychol. 2017;46(2):284-294. doi:10.1080/15374416.2016.1183499
23. Weiss MD, Salpekar J. Sleep problems in the child with attention-deficit hyperactivity disorder: Defining aetiology and appropriate treatments. CNS Drugs. 2010;24(10):811-828. doi:10.2165/11538990-000000000-00000
24. Galland BC, Tripp EG, Taylor BJ. The sleep of children with attention deficit hyperactivity disorder on and off methylphenidate: a matched case-control study. J Sleep Res. 2010;19(2):366-373. doi:10.1111/j.1365-2869.2009.00795.x
25. Becker SP, Froehlich TE, Epstein JN. Effects of methylphenidate on sleep functioning in children with attention-deficit/hyperactivity disorder. J Dev Behav Pediatr. 2016;37(5):395-404. doi:10.1097/DBP.0000000000000285
26. Roberts RE, Duong HT. Depression and insomnia among adolescents: a prospective perspective. J Affect Disord. 2013;148(1):66-71. doi:10.1016/j.jad.2012.11.049
27. Emslie GJ, Rush AJ, Weinberg WA, et al. Sleep EEG features of adolescents with major depression. Biol Psychiatry. 1994;36(9):573-581. doi:10.1016/0006-3223(94)90067-1
28. Alfano CA, Zakem AH, Costa NM, et al. Sleep problems and their relation to cognitive factors, anxiety, and depressive symptoms in children and adolescents. Depress Anxiety. 2009;26(6):503-512. doi:10.1002/da.20443
29. Alfano CA, Ginsburg GS, Kingery JN. Sleep-related problems among children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):224-232. doi:10.1097/01.chi.0000242233.06011.8e
30. Gregory AM, Caspi A, Eley TC, et al. Prospective longitudinal associations between persistent sleep problems in childhood and anxiety and depression disorders in adulthood. J Abnorm Child Psychol. 2005;33(2):157-163. doi: 10.1007/s10802-005-1824-0
31. Chorney DB, Detweiler MF, Morris TL, et al. The interplay of sleep disturbance, anxiety, and depression in children. J Pediatr Psychol. 2008;33(4):339-348. doi:10.1093/jpepsy/jsm105
32. Sadeh A. Stress, trauma, and sleep in children. Child Adolesc Psychiatr Clin N Am. 1996;5(3):685-700. doi:10.1016/S1056-4993(18)30356-0
33. Glod CA, Teicher MH, Hartman CR, et al. Increased nocturnal activity and impaired sleep maintenance in abused children. J Am Acad Child Adolesc Psychiatry. 1997;36(9):1236-1243. doi:10.1097/00004583-199709000-00016
34. Strawn JR, Lu L, Peris TS, et al. Research review: pediatric anxiety disorders: what have we learnt in the last 10 years? J Child Psychol Psychiatry. 2021;62(2):114-139. doi:10.1111/jcpp.13262
35. Wehry AM, Beesdo-Baum K, Hennelly MM, et al. Assessment and treatment of anxiety disorders in children and adolescents. Curr Psychiatry Rep. 2015;17(7):52. doi:10.1007/s11920-015-0591-z
36. Hamill Skoch S, Mills JA, Ramsey L, et al. Letter to editor: sleep disturbances in selective serotonin reuptake inhibitor-treated youth with anxiety disorders and obsessive compulsive disorder— a bayesian hierarchical modeling meta-analysis. J Child Adolesc Psychopharmacol. 2021;31(5):387-388. doi:10.1089/cap.2020.0169
1. Meltzer LJ, Mindell JA. Systematic review and meta-analysis of behavioral interventions for pediatric insomnia. J Pediatr Psychol. 2014;39(8):932-948. doi:10.1093/jpepsy/jsu041
2. Owens JA, Mindell JA. Pediatric insomnia. Pediatr Clin North Am. 2011;58(3):555-569. doi:10.1016/j.pcl.2011.03.011
3. Meltzer LJ, Plaufcan MR, Thomas JH, et al. Sleep problems and sleep disorders in pediatric primary care: treatment recommendations, persistence, and health care utilization. J Clin Sleep Med. 2014;10(4):421-426. doi:10.5664/jcsm.3620
4. Moore M, Meltzer LJ, Mindell JA. Bedtime problems and night wakings in children. Prim Care. 2008;35(3):569-581, viii. doi:10.1016/j.pop.2008.06.002
5. Williamson AA, Mindell JA, Hiscock H, et al. Longitudinal sleep problem trajectories are associated with multiple impairments in child well-being. J Child Psychol Psychiatry. 2020;61(10):1092-1103. doi:10.1111/jcpp.13303
6. Roberts RE, Roberts CR, Chen IG. Impact of insomnia on future functioning of adolescents. J Psychosom Res. 2002; 53(1):561-569. doi:10.1016/s0022-3999(02)00446-4
7. Singareddy R, Krishnamurthy VB, Vgontzas AN, et al. Subjective and objective sleep and self-harm behaviors in young children: a general population study. Psychiatry Res. 2013;209(3):549-553. doi:10.1016/j.psychres.2013.03.036
8. Hirshkowitz M, Whiton K, Albert SM, et al. National Sleep Foundation’s updated sleep duration recommendations: final report. Sleep Health. 2015;1(4):233-243. doi:10.1016/j.sleh.2015.10.004
9. Calamaro CJ, Mason TBA, Ratcliffe SJ. Adolescents living the 24/7 lifestyle: Effects of caffeine and technology on sleep duration and daytime functioning. Pediatrics. 2009;123(6):e1005-1010. doi:10.1542/peds.2008-3641
10. Mindell JA, Owens JA, Carskadon MA. Developmental features of sleep. Child Adolesc Psychiatr Clin N Am. 1999;8(4):695-725.
11. Moore M, Meltzer LJ. The sleepy adolescent: causes and consequences of sleepiness in teens. Paediatr Respir Rev. 2008;9(2):114-120. doi:10.1016/j.prrv.2008.01.001
12. Crowley SJ, Acebo C, Carskadon MA. Sleep, circadian rhythms, and delayed phase in adolescence. Sleep Med. 2007;8(6):602-612. doi:10.1016/j.sleep.2006.12.002
13. Millman RP; Working Group on Sleepiness in Adolescents/Young Adults; AAP Committee on Adolescence. Excessive sleepiness in adolescents and young adults: causes, consequences, and treatment strategies. Pediatrics. 2005;115(6):1774-1786. doi:10.1542/peds.2005-0772
14. Kaczor M, Skalski M. Prevalence and consequences of insomnia in pediatric population. Psychiatr Pol. 2016;50(3):555-569. doi:10.12740/PP/61226
15. Gomes TN, Dos Santos FK, Santos D, et al. Correlates of sedentary time in children: a multilevel modelling approach. BMC Public Health. 2014;14:890. doi:10.1186/1471-2458-14-890
16. Stone MR, Stevens D, Faulkner GEJ. Maintaining recommended sleep throughout the week is associated with increased physical activity in children. Prev Med. 2013;56(2):112-117. doi:10.1016/j.ypmed.2012.11.015
17. Hart CN, Fava JL, Subak LL, et al. Time in bed is associated with decreased physical activity and higher BMI in women seeking weight loss treatment. ISRN Obes. 2012;2012:320157. doi:10.5402/2012/320157
18. Tasali E, Leproult R, Ehrmann DA, et al. Slow-wave sleep and the risk of type 2 diabetes in humans. Proc Natl Acad Sci U S A. 2008;105(3):1044-1049. doi:10.1073/pnas.0706446105
19. de Zambotti M, Goldstone A, Colrain IM, et al. Insomnia disorder in adolescence: diagnosis, impact, and treatment. Sleep Med Rev. 2018;39:12-24. doi:10.1016/j.smrv.2017.06.009
20. Mindell JA, Owens JA. A clinical guide to pediatric sleep: diagnosis and management of sleep problems. 3rd ed. Lippincott Williams & Wilkins; 2015.
21. Zhang J, Paksarian D, Lamers F, et al. Sleep patterns and mental health correlates in US adolescents. J Pediatr. 2017;182:137-143. doi:10.1016/j.jpeds.2016.11.007
22. Gregory AM, Agnew-Blais JC, Matthews T, et al. ADHD and sleep quality: longitudinal analyses from childhood to early adulthood in a twin cohort. J Clin Child Adolesc Psychol. 2017;46(2):284-294. doi:10.1080/15374416.2016.1183499
23. Weiss MD, Salpekar J. Sleep problems in the child with attention-deficit hyperactivity disorder: Defining aetiology and appropriate treatments. CNS Drugs. 2010;24(10):811-828. doi:10.2165/11538990-000000000-00000
24. Galland BC, Tripp EG, Taylor BJ. The sleep of children with attention deficit hyperactivity disorder on and off methylphenidate: a matched case-control study. J Sleep Res. 2010;19(2):366-373. doi:10.1111/j.1365-2869.2009.00795.x
25. Becker SP, Froehlich TE, Epstein JN. Effects of methylphenidate on sleep functioning in children with attention-deficit/hyperactivity disorder. J Dev Behav Pediatr. 2016;37(5):395-404. doi:10.1097/DBP.0000000000000285
26. Roberts RE, Duong HT. Depression and insomnia among adolescents: a prospective perspective. J Affect Disord. 2013;148(1):66-71. doi:10.1016/j.jad.2012.11.049
27. Emslie GJ, Rush AJ, Weinberg WA, et al. Sleep EEG features of adolescents with major depression. Biol Psychiatry. 1994;36(9):573-581. doi:10.1016/0006-3223(94)90067-1
28. Alfano CA, Zakem AH, Costa NM, et al. Sleep problems and their relation to cognitive factors, anxiety, and depressive symptoms in children and adolescents. Depress Anxiety. 2009;26(6):503-512. doi:10.1002/da.20443
29. Alfano CA, Ginsburg GS, Kingery JN. Sleep-related problems among children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):224-232. doi:10.1097/01.chi.0000242233.06011.8e
30. Gregory AM, Caspi A, Eley TC, et al. Prospective longitudinal associations between persistent sleep problems in childhood and anxiety and depression disorders in adulthood. J Abnorm Child Psychol. 2005;33(2):157-163. doi: 10.1007/s10802-005-1824-0
31. Chorney DB, Detweiler MF, Morris TL, et al. The interplay of sleep disturbance, anxiety, and depression in children. J Pediatr Psychol. 2008;33(4):339-348. doi:10.1093/jpepsy/jsm105
32. Sadeh A. Stress, trauma, and sleep in children. Child Adolesc Psychiatr Clin N Am. 1996;5(3):685-700. doi:10.1016/S1056-4993(18)30356-0
33. Glod CA, Teicher MH, Hartman CR, et al. Increased nocturnal activity and impaired sleep maintenance in abused children. J Am Acad Child Adolesc Psychiatry. 1997;36(9):1236-1243. doi:10.1097/00004583-199709000-00016
34. Strawn JR, Lu L, Peris TS, et al. Research review: pediatric anxiety disorders: what have we learnt in the last 10 years? J Child Psychol Psychiatry. 2021;62(2):114-139. doi:10.1111/jcpp.13262
35. Wehry AM, Beesdo-Baum K, Hennelly MM, et al. Assessment and treatment of anxiety disorders in children and adolescents. Curr Psychiatry Rep. 2015;17(7):52. doi:10.1007/s11920-015-0591-z
36. Hamill Skoch S, Mills JA, Ramsey L, et al. Letter to editor: sleep disturbances in selective serotonin reuptake inhibitor-treated youth with anxiety disorders and obsessive compulsive disorder— a bayesian hierarchical modeling meta-analysis. J Child Adolesc Psychopharmacol. 2021;31(5):387-388. doi:10.1089/cap.2020.0169

A pandemic of pediatric panic
Seventy-three. That is the average number of questions asked daily by preschool-aged children.
Children ask questions to make sense of their world, to learn how things work, to verify their safety, and to interact with others. As a physician, a child and adolescent psychiatrist, and a father to 6-year-old twin daughters, I too am asking more questions these days. Both professionally and personally, these questions are prompted by shifts in routines, uncertainty, and anxiety brought on by the ongoing coronavirus disease 2019 (COVID-19) pandemic. In parallel, I find myself reflecting on my twin daughters’ questions; their questions reverberate with my own, and with the increased anxiety and fears of my patients and their parents.
With this in mind, I’d like to share 2 questions related to pediatric anxiety that may sculpt our clinical work—whether with children, adolescents, or adults—as we provide treatment and comfort to our patients during this pandemic of anxiety.
How do parents affect children’s anxiety?
First, children take cues from their parents. Almost a half century ago, child and adolescent psychiatrist Robert Emde, MD, and others, using elegantly designed experimental settings, documented that a mother’s response strongly influences her young son or daughter’s emotional reaction to a stranger, or to new situations.1 Specifically, very young children were less afraid and interacted more with a stranger and did so more quickly when their mother had a positive (as opposed to neutral or fearful) reaction to the situation.2 Further, in these studies, when the parent’s face was partially covered, very young children became more fearful. Taken together, these findings remind us that children actively seek to read the affective states of those who care for them, and use these reactions to anchor their responses to shifts in routine, such as those brought on by the ongoing COVID-19 pandemic.
Second, in reacting to the pandemic, parents model emotional regulation—an important skill that children and adolescents must develop as they experience intense affect and anxiety. As mental health clinicians, we know that emotional regulation is an essential component of mental health, and problems with it are a hallmark characteristic of several disorders, including anxiety disorders. Further, neuroimaging studies over the past decade have demonstrated that the way in which the medial prefrontal cortex and lower limbic structures (eg, the amygdala) are connected shifts from early childhood through adolescence and into early adulthood.3 It is likely that these shifts in functional connectivity are shaped by the environment as well as intrinsic aspects of the patient’s biology, and that these shifts subtend the developmental expression of anxiety, particularly in times of stress.
How should we talk to children about the pandemic?
Trust is not only the scaffold of our therapeutic relationships, but also a critical component of our conversations with children about the pandemic. Having established a trusting relationship prior to talking with children about their anxiety and about the pandemic, we will do well to remember that there is often more to a question than the actual direct interrogative. From a developmental standpoint, children may repeatedly ask the same question because they are struggling to understand an abstract concept, or are unable to make the same implicit causal link that we—as adults—have made. Also, children may ask the same question multiple times as a way of seeking reassurance. Finally, when a child asks her father “How many people are going to die?” she may actually be asking whether her parents, grandparents, or friends will be safe and healthy. Thus, as we talk with children, we must remember that they may be implicitly asking for more than a number, date, or mechanism. We must think about the motivation for their questions vis a vis their specific fears and past experiences.
For children, adolescents, and adults, the anxiety created by the pandemic constantly shifts, is hard-to-define, and pervades their lives. This ensuing chronic variable stress can worsen both physical and mental health.4 But, it also creates an opportunity for resiliency which—like the coronavirus—can be contagious.5,6 Knowing this, I’d like to ask 4 questions, based on David Brooks’ recent Op-Ed in the New York Times7:
- Can we become “softer and wiser” as a result of the pandemic?
- How can we inoculate our patients against the loneliness and isolation that worsen most psychiatric disorders?
- How can we “see deeper into [our]selves” to provide comfort to our patients, families, and each other as we confront this viral pandemic of anxiety?
- Following “social distancing,” how do we rekindle “social trust”?
1. Emde RN, Gaensbauer TJ, Harmon RJ. Emotional expression in infancy; a biobehavioral study. Psychol Issues. 1976;10(01):1-200.
2. Feinman S, Lewis M. Social referencing at ten months: a second-order effect on infants’ responses to strangers. Child Dev. 1983;54(4):878-887.
3. Gee DG, Gabard-Durnam LJ, Flannery J, et al. Early developmental emergence of human amygdala-prefrontal connectivity after maternal deprivation. Proc Natl Acad Sci U S A. 2013;110(39):15638-15643.
4. Keeshin BR, Cronholm PF, Strawn JR. Physiologic changes associated with violence and abuse exposure: an examination of related medical conditions. Trauma Violence Abuse. 2012;13(1):41-56.
5. Malhi GS, Das P, Bell E, et al. Modelling resilience in adolescence and adversity: a novel framework to inform research and practice. Transl Psychiatry. 2019;9(1):316. doi: 10.1038/s41398-019-0651-y.
6. Rutter M. Annual Research Review: resilience--clinical implications. J Child Psychol Psychiatry. 2013;54(4):474-487.
7. Brooks D. The pandemic of fear and agony. New York Times. April 9, 2020. https://www.nytimes.com/2020/04/09/opinion/covid-anxiety.html. Accessed April 14, 2020.
Seventy-three. That is the average number of questions asked daily by preschool-aged children.
Children ask questions to make sense of their world, to learn how things work, to verify their safety, and to interact with others. As a physician, a child and adolescent psychiatrist, and a father to 6-year-old twin daughters, I too am asking more questions these days. Both professionally and personally, these questions are prompted by shifts in routines, uncertainty, and anxiety brought on by the ongoing coronavirus disease 2019 (COVID-19) pandemic. In parallel, I find myself reflecting on my twin daughters’ questions; their questions reverberate with my own, and with the increased anxiety and fears of my patients and their parents.
With this in mind, I’d like to share 2 questions related to pediatric anxiety that may sculpt our clinical work—whether with children, adolescents, or adults—as we provide treatment and comfort to our patients during this pandemic of anxiety.
How do parents affect children’s anxiety?
First, children take cues from their parents. Almost a half century ago, child and adolescent psychiatrist Robert Emde, MD, and others, using elegantly designed experimental settings, documented that a mother’s response strongly influences her young son or daughter’s emotional reaction to a stranger, or to new situations.1 Specifically, very young children were less afraid and interacted more with a stranger and did so more quickly when their mother had a positive (as opposed to neutral or fearful) reaction to the situation.2 Further, in these studies, when the parent’s face was partially covered, very young children became more fearful. Taken together, these findings remind us that children actively seek to read the affective states of those who care for them, and use these reactions to anchor their responses to shifts in routine, such as those brought on by the ongoing COVID-19 pandemic.
Second, in reacting to the pandemic, parents model emotional regulation—an important skill that children and adolescents must develop as they experience intense affect and anxiety. As mental health clinicians, we know that emotional regulation is an essential component of mental health, and problems with it are a hallmark characteristic of several disorders, including anxiety disorders. Further, neuroimaging studies over the past decade have demonstrated that the way in which the medial prefrontal cortex and lower limbic structures (eg, the amygdala) are connected shifts from early childhood through adolescence and into early adulthood.3 It is likely that these shifts in functional connectivity are shaped by the environment as well as intrinsic aspects of the patient’s biology, and that these shifts subtend the developmental expression of anxiety, particularly in times of stress.
How should we talk to children about the pandemic?
Trust is not only the scaffold of our therapeutic relationships, but also a critical component of our conversations with children about the pandemic. Having established a trusting relationship prior to talking with children about their anxiety and about the pandemic, we will do well to remember that there is often more to a question than the actual direct interrogative. From a developmental standpoint, children may repeatedly ask the same question because they are struggling to understand an abstract concept, or are unable to make the same implicit causal link that we—as adults—have made. Also, children may ask the same question multiple times as a way of seeking reassurance. Finally, when a child asks her father “How many people are going to die?” she may actually be asking whether her parents, grandparents, or friends will be safe and healthy. Thus, as we talk with children, we must remember that they may be implicitly asking for more than a number, date, or mechanism. We must think about the motivation for their questions vis a vis their specific fears and past experiences.
For children, adolescents, and adults, the anxiety created by the pandemic constantly shifts, is hard-to-define, and pervades their lives. This ensuing chronic variable stress can worsen both physical and mental health.4 But, it also creates an opportunity for resiliency which—like the coronavirus—can be contagious.5,6 Knowing this, I’d like to ask 4 questions, based on David Brooks’ recent Op-Ed in the New York Times7:
- Can we become “softer and wiser” as a result of the pandemic?
- How can we inoculate our patients against the loneliness and isolation that worsen most psychiatric disorders?
- How can we “see deeper into [our]selves” to provide comfort to our patients, families, and each other as we confront this viral pandemic of anxiety?
- Following “social distancing,” how do we rekindle “social trust”?
Seventy-three. That is the average number of questions asked daily by preschool-aged children.
Children ask questions to make sense of their world, to learn how things work, to verify their safety, and to interact with others. As a physician, a child and adolescent psychiatrist, and a father to 6-year-old twin daughters, I too am asking more questions these days. Both professionally and personally, these questions are prompted by shifts in routines, uncertainty, and anxiety brought on by the ongoing coronavirus disease 2019 (COVID-19) pandemic. In parallel, I find myself reflecting on my twin daughters’ questions; their questions reverberate with my own, and with the increased anxiety and fears of my patients and their parents.
With this in mind, I’d like to share 2 questions related to pediatric anxiety that may sculpt our clinical work—whether with children, adolescents, or adults—as we provide treatment and comfort to our patients during this pandemic of anxiety.
How do parents affect children’s anxiety?
First, children take cues from their parents. Almost a half century ago, child and adolescent psychiatrist Robert Emde, MD, and others, using elegantly designed experimental settings, documented that a mother’s response strongly influences her young son or daughter’s emotional reaction to a stranger, or to new situations.1 Specifically, very young children were less afraid and interacted more with a stranger and did so more quickly when their mother had a positive (as opposed to neutral or fearful) reaction to the situation.2 Further, in these studies, when the parent’s face was partially covered, very young children became more fearful. Taken together, these findings remind us that children actively seek to read the affective states of those who care for them, and use these reactions to anchor their responses to shifts in routine, such as those brought on by the ongoing COVID-19 pandemic.
Second, in reacting to the pandemic, parents model emotional regulation—an important skill that children and adolescents must develop as they experience intense affect and anxiety. As mental health clinicians, we know that emotional regulation is an essential component of mental health, and problems with it are a hallmark characteristic of several disorders, including anxiety disorders. Further, neuroimaging studies over the past decade have demonstrated that the way in which the medial prefrontal cortex and lower limbic structures (eg, the amygdala) are connected shifts from early childhood through adolescence and into early adulthood.3 It is likely that these shifts in functional connectivity are shaped by the environment as well as intrinsic aspects of the patient’s biology, and that these shifts subtend the developmental expression of anxiety, particularly in times of stress.
How should we talk to children about the pandemic?
Trust is not only the scaffold of our therapeutic relationships, but also a critical component of our conversations with children about the pandemic. Having established a trusting relationship prior to talking with children about their anxiety and about the pandemic, we will do well to remember that there is often more to a question than the actual direct interrogative. From a developmental standpoint, children may repeatedly ask the same question because they are struggling to understand an abstract concept, or are unable to make the same implicit causal link that we—as adults—have made. Also, children may ask the same question multiple times as a way of seeking reassurance. Finally, when a child asks her father “How many people are going to die?” she may actually be asking whether her parents, grandparents, or friends will be safe and healthy. Thus, as we talk with children, we must remember that they may be implicitly asking for more than a number, date, or mechanism. We must think about the motivation for their questions vis a vis their specific fears and past experiences.
For children, adolescents, and adults, the anxiety created by the pandemic constantly shifts, is hard-to-define, and pervades their lives. This ensuing chronic variable stress can worsen both physical and mental health.4 But, it also creates an opportunity for resiliency which—like the coronavirus—can be contagious.5,6 Knowing this, I’d like to ask 4 questions, based on David Brooks’ recent Op-Ed in the New York Times7:
- Can we become “softer and wiser” as a result of the pandemic?
- How can we inoculate our patients against the loneliness and isolation that worsen most psychiatric disorders?
- How can we “see deeper into [our]selves” to provide comfort to our patients, families, and each other as we confront this viral pandemic of anxiety?
- Following “social distancing,” how do we rekindle “social trust”?
1. Emde RN, Gaensbauer TJ, Harmon RJ. Emotional expression in infancy; a biobehavioral study. Psychol Issues. 1976;10(01):1-200.
2. Feinman S, Lewis M. Social referencing at ten months: a second-order effect on infants’ responses to strangers. Child Dev. 1983;54(4):878-887.
3. Gee DG, Gabard-Durnam LJ, Flannery J, et al. Early developmental emergence of human amygdala-prefrontal connectivity after maternal deprivation. Proc Natl Acad Sci U S A. 2013;110(39):15638-15643.
4. Keeshin BR, Cronholm PF, Strawn JR. Physiologic changes associated with violence and abuse exposure: an examination of related medical conditions. Trauma Violence Abuse. 2012;13(1):41-56.
5. Malhi GS, Das P, Bell E, et al. Modelling resilience in adolescence and adversity: a novel framework to inform research and practice. Transl Psychiatry. 2019;9(1):316. doi: 10.1038/s41398-019-0651-y.
6. Rutter M. Annual Research Review: resilience--clinical implications. J Child Psychol Psychiatry. 2013;54(4):474-487.
7. Brooks D. The pandemic of fear and agony. New York Times. April 9, 2020. https://www.nytimes.com/2020/04/09/opinion/covid-anxiety.html. Accessed April 14, 2020.
1. Emde RN, Gaensbauer TJ, Harmon RJ. Emotional expression in infancy; a biobehavioral study. Psychol Issues. 1976;10(01):1-200.
2. Feinman S, Lewis M. Social referencing at ten months: a second-order effect on infants’ responses to strangers. Child Dev. 1983;54(4):878-887.
3. Gee DG, Gabard-Durnam LJ, Flannery J, et al. Early developmental emergence of human amygdala-prefrontal connectivity after maternal deprivation. Proc Natl Acad Sci U S A. 2013;110(39):15638-15643.
4. Keeshin BR, Cronholm PF, Strawn JR. Physiologic changes associated with violence and abuse exposure: an examination of related medical conditions. Trauma Violence Abuse. 2012;13(1):41-56.
5. Malhi GS, Das P, Bell E, et al. Modelling resilience in adolescence and adversity: a novel framework to inform research and practice. Transl Psychiatry. 2019;9(1):316. doi: 10.1038/s41398-019-0651-y.
6. Rutter M. Annual Research Review: resilience--clinical implications. J Child Psychol Psychiatry. 2013;54(4):474-487.
7. Brooks D. The pandemic of fear and agony. New York Times. April 9, 2020. https://www.nytimes.com/2020/04/09/opinion/covid-anxiety.html. Accessed April 14, 2020.

Anxiety disorders in children and adolescents

Triple-bead mixed amphetamine salt for ADHD
Stimulants are first-line psychopharmacologic interventions for attention-deficit/hyperactivity disorder (ADHD), and their efficacy is supported by clinical trials and meta-analyses in children and adolescents1 as well as adults.2 Despite decades of tolerability and efficacy data supporting their use, a major drawback of stimulants is that their salutary therapeutic effects wane once the medication is cleared or metabolized. Both mixed amphetamine- and methylphenidate-based preparations have short half-lives, necessitating multiple doses per day (eg, 3 or 4 times a day) when short-acting preparations are used. Over the past 15 years, nearly a dozen formulations were developed that extend the duration of action through delayed release, delayed absorption, or utilizing prodrugs.

The encapsulated preparation contains 3 MAS beads: an immediate-release amphetamine salt bead, a pulsed-delayed release bead, and an extended-release bead (Figure 1), which give rise to a unique pharmacokinetic profile (Figure 2).3
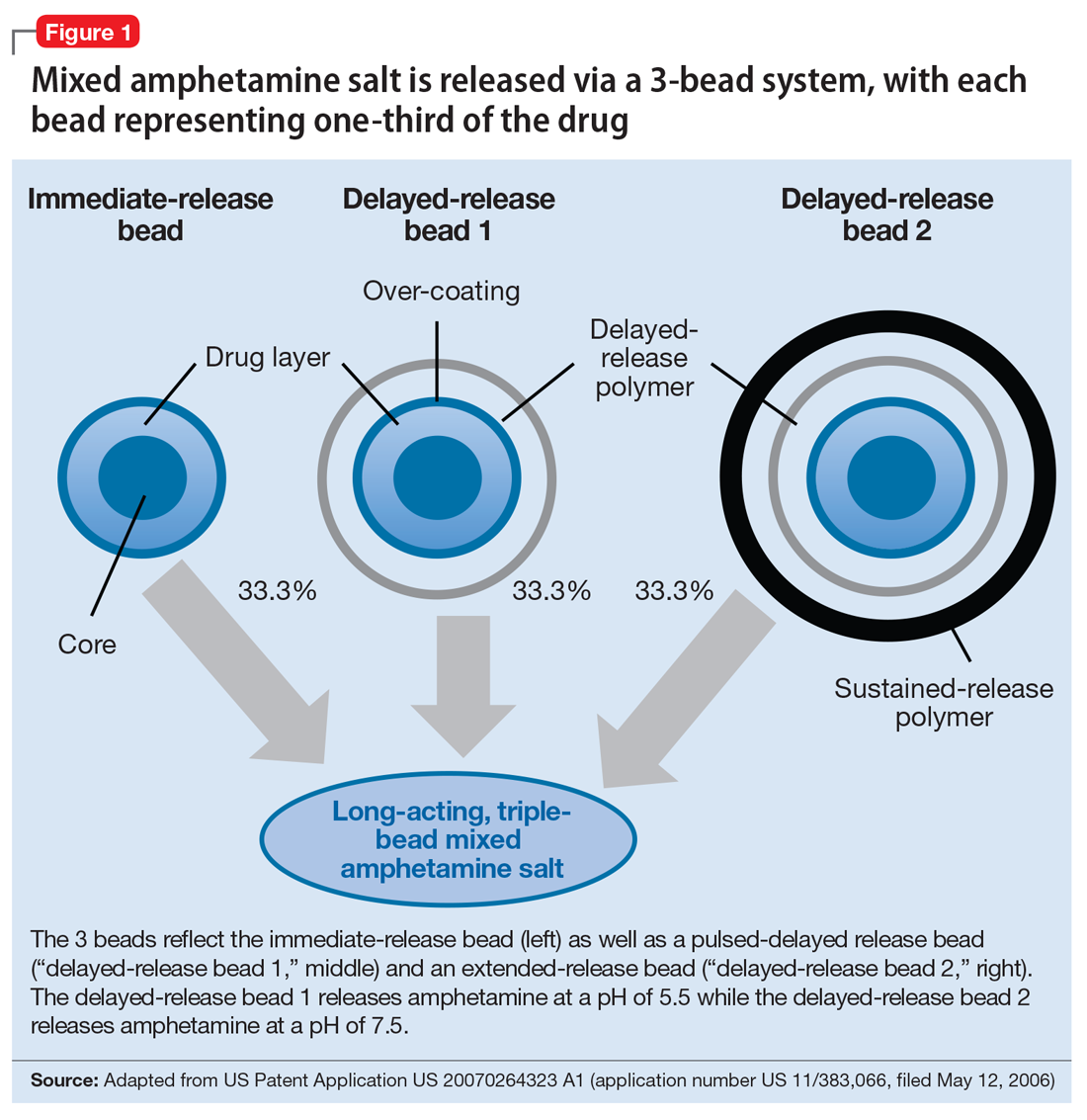
Mechanism of action
Like all MAS, this formulation blocks the reuptake of norepinephrine and dopamine, increasing synaptic concentrations of these monoamine neurotransmitters. Additionally, amphetamine salts may inhibit the activity of monoamine oxidase (MAO), further increasing synaptic levels of monoamines.4 Enhancing noradrenergic, dopaminergic neurotransmission, particularly within the prefrontal cortex, increases attention, working memory, and processing speed in patients with ADHD.4
Pharmacokinetics
Cmax occurs approximately 7 to 10 hours and 8 hours following administration in adolescent and adult patients, respectively (Figure 2).3 In adolescents who were administered a single dose of long-acting, triple-bead MAS, Cmax and area under the curve (AUC) for d- and l-amphetamine were both 21% to 31% higher compared with adults3 and did not appear to be affected by sex or race.3
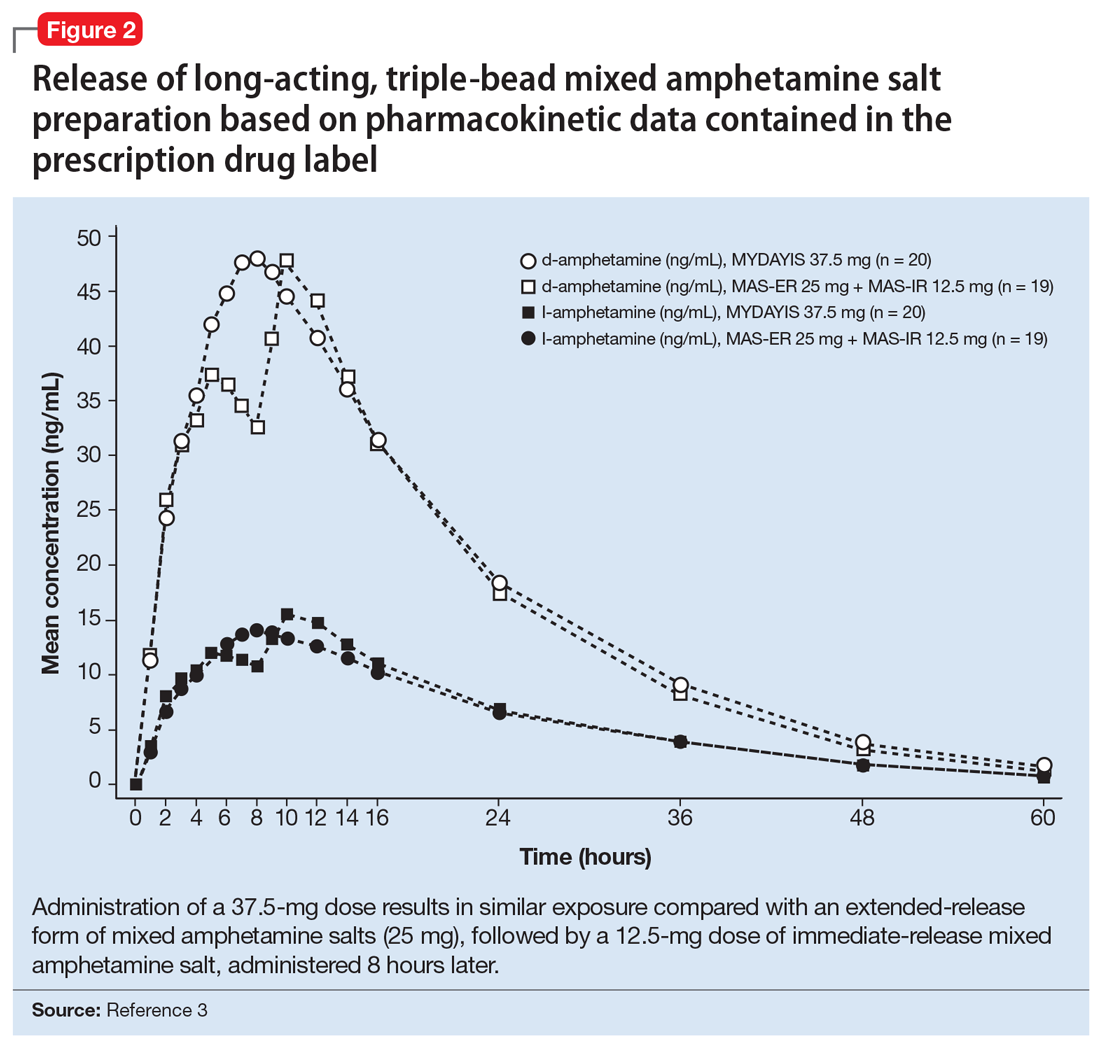
Half-life is 10 to 11 hours for d-amphetamine and 10 to 13 hours for l-amphetamine and does not statistically differ between pediatric and adult studies.3
Metabolism and elimination. Amphetamines are partially metabolized through cytochrome 450 (CYP) 2D6-dependent mechanisms, and thus in CYP2D6 poor metabolizers medication exposure may be increased, while decreased exposure may occur in ultra-rapid metabolizers; however, there are no guidelines from the Clinical Pharmacogenetics Implementation Consortium regarding alternate dosing strategies for patients based on CYP2D6 genotype or activity phenotype.5 Because amphetamines are renally excreted, dosages should be adjusted in patients with renal impairment.
Drug interactions. Medications that affect gastrointestinal and urinary pH may affect serum concentrations of amphetamine. Specifically, agents that increase gastric pH (eg, proton pump inhibitors) as well as urinary alkalinizing agents (eg, acetazolamide, some thiazide diuretics) increase serum amphetamine concentrations.3 Because amphetamine is a weak MAOI, there is a theoretical risk of serotonin syndrome when amphetamine-based preparations are used concurrently with SSRIs, TCAs, and MAOIs. However, the concurrent use of MAS and SSRIs generally is considered safe and common practice in patients with ADHD and co-occurring anxiety6,7 or depressive disorders.
Dosing
Long-acting, triple-bead MAS is available in 12.5-, 25-, 37.5-, and 50-mg capsules. The capsule may be opened and sprinkled in food for patients who cannot swallow capsules. Opening of the capsule results in similar absorption relative to oral administration of the intact capsule.3
In adults with ADHD, long-acting, triple-bead MAS should be initiated at 12.5 mg in the morning (Table 2). However, in some individuals, long-acting, triple-bead MAS may be initiated at 25 mg each morning. Titration should occur in 12.5-mg weekly increments to a maximum dosage of 50 mg/d.3

In adults with severe renal impairment (glomerular filtrate rate, 15 to 30 mL/min/1.73 m2), the recommended starting dose is 12.5 mg/d, with a maximum dosage of 25 mg/d.3
The efficacy of long-acting, triple-bead MAS in adults with ADHD was demonstrated in 3 studies involving adults ages 18 to 55, and the effectiveness of the medication, with regard to duration of action, was assessed using the Time-Sensitive ADHD Symptom Scale—a self-report scale that consists of items indexed by the ADHD Rating Scale-IV (ADHD-RS-IV) which assesses ADHD symptom severity. Doses up to 75 mg/d were studied; however, there were no significant effects. It should be noted that this maximum daily dose was not determined by any safety parameter.
Study 1 (dose-optimization, triple-bead MAS, n = 137; placebo, n = 135, dosing: 12.5 to 75 mg) and Study 2 (forced dose-titration study, triple-bead MAS, n = 308; placebo, n = 104, dosing: 25 mg, 50 mg, 75 mg) demonstrated efficacy of triple-bead MAS for treating ADHD in adults. Despite differences in study designs, statistically significant and similar clinically relevant improvement was observed with triple-bead MAS (vs placebo) on ADHD-RS-IV total scores in both Study 1 and Study 2.8 An additional study in adults ages 18 to 55 (N = 275) with ADHD (DSM-5 criteria) involved randomization to either 12.5 mg (fixed dose) or forced titration (12.5 to 37.5 mg) or placebo and, as with the first 2 studies, improvement in ADHD symptoms was observed in triple-bead MAS-treated patients relative to those who had received placebo. (See Reference 3 for a summary of the clinical trials of triple-bead MAS in adults with ADHD.)
The tolerability of this medication was evaluated in a 12-month open-label study of adults with ADHD (DSM-IV-TR criteria) in which discontinuation was higher at doses >25 mg/d.7 Treatment-related increases in blood pressure and heart rate were consistent with the known hemodynamic adverse effect profile of stimulants.9
In adolescents with ADHD ages 13 to 17, long-acting, triple-bead MAS should be initiated at 12.5 mg/d and may be increased to 25 mg/d (Table 2). Importantly, in younger patients, including those younger than age 12, triple-bead MAS was associated with an increased risk of adverse events including insomnia and anorexia, and this was thought to be related to increased drug exposure (ie, AUC).
The efficacy of long-acting, triple-bead MAS was evaluated in 2 studies of adolescents ages 13 to 17, including 1 fixed-dose trial (25 mg/d) and 1 flexibly-dosed trial (12.5 to 25 mg/d). These unpublished studies utilized the ADHD-RS-IV score and the Average Permanent Product Measure of Performance, an age-adjusted math test and measure of sustained attention, and revealed statistically significant differences between medication and placebo in the primary outcomes.3
Adverse effects
Long-acting, triple-bead MAS was developed to treat ADHD symptoms throughout the day, and serum concentrations of the medication may be higher with this formulation compared with other long-acting preparations. Therefore, adverse effects that are directly related to plasma exposure (eg, insomnia and appetite suppression) may occur at higher rates with this preparation compared with alternatives. For example, in some of the registration trials, insomnia occurred in more than one-third of patients receiving the active medication (38%).9 Although insomnia was the most frequently reported adverse event in adults with ADHD, most reports of insomnia occurred early in the course of treatment. Of these insomnia-related adverse events, 94% were mild to moderate and resulted in discontinuation of the medication in approximately 2% of patients. Further, 73.9% of treatment-emergent, insomnia–related adverse events resolved during the course of the study. It is also important to note that the Pittsburgh Sleep Quality Index did not differ from placebo in studies of triple-bead MAS in adults with ADHD.10 Similarly, rates of stimulant-induced appetite suppression may be higher with this preparation compared with other long-acting preparations.9
Adverse effects observed in adults with ADHD that occurred in ≥2% of patients receiving triple-bead MAS and at least twice the incidence in patients randomized to placebo included:
- anxiety (7% vs 3%)
- feeling jittery (2% vs 1%)
- agitation (2% vs 0%)
- insomnia (31% vs 8%)
- depression (3% vs 0%)
- decreased appetite (30% vs 4%)
- weight loss (9% vs 0%)
- xerostomia (23% vs 4%)
- diarrhea (3% vs 0%)
- increased heart rate (9% vs 0%)
- palpitations (4% vs 2%)
- dysmenorrhea (4% vs 2%)
- erectile dysfunction (2% vs 1%).
In adolescents receiving triple-bea
1. Punja S, Shamseer L, Hartling L, et al. Amphetamines for attention deficit hyperactivity disorder (ADHD) in children and adolescents. Cochrane Database Syst Rev. 2016;2016(2):CD009996.
2. Castells X, Ramos-Quiroga J, Bosch R, et al. Amphetamines for attention deficit hyperactivity disorder (ADHD) in adults. Cochrane Database Syst Rev. 2011;(6):CD007813.
3. Mydayis [package insert]. Lexington, MA: Shire; 2017.
4. Heal DJ, Smith SL, Gosden J, et al. Amphetamine, past and present—a pharmacological and clinical perspective. J Psychopharmacol. 2013;27(6):479-496.
5. Hoffman JM, Dunnenberger HM, Kevin Hicks J, et al. Developing knowledge resources to support precision medicine: principles from the Clinical Pharmacogenetics Implementation Consortium (CPIC). J Am Med Inform Assoc. 2016;23(4):766-801.
6. Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359(26):2753-2766.
7. Connolly SD, Bernstein GA; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
8. Goodman DW, Spencer TJ, Adler LA, et al. Clinical evaluation of triple-bead mixed amphetamine salts in adult ADHD. Presented at: 54th Annual Meeting of the American Academy of Child and Adolescent Psychiatry; October 25, 2007; Boston, MA.
9. Adler LA, Frick G, Yan B. A Long-term, open-label, safety study of triple-bead mixed amphetamine salts (SHP465) in adults with ADHD [published online April 1, 2017]. J Atten Disord. doi: 10.1177/1087054717696770.
10. Backhaus J, Junghanns K, Broocks A, et al. test-retest reliability and validity of the Pittsburgh Sleep Quality Index in primary insomnia. J Psychosom Res. 2002;53(3):737-740.
Stimulants are first-line psychopharmacologic interventions for attention-deficit/hyperactivity disorder (ADHD), and their efficacy is supported by clinical trials and meta-analyses in children and adolescents1 as well as adults.2 Despite decades of tolerability and efficacy data supporting their use, a major drawback of stimulants is that their salutary therapeutic effects wane once the medication is cleared or metabolized. Both mixed amphetamine- and methylphenidate-based preparations have short half-lives, necessitating multiple doses per day (eg, 3 or 4 times a day) when short-acting preparations are used. Over the past 15 years, nearly a dozen formulations were developed that extend the duration of action through delayed release, delayed absorption, or utilizing prodrugs.
The encapsulated preparation contains 3 MAS beads: an immediate-release amphetamine salt bead, a pulsed-delayed release bead, and an extended-release bead (Figure 1), which give rise to a unique pharmacokinetic profile (Figure 2).3
Mechanism of action
Like all MAS, this formulation blocks the reuptake of norepinephrine and dopamine, increasing synaptic concentrations of these monoamine neurotransmitters. Additionally, amphetamine salts may inhibit the activity of monoamine oxidase (MAO), further increasing synaptic levels of monoamines.4 Enhancing noradrenergic, dopaminergic neurotransmission, particularly within the prefrontal cortex, increases attention, working memory, and processing speed in patients with ADHD.4
Pharmacokinetics
Cmax occurs approximately 7 to 10 hours and 8 hours following administration in adolescent and adult patients, respectively (Figure 2).3 In adolescents who were administered a single dose of long-acting, triple-bead MAS, Cmax and area under the curve (AUC) for d- and l-amphetamine were both 21% to 31% higher compared with adults3 and did not appear to be affected by sex or race.3
Half-life is 10 to 11 hours for d-amphetamine and 10 to 13 hours for l-amphetamine and does not statistically differ between pediatric and adult studies.3
Metabolism and elimination. Amphetamines are partially metabolized through cytochrome 450 (CYP) 2D6-dependent mechanisms, and thus in CYP2D6 poor metabolizers medication exposure may be increased, while decreased exposure may occur in ultra-rapid metabolizers; however, there are no guidelines from the Clinical Pharmacogenetics Implementation Consortium regarding alternate dosing strategies for patients based on CYP2D6 genotype or activity phenotype.5 Because amphetamines are renally excreted, dosages should be adjusted in patients with renal impairment.
Drug interactions. Medications that affect gastrointestinal and urinary pH may affect serum concentrations of amphetamine. Specifically, agents that increase gastric pH (eg, proton pump inhibitors) as well as urinary alkalinizing agents (eg, acetazolamide, some thiazide diuretics) increase serum amphetamine concentrations.3 Because amphetamine is a weak MAOI, there is a theoretical risk of serotonin syndrome when amphetamine-based preparations are used concurrently with SSRIs, TCAs, and MAOIs. However, the concurrent use of MAS and SSRIs generally is considered safe and common practice in patients with ADHD and co-occurring anxiety6,7 or depressive disorders.
Dosing
Long-acting, triple-bead MAS is available in 12.5-, 25-, 37.5-, and 50-mg capsules. The capsule may be opened and sprinkled in food for patients who cannot swallow capsules. Opening of the capsule results in similar absorption relative to oral administration of the intact capsule.3
In adults with ADHD, long-acting, triple-bead MAS should be initiated at 12.5 mg in the morning (Table 2). However, in some individuals, long-acting, triple-bead MAS may be initiated at 25 mg each morning. Titration should occur in 12.5-mg weekly increments to a maximum dosage of 50 mg/d.3
In adults with severe renal impairment (glomerular filtrate rate, 15 to 30 mL/min/1.73 m2), the recommended starting dose is 12.5 mg/d, with a maximum dosage of 25 mg/d.3
The efficacy of long-acting, triple-bead MAS in adults with ADHD was demonstrated in 3 studies involving adults ages 18 to 55, and the effectiveness of the medication, with regard to duration of action, was assessed using the Time-Sensitive ADHD Symptom Scale—a self-report scale that consists of items indexed by the ADHD Rating Scale-IV (ADHD-RS-IV) which assesses ADHD symptom severity. Doses up to 75 mg/d were studied; however, there were no significant effects. It should be noted that this maximum daily dose was not determined by any safety parameter.
Study 1 (dose-optimization, triple-bead MAS, n = 137; placebo, n = 135, dosing: 12.5 to 75 mg) and Study 2 (forced dose-titration study, triple-bead MAS, n = 308; placebo, n = 104, dosing: 25 mg, 50 mg, 75 mg) demonstrated efficacy of triple-bead MAS for treating ADHD in adults. Despite differences in study designs, statistically significant and similar clinically relevant improvement was observed with triple-bead MAS (vs placebo) on ADHD-RS-IV total scores in both Study 1 and Study 2.8 An additional study in adults ages 18 to 55 (N = 275) with ADHD (DSM-5 criteria) involved randomization to either 12.5 mg (fixed dose) or forced titration (12.5 to 37.5 mg) or placebo and, as with the first 2 studies, improvement in ADHD symptoms was observed in triple-bead MAS-treated patients relative to those who had received placebo. (See Reference 3 for a summary of the clinical trials of triple-bead MAS in adults with ADHD.)
The tolerability of this medication was evaluated in a 12-month open-label study of adults with ADHD (DSM-IV-TR criteria) in which discontinuation was higher at doses >25 mg/d.7 Treatment-related increases in blood pressure and heart rate were consistent with the known hemodynamic adverse effect profile of stimulants.9
In adolescents with ADHD ages 13 to 17, long-acting, triple-bead MAS should be initiated at 12.5 mg/d and may be increased to 25 mg/d (Table 2). Importantly, in younger patients, including those younger than age 12, triple-bead MAS was associated with an increased risk of adverse events including insomnia and anorexia, and this was thought to be related to increased drug exposure (ie, AUC).
The efficacy of long-acting, triple-bead MAS was evaluated in 2 studies of adolescents ages 13 to 17, including 1 fixed-dose trial (25 mg/d) and 1 flexibly-dosed trial (12.5 to 25 mg/d). These unpublished studies utilized the ADHD-RS-IV score and the Average Permanent Product Measure of Performance, an age-adjusted math test and measure of sustained attention, and revealed statistically significant differences between medication and placebo in the primary outcomes.3
Adverse effects
Long-acting, triple-bead MAS was developed to treat ADHD symptoms throughout the day, and serum concentrations of the medication may be higher with this formulation compared with other long-acting preparations. Therefore, adverse effects that are directly related to plasma exposure (eg, insomnia and appetite suppression) may occur at higher rates with this preparation compared with alternatives. For example, in some of the registration trials, insomnia occurred in more than one-third of patients receiving the active medication (38%).9 Although insomnia was the most frequently reported adverse event in adults with ADHD, most reports of insomnia occurred early in the course of treatment. Of these insomnia-related adverse events, 94% were mild to moderate and resulted in discontinuation of the medication in approximately 2% of patients. Further, 73.9% of treatment-emergent, insomnia–related adverse events resolved during the course of the study. It is also important to note that the Pittsburgh Sleep Quality Index did not differ from placebo in studies of triple-bead MAS in adults with ADHD.10 Similarly, rates of stimulant-induced appetite suppression may be higher with this preparation compared with other long-acting preparations.9
Adverse effects observed in adults with ADHD that occurred in ≥2% of patients receiving triple-bead MAS and at least twice the incidence in patients randomized to placebo included:
- anxiety (7% vs 3%)
- feeling jittery (2% vs 1%)
- agitation (2% vs 0%)
- insomnia (31% vs 8%)
- depression (3% vs 0%)
- decreased appetite (30% vs 4%)
- weight loss (9% vs 0%)
- xerostomia (23% vs 4%)
- diarrhea (3% vs 0%)
- increased heart rate (9% vs 0%)
- palpitations (4% vs 2%)
- dysmenorrhea (4% vs 2%)
- erectile dysfunction (2% vs 1%).
In adolescents receiving triple-bea
Stimulants are first-line psychopharmacologic interventions for attention-deficit/hyperactivity disorder (ADHD), and their efficacy is supported by clinical trials and meta-analyses in children and adolescents1 as well as adults.2 Despite decades of tolerability and efficacy data supporting their use, a major drawback of stimulants is that their salutary therapeutic effects wane once the medication is cleared or metabolized. Both mixed amphetamine- and methylphenidate-based preparations have short half-lives, necessitating multiple doses per day (eg, 3 or 4 times a day) when short-acting preparations are used. Over the past 15 years, nearly a dozen formulations were developed that extend the duration of action through delayed release, delayed absorption, or utilizing prodrugs.
The encapsulated preparation contains 3 MAS beads: an immediate-release amphetamine salt bead, a pulsed-delayed release bead, and an extended-release bead (Figure 1), which give rise to a unique pharmacokinetic profile (Figure 2).3
Mechanism of action
Like all MAS, this formulation blocks the reuptake of norepinephrine and dopamine, increasing synaptic concentrations of these monoamine neurotransmitters. Additionally, amphetamine salts may inhibit the activity of monoamine oxidase (MAO), further increasing synaptic levels of monoamines.4 Enhancing noradrenergic, dopaminergic neurotransmission, particularly within the prefrontal cortex, increases attention, working memory, and processing speed in patients with ADHD.4
Pharmacokinetics
Cmax occurs approximately 7 to 10 hours and 8 hours following administration in adolescent and adult patients, respectively (Figure 2).3 In adolescents who were administered a single dose of long-acting, triple-bead MAS, Cmax and area under the curve (AUC) for d- and l-amphetamine were both 21% to 31% higher compared with adults3 and did not appear to be affected by sex or race.3
Half-life is 10 to 11 hours for d-amphetamine and 10 to 13 hours for l-amphetamine and does not statistically differ between pediatric and adult studies.3
Metabolism and elimination. Amphetamines are partially metabolized through cytochrome 450 (CYP) 2D6-dependent mechanisms, and thus in CYP2D6 poor metabolizers medication exposure may be increased, while decreased exposure may occur in ultra-rapid metabolizers; however, there are no guidelines from the Clinical Pharmacogenetics Implementation Consortium regarding alternate dosing strategies for patients based on CYP2D6 genotype or activity phenotype.5 Because amphetamines are renally excreted, dosages should be adjusted in patients with renal impairment.
Drug interactions. Medications that affect gastrointestinal and urinary pH may affect serum concentrations of amphetamine. Specifically, agents that increase gastric pH (eg, proton pump inhibitors) as well as urinary alkalinizing agents (eg, acetazolamide, some thiazide diuretics) increase serum amphetamine concentrations.3 Because amphetamine is a weak MAOI, there is a theoretical risk of serotonin syndrome when amphetamine-based preparations are used concurrently with SSRIs, TCAs, and MAOIs. However, the concurrent use of MAS and SSRIs generally is considered safe and common practice in patients with ADHD and co-occurring anxiety6,7 or depressive disorders.
Dosing
Long-acting, triple-bead MAS is available in 12.5-, 25-, 37.5-, and 50-mg capsules. The capsule may be opened and sprinkled in food for patients who cannot swallow capsules. Opening of the capsule results in similar absorption relative to oral administration of the intact capsule.3
In adults with ADHD, long-acting, triple-bead MAS should be initiated at 12.5 mg in the morning (Table 2). However, in some individuals, long-acting, triple-bead MAS may be initiated at 25 mg each morning. Titration should occur in 12.5-mg weekly increments to a maximum dosage of 50 mg/d.3
In adults with severe renal impairment (glomerular filtrate rate, 15 to 30 mL/min/1.73 m2), the recommended starting dose is 12.5 mg/d, with a maximum dosage of 25 mg/d.3
The efficacy of long-acting, triple-bead MAS in adults with ADHD was demonstrated in 3 studies involving adults ages 18 to 55, and the effectiveness of the medication, with regard to duration of action, was assessed using the Time-Sensitive ADHD Symptom Scale—a self-report scale that consists of items indexed by the ADHD Rating Scale-IV (ADHD-RS-IV) which assesses ADHD symptom severity. Doses up to 75 mg/d were studied; however, there were no significant effects. It should be noted that this maximum daily dose was not determined by any safety parameter.
Study 1 (dose-optimization, triple-bead MAS, n = 137; placebo, n = 135, dosing: 12.5 to 75 mg) and Study 2 (forced dose-titration study, triple-bead MAS, n = 308; placebo, n = 104, dosing: 25 mg, 50 mg, 75 mg) demonstrated efficacy of triple-bead MAS for treating ADHD in adults. Despite differences in study designs, statistically significant and similar clinically relevant improvement was observed with triple-bead MAS (vs placebo) on ADHD-RS-IV total scores in both Study 1 and Study 2.8 An additional study in adults ages 18 to 55 (N = 275) with ADHD (DSM-5 criteria) involved randomization to either 12.5 mg (fixed dose) or forced titration (12.5 to 37.5 mg) or placebo and, as with the first 2 studies, improvement in ADHD symptoms was observed in triple-bead MAS-treated patients relative to those who had received placebo. (See Reference 3 for a summary of the clinical trials of triple-bead MAS in adults with ADHD.)
The tolerability of this medication was evaluated in a 12-month open-label study of adults with ADHD (DSM-IV-TR criteria) in which discontinuation was higher at doses >25 mg/d.7 Treatment-related increases in blood pressure and heart rate were consistent with the known hemodynamic adverse effect profile of stimulants.9
In adolescents with ADHD ages 13 to 17, long-acting, triple-bead MAS should be initiated at 12.5 mg/d and may be increased to 25 mg/d (Table 2). Importantly, in younger patients, including those younger than age 12, triple-bead MAS was associated with an increased risk of adverse events including insomnia and anorexia, and this was thought to be related to increased drug exposure (ie, AUC).
The efficacy of long-acting, triple-bead MAS was evaluated in 2 studies of adolescents ages 13 to 17, including 1 fixed-dose trial (25 mg/d) and 1 flexibly-dosed trial (12.5 to 25 mg/d). These unpublished studies utilized the ADHD-RS-IV score and the Average Permanent Product Measure of Performance, an age-adjusted math test and measure of sustained attention, and revealed statistically significant differences between medication and placebo in the primary outcomes.3
Adverse effects
Long-acting, triple-bead MAS was developed to treat ADHD symptoms throughout the day, and serum concentrations of the medication may be higher with this formulation compared with other long-acting preparations. Therefore, adverse effects that are directly related to plasma exposure (eg, insomnia and appetite suppression) may occur at higher rates with this preparation compared with alternatives. For example, in some of the registration trials, insomnia occurred in more than one-third of patients receiving the active medication (38%).9 Although insomnia was the most frequently reported adverse event in adults with ADHD, most reports of insomnia occurred early in the course of treatment. Of these insomnia-related adverse events, 94% were mild to moderate and resulted in discontinuation of the medication in approximately 2% of patients. Further, 73.9% of treatment-emergent, insomnia–related adverse events resolved during the course of the study. It is also important to note that the Pittsburgh Sleep Quality Index did not differ from placebo in studies of triple-bead MAS in adults with ADHD.10 Similarly, rates of stimulant-induced appetite suppression may be higher with this preparation compared with other long-acting preparations.9
Adverse effects observed in adults with ADHD that occurred in ≥2% of patients receiving triple-bead MAS and at least twice the incidence in patients randomized to placebo included:
- anxiety (7% vs 3%)
- feeling jittery (2% vs 1%)
- agitation (2% vs 0%)
- insomnia (31% vs 8%)
- depression (3% vs 0%)
- decreased appetite (30% vs 4%)
- weight loss (9% vs 0%)
- xerostomia (23% vs 4%)
- diarrhea (3% vs 0%)
- increased heart rate (9% vs 0%)
- palpitations (4% vs 2%)
- dysmenorrhea (4% vs 2%)
- erectile dysfunction (2% vs 1%).
In adolescents receiving triple-bea
1. Punja S, Shamseer L, Hartling L, et al. Amphetamines for attention deficit hyperactivity disorder (ADHD) in children and adolescents. Cochrane Database Syst Rev. 2016;2016(2):CD009996.
2. Castells X, Ramos-Quiroga J, Bosch R, et al. Amphetamines for attention deficit hyperactivity disorder (ADHD) in adults. Cochrane Database Syst Rev. 2011;(6):CD007813.
3. Mydayis [package insert]. Lexington, MA: Shire; 2017.
4. Heal DJ, Smith SL, Gosden J, et al. Amphetamine, past and present—a pharmacological and clinical perspective. J Psychopharmacol. 2013;27(6):479-496.
5. Hoffman JM, Dunnenberger HM, Kevin Hicks J, et al. Developing knowledge resources to support precision medicine: principles from the Clinical Pharmacogenetics Implementation Consortium (CPIC). J Am Med Inform Assoc. 2016;23(4):766-801.
6. Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359(26):2753-2766.
7. Connolly SD, Bernstein GA; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
8. Goodman DW, Spencer TJ, Adler LA, et al. Clinical evaluation of triple-bead mixed amphetamine salts in adult ADHD. Presented at: 54th Annual Meeting of the American Academy of Child and Adolescent Psychiatry; October 25, 2007; Boston, MA.
9. Adler LA, Frick G, Yan B. A Long-term, open-label, safety study of triple-bead mixed amphetamine salts (SHP465) in adults with ADHD [published online April 1, 2017]. J Atten Disord. doi: 10.1177/1087054717696770.
10. Backhaus J, Junghanns K, Broocks A, et al. test-retest reliability and validity of the Pittsburgh Sleep Quality Index in primary insomnia. J Psychosom Res. 2002;53(3):737-740.
1. Punja S, Shamseer L, Hartling L, et al. Amphetamines for attention deficit hyperactivity disorder (ADHD) in children and adolescents. Cochrane Database Syst Rev. 2016;2016(2):CD009996.
2. Castells X, Ramos-Quiroga J, Bosch R, et al. Amphetamines for attention deficit hyperactivity disorder (ADHD) in adults. Cochrane Database Syst Rev. 2011;(6):CD007813.
3. Mydayis [package insert]. Lexington, MA: Shire; 2017.
4. Heal DJ, Smith SL, Gosden J, et al. Amphetamine, past and present—a pharmacological and clinical perspective. J Psychopharmacol. 2013;27(6):479-496.
5. Hoffman JM, Dunnenberger HM, Kevin Hicks J, et al. Developing knowledge resources to support precision medicine: principles from the Clinical Pharmacogenetics Implementation Consortium (CPIC). J Am Med Inform Assoc. 2016;23(4):766-801.
6. Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359(26):2753-2766.
7. Connolly SD, Bernstein GA; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
8. Goodman DW, Spencer TJ, Adler LA, et al. Clinical evaluation of triple-bead mixed amphetamine salts in adult ADHD. Presented at: 54th Annual Meeting of the American Academy of Child and Adolescent Psychiatry; October 25, 2007; Boston, MA.
9. Adler LA, Frick G, Yan B. A Long-term, open-label, safety study of triple-bead mixed amphetamine salts (SHP465) in adults with ADHD [published online April 1, 2017]. J Atten Disord. doi: 10.1177/1087054717696770.
10. Backhaus J, Junghanns K, Broocks A, et al. test-retest reliability and validity of the Pittsburgh Sleep Quality Index in primary insomnia. J Psychosom Res. 2002;53(3):737-740.
Hunting for ‘Woozles’ in the Hundred Acre Wood of ADHD
One fine winter’s day when Piglet was brushing away the snow
in front of his house he happened to look up, and there was
Winnie-the-Pooh. Pooh was walking round and round in a circle,
thinking of something else…
So begins the 1926 Winnie-the-Pooh story.1 In this chapter, the well-meaning yellow bear, Winnie-the-Pooh, has found strange tracks in the snow, which he believes belong to a “Woozle.” Pooh follows the tracks, not realizing that he’s walking in a circle. As such, he begins to notice that the tracks have multiplied, which he interprets as evidence of several Woozles.
This “Woozle Effect” has been well described in research settings and is believed to have resulted in conclusions that are not supported by or are inconsistent with the original data, which are then propagated through successive citations, resulting in a scientific “urban legend.”2
Throughout my training from medical school, through fellowship, and during my tenure as a faculty member, I have found myself, at times, searching for Woozles and often have joined my colleagues on these hunts. Herein, I would like to share with you 3 Woozles that have resulted in current false dogmas related to attention-deficit/hyperactivity disorder (ADHD) and stimulant psychopharmacology.
Stimulants worsen anxiety
FDA-required labeling for stimulants includes strong language noting that these drugs are “contraindicated in marked anxiety, tension, and agitation, since the drug may aggravate these symptoms.”3 However, data from randomized controlled trials and meta-analyses consistently have failed to demonstrate this effect. Moreover, sequenced treatment trials involving adolescents with anxiety disorders and co-occurring ADHD suggest that stimulants actually could reduce anxiety symptoms.
A recent meta-analysis4 that evaluated nearly 2 dozen studies involving approximately 3,000 pediatric patients with ADHD reported that stimulant treatment was associated with a decreased relative risk of anxiety (relative risk: 0.86). The study also observed a dose-response relationship between stimulant dosage and anxiety (Figure, page 6).4 Although the authors note that it is possible that some individuals might experience increased anxiety with stimulants, many patients could show improvement in anxiety symptoms when treated with stimulants, and the authors also advise us, as clinicians, to “consider re-challenging children with ADHD who report … anxiety with psychostimulants, as these symptoms are much more likely to be coincidental rather than caused by psychostimulants.”4
More evidence of a lack of stimulant-induced anxiety comes from a large randomized controlled trial of pediatric patients (age 6 to 17) who met DSM-IV criteria for ADHD and a co-occurring anxiety disorder who were treated with methylphenidate (open-label) and then randomized to fluvoxamine or placebo for treatment of anxiety symptoms.5 However, in this trial >80% of the 32 medication-naïve youth improved after stimulant treatment to the point that they no longer had anxiety symptoms severe enough to be eligible for randomization to adjunctive fluvoxamine or placebo.
Stimulants are contraindicated in patients with tic disorders
The package inserts for most stimulant medications warn clinicians that stimulants are “contraindicated in patients with motor tics or with a family history or diagnosis of Tourette’s syndrome.” This is particularly concerning, especially because of the medicolegal implications of the term “contraindicated” and given that as many as 1 in 5 pediatric patients with ADHD have a tic disorder.6 Therefore, labels that list motor tics as a contraindication to stimulant use potentially eliminate the choice of stimulant pharmacotherapy—the most effective treatment for ADHD—for a large number of patients.
When hunting for the Woozle that linked stimulants and tics and led to this language in the package insert, it is worthwhile to review a recent meta-analysis of 22 studies (involving nearly 2,400 youths with ADHD) that suggested new-onset tics or worsening of tics to be present in 5.7% of patients receiving stimulants and in 6.5% of patients receiving placebo. In addition, in this meta-analysis the class of stimulant, dosage, treatment duration, or patient age did not seem to be associated with onset or worsening of tics.7
Polypharmacy represents a therapeutic failure and is not evidence-based
Although treatment guidelines generally have discouraged combination therapy for treating ADHD, there are—on the basis of efficacy—insufficient data to support this prohibition. Moreover, over the last decade, several studies have suggested benefits for combining ADHD medications that have complimentary mechanisms. In this regard, 2 extended-release formulations of α2 agonists have received FDA approval for as adjunctive treatments in pediatric patients with ADHD (extended-release guanfacine and extended-release clonidine). However, despite these FDA indications as adjunctive treatments, many clinicians remain concerned about combination therapy.
Several months ago, a large, 8-week, National Institutes of Health–sponsored trial shed more light on the use of α2agonist + stimulant combinations. Patients age 7 to 17 (N = 179) were randomized to (1) guanfacine + d-methylphenidate, (2) guanfacine monotherapy, or (3) d-methylphenidate monotherapy.8 In addition to clinical outcomes, the authors evaluated the effects of the medication on background cortical activity. Of interest, monotherapies differed between one another and the combination treatment in their effects on cortical activity. Guanfacine decreased alpha band power and methylphenidate administration was associated with an increase in frontal/central beta power, while combination treatment dampened theta band power and was associated with specific, focal increases in beta power.8 These results, although preliminary, suggest not only that medication results in changes in cortical activity that correlate with symptomatic improvement, but that combination treatment may be associated with a distinct cortical activity pattern that is more than the summation of the effects of the monotherapies. Moreover, these data raise the possibility that this synergistic effect on cortical activity may subtend—or at least—relate to the synergistic clinical effects of the 2 medications.
‘Think it over, think it under’
Having discussed several important Woozles that have inhabited the Hundred Acre Wood of ADHD for decades, it is important to remember there are countless Woozles in the larger “Thousand Acre Wood” of psychiatry and medicine. As we evaluate evidence for our interventions, whether psychopharmacologic or psychotherapeutic, we will do w
1. Milne AA. Winnie-the-Pooh. London, United Kingdom: Methuen & Co. Ltd.; 1926.
2. Strauss MA. Processes Explaining the concealment and distortion of evidence on gender symmetry in partner violence. Eur J Crim Pol Res. 1980;74:227-232.
3. Ritalin LA [package insert]. East Hanover, NJ: Novartis; 2015.
4. Coughlin CG, Cohen SC, Mulqueen JM, et al. Meta-analysis: reduced risk of anxiety with psychostimulant treatment in children with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2015;25(8):611-617.
5. Abikoff H, McGough J, Vitiello B, et al; RUPP ADHD/Anxiety Study Group. Sequential pharmacotherapy for children with comorbid attention-deficit/hyperactivity and anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2005;44(5):418-427.
6. Bloch MH, Panza KE, Landeros-Weisenberger A, et al. Meta-analysis: treatment of attention-deficit/hyperactivity disorder in children with comorbid tic disorders. J Am Acad Child Adolesc Psychiatry. 2009;48(9):884-893.
7. Cohen SC, Mulqueen JM, Ferracioli-Oda E, et al. Meta-analysis: risk of tics associated with psychostimulant use in randomized, placebo-controlled trials. J Am Acad Child Adolesc Psychiatry. 2015;54(9):728-736.
8. Loo SK, Bilder RM, Cho AL, et al. Effects of d-methylphenidate, guanfacine, and their combination on electroencephalogram resting state spectral power in attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2016;55(8):674-682.e1.
One fine winter’s day when Piglet was brushing away the snow
in front of his house he happened to look up, and there was
Winnie-the-Pooh. Pooh was walking round and round in a circle,
thinking of something else…
So begins the 1926 Winnie-the-Pooh story.1 In this chapter, the well-meaning yellow bear, Winnie-the-Pooh, has found strange tracks in the snow, which he believes belong to a “Woozle.” Pooh follows the tracks, not realizing that he’s walking in a circle. As such, he begins to notice that the tracks have multiplied, which he interprets as evidence of several Woozles.
This “Woozle Effect” has been well described in research settings and is believed to have resulted in conclusions that are not supported by or are inconsistent with the original data, which are then propagated through successive citations, resulting in a scientific “urban legend.”2
Throughout my training from medical school, through fellowship, and during my tenure as a faculty member, I have found myself, at times, searching for Woozles and often have joined my colleagues on these hunts. Herein, I would like to share with you 3 Woozles that have resulted in current false dogmas related to attention-deficit/hyperactivity disorder (ADHD) and stimulant psychopharmacology.
Stimulants worsen anxiety
FDA-required labeling for stimulants includes strong language noting that these drugs are “contraindicated in marked anxiety, tension, and agitation, since the drug may aggravate these symptoms.”3 However, data from randomized controlled trials and meta-analyses consistently have failed to demonstrate this effect. Moreover, sequenced treatment trials involving adolescents with anxiety disorders and co-occurring ADHD suggest that stimulants actually could reduce anxiety symptoms.
A recent meta-analysis4 that evaluated nearly 2 dozen studies involving approximately 3,000 pediatric patients with ADHD reported that stimulant treatment was associated with a decreased relative risk of anxiety (relative risk: 0.86). The study also observed a dose-response relationship between stimulant dosage and anxiety (Figure, page 6).4 Although the authors note that it is possible that some individuals might experience increased anxiety with stimulants, many patients could show improvement in anxiety symptoms when treated with stimulants, and the authors also advise us, as clinicians, to “consider re-challenging children with ADHD who report … anxiety with psychostimulants, as these symptoms are much more likely to be coincidental rather than caused by psychostimulants.”4
More evidence of a lack of stimulant-induced anxiety comes from a large randomized controlled trial of pediatric patients (age 6 to 17) who met DSM-IV criteria for ADHD and a co-occurring anxiety disorder who were treated with methylphenidate (open-label) and then randomized to fluvoxamine or placebo for treatment of anxiety symptoms.5 However, in this trial >80% of the 32 medication-naïve youth improved after stimulant treatment to the point that they no longer had anxiety symptoms severe enough to be eligible for randomization to adjunctive fluvoxamine or placebo.
Stimulants are contraindicated in patients with tic disorders
The package inserts for most stimulant medications warn clinicians that stimulants are “contraindicated in patients with motor tics or with a family history or diagnosis of Tourette’s syndrome.” This is particularly concerning, especially because of the medicolegal implications of the term “contraindicated” and given that as many as 1 in 5 pediatric patients with ADHD have a tic disorder.6 Therefore, labels that list motor tics as a contraindication to stimulant use potentially eliminate the choice of stimulant pharmacotherapy—the most effective treatment for ADHD—for a large number of patients.
When hunting for the Woozle that linked stimulants and tics and led to this language in the package insert, it is worthwhile to review a recent meta-analysis of 22 studies (involving nearly 2,400 youths with ADHD) that suggested new-onset tics or worsening of tics to be present in 5.7% of patients receiving stimulants and in 6.5% of patients receiving placebo. In addition, in this meta-analysis the class of stimulant, dosage, treatment duration, or patient age did not seem to be associated with onset or worsening of tics.7
Polypharmacy represents a therapeutic failure and is not evidence-based
Although treatment guidelines generally have discouraged combination therapy for treating ADHD, there are—on the basis of efficacy—insufficient data to support this prohibition. Moreover, over the last decade, several studies have suggested benefits for combining ADHD medications that have complimentary mechanisms. In this regard, 2 extended-release formulations of α2 agonists have received FDA approval for as adjunctive treatments in pediatric patients with ADHD (extended-release guanfacine and extended-release clonidine). However, despite these FDA indications as adjunctive treatments, many clinicians remain concerned about combination therapy.
Several months ago, a large, 8-week, National Institutes of Health–sponsored trial shed more light on the use of α2agonist + stimulant combinations. Patients age 7 to 17 (N = 179) were randomized to (1) guanfacine + d-methylphenidate, (2) guanfacine monotherapy, or (3) d-methylphenidate monotherapy.8 In addition to clinical outcomes, the authors evaluated the effects of the medication on background cortical activity. Of interest, monotherapies differed between one another and the combination treatment in their effects on cortical activity. Guanfacine decreased alpha band power and methylphenidate administration was associated with an increase in frontal/central beta power, while combination treatment dampened theta band power and was associated with specific, focal increases in beta power.8 These results, although preliminary, suggest not only that medication results in changes in cortical activity that correlate with symptomatic improvement, but that combination treatment may be associated with a distinct cortical activity pattern that is more than the summation of the effects of the monotherapies. Moreover, these data raise the possibility that this synergistic effect on cortical activity may subtend—or at least—relate to the synergistic clinical effects of the 2 medications.
‘Think it over, think it under’
Having discussed several important Woozles that have inhabited the Hundred Acre Wood of ADHD for decades, it is important to remember there are countless Woozles in the larger “Thousand Acre Wood” of psychiatry and medicine. As we evaluate evidence for our interventions, whether psychopharmacologic or psychotherapeutic, we will do w
One fine winter’s day when Piglet was brushing away the snow
in front of his house he happened to look up, and there was
Winnie-the-Pooh. Pooh was walking round and round in a circle,
thinking of something else…
So begins the 1926 Winnie-the-Pooh story.1 In this chapter, the well-meaning yellow bear, Winnie-the-Pooh, has found strange tracks in the snow, which he believes belong to a “Woozle.” Pooh follows the tracks, not realizing that he’s walking in a circle. As such, he begins to notice that the tracks have multiplied, which he interprets as evidence of several Woozles.
This “Woozle Effect” has been well described in research settings and is believed to have resulted in conclusions that are not supported by or are inconsistent with the original data, which are then propagated through successive citations, resulting in a scientific “urban legend.”2
Throughout my training from medical school, through fellowship, and during my tenure as a faculty member, I have found myself, at times, searching for Woozles and often have joined my colleagues on these hunts. Herein, I would like to share with you 3 Woozles that have resulted in current false dogmas related to attention-deficit/hyperactivity disorder (ADHD) and stimulant psychopharmacology.
Stimulants worsen anxiety
FDA-required labeling for stimulants includes strong language noting that these drugs are “contraindicated in marked anxiety, tension, and agitation, since the drug may aggravate these symptoms.”3 However, data from randomized controlled trials and meta-analyses consistently have failed to demonstrate this effect. Moreover, sequenced treatment trials involving adolescents with anxiety disorders and co-occurring ADHD suggest that stimulants actually could reduce anxiety symptoms.
A recent meta-analysis4 that evaluated nearly 2 dozen studies involving approximately 3,000 pediatric patients with ADHD reported that stimulant treatment was associated with a decreased relative risk of anxiety (relative risk: 0.86). The study also observed a dose-response relationship between stimulant dosage and anxiety (Figure, page 6).4 Although the authors note that it is possible that some individuals might experience increased anxiety with stimulants, many patients could show improvement in anxiety symptoms when treated with stimulants, and the authors also advise us, as clinicians, to “consider re-challenging children with ADHD who report … anxiety with psychostimulants, as these symptoms are much more likely to be coincidental rather than caused by psychostimulants.”4
More evidence of a lack of stimulant-induced anxiety comes from a large randomized controlled trial of pediatric patients (age 6 to 17) who met DSM-IV criteria for ADHD and a co-occurring anxiety disorder who were treated with methylphenidate (open-label) and then randomized to fluvoxamine or placebo for treatment of anxiety symptoms.5 However, in this trial >80% of the 32 medication-naïve youth improved after stimulant treatment to the point that they no longer had anxiety symptoms severe enough to be eligible for randomization to adjunctive fluvoxamine or placebo.
Stimulants are contraindicated in patients with tic disorders
The package inserts for most stimulant medications warn clinicians that stimulants are “contraindicated in patients with motor tics or with a family history or diagnosis of Tourette’s syndrome.” This is particularly concerning, especially because of the medicolegal implications of the term “contraindicated” and given that as many as 1 in 5 pediatric patients with ADHD have a tic disorder.6 Therefore, labels that list motor tics as a contraindication to stimulant use potentially eliminate the choice of stimulant pharmacotherapy—the most effective treatment for ADHD—for a large number of patients.
When hunting for the Woozle that linked stimulants and tics and led to this language in the package insert, it is worthwhile to review a recent meta-analysis of 22 studies (involving nearly 2,400 youths with ADHD) that suggested new-onset tics or worsening of tics to be present in 5.7% of patients receiving stimulants and in 6.5% of patients receiving placebo. In addition, in this meta-analysis the class of stimulant, dosage, treatment duration, or patient age did not seem to be associated with onset or worsening of tics.7
Polypharmacy represents a therapeutic failure and is not evidence-based
Although treatment guidelines generally have discouraged combination therapy for treating ADHD, there are—on the basis of efficacy—insufficient data to support this prohibition. Moreover, over the last decade, several studies have suggested benefits for combining ADHD medications that have complimentary mechanisms. In this regard, 2 extended-release formulations of α2 agonists have received FDA approval for as adjunctive treatments in pediatric patients with ADHD (extended-release guanfacine and extended-release clonidine). However, despite these FDA indications as adjunctive treatments, many clinicians remain concerned about combination therapy.
Several months ago, a large, 8-week, National Institutes of Health–sponsored trial shed more light on the use of α2agonist + stimulant combinations. Patients age 7 to 17 (N = 179) were randomized to (1) guanfacine + d-methylphenidate, (2) guanfacine monotherapy, or (3) d-methylphenidate monotherapy.8 In addition to clinical outcomes, the authors evaluated the effects of the medication on background cortical activity. Of interest, monotherapies differed between one another and the combination treatment in their effects on cortical activity. Guanfacine decreased alpha band power and methylphenidate administration was associated with an increase in frontal/central beta power, while combination treatment dampened theta band power and was associated with specific, focal increases in beta power.8 These results, although preliminary, suggest not only that medication results in changes in cortical activity that correlate with symptomatic improvement, but that combination treatment may be associated with a distinct cortical activity pattern that is more than the summation of the effects of the monotherapies. Moreover, these data raise the possibility that this synergistic effect on cortical activity may subtend—or at least—relate to the synergistic clinical effects of the 2 medications.
‘Think it over, think it under’
Having discussed several important Woozles that have inhabited the Hundred Acre Wood of ADHD for decades, it is important to remember there are countless Woozles in the larger “Thousand Acre Wood” of psychiatry and medicine. As we evaluate evidence for our interventions, whether psychopharmacologic or psychotherapeutic, we will do w
1. Milne AA. Winnie-the-Pooh. London, United Kingdom: Methuen & Co. Ltd.; 1926.
2. Strauss MA. Processes Explaining the concealment and distortion of evidence on gender symmetry in partner violence. Eur J Crim Pol Res. 1980;74:227-232.
3. Ritalin LA [package insert]. East Hanover, NJ: Novartis; 2015.
4. Coughlin CG, Cohen SC, Mulqueen JM, et al. Meta-analysis: reduced risk of anxiety with psychostimulant treatment in children with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2015;25(8):611-617.
5. Abikoff H, McGough J, Vitiello B, et al; RUPP ADHD/Anxiety Study Group. Sequential pharmacotherapy for children with comorbid attention-deficit/hyperactivity and anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2005;44(5):418-427.
6. Bloch MH, Panza KE, Landeros-Weisenberger A, et al. Meta-analysis: treatment of attention-deficit/hyperactivity disorder in children with comorbid tic disorders. J Am Acad Child Adolesc Psychiatry. 2009;48(9):884-893.
7. Cohen SC, Mulqueen JM, Ferracioli-Oda E, et al. Meta-analysis: risk of tics associated with psychostimulant use in randomized, placebo-controlled trials. J Am Acad Child Adolesc Psychiatry. 2015;54(9):728-736.
8. Loo SK, Bilder RM, Cho AL, et al. Effects of d-methylphenidate, guanfacine, and their combination on electroencephalogram resting state spectral power in attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2016;55(8):674-682.e1.
1. Milne AA. Winnie-the-Pooh. London, United Kingdom: Methuen & Co. Ltd.; 1926.
2. Strauss MA. Processes Explaining the concealment and distortion of evidence on gender symmetry in partner violence. Eur J Crim Pol Res. 1980;74:227-232.
3. Ritalin LA [package insert]. East Hanover, NJ: Novartis; 2015.
4. Coughlin CG, Cohen SC, Mulqueen JM, et al. Meta-analysis: reduced risk of anxiety with psychostimulant treatment in children with attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2015;25(8):611-617.
5. Abikoff H, McGough J, Vitiello B, et al; RUPP ADHD/Anxiety Study Group. Sequential pharmacotherapy for children with comorbid attention-deficit/hyperactivity and anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2005;44(5):418-427.
6. Bloch MH, Panza KE, Landeros-Weisenberger A, et al. Meta-analysis: treatment of attention-deficit/hyperactivity disorder in children with comorbid tic disorders. J Am Acad Child Adolesc Psychiatry. 2009;48(9):884-893.
7. Cohen SC, Mulqueen JM, Ferracioli-Oda E, et al. Meta-analysis: risk of tics associated with psychostimulant use in randomized, placebo-controlled trials. J Am Acad Child Adolesc Psychiatry. 2015;54(9):728-736.
8. Loo SK, Bilder RM, Cho AL, et al. Effects of d-methylphenidate, guanfacine, and their combination on electroencephalogram resting state spectral power in attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2016;55(8):674-682.e1.

Neuroimaging in children and adolescents: When do you scan? With which modalities?
The first 15 years of the new millennium have seen a great increase in research on neuroimaging in children and adolescents who have a psychiatric disorder. In addition, imaging modalities continue to evolve, and are becoming increasingly accessible and informative. The literature is now replete with reports of neurostructural differences between patients and healthy subjects in a variety of common pediatric psychiatric conditions, including anxiety disorders, mood disorders, autism spectrum disorder (ASD), and attention-deficit/hyperactivity disorder (ADHD).
Historically, the clinical utility of neuroimaging was restricted to the identification of structural pathology. Today, accumulating data reveal novel roles for neuroimaging; these revelations are supported by studies demonstrating that treatment response for psychotherapeutic and psychopharmacotherapeutic interventions can be predicted by neurochemical and neurofunctional characteristics assessed by advanced imaging technologies, such as magnetic resonance spectroscopy (MRS) and functional MRI.
However, such advanced techniques are (at least at present) not ready for routine clinical use for this purpose. Instead, neuroimaging in the child and adolescent psychiatric clinic remains largely focused on ruling out neurostructural, neurologic, “nonpsychiatric” causes of our patients’ symptoms.
Understanding the role and limitations of major imaging modalities is key to guiding efficient and appropriate neuroimaging selection for pediatric patients. In this article, we describe and review:
- neuroimaging approaches for children and adolescents with psychiatric disorders
- the role of neuroimaging in (1) the differential diagnosis and workup of common psychiatric disorders and (2) urgent clinical situations
- how to determine what type of imaging to obtain.
Computed tomography
CT, which utilizes ionizing radiation, often is reserved, in the pediatric setting, for (1) emergency evaluation and (2) excluding potentially catastrophic neurologic injury resulting from:
- ischemic or hemorrhagic stroke
- herniation
- intracerebral hemorrhage
- subdural and epidural hematoma
- large intracranial mass with mass effect
- increased intracranial pressure
- acute skull fracture.
Although a CT scan is, typically, quick and has excellent sensitivity for acute bleeding and bony pathology, it exposes the patient to radiation and provides poor resolution compared with MRI.
In pediatrics, there has been practice-changing recognition of the importance of limiting lifetime radiation exposure incurred from medical procedures and imaging. As a result, most providers now agree that use of MRI in lieu of CT is appropriate in many, if not most, non-emergent situations. In an emergent situation, however, CT imaging is appropriate and should not be delayed. Moreover, in an emergent situation, you should not hesitate to use head CT in children, although timely discussion with the radiologist is recommended to review your differential diagnosis to better determine the preferred imaging modality.
Magnetic resonance imaging
Over the past several decades, MRI has been increasingly available in most pediatric health care facilities. The modality offers specific advantages for pediatric patients, including:
- better spatial resolution
- the ability to concurrently assess multiple pathologic processes
- lack of exposure to ionizing radiation.1
A number of MRI sequences, described below, can be used to assess vascular, inflammatory, structural, and metabolic processes.
A look inside. Comprehensive review of the physics that underlies MRI is beyond the scope of this article; several important principles are relevant to clinicians, however. Image contrast is dependent on intrinsic properties of tissue with regard to proton density, longitudinal relaxation time (T1), and transverse relaxation time (T2). Pulse sequences, which describe the strength and timing of the radiofrequency pulse and gradient pulses, define imaging acquisition parameters (eg, repetition time between the radio frequency pulse and echo time).
In turn, the intensity of the signal that is “seen” with various pulse sequences is differentially affected by intrinsic properties of tissue. At most pediatric institutions, the standard MRI-examination protocol includes: a T1-weighted image (Figure 1A); a T2-weighted scan (Figure 1B); fluid attenuated inversion recovery (FLAIR) (Figure 1C); and diffusion-weighted imaging (DWI) (Figure 1D).
Specific MRI sequences
T1 images. T1 sequences, or so-called anatomy sequences, are ideally suited for detailed neuroanatomic evaluations. They are generated in such a way that structures containing fluid are dark (hypo-intense), whereas other structures, with higher fat or protein content, are brighter (iso-intense, even hyper-intense). For this reason, CSF in the intracranial ventricles is dark, and white matter is brighter than the cortex because of lipid in myelin sheaths.
In addition, to view structural abnormalities that are characterized by altered vascular supply or flow, such as tumors and infections (abscesses), contrast imaging can be particularly helpful; such images generally are obtained as T1 sequences.
T2 images. By contrast to the T1-weighted sequence, the T2-weighted sequences emphasize fluid signal; structures such as the ventricles, which contain CSF, therefore will be bright (hyper-intense). Pathology that produces edema or fluid, such as edema surrounding demyelinating lesions or infections, also will show bright hyper-intense signal. In T2-weighted images of the brain, white matter shows lower signal intensity than the cortex because of the relatively lower water content in white matter tracts and myelin sheaths.
Fluid attenuation inversion recovery. FLAIR images are generated so that the baseline bright T2 signal seen in normal structures, such as the CSF, containing ventricles is cancelled out, or attenuated. In effect, this subtraction of typical background hyper-intense fluid signal leaves only abnormal T2 bright hyper-intense signal, such as vasogenic edema surrounding tumors, cytotoxic edema within an infarction, or extra-axial fluid collections such as a subarachnoid or subdural hemorrhage.
Diffusion-weighted imaging. DWI utilizes the random motion (ie, diffusion) of water molecules to generate contrast. In this regard, the diffusion of any molecule is influenced by its interaction with other molecules (eg, white-matter fibers and membranes, and macromolecules). Diffusion patterns therefore reflect details about tissue boundaries; as such, DWI is sensitive to a number of neurologic processes, such as ischemia, demyelinating disease, and some tumors, which restrict the free motion of water. DWI detects this so-called restricted diffusion and displays an area of bright signal.
Susceptibility-weighted imaging (SWI). In the pediatric population, SWI (Figure 2) utilizes a long-echo, 3-dimensional, velocity-compensated gradient recalled echo for image acquisition2 and, ultimately, leverages susceptibility differences across tissues by employing the phase image to identify these differences. SWI, which uses both magnitude and phase images and is remarkably sensitive to venous blood (and blood products), iron, and calcifications, therefore might be of increasing utility in pediatric patients with traumatic brain injury (TBI) (Figure 2B). As such, SWI has become a critical component of many pediatric MRI studies.3
Magnetic resonance angiography (MRA) (Figure 3A) is helpful for assessing intracranial arteries and may be employed in the evaluation of:
- vessel pathology and injury underlying stroke, such as vessel occlusion or injury
- patterns of vessel involvement suggestive of vasculitis
- developmental or acquired structural vascular abnormalities, such as aneurysm or vascular malformations
- determination of tumor blood supply.

MRA can be performed without or with contrast, although MRA with contrast might provide a higher quality study and therefore be of greater utility. Of note: The spatial resolution of MRA is not as good as CT angiography; abnormalities, such as a small aneurysm, might not be apparent.
Magnetic resonance venography (MRV) (Figure 3B) is most commonly performed when the possibility of thrombosis of the dural venous sinuses is being considered; it also is employed to evaluate vascular malformations, tumor drainage patterns, and other pathologic states. As with MRA, MRV can be performed without or with contrast, although post-contrast MRV is generally of higher quality and might be preferred when assessing for sinus thrombosis.
Magnetic resonance spectroscopy (MRS) resides at the border between research and clinical practice. In children and adolescents, MRS provides data on neuronal and axonal viability as well as energetics and cell membranes.4 Pediatric neurologists often use MRS to evaluate for congenital neurometabolic disease; this modality also can help distinguish between an active intracranial tumor from an abscess or gliosis.5
Neuroimaging in pediatric neuropsychiatric conditions: Evidence, guidance
Delirium (altered mental status). The acute neuropsychiatric syndrome characterized by impaired attention and sensorium might have a broad underlying etiology, but it is always associated with alteration of CNS neurophysiology. Children with neurostructural abnormalities might have increased vulnerability to CNS insult and therefore be at increased risk of delirium.6 Additionally, delirium can present subtly in children, with the precise signs dependent on the individual patient’s developmental stage.
Neuroimaging may be helpful when an infectious, inflammatory, toxic, or a metabolic basis for delirium is suspected, or when a patient has new focal neurologic findings. In this regard, focal neurologic findings suggest an underlying localizable lesion and warrant dedicated neuroimaging to localize the lesion.
In general, the differential diagnosis should guide consideration of neuroimaging. When considering the possibility of an unwitnessed seizure in a child who presents with altered mental status, neuroimaging certainly is an important component of the workup.
As another example, when underlying trauma, intracranial hemorrhage, or mass is a possibility in acute delirium, urgent head CT is appropriate. In non-emergent cases, MRI is the modality of choice. In immunocompromised patients presenting with delirium, maintain a low threshold for neuroimaging with contrast to rule out opportunistic intracranial infection.
Last, in children who have hydrocephalus with a shunt, delirium could be a harbinger of underlying shunt malfunction, warranting a “shunt series.”
ADHD. The diagnosis of ADHD remains a clinical one; for the typical pediatric patient with ADHD but who does not have focal neurologic deficits, neuroimaging is unnecessary. Some structural MRI studies of youth with ADHD suggest diminished volume of the globus pallidus, putamen, and caudate7; other studies reveal changes in gyrification and cortical thickness8 in regions subserving attentional processes. However, intra-individual and developmental-related variability preclude routine use of neuroimaging in the standard diagnostic work-up of ADHD.
Nevertheless, neuroimaging should be strongly considered in a child with progressive worsening of inattention, especially if combined with other psychiatric or neurologic findings. In such a case, MRI should be obtained to evaluate for a progressive neurodegenerative leukoencephalopathy (eg, adrenoleukodystrophy).9
Depressive and anxiety disorders. In pediatric patients who exhibit depressive or anxiety symptoms, abnormalities have been observed in cortical thickness10 and gray matter volume,11,12 and functional signatures13 have been identified in the circuitry of the prefrontal amygdala. No data suggest that, in an individual patient, neuroimaging can be of diagnostic utility—particularly in the absence of focal neurologic findings.
That being said, headache and other somatic symptoms are common in pediatric patients with a mood or anxiety disorder. Evidence for neuroimaging in the context of pediatric headache suggests that MRI should be considered when headache is associated with neurologic signs or symptoms, such as aura, or accompanied by focal neurologic deficit.14
Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS) and pediatric acute-onset neuropsychiatric syndrome (PANS). Neuroimaging studies of patients with confirmed PANDAS or PANS are rare, but group analyses suggest a decreased average volume of the caudate, putamen, and globus pallidus in patients with PANDAS compared with healthy comparison subjects, although total cerebral volume does not appear to differ.15 Moreover, thalamic findings in patients with PANDAS have been noted to be similar to what is seen in patients with Sydenham’s chorea.
The most recent consensus statement regarding the treatment and assessment of PANDAS and PANS recommends ordering brain MRI when other conditions are suspected (eg, CNS, small vessel vasculitis, limbic encephalitis) or when the patient has severe headache, gait disturbance, cognitive deterioration, or psychosis.16 Furthermore, the consensus statement notes the potential utility of T2-weighted imaging with contrast to evaluate inflammatory changes in the basal ganglia.16
Autism spectrum disorder. Significant progress has been made during the past decade on the neuroanatomic characterization of ASD. Accumulating data indicate that, in pediatric patients with ASD, (1) development of white matter and gray matter is disrupted early in the course of the disorder and (2) cortical thickness is increased in regions subserving social cognition.17,18
Several studies have examined the presence of patient-level findings in samples of pediatric patients with ASD. Approximately 8% of pediatric patients with ASD were found to have some abnormality on routine brain MRI, the most common being white-matter signal abnormalities, dilated Virchow-Robin space, and temporal lobe abnormalities.19
Although abnormalities might be present in a large percentage of individual scans, routine screening MRI is unlikely to be of clinical utility in youth with ASD. In fact, no recommendation for routine MRI screening in patients with ASD has been made by the American Academy of Child & Adolescent Psychiatry, the Child Neurology Society of the American Academy of Neurology, or the American Academy of Pediatrics.
However, in patients with an underlying neurostructural disease that is phenotypically associated with ASD-like symptoms, imaging might be of use. In tuberous sclerosis, for example, MRI is especially important to classify intracranial lesions; determine burden and location; or identify treatment options (Figure 4). For patients with tuberous sclerosis—of whom more than one-third meet diagnostic criteria for ASD20—MRI study should include FLAIR, spin-echo, and gradient-echo sequences.

Movement disorders. Hyperkinetic movement disorders, including tic disorders and drug-induced movement disorders (eg, tremor) are common in pediatric patients. In pediatric patients with a tic disorder or Tourette’s disorder (TD), neuroimaging typically is unnecessary, despite the suggestion that the caudate nucleus volume is reduced in groups of patients with TD.
Many CNS-acting medications can exacerbate physiologic tremor; in pediatric patients with symptoms of a movement disorder, home medications should be carefully reviewed for potentially offending agents. When the patient is clinically and biochemically euthyroid and medication-induced movement disorder has been ruled out, or when the patient meets clinical diagnostic criteria for TD or a tic disorder, routine neuroimaging generally is unnecessary.
When tremor accompanies other cerebellar signs, such as ataxia or dysmetria, strongly consider MRI of the brain to evaluate pathology in the posterior fossa. In addition, neuroimaging should be considered for children with a new-onset abnormality on neurologic exam, including rapid onset of abnormal movements (other than common tics), continuous progressive worsening of symptoms, or any loss of developmental milestones.
Last, although tics and stereotypies often are transient and wane with age, other abnormal movements, such as dystonia, chorea, and parkinsonism (aside from those potentially associated with antipsychotic use), are never expected during typical development and warrant MRI.
Traumatic brain injury. Prompt evaluation and intervention for TBI can significantly affect overall outcome. Moreover, there has been increased enthusiasm around the pre-hospital assessment of TBI severity using (1) any of several proprietary testing systems (eg, Immediate Post-Concussion Assessment and Cognitive Testing [ImPACT]) plus (2) standard clinical staging, which is based on duration of loss of consciousness, persistence of memory loss, and the Glasgow Coma Scale score.
The goal for any TBI patient during the acute post-injury phase is to minimize continued neuronal injury from secondary effects of TBI, such as cerebral edema and herniation, and to optimize protection of surviving brain tissue; neuroimaging is a critical component of assessment during both acute and chronic recovery periods of TBI.21
The optimal imaging modality varies with the amount of time that has passed since initial injury.22 Urgent neuroimaging (the first 24 hours after brain injury) is typically obtained using head CT to assist decision-making in acute neurosurgical management. In this setting, head CT is fast and efficient; minimizes the amount of time that the patient is in the scanner; and provides valuable information on the acuity and extent of injury, degree of cerebral edema, and evidence or risk of pending herniation.
On the other hand, MRI is superior to CT during 48 to 72 hours after injury, given its higher resolution; superior imaging of the brainstem and deep gray nuclei; and ability to detect axonal injury, small contusions, and subtle neuronal damage. Specifically, SWI sequences can be particularly helpful in TBI for detecting diffuse axonal injury and micro-hemorrhages; several recent studies also suggest that SWI may be of particular value in pediatric patients with TBI.23
Additionally, given the increased sensitivity of MRI to detect subtle injuries, this modality can assist in identifying chronic sequelae of brain injury—thus contributing to determining of chronic therapy options and assisting with long-term prognosis. Gross structural changes resulting from TBI often are evident even in the acute post-injury phase; synaptic remodeling continues, however, for an indefinite period after injury, and this remodeling capacity is even more pronounced in the highly plastic brain of a young child.
Microstructural changes might not be detectable using traditional, readily available imaging sequences (CT, MRI). When those traditional modalities are used in concert with functional imaging techniques (eg, PET to evaluate cerebral metabolism and SPECT imaging which can detect abnormalities in cerebral blood flow), the combination of older and newer might provide a more complete picture of recovery after TBI.24
The important role of neuroimaging in severe TBI is intuitive. However, it is important to consider the role of neuroimaging in mild TBI in children, especially in the setting of repetitive mild injury.25 A growing body of evidence supports close, serial monitoring of children after even mild closed head injury for neurologic and psychiatric sequelae. Although it is rare that a child who is awake, interactive, and lacking focal neurologic deficits would need emergent (ie, CT) imaging after mild closed head injury, there might be a role for MRI later in the course of that child’s recovery—especially if recovery is complicated by clinical sequelae of mild TBI, such as cognitive impairment, headaches, or altered behavior.
When is additional neuroimaging needed?
It’s worthwhile briefly reviewing 4 scenarios that you might encounter, when you work with children and adolescents, in which urgent or emergent neuroimaging (often with consultation) should be obtained. The Table describes these situations and appropriate first- and second-line interventions.
1. When the presentation of your patient is consistent with an acute neurologic deficit, acute TBI, progressive neuropsychiatric decline, CNS infection, mass, demyelinating process, or toxic exposure, neuroimaging is likely critical.
2. In patients with progressive neurologic decline, including loss of developmental milestones, MRI, MRS, and referral to neurology should be part of the comprehensive evaluation.
3. In young children who exhibit a decrease in head circumference on growth curves, MRI is important to evaluate for underlying structural causes.
4. Pediatric patients with symptoms consistent with either stroke (ie, a new, persistent neurologic deficit) or a demyelinating process (eg, multiple episodes of variable transient focal neurologic symptoms), MRI should be obtained without compunction.

Consultation with pediatric neuroradiology
In deciding whether to obtain neuroimaging for a particular case, you should discuss your concerns with the pediatric radiologist or pediatric neuroradiologist, who will likely provide important guidance on key aspects of the study (eg, modifying slice thickness in a particular scan; recommending the use of contrast; including MRS in the order for imaging; performing appropriate vessel imaging). Consider asking 1 or more important questions when you discuss a patient’s presentation with the pediatric radiologist or pediatric neuroradiologist:
- “What neuroimaging studies are appropriate, based on my differential diagnosis?”
- “Are there specific imaging sequences that we should consider?”
- “Are there contraindications to the imaging modality for my patient?”
- “Is my patient likely to have difficulty tolerating the imaging procedure?”
- “Does my patient need sedation to tolerate this procedure?”
- “Should additional regions be included in the scan?” (Examples: In a child with stroke it might be important to include neck and chest vasculature and the heart. Other conditions might warrant imaging of the spinal cord.)
1. Abdelhalim AN, Alberico RA. Pediatric neuroimaging. Neurol Clin. 2009;27(1):285-301, x.
2. Sehgal V, Delproposto Z, Haacke EM, et al. Clinical applications of neuroimaging with susceptibility-weighted imaging. J Magn Reson Imaging. 2005;22(4):439-450.
3. Bosemani T, Poretti A, Huisman TA. Susceptibility-weighted imaging in pediatric neuroimaging. J Magn Reson Imaging. 2014;40(3):530-544.
4. Cecil KM. Proton magnetic resonance spectroscopy: technique for the neuroradiologist. Neuroimaging Clin N Am. 2013;23(3):381-392.
5. Panigrahy A, Nelson MD Jr, Blüml S. Magnetic resonance spectroscopy in pediatric neuroradiology: clinical and research applications. Pediatr Radiol. 2010;40(1):3-30.
6. Leentjens AF, Schieveld JN, Leonard M, et al. A comparison of the phenomenology of pediatric, adult, and geriatric delirium. J Psychosom Res. 2008;64(2):219-223.
7. Frodl T, Skokauskas N. Meta-analysis of structural MRI studies in children and adults with attention deficit hyperactivity disorder indicates treatment effects. Acta Psychiatr Scand. 2012;125(2):114-126.
8. Shaw P, Malek M, Watson B, et al. Development of cortical surface area and gyrification in attention-deficit/hyperactivity disorder. Biol Psychiatry. 2012;72(3):191-197.
9. Phelan JA, Lowe LH, Glasier CM. Pediatric neurodegenerative white matter processes: leukodystrophies and beyond. Pediatr Radiol. 2008;38(7):729-749.
10. Strawn JR, Wegman CJ, Dominick KC, et al. Cortical surface anatomy in pediatric patients with generalized anxiety disorder. J Anxiety Disord. 2014;28(7):717-723.
11. Mueller SC, Aouidad A, Gorodetsky E, et al. Gray matter volume in adolescent anxiety: an impact of the brain-derived neurotrophic factor Val(66)Met polymorphism [Erratum in J Am Acad Child Adolesc Psychiatry. 2013;52(2):184-195]? J Am Acad Child Adolesc Psychiatry. 2013;52(2):184-195.
12. Strawn JR, Hamm L, Fitzgerald DA, et al. Neurostructural abnormalities in pediatric anxiety disorders. J Anxiety Disord. 2015;32:81-88.
13. Strawn JR, Dominick KC, Patino LR, et al. Neurobiology of pediatric anxiety disorders. Curr Behav Neurosci Reports. 2014;1(3):154-160.
14. Alexiou GA, Argyropoulou MI. Neuroimaging in childhood headache: a systematic review. Pediatr Radiol. 2013;43(7):777-784.
15. Giedd JN, Rapoport JL, Garvey MA, et al. MRI assessment of children with obsessive-compulsive disorder or tics associated with streptococcal infection. Am J Psychiatry. 2000;157(2):281-283.
16. Chang K, Frankovich J, Cooperstock M, et al; PANS Collaborative Consortium. Clinical evaluation of youth with pediatric acute-onset neuropsychiatric syndrome (PANS): recommendations from the 2013 PANS Consensus Conference. J Child Adolesc Psychopharmacol. 2014;25(1):3-13.
17. Wallace GL, Robustelli B, Dankner N, et al. Increased gyrification, but comparable surface area in adolescents with autism spectrum disorders. Brain. 2013;136(pt 6):1956-1967.
18. Libero LE, DeRamus TP, Deshpande HD, et al. Surface-based morphometry of the cortical architecture of autism spectrum disorders: volume, thickness, area, and gyrification. Neuropsychologia. 2014;62:1-10.
19. Boddaert N, Zilbovicius M, Philipe A, et al. MRI findings in 77 children with non-syndromic autistic disorder. PLoS One. 2009;4:e445. doi: 10.1371/journal.pone.0004415.
20. Richards C, Jones C, Groves L, et al. Prevalence of autism spectrum disorder phenomenology in genetic disorders: a systematic review and meta-analysis. Lancet Psychiatry. 2015;2(10):909-916.
21. Wilde EA, Hunter JV, Bigler ED. Pediatric traumatic brain injury: neuroimaging and neurorehabilitation outcome. NeuroRehabilitation. 2012;31(3):245-260.
22. Mechtler LL, Shastri KK, Crutchfield KE. Advanced neuroimaging of mild traumatic brain injury. Neurol Clin. 2014;32(1):31-58.
23. Ashwal S, Tong KA, Ghosh N, et al. Application of advanced neuroimaging modalities in pediatric traumatic brain injury. J Child Neurol. 2014;29(12):1704-1717.
24. Munson S, Schroth E, Ernst M. The role of functional neuroimaging in pediatric brain injury. Pediatrics. 2006;117(4):1372-1381.
25. Wozniak JR, Krach L, Ward E, et al. Neurocognitive and neuroimaging correlates of pediatric traumatic brain injury: a diffusion tensor imaging (DTI) study. Arch Clin Neuropsychol. 2007;22(5):555-568.
The first 15 years of the new millennium have seen a great increase in research on neuroimaging in children and adolescents who have a psychiatric disorder. In addition, imaging modalities continue to evolve, and are becoming increasingly accessible and informative. The literature is now replete with reports of neurostructural differences between patients and healthy subjects in a variety of common pediatric psychiatric conditions, including anxiety disorders, mood disorders, autism spectrum disorder (ASD), and attention-deficit/hyperactivity disorder (ADHD).
Historically, the clinical utility of neuroimaging was restricted to the identification of structural pathology. Today, accumulating data reveal novel roles for neuroimaging; these revelations are supported by studies demonstrating that treatment response for psychotherapeutic and psychopharmacotherapeutic interventions can be predicted by neurochemical and neurofunctional characteristics assessed by advanced imaging technologies, such as magnetic resonance spectroscopy (MRS) and functional MRI.
However, such advanced techniques are (at least at present) not ready for routine clinical use for this purpose. Instead, neuroimaging in the child and adolescent psychiatric clinic remains largely focused on ruling out neurostructural, neurologic, “nonpsychiatric” causes of our patients’ symptoms.
Understanding the role and limitations of major imaging modalities is key to guiding efficient and appropriate neuroimaging selection for pediatric patients. In this article, we describe and review:
- neuroimaging approaches for children and adolescents with psychiatric disorders
- the role of neuroimaging in (1) the differential diagnosis and workup of common psychiatric disorders and (2) urgent clinical situations
- how to determine what type of imaging to obtain.
Computed tomography
CT, which utilizes ionizing radiation, often is reserved, in the pediatric setting, for (1) emergency evaluation and (2) excluding potentially catastrophic neurologic injury resulting from:
- ischemic or hemorrhagic stroke
- herniation
- intracerebral hemorrhage
- subdural and epidural hematoma
- large intracranial mass with mass effect
- increased intracranial pressure
- acute skull fracture.
Although a CT scan is, typically, quick and has excellent sensitivity for acute bleeding and bony pathology, it exposes the patient to radiation and provides poor resolution compared with MRI.
In pediatrics, there has been practice-changing recognition of the importance of limiting lifetime radiation exposure incurred from medical procedures and imaging. As a result, most providers now agree that use of MRI in lieu of CT is appropriate in many, if not most, non-emergent situations. In an emergent situation, however, CT imaging is appropriate and should not be delayed. Moreover, in an emergent situation, you should not hesitate to use head CT in children, although timely discussion with the radiologist is recommended to review your differential diagnosis to better determine the preferred imaging modality.
Magnetic resonance imaging
Over the past several decades, MRI has been increasingly available in most pediatric health care facilities. The modality offers specific advantages for pediatric patients, including:
- better spatial resolution
- the ability to concurrently assess multiple pathologic processes
- lack of exposure to ionizing radiation.1
A number of MRI sequences, described below, can be used to assess vascular, inflammatory, structural, and metabolic processes.
A look inside. Comprehensive review of the physics that underlies MRI is beyond the scope of this article; several important principles are relevant to clinicians, however. Image contrast is dependent on intrinsic properties of tissue with regard to proton density, longitudinal relaxation time (T1), and transverse relaxation time (T2). Pulse sequences, which describe the strength and timing of the radiofrequency pulse and gradient pulses, define imaging acquisition parameters (eg, repetition time between the radio frequency pulse and echo time).
In turn, the intensity of the signal that is “seen” with various pulse sequences is differentially affected by intrinsic properties of tissue. At most pediatric institutions, the standard MRI-examination protocol includes: a T1-weighted image (Figure 1A); a T2-weighted scan (Figure 1B); fluid attenuated inversion recovery (FLAIR) (Figure 1C); and diffusion-weighted imaging (DWI) (Figure 1D).
Specific MRI sequences
T1 images. T1 sequences, or so-called anatomy sequences, are ideally suited for detailed neuroanatomic evaluations. They are generated in such a way that structures containing fluid are dark (hypo-intense), whereas other structures, with higher fat or protein content, are brighter (iso-intense, even hyper-intense). For this reason, CSF in the intracranial ventricles is dark, and white matter is brighter than the cortex because of lipid in myelin sheaths.
In addition, to view structural abnormalities that are characterized by altered vascular supply or flow, such as tumors and infections (abscesses), contrast imaging can be particularly helpful; such images generally are obtained as T1 sequences.
T2 images. By contrast to the T1-weighted sequence, the T2-weighted sequences emphasize fluid signal; structures such as the ventricles, which contain CSF, therefore will be bright (hyper-intense). Pathology that produces edema or fluid, such as edema surrounding demyelinating lesions or infections, also will show bright hyper-intense signal. In T2-weighted images of the brain, white matter shows lower signal intensity than the cortex because of the relatively lower water content in white matter tracts and myelin sheaths.
Fluid attenuation inversion recovery. FLAIR images are generated so that the baseline bright T2 signal seen in normal structures, such as the CSF, containing ventricles is cancelled out, or attenuated. In effect, this subtraction of typical background hyper-intense fluid signal leaves only abnormal T2 bright hyper-intense signal, such as vasogenic edema surrounding tumors, cytotoxic edema within an infarction, or extra-axial fluid collections such as a subarachnoid or subdural hemorrhage.
Diffusion-weighted imaging. DWI utilizes the random motion (ie, diffusion) of water molecules to generate contrast. In this regard, the diffusion of any molecule is influenced by its interaction with other molecules (eg, white-matter fibers and membranes, and macromolecules). Diffusion patterns therefore reflect details about tissue boundaries; as such, DWI is sensitive to a number of neurologic processes, such as ischemia, demyelinating disease, and some tumors, which restrict the free motion of water. DWI detects this so-called restricted diffusion and displays an area of bright signal.
Susceptibility-weighted imaging (SWI). In the pediatric population, SWI (Figure 2) utilizes a long-echo, 3-dimensional, velocity-compensated gradient recalled echo for image acquisition2 and, ultimately, leverages susceptibility differences across tissues by employing the phase image to identify these differences. SWI, which uses both magnitude and phase images and is remarkably sensitive to venous blood (and blood products), iron, and calcifications, therefore might be of increasing utility in pediatric patients with traumatic brain injury (TBI) (Figure 2B). As such, SWI has become a critical component of many pediatric MRI studies.3
Magnetic resonance angiography (MRA) (Figure 3A) is helpful for assessing intracranial arteries and may be employed in the evaluation of:
- vessel pathology and injury underlying stroke, such as vessel occlusion or injury
- patterns of vessel involvement suggestive of vasculitis
- developmental or acquired structural vascular abnormalities, such as aneurysm or vascular malformations
- determination of tumor blood supply.
MRA can be performed without or with contrast, although MRA with contrast might provide a higher quality study and therefore be of greater utility. Of note: The spatial resolution of MRA is not as good as CT angiography; abnormalities, such as a small aneurysm, might not be apparent.
Magnetic resonance venography (MRV) (Figure 3B) is most commonly performed when the possibility of thrombosis of the dural venous sinuses is being considered; it also is employed to evaluate vascular malformations, tumor drainage patterns, and other pathologic states. As with MRA, MRV can be performed without or with contrast, although post-contrast MRV is generally of higher quality and might be preferred when assessing for sinus thrombosis.
Magnetic resonance spectroscopy (MRS) resides at the border between research and clinical practice. In children and adolescents, MRS provides data on neuronal and axonal viability as well as energetics and cell membranes.4 Pediatric neurologists often use MRS to evaluate for congenital neurometabolic disease; this modality also can help distinguish between an active intracranial tumor from an abscess or gliosis.5
Neuroimaging in pediatric neuropsychiatric conditions: Evidence, guidance
Delirium (altered mental status). The acute neuropsychiatric syndrome characterized by impaired attention and sensorium might have a broad underlying etiology, but it is always associated with alteration of CNS neurophysiology. Children with neurostructural abnormalities might have increased vulnerability to CNS insult and therefore be at increased risk of delirium.6 Additionally, delirium can present subtly in children, with the precise signs dependent on the individual patient’s developmental stage.
Neuroimaging may be helpful when an infectious, inflammatory, toxic, or a metabolic basis for delirium is suspected, or when a patient has new focal neurologic findings. In this regard, focal neurologic findings suggest an underlying localizable lesion and warrant dedicated neuroimaging to localize the lesion.
In general, the differential diagnosis should guide consideration of neuroimaging. When considering the possibility of an unwitnessed seizure in a child who presents with altered mental status, neuroimaging certainly is an important component of the workup.
As another example, when underlying trauma, intracranial hemorrhage, or mass is a possibility in acute delirium, urgent head CT is appropriate. In non-emergent cases, MRI is the modality of choice. In immunocompromised patients presenting with delirium, maintain a low threshold for neuroimaging with contrast to rule out opportunistic intracranial infection.
Last, in children who have hydrocephalus with a shunt, delirium could be a harbinger of underlying shunt malfunction, warranting a “shunt series.”
ADHD. The diagnosis of ADHD remains a clinical one; for the typical pediatric patient with ADHD but who does not have focal neurologic deficits, neuroimaging is unnecessary. Some structural MRI studies of youth with ADHD suggest diminished volume of the globus pallidus, putamen, and caudate7; other studies reveal changes in gyrification and cortical thickness8 in regions subserving attentional processes. However, intra-individual and developmental-related variability preclude routine use of neuroimaging in the standard diagnostic work-up of ADHD.
Nevertheless, neuroimaging should be strongly considered in a child with progressive worsening of inattention, especially if combined with other psychiatric or neurologic findings. In such a case, MRI should be obtained to evaluate for a progressive neurodegenerative leukoencephalopathy (eg, adrenoleukodystrophy).9
Depressive and anxiety disorders. In pediatric patients who exhibit depressive or anxiety symptoms, abnormalities have been observed in cortical thickness10 and gray matter volume,11,12 and functional signatures13 have been identified in the circuitry of the prefrontal amygdala. No data suggest that, in an individual patient, neuroimaging can be of diagnostic utility—particularly in the absence of focal neurologic findings.
That being said, headache and other somatic symptoms are common in pediatric patients with a mood or anxiety disorder. Evidence for neuroimaging in the context of pediatric headache suggests that MRI should be considered when headache is associated with neurologic signs or symptoms, such as aura, or accompanied by focal neurologic deficit.14
Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS) and pediatric acute-onset neuropsychiatric syndrome (PANS). Neuroimaging studies of patients with confirmed PANDAS or PANS are rare, but group analyses suggest a decreased average volume of the caudate, putamen, and globus pallidus in patients with PANDAS compared with healthy comparison subjects, although total cerebral volume does not appear to differ.15 Moreover, thalamic findings in patients with PANDAS have been noted to be similar to what is seen in patients with Sydenham’s chorea.
The most recent consensus statement regarding the treatment and assessment of PANDAS and PANS recommends ordering brain MRI when other conditions are suspected (eg, CNS, small vessel vasculitis, limbic encephalitis) or when the patient has severe headache, gait disturbance, cognitive deterioration, or psychosis.16 Furthermore, the consensus statement notes the potential utility of T2-weighted imaging with contrast to evaluate inflammatory changes in the basal ganglia.16
Autism spectrum disorder. Significant progress has been made during the past decade on the neuroanatomic characterization of ASD. Accumulating data indicate that, in pediatric patients with ASD, (1) development of white matter and gray matter is disrupted early in the course of the disorder and (2) cortical thickness is increased in regions subserving social cognition.17,18
Several studies have examined the presence of patient-level findings in samples of pediatric patients with ASD. Approximately 8% of pediatric patients with ASD were found to have some abnormality on routine brain MRI, the most common being white-matter signal abnormalities, dilated Virchow-Robin space, and temporal lobe abnormalities.19
Although abnormalities might be present in a large percentage of individual scans, routine screening MRI is unlikely to be of clinical utility in youth with ASD. In fact, no recommendation for routine MRI screening in patients with ASD has been made by the American Academy of Child & Adolescent Psychiatry, the Child Neurology Society of the American Academy of Neurology, or the American Academy of Pediatrics.
However, in patients with an underlying neurostructural disease that is phenotypically associated with ASD-like symptoms, imaging might be of use. In tuberous sclerosis, for example, MRI is especially important to classify intracranial lesions; determine burden and location; or identify treatment options (Figure 4). For patients with tuberous sclerosis—of whom more than one-third meet diagnostic criteria for ASD20—MRI study should include FLAIR, spin-echo, and gradient-echo sequences.
Movement disorders. Hyperkinetic movement disorders, including tic disorders and drug-induced movement disorders (eg, tremor) are common in pediatric patients. In pediatric patients with a tic disorder or Tourette’s disorder (TD), neuroimaging typically is unnecessary, despite the suggestion that the caudate nucleus volume is reduced in groups of patients with TD.
Many CNS-acting medications can exacerbate physiologic tremor; in pediatric patients with symptoms of a movement disorder, home medications should be carefully reviewed for potentially offending agents. When the patient is clinically and biochemically euthyroid and medication-induced movement disorder has been ruled out, or when the patient meets clinical diagnostic criteria for TD or a tic disorder, routine neuroimaging generally is unnecessary.
When tremor accompanies other cerebellar signs, such as ataxia or dysmetria, strongly consider MRI of the brain to evaluate pathology in the posterior fossa. In addition, neuroimaging should be considered for children with a new-onset abnormality on neurologic exam, including rapid onset of abnormal movements (other than common tics), continuous progressive worsening of symptoms, or any loss of developmental milestones.
Last, although tics and stereotypies often are transient and wane with age, other abnormal movements, such as dystonia, chorea, and parkinsonism (aside from those potentially associated with antipsychotic use), are never expected during typical development and warrant MRI.
Traumatic brain injury. Prompt evaluation and intervention for TBI can significantly affect overall outcome. Moreover, there has been increased enthusiasm around the pre-hospital assessment of TBI severity using (1) any of several proprietary testing systems (eg, Immediate Post-Concussion Assessment and Cognitive Testing [ImPACT]) plus (2) standard clinical staging, which is based on duration of loss of consciousness, persistence of memory loss, and the Glasgow Coma Scale score.
The goal for any TBI patient during the acute post-injury phase is to minimize continued neuronal injury from secondary effects of TBI, such as cerebral edema and herniation, and to optimize protection of surviving brain tissue; neuroimaging is a critical component of assessment during both acute and chronic recovery periods of TBI.21
The optimal imaging modality varies with the amount of time that has passed since initial injury.22 Urgent neuroimaging (the first 24 hours after brain injury) is typically obtained using head CT to assist decision-making in acute neurosurgical management. In this setting, head CT is fast and efficient; minimizes the amount of time that the patient is in the scanner; and provides valuable information on the acuity and extent of injury, degree of cerebral edema, and evidence or risk of pending herniation.
On the other hand, MRI is superior to CT during 48 to 72 hours after injury, given its higher resolution; superior imaging of the brainstem and deep gray nuclei; and ability to detect axonal injury, small contusions, and subtle neuronal damage. Specifically, SWI sequences can be particularly helpful in TBI for detecting diffuse axonal injury and micro-hemorrhages; several recent studies also suggest that SWI may be of particular value in pediatric patients with TBI.23
Additionally, given the increased sensitivity of MRI to detect subtle injuries, this modality can assist in identifying chronic sequelae of brain injury—thus contributing to determining of chronic therapy options and assisting with long-term prognosis. Gross structural changes resulting from TBI often are evident even in the acute post-injury phase; synaptic remodeling continues, however, for an indefinite period after injury, and this remodeling capacity is even more pronounced in the highly plastic brain of a young child.
Microstructural changes might not be detectable using traditional, readily available imaging sequences (CT, MRI). When those traditional modalities are used in concert with functional imaging techniques (eg, PET to evaluate cerebral metabolism and SPECT imaging which can detect abnormalities in cerebral blood flow), the combination of older and newer might provide a more complete picture of recovery after TBI.24
The important role of neuroimaging in severe TBI is intuitive. However, it is important to consider the role of neuroimaging in mild TBI in children, especially in the setting of repetitive mild injury.25 A growing body of evidence supports close, serial monitoring of children after even mild closed head injury for neurologic and psychiatric sequelae. Although it is rare that a child who is awake, interactive, and lacking focal neurologic deficits would need emergent (ie, CT) imaging after mild closed head injury, there might be a role for MRI later in the course of that child’s recovery—especially if recovery is complicated by clinical sequelae of mild TBI, such as cognitive impairment, headaches, or altered behavior.
When is additional neuroimaging needed?
It’s worthwhile briefly reviewing 4 scenarios that you might encounter, when you work with children and adolescents, in which urgent or emergent neuroimaging (often with consultation) should be obtained. The Table describes these situations and appropriate first- and second-line interventions.
1. When the presentation of your patient is consistent with an acute neurologic deficit, acute TBI, progressive neuropsychiatric decline, CNS infection, mass, demyelinating process, or toxic exposure, neuroimaging is likely critical.
2. In patients with progressive neurologic decline, including loss of developmental milestones, MRI, MRS, and referral to neurology should be part of the comprehensive evaluation.
3. In young children who exhibit a decrease in head circumference on growth curves, MRI is important to evaluate for underlying structural causes.
4. Pediatric patients with symptoms consistent with either stroke (ie, a new, persistent neurologic deficit) or a demyelinating process (eg, multiple episodes of variable transient focal neurologic symptoms), MRI should be obtained without compunction.
Consultation with pediatric neuroradiology
In deciding whether to obtain neuroimaging for a particular case, you should discuss your concerns with the pediatric radiologist or pediatric neuroradiologist, who will likely provide important guidance on key aspects of the study (eg, modifying slice thickness in a particular scan; recommending the use of contrast; including MRS in the order for imaging; performing appropriate vessel imaging). Consider asking 1 or more important questions when you discuss a patient’s presentation with the pediatric radiologist or pediatric neuroradiologist:
- “What neuroimaging studies are appropriate, based on my differential diagnosis?”
- “Are there specific imaging sequences that we should consider?”
- “Are there contraindications to the imaging modality for my patient?”
- “Is my patient likely to have difficulty tolerating the imaging procedure?”
- “Does my patient need sedation to tolerate this procedure?”
- “Should additional regions be included in the scan?” (Examples: In a child with stroke it might be important to include neck and chest vasculature and the heart. Other conditions might warrant imaging of the spinal cord.)
The first 15 years of the new millennium have seen a great increase in research on neuroimaging in children and adolescents who have a psychiatric disorder. In addition, imaging modalities continue to evolve, and are becoming increasingly accessible and informative. The literature is now replete with reports of neurostructural differences between patients and healthy subjects in a variety of common pediatric psychiatric conditions, including anxiety disorders, mood disorders, autism spectrum disorder (ASD), and attention-deficit/hyperactivity disorder (ADHD).
Historically, the clinical utility of neuroimaging was restricted to the identification of structural pathology. Today, accumulating data reveal novel roles for neuroimaging; these revelations are supported by studies demonstrating that treatment response for psychotherapeutic and psychopharmacotherapeutic interventions can be predicted by neurochemical and neurofunctional characteristics assessed by advanced imaging technologies, such as magnetic resonance spectroscopy (MRS) and functional MRI.
However, such advanced techniques are (at least at present) not ready for routine clinical use for this purpose. Instead, neuroimaging in the child and adolescent psychiatric clinic remains largely focused on ruling out neurostructural, neurologic, “nonpsychiatric” causes of our patients’ symptoms.
Understanding the role and limitations of major imaging modalities is key to guiding efficient and appropriate neuroimaging selection for pediatric patients. In this article, we describe and review:
- neuroimaging approaches for children and adolescents with psychiatric disorders
- the role of neuroimaging in (1) the differential diagnosis and workup of common psychiatric disorders and (2) urgent clinical situations
- how to determine what type of imaging to obtain.
Computed tomography
CT, which utilizes ionizing radiation, often is reserved, in the pediatric setting, for (1) emergency evaluation and (2) excluding potentially catastrophic neurologic injury resulting from:
- ischemic or hemorrhagic stroke
- herniation
- intracerebral hemorrhage
- subdural and epidural hematoma
- large intracranial mass with mass effect
- increased intracranial pressure
- acute skull fracture.
Although a CT scan is, typically, quick and has excellent sensitivity for acute bleeding and bony pathology, it exposes the patient to radiation and provides poor resolution compared with MRI.
In pediatrics, there has been practice-changing recognition of the importance of limiting lifetime radiation exposure incurred from medical procedures and imaging. As a result, most providers now agree that use of MRI in lieu of CT is appropriate in many, if not most, non-emergent situations. In an emergent situation, however, CT imaging is appropriate and should not be delayed. Moreover, in an emergent situation, you should not hesitate to use head CT in children, although timely discussion with the radiologist is recommended to review your differential diagnosis to better determine the preferred imaging modality.
Magnetic resonance imaging
Over the past several decades, MRI has been increasingly available in most pediatric health care facilities. The modality offers specific advantages for pediatric patients, including:
- better spatial resolution
- the ability to concurrently assess multiple pathologic processes
- lack of exposure to ionizing radiation.1
A number of MRI sequences, described below, can be used to assess vascular, inflammatory, structural, and metabolic processes.
A look inside. Comprehensive review of the physics that underlies MRI is beyond the scope of this article; several important principles are relevant to clinicians, however. Image contrast is dependent on intrinsic properties of tissue with regard to proton density, longitudinal relaxation time (T1), and transverse relaxation time (T2). Pulse sequences, which describe the strength and timing of the radiofrequency pulse and gradient pulses, define imaging acquisition parameters (eg, repetition time between the radio frequency pulse and echo time).
In turn, the intensity of the signal that is “seen” with various pulse sequences is differentially affected by intrinsic properties of tissue. At most pediatric institutions, the standard MRI-examination protocol includes: a T1-weighted image (Figure 1A); a T2-weighted scan (Figure 1B); fluid attenuated inversion recovery (FLAIR) (Figure 1C); and diffusion-weighted imaging (DWI) (Figure 1D).
Specific MRI sequences
T1 images. T1 sequences, or so-called anatomy sequences, are ideally suited for detailed neuroanatomic evaluations. They are generated in such a way that structures containing fluid are dark (hypo-intense), whereas other structures, with higher fat or protein content, are brighter (iso-intense, even hyper-intense). For this reason, CSF in the intracranial ventricles is dark, and white matter is brighter than the cortex because of lipid in myelin sheaths.
In addition, to view structural abnormalities that are characterized by altered vascular supply or flow, such as tumors and infections (abscesses), contrast imaging can be particularly helpful; such images generally are obtained as T1 sequences.
T2 images. By contrast to the T1-weighted sequence, the T2-weighted sequences emphasize fluid signal; structures such as the ventricles, which contain CSF, therefore will be bright (hyper-intense). Pathology that produces edema or fluid, such as edema surrounding demyelinating lesions or infections, also will show bright hyper-intense signal. In T2-weighted images of the brain, white matter shows lower signal intensity than the cortex because of the relatively lower water content in white matter tracts and myelin sheaths.
Fluid attenuation inversion recovery. FLAIR images are generated so that the baseline bright T2 signal seen in normal structures, such as the CSF, containing ventricles is cancelled out, or attenuated. In effect, this subtraction of typical background hyper-intense fluid signal leaves only abnormal T2 bright hyper-intense signal, such as vasogenic edema surrounding tumors, cytotoxic edema within an infarction, or extra-axial fluid collections such as a subarachnoid or subdural hemorrhage.
Diffusion-weighted imaging. DWI utilizes the random motion (ie, diffusion) of water molecules to generate contrast. In this regard, the diffusion of any molecule is influenced by its interaction with other molecules (eg, white-matter fibers and membranes, and macromolecules). Diffusion patterns therefore reflect details about tissue boundaries; as such, DWI is sensitive to a number of neurologic processes, such as ischemia, demyelinating disease, and some tumors, which restrict the free motion of water. DWI detects this so-called restricted diffusion and displays an area of bright signal.
Susceptibility-weighted imaging (SWI). In the pediatric population, SWI (Figure 2) utilizes a long-echo, 3-dimensional, velocity-compensated gradient recalled echo for image acquisition2 and, ultimately, leverages susceptibility differences across tissues by employing the phase image to identify these differences. SWI, which uses both magnitude and phase images and is remarkably sensitive to venous blood (and blood products), iron, and calcifications, therefore might be of increasing utility in pediatric patients with traumatic brain injury (TBI) (Figure 2B). As such, SWI has become a critical component of many pediatric MRI studies.3
Magnetic resonance angiography (MRA) (Figure 3A) is helpful for assessing intracranial arteries and may be employed in the evaluation of:
- vessel pathology and injury underlying stroke, such as vessel occlusion or injury
- patterns of vessel involvement suggestive of vasculitis
- developmental or acquired structural vascular abnormalities, such as aneurysm or vascular malformations
- determination of tumor blood supply.
MRA can be performed without or with contrast, although MRA with contrast might provide a higher quality study and therefore be of greater utility. Of note: The spatial resolution of MRA is not as good as CT angiography; abnormalities, such as a small aneurysm, might not be apparent.
Magnetic resonance venography (MRV) (Figure 3B) is most commonly performed when the possibility of thrombosis of the dural venous sinuses is being considered; it also is employed to evaluate vascular malformations, tumor drainage patterns, and other pathologic states. As with MRA, MRV can be performed without or with contrast, although post-contrast MRV is generally of higher quality and might be preferred when assessing for sinus thrombosis.
Magnetic resonance spectroscopy (MRS) resides at the border between research and clinical practice. In children and adolescents, MRS provides data on neuronal and axonal viability as well as energetics and cell membranes.4 Pediatric neurologists often use MRS to evaluate for congenital neurometabolic disease; this modality also can help distinguish between an active intracranial tumor from an abscess or gliosis.5
Neuroimaging in pediatric neuropsychiatric conditions: Evidence, guidance
Delirium (altered mental status). The acute neuropsychiatric syndrome characterized by impaired attention and sensorium might have a broad underlying etiology, but it is always associated with alteration of CNS neurophysiology. Children with neurostructural abnormalities might have increased vulnerability to CNS insult and therefore be at increased risk of delirium.6 Additionally, delirium can present subtly in children, with the precise signs dependent on the individual patient’s developmental stage.
Neuroimaging may be helpful when an infectious, inflammatory, toxic, or a metabolic basis for delirium is suspected, or when a patient has new focal neurologic findings. In this regard, focal neurologic findings suggest an underlying localizable lesion and warrant dedicated neuroimaging to localize the lesion.
In general, the differential diagnosis should guide consideration of neuroimaging. When considering the possibility of an unwitnessed seizure in a child who presents with altered mental status, neuroimaging certainly is an important component of the workup.
As another example, when underlying trauma, intracranial hemorrhage, or mass is a possibility in acute delirium, urgent head CT is appropriate. In non-emergent cases, MRI is the modality of choice. In immunocompromised patients presenting with delirium, maintain a low threshold for neuroimaging with contrast to rule out opportunistic intracranial infection.
Last, in children who have hydrocephalus with a shunt, delirium could be a harbinger of underlying shunt malfunction, warranting a “shunt series.”
ADHD. The diagnosis of ADHD remains a clinical one; for the typical pediatric patient with ADHD but who does not have focal neurologic deficits, neuroimaging is unnecessary. Some structural MRI studies of youth with ADHD suggest diminished volume of the globus pallidus, putamen, and caudate7; other studies reveal changes in gyrification and cortical thickness8 in regions subserving attentional processes. However, intra-individual and developmental-related variability preclude routine use of neuroimaging in the standard diagnostic work-up of ADHD.
Nevertheless, neuroimaging should be strongly considered in a child with progressive worsening of inattention, especially if combined with other psychiatric or neurologic findings. In such a case, MRI should be obtained to evaluate for a progressive neurodegenerative leukoencephalopathy (eg, adrenoleukodystrophy).9
Depressive and anxiety disorders. In pediatric patients who exhibit depressive or anxiety symptoms, abnormalities have been observed in cortical thickness10 and gray matter volume,11,12 and functional signatures13 have been identified in the circuitry of the prefrontal amygdala. No data suggest that, in an individual patient, neuroimaging can be of diagnostic utility—particularly in the absence of focal neurologic findings.
That being said, headache and other somatic symptoms are common in pediatric patients with a mood or anxiety disorder. Evidence for neuroimaging in the context of pediatric headache suggests that MRI should be considered when headache is associated with neurologic signs or symptoms, such as aura, or accompanied by focal neurologic deficit.14
Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS) and pediatric acute-onset neuropsychiatric syndrome (PANS). Neuroimaging studies of patients with confirmed PANDAS or PANS are rare, but group analyses suggest a decreased average volume of the caudate, putamen, and globus pallidus in patients with PANDAS compared with healthy comparison subjects, although total cerebral volume does not appear to differ.15 Moreover, thalamic findings in patients with PANDAS have been noted to be similar to what is seen in patients with Sydenham’s chorea.
The most recent consensus statement regarding the treatment and assessment of PANDAS and PANS recommends ordering brain MRI when other conditions are suspected (eg, CNS, small vessel vasculitis, limbic encephalitis) or when the patient has severe headache, gait disturbance, cognitive deterioration, or psychosis.16 Furthermore, the consensus statement notes the potential utility of T2-weighted imaging with contrast to evaluate inflammatory changes in the basal ganglia.16
Autism spectrum disorder. Significant progress has been made during the past decade on the neuroanatomic characterization of ASD. Accumulating data indicate that, in pediatric patients with ASD, (1) development of white matter and gray matter is disrupted early in the course of the disorder and (2) cortical thickness is increased in regions subserving social cognition.17,18
Several studies have examined the presence of patient-level findings in samples of pediatric patients with ASD. Approximately 8% of pediatric patients with ASD were found to have some abnormality on routine brain MRI, the most common being white-matter signal abnormalities, dilated Virchow-Robin space, and temporal lobe abnormalities.19
Although abnormalities might be present in a large percentage of individual scans, routine screening MRI is unlikely to be of clinical utility in youth with ASD. In fact, no recommendation for routine MRI screening in patients with ASD has been made by the American Academy of Child & Adolescent Psychiatry, the Child Neurology Society of the American Academy of Neurology, or the American Academy of Pediatrics.
However, in patients with an underlying neurostructural disease that is phenotypically associated with ASD-like symptoms, imaging might be of use. In tuberous sclerosis, for example, MRI is especially important to classify intracranial lesions; determine burden and location; or identify treatment options (Figure 4). For patients with tuberous sclerosis—of whom more than one-third meet diagnostic criteria for ASD20—MRI study should include FLAIR, spin-echo, and gradient-echo sequences.
Movement disorders. Hyperkinetic movement disorders, including tic disorders and drug-induced movement disorders (eg, tremor) are common in pediatric patients. In pediatric patients with a tic disorder or Tourette’s disorder (TD), neuroimaging typically is unnecessary, despite the suggestion that the caudate nucleus volume is reduced in groups of patients with TD.
Many CNS-acting medications can exacerbate physiologic tremor; in pediatric patients with symptoms of a movement disorder, home medications should be carefully reviewed for potentially offending agents. When the patient is clinically and biochemically euthyroid and medication-induced movement disorder has been ruled out, or when the patient meets clinical diagnostic criteria for TD or a tic disorder, routine neuroimaging generally is unnecessary.
When tremor accompanies other cerebellar signs, such as ataxia or dysmetria, strongly consider MRI of the brain to evaluate pathology in the posterior fossa. In addition, neuroimaging should be considered for children with a new-onset abnormality on neurologic exam, including rapid onset of abnormal movements (other than common tics), continuous progressive worsening of symptoms, or any loss of developmental milestones.
Last, although tics and stereotypies often are transient and wane with age, other abnormal movements, such as dystonia, chorea, and parkinsonism (aside from those potentially associated with antipsychotic use), are never expected during typical development and warrant MRI.
Traumatic brain injury. Prompt evaluation and intervention for TBI can significantly affect overall outcome. Moreover, there has been increased enthusiasm around the pre-hospital assessment of TBI severity using (1) any of several proprietary testing systems (eg, Immediate Post-Concussion Assessment and Cognitive Testing [ImPACT]) plus (2) standard clinical staging, which is based on duration of loss of consciousness, persistence of memory loss, and the Glasgow Coma Scale score.
The goal for any TBI patient during the acute post-injury phase is to minimize continued neuronal injury from secondary effects of TBI, such as cerebral edema and herniation, and to optimize protection of surviving brain tissue; neuroimaging is a critical component of assessment during both acute and chronic recovery periods of TBI.21
The optimal imaging modality varies with the amount of time that has passed since initial injury.22 Urgent neuroimaging (the first 24 hours after brain injury) is typically obtained using head CT to assist decision-making in acute neurosurgical management. In this setting, head CT is fast and efficient; minimizes the amount of time that the patient is in the scanner; and provides valuable information on the acuity and extent of injury, degree of cerebral edema, and evidence or risk of pending herniation.
On the other hand, MRI is superior to CT during 48 to 72 hours after injury, given its higher resolution; superior imaging of the brainstem and deep gray nuclei; and ability to detect axonal injury, small contusions, and subtle neuronal damage. Specifically, SWI sequences can be particularly helpful in TBI for detecting diffuse axonal injury and micro-hemorrhages; several recent studies also suggest that SWI may be of particular value in pediatric patients with TBI.23
Additionally, given the increased sensitivity of MRI to detect subtle injuries, this modality can assist in identifying chronic sequelae of brain injury—thus contributing to determining of chronic therapy options and assisting with long-term prognosis. Gross structural changes resulting from TBI often are evident even in the acute post-injury phase; synaptic remodeling continues, however, for an indefinite period after injury, and this remodeling capacity is even more pronounced in the highly plastic brain of a young child.
Microstructural changes might not be detectable using traditional, readily available imaging sequences (CT, MRI). When those traditional modalities are used in concert with functional imaging techniques (eg, PET to evaluate cerebral metabolism and SPECT imaging which can detect abnormalities in cerebral blood flow), the combination of older and newer might provide a more complete picture of recovery after TBI.24
The important role of neuroimaging in severe TBI is intuitive. However, it is important to consider the role of neuroimaging in mild TBI in children, especially in the setting of repetitive mild injury.25 A growing body of evidence supports close, serial monitoring of children after even mild closed head injury for neurologic and psychiatric sequelae. Although it is rare that a child who is awake, interactive, and lacking focal neurologic deficits would need emergent (ie, CT) imaging after mild closed head injury, there might be a role for MRI later in the course of that child’s recovery—especially if recovery is complicated by clinical sequelae of mild TBI, such as cognitive impairment, headaches, or altered behavior.
When is additional neuroimaging needed?
It’s worthwhile briefly reviewing 4 scenarios that you might encounter, when you work with children and adolescents, in which urgent or emergent neuroimaging (often with consultation) should be obtained. The Table describes these situations and appropriate first- and second-line interventions.
1. When the presentation of your patient is consistent with an acute neurologic deficit, acute TBI, progressive neuropsychiatric decline, CNS infection, mass, demyelinating process, or toxic exposure, neuroimaging is likely critical.
2. In patients with progressive neurologic decline, including loss of developmental milestones, MRI, MRS, and referral to neurology should be part of the comprehensive evaluation.
3. In young children who exhibit a decrease in head circumference on growth curves, MRI is important to evaluate for underlying structural causes.
4. Pediatric patients with symptoms consistent with either stroke (ie, a new, persistent neurologic deficit) or a demyelinating process (eg, multiple episodes of variable transient focal neurologic symptoms), MRI should be obtained without compunction.
Consultation with pediatric neuroradiology
In deciding whether to obtain neuroimaging for a particular case, you should discuss your concerns with the pediatric radiologist or pediatric neuroradiologist, who will likely provide important guidance on key aspects of the study (eg, modifying slice thickness in a particular scan; recommending the use of contrast; including MRS in the order for imaging; performing appropriate vessel imaging). Consider asking 1 or more important questions when you discuss a patient’s presentation with the pediatric radiologist or pediatric neuroradiologist:
- “What neuroimaging studies are appropriate, based on my differential diagnosis?”
- “Are there specific imaging sequences that we should consider?”
- “Are there contraindications to the imaging modality for my patient?”
- “Is my patient likely to have difficulty tolerating the imaging procedure?”
- “Does my patient need sedation to tolerate this procedure?”
- “Should additional regions be included in the scan?” (Examples: In a child with stroke it might be important to include neck and chest vasculature and the heart. Other conditions might warrant imaging of the spinal cord.)
1. Abdelhalim AN, Alberico RA. Pediatric neuroimaging. Neurol Clin. 2009;27(1):285-301, x.
2. Sehgal V, Delproposto Z, Haacke EM, et al. Clinical applications of neuroimaging with susceptibility-weighted imaging. J Magn Reson Imaging. 2005;22(4):439-450.
3. Bosemani T, Poretti A, Huisman TA. Susceptibility-weighted imaging in pediatric neuroimaging. J Magn Reson Imaging. 2014;40(3):530-544.
4. Cecil KM. Proton magnetic resonance spectroscopy: technique for the neuroradiologist. Neuroimaging Clin N Am. 2013;23(3):381-392.
5. Panigrahy A, Nelson MD Jr, Blüml S. Magnetic resonance spectroscopy in pediatric neuroradiology: clinical and research applications. Pediatr Radiol. 2010;40(1):3-30.
6. Leentjens AF, Schieveld JN, Leonard M, et al. A comparison of the phenomenology of pediatric, adult, and geriatric delirium. J Psychosom Res. 2008;64(2):219-223.
7. Frodl T, Skokauskas N. Meta-analysis of structural MRI studies in children and adults with attention deficit hyperactivity disorder indicates treatment effects. Acta Psychiatr Scand. 2012;125(2):114-126.
8. Shaw P, Malek M, Watson B, et al. Development of cortical surface area and gyrification in attention-deficit/hyperactivity disorder. Biol Psychiatry. 2012;72(3):191-197.
9. Phelan JA, Lowe LH, Glasier CM. Pediatric neurodegenerative white matter processes: leukodystrophies and beyond. Pediatr Radiol. 2008;38(7):729-749.
10. Strawn JR, Wegman CJ, Dominick KC, et al. Cortical surface anatomy in pediatric patients with generalized anxiety disorder. J Anxiety Disord. 2014;28(7):717-723.
11. Mueller SC, Aouidad A, Gorodetsky E, et al. Gray matter volume in adolescent anxiety: an impact of the brain-derived neurotrophic factor Val(66)Met polymorphism [Erratum in J Am Acad Child Adolesc Psychiatry. 2013;52(2):184-195]? J Am Acad Child Adolesc Psychiatry. 2013;52(2):184-195.
12. Strawn JR, Hamm L, Fitzgerald DA, et al. Neurostructural abnormalities in pediatric anxiety disorders. J Anxiety Disord. 2015;32:81-88.
13. Strawn JR, Dominick KC, Patino LR, et al. Neurobiology of pediatric anxiety disorders. Curr Behav Neurosci Reports. 2014;1(3):154-160.
14. Alexiou GA, Argyropoulou MI. Neuroimaging in childhood headache: a systematic review. Pediatr Radiol. 2013;43(7):777-784.
15. Giedd JN, Rapoport JL, Garvey MA, et al. MRI assessment of children with obsessive-compulsive disorder or tics associated with streptococcal infection. Am J Psychiatry. 2000;157(2):281-283.
16. Chang K, Frankovich J, Cooperstock M, et al; PANS Collaborative Consortium. Clinical evaluation of youth with pediatric acute-onset neuropsychiatric syndrome (PANS): recommendations from the 2013 PANS Consensus Conference. J Child Adolesc Psychopharmacol. 2014;25(1):3-13.
17. Wallace GL, Robustelli B, Dankner N, et al. Increased gyrification, but comparable surface area in adolescents with autism spectrum disorders. Brain. 2013;136(pt 6):1956-1967.
18. Libero LE, DeRamus TP, Deshpande HD, et al. Surface-based morphometry of the cortical architecture of autism spectrum disorders: volume, thickness, area, and gyrification. Neuropsychologia. 2014;62:1-10.
19. Boddaert N, Zilbovicius M, Philipe A, et al. MRI findings in 77 children with non-syndromic autistic disorder. PLoS One. 2009;4:e445. doi: 10.1371/journal.pone.0004415.
20. Richards C, Jones C, Groves L, et al. Prevalence of autism spectrum disorder phenomenology in genetic disorders: a systematic review and meta-analysis. Lancet Psychiatry. 2015;2(10):909-916.
21. Wilde EA, Hunter JV, Bigler ED. Pediatric traumatic brain injury: neuroimaging and neurorehabilitation outcome. NeuroRehabilitation. 2012;31(3):245-260.
22. Mechtler LL, Shastri KK, Crutchfield KE. Advanced neuroimaging of mild traumatic brain injury. Neurol Clin. 2014;32(1):31-58.
23. Ashwal S, Tong KA, Ghosh N, et al. Application of advanced neuroimaging modalities in pediatric traumatic brain injury. J Child Neurol. 2014;29(12):1704-1717.
24. Munson S, Schroth E, Ernst M. The role of functional neuroimaging in pediatric brain injury. Pediatrics. 2006;117(4):1372-1381.
25. Wozniak JR, Krach L, Ward E, et al. Neurocognitive and neuroimaging correlates of pediatric traumatic brain injury: a diffusion tensor imaging (DTI) study. Arch Clin Neuropsychol. 2007;22(5):555-568.
1. Abdelhalim AN, Alberico RA. Pediatric neuroimaging. Neurol Clin. 2009;27(1):285-301, x.
2. Sehgal V, Delproposto Z, Haacke EM, et al. Clinical applications of neuroimaging with susceptibility-weighted imaging. J Magn Reson Imaging. 2005;22(4):439-450.
3. Bosemani T, Poretti A, Huisman TA. Susceptibility-weighted imaging in pediatric neuroimaging. J Magn Reson Imaging. 2014;40(3):530-544.
4. Cecil KM. Proton magnetic resonance spectroscopy: technique for the neuroradiologist. Neuroimaging Clin N Am. 2013;23(3):381-392.
5. Panigrahy A, Nelson MD Jr, Blüml S. Magnetic resonance spectroscopy in pediatric neuroradiology: clinical and research applications. Pediatr Radiol. 2010;40(1):3-30.
6. Leentjens AF, Schieveld JN, Leonard M, et al. A comparison of the phenomenology of pediatric, adult, and geriatric delirium. J Psychosom Res. 2008;64(2):219-223.
7. Frodl T, Skokauskas N. Meta-analysis of structural MRI studies in children and adults with attention deficit hyperactivity disorder indicates treatment effects. Acta Psychiatr Scand. 2012;125(2):114-126.
8. Shaw P, Malek M, Watson B, et al. Development of cortical surface area and gyrification in attention-deficit/hyperactivity disorder. Biol Psychiatry. 2012;72(3):191-197.
9. Phelan JA, Lowe LH, Glasier CM. Pediatric neurodegenerative white matter processes: leukodystrophies and beyond. Pediatr Radiol. 2008;38(7):729-749.
10. Strawn JR, Wegman CJ, Dominick KC, et al. Cortical surface anatomy in pediatric patients with generalized anxiety disorder. J Anxiety Disord. 2014;28(7):717-723.
11. Mueller SC, Aouidad A, Gorodetsky E, et al. Gray matter volume in adolescent anxiety: an impact of the brain-derived neurotrophic factor Val(66)Met polymorphism [Erratum in J Am Acad Child Adolesc Psychiatry. 2013;52(2):184-195]? J Am Acad Child Adolesc Psychiatry. 2013;52(2):184-195.
12. Strawn JR, Hamm L, Fitzgerald DA, et al. Neurostructural abnormalities in pediatric anxiety disorders. J Anxiety Disord. 2015;32:81-88.
13. Strawn JR, Dominick KC, Patino LR, et al. Neurobiology of pediatric anxiety disorders. Curr Behav Neurosci Reports. 2014;1(3):154-160.
14. Alexiou GA, Argyropoulou MI. Neuroimaging in childhood headache: a systematic review. Pediatr Radiol. 2013;43(7):777-784.
15. Giedd JN, Rapoport JL, Garvey MA, et al. MRI assessment of children with obsessive-compulsive disorder or tics associated with streptococcal infection. Am J Psychiatry. 2000;157(2):281-283.
16. Chang K, Frankovich J, Cooperstock M, et al; PANS Collaborative Consortium. Clinical evaluation of youth with pediatric acute-onset neuropsychiatric syndrome (PANS): recommendations from the 2013 PANS Consensus Conference. J Child Adolesc Psychopharmacol. 2014;25(1):3-13.
17. Wallace GL, Robustelli B, Dankner N, et al. Increased gyrification, but comparable surface area in adolescents with autism spectrum disorders. Brain. 2013;136(pt 6):1956-1967.
18. Libero LE, DeRamus TP, Deshpande HD, et al. Surface-based morphometry of the cortical architecture of autism spectrum disorders: volume, thickness, area, and gyrification. Neuropsychologia. 2014;62:1-10.
19. Boddaert N, Zilbovicius M, Philipe A, et al. MRI findings in 77 children with non-syndromic autistic disorder. PLoS One. 2009;4:e445. doi: 10.1371/journal.pone.0004415.
20. Richards C, Jones C, Groves L, et al. Prevalence of autism spectrum disorder phenomenology in genetic disorders: a systematic review and meta-analysis. Lancet Psychiatry. 2015;2(10):909-916.
21. Wilde EA, Hunter JV, Bigler ED. Pediatric traumatic brain injury: neuroimaging and neurorehabilitation outcome. NeuroRehabilitation. 2012;31(3):245-260.
22. Mechtler LL, Shastri KK, Crutchfield KE. Advanced neuroimaging of mild traumatic brain injury. Neurol Clin. 2014;32(1):31-58.
23. Ashwal S, Tong KA, Ghosh N, et al. Application of advanced neuroimaging modalities in pediatric traumatic brain injury. J Child Neurol. 2014;29(12):1704-1717.
24. Munson S, Schroth E, Ernst M. The role of functional neuroimaging in pediatric brain injury. Pediatrics. 2006;117(4):1372-1381.
25. Wozniak JR, Krach L, Ward E, et al. Neurocognitive and neuroimaging correlates of pediatric traumatic brain injury: a diffusion tensor imaging (DTI) study. Arch Clin Neuropsychol. 2007;22(5):555-568.

