User login
Screen for portopulmonary hypertension, especially in liver transplant candidates
Portopulmonary hypertension poses difficulties for patients with liver disease. The elevated pulmonary artery pressure in this disorder makes liver transplantation more dangerous and in fact may rule out the procedure, although in a selected few patients, medical treatment may enable transplantation to proceed. In any event, portopulmonary hypertension should be looked for in patients with liver disease, especially if liver transplantation is being considered.
In this article we discuss the definition, pathophysiology, clinical features, diagnosis, and management of portopulmonary hypertension.
DEFINED BY HEMODYNAMIC CRITERIA
Portopulmonary hypertension—elevated pulmonary artery pressure due to increased resistance to blood flow in patients with portal hypertension—is one of several pulmonary complications of liver disease. A few others to be aware of are pleural effusions (hepatic hydrothorax), dilatation of the pulmonary vasculature with shunting and hypoxemia (hepatopulmonary syndrome), and elevation in pulmonary pressures due to the high cardiac output usually seen in liver disease (flow phenomenon).
The definition of portopulmonary hypertension has evolved as the various hemodynamic profiles that occur in liver disease and their consequences have been described. Currently, it is defined by the following criteria (obtained by right heart catheterization) in a patient with portal hypertension1:
- Elevated mean pulmonary artery pressure (> 25 mm Hg at rest, > 30 mm Hg with exercise);
- Increased pulmonary vascular resistance (> 240 dynes.s.cm−5; pulmonary vascular resistance = [(mean pulmonary artery pressure minus pulmonary artery occlusion pressure) /cardiac output] times 80); and
- Normal pulmonary artery occlusion pressure (< 15 mm Hg) or an elevated transpulmonary gradient (the mean pulmonary artery pressure minus the pulmonary artery occlusion pressure; abnormal is > 12 mm Hg).
The transpulmonary gradient sometimes helps in further assessing the resistance to blood flow in cases that do not meet the other criteria.2 For example, how should we classify a patient whose mean pulmonary artery pressure is 45 mm Hg but whose pulmonary vascular resistance is only 432 dynes.s.cm−5 and whose pulmonary artery occlusion pressure is slightly high at 18 mm Hg? Although this patient does not meet the hemodynamic criteria for portopulmonary hypertension listed above, intuitively, we should not exclude the diagnosis, as the transpulmonary gradient is high at 27 mm Hg.
FLOW PHENOMENON VS TRUE PORTOPULMONARY HYPERTENSION
The cardiopulmonary hemodynamic profile is different in patients with liver disease than in those without liver disease. Understanding the “normal” hemodynamics in liver disease is paramount in understanding the abnormal hemodynamics that occur in portopulmonary hypertension. In general, patients with liver disease have a high cardiac output at baseline (high flow). They may also have an increased blood volume due to fluid shifts (elevated pulmonary artery occlusion pressure).
Right heart catheterization is necessary to make the diagnosis of portopulmonary hypertension, as pulmonary artery pressures may be increased simply from increases in cardiac output and blood volume without an increase in pulmonary vascular resistance.
Consider, for example, a patient whose mean pulmonary artery pressure is 38 mm Hg, pulmonary artery occlusion pressure 14 mm Hg, and cardiac output 8.8 L/minute. In this case, the pulmonary vascular resistance is 218 dynes.s.cm−5. About 30% to 50% of patients with cirrhosis have this type of hyperdynamic pattern, with high cardiac output, low systemic vascular resistance, and low pulmonary vascular resistance.1,3,4 These patients typically have a much better prognosis than those with portopulmonary hypertension and do well with liver transplantation.
Right heart catheterization is also helpful in assessing whether elevated pulmonary pressures are due to increased volume (increased pulmonary artery occlusion pressure), in which case the patient might benefit from more aggressive diuresis.
In true portopulmonary hypertension, the pulmonary vascular resistance is increased due to obstruction of arterial blood flow. Cardiac output may be elevated initially and then decline as pulmonary hypertension becomes more severe. These hemodynamic patterns have different treatment implications and are important when liver transplantation is being considered.5
HOW COMMON IS PORTOPULMONARY HYPERTENSION?
The incidence and prevalence of portopulmonary hypertension is difficult to assess, as many of the estimates are in patients with severe liver disease undergoing evaluation for liver transplantation. Its prevalence in patients with cirrhosis and refractory ascites has been documented at 16.1%,6 while its prevalence in patients with cirrhosis without refractory ascites has been in the range of 0.25% to 4%.7–9
Overall, about 8% of candidates for liver transplantation have portopulmonary hypertension and are at risk of its complications.10 In view of this figure, screening for it should be performed before proceeding with liver transplantation.
VASOCONSTRICTION, REMODELING, THROMBOSIS
The pathogenesis of portopulmonary hypertension is not completely understood but likely involves a complex interaction of several mechanisms, including an imbalance of vascular mediators favoring vasoconstriction,11–13 endothelial damage with vascular remodeling due to excessive pulmonary blood flow,14,15 smooth muscle proliferation, and microvascular thrombosis.16,17
The pulmonary endothelium is a complex, dynamic organ capable of influencing a variety of vascular mediators and adapting to changes in pulmonary volume as necessary. Endothelial dysfunction may initiate the vascular changes seen in portopulmonary hypertension.
Endothelin-1 (ET-1) is a potent vasoconstrictor that has been implicated in the pathogenesis of idiopathic pulmonary artery hypertension. ET-1 levels are also increased in cirrhotic patients with refractory ascites.6
Other mediators favoring vasoconstriction include serotonin, angiotensin II, and norepinephrine. Whether these mediators influence the development of portopulmonary hypertension is not clear.
At the same time, production of vasodilatory mediators such as nitric oxide and prostacyclin may be decreased in portopulmonary hypertension, facilitating vascular remodeling and a proliferative vascular response. Prostacyclin is a potent vasodilator normally found in high concentrations in the lungs. Prostacyclin synthase is the precursor enzyme for the production of prostacyclin and is decreased in the lungs of patients with portopulmonary hypertension.18
Another way that portal hypertension may influence lung vascular tone is that endotoxin, cytokines, or both, released from the splanchnic circulation, may bypass the liver and get into the lungs.19 Evidence in support of this is that patients with portosystemic shunting can develop similar pathologic changes in the pulmonary vascular bed that normalize when the shunt is reversed. To date, however, no substance has been definitively identified.
Yet another proposed mechanism is shear stress on the pulmonary endothelium from the hyperdynamic cardiac output, with resultant vascular remodeling; however, other mechanisms must be involved, as not everyone with liver disease develops portopulmonary hypertension (see below).
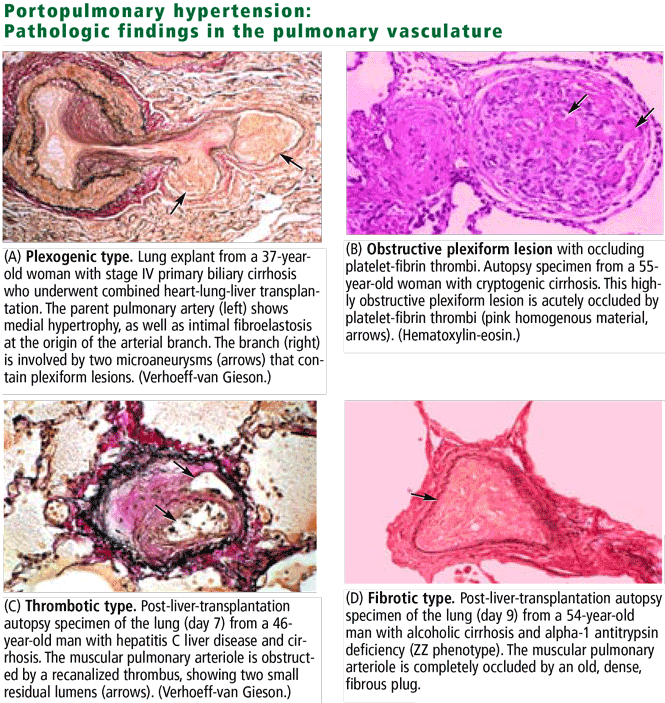
These changes are identical to those in idiopathic and familial pulmonary arterial hypertension,21 and indeed, the World Health Organization now classifies portopulmonary hypertension in the same category as these primary forms of pulmonary hypertension rather than in the secondary forms.3
Why doesn’t everyone with liver disease develop portopulmonary hypertension?
The severity of liver disease or degree of portal hypertension does not appear to correlate with the severity of pulmonary hypertension,4 and portopulmonary hypertension does not develop in all patients with portal hypertension. Therefore, it is likely that some patients have a genetic or environmental susceptibility or suffer a “second hit” that triggers dysregulated pulmonary vascular proliferation and contributes to the development of pulmonary hypertension.
Whether genetic mutations play a role in portopulmonary hypertension remains unknown. Such a mutation could be similar to the one identified in the bone morphogenetic protein receptor type 2 gene (BMPR2) in familial pulmonary artery hypertension or the mutation in the activin-like kinase gene (ALK1) seen in pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia.22
Current studies are investigating the role that bone-marrow-derived progenitor cells might play in the pathogenesis of portopulmonary hypertension.
CLINICAL FEATURES MAY NOT BE OBVIOUS AT FIRST
In the early stages of portopulmonary hypertension, patients may have no symptoms or only symptoms of liver disease, so it is important to have a high index of suspicion and screen for pulmonary hypertension. As its severity increases, symptoms may include fatigue, dyspnea, abdominal bloating, palpitations, chest pain or pressure, and syncope. The most common presenting symptom is dyspnea on exertion.
Similarly, the findings on physical examination also depend on the severity of pulmonary hypertension. Patients with mild portopulmonary hypertension may have only signs suggesting liver disease, such as spider telangiectases, jaundice, mild lower extremity edema, and ascites. As the severity of portopulmonary hypertension increases, however, findings of right heart pressure-and-volume overload become more obvious. These include peripheral edema, elevation of the jugular venous pressure, a right ventricular lift, a loud pulmonic valve closure, increased split of the second heart sound, a pulsatile liver, or a right-sided third or fourth heart sound.
SCREEN LIVER TRANSPLANT CANDIDATES
Screening for portopulmonary hypertension should be mandatory in patients undergoing evaluation for liver transplantation. This condition increases the risk of perioperative death, so it is not acceptable to make the diagnosis in the operating room!5
Electrocardiographic abnormalities that may raise the suspicion of portopulmonary hypertension include right atrial or ventricular enlargement and a right bundle branch pattern.
Chest radiographic signs are enlarged central pulmonary arteries and cardiomegaly. These electrocardiographic and radiographic signs tend to reflect advanced pulmonary hypertension.
Pulmonary function testing is not generally helpful, but the diffusing capacity may be decreased.
B-type natriuretic peptide (BNP) measurement may be helpful. BNP is released from the ventricles when the ventricles become dilated (due to pressure or volume overload), as in left or right heart failure. BNP testing is clinically useful in monitoring the severity of disease and the efficacy of treatment in patients with pulmonary hypertension; its role in portopulmonary hypertension requires prospective study.23
Transthoracic Doppler echocardiography is an excellent screening test and should be performed in patients undergoing evaluation for liver transplantation to exclude pulmonary hypertension.1 Findings on echocardiography that suggest portopulmonary hypertension include elevation of right ventricular systolic pressure (RVSP), which is calculated from the peak tricuspid regurgitant velocity (TRV) using the modified Bernoulli equation and an estimate of right atrial pressure (RAP):
RVSP = 4(TRV)2 + RAP.
Right atrial pressure is estimated from the filling characteristics of the inferior vena cava.
Transthoracic Doppler echocardiography has a sensitivity of 97% and a specificity of 77% in diagnosing moderate to severe pulmonary hypertension in patients undergoing evaluation for liver transplantation.24 Using an RVSP cutoff of 40 mm Hg, the sensitivity of Doppler echocardiography is about 80%, specificity 96%, positive predictive value 60%, and negative predictive value 98%.25
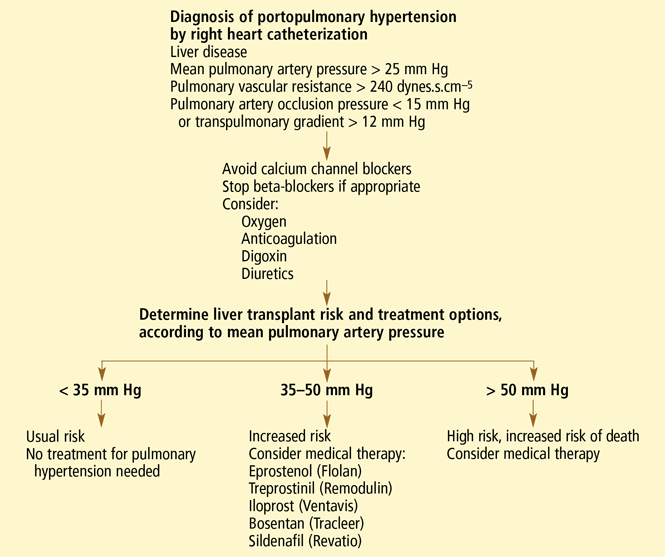
At Mayo Clinic, patients with an estimated RVSP greater than 50 mm Hg undergo right heart catheterization (see below). Such patients should also have repeat echocardiography at 1-year intervals to monitor for increasing pulmonary artery pressures5; for those on the waiting list for liver transplantation, the interval should probably be every 6 to 12 months.
RIGHT HEART CATHETERIZATION CONFIRMS THE DIAGNOSIS
The diagnosis of portopulmonary hypertension is confirmed with right heart catheterization to accurately measure pulmonary artery pressures, pulmonary artery occlusion pressure (to exclude volume overload), cardiac output (to exclude high-output pulmonary hypertension), and pulmonary vascular resistance. One study in patients with decompensated cirrhosis and refractory ascites found that a right atrial pressure of 14 mm Hg or greater had a positive predictive value of 83% for pulmonary hypertension.6
Other, potentially treatable causes of pulmonary hypertension must be excluded before diagnosing portopulmonary hypertension. These include thromboembolic disease, interstitial lung disease, connective tissue disease, untreated obstructive sleep apnea, and elevated pulmonary artery pressures due to increased cardiac output.
Vasodilator studies are being done less frequently in patients with portopulmonary hypertension, as they generally cannot tolerate calcium channel blocker therapy. Calcium channel blocker therapy is usually started in patients with idiopathic pulmonary artery hypertension who exhibit a positive vasodilator response. A positive vasodilator response also does not predict survival with or without liver transplantation. Unlike those with idiopathic pulmonary artery hypertension, many patients with portopulmonary hypertension cannot tolerate calcium channel blockers, as some of these drugs can exacerbate edema and portal hypertension.
GENERAL MANAGEMENT
Treatment of mild portopulmonary hypertension (mean pulmonary artery pressure < 35 mm Hg) is debatable. In these cases many patients do not have any symptoms attributable to portopulmonary hypertension, but only symptoms of liver disease, and they have a good functional status. As a group, such patients have not been formally studied to date.
Anticoagulation is often contraindicated in portopulmonary hypertension because of gastroesophageal varices, thrombocytopenia, or other coagulation abnormalities related to liver disease. If contraindications to anticoagulation do not exist, it should be considered.
Diuretics are a mainstay in the treatment of portopulmonary hypertension, both for the pulmonary hypertension and for the liver disease, especially if ascites or peripheral edema is present.
Oxygen should be given to patients with hypoxemia to keep the saturation greater than 90%.
Beta-blockers: A dilemma
Beta-blockers are used in many patients with liver disease as both primary and secondary prophylaxis of variceal bleeding.
However, one study has shown that in patients with moderate to severe portopulmonary hypertension, beta-blockers are associated with significant worsening of exercise capacity and pulmonary hemodynamic measurements.26 After beta-blockers were withdrawn, the 6-minute walking distance increased in 9 of 10 patients, and cardiac output increased with no change in mean pulmonary artery pressure, resulting in a 19% decrease in pulmonary vascular resistance. The increases in cardiac output were related to a 25% increase in heart rate. Long-term follow-up was not reported, and it remains unclear whether rates of gastrointestinal bleeding may increase when beta-blockers are withdrawn.
Beta-blocker therapy in portopulmonary hypertension needs to be carefully considered and if at all possible should be avoided.
VASODILATOR THERAPY
Several vasodilating or vasomodulating drugs are available. However, much of the information about them comes from studies in patients with idiopathic pulmonary artery hypertension or pulmonary hypertension due to connective tissue disease, and no randomized controlled trials in portopulmonary hypertension have been performed.
Prostanoids
Prostanoids have been used successfully to lower pulmonary pressures in portopulmonary hypertension.
Epoprostenol (Flolan) is a pulmonary and systemic vasodilator as well as an inhibitor of platelet aggregation. It is given as a continuous intravenous infusion via an indwelling central venous catheter and a portable infusion pump. It has a very short half-life, requires mixing, and must be kept cold with ice packs, making it somewhat cumbersome to administer.
This medication has been shown to improve cardiopulmonary hemodynamics and exercise capacity in portopulmonary hypertension, although a survival advantage has not been documented to date.27 In several case series, some patients with portopulmonary hypertension treated with intravenous epoprostenol responded with a reduction in pulmonary pressures and successfully underwent liver transplantation.28–31
Complications of intravenous epoprostenol therapy include central venous catheter thrombosis, infection, and infusion pump failure; a backup pump must be available at all times. Patients with portopulmonary hypertension may also develop progressive splenomegaly and thrombocytopenia that may be due to increased blood flow in the splanchnic circulation.32
Treprostinil (Remodulin) has a longer half-life and does not have to be kept cold. It is given as a 24-hour intravenous or subcutaneous infusion, using an infusion pump that is smaller than that used with epoprostenol.
Although treprostinil is easier for patients to use, larger doses are necessary to achieve the same effect as with epoprostenol. With subcutaneous administration, the biggest drawback is site pain. Prostacyclin-related side effects include flushing, diarrhea, jaw discomfort, and lower extremity pain.
Iloprost (Ventavis) has the advantage of being given by inhalation. It is very short-acting, however, and requires six to nine inhalations per day.
Endothelin receptor blockers
Bosentan (Tracleer) is an oral agent that has been approved by the US Food and Drug Administration (FDA) for the treatment of pulmonary hypertension, including in patients with portopulmonary hypertension who have mild hepatic derangement. This medication is a dual endothelin receptor antagonist, nonselectively blocking the endothelin A and B receptors on the endothelial and vascular smooth muscle cells so that ET-1 cannot bind and cause vasoconstriction.
In approximately 10% of patients, bosentan can cause elevations in aminotransferase, alkaline phosphatase, and bilirubin levels, which therefore must be checked monthly.33 Irreversible hepatic toxicity is uncommon; in most cases, liver function abnormalities return to baseline levels when the medication is stopped. The presumed mechanism is impairment of bile-salt transporters, leading to bile-salt accumulation in the liver.34 Bosentan’s use in patients with liver disease has not been well studied, although several case reports have described its use in patients with portopulmonary hypertension.35–38
Ambrisentan (Letairis) is a selective endothelin receptor-A blocker that has just received FDA approval for the treatment of pulmonary artery hypertension. It has not yet been studied in portopulmonary hypertension. Elevations in liver enzymes and bilirubin may also occur, and monthly monitoring is indicated.
Sildenafil
Another oral agent that might be effective in portopulmonary hypertension is sildenafil (Revatio). A phosphodiesterase-5 inhibitor, it selectively inhibits the cyclic guanosine monophosphatase-specific phosphodiesterase type 5 enzyme that is found in large concentrations in pulmonary artery smooth muscle cells.
In other forms of pulmonary hypertension, sildenafil has been shown to increase cardiac output and decrease pulmonary artery pressures and pulmonary vascular resistance without serious adverse events.39–41
In one reported case, treatment with sildenafil in a patient with portopulmonary hypertension decreased the mean pulmonary artery pressure from 56 mm Hg to 28 to 31 mm Hg, and the patient underwent successful liver transplantation.42 A recent case series of 14 patients with portopulmonary hypertension treated with sildenafil documents some improvement in 6-minute walking distance, suggesting that sildenafil as monotherapy or in combination therapy might be effective in portopulmonary hypertension.43 However, in 3 of these patients, the cardiac index decreased and pulmonary vascular resistance increased.44
We must emphasize that controlled studies in portopulmonary hypertension need to be done to find the optimal therapy.
LIVER TRANSPLANTATION MAY BENEFIT A FEW PATIENTS
Liver transplantation may be beneficial in highly selected patients with portopulmonary hypertension. However, this condition increases the risk of intraoperative and immediate postoperative complications of liver transplantation, so patients should be carefully evaluated5,45 at a liver transplantation center experienced in its management, including medical treatment with well-defined protocols regarding timing of liver transplantation.
Patients with mean pulmonary artery pressures greater than 50 mm Hg should not undergo liver transplantation. Those with mean pulmonary artery pressure between 35 and 50 mm Hg also have an increased mortality rate and may benefit from prolonged treatment for pulmonary hypertension.5,46
One successful case of living-related liver transplantation in a patient with portopulmonary hypertension has been published.47 (Most other successful transplants were from unrelated cadaver donors.)
Some patients who initially cannot undergo liver transplantation owing to severe pulmonary hypertension may eventually be able to do so if they receive medical therapy that improves their pulmonary hemodynamic profile, decreasing their mean pulmonary artery pressure and pulmonary vascular resistance. This would apply to a small subset of patients with portopulmonary hypertension.
When patients without pulmonary hypertension undergo liver transplantation, right ventricular function is preserved throughout all phases of the surgery.48 Patients with portopulmonary hypertension, however, may develop hemodynamic instability during liver transplantation. The most critical times are the induction of anesthesia, during and after graft reperfusion, and the immediate postoperative period.49,50
During the surgery, patients may require vasodilators if they have worsening pulmonary hypertension, or inotropic medications if they have right ventricular dysfunction and heart failure. In one study,51 eight patients with portopulmonary hypertension diagnosed at anesthesia induction for liver transplantation all required intraoperative vasodilator therapy after graft reperfusion because of marked increases in pulmonary artery pressures and pulmonary vascular resistance.
The increase in blood flow following reperfusion or necessary fluid challenges may exacerbate pulmonary hypertension, resulting in worsening right heart function and backup into the transplanted liver. Infusion of 1 liter of crystalloid over 10 minutes has been shown to increase mean pulmonary artery pressure and pulmonary artery occlusion pressure in liver transplantation candidates without pulmonary hypertension52; this response may be exaggerated in portopulmonary hypertension.
PROGNOSIS VARIES WITH SEVERITY OF DISEASE
The natural history of untreated portopulmonary hypertension varies with the degree of liver disease and the severity of pulmonary hypertension. Transplant-free survival was 85% at 1 year and 38% at 3 years in one study.45 The cardiac index appears to be the most significant prognostic variable.20
In a retrospective study of 78 patients with portopulmonary hypertension treated conservatively (before prostanoids were available) the median survival was 6 months (range 0–84 months) from the time of diagnosis.53 Causes of death included right heart failure, sudden death, gastrointestinal bleeding, and small bowel perforation.
Most of the data on outcomes of drug treatment and liver transplantation in patients with portopulmonary hypertension come from case series and retrospective reviews; prospective trials have been lacking.
If right ventricular function is normal and pulmonary hypertension is mild (mean pulmonary artery pressure < 35 mm Hg), patients tend to do well with liver transplantation.9
Outcomes are worse if pulmonary hypertension is more severe. In a database54 from 10 liver transplant centers from 1996 to 2001, 13 (36%) of 36 patients undergoing liver transplantation died in the hospital, emphasizing the importance of accurately assessing the severity of pulmonary hypertension before attempting liver transplantation.46 The rate was even higher—92%—in those with a mean pulmonary artery pressure greater than 35 mm Hg. The cause of death in severe pulmonary hypertension was failure of the right ventricle.
However, some patients with moderate to severe portopulmonary hypertension have been bridged with medications to lower pulmonary artery pressures and pulmonary vascular resistance so that liver transplantation can be safely done, and some have even been able to discontinue medications because their pulmonary hypertension resolved.29,31,41,42,47
Unlike in hepatopulmonary syndrome, liver transplantation is not the treatment of choice for portopulmonary hypertension, and pulmonary hypertension does not always resolve after liver transplantation. Many patients continue therapy for pulmonary hypertension after liver transplantation. Pulmonary hypertension may resolve, persist, or even develop de novo after liver transplantation.1 If pulmonary hypertension resolves, it does so over a prolonged time—months to years—favoring a vascular remodeling hypothesis as opposed to simply reversing vasoconstriction.
- Rodriguez-Roisin R, Krowka MJ, Hervé P, Fallon MB; ERS Task Force Pulmonary-Hepatic Vascular Disorders (PHD) Scientific Committee. Pulmonary-hepatic vascular disorders (PHD). Eur Respir J 2004; 24:861–880.
- Krowka MJ, Swanson KL, Frantz RP, et al. Portopulmonary hypertension: results from a 10-year screening algorithm. Hepatology 2006; 44:1502–1510.
- Simonneau G, Galie N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004; 43:5S–12S.
- Hadengue A, Benhayoun MK, Lebrec D, et al. Pulmonary hypertension complicating portal hypertension: prevalence and relation to splanchnic hemodynamics. Gastroenterology 1991; 100:520–528.
- Krowka MJ, Plevak DJ, Findlay JY, et al. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transplant 2000; 6:443–450.
- Benjaminov FS, Prentice M, Sniderman KW, et al. Portopulmonary hypertension in decompensated cirrhosis with refractory ascites. Gut 2003; 52:1355–1362.
- McDonnell PJ, Toye PA, Hutchins GM. Primary pulmonary hypertension and cirrhosis: are they related? Am Rev Respir Dis 1983; 127:437–441.
- Cheng EY, Woehlck H. Pulmonary artery hypertension complicating anesthesia for liver transplantation. Anesthesiology 1992; 77:375–378.
- Castro M, Krowka MJ, Schroeder DR, et al. Frequency and clinical implications of increased pulmonary artery pressures in liver transplantation. Mayo Clin Proc 1996; 71:543–551.
- Ramsay MA, Simpson BR, Nguyen AT, et al. Severe pulmonary hypertension in liver transplant candidates. Liver Transplant Surg 1997; 3:494–500.
- Kiely DG, Cargill RI, Struthers AD, et al. Cardiopulmonary effects of endothelin-1 in man. Cardiovasc Res 1997; 33:378–386.
- Panos RJ, Baker SK. Mediators, cytokines, and growth factors in liver-lung interactions. Clin Chest Med 1996; 17:151–169.
- Higgenbottam T. Pathophysiology of pulmonary hypertension. Chest 1994; 105:7S–12S.
- Krowka MJ. Hepatopulmonary syndrome and portopulmonary hypertension: distinction and dilemmas. Hepatology 1997; 25:1282–1284.
- Hongqun L, Lee SS. Cardiopulmonary dysfunction in cirrhosis. Hepatology 2000; 14:600–608.
- Lebrec D, Brenot F, Simonneau G, et al. Pulmonary arterial hypertension in portal hypertension. Eur Respir J 1998; 11:1153–1166.
- Herve P, Lebrec D, Brenot F, et al. Pulmonary vascular disorders in portal hypertension. Eur Respir J 1998; 11:1153–1166.
- Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 1999; 159:1925–1932.
- Hoeper MM, Krowka MJ, Strassburg CP. Portopulmonary hypertension and hepatopulmonary syndrome. Lancet 2004; 363:1461–1468.
- Edwards B, Weir K, Edwards WD, et al. Coexistent pulmonary and portal hypertension: morphologic and clinical features. J Am Coll Cardiol 1987; 10:1233–1238.
- Ramsay MAE, Simpson BR, Nguyen AT, Ramsay KJ, East C, Klintmalm GB. Severe pulmonary hypertension in liver transplant candidates. Liver Transplant Surg 1997; 3:494–500.
- Trembath RC. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001; 345:325–334.
- Leuchte HH, Holzapfel M, Baumgartner RA, et al. Clinical significance of brain natriuretic peptide in primary pulmonary hypertension. J Am Coll Cardiol 2004; 43:764–770.
- Kim WR, Krowka MJ, Plevak DJ, et al. Accuracy of Doppler echocardiography in the assessment of pulmonary hypertension in liver transplant candidates. Liver Transplant 2000; 6:453–458.
- Colle IO, Moreau R, Godinho E, et al. Diagnosis of portopulmonary hypertension in candidates for liver transplantation: a prospective study. Hepatology 2003; 37:401–409.
- Provencher S, Herve P, Jais X, et al. Deleterious effects of beta-blockers on exercise capacity and hemodynamics in patients with portopulmonary hypertension. Gastroenterology 2006; 130:120–126.
- Swanson KL, McGoon MD, Krowka MJ. Survival in patients with portopulmonary hypertension [abstract]. Am J Respir Crit Care Med 2003; 167:A693.
- Kuo PC, Johnson LB, Plotkin JS, et al. Continuous intravenous infusion of epoprostenol for the treatment of portopulmonary hypertension. Transplantation 1997; 63:604–616.
- Krowka MJ, Frantz RP, McGoon MD, et al. Improvement in pulmonary hemodynamics during intravenous epoprostenol (prostacyclin): A study of 15 patients with moderate to severe portopulmonary hypertension. Hepatology 1999; 30:641–648.
- Kähler CM, Graziadei I, Wiedermann CJ, Kneussl MP, Vogel W. Successful use of continuous intravenous prostacyclin in a patient with severe portopulmonary hypertension. Wien Klin Wochenschr 2000; 112:637–640.
- Sussman N, Kaza V, Barshes N, et al. Successful liver transplantation following medical management of portopulmonary hypertension: a single-center series. Am J Transplant 2006; 6:2177–2182.
- Findlay JY, Plevak DJ, Krowka MJ, et al. Progressive splenomegaly after epoprostenol therapy in portopulmonary hypertension. Liver Transplant Surg 1999; 5:381–387.
- Rubin LJ, Roux S. Bosentan: a dual endothelin receptor antagonist. Expert Opin Invest Drugs 2002; 11:991–1002.
- Fattinger K, Funk C, Pantze M, et al. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther 2001; 69:223–231.
- Hinterhuber L, Graziadei IW, Kahler CM, et al. Endothelin-receptor anatgonist treatment of portopulmonary hypertension. Clin Gastroenterol Hepatol 2004; 2:1039–1042.
- Clift PF, Townend JN, Bramhall S, et al. Successful treatment of severe portopulmonary hypertension after liver transplantation by bosentan. Transplantation 2004; 77:1774–1775.
- Halank M, Miehlke S, Hoeffken G, et al. Use of oral endothelin-receptor antagonist bosentan in the treatment of portopulmonary hypertension. Transplantation 2004; 77:1775–1776.
- Kuntzen C, Gulberg V, Gerbes AL. Use of a mixed endothelin receptor antagonist in portopulmonary hypertension: a safe and effective therapy? Gastroenterology 2005; 128:164–168.
- Watanabe H, Ohashi K, Takeuchi K, et al. Sildenafil for primary and secondary pulmonary hypertension. Clin Pharmacol Ther 2002; 71:398–402.
- Michelakis E, Tymchak W, Lien D, et al. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: comparison with inhaled nitric oxide. Circulation 2002; 105:2398–2403.
- Ghofrani HA, Wiedemann R, Rose F, et al. Sildenafil for treatment of lung fibrosis and pulmonary hypertension: a randomised controlled trial. Lancet 2002; 360:895–900.
- Makisalo H, Koivusalo A, Vakkuri A, et al. Sildenafil for portopulmonary hypertension in a patient undergoing liver transplantation. Liver Transplant 2004; 10:945–950.
- Reichengerger F, Voswinckel R, Steveling E, et al. Sildenafil treatment for portopulmonary hypertension. Eur Respir J 2006; 28:563–567.
- Krowka MJ, Swanson KL. How should we treat portopulmonary hypertension? Eur Respir J 2006; 28:466–467.
- Kawut SM, Taichman DB, Ahya VN, et al. Hemodynamics and survival of patients with portopulmonary hypertension. Liver Transplant 2005; 11:1107–1111.
- Krowka MJ, Mandell MS, Ramsay MA, et al. Hepatopulmonary syndrome and portopulmonary hypertension: a report of the multicenter liver transplant database. Liver Transplant 2004; 10:174–182.
- Sulica R, Emre S, Poon M. Medical management of portopulmonary hypertension and right heart failure prior to living-related liver transplantation. Congest Heart Fail 2004; 10:192–194.
- De Wolf AM, Begliomini B, Gasior TA, et al. Right ventricular function during orthotopic liver transplantation. Anesthes Analges 1993; 76:562–568.
- Csete M. Intraoperative management of liver transplant patients with pulmonary hypertension. Liver Transplant Surg 1997; 3:454–455.
- Acosta F, Sansano T, Palenciano CG, et al. Portopulmonary hypertension and liver transplantation: hemodynamic consequences at reperfusion. Transplant Proc 2005; 37:3865–3866.
- Taura P, Garcia-Valdecasas JC, Beltran J, et al. Moderate primary pulmonary hypertension in patients undergoing liver transplantation. Anesthes Analges 1996; 83:675–680.
- Kuo PC, Schroeder RA, Vagelos RH, et al. Volume-mediated pulmonary responses in liver transplant candidates. Clin Transplant 1996; 10:521–527.
- Robalino BD, Moodie DS. Association between primary pulmonary hypertension and portal hypertension: analysis of its pathophysiology and clinical, laboratory and hemodynamic manifestations. J Am Coll Cardiol 1991; 17:492–498.
- Mandell MS, Krowka MJ. Formation of a national database on pulmonary hypertension and hepatopulmonary syndrome in chronic liver disease. Anesthesiology 1997; 87:450–451.
Portopulmonary hypertension poses difficulties for patients with liver disease. The elevated pulmonary artery pressure in this disorder makes liver transplantation more dangerous and in fact may rule out the procedure, although in a selected few patients, medical treatment may enable transplantation to proceed. In any event, portopulmonary hypertension should be looked for in patients with liver disease, especially if liver transplantation is being considered.
In this article we discuss the definition, pathophysiology, clinical features, diagnosis, and management of portopulmonary hypertension.
DEFINED BY HEMODYNAMIC CRITERIA
Portopulmonary hypertension—elevated pulmonary artery pressure due to increased resistance to blood flow in patients with portal hypertension—is one of several pulmonary complications of liver disease. A few others to be aware of are pleural effusions (hepatic hydrothorax), dilatation of the pulmonary vasculature with shunting and hypoxemia (hepatopulmonary syndrome), and elevation in pulmonary pressures due to the high cardiac output usually seen in liver disease (flow phenomenon).
The definition of portopulmonary hypertension has evolved as the various hemodynamic profiles that occur in liver disease and their consequences have been described. Currently, it is defined by the following criteria (obtained by right heart catheterization) in a patient with portal hypertension1:
- Elevated mean pulmonary artery pressure (> 25 mm Hg at rest, > 30 mm Hg with exercise);
- Increased pulmonary vascular resistance (> 240 dynes.s.cm−5; pulmonary vascular resistance = [(mean pulmonary artery pressure minus pulmonary artery occlusion pressure) /cardiac output] times 80); and
- Normal pulmonary artery occlusion pressure (< 15 mm Hg) or an elevated transpulmonary gradient (the mean pulmonary artery pressure minus the pulmonary artery occlusion pressure; abnormal is > 12 mm Hg).
The transpulmonary gradient sometimes helps in further assessing the resistance to blood flow in cases that do not meet the other criteria.2 For example, how should we classify a patient whose mean pulmonary artery pressure is 45 mm Hg but whose pulmonary vascular resistance is only 432 dynes.s.cm−5 and whose pulmonary artery occlusion pressure is slightly high at 18 mm Hg? Although this patient does not meet the hemodynamic criteria for portopulmonary hypertension listed above, intuitively, we should not exclude the diagnosis, as the transpulmonary gradient is high at 27 mm Hg.
FLOW PHENOMENON VS TRUE PORTOPULMONARY HYPERTENSION
The cardiopulmonary hemodynamic profile is different in patients with liver disease than in those without liver disease. Understanding the “normal” hemodynamics in liver disease is paramount in understanding the abnormal hemodynamics that occur in portopulmonary hypertension. In general, patients with liver disease have a high cardiac output at baseline (high flow). They may also have an increased blood volume due to fluid shifts (elevated pulmonary artery occlusion pressure).
Right heart catheterization is necessary to make the diagnosis of portopulmonary hypertension, as pulmonary artery pressures may be increased simply from increases in cardiac output and blood volume without an increase in pulmonary vascular resistance.
Consider, for example, a patient whose mean pulmonary artery pressure is 38 mm Hg, pulmonary artery occlusion pressure 14 mm Hg, and cardiac output 8.8 L/minute. In this case, the pulmonary vascular resistance is 218 dynes.s.cm−5. About 30% to 50% of patients with cirrhosis have this type of hyperdynamic pattern, with high cardiac output, low systemic vascular resistance, and low pulmonary vascular resistance.1,3,4 These patients typically have a much better prognosis than those with portopulmonary hypertension and do well with liver transplantation.
Right heart catheterization is also helpful in assessing whether elevated pulmonary pressures are due to increased volume (increased pulmonary artery occlusion pressure), in which case the patient might benefit from more aggressive diuresis.
In true portopulmonary hypertension, the pulmonary vascular resistance is increased due to obstruction of arterial blood flow. Cardiac output may be elevated initially and then decline as pulmonary hypertension becomes more severe. These hemodynamic patterns have different treatment implications and are important when liver transplantation is being considered.5
HOW COMMON IS PORTOPULMONARY HYPERTENSION?
The incidence and prevalence of portopulmonary hypertension is difficult to assess, as many of the estimates are in patients with severe liver disease undergoing evaluation for liver transplantation. Its prevalence in patients with cirrhosis and refractory ascites has been documented at 16.1%,6 while its prevalence in patients with cirrhosis without refractory ascites has been in the range of 0.25% to 4%.7–9
Overall, about 8% of candidates for liver transplantation have portopulmonary hypertension and are at risk of its complications.10 In view of this figure, screening for it should be performed before proceeding with liver transplantation.
VASOCONSTRICTION, REMODELING, THROMBOSIS
The pathogenesis of portopulmonary hypertension is not completely understood but likely involves a complex interaction of several mechanisms, including an imbalance of vascular mediators favoring vasoconstriction,11–13 endothelial damage with vascular remodeling due to excessive pulmonary blood flow,14,15 smooth muscle proliferation, and microvascular thrombosis.16,17
The pulmonary endothelium is a complex, dynamic organ capable of influencing a variety of vascular mediators and adapting to changes in pulmonary volume as necessary. Endothelial dysfunction may initiate the vascular changes seen in portopulmonary hypertension.
Endothelin-1 (ET-1) is a potent vasoconstrictor that has been implicated in the pathogenesis of idiopathic pulmonary artery hypertension. ET-1 levels are also increased in cirrhotic patients with refractory ascites.6
Other mediators favoring vasoconstriction include serotonin, angiotensin II, and norepinephrine. Whether these mediators influence the development of portopulmonary hypertension is not clear.
At the same time, production of vasodilatory mediators such as nitric oxide and prostacyclin may be decreased in portopulmonary hypertension, facilitating vascular remodeling and a proliferative vascular response. Prostacyclin is a potent vasodilator normally found in high concentrations in the lungs. Prostacyclin synthase is the precursor enzyme for the production of prostacyclin and is decreased in the lungs of patients with portopulmonary hypertension.18
Another way that portal hypertension may influence lung vascular tone is that endotoxin, cytokines, or both, released from the splanchnic circulation, may bypass the liver and get into the lungs.19 Evidence in support of this is that patients with portosystemic shunting can develop similar pathologic changes in the pulmonary vascular bed that normalize when the shunt is reversed. To date, however, no substance has been definitively identified.
Yet another proposed mechanism is shear stress on the pulmonary endothelium from the hyperdynamic cardiac output, with resultant vascular remodeling; however, other mechanisms must be involved, as not everyone with liver disease develops portopulmonary hypertension (see below).
These changes are identical to those in idiopathic and familial pulmonary arterial hypertension,21 and indeed, the World Health Organization now classifies portopulmonary hypertension in the same category as these primary forms of pulmonary hypertension rather than in the secondary forms.3
Why doesn’t everyone with liver disease develop portopulmonary hypertension?
The severity of liver disease or degree of portal hypertension does not appear to correlate with the severity of pulmonary hypertension,4 and portopulmonary hypertension does not develop in all patients with portal hypertension. Therefore, it is likely that some patients have a genetic or environmental susceptibility or suffer a “second hit” that triggers dysregulated pulmonary vascular proliferation and contributes to the development of pulmonary hypertension.
Whether genetic mutations play a role in portopulmonary hypertension remains unknown. Such a mutation could be similar to the one identified in the bone morphogenetic protein receptor type 2 gene (BMPR2) in familial pulmonary artery hypertension or the mutation in the activin-like kinase gene (ALK1) seen in pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia.22
Current studies are investigating the role that bone-marrow-derived progenitor cells might play in the pathogenesis of portopulmonary hypertension.
CLINICAL FEATURES MAY NOT BE OBVIOUS AT FIRST
In the early stages of portopulmonary hypertension, patients may have no symptoms or only symptoms of liver disease, so it is important to have a high index of suspicion and screen for pulmonary hypertension. As its severity increases, symptoms may include fatigue, dyspnea, abdominal bloating, palpitations, chest pain or pressure, and syncope. The most common presenting symptom is dyspnea on exertion.
Similarly, the findings on physical examination also depend on the severity of pulmonary hypertension. Patients with mild portopulmonary hypertension may have only signs suggesting liver disease, such as spider telangiectases, jaundice, mild lower extremity edema, and ascites. As the severity of portopulmonary hypertension increases, however, findings of right heart pressure-and-volume overload become more obvious. These include peripheral edema, elevation of the jugular venous pressure, a right ventricular lift, a loud pulmonic valve closure, increased split of the second heart sound, a pulsatile liver, or a right-sided third or fourth heart sound.
SCREEN LIVER TRANSPLANT CANDIDATES
Screening for portopulmonary hypertension should be mandatory in patients undergoing evaluation for liver transplantation. This condition increases the risk of perioperative death, so it is not acceptable to make the diagnosis in the operating room!5
Electrocardiographic abnormalities that may raise the suspicion of portopulmonary hypertension include right atrial or ventricular enlargement and a right bundle branch pattern.
Chest radiographic signs are enlarged central pulmonary arteries and cardiomegaly. These electrocardiographic and radiographic signs tend to reflect advanced pulmonary hypertension.
Pulmonary function testing is not generally helpful, but the diffusing capacity may be decreased.
B-type natriuretic peptide (BNP) measurement may be helpful. BNP is released from the ventricles when the ventricles become dilated (due to pressure or volume overload), as in left or right heart failure. BNP testing is clinically useful in monitoring the severity of disease and the efficacy of treatment in patients with pulmonary hypertension; its role in portopulmonary hypertension requires prospective study.23
Transthoracic Doppler echocardiography is an excellent screening test and should be performed in patients undergoing evaluation for liver transplantation to exclude pulmonary hypertension.1 Findings on echocardiography that suggest portopulmonary hypertension include elevation of right ventricular systolic pressure (RVSP), which is calculated from the peak tricuspid regurgitant velocity (TRV) using the modified Bernoulli equation and an estimate of right atrial pressure (RAP):
RVSP = 4(TRV)2 + RAP.
Right atrial pressure is estimated from the filling characteristics of the inferior vena cava.
Transthoracic Doppler echocardiography has a sensitivity of 97% and a specificity of 77% in diagnosing moderate to severe pulmonary hypertension in patients undergoing evaluation for liver transplantation.24 Using an RVSP cutoff of 40 mm Hg, the sensitivity of Doppler echocardiography is about 80%, specificity 96%, positive predictive value 60%, and negative predictive value 98%.25
At Mayo Clinic, patients with an estimated RVSP greater than 50 mm Hg undergo right heart catheterization (see below). Such patients should also have repeat echocardiography at 1-year intervals to monitor for increasing pulmonary artery pressures5; for those on the waiting list for liver transplantation, the interval should probably be every 6 to 12 months.
RIGHT HEART CATHETERIZATION CONFIRMS THE DIAGNOSIS
The diagnosis of portopulmonary hypertension is confirmed with right heart catheterization to accurately measure pulmonary artery pressures, pulmonary artery occlusion pressure (to exclude volume overload), cardiac output (to exclude high-output pulmonary hypertension), and pulmonary vascular resistance. One study in patients with decompensated cirrhosis and refractory ascites found that a right atrial pressure of 14 mm Hg or greater had a positive predictive value of 83% for pulmonary hypertension.6
Other, potentially treatable causes of pulmonary hypertension must be excluded before diagnosing portopulmonary hypertension. These include thromboembolic disease, interstitial lung disease, connective tissue disease, untreated obstructive sleep apnea, and elevated pulmonary artery pressures due to increased cardiac output.
Vasodilator studies are being done less frequently in patients with portopulmonary hypertension, as they generally cannot tolerate calcium channel blocker therapy. Calcium channel blocker therapy is usually started in patients with idiopathic pulmonary artery hypertension who exhibit a positive vasodilator response. A positive vasodilator response also does not predict survival with or without liver transplantation. Unlike those with idiopathic pulmonary artery hypertension, many patients with portopulmonary hypertension cannot tolerate calcium channel blockers, as some of these drugs can exacerbate edema and portal hypertension.
GENERAL MANAGEMENT
Treatment of mild portopulmonary hypertension (mean pulmonary artery pressure < 35 mm Hg) is debatable. In these cases many patients do not have any symptoms attributable to portopulmonary hypertension, but only symptoms of liver disease, and they have a good functional status. As a group, such patients have not been formally studied to date.
Anticoagulation is often contraindicated in portopulmonary hypertension because of gastroesophageal varices, thrombocytopenia, or other coagulation abnormalities related to liver disease. If contraindications to anticoagulation do not exist, it should be considered.
Diuretics are a mainstay in the treatment of portopulmonary hypertension, both for the pulmonary hypertension and for the liver disease, especially if ascites or peripheral edema is present.
Oxygen should be given to patients with hypoxemia to keep the saturation greater than 90%.
Beta-blockers: A dilemma
Beta-blockers are used in many patients with liver disease as both primary and secondary prophylaxis of variceal bleeding.
However, one study has shown that in patients with moderate to severe portopulmonary hypertension, beta-blockers are associated with significant worsening of exercise capacity and pulmonary hemodynamic measurements.26 After beta-blockers were withdrawn, the 6-minute walking distance increased in 9 of 10 patients, and cardiac output increased with no change in mean pulmonary artery pressure, resulting in a 19% decrease in pulmonary vascular resistance. The increases in cardiac output were related to a 25% increase in heart rate. Long-term follow-up was not reported, and it remains unclear whether rates of gastrointestinal bleeding may increase when beta-blockers are withdrawn.
Beta-blocker therapy in portopulmonary hypertension needs to be carefully considered and if at all possible should be avoided.
VASODILATOR THERAPY
Several vasodilating or vasomodulating drugs are available. However, much of the information about them comes from studies in patients with idiopathic pulmonary artery hypertension or pulmonary hypertension due to connective tissue disease, and no randomized controlled trials in portopulmonary hypertension have been performed.
Prostanoids
Prostanoids have been used successfully to lower pulmonary pressures in portopulmonary hypertension.
Epoprostenol (Flolan) is a pulmonary and systemic vasodilator as well as an inhibitor of platelet aggregation. It is given as a continuous intravenous infusion via an indwelling central venous catheter and a portable infusion pump. It has a very short half-life, requires mixing, and must be kept cold with ice packs, making it somewhat cumbersome to administer.
This medication has been shown to improve cardiopulmonary hemodynamics and exercise capacity in portopulmonary hypertension, although a survival advantage has not been documented to date.27 In several case series, some patients with portopulmonary hypertension treated with intravenous epoprostenol responded with a reduction in pulmonary pressures and successfully underwent liver transplantation.28–31
Complications of intravenous epoprostenol therapy include central venous catheter thrombosis, infection, and infusion pump failure; a backup pump must be available at all times. Patients with portopulmonary hypertension may also develop progressive splenomegaly and thrombocytopenia that may be due to increased blood flow in the splanchnic circulation.32
Treprostinil (Remodulin) has a longer half-life and does not have to be kept cold. It is given as a 24-hour intravenous or subcutaneous infusion, using an infusion pump that is smaller than that used with epoprostenol.
Although treprostinil is easier for patients to use, larger doses are necessary to achieve the same effect as with epoprostenol. With subcutaneous administration, the biggest drawback is site pain. Prostacyclin-related side effects include flushing, diarrhea, jaw discomfort, and lower extremity pain.
Iloprost (Ventavis) has the advantage of being given by inhalation. It is very short-acting, however, and requires six to nine inhalations per day.
Endothelin receptor blockers
Bosentan (Tracleer) is an oral agent that has been approved by the US Food and Drug Administration (FDA) for the treatment of pulmonary hypertension, including in patients with portopulmonary hypertension who have mild hepatic derangement. This medication is a dual endothelin receptor antagonist, nonselectively blocking the endothelin A and B receptors on the endothelial and vascular smooth muscle cells so that ET-1 cannot bind and cause vasoconstriction.
In approximately 10% of patients, bosentan can cause elevations in aminotransferase, alkaline phosphatase, and bilirubin levels, which therefore must be checked monthly.33 Irreversible hepatic toxicity is uncommon; in most cases, liver function abnormalities return to baseline levels when the medication is stopped. The presumed mechanism is impairment of bile-salt transporters, leading to bile-salt accumulation in the liver.34 Bosentan’s use in patients with liver disease has not been well studied, although several case reports have described its use in patients with portopulmonary hypertension.35–38
Ambrisentan (Letairis) is a selective endothelin receptor-A blocker that has just received FDA approval for the treatment of pulmonary artery hypertension. It has not yet been studied in portopulmonary hypertension. Elevations in liver enzymes and bilirubin may also occur, and monthly monitoring is indicated.
Sildenafil
Another oral agent that might be effective in portopulmonary hypertension is sildenafil (Revatio). A phosphodiesterase-5 inhibitor, it selectively inhibits the cyclic guanosine monophosphatase-specific phosphodiesterase type 5 enzyme that is found in large concentrations in pulmonary artery smooth muscle cells.
In other forms of pulmonary hypertension, sildenafil has been shown to increase cardiac output and decrease pulmonary artery pressures and pulmonary vascular resistance without serious adverse events.39–41
In one reported case, treatment with sildenafil in a patient with portopulmonary hypertension decreased the mean pulmonary artery pressure from 56 mm Hg to 28 to 31 mm Hg, and the patient underwent successful liver transplantation.42 A recent case series of 14 patients with portopulmonary hypertension treated with sildenafil documents some improvement in 6-minute walking distance, suggesting that sildenafil as monotherapy or in combination therapy might be effective in portopulmonary hypertension.43 However, in 3 of these patients, the cardiac index decreased and pulmonary vascular resistance increased.44
We must emphasize that controlled studies in portopulmonary hypertension need to be done to find the optimal therapy.
LIVER TRANSPLANTATION MAY BENEFIT A FEW PATIENTS
Liver transplantation may be beneficial in highly selected patients with portopulmonary hypertension. However, this condition increases the risk of intraoperative and immediate postoperative complications of liver transplantation, so patients should be carefully evaluated5,45 at a liver transplantation center experienced in its management, including medical treatment with well-defined protocols regarding timing of liver transplantation.
Patients with mean pulmonary artery pressures greater than 50 mm Hg should not undergo liver transplantation. Those with mean pulmonary artery pressure between 35 and 50 mm Hg also have an increased mortality rate and may benefit from prolonged treatment for pulmonary hypertension.5,46
One successful case of living-related liver transplantation in a patient with portopulmonary hypertension has been published.47 (Most other successful transplants were from unrelated cadaver donors.)
Some patients who initially cannot undergo liver transplantation owing to severe pulmonary hypertension may eventually be able to do so if they receive medical therapy that improves their pulmonary hemodynamic profile, decreasing their mean pulmonary artery pressure and pulmonary vascular resistance. This would apply to a small subset of patients with portopulmonary hypertension.
When patients without pulmonary hypertension undergo liver transplantation, right ventricular function is preserved throughout all phases of the surgery.48 Patients with portopulmonary hypertension, however, may develop hemodynamic instability during liver transplantation. The most critical times are the induction of anesthesia, during and after graft reperfusion, and the immediate postoperative period.49,50
During the surgery, patients may require vasodilators if they have worsening pulmonary hypertension, or inotropic medications if they have right ventricular dysfunction and heart failure. In one study,51 eight patients with portopulmonary hypertension diagnosed at anesthesia induction for liver transplantation all required intraoperative vasodilator therapy after graft reperfusion because of marked increases in pulmonary artery pressures and pulmonary vascular resistance.
The increase in blood flow following reperfusion or necessary fluid challenges may exacerbate pulmonary hypertension, resulting in worsening right heart function and backup into the transplanted liver. Infusion of 1 liter of crystalloid over 10 minutes has been shown to increase mean pulmonary artery pressure and pulmonary artery occlusion pressure in liver transplantation candidates without pulmonary hypertension52; this response may be exaggerated in portopulmonary hypertension.
PROGNOSIS VARIES WITH SEVERITY OF DISEASE
The natural history of untreated portopulmonary hypertension varies with the degree of liver disease and the severity of pulmonary hypertension. Transplant-free survival was 85% at 1 year and 38% at 3 years in one study.45 The cardiac index appears to be the most significant prognostic variable.20
In a retrospective study of 78 patients with portopulmonary hypertension treated conservatively (before prostanoids were available) the median survival was 6 months (range 0–84 months) from the time of diagnosis.53 Causes of death included right heart failure, sudden death, gastrointestinal bleeding, and small bowel perforation.
Most of the data on outcomes of drug treatment and liver transplantation in patients with portopulmonary hypertension come from case series and retrospective reviews; prospective trials have been lacking.
If right ventricular function is normal and pulmonary hypertension is mild (mean pulmonary artery pressure < 35 mm Hg), patients tend to do well with liver transplantation.9
Outcomes are worse if pulmonary hypertension is more severe. In a database54 from 10 liver transplant centers from 1996 to 2001, 13 (36%) of 36 patients undergoing liver transplantation died in the hospital, emphasizing the importance of accurately assessing the severity of pulmonary hypertension before attempting liver transplantation.46 The rate was even higher—92%—in those with a mean pulmonary artery pressure greater than 35 mm Hg. The cause of death in severe pulmonary hypertension was failure of the right ventricle.
However, some patients with moderate to severe portopulmonary hypertension have been bridged with medications to lower pulmonary artery pressures and pulmonary vascular resistance so that liver transplantation can be safely done, and some have even been able to discontinue medications because their pulmonary hypertension resolved.29,31,41,42,47
Unlike in hepatopulmonary syndrome, liver transplantation is not the treatment of choice for portopulmonary hypertension, and pulmonary hypertension does not always resolve after liver transplantation. Many patients continue therapy for pulmonary hypertension after liver transplantation. Pulmonary hypertension may resolve, persist, or even develop de novo after liver transplantation.1 If pulmonary hypertension resolves, it does so over a prolonged time—months to years—favoring a vascular remodeling hypothesis as opposed to simply reversing vasoconstriction.
Portopulmonary hypertension poses difficulties for patients with liver disease. The elevated pulmonary artery pressure in this disorder makes liver transplantation more dangerous and in fact may rule out the procedure, although in a selected few patients, medical treatment may enable transplantation to proceed. In any event, portopulmonary hypertension should be looked for in patients with liver disease, especially if liver transplantation is being considered.
In this article we discuss the definition, pathophysiology, clinical features, diagnosis, and management of portopulmonary hypertension.
DEFINED BY HEMODYNAMIC CRITERIA
Portopulmonary hypertension—elevated pulmonary artery pressure due to increased resistance to blood flow in patients with portal hypertension—is one of several pulmonary complications of liver disease. A few others to be aware of are pleural effusions (hepatic hydrothorax), dilatation of the pulmonary vasculature with shunting and hypoxemia (hepatopulmonary syndrome), and elevation in pulmonary pressures due to the high cardiac output usually seen in liver disease (flow phenomenon).
The definition of portopulmonary hypertension has evolved as the various hemodynamic profiles that occur in liver disease and their consequences have been described. Currently, it is defined by the following criteria (obtained by right heart catheterization) in a patient with portal hypertension1:
- Elevated mean pulmonary artery pressure (> 25 mm Hg at rest, > 30 mm Hg with exercise);
- Increased pulmonary vascular resistance (> 240 dynes.s.cm−5; pulmonary vascular resistance = [(mean pulmonary artery pressure minus pulmonary artery occlusion pressure) /cardiac output] times 80); and
- Normal pulmonary artery occlusion pressure (< 15 mm Hg) or an elevated transpulmonary gradient (the mean pulmonary artery pressure minus the pulmonary artery occlusion pressure; abnormal is > 12 mm Hg).
The transpulmonary gradient sometimes helps in further assessing the resistance to blood flow in cases that do not meet the other criteria.2 For example, how should we classify a patient whose mean pulmonary artery pressure is 45 mm Hg but whose pulmonary vascular resistance is only 432 dynes.s.cm−5 and whose pulmonary artery occlusion pressure is slightly high at 18 mm Hg? Although this patient does not meet the hemodynamic criteria for portopulmonary hypertension listed above, intuitively, we should not exclude the diagnosis, as the transpulmonary gradient is high at 27 mm Hg.
FLOW PHENOMENON VS TRUE PORTOPULMONARY HYPERTENSION
The cardiopulmonary hemodynamic profile is different in patients with liver disease than in those without liver disease. Understanding the “normal” hemodynamics in liver disease is paramount in understanding the abnormal hemodynamics that occur in portopulmonary hypertension. In general, patients with liver disease have a high cardiac output at baseline (high flow). They may also have an increased blood volume due to fluid shifts (elevated pulmonary artery occlusion pressure).
Right heart catheterization is necessary to make the diagnosis of portopulmonary hypertension, as pulmonary artery pressures may be increased simply from increases in cardiac output and blood volume without an increase in pulmonary vascular resistance.
Consider, for example, a patient whose mean pulmonary artery pressure is 38 mm Hg, pulmonary artery occlusion pressure 14 mm Hg, and cardiac output 8.8 L/minute. In this case, the pulmonary vascular resistance is 218 dynes.s.cm−5. About 30% to 50% of patients with cirrhosis have this type of hyperdynamic pattern, with high cardiac output, low systemic vascular resistance, and low pulmonary vascular resistance.1,3,4 These patients typically have a much better prognosis than those with portopulmonary hypertension and do well with liver transplantation.
Right heart catheterization is also helpful in assessing whether elevated pulmonary pressures are due to increased volume (increased pulmonary artery occlusion pressure), in which case the patient might benefit from more aggressive diuresis.
In true portopulmonary hypertension, the pulmonary vascular resistance is increased due to obstruction of arterial blood flow. Cardiac output may be elevated initially and then decline as pulmonary hypertension becomes more severe. These hemodynamic patterns have different treatment implications and are important when liver transplantation is being considered.5
HOW COMMON IS PORTOPULMONARY HYPERTENSION?
The incidence and prevalence of portopulmonary hypertension is difficult to assess, as many of the estimates are in patients with severe liver disease undergoing evaluation for liver transplantation. Its prevalence in patients with cirrhosis and refractory ascites has been documented at 16.1%,6 while its prevalence in patients with cirrhosis without refractory ascites has been in the range of 0.25% to 4%.7–9
Overall, about 8% of candidates for liver transplantation have portopulmonary hypertension and are at risk of its complications.10 In view of this figure, screening for it should be performed before proceeding with liver transplantation.
VASOCONSTRICTION, REMODELING, THROMBOSIS
The pathogenesis of portopulmonary hypertension is not completely understood but likely involves a complex interaction of several mechanisms, including an imbalance of vascular mediators favoring vasoconstriction,11–13 endothelial damage with vascular remodeling due to excessive pulmonary blood flow,14,15 smooth muscle proliferation, and microvascular thrombosis.16,17
The pulmonary endothelium is a complex, dynamic organ capable of influencing a variety of vascular mediators and adapting to changes in pulmonary volume as necessary. Endothelial dysfunction may initiate the vascular changes seen in portopulmonary hypertension.
Endothelin-1 (ET-1) is a potent vasoconstrictor that has been implicated in the pathogenesis of idiopathic pulmonary artery hypertension. ET-1 levels are also increased in cirrhotic patients with refractory ascites.6
Other mediators favoring vasoconstriction include serotonin, angiotensin II, and norepinephrine. Whether these mediators influence the development of portopulmonary hypertension is not clear.
At the same time, production of vasodilatory mediators such as nitric oxide and prostacyclin may be decreased in portopulmonary hypertension, facilitating vascular remodeling and a proliferative vascular response. Prostacyclin is a potent vasodilator normally found in high concentrations in the lungs. Prostacyclin synthase is the precursor enzyme for the production of prostacyclin and is decreased in the lungs of patients with portopulmonary hypertension.18
Another way that portal hypertension may influence lung vascular tone is that endotoxin, cytokines, or both, released from the splanchnic circulation, may bypass the liver and get into the lungs.19 Evidence in support of this is that patients with portosystemic shunting can develop similar pathologic changes in the pulmonary vascular bed that normalize when the shunt is reversed. To date, however, no substance has been definitively identified.
Yet another proposed mechanism is shear stress on the pulmonary endothelium from the hyperdynamic cardiac output, with resultant vascular remodeling; however, other mechanisms must be involved, as not everyone with liver disease develops portopulmonary hypertension (see below).
These changes are identical to those in idiopathic and familial pulmonary arterial hypertension,21 and indeed, the World Health Organization now classifies portopulmonary hypertension in the same category as these primary forms of pulmonary hypertension rather than in the secondary forms.3
Why doesn’t everyone with liver disease develop portopulmonary hypertension?
The severity of liver disease or degree of portal hypertension does not appear to correlate with the severity of pulmonary hypertension,4 and portopulmonary hypertension does not develop in all patients with portal hypertension. Therefore, it is likely that some patients have a genetic or environmental susceptibility or suffer a “second hit” that triggers dysregulated pulmonary vascular proliferation and contributes to the development of pulmonary hypertension.
Whether genetic mutations play a role in portopulmonary hypertension remains unknown. Such a mutation could be similar to the one identified in the bone morphogenetic protein receptor type 2 gene (BMPR2) in familial pulmonary artery hypertension or the mutation in the activin-like kinase gene (ALK1) seen in pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia.22
Current studies are investigating the role that bone-marrow-derived progenitor cells might play in the pathogenesis of portopulmonary hypertension.
CLINICAL FEATURES MAY NOT BE OBVIOUS AT FIRST
In the early stages of portopulmonary hypertension, patients may have no symptoms or only symptoms of liver disease, so it is important to have a high index of suspicion and screen for pulmonary hypertension. As its severity increases, symptoms may include fatigue, dyspnea, abdominal bloating, palpitations, chest pain or pressure, and syncope. The most common presenting symptom is dyspnea on exertion.
Similarly, the findings on physical examination also depend on the severity of pulmonary hypertension. Patients with mild portopulmonary hypertension may have only signs suggesting liver disease, such as spider telangiectases, jaundice, mild lower extremity edema, and ascites. As the severity of portopulmonary hypertension increases, however, findings of right heart pressure-and-volume overload become more obvious. These include peripheral edema, elevation of the jugular venous pressure, a right ventricular lift, a loud pulmonic valve closure, increased split of the second heart sound, a pulsatile liver, or a right-sided third or fourth heart sound.
SCREEN LIVER TRANSPLANT CANDIDATES
Screening for portopulmonary hypertension should be mandatory in patients undergoing evaluation for liver transplantation. This condition increases the risk of perioperative death, so it is not acceptable to make the diagnosis in the operating room!5
Electrocardiographic abnormalities that may raise the suspicion of portopulmonary hypertension include right atrial or ventricular enlargement and a right bundle branch pattern.
Chest radiographic signs are enlarged central pulmonary arteries and cardiomegaly. These electrocardiographic and radiographic signs tend to reflect advanced pulmonary hypertension.
Pulmonary function testing is not generally helpful, but the diffusing capacity may be decreased.
B-type natriuretic peptide (BNP) measurement may be helpful. BNP is released from the ventricles when the ventricles become dilated (due to pressure or volume overload), as in left or right heart failure. BNP testing is clinically useful in monitoring the severity of disease and the efficacy of treatment in patients with pulmonary hypertension; its role in portopulmonary hypertension requires prospective study.23
Transthoracic Doppler echocardiography is an excellent screening test and should be performed in patients undergoing evaluation for liver transplantation to exclude pulmonary hypertension.1 Findings on echocardiography that suggest portopulmonary hypertension include elevation of right ventricular systolic pressure (RVSP), which is calculated from the peak tricuspid regurgitant velocity (TRV) using the modified Bernoulli equation and an estimate of right atrial pressure (RAP):
RVSP = 4(TRV)2 + RAP.
Right atrial pressure is estimated from the filling characteristics of the inferior vena cava.
Transthoracic Doppler echocardiography has a sensitivity of 97% and a specificity of 77% in diagnosing moderate to severe pulmonary hypertension in patients undergoing evaluation for liver transplantation.24 Using an RVSP cutoff of 40 mm Hg, the sensitivity of Doppler echocardiography is about 80%, specificity 96%, positive predictive value 60%, and negative predictive value 98%.25
At Mayo Clinic, patients with an estimated RVSP greater than 50 mm Hg undergo right heart catheterization (see below). Such patients should also have repeat echocardiography at 1-year intervals to monitor for increasing pulmonary artery pressures5; for those on the waiting list for liver transplantation, the interval should probably be every 6 to 12 months.
RIGHT HEART CATHETERIZATION CONFIRMS THE DIAGNOSIS
The diagnosis of portopulmonary hypertension is confirmed with right heart catheterization to accurately measure pulmonary artery pressures, pulmonary artery occlusion pressure (to exclude volume overload), cardiac output (to exclude high-output pulmonary hypertension), and pulmonary vascular resistance. One study in patients with decompensated cirrhosis and refractory ascites found that a right atrial pressure of 14 mm Hg or greater had a positive predictive value of 83% for pulmonary hypertension.6
Other, potentially treatable causes of pulmonary hypertension must be excluded before diagnosing portopulmonary hypertension. These include thromboembolic disease, interstitial lung disease, connective tissue disease, untreated obstructive sleep apnea, and elevated pulmonary artery pressures due to increased cardiac output.
Vasodilator studies are being done less frequently in patients with portopulmonary hypertension, as they generally cannot tolerate calcium channel blocker therapy. Calcium channel blocker therapy is usually started in patients with idiopathic pulmonary artery hypertension who exhibit a positive vasodilator response. A positive vasodilator response also does not predict survival with or without liver transplantation. Unlike those with idiopathic pulmonary artery hypertension, many patients with portopulmonary hypertension cannot tolerate calcium channel blockers, as some of these drugs can exacerbate edema and portal hypertension.
GENERAL MANAGEMENT
Treatment of mild portopulmonary hypertension (mean pulmonary artery pressure < 35 mm Hg) is debatable. In these cases many patients do not have any symptoms attributable to portopulmonary hypertension, but only symptoms of liver disease, and they have a good functional status. As a group, such patients have not been formally studied to date.
Anticoagulation is often contraindicated in portopulmonary hypertension because of gastroesophageal varices, thrombocytopenia, or other coagulation abnormalities related to liver disease. If contraindications to anticoagulation do not exist, it should be considered.
Diuretics are a mainstay in the treatment of portopulmonary hypertension, both for the pulmonary hypertension and for the liver disease, especially if ascites or peripheral edema is present.
Oxygen should be given to patients with hypoxemia to keep the saturation greater than 90%.
Beta-blockers: A dilemma
Beta-blockers are used in many patients with liver disease as both primary and secondary prophylaxis of variceal bleeding.
However, one study has shown that in patients with moderate to severe portopulmonary hypertension, beta-blockers are associated with significant worsening of exercise capacity and pulmonary hemodynamic measurements.26 After beta-blockers were withdrawn, the 6-minute walking distance increased in 9 of 10 patients, and cardiac output increased with no change in mean pulmonary artery pressure, resulting in a 19% decrease in pulmonary vascular resistance. The increases in cardiac output were related to a 25% increase in heart rate. Long-term follow-up was not reported, and it remains unclear whether rates of gastrointestinal bleeding may increase when beta-blockers are withdrawn.
Beta-blocker therapy in portopulmonary hypertension needs to be carefully considered and if at all possible should be avoided.
VASODILATOR THERAPY
Several vasodilating or vasomodulating drugs are available. However, much of the information about them comes from studies in patients with idiopathic pulmonary artery hypertension or pulmonary hypertension due to connective tissue disease, and no randomized controlled trials in portopulmonary hypertension have been performed.
Prostanoids
Prostanoids have been used successfully to lower pulmonary pressures in portopulmonary hypertension.
Epoprostenol (Flolan) is a pulmonary and systemic vasodilator as well as an inhibitor of platelet aggregation. It is given as a continuous intravenous infusion via an indwelling central venous catheter and a portable infusion pump. It has a very short half-life, requires mixing, and must be kept cold with ice packs, making it somewhat cumbersome to administer.
This medication has been shown to improve cardiopulmonary hemodynamics and exercise capacity in portopulmonary hypertension, although a survival advantage has not been documented to date.27 In several case series, some patients with portopulmonary hypertension treated with intravenous epoprostenol responded with a reduction in pulmonary pressures and successfully underwent liver transplantation.28–31
Complications of intravenous epoprostenol therapy include central venous catheter thrombosis, infection, and infusion pump failure; a backup pump must be available at all times. Patients with portopulmonary hypertension may also develop progressive splenomegaly and thrombocytopenia that may be due to increased blood flow in the splanchnic circulation.32
Treprostinil (Remodulin) has a longer half-life and does not have to be kept cold. It is given as a 24-hour intravenous or subcutaneous infusion, using an infusion pump that is smaller than that used with epoprostenol.
Although treprostinil is easier for patients to use, larger doses are necessary to achieve the same effect as with epoprostenol. With subcutaneous administration, the biggest drawback is site pain. Prostacyclin-related side effects include flushing, diarrhea, jaw discomfort, and lower extremity pain.
Iloprost (Ventavis) has the advantage of being given by inhalation. It is very short-acting, however, and requires six to nine inhalations per day.
Endothelin receptor blockers
Bosentan (Tracleer) is an oral agent that has been approved by the US Food and Drug Administration (FDA) for the treatment of pulmonary hypertension, including in patients with portopulmonary hypertension who have mild hepatic derangement. This medication is a dual endothelin receptor antagonist, nonselectively blocking the endothelin A and B receptors on the endothelial and vascular smooth muscle cells so that ET-1 cannot bind and cause vasoconstriction.
In approximately 10% of patients, bosentan can cause elevations in aminotransferase, alkaline phosphatase, and bilirubin levels, which therefore must be checked monthly.33 Irreversible hepatic toxicity is uncommon; in most cases, liver function abnormalities return to baseline levels when the medication is stopped. The presumed mechanism is impairment of bile-salt transporters, leading to bile-salt accumulation in the liver.34 Bosentan’s use in patients with liver disease has not been well studied, although several case reports have described its use in patients with portopulmonary hypertension.35–38
Ambrisentan (Letairis) is a selective endothelin receptor-A blocker that has just received FDA approval for the treatment of pulmonary artery hypertension. It has not yet been studied in portopulmonary hypertension. Elevations in liver enzymes and bilirubin may also occur, and monthly monitoring is indicated.
Sildenafil
Another oral agent that might be effective in portopulmonary hypertension is sildenafil (Revatio). A phosphodiesterase-5 inhibitor, it selectively inhibits the cyclic guanosine monophosphatase-specific phosphodiesterase type 5 enzyme that is found in large concentrations in pulmonary artery smooth muscle cells.
In other forms of pulmonary hypertension, sildenafil has been shown to increase cardiac output and decrease pulmonary artery pressures and pulmonary vascular resistance without serious adverse events.39–41
In one reported case, treatment with sildenafil in a patient with portopulmonary hypertension decreased the mean pulmonary artery pressure from 56 mm Hg to 28 to 31 mm Hg, and the patient underwent successful liver transplantation.42 A recent case series of 14 patients with portopulmonary hypertension treated with sildenafil documents some improvement in 6-minute walking distance, suggesting that sildenafil as monotherapy or in combination therapy might be effective in portopulmonary hypertension.43 However, in 3 of these patients, the cardiac index decreased and pulmonary vascular resistance increased.44
We must emphasize that controlled studies in portopulmonary hypertension need to be done to find the optimal therapy.
LIVER TRANSPLANTATION MAY BENEFIT A FEW PATIENTS
Liver transplantation may be beneficial in highly selected patients with portopulmonary hypertension. However, this condition increases the risk of intraoperative and immediate postoperative complications of liver transplantation, so patients should be carefully evaluated5,45 at a liver transplantation center experienced in its management, including medical treatment with well-defined protocols regarding timing of liver transplantation.
Patients with mean pulmonary artery pressures greater than 50 mm Hg should not undergo liver transplantation. Those with mean pulmonary artery pressure between 35 and 50 mm Hg also have an increased mortality rate and may benefit from prolonged treatment for pulmonary hypertension.5,46
One successful case of living-related liver transplantation in a patient with portopulmonary hypertension has been published.47 (Most other successful transplants were from unrelated cadaver donors.)
Some patients who initially cannot undergo liver transplantation owing to severe pulmonary hypertension may eventually be able to do so if they receive medical therapy that improves their pulmonary hemodynamic profile, decreasing their mean pulmonary artery pressure and pulmonary vascular resistance. This would apply to a small subset of patients with portopulmonary hypertension.
When patients without pulmonary hypertension undergo liver transplantation, right ventricular function is preserved throughout all phases of the surgery.48 Patients with portopulmonary hypertension, however, may develop hemodynamic instability during liver transplantation. The most critical times are the induction of anesthesia, during and after graft reperfusion, and the immediate postoperative period.49,50
During the surgery, patients may require vasodilators if they have worsening pulmonary hypertension, or inotropic medications if they have right ventricular dysfunction and heart failure. In one study,51 eight patients with portopulmonary hypertension diagnosed at anesthesia induction for liver transplantation all required intraoperative vasodilator therapy after graft reperfusion because of marked increases in pulmonary artery pressures and pulmonary vascular resistance.
The increase in blood flow following reperfusion or necessary fluid challenges may exacerbate pulmonary hypertension, resulting in worsening right heart function and backup into the transplanted liver. Infusion of 1 liter of crystalloid over 10 minutes has been shown to increase mean pulmonary artery pressure and pulmonary artery occlusion pressure in liver transplantation candidates without pulmonary hypertension52; this response may be exaggerated in portopulmonary hypertension.
PROGNOSIS VARIES WITH SEVERITY OF DISEASE
The natural history of untreated portopulmonary hypertension varies with the degree of liver disease and the severity of pulmonary hypertension. Transplant-free survival was 85% at 1 year and 38% at 3 years in one study.45 The cardiac index appears to be the most significant prognostic variable.20
In a retrospective study of 78 patients with portopulmonary hypertension treated conservatively (before prostanoids were available) the median survival was 6 months (range 0–84 months) from the time of diagnosis.53 Causes of death included right heart failure, sudden death, gastrointestinal bleeding, and small bowel perforation.
Most of the data on outcomes of drug treatment and liver transplantation in patients with portopulmonary hypertension come from case series and retrospective reviews; prospective trials have been lacking.
If right ventricular function is normal and pulmonary hypertension is mild (mean pulmonary artery pressure < 35 mm Hg), patients tend to do well with liver transplantation.9
Outcomes are worse if pulmonary hypertension is more severe. In a database54 from 10 liver transplant centers from 1996 to 2001, 13 (36%) of 36 patients undergoing liver transplantation died in the hospital, emphasizing the importance of accurately assessing the severity of pulmonary hypertension before attempting liver transplantation.46 The rate was even higher—92%—in those with a mean pulmonary artery pressure greater than 35 mm Hg. The cause of death in severe pulmonary hypertension was failure of the right ventricle.
However, some patients with moderate to severe portopulmonary hypertension have been bridged with medications to lower pulmonary artery pressures and pulmonary vascular resistance so that liver transplantation can be safely done, and some have even been able to discontinue medications because their pulmonary hypertension resolved.29,31,41,42,47
Unlike in hepatopulmonary syndrome, liver transplantation is not the treatment of choice for portopulmonary hypertension, and pulmonary hypertension does not always resolve after liver transplantation. Many patients continue therapy for pulmonary hypertension after liver transplantation. Pulmonary hypertension may resolve, persist, or even develop de novo after liver transplantation.1 If pulmonary hypertension resolves, it does so over a prolonged time—months to years—favoring a vascular remodeling hypothesis as opposed to simply reversing vasoconstriction.
- Rodriguez-Roisin R, Krowka MJ, Hervé P, Fallon MB; ERS Task Force Pulmonary-Hepatic Vascular Disorders (PHD) Scientific Committee. Pulmonary-hepatic vascular disorders (PHD). Eur Respir J 2004; 24:861–880.
- Krowka MJ, Swanson KL, Frantz RP, et al. Portopulmonary hypertension: results from a 10-year screening algorithm. Hepatology 2006; 44:1502–1510.
- Simonneau G, Galie N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004; 43:5S–12S.
- Hadengue A, Benhayoun MK, Lebrec D, et al. Pulmonary hypertension complicating portal hypertension: prevalence and relation to splanchnic hemodynamics. Gastroenterology 1991; 100:520–528.
- Krowka MJ, Plevak DJ, Findlay JY, et al. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transplant 2000; 6:443–450.
- Benjaminov FS, Prentice M, Sniderman KW, et al. Portopulmonary hypertension in decompensated cirrhosis with refractory ascites. Gut 2003; 52:1355–1362.
- McDonnell PJ, Toye PA, Hutchins GM. Primary pulmonary hypertension and cirrhosis: are they related? Am Rev Respir Dis 1983; 127:437–441.
- Cheng EY, Woehlck H. Pulmonary artery hypertension complicating anesthesia for liver transplantation. Anesthesiology 1992; 77:375–378.
- Castro M, Krowka MJ, Schroeder DR, et al. Frequency and clinical implications of increased pulmonary artery pressures in liver transplantation. Mayo Clin Proc 1996; 71:543–551.
- Ramsay MA, Simpson BR, Nguyen AT, et al. Severe pulmonary hypertension in liver transplant candidates. Liver Transplant Surg 1997; 3:494–500.
- Kiely DG, Cargill RI, Struthers AD, et al. Cardiopulmonary effects of endothelin-1 in man. Cardiovasc Res 1997; 33:378–386.
- Panos RJ, Baker SK. Mediators, cytokines, and growth factors in liver-lung interactions. Clin Chest Med 1996; 17:151–169.
- Higgenbottam T. Pathophysiology of pulmonary hypertension. Chest 1994; 105:7S–12S.
- Krowka MJ. Hepatopulmonary syndrome and portopulmonary hypertension: distinction and dilemmas. Hepatology 1997; 25:1282–1284.
- Hongqun L, Lee SS. Cardiopulmonary dysfunction in cirrhosis. Hepatology 2000; 14:600–608.
- Lebrec D, Brenot F, Simonneau G, et al. Pulmonary arterial hypertension in portal hypertension. Eur Respir J 1998; 11:1153–1166.
- Herve P, Lebrec D, Brenot F, et al. Pulmonary vascular disorders in portal hypertension. Eur Respir J 1998; 11:1153–1166.
- Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 1999; 159:1925–1932.
- Hoeper MM, Krowka MJ, Strassburg CP. Portopulmonary hypertension and hepatopulmonary syndrome. Lancet 2004; 363:1461–1468.
- Edwards B, Weir K, Edwards WD, et al. Coexistent pulmonary and portal hypertension: morphologic and clinical features. J Am Coll Cardiol 1987; 10:1233–1238.
- Ramsay MAE, Simpson BR, Nguyen AT, Ramsay KJ, East C, Klintmalm GB. Severe pulmonary hypertension in liver transplant candidates. Liver Transplant Surg 1997; 3:494–500.
- Trembath RC. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001; 345:325–334.
- Leuchte HH, Holzapfel M, Baumgartner RA, et al. Clinical significance of brain natriuretic peptide in primary pulmonary hypertension. J Am Coll Cardiol 2004; 43:764–770.
- Kim WR, Krowka MJ, Plevak DJ, et al. Accuracy of Doppler echocardiography in the assessment of pulmonary hypertension in liver transplant candidates. Liver Transplant 2000; 6:453–458.
- Colle IO, Moreau R, Godinho E, et al. Diagnosis of portopulmonary hypertension in candidates for liver transplantation: a prospective study. Hepatology 2003; 37:401–409.
- Provencher S, Herve P, Jais X, et al. Deleterious effects of beta-blockers on exercise capacity and hemodynamics in patients with portopulmonary hypertension. Gastroenterology 2006; 130:120–126.
- Swanson KL, McGoon MD, Krowka MJ. Survival in patients with portopulmonary hypertension [abstract]. Am J Respir Crit Care Med 2003; 167:A693.
- Kuo PC, Johnson LB, Plotkin JS, et al. Continuous intravenous infusion of epoprostenol for the treatment of portopulmonary hypertension. Transplantation 1997; 63:604–616.
- Krowka MJ, Frantz RP, McGoon MD, et al. Improvement in pulmonary hemodynamics during intravenous epoprostenol (prostacyclin): A study of 15 patients with moderate to severe portopulmonary hypertension. Hepatology 1999; 30:641–648.
- Kähler CM, Graziadei I, Wiedermann CJ, Kneussl MP, Vogel W. Successful use of continuous intravenous prostacyclin in a patient with severe portopulmonary hypertension. Wien Klin Wochenschr 2000; 112:637–640.
- Sussman N, Kaza V, Barshes N, et al. Successful liver transplantation following medical management of portopulmonary hypertension: a single-center series. Am J Transplant 2006; 6:2177–2182.
- Findlay JY, Plevak DJ, Krowka MJ, et al. Progressive splenomegaly after epoprostenol therapy in portopulmonary hypertension. Liver Transplant Surg 1999; 5:381–387.
- Rubin LJ, Roux S. Bosentan: a dual endothelin receptor antagonist. Expert Opin Invest Drugs 2002; 11:991–1002.
- Fattinger K, Funk C, Pantze M, et al. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther 2001; 69:223–231.
- Hinterhuber L, Graziadei IW, Kahler CM, et al. Endothelin-receptor anatgonist treatment of portopulmonary hypertension. Clin Gastroenterol Hepatol 2004; 2:1039–1042.
- Clift PF, Townend JN, Bramhall S, et al. Successful treatment of severe portopulmonary hypertension after liver transplantation by bosentan. Transplantation 2004; 77:1774–1775.
- Halank M, Miehlke S, Hoeffken G, et al. Use of oral endothelin-receptor antagonist bosentan in the treatment of portopulmonary hypertension. Transplantation 2004; 77:1775–1776.
- Kuntzen C, Gulberg V, Gerbes AL. Use of a mixed endothelin receptor antagonist in portopulmonary hypertension: a safe and effective therapy? Gastroenterology 2005; 128:164–168.
- Watanabe H, Ohashi K, Takeuchi K, et al. Sildenafil for primary and secondary pulmonary hypertension. Clin Pharmacol Ther 2002; 71:398–402.
- Michelakis E, Tymchak W, Lien D, et al. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: comparison with inhaled nitric oxide. Circulation 2002; 105:2398–2403.
- Ghofrani HA, Wiedemann R, Rose F, et al. Sildenafil for treatment of lung fibrosis and pulmonary hypertension: a randomised controlled trial. Lancet 2002; 360:895–900.
- Makisalo H, Koivusalo A, Vakkuri A, et al. Sildenafil for portopulmonary hypertension in a patient undergoing liver transplantation. Liver Transplant 2004; 10:945–950.
- Reichengerger F, Voswinckel R, Steveling E, et al. Sildenafil treatment for portopulmonary hypertension. Eur Respir J 2006; 28:563–567.
- Krowka MJ, Swanson KL. How should we treat portopulmonary hypertension? Eur Respir J 2006; 28:466–467.
- Kawut SM, Taichman DB, Ahya VN, et al. Hemodynamics and survival of patients with portopulmonary hypertension. Liver Transplant 2005; 11:1107–1111.
- Krowka MJ, Mandell MS, Ramsay MA, et al. Hepatopulmonary syndrome and portopulmonary hypertension: a report of the multicenter liver transplant database. Liver Transplant 2004; 10:174–182.
- Sulica R, Emre S, Poon M. Medical management of portopulmonary hypertension and right heart failure prior to living-related liver transplantation. Congest Heart Fail 2004; 10:192–194.
- De Wolf AM, Begliomini B, Gasior TA, et al. Right ventricular function during orthotopic liver transplantation. Anesthes Analges 1993; 76:562–568.
- Csete M. Intraoperative management of liver transplant patients with pulmonary hypertension. Liver Transplant Surg 1997; 3:454–455.
- Acosta F, Sansano T, Palenciano CG, et al. Portopulmonary hypertension and liver transplantation: hemodynamic consequences at reperfusion. Transplant Proc 2005; 37:3865–3866.
- Taura P, Garcia-Valdecasas JC, Beltran J, et al. Moderate primary pulmonary hypertension in patients undergoing liver transplantation. Anesthes Analges 1996; 83:675–680.
- Kuo PC, Schroeder RA, Vagelos RH, et al. Volume-mediated pulmonary responses in liver transplant candidates. Clin Transplant 1996; 10:521–527.
- Robalino BD, Moodie DS. Association between primary pulmonary hypertension and portal hypertension: analysis of its pathophysiology and clinical, laboratory and hemodynamic manifestations. J Am Coll Cardiol 1991; 17:492–498.
- Mandell MS, Krowka MJ. Formation of a national database on pulmonary hypertension and hepatopulmonary syndrome in chronic liver disease. Anesthesiology 1997; 87:450–451.
- Rodriguez-Roisin R, Krowka MJ, Hervé P, Fallon MB; ERS Task Force Pulmonary-Hepatic Vascular Disorders (PHD) Scientific Committee. Pulmonary-hepatic vascular disorders (PHD). Eur Respir J 2004; 24:861–880.
- Krowka MJ, Swanson KL, Frantz RP, et al. Portopulmonary hypertension: results from a 10-year screening algorithm. Hepatology 2006; 44:1502–1510.
- Simonneau G, Galie N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004; 43:5S–12S.
- Hadengue A, Benhayoun MK, Lebrec D, et al. Pulmonary hypertension complicating portal hypertension: prevalence and relation to splanchnic hemodynamics. Gastroenterology 1991; 100:520–528.
- Krowka MJ, Plevak DJ, Findlay JY, et al. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transplant 2000; 6:443–450.
- Benjaminov FS, Prentice M, Sniderman KW, et al. Portopulmonary hypertension in decompensated cirrhosis with refractory ascites. Gut 2003; 52:1355–1362.
- McDonnell PJ, Toye PA, Hutchins GM. Primary pulmonary hypertension and cirrhosis: are they related? Am Rev Respir Dis 1983; 127:437–441.
- Cheng EY, Woehlck H. Pulmonary artery hypertension complicating anesthesia for liver transplantation. Anesthesiology 1992; 77:375–378.
- Castro M, Krowka MJ, Schroeder DR, et al. Frequency and clinical implications of increased pulmonary artery pressures in liver transplantation. Mayo Clin Proc 1996; 71:543–551.
- Ramsay MA, Simpson BR, Nguyen AT, et al. Severe pulmonary hypertension in liver transplant candidates. Liver Transplant Surg 1997; 3:494–500.
- Kiely DG, Cargill RI, Struthers AD, et al. Cardiopulmonary effects of endothelin-1 in man. Cardiovasc Res 1997; 33:378–386.
- Panos RJ, Baker SK. Mediators, cytokines, and growth factors in liver-lung interactions. Clin Chest Med 1996; 17:151–169.
- Higgenbottam T. Pathophysiology of pulmonary hypertension. Chest 1994; 105:7S–12S.
- Krowka MJ. Hepatopulmonary syndrome and portopulmonary hypertension: distinction and dilemmas. Hepatology 1997; 25:1282–1284.
- Hongqun L, Lee SS. Cardiopulmonary dysfunction in cirrhosis. Hepatology 2000; 14:600–608.
- Lebrec D, Brenot F, Simonneau G, et al. Pulmonary arterial hypertension in portal hypertension. Eur Respir J 1998; 11:1153–1166.
- Herve P, Lebrec D, Brenot F, et al. Pulmonary vascular disorders in portal hypertension. Eur Respir J 1998; 11:1153–1166.
- Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 1999; 159:1925–1932.
- Hoeper MM, Krowka MJ, Strassburg CP. Portopulmonary hypertension and hepatopulmonary syndrome. Lancet 2004; 363:1461–1468.
- Edwards B, Weir K, Edwards WD, et al. Coexistent pulmonary and portal hypertension: morphologic and clinical features. J Am Coll Cardiol 1987; 10:1233–1238.
- Ramsay MAE, Simpson BR, Nguyen AT, Ramsay KJ, East C, Klintmalm GB. Severe pulmonary hypertension in liver transplant candidates. Liver Transplant Surg 1997; 3:494–500.
- Trembath RC. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001; 345:325–334.
- Leuchte HH, Holzapfel M, Baumgartner RA, et al. Clinical significance of brain natriuretic peptide in primary pulmonary hypertension. J Am Coll Cardiol 2004; 43:764–770.
- Kim WR, Krowka MJ, Plevak DJ, et al. Accuracy of Doppler echocardiography in the assessment of pulmonary hypertension in liver transplant candidates. Liver Transplant 2000; 6:453–458.
- Colle IO, Moreau R, Godinho E, et al. Diagnosis of portopulmonary hypertension in candidates for liver transplantation: a prospective study. Hepatology 2003; 37:401–409.
- Provencher S, Herve P, Jais X, et al. Deleterious effects of beta-blockers on exercise capacity and hemodynamics in patients with portopulmonary hypertension. Gastroenterology 2006; 130:120–126.
- Swanson KL, McGoon MD, Krowka MJ. Survival in patients with portopulmonary hypertension [abstract]. Am J Respir Crit Care Med 2003; 167:A693.
- Kuo PC, Johnson LB, Plotkin JS, et al. Continuous intravenous infusion of epoprostenol for the treatment of portopulmonary hypertension. Transplantation 1997; 63:604–616.
- Krowka MJ, Frantz RP, McGoon MD, et al. Improvement in pulmonary hemodynamics during intravenous epoprostenol (prostacyclin): A study of 15 patients with moderate to severe portopulmonary hypertension. Hepatology 1999; 30:641–648.
- Kähler CM, Graziadei I, Wiedermann CJ, Kneussl MP, Vogel W. Successful use of continuous intravenous prostacyclin in a patient with severe portopulmonary hypertension. Wien Klin Wochenschr 2000; 112:637–640.
- Sussman N, Kaza V, Barshes N, et al. Successful liver transplantation following medical management of portopulmonary hypertension: a single-center series. Am J Transplant 2006; 6:2177–2182.
- Findlay JY, Plevak DJ, Krowka MJ, et al. Progressive splenomegaly after epoprostenol therapy in portopulmonary hypertension. Liver Transplant Surg 1999; 5:381–387.
- Rubin LJ, Roux S. Bosentan: a dual endothelin receptor antagonist. Expert Opin Invest Drugs 2002; 11:991–1002.
- Fattinger K, Funk C, Pantze M, et al. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther 2001; 69:223–231.
- Hinterhuber L, Graziadei IW, Kahler CM, et al. Endothelin-receptor anatgonist treatment of portopulmonary hypertension. Clin Gastroenterol Hepatol 2004; 2:1039–1042.
- Clift PF, Townend JN, Bramhall S, et al. Successful treatment of severe portopulmonary hypertension after liver transplantation by bosentan. Transplantation 2004; 77:1774–1775.
- Halank M, Miehlke S, Hoeffken G, et al. Use of oral endothelin-receptor antagonist bosentan in the treatment of portopulmonary hypertension. Transplantation 2004; 77:1775–1776.
- Kuntzen C, Gulberg V, Gerbes AL. Use of a mixed endothelin receptor antagonist in portopulmonary hypertension: a safe and effective therapy? Gastroenterology 2005; 128:164–168.
- Watanabe H, Ohashi K, Takeuchi K, et al. Sildenafil for primary and secondary pulmonary hypertension. Clin Pharmacol Ther 2002; 71:398–402.
- Michelakis E, Tymchak W, Lien D, et al. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: comparison with inhaled nitric oxide. Circulation 2002; 105:2398–2403.
- Ghofrani HA, Wiedemann R, Rose F, et al. Sildenafil for treatment of lung fibrosis and pulmonary hypertension: a randomised controlled trial. Lancet 2002; 360:895–900.
- Makisalo H, Koivusalo A, Vakkuri A, et al. Sildenafil for portopulmonary hypertension in a patient undergoing liver transplantation. Liver Transplant 2004; 10:945–950.
- Reichengerger F, Voswinckel R, Steveling E, et al. Sildenafil treatment for portopulmonary hypertension. Eur Respir J 2006; 28:563–567.
- Krowka MJ, Swanson KL. How should we treat portopulmonary hypertension? Eur Respir J 2006; 28:466–467.
- Kawut SM, Taichman DB, Ahya VN, et al. Hemodynamics and survival of patients with portopulmonary hypertension. Liver Transplant 2005; 11:1107–1111.
- Krowka MJ, Mandell MS, Ramsay MA, et al. Hepatopulmonary syndrome and portopulmonary hypertension: a report of the multicenter liver transplant database. Liver Transplant 2004; 10:174–182.
- Sulica R, Emre S, Poon M. Medical management of portopulmonary hypertension and right heart failure prior to living-related liver transplantation. Congest Heart Fail 2004; 10:192–194.
- De Wolf AM, Begliomini B, Gasior TA, et al. Right ventricular function during orthotopic liver transplantation. Anesthes Analges 1993; 76:562–568.
- Csete M. Intraoperative management of liver transplant patients with pulmonary hypertension. Liver Transplant Surg 1997; 3:454–455.
- Acosta F, Sansano T, Palenciano CG, et al. Portopulmonary hypertension and liver transplantation: hemodynamic consequences at reperfusion. Transplant Proc 2005; 37:3865–3866.
- Taura P, Garcia-Valdecasas JC, Beltran J, et al. Moderate primary pulmonary hypertension in patients undergoing liver transplantation. Anesthes Analges 1996; 83:675–680.
- Kuo PC, Schroeder RA, Vagelos RH, et al. Volume-mediated pulmonary responses in liver transplant candidates. Clin Transplant 1996; 10:521–527.
- Robalino BD, Moodie DS. Association between primary pulmonary hypertension and portal hypertension: analysis of its pathophysiology and clinical, laboratory and hemodynamic manifestations. J Am Coll Cardiol 1991; 17:492–498.
- Mandell MS, Krowka MJ. Formation of a national database on pulmonary hypertension and hepatopulmonary syndrome in chronic liver disease. Anesthesiology 1997; 87:450–451.
KEY POINTS
- In portopulmonary hypertension, the pulmonary artery pressures, pulmonary vascular resistance, and portal venous pressure are all elevated.
- All candidates for liver transplantation should undergo echocardiography to screen for portopulmonary hypertension. If the echocardiogram shows elevated pulmonary pressures, right heart catheterization must be performed to confirm the diagnosis.
- The ideal medical regimen remains to be determined. Although drug treatment may lower pulmonary artery pressures in selected patients so that liver transplantation can be safely done, morbidity and mortality rates remain higher in patients with moderate to severe portopulmonary hypertension.
- Liver transplantation is not the treatment of choice for portopulmonary hypertension.