User login
Squamoid Eccrine Ductal Carcinoma
Squamoid eccrine ductal carcinoma (SEDC) is an aggressive underrecognized cutaneous malignancy of unknown etiology.1 It is most likely to occur in sun-exposed areas of the body, most commonly the head and neck. Risk factors include male sex, increased age, and chronic immunosuppression.1-4 Current reports suggest that SEDC is likely a high-grade subtype of squamous cell carcinoma (SCC) with a high risk for local recurrence (25%) and metastasis (13%).1,3,5,6 There are as few as 56 cases of SEDC reported in the literature; however, the number of cases may be closer to 100 due to SEDC being classified as either adenosquamous carcinoma of the skin or ductal eccrine carcinoma with squamous differentiation.1
Clinically, SEDC mimics keratinocyte carcinomas. Histologically, SEDC is biphasic, with a superficial portion resembling well-differentiated SCC and a deeply invasive portion having infiltrative irregular cords with ductal differentiation. Perineural invasion (PNI) frequently is present. Multiple connections to the overlying epidermis also can be seen, serving as a subtle clue to the diagnosis on broad superficial specimens.1-3 Due to superficial sampling, approximately 50% of reported cases are misdiagnosed as SCC during the initial biopsy.4 The diagnosis of SEDC often is made during complete excision when deeper tissue is sampled. Establishing an accurate diagnosis is important given the more aggressive nature of SEDC compared with SCC and its proclivity for PNI.1,3,6 The purpose of this review is to increase awareness of this underrecognized entity and describe the histologic findings that help distinguish SEDC from SCC.
Patient Chart Review
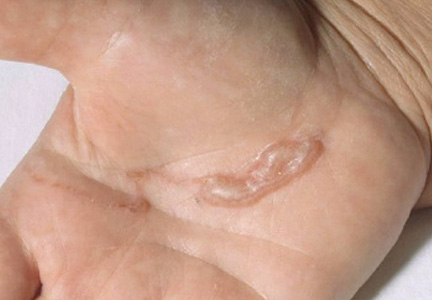
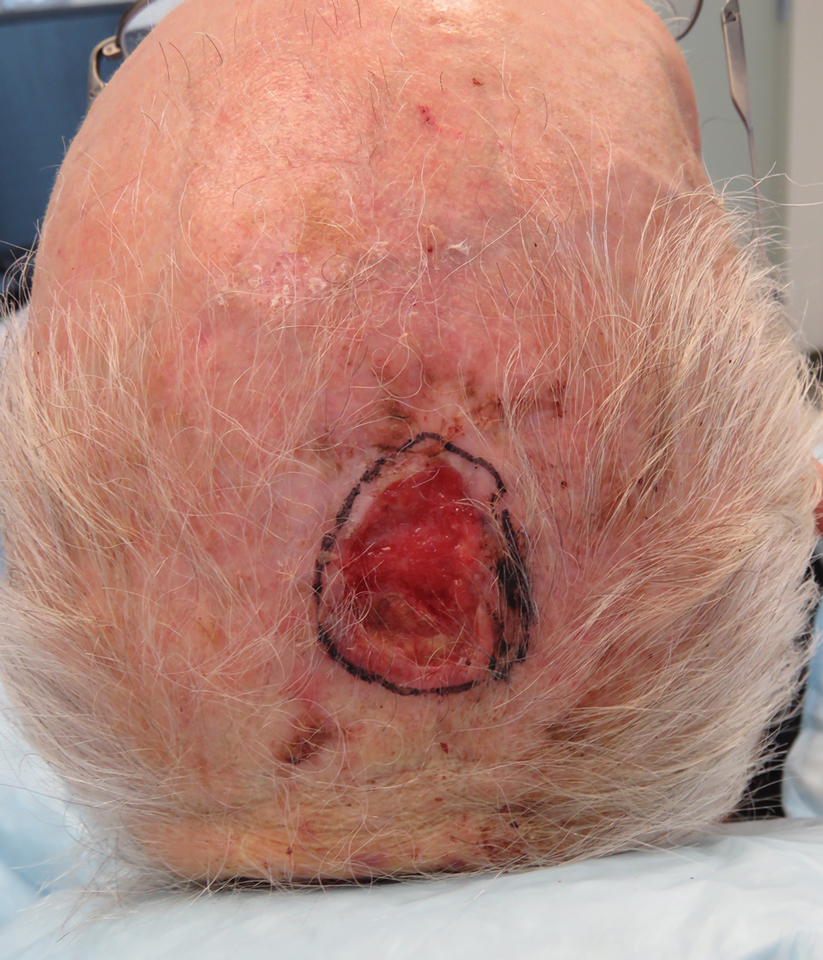
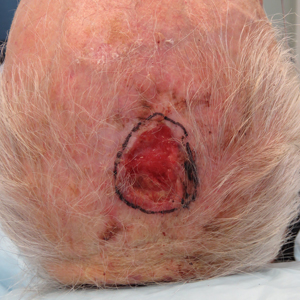
We reviewed chart notes as well as frozen and formalin-fixed paraffin-embedded tissue sections from all 5 patients diagnosed with SEDC at a single institution between November 2018 and May 2020. The mean age of patients was 81 years, and 4 were male. Four of the patients presented for MMS with a preoperative diagnosis of SCC per the original biopsy results. Only 1 patient had a preoperative diagnosis of SEDC. The details of each case are recorded in the Table. All tumors were greater than 2 cm in diameter on initial presentation, were located on the head, and clinically resembled keratinocyte carcinoma with either a nodular or plaquelike appearance (Figure 1).
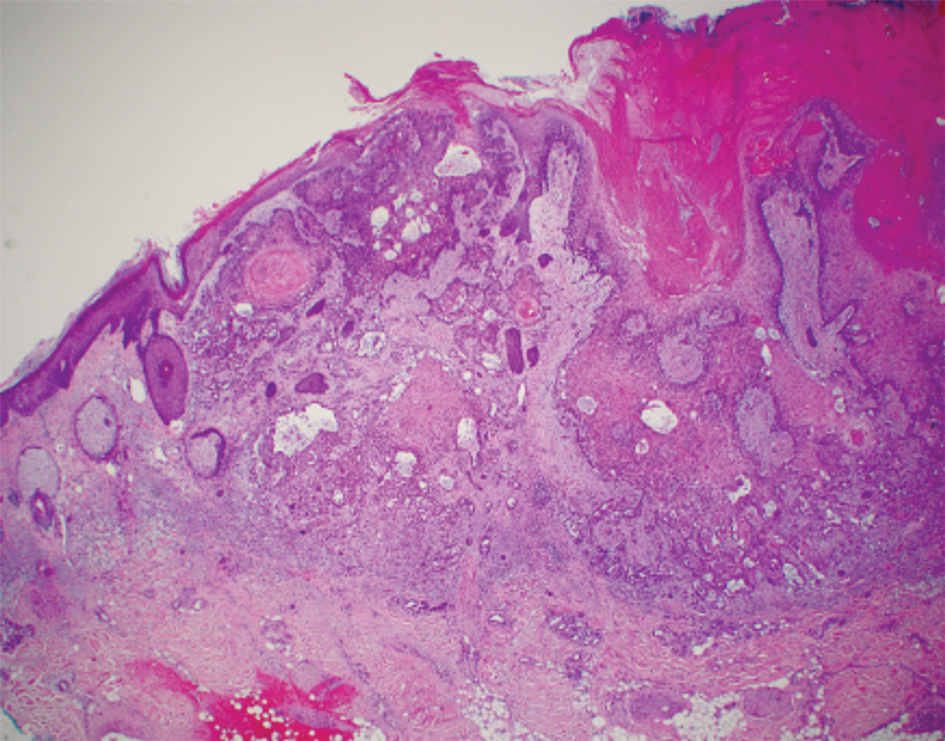
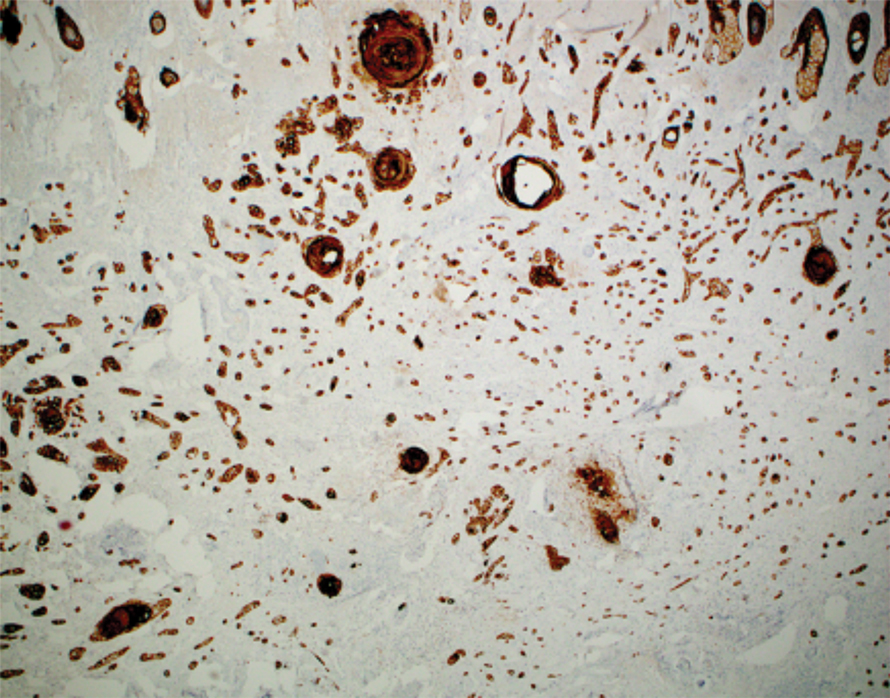
Intraoperative histologic examination of the excised tissue revealed a biphasic pattern consisting of superficial SCC features overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation in all 5 patients (Figures 2–4). Immunohistochemical staining with cytokeratin AE1/AE3 revealed thin strands of carcinoma in the mid to deeper dermis with squamous differentiation and eccrine ductal differentiation (Figure 5), thus confirming the diagnosis in all 5 patients.


The median depth of tumor invasion was 4.1 mm (range, 2.2–5.45 mm). Ulceration was seen in 3 of the patients, and PNI of large-caliber nerves was observed in all 5 patients. A connection with the overlying epidermis was present in all 5 patients. All 5 patients required more than 1 Mohs stage for complete tumor clearance (Table).
In 4 of the patients, nodal imaging performed at the time of diagnosis revealed no evidence of metastasis. Two patients received adjuvant radiation therapy, and none demonstrated evidence of recurrence. The mean follow-up time was 11 months (range, 6.5–18 months) for the 4 cases with available follow-up data (Table).
Literature Review
A PubMed review of the literature using the search term squamoid eccrine ductal carcinoma resulted in 28 articles, 19 of which were included in the review based on inclusion criteria (original articles available in English, in full text, and pertained to SEDC). Our review yielded 56 cases of SEDC.1-19 The mean age of patients with SEDC was 72 years. The number of male and female cases was 52% (29/56) and 48% (27/56), respectively. The most common location of SEDC was on the head or neck (71% [40/56]), followed by the extremities (19% [11/56]). Immunosuppression was noted in 9% (5/56) of cases. Wide local excision was the most commonly employed treatment modality (91% [51/56]), with MMS being used in 4 patients (7%). Adjuvant radiation was reported in 5% (3/56) of cases. Perineural invasion was reported in 34% (19/56) of cases. Recurrence was seen in 23% (13/56) of cases, with a mean time to recurrence of 10.4 months. Metastasis to regional lymph nodes was observed in 13% (7/56) of cases, with 7% (4/56) of those cases having distant metastases.
Comment
Squamoid eccrine ductal carcinoma was successfully treated with MMS in all 5 of the patients we reviewed. Recognition of a distinct biphasic pattern consisting of squamous differentiation superficially with epidermal connection overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation should lead to consideration of this diagnosis. A thorough inspection for PNI also should be performed, as this finding was present in all of 5 cases and in 34% of reported cases in our literature review.
The differential diagnosis for SEDC includes SCC, metastatic adenocarcinoma with squamoid features, and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma (MAC), and porocarcinoma with squamous differentiation. The combination of histologic features with the immunoexpression profile of carcinoembryonic antigen (CEA), epithelial membrane antigen (EMA), cytokeratin (CK) 5/6, and p63 can effectively exclude the other entities in the differential and confirm the diagnosis of SEDC.1,3,4 While the diagnosis of SEDC relies on the specific histologic features of multiple surface attachments and superficial squamoid changes with deep ductular elements, immunohistochemistry can nonetheless be adjunctive in difficult cases. Positive immunohistochemical staining for CEA and EMA can help to highlight and delineate true glandular elements, whereas CK5/6 highlights the overall contour of the tumor, displaying more clearly the multiple epidermal attachments and the subtle infiltrative nature of the deeper components of invasive cords and ducts. In addition, the combination of CK5/6 and p63 positivity supports the primary cutaneous nature of the lesion rather than metastatic adenocarcinoma.13,20 Other markers of eccrine secretory coils, such as CK7, CAM5.2, and S100, also are sometimes used for confirmation, some of which can aid in distinction from noneccrine sweat gland differentiation, as CK7 and CAM5.2 are negative in both luminal and basal cells of the dermal duct while being positive within the secretory coil, and S100 protein is expressed within eccrine secretory coil but negative within the apocrine sweat glands.2,4,21
The clinical findings from our chart review corroborated those reported in the literature. The mean age of SEDC in the 5 patients we reviewed was 81 years, and all cases presented on the head, consistent with the findings observed in the literature. Although 4 of our cases were male, there may not be a difference in risk based on sex as previously thought.1 Our literature review revealed an almost equivalent percentage of male and female cases, with 52% being male.
Immunosuppression has been associated with an increased risk for SEDC. Our literature review revealed that approximately 9% (5/56) of cases occurred in immunosuppressed individuals. Two of these reported cases were in the setting of underlying chronic lymphocytic leukemia, 2 in individuals with a history of organ transplant, and 1 treated with azathioprine for myasthenia gravis.2,4,10,12,13 Our chart review supported this correlation, as all 5 patients had a medical history potentially consistent with being in an immunocompromised state (Table). Notably, patient 5 represents a unique case of SEDC occurring in the setting of HIV. The patient had HIV for 33 years, with his most recent CD4+ count of 794 mm3 and HIV-1 RNA load of 35 copies/mL. Given that HIV-positive individuals may have more than a 2-fold increased risk of SCC, a greater degree of suspicion for SEDC should be maintained for these patients.22,23
The etiology of SEDC is controversial but is thought to be either an SCC arising from eccrine glands or a variant of eccrine carcinoma with extensive squamoid differentiation.4,6,13,14,17,24 While SEDC certainly appears to share the proclivity for PNI with the malignant eccrine tumor MAC, it is simultaneously quite distinct, demonstrating nuclear pleomorphism and mitotic activity, both of which are lacking in the bland nature of MACs.12,25
The exact prevalence of SEDC is difficult to ascertain because of its frequent misdiagnosis and variable nomenclature used within the literature. Most reported cases of SEDC are mistakenly diagnosed as SCC on the initial shave or punch biopsy because of superficial sampling. This also was the case in 4 of the patients we reviewed. In addition, there are reported cases of SEDC that were referred to by the investigators as cutaneous adenosquamous carcinoma (cASC), among other descriptors, such as ductal eccrine carcinoma with squamous differentiation, adnexal carcinoma with squamous and ductal differentiation, and syringoid eccrine carcinoma.26-32 While the World Health Organization classifies SEDC as a distinct variant of cASC, which is a rare variant of SCC in itself, the 2 can be differentiated. Despite the similar clinical and histologic features shared between cASC and SEDC, the neoplastic aggregates in SEDC exhibit ductal differentiation containing lumina positive for CEA and EMA.4 Overall, we favor the term squamoid eccrine ductal carcinoma, as there has recently been more uniformity for the designation of this disease entity as such.
It is unclear whether the high incidence of local recurrence (23% [13/56]) of SEDC reported in the literature is related to the treatment modality employed (ie, wide local excision) or due to the innate aggressiveness of SEDC.1,3,5 The literature has shown that MMS has lower recurrence rates than other treatments at 5-year follow-up for SCC (3.1%–5%) and eccrine carcinomas (0%–5%).33,34 Although studies assessing tumor behavior or comparing treatment modalities are limited because of the rarity and underrecognition of SEDC, MMS has been used several times for SEDC with only 1 recurrence reported.4,13,17,24 Given that all 5 of the patients we reviewed required more than 1 Mohs stage for complete tumor clearance and none demonstrated evidence of recurrence or metastasis (Table), we recommend MMS as the treatment of choice for SEDC.
Conclusion
Squamoid eccrine ductal carcinoma is a rare but likely underdiagnosed cutaneous tumor of uncertain etiology. Because of its propensity for recurrence and metastasis, excision of SEDC with complete circumferential peripheral and deep margin assessment with close follow-up is recommended.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Jacob J, Kugelman L. Squamoid eccrine ductal carcinoma. Cutis. 2018;101:378-380, 385.
- Yim S, Lee YH, Chae SW, et al. Squamoid eccrine ductal carcinoma of the ear helix. Clin Case Rep. 2019;7:1409-1411.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Jung YH, Jo HJ, Kang MS. Squamoid eccrine ductal carcinoma of the scalp. Korean J Pathol. 2012;46:278-281.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;91:799-802.
- Phan K, Kim L, Lim P, et al. A case report of temple squamoid eccrine ductal carcinoma: a diagnostic challenge beneath the tip of the iceberg. Dermatol Ther. 2020;33:E13213.
- McKissack SS, Wohltmann W, Dalton SR, et al. Squamoid eccrine ductal carcinoma: an aggressive mimicker of squamous cell carcinoma. Am J Dermatopathol. 2019;41:140-143.
- Lobo-Jardim MM, Souza BdCE, Kakizaki P, et al. Dermoscopy of squamoid eccrine ductal carcinoma: an aid for early diagnosis. An Bras Dermatol. 2018;93:893-895.
- Chan H, Howard V, Moir D, et al. Squamoid eccrine ductal carcinoma of the scalp. Aust J Dermatol. 2016;57:E117-E119.
- Wang B, Jarell AD, Bingham JL, et al. PET/CT imaging of squamoid eccrine ductal carcinoma. Clin Nucl Med. 2015;40:322-324.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma). J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. J Clin Aesthet Dermatol. 2013;6:33-36.
- Pusiol T, Morichetti D, Zorzi MG, et al. Squamoid eccrine ductal carcinoma: inappropriate diagnosis. Dermatol Surg. 2011;37:1819-1820.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Wasserman DI, Sack J, Gonzalez-Serva A, et al. Sentinel lymph node biopsy for a squamoid eccrine carcinoma with lymphatic invasion. Dermatol Surg. 2007;33:1126-1129.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Herrero J, Monteagudo C, Jorda E, et al. Squamoid eccrine ductal carcinoma. Histopathology. 1998;32:478-480.
- Wong TY, Suster S, Mihm MC. Squamoid eccrine ductal carcinoma. Histopathology. 1997;30:288-293.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Dabbs DJ. Diagnostic Immunohistochemistry: Theranostic and Genomic Applications. 4th ed. Elsevier/Saunders; 2014.
- Silverberg MJ, Leyden W, Warton EM, et al. HIV infection status, immunodeficiency, and the incidence of non-melanoma skin cancer. J Natl Cancer Inst. 2013;105:350-360.
- Asgari MM, Ray GT, Quesenberry CP Jr, et al. Association of multiple primary skin cancers with human immunodeficiency virus infection, CD4 count, and viral load. JAMA Dermatol. 2017;153:892-896.
- Tolkachjov SN. Adnexal carcinomas treated with Mohs micrographic surgery: a comprehensive review. Dermatol Surg. 2017;43:1199-1207.
- Kazakov DV. Cutaneous Adnexal Tumors. Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2012.
- Weidner N, Foucar E. Adenosquamous carcinoma of the skin. an aggressive mucin- and gland-forming squamous carcinoma. Arch Dermatol. 1985;121:775-779.
- Banks ER, Cooper PH. Adenosquamous carcinoma of the skin: a report of 10 cases. J Cutan Pathol. 1991;18:227-234.
- Ko CJ, Leffell DJ, McNiff JM. Adenosquamous carcinoma: a report of nine cases with p63 and cytokeratin 5/6 staining. J Cutan Pathol. 2009;36:448-452.
- Patel V, Squires SM, Liu DY, et al. Cutaneous adenosquamous carcinoma: a rare neoplasm with biphasic differentiation. Cutis. 2014;94:231-233.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
- Azorín D, López-Ríos F, Ballestín C, et al. Primary cutaneous adenosquamous carcinoma: a case report and review of the literature. J Cutan Pathol. 2001;28:542-545.
- Wildemore JK, Lee JB, Humphreys TR. Mohs surgery for malignant eccrine neoplasms. Dermatol Surg. 2004;30(12 pt 2):1574-1579.
- Garcia-Zuazaga J, Olbricht SM. Cutaneous squamous cell carcinoma. Adv Dermatol. 2008;24:33-57.
Squamoid eccrine ductal carcinoma (SEDC) is an aggressive underrecognized cutaneous malignancy of unknown etiology.1 It is most likely to occur in sun-exposed areas of the body, most commonly the head and neck. Risk factors include male sex, increased age, and chronic immunosuppression.1-4 Current reports suggest that SEDC is likely a high-grade subtype of squamous cell carcinoma (SCC) with a high risk for local recurrence (25%) and metastasis (13%).1,3,5,6 There are as few as 56 cases of SEDC reported in the literature; however, the number of cases may be closer to 100 due to SEDC being classified as either adenosquamous carcinoma of the skin or ductal eccrine carcinoma with squamous differentiation.1
Clinically, SEDC mimics keratinocyte carcinomas. Histologically, SEDC is biphasic, with a superficial portion resembling well-differentiated SCC and a deeply invasive portion having infiltrative irregular cords with ductal differentiation. Perineural invasion (PNI) frequently is present. Multiple connections to the overlying epidermis also can be seen, serving as a subtle clue to the diagnosis on broad superficial specimens.1-3 Due to superficial sampling, approximately 50% of reported cases are misdiagnosed as SCC during the initial biopsy.4 The diagnosis of SEDC often is made during complete excision when deeper tissue is sampled. Establishing an accurate diagnosis is important given the more aggressive nature of SEDC compared with SCC and its proclivity for PNI.1,3,6 The purpose of this review is to increase awareness of this underrecognized entity and describe the histologic findings that help distinguish SEDC from SCC.
Patient Chart Review
We reviewed chart notes as well as frozen and formalin-fixed paraffin-embedded tissue sections from all 5 patients diagnosed with SEDC at a single institution between November 2018 and May 2020. The mean age of patients was 81 years, and 4 were male. Four of the patients presented for MMS with a preoperative diagnosis of SCC per the original biopsy results. Only 1 patient had a preoperative diagnosis of SEDC. The details of each case are recorded in the Table. All tumors were greater than 2 cm in diameter on initial presentation, were located on the head, and clinically resembled keratinocyte carcinoma with either a nodular or plaquelike appearance (Figure 1).
Intraoperative histologic examination of the excised tissue revealed a biphasic pattern consisting of superficial SCC features overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation in all 5 patients (Figures 2–4). Immunohistochemical staining with cytokeratin AE1/AE3 revealed thin strands of carcinoma in the mid to deeper dermis with squamous differentiation and eccrine ductal differentiation (Figure 5), thus confirming the diagnosis in all 5 patients.
The median depth of tumor invasion was 4.1 mm (range, 2.2–5.45 mm). Ulceration was seen in 3 of the patients, and PNI of large-caliber nerves was observed in all 5 patients. A connection with the overlying epidermis was present in all 5 patients. All 5 patients required more than 1 Mohs stage for complete tumor clearance (Table).
In 4 of the patients, nodal imaging performed at the time of diagnosis revealed no evidence of metastasis. Two patients received adjuvant radiation therapy, and none demonstrated evidence of recurrence. The mean follow-up time was 11 months (range, 6.5–18 months) for the 4 cases with available follow-up data (Table).
Literature Review
A PubMed review of the literature using the search term squamoid eccrine ductal carcinoma resulted in 28 articles, 19 of which were included in the review based on inclusion criteria (original articles available in English, in full text, and pertained to SEDC). Our review yielded 56 cases of SEDC.1-19 The mean age of patients with SEDC was 72 years. The number of male and female cases was 52% (29/56) and 48% (27/56), respectively. The most common location of SEDC was on the head or neck (71% [40/56]), followed by the extremities (19% [11/56]). Immunosuppression was noted in 9% (5/56) of cases. Wide local excision was the most commonly employed treatment modality (91% [51/56]), with MMS being used in 4 patients (7%). Adjuvant radiation was reported in 5% (3/56) of cases. Perineural invasion was reported in 34% (19/56) of cases. Recurrence was seen in 23% (13/56) of cases, with a mean time to recurrence of 10.4 months. Metastasis to regional lymph nodes was observed in 13% (7/56) of cases, with 7% (4/56) of those cases having distant metastases.
Comment
Squamoid eccrine ductal carcinoma was successfully treated with MMS in all 5 of the patients we reviewed. Recognition of a distinct biphasic pattern consisting of squamous differentiation superficially with epidermal connection overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation should lead to consideration of this diagnosis. A thorough inspection for PNI also should be performed, as this finding was present in all of 5 cases and in 34% of reported cases in our literature review.
The differential diagnosis for SEDC includes SCC, metastatic adenocarcinoma with squamoid features, and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma (MAC), and porocarcinoma with squamous differentiation. The combination of histologic features with the immunoexpression profile of carcinoembryonic antigen (CEA), epithelial membrane antigen (EMA), cytokeratin (CK) 5/6, and p63 can effectively exclude the other entities in the differential and confirm the diagnosis of SEDC.1,3,4 While the diagnosis of SEDC relies on the specific histologic features of multiple surface attachments and superficial squamoid changes with deep ductular elements, immunohistochemistry can nonetheless be adjunctive in difficult cases. Positive immunohistochemical staining for CEA and EMA can help to highlight and delineate true glandular elements, whereas CK5/6 highlights the overall contour of the tumor, displaying more clearly the multiple epidermal attachments and the subtle infiltrative nature of the deeper components of invasive cords and ducts. In addition, the combination of CK5/6 and p63 positivity supports the primary cutaneous nature of the lesion rather than metastatic adenocarcinoma.13,20 Other markers of eccrine secretory coils, such as CK7, CAM5.2, and S100, also are sometimes used for confirmation, some of which can aid in distinction from noneccrine sweat gland differentiation, as CK7 and CAM5.2 are negative in both luminal and basal cells of the dermal duct while being positive within the secretory coil, and S100 protein is expressed within eccrine secretory coil but negative within the apocrine sweat glands.2,4,21
The clinical findings from our chart review corroborated those reported in the literature. The mean age of SEDC in the 5 patients we reviewed was 81 years, and all cases presented on the head, consistent with the findings observed in the literature. Although 4 of our cases were male, there may not be a difference in risk based on sex as previously thought.1 Our literature review revealed an almost equivalent percentage of male and female cases, with 52% being male.
Immunosuppression has been associated with an increased risk for SEDC. Our literature review revealed that approximately 9% (5/56) of cases occurred in immunosuppressed individuals. Two of these reported cases were in the setting of underlying chronic lymphocytic leukemia, 2 in individuals with a history of organ transplant, and 1 treated with azathioprine for myasthenia gravis.2,4,10,12,13 Our chart review supported this correlation, as all 5 patients had a medical history potentially consistent with being in an immunocompromised state (Table). Notably, patient 5 represents a unique case of SEDC occurring in the setting of HIV. The patient had HIV for 33 years, with his most recent CD4+ count of 794 mm3 and HIV-1 RNA load of 35 copies/mL. Given that HIV-positive individuals may have more than a 2-fold increased risk of SCC, a greater degree of suspicion for SEDC should be maintained for these patients.22,23
The etiology of SEDC is controversial but is thought to be either an SCC arising from eccrine glands or a variant of eccrine carcinoma with extensive squamoid differentiation.4,6,13,14,17,24 While SEDC certainly appears to share the proclivity for PNI with the malignant eccrine tumor MAC, it is simultaneously quite distinct, demonstrating nuclear pleomorphism and mitotic activity, both of which are lacking in the bland nature of MACs.12,25
The exact prevalence of SEDC is difficult to ascertain because of its frequent misdiagnosis and variable nomenclature used within the literature. Most reported cases of SEDC are mistakenly diagnosed as SCC on the initial shave or punch biopsy because of superficial sampling. This also was the case in 4 of the patients we reviewed. In addition, there are reported cases of SEDC that were referred to by the investigators as cutaneous adenosquamous carcinoma (cASC), among other descriptors, such as ductal eccrine carcinoma with squamous differentiation, adnexal carcinoma with squamous and ductal differentiation, and syringoid eccrine carcinoma.26-32 While the World Health Organization classifies SEDC as a distinct variant of cASC, which is a rare variant of SCC in itself, the 2 can be differentiated. Despite the similar clinical and histologic features shared between cASC and SEDC, the neoplastic aggregates in SEDC exhibit ductal differentiation containing lumina positive for CEA and EMA.4 Overall, we favor the term squamoid eccrine ductal carcinoma, as there has recently been more uniformity for the designation of this disease entity as such.
It is unclear whether the high incidence of local recurrence (23% [13/56]) of SEDC reported in the literature is related to the treatment modality employed (ie, wide local excision) or due to the innate aggressiveness of SEDC.1,3,5 The literature has shown that MMS has lower recurrence rates than other treatments at 5-year follow-up for SCC (3.1%–5%) and eccrine carcinomas (0%–5%).33,34 Although studies assessing tumor behavior or comparing treatment modalities are limited because of the rarity and underrecognition of SEDC, MMS has been used several times for SEDC with only 1 recurrence reported.4,13,17,24 Given that all 5 of the patients we reviewed required more than 1 Mohs stage for complete tumor clearance and none demonstrated evidence of recurrence or metastasis (Table), we recommend MMS as the treatment of choice for SEDC.
Conclusion
Squamoid eccrine ductal carcinoma is a rare but likely underdiagnosed cutaneous tumor of uncertain etiology. Because of its propensity for recurrence and metastasis, excision of SEDC with complete circumferential peripheral and deep margin assessment with close follow-up is recommended.
Squamoid eccrine ductal carcinoma (SEDC) is an aggressive underrecognized cutaneous malignancy of unknown etiology.1 It is most likely to occur in sun-exposed areas of the body, most commonly the head and neck. Risk factors include male sex, increased age, and chronic immunosuppression.1-4 Current reports suggest that SEDC is likely a high-grade subtype of squamous cell carcinoma (SCC) with a high risk for local recurrence (25%) and metastasis (13%).1,3,5,6 There are as few as 56 cases of SEDC reported in the literature; however, the number of cases may be closer to 100 due to SEDC being classified as either adenosquamous carcinoma of the skin or ductal eccrine carcinoma with squamous differentiation.1
Clinically, SEDC mimics keratinocyte carcinomas. Histologically, SEDC is biphasic, with a superficial portion resembling well-differentiated SCC and a deeply invasive portion having infiltrative irregular cords with ductal differentiation. Perineural invasion (PNI) frequently is present. Multiple connections to the overlying epidermis also can be seen, serving as a subtle clue to the diagnosis on broad superficial specimens.1-3 Due to superficial sampling, approximately 50% of reported cases are misdiagnosed as SCC during the initial biopsy.4 The diagnosis of SEDC often is made during complete excision when deeper tissue is sampled. Establishing an accurate diagnosis is important given the more aggressive nature of SEDC compared with SCC and its proclivity for PNI.1,3,6 The purpose of this review is to increase awareness of this underrecognized entity and describe the histologic findings that help distinguish SEDC from SCC.
Patient Chart Review
We reviewed chart notes as well as frozen and formalin-fixed paraffin-embedded tissue sections from all 5 patients diagnosed with SEDC at a single institution between November 2018 and May 2020. The mean age of patients was 81 years, and 4 were male. Four of the patients presented for MMS with a preoperative diagnosis of SCC per the original biopsy results. Only 1 patient had a preoperative diagnosis of SEDC. The details of each case are recorded in the Table. All tumors were greater than 2 cm in diameter on initial presentation, were located on the head, and clinically resembled keratinocyte carcinoma with either a nodular or plaquelike appearance (Figure 1).
Intraoperative histologic examination of the excised tissue revealed a biphasic pattern consisting of superficial SCC features overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation in all 5 patients (Figures 2–4). Immunohistochemical staining with cytokeratin AE1/AE3 revealed thin strands of carcinoma in the mid to deeper dermis with squamous differentiation and eccrine ductal differentiation (Figure 5), thus confirming the diagnosis in all 5 patients.
The median depth of tumor invasion was 4.1 mm (range, 2.2–5.45 mm). Ulceration was seen in 3 of the patients, and PNI of large-caliber nerves was observed in all 5 patients. A connection with the overlying epidermis was present in all 5 patients. All 5 patients required more than 1 Mohs stage for complete tumor clearance (Table).
In 4 of the patients, nodal imaging performed at the time of diagnosis revealed no evidence of metastasis. Two patients received adjuvant radiation therapy, and none demonstrated evidence of recurrence. The mean follow-up time was 11 months (range, 6.5–18 months) for the 4 cases with available follow-up data (Table).
Literature Review
A PubMed review of the literature using the search term squamoid eccrine ductal carcinoma resulted in 28 articles, 19 of which were included in the review based on inclusion criteria (original articles available in English, in full text, and pertained to SEDC). Our review yielded 56 cases of SEDC.1-19 The mean age of patients with SEDC was 72 years. The number of male and female cases was 52% (29/56) and 48% (27/56), respectively. The most common location of SEDC was on the head or neck (71% [40/56]), followed by the extremities (19% [11/56]). Immunosuppression was noted in 9% (5/56) of cases. Wide local excision was the most commonly employed treatment modality (91% [51/56]), with MMS being used in 4 patients (7%). Adjuvant radiation was reported in 5% (3/56) of cases. Perineural invasion was reported in 34% (19/56) of cases. Recurrence was seen in 23% (13/56) of cases, with a mean time to recurrence of 10.4 months. Metastasis to regional lymph nodes was observed in 13% (7/56) of cases, with 7% (4/56) of those cases having distant metastases.
Comment
Squamoid eccrine ductal carcinoma was successfully treated with MMS in all 5 of the patients we reviewed. Recognition of a distinct biphasic pattern consisting of squamous differentiation superficially with epidermal connection overlying deeper dermal and subcutaneous infiltrative malignant ductal elements with gland formation should lead to consideration of this diagnosis. A thorough inspection for PNI also should be performed, as this finding was present in all of 5 cases and in 34% of reported cases in our literature review.
The differential diagnosis for SEDC includes SCC, metastatic adenocarcinoma with squamoid features, and eccrine tumors, including eccrine poroma, microcystic adnexal carcinoma (MAC), and porocarcinoma with squamous differentiation. The combination of histologic features with the immunoexpression profile of carcinoembryonic antigen (CEA), epithelial membrane antigen (EMA), cytokeratin (CK) 5/6, and p63 can effectively exclude the other entities in the differential and confirm the diagnosis of SEDC.1,3,4 While the diagnosis of SEDC relies on the specific histologic features of multiple surface attachments and superficial squamoid changes with deep ductular elements, immunohistochemistry can nonetheless be adjunctive in difficult cases. Positive immunohistochemical staining for CEA and EMA can help to highlight and delineate true glandular elements, whereas CK5/6 highlights the overall contour of the tumor, displaying more clearly the multiple epidermal attachments and the subtle infiltrative nature of the deeper components of invasive cords and ducts. In addition, the combination of CK5/6 and p63 positivity supports the primary cutaneous nature of the lesion rather than metastatic adenocarcinoma.13,20 Other markers of eccrine secretory coils, such as CK7, CAM5.2, and S100, also are sometimes used for confirmation, some of which can aid in distinction from noneccrine sweat gland differentiation, as CK7 and CAM5.2 are negative in both luminal and basal cells of the dermal duct while being positive within the secretory coil, and S100 protein is expressed within eccrine secretory coil but negative within the apocrine sweat glands.2,4,21
The clinical findings from our chart review corroborated those reported in the literature. The mean age of SEDC in the 5 patients we reviewed was 81 years, and all cases presented on the head, consistent with the findings observed in the literature. Although 4 of our cases were male, there may not be a difference in risk based on sex as previously thought.1 Our literature review revealed an almost equivalent percentage of male and female cases, with 52% being male.
Immunosuppression has been associated with an increased risk for SEDC. Our literature review revealed that approximately 9% (5/56) of cases occurred in immunosuppressed individuals. Two of these reported cases were in the setting of underlying chronic lymphocytic leukemia, 2 in individuals with a history of organ transplant, and 1 treated with azathioprine for myasthenia gravis.2,4,10,12,13 Our chart review supported this correlation, as all 5 patients had a medical history potentially consistent with being in an immunocompromised state (Table). Notably, patient 5 represents a unique case of SEDC occurring in the setting of HIV. The patient had HIV for 33 years, with his most recent CD4+ count of 794 mm3 and HIV-1 RNA load of 35 copies/mL. Given that HIV-positive individuals may have more than a 2-fold increased risk of SCC, a greater degree of suspicion for SEDC should be maintained for these patients.22,23
The etiology of SEDC is controversial but is thought to be either an SCC arising from eccrine glands or a variant of eccrine carcinoma with extensive squamoid differentiation.4,6,13,14,17,24 While SEDC certainly appears to share the proclivity for PNI with the malignant eccrine tumor MAC, it is simultaneously quite distinct, demonstrating nuclear pleomorphism and mitotic activity, both of which are lacking in the bland nature of MACs.12,25
The exact prevalence of SEDC is difficult to ascertain because of its frequent misdiagnosis and variable nomenclature used within the literature. Most reported cases of SEDC are mistakenly diagnosed as SCC on the initial shave or punch biopsy because of superficial sampling. This also was the case in 4 of the patients we reviewed. In addition, there are reported cases of SEDC that were referred to by the investigators as cutaneous adenosquamous carcinoma (cASC), among other descriptors, such as ductal eccrine carcinoma with squamous differentiation, adnexal carcinoma with squamous and ductal differentiation, and syringoid eccrine carcinoma.26-32 While the World Health Organization classifies SEDC as a distinct variant of cASC, which is a rare variant of SCC in itself, the 2 can be differentiated. Despite the similar clinical and histologic features shared between cASC and SEDC, the neoplastic aggregates in SEDC exhibit ductal differentiation containing lumina positive for CEA and EMA.4 Overall, we favor the term squamoid eccrine ductal carcinoma, as there has recently been more uniformity for the designation of this disease entity as such.
It is unclear whether the high incidence of local recurrence (23% [13/56]) of SEDC reported in the literature is related to the treatment modality employed (ie, wide local excision) or due to the innate aggressiveness of SEDC.1,3,5 The literature has shown that MMS has lower recurrence rates than other treatments at 5-year follow-up for SCC (3.1%–5%) and eccrine carcinomas (0%–5%).33,34 Although studies assessing tumor behavior or comparing treatment modalities are limited because of the rarity and underrecognition of SEDC, MMS has been used several times for SEDC with only 1 recurrence reported.4,13,17,24 Given that all 5 of the patients we reviewed required more than 1 Mohs stage for complete tumor clearance and none demonstrated evidence of recurrence or metastasis (Table), we recommend MMS as the treatment of choice for SEDC.
Conclusion
Squamoid eccrine ductal carcinoma is a rare but likely underdiagnosed cutaneous tumor of uncertain etiology. Because of its propensity for recurrence and metastasis, excision of SEDC with complete circumferential peripheral and deep margin assessment with close follow-up is recommended.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Jacob J, Kugelman L. Squamoid eccrine ductal carcinoma. Cutis. 2018;101:378-380, 385.
- Yim S, Lee YH, Chae SW, et al. Squamoid eccrine ductal carcinoma of the ear helix. Clin Case Rep. 2019;7:1409-1411.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Jung YH, Jo HJ, Kang MS. Squamoid eccrine ductal carcinoma of the scalp. Korean J Pathol. 2012;46:278-281.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;91:799-802.
- Phan K, Kim L, Lim P, et al. A case report of temple squamoid eccrine ductal carcinoma: a diagnostic challenge beneath the tip of the iceberg. Dermatol Ther. 2020;33:E13213.
- McKissack SS, Wohltmann W, Dalton SR, et al. Squamoid eccrine ductal carcinoma: an aggressive mimicker of squamous cell carcinoma. Am J Dermatopathol. 2019;41:140-143.
- Lobo-Jardim MM, Souza BdCE, Kakizaki P, et al. Dermoscopy of squamoid eccrine ductal carcinoma: an aid for early diagnosis. An Bras Dermatol. 2018;93:893-895.
- Chan H, Howard V, Moir D, et al. Squamoid eccrine ductal carcinoma of the scalp. Aust J Dermatol. 2016;57:E117-E119.
- Wang B, Jarell AD, Bingham JL, et al. PET/CT imaging of squamoid eccrine ductal carcinoma. Clin Nucl Med. 2015;40:322-324.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma). J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. J Clin Aesthet Dermatol. 2013;6:33-36.
- Pusiol T, Morichetti D, Zorzi MG, et al. Squamoid eccrine ductal carcinoma: inappropriate diagnosis. Dermatol Surg. 2011;37:1819-1820.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Wasserman DI, Sack J, Gonzalez-Serva A, et al. Sentinel lymph node biopsy for a squamoid eccrine carcinoma with lymphatic invasion. Dermatol Surg. 2007;33:1126-1129.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Herrero J, Monteagudo C, Jorda E, et al. Squamoid eccrine ductal carcinoma. Histopathology. 1998;32:478-480.
- Wong TY, Suster S, Mihm MC. Squamoid eccrine ductal carcinoma. Histopathology. 1997;30:288-293.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Dabbs DJ. Diagnostic Immunohistochemistry: Theranostic and Genomic Applications. 4th ed. Elsevier/Saunders; 2014.
- Silverberg MJ, Leyden W, Warton EM, et al. HIV infection status, immunodeficiency, and the incidence of non-melanoma skin cancer. J Natl Cancer Inst. 2013;105:350-360.
- Asgari MM, Ray GT, Quesenberry CP Jr, et al. Association of multiple primary skin cancers with human immunodeficiency virus infection, CD4 count, and viral load. JAMA Dermatol. 2017;153:892-896.
- Tolkachjov SN. Adnexal carcinomas treated with Mohs micrographic surgery: a comprehensive review. Dermatol Surg. 2017;43:1199-1207.
- Kazakov DV. Cutaneous Adnexal Tumors. Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2012.
- Weidner N, Foucar E. Adenosquamous carcinoma of the skin. an aggressive mucin- and gland-forming squamous carcinoma. Arch Dermatol. 1985;121:775-779.
- Banks ER, Cooper PH. Adenosquamous carcinoma of the skin: a report of 10 cases. J Cutan Pathol. 1991;18:227-234.
- Ko CJ, Leffell DJ, McNiff JM. Adenosquamous carcinoma: a report of nine cases with p63 and cytokeratin 5/6 staining. J Cutan Pathol. 2009;36:448-452.
- Patel V, Squires SM, Liu DY, et al. Cutaneous adenosquamous carcinoma: a rare neoplasm with biphasic differentiation. Cutis. 2014;94:231-233.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
- Azorín D, López-Ríos F, Ballestín C, et al. Primary cutaneous adenosquamous carcinoma: a case report and review of the literature. J Cutan Pathol. 2001;28:542-545.
- Wildemore JK, Lee JB, Humphreys TR. Mohs surgery for malignant eccrine neoplasms. Dermatol Surg. 2004;30(12 pt 2):1574-1579.
- Garcia-Zuazaga J, Olbricht SM. Cutaneous squamous cell carcinoma. Adv Dermatol. 2008;24:33-57.
- van der Horst MP, Garcia-Herrera A, Markiewicz D, et al. Squamoid eccrine ductal carcinoma: a clinicopathologic study of 30 cases. Am J Surg Pathol. 2016;40:755-760.
- Jacob J, Kugelman L. Squamoid eccrine ductal carcinoma. Cutis. 2018;101:378-380, 385.
- Yim S, Lee YH, Chae SW, et al. Squamoid eccrine ductal carcinoma of the ear helix. Clin Case Rep. 2019;7:1409-1411.
- Terushkin E, Leffell DJ, Futoryan T, et al. Squamoid eccrine ductal carcinoma: a case report and review of the literature. Am J Dermatopathol. 2010;32:287-292.
- Jung YH, Jo HJ, Kang MS. Squamoid eccrine ductal carcinoma of the scalp. Korean J Pathol. 2012;46:278-281.
- Saraiva MI, Vieira MA, Portocarrero LK, et al. Squamoid eccrine ductal carcinoma. An Bras Dermatol. 2016;91:799-802.
- Phan K, Kim L, Lim P, et al. A case report of temple squamoid eccrine ductal carcinoma: a diagnostic challenge beneath the tip of the iceberg. Dermatol Ther. 2020;33:E13213.
- McKissack SS, Wohltmann W, Dalton SR, et al. Squamoid eccrine ductal carcinoma: an aggressive mimicker of squamous cell carcinoma. Am J Dermatopathol. 2019;41:140-143.
- Lobo-Jardim MM, Souza BdCE, Kakizaki P, et al. Dermoscopy of squamoid eccrine ductal carcinoma: an aid for early diagnosis. An Bras Dermatol. 2018;93:893-895.
- Chan H, Howard V, Moir D, et al. Squamoid eccrine ductal carcinoma of the scalp. Aust J Dermatol. 2016;57:E117-E119.
- Wang B, Jarell AD, Bingham JL, et al. PET/CT imaging of squamoid eccrine ductal carcinoma. Clin Nucl Med. 2015;40:322-324.
- Frouin E, Vignon-Pennamen MD, Balme B, et al. Anatomoclinical study of 30 cases of sclerosing sweat duct carcinomas (microcystic adnexal carcinoma, syringomatous carcinoma and squamoid eccrine ductal carcinoma). J Eur Acad Dermatol Venereol. 2015;29:1978-1994.
- Clark S, Young A, Piatigorsky E, et al. Mohs micrographic surgery in the setting of squamoid eccrine ductal carcinoma: addressing a diagnostic and therapeutic challenge. J Clin Aesthet Dermatol. 2013;6:33-36.
- Pusiol T, Morichetti D, Zorzi MG, et al. Squamoid eccrine ductal carcinoma: inappropriate diagnosis. Dermatol Surg. 2011;37:1819-1820.
- Kavand S, Cassarino DS. “Squamoid eccrine ductal carcinoma”: an unusual low-grade case with follicular differentiation. are these tumors squamoid variants of microcystic adnexal carcinoma? Am J Dermatopathol. 2009;31:849-852.
- Wasserman DI, Sack J, Gonzalez-Serva A, et al. Sentinel lymph node biopsy for a squamoid eccrine carcinoma with lymphatic invasion. Dermatol Surg. 2007;33:1126-1129.
- Kim YJ, Kim AR, Yu DS. Mohs micrographic surgery for squamoid eccrine ductal carcinoma. Dermatol Surg. 2005;31:1462-1464.
- Herrero J, Monteagudo C, Jorda E, et al. Squamoid eccrine ductal carcinoma. Histopathology. 1998;32:478-480.
- Wong TY, Suster S, Mihm MC. Squamoid eccrine ductal carcinoma. Histopathology. 1997;30:288-293.
- Qureshi HS, Ormsby AH, Lee MW, et al. The diagnostic utility of p63, CK5/6, CK 7, and CK 20 in distinguishing primary cutaneous adnexal neoplasms from metastatic carcinomas. J Cutan Pathol. 2004;31:145-152.
- Dabbs DJ. Diagnostic Immunohistochemistry: Theranostic and Genomic Applications. 4th ed. Elsevier/Saunders; 2014.
- Silverberg MJ, Leyden W, Warton EM, et al. HIV infection status, immunodeficiency, and the incidence of non-melanoma skin cancer. J Natl Cancer Inst. 2013;105:350-360.
- Asgari MM, Ray GT, Quesenberry CP Jr, et al. Association of multiple primary skin cancers with human immunodeficiency virus infection, CD4 count, and viral load. JAMA Dermatol. 2017;153:892-896.
- Tolkachjov SN. Adnexal carcinomas treated with Mohs micrographic surgery: a comprehensive review. Dermatol Surg. 2017;43:1199-1207.
- Kazakov DV. Cutaneous Adnexal Tumors. Wolters Kluwer Health/ Lippincott Williams & Wilkins; 2012.
- Weidner N, Foucar E. Adenosquamous carcinoma of the skin. an aggressive mucin- and gland-forming squamous carcinoma. Arch Dermatol. 1985;121:775-779.
- Banks ER, Cooper PH. Adenosquamous carcinoma of the skin: a report of 10 cases. J Cutan Pathol. 1991;18:227-234.
- Ko CJ, Leffell DJ, McNiff JM. Adenosquamous carcinoma: a report of nine cases with p63 and cytokeratin 5/6 staining. J Cutan Pathol. 2009;36:448-452.
- Patel V, Squires SM, Liu DY, et al. Cutaneous adenosquamous carcinoma: a rare neoplasm with biphasic differentiation. Cutis. 2014;94:231-233.
- Chhibber V, Lyle S, Mahalingam M. Ductal eccrine carcinoma with squamous differentiation: apropos a case. J Cutan Pathol. 2007;34:503-507.
- Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
- Azorín D, López-Ríos F, Ballestín C, et al. Primary cutaneous adenosquamous carcinoma: a case report and review of the literature. J Cutan Pathol. 2001;28:542-545.
- Wildemore JK, Lee JB, Humphreys TR. Mohs surgery for malignant eccrine neoplasms. Dermatol Surg. 2004;30(12 pt 2):1574-1579.
- Garcia-Zuazaga J, Olbricht SM. Cutaneous squamous cell carcinoma. Adv Dermatol. 2008;24:33-57.
PRACTICE POINTS
- Squamoid eccrine ductal carcinoma is an aggressive underrecognized cutaneous malignancy that often is misdiagnosed as squamous cell carcinoma (SCC) during initial biopsy.
- Squamoid eccrine ductal carcinoma has a biphasic histologic appearance with a superficial portion resembling well-differentiated SCC and a deeply invasive portion comprised of infiltrative irregular cords with ductal differentiation.
- Excision with complete circumferential peripheral and deep margin assessment with close follow-up is recommended for these patients because of the high risk for recurrence and metastasis.
Systemic Medications Linked to an Increased Risk for Skin Malignancy
Dermatologists are increasingly called on to evaluate patients with complex medical problems who are often taking many medications. Over the last several decades, many new drugs that target molecular pathways in carcinogenesis and the inflammatory immune system have been developed. Increased skin cancer risk has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, Janus kinase (JAK) inhibitors, and phosphodiesterase 5 (PDE-5) inhibitors. We review the literature and data regarding the significance and strength of these associations and the molecular pathways by which these medications promote cutaneous tumorigenesis. The association of skin cancer with drugs that either induce photosensitivity—nonsteroidal anti-inflammatory drugs, antibiotics (eg, tetracyclines, fluoroquinolones, trimethoprim-sulfamethoxazole), voriconazole, thiazides—or suppress the immune system—certain biologics (eg, anti–tumor necrosis factor agents), calcineurin inhibitors, thiopurines, methotrexate, cyclosporine—is well known and is therefore not reviewed in this discussion.
BRAF Inhibitors
The mitogen-activated protein kinase (MAPK) pathway (also known as the RAS/RAF/MAPK signaling pathway) is important in growth factor–receptor signaling and plays a key role in cell differentiation, survival, and proliferation. Activating mutations in this pathway allow cells to grow and proliferate in a growth factor–independent manner. Twenty percent of human cancers harbor a mutation in the RAS oncogene, an upstream mediator of the pathway.1 Activating mutations in BRAF, a serine/threonine kinase, predominate in cutaneous melanoma and also have been found in 40% to 70% of papillary thyroid malignancies, 10% to 20% of cholangiocarcinomas, and 5% to 20% of colorectal carcinomas. The most common BRAF mutation in cutaneous melanoma is V600E, which involves a glutamic acid for valine substitution at codon 600. This mutation activates BRAF 500-fold and is present in approximately 50% of melanomas.1,2
Vemurafenib, a selective BRAF inhibitor, was approved by the US Food and Drug Administration (FDA) for the treatment of metastatic melanoma in the United States in 2011. Phase 3 trial data demonstrated that vemurafenib resulted in improved survival and decreased risk for disease progression compared to dacarbazine, the former best treatment.3 During phase 1 testing, it became apparent that vemurafenib treatment was associated with a 31% increased risk for squamous cell carcinoma (SCC), most commonly well-differentiated SCC, and keratoacanthomas (KAs).4 This association was confirmed in phase 2 and 3 studies, though the incidence was lower. McArthur et al5 reported a 19% incidence of cutaneous SCC with extended follow-up analysis of the phase 3 trial. Dabrafenib, another BRAF inhibitor, has been similarly associated with increasing the risk for SCC and KA.
In one study, the mean time to development of SCC after initiating vemurafenib therapy was 10 weeks, with lesions reported as early as 3 weeks. Most patients had clinical signs of chronically sun damaged skin; however, a history of SCC was present in only 17%. Most lesions (63%) were characterized as KAs.6
The mechanism for BRAF inhibitor–induced squamoproliferative growth is due to paradoxical activation of the MAPK pathway in cells with wild-type BRAF that harbor upstream-activating mutations in RAS or tyrosine kinase receptors.7 In the presence of a BRAF inhibitor, inactivated BRAF forms heterodimers with wild-type CRAF (a BRAF-CRAF heterodimer). The heterodimer forms a complex with the mutant RAS that leads to transactivation of the CRAF molecule,8,9 resulting in a paradoxical increase in MAPK signaling and consequent ERK phosphorylation and activation through CRAF signaling. RAS, particularly HRAS, mutations have been found in 60% of all vemurafenib-associated SCCs and KAs. For this reason, it is thought that vemurafenib potentiates tumorigenesis in subclinical lesions harboring upstream MAPK pathway mutations as opposed to inducing de novo lesions.6
Because BRAF inhibitors are remarkably efficacious in the treatment of metastatic melanomas harboring the V600E BRAF mutation, there are no restrictions on their use, despite the known increased risk for SCC. Squamous cell carcinomas tend to be low grade, and all tumors that developed in phase 1 to 3 trials were treated with simple excision. The development of SCC did not necessitate interruption of treatment. Furthermore, the addition of MEK inhibition to BRAF inhibitor therapy reduces the risk for SCC from 19% to 7%.7,10,11
In addition to SCC, second primary melanomas (SPMs) have been reported in patients treated with BRAF inhibitors. It has been shown that these melanomas occur in melanocytes with wild-type BRAF. It has been postulated that some of these tumors occur in cells that harbor upstream mutations in RAS, whereas others might result from alternate signaling through non-RAF oncogenic pathways.9,12
Zimmer et al1 reported 12 SPMs in 11 patients treated with BRAF inhibitor therapy. They reported a median delay of 8 weeks (range, 4–27 weeks) for SPM development. Tumors were detected in early stages; 1 tumor harbored an NRAS mutation.1
Dalle et al13 reported 25 SPMs in 120 vemurafenib-treated patients. Median delay in SPM development was 14 weeks (range, 4–42 weeks). All tumors were thin, ranging from in situ to 0.45-mm thick. Wild-type BRAF was detected in the 21 melanomas sampled; 1 lesion showed mutated NRAS.13
The exact incidence of SPM in the setting of BRAF inhibition is thought to be at least 10-fold less than SCC and KA.2 Patients on BRAF inhibitor therapy should have routine full-body skin examinations, given the increased risk for SPM and SCC.
Another drug belonging to the tyrosine kinase inhibitor family, sorafenib, is used in the treatment of solid tumors, particularly hepatocellular and renal cell carcinomas, and also has been associated with development of cutaneous SCC and KAs.14 Sorafenib is a multiple tyrosine kinase inhibitor that also inhibits the RAF serine/threonine kinases. Similar to vemurafenib and dabrafenib, SCCs and KAs associated with sorafenib tend to arise in patients with chronic actinic damage during the first 2 months of treatment. It has been hypothesized that inhibition of RAF kinases is pathogenic in inducing SCCs because these lesions have not been reported with sunitinib, another multiple tyrosine kinase inhibitor that lacks the ability to inhibit serine/threonine kinases.15,16 Although SCCs and KAs associated with sorafenib tend to be low grade, it is reasonable to consider sunitinib or an alternative tyrosine kinase inhibitor in patients who develop multiple SCCs while taking sorafenib.16
Sonic Hedgehog–Inhibiting Agents
Vismodegib, the first small molecule inhibitor of the signaling protein smoothened, gained FDA approval for the treatment of metastatic or locally advanced basal cell carcinoma (BCC) in 2012. A second agent with an identical mechanism of action, sonidegib, was approved by the FDA for locally advanced BCC in 2015. Approximately 90% of BCCs contain mutations in the sonic hedgehog pathway, which lead to constitutive smoothened activation and uncontrolled cell proliferation.17 The development of smoothened inhibitors introduced a much-needed treatment for inoperable or metastatic BCC,17,18 though long-term utility is limited by drug resistance with extended use in this patient population.19,20 Several case reports have documented the emergence of KA21 and cutaneous SCC following vismodegib treatment of advanced or metastatic BCC.22-24 A larger case-control study by Mohan et al25 showed that patients with BCC treated with vismodegib had an increased risk for non-BCC malignancy (hazard ratio [HR]=6.37), most of which were cutaneous SCC (HR=8.12).
The mechanism by which selective inhibition of smoothened leads to cutaneous SCC is unclear. A study found that patients on vismodegib who developed SCC within the original BCC site had elevated ERK levels within tumor tissue, suggesting that the RAS/RAF/MAPK pathway can become upregulated during hedgehog inhibition.26 Other studies looking at hedgehog inhibition in medulloblastoma models also have shown activated RAS/RAF/MAPK pathways.25 These findings suggest that tumors under smoothened inhibition might be able to bypass the sonic hedgehog pathway and continue to grow by upregulating alternative growth pathways, such as RAS/RAF/MAPK.25,26
The incidence of cutaneous SCC following vismodegib treatment is unknown. Chang and Oro27 examined BCC tumor regrowth from secondary (acquired) resistance to vismodegib and noted that lesions recurred within 1 cm of the original tumor 21% of the time. Although none of the 12 patients whose tumors regrew during treatment were reported to have developed SCC, several demonstrated different BCC subtypes than the pretreatment specimen. The authors proposed that regrowth of BCC was due to upregulated alternative pathways allowing tumors to bypass smoothened inhibition, which is similar to the proposed mechanism for SCC development in vismodegib patients.27
Prospective studies are needed to confirm the link between vismodegib and cutaneous SCC; establish the incidence of SCC development; and identify any pretreatment factors, tumor characteristics, or treatment details (eg, dosage, duration) that might contribute to SCC development. Furthermore, because Mohan et al25 observed that vismodegib-treated patients were less likely to develop SCC in situ than controls, it is unknown if these tumors are more aggressive than traditional SCC. At this point, careful surveillance and regular full-body skin examinations are advised for patients on vismodegib for treatment of advanced BCC.
JAK Inhibitors
Another class of medications potentially associated with increased development of nonmelanoma skin cancer (NMSC) is the JAK inhibitors (also known as jakinibs). Many proinflammatory signaling pathways converge on the JAK family of enzymes—JAK1, JAK2, JAK3, and TYK2. These enzymes operate in cytokine signal transduction by phosphorylating activated cytokine receptors, which allows for recruitment and activation by means of phosphorylation of transcription factors collectively known as signal transducers and activators of transcription (STATs). Phosphorylated STATs dimerize and translocate to the nucleus, acting as direct transcription promoters. Janus kinase inhibitors modulate the immune response by reducing the effect of interleukin and interferon signaling.
Ruxolitinib, a JAK1/JAK2 inhibitor, was the first JAK inhibitor approved by the FDA and is indicated for the treatment of myelofibrosis and polycythemia vera. Additionally, oral and topical JAK inhibitors have shown efficacy in the treatment of psoriasis, rheumatoid arthritis, alopecia areata, vitiligo, and pruritus from atopic dermatitis.28
The JAK-STAT pathway is complex, and the biological activity of the pathway is both proinflammatory and pro–cell survival and proliferation. Because signaling through the pathway can increase angiogenesis and inhibit apoptosis, inhibition of this pathway has been exploited for the treatment of some tumors. However, inhibition of interferon and proinflammatory interleukin signaling also can potentially promote tumor growth by means of inhibition of downstream cytotoxic T-cell signaling, theoretically increasing the risk for NMSC. A study examining the 5-year efficacy of ruxolitinib in myelofibrosis patients (COMFORT-II trial) found that 17.1% of patients developed NMSC compared to only 2.7% of those on the best available therapy. After adjustment by patient exposure, the NMSC rate was still doubled for ruxolitinib-treated patients compared to controls (6.1/100 patient-years and 3.0/100 patient-years, respectively).29 Eighty-week follow-up of the phase 3 clinical trial of ruxolitinib for the treatment of polycythemia vera also noted an increased incidence of NMSC, albeit a more conservative increase. Patients randomized to the ruxolitinib treatment group developed NMSC at a rate of 4.4/100 patient-years, whereas the rate for controls treated with best available therapy was 2.7/100 patient-years.30 In contrast, 5-year follow-up of the COMFORT-I trial, also examining the efficacy of ruxolitinib in myelofibrosis, showed no increased risk for NMSC between ruxolitinib-treated patients and placebo (2.7/100 patient-years and 3.9/100 patient-years, respectively).31
A 2017 case series described 5 patients with myelofibrosis who developed multiple skin cancers with aggressive features while receiving ruxolitinib.32 Duration of ruxolitinib therapy ranged from 4 months to 4 years; 3 patients had a history of hydroxyurea exposure, and only 1 patient had a history of NMSC. High-risk cutaneous SCC, undifferentiated pleomorphic sarcoma, and lentigo maligna melanoma (Breslow thickness, 0.45 mm) were among the tumors reported in this series. Although no definitive conclusion can be made regarding the causality of JAK inhibitors in promoting these tumors, the association warrants further investigation. Clinicians should be aware that ruxolitinib might amplify the risk for NMSC in patients with pre-existing genetic or exposure-related susceptibility. Interruption of drug therapy may be necessary in managing patients who develop an aggressive tumor.32
In contrast, tofacitinib, which specifically inhibits JAK3, carries very low risk, if any, for NMSC when used for the treatment of psoriasis and rheumatoid arthritis. Results from 2 phase 3 trials analyzing the efficacy of tofacitinib in psoriasis demonstrated that only 2 of 1486 patients treated developed NMSC compared to none in the control group.33 Furthermore, analysis of NMSC across the tofacitinib rheumatoid arthritis clinical program, which included a total of 15,103 patient-years of exposure, demonstrated that the overall NMSC incidence was 0.55 for every 100 patient-years. Of note, the risk in patients receiving high-dose treatment (10 mg vs 5 mg) was nearly doubled in long-term follow-up studies (0.79/100 patient-years and 0.41/100 patient-years, respectively). Overall, the study concluded that treatment with tofacitinib presents no greater increased risk for NMSC than treatment with tumor necrosis factor inhibitors.33
PDE-5 Inhibitors
Phosphodiesterase 5 inhibitors, such as sildenafil citrate, have been widely prescribed for the treatment of erectile dysfunction. Studies have shown that BRAF-activated melanomas, which occur in approximately 50% to 70% of melanomas, also result in reduced PDE-5 expression.34-36 In these melanomas, downregulation of PDE-5 results in increased intracellular calcium,36 which has been shown to induce melanoma invasion.36,37 Given this similarity in molecular pathway between BRAF-activated melanomas and PDE-5 inhibitors, there has been increased concern that PDE-5 inhibitors might be associated with an increased risk for melanoma.
In 2014, Li et al38 published a retrospective analysis suggesting an association with sildenafil and an increased risk for melanoma. Their study utilized the Health Professionals Follow-up Study to identify a statistically significant elevation in the risk for invasive melanoma with both recent sildenafil use (multivariate-adjusted HR=2.24) and use at any time (HR=1.92). These results controlled for confounding variables, such as presence of major chronic disease, use of other erectile dysfunction treatments, family history of melanoma, history of sun exposure, and UV index of the patient’s residence. Notably, the study also found that sildenafil did not affect the incidence of BCC or SCC.38
In 2015, Loeb et al39 also examined the potential association between PDE-5 inhibitors and melanoma. Review of several Swedish drug and cancer registries allowed for analysis of melanoma risk and PDE-5 inhibitor use, based on number of prescriptions filled and type of PDE-5 inhibitor prescribed. Their analysis showed that men developing melanoma were more likely than nonmelanoma controls to have taken a PDE-5 inhibitor (11% vs 8%). In a subgroup analysis, however, statistical significance was shown for men with only a single prescription filled (34% of cases; P<.05), whereas the difference for men with multiple filled prescriptions did not meet statistical significance. Furthermore, the study did not find increased risk with longer-acting tadalafil and vardenafil (odds ratio [OR]=1.16) compared to sildenafil (OR=1.14). Last, use of PDE-5 inhibitors was only associated with stage 0 (OR=1.49) and stage I (OR=1.21) tumors, not with stages II to IV (OR=0.83) tumors. Although there was a statistically significant association between PDE-5 inhibitors and malignant melanoma (P<.05), the subgroup analysis findings pointed away from a causal relationship and likely toward a confounding of variable(s).39
A 2016 study by Lian et al40 looked at the risk for melanoma in a cohort of patients diagnosed with erectile dysfunction. No association between PDE-5 inhibitors and melanoma risk was shown when comparing patients who received a PDE-5 inhibitor and those who did not receive a PDE-5 inhibitor. However, secondary analysis did show that melanoma risk was increased among patients receiving more pills (34%) and prescriptions (30%). The authors concluded that there was no association between PDE-5 inhibitor use and overall increased risk for melanoma, and the increased risk associated with a greater number of pills and prescriptions would require further study.40
In contrast, a 2017 meta-analysis by Tang et al41 of 5 studies (3 of which were the aforementioned trials38-40) concluded that use of PDE-5 inhibitors was associated with a small but significantly increased risk for melanoma (OR=1.12) and BCC (OR=1.14) but not SCC. Furthermore, the study found no evidence of dosage-dependent association between PDE-5 inhibitor use and melanoma risk.41
Overall, clinical studies have been inconclusive in determining the risk for melanoma in the setting of PDE-5 inhibitor use. Studies showing an increased rate of melanoma within patient cohorts receiving PDE-5 inhibitors are limited; results might be affected by confounding variables. However, given the similarity in mechanism between PDE-5 inhibitors and HRAS-activated melanomas, it is reasonable to continue research into this potential association.
Conclusion
Since the turn of the century, drugs targeting cell-signaling pathways have been developed to treat inflammatory, oncologic, and immune conditions. The role of immunosuppressants in promoting skin cancer is well established and supported by a vast literature base. However, associations are less clear with newer immunomodulatory and antineoplastic medications. Skin cancer has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, JAK inhibitors, and PDE-5 inhibitors. In the case of JAK and PDE-5 inhibitors, the increased risk for melanoma and NMSC is somewhat inconclusive; risk is more firmly established for BRAF inhibitors and smoothened inhibitors. For the antineoplastic agents reviewed, the therapeutic effect of cancer regression is well documented, and benefits of continued therapy outweigh the increased risk for skin cancer promotion in nearly all cases. The value of early detection has been well documented for skin malignancy; therefore, increased skin surveillance and prompt management of suspicious lesions should be a priority for physicians treating patients undergoing therapy with these medications
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanoma in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
- Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA Dermatol. 2015;151:1103-1109.
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427-430.
- Ryan MB, Der CJ, Wang-Gillam A, et al. Targeting RAS-mutant cancers: is ERK the key? Trends Cancer. 2015;1:183-198.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
- Holderfield M, Nagel TE, Stuart DD. Mechanism and consequence of RAF kinase activation by small-molecule inhibitors. Br J Cancer. 2014;111:640-645.
- Dalle S, Poulalhon N, Debarbieux S, et al. Tracking of second primary melanomas in vemurafenib-treated patients. JAMA Dermatol. 2013;149:488-490.
- Williams VL, Cohen PR, Stewart DJ. Sorafenib-induced premalignant and malignant skin lesions. Int J Dermatol. 2011;50:396-402.
- Arnault JP, Wechsler J, Escudier B, et al. Keratoacanthomas and squamous cell carcinomas in patients receiving sorafenib. J Clin Oncol. 2009;27:e59-e61.
- Smith KJ, Haley H, Hamza S, et al. Eruptive keratoacanthoma-type squamous cell carcinomas in patients taking sorafenib for the treatment of solid tumors. Dermatol Surg. 2009;35:1766-1770.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Demirci H, Worden F, Nelson CC, et al. Efficacy of vismodegib (Erivedge) for basal cell carcinoma involving the orbit and periocular area. Ophthalmic Plast Reconstr Surg. 2015;31:463-466.
- Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342-353.
- Ridky TW, Cotsarelis G. Vismodegib resistance in basal cell carcinoma: not a smooth fit. Cancer Cell. 2015;27:315-316.
- Aasi S, Silkiss R, Tang JY, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242-243.
- Orouji A, Goerdt S, Utikal J, et al. Multiple highly and moderately differentiated squamous cell carcinomas of the skin during vismodegib treatment of inoperable basal cell carcinoma. Br J Dermatol. 2014;171:431-433.
- Iarrobino A, Messina JL, Kudchadkar R, et al. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2013;69:e33-e34.
- Saintes C, Saint-Jean M, Brocard A, et al. Development of squamous cell carcinoma into basal cell carcinoma under treatment with vismodegib. J Eur Acad Dermatol Venereol. 2015;29:1006-1009.
- Mohan SV, Chang J, Li S, et al. Increased risk of cutaneous squamous cell carcinoma after vismodegib therapy for basal cell carcinoma. JAMA Dermatol. 2016;152:527-532.
- Zhao X, Ponomaryov T, Ornell KJ, et al. RAS/MAPK activation drives resistance to Smo inhibition, metastasis, and tumor evolution in Shh pathway-dependent tumors. Cancer Res. 2015;75:3623-3635.
- Chang AL, Oro AE. Initial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma. Arch Dermatol. 2012;148:1324-1325.
- Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76:736-744.
- Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30:1701-1707.
- Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica. 2016;101:821-829.
- Verstovsek S, Mesa RA, Gotlib J, et al; COMFORT-I investigators. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10:55.
- Blechman AB, Cabell CE, Weinberger CH, et al. Aggressive skin cancers occurring in patients treated with the Janus kinase inhibitor ruxolitinib. J Drugs Dermatol. 2017;16:508-511.
- Papp KA, Menter MA, Abe M, et al; OPT Pivotal 1 and OPT Pivotal 2 investigators. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two randomized, placebo-controlled, phase III trials. Br J Dermatol. 2015;173:949-961.
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875-885.
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851-857.
- Arozarena I, Sanchez-Laorden B, Packer L, et al. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell. 2011;19:45-57.
- Houslay MD. Hard times for oncogenic BRAF-expressing melanoma cells. Cancer Cell. 2011;19:3-4.
- Li WQ, Qureshi AA, Robinson KC, et al. Sildenafil use and increased risk of incident melanoma in US men: a prospective cohort study. JAMA Intern Med. 2014;174:964-970.
- Loeb S, Folkvaljon Y, Lambe M, et al. Use of phosphodiesterase type 5 inhibitors for erectile dysfunction and risk of malignant melanoma. JAMA. 2015;313:2449-2455.
- Lian Y, Yin H, Pollak MN, et al. Phosphodiesterase type 5 inhibitors and the risk of melanoma skin cancer. Eur Urol. 2016;70:808-815.
- Tang H, Wu W, Fu S, et al. Phosphodiesterase type 5 inhibitors and risk of melanoma: a meta-analysis. J Am Acad Dermatol. 2017;77:480.e9-488.e9.
Dermatologists are increasingly called on to evaluate patients with complex medical problems who are often taking many medications. Over the last several decades, many new drugs that target molecular pathways in carcinogenesis and the inflammatory immune system have been developed. Increased skin cancer risk has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, Janus kinase (JAK) inhibitors, and phosphodiesterase 5 (PDE-5) inhibitors. We review the literature and data regarding the significance and strength of these associations and the molecular pathways by which these medications promote cutaneous tumorigenesis. The association of skin cancer with drugs that either induce photosensitivity—nonsteroidal anti-inflammatory drugs, antibiotics (eg, tetracyclines, fluoroquinolones, trimethoprim-sulfamethoxazole), voriconazole, thiazides—or suppress the immune system—certain biologics (eg, anti–tumor necrosis factor agents), calcineurin inhibitors, thiopurines, methotrexate, cyclosporine—is well known and is therefore not reviewed in this discussion.
BRAF Inhibitors
The mitogen-activated protein kinase (MAPK) pathway (also known as the RAS/RAF/MAPK signaling pathway) is important in growth factor–receptor signaling and plays a key role in cell differentiation, survival, and proliferation. Activating mutations in this pathway allow cells to grow and proliferate in a growth factor–independent manner. Twenty percent of human cancers harbor a mutation in the RAS oncogene, an upstream mediator of the pathway.1 Activating mutations in BRAF, a serine/threonine kinase, predominate in cutaneous melanoma and also have been found in 40% to 70% of papillary thyroid malignancies, 10% to 20% of cholangiocarcinomas, and 5% to 20% of colorectal carcinomas. The most common BRAF mutation in cutaneous melanoma is V600E, which involves a glutamic acid for valine substitution at codon 600. This mutation activates BRAF 500-fold and is present in approximately 50% of melanomas.1,2
Vemurafenib, a selective BRAF inhibitor, was approved by the US Food and Drug Administration (FDA) for the treatment of metastatic melanoma in the United States in 2011. Phase 3 trial data demonstrated that vemurafenib resulted in improved survival and decreased risk for disease progression compared to dacarbazine, the former best treatment.3 During phase 1 testing, it became apparent that vemurafenib treatment was associated with a 31% increased risk for squamous cell carcinoma (SCC), most commonly well-differentiated SCC, and keratoacanthomas (KAs).4 This association was confirmed in phase 2 and 3 studies, though the incidence was lower. McArthur et al5 reported a 19% incidence of cutaneous SCC with extended follow-up analysis of the phase 3 trial. Dabrafenib, another BRAF inhibitor, has been similarly associated with increasing the risk for SCC and KA.
In one study, the mean time to development of SCC after initiating vemurafenib therapy was 10 weeks, with lesions reported as early as 3 weeks. Most patients had clinical signs of chronically sun damaged skin; however, a history of SCC was present in only 17%. Most lesions (63%) were characterized as KAs.6
The mechanism for BRAF inhibitor–induced squamoproliferative growth is due to paradoxical activation of the MAPK pathway in cells with wild-type BRAF that harbor upstream-activating mutations in RAS or tyrosine kinase receptors.7 In the presence of a BRAF inhibitor, inactivated BRAF forms heterodimers with wild-type CRAF (a BRAF-CRAF heterodimer). The heterodimer forms a complex with the mutant RAS that leads to transactivation of the CRAF molecule,8,9 resulting in a paradoxical increase in MAPK signaling and consequent ERK phosphorylation and activation through CRAF signaling. RAS, particularly HRAS, mutations have been found in 60% of all vemurafenib-associated SCCs and KAs. For this reason, it is thought that vemurafenib potentiates tumorigenesis in subclinical lesions harboring upstream MAPK pathway mutations as opposed to inducing de novo lesions.6
Because BRAF inhibitors are remarkably efficacious in the treatment of metastatic melanomas harboring the V600E BRAF mutation, there are no restrictions on their use, despite the known increased risk for SCC. Squamous cell carcinomas tend to be low grade, and all tumors that developed in phase 1 to 3 trials were treated with simple excision. The development of SCC did not necessitate interruption of treatment. Furthermore, the addition of MEK inhibition to BRAF inhibitor therapy reduces the risk for SCC from 19% to 7%.7,10,11
In addition to SCC, second primary melanomas (SPMs) have been reported in patients treated with BRAF inhibitors. It has been shown that these melanomas occur in melanocytes with wild-type BRAF. It has been postulated that some of these tumors occur in cells that harbor upstream mutations in RAS, whereas others might result from alternate signaling through non-RAF oncogenic pathways.9,12
Zimmer et al1 reported 12 SPMs in 11 patients treated with BRAF inhibitor therapy. They reported a median delay of 8 weeks (range, 4–27 weeks) for SPM development. Tumors were detected in early stages; 1 tumor harbored an NRAS mutation.1
Dalle et al13 reported 25 SPMs in 120 vemurafenib-treated patients. Median delay in SPM development was 14 weeks (range, 4–42 weeks). All tumors were thin, ranging from in situ to 0.45-mm thick. Wild-type BRAF was detected in the 21 melanomas sampled; 1 lesion showed mutated NRAS.13
The exact incidence of SPM in the setting of BRAF inhibition is thought to be at least 10-fold less than SCC and KA.2 Patients on BRAF inhibitor therapy should have routine full-body skin examinations, given the increased risk for SPM and SCC.
Another drug belonging to the tyrosine kinase inhibitor family, sorafenib, is used in the treatment of solid tumors, particularly hepatocellular and renal cell carcinomas, and also has been associated with development of cutaneous SCC and KAs.14 Sorafenib is a multiple tyrosine kinase inhibitor that also inhibits the RAF serine/threonine kinases. Similar to vemurafenib and dabrafenib, SCCs and KAs associated with sorafenib tend to arise in patients with chronic actinic damage during the first 2 months of treatment. It has been hypothesized that inhibition of RAF kinases is pathogenic in inducing SCCs because these lesions have not been reported with sunitinib, another multiple tyrosine kinase inhibitor that lacks the ability to inhibit serine/threonine kinases.15,16 Although SCCs and KAs associated with sorafenib tend to be low grade, it is reasonable to consider sunitinib or an alternative tyrosine kinase inhibitor in patients who develop multiple SCCs while taking sorafenib.16
Sonic Hedgehog–Inhibiting Agents
Vismodegib, the first small molecule inhibitor of the signaling protein smoothened, gained FDA approval for the treatment of metastatic or locally advanced basal cell carcinoma (BCC) in 2012. A second agent with an identical mechanism of action, sonidegib, was approved by the FDA for locally advanced BCC in 2015. Approximately 90% of BCCs contain mutations in the sonic hedgehog pathway, which lead to constitutive smoothened activation and uncontrolled cell proliferation.17 The development of smoothened inhibitors introduced a much-needed treatment for inoperable or metastatic BCC,17,18 though long-term utility is limited by drug resistance with extended use in this patient population.19,20 Several case reports have documented the emergence of KA21 and cutaneous SCC following vismodegib treatment of advanced or metastatic BCC.22-24 A larger case-control study by Mohan et al25 showed that patients with BCC treated with vismodegib had an increased risk for non-BCC malignancy (hazard ratio [HR]=6.37), most of which were cutaneous SCC (HR=8.12).
The mechanism by which selective inhibition of smoothened leads to cutaneous SCC is unclear. A study found that patients on vismodegib who developed SCC within the original BCC site had elevated ERK levels within tumor tissue, suggesting that the RAS/RAF/MAPK pathway can become upregulated during hedgehog inhibition.26 Other studies looking at hedgehog inhibition in medulloblastoma models also have shown activated RAS/RAF/MAPK pathways.25 These findings suggest that tumors under smoothened inhibition might be able to bypass the sonic hedgehog pathway and continue to grow by upregulating alternative growth pathways, such as RAS/RAF/MAPK.25,26
The incidence of cutaneous SCC following vismodegib treatment is unknown. Chang and Oro27 examined BCC tumor regrowth from secondary (acquired) resistance to vismodegib and noted that lesions recurred within 1 cm of the original tumor 21% of the time. Although none of the 12 patients whose tumors regrew during treatment were reported to have developed SCC, several demonstrated different BCC subtypes than the pretreatment specimen. The authors proposed that regrowth of BCC was due to upregulated alternative pathways allowing tumors to bypass smoothened inhibition, which is similar to the proposed mechanism for SCC development in vismodegib patients.27
Prospective studies are needed to confirm the link between vismodegib and cutaneous SCC; establish the incidence of SCC development; and identify any pretreatment factors, tumor characteristics, or treatment details (eg, dosage, duration) that might contribute to SCC development. Furthermore, because Mohan et al25 observed that vismodegib-treated patients were less likely to develop SCC in situ than controls, it is unknown if these tumors are more aggressive than traditional SCC. At this point, careful surveillance and regular full-body skin examinations are advised for patients on vismodegib for treatment of advanced BCC.
JAK Inhibitors
Another class of medications potentially associated with increased development of nonmelanoma skin cancer (NMSC) is the JAK inhibitors (also known as jakinibs). Many proinflammatory signaling pathways converge on the JAK family of enzymes—JAK1, JAK2, JAK3, and TYK2. These enzymes operate in cytokine signal transduction by phosphorylating activated cytokine receptors, which allows for recruitment and activation by means of phosphorylation of transcription factors collectively known as signal transducers and activators of transcription (STATs). Phosphorylated STATs dimerize and translocate to the nucleus, acting as direct transcription promoters. Janus kinase inhibitors modulate the immune response by reducing the effect of interleukin and interferon signaling.
Ruxolitinib, a JAK1/JAK2 inhibitor, was the first JAK inhibitor approved by the FDA and is indicated for the treatment of myelofibrosis and polycythemia vera. Additionally, oral and topical JAK inhibitors have shown efficacy in the treatment of psoriasis, rheumatoid arthritis, alopecia areata, vitiligo, and pruritus from atopic dermatitis.28
The JAK-STAT pathway is complex, and the biological activity of the pathway is both proinflammatory and pro–cell survival and proliferation. Because signaling through the pathway can increase angiogenesis and inhibit apoptosis, inhibition of this pathway has been exploited for the treatment of some tumors. However, inhibition of interferon and proinflammatory interleukin signaling also can potentially promote tumor growth by means of inhibition of downstream cytotoxic T-cell signaling, theoretically increasing the risk for NMSC. A study examining the 5-year efficacy of ruxolitinib in myelofibrosis patients (COMFORT-II trial) found that 17.1% of patients developed NMSC compared to only 2.7% of those on the best available therapy. After adjustment by patient exposure, the NMSC rate was still doubled for ruxolitinib-treated patients compared to controls (6.1/100 patient-years and 3.0/100 patient-years, respectively).29 Eighty-week follow-up of the phase 3 clinical trial of ruxolitinib for the treatment of polycythemia vera also noted an increased incidence of NMSC, albeit a more conservative increase. Patients randomized to the ruxolitinib treatment group developed NMSC at a rate of 4.4/100 patient-years, whereas the rate for controls treated with best available therapy was 2.7/100 patient-years.30 In contrast, 5-year follow-up of the COMFORT-I trial, also examining the efficacy of ruxolitinib in myelofibrosis, showed no increased risk for NMSC between ruxolitinib-treated patients and placebo (2.7/100 patient-years and 3.9/100 patient-years, respectively).31
A 2017 case series described 5 patients with myelofibrosis who developed multiple skin cancers with aggressive features while receiving ruxolitinib.32 Duration of ruxolitinib therapy ranged from 4 months to 4 years; 3 patients had a history of hydroxyurea exposure, and only 1 patient had a history of NMSC. High-risk cutaneous SCC, undifferentiated pleomorphic sarcoma, and lentigo maligna melanoma (Breslow thickness, 0.45 mm) were among the tumors reported in this series. Although no definitive conclusion can be made regarding the causality of JAK inhibitors in promoting these tumors, the association warrants further investigation. Clinicians should be aware that ruxolitinib might amplify the risk for NMSC in patients with pre-existing genetic or exposure-related susceptibility. Interruption of drug therapy may be necessary in managing patients who develop an aggressive tumor.32
In contrast, tofacitinib, which specifically inhibits JAK3, carries very low risk, if any, for NMSC when used for the treatment of psoriasis and rheumatoid arthritis. Results from 2 phase 3 trials analyzing the efficacy of tofacitinib in psoriasis demonstrated that only 2 of 1486 patients treated developed NMSC compared to none in the control group.33 Furthermore, analysis of NMSC across the tofacitinib rheumatoid arthritis clinical program, which included a total of 15,103 patient-years of exposure, demonstrated that the overall NMSC incidence was 0.55 for every 100 patient-years. Of note, the risk in patients receiving high-dose treatment (10 mg vs 5 mg) was nearly doubled in long-term follow-up studies (0.79/100 patient-years and 0.41/100 patient-years, respectively). Overall, the study concluded that treatment with tofacitinib presents no greater increased risk for NMSC than treatment with tumor necrosis factor inhibitors.33
PDE-5 Inhibitors
Phosphodiesterase 5 inhibitors, such as sildenafil citrate, have been widely prescribed for the treatment of erectile dysfunction. Studies have shown that BRAF-activated melanomas, which occur in approximately 50% to 70% of melanomas, also result in reduced PDE-5 expression.34-36 In these melanomas, downregulation of PDE-5 results in increased intracellular calcium,36 which has been shown to induce melanoma invasion.36,37 Given this similarity in molecular pathway between BRAF-activated melanomas and PDE-5 inhibitors, there has been increased concern that PDE-5 inhibitors might be associated with an increased risk for melanoma.
In 2014, Li et al38 published a retrospective analysis suggesting an association with sildenafil and an increased risk for melanoma. Their study utilized the Health Professionals Follow-up Study to identify a statistically significant elevation in the risk for invasive melanoma with both recent sildenafil use (multivariate-adjusted HR=2.24) and use at any time (HR=1.92). These results controlled for confounding variables, such as presence of major chronic disease, use of other erectile dysfunction treatments, family history of melanoma, history of sun exposure, and UV index of the patient’s residence. Notably, the study also found that sildenafil did not affect the incidence of BCC or SCC.38
In 2015, Loeb et al39 also examined the potential association between PDE-5 inhibitors and melanoma. Review of several Swedish drug and cancer registries allowed for analysis of melanoma risk and PDE-5 inhibitor use, based on number of prescriptions filled and type of PDE-5 inhibitor prescribed. Their analysis showed that men developing melanoma were more likely than nonmelanoma controls to have taken a PDE-5 inhibitor (11% vs 8%). In a subgroup analysis, however, statistical significance was shown for men with only a single prescription filled (34% of cases; P<.05), whereas the difference for men with multiple filled prescriptions did not meet statistical significance. Furthermore, the study did not find increased risk with longer-acting tadalafil and vardenafil (odds ratio [OR]=1.16) compared to sildenafil (OR=1.14). Last, use of PDE-5 inhibitors was only associated with stage 0 (OR=1.49) and stage I (OR=1.21) tumors, not with stages II to IV (OR=0.83) tumors. Although there was a statistically significant association between PDE-5 inhibitors and malignant melanoma (P<.05), the subgroup analysis findings pointed away from a causal relationship and likely toward a confounding of variable(s).39
A 2016 study by Lian et al40 looked at the risk for melanoma in a cohort of patients diagnosed with erectile dysfunction. No association between PDE-5 inhibitors and melanoma risk was shown when comparing patients who received a PDE-5 inhibitor and those who did not receive a PDE-5 inhibitor. However, secondary analysis did show that melanoma risk was increased among patients receiving more pills (34%) and prescriptions (30%). The authors concluded that there was no association between PDE-5 inhibitor use and overall increased risk for melanoma, and the increased risk associated with a greater number of pills and prescriptions would require further study.40
In contrast, a 2017 meta-analysis by Tang et al41 of 5 studies (3 of which were the aforementioned trials38-40) concluded that use of PDE-5 inhibitors was associated with a small but significantly increased risk for melanoma (OR=1.12) and BCC (OR=1.14) but not SCC. Furthermore, the study found no evidence of dosage-dependent association between PDE-5 inhibitor use and melanoma risk.41
Overall, clinical studies have been inconclusive in determining the risk for melanoma in the setting of PDE-5 inhibitor use. Studies showing an increased rate of melanoma within patient cohorts receiving PDE-5 inhibitors are limited; results might be affected by confounding variables. However, given the similarity in mechanism between PDE-5 inhibitors and HRAS-activated melanomas, it is reasonable to continue research into this potential association.
Conclusion
Since the turn of the century, drugs targeting cell-signaling pathways have been developed to treat inflammatory, oncologic, and immune conditions. The role of immunosuppressants in promoting skin cancer is well established and supported by a vast literature base. However, associations are less clear with newer immunomodulatory and antineoplastic medications. Skin cancer has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, JAK inhibitors, and PDE-5 inhibitors. In the case of JAK and PDE-5 inhibitors, the increased risk for melanoma and NMSC is somewhat inconclusive; risk is more firmly established for BRAF inhibitors and smoothened inhibitors. For the antineoplastic agents reviewed, the therapeutic effect of cancer regression is well documented, and benefits of continued therapy outweigh the increased risk for skin cancer promotion in nearly all cases. The value of early detection has been well documented for skin malignancy; therefore, increased skin surveillance and prompt management of suspicious lesions should be a priority for physicians treating patients undergoing therapy with these medications
Dermatologists are increasingly called on to evaluate patients with complex medical problems who are often taking many medications. Over the last several decades, many new drugs that target molecular pathways in carcinogenesis and the inflammatory immune system have been developed. Increased skin cancer risk has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, Janus kinase (JAK) inhibitors, and phosphodiesterase 5 (PDE-5) inhibitors. We review the literature and data regarding the significance and strength of these associations and the molecular pathways by which these medications promote cutaneous tumorigenesis. The association of skin cancer with drugs that either induce photosensitivity—nonsteroidal anti-inflammatory drugs, antibiotics (eg, tetracyclines, fluoroquinolones, trimethoprim-sulfamethoxazole), voriconazole, thiazides—or suppress the immune system—certain biologics (eg, anti–tumor necrosis factor agents), calcineurin inhibitors, thiopurines, methotrexate, cyclosporine—is well known and is therefore not reviewed in this discussion.
BRAF Inhibitors
The mitogen-activated protein kinase (MAPK) pathway (also known as the RAS/RAF/MAPK signaling pathway) is important in growth factor–receptor signaling and plays a key role in cell differentiation, survival, and proliferation. Activating mutations in this pathway allow cells to grow and proliferate in a growth factor–independent manner. Twenty percent of human cancers harbor a mutation in the RAS oncogene, an upstream mediator of the pathway.1 Activating mutations in BRAF, a serine/threonine kinase, predominate in cutaneous melanoma and also have been found in 40% to 70% of papillary thyroid malignancies, 10% to 20% of cholangiocarcinomas, and 5% to 20% of colorectal carcinomas. The most common BRAF mutation in cutaneous melanoma is V600E, which involves a glutamic acid for valine substitution at codon 600. This mutation activates BRAF 500-fold and is present in approximately 50% of melanomas.1,2
Vemurafenib, a selective BRAF inhibitor, was approved by the US Food and Drug Administration (FDA) for the treatment of metastatic melanoma in the United States in 2011. Phase 3 trial data demonstrated that vemurafenib resulted in improved survival and decreased risk for disease progression compared to dacarbazine, the former best treatment.3 During phase 1 testing, it became apparent that vemurafenib treatment was associated with a 31% increased risk for squamous cell carcinoma (SCC), most commonly well-differentiated SCC, and keratoacanthomas (KAs).4 This association was confirmed in phase 2 and 3 studies, though the incidence was lower. McArthur et al5 reported a 19% incidence of cutaneous SCC with extended follow-up analysis of the phase 3 trial. Dabrafenib, another BRAF inhibitor, has been similarly associated with increasing the risk for SCC and KA.
In one study, the mean time to development of SCC after initiating vemurafenib therapy was 10 weeks, with lesions reported as early as 3 weeks. Most patients had clinical signs of chronically sun damaged skin; however, a history of SCC was present in only 17%. Most lesions (63%) were characterized as KAs.6
The mechanism for BRAF inhibitor–induced squamoproliferative growth is due to paradoxical activation of the MAPK pathway in cells with wild-type BRAF that harbor upstream-activating mutations in RAS or tyrosine kinase receptors.7 In the presence of a BRAF inhibitor, inactivated BRAF forms heterodimers with wild-type CRAF (a BRAF-CRAF heterodimer). The heterodimer forms a complex with the mutant RAS that leads to transactivation of the CRAF molecule,8,9 resulting in a paradoxical increase in MAPK signaling and consequent ERK phosphorylation and activation through CRAF signaling. RAS, particularly HRAS, mutations have been found in 60% of all vemurafenib-associated SCCs and KAs. For this reason, it is thought that vemurafenib potentiates tumorigenesis in subclinical lesions harboring upstream MAPK pathway mutations as opposed to inducing de novo lesions.6
Because BRAF inhibitors are remarkably efficacious in the treatment of metastatic melanomas harboring the V600E BRAF mutation, there are no restrictions on their use, despite the known increased risk for SCC. Squamous cell carcinomas tend to be low grade, and all tumors that developed in phase 1 to 3 trials were treated with simple excision. The development of SCC did not necessitate interruption of treatment. Furthermore, the addition of MEK inhibition to BRAF inhibitor therapy reduces the risk for SCC from 19% to 7%.7,10,11
In addition to SCC, second primary melanomas (SPMs) have been reported in patients treated with BRAF inhibitors. It has been shown that these melanomas occur in melanocytes with wild-type BRAF. It has been postulated that some of these tumors occur in cells that harbor upstream mutations in RAS, whereas others might result from alternate signaling through non-RAF oncogenic pathways.9,12
Zimmer et al1 reported 12 SPMs in 11 patients treated with BRAF inhibitor therapy. They reported a median delay of 8 weeks (range, 4–27 weeks) for SPM development. Tumors were detected in early stages; 1 tumor harbored an NRAS mutation.1
Dalle et al13 reported 25 SPMs in 120 vemurafenib-treated patients. Median delay in SPM development was 14 weeks (range, 4–42 weeks). All tumors were thin, ranging from in situ to 0.45-mm thick. Wild-type BRAF was detected in the 21 melanomas sampled; 1 lesion showed mutated NRAS.13
The exact incidence of SPM in the setting of BRAF inhibition is thought to be at least 10-fold less than SCC and KA.2 Patients on BRAF inhibitor therapy should have routine full-body skin examinations, given the increased risk for SPM and SCC.
Another drug belonging to the tyrosine kinase inhibitor family, sorafenib, is used in the treatment of solid tumors, particularly hepatocellular and renal cell carcinomas, and also has been associated with development of cutaneous SCC and KAs.14 Sorafenib is a multiple tyrosine kinase inhibitor that also inhibits the RAF serine/threonine kinases. Similar to vemurafenib and dabrafenib, SCCs and KAs associated with sorafenib tend to arise in patients with chronic actinic damage during the first 2 months of treatment. It has been hypothesized that inhibition of RAF kinases is pathogenic in inducing SCCs because these lesions have not been reported with sunitinib, another multiple tyrosine kinase inhibitor that lacks the ability to inhibit serine/threonine kinases.15,16 Although SCCs and KAs associated with sorafenib tend to be low grade, it is reasonable to consider sunitinib or an alternative tyrosine kinase inhibitor in patients who develop multiple SCCs while taking sorafenib.16
Sonic Hedgehog–Inhibiting Agents
Vismodegib, the first small molecule inhibitor of the signaling protein smoothened, gained FDA approval for the treatment of metastatic or locally advanced basal cell carcinoma (BCC) in 2012. A second agent with an identical mechanism of action, sonidegib, was approved by the FDA for locally advanced BCC in 2015. Approximately 90% of BCCs contain mutations in the sonic hedgehog pathway, which lead to constitutive smoothened activation and uncontrolled cell proliferation.17 The development of smoothened inhibitors introduced a much-needed treatment for inoperable or metastatic BCC,17,18 though long-term utility is limited by drug resistance with extended use in this patient population.19,20 Several case reports have documented the emergence of KA21 and cutaneous SCC following vismodegib treatment of advanced or metastatic BCC.22-24 A larger case-control study by Mohan et al25 showed that patients with BCC treated with vismodegib had an increased risk for non-BCC malignancy (hazard ratio [HR]=6.37), most of which were cutaneous SCC (HR=8.12).
The mechanism by which selective inhibition of smoothened leads to cutaneous SCC is unclear. A study found that patients on vismodegib who developed SCC within the original BCC site had elevated ERK levels within tumor tissue, suggesting that the RAS/RAF/MAPK pathway can become upregulated during hedgehog inhibition.26 Other studies looking at hedgehog inhibition in medulloblastoma models also have shown activated RAS/RAF/MAPK pathways.25 These findings suggest that tumors under smoothened inhibition might be able to bypass the sonic hedgehog pathway and continue to grow by upregulating alternative growth pathways, such as RAS/RAF/MAPK.25,26
The incidence of cutaneous SCC following vismodegib treatment is unknown. Chang and Oro27 examined BCC tumor regrowth from secondary (acquired) resistance to vismodegib and noted that lesions recurred within 1 cm of the original tumor 21% of the time. Although none of the 12 patients whose tumors regrew during treatment were reported to have developed SCC, several demonstrated different BCC subtypes than the pretreatment specimen. The authors proposed that regrowth of BCC was due to upregulated alternative pathways allowing tumors to bypass smoothened inhibition, which is similar to the proposed mechanism for SCC development in vismodegib patients.27
Prospective studies are needed to confirm the link between vismodegib and cutaneous SCC; establish the incidence of SCC development; and identify any pretreatment factors, tumor characteristics, or treatment details (eg, dosage, duration) that might contribute to SCC development. Furthermore, because Mohan et al25 observed that vismodegib-treated patients were less likely to develop SCC in situ than controls, it is unknown if these tumors are more aggressive than traditional SCC. At this point, careful surveillance and regular full-body skin examinations are advised for patients on vismodegib for treatment of advanced BCC.
JAK Inhibitors
Another class of medications potentially associated with increased development of nonmelanoma skin cancer (NMSC) is the JAK inhibitors (also known as jakinibs). Many proinflammatory signaling pathways converge on the JAK family of enzymes—JAK1, JAK2, JAK3, and TYK2. These enzymes operate in cytokine signal transduction by phosphorylating activated cytokine receptors, which allows for recruitment and activation by means of phosphorylation of transcription factors collectively known as signal transducers and activators of transcription (STATs). Phosphorylated STATs dimerize and translocate to the nucleus, acting as direct transcription promoters. Janus kinase inhibitors modulate the immune response by reducing the effect of interleukin and interferon signaling.
Ruxolitinib, a JAK1/JAK2 inhibitor, was the first JAK inhibitor approved by the FDA and is indicated for the treatment of myelofibrosis and polycythemia vera. Additionally, oral and topical JAK inhibitors have shown efficacy in the treatment of psoriasis, rheumatoid arthritis, alopecia areata, vitiligo, and pruritus from atopic dermatitis.28
The JAK-STAT pathway is complex, and the biological activity of the pathway is both proinflammatory and pro–cell survival and proliferation. Because signaling through the pathway can increase angiogenesis and inhibit apoptosis, inhibition of this pathway has been exploited for the treatment of some tumors. However, inhibition of interferon and proinflammatory interleukin signaling also can potentially promote tumor growth by means of inhibition of downstream cytotoxic T-cell signaling, theoretically increasing the risk for NMSC. A study examining the 5-year efficacy of ruxolitinib in myelofibrosis patients (COMFORT-II trial) found that 17.1% of patients developed NMSC compared to only 2.7% of those on the best available therapy. After adjustment by patient exposure, the NMSC rate was still doubled for ruxolitinib-treated patients compared to controls (6.1/100 patient-years and 3.0/100 patient-years, respectively).29 Eighty-week follow-up of the phase 3 clinical trial of ruxolitinib for the treatment of polycythemia vera also noted an increased incidence of NMSC, albeit a more conservative increase. Patients randomized to the ruxolitinib treatment group developed NMSC at a rate of 4.4/100 patient-years, whereas the rate for controls treated with best available therapy was 2.7/100 patient-years.30 In contrast, 5-year follow-up of the COMFORT-I trial, also examining the efficacy of ruxolitinib in myelofibrosis, showed no increased risk for NMSC between ruxolitinib-treated patients and placebo (2.7/100 patient-years and 3.9/100 patient-years, respectively).31
A 2017 case series described 5 patients with myelofibrosis who developed multiple skin cancers with aggressive features while receiving ruxolitinib.32 Duration of ruxolitinib therapy ranged from 4 months to 4 years; 3 patients had a history of hydroxyurea exposure, and only 1 patient had a history of NMSC. High-risk cutaneous SCC, undifferentiated pleomorphic sarcoma, and lentigo maligna melanoma (Breslow thickness, 0.45 mm) were among the tumors reported in this series. Although no definitive conclusion can be made regarding the causality of JAK inhibitors in promoting these tumors, the association warrants further investigation. Clinicians should be aware that ruxolitinib might amplify the risk for NMSC in patients with pre-existing genetic or exposure-related susceptibility. Interruption of drug therapy may be necessary in managing patients who develop an aggressive tumor.32
In contrast, tofacitinib, which specifically inhibits JAK3, carries very low risk, if any, for NMSC when used for the treatment of psoriasis and rheumatoid arthritis. Results from 2 phase 3 trials analyzing the efficacy of tofacitinib in psoriasis demonstrated that only 2 of 1486 patients treated developed NMSC compared to none in the control group.33 Furthermore, analysis of NMSC across the tofacitinib rheumatoid arthritis clinical program, which included a total of 15,103 patient-years of exposure, demonstrated that the overall NMSC incidence was 0.55 for every 100 patient-years. Of note, the risk in patients receiving high-dose treatment (10 mg vs 5 mg) was nearly doubled in long-term follow-up studies (0.79/100 patient-years and 0.41/100 patient-years, respectively). Overall, the study concluded that treatment with tofacitinib presents no greater increased risk for NMSC than treatment with tumor necrosis factor inhibitors.33
PDE-5 Inhibitors
Phosphodiesterase 5 inhibitors, such as sildenafil citrate, have been widely prescribed for the treatment of erectile dysfunction. Studies have shown that BRAF-activated melanomas, which occur in approximately 50% to 70% of melanomas, also result in reduced PDE-5 expression.34-36 In these melanomas, downregulation of PDE-5 results in increased intracellular calcium,36 which has been shown to induce melanoma invasion.36,37 Given this similarity in molecular pathway between BRAF-activated melanomas and PDE-5 inhibitors, there has been increased concern that PDE-5 inhibitors might be associated with an increased risk for melanoma.
In 2014, Li et al38 published a retrospective analysis suggesting an association with sildenafil and an increased risk for melanoma. Their study utilized the Health Professionals Follow-up Study to identify a statistically significant elevation in the risk for invasive melanoma with both recent sildenafil use (multivariate-adjusted HR=2.24) and use at any time (HR=1.92). These results controlled for confounding variables, such as presence of major chronic disease, use of other erectile dysfunction treatments, family history of melanoma, history of sun exposure, and UV index of the patient’s residence. Notably, the study also found that sildenafil did not affect the incidence of BCC or SCC.38
In 2015, Loeb et al39 also examined the potential association between PDE-5 inhibitors and melanoma. Review of several Swedish drug and cancer registries allowed for analysis of melanoma risk and PDE-5 inhibitor use, based on number of prescriptions filled and type of PDE-5 inhibitor prescribed. Their analysis showed that men developing melanoma were more likely than nonmelanoma controls to have taken a PDE-5 inhibitor (11% vs 8%). In a subgroup analysis, however, statistical significance was shown for men with only a single prescription filled (34% of cases; P<.05), whereas the difference for men with multiple filled prescriptions did not meet statistical significance. Furthermore, the study did not find increased risk with longer-acting tadalafil and vardenafil (odds ratio [OR]=1.16) compared to sildenafil (OR=1.14). Last, use of PDE-5 inhibitors was only associated with stage 0 (OR=1.49) and stage I (OR=1.21) tumors, not with stages II to IV (OR=0.83) tumors. Although there was a statistically significant association between PDE-5 inhibitors and malignant melanoma (P<.05), the subgroup analysis findings pointed away from a causal relationship and likely toward a confounding of variable(s).39
A 2016 study by Lian et al40 looked at the risk for melanoma in a cohort of patients diagnosed with erectile dysfunction. No association between PDE-5 inhibitors and melanoma risk was shown when comparing patients who received a PDE-5 inhibitor and those who did not receive a PDE-5 inhibitor. However, secondary analysis did show that melanoma risk was increased among patients receiving more pills (34%) and prescriptions (30%). The authors concluded that there was no association between PDE-5 inhibitor use and overall increased risk for melanoma, and the increased risk associated with a greater number of pills and prescriptions would require further study.40
In contrast, a 2017 meta-analysis by Tang et al41 of 5 studies (3 of which were the aforementioned trials38-40) concluded that use of PDE-5 inhibitors was associated with a small but significantly increased risk for melanoma (OR=1.12) and BCC (OR=1.14) but not SCC. Furthermore, the study found no evidence of dosage-dependent association between PDE-5 inhibitor use and melanoma risk.41
Overall, clinical studies have been inconclusive in determining the risk for melanoma in the setting of PDE-5 inhibitor use. Studies showing an increased rate of melanoma within patient cohorts receiving PDE-5 inhibitors are limited; results might be affected by confounding variables. However, given the similarity in mechanism between PDE-5 inhibitors and HRAS-activated melanomas, it is reasonable to continue research into this potential association.
Conclusion
Since the turn of the century, drugs targeting cell-signaling pathways have been developed to treat inflammatory, oncologic, and immune conditions. The role of immunosuppressants in promoting skin cancer is well established and supported by a vast literature base. However, associations are less clear with newer immunomodulatory and antineoplastic medications. Skin cancer has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, JAK inhibitors, and PDE-5 inhibitors. In the case of JAK and PDE-5 inhibitors, the increased risk for melanoma and NMSC is somewhat inconclusive; risk is more firmly established for BRAF inhibitors and smoothened inhibitors. For the antineoplastic agents reviewed, the therapeutic effect of cancer regression is well documented, and benefits of continued therapy outweigh the increased risk for skin cancer promotion in nearly all cases. The value of early detection has been well documented for skin malignancy; therefore, increased skin surveillance and prompt management of suspicious lesions should be a priority for physicians treating patients undergoing therapy with these medications
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanoma in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
- Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA Dermatol. 2015;151:1103-1109.
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427-430.
- Ryan MB, Der CJ, Wang-Gillam A, et al. Targeting RAS-mutant cancers: is ERK the key? Trends Cancer. 2015;1:183-198.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
- Holderfield M, Nagel TE, Stuart DD. Mechanism and consequence of RAF kinase activation by small-molecule inhibitors. Br J Cancer. 2014;111:640-645.
- Dalle S, Poulalhon N, Debarbieux S, et al. Tracking of second primary melanomas in vemurafenib-treated patients. JAMA Dermatol. 2013;149:488-490.
- Williams VL, Cohen PR, Stewart DJ. Sorafenib-induced premalignant and malignant skin lesions. Int J Dermatol. 2011;50:396-402.
- Arnault JP, Wechsler J, Escudier B, et al. Keratoacanthomas and squamous cell carcinomas in patients receiving sorafenib. J Clin Oncol. 2009;27:e59-e61.
- Smith KJ, Haley H, Hamza S, et al. Eruptive keratoacanthoma-type squamous cell carcinomas in patients taking sorafenib for the treatment of solid tumors. Dermatol Surg. 2009;35:1766-1770.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Demirci H, Worden F, Nelson CC, et al. Efficacy of vismodegib (Erivedge) for basal cell carcinoma involving the orbit and periocular area. Ophthalmic Plast Reconstr Surg. 2015;31:463-466.
- Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342-353.
- Ridky TW, Cotsarelis G. Vismodegib resistance in basal cell carcinoma: not a smooth fit. Cancer Cell. 2015;27:315-316.
- Aasi S, Silkiss R, Tang JY, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242-243.
- Orouji A, Goerdt S, Utikal J, et al. Multiple highly and moderately differentiated squamous cell carcinomas of the skin during vismodegib treatment of inoperable basal cell carcinoma. Br J Dermatol. 2014;171:431-433.
- Iarrobino A, Messina JL, Kudchadkar R, et al. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2013;69:e33-e34.
- Saintes C, Saint-Jean M, Brocard A, et al. Development of squamous cell carcinoma into basal cell carcinoma under treatment with vismodegib. J Eur Acad Dermatol Venereol. 2015;29:1006-1009.
- Mohan SV, Chang J, Li S, et al. Increased risk of cutaneous squamous cell carcinoma after vismodegib therapy for basal cell carcinoma. JAMA Dermatol. 2016;152:527-532.
- Zhao X, Ponomaryov T, Ornell KJ, et al. RAS/MAPK activation drives resistance to Smo inhibition, metastasis, and tumor evolution in Shh pathway-dependent tumors. Cancer Res. 2015;75:3623-3635.
- Chang AL, Oro AE. Initial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma. Arch Dermatol. 2012;148:1324-1325.
- Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76:736-744.
- Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30:1701-1707.
- Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica. 2016;101:821-829.
- Verstovsek S, Mesa RA, Gotlib J, et al; COMFORT-I investigators. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10:55.
- Blechman AB, Cabell CE, Weinberger CH, et al. Aggressive skin cancers occurring in patients treated with the Janus kinase inhibitor ruxolitinib. J Drugs Dermatol. 2017;16:508-511.
- Papp KA, Menter MA, Abe M, et al; OPT Pivotal 1 and OPT Pivotal 2 investigators. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two randomized, placebo-controlled, phase III trials. Br J Dermatol. 2015;173:949-961.
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875-885.
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851-857.
- Arozarena I, Sanchez-Laorden B, Packer L, et al. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell. 2011;19:45-57.
- Houslay MD. Hard times for oncogenic BRAF-expressing melanoma cells. Cancer Cell. 2011;19:3-4.
- Li WQ, Qureshi AA, Robinson KC, et al. Sildenafil use and increased risk of incident melanoma in US men: a prospective cohort study. JAMA Intern Med. 2014;174:964-970.
- Loeb S, Folkvaljon Y, Lambe M, et al. Use of phosphodiesterase type 5 inhibitors for erectile dysfunction and risk of malignant melanoma. JAMA. 2015;313:2449-2455.
- Lian Y, Yin H, Pollak MN, et al. Phosphodiesterase type 5 inhibitors and the risk of melanoma skin cancer. Eur Urol. 2016;70:808-815.
- Tang H, Wu W, Fu S, et al. Phosphodiesterase type 5 inhibitors and risk of melanoma: a meta-analysis. J Am Acad Dermatol. 2017;77:480.e9-488.e9.
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanoma in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
- Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA Dermatol. 2015;151:1103-1109.
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427-430.
- Ryan MB, Der CJ, Wang-Gillam A, et al. Targeting RAS-mutant cancers: is ERK the key? Trends Cancer. 2015;1:183-198.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
- Holderfield M, Nagel TE, Stuart DD. Mechanism and consequence of RAF kinase activation by small-molecule inhibitors. Br J Cancer. 2014;111:640-645.
- Dalle S, Poulalhon N, Debarbieux S, et al. Tracking of second primary melanomas in vemurafenib-treated patients. JAMA Dermatol. 2013;149:488-490.
- Williams VL, Cohen PR, Stewart DJ. Sorafenib-induced premalignant and malignant skin lesions. Int J Dermatol. 2011;50:396-402.
- Arnault JP, Wechsler J, Escudier B, et al. Keratoacanthomas and squamous cell carcinomas in patients receiving sorafenib. J Clin Oncol. 2009;27:e59-e61.
- Smith KJ, Haley H, Hamza S, et al. Eruptive keratoacanthoma-type squamous cell carcinomas in patients taking sorafenib for the treatment of solid tumors. Dermatol Surg. 2009;35:1766-1770.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Demirci H, Worden F, Nelson CC, et al. Efficacy of vismodegib (Erivedge) for basal cell carcinoma involving the orbit and periocular area. Ophthalmic Plast Reconstr Surg. 2015;31:463-466.
- Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342-353.
- Ridky TW, Cotsarelis G. Vismodegib resistance in basal cell carcinoma: not a smooth fit. Cancer Cell. 2015;27:315-316.
- Aasi S, Silkiss R, Tang JY, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242-243.
- Orouji A, Goerdt S, Utikal J, et al. Multiple highly and moderately differentiated squamous cell carcinomas of the skin during vismodegib treatment of inoperable basal cell carcinoma. Br J Dermatol. 2014;171:431-433.
- Iarrobino A, Messina JL, Kudchadkar R, et al. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2013;69:e33-e34.
- Saintes C, Saint-Jean M, Brocard A, et al. Development of squamous cell carcinoma into basal cell carcinoma under treatment with vismodegib. J Eur Acad Dermatol Venereol. 2015;29:1006-1009.
- Mohan SV, Chang J, Li S, et al. Increased risk of cutaneous squamous cell carcinoma after vismodegib therapy for basal cell carcinoma. JAMA Dermatol. 2016;152:527-532.
- Zhao X, Ponomaryov T, Ornell KJ, et al. RAS/MAPK activation drives resistance to Smo inhibition, metastasis, and tumor evolution in Shh pathway-dependent tumors. Cancer Res. 2015;75:3623-3635.
- Chang AL, Oro AE. Initial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma. Arch Dermatol. 2012;148:1324-1325.
- Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76:736-744.
- Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30:1701-1707.
- Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica. 2016;101:821-829.
- Verstovsek S, Mesa RA, Gotlib J, et al; COMFORT-I investigators. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10:55.
- Blechman AB, Cabell CE, Weinberger CH, et al. Aggressive skin cancers occurring in patients treated with the Janus kinase inhibitor ruxolitinib. J Drugs Dermatol. 2017;16:508-511.
- Papp KA, Menter MA, Abe M, et al; OPT Pivotal 1 and OPT Pivotal 2 investigators. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two randomized, placebo-controlled, phase III trials. Br J Dermatol. 2015;173:949-961.
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875-885.
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851-857.
- Arozarena I, Sanchez-Laorden B, Packer L, et al. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell. 2011;19:45-57.
- Houslay MD. Hard times for oncogenic BRAF-expressing melanoma cells. Cancer Cell. 2011;19:3-4.
- Li WQ, Qureshi AA, Robinson KC, et al. Sildenafil use and increased risk of incident melanoma in US men: a prospective cohort study. JAMA Intern Med. 2014;174:964-970.
- Loeb S, Folkvaljon Y, Lambe M, et al. Use of phosphodiesterase type 5 inhibitors for erectile dysfunction and risk of malignant melanoma. JAMA. 2015;313:2449-2455.
- Lian Y, Yin H, Pollak MN, et al. Phosphodiesterase type 5 inhibitors and the risk of melanoma skin cancer. Eur Urol. 2016;70:808-815.
- Tang H, Wu W, Fu S, et al. Phosphodiesterase type 5 inhibitors and risk of melanoma: a meta-analysis. J Am Acad Dermatol. 2017;77:480.e9-488.e9.
Practice Points
- Patients should be educated about the increased risk for skin malignancy while undergoing treatment with BRAF inhibitors, sonic hedgehog–inhibiting agents, Janus kinase (JAK) inhibitors, and phosphodiesterase 5 (PDE-5) inhibitors.
- For BRAF inhibitors, sonic hedgehog–inhibiting agents, and JAK inhibitors, the increased risk for skin cancer warrants regular surveillance; however, given the indications for these medications, many patients will already be receiving regular skin screenings.
- The association between PDE-5 inhibitors and melanoma as well as nonmelanoma skin cancer remains questionable, and increased skin surveillance is not recommended at this time, unless patients have other risk factors for cutaneous malignancy.

What’s Eating You? Cutaneous Larva Migrans
Cutaneous larva migrans (CLM), also known as creeping eruption, is a pruritic serpiginous eruption caused by the migration of animal hookworm larvae through the epidermis.1,2 The most common parasites are Ancylostoma braziliense (common in dogs and cats) and Ancylostoma caninum (common in dogs).1
Disease Transmission
The infection is typically acquired in warm climates and tropical areas after coming in direct contact with sand or soil that is contaminated with animal feces. Therefore, the eruption most commonly occurs as a single or unilateral erythematous, pruritic, serpiginous tract on the feet, hands, or buttocks (Figure).2 The larval tract typically migrates at a rate of 1 to 2 cm per day,3 which is in contrast to the serpiginous urticarial rash of larva currens of strongyloidiasis that can travel up to 10 cm per hour.4
  |
Clinical Presentation
Rarely, CLM can present with bilateral lesions5; in severe cases a single patient can have hundreds of lesions. It also may present as folliculitis and urticarial papules.6 Shih et al7 reported a patient with CLM that presented as a diffuse papular urticarialike eruption following a trip to Thailand. This case may represent an underdiagnosed presentation of CLM. Patients with a history of exposure to contaminated sand or soil diffusely on the body may exhibit lesions in less classic locations, such as the trunk and upper proximal extremities.3
Cutaneous larva migrans is a self-limited eruption, as the larvae cannot complete their lifecycles in the human body and typically die within 2 to 8 weeks.2 However, rare cases lasting up to a year have been reported.3 Sarasombath and Young2 reported a case of CLM that persisted for 4 months with intermittent symptoms characterized by several weeklong intervals with no symptoms or visible rash.
Cutaneous larva migrans typically presents with isolated dermatologic symptoms. Rare cases associated with Löffler syndrome characterized by migratory pulmonary infiltrates and peripheral eosinophilia have been reported.8 Two proposed mechanisms for pulmonary involvement include direct invasion of the lungs by the helminths and a systemic immunologic process triggered by the helminths, resulting in eosinophilic pulmonary infiltration.9
Diagnosis
Cutaneous larva migrans is a clinical diagnosis and skin biopsy usually is not obtained because the larvae often are located 1 to 2 cm beyond the visible erythematous border.3,5 Rarely, the parasites are found on biopsy, revealing larvae that are 0.5-mm thick and up to 10-mm long.10 The larvae typically are confined to the deep epidermis because the parasite lacks the collagenase required to penetrate the basement membrane.2
Langley et al11 showed that confocal scanning laser microscopy can be an effective method for identifying the highly refractile oval larva that disrupt the normal honeycomb pattern of the epidermis. Performing a 4-mm punch biopsy over the identified site can allow for precise excision and treatment of the intact hookworm larvae of CLM. There also are limited reports of dermoscopy being used to facilitate diagnosis of CLM.12 Dermoscopic features of CLM include translucent, brown, structureless areas in a segmental arrangement corresponding to the larval bodies and red-dotted vessels corresponding to an empty burrow.13 However, Zalaudek et al13 concluded that the efficacy of dermoscopy in aiding in the diagnosis of CLM has not been fully established.
Treatment
Cutaneous larva migrans is a self-limited condition that often resolves within 2 to 8 weeks; however, pruritus can be intense and patients therefore are seldom willing to forego treatment. Treatment options include a single oral dose of albendazole 400 mg in adults, with increased efficacy if administered daily for 3 to 5 days (or 10–15 mg/kg, with a maximum dose of 800 mg daily in children), a single oral dose of ivermectin 12 mg in adults (or 150 µg/kg in children), or topical application of thiabendazole 10% to 15% three times daily for at least 15 days.14 Cases of CLM complicated by Löffler syndrome may require a longer treatment course, such as a 7-day course of albendazole 400 mg daily. Tan and Liu9 reported a case of CLM complicated by Löffler syndrome that was successfully treated with albendazole. In this patient, initial treatment with 2 courses of mebendazole (3 days each for a total of 6 days) resulted in improvement of cutaneous lesions but not the pulmonary infiltrate. A subsequent prolonged course of albendazole and intravenous hydrocortisone for 5 days resulted in complete resolution of the pulmonary infiltrate and peripheral eosinophilia. The authors concluded that inadequacy of treatment with mebendazole may be related to differences in the rate of absorption and efficacy when compared to albendazole.9
Conclusion
Cutaneous larva migrans is a self-limited and pruritic skin eruption that is acquired after direct inoculation with sand or soil that is contaminated with feces containing A braziliense or A caninum. Although the classic presentation is readily identifiable, there are a variety of atypical presentations that may go undiagnosed. Symptomatic relief usually can be achieved with short courses of oral or topical antihelminth medications.
1. Berlin JM, Goldberg SJ, McDonough RD, et al. JAAD grand rounds quiz. serpiginous eruption on the leg. J Am Acad Dermatol. 2010;63:921-922.
2. Sarasombath PA, Young PK. An unusual presentation of cutaneous larva migrans. Arch Dermatol. 2007;143:955.
3. Patel S, Aboutalebi S, Vindhya PL, et al. What’s eating you? extensive cutaneous larva migrans (Ancylostoma braziliense). Cutis. 2008;82:239-240.
4. Elston DM, Czarnik K, Brockett R, et al. What’s eating you? Strongyloides stercoralis. Cutis. 2003;71:22-24.
5. Duarte De Sousa ICV, De La Pascua L. Bilateral cutaneous larva migrans [poster reference number 4677]. J Am Acad Dermatol. 2012;66(4, suppl 1):AB106.
6. Caumes E, Ly F, Bricaire F. Cutaneous larva migrans with folliculitis: report of seven cases and review of the literature. Br J Dermatol. 2002;146:314-316.
7. Shih PY, Hsieh MY, Huang YH, et al. Multiple pruritic erythematous papules on the trunk after a trip to Thailand–quiz case. Arch Dermatol. 2010;146:557-562.
8. Wright DO, Gold ED. Löffler’s syndrome associated with creeping eruption (cutaneous helminthiasis): report of twenty-six cases. Arch Intern Med. 1946;78:303-312.
9. Tan SK, Liu TT. Cutaneous larva migrans complicated by Löffler’s syndrome. Arch Dermatol. 2010;146:210-212.
10. Rapini RP, ed. Practical Dermatopathology. Philadelphia, PA: Elsevier; 2005.
11. Langley R, Webb A, Haldane D, et al. Confocal microscopy of cutaneous larva migrans. J Am Acad Dermatol. 2011;64(2, suppl 1):AB100.
12. Aljasser MI, Lui H, Zeng H, et al. Dermoscopy and near-infrared fluorescence imaging of cutaneous larva migrans. Photodermatol Photoimmunol Photomed. 2013;29:337-338.
13. Zalaudek I, Giacomel J, Cabo H, et al. Entodermoscopy: a new tool for diagnosing skin infections and infestations. Dermatology. 2008;216:14-23.
14. Caumes E. Treatment of cutaneous larva migrans. Clin Infect Dis. 2000;30:811-814.
Cutaneous larva migrans (CLM), also known as creeping eruption, is a pruritic serpiginous eruption caused by the migration of animal hookworm larvae through the epidermis.1,2 The most common parasites are Ancylostoma braziliense (common in dogs and cats) and Ancylostoma caninum (common in dogs).1
Disease Transmission
The infection is typically acquired in warm climates and tropical areas after coming in direct contact with sand or soil that is contaminated with animal feces. Therefore, the eruption most commonly occurs as a single or unilateral erythematous, pruritic, serpiginous tract on the feet, hands, or buttocks (Figure).2 The larval tract typically migrates at a rate of 1 to 2 cm per day,3 which is in contrast to the serpiginous urticarial rash of larva currens of strongyloidiasis that can travel up to 10 cm per hour.4
|
Clinical Presentation
Rarely, CLM can present with bilateral lesions5; in severe cases a single patient can have hundreds of lesions. It also may present as folliculitis and urticarial papules.6 Shih et al7 reported a patient with CLM that presented as a diffuse papular urticarialike eruption following a trip to Thailand. This case may represent an underdiagnosed presentation of CLM. Patients with a history of exposure to contaminated sand or soil diffusely on the body may exhibit lesions in less classic locations, such as the trunk and upper proximal extremities.3
Cutaneous larva migrans is a self-limited eruption, as the larvae cannot complete their lifecycles in the human body and typically die within 2 to 8 weeks.2 However, rare cases lasting up to a year have been reported.3 Sarasombath and Young2 reported a case of CLM that persisted for 4 months with intermittent symptoms characterized by several weeklong intervals with no symptoms or visible rash.
Cutaneous larva migrans typically presents with isolated dermatologic symptoms. Rare cases associated with Löffler syndrome characterized by migratory pulmonary infiltrates and peripheral eosinophilia have been reported.8 Two proposed mechanisms for pulmonary involvement include direct invasion of the lungs by the helminths and a systemic immunologic process triggered by the helminths, resulting in eosinophilic pulmonary infiltration.9
Diagnosis
Cutaneous larva migrans is a clinical diagnosis and skin biopsy usually is not obtained because the larvae often are located 1 to 2 cm beyond the visible erythematous border.3,5 Rarely, the parasites are found on biopsy, revealing larvae that are 0.5-mm thick and up to 10-mm long.10 The larvae typically are confined to the deep epidermis because the parasite lacks the collagenase required to penetrate the basement membrane.2
Langley et al11 showed that confocal scanning laser microscopy can be an effective method for identifying the highly refractile oval larva that disrupt the normal honeycomb pattern of the epidermis. Performing a 4-mm punch biopsy over the identified site can allow for precise excision and treatment of the intact hookworm larvae of CLM. There also are limited reports of dermoscopy being used to facilitate diagnosis of CLM.12 Dermoscopic features of CLM include translucent, brown, structureless areas in a segmental arrangement corresponding to the larval bodies and red-dotted vessels corresponding to an empty burrow.13 However, Zalaudek et al13 concluded that the efficacy of dermoscopy in aiding in the diagnosis of CLM has not been fully established.
Treatment
Cutaneous larva migrans is a self-limited condition that often resolves within 2 to 8 weeks; however, pruritus can be intense and patients therefore are seldom willing to forego treatment. Treatment options include a single oral dose of albendazole 400 mg in adults, with increased efficacy if administered daily for 3 to 5 days (or 10–15 mg/kg, with a maximum dose of 800 mg daily in children), a single oral dose of ivermectin 12 mg in adults (or 150 µg/kg in children), or topical application of thiabendazole 10% to 15% three times daily for at least 15 days.14 Cases of CLM complicated by Löffler syndrome may require a longer treatment course, such as a 7-day course of albendazole 400 mg daily. Tan and Liu9 reported a case of CLM complicated by Löffler syndrome that was successfully treated with albendazole. In this patient, initial treatment with 2 courses of mebendazole (3 days each for a total of 6 days) resulted in improvement of cutaneous lesions but not the pulmonary infiltrate. A subsequent prolonged course of albendazole and intravenous hydrocortisone for 5 days resulted in complete resolution of the pulmonary infiltrate and peripheral eosinophilia. The authors concluded that inadequacy of treatment with mebendazole may be related to differences in the rate of absorption and efficacy when compared to albendazole.9
Conclusion
Cutaneous larva migrans is a self-limited and pruritic skin eruption that is acquired after direct inoculation with sand or soil that is contaminated with feces containing A braziliense or A caninum. Although the classic presentation is readily identifiable, there are a variety of atypical presentations that may go undiagnosed. Symptomatic relief usually can be achieved with short courses of oral or topical antihelminth medications.
Cutaneous larva migrans (CLM), also known as creeping eruption, is a pruritic serpiginous eruption caused by the migration of animal hookworm larvae through the epidermis.1,2 The most common parasites are Ancylostoma braziliense (common in dogs and cats) and Ancylostoma caninum (common in dogs).1
Disease Transmission
The infection is typically acquired in warm climates and tropical areas after coming in direct contact with sand or soil that is contaminated with animal feces. Therefore, the eruption most commonly occurs as a single or unilateral erythematous, pruritic, serpiginous tract on the feet, hands, or buttocks (Figure).2 The larval tract typically migrates at a rate of 1 to 2 cm per day,3 which is in contrast to the serpiginous urticarial rash of larva currens of strongyloidiasis that can travel up to 10 cm per hour.4
|
Clinical Presentation
Rarely, CLM can present with bilateral lesions5; in severe cases a single patient can have hundreds of lesions. It also may present as folliculitis and urticarial papules.6 Shih et al7 reported a patient with CLM that presented as a diffuse papular urticarialike eruption following a trip to Thailand. This case may represent an underdiagnosed presentation of CLM. Patients with a history of exposure to contaminated sand or soil diffusely on the body may exhibit lesions in less classic locations, such as the trunk and upper proximal extremities.3
Cutaneous larva migrans is a self-limited eruption, as the larvae cannot complete their lifecycles in the human body and typically die within 2 to 8 weeks.2 However, rare cases lasting up to a year have been reported.3 Sarasombath and Young2 reported a case of CLM that persisted for 4 months with intermittent symptoms characterized by several weeklong intervals with no symptoms or visible rash.
Cutaneous larva migrans typically presents with isolated dermatologic symptoms. Rare cases associated with Löffler syndrome characterized by migratory pulmonary infiltrates and peripheral eosinophilia have been reported.8 Two proposed mechanisms for pulmonary involvement include direct invasion of the lungs by the helminths and a systemic immunologic process triggered by the helminths, resulting in eosinophilic pulmonary infiltration.9
Diagnosis
Cutaneous larva migrans is a clinical diagnosis and skin biopsy usually is not obtained because the larvae often are located 1 to 2 cm beyond the visible erythematous border.3,5 Rarely, the parasites are found on biopsy, revealing larvae that are 0.5-mm thick and up to 10-mm long.10 The larvae typically are confined to the deep epidermis because the parasite lacks the collagenase required to penetrate the basement membrane.2
Langley et al11 showed that confocal scanning laser microscopy can be an effective method for identifying the highly refractile oval larva that disrupt the normal honeycomb pattern of the epidermis. Performing a 4-mm punch biopsy over the identified site can allow for precise excision and treatment of the intact hookworm larvae of CLM. There also are limited reports of dermoscopy being used to facilitate diagnosis of CLM.12 Dermoscopic features of CLM include translucent, brown, structureless areas in a segmental arrangement corresponding to the larval bodies and red-dotted vessels corresponding to an empty burrow.13 However, Zalaudek et al13 concluded that the efficacy of dermoscopy in aiding in the diagnosis of CLM has not been fully established.
Treatment
Cutaneous larva migrans is a self-limited condition that often resolves within 2 to 8 weeks; however, pruritus can be intense and patients therefore are seldom willing to forego treatment. Treatment options include a single oral dose of albendazole 400 mg in adults, with increased efficacy if administered daily for 3 to 5 days (or 10–15 mg/kg, with a maximum dose of 800 mg daily in children), a single oral dose of ivermectin 12 mg in adults (or 150 µg/kg in children), or topical application of thiabendazole 10% to 15% three times daily for at least 15 days.14 Cases of CLM complicated by Löffler syndrome may require a longer treatment course, such as a 7-day course of albendazole 400 mg daily. Tan and Liu9 reported a case of CLM complicated by Löffler syndrome that was successfully treated with albendazole. In this patient, initial treatment with 2 courses of mebendazole (3 days each for a total of 6 days) resulted in improvement of cutaneous lesions but not the pulmonary infiltrate. A subsequent prolonged course of albendazole and intravenous hydrocortisone for 5 days resulted in complete resolution of the pulmonary infiltrate and peripheral eosinophilia. The authors concluded that inadequacy of treatment with mebendazole may be related to differences in the rate of absorption and efficacy when compared to albendazole.9
Conclusion
Cutaneous larva migrans is a self-limited and pruritic skin eruption that is acquired after direct inoculation with sand or soil that is contaminated with feces containing A braziliense or A caninum. Although the classic presentation is readily identifiable, there are a variety of atypical presentations that may go undiagnosed. Symptomatic relief usually can be achieved with short courses of oral or topical antihelminth medications.
1. Berlin JM, Goldberg SJ, McDonough RD, et al. JAAD grand rounds quiz. serpiginous eruption on the leg. J Am Acad Dermatol. 2010;63:921-922.
2. Sarasombath PA, Young PK. An unusual presentation of cutaneous larva migrans. Arch Dermatol. 2007;143:955.
3. Patel S, Aboutalebi S, Vindhya PL, et al. What’s eating you? extensive cutaneous larva migrans (Ancylostoma braziliense). Cutis. 2008;82:239-240.
4. Elston DM, Czarnik K, Brockett R, et al. What’s eating you? Strongyloides stercoralis. Cutis. 2003;71:22-24.
5. Duarte De Sousa ICV, De La Pascua L. Bilateral cutaneous larva migrans [poster reference number 4677]. J Am Acad Dermatol. 2012;66(4, suppl 1):AB106.
6. Caumes E, Ly F, Bricaire F. Cutaneous larva migrans with folliculitis: report of seven cases and review of the literature. Br J Dermatol. 2002;146:314-316.
7. Shih PY, Hsieh MY, Huang YH, et al. Multiple pruritic erythematous papules on the trunk after a trip to Thailand–quiz case. Arch Dermatol. 2010;146:557-562.
8. Wright DO, Gold ED. Löffler’s syndrome associated with creeping eruption (cutaneous helminthiasis): report of twenty-six cases. Arch Intern Med. 1946;78:303-312.
9. Tan SK, Liu TT. Cutaneous larva migrans complicated by Löffler’s syndrome. Arch Dermatol. 2010;146:210-212.
10. Rapini RP, ed. Practical Dermatopathology. Philadelphia, PA: Elsevier; 2005.
11. Langley R, Webb A, Haldane D, et al. Confocal microscopy of cutaneous larva migrans. J Am Acad Dermatol. 2011;64(2, suppl 1):AB100.
12. Aljasser MI, Lui H, Zeng H, et al. Dermoscopy and near-infrared fluorescence imaging of cutaneous larva migrans. Photodermatol Photoimmunol Photomed. 2013;29:337-338.
13. Zalaudek I, Giacomel J, Cabo H, et al. Entodermoscopy: a new tool for diagnosing skin infections and infestations. Dermatology. 2008;216:14-23.
14. Caumes E. Treatment of cutaneous larva migrans. Clin Infect Dis. 2000;30:811-814.
1. Berlin JM, Goldberg SJ, McDonough RD, et al. JAAD grand rounds quiz. serpiginous eruption on the leg. J Am Acad Dermatol. 2010;63:921-922.
2. Sarasombath PA, Young PK. An unusual presentation of cutaneous larva migrans. Arch Dermatol. 2007;143:955.
3. Patel S, Aboutalebi S, Vindhya PL, et al. What’s eating you? extensive cutaneous larva migrans (Ancylostoma braziliense). Cutis. 2008;82:239-240.
4. Elston DM, Czarnik K, Brockett R, et al. What’s eating you? Strongyloides stercoralis. Cutis. 2003;71:22-24.
5. Duarte De Sousa ICV, De La Pascua L. Bilateral cutaneous larva migrans [poster reference number 4677]. J Am Acad Dermatol. 2012;66(4, suppl 1):AB106.
6. Caumes E, Ly F, Bricaire F. Cutaneous larva migrans with folliculitis: report of seven cases and review of the literature. Br J Dermatol. 2002;146:314-316.
7. Shih PY, Hsieh MY, Huang YH, et al. Multiple pruritic erythematous papules on the trunk after a trip to Thailand–quiz case. Arch Dermatol. 2010;146:557-562.
8. Wright DO, Gold ED. Löffler’s syndrome associated with creeping eruption (cutaneous helminthiasis): report of twenty-six cases. Arch Intern Med. 1946;78:303-312.
9. Tan SK, Liu TT. Cutaneous larva migrans complicated by Löffler’s syndrome. Arch Dermatol. 2010;146:210-212.
10. Rapini RP, ed. Practical Dermatopathology. Philadelphia, PA: Elsevier; 2005.
11. Langley R, Webb A, Haldane D, et al. Confocal microscopy of cutaneous larva migrans. J Am Acad Dermatol. 2011;64(2, suppl 1):AB100.
12. Aljasser MI, Lui H, Zeng H, et al. Dermoscopy and near-infrared fluorescence imaging of cutaneous larva migrans. Photodermatol Photoimmunol Photomed. 2013;29:337-338.
13. Zalaudek I, Giacomel J, Cabo H, et al. Entodermoscopy: a new tool for diagnosing skin infections and infestations. Dermatology. 2008;216:14-23.
14. Caumes E. Treatment of cutaneous larva migrans. Clin Infect Dis. 2000;30:811-814.
Practice Points
- Classic cutaneous larva migrans (CLM) presents with a unilateral, serpiginous, pruritic eruption on the hands, feet, or buttocks following direct contact with sand or soil that is contaminated with Ancylostoma braziliense or Ancylostoma caninum.
- Atypical presentations of CLM include bilateral distribution; folliculitis and urticarial plaques; prolonged cases lasting up to 1 year; and Löffler syndrome characterized by migratory pulmonary infiltrates and peripheral eosinophilia.
- Cutaneous larva migrans is self-limited, but treatment often is necessary due to intense pruritus. Treatment options include a single oral dose of albendazole or ivermectin, topical thiabendazole, and prolonged courses of oral albendazole in cases complicated by Löffler syndrome.