User login
Epileptic and depressed
CASE: New-onset seizures
Ms. R, age 33, is referred by her neurologist for treatment of depressive symptoms that have intensified after she was diagnosed with epilepsy 1 year ago. She has a history of bulimia and ongoing anxiety and depression. She also has long-standing neuropathic pain in her left lateral shin and ankle that started after her foot was amputated in a lawn mower accident at age 5. Ms. R says she didn’t take pain medication until age 24, when her pain specialist prescribed tramadol, 300 to 400 mg/d, which she continues to take.
Ms. R’s first seizure occurred 1 year ago. Despite trials of several antiepileptics, her seizures persist; she is taking lamotrigine, 200 mg/d, when she presents for treatment. She has no history of brain injuries or strokes to explain her epilepsy. An MRI and 3 electroencephalograms show no signs of focal, potentially epileptogenic lesions.
Ms. R reports worsening depressive symptoms—particularly impaired attention and concentration—over several months that interfere with her housekeeping and ability to finish simple tasks at work. She says she drinks alcohol occasionally, but denies substance abuse. We initiate venlafaxine, titrated to 300 mg/d, because Ms. R has a history of intolerable side effects with fluoxetine (gastrointestinal distress) and citalopram (weight gain).
The authors’ observations
Tramadol, a centrally acting synthetic analgesic, consists of 2 enantiomers that act as weak agonists at μ-opioid receptors while also inhibiting serotonin and norepinephrine reuptake.1 Euphoria associated with μ receptor activation often is considered a “high.” Most abused opioids are prototypical μ agonists. When opioids are injected or inhaled, drug levels in the brain rise rapidly, causing a “rush”—a brief, intense, pleasurable sensation—followed by a longer-lasting high. Tolerance and physical dependence occur when opioids are used chronically.
Despite tramadol’s μ-opioid activity, the FDA approved it as an unscheduled analgesic in 1994 based on several human studies.2 Experience with tramadol has confirmed it has low abuse potential, yet human laboratory data—and some epidemiologic data—show that repeated use can lead to physical dependence. Although tramadol is considered a relatively weak opioid, human studies suggest that it possesses μ-agonist activity. The Drug Abuse Warning Network reported >15,000 emergency department (ED) visits for nonmedical tramadol use in 2009, which was more than the number of ED visits for codeine products (7,958) or propoxyphene products (9,526), but much fewer than visits for hydrocodone (86,258) or oxycodone (148,449) products.3
The recommended tramadol dose is 50 to 100 mg every 4 to 6 hours (maximum 400 mg/d). Adverse effects range from dysphoria, constipation, and nausea to agitation, seizures, respiratory depression, and coma.4 Tramadol withdrawal is similar to opioid withdrawal, and is characterized by anxiety, restlessness, insomnia, yawning, rhinorrhea, lacrimation, diaphoresis, tremor, muscle spasms, vomiting, diarrhea, and tachycardia. Rarely, psychomotor agitation and confusion may occur.5
Tramadol and seizures
At clinically appropriate doses, tramadol slightly suppresses seizure severity,6 but higher doses can induce seizures.7-12 This paradox is explained by tramadol’s effect on γ-aminobutyric acid (GABA) receptors. Although at clinical doses tramadol does not affect GABA, which could precipitate seizures, at higher doses it has been shown to have an inhibitory effect on GABA receptors.13,14 No prospective studies have assessed how often tramadol-induced seizures occur. Case reports12,15 suggest that seizures are more likely with acute tramadol intoxication, in patients with a history of alcohol abuse, or with pharmacologic regimens that include other medications that may cause seizures. Tramadol-induced seizures are generalized tonic-clonic in nature, and typically occur within 24 hours of the last dose.16
HISTORY: Worsening seizures
Two months after she presents for psychiatric evaluation, Ms. R experiences 6 generalized convulsions lasting from 15 minutes to 1 hour with no identifiable precipitant. Because oxcarbazepine and lamotrigine have failed to suppress her seizures, her neurologist adds phenytoin, 200 mg/d, and increases lamotrigine from 200 to 300 mg/d. Her depression continues to worsen. She reports severe insomnia, anhedonia, restlessness, and hopelessness, so we add sertraline, 50 mg/d, to venlafaxine. Ms. R says the seizures are terrifying and she cannot work. She moves in with her parents because she is unable to care for herself.
During a psychiatric appointment, Ms. R confesses that for 2 years her pain has been so unbearable that she has been buying extra tramadol from Internet retailers and taking 600 to 800 mg/d in addition to the prescribed 400 mg/d.
The authors’ observations
Ms. R had a history of chronic pain Table 117 and developed seizures after escalating her tramadol use. After her first epilepsy attack, she did not tell her physicians she was taking additional tramadol nor did she stop taking it. Treatment with several antiepileptics was unsuccessful. Her seizures persisted as long as her tramadol addiction continued.
Table 1
DSM-IV-TR criteria for pain disorder
|
| Source: Reference 17 |
Spiller et al18 reported the lowest daily tramadol dose associated with seizures is 500 mg/d, although Talaie et al16 observed seizures at doses as low as 100 mg/d. Additionally, seizure risk may increase through tramadol’s interactions with several medications, including tricyclic antidepressants, selective serotonin reuptake inhibitors, phenothiazines, fluoroquinolone antibiotics, meperidine, clozapine, buspirone, bupropion, phenylephrine, guaifenesin, tripelennamine, thioridazine, theophylline, and acetaminophen, butalbital, and caffeine combination (Table 2).19 Transdermal selegiline is contraindicated with tramadol. For Ms. R, the sertraline and venlafaxine she was taking may have augmented tramadol’s seizure potential.
Table 2
Tramadol: Major drug-drug interactions
| Drug | Symptoms |
|---|---|
| Selegiline | Nausea, vomiting, cardiovascular collapse, respiratory depression, seizures, or serotonin syndrome (hypertension, hyperthermia, myoclonus, mental status changes); use of the transdermal formulation with tramadol is contraindicated |
| Carbamazepine | Decreased tramadol efficacy and increased seizure risk |
| Venlafaxine | Increased risk of serotonin syndrome |
| Linezolid | Increased risk of serotonin syndrome |
| Fluoxetine | Increased risk of seizures and serotonin syndrome; increased concentrations of tramadol and decreased concentrations of tramadol active metabolite, O-desmethyltramadol (M1) |
| Olanzapine | Increased risk of serotonin syndrome |
| Mirtazapine | Increased risk of serotonin syndrome |
| Haloperidol | Increased risk of seizures |
| Escitalopram | Increased risk of seizures and serotonin syndrome |
| Clomipramine | Increased risk of seizures |
| Risperidone | Increased risk of seizures |
| Ketamine | Increased risk of respiratory depression and excessive CNS depression |
| Imipramine | Increased risk of seizures |
| Duloxetine | Increased risk of serotonin syndrome |
| Nortriptyline | Increased risk of seizures |
| Clozapine | Increased risk of seizures |
| Sertraline | Increased risk of seizures and serotonin syndrome |
| Paroxetine | Increased risk of seizures and serotonin syndrome; decrease in the analgesic effect of tramadol |
| Amitriptyline | Increased risk of seizures; increased concentrations of tramadol and decreased concentrations of tramadol active metabolite, M1 |
| Desipramine | Increased risk of seizures |
| Doxepin | Increased risk of seizures |
| Citalopram | Increased risk of seizures and serotonin syndrome |
| Fluvoxamine | Increased risk of seizures and serotonin syndrome |
| Source: Reference 19 | |
It is important to avoid polypharmacy in patients taking tramadol.20 Most psychiatrists are aware of the risk of serotonin syndrome with antidepressants, but may be less likely to attribute serotonergic additive effects from other medication classes such as analgesics. Recognizing tramadol’s potential to contribute to serotonin syndrome—especially in light of concomitant usage with other serotonergic medications such as antidepressants—is essential.
Tramadol toxicity appears to be caused by monoamine uptake inhibition rather than its opioid effects.21 The most frequent pharmacokinetic drug-drug interactions that lead to side effects such as serotonin syndrome or seizures involve several isoenzymes of the hepatic cytochrome P450 (CYP). The isoenzymes CYP2D6 (substrates—eg, amitriptyline, tramadol, and venlafaxine; inhibitors—eg, fluoxetine and duloxetine) and CYP3A4 (substrates—eg, carbamazepine, oxycodone, and venlafaxine; inductors—eg, carbamazepine; inhibitors, eg—grapefruit juice) are most important clinically.22
Ms. R readily obtained tramadol from Internet retailers. In a 2004 report, a Google search yielded 2,150,000 sources for acquiring tramadol, most of which did not require a prescription.23 Chronic pain patients have a higher prevalence of substance abuse than the general population.24 Because Ms. R did not have a documented substance abuse history, none of her physicians screened her for drug abuse, although toxicology screening wouldn’t have helped because the tramadol had been prescribed. We didn’t think to directly ask Ms. R about medication misuse, but if we had, she might have revealed it sooner.
OUTCOME: Seizure free
With Ms. R’s permission, we speak to her neurologist, who agrees that excess tramadol likely induced her seizures. The seizures stop after Ms. R discontinues tramadol. After 3 months without seizures, phenytoin is discontinued and lamotrigine is tapered to 200 mg/d. Ms. R participates in a pain rehabilitation program and continues to take venlafaxine, 300 mg/d, and sertraline, 50 mg/d. Her mood improves and she returns to work. Her pain is managed by non-steroidal anti-inflammatory drugs because she decides to decrease her activity level. Ms. R also is trying alternative medicine modalities such as acupuncture and acupressure.
Related Resource
- Clark MR, Treisman GJ. Chronic pain and addiction. Basel, Switzerland: Karger; 2011.
Drug Brand Names
- Acetaminophen, butalbital, and caffeine • Fioricet
- Amitriptyline • Elavil
- Bupropion • Wellbutrin
- Buspirone • Buspar
- Carbamazepine • Tegretol, Carbatrol
- Citalopram • Celexa
- Clomipramine • Anafranil
- Clozapine • Clozaril
- Desipramine • Norpramin
- Doxepin • Adapin, Silenor
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Guaifenesin • Tenex
- Haloperidol • Haldol
- Imipramine • Tofranil
- Ketamine • Ketalar
- Lamotrigine • Lamictal
- Linezolid • Zyvox
- Meperidine • Demerol
- Mirtazapine • Remeron
- Nortriptyline • Aventyl
- Olanzapine • Zyprexa
- Oxcarbazepine • Trileptal
- Oxycodone • Percolone, OxyContin
- Paroxetine • Paxil
- Phenylephrine • Lusonal
- Phenytoin • Dilantin
- Propoxyphene • Darvon
- Risperidone • Risperdal
- Selegiline • Eldepryl, EMSAM
- Sertraline • Zoloft
- Theophylline • Aerolate
- Thioridazine • Mellaril
- Tramadol • Ultram
- Tripelennamine • Pyribenzamine
- Venlafaxine • Effexor
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufactur-ers of competing products.
1. Katz KD. Tramadol is an opioid. J Med Toxicol. 2008;4(2):145-
2. Preston KL, Jasinski DR, Testa M. Abuse potential and pharmacological comparison of tramadol and morphine. Drug Alcohol Depend. 1991;27(1):7-17.
3. U.S. Department of Health and Human Services. Substance Abuse and Mental Health Services Administration. Drug abuse warning network, 2009: national estimates of drug-related emergency department visits. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2011. HHS publication (SMA) 11-4659, DAWN Series D-35.
4. Afshari R, Ghooshkhanehee H. Tramadol overdose induced seizure dramatic rise of CPK and acute renal failure. J Pak Med Assoc. 2009;59(3):178.-
5. Rodriguez Villamañan JC, Albaladejo Blanco C, Sanchez Sanchez A, et al. Withdrawal syndrome after long-term treatment with tramadol. Br J Gen Pract. 2000;50(454):406.-
6. Manocha A, Sharma KK, Mediratta PK. On the mechanism of anticonvulsant effect of tramadol in mice. Pharmacol Biochem Behav. 2005;82(1):74-81.
7. Boyd IW. Tramadol and seizures. Med J Aust. 2005;182(11):595-596.
8. Labate A, Newton MR, Vernon GM, et al. Tramadol and new-onset seizures. Med J Aust. 2005;182(1):42-43.
9. Gasse C, Derby L, Vasilakis-Scaramozza C, et al. Incidence of first-time idiopathic seizures in users of tramadol. Pharmacotherapy. 2000;20(6):629-634.
10. Kahn LH, Alderfer RJ, Graham DJ. Seizures reported with tramadol. JAMA. 1997;278(20):1661.-
11. Mazor SS, Feldman KW, Sugar NF, et al. Pediatric tramadol ingestion resulting in seizurelike activity: a case series. Pediatr Emerg Care. 2008;24(6):380-381.
12. Raffa RB, Stone DJ, Jr. Unexceptional seizure potential of tramadol or its enantiomers or metabolites in mice. J Pharmacol Exp Ther. 2008;325(2):500-506.
13. Rehni AK, Singh TG, Singh N, et al. Tramadol-induced seizurogenic effect: a possible role of opioid-dependent histamine H1 receptor activation-linked mechanism. Naunyn Schmiedebergs Arch Pharmacol. 2010;381(1):11-19.
14. Rehni AK, Singh I, Kumar M. Tramadol-induced seizurogenic effect: a possible role of opioid-dependent gamma-aminobutyric acid inhibitory pathway. Basic Clin Pharmacol Toxicol. 2008;103(3):262-266.
15. Jovanović-Cupić V, Martinović Z, Nesić N. Seizures associated with intoxication and abuse of tramadol. Clin Toxicol (Phila). 2006;44(2):143-146.
16. Talaie H, Panahandeh R, Fayaznouri M, et al. Dose-independent occurrence of seizure with tramadol. J Med Toxicol. 2009;5(2):63-67.
17. Diagnostic and statistical manual of mental disorders 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
18. Spiller HA, Gorman SE, Villalobos D, et al. Prospective multicenter evaluation of tramadol exposure. J Toxicol Clin Toxicol. 1997;35(4):361-364.
19. Reus VI, Rawitscher L. Possible interaction of tramadol and antidepressants. Am J Psychiatry. 2000;157(5):839.-
20. Thundiyil JG, Kearney TE, Olson KR. Evolving epidemiology of drug-induced seizures reported to a Poison Control Center System. J Med Toxicol. 2007;3(1):15-19.
21. Looper KJ. Potential medical and surgical complications of serotonergic antidepressant medications. Psychosomatics. 2007;48(1):1-9.
22. Grond S, Sablotzki A. Clinical pharmacology of tramadol. Clin Pharmacokinet. 2004;43(13):879-923.
23. Lineberry TW, Bostwick JM. Taking the physician out of “physician shopping”: a case series of clinical problems associated with Internet purchases of medication. Mayo Clin Proc. 2004;79(8):1031-1034.
24. Savage SR. Assessment for addiction in pain-treatment settings. Clin J Pain. 2002;18(4 suppl):S28-S38.
CASE: New-onset seizures
Ms. R, age 33, is referred by her neurologist for treatment of depressive symptoms that have intensified after she was diagnosed with epilepsy 1 year ago. She has a history of bulimia and ongoing anxiety and depression. She also has long-standing neuropathic pain in her left lateral shin and ankle that started after her foot was amputated in a lawn mower accident at age 5. Ms. R says she didn’t take pain medication until age 24, when her pain specialist prescribed tramadol, 300 to 400 mg/d, which she continues to take.
Ms. R’s first seizure occurred 1 year ago. Despite trials of several antiepileptics, her seizures persist; she is taking lamotrigine, 200 mg/d, when she presents for treatment. She has no history of brain injuries or strokes to explain her epilepsy. An MRI and 3 electroencephalograms show no signs of focal, potentially epileptogenic lesions.
Ms. R reports worsening depressive symptoms—particularly impaired attention and concentration—over several months that interfere with her housekeeping and ability to finish simple tasks at work. She says she drinks alcohol occasionally, but denies substance abuse. We initiate venlafaxine, titrated to 300 mg/d, because Ms. R has a history of intolerable side effects with fluoxetine (gastrointestinal distress) and citalopram (weight gain).
The authors’ observations
Tramadol, a centrally acting synthetic analgesic, consists of 2 enantiomers that act as weak agonists at μ-opioid receptors while also inhibiting serotonin and norepinephrine reuptake.1 Euphoria associated with μ receptor activation often is considered a “high.” Most abused opioids are prototypical μ agonists. When opioids are injected or inhaled, drug levels in the brain rise rapidly, causing a “rush”—a brief, intense, pleasurable sensation—followed by a longer-lasting high. Tolerance and physical dependence occur when opioids are used chronically.
Despite tramadol’s μ-opioid activity, the FDA approved it as an unscheduled analgesic in 1994 based on several human studies.2 Experience with tramadol has confirmed it has low abuse potential, yet human laboratory data—and some epidemiologic data—show that repeated use can lead to physical dependence. Although tramadol is considered a relatively weak opioid, human studies suggest that it possesses μ-agonist activity. The Drug Abuse Warning Network reported >15,000 emergency department (ED) visits for nonmedical tramadol use in 2009, which was more than the number of ED visits for codeine products (7,958) or propoxyphene products (9,526), but much fewer than visits for hydrocodone (86,258) or oxycodone (148,449) products.3
The recommended tramadol dose is 50 to 100 mg every 4 to 6 hours (maximum 400 mg/d). Adverse effects range from dysphoria, constipation, and nausea to agitation, seizures, respiratory depression, and coma.4 Tramadol withdrawal is similar to opioid withdrawal, and is characterized by anxiety, restlessness, insomnia, yawning, rhinorrhea, lacrimation, diaphoresis, tremor, muscle spasms, vomiting, diarrhea, and tachycardia. Rarely, psychomotor agitation and confusion may occur.5
Tramadol and seizures
At clinically appropriate doses, tramadol slightly suppresses seizure severity,6 but higher doses can induce seizures.7-12 This paradox is explained by tramadol’s effect on γ-aminobutyric acid (GABA) receptors. Although at clinical doses tramadol does not affect GABA, which could precipitate seizures, at higher doses it has been shown to have an inhibitory effect on GABA receptors.13,14 No prospective studies have assessed how often tramadol-induced seizures occur. Case reports12,15 suggest that seizures are more likely with acute tramadol intoxication, in patients with a history of alcohol abuse, or with pharmacologic regimens that include other medications that may cause seizures. Tramadol-induced seizures are generalized tonic-clonic in nature, and typically occur within 24 hours of the last dose.16
HISTORY: Worsening seizures
Two months after she presents for psychiatric evaluation, Ms. R experiences 6 generalized convulsions lasting from 15 minutes to 1 hour with no identifiable precipitant. Because oxcarbazepine and lamotrigine have failed to suppress her seizures, her neurologist adds phenytoin, 200 mg/d, and increases lamotrigine from 200 to 300 mg/d. Her depression continues to worsen. She reports severe insomnia, anhedonia, restlessness, and hopelessness, so we add sertraline, 50 mg/d, to venlafaxine. Ms. R says the seizures are terrifying and she cannot work. She moves in with her parents because she is unable to care for herself.
During a psychiatric appointment, Ms. R confesses that for 2 years her pain has been so unbearable that she has been buying extra tramadol from Internet retailers and taking 600 to 800 mg/d in addition to the prescribed 400 mg/d.
The authors’ observations
Ms. R had a history of chronic pain Table 117 and developed seizures after escalating her tramadol use. After her first epilepsy attack, she did not tell her physicians she was taking additional tramadol nor did she stop taking it. Treatment with several antiepileptics was unsuccessful. Her seizures persisted as long as her tramadol addiction continued.
Table 1
DSM-IV-TR criteria for pain disorder
|
| Source: Reference 17 |
Spiller et al18 reported the lowest daily tramadol dose associated with seizures is 500 mg/d, although Talaie et al16 observed seizures at doses as low as 100 mg/d. Additionally, seizure risk may increase through tramadol’s interactions with several medications, including tricyclic antidepressants, selective serotonin reuptake inhibitors, phenothiazines, fluoroquinolone antibiotics, meperidine, clozapine, buspirone, bupropion, phenylephrine, guaifenesin, tripelennamine, thioridazine, theophylline, and acetaminophen, butalbital, and caffeine combination (Table 2).19 Transdermal selegiline is contraindicated with tramadol. For Ms. R, the sertraline and venlafaxine she was taking may have augmented tramadol’s seizure potential.
Table 2
Tramadol: Major drug-drug interactions
| Drug | Symptoms |
|---|---|
| Selegiline | Nausea, vomiting, cardiovascular collapse, respiratory depression, seizures, or serotonin syndrome (hypertension, hyperthermia, myoclonus, mental status changes); use of the transdermal formulation with tramadol is contraindicated |
| Carbamazepine | Decreased tramadol efficacy and increased seizure risk |
| Venlafaxine | Increased risk of serotonin syndrome |
| Linezolid | Increased risk of serotonin syndrome |
| Fluoxetine | Increased risk of seizures and serotonin syndrome; increased concentrations of tramadol and decreased concentrations of tramadol active metabolite, O-desmethyltramadol (M1) |
| Olanzapine | Increased risk of serotonin syndrome |
| Mirtazapine | Increased risk of serotonin syndrome |
| Haloperidol | Increased risk of seizures |
| Escitalopram | Increased risk of seizures and serotonin syndrome |
| Clomipramine | Increased risk of seizures |
| Risperidone | Increased risk of seizures |
| Ketamine | Increased risk of respiratory depression and excessive CNS depression |
| Imipramine | Increased risk of seizures |
| Duloxetine | Increased risk of serotonin syndrome |
| Nortriptyline | Increased risk of seizures |
| Clozapine | Increased risk of seizures |
| Sertraline | Increased risk of seizures and serotonin syndrome |
| Paroxetine | Increased risk of seizures and serotonin syndrome; decrease in the analgesic effect of tramadol |
| Amitriptyline | Increased risk of seizures; increased concentrations of tramadol and decreased concentrations of tramadol active metabolite, M1 |
| Desipramine | Increased risk of seizures |
| Doxepin | Increased risk of seizures |
| Citalopram | Increased risk of seizures and serotonin syndrome |
| Fluvoxamine | Increased risk of seizures and serotonin syndrome |
| Source: Reference 19 | |
It is important to avoid polypharmacy in patients taking tramadol.20 Most psychiatrists are aware of the risk of serotonin syndrome with antidepressants, but may be less likely to attribute serotonergic additive effects from other medication classes such as analgesics. Recognizing tramadol’s potential to contribute to serotonin syndrome—especially in light of concomitant usage with other serotonergic medications such as antidepressants—is essential.
Tramadol toxicity appears to be caused by monoamine uptake inhibition rather than its opioid effects.21 The most frequent pharmacokinetic drug-drug interactions that lead to side effects such as serotonin syndrome or seizures involve several isoenzymes of the hepatic cytochrome P450 (CYP). The isoenzymes CYP2D6 (substrates—eg, amitriptyline, tramadol, and venlafaxine; inhibitors—eg, fluoxetine and duloxetine) and CYP3A4 (substrates—eg, carbamazepine, oxycodone, and venlafaxine; inductors—eg, carbamazepine; inhibitors, eg—grapefruit juice) are most important clinically.22
Ms. R readily obtained tramadol from Internet retailers. In a 2004 report, a Google search yielded 2,150,000 sources for acquiring tramadol, most of which did not require a prescription.23 Chronic pain patients have a higher prevalence of substance abuse than the general population.24 Because Ms. R did not have a documented substance abuse history, none of her physicians screened her for drug abuse, although toxicology screening wouldn’t have helped because the tramadol had been prescribed. We didn’t think to directly ask Ms. R about medication misuse, but if we had, she might have revealed it sooner.
OUTCOME: Seizure free
With Ms. R’s permission, we speak to her neurologist, who agrees that excess tramadol likely induced her seizures. The seizures stop after Ms. R discontinues tramadol. After 3 months without seizures, phenytoin is discontinued and lamotrigine is tapered to 200 mg/d. Ms. R participates in a pain rehabilitation program and continues to take venlafaxine, 300 mg/d, and sertraline, 50 mg/d. Her mood improves and she returns to work. Her pain is managed by non-steroidal anti-inflammatory drugs because she decides to decrease her activity level. Ms. R also is trying alternative medicine modalities such as acupuncture and acupressure.
Related Resource
- Clark MR, Treisman GJ. Chronic pain and addiction. Basel, Switzerland: Karger; 2011.
Drug Brand Names
- Acetaminophen, butalbital, and caffeine • Fioricet
- Amitriptyline • Elavil
- Bupropion • Wellbutrin
- Buspirone • Buspar
- Carbamazepine • Tegretol, Carbatrol
- Citalopram • Celexa
- Clomipramine • Anafranil
- Clozapine • Clozaril
- Desipramine • Norpramin
- Doxepin • Adapin, Silenor
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Guaifenesin • Tenex
- Haloperidol • Haldol
- Imipramine • Tofranil
- Ketamine • Ketalar
- Lamotrigine • Lamictal
- Linezolid • Zyvox
- Meperidine • Demerol
- Mirtazapine • Remeron
- Nortriptyline • Aventyl
- Olanzapine • Zyprexa
- Oxcarbazepine • Trileptal
- Oxycodone • Percolone, OxyContin
- Paroxetine • Paxil
- Phenylephrine • Lusonal
- Phenytoin • Dilantin
- Propoxyphene • Darvon
- Risperidone • Risperdal
- Selegiline • Eldepryl, EMSAM
- Sertraline • Zoloft
- Theophylline • Aerolate
- Thioridazine • Mellaril
- Tramadol • Ultram
- Tripelennamine • Pyribenzamine
- Venlafaxine • Effexor
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufactur-ers of competing products.
CASE: New-onset seizures
Ms. R, age 33, is referred by her neurologist for treatment of depressive symptoms that have intensified after she was diagnosed with epilepsy 1 year ago. She has a history of bulimia and ongoing anxiety and depression. She also has long-standing neuropathic pain in her left lateral shin and ankle that started after her foot was amputated in a lawn mower accident at age 5. Ms. R says she didn’t take pain medication until age 24, when her pain specialist prescribed tramadol, 300 to 400 mg/d, which she continues to take.
Ms. R’s first seizure occurred 1 year ago. Despite trials of several antiepileptics, her seizures persist; she is taking lamotrigine, 200 mg/d, when she presents for treatment. She has no history of brain injuries or strokes to explain her epilepsy. An MRI and 3 electroencephalograms show no signs of focal, potentially epileptogenic lesions.
Ms. R reports worsening depressive symptoms—particularly impaired attention and concentration—over several months that interfere with her housekeeping and ability to finish simple tasks at work. She says she drinks alcohol occasionally, but denies substance abuse. We initiate venlafaxine, titrated to 300 mg/d, because Ms. R has a history of intolerable side effects with fluoxetine (gastrointestinal distress) and citalopram (weight gain).
The authors’ observations
Tramadol, a centrally acting synthetic analgesic, consists of 2 enantiomers that act as weak agonists at μ-opioid receptors while also inhibiting serotonin and norepinephrine reuptake.1 Euphoria associated with μ receptor activation often is considered a “high.” Most abused opioids are prototypical μ agonists. When opioids are injected or inhaled, drug levels in the brain rise rapidly, causing a “rush”—a brief, intense, pleasurable sensation—followed by a longer-lasting high. Tolerance and physical dependence occur when opioids are used chronically.
Despite tramadol’s μ-opioid activity, the FDA approved it as an unscheduled analgesic in 1994 based on several human studies.2 Experience with tramadol has confirmed it has low abuse potential, yet human laboratory data—and some epidemiologic data—show that repeated use can lead to physical dependence. Although tramadol is considered a relatively weak opioid, human studies suggest that it possesses μ-agonist activity. The Drug Abuse Warning Network reported >15,000 emergency department (ED) visits for nonmedical tramadol use in 2009, which was more than the number of ED visits for codeine products (7,958) or propoxyphene products (9,526), but much fewer than visits for hydrocodone (86,258) or oxycodone (148,449) products.3
The recommended tramadol dose is 50 to 100 mg every 4 to 6 hours (maximum 400 mg/d). Adverse effects range from dysphoria, constipation, and nausea to agitation, seizures, respiratory depression, and coma.4 Tramadol withdrawal is similar to opioid withdrawal, and is characterized by anxiety, restlessness, insomnia, yawning, rhinorrhea, lacrimation, diaphoresis, tremor, muscle spasms, vomiting, diarrhea, and tachycardia. Rarely, psychomotor agitation and confusion may occur.5
Tramadol and seizures
At clinically appropriate doses, tramadol slightly suppresses seizure severity,6 but higher doses can induce seizures.7-12 This paradox is explained by tramadol’s effect on γ-aminobutyric acid (GABA) receptors. Although at clinical doses tramadol does not affect GABA, which could precipitate seizures, at higher doses it has been shown to have an inhibitory effect on GABA receptors.13,14 No prospective studies have assessed how often tramadol-induced seizures occur. Case reports12,15 suggest that seizures are more likely with acute tramadol intoxication, in patients with a history of alcohol abuse, or with pharmacologic regimens that include other medications that may cause seizures. Tramadol-induced seizures are generalized tonic-clonic in nature, and typically occur within 24 hours of the last dose.16
HISTORY: Worsening seizures
Two months after she presents for psychiatric evaluation, Ms. R experiences 6 generalized convulsions lasting from 15 minutes to 1 hour with no identifiable precipitant. Because oxcarbazepine and lamotrigine have failed to suppress her seizures, her neurologist adds phenytoin, 200 mg/d, and increases lamotrigine from 200 to 300 mg/d. Her depression continues to worsen. She reports severe insomnia, anhedonia, restlessness, and hopelessness, so we add sertraline, 50 mg/d, to venlafaxine. Ms. R says the seizures are terrifying and she cannot work. She moves in with her parents because she is unable to care for herself.
During a psychiatric appointment, Ms. R confesses that for 2 years her pain has been so unbearable that she has been buying extra tramadol from Internet retailers and taking 600 to 800 mg/d in addition to the prescribed 400 mg/d.
The authors’ observations
Ms. R had a history of chronic pain Table 117 and developed seizures after escalating her tramadol use. After her first epilepsy attack, she did not tell her physicians she was taking additional tramadol nor did she stop taking it. Treatment with several antiepileptics was unsuccessful. Her seizures persisted as long as her tramadol addiction continued.
Table 1
DSM-IV-TR criteria for pain disorder
|
| Source: Reference 17 |
Spiller et al18 reported the lowest daily tramadol dose associated with seizures is 500 mg/d, although Talaie et al16 observed seizures at doses as low as 100 mg/d. Additionally, seizure risk may increase through tramadol’s interactions with several medications, including tricyclic antidepressants, selective serotonin reuptake inhibitors, phenothiazines, fluoroquinolone antibiotics, meperidine, clozapine, buspirone, bupropion, phenylephrine, guaifenesin, tripelennamine, thioridazine, theophylline, and acetaminophen, butalbital, and caffeine combination (Table 2).19 Transdermal selegiline is contraindicated with tramadol. For Ms. R, the sertraline and venlafaxine she was taking may have augmented tramadol’s seizure potential.
Table 2
Tramadol: Major drug-drug interactions
| Drug | Symptoms |
|---|---|
| Selegiline | Nausea, vomiting, cardiovascular collapse, respiratory depression, seizures, or serotonin syndrome (hypertension, hyperthermia, myoclonus, mental status changes); use of the transdermal formulation with tramadol is contraindicated |
| Carbamazepine | Decreased tramadol efficacy and increased seizure risk |
| Venlafaxine | Increased risk of serotonin syndrome |
| Linezolid | Increased risk of serotonin syndrome |
| Fluoxetine | Increased risk of seizures and serotonin syndrome; increased concentrations of tramadol and decreased concentrations of tramadol active metabolite, O-desmethyltramadol (M1) |
| Olanzapine | Increased risk of serotonin syndrome |
| Mirtazapine | Increased risk of serotonin syndrome |
| Haloperidol | Increased risk of seizures |
| Escitalopram | Increased risk of seizures and serotonin syndrome |
| Clomipramine | Increased risk of seizures |
| Risperidone | Increased risk of seizures |
| Ketamine | Increased risk of respiratory depression and excessive CNS depression |
| Imipramine | Increased risk of seizures |
| Duloxetine | Increased risk of serotonin syndrome |
| Nortriptyline | Increased risk of seizures |
| Clozapine | Increased risk of seizures |
| Sertraline | Increased risk of seizures and serotonin syndrome |
| Paroxetine | Increased risk of seizures and serotonin syndrome; decrease in the analgesic effect of tramadol |
| Amitriptyline | Increased risk of seizures; increased concentrations of tramadol and decreased concentrations of tramadol active metabolite, M1 |
| Desipramine | Increased risk of seizures |
| Doxepin | Increased risk of seizures |
| Citalopram | Increased risk of seizures and serotonin syndrome |
| Fluvoxamine | Increased risk of seizures and serotonin syndrome |
| Source: Reference 19 | |
It is important to avoid polypharmacy in patients taking tramadol.20 Most psychiatrists are aware of the risk of serotonin syndrome with antidepressants, but may be less likely to attribute serotonergic additive effects from other medication classes such as analgesics. Recognizing tramadol’s potential to contribute to serotonin syndrome—especially in light of concomitant usage with other serotonergic medications such as antidepressants—is essential.
Tramadol toxicity appears to be caused by monoamine uptake inhibition rather than its opioid effects.21 The most frequent pharmacokinetic drug-drug interactions that lead to side effects such as serotonin syndrome or seizures involve several isoenzymes of the hepatic cytochrome P450 (CYP). The isoenzymes CYP2D6 (substrates—eg, amitriptyline, tramadol, and venlafaxine; inhibitors—eg, fluoxetine and duloxetine) and CYP3A4 (substrates—eg, carbamazepine, oxycodone, and venlafaxine; inductors—eg, carbamazepine; inhibitors, eg—grapefruit juice) are most important clinically.22
Ms. R readily obtained tramadol from Internet retailers. In a 2004 report, a Google search yielded 2,150,000 sources for acquiring tramadol, most of which did not require a prescription.23 Chronic pain patients have a higher prevalence of substance abuse than the general population.24 Because Ms. R did not have a documented substance abuse history, none of her physicians screened her for drug abuse, although toxicology screening wouldn’t have helped because the tramadol had been prescribed. We didn’t think to directly ask Ms. R about medication misuse, but if we had, she might have revealed it sooner.
OUTCOME: Seizure free
With Ms. R’s permission, we speak to her neurologist, who agrees that excess tramadol likely induced her seizures. The seizures stop after Ms. R discontinues tramadol. After 3 months without seizures, phenytoin is discontinued and lamotrigine is tapered to 200 mg/d. Ms. R participates in a pain rehabilitation program and continues to take venlafaxine, 300 mg/d, and sertraline, 50 mg/d. Her mood improves and she returns to work. Her pain is managed by non-steroidal anti-inflammatory drugs because she decides to decrease her activity level. Ms. R also is trying alternative medicine modalities such as acupuncture and acupressure.
Related Resource
- Clark MR, Treisman GJ. Chronic pain and addiction. Basel, Switzerland: Karger; 2011.
Drug Brand Names
- Acetaminophen, butalbital, and caffeine • Fioricet
- Amitriptyline • Elavil
- Bupropion • Wellbutrin
- Buspirone • Buspar
- Carbamazepine • Tegretol, Carbatrol
- Citalopram • Celexa
- Clomipramine • Anafranil
- Clozapine • Clozaril
- Desipramine • Norpramin
- Doxepin • Adapin, Silenor
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Guaifenesin • Tenex
- Haloperidol • Haldol
- Imipramine • Tofranil
- Ketamine • Ketalar
- Lamotrigine • Lamictal
- Linezolid • Zyvox
- Meperidine • Demerol
- Mirtazapine • Remeron
- Nortriptyline • Aventyl
- Olanzapine • Zyprexa
- Oxcarbazepine • Trileptal
- Oxycodone • Percolone, OxyContin
- Paroxetine • Paxil
- Phenylephrine • Lusonal
- Phenytoin • Dilantin
- Propoxyphene • Darvon
- Risperidone • Risperdal
- Selegiline • Eldepryl, EMSAM
- Sertraline • Zoloft
- Theophylline • Aerolate
- Thioridazine • Mellaril
- Tramadol • Ultram
- Tripelennamine • Pyribenzamine
- Venlafaxine • Effexor
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufactur-ers of competing products.
1. Katz KD. Tramadol is an opioid. J Med Toxicol. 2008;4(2):145-
2. Preston KL, Jasinski DR, Testa M. Abuse potential and pharmacological comparison of tramadol and morphine. Drug Alcohol Depend. 1991;27(1):7-17.
3. U.S. Department of Health and Human Services. Substance Abuse and Mental Health Services Administration. Drug abuse warning network, 2009: national estimates of drug-related emergency department visits. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2011. HHS publication (SMA) 11-4659, DAWN Series D-35.
4. Afshari R, Ghooshkhanehee H. Tramadol overdose induced seizure dramatic rise of CPK and acute renal failure. J Pak Med Assoc. 2009;59(3):178.-
5. Rodriguez Villamañan JC, Albaladejo Blanco C, Sanchez Sanchez A, et al. Withdrawal syndrome after long-term treatment with tramadol. Br J Gen Pract. 2000;50(454):406.-
6. Manocha A, Sharma KK, Mediratta PK. On the mechanism of anticonvulsant effect of tramadol in mice. Pharmacol Biochem Behav. 2005;82(1):74-81.
7. Boyd IW. Tramadol and seizures. Med J Aust. 2005;182(11):595-596.
8. Labate A, Newton MR, Vernon GM, et al. Tramadol and new-onset seizures. Med J Aust. 2005;182(1):42-43.
9. Gasse C, Derby L, Vasilakis-Scaramozza C, et al. Incidence of first-time idiopathic seizures in users of tramadol. Pharmacotherapy. 2000;20(6):629-634.
10. Kahn LH, Alderfer RJ, Graham DJ. Seizures reported with tramadol. JAMA. 1997;278(20):1661.-
11. Mazor SS, Feldman KW, Sugar NF, et al. Pediatric tramadol ingestion resulting in seizurelike activity: a case series. Pediatr Emerg Care. 2008;24(6):380-381.
12. Raffa RB, Stone DJ, Jr. Unexceptional seizure potential of tramadol or its enantiomers or metabolites in mice. J Pharmacol Exp Ther. 2008;325(2):500-506.
13. Rehni AK, Singh TG, Singh N, et al. Tramadol-induced seizurogenic effect: a possible role of opioid-dependent histamine H1 receptor activation-linked mechanism. Naunyn Schmiedebergs Arch Pharmacol. 2010;381(1):11-19.
14. Rehni AK, Singh I, Kumar M. Tramadol-induced seizurogenic effect: a possible role of opioid-dependent gamma-aminobutyric acid inhibitory pathway. Basic Clin Pharmacol Toxicol. 2008;103(3):262-266.
15. Jovanović-Cupić V, Martinović Z, Nesić N. Seizures associated with intoxication and abuse of tramadol. Clin Toxicol (Phila). 2006;44(2):143-146.
16. Talaie H, Panahandeh R, Fayaznouri M, et al. Dose-independent occurrence of seizure with tramadol. J Med Toxicol. 2009;5(2):63-67.
17. Diagnostic and statistical manual of mental disorders 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
18. Spiller HA, Gorman SE, Villalobos D, et al. Prospective multicenter evaluation of tramadol exposure. J Toxicol Clin Toxicol. 1997;35(4):361-364.
19. Reus VI, Rawitscher L. Possible interaction of tramadol and antidepressants. Am J Psychiatry. 2000;157(5):839.-
20. Thundiyil JG, Kearney TE, Olson KR. Evolving epidemiology of drug-induced seizures reported to a Poison Control Center System. J Med Toxicol. 2007;3(1):15-19.
21. Looper KJ. Potential medical and surgical complications of serotonergic antidepressant medications. Psychosomatics. 2007;48(1):1-9.
22. Grond S, Sablotzki A. Clinical pharmacology of tramadol. Clin Pharmacokinet. 2004;43(13):879-923.
23. Lineberry TW, Bostwick JM. Taking the physician out of “physician shopping”: a case series of clinical problems associated with Internet purchases of medication. Mayo Clin Proc. 2004;79(8):1031-1034.
24. Savage SR. Assessment for addiction in pain-treatment settings. Clin J Pain. 2002;18(4 suppl):S28-S38.
1. Katz KD. Tramadol is an opioid. J Med Toxicol. 2008;4(2):145-
2. Preston KL, Jasinski DR, Testa M. Abuse potential and pharmacological comparison of tramadol and morphine. Drug Alcohol Depend. 1991;27(1):7-17.
3. U.S. Department of Health and Human Services. Substance Abuse and Mental Health Services Administration. Drug abuse warning network, 2009: national estimates of drug-related emergency department visits. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2011. HHS publication (SMA) 11-4659, DAWN Series D-35.
4. Afshari R, Ghooshkhanehee H. Tramadol overdose induced seizure dramatic rise of CPK and acute renal failure. J Pak Med Assoc. 2009;59(3):178.-
5. Rodriguez Villamañan JC, Albaladejo Blanco C, Sanchez Sanchez A, et al. Withdrawal syndrome after long-term treatment with tramadol. Br J Gen Pract. 2000;50(454):406.-
6. Manocha A, Sharma KK, Mediratta PK. On the mechanism of anticonvulsant effect of tramadol in mice. Pharmacol Biochem Behav. 2005;82(1):74-81.
7. Boyd IW. Tramadol and seizures. Med J Aust. 2005;182(11):595-596.
8. Labate A, Newton MR, Vernon GM, et al. Tramadol and new-onset seizures. Med J Aust. 2005;182(1):42-43.
9. Gasse C, Derby L, Vasilakis-Scaramozza C, et al. Incidence of first-time idiopathic seizures in users of tramadol. Pharmacotherapy. 2000;20(6):629-634.
10. Kahn LH, Alderfer RJ, Graham DJ. Seizures reported with tramadol. JAMA. 1997;278(20):1661.-
11. Mazor SS, Feldman KW, Sugar NF, et al. Pediatric tramadol ingestion resulting in seizurelike activity: a case series. Pediatr Emerg Care. 2008;24(6):380-381.
12. Raffa RB, Stone DJ, Jr. Unexceptional seizure potential of tramadol or its enantiomers or metabolites in mice. J Pharmacol Exp Ther. 2008;325(2):500-506.
13. Rehni AK, Singh TG, Singh N, et al. Tramadol-induced seizurogenic effect: a possible role of opioid-dependent histamine H1 receptor activation-linked mechanism. Naunyn Schmiedebergs Arch Pharmacol. 2010;381(1):11-19.
14. Rehni AK, Singh I, Kumar M. Tramadol-induced seizurogenic effect: a possible role of opioid-dependent gamma-aminobutyric acid inhibitory pathway. Basic Clin Pharmacol Toxicol. 2008;103(3):262-266.
15. Jovanović-Cupić V, Martinović Z, Nesić N. Seizures associated with intoxication and abuse of tramadol. Clin Toxicol (Phila). 2006;44(2):143-146.
16. Talaie H, Panahandeh R, Fayaznouri M, et al. Dose-independent occurrence of seizure with tramadol. J Med Toxicol. 2009;5(2):63-67.
17. Diagnostic and statistical manual of mental disorders 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
18. Spiller HA, Gorman SE, Villalobos D, et al. Prospective multicenter evaluation of tramadol exposure. J Toxicol Clin Toxicol. 1997;35(4):361-364.
19. Reus VI, Rawitscher L. Possible interaction of tramadol and antidepressants. Am J Psychiatry. 2000;157(5):839.-
20. Thundiyil JG, Kearney TE, Olson KR. Evolving epidemiology of drug-induced seizures reported to a Poison Control Center System. J Med Toxicol. 2007;3(1):15-19.
21. Looper KJ. Potential medical and surgical complications of serotonergic antidepressant medications. Psychosomatics. 2007;48(1):1-9.
22. Grond S, Sablotzki A. Clinical pharmacology of tramadol. Clin Pharmacokinet. 2004;43(13):879-923.
23. Lineberry TW, Bostwick JM. Taking the physician out of “physician shopping”: a case series of clinical problems associated with Internet purchases of medication. Mayo Clin Proc. 2004;79(8):1031-1034.
24. Savage SR. Assessment for addiction in pain-treatment settings. Clin J Pain. 2002;18(4 suppl):S28-S38.
Diabetic and depressed
CASE: Worsening depression
Mr. N, age 64, is a disabled factory worker with a complicated medical history. He has poorly controlled type II diabetes mellitus; obesity (body mass index 40 kg/m2); complicated cryptogenic cirrhosis with prior esophageal varices, portal gastropathy, splenomegaly, and no encephalopathy; surgically treated colon adenocarcinoma; and bilateral thalamic and right occipital infarcts with residual left homonymous hemianopsia and vertical gaze paresis. Mr. N sustained a perioperative stroke 18 months ago while undergoing a colectomy procedure for colon adenocarcinoma; an MRI done at that time showed the bilateral thalamic and right occipital infarcts. While in the internal medicine consultation clinic, Mr. N expresses suicidal and homicidal thoughts, which prompted the internal medicine team to refer him to the emergency department (ED). The team deems Mr. N’s medical problems stable except for diabetic dyscontrol.
In the ED, Mr. N says he feels sad, worthless, and “tired” of his complex family issues and multiple medical conditions. He says he’s had these feeling for at least a year, but his depression has worsened in the last few days. Mr. N is tearful while explaining his discouragement with following a diet for diabetes; earlier that day he ate an entire chocolate cake. He says all 3 of his children have ongoing substance abuse and relationship problems, but he is particularly focused on his youngest daughter, who is involved with a man who is addicted to drugs and physically abuses her and her children. Mr. N describes a detailed plan to shoot him and then commit suicide. This disclosure prompts the ED physician to admit Mr. N to ensure his safety and stabilize his mood.
Mr. N’s temperature is 36. 4°C (97. 5°F), blood pressure is 123/60 mm Hg, pulse is 81 beats per minute, respiratory rate is 24 breaths per minute, and oxygen saturation is 96% on ambient air. His physical exam is notable only for dysphoria and mild gynecomastia. He shows no evidence of acute cardiopulmonary, gastrointestinal, or other neurologic changes. His serum glucose is 650 mg/dL, and his recent hemoglobin A1c (HbA1c) is 10. 9%. His other laboratory tests include a hemoglobin of 11. 7 g/dL; white cell count, 3500/mm3; platelet count, 41, 000/mm3; sodium, 129 mEq/L; potassium, 5. 0 mEq/L; alkaline phosphatase, 90 U/L; aspartate aminotransferase, 23 U/L; alanine aminotransferase, 21 U/L; total bilirubin, 1. 8 mg/dL; creatinine, 1. 2 mg/dL; prothrombin time, 10. 4 sec; and arterial ammonia, <50 ?g/dL. Arterial blood gases are normal.
A year ago, his primary care physician prescribed fluoxetine, 20 mg/d, for fatigue and chronic back pain and neuropathic pain related to diabetes. We continue Mr. N’s outpatient prescription of fluoxetine, 20 mg/d, and low-dose acetaminophen as needed for pain. Furosemide, 40 mg/d, spironolactone, 100 mg/d, and propranolol sustained release, 60 mg/d, are maintained for complications of cirrhosis. Insulin aspart, 22 units with breakfast, 24 units with lunch, and 24 units with supper, also are administered routinely.
We consult with the internal medicine, ophthalmology, neurology, endocrinology, and diabetes services to assist in evaluating and managing Mr. N’s multiple medical conditions.
The authors’ observations
Depression and other forms of psychopathology may be underrecognized in geriatric patients because older adults may not report psychiatric symptoms that are secondary to physical conditions. Cognitive impairment in some older adults also may lead to underreporting of symptoms. Mr. N denies a history of depression, which we confirmed with his wife, daughter, and primary care physician. The late onset of his initial presentation prompted close investigation for a potential medical etiology (Table 1).1,2
We considered post-stroke depression because shortly after Mr. N’s stroke, his neurologist described emotional lability and frustration related to his poor vision. Depression occurs in one-third of chronic stroke survivors and is prevalent among patients referred for neurologic rehabilitation.1 Premorbid neuroticism3 and a history of mental illness are predictors of post-stroke depression. Stroke laterality is not related to risk of post-stroke depressive symptoms,3 but women have a higher risk of developing post-stroke depression.3
Table 1
When to consider medical causes of depressive symptoms
| Late onset of initial depressive presentation |
| Known underlying medical condition, such as cancer, diabetes, or stroke |
| Atypical symptoms and signs of depression, such as hypersomnia, hyperphagia, or agitation |
| Absence of personal or family history of psychiatric illnesses |
| Illicit substance use |
| Medication use (eg, opioids, reserpine, methyldopa, chemotherapy agents, steroids, and oral contraceptives) |
| Treatment resistance or unusual response to treatment |
| Sudden onset of mental symptoms (eg, sudden episode of uncontrollable crying) |
| Source: References 1,2 |
Diabetes and depression
Up to 30% of patients with type 2 diabetes mellitus report a lifetime history of major depression.2 Depression increases the risk of hyperglycemia and accompanying long-term metabolic complications.4,5 Few studies have explored the effects of poor glycemic control on depressive symptoms among diabetic patients.6-9 A literature review revealed no large-scale study investigating worsened depressive symptoms in patients with poor glycemic control.10,11
The cross-sectional difference between a single episode of major depression and adjustment disorder can be subtle. DSM-IV-TR describes the latter as a maladaptive reaction to an identifiable psychosocial stressor, or stressors, that occurs within 3 months of onset of that stressor (Table 2).12 Because we did not deem Mr. N’s depressive symptoms, which were evident only when he was hyperglycemic, to be grossly disproportionate to his stressors, we diagnose him with major depression rather than adjustment disorder.
Table 2
DSM-IV-TR diagnostic criteria for adjustment disorder
| A. The development of emotional or behavioral symptoms in response to an identifiable stressor(s) that occurs within 3 months of the onset of the stressor(s) |
B. These symptoms or behaviors are clinically significant, as evidenced by either of the following:
|
| C. The stress-related disturbance does not meet criteria for another specific axis I disorder and is not merely an exacerbation of a pre-existing axis I or axis II disorder |
| D. The symptoms do not represent bereavement |
| E. Once the stressor (or its consequences) has terminated, the symptoms do not persist for more than an additional 6 months |
Specify whether the condition is acute or chronic, as follows:
|
| Source: Reference 12 |
EVALUATION: No psychiatric history
On admission, Mr. N is overwhelmed, tearful, and dysphoric when describing his situation. He displays no evidence of psychosis, but his judgment and insight are impaired. He shows no change in consciousness or ability to stay awake. Mr. N acknowledges hypersomnolence, anhedonia, anergia, and decreased concentration and continues to express suicidal and homicidal thoughts. He repeatedly denies any personal or family psychiatric history or personal substance abuse, including alcohol and nicotine.
TREATMENT: Glycemic control
Mr. N receives 1 L of saline in the ED and is encouraged to drink more water during hospitalization. With appropriate insulin dosing, Mr. N’s serum glucose levels improve from 650 to 426 mg/dL by the next morning. On his third hospital day, Mr. N’s glucose level is 155 mg/dL in the morning. With tighter glycemic control, his dysphoria improves. He is future-oriented, markedly less dysphoric, and denies homicidal or suicidal ideation. Mr. N is interested in participating in group therapy, and his insight and judgment regarding his homicidal and suicidal thoughts improve. He still doesn’t fully understand the importance of diabetic control, and he struggles with his diet.
On the fourth hospital day, Mr. N’s glucose level rises to 325 mg/dL in the early evening. Subsequently, his mood deteriorates; he becomes increasingly withdrawn and somnolent. With appropriate attention to his dietary intake and supplemental insulin, his serum glucose improves to the 100 to 200 mg/dL range overnight, and his mood improves. When the glucose is controlled, he attends group therapy, tends to his hygiene, denies feeling hopeless, and offers several ideas about how to manage his family situation. After his glucose rises, Mr. N becomes isolative, hopeless, and unable to cope with stressors. With considerable education about the importance of glycemic control, Mr. N is hopeful and future-oriented when he is discharged after 9 days of hospitalization. At outpatient evaluation, he continues to report euthymia with adequate glycemic control.
The authors’ observations
Patients with hyperglycemia may experience symptoms secondary to volume depletion and hyperosmolality. The severity of these symptoms generally is proportional to the extent and duration of the hyperosmolar state. Initially, most patients complain of polyuria and polydipsia, but in more severe cases, mental status changes may evolve and include lethargy, twitching, cloudiness, motor or sensory defects, seizures, and coma. Some evidence shows that hyperglycemic patients with hyperosmolality are symptomatic only if hypernatremia is present.13 Mr. N was hyponatremic, which resolved with aggressive hydration and insulin administration.
Traditionally, depression has been observed to worsen glycemic control in diabetic patients; discussion of increased glucose levels leading to worsened depression rarely has been reported. In a meta-analysis, Lustman et al7 revealed that depression is significantly associated with hyperglycemia. A review by Li et al14 demonstrated that depression is much more common in patients with diabetes compared with general population and 45% of diabetes patients with depression were undiagnosed. Calhoun et al15 showed that for every 1-unit increase in HbA1c the odds of depressive symptoms increase by 22%. Researchers also found a positive relationship between depression and glycemic control in American Indians.15
Mr. N’s case is further evidence that the relationship between diabetes and depression is bidirectional and diagnosis and treatment of each illness impacts the other. Although this case does not confirm causality, it highlights the importance of aggressive approaches to screening and treatment of depression in patients with diabetes, and vice versa.
Related Resources
- Katon W, Russo J, Lin EH, et al. Depression and diabetes: factors associated with major depression at five-year follow-up. Psychosomatics. 2009; 50(6): 570-579.
- Biessels GJ, Luchsinger JA. Diabetes and the brain. New York, NY: Humana Press; 2009.
Drug Brand Names
- Fluoxetine • Prozac
- Furosemide • Lasix
- Insulin aspart • NovoLog
- Insulin glargine • Lantus
- Methyldopa • Aldomet
- Propranolol • Inderal
- Reserpine • Serpasil
- Spironolactone • Aldactone
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Srivastava A, Taly AB, Gupta A, et al. Post-stroke depression: prevalence and relationship with disability in chronic stroke survivors. Ann Indian Acad Neurol. 2010;13(2):123-127.
2. Marcus MD, Wing RR, Guare J, et al. Lifetime prevalence of major depression and its effect on treatment outcome in obese type II diabetic patients. Diabetes Care. 1992;15(2):253-255.
3. Storor DL, Byrne GJ. Pre-morbid personality and depression following stroke. Int Psychogeriatr. 2006;18(3):457-469.
4. Songar A, Kocabasoglu N, Balcioglu I, et al. The relationship between diabetics’ metabolic control levels and psychiatric symptomatology. Integrative Psychiatry. 1993;9:34-40.
5. Von Dras DD, Lichty W. Correlates of depression in diabetic adults. Behav Health Aging. 1990;1:79-84.
6. Lustman PJ, Clouse RE. Depression in diabetic patients: the relationship between mood and glycemic control. J Diabetes Complications. 2005;19(2):113-122.
7. Lustman PJ, Anderson RJ, Freedland KE, et al. Depression and poor glycemic control: a meta-analytic review of the literature. Diabetes Care. 2000;23(7):934-942.
8. Lustman PJ, Griffith LS, Clouse RE. Depression in adults with diabetes: results of a 5-yr follow-up study. Diabetes Care. 1988;11:605-612.
9. Van der Does FE, De Neeling JN, Snoek FJ, et al. Symptoms and well-being in relation to glycemic control in type II diabetes. Diabetes Care. 1996;19:204-210.
10. Genuth S. A case for blood glucose control. Adv Intern Med. 1995;40:573-623.
11. Wrigley M, Mayou R. Psychological factors and admission for poor glycaemic control: a study of psychological and social factors in poorly controlled insulin dependent diabetic patients. J Psychosom Res. 1991;35:335-343.
12. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
13. Magee MF, Bhatt BA. Management of decompensated diabetes. Diabetic ketoacidosis and hyperglycemic hyperosmolar syndrome. Crit Care Clin. 2001;17(1):75-106.
14. Li C, Ford ES, Zhao G, et al. Prevalence and correlates of undiagnosed depression among U.S. adults with diabetes: the Behavioral Risk Factor Surveillance System, 2006. Diabetes Res Clin Pract. 2009;83(2):268-279.
15. Calhoun D, Beals J, Carter EA, et al. Relationship between glycemic control and depression among American Indians in the Strong Heart Study. J Diabetes Complications. 2010;24:217-222.
CASE: Worsening depression
Mr. N, age 64, is a disabled factory worker with a complicated medical history. He has poorly controlled type II diabetes mellitus; obesity (body mass index 40 kg/m2); complicated cryptogenic cirrhosis with prior esophageal varices, portal gastropathy, splenomegaly, and no encephalopathy; surgically treated colon adenocarcinoma; and bilateral thalamic and right occipital infarcts with residual left homonymous hemianopsia and vertical gaze paresis. Mr. N sustained a perioperative stroke 18 months ago while undergoing a colectomy procedure for colon adenocarcinoma; an MRI done at that time showed the bilateral thalamic and right occipital infarcts. While in the internal medicine consultation clinic, Mr. N expresses suicidal and homicidal thoughts, which prompted the internal medicine team to refer him to the emergency department (ED). The team deems Mr. N’s medical problems stable except for diabetic dyscontrol.
In the ED, Mr. N says he feels sad, worthless, and “tired” of his complex family issues and multiple medical conditions. He says he’s had these feeling for at least a year, but his depression has worsened in the last few days. Mr. N is tearful while explaining his discouragement with following a diet for diabetes; earlier that day he ate an entire chocolate cake. He says all 3 of his children have ongoing substance abuse and relationship problems, but he is particularly focused on his youngest daughter, who is involved with a man who is addicted to drugs and physically abuses her and her children. Mr. N describes a detailed plan to shoot him and then commit suicide. This disclosure prompts the ED physician to admit Mr. N to ensure his safety and stabilize his mood.
Mr. N’s temperature is 36. 4°C (97. 5°F), blood pressure is 123/60 mm Hg, pulse is 81 beats per minute, respiratory rate is 24 breaths per minute, and oxygen saturation is 96% on ambient air. His physical exam is notable only for dysphoria and mild gynecomastia. He shows no evidence of acute cardiopulmonary, gastrointestinal, or other neurologic changes. His serum glucose is 650 mg/dL, and his recent hemoglobin A1c (HbA1c) is 10. 9%. His other laboratory tests include a hemoglobin of 11. 7 g/dL; white cell count, 3500/mm3; platelet count, 41, 000/mm3; sodium, 129 mEq/L; potassium, 5. 0 mEq/L; alkaline phosphatase, 90 U/L; aspartate aminotransferase, 23 U/L; alanine aminotransferase, 21 U/L; total bilirubin, 1. 8 mg/dL; creatinine, 1. 2 mg/dL; prothrombin time, 10. 4 sec; and arterial ammonia, <50 ?g/dL. Arterial blood gases are normal.
A year ago, his primary care physician prescribed fluoxetine, 20 mg/d, for fatigue and chronic back pain and neuropathic pain related to diabetes. We continue Mr. N’s outpatient prescription of fluoxetine, 20 mg/d, and low-dose acetaminophen as needed for pain. Furosemide, 40 mg/d, spironolactone, 100 mg/d, and propranolol sustained release, 60 mg/d, are maintained for complications of cirrhosis. Insulin aspart, 22 units with breakfast, 24 units with lunch, and 24 units with supper, also are administered routinely.
We consult with the internal medicine, ophthalmology, neurology, endocrinology, and diabetes services to assist in evaluating and managing Mr. N’s multiple medical conditions.
The authors’ observations
Depression and other forms of psychopathology may be underrecognized in geriatric patients because older adults may not report psychiatric symptoms that are secondary to physical conditions. Cognitive impairment in some older adults also may lead to underreporting of symptoms. Mr. N denies a history of depression, which we confirmed with his wife, daughter, and primary care physician. The late onset of his initial presentation prompted close investigation for a potential medical etiology (Table 1).1,2
We considered post-stroke depression because shortly after Mr. N’s stroke, his neurologist described emotional lability and frustration related to his poor vision. Depression occurs in one-third of chronic stroke survivors and is prevalent among patients referred for neurologic rehabilitation.1 Premorbid neuroticism3 and a history of mental illness are predictors of post-stroke depression. Stroke laterality is not related to risk of post-stroke depressive symptoms,3 but women have a higher risk of developing post-stroke depression.3
Table 1
When to consider medical causes of depressive symptoms
| Late onset of initial depressive presentation |
| Known underlying medical condition, such as cancer, diabetes, or stroke |
| Atypical symptoms and signs of depression, such as hypersomnia, hyperphagia, or agitation |
| Absence of personal or family history of psychiatric illnesses |
| Illicit substance use |
| Medication use (eg, opioids, reserpine, methyldopa, chemotherapy agents, steroids, and oral contraceptives) |
| Treatment resistance or unusual response to treatment |
| Sudden onset of mental symptoms (eg, sudden episode of uncontrollable crying) |
| Source: References 1,2 |
Diabetes and depression
Up to 30% of patients with type 2 diabetes mellitus report a lifetime history of major depression.2 Depression increases the risk of hyperglycemia and accompanying long-term metabolic complications.4,5 Few studies have explored the effects of poor glycemic control on depressive symptoms among diabetic patients.6-9 A literature review revealed no large-scale study investigating worsened depressive symptoms in patients with poor glycemic control.10,11
The cross-sectional difference between a single episode of major depression and adjustment disorder can be subtle. DSM-IV-TR describes the latter as a maladaptive reaction to an identifiable psychosocial stressor, or stressors, that occurs within 3 months of onset of that stressor (Table 2).12 Because we did not deem Mr. N’s depressive symptoms, which were evident only when he was hyperglycemic, to be grossly disproportionate to his stressors, we diagnose him with major depression rather than adjustment disorder.
Table 2
DSM-IV-TR diagnostic criteria for adjustment disorder
| A. The development of emotional or behavioral symptoms in response to an identifiable stressor(s) that occurs within 3 months of the onset of the stressor(s) |
B. These symptoms or behaviors are clinically significant, as evidenced by either of the following:
|
| C. The stress-related disturbance does not meet criteria for another specific axis I disorder and is not merely an exacerbation of a pre-existing axis I or axis II disorder |
| D. The symptoms do not represent bereavement |
| E. Once the stressor (or its consequences) has terminated, the symptoms do not persist for more than an additional 6 months |
Specify whether the condition is acute or chronic, as follows:
|
| Source: Reference 12 |
EVALUATION: No psychiatric history
On admission, Mr. N is overwhelmed, tearful, and dysphoric when describing his situation. He displays no evidence of psychosis, but his judgment and insight are impaired. He shows no change in consciousness or ability to stay awake. Mr. N acknowledges hypersomnolence, anhedonia, anergia, and decreased concentration and continues to express suicidal and homicidal thoughts. He repeatedly denies any personal or family psychiatric history or personal substance abuse, including alcohol and nicotine.
TREATMENT: Glycemic control
Mr. N receives 1 L of saline in the ED and is encouraged to drink more water during hospitalization. With appropriate insulin dosing, Mr. N’s serum glucose levels improve from 650 to 426 mg/dL by the next morning. On his third hospital day, Mr. N’s glucose level is 155 mg/dL in the morning. With tighter glycemic control, his dysphoria improves. He is future-oriented, markedly less dysphoric, and denies homicidal or suicidal ideation. Mr. N is interested in participating in group therapy, and his insight and judgment regarding his homicidal and suicidal thoughts improve. He still doesn’t fully understand the importance of diabetic control, and he struggles with his diet.
On the fourth hospital day, Mr. N’s glucose level rises to 325 mg/dL in the early evening. Subsequently, his mood deteriorates; he becomes increasingly withdrawn and somnolent. With appropriate attention to his dietary intake and supplemental insulin, his serum glucose improves to the 100 to 200 mg/dL range overnight, and his mood improves. When the glucose is controlled, he attends group therapy, tends to his hygiene, denies feeling hopeless, and offers several ideas about how to manage his family situation. After his glucose rises, Mr. N becomes isolative, hopeless, and unable to cope with stressors. With considerable education about the importance of glycemic control, Mr. N is hopeful and future-oriented when he is discharged after 9 days of hospitalization. At outpatient evaluation, he continues to report euthymia with adequate glycemic control.
The authors’ observations
Patients with hyperglycemia may experience symptoms secondary to volume depletion and hyperosmolality. The severity of these symptoms generally is proportional to the extent and duration of the hyperosmolar state. Initially, most patients complain of polyuria and polydipsia, but in more severe cases, mental status changes may evolve and include lethargy, twitching, cloudiness, motor or sensory defects, seizures, and coma. Some evidence shows that hyperglycemic patients with hyperosmolality are symptomatic only if hypernatremia is present.13 Mr. N was hyponatremic, which resolved with aggressive hydration and insulin administration.
Traditionally, depression has been observed to worsen glycemic control in diabetic patients; discussion of increased glucose levels leading to worsened depression rarely has been reported. In a meta-analysis, Lustman et al7 revealed that depression is significantly associated with hyperglycemia. A review by Li et al14 demonstrated that depression is much more common in patients with diabetes compared with general population and 45% of diabetes patients with depression were undiagnosed. Calhoun et al15 showed that for every 1-unit increase in HbA1c the odds of depressive symptoms increase by 22%. Researchers also found a positive relationship between depression and glycemic control in American Indians.15
Mr. N’s case is further evidence that the relationship between diabetes and depression is bidirectional and diagnosis and treatment of each illness impacts the other. Although this case does not confirm causality, it highlights the importance of aggressive approaches to screening and treatment of depression in patients with diabetes, and vice versa.
Related Resources
- Katon W, Russo J, Lin EH, et al. Depression and diabetes: factors associated with major depression at five-year follow-up. Psychosomatics. 2009; 50(6): 570-579.
- Biessels GJ, Luchsinger JA. Diabetes and the brain. New York, NY: Humana Press; 2009.
Drug Brand Names
- Fluoxetine • Prozac
- Furosemide • Lasix
- Insulin aspart • NovoLog
- Insulin glargine • Lantus
- Methyldopa • Aldomet
- Propranolol • Inderal
- Reserpine • Serpasil
- Spironolactone • Aldactone
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE: Worsening depression
Mr. N, age 64, is a disabled factory worker with a complicated medical history. He has poorly controlled type II diabetes mellitus; obesity (body mass index 40 kg/m2); complicated cryptogenic cirrhosis with prior esophageal varices, portal gastropathy, splenomegaly, and no encephalopathy; surgically treated colon adenocarcinoma; and bilateral thalamic and right occipital infarcts with residual left homonymous hemianopsia and vertical gaze paresis. Mr. N sustained a perioperative stroke 18 months ago while undergoing a colectomy procedure for colon adenocarcinoma; an MRI done at that time showed the bilateral thalamic and right occipital infarcts. While in the internal medicine consultation clinic, Mr. N expresses suicidal and homicidal thoughts, which prompted the internal medicine team to refer him to the emergency department (ED). The team deems Mr. N’s medical problems stable except for diabetic dyscontrol.
In the ED, Mr. N says he feels sad, worthless, and “tired” of his complex family issues and multiple medical conditions. He says he’s had these feeling for at least a year, but his depression has worsened in the last few days. Mr. N is tearful while explaining his discouragement with following a diet for diabetes; earlier that day he ate an entire chocolate cake. He says all 3 of his children have ongoing substance abuse and relationship problems, but he is particularly focused on his youngest daughter, who is involved with a man who is addicted to drugs and physically abuses her and her children. Mr. N describes a detailed plan to shoot him and then commit suicide. This disclosure prompts the ED physician to admit Mr. N to ensure his safety and stabilize his mood.
Mr. N’s temperature is 36. 4°C (97. 5°F), blood pressure is 123/60 mm Hg, pulse is 81 beats per minute, respiratory rate is 24 breaths per minute, and oxygen saturation is 96% on ambient air. His physical exam is notable only for dysphoria and mild gynecomastia. He shows no evidence of acute cardiopulmonary, gastrointestinal, or other neurologic changes. His serum glucose is 650 mg/dL, and his recent hemoglobin A1c (HbA1c) is 10. 9%. His other laboratory tests include a hemoglobin of 11. 7 g/dL; white cell count, 3500/mm3; platelet count, 41, 000/mm3; sodium, 129 mEq/L; potassium, 5. 0 mEq/L; alkaline phosphatase, 90 U/L; aspartate aminotransferase, 23 U/L; alanine aminotransferase, 21 U/L; total bilirubin, 1. 8 mg/dL; creatinine, 1. 2 mg/dL; prothrombin time, 10. 4 sec; and arterial ammonia, <50 ?g/dL. Arterial blood gases are normal.
A year ago, his primary care physician prescribed fluoxetine, 20 mg/d, for fatigue and chronic back pain and neuropathic pain related to diabetes. We continue Mr. N’s outpatient prescription of fluoxetine, 20 mg/d, and low-dose acetaminophen as needed for pain. Furosemide, 40 mg/d, spironolactone, 100 mg/d, and propranolol sustained release, 60 mg/d, are maintained for complications of cirrhosis. Insulin aspart, 22 units with breakfast, 24 units with lunch, and 24 units with supper, also are administered routinely.
We consult with the internal medicine, ophthalmology, neurology, endocrinology, and diabetes services to assist in evaluating and managing Mr. N’s multiple medical conditions.
The authors’ observations
Depression and other forms of psychopathology may be underrecognized in geriatric patients because older adults may not report psychiatric symptoms that are secondary to physical conditions. Cognitive impairment in some older adults also may lead to underreporting of symptoms. Mr. N denies a history of depression, which we confirmed with his wife, daughter, and primary care physician. The late onset of his initial presentation prompted close investigation for a potential medical etiology (Table 1).1,2
We considered post-stroke depression because shortly after Mr. N’s stroke, his neurologist described emotional lability and frustration related to his poor vision. Depression occurs in one-third of chronic stroke survivors and is prevalent among patients referred for neurologic rehabilitation.1 Premorbid neuroticism3 and a history of mental illness are predictors of post-stroke depression. Stroke laterality is not related to risk of post-stroke depressive symptoms,3 but women have a higher risk of developing post-stroke depression.3
Table 1
When to consider medical causes of depressive symptoms
| Late onset of initial depressive presentation |
| Known underlying medical condition, such as cancer, diabetes, or stroke |
| Atypical symptoms and signs of depression, such as hypersomnia, hyperphagia, or agitation |
| Absence of personal or family history of psychiatric illnesses |
| Illicit substance use |
| Medication use (eg, opioids, reserpine, methyldopa, chemotherapy agents, steroids, and oral contraceptives) |
| Treatment resistance or unusual response to treatment |
| Sudden onset of mental symptoms (eg, sudden episode of uncontrollable crying) |
| Source: References 1,2 |
Diabetes and depression
Up to 30% of patients with type 2 diabetes mellitus report a lifetime history of major depression.2 Depression increases the risk of hyperglycemia and accompanying long-term metabolic complications.4,5 Few studies have explored the effects of poor glycemic control on depressive symptoms among diabetic patients.6-9 A literature review revealed no large-scale study investigating worsened depressive symptoms in patients with poor glycemic control.10,11
The cross-sectional difference between a single episode of major depression and adjustment disorder can be subtle. DSM-IV-TR describes the latter as a maladaptive reaction to an identifiable psychosocial stressor, or stressors, that occurs within 3 months of onset of that stressor (Table 2).12 Because we did not deem Mr. N’s depressive symptoms, which were evident only when he was hyperglycemic, to be grossly disproportionate to his stressors, we diagnose him with major depression rather than adjustment disorder.
Table 2
DSM-IV-TR diagnostic criteria for adjustment disorder
| A. The development of emotional or behavioral symptoms in response to an identifiable stressor(s) that occurs within 3 months of the onset of the stressor(s) |
B. These symptoms or behaviors are clinically significant, as evidenced by either of the following:
|
| C. The stress-related disturbance does not meet criteria for another specific axis I disorder and is not merely an exacerbation of a pre-existing axis I or axis II disorder |
| D. The symptoms do not represent bereavement |
| E. Once the stressor (or its consequences) has terminated, the symptoms do not persist for more than an additional 6 months |
Specify whether the condition is acute or chronic, as follows:
|
| Source: Reference 12 |
EVALUATION: No psychiatric history
On admission, Mr. N is overwhelmed, tearful, and dysphoric when describing his situation. He displays no evidence of psychosis, but his judgment and insight are impaired. He shows no change in consciousness or ability to stay awake. Mr. N acknowledges hypersomnolence, anhedonia, anergia, and decreased concentration and continues to express suicidal and homicidal thoughts. He repeatedly denies any personal or family psychiatric history or personal substance abuse, including alcohol and nicotine.
TREATMENT: Glycemic control
Mr. N receives 1 L of saline in the ED and is encouraged to drink more water during hospitalization. With appropriate insulin dosing, Mr. N’s serum glucose levels improve from 650 to 426 mg/dL by the next morning. On his third hospital day, Mr. N’s glucose level is 155 mg/dL in the morning. With tighter glycemic control, his dysphoria improves. He is future-oriented, markedly less dysphoric, and denies homicidal or suicidal ideation. Mr. N is interested in participating in group therapy, and his insight and judgment regarding his homicidal and suicidal thoughts improve. He still doesn’t fully understand the importance of diabetic control, and he struggles with his diet.
On the fourth hospital day, Mr. N’s glucose level rises to 325 mg/dL in the early evening. Subsequently, his mood deteriorates; he becomes increasingly withdrawn and somnolent. With appropriate attention to his dietary intake and supplemental insulin, his serum glucose improves to the 100 to 200 mg/dL range overnight, and his mood improves. When the glucose is controlled, he attends group therapy, tends to his hygiene, denies feeling hopeless, and offers several ideas about how to manage his family situation. After his glucose rises, Mr. N becomes isolative, hopeless, and unable to cope with stressors. With considerable education about the importance of glycemic control, Mr. N is hopeful and future-oriented when he is discharged after 9 days of hospitalization. At outpatient evaluation, he continues to report euthymia with adequate glycemic control.
The authors’ observations
Patients with hyperglycemia may experience symptoms secondary to volume depletion and hyperosmolality. The severity of these symptoms generally is proportional to the extent and duration of the hyperosmolar state. Initially, most patients complain of polyuria and polydipsia, but in more severe cases, mental status changes may evolve and include lethargy, twitching, cloudiness, motor or sensory defects, seizures, and coma. Some evidence shows that hyperglycemic patients with hyperosmolality are symptomatic only if hypernatremia is present.13 Mr. N was hyponatremic, which resolved with aggressive hydration and insulin administration.
Traditionally, depression has been observed to worsen glycemic control in diabetic patients; discussion of increased glucose levels leading to worsened depression rarely has been reported. In a meta-analysis, Lustman et al7 revealed that depression is significantly associated with hyperglycemia. A review by Li et al14 demonstrated that depression is much more common in patients with diabetes compared with general population and 45% of diabetes patients with depression were undiagnosed. Calhoun et al15 showed that for every 1-unit increase in HbA1c the odds of depressive symptoms increase by 22%. Researchers also found a positive relationship between depression and glycemic control in American Indians.15
Mr. N’s case is further evidence that the relationship between diabetes and depression is bidirectional and diagnosis and treatment of each illness impacts the other. Although this case does not confirm causality, it highlights the importance of aggressive approaches to screening and treatment of depression in patients with diabetes, and vice versa.
Related Resources
- Katon W, Russo J, Lin EH, et al. Depression and diabetes: factors associated with major depression at five-year follow-up. Psychosomatics. 2009; 50(6): 570-579.
- Biessels GJ, Luchsinger JA. Diabetes and the brain. New York, NY: Humana Press; 2009.
Drug Brand Names
- Fluoxetine • Prozac
- Furosemide • Lasix
- Insulin aspart • NovoLog
- Insulin glargine • Lantus
- Methyldopa • Aldomet
- Propranolol • Inderal
- Reserpine • Serpasil
- Spironolactone • Aldactone
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Srivastava A, Taly AB, Gupta A, et al. Post-stroke depression: prevalence and relationship with disability in chronic stroke survivors. Ann Indian Acad Neurol. 2010;13(2):123-127.
2. Marcus MD, Wing RR, Guare J, et al. Lifetime prevalence of major depression and its effect on treatment outcome in obese type II diabetic patients. Diabetes Care. 1992;15(2):253-255.
3. Storor DL, Byrne GJ. Pre-morbid personality and depression following stroke. Int Psychogeriatr. 2006;18(3):457-469.
4. Songar A, Kocabasoglu N, Balcioglu I, et al. The relationship between diabetics’ metabolic control levels and psychiatric symptomatology. Integrative Psychiatry. 1993;9:34-40.
5. Von Dras DD, Lichty W. Correlates of depression in diabetic adults. Behav Health Aging. 1990;1:79-84.
6. Lustman PJ, Clouse RE. Depression in diabetic patients: the relationship between mood and glycemic control. J Diabetes Complications. 2005;19(2):113-122.
7. Lustman PJ, Anderson RJ, Freedland KE, et al. Depression and poor glycemic control: a meta-analytic review of the literature. Diabetes Care. 2000;23(7):934-942.
8. Lustman PJ, Griffith LS, Clouse RE. Depression in adults with diabetes: results of a 5-yr follow-up study. Diabetes Care. 1988;11:605-612.
9. Van der Does FE, De Neeling JN, Snoek FJ, et al. Symptoms and well-being in relation to glycemic control in type II diabetes. Diabetes Care. 1996;19:204-210.
10. Genuth S. A case for blood glucose control. Adv Intern Med. 1995;40:573-623.
11. Wrigley M, Mayou R. Psychological factors and admission for poor glycaemic control: a study of psychological and social factors in poorly controlled insulin dependent diabetic patients. J Psychosom Res. 1991;35:335-343.
12. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
13. Magee MF, Bhatt BA. Management of decompensated diabetes. Diabetic ketoacidosis and hyperglycemic hyperosmolar syndrome. Crit Care Clin. 2001;17(1):75-106.
14. Li C, Ford ES, Zhao G, et al. Prevalence and correlates of undiagnosed depression among U.S. adults with diabetes: the Behavioral Risk Factor Surveillance System, 2006. Diabetes Res Clin Pract. 2009;83(2):268-279.
15. Calhoun D, Beals J, Carter EA, et al. Relationship between glycemic control and depression among American Indians in the Strong Heart Study. J Diabetes Complications. 2010;24:217-222.
1. Srivastava A, Taly AB, Gupta A, et al. Post-stroke depression: prevalence and relationship with disability in chronic stroke survivors. Ann Indian Acad Neurol. 2010;13(2):123-127.
2. Marcus MD, Wing RR, Guare J, et al. Lifetime prevalence of major depression and its effect on treatment outcome in obese type II diabetic patients. Diabetes Care. 1992;15(2):253-255.
3. Storor DL, Byrne GJ. Pre-morbid personality and depression following stroke. Int Psychogeriatr. 2006;18(3):457-469.
4. Songar A, Kocabasoglu N, Balcioglu I, et al. The relationship between diabetics’ metabolic control levels and psychiatric symptomatology. Integrative Psychiatry. 1993;9:34-40.
5. Von Dras DD, Lichty W. Correlates of depression in diabetic adults. Behav Health Aging. 1990;1:79-84.
6. Lustman PJ, Clouse RE. Depression in diabetic patients: the relationship between mood and glycemic control. J Diabetes Complications. 2005;19(2):113-122.
7. Lustman PJ, Anderson RJ, Freedland KE, et al. Depression and poor glycemic control: a meta-analytic review of the literature. Diabetes Care. 2000;23(7):934-942.
8. Lustman PJ, Griffith LS, Clouse RE. Depression in adults with diabetes: results of a 5-yr follow-up study. Diabetes Care. 1988;11:605-612.
9. Van der Does FE, De Neeling JN, Snoek FJ, et al. Symptoms and well-being in relation to glycemic control in type II diabetes. Diabetes Care. 1996;19:204-210.
10. Genuth S. A case for blood glucose control. Adv Intern Med. 1995;40:573-623.
11. Wrigley M, Mayou R. Psychological factors and admission for poor glycaemic control: a study of psychological and social factors in poorly controlled insulin dependent diabetic patients. J Psychosom Res. 1991;35:335-343.
12. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
13. Magee MF, Bhatt BA. Management of decompensated diabetes. Diabetic ketoacidosis and hyperglycemic hyperosmolar syndrome. Crit Care Clin. 2001;17(1):75-106.
14. Li C, Ford ES, Zhao G, et al. Prevalence and correlates of undiagnosed depression among U.S. adults with diabetes: the Behavioral Risk Factor Surveillance System, 2006. Diabetes Res Clin Pract. 2009;83(2):268-279.
15. Calhoun D, Beals J, Carter EA, et al. Relationship between glycemic control and depression among American Indians in the Strong Heart Study. J Diabetes Complications. 2010;24:217-222.
A mysterious case of mania
CASE: First-episode mania
Mrs. P, age 47, is brought to the emergency department (ED) because her family is concerned about her behavioral changes over the last week. Her husband reports that Mrs. P has become hyper-religious and talkative. She has been perseverating on numbers and dates and incessantly calling people. Mrs. P reports increased energy and decreased need for sleep. On examination, she has pressured speech. She has no psychiatric history; however, for the past year, she has been taking sertraline, 100 mg/d, and desipramine, 25 mg/d, which her primary care physician prescribed for unknown reasons.
Mrs. P has struggled with chronic back pain for years, but an MRI of her spine is negative. Her family strongly believes that for the past 3 years Mrs. P has been receiving too many medications from her pain management specialist. Six weeks before her current presentation, she was receiving methadone, 40 mg/d, hydrocodone, at least 20 mg/d, and tramadol, 400 mg/d in divided doses. She also was taking an unknown dose of at least 1 benzodiazepine.
Mrs. P’s husband notes she stopped taking methadone abruptly approximately 5 weeks ago. However, about 3 weeks ago, Mrs. P accidentally overdosed on opioids and was hospitalized for several days. Urine drug screen at the time was positive for acetaminophen, salicylate, propoxyphene, opiate, benzodiazepine, and tricyclic antidepressant.
Mrs. P’s medical history includes auditory nerve loss from birth; her mother had German measles (rubella). Mrs. P never learned American Sign Language. She underwent cochlear implant surgery 1 year ago and now has only mild difficulties speaking.
The authors’ observations
Manic symptoms are common in patients with comorbid medical disorders and present a diagnostic challenge. Obtaining an accurate history from the patient may be difficult. Such evaluations often require extensive investigation and collection of data from multiple sources, including:
- medical records
- family members
- patient observation.
Mrs. P’s history is marked by contradicting data from these sources. For example, her family says she stopped taking “pain medications” 5 weeks ago, but 2 weeks later her urine drug screen showed opioids.
Both illicit drugs and prescribed medications can precipitate manic symptoms. From medical records and drug testing, it was evident that Mrs. P had a history of medication abuse/overdose/misuse.
Mania also has been associated with substance withdrawal. Mrs. P allegedly stopped taking methadone 4 weeks before the onset of manic symptoms. Methadone is a synthetic opioid with a pharmacokinetic and pharmacodynamic profile that presents clinical challenges, including:
- large interindividual variability in methadone pharmacokinetics
- lack of reliable equianalgesic conversion ratio to and from other opioids
- potential for multiple drug interactions and complex pharmacodynamics.
An opioid’s half-life determines the onset and duration of withdrawal syndrome symptoms.1 Methadone metabolism is predominantly mediated by CYP3A4, CYP2B6, CYP2D6, and to some extent by CYP2C19.1 We performed genetic testing to help evaluate how Mrs. P metabolized medications. Mrs. P had a normal genotype for CYP2D6, which meant that she should process opioids at a normal rate; however, she was heterozygous for CYP2C19*2 polymorphism, so it is possible that methadone stayed in her system longer than average.
Evidence documenting methadone drug interactions is limited (Table 1).1 Mrs. P was taking sertraline and desipramine; both have potent effects via 2D6 inhibition that could increase plasma methadone concentration. Other evidence indicates that benzodiazepines and methadone may have synergistic interactions that could increase opioid sedation or respiratory depression.1
Table 1
Is a drug interaction with methadone causing Mrs. P’s mania?
| Medication class/agent | Effect on methadone level | Effect on methadone metabolism | Additional effects of interaction |
|---|---|---|---|
| Selective serotonin reuptake inhibitors | |||
| Fluvoxamine | Increase | Inhibition | Opioid toxicity |
| Fluoxetine | Increase | Inhibition | Torsades de pointes |
| Paroxetine | Increase | Inhibition | Decreased hepatic metabolism |
| Sertraline* | Increase | Autoinduction | Torsades de pointes |
| Citalopram | — | — | Torsades de pointes |
| Tricyclic antidepressants | |||
| Desipramine* | — | Inhibition | Increased desipramine levels/inhibition of desipramine metabolism |
| Amitriptyline | Increase methadone clearance | — | Torsades de pointes/prolonged QT interval |
| Anti-inflammatory drugs | |||
| NSAIDs* | — | — | Enhanced analgesia/opioid-sparing effect |
| Aspirin* | — | — | Paradoxical activation of platelet receptors |
| Benzodiazepines | |||
| Alprazolam | — | — | CNS depression/sedation/overdose |
| Diazepam* | — | Inhibition | Additive depressant effects |
| Opioid agonists | |||
| Dextromethorphan | — | Inhibition (not significant) | Increased side effects, especially sleepiness and drowsiness |
| Tramadol* | — | — | Well tolerated |
| Nicotine | Decrease | Can increase smoking rate | |
| *Medications taken by Mrs. P | |||
| NSAIDs: nonsteroidal anti-inflammatory drugs | |||
| Source: Reference 1 | |||
EVALUATION: Few clues
In our ED, Mrs. P’s urine drug abuse screen is positive for salicylate and benzodiazepine only. Findings from physical examination, vital signs, ECG, and chest radiography are within normal limits. Internal medicine consultation is unremarkable. Mrs. P’s laboratory investigation is notable for an elevated white blood cell count, but this normalizes over a week.
Mrs. P shows no evidence of infection and is normoglycemic. B12 and folate are within normal limits. Serum electrolytes, liver function testing, sensitive thyroid stimulating hormone, and C-reactive protein are within normal limits. Urinalysis is negative except for a small amount of hemoglobin. Her creatine kinase (CK) is in the upper normal range. Human immunodeficiency virus (HIV) and syphilis testing is negative. Ceruloplasmin level also is normal. Heavy metal screen is negative. Head MRI and CT from previous hospitalizations were unremarkable.
The authors’ observations
Our first step was to clarify Mrs. P’s diagnosis. In reviewing differential diagnoses, we considered:
- serotonin syndrome
- benzodiazepine withdrawal syndrome
- antidepressant-induced mania
- adrenergic toxicity
- malignant hyperthermia
- heat stroke
- infectious causes.
Our index of suspicion for serotonin syndrome was low because Mrs. P didn’t meet criteria required for diagnosis. Relevant signs and symptoms included confusion, elevated mood (major) and agitation, nervousness, insomnia, and low blood pressure (minor).
Based on concerns about medication interactions, we discontinued sertraline and desipramine. According to the patient’s sister, Mrs. P’s manic symptoms markedly responded to PRN doses of lorazepam. We prescribed lorazepam, 1 mg every 6 hours, and observed Mrs. P for signs and symptoms of benzodiazepine withdrawal.
HISTORY: OTC drug use
According to Mrs. P’s mother, after her daughter abruptly discontinued methadone, she began to have very strong headaches, which she treated with Excedrin or Excedrin Sinus. The mother said that 4 days before Mrs. P came to the ED, she found her daughter holding 4 tablets of Excedrin and an empty bottle. Unfortunately her mother was unable to say what type of Excedrin it was. When the treatment team asks Mrs. P how many pills she usually takes, she says she doesn’t know but usually until the pain stops.
The authors’ observations
Management of secondary mania should focus on treating the underlying condition (Algorithm). Neurology categorizes mania into 3 categories:2
- confusional-delirious states
- manic symptoms associated with focal or multifocal cerebral lesions
- affective disorders (manic-depressive and depressive psychoses).
Medical workup ruled out common secondary causes of psychosis. Collaborative information from relatives revealed no family history of mental illness.
Patients with hearing loss and deafness have been shown to be at increased risk for psychotic disorders compared with the general population. Severe sensory deficits early in Mrs. P’s life may have influenced the orderly development of neural connections in her sensory cortex and association areas.3 Mrs. P was deaf for the first 45 years of life. It could be hypothesized that her sensory deficits significantly influenced her ability to reality test. After receiving a cochlear implant, Mrs. P rapidly went from no auditory stimulation to marked improvement. This stressor might precipitate psychotic symptoms. However, her presentation seemed to be characterized more by manic symptoms or an agitated delirium. It also did not fit temporally with her presentation.
We begin to suspect that Mrs. P’s mania is substance-induced. Excedrin, an over-the-counter medication, contains aspirin and caffeine. Excedrin Sinus also contains phenylephrine. Amphetamines, caffeine, ephedrine, pseudoephedrine, and phenylpropanolamine have all been linked to manic-like psychotic episodes.
Concerns about the illicit conversion of pseudoephedrine into methamphetamine obliged pharmaceutical companies in the United States to switch product formulations to phenylephrine in 2005,4 although some “behind-the-counter” medications may contain pseudoephedrine. Phenylephrine is a relatively selective α1 agonist with weak α2 adrenoceptor agonist activity and low β agonist activity. It is very similar to pseudo-ephedrine, which is known to be implicated in the development of manic symptoms.5,6
Pseudoephedrine can raise CK levels and cause rhabdomyolysis.7,8 Mrs. P’s CK level was 176 (normal range 36 to 176 U/L) 4 days after her initial presentation, and she had a moderate amount of myoglobin in her urine. Her creatinine was normal. The patient was taking excessive amounts of caffeine and—if she was using Excedrin Sinus—pseudoephedrine or phenylephrine. We were unable to determine whether her Excedrin contained pseudoephedrine or phenylephrine. In addition, she was going through opioid withdrawal and reported problems with her sleep. There was also a question of Mrs. P’s unknown methadone use combined with its decreased clearance secondary to medication interactions.
While previously hospitalized for overdose, Mrs. P tested positive for propoxyphene. Excessive use of propoxyphene also can cause numerous adverse reactions. Some of that could have explained why Mrs. P’s presentation includes nervousness, CNS stimulation, excitement, insomnia, and restlessness.5
Based on multiple factors, we believe Mrs. P meets DSM-IV-TR criteria for substance-induced mood disorder (Table 2).9 This diagnosis is supported by Mrs. P’s history of complex polypharmacy, excessive caffeine use, sleep deprivation, and possible opioid withdrawal.
Algorithm: Managing substance-induced manic disorder
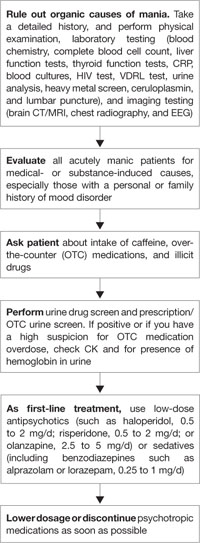
CK: creatine kinase; CRP: C-reactive protein; CT: computed tomography; EEG: electroencephalogram; HIV: human immunodeficiency virus; MRI: magnetic resonance imaging; VDRL: venereal disease research laboratoryTable 2
DSM-IV-TR criteria for substance-induced mood disorder*
| A. A prominent and persistent disturbance in mood predominates in the clinical picture and is characterized by either (or both) of the following: 1. depressed mood or markedly diminished interest or pleasure in all, or almost all, activities 2. elevated, expansive, or irritable mood |
| B. There is evidence from the history, physical examination, or laboratory findings of: 1. the symptoms in Criterion A developed during, or within 1 month of, substance intoxication or withdrawal, or 2. medication use is etiologically related to the disturbance |
| C. The disturbance is not better accounted for by a mood disorder that is not substance-induced |
| D. The disturbance does not occur exclusively during the course of a delirium |
| E. The symptoms cause clinically significant distress or impairment in social, occupational, or other important areas of functioning |
| Minimal criteria are A plus B plus E |
| *Make this diagnosis only when mood symptoms are in excess of those usually associated with substance intoxication or substance withdrawal syndrome and when symptoms are sufficiently severe to warrant independent clinical attention |
| Source: Reference 9 |
TREATMENT: Escalating symptoms
While hospitalized, Mrs. P focuses solely on receiving pain medication. She does not know why she is in the hospital. She is easily distractible, intermittently intrusive, and disorganized and tangential in her thought process.
Two days after admission, her uncontrolled behavior escalates and she has marked psychomotor agitation. She is confused but remains oriented to time, place, and person. We start treatment with risperidone, 0.5 mg each morning and 1 mg at bedtime, because this agent is well tolerated, efficacious, and easily titrated to symptom response. Mrs. P’s symptoms improve, but she does not return to her reported baseline. Two days later, we increase risperidone to 1 mg every morning and 2 mg at bedtime. On the 6th day of hospitalization, Mrs. P is more organized and able to follow simple commands. She denies auditory or visual hallucinations. On the 10th day, she improves markedly and is back to her baseline level of functioning.
We perform psychological testing, including the Wechsler Adult Intelligence Scale (WAIS III) and the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS, Form A). The results show global neurocognitive deficits. Mrs. P’s intellectual skill is significantly below average, with verbal abilities reflecting functioning in the mildly retarded range. Nonverbal skills were stronger but still below average. Mrs. P’s capacity to learn and retain new information and to understand even modestly complex concepts is quite limited.
Because of Mrs. P’s long history of poly-substance abuse, inability to process information, and chronic back pain, we judge her to be at high risk for relapse. However, Mrs. P and her family are not interested in chemical dependence treatment.
This left us facing a difficult clinical situation. Mrs. P had a pattern of presenting to multiple physicians and eventually receiving narcotics. Her family provided transportation for her to these appointments but also was concerned about her drug use. With the patient and her family, we carefully outline Mrs. P’s treatment needs, including:
- medication monitoring by a psychiatrist after discharge
- a single, consistent primary care physician to manage her care
- a treatment plan shared by all clinicians involved in her care.
We review with Mrs. P and her family the benefits of behavioral approaches to chronic pain management. They agree to our recommendation that the family control Mrs. P’s medication supply. We discharge her on risperidone, 0.5 mg each morning and 1 mg at bedtime, and she is scheduled for follow-up with a local psychiatrist.
Related resource
- Krauthammer C, Klerman GL. Manic syndromes associated with antecedent physical illness or drugs. Arch Gen Psychiatry. 1978;35(11):1333-1339.
Drug brand names
- Alprazolam • Xanax
- Amitriptyline • Elavil
- Citalopram • Celexa
- Desipramine • Norpramin
- Diazepam • Valium
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Hydrocodone • Vicodin, Lortab, others
- Lorazepam • Ativan
- Methadone • Dolophine, Methadose
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Propoxyphene • Darvon, Darvocet, others
- Risperidone • Risperdal
- Sertraline • Zoloft
- Tramadol • Ultram
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. De Fazio S, Gallelli L, De Siena A, et al. Role of CYP3A5 in abnormal clearance methadone. Ann Pharmacother. 2008;42(6):893-897.
2. Ropper AH, Brown RH. Adams and Victor’s principles of neurology. 8th ed. New York, NY: McGraw-Hill Professional; 2005.
3. Thewissen V, Myin-Germeys I, Bentall R, et al. Hearing impairment and psychosis revisited. Schizophr Res. 2005;76(1):99-103.
4. Eccles R. Substitution of phenylephrine for pseudoephedrine as a nasal decongeststant. An illogical way to control methamphetamine abuse. Br J Clin Pharmacol. 2007;63(1):10-14.
5. Wilson H, Woods D. Pseudoephedrine causing mania-like symptoms. N Z Med J. 2002;115(1148):86.-
6. Dalton R. Mixed bipolar disorder precipitated by pseudoephedrine hydrochloride. South Med J. 1990;83(1):64-65.
7. Mansi IA, Huang J. Rhabdomyolysis in response to weight-loss herbal medicine. Am J Med Sci. 2004;327:356-357.
8. Sandhu RS, Como JJ, Scalea TS. Renal failure and exercise-induced rhabdomyolysis in patients taking performance-enhancing compounds. J Trauma. 2002;53:761-764.
9. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
CASE: First-episode mania
Mrs. P, age 47, is brought to the emergency department (ED) because her family is concerned about her behavioral changes over the last week. Her husband reports that Mrs. P has become hyper-religious and talkative. She has been perseverating on numbers and dates and incessantly calling people. Mrs. P reports increased energy and decreased need for sleep. On examination, she has pressured speech. She has no psychiatric history; however, for the past year, she has been taking sertraline, 100 mg/d, and desipramine, 25 mg/d, which her primary care physician prescribed for unknown reasons.
Mrs. P has struggled with chronic back pain for years, but an MRI of her spine is negative. Her family strongly believes that for the past 3 years Mrs. P has been receiving too many medications from her pain management specialist. Six weeks before her current presentation, she was receiving methadone, 40 mg/d, hydrocodone, at least 20 mg/d, and tramadol, 400 mg/d in divided doses. She also was taking an unknown dose of at least 1 benzodiazepine.
Mrs. P’s husband notes she stopped taking methadone abruptly approximately 5 weeks ago. However, about 3 weeks ago, Mrs. P accidentally overdosed on opioids and was hospitalized for several days. Urine drug screen at the time was positive for acetaminophen, salicylate, propoxyphene, opiate, benzodiazepine, and tricyclic antidepressant.
Mrs. P’s medical history includes auditory nerve loss from birth; her mother had German measles (rubella). Mrs. P never learned American Sign Language. She underwent cochlear implant surgery 1 year ago and now has only mild difficulties speaking.
The authors’ observations
Manic symptoms are common in patients with comorbid medical disorders and present a diagnostic challenge. Obtaining an accurate history from the patient may be difficult. Such evaluations often require extensive investigation and collection of data from multiple sources, including:
- medical records
- family members
- patient observation.
Mrs. P’s history is marked by contradicting data from these sources. For example, her family says she stopped taking “pain medications” 5 weeks ago, but 2 weeks later her urine drug screen showed opioids.
Both illicit drugs and prescribed medications can precipitate manic symptoms. From medical records and drug testing, it was evident that Mrs. P had a history of medication abuse/overdose/misuse.
Mania also has been associated with substance withdrawal. Mrs. P allegedly stopped taking methadone 4 weeks before the onset of manic symptoms. Methadone is a synthetic opioid with a pharmacokinetic and pharmacodynamic profile that presents clinical challenges, including:
- large interindividual variability in methadone pharmacokinetics
- lack of reliable equianalgesic conversion ratio to and from other opioids
- potential for multiple drug interactions and complex pharmacodynamics.
An opioid’s half-life determines the onset and duration of withdrawal syndrome symptoms.1 Methadone metabolism is predominantly mediated by CYP3A4, CYP2B6, CYP2D6, and to some extent by CYP2C19.1 We performed genetic testing to help evaluate how Mrs. P metabolized medications. Mrs. P had a normal genotype for CYP2D6, which meant that she should process opioids at a normal rate; however, she was heterozygous for CYP2C19*2 polymorphism, so it is possible that methadone stayed in her system longer than average.
Evidence documenting methadone drug interactions is limited (Table 1).1 Mrs. P was taking sertraline and desipramine; both have potent effects via 2D6 inhibition that could increase plasma methadone concentration. Other evidence indicates that benzodiazepines and methadone may have synergistic interactions that could increase opioid sedation or respiratory depression.1
Table 1
Is a drug interaction with methadone causing Mrs. P’s mania?
| Medication class/agent | Effect on methadone level | Effect on methadone metabolism | Additional effects of interaction |
|---|---|---|---|
| Selective serotonin reuptake inhibitors | |||
| Fluvoxamine | Increase | Inhibition | Opioid toxicity |
| Fluoxetine | Increase | Inhibition | Torsades de pointes |
| Paroxetine | Increase | Inhibition | Decreased hepatic metabolism |
| Sertraline* | Increase | Autoinduction | Torsades de pointes |
| Citalopram | — | — | Torsades de pointes |
| Tricyclic antidepressants | |||
| Desipramine* | — | Inhibition | Increased desipramine levels/inhibition of desipramine metabolism |
| Amitriptyline | Increase methadone clearance | — | Torsades de pointes/prolonged QT interval |
| Anti-inflammatory drugs | |||
| NSAIDs* | — | — | Enhanced analgesia/opioid-sparing effect |
| Aspirin* | — | — | Paradoxical activation of platelet receptors |
| Benzodiazepines | |||
| Alprazolam | — | — | CNS depression/sedation/overdose |
| Diazepam* | — | Inhibition | Additive depressant effects |
| Opioid agonists | |||
| Dextromethorphan | — | Inhibition (not significant) | Increased side effects, especially sleepiness and drowsiness |
| Tramadol* | — | — | Well tolerated |
| Nicotine | Decrease | Can increase smoking rate | |
| *Medications taken by Mrs. P | |||
| NSAIDs: nonsteroidal anti-inflammatory drugs | |||
| Source: Reference 1 | |||
EVALUATION: Few clues
In our ED, Mrs. P’s urine drug abuse screen is positive for salicylate and benzodiazepine only. Findings from physical examination, vital signs, ECG, and chest radiography are within normal limits. Internal medicine consultation is unremarkable. Mrs. P’s laboratory investigation is notable for an elevated white blood cell count, but this normalizes over a week.
Mrs. P shows no evidence of infection and is normoglycemic. B12 and folate are within normal limits. Serum electrolytes, liver function testing, sensitive thyroid stimulating hormone, and C-reactive protein are within normal limits. Urinalysis is negative except for a small amount of hemoglobin. Her creatine kinase (CK) is in the upper normal range. Human immunodeficiency virus (HIV) and syphilis testing is negative. Ceruloplasmin level also is normal. Heavy metal screen is negative. Head MRI and CT from previous hospitalizations were unremarkable.
The authors’ observations
Our first step was to clarify Mrs. P’s diagnosis. In reviewing differential diagnoses, we considered:
- serotonin syndrome
- benzodiazepine withdrawal syndrome
- antidepressant-induced mania
- adrenergic toxicity
- malignant hyperthermia
- heat stroke
- infectious causes.
Our index of suspicion for serotonin syndrome was low because Mrs. P didn’t meet criteria required for diagnosis. Relevant signs and symptoms included confusion, elevated mood (major) and agitation, nervousness, insomnia, and low blood pressure (minor).
Based on concerns about medication interactions, we discontinued sertraline and desipramine. According to the patient’s sister, Mrs. P’s manic symptoms markedly responded to PRN doses of lorazepam. We prescribed lorazepam, 1 mg every 6 hours, and observed Mrs. P for signs and symptoms of benzodiazepine withdrawal.
HISTORY: OTC drug use
According to Mrs. P’s mother, after her daughter abruptly discontinued methadone, she began to have very strong headaches, which she treated with Excedrin or Excedrin Sinus. The mother said that 4 days before Mrs. P came to the ED, she found her daughter holding 4 tablets of Excedrin and an empty bottle. Unfortunately her mother was unable to say what type of Excedrin it was. When the treatment team asks Mrs. P how many pills she usually takes, she says she doesn’t know but usually until the pain stops.
The authors’ observations
Management of secondary mania should focus on treating the underlying condition (Algorithm). Neurology categorizes mania into 3 categories:2
- confusional-delirious states
- manic symptoms associated with focal or multifocal cerebral lesions
- affective disorders (manic-depressive and depressive psychoses).
Medical workup ruled out common secondary causes of psychosis. Collaborative information from relatives revealed no family history of mental illness.
Patients with hearing loss and deafness have been shown to be at increased risk for psychotic disorders compared with the general population. Severe sensory deficits early in Mrs. P’s life may have influenced the orderly development of neural connections in her sensory cortex and association areas.3 Mrs. P was deaf for the first 45 years of life. It could be hypothesized that her sensory deficits significantly influenced her ability to reality test. After receiving a cochlear implant, Mrs. P rapidly went from no auditory stimulation to marked improvement. This stressor might precipitate psychotic symptoms. However, her presentation seemed to be characterized more by manic symptoms or an agitated delirium. It also did not fit temporally with her presentation.
We begin to suspect that Mrs. P’s mania is substance-induced. Excedrin, an over-the-counter medication, contains aspirin and caffeine. Excedrin Sinus also contains phenylephrine. Amphetamines, caffeine, ephedrine, pseudoephedrine, and phenylpropanolamine have all been linked to manic-like psychotic episodes.
Concerns about the illicit conversion of pseudoephedrine into methamphetamine obliged pharmaceutical companies in the United States to switch product formulations to phenylephrine in 2005,4 although some “behind-the-counter” medications may contain pseudoephedrine. Phenylephrine is a relatively selective α1 agonist with weak α2 adrenoceptor agonist activity and low β agonist activity. It is very similar to pseudo-ephedrine, which is known to be implicated in the development of manic symptoms.5,6
Pseudoephedrine can raise CK levels and cause rhabdomyolysis.7,8 Mrs. P’s CK level was 176 (normal range 36 to 176 U/L) 4 days after her initial presentation, and she had a moderate amount of myoglobin in her urine. Her creatinine was normal. The patient was taking excessive amounts of caffeine and—if she was using Excedrin Sinus—pseudoephedrine or phenylephrine. We were unable to determine whether her Excedrin contained pseudoephedrine or phenylephrine. In addition, she was going through opioid withdrawal and reported problems with her sleep. There was also a question of Mrs. P’s unknown methadone use combined with its decreased clearance secondary to medication interactions.
While previously hospitalized for overdose, Mrs. P tested positive for propoxyphene. Excessive use of propoxyphene also can cause numerous adverse reactions. Some of that could have explained why Mrs. P’s presentation includes nervousness, CNS stimulation, excitement, insomnia, and restlessness.5
Based on multiple factors, we believe Mrs. P meets DSM-IV-TR criteria for substance-induced mood disorder (Table 2).9 This diagnosis is supported by Mrs. P’s history of complex polypharmacy, excessive caffeine use, sleep deprivation, and possible opioid withdrawal.
Algorithm: Managing substance-induced manic disorder
CK: creatine kinase; CRP: C-reactive protein; CT: computed tomography; EEG: electroencephalogram; HIV: human immunodeficiency virus; MRI: magnetic resonance imaging; VDRL: venereal disease research laboratoryTable 2
DSM-IV-TR criteria for substance-induced mood disorder*
| A. A prominent and persistent disturbance in mood predominates in the clinical picture and is characterized by either (or both) of the following: 1. depressed mood or markedly diminished interest or pleasure in all, or almost all, activities 2. elevated, expansive, or irritable mood |
| B. There is evidence from the history, physical examination, or laboratory findings of: 1. the symptoms in Criterion A developed during, or within 1 month of, substance intoxication or withdrawal, or 2. medication use is etiologically related to the disturbance |
| C. The disturbance is not better accounted for by a mood disorder that is not substance-induced |
| D. The disturbance does not occur exclusively during the course of a delirium |
| E. The symptoms cause clinically significant distress or impairment in social, occupational, or other important areas of functioning |
| Minimal criteria are A plus B plus E |
| *Make this diagnosis only when mood symptoms are in excess of those usually associated with substance intoxication or substance withdrawal syndrome and when symptoms are sufficiently severe to warrant independent clinical attention |
| Source: Reference 9 |
TREATMENT: Escalating symptoms
While hospitalized, Mrs. P focuses solely on receiving pain medication. She does not know why she is in the hospital. She is easily distractible, intermittently intrusive, and disorganized and tangential in her thought process.
Two days after admission, her uncontrolled behavior escalates and she has marked psychomotor agitation. She is confused but remains oriented to time, place, and person. We start treatment with risperidone, 0.5 mg each morning and 1 mg at bedtime, because this agent is well tolerated, efficacious, and easily titrated to symptom response. Mrs. P’s symptoms improve, but she does not return to her reported baseline. Two days later, we increase risperidone to 1 mg every morning and 2 mg at bedtime. On the 6th day of hospitalization, Mrs. P is more organized and able to follow simple commands. She denies auditory or visual hallucinations. On the 10th day, she improves markedly and is back to her baseline level of functioning.
We perform psychological testing, including the Wechsler Adult Intelligence Scale (WAIS III) and the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS, Form A). The results show global neurocognitive deficits. Mrs. P’s intellectual skill is significantly below average, with verbal abilities reflecting functioning in the mildly retarded range. Nonverbal skills were stronger but still below average. Mrs. P’s capacity to learn and retain new information and to understand even modestly complex concepts is quite limited.
Because of Mrs. P’s long history of poly-substance abuse, inability to process information, and chronic back pain, we judge her to be at high risk for relapse. However, Mrs. P and her family are not interested in chemical dependence treatment.
This left us facing a difficult clinical situation. Mrs. P had a pattern of presenting to multiple physicians and eventually receiving narcotics. Her family provided transportation for her to these appointments but also was concerned about her drug use. With the patient and her family, we carefully outline Mrs. P’s treatment needs, including:
- medication monitoring by a psychiatrist after discharge
- a single, consistent primary care physician to manage her care
- a treatment plan shared by all clinicians involved in her care.
We review with Mrs. P and her family the benefits of behavioral approaches to chronic pain management. They agree to our recommendation that the family control Mrs. P’s medication supply. We discharge her on risperidone, 0.5 mg each morning and 1 mg at bedtime, and she is scheduled for follow-up with a local psychiatrist.
Related resource
- Krauthammer C, Klerman GL. Manic syndromes associated with antecedent physical illness or drugs. Arch Gen Psychiatry. 1978;35(11):1333-1339.
Drug brand names
- Alprazolam • Xanax
- Amitriptyline • Elavil
- Citalopram • Celexa
- Desipramine • Norpramin
- Diazepam • Valium
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Hydrocodone • Vicodin, Lortab, others
- Lorazepam • Ativan
- Methadone • Dolophine, Methadose
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Propoxyphene • Darvon, Darvocet, others
- Risperidone • Risperdal
- Sertraline • Zoloft
- Tramadol • Ultram
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE: First-episode mania
Mrs. P, age 47, is brought to the emergency department (ED) because her family is concerned about her behavioral changes over the last week. Her husband reports that Mrs. P has become hyper-religious and talkative. She has been perseverating on numbers and dates and incessantly calling people. Mrs. P reports increased energy and decreased need for sleep. On examination, she has pressured speech. She has no psychiatric history; however, for the past year, she has been taking sertraline, 100 mg/d, and desipramine, 25 mg/d, which her primary care physician prescribed for unknown reasons.
Mrs. P has struggled with chronic back pain for years, but an MRI of her spine is negative. Her family strongly believes that for the past 3 years Mrs. P has been receiving too many medications from her pain management specialist. Six weeks before her current presentation, she was receiving methadone, 40 mg/d, hydrocodone, at least 20 mg/d, and tramadol, 400 mg/d in divided doses. She also was taking an unknown dose of at least 1 benzodiazepine.
Mrs. P’s husband notes she stopped taking methadone abruptly approximately 5 weeks ago. However, about 3 weeks ago, Mrs. P accidentally overdosed on opioids and was hospitalized for several days. Urine drug screen at the time was positive for acetaminophen, salicylate, propoxyphene, opiate, benzodiazepine, and tricyclic antidepressant.
Mrs. P’s medical history includes auditory nerve loss from birth; her mother had German measles (rubella). Mrs. P never learned American Sign Language. She underwent cochlear implant surgery 1 year ago and now has only mild difficulties speaking.
The authors’ observations
Manic symptoms are common in patients with comorbid medical disorders and present a diagnostic challenge. Obtaining an accurate history from the patient may be difficult. Such evaluations often require extensive investigation and collection of data from multiple sources, including:
- medical records
- family members
- patient observation.
Mrs. P’s history is marked by contradicting data from these sources. For example, her family says she stopped taking “pain medications” 5 weeks ago, but 2 weeks later her urine drug screen showed opioids.
Both illicit drugs and prescribed medications can precipitate manic symptoms. From medical records and drug testing, it was evident that Mrs. P had a history of medication abuse/overdose/misuse.
Mania also has been associated with substance withdrawal. Mrs. P allegedly stopped taking methadone 4 weeks before the onset of manic symptoms. Methadone is a synthetic opioid with a pharmacokinetic and pharmacodynamic profile that presents clinical challenges, including:
- large interindividual variability in methadone pharmacokinetics
- lack of reliable equianalgesic conversion ratio to and from other opioids
- potential for multiple drug interactions and complex pharmacodynamics.
An opioid’s half-life determines the onset and duration of withdrawal syndrome symptoms.1 Methadone metabolism is predominantly mediated by CYP3A4, CYP2B6, CYP2D6, and to some extent by CYP2C19.1 We performed genetic testing to help evaluate how Mrs. P metabolized medications. Mrs. P had a normal genotype for CYP2D6, which meant that she should process opioids at a normal rate; however, she was heterozygous for CYP2C19*2 polymorphism, so it is possible that methadone stayed in her system longer than average.
Evidence documenting methadone drug interactions is limited (Table 1).1 Mrs. P was taking sertraline and desipramine; both have potent effects via 2D6 inhibition that could increase plasma methadone concentration. Other evidence indicates that benzodiazepines and methadone may have synergistic interactions that could increase opioid sedation or respiratory depression.1
Table 1
Is a drug interaction with methadone causing Mrs. P’s mania?
| Medication class/agent | Effect on methadone level | Effect on methadone metabolism | Additional effects of interaction |
|---|---|---|---|
| Selective serotonin reuptake inhibitors | |||
| Fluvoxamine | Increase | Inhibition | Opioid toxicity |
| Fluoxetine | Increase | Inhibition | Torsades de pointes |
| Paroxetine | Increase | Inhibition | Decreased hepatic metabolism |
| Sertraline* | Increase | Autoinduction | Torsades de pointes |
| Citalopram | — | — | Torsades de pointes |
| Tricyclic antidepressants | |||
| Desipramine* | — | Inhibition | Increased desipramine levels/inhibition of desipramine metabolism |
| Amitriptyline | Increase methadone clearance | — | Torsades de pointes/prolonged QT interval |
| Anti-inflammatory drugs | |||
| NSAIDs* | — | — | Enhanced analgesia/opioid-sparing effect |
| Aspirin* | — | — | Paradoxical activation of platelet receptors |
| Benzodiazepines | |||
| Alprazolam | — | — | CNS depression/sedation/overdose |
| Diazepam* | — | Inhibition | Additive depressant effects |
| Opioid agonists | |||
| Dextromethorphan | — | Inhibition (not significant) | Increased side effects, especially sleepiness and drowsiness |
| Tramadol* | — | — | Well tolerated |
| Nicotine | Decrease | Can increase smoking rate | |
| *Medications taken by Mrs. P | |||
| NSAIDs: nonsteroidal anti-inflammatory drugs | |||
| Source: Reference 1 | |||
EVALUATION: Few clues
In our ED, Mrs. P’s urine drug abuse screen is positive for salicylate and benzodiazepine only. Findings from physical examination, vital signs, ECG, and chest radiography are within normal limits. Internal medicine consultation is unremarkable. Mrs. P’s laboratory investigation is notable for an elevated white blood cell count, but this normalizes over a week.
Mrs. P shows no evidence of infection and is normoglycemic. B12 and folate are within normal limits. Serum electrolytes, liver function testing, sensitive thyroid stimulating hormone, and C-reactive protein are within normal limits. Urinalysis is negative except for a small amount of hemoglobin. Her creatine kinase (CK) is in the upper normal range. Human immunodeficiency virus (HIV) and syphilis testing is negative. Ceruloplasmin level also is normal. Heavy metal screen is negative. Head MRI and CT from previous hospitalizations were unremarkable.
The authors’ observations
Our first step was to clarify Mrs. P’s diagnosis. In reviewing differential diagnoses, we considered:
- serotonin syndrome
- benzodiazepine withdrawal syndrome
- antidepressant-induced mania
- adrenergic toxicity
- malignant hyperthermia
- heat stroke
- infectious causes.
Our index of suspicion for serotonin syndrome was low because Mrs. P didn’t meet criteria required for diagnosis. Relevant signs and symptoms included confusion, elevated mood (major) and agitation, nervousness, insomnia, and low blood pressure (minor).
Based on concerns about medication interactions, we discontinued sertraline and desipramine. According to the patient’s sister, Mrs. P’s manic symptoms markedly responded to PRN doses of lorazepam. We prescribed lorazepam, 1 mg every 6 hours, and observed Mrs. P for signs and symptoms of benzodiazepine withdrawal.
HISTORY: OTC drug use
According to Mrs. P’s mother, after her daughter abruptly discontinued methadone, she began to have very strong headaches, which she treated with Excedrin or Excedrin Sinus. The mother said that 4 days before Mrs. P came to the ED, she found her daughter holding 4 tablets of Excedrin and an empty bottle. Unfortunately her mother was unable to say what type of Excedrin it was. When the treatment team asks Mrs. P how many pills she usually takes, she says she doesn’t know but usually until the pain stops.
The authors’ observations
Management of secondary mania should focus on treating the underlying condition (Algorithm). Neurology categorizes mania into 3 categories:2
- confusional-delirious states
- manic symptoms associated with focal or multifocal cerebral lesions
- affective disorders (manic-depressive and depressive psychoses).
Medical workup ruled out common secondary causes of psychosis. Collaborative information from relatives revealed no family history of mental illness.
Patients with hearing loss and deafness have been shown to be at increased risk for psychotic disorders compared with the general population. Severe sensory deficits early in Mrs. P’s life may have influenced the orderly development of neural connections in her sensory cortex and association areas.3 Mrs. P was deaf for the first 45 years of life. It could be hypothesized that her sensory deficits significantly influenced her ability to reality test. After receiving a cochlear implant, Mrs. P rapidly went from no auditory stimulation to marked improvement. This stressor might precipitate psychotic symptoms. However, her presentation seemed to be characterized more by manic symptoms or an agitated delirium. It also did not fit temporally with her presentation.
We begin to suspect that Mrs. P’s mania is substance-induced. Excedrin, an over-the-counter medication, contains aspirin and caffeine. Excedrin Sinus also contains phenylephrine. Amphetamines, caffeine, ephedrine, pseudoephedrine, and phenylpropanolamine have all been linked to manic-like psychotic episodes.
Concerns about the illicit conversion of pseudoephedrine into methamphetamine obliged pharmaceutical companies in the United States to switch product formulations to phenylephrine in 2005,4 although some “behind-the-counter” medications may contain pseudoephedrine. Phenylephrine is a relatively selective α1 agonist with weak α2 adrenoceptor agonist activity and low β agonist activity. It is very similar to pseudo-ephedrine, which is known to be implicated in the development of manic symptoms.5,6
Pseudoephedrine can raise CK levels and cause rhabdomyolysis.7,8 Mrs. P’s CK level was 176 (normal range 36 to 176 U/L) 4 days after her initial presentation, and she had a moderate amount of myoglobin in her urine. Her creatinine was normal. The patient was taking excessive amounts of caffeine and—if she was using Excedrin Sinus—pseudoephedrine or phenylephrine. We were unable to determine whether her Excedrin contained pseudoephedrine or phenylephrine. In addition, she was going through opioid withdrawal and reported problems with her sleep. There was also a question of Mrs. P’s unknown methadone use combined with its decreased clearance secondary to medication interactions.
While previously hospitalized for overdose, Mrs. P tested positive for propoxyphene. Excessive use of propoxyphene also can cause numerous adverse reactions. Some of that could have explained why Mrs. P’s presentation includes nervousness, CNS stimulation, excitement, insomnia, and restlessness.5
Based on multiple factors, we believe Mrs. P meets DSM-IV-TR criteria for substance-induced mood disorder (Table 2).9 This diagnosis is supported by Mrs. P’s history of complex polypharmacy, excessive caffeine use, sleep deprivation, and possible opioid withdrawal.
Algorithm: Managing substance-induced manic disorder
CK: creatine kinase; CRP: C-reactive protein; CT: computed tomography; EEG: electroencephalogram; HIV: human immunodeficiency virus; MRI: magnetic resonance imaging; VDRL: venereal disease research laboratoryTable 2
DSM-IV-TR criteria for substance-induced mood disorder*
| A. A prominent and persistent disturbance in mood predominates in the clinical picture and is characterized by either (or both) of the following: 1. depressed mood or markedly diminished interest or pleasure in all, or almost all, activities 2. elevated, expansive, or irritable mood |
| B. There is evidence from the history, physical examination, or laboratory findings of: 1. the symptoms in Criterion A developed during, or within 1 month of, substance intoxication or withdrawal, or 2. medication use is etiologically related to the disturbance |
| C. The disturbance is not better accounted for by a mood disorder that is not substance-induced |
| D. The disturbance does not occur exclusively during the course of a delirium |
| E. The symptoms cause clinically significant distress or impairment in social, occupational, or other important areas of functioning |
| Minimal criteria are A plus B plus E |
| *Make this diagnosis only when mood symptoms are in excess of those usually associated with substance intoxication or substance withdrawal syndrome and when symptoms are sufficiently severe to warrant independent clinical attention |
| Source: Reference 9 |
TREATMENT: Escalating symptoms
While hospitalized, Mrs. P focuses solely on receiving pain medication. She does not know why she is in the hospital. She is easily distractible, intermittently intrusive, and disorganized and tangential in her thought process.
Two days after admission, her uncontrolled behavior escalates and she has marked psychomotor agitation. She is confused but remains oriented to time, place, and person. We start treatment with risperidone, 0.5 mg each morning and 1 mg at bedtime, because this agent is well tolerated, efficacious, and easily titrated to symptom response. Mrs. P’s symptoms improve, but she does not return to her reported baseline. Two days later, we increase risperidone to 1 mg every morning and 2 mg at bedtime. On the 6th day of hospitalization, Mrs. P is more organized and able to follow simple commands. She denies auditory or visual hallucinations. On the 10th day, she improves markedly and is back to her baseline level of functioning.
We perform psychological testing, including the Wechsler Adult Intelligence Scale (WAIS III) and the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS, Form A). The results show global neurocognitive deficits. Mrs. P’s intellectual skill is significantly below average, with verbal abilities reflecting functioning in the mildly retarded range. Nonverbal skills were stronger but still below average. Mrs. P’s capacity to learn and retain new information and to understand even modestly complex concepts is quite limited.
Because of Mrs. P’s long history of poly-substance abuse, inability to process information, and chronic back pain, we judge her to be at high risk for relapse. However, Mrs. P and her family are not interested in chemical dependence treatment.
This left us facing a difficult clinical situation. Mrs. P had a pattern of presenting to multiple physicians and eventually receiving narcotics. Her family provided transportation for her to these appointments but also was concerned about her drug use. With the patient and her family, we carefully outline Mrs. P’s treatment needs, including:
- medication monitoring by a psychiatrist after discharge
- a single, consistent primary care physician to manage her care
- a treatment plan shared by all clinicians involved in her care.
We review with Mrs. P and her family the benefits of behavioral approaches to chronic pain management. They agree to our recommendation that the family control Mrs. P’s medication supply. We discharge her on risperidone, 0.5 mg each morning and 1 mg at bedtime, and she is scheduled for follow-up with a local psychiatrist.
Related resource
- Krauthammer C, Klerman GL. Manic syndromes associated with antecedent physical illness or drugs. Arch Gen Psychiatry. 1978;35(11):1333-1339.
Drug brand names
- Alprazolam • Xanax
- Amitriptyline • Elavil
- Citalopram • Celexa
- Desipramine • Norpramin
- Diazepam • Valium
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Hydrocodone • Vicodin, Lortab, others
- Lorazepam • Ativan
- Methadone • Dolophine, Methadose
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Propoxyphene • Darvon, Darvocet, others
- Risperidone • Risperdal
- Sertraline • Zoloft
- Tramadol • Ultram
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. De Fazio S, Gallelli L, De Siena A, et al. Role of CYP3A5 in abnormal clearance methadone. Ann Pharmacother. 2008;42(6):893-897.
2. Ropper AH, Brown RH. Adams and Victor’s principles of neurology. 8th ed. New York, NY: McGraw-Hill Professional; 2005.
3. Thewissen V, Myin-Germeys I, Bentall R, et al. Hearing impairment and psychosis revisited. Schizophr Res. 2005;76(1):99-103.
4. Eccles R. Substitution of phenylephrine for pseudoephedrine as a nasal decongeststant. An illogical way to control methamphetamine abuse. Br J Clin Pharmacol. 2007;63(1):10-14.
5. Wilson H, Woods D. Pseudoephedrine causing mania-like symptoms. N Z Med J. 2002;115(1148):86.-
6. Dalton R. Mixed bipolar disorder precipitated by pseudoephedrine hydrochloride. South Med J. 1990;83(1):64-65.
7. Mansi IA, Huang J. Rhabdomyolysis in response to weight-loss herbal medicine. Am J Med Sci. 2004;327:356-357.
8. Sandhu RS, Como JJ, Scalea TS. Renal failure and exercise-induced rhabdomyolysis in patients taking performance-enhancing compounds. J Trauma. 2002;53:761-764.
9. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
1. De Fazio S, Gallelli L, De Siena A, et al. Role of CYP3A5 in abnormal clearance methadone. Ann Pharmacother. 2008;42(6):893-897.
2. Ropper AH, Brown RH. Adams and Victor’s principles of neurology. 8th ed. New York, NY: McGraw-Hill Professional; 2005.
3. Thewissen V, Myin-Germeys I, Bentall R, et al. Hearing impairment and psychosis revisited. Schizophr Res. 2005;76(1):99-103.
4. Eccles R. Substitution of phenylephrine for pseudoephedrine as a nasal decongeststant. An illogical way to control methamphetamine abuse. Br J Clin Pharmacol. 2007;63(1):10-14.
5. Wilson H, Woods D. Pseudoephedrine causing mania-like symptoms. N Z Med J. 2002;115(1148):86.-
6. Dalton R. Mixed bipolar disorder precipitated by pseudoephedrine hydrochloride. South Med J. 1990;83(1):64-65.
7. Mansi IA, Huang J. Rhabdomyolysis in response to weight-loss herbal medicine. Am J Med Sci. 2004;327:356-357.
8. Sandhu RS, Como JJ, Scalea TS. Renal failure and exercise-induced rhabdomyolysis in patients taking performance-enhancing compounds. J Trauma. 2002;53:761-764.
9. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
A pacemaker patient’s electrical dilemma
CASE: Relapsing depression
Mrs. A, age 41, presents with worsening depression and suicidal ideation with a plan to take an overdose of her medications. She describes herself as “tense, anxious, and worrying all the time.” She reports worsening mood, loss of interest in previously pleasurable activities, lack of energy and drive, and difficulties performing routine household tasks. She also endorses a combination of initial and middle insomnia. According to her husband, the patient has been slow in movement and speech and has not been taking adequate care of herself.
Mrs. A denies auditory or visual hallucinations, thought insertion, thought withdrawal, thought broadcast, ideas of reference, or paranoid ideation. She also denies recent or past symptoms of mania or hypomania.
Mrs. A has a history of alcohol abuse and major depressive disorder. For her first depressive episode 5 years ago, she was treated with paroxetine, 20 to 80 mg/d, with good results. Following a depressive relapse, she was switched to fluoxetine, 80 mg/d, which improved her depressive symptoms. Approximately 2 years later, she experienced another depressive relapse that resulted in hospitalization. During hospitalization and subsequent outpatient visits, she was treated with citalopram, 20 mg/d, ziprasidone, 80 mg bid, and lorazepam, 1 mg tid. Her depressive symptoms were in partial remission for 2 years until her current relapse.
Her medical history includes syncope of unexplained origin, for which she received an implanted cardiac pacemaker 3 years ago. She takes sertraline, 150 mg/d, methylphenidate, 15 mg/d, and trazodone, 200 mg at night. Laboratory testing is unremarkable.
On mental status examination, Mrs. A’s mood is sad and her affect constricted. Her speech is fluent but slow, and she speaks only when spoken to. We note that Mrs. A has thought blocking but no hallucinations or delusions. She is alert and oriented, but her attention and concentration are impaired. Her insight is fair, and judgment is poor.
The authors’ observations
Somatic therapy for severe major depressive disorders has been limited principally to pharmacotherapy. Despite the availability of effective antidepressants and aggressive treatment, for many patients—such as Mrs. A—the course of depression is characterized by relapse, recurrence, and chronicity.1,2
Because Mrs. A has treatment-refractory depression, we decide to treat her with ECT. ECT has few contraindications and typically is well tolerated. It commonly is used to treat depression in patients with cardiac conditions and generally is quite safe in this population.3,4
ECT in patients with cardiac pacemakers in situ theoretically presents an increased risk of complications, however.5 Specific concerns of administering ECT to pacemaker patients include electrical interference from ECT stimulus and pacemaker sensing of:
- myopotentials that originate from succinylcholine-induced fasciculation (muscular twitching of contiguous groups of muscle fibers)
- muscle contractions that result in incomplete muscle paralysis
- dysrhythmias during the seizure.
Skeletal muscle can generate significant electrical potentials that are well within the sensing capabilities of most newer pulse generators. This happens most frequently in some dual-chamber pacemakers that can automatically perform mode switching or adapt their sensing and pacing thresholds to new situations, which might make them more sensitive to interference by ECT.
Similar concerns apply to administering ECT to patients receiving vagus nerve stimulation (VNS) therapy, as both VNS pulse generators and cardiac pacemakers are battery-powered, electrical signal-producing mechanisms housed in a metal case. The safety of concurrent ECT and VNS therapy is unknown (Box).6,7
Although vagus nerve stimulation (VNS) and electroconvulsive therapy (ECT) are not mutually exclusive, the safety of concurrent use of these 2 therapies is uncertain.6 The manufacturer of the VNS device recommends turning off the VNS pulse generator before administering ECT. In at least 1 case report, however, ECT was administered safely without the VNS pulse generator turned off.7
No case reports describe the safety of VNS in patients with an implanted device such as a pacemaker or automatic cardioverter defibrillator. According to the manufacturer, the VNS system may affect the operation of other devices. For VNS patients who require an implantable pacemaker, defibrillator therapy, or other types of stimulators, the VNS manufacturer advises careful programming of each system and implanting the 2 stimulators at least 10 centimeters (4 inches) apart to avoid communication interference.
What the evidence says
In evidence-based medicine, we tend to say: “In God we trust; all the others have to bring their data.” Unfortunately, it is difficult to conduct a trial of patients with multiple medical issues. Based on anecdotal reports, it appears that ECT use in patients with an implanted cardiac device such as a pacemaker or automatic internal cardioverter-defibrillator (AICD) generally is safe.8-12
One case report describes successful administration of ECT in a treatment-refractory depressed patient with an AICD. The AICD was deactivated during ECT and re-activated immediately upon completion of each treatment. The case report’s authors concluded that the presence of an AICD should not be a contraindication to ECT.13
A chart review of 3 patients with ICDs who received concurrent ECT found treatment was generally uneventful.12 One patient developed tachycardia with a rate-dependent left bundle branch block and hypotension in the recovery room, which responded promptly to esmolol. She did not experience similar events after subsequent ECT treatments.
Minimizing risk
In the absence of controlled data about the use of ECT in patients with implanted cardiac devices, crucial therapeutic decisions depend on the physician’s skill and judgment. Risk strategies can minimize complications (Algorithm).12 An internist or cardiologist experienced in pacemaker management should conduct a device interrogation—evaluating thresholds, lead impedance, and battery voltage and reviewing histograms, mode switch episodes, and stored electrograms—before the first ECT session and after the final one.
Most modern implantable pacemakers work in the synchronous (demand), rate-adaptive mode. In a patient in whom non-cardiac electrical signals cause bradycardia or asystole during ECT, the pacemaker can be reprogrammed to be less sensitive by placing a magnet over the pulse generator, which converts the pacemaker to an asynchronous (fixed), non-sensing mode. It is important to keep in mind that magnet application will not “turn off” a pacemaker; although each pacemaker is programmed to respond to a magnet in a specific fashion, the main response is asynchronous pacing.
Careful cardiac monitoring during ECT is essential (Table). The cardiologist or internist should be available during the first few ECT sessions to monitor for potential pacemaker interference or malfunction. This physician should be familiar with the pacemaker model and type of lead system so he or she can deactivate, reactivate, or reprogram the device.
Algorithm
Reducing risk when administering ECT to cardiac pacemaker patients
| Step 1 | |
| Evaluate the patient to ensure medical suitability for ECT and associated anesthesia | |
| Step 2 | ↓ |
| Conduct pacemaker interrogation (evaluating thresholds, lead impedance, and battery voltage and reviewing histograms, mode switch episodes, and stored electrograms) prior to first ECT treatment and after completion of full ECT course | |
| Step 3 | ↓ |
| Perform cardiac monitoring during and immediately after administering ECT | |
| Step 4 | ↓ |
| Have a magnet available to reprogram the pacemaker in the event of pacemaker inhibition or symptomatic bradycardia during ECT | |
| Step 5 | ↓ |
| Check that all monitoring devices are properly grounded, insulate the patient’s stretcher, and ensure that the patient does not touch anyone who is in contact with the ground during presentation of the ECT electrical stimulus | |
| ECT: electroconvulsive therapy | |
| Source: Reference 12 | |
Guidelines for monitoring cardiac pacemaker patients during ECT
| Use multilead ECG monitoring |
| Have equipment available to rapidly obtain central access (if vasoactive medications or transvenous pacing is needed) |
| Assess the plethysmography tracing of the pulse oximeter (a useful surrogate if the patient experiences dysrhythmias) |
| Have ready an external defibrillator |
TREATMENT: Successful ECT
We seek a medical consultation before initiating ECT. An internist performs device interrogation before the first ECT treatment and is present in the ECT treatment suite to ensure proper pacemaker conversion and to monitor for cardiac complications. The internist conducts another device interrogation after the acute series of ECT treatments.
Mrs. A tolerates the ECT sessions without cardiac complications. Her depressive symptoms respond well to 12 ECT sessions. She is more interactive and reports better attention and concentration. Although Mrs. A still has middle and initial insomnia, she denies thoughts of harming herself or anyone else.
Related resources
- Yarlagadda C. Pacemaker failure. www.emedicine.com/med/TOPIC1704.HTM.
- Atracurium • Tracrium
- Citalopram • Celexa
- Esmolol • Brevibloc
- Fluoxetine • Prozac
- Lorazepam • Ativan
- Methylphenidate • Ritalin, Concerta, others
- Nortriptyline • Aventyl, Pamelor, others
- Paroxetine • Paxil
- Sertraline • Zoloft
- Succinylcholine • Anectine
- Trazodone • Desyrel
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Dr. Romanowicz reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Ramaswamy receives research support from Bristol-Myers Squibb, Shire, and Forest Pharmaceuticals and is a consultant to Dainippon Sumitomo Pharma.
1. American Psychiatric Association Committee on ECT. The practice of electroconvulsive therapy: recommendations for treatment, training, and privileging. 2nd ed. Washington, D.C: American Psychiatric Association; 2001.
2. Russell JC, Rasmussen KG, O’Connor MK, et al. Long-term maintenance ECT: a retrospective review of efficacy and cognitive outcome. J ECT. 2003;19(1):4-9.
3. Alexopoulos GS, Shamoian CJ, Lucas J, et al. Medical problems of geriatric psychiatric patients and younger controls during electroconvulsive therapy. J Am Geriatr Soc. 1984;32(9):651-654.
4. Rasmussen KG, Rummans TA, Richardson JR. Electroconvulsive therapy in the medically ill. Psychiatric Clin North Am. 2002;25:177-193.
5. MacPherson RD, Loo CK, Barrett N. Electroconvulsive therapy in patients with cardiac pacemakers. Anaesth Intensive Care. 2006;34(4):470-474.
6. Burke MJ, Husain MM. Concomitant use of vagus nerve stimulation and electroconvulsive therapy for treatment-resistant depression. J ECT. 2006;22(3):218-222.
7. Husain MM, Montgomery JH, Fernandes P, et al. Safety of vagus nerve stimulation with ECT. Am J Psychiatry. 2002;159:1243.-
8. Alexopoulos GS, Frances RJ. ECT and cardiac patients with pacemakers. Am J Psychiatry. 1980;137(9):1111-1112.
9. Stone KR, McPherson CA. Assessment and management of patients with pacemakers and implantable cardioverter defibrillators. Crit Care Med. 2004;32(4 suppl):S155-S165.
10. Maisel WH, Sweeney MO, Stevenson WG, et al. Recalls and safety alerts involving pacemakers and implantable cardioverter-defibrillator generators. JAMA. 2001;286(7):793-799.
11. Gibson TC, Leaman DM, Devors J, et al. Pacemaker function in relation to electroconvulsive therapy. Chest. 1973;63(6):1025-1027.
12. Dolenc TJ, Barnes RD, Hayes DL, et al. Electroconvulsive therapy in patients with cardiac pacemakers and implantable cardioverter defibrillators. Pacing Clin Electrophysiol. 2004;27(9):1257-1263.
13. Lapid MI, Rummans TA, Hofmann VE, et al. ECT and automatic internal cardioverter-defibrillator. J ECT. 2001;17(2):146-148.
CASE: Relapsing depression
Mrs. A, age 41, presents with worsening depression and suicidal ideation with a plan to take an overdose of her medications. She describes herself as “tense, anxious, and worrying all the time.” She reports worsening mood, loss of interest in previously pleasurable activities, lack of energy and drive, and difficulties performing routine household tasks. She also endorses a combination of initial and middle insomnia. According to her husband, the patient has been slow in movement and speech and has not been taking adequate care of herself.
Mrs. A denies auditory or visual hallucinations, thought insertion, thought withdrawal, thought broadcast, ideas of reference, or paranoid ideation. She also denies recent or past symptoms of mania or hypomania.
Mrs. A has a history of alcohol abuse and major depressive disorder. For her first depressive episode 5 years ago, she was treated with paroxetine, 20 to 80 mg/d, with good results. Following a depressive relapse, she was switched to fluoxetine, 80 mg/d, which improved her depressive symptoms. Approximately 2 years later, she experienced another depressive relapse that resulted in hospitalization. During hospitalization and subsequent outpatient visits, she was treated with citalopram, 20 mg/d, ziprasidone, 80 mg bid, and lorazepam, 1 mg tid. Her depressive symptoms were in partial remission for 2 years until her current relapse.
Her medical history includes syncope of unexplained origin, for which she received an implanted cardiac pacemaker 3 years ago. She takes sertraline, 150 mg/d, methylphenidate, 15 mg/d, and trazodone, 200 mg at night. Laboratory testing is unremarkable.
On mental status examination, Mrs. A’s mood is sad and her affect constricted. Her speech is fluent but slow, and she speaks only when spoken to. We note that Mrs. A has thought blocking but no hallucinations or delusions. She is alert and oriented, but her attention and concentration are impaired. Her insight is fair, and judgment is poor.
The authors’ observations
Somatic therapy for severe major depressive disorders has been limited principally to pharmacotherapy. Despite the availability of effective antidepressants and aggressive treatment, for many patients—such as Mrs. A—the course of depression is characterized by relapse, recurrence, and chronicity.1,2
Because Mrs. A has treatment-refractory depression, we decide to treat her with ECT. ECT has few contraindications and typically is well tolerated. It commonly is used to treat depression in patients with cardiac conditions and generally is quite safe in this population.3,4
ECT in patients with cardiac pacemakers in situ theoretically presents an increased risk of complications, however.5 Specific concerns of administering ECT to pacemaker patients include electrical interference from ECT stimulus and pacemaker sensing of:
- myopotentials that originate from succinylcholine-induced fasciculation (muscular twitching of contiguous groups of muscle fibers)
- muscle contractions that result in incomplete muscle paralysis
- dysrhythmias during the seizure.
Skeletal muscle can generate significant electrical potentials that are well within the sensing capabilities of most newer pulse generators. This happens most frequently in some dual-chamber pacemakers that can automatically perform mode switching or adapt their sensing and pacing thresholds to new situations, which might make them more sensitive to interference by ECT.
Similar concerns apply to administering ECT to patients receiving vagus nerve stimulation (VNS) therapy, as both VNS pulse generators and cardiac pacemakers are battery-powered, electrical signal-producing mechanisms housed in a metal case. The safety of concurrent ECT and VNS therapy is unknown (Box).6,7
Although vagus nerve stimulation (VNS) and electroconvulsive therapy (ECT) are not mutually exclusive, the safety of concurrent use of these 2 therapies is uncertain.6 The manufacturer of the VNS device recommends turning off the VNS pulse generator before administering ECT. In at least 1 case report, however, ECT was administered safely without the VNS pulse generator turned off.7
No case reports describe the safety of VNS in patients with an implanted device such as a pacemaker or automatic cardioverter defibrillator. According to the manufacturer, the VNS system may affect the operation of other devices. For VNS patients who require an implantable pacemaker, defibrillator therapy, or other types of stimulators, the VNS manufacturer advises careful programming of each system and implanting the 2 stimulators at least 10 centimeters (4 inches) apart to avoid communication interference.
What the evidence says
In evidence-based medicine, we tend to say: “In God we trust; all the others have to bring their data.” Unfortunately, it is difficult to conduct a trial of patients with multiple medical issues. Based on anecdotal reports, it appears that ECT use in patients with an implanted cardiac device such as a pacemaker or automatic internal cardioverter-defibrillator (AICD) generally is safe.8-12
One case report describes successful administration of ECT in a treatment-refractory depressed patient with an AICD. The AICD was deactivated during ECT and re-activated immediately upon completion of each treatment. The case report’s authors concluded that the presence of an AICD should not be a contraindication to ECT.13
A chart review of 3 patients with ICDs who received concurrent ECT found treatment was generally uneventful.12 One patient developed tachycardia with a rate-dependent left bundle branch block and hypotension in the recovery room, which responded promptly to esmolol. She did not experience similar events after subsequent ECT treatments.
Minimizing risk
In the absence of controlled data about the use of ECT in patients with implanted cardiac devices, crucial therapeutic decisions depend on the physician’s skill and judgment. Risk strategies can minimize complications (Algorithm).12 An internist or cardiologist experienced in pacemaker management should conduct a device interrogation—evaluating thresholds, lead impedance, and battery voltage and reviewing histograms, mode switch episodes, and stored electrograms—before the first ECT session and after the final one.
Most modern implantable pacemakers work in the synchronous (demand), rate-adaptive mode. In a patient in whom non-cardiac electrical signals cause bradycardia or asystole during ECT, the pacemaker can be reprogrammed to be less sensitive by placing a magnet over the pulse generator, which converts the pacemaker to an asynchronous (fixed), non-sensing mode. It is important to keep in mind that magnet application will not “turn off” a pacemaker; although each pacemaker is programmed to respond to a magnet in a specific fashion, the main response is asynchronous pacing.
Careful cardiac monitoring during ECT is essential (Table). The cardiologist or internist should be available during the first few ECT sessions to monitor for potential pacemaker interference or malfunction. This physician should be familiar with the pacemaker model and type of lead system so he or she can deactivate, reactivate, or reprogram the device.
Algorithm
Reducing risk when administering ECT to cardiac pacemaker patients
| Step 1 | |
| Evaluate the patient to ensure medical suitability for ECT and associated anesthesia | |
| Step 2 | ↓ |
| Conduct pacemaker interrogation (evaluating thresholds, lead impedance, and battery voltage and reviewing histograms, mode switch episodes, and stored electrograms) prior to first ECT treatment and after completion of full ECT course | |
| Step 3 | ↓ |
| Perform cardiac monitoring during and immediately after administering ECT | |
| Step 4 | ↓ |
| Have a magnet available to reprogram the pacemaker in the event of pacemaker inhibition or symptomatic bradycardia during ECT | |
| Step 5 | ↓ |
| Check that all monitoring devices are properly grounded, insulate the patient’s stretcher, and ensure that the patient does not touch anyone who is in contact with the ground during presentation of the ECT electrical stimulus | |
| ECT: electroconvulsive therapy | |
| Source: Reference 12 | |
Guidelines for monitoring cardiac pacemaker patients during ECT
| Use multilead ECG monitoring |
| Have equipment available to rapidly obtain central access (if vasoactive medications or transvenous pacing is needed) |
| Assess the plethysmography tracing of the pulse oximeter (a useful surrogate if the patient experiences dysrhythmias) |
| Have ready an external defibrillator |
TREATMENT: Successful ECT
We seek a medical consultation before initiating ECT. An internist performs device interrogation before the first ECT treatment and is present in the ECT treatment suite to ensure proper pacemaker conversion and to monitor for cardiac complications. The internist conducts another device interrogation after the acute series of ECT treatments.
Mrs. A tolerates the ECT sessions without cardiac complications. Her depressive symptoms respond well to 12 ECT sessions. She is more interactive and reports better attention and concentration. Although Mrs. A still has middle and initial insomnia, she denies thoughts of harming herself or anyone else.
Related resources
- Yarlagadda C. Pacemaker failure. www.emedicine.com/med/TOPIC1704.HTM.
- Atracurium • Tracrium
- Citalopram • Celexa
- Esmolol • Brevibloc
- Fluoxetine • Prozac
- Lorazepam • Ativan
- Methylphenidate • Ritalin, Concerta, others
- Nortriptyline • Aventyl, Pamelor, others
- Paroxetine • Paxil
- Sertraline • Zoloft
- Succinylcholine • Anectine
- Trazodone • Desyrel
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Dr. Romanowicz reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Ramaswamy receives research support from Bristol-Myers Squibb, Shire, and Forest Pharmaceuticals and is a consultant to Dainippon Sumitomo Pharma.
CASE: Relapsing depression
Mrs. A, age 41, presents with worsening depression and suicidal ideation with a plan to take an overdose of her medications. She describes herself as “tense, anxious, and worrying all the time.” She reports worsening mood, loss of interest in previously pleasurable activities, lack of energy and drive, and difficulties performing routine household tasks. She also endorses a combination of initial and middle insomnia. According to her husband, the patient has been slow in movement and speech and has not been taking adequate care of herself.
Mrs. A denies auditory or visual hallucinations, thought insertion, thought withdrawal, thought broadcast, ideas of reference, or paranoid ideation. She also denies recent or past symptoms of mania or hypomania.
Mrs. A has a history of alcohol abuse and major depressive disorder. For her first depressive episode 5 years ago, she was treated with paroxetine, 20 to 80 mg/d, with good results. Following a depressive relapse, she was switched to fluoxetine, 80 mg/d, which improved her depressive symptoms. Approximately 2 years later, she experienced another depressive relapse that resulted in hospitalization. During hospitalization and subsequent outpatient visits, she was treated with citalopram, 20 mg/d, ziprasidone, 80 mg bid, and lorazepam, 1 mg tid. Her depressive symptoms were in partial remission for 2 years until her current relapse.
Her medical history includes syncope of unexplained origin, for which she received an implanted cardiac pacemaker 3 years ago. She takes sertraline, 150 mg/d, methylphenidate, 15 mg/d, and trazodone, 200 mg at night. Laboratory testing is unremarkable.
On mental status examination, Mrs. A’s mood is sad and her affect constricted. Her speech is fluent but slow, and she speaks only when spoken to. We note that Mrs. A has thought blocking but no hallucinations or delusions. She is alert and oriented, but her attention and concentration are impaired. Her insight is fair, and judgment is poor.
The authors’ observations
Somatic therapy for severe major depressive disorders has been limited principally to pharmacotherapy. Despite the availability of effective antidepressants and aggressive treatment, for many patients—such as Mrs. A—the course of depression is characterized by relapse, recurrence, and chronicity.1,2
Because Mrs. A has treatment-refractory depression, we decide to treat her with ECT. ECT has few contraindications and typically is well tolerated. It commonly is used to treat depression in patients with cardiac conditions and generally is quite safe in this population.3,4
ECT in patients with cardiac pacemakers in situ theoretically presents an increased risk of complications, however.5 Specific concerns of administering ECT to pacemaker patients include electrical interference from ECT stimulus and pacemaker sensing of:
- myopotentials that originate from succinylcholine-induced fasciculation (muscular twitching of contiguous groups of muscle fibers)
- muscle contractions that result in incomplete muscle paralysis
- dysrhythmias during the seizure.
Skeletal muscle can generate significant electrical potentials that are well within the sensing capabilities of most newer pulse generators. This happens most frequently in some dual-chamber pacemakers that can automatically perform mode switching or adapt their sensing and pacing thresholds to new situations, which might make them more sensitive to interference by ECT.
Similar concerns apply to administering ECT to patients receiving vagus nerve stimulation (VNS) therapy, as both VNS pulse generators and cardiac pacemakers are battery-powered, electrical signal-producing mechanisms housed in a metal case. The safety of concurrent ECT and VNS therapy is unknown (Box).6,7
Although vagus nerve stimulation (VNS) and electroconvulsive therapy (ECT) are not mutually exclusive, the safety of concurrent use of these 2 therapies is uncertain.6 The manufacturer of the VNS device recommends turning off the VNS pulse generator before administering ECT. In at least 1 case report, however, ECT was administered safely without the VNS pulse generator turned off.7
No case reports describe the safety of VNS in patients with an implanted device such as a pacemaker or automatic cardioverter defibrillator. According to the manufacturer, the VNS system may affect the operation of other devices. For VNS patients who require an implantable pacemaker, defibrillator therapy, or other types of stimulators, the VNS manufacturer advises careful programming of each system and implanting the 2 stimulators at least 10 centimeters (4 inches) apart to avoid communication interference.
What the evidence says
In evidence-based medicine, we tend to say: “In God we trust; all the others have to bring their data.” Unfortunately, it is difficult to conduct a trial of patients with multiple medical issues. Based on anecdotal reports, it appears that ECT use in patients with an implanted cardiac device such as a pacemaker or automatic internal cardioverter-defibrillator (AICD) generally is safe.8-12
One case report describes successful administration of ECT in a treatment-refractory depressed patient with an AICD. The AICD was deactivated during ECT and re-activated immediately upon completion of each treatment. The case report’s authors concluded that the presence of an AICD should not be a contraindication to ECT.13
A chart review of 3 patients with ICDs who received concurrent ECT found treatment was generally uneventful.12 One patient developed tachycardia with a rate-dependent left bundle branch block and hypotension in the recovery room, which responded promptly to esmolol. She did not experience similar events after subsequent ECT treatments.
Minimizing risk
In the absence of controlled data about the use of ECT in patients with implanted cardiac devices, crucial therapeutic decisions depend on the physician’s skill and judgment. Risk strategies can minimize complications (Algorithm).12 An internist or cardiologist experienced in pacemaker management should conduct a device interrogation—evaluating thresholds, lead impedance, and battery voltage and reviewing histograms, mode switch episodes, and stored electrograms—before the first ECT session and after the final one.
Most modern implantable pacemakers work in the synchronous (demand), rate-adaptive mode. In a patient in whom non-cardiac electrical signals cause bradycardia or asystole during ECT, the pacemaker can be reprogrammed to be less sensitive by placing a magnet over the pulse generator, which converts the pacemaker to an asynchronous (fixed), non-sensing mode. It is important to keep in mind that magnet application will not “turn off” a pacemaker; although each pacemaker is programmed to respond to a magnet in a specific fashion, the main response is asynchronous pacing.
Careful cardiac monitoring during ECT is essential (Table). The cardiologist or internist should be available during the first few ECT sessions to monitor for potential pacemaker interference or malfunction. This physician should be familiar with the pacemaker model and type of lead system so he or she can deactivate, reactivate, or reprogram the device.
Algorithm
Reducing risk when administering ECT to cardiac pacemaker patients
| Step 1 | |
| Evaluate the patient to ensure medical suitability for ECT and associated anesthesia | |
| Step 2 | ↓ |
| Conduct pacemaker interrogation (evaluating thresholds, lead impedance, and battery voltage and reviewing histograms, mode switch episodes, and stored electrograms) prior to first ECT treatment and after completion of full ECT course | |
| Step 3 | ↓ |
| Perform cardiac monitoring during and immediately after administering ECT | |
| Step 4 | ↓ |
| Have a magnet available to reprogram the pacemaker in the event of pacemaker inhibition or symptomatic bradycardia during ECT | |
| Step 5 | ↓ |
| Check that all monitoring devices are properly grounded, insulate the patient’s stretcher, and ensure that the patient does not touch anyone who is in contact with the ground during presentation of the ECT electrical stimulus | |
| ECT: electroconvulsive therapy | |
| Source: Reference 12 | |
Guidelines for monitoring cardiac pacemaker patients during ECT
| Use multilead ECG monitoring |
| Have equipment available to rapidly obtain central access (if vasoactive medications or transvenous pacing is needed) |
| Assess the plethysmography tracing of the pulse oximeter (a useful surrogate if the patient experiences dysrhythmias) |
| Have ready an external defibrillator |
TREATMENT: Successful ECT
We seek a medical consultation before initiating ECT. An internist performs device interrogation before the first ECT treatment and is present in the ECT treatment suite to ensure proper pacemaker conversion and to monitor for cardiac complications. The internist conducts another device interrogation after the acute series of ECT treatments.
Mrs. A tolerates the ECT sessions without cardiac complications. Her depressive symptoms respond well to 12 ECT sessions. She is more interactive and reports better attention and concentration. Although Mrs. A still has middle and initial insomnia, she denies thoughts of harming herself or anyone else.
Related resources
- Yarlagadda C. Pacemaker failure. www.emedicine.com/med/TOPIC1704.HTM.
- Atracurium • Tracrium
- Citalopram • Celexa
- Esmolol • Brevibloc
- Fluoxetine • Prozac
- Lorazepam • Ativan
- Methylphenidate • Ritalin, Concerta, others
- Nortriptyline • Aventyl, Pamelor, others
- Paroxetine • Paxil
- Sertraline • Zoloft
- Succinylcholine • Anectine
- Trazodone • Desyrel
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Dr. Romanowicz reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Ramaswamy receives research support from Bristol-Myers Squibb, Shire, and Forest Pharmaceuticals and is a consultant to Dainippon Sumitomo Pharma.
1. American Psychiatric Association Committee on ECT. The practice of electroconvulsive therapy: recommendations for treatment, training, and privileging. 2nd ed. Washington, D.C: American Psychiatric Association; 2001.
2. Russell JC, Rasmussen KG, O’Connor MK, et al. Long-term maintenance ECT: a retrospective review of efficacy and cognitive outcome. J ECT. 2003;19(1):4-9.
3. Alexopoulos GS, Shamoian CJ, Lucas J, et al. Medical problems of geriatric psychiatric patients and younger controls during electroconvulsive therapy. J Am Geriatr Soc. 1984;32(9):651-654.
4. Rasmussen KG, Rummans TA, Richardson JR. Electroconvulsive therapy in the medically ill. Psychiatric Clin North Am. 2002;25:177-193.
5. MacPherson RD, Loo CK, Barrett N. Electroconvulsive therapy in patients with cardiac pacemakers. Anaesth Intensive Care. 2006;34(4):470-474.
6. Burke MJ, Husain MM. Concomitant use of vagus nerve stimulation and electroconvulsive therapy for treatment-resistant depression. J ECT. 2006;22(3):218-222.
7. Husain MM, Montgomery JH, Fernandes P, et al. Safety of vagus nerve stimulation with ECT. Am J Psychiatry. 2002;159:1243.-
8. Alexopoulos GS, Frances RJ. ECT and cardiac patients with pacemakers. Am J Psychiatry. 1980;137(9):1111-1112.
9. Stone KR, McPherson CA. Assessment and management of patients with pacemakers and implantable cardioverter defibrillators. Crit Care Med. 2004;32(4 suppl):S155-S165.
10. Maisel WH, Sweeney MO, Stevenson WG, et al. Recalls and safety alerts involving pacemakers and implantable cardioverter-defibrillator generators. JAMA. 2001;286(7):793-799.
11. Gibson TC, Leaman DM, Devors J, et al. Pacemaker function in relation to electroconvulsive therapy. Chest. 1973;63(6):1025-1027.
12. Dolenc TJ, Barnes RD, Hayes DL, et al. Electroconvulsive therapy in patients with cardiac pacemakers and implantable cardioverter defibrillators. Pacing Clin Electrophysiol. 2004;27(9):1257-1263.
13. Lapid MI, Rummans TA, Hofmann VE, et al. ECT and automatic internal cardioverter-defibrillator. J ECT. 2001;17(2):146-148.
1. American Psychiatric Association Committee on ECT. The practice of electroconvulsive therapy: recommendations for treatment, training, and privileging. 2nd ed. Washington, D.C: American Psychiatric Association; 2001.
2. Russell JC, Rasmussen KG, O’Connor MK, et al. Long-term maintenance ECT: a retrospective review of efficacy and cognitive outcome. J ECT. 2003;19(1):4-9.
3. Alexopoulos GS, Shamoian CJ, Lucas J, et al. Medical problems of geriatric psychiatric patients and younger controls during electroconvulsive therapy. J Am Geriatr Soc. 1984;32(9):651-654.
4. Rasmussen KG, Rummans TA, Richardson JR. Electroconvulsive therapy in the medically ill. Psychiatric Clin North Am. 2002;25:177-193.
5. MacPherson RD, Loo CK, Barrett N. Electroconvulsive therapy in patients with cardiac pacemakers. Anaesth Intensive Care. 2006;34(4):470-474.
6. Burke MJ, Husain MM. Concomitant use of vagus nerve stimulation and electroconvulsive therapy for treatment-resistant depression. J ECT. 2006;22(3):218-222.
7. Husain MM, Montgomery JH, Fernandes P, et al. Safety of vagus nerve stimulation with ECT. Am J Psychiatry. 2002;159:1243.-
8. Alexopoulos GS, Frances RJ. ECT and cardiac patients with pacemakers. Am J Psychiatry. 1980;137(9):1111-1112.
9. Stone KR, McPherson CA. Assessment and management of patients with pacemakers and implantable cardioverter defibrillators. Crit Care Med. 2004;32(4 suppl):S155-S165.
10. Maisel WH, Sweeney MO, Stevenson WG, et al. Recalls and safety alerts involving pacemakers and implantable cardioverter-defibrillator generators. JAMA. 2001;286(7):793-799.
11. Gibson TC, Leaman DM, Devors J, et al. Pacemaker function in relation to electroconvulsive therapy. Chest. 1973;63(6):1025-1027.
12. Dolenc TJ, Barnes RD, Hayes DL, et al. Electroconvulsive therapy in patients with cardiac pacemakers and implantable cardioverter defibrillators. Pacing Clin Electrophysiol. 2004;27(9):1257-1263.
13. Lapid MI, Rummans TA, Hofmann VE, et al. ECT and automatic internal cardioverter-defibrillator. J ECT. 2001;17(2):146-148.
The patient who ‘spilled salt’
HISTORY: ‘They’re out to get me’
Mrs. V, age 64, tells her primary care physician she has felt “bad” for 2 weeks. She complains of depressed mood, middle insomnia, diminished appetite, poor concentration, and poor energy. She denies suicidal thoughts but reports feeling alone, overwhelmed, and unable to manage her daily life.
Mrs. V is very concerned about losing her job because she cannot function at work. She believes her coworkers may be plotting to get her fired. The primary care physician refers Mrs. V to us to evaluate her mood.
According to her daughter, Mrs. V has had multiple psychiatric hospitalizations; the most recent occurred 2 years ago when she was admitted for paranoia and disorganized behavior. The daughter also mentions that her mother has a remote history of daily alcohol use, drinking until she was intoxicated. Mrs. V says she occasionally drinks beer and she scores 2 out of 4 on the CAGE questionnaire, which may indicate alcohol dependence.
During mental status examination, Mrs. V is alert and oriented to person, place, and date. She is pleasant and cooperative but shows apparent thought blocking and some tangentiality. She has substantial difficulty answering questions and articulating symptoms. Speech is slow in rate and rhythm. Mrs. V’s mood is severely depressed and her affect constricted.
She denies suicidal or homicidal ideations or visual or auditory hallucinations. Cognitive testing reveals mild deficits in recall memory and poor concentration. Her insight is limited and her judgment fair.
Her medical history includes hypertension, hyperlipidemia, coronary artery disease, cardiac catheterization, and hyponatremia. Her medication regimen consists of aripiprazole, 15 mg/d; diltiazem, 180 mg/d; atenolol, 25 mg/d; aspirin, 325 mg/d; atorvastatin, 10 mg/d; sertraline, 50 mg/d; and ibuprofen, 600 mg as needed for hip pain. She also reports taking diuretics in the past.
Vital signs include blood pressure, 125/95 mm Hg; respirations, 16/min; temperature, 98.2° F; and pulse rate, 72/min. Serum investigations reveal sodium, 119 mEq/L (normal range: 135 to 145 mEq/L) and random blood sugar, 160 mg/dL (normal range: 60 to 114 mg/dL).
The authors’ observations
The combination of major depression with psychosis and hyponatremia makes Mrs. V’s case challenging. Hyponatremia in psychiatric inpatients can prompt medical consultation, thus possibly halting or delaying psychiatric treatment.
Hyponatremia has been associated with the use of:
- diuretics
- selective serotonin reuptake inhibitors (SSRIs)
- serotonin-norepinephrine reuptake inhibitors (SNRIs)
- tricyclic antidepressants
- calcium antagonists.
Among psychiatric inpatients, the risk of hyponatremia is doubled in women.1 It is unclear, however, if female gender is an independent risk factor for hyponatremia. Sharabi et al2 reported that patients of both sexes age >65 have a 9-fold greater risk of developing hyponatremia than younger counterparts.
In addition, hyponatremia risk during any antidepressant treatment is highest:
- in the summer
- during the first weeks of treatment
- with concomitant drug use, especially with diuretics.3
The authors’ observations
Based on Mrs. V’s initial lab results (Table 1), we classify her hyponatremia as euvolemic, with high urine osmolarity (≥100 mOsm/L). That helps narrow our differential diagnosis to glucocorticoid deficiency, hypothyroidism, and SIADH (Table 2).5 We exclude psychogenic polydipsia, “tea and toast” syndrome, or beer potomania because they usually present as euvolemic hyponatremia with low urinary osmolality.
SSRI use in elderly persons has been associated with hyponatremia, which in some cases may be consistent with SIADH. Unfortunately, few psychiatrists are aware of this potentially fatal side effect.
SIADH occurs in association with reduced serum osmolality. It is characterized by:
- hypotonic hyponatremia (serum sodium
- inappropriately elevated urine osmolarity (>200 mOsm/L) relative to plasma osmolarity
- elevated urine sodium (typically >20 mEq/L).4
The key to the pathophysiology, signs, symptoms, and treatment of SIADH is understanding that the hyponatremia is a result of excess water and not a sodium deficiency. Hyponatremia’s signs and symptoms primarily are related to CNS dysfunction and correlate with how rapidly and severely the condition develops.
We monitor Mrs. V for anorexia, nausea, and malaise because they would be the earliest findings, followed by headache, irritability, confusion, muscle cramps, weakness, obtundation, seizures, and coma. These occur as osmotic fluid shifts and results in cerebral edema and increased intracranial pressure. When sodium concentration drops below 105 mEq/L, life-threatening complications are likely.
Table 1
Mrs. V’s laboratory results
| Mrs. V’s results | |||
|---|---|---|---|
| Normal range | Before Tx | After Tx | |
| Serum sodium (mEq/L) | 135 to 145 | 119 | 127 |
| Serum potassium (mEq/L) | 3.5 to 5.0 | 3.6 | 3.8 |
| Creatinine (mg/dL) | 0.5 to 1.7 | 0.74 | 0.84 |
| Glucose (mg/dL) | 60 to 114 | 160 | 150 |
| Osmolarity | |||
| Serum (measured; mOsm/L) | 275 to 300 | 258 | 242 |
| Urine (mOsm/L) | 257 | 180 | |
| Urine sodium (mEq/L) | 20 to 40 | 48 | 42 |
Mrs. V’s laboratory results
| Hypovolemic hyponatremia | Euvolemic hyponatremia | Hypervolemic hyponatremia |
|---|---|---|
| Vomiting Diarrhea Laxative abuse Renal disease Nasogastric suction Salt-wasting nephropathy Addison’s disease | Normal urinary sodium Glucocorticoid deficiency Hypothyroidism Certain drugs SIADH | Congestive heart failure Nephrotic syndrome Cirrhosis |
| Low urinary osmolality Psychogenic polydipsia ‘Tea and toast’ syndrome Beer potomania | ||
| SIADH: syndrome of inappropriate antidiuretic hormone | ||
| Source: Reference 5 | ||
SSRIs and SIADH
Bouman et al6 estimated that the incidence of SSRI-induced SIADH in elderly patients is 12%. Liu et al7 described 706 cases of hyponatremia associated with SSRI use in unpublished reports. Fluoxetine was most commonly the cause (75.3% cases), followed by paroxetine (12.4%), sertraline (11.7%), and fluvoxamine (1.5%). Resuming the same drug resulted in hyponatremia in 16 of 24 of these cases (66.7%).
Kirby et al,8 however, found no clear advantages in different SSRIs’ propensity to cause hyponatremia. Seventy-one percent of patients treated with the SNRI venlafaxine developed hyponatremia, compared with 32% taking paroxetine and 29% receiving sertraline. It is unclear whether a specific SSRI or venlafaxine has a stronger association with hyponatremia than any other antidepressant.
Hyponatremia’s nonspecific symptoms and wide range of time to detection (1 to 253 days) suggest clinicians usually detect the condition by chance rather than specifically assessing for it.9
TREATMENT: Medication change?
Coordinating Mrs. V’s depression and hyponatremia treatment is critical. We propose discontinuing sertraline and treating Mrs. V’s symptoms with electroconvulsive therapy (ECT). She refuses ECT, stating “I don’t feel that bad. My father was treated with ECT and I am scared of it.”
The authors’ observations
SSRI-induced hyponatremia can be transient or persistent and recurrent. The usual approach is to discontinue the SSRI and try a different antidepressant. Because hyponatremia has been associated with all SSRIs and SNRIs, it would be prudent to choose an alternate antidepressant agent outside these classes. If patients must continue taking an antidepressant that causes hyponatremia, avoid concurrent use of drugs that cause hyponatremia, restrict fluid intake, and consider adding a medication that prevents hyponatremia, such as demeclocycline or fludrocortisone.
SSRI-induced hyponatremia may resolve:
- with SSRI discontinuation alone11
- with fluid restriction and without discontinuation of the SSRI11
- with drug discontinuation, fluid restriction, and sodium chloride and potassium supplementation.12
FOLLOW-UP: Analysis error?
Despite modifications to Mrs. V’s diet, her fasting serum glucose level remains >100. She is diagnosed with diabetes mellitus type 2 and treated with metformin. We continue mirtazapine, which has successfully controlled Mrs. V’s depressive symptoms. Her serum sodium levels start normalizing.
The authors’ observations
Related resources
- Siegel AJ. Hyponatremia in psychiatric patients: update on evaluation and management. Harv Rev Psychiatry 2008;16(1):13-24.
- Atalay A, Turhan N, Aki OE. A challenging case of syndrome of inappropriate secretion of antidiuretic hormone in an elderly patient secondary to quetiapine. South Med J 2007;100(8):832-3.
- Aripiprazole • Abilify
- Atenolol • Tenormin
- Atorvastatin • Lipitor
- Demeclocycline • Declomycin, Declostatin, others
- Diltiazem • Cardizem, Dilacor, others
- Fludrocortisone • Florinef
- Fluvoxamine • Luvox
- Ibuprofen • Advil, Motrin, others
- Metformin • Glucophage, Diabex, others
- Mirtazapine • Remeron
- Paroxetine • Paxil
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosures
Dr. Romanowicz reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Ramaswamy receives research support from Bristol-Myers Squibb, Shire, and Forest Pharmaceuticals and is a consultant to Dainippon Sumitomo Pharma.
Dr. Wilson receives research support from the National Institute of Mental Health, the Veterans Administration, the State of Nebraska, Health Futures Foundation, Inc., AstraZeneca, Dainippon Sumitomo Pharma, Eli Lilly and Company, and Pfizer Inc. and serves as a consultant to the Substance Abuse and Mental Health Services Administration and the State of Nebraska.
Reference
1. Siegler EL, Tamres D, Berlin JA, et al. Risk factors for the development of hyponatremia in psychiatric inpatients. Arch Intern Med 1995;155(9):953-7.
2. Sharabi Y, Illan R, Kamari Y, et al. Diuretic induced hyponatraemia in elderly hypertensive women. J Hum Hypertens 2002;16(9):631-5.
3. Rosner MH. Severe hyponatremia associated with the combined use of thiazide diuretics and selective serotonin reuptake inhibitors. Am J Med Sci 2004;327(2):109-11.
4. Buff DD, Markowitz S. Hyponatremia in the psychiatric patient: a review of diagnostic and management strategies. Psychiatr Ann 2003;33(5):318-25.
5. Levitan A. Hyponatremia: how to recognize the cause promptly—and avoid treatment pitfalls. Consultant 2003;43(7):861-70.
6. Bouman WP, Pinner G, Johnson H. Incidence of selective serotonin reuptake inhibitor (SSRI) induced hyponatraemia due to the syndrome of inappropriate antidiuretic hormone (SIADH) secretion in the elderly. Int J Geriatr Psychiatry 1998;13(1):12-5
7. Liu BA, Mittmann N, Knowles SR, et al. Hyponatremia and the syndrome of inappropriate secretion of antidiuretic hormone associated with the use of selective serotonin reuptake inhibitors: a review of spontaneous reports. CMAJ 1996;155(5):519-27
8. Kirby D, Ames D. Hyponatraemia and selective serotonin re-uptake inhibitors in elderly patients. Int J Geriatr Psychiatry 2001;16(5):484-93
9. Kirchner V, Silver LE, Kelly CA. Selective serotonin reuptake inhibitors and hyponatraemia: review and proposed mechanisms in the elderly. J Psychopharmacol 1998;12(4):396-400.
10. Jagsch C, Marksteiner J, Seiringer E, Windhager E. Successful mirtazapine treatment of an 81-year-old patient with syndrome of inappropriate antidiuretic hormone secretion. Pharmacopsychiatry 2007;40(3):129-31.
11. Bigaillon C, El Jahiri Y, Garcia C, et al. Inappropriate ADH secretion-induced hyponatremia and associated with paroxetine use. Rev Med Interne 2007;28(9):642-4.
12. Blacksten JV, Birt JA. Syndrome of inappropriate secretion of antidiuretic hormone secondary to fluoxetine. Ann Pharmacother 1993;27(6):723-4.
HISTORY: ‘They’re out to get me’
Mrs. V, age 64, tells her primary care physician she has felt “bad” for 2 weeks. She complains of depressed mood, middle insomnia, diminished appetite, poor concentration, and poor energy. She denies suicidal thoughts but reports feeling alone, overwhelmed, and unable to manage her daily life.
Mrs. V is very concerned about losing her job because she cannot function at work. She believes her coworkers may be plotting to get her fired. The primary care physician refers Mrs. V to us to evaluate her mood.
According to her daughter, Mrs. V has had multiple psychiatric hospitalizations; the most recent occurred 2 years ago when she was admitted for paranoia and disorganized behavior. The daughter also mentions that her mother has a remote history of daily alcohol use, drinking until she was intoxicated. Mrs. V says she occasionally drinks beer and she scores 2 out of 4 on the CAGE questionnaire, which may indicate alcohol dependence.
During mental status examination, Mrs. V is alert and oriented to person, place, and date. She is pleasant and cooperative but shows apparent thought blocking and some tangentiality. She has substantial difficulty answering questions and articulating symptoms. Speech is slow in rate and rhythm. Mrs. V’s mood is severely depressed and her affect constricted.
She denies suicidal or homicidal ideations or visual or auditory hallucinations. Cognitive testing reveals mild deficits in recall memory and poor concentration. Her insight is limited and her judgment fair.
Her medical history includes hypertension, hyperlipidemia, coronary artery disease, cardiac catheterization, and hyponatremia. Her medication regimen consists of aripiprazole, 15 mg/d; diltiazem, 180 mg/d; atenolol, 25 mg/d; aspirin, 325 mg/d; atorvastatin, 10 mg/d; sertraline, 50 mg/d; and ibuprofen, 600 mg as needed for hip pain. She also reports taking diuretics in the past.
Vital signs include blood pressure, 125/95 mm Hg; respirations, 16/min; temperature, 98.2° F; and pulse rate, 72/min. Serum investigations reveal sodium, 119 mEq/L (normal range: 135 to 145 mEq/L) and random blood sugar, 160 mg/dL (normal range: 60 to 114 mg/dL).
The authors’ observations
The combination of major depression with psychosis and hyponatremia makes Mrs. V’s case challenging. Hyponatremia in psychiatric inpatients can prompt medical consultation, thus possibly halting or delaying psychiatric treatment.
Hyponatremia has been associated with the use of:
- diuretics
- selective serotonin reuptake inhibitors (SSRIs)
- serotonin-norepinephrine reuptake inhibitors (SNRIs)
- tricyclic antidepressants
- calcium antagonists.
Among psychiatric inpatients, the risk of hyponatremia is doubled in women.1 It is unclear, however, if female gender is an independent risk factor for hyponatremia. Sharabi et al2 reported that patients of both sexes age >65 have a 9-fold greater risk of developing hyponatremia than younger counterparts.
In addition, hyponatremia risk during any antidepressant treatment is highest:
- in the summer
- during the first weeks of treatment
- with concomitant drug use, especially with diuretics.3
The authors’ observations
Based on Mrs. V’s initial lab results (Table 1), we classify her hyponatremia as euvolemic, with high urine osmolarity (≥100 mOsm/L). That helps narrow our differential diagnosis to glucocorticoid deficiency, hypothyroidism, and SIADH (Table 2).5 We exclude psychogenic polydipsia, “tea and toast” syndrome, or beer potomania because they usually present as euvolemic hyponatremia with low urinary osmolality.
SSRI use in elderly persons has been associated with hyponatremia, which in some cases may be consistent with SIADH. Unfortunately, few psychiatrists are aware of this potentially fatal side effect.
SIADH occurs in association with reduced serum osmolality. It is characterized by:
- hypotonic hyponatremia (serum sodium
- inappropriately elevated urine osmolarity (>200 mOsm/L) relative to plasma osmolarity
- elevated urine sodium (typically >20 mEq/L).4
The key to the pathophysiology, signs, symptoms, and treatment of SIADH is understanding that the hyponatremia is a result of excess water and not a sodium deficiency. Hyponatremia’s signs and symptoms primarily are related to CNS dysfunction and correlate with how rapidly and severely the condition develops.
We monitor Mrs. V for anorexia, nausea, and malaise because they would be the earliest findings, followed by headache, irritability, confusion, muscle cramps, weakness, obtundation, seizures, and coma. These occur as osmotic fluid shifts and results in cerebral edema and increased intracranial pressure. When sodium concentration drops below 105 mEq/L, life-threatening complications are likely.
Table 1
Mrs. V’s laboratory results
| Mrs. V’s results | |||
|---|---|---|---|
| Normal range | Before Tx | After Tx | |
| Serum sodium (mEq/L) | 135 to 145 | 119 | 127 |
| Serum potassium (mEq/L) | 3.5 to 5.0 | 3.6 | 3.8 |
| Creatinine (mg/dL) | 0.5 to 1.7 | 0.74 | 0.84 |
| Glucose (mg/dL) | 60 to 114 | 160 | 150 |
| Osmolarity | |||
| Serum (measured; mOsm/L) | 275 to 300 | 258 | 242 |
| Urine (mOsm/L) | 257 | 180 | |
| Urine sodium (mEq/L) | 20 to 40 | 48 | 42 |
Mrs. V’s laboratory results
| Hypovolemic hyponatremia | Euvolemic hyponatremia | Hypervolemic hyponatremia |
|---|---|---|
| Vomiting Diarrhea Laxative abuse Renal disease Nasogastric suction Salt-wasting nephropathy Addison’s disease | Normal urinary sodium Glucocorticoid deficiency Hypothyroidism Certain drugs SIADH | Congestive heart failure Nephrotic syndrome Cirrhosis |
| Low urinary osmolality Psychogenic polydipsia ‘Tea and toast’ syndrome Beer potomania | ||
| SIADH: syndrome of inappropriate antidiuretic hormone | ||
| Source: Reference 5 | ||
SSRIs and SIADH
Bouman et al6 estimated that the incidence of SSRI-induced SIADH in elderly patients is 12%. Liu et al7 described 706 cases of hyponatremia associated with SSRI use in unpublished reports. Fluoxetine was most commonly the cause (75.3% cases), followed by paroxetine (12.4%), sertraline (11.7%), and fluvoxamine (1.5%). Resuming the same drug resulted in hyponatremia in 16 of 24 of these cases (66.7%).
Kirby et al,8 however, found no clear advantages in different SSRIs’ propensity to cause hyponatremia. Seventy-one percent of patients treated with the SNRI venlafaxine developed hyponatremia, compared with 32% taking paroxetine and 29% receiving sertraline. It is unclear whether a specific SSRI or venlafaxine has a stronger association with hyponatremia than any other antidepressant.
Hyponatremia’s nonspecific symptoms and wide range of time to detection (1 to 253 days) suggest clinicians usually detect the condition by chance rather than specifically assessing for it.9
TREATMENT: Medication change?
Coordinating Mrs. V’s depression and hyponatremia treatment is critical. We propose discontinuing sertraline and treating Mrs. V’s symptoms with electroconvulsive therapy (ECT). She refuses ECT, stating “I don’t feel that bad. My father was treated with ECT and I am scared of it.”
The authors’ observations
SSRI-induced hyponatremia can be transient or persistent and recurrent. The usual approach is to discontinue the SSRI and try a different antidepressant. Because hyponatremia has been associated with all SSRIs and SNRIs, it would be prudent to choose an alternate antidepressant agent outside these classes. If patients must continue taking an antidepressant that causes hyponatremia, avoid concurrent use of drugs that cause hyponatremia, restrict fluid intake, and consider adding a medication that prevents hyponatremia, such as demeclocycline or fludrocortisone.
SSRI-induced hyponatremia may resolve:
- with SSRI discontinuation alone11
- with fluid restriction and without discontinuation of the SSRI11
- with drug discontinuation, fluid restriction, and sodium chloride and potassium supplementation.12
FOLLOW-UP: Analysis error?
Despite modifications to Mrs. V’s diet, her fasting serum glucose level remains >100. She is diagnosed with diabetes mellitus type 2 and treated with metformin. We continue mirtazapine, which has successfully controlled Mrs. V’s depressive symptoms. Her serum sodium levels start normalizing.
The authors’ observations
Related resources
- Siegel AJ. Hyponatremia in psychiatric patients: update on evaluation and management. Harv Rev Psychiatry 2008;16(1):13-24.
- Atalay A, Turhan N, Aki OE. A challenging case of syndrome of inappropriate secretion of antidiuretic hormone in an elderly patient secondary to quetiapine. South Med J 2007;100(8):832-3.
- Aripiprazole • Abilify
- Atenolol • Tenormin
- Atorvastatin • Lipitor
- Demeclocycline • Declomycin, Declostatin, others
- Diltiazem • Cardizem, Dilacor, others
- Fludrocortisone • Florinef
- Fluvoxamine • Luvox
- Ibuprofen • Advil, Motrin, others
- Metformin • Glucophage, Diabex, others
- Mirtazapine • Remeron
- Paroxetine • Paxil
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosures
Dr. Romanowicz reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Ramaswamy receives research support from Bristol-Myers Squibb, Shire, and Forest Pharmaceuticals and is a consultant to Dainippon Sumitomo Pharma.
Dr. Wilson receives research support from the National Institute of Mental Health, the Veterans Administration, the State of Nebraska, Health Futures Foundation, Inc., AstraZeneca, Dainippon Sumitomo Pharma, Eli Lilly and Company, and Pfizer Inc. and serves as a consultant to the Substance Abuse and Mental Health Services Administration and the State of Nebraska.
HISTORY: ‘They’re out to get me’
Mrs. V, age 64, tells her primary care physician she has felt “bad” for 2 weeks. She complains of depressed mood, middle insomnia, diminished appetite, poor concentration, and poor energy. She denies suicidal thoughts but reports feeling alone, overwhelmed, and unable to manage her daily life.
Mrs. V is very concerned about losing her job because she cannot function at work. She believes her coworkers may be plotting to get her fired. The primary care physician refers Mrs. V to us to evaluate her mood.
According to her daughter, Mrs. V has had multiple psychiatric hospitalizations; the most recent occurred 2 years ago when she was admitted for paranoia and disorganized behavior. The daughter also mentions that her mother has a remote history of daily alcohol use, drinking until she was intoxicated. Mrs. V says she occasionally drinks beer and she scores 2 out of 4 on the CAGE questionnaire, which may indicate alcohol dependence.
During mental status examination, Mrs. V is alert and oriented to person, place, and date. She is pleasant and cooperative but shows apparent thought blocking and some tangentiality. She has substantial difficulty answering questions and articulating symptoms. Speech is slow in rate and rhythm. Mrs. V’s mood is severely depressed and her affect constricted.
She denies suicidal or homicidal ideations or visual or auditory hallucinations. Cognitive testing reveals mild deficits in recall memory and poor concentration. Her insight is limited and her judgment fair.
Her medical history includes hypertension, hyperlipidemia, coronary artery disease, cardiac catheterization, and hyponatremia. Her medication regimen consists of aripiprazole, 15 mg/d; diltiazem, 180 mg/d; atenolol, 25 mg/d; aspirin, 325 mg/d; atorvastatin, 10 mg/d; sertraline, 50 mg/d; and ibuprofen, 600 mg as needed for hip pain. She also reports taking diuretics in the past.
Vital signs include blood pressure, 125/95 mm Hg; respirations, 16/min; temperature, 98.2° F; and pulse rate, 72/min. Serum investigations reveal sodium, 119 mEq/L (normal range: 135 to 145 mEq/L) and random blood sugar, 160 mg/dL (normal range: 60 to 114 mg/dL).
The authors’ observations
The combination of major depression with psychosis and hyponatremia makes Mrs. V’s case challenging. Hyponatremia in psychiatric inpatients can prompt medical consultation, thus possibly halting or delaying psychiatric treatment.
Hyponatremia has been associated with the use of:
- diuretics
- selective serotonin reuptake inhibitors (SSRIs)
- serotonin-norepinephrine reuptake inhibitors (SNRIs)
- tricyclic antidepressants
- calcium antagonists.
Among psychiatric inpatients, the risk of hyponatremia is doubled in women.1 It is unclear, however, if female gender is an independent risk factor for hyponatremia. Sharabi et al2 reported that patients of both sexes age >65 have a 9-fold greater risk of developing hyponatremia than younger counterparts.
In addition, hyponatremia risk during any antidepressant treatment is highest:
- in the summer
- during the first weeks of treatment
- with concomitant drug use, especially with diuretics.3
The authors’ observations
Based on Mrs. V’s initial lab results (Table 1), we classify her hyponatremia as euvolemic, with high urine osmolarity (≥100 mOsm/L). That helps narrow our differential diagnosis to glucocorticoid deficiency, hypothyroidism, and SIADH (Table 2).5 We exclude psychogenic polydipsia, “tea and toast” syndrome, or beer potomania because they usually present as euvolemic hyponatremia with low urinary osmolality.
SSRI use in elderly persons has been associated with hyponatremia, which in some cases may be consistent with SIADH. Unfortunately, few psychiatrists are aware of this potentially fatal side effect.
SIADH occurs in association with reduced serum osmolality. It is characterized by:
- hypotonic hyponatremia (serum sodium
- inappropriately elevated urine osmolarity (>200 mOsm/L) relative to plasma osmolarity
- elevated urine sodium (typically >20 mEq/L).4
The key to the pathophysiology, signs, symptoms, and treatment of SIADH is understanding that the hyponatremia is a result of excess water and not a sodium deficiency. Hyponatremia’s signs and symptoms primarily are related to CNS dysfunction and correlate with how rapidly and severely the condition develops.
We monitor Mrs. V for anorexia, nausea, and malaise because they would be the earliest findings, followed by headache, irritability, confusion, muscle cramps, weakness, obtundation, seizures, and coma. These occur as osmotic fluid shifts and results in cerebral edema and increased intracranial pressure. When sodium concentration drops below 105 mEq/L, life-threatening complications are likely.
Table 1
Mrs. V’s laboratory results
| Mrs. V’s results | |||
|---|---|---|---|
| Normal range | Before Tx | After Tx | |
| Serum sodium (mEq/L) | 135 to 145 | 119 | 127 |
| Serum potassium (mEq/L) | 3.5 to 5.0 | 3.6 | 3.8 |
| Creatinine (mg/dL) | 0.5 to 1.7 | 0.74 | 0.84 |
| Glucose (mg/dL) | 60 to 114 | 160 | 150 |
| Osmolarity | |||
| Serum (measured; mOsm/L) | 275 to 300 | 258 | 242 |
| Urine (mOsm/L) | 257 | 180 | |
| Urine sodium (mEq/L) | 20 to 40 | 48 | 42 |
Mrs. V’s laboratory results
| Hypovolemic hyponatremia | Euvolemic hyponatremia | Hypervolemic hyponatremia |
|---|---|---|
| Vomiting Diarrhea Laxative abuse Renal disease Nasogastric suction Salt-wasting nephropathy Addison’s disease | Normal urinary sodium Glucocorticoid deficiency Hypothyroidism Certain drugs SIADH | Congestive heart failure Nephrotic syndrome Cirrhosis |
| Low urinary osmolality Psychogenic polydipsia ‘Tea and toast’ syndrome Beer potomania | ||
| SIADH: syndrome of inappropriate antidiuretic hormone | ||
| Source: Reference 5 | ||
SSRIs and SIADH
Bouman et al6 estimated that the incidence of SSRI-induced SIADH in elderly patients is 12%. Liu et al7 described 706 cases of hyponatremia associated with SSRI use in unpublished reports. Fluoxetine was most commonly the cause (75.3% cases), followed by paroxetine (12.4%), sertraline (11.7%), and fluvoxamine (1.5%). Resuming the same drug resulted in hyponatremia in 16 of 24 of these cases (66.7%).
Kirby et al,8 however, found no clear advantages in different SSRIs’ propensity to cause hyponatremia. Seventy-one percent of patients treated with the SNRI venlafaxine developed hyponatremia, compared with 32% taking paroxetine and 29% receiving sertraline. It is unclear whether a specific SSRI or venlafaxine has a stronger association with hyponatremia than any other antidepressant.
Hyponatremia’s nonspecific symptoms and wide range of time to detection (1 to 253 days) suggest clinicians usually detect the condition by chance rather than specifically assessing for it.9
TREATMENT: Medication change?
Coordinating Mrs. V’s depression and hyponatremia treatment is critical. We propose discontinuing sertraline and treating Mrs. V’s symptoms with electroconvulsive therapy (ECT). She refuses ECT, stating “I don’t feel that bad. My father was treated with ECT and I am scared of it.”
The authors’ observations
SSRI-induced hyponatremia can be transient or persistent and recurrent. The usual approach is to discontinue the SSRI and try a different antidepressant. Because hyponatremia has been associated with all SSRIs and SNRIs, it would be prudent to choose an alternate antidepressant agent outside these classes. If patients must continue taking an antidepressant that causes hyponatremia, avoid concurrent use of drugs that cause hyponatremia, restrict fluid intake, and consider adding a medication that prevents hyponatremia, such as demeclocycline or fludrocortisone.
SSRI-induced hyponatremia may resolve:
- with SSRI discontinuation alone11
- with fluid restriction and without discontinuation of the SSRI11
- with drug discontinuation, fluid restriction, and sodium chloride and potassium supplementation.12
FOLLOW-UP: Analysis error?
Despite modifications to Mrs. V’s diet, her fasting serum glucose level remains >100. She is diagnosed with diabetes mellitus type 2 and treated with metformin. We continue mirtazapine, which has successfully controlled Mrs. V’s depressive symptoms. Her serum sodium levels start normalizing.
The authors’ observations
Related resources
- Siegel AJ. Hyponatremia in psychiatric patients: update on evaluation and management. Harv Rev Psychiatry 2008;16(1):13-24.
- Atalay A, Turhan N, Aki OE. A challenging case of syndrome of inappropriate secretion of antidiuretic hormone in an elderly patient secondary to quetiapine. South Med J 2007;100(8):832-3.
- Aripiprazole • Abilify
- Atenolol • Tenormin
- Atorvastatin • Lipitor
- Demeclocycline • Declomycin, Declostatin, others
- Diltiazem • Cardizem, Dilacor, others
- Fludrocortisone • Florinef
- Fluvoxamine • Luvox
- Ibuprofen • Advil, Motrin, others
- Metformin • Glucophage, Diabex, others
- Mirtazapine • Remeron
- Paroxetine • Paxil
- Sertraline • Zoloft
- Venlafaxine • Effexor
Disclosures
Dr. Romanowicz reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Ramaswamy receives research support from Bristol-Myers Squibb, Shire, and Forest Pharmaceuticals and is a consultant to Dainippon Sumitomo Pharma.
Dr. Wilson receives research support from the National Institute of Mental Health, the Veterans Administration, the State of Nebraska, Health Futures Foundation, Inc., AstraZeneca, Dainippon Sumitomo Pharma, Eli Lilly and Company, and Pfizer Inc. and serves as a consultant to the Substance Abuse and Mental Health Services Administration and the State of Nebraska.
Reference
1. Siegler EL, Tamres D, Berlin JA, et al. Risk factors for the development of hyponatremia in psychiatric inpatients. Arch Intern Med 1995;155(9):953-7.
2. Sharabi Y, Illan R, Kamari Y, et al. Diuretic induced hyponatraemia in elderly hypertensive women. J Hum Hypertens 2002;16(9):631-5.
3. Rosner MH. Severe hyponatremia associated with the combined use of thiazide diuretics and selective serotonin reuptake inhibitors. Am J Med Sci 2004;327(2):109-11.
4. Buff DD, Markowitz S. Hyponatremia in the psychiatric patient: a review of diagnostic and management strategies. Psychiatr Ann 2003;33(5):318-25.
5. Levitan A. Hyponatremia: how to recognize the cause promptly—and avoid treatment pitfalls. Consultant 2003;43(7):861-70.
6. Bouman WP, Pinner G, Johnson H. Incidence of selective serotonin reuptake inhibitor (SSRI) induced hyponatraemia due to the syndrome of inappropriate antidiuretic hormone (SIADH) secretion in the elderly. Int J Geriatr Psychiatry 1998;13(1):12-5
7. Liu BA, Mittmann N, Knowles SR, et al. Hyponatremia and the syndrome of inappropriate secretion of antidiuretic hormone associated with the use of selective serotonin reuptake inhibitors: a review of spontaneous reports. CMAJ 1996;155(5):519-27
8. Kirby D, Ames D. Hyponatraemia and selective serotonin re-uptake inhibitors in elderly patients. Int J Geriatr Psychiatry 2001;16(5):484-93
9. Kirchner V, Silver LE, Kelly CA. Selective serotonin reuptake inhibitors and hyponatraemia: review and proposed mechanisms in the elderly. J Psychopharmacol 1998;12(4):396-400.
10. Jagsch C, Marksteiner J, Seiringer E, Windhager E. Successful mirtazapine treatment of an 81-year-old patient with syndrome of inappropriate antidiuretic hormone secretion. Pharmacopsychiatry 2007;40(3):129-31.
11. Bigaillon C, El Jahiri Y, Garcia C, et al. Inappropriate ADH secretion-induced hyponatremia and associated with paroxetine use. Rev Med Interne 2007;28(9):642-4.
12. Blacksten JV, Birt JA. Syndrome of inappropriate secretion of antidiuretic hormone secondary to fluoxetine. Ann Pharmacother 1993;27(6):723-4.
Reference
1. Siegler EL, Tamres D, Berlin JA, et al. Risk factors for the development of hyponatremia in psychiatric inpatients. Arch Intern Med 1995;155(9):953-7.
2. Sharabi Y, Illan R, Kamari Y, et al. Diuretic induced hyponatraemia in elderly hypertensive women. J Hum Hypertens 2002;16(9):631-5.
3. Rosner MH. Severe hyponatremia associated with the combined use of thiazide diuretics and selective serotonin reuptake inhibitors. Am J Med Sci 2004;327(2):109-11.
4. Buff DD, Markowitz S. Hyponatremia in the psychiatric patient: a review of diagnostic and management strategies. Psychiatr Ann 2003;33(5):318-25.
5. Levitan A. Hyponatremia: how to recognize the cause promptly—and avoid treatment pitfalls. Consultant 2003;43(7):861-70.
6. Bouman WP, Pinner G, Johnson H. Incidence of selective serotonin reuptake inhibitor (SSRI) induced hyponatraemia due to the syndrome of inappropriate antidiuretic hormone (SIADH) secretion in the elderly. Int J Geriatr Psychiatry 1998;13(1):12-5
7. Liu BA, Mittmann N, Knowles SR, et al. Hyponatremia and the syndrome of inappropriate secretion of antidiuretic hormone associated with the use of selective serotonin reuptake inhibitors: a review of spontaneous reports. CMAJ 1996;155(5):519-27
8. Kirby D, Ames D. Hyponatraemia and selective serotonin re-uptake inhibitors in elderly patients. Int J Geriatr Psychiatry 2001;16(5):484-93
9. Kirchner V, Silver LE, Kelly CA. Selective serotonin reuptake inhibitors and hyponatraemia: review and proposed mechanisms in the elderly. J Psychopharmacol 1998;12(4):396-400.
10. Jagsch C, Marksteiner J, Seiringer E, Windhager E. Successful mirtazapine treatment of an 81-year-old patient with syndrome of inappropriate antidiuretic hormone secretion. Pharmacopsychiatry 2007;40(3):129-31.
11. Bigaillon C, El Jahiri Y, Garcia C, et al. Inappropriate ADH secretion-induced hyponatremia and associated with paroxetine use. Rev Med Interne 2007;28(9):642-4.
12. Blacksten JV, Birt JA. Syndrome of inappropriate secretion of antidiuretic hormone secondary to fluoxetine. Ann Pharmacother 1993;27(6):723-4.