User login
Genetic variants account for up to one-third of cases of cerebral palsy
Cerebral palsy (CP) is the most common cause of severe neurodisability in children, and it occurs in about 2 to 3 per 1,000 births worldwide.1 This nonprogressive disorder is characterized by symptoms that include spasticity, dystonia, choreoathetosis, and/or ataxia that are evident in the first few years of life. While many perinatal variables have been associated with CP, in most cases a specific cause is not identified.
Other neurodevelopmental disorders, such as intellectual disability, epilepsy, and autism spectrum disorder, are often associated with CP.2 These other neurodevelopmental disorders are often genetic, and this has raised the question as to whether CP also might have a substantial genetic component, although this has not been investigated in any significant way until recently. This topic is of great interest to the obstetric community, given that CP often is attributed to obstetric events, including mismanagement of labor and delivery.
Emerging evidence of a genetic-CP association
In an article published recently in JAMA, Moreno-De-Luca and colleagues sought to determine the diagnostic yield of exome sequencing for CP.3 This large cross-sectional study included results of exome sequencing performed in 2 settings. The first setting was a commercial laboratory in which samples were sent for analysis due to a diagnosis of CP, primarily in children (n = 1,345) with a median age of 8.8 years. A second cohort, recruited from a neurodevelopmental disorders clinic at Geisinger, included primarily adults (n = 181) with a median age of 41.9 years.
As is standard in exome sequencing, results were considered likely causative if they were classified as pathogenic or likely pathogenic based on criteria of the American College of Genetics and Genomics. In the laboratory group, 32.7% (440 of 1,345) had a genetic cause of the CP identified, while in the clinic group, 10.5% (19 of 181) had a genetic etiology found. Although most of the identified genetic variants were de novo (that is, they arose in the affected individual and were not clearly inherited), some were inherited from carrier parents.3
A number of other recent studies also have investigated genetic causes of CP and similarly have reported that a substantial number of cases are genetic. Several studies that performed chromosomal microarray analysis in individuals with CP found deleterious copy number variants in 10% to 31% of cases.4-6 Genomic variants detectable by exome sequencing have been reported in 15% to 20% of cases.3 In a recent study in Nature Genetics, researchers performed exome sequencing on 250 parent-child “trios” in which the child had CP, and they found that 14% of cases had an associated genetic variant that was thought to be causative.4 These studies all provide consistent evidence that a substantial proportion of CP cases are due to genetic causes.
Contributors to CP risk
Historically, CP was considered to occur largely as a result of perinatal anoxia. In 1862, the British orthopedic surgeon William John Little first reported an association between prematurity, asphyxia, difficult delivery, and CP in a paper presented to the Obstetrical Society of London.7 Subsequently, much effort has gone into the prevention of perinatal asphyxia and birth injury, although our ability to monitor fetal well-being remains limited. Nonreassuring fetal heart rate patterns are nonspecific and can occur for many reasons other than fetal asphyxia. Studies of electronic fetal monitoring have found that continuous monitoring primarily leads to an increase in cesarean delivery with no decrease in CP or infant mortality.8
While some have attributed this to failure to accurately interpret the fetal heart rate tracing, it also may be because a substantial number of CP cases are due to genetic and other causes, and that very few in fact result from preventable intrapartum injury.
The American College of Obstetricians and Gynecologists and the American Academy of Pediatrics agree that knowledge gaps preclude definitive determination that a given case of neonatal encephalopathy is attributable to an acute intrapartum event, and they provide criteria that must be fulfilled to establish a reasonable causal link between an intrapartum event and subsequent long-term neurologic disability.9 However, there continues to be a belief in the medical, scientific, and lay communities that birth asphyxia, secondary to adverse intrapartum events, is the leading cause of CP. A “brain-damaged infant” remains one of the most common malpractice claims, and birth injury one of the highest paid claims. Such claims generally allege that intrapartum asphyxia has caused long-term neurologic sequelae, including CP.
While it is true that prematurity, infection, hypoxia-ischemia, and pre- and perinatal stroke all have been implicated as contributing to CP risk, large population-based studies have shown that birth asphyxia accounts for less than 12% of CP cases.10 Specifically, recent data indicate that acute intrapartum hypoxia-ischemia occurs only in about 6% of CP cases. In other words, it does occur and may contribute to some cases, but this is likely a smaller percent than previously thought, and genetic factors now appear to be far more significant contributors.11
Continue to: Exploring a genetic etiology...
Exploring a genetic etiology
In considering the etiologies of CP, it is important to note that 21% to 40% of individuals with CP have an associated congenital anomaly, suggesting a genetic origin in at least some individuals. Moreover, a 40% heritability has been estimated in CP, which is comparable to the heritability rate for autism spectrum disorders.12
In the recent study by Moreno-De-Luca and colleagues, some of the gene variants detected were previously associated with other forms of neurodevelopmental disability, such as epilepsy and autism spectrum disorder.3 Many individuals in the study cohort were found to have multiple neurologic comorbidities, for example, CP as well as epilepsy, autism spectrum disorder, and/or intellectual disability. The presence of these additional comorbidities increased the likelihood of finding a genetic cause; the authors found that the diagnostic yield ranged from 11.2% with isolated CP to 32.9% with all 3 comorbidities. The yield was highest with CP and intellectual disability and CP with all 3 comorbidities. A few genes were particularly common, and some were reported previously in association with CP and/or other neurodevelopmental disorders. In some patients, variants were found in genes or gene regions associated with disorders that do not frequently include CP, such as Rett syndrome.3
Implications for ObGyns
The data from the study by Moreno-De-Luca and colleagues are interesting and relevant to pediatricians, neurologists, and geneticists, as well as obstetricians. Understanding the cause of any disease or disorder improves care, including counseling regarding the cause, the appropriate interventions or therapy, and in some families, the recurrence risk in another pregnancy. The treatment for CP has not changed significantly in many years. Increasingly, detection of an underlying genetic cause can guide precision treatments; thus, the detection of specific gene variants allows a targeted approach to therapy.
Identification of a genetic cause also can significantly impact recurrence risk counseling and prenatal diagnosis options in another pregnancy. In general, the empiric recurrence risk of CP is quoted as 1% to 2%,13 and with de novo variants this does not change. However, with inherited variants the recurrence risk in future children is substantially higher. While 72% of the genetic variants associated with CP in the Moreno-De-Luca study were de novo with a low recurrence risk, in the other 28% the mode of inheritance indicated a substantial risk of recurrence (25%–50%) in another pregnancy.3 Detecting such causative variant(s) allows not only accurate counseling about recurrence risk but also preimplantation genetic testing or prenatal diagnosis when recurrence risk is high.
In the field of obstetrics, the debate about the etiology of CP is important largely due to the medicolegal implications. Patient-oriented information on the internet often states that CP is caused by damage to the child’s brain just before, during, or soon after birth, supporting potential blame of those providing care during those times. Patient-oriented websites regarding CP do not list genetic disorders among the causes but rather include primarily environmental factors, such as prematurity, low birth weight, in utero infections, anoxia or other brain injury, or perinatal stroke. Even the Centers for Disease Control and Prevention website lists brain damage as the primary etiology of CP.14 Hopefully, these new data will increase a broader understanding of this condition.
Exome sequencing is now recommended as a first-tier test for individuals with many neurodevelopmental disorders, including epilepsy, intellectual disability, and autism spectrum disorder.15 However, comprehensive genetic testing is not typically recommended or performed in cases of CP. Based on recent data, including the report by Moreno-De-Luca and colleagues, it would seem that CP should be added to the list of disorders for which exome sequencing is ordered, given the similar prevalence and diagnostic yield. ●
- Oskoui M, Coutinho F, Dykeman J, et al. An update on the prevalence of cerebral palsy: a systematic review and meta-analysis. Dev Med Child Neurol. 2013;55:509-519.
- Rosenbaum P, Paneth N, Leviton A, et al. A report: the definition and classification of cerebral palsy April 2006. Dev Med Child Neurol Suppl. 2007;109:8-14.
- Moreno-De-Luca A, Millan F, Pesacreta DR, et al. Molecular diagnostic yield of exome sequencing in patients with cerebral palsy. JAMA. 2021;325:467-475.
- Jin SC, Lewis SA, Bakhtiari S, et al. Mutations disrupting neuritogenesis genes confer risk for cerebral palsy. Nat Genet. 2020;52:1046-1056.
- Segel R, Ben-Pazi H, Zeligson S, et al. Copy number variations in cryptogenic cerebral palsy. Neurology. 2015;84:1660-1668.
- McMichael G, Girirrajan S, Moreno-De-Luca A, et al. Rare copy number variation in cerebral palsy. Eur J Hum Genet. 2014;22:40-45.
- Little WJ. On the influence of abnormal parturition, difficult labours, premature births, and asphyxia neonatorum, on the mental and physical condition of the child, especially in relation to deformities. Trans Obstet Soc Lond. 1862;3:293-344.
- Alfirevic Z, Devane D, Gyte GM. Continuous cardiotocography (CTG) as a form of electronic fetal monitoring (EFM) for fetal assessment during labour. Cochrane Database Syst Rev. 2013;5;CD006066.
- American College of Obstetricians and Gynecologists. Executive summary: neonatal encephalopathy and neurologic outcome second edition. Report of the American College of Obstetricians and Gynecologists’ Task Force on Neonatal Encephalopathy. Obstet Gynecol. 2014;123:896- 901.
- Ellenberg JH, Nelson KB. The association of cerebral palsy with birth asphyxia: a definitional quagmire. Dev Med Child Neurol. 2013;55:210- 216.
- Himmelmann K, Uvebrant P. The panorama of cerebral palsy in Sweden part XII shows that patterns changed in the birth years 2007–2010. Acta Paediatr. 2018;107: 462-468.
- Petterson B, Stanley F, Henderson D. Cerebral palsy in multiple births in Western Australia: genetic aspects. Am J Med Genet. 1990;37:346- 351.
- Korzeniewski SJ, Slaughter J, Lenski M, et al. The complex aetiology of cerebral palsy. Nat Rev Neurol. 2018;14:528-543.
- Centers for Disease Control and Prevention. Causes and risk factors of cerebral palsy. https:// www.cdc.gov/ncbddd/cp/causes.html. Accessed March 23, 2021.
- Srivastava S, Love-Nichols JA, Dies KA, et al; NDD Exome Scoping Review Work Group. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. 2019;21:2413-2421.

Mary E. Norton, MD
Professor of Obstetrics, Gynecology, and Reproductive Sciences, Director, Division of Maternal-Fetal Medicine, University of California, San Francisco
The author reports no financial relationships relevant to this article.
Mary E. Norton, MD
Professor of Obstetrics, Gynecology, and Reproductive Sciences, Director, Division of Maternal-Fetal Medicine, University of California, San Francisco
The author reports no financial relationships relevant to this article.
Mary E. Norton, MD
Professor of Obstetrics, Gynecology, and Reproductive Sciences, Director, Division of Maternal-Fetal Medicine, University of California, San Francisco
The author reports no financial relationships relevant to this article.
Cerebral palsy (CP) is the most common cause of severe neurodisability in children, and it occurs in about 2 to 3 per 1,000 births worldwide.1 This nonprogressive disorder is characterized by symptoms that include spasticity, dystonia, choreoathetosis, and/or ataxia that are evident in the first few years of life. While many perinatal variables have been associated with CP, in most cases a specific cause is not identified.
Other neurodevelopmental disorders, such as intellectual disability, epilepsy, and autism spectrum disorder, are often associated with CP.2 These other neurodevelopmental disorders are often genetic, and this has raised the question as to whether CP also might have a substantial genetic component, although this has not been investigated in any significant way until recently. This topic is of great interest to the obstetric community, given that CP often is attributed to obstetric events, including mismanagement of labor and delivery.
Emerging evidence of a genetic-CP association
In an article published recently in JAMA, Moreno-De-Luca and colleagues sought to determine the diagnostic yield of exome sequencing for CP.3 This large cross-sectional study included results of exome sequencing performed in 2 settings. The first setting was a commercial laboratory in which samples were sent for analysis due to a diagnosis of CP, primarily in children (n = 1,345) with a median age of 8.8 years. A second cohort, recruited from a neurodevelopmental disorders clinic at Geisinger, included primarily adults (n = 181) with a median age of 41.9 years.
As is standard in exome sequencing, results were considered likely causative if they were classified as pathogenic or likely pathogenic based on criteria of the American College of Genetics and Genomics. In the laboratory group, 32.7% (440 of 1,345) had a genetic cause of the CP identified, while in the clinic group, 10.5% (19 of 181) had a genetic etiology found. Although most of the identified genetic variants were de novo (that is, they arose in the affected individual and were not clearly inherited), some were inherited from carrier parents.3
A number of other recent studies also have investigated genetic causes of CP and similarly have reported that a substantial number of cases are genetic. Several studies that performed chromosomal microarray analysis in individuals with CP found deleterious copy number variants in 10% to 31% of cases.4-6 Genomic variants detectable by exome sequencing have been reported in 15% to 20% of cases.3 In a recent study in Nature Genetics, researchers performed exome sequencing on 250 parent-child “trios” in which the child had CP, and they found that 14% of cases had an associated genetic variant that was thought to be causative.4 These studies all provide consistent evidence that a substantial proportion of CP cases are due to genetic causes.
Contributors to CP risk
Historically, CP was considered to occur largely as a result of perinatal anoxia. In 1862, the British orthopedic surgeon William John Little first reported an association between prematurity, asphyxia, difficult delivery, and CP in a paper presented to the Obstetrical Society of London.7 Subsequently, much effort has gone into the prevention of perinatal asphyxia and birth injury, although our ability to monitor fetal well-being remains limited. Nonreassuring fetal heart rate patterns are nonspecific and can occur for many reasons other than fetal asphyxia. Studies of electronic fetal monitoring have found that continuous monitoring primarily leads to an increase in cesarean delivery with no decrease in CP or infant mortality.8
While some have attributed this to failure to accurately interpret the fetal heart rate tracing, it also may be because a substantial number of CP cases are due to genetic and other causes, and that very few in fact result from preventable intrapartum injury.
The American College of Obstetricians and Gynecologists and the American Academy of Pediatrics agree that knowledge gaps preclude definitive determination that a given case of neonatal encephalopathy is attributable to an acute intrapartum event, and they provide criteria that must be fulfilled to establish a reasonable causal link between an intrapartum event and subsequent long-term neurologic disability.9 However, there continues to be a belief in the medical, scientific, and lay communities that birth asphyxia, secondary to adverse intrapartum events, is the leading cause of CP. A “brain-damaged infant” remains one of the most common malpractice claims, and birth injury one of the highest paid claims. Such claims generally allege that intrapartum asphyxia has caused long-term neurologic sequelae, including CP.
While it is true that prematurity, infection, hypoxia-ischemia, and pre- and perinatal stroke all have been implicated as contributing to CP risk, large population-based studies have shown that birth asphyxia accounts for less than 12% of CP cases.10 Specifically, recent data indicate that acute intrapartum hypoxia-ischemia occurs only in about 6% of CP cases. In other words, it does occur and may contribute to some cases, but this is likely a smaller percent than previously thought, and genetic factors now appear to be far more significant contributors.11
Continue to: Exploring a genetic etiology...
Exploring a genetic etiology
In considering the etiologies of CP, it is important to note that 21% to 40% of individuals with CP have an associated congenital anomaly, suggesting a genetic origin in at least some individuals. Moreover, a 40% heritability has been estimated in CP, which is comparable to the heritability rate for autism spectrum disorders.12
In the recent study by Moreno-De-Luca and colleagues, some of the gene variants detected were previously associated with other forms of neurodevelopmental disability, such as epilepsy and autism spectrum disorder.3 Many individuals in the study cohort were found to have multiple neurologic comorbidities, for example, CP as well as epilepsy, autism spectrum disorder, and/or intellectual disability. The presence of these additional comorbidities increased the likelihood of finding a genetic cause; the authors found that the diagnostic yield ranged from 11.2% with isolated CP to 32.9% with all 3 comorbidities. The yield was highest with CP and intellectual disability and CP with all 3 comorbidities. A few genes were particularly common, and some were reported previously in association with CP and/or other neurodevelopmental disorders. In some patients, variants were found in genes or gene regions associated with disorders that do not frequently include CP, such as Rett syndrome.3
Implications for ObGyns
The data from the study by Moreno-De-Luca and colleagues are interesting and relevant to pediatricians, neurologists, and geneticists, as well as obstetricians. Understanding the cause of any disease or disorder improves care, including counseling regarding the cause, the appropriate interventions or therapy, and in some families, the recurrence risk in another pregnancy. The treatment for CP has not changed significantly in many years. Increasingly, detection of an underlying genetic cause can guide precision treatments; thus, the detection of specific gene variants allows a targeted approach to therapy.
Identification of a genetic cause also can significantly impact recurrence risk counseling and prenatal diagnosis options in another pregnancy. In general, the empiric recurrence risk of CP is quoted as 1% to 2%,13 and with de novo variants this does not change. However, with inherited variants the recurrence risk in future children is substantially higher. While 72% of the genetic variants associated with CP in the Moreno-De-Luca study were de novo with a low recurrence risk, in the other 28% the mode of inheritance indicated a substantial risk of recurrence (25%–50%) in another pregnancy.3 Detecting such causative variant(s) allows not only accurate counseling about recurrence risk but also preimplantation genetic testing or prenatal diagnosis when recurrence risk is high.
In the field of obstetrics, the debate about the etiology of CP is important largely due to the medicolegal implications. Patient-oriented information on the internet often states that CP is caused by damage to the child’s brain just before, during, or soon after birth, supporting potential blame of those providing care during those times. Patient-oriented websites regarding CP do not list genetic disorders among the causes but rather include primarily environmental factors, such as prematurity, low birth weight, in utero infections, anoxia or other brain injury, or perinatal stroke. Even the Centers for Disease Control and Prevention website lists brain damage as the primary etiology of CP.14 Hopefully, these new data will increase a broader understanding of this condition.
Exome sequencing is now recommended as a first-tier test for individuals with many neurodevelopmental disorders, including epilepsy, intellectual disability, and autism spectrum disorder.15 However, comprehensive genetic testing is not typically recommended or performed in cases of CP. Based on recent data, including the report by Moreno-De-Luca and colleagues, it would seem that CP should be added to the list of disorders for which exome sequencing is ordered, given the similar prevalence and diagnostic yield. ●
Cerebral palsy (CP) is the most common cause of severe neurodisability in children, and it occurs in about 2 to 3 per 1,000 births worldwide.1 This nonprogressive disorder is characterized by symptoms that include spasticity, dystonia, choreoathetosis, and/or ataxia that are evident in the first few years of life. While many perinatal variables have been associated with CP, in most cases a specific cause is not identified.
Other neurodevelopmental disorders, such as intellectual disability, epilepsy, and autism spectrum disorder, are often associated with CP.2 These other neurodevelopmental disorders are often genetic, and this has raised the question as to whether CP also might have a substantial genetic component, although this has not been investigated in any significant way until recently. This topic is of great interest to the obstetric community, given that CP often is attributed to obstetric events, including mismanagement of labor and delivery.
Emerging evidence of a genetic-CP association
In an article published recently in JAMA, Moreno-De-Luca and colleagues sought to determine the diagnostic yield of exome sequencing for CP.3 This large cross-sectional study included results of exome sequencing performed in 2 settings. The first setting was a commercial laboratory in which samples were sent for analysis due to a diagnosis of CP, primarily in children (n = 1,345) with a median age of 8.8 years. A second cohort, recruited from a neurodevelopmental disorders clinic at Geisinger, included primarily adults (n = 181) with a median age of 41.9 years.
As is standard in exome sequencing, results were considered likely causative if they were classified as pathogenic or likely pathogenic based on criteria of the American College of Genetics and Genomics. In the laboratory group, 32.7% (440 of 1,345) had a genetic cause of the CP identified, while in the clinic group, 10.5% (19 of 181) had a genetic etiology found. Although most of the identified genetic variants were de novo (that is, they arose in the affected individual and were not clearly inherited), some were inherited from carrier parents.3
A number of other recent studies also have investigated genetic causes of CP and similarly have reported that a substantial number of cases are genetic. Several studies that performed chromosomal microarray analysis in individuals with CP found deleterious copy number variants in 10% to 31% of cases.4-6 Genomic variants detectable by exome sequencing have been reported in 15% to 20% of cases.3 In a recent study in Nature Genetics, researchers performed exome sequencing on 250 parent-child “trios” in which the child had CP, and they found that 14% of cases had an associated genetic variant that was thought to be causative.4 These studies all provide consistent evidence that a substantial proportion of CP cases are due to genetic causes.
Contributors to CP risk
Historically, CP was considered to occur largely as a result of perinatal anoxia. In 1862, the British orthopedic surgeon William John Little first reported an association between prematurity, asphyxia, difficult delivery, and CP in a paper presented to the Obstetrical Society of London.7 Subsequently, much effort has gone into the prevention of perinatal asphyxia and birth injury, although our ability to monitor fetal well-being remains limited. Nonreassuring fetal heart rate patterns are nonspecific and can occur for many reasons other than fetal asphyxia. Studies of electronic fetal monitoring have found that continuous monitoring primarily leads to an increase in cesarean delivery with no decrease in CP or infant mortality.8
While some have attributed this to failure to accurately interpret the fetal heart rate tracing, it also may be because a substantial number of CP cases are due to genetic and other causes, and that very few in fact result from preventable intrapartum injury.
The American College of Obstetricians and Gynecologists and the American Academy of Pediatrics agree that knowledge gaps preclude definitive determination that a given case of neonatal encephalopathy is attributable to an acute intrapartum event, and they provide criteria that must be fulfilled to establish a reasonable causal link between an intrapartum event and subsequent long-term neurologic disability.9 However, there continues to be a belief in the medical, scientific, and lay communities that birth asphyxia, secondary to adverse intrapartum events, is the leading cause of CP. A “brain-damaged infant” remains one of the most common malpractice claims, and birth injury one of the highest paid claims. Such claims generally allege that intrapartum asphyxia has caused long-term neurologic sequelae, including CP.
While it is true that prematurity, infection, hypoxia-ischemia, and pre- and perinatal stroke all have been implicated as contributing to CP risk, large population-based studies have shown that birth asphyxia accounts for less than 12% of CP cases.10 Specifically, recent data indicate that acute intrapartum hypoxia-ischemia occurs only in about 6% of CP cases. In other words, it does occur and may contribute to some cases, but this is likely a smaller percent than previously thought, and genetic factors now appear to be far more significant contributors.11
Continue to: Exploring a genetic etiology...
Exploring a genetic etiology
In considering the etiologies of CP, it is important to note that 21% to 40% of individuals with CP have an associated congenital anomaly, suggesting a genetic origin in at least some individuals. Moreover, a 40% heritability has been estimated in CP, which is comparable to the heritability rate for autism spectrum disorders.12
In the recent study by Moreno-De-Luca and colleagues, some of the gene variants detected were previously associated with other forms of neurodevelopmental disability, such as epilepsy and autism spectrum disorder.3 Many individuals in the study cohort were found to have multiple neurologic comorbidities, for example, CP as well as epilepsy, autism spectrum disorder, and/or intellectual disability. The presence of these additional comorbidities increased the likelihood of finding a genetic cause; the authors found that the diagnostic yield ranged from 11.2% with isolated CP to 32.9% with all 3 comorbidities. The yield was highest with CP and intellectual disability and CP with all 3 comorbidities. A few genes were particularly common, and some were reported previously in association with CP and/or other neurodevelopmental disorders. In some patients, variants were found in genes or gene regions associated with disorders that do not frequently include CP, such as Rett syndrome.3
Implications for ObGyns
The data from the study by Moreno-De-Luca and colleagues are interesting and relevant to pediatricians, neurologists, and geneticists, as well as obstetricians. Understanding the cause of any disease or disorder improves care, including counseling regarding the cause, the appropriate interventions or therapy, and in some families, the recurrence risk in another pregnancy. The treatment for CP has not changed significantly in many years. Increasingly, detection of an underlying genetic cause can guide precision treatments; thus, the detection of specific gene variants allows a targeted approach to therapy.
Identification of a genetic cause also can significantly impact recurrence risk counseling and prenatal diagnosis options in another pregnancy. In general, the empiric recurrence risk of CP is quoted as 1% to 2%,13 and with de novo variants this does not change. However, with inherited variants the recurrence risk in future children is substantially higher. While 72% of the genetic variants associated with CP in the Moreno-De-Luca study were de novo with a low recurrence risk, in the other 28% the mode of inheritance indicated a substantial risk of recurrence (25%–50%) in another pregnancy.3 Detecting such causative variant(s) allows not only accurate counseling about recurrence risk but also preimplantation genetic testing or prenatal diagnosis when recurrence risk is high.
In the field of obstetrics, the debate about the etiology of CP is important largely due to the medicolegal implications. Patient-oriented information on the internet often states that CP is caused by damage to the child’s brain just before, during, or soon after birth, supporting potential blame of those providing care during those times. Patient-oriented websites regarding CP do not list genetic disorders among the causes but rather include primarily environmental factors, such as prematurity, low birth weight, in utero infections, anoxia or other brain injury, or perinatal stroke. Even the Centers for Disease Control and Prevention website lists brain damage as the primary etiology of CP.14 Hopefully, these new data will increase a broader understanding of this condition.
Exome sequencing is now recommended as a first-tier test for individuals with many neurodevelopmental disorders, including epilepsy, intellectual disability, and autism spectrum disorder.15 However, comprehensive genetic testing is not typically recommended or performed in cases of CP. Based on recent data, including the report by Moreno-De-Luca and colleagues, it would seem that CP should be added to the list of disorders for which exome sequencing is ordered, given the similar prevalence and diagnostic yield. ●
- Oskoui M, Coutinho F, Dykeman J, et al. An update on the prevalence of cerebral palsy: a systematic review and meta-analysis. Dev Med Child Neurol. 2013;55:509-519.
- Rosenbaum P, Paneth N, Leviton A, et al. A report: the definition and classification of cerebral palsy April 2006. Dev Med Child Neurol Suppl. 2007;109:8-14.
- Moreno-De-Luca A, Millan F, Pesacreta DR, et al. Molecular diagnostic yield of exome sequencing in patients with cerebral palsy. JAMA. 2021;325:467-475.
- Jin SC, Lewis SA, Bakhtiari S, et al. Mutations disrupting neuritogenesis genes confer risk for cerebral palsy. Nat Genet. 2020;52:1046-1056.
- Segel R, Ben-Pazi H, Zeligson S, et al. Copy number variations in cryptogenic cerebral palsy. Neurology. 2015;84:1660-1668.
- McMichael G, Girirrajan S, Moreno-De-Luca A, et al. Rare copy number variation in cerebral palsy. Eur J Hum Genet. 2014;22:40-45.
- Little WJ. On the influence of abnormal parturition, difficult labours, premature births, and asphyxia neonatorum, on the mental and physical condition of the child, especially in relation to deformities. Trans Obstet Soc Lond. 1862;3:293-344.
- Alfirevic Z, Devane D, Gyte GM. Continuous cardiotocography (CTG) as a form of electronic fetal monitoring (EFM) for fetal assessment during labour. Cochrane Database Syst Rev. 2013;5;CD006066.
- American College of Obstetricians and Gynecologists. Executive summary: neonatal encephalopathy and neurologic outcome second edition. Report of the American College of Obstetricians and Gynecologists’ Task Force on Neonatal Encephalopathy. Obstet Gynecol. 2014;123:896- 901.
- Ellenberg JH, Nelson KB. The association of cerebral palsy with birth asphyxia: a definitional quagmire. Dev Med Child Neurol. 2013;55:210- 216.
- Himmelmann K, Uvebrant P. The panorama of cerebral palsy in Sweden part XII shows that patterns changed in the birth years 2007–2010. Acta Paediatr. 2018;107: 462-468.
- Petterson B, Stanley F, Henderson D. Cerebral palsy in multiple births in Western Australia: genetic aspects. Am J Med Genet. 1990;37:346- 351.
- Korzeniewski SJ, Slaughter J, Lenski M, et al. The complex aetiology of cerebral palsy. Nat Rev Neurol. 2018;14:528-543.
- Centers for Disease Control and Prevention. Causes and risk factors of cerebral palsy. https:// www.cdc.gov/ncbddd/cp/causes.html. Accessed March 23, 2021.
- Srivastava S, Love-Nichols JA, Dies KA, et al; NDD Exome Scoping Review Work Group. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. 2019;21:2413-2421.
- Oskoui M, Coutinho F, Dykeman J, et al. An update on the prevalence of cerebral palsy: a systematic review and meta-analysis. Dev Med Child Neurol. 2013;55:509-519.
- Rosenbaum P, Paneth N, Leviton A, et al. A report: the definition and classification of cerebral palsy April 2006. Dev Med Child Neurol Suppl. 2007;109:8-14.
- Moreno-De-Luca A, Millan F, Pesacreta DR, et al. Molecular diagnostic yield of exome sequencing in patients with cerebral palsy. JAMA. 2021;325:467-475.
- Jin SC, Lewis SA, Bakhtiari S, et al. Mutations disrupting neuritogenesis genes confer risk for cerebral palsy. Nat Genet. 2020;52:1046-1056.
- Segel R, Ben-Pazi H, Zeligson S, et al. Copy number variations in cryptogenic cerebral palsy. Neurology. 2015;84:1660-1668.
- McMichael G, Girirrajan S, Moreno-De-Luca A, et al. Rare copy number variation in cerebral palsy. Eur J Hum Genet. 2014;22:40-45.
- Little WJ. On the influence of abnormal parturition, difficult labours, premature births, and asphyxia neonatorum, on the mental and physical condition of the child, especially in relation to deformities. Trans Obstet Soc Lond. 1862;3:293-344.
- Alfirevic Z, Devane D, Gyte GM. Continuous cardiotocography (CTG) as a form of electronic fetal monitoring (EFM) for fetal assessment during labour. Cochrane Database Syst Rev. 2013;5;CD006066.
- American College of Obstetricians and Gynecologists. Executive summary: neonatal encephalopathy and neurologic outcome second edition. Report of the American College of Obstetricians and Gynecologists’ Task Force on Neonatal Encephalopathy. Obstet Gynecol. 2014;123:896- 901.
- Ellenberg JH, Nelson KB. The association of cerebral palsy with birth asphyxia: a definitional quagmire. Dev Med Child Neurol. 2013;55:210- 216.
- Himmelmann K, Uvebrant P. The panorama of cerebral palsy in Sweden part XII shows that patterns changed in the birth years 2007–2010. Acta Paediatr. 2018;107: 462-468.
- Petterson B, Stanley F, Henderson D. Cerebral palsy in multiple births in Western Australia: genetic aspects. Am J Med Genet. 1990;37:346- 351.
- Korzeniewski SJ, Slaughter J, Lenski M, et al. The complex aetiology of cerebral palsy. Nat Rev Neurol. 2018;14:528-543.
- Centers for Disease Control and Prevention. Causes and risk factors of cerebral palsy. https:// www.cdc.gov/ncbddd/cp/causes.html. Accessed March 23, 2021.
- Srivastava S, Love-Nichols JA, Dies KA, et al; NDD Exome Scoping Review Work Group. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. 2019;21:2413-2421.
2019 Update on prenatal exome sequencing
Prenatal diagnosis of genetic anomalies is important for diagnosing lethal genetic conditions before birth. It can provide information for parents regarding pregnancy options and allow for recurrence risk counseling and the potential use of preimplantation genetic testing in the next pregnancy. For decades, a karyotype was used to analyze amniocentesis and chorionic villus sampling specimens; in recent years, chromosomal microarray analysis provides more information about significant chromosomal abnormalities, including microdeletions and microduplications. However, microarrays also have limitations, as they do not identify base pair changes associated with single-gene disorders.
The advent of next-generation sequencing has substantially reduced the cost of DNA sequencing. Whole genome sequencing (WGS) can sequence the entire genome— both the coding (exonic) and noncoding (intronic) regions—while exome sequencing analyzes only the protein-coding exons, which make up 1% to 2% of the genome and about 85% of the protein-coding genes associated with known human disease. Exome sequencing increasingly is used in cases of suspected genetic disorders when other tests have been unrevealing.
In this Update, we review recent reports of prenatal exome sequencing, including studies exploring the yield in fetuses with structural anomalies; the importance of prenatal phenotyping; the perspectives of parents and health care professionals who were involved in prenatal exome sequencing studies; and a summary of a joint position statement from 3 societies regarding prenatal sequencing.
Prenatal whole exome sequencing has potential utility, with some limitations
Petrovski S, Aggarwal V, Giordano JL, et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet. 2019;393:758-767.
Lord J, McMullan DJ, Eberhardt RY, et al; for the Prenatal Assessment of Genomes and Exomes Consortium. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet. 2019;393:747-757.
Exome sequencing has been shown to identify an underlying genetic cause in 25% to 30% of children with an undiagnosed suspected genetic disorder. Two studies recently published in the Lancet sought to determine the incremental diagnostic yield of prenatal whole exome sequencing (WES) in the setting of fetal structural anomalies when karyotype and microarray results were normal.
Continue to: Details of the studies...
Details of the studies
In a prospective cohort study by Petrovski and colleagues, DNA samples from 234 fetuses with a structural anomaly (identified on ultrasonography) and both parents (parent-fetus "trios") were used for analysis. WES identified diagnostic genetic variants in 24 trios (10%). An additional 46 (20%) had variants that indicated pathogenicity but without sufficient evidence to be considered diagnostic.
The anomalies with the highest frequency of a genetic diagnosis were lymphatic, 24%; skeletal, 24%; central nervous system, 22%; and renal, 16%; while cardiac anomalies had the lowest yield at 5%.
In another prospective cohort study, known as the Prenatal Assessment of Genomes and Exomes (PAGE), Lord and colleagues sequenced DNA samples from 610 parent-fetus trios, but they restricted sequencing to a predefined list of 1,628 genes. Diagnostic genetic variants were identified in 52 fetuses (8.5%), while 24 (3.9%) had a variant of uncertain significance that was thought to be of potential clinical usefulness.
Fetuses with multiple anomalies had the highest genetic yield (15.4%), followed by skeletal (15.4%) and cardiac anomalies (11.1%), with the lowest yield in fetuses with isolated increased nuchal translucency (3.2%).
Diagnostic yield is high, but prenatal utility is limited
Both studies showed a clinically significant diagnostic yield of 8% to 10% for prenatal exome sequencing in cases of fetal structural anomalies with normal karyotype and microarray testing. While this yield demonstrates the utility of prenatal exome sequencing, it is significantly lower than what has been reported in postnatal studies. One of the reasons for this is the inherent limitation of prenatal phenotyping (discussed below).
The cohort studies by both Petrovski and Lord and their colleagues show the feasibility and potential diagnostic utility of exome sequencing in cases of fetal structural anomalies where karyotype and microarray are not diagnostic. However, the lower yield found in these studies compared with those in postnatal studies highlights in part the limitations of prenatal phenotyping.
The importance of prenatal phenotyping
Aarabi M, Sniezek O, Jiang H, et al. Importance of complete phenotyping in prenatal whole exome sequencing. Hum Genet. 2018;137:175-181.
In postnatal exome sequencing, the physical exam, imaging findings, and laboratory results are components of the phenotype that are used to interpret the sequencing data. Prenatal phenotyping, however, is limited to the use of fetal ultrasonography and, occasionally, the addition of magnetic resonance imaging. Prenatal phenotyping is without the benefit of an exam to detect more subtle anomalies or functional status, such as developmental delay, seizures, or failure to thrive.
When a structural anomaly is identified on prenatal ultrasonography, it is especially important that detailed imaging be undertaken to detect other anomalies, including more subtle facial features and dysmorphology.
Value of reanalyzing exome sequencing data
Aarabi and colleagues conducted a retrospective study of 20 fetuses with structural anomalies and normal karyotype and microarray. They performed trio exome sequencing first using information available only prenatally and then conducted a reanalysis using information available after delivery.
With prenatal phenotyping only, the investigators identified no pathogenic, or likely pathogenic, variants. On reanalysis of combined prenatal and postnatal findings, however, they identified pathogenic variants in 20% of cases.
Significance of the findings
This study highlights both the importance of a careful, detailed fetal ultrasonography study and the possible additional benefit of a postnatal examination (such as an autopsy) in order to yield improved results. In addition, the authors noted that the development of a prenatal phenotype-genotype database would significantly help exome sequencing interpretation in the prenatal setting.
Careful prenatal ultrasonography is crucial to help in the interpretation of prenatal exome sequencing. Patients who have undergone prenatal clinical exome sequencing may benefit from reanalysis of the genetic data based on detailed postnatal findings.
Social impact of WES: Parent and provider perspectives
Wou K, Weitz T, McCormack C, et al. Parental perceptions of prenatal whole exome sequencing (PPPWES) study. Prenat Diagn. 2018;38:801-811.
Horn R, Parker M. Health professionals' and researchers' perspectives on prenatal whole genome and exome sequencing: 'We can't shut the door now, the genie's out, we need to refine it.' PLoS One. 2018;13:e0204158.
As health care providers enter a new era of prenatal genetic testing with exome sequencing, it is crucial to the path forward that we obtain perspectives from the parents and providers who participated in these studies. Notably, in both of the previously discussed Lancet reports, the authors interviewed the participants to discuss the challenges involved and identify strategies for improving future testing.
Continue to: What parents want...
What parents want
To ascertain the perceptions of couples who underwent prenatal WES, Wou and colleagues conducted semi-structured interviews with participants from the Fetal Sequencing Study regarding their experience. They interviewed 29 parents from 17 pregnancies, including a mix of those who had pathogenic prenatal results, terminated prior to receiving the results, and had normal results.
Expressed feelings and desires. Parents recalled feelings of anxiety and stress around the time of diagnosis and the need for help with coping while awaiting results. The majority of parents reported that they would like to be told about uncertain results, but that desire decreased as the certainty of results decreased.
Parents were overall satisfied with the prenatal genetic testing experience, but they added that they would have liked to receive written materials beforehand and a written report of the test results (including negative cases). They also would like to have connected with other families with similar experiences, to have received results sooner, and to have an in-person meeting after telephone disclosure of the results.
Health professionals articulate complexity of prenatal genomics
In a qualitative interview study to explore critical issues involved in the clinical practice use of prenatal genomics, Horn and Parker conducted interviews with 20 health care professionals who were involved in the previously described PAGE trial. Patient recruiters, midwives, genetic counselors, research assistants, and laboratory staff were included.
Interviewees cited numerous challenges involved in their day-to-day work with prenatal whole genome and exome sequencing, including:
- the complexity of achieving valid parental consent at a time of vulnerability
- management of parent expectations
- transmitting and comprehending complex information
- the usefulness of information
- the difficulty of a long turnaround time for study results.
All the interviewees agreed that prenatal exome sequencing studies contribute to knowledge generation and the advancement of technology.
The authors concluded that an appropriate next step would be the development of appropriate guidelines for good ethical practice that address the concerns encountered in genomics clinical practice.
The prenatal experience can be overwhelming for parents. Pretest and posttest counseling on genetic testing and results are of the utmost importance, as is finding ways to help support parents through this anxious time.
Societies offer guidance on using genome and exome sequencing
International Society for Prenatal Diagnosis, Society for Maternal and Fetal Medicine, Perinatal Quality Foundation. Joint Position Statement from the International Society for Prenatal Diagnosis (ISPD), the Society for Maternal Fetal Medicine (SMFM), and the Perinatal Quality Foundation (PQF) on the use of genome-wide sequencing for fetal diagnosis. Prenat Diagn. 2018;38:6-9.
In response to the rapid integration of exome sequencing for genetic diagnosis, several professional societies—the International Society for Prenatal Diagnosis, Society for Maternal Fetal Medicine, and Perinatal Quality Foundation—issued a joint statement addressing the clinical use of prenatal diagnostic genome wide sequencing, including exome sequencing.
Continue to: Guidance at a glance...
Guidance at a glance
The societies' recommendations are summarized as follows:
- Exome sequencing is best done as a trio analysis, with fetal and both parental samples sequenced and analyzed together.
- Extensive pretest education, counseling, and informed consent, as well as posttest counseling, are essential. This should include:
—the types of results to be conveyed (variants that are pathogenic, likely pathogenic, of uncertain significance, likely benign, and benign)
—the possibility that results will not be obtained or may not be available before the birth of the fetus
—realistic expectations regarding the likelihood that a significant result will be obtained
—the timeframe to results
—the option to include or exclude in the results incidental or secondary findings (such as an unexpected childhood disorder, cancer susceptibility genes, adult-onset disorders)
—the possibility of uncovering nonpaternity or consanguinity
—the potential reanalysis of results over time
—how data are stored, who has access, and for what purpose.
- Fetal sequencing may be beneficial in the following scenarios:
—multiple fetal anomalies or a single major anomaly suggestive of a genetic disorder, when the microarray is negative
—no microarray result is available, but the fetus exhibits a pattern of anomalies strongly suggestive of a single-gene disorder
—a prior undiagnosed fetus (or child) with anomalies suggestive of a genetic etiology, and with similar anomalies in the current pregnancy, with normal karyotype or microarray. Providers also can consider sequencing samples from both parents prior to preimplantation genetic testing to check for shared carrier status for autosomal recessive mutations, although obtaining exome sequencing from the prior affected fetus (or child) is ideal.
—history of recurrent stillbirths of unknown etiology, with a recurrent pattern of anomalies in the current pregnancy, with normal karyotype or microarray.
- Interpretation of results should be done using a multidisciplinary team-based approach, including clinical scientists, geneticists, genetic counselors, and experts in prenatal diagnosis.
- Where possible and after informed consent, reanalysis of results should be undertaken if a future pregnancy is planned or ongoing, and a significant amount of time has elapsed since the time the result was last reported.
- Parents should be given a written report of test results.
Three professional societies have convened to issue consensus opinion that includes current indications for prenatal exome sequencing and important factors to include in the consent process. We follow these guidelines in our own practice.
Summary
Exome sequencing is increasingly becoming mainstream in postnatal genetic testing, and it is emerging as the newest diagnostic frontier in prenatal genetic testing. However, there are limitations to prenatal exome sequencing, including issues with consent at a vulnerable time for parents, limited information available regarding the phenotype, and results that may not be available before the birth of a fetus. Providers should be familiar with the indications for testing, the possible results, the limitations of prenatal phenotyping, and the implications for future pregnancies.

Anne H. Mardy, MD
Dr. Mardy is Fellow in Maternal Fetal Medicine and Clinical Genetics, University of California, San Francisco.

Mary E. Norton, MD
Dr. Norton is Professor of Obstetrics, Gynecology, and Reproductive Sciences, Division of Maternal Fetal Medicine, University of California, San Francisco.
Dr. Norton reports that she has received grant or research support from Natera and that she is a consultant to Invitae. Dr. Mardy reports no financial relationships relevant to this article.
Anne H. Mardy, MD
Dr. Mardy is Fellow in Maternal Fetal Medicine and Clinical Genetics, University of California, San Francisco.
Mary E. Norton, MD
Dr. Norton is Professor of Obstetrics, Gynecology, and Reproductive Sciences, Division of Maternal Fetal Medicine, University of California, San Francisco.
Dr. Norton reports that she has received grant or research support from Natera and that she is a consultant to Invitae. Dr. Mardy reports no financial relationships relevant to this article.
Anne H. Mardy, MD
Dr. Mardy is Fellow in Maternal Fetal Medicine and Clinical Genetics, University of California, San Francisco.
Mary E. Norton, MD
Dr. Norton is Professor of Obstetrics, Gynecology, and Reproductive Sciences, Division of Maternal Fetal Medicine, University of California, San Francisco.
Dr. Norton reports that she has received grant or research support from Natera and that she is a consultant to Invitae. Dr. Mardy reports no financial relationships relevant to this article.
Prenatal diagnosis of genetic anomalies is important for diagnosing lethal genetic conditions before birth. It can provide information for parents regarding pregnancy options and allow for recurrence risk counseling and the potential use of preimplantation genetic testing in the next pregnancy. For decades, a karyotype was used to analyze amniocentesis and chorionic villus sampling specimens; in recent years, chromosomal microarray analysis provides more information about significant chromosomal abnormalities, including microdeletions and microduplications. However, microarrays also have limitations, as they do not identify base pair changes associated with single-gene disorders.
The advent of next-generation sequencing has substantially reduced the cost of DNA sequencing. Whole genome sequencing (WGS) can sequence the entire genome— both the coding (exonic) and noncoding (intronic) regions—while exome sequencing analyzes only the protein-coding exons, which make up 1% to 2% of the genome and about 85% of the protein-coding genes associated with known human disease. Exome sequencing increasingly is used in cases of suspected genetic disorders when other tests have been unrevealing.
In this Update, we review recent reports of prenatal exome sequencing, including studies exploring the yield in fetuses with structural anomalies; the importance of prenatal phenotyping; the perspectives of parents and health care professionals who were involved in prenatal exome sequencing studies; and a summary of a joint position statement from 3 societies regarding prenatal sequencing.
Prenatal whole exome sequencing has potential utility, with some limitations
Petrovski S, Aggarwal V, Giordano JL, et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet. 2019;393:758-767.
Lord J, McMullan DJ, Eberhardt RY, et al; for the Prenatal Assessment of Genomes and Exomes Consortium. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet. 2019;393:747-757.
Exome sequencing has been shown to identify an underlying genetic cause in 25% to 30% of children with an undiagnosed suspected genetic disorder. Two studies recently published in the Lancet sought to determine the incremental diagnostic yield of prenatal whole exome sequencing (WES) in the setting of fetal structural anomalies when karyotype and microarray results were normal.
Continue to: Details of the studies...
Details of the studies
In a prospective cohort study by Petrovski and colleagues, DNA samples from 234 fetuses with a structural anomaly (identified on ultrasonography) and both parents (parent-fetus "trios") were used for analysis. WES identified diagnostic genetic variants in 24 trios (10%). An additional 46 (20%) had variants that indicated pathogenicity but without sufficient evidence to be considered diagnostic.
The anomalies with the highest frequency of a genetic diagnosis were lymphatic, 24%; skeletal, 24%; central nervous system, 22%; and renal, 16%; while cardiac anomalies had the lowest yield at 5%.
In another prospective cohort study, known as the Prenatal Assessment of Genomes and Exomes (PAGE), Lord and colleagues sequenced DNA samples from 610 parent-fetus trios, but they restricted sequencing to a predefined list of 1,628 genes. Diagnostic genetic variants were identified in 52 fetuses (8.5%), while 24 (3.9%) had a variant of uncertain significance that was thought to be of potential clinical usefulness.
Fetuses with multiple anomalies had the highest genetic yield (15.4%), followed by skeletal (15.4%) and cardiac anomalies (11.1%), with the lowest yield in fetuses with isolated increased nuchal translucency (3.2%).
Diagnostic yield is high, but prenatal utility is limited
Both studies showed a clinically significant diagnostic yield of 8% to 10% for prenatal exome sequencing in cases of fetal structural anomalies with normal karyotype and microarray testing. While this yield demonstrates the utility of prenatal exome sequencing, it is significantly lower than what has been reported in postnatal studies. One of the reasons for this is the inherent limitation of prenatal phenotyping (discussed below).
The cohort studies by both Petrovski and Lord and their colleagues show the feasibility and potential diagnostic utility of exome sequencing in cases of fetal structural anomalies where karyotype and microarray are not diagnostic. However, the lower yield found in these studies compared with those in postnatal studies highlights in part the limitations of prenatal phenotyping.
The importance of prenatal phenotyping
Aarabi M, Sniezek O, Jiang H, et al. Importance of complete phenotyping in prenatal whole exome sequencing. Hum Genet. 2018;137:175-181.
In postnatal exome sequencing, the physical exam, imaging findings, and laboratory results are components of the phenotype that are used to interpret the sequencing data. Prenatal phenotyping, however, is limited to the use of fetal ultrasonography and, occasionally, the addition of magnetic resonance imaging. Prenatal phenotyping is without the benefit of an exam to detect more subtle anomalies or functional status, such as developmental delay, seizures, or failure to thrive.
When a structural anomaly is identified on prenatal ultrasonography, it is especially important that detailed imaging be undertaken to detect other anomalies, including more subtle facial features and dysmorphology.
Value of reanalyzing exome sequencing data
Aarabi and colleagues conducted a retrospective study of 20 fetuses with structural anomalies and normal karyotype and microarray. They performed trio exome sequencing first using information available only prenatally and then conducted a reanalysis using information available after delivery.
With prenatal phenotyping only, the investigators identified no pathogenic, or likely pathogenic, variants. On reanalysis of combined prenatal and postnatal findings, however, they identified pathogenic variants in 20% of cases.
Significance of the findings
This study highlights both the importance of a careful, detailed fetal ultrasonography study and the possible additional benefit of a postnatal examination (such as an autopsy) in order to yield improved results. In addition, the authors noted that the development of a prenatal phenotype-genotype database would significantly help exome sequencing interpretation in the prenatal setting.
Careful prenatal ultrasonography is crucial to help in the interpretation of prenatal exome sequencing. Patients who have undergone prenatal clinical exome sequencing may benefit from reanalysis of the genetic data based on detailed postnatal findings.
Social impact of WES: Parent and provider perspectives
Wou K, Weitz T, McCormack C, et al. Parental perceptions of prenatal whole exome sequencing (PPPWES) study. Prenat Diagn. 2018;38:801-811.
Horn R, Parker M. Health professionals' and researchers' perspectives on prenatal whole genome and exome sequencing: 'We can't shut the door now, the genie's out, we need to refine it.' PLoS One. 2018;13:e0204158.
As health care providers enter a new era of prenatal genetic testing with exome sequencing, it is crucial to the path forward that we obtain perspectives from the parents and providers who participated in these studies. Notably, in both of the previously discussed Lancet reports, the authors interviewed the participants to discuss the challenges involved and identify strategies for improving future testing.
Continue to: What parents want...
What parents want
To ascertain the perceptions of couples who underwent prenatal WES, Wou and colleagues conducted semi-structured interviews with participants from the Fetal Sequencing Study regarding their experience. They interviewed 29 parents from 17 pregnancies, including a mix of those who had pathogenic prenatal results, terminated prior to receiving the results, and had normal results.
Expressed feelings and desires. Parents recalled feelings of anxiety and stress around the time of diagnosis and the need for help with coping while awaiting results. The majority of parents reported that they would like to be told about uncertain results, but that desire decreased as the certainty of results decreased.
Parents were overall satisfied with the prenatal genetic testing experience, but they added that they would have liked to receive written materials beforehand and a written report of the test results (including negative cases). They also would like to have connected with other families with similar experiences, to have received results sooner, and to have an in-person meeting after telephone disclosure of the results.
Health professionals articulate complexity of prenatal genomics
In a qualitative interview study to explore critical issues involved in the clinical practice use of prenatal genomics, Horn and Parker conducted interviews with 20 health care professionals who were involved in the previously described PAGE trial. Patient recruiters, midwives, genetic counselors, research assistants, and laboratory staff were included.
Interviewees cited numerous challenges involved in their day-to-day work with prenatal whole genome and exome sequencing, including:
- the complexity of achieving valid parental consent at a time of vulnerability
- management of parent expectations
- transmitting and comprehending complex information
- the usefulness of information
- the difficulty of a long turnaround time for study results.
All the interviewees agreed that prenatal exome sequencing studies contribute to knowledge generation and the advancement of technology.
The authors concluded that an appropriate next step would be the development of appropriate guidelines for good ethical practice that address the concerns encountered in genomics clinical practice.
The prenatal experience can be overwhelming for parents. Pretest and posttest counseling on genetic testing and results are of the utmost importance, as is finding ways to help support parents through this anxious time.
Societies offer guidance on using genome and exome sequencing
International Society for Prenatal Diagnosis, Society for Maternal and Fetal Medicine, Perinatal Quality Foundation. Joint Position Statement from the International Society for Prenatal Diagnosis (ISPD), the Society for Maternal Fetal Medicine (SMFM), and the Perinatal Quality Foundation (PQF) on the use of genome-wide sequencing for fetal diagnosis. Prenat Diagn. 2018;38:6-9.
In response to the rapid integration of exome sequencing for genetic diagnosis, several professional societies—the International Society for Prenatal Diagnosis, Society for Maternal Fetal Medicine, and Perinatal Quality Foundation—issued a joint statement addressing the clinical use of prenatal diagnostic genome wide sequencing, including exome sequencing.
Continue to: Guidance at a glance...
Guidance at a glance
The societies' recommendations are summarized as follows:
- Exome sequencing is best done as a trio analysis, with fetal and both parental samples sequenced and analyzed together.
- Extensive pretest education, counseling, and informed consent, as well as posttest counseling, are essential. This should include:
—the types of results to be conveyed (variants that are pathogenic, likely pathogenic, of uncertain significance, likely benign, and benign)
—the possibility that results will not be obtained or may not be available before the birth of the fetus
—realistic expectations regarding the likelihood that a significant result will be obtained
—the timeframe to results
—the option to include or exclude in the results incidental or secondary findings (such as an unexpected childhood disorder, cancer susceptibility genes, adult-onset disorders)
—the possibility of uncovering nonpaternity or consanguinity
—the potential reanalysis of results over time
—how data are stored, who has access, and for what purpose.
- Fetal sequencing may be beneficial in the following scenarios:
—multiple fetal anomalies or a single major anomaly suggestive of a genetic disorder, when the microarray is negative
—no microarray result is available, but the fetus exhibits a pattern of anomalies strongly suggestive of a single-gene disorder
—a prior undiagnosed fetus (or child) with anomalies suggestive of a genetic etiology, and with similar anomalies in the current pregnancy, with normal karyotype or microarray. Providers also can consider sequencing samples from both parents prior to preimplantation genetic testing to check for shared carrier status for autosomal recessive mutations, although obtaining exome sequencing from the prior affected fetus (or child) is ideal.
—history of recurrent stillbirths of unknown etiology, with a recurrent pattern of anomalies in the current pregnancy, with normal karyotype or microarray.
- Interpretation of results should be done using a multidisciplinary team-based approach, including clinical scientists, geneticists, genetic counselors, and experts in prenatal diagnosis.
- Where possible and after informed consent, reanalysis of results should be undertaken if a future pregnancy is planned or ongoing, and a significant amount of time has elapsed since the time the result was last reported.
- Parents should be given a written report of test results.
Three professional societies have convened to issue consensus opinion that includes current indications for prenatal exome sequencing and important factors to include in the consent process. We follow these guidelines in our own practice.
Summary
Exome sequencing is increasingly becoming mainstream in postnatal genetic testing, and it is emerging as the newest diagnostic frontier in prenatal genetic testing. However, there are limitations to prenatal exome sequencing, including issues with consent at a vulnerable time for parents, limited information available regarding the phenotype, and results that may not be available before the birth of a fetus. Providers should be familiar with the indications for testing, the possible results, the limitations of prenatal phenotyping, and the implications for future pregnancies.
Prenatal diagnosis of genetic anomalies is important for diagnosing lethal genetic conditions before birth. It can provide information for parents regarding pregnancy options and allow for recurrence risk counseling and the potential use of preimplantation genetic testing in the next pregnancy. For decades, a karyotype was used to analyze amniocentesis and chorionic villus sampling specimens; in recent years, chromosomal microarray analysis provides more information about significant chromosomal abnormalities, including microdeletions and microduplications. However, microarrays also have limitations, as they do not identify base pair changes associated with single-gene disorders.
The advent of next-generation sequencing has substantially reduced the cost of DNA sequencing. Whole genome sequencing (WGS) can sequence the entire genome— both the coding (exonic) and noncoding (intronic) regions—while exome sequencing analyzes only the protein-coding exons, which make up 1% to 2% of the genome and about 85% of the protein-coding genes associated with known human disease. Exome sequencing increasingly is used in cases of suspected genetic disorders when other tests have been unrevealing.
In this Update, we review recent reports of prenatal exome sequencing, including studies exploring the yield in fetuses with structural anomalies; the importance of prenatal phenotyping; the perspectives of parents and health care professionals who were involved in prenatal exome sequencing studies; and a summary of a joint position statement from 3 societies regarding prenatal sequencing.
Prenatal whole exome sequencing has potential utility, with some limitations
Petrovski S, Aggarwal V, Giordano JL, et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet. 2019;393:758-767.
Lord J, McMullan DJ, Eberhardt RY, et al; for the Prenatal Assessment of Genomes and Exomes Consortium. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet. 2019;393:747-757.
Exome sequencing has been shown to identify an underlying genetic cause in 25% to 30% of children with an undiagnosed suspected genetic disorder. Two studies recently published in the Lancet sought to determine the incremental diagnostic yield of prenatal whole exome sequencing (WES) in the setting of fetal structural anomalies when karyotype and microarray results were normal.
Continue to: Details of the studies...
Details of the studies
In a prospective cohort study by Petrovski and colleagues, DNA samples from 234 fetuses with a structural anomaly (identified on ultrasonography) and both parents (parent-fetus "trios") were used for analysis. WES identified diagnostic genetic variants in 24 trios (10%). An additional 46 (20%) had variants that indicated pathogenicity but without sufficient evidence to be considered diagnostic.
The anomalies with the highest frequency of a genetic diagnosis were lymphatic, 24%; skeletal, 24%; central nervous system, 22%; and renal, 16%; while cardiac anomalies had the lowest yield at 5%.
In another prospective cohort study, known as the Prenatal Assessment of Genomes and Exomes (PAGE), Lord and colleagues sequenced DNA samples from 610 parent-fetus trios, but they restricted sequencing to a predefined list of 1,628 genes. Diagnostic genetic variants were identified in 52 fetuses (8.5%), while 24 (3.9%) had a variant of uncertain significance that was thought to be of potential clinical usefulness.
Fetuses with multiple anomalies had the highest genetic yield (15.4%), followed by skeletal (15.4%) and cardiac anomalies (11.1%), with the lowest yield in fetuses with isolated increased nuchal translucency (3.2%).
Diagnostic yield is high, but prenatal utility is limited
Both studies showed a clinically significant diagnostic yield of 8% to 10% for prenatal exome sequencing in cases of fetal structural anomalies with normal karyotype and microarray testing. While this yield demonstrates the utility of prenatal exome sequencing, it is significantly lower than what has been reported in postnatal studies. One of the reasons for this is the inherent limitation of prenatal phenotyping (discussed below).
The cohort studies by both Petrovski and Lord and their colleagues show the feasibility and potential diagnostic utility of exome sequencing in cases of fetal structural anomalies where karyotype and microarray are not diagnostic. However, the lower yield found in these studies compared with those in postnatal studies highlights in part the limitations of prenatal phenotyping.
The importance of prenatal phenotyping
Aarabi M, Sniezek O, Jiang H, et al. Importance of complete phenotyping in prenatal whole exome sequencing. Hum Genet. 2018;137:175-181.
In postnatal exome sequencing, the physical exam, imaging findings, and laboratory results are components of the phenotype that are used to interpret the sequencing data. Prenatal phenotyping, however, is limited to the use of fetal ultrasonography and, occasionally, the addition of magnetic resonance imaging. Prenatal phenotyping is without the benefit of an exam to detect more subtle anomalies or functional status, such as developmental delay, seizures, or failure to thrive.
When a structural anomaly is identified on prenatal ultrasonography, it is especially important that detailed imaging be undertaken to detect other anomalies, including more subtle facial features and dysmorphology.
Value of reanalyzing exome sequencing data
Aarabi and colleagues conducted a retrospective study of 20 fetuses with structural anomalies and normal karyotype and microarray. They performed trio exome sequencing first using information available only prenatally and then conducted a reanalysis using information available after delivery.
With prenatal phenotyping only, the investigators identified no pathogenic, or likely pathogenic, variants. On reanalysis of combined prenatal and postnatal findings, however, they identified pathogenic variants in 20% of cases.
Significance of the findings
This study highlights both the importance of a careful, detailed fetal ultrasonography study and the possible additional benefit of a postnatal examination (such as an autopsy) in order to yield improved results. In addition, the authors noted that the development of a prenatal phenotype-genotype database would significantly help exome sequencing interpretation in the prenatal setting.
Careful prenatal ultrasonography is crucial to help in the interpretation of prenatal exome sequencing. Patients who have undergone prenatal clinical exome sequencing may benefit from reanalysis of the genetic data based on detailed postnatal findings.
Social impact of WES: Parent and provider perspectives
Wou K, Weitz T, McCormack C, et al. Parental perceptions of prenatal whole exome sequencing (PPPWES) study. Prenat Diagn. 2018;38:801-811.
Horn R, Parker M. Health professionals' and researchers' perspectives on prenatal whole genome and exome sequencing: 'We can't shut the door now, the genie's out, we need to refine it.' PLoS One. 2018;13:e0204158.
As health care providers enter a new era of prenatal genetic testing with exome sequencing, it is crucial to the path forward that we obtain perspectives from the parents and providers who participated in these studies. Notably, in both of the previously discussed Lancet reports, the authors interviewed the participants to discuss the challenges involved and identify strategies for improving future testing.
Continue to: What parents want...
What parents want
To ascertain the perceptions of couples who underwent prenatal WES, Wou and colleagues conducted semi-structured interviews with participants from the Fetal Sequencing Study regarding their experience. They interviewed 29 parents from 17 pregnancies, including a mix of those who had pathogenic prenatal results, terminated prior to receiving the results, and had normal results.
Expressed feelings and desires. Parents recalled feelings of anxiety and stress around the time of diagnosis and the need for help with coping while awaiting results. The majority of parents reported that they would like to be told about uncertain results, but that desire decreased as the certainty of results decreased.
Parents were overall satisfied with the prenatal genetic testing experience, but they added that they would have liked to receive written materials beforehand and a written report of the test results (including negative cases). They also would like to have connected with other families with similar experiences, to have received results sooner, and to have an in-person meeting after telephone disclosure of the results.
Health professionals articulate complexity of prenatal genomics
In a qualitative interview study to explore critical issues involved in the clinical practice use of prenatal genomics, Horn and Parker conducted interviews with 20 health care professionals who were involved in the previously described PAGE trial. Patient recruiters, midwives, genetic counselors, research assistants, and laboratory staff were included.
Interviewees cited numerous challenges involved in their day-to-day work with prenatal whole genome and exome sequencing, including:
- the complexity of achieving valid parental consent at a time of vulnerability
- management of parent expectations
- transmitting and comprehending complex information
- the usefulness of information
- the difficulty of a long turnaround time for study results.
All the interviewees agreed that prenatal exome sequencing studies contribute to knowledge generation and the advancement of technology.
The authors concluded that an appropriate next step would be the development of appropriate guidelines for good ethical practice that address the concerns encountered in genomics clinical practice.
The prenatal experience can be overwhelming for parents. Pretest and posttest counseling on genetic testing and results are of the utmost importance, as is finding ways to help support parents through this anxious time.
Societies offer guidance on using genome and exome sequencing
International Society for Prenatal Diagnosis, Society for Maternal and Fetal Medicine, Perinatal Quality Foundation. Joint Position Statement from the International Society for Prenatal Diagnosis (ISPD), the Society for Maternal Fetal Medicine (SMFM), and the Perinatal Quality Foundation (PQF) on the use of genome-wide sequencing for fetal diagnosis. Prenat Diagn. 2018;38:6-9.
In response to the rapid integration of exome sequencing for genetic diagnosis, several professional societies—the International Society for Prenatal Diagnosis, Society for Maternal Fetal Medicine, and Perinatal Quality Foundation—issued a joint statement addressing the clinical use of prenatal diagnostic genome wide sequencing, including exome sequencing.
Continue to: Guidance at a glance...
Guidance at a glance
The societies' recommendations are summarized as follows:
- Exome sequencing is best done as a trio analysis, with fetal and both parental samples sequenced and analyzed together.
- Extensive pretest education, counseling, and informed consent, as well as posttest counseling, are essential. This should include:
—the types of results to be conveyed (variants that are pathogenic, likely pathogenic, of uncertain significance, likely benign, and benign)
—the possibility that results will not be obtained or may not be available before the birth of the fetus
—realistic expectations regarding the likelihood that a significant result will be obtained
—the timeframe to results
—the option to include or exclude in the results incidental or secondary findings (such as an unexpected childhood disorder, cancer susceptibility genes, adult-onset disorders)
—the possibility of uncovering nonpaternity or consanguinity
—the potential reanalysis of results over time
—how data are stored, who has access, and for what purpose.
- Fetal sequencing may be beneficial in the following scenarios:
—multiple fetal anomalies or a single major anomaly suggestive of a genetic disorder, when the microarray is negative
—no microarray result is available, but the fetus exhibits a pattern of anomalies strongly suggestive of a single-gene disorder
—a prior undiagnosed fetus (or child) with anomalies suggestive of a genetic etiology, and with similar anomalies in the current pregnancy, with normal karyotype or microarray. Providers also can consider sequencing samples from both parents prior to preimplantation genetic testing to check for shared carrier status for autosomal recessive mutations, although obtaining exome sequencing from the prior affected fetus (or child) is ideal.
—history of recurrent stillbirths of unknown etiology, with a recurrent pattern of anomalies in the current pregnancy, with normal karyotype or microarray.
- Interpretation of results should be done using a multidisciplinary team-based approach, including clinical scientists, geneticists, genetic counselors, and experts in prenatal diagnosis.
- Where possible and after informed consent, reanalysis of results should be undertaken if a future pregnancy is planned or ongoing, and a significant amount of time has elapsed since the time the result was last reported.
- Parents should be given a written report of test results.
Three professional societies have convened to issue consensus opinion that includes current indications for prenatal exome sequencing and important factors to include in the consent process. We follow these guidelines in our own practice.
Summary
Exome sequencing is increasingly becoming mainstream in postnatal genetic testing, and it is emerging as the newest diagnostic frontier in prenatal genetic testing. However, there are limitations to prenatal exome sequencing, including issues with consent at a vulnerable time for parents, limited information available regarding the phenotype, and results that may not be available before the birth of a fetus. Providers should be familiar with the indications for testing, the possible results, the limitations of prenatal phenotyping, and the implications for future pregnancies.

2018 Update on prenatal carrier screening
Prenatal care has long included carrier screening for genetic diseases, such as cystic fibrosis and Tay-Sachs disease. Recently, advances in genetics technologies led to the development of multiplex panels that can be used to test for hundreds of genetic disorders simultaneously, and can be used to assess carrier status for expectant couples or those planning a pregnancy. Although such screening covers many more conditions than those recommended in traditional guidelines, the benefit of expanded carrier screening (ECS) over standard gene-by-gene testing is not clear.
In this Update, I review recent ECS research that can be helpful to those who practice reproductive endocrinology and infertility medicine, maternal–fetal medicine, and general ObGyn. This research considered some of the many complexities of ECS:
- number and type of severe autosomal recessive conditions identified by an ECS panel, or by panethnic screening for 3 common conditions (cystic fibrosis, fragile X syndrome, spinal muscular atrophy)
- whether the disorders covered by ECS panels meet recommended criteria regarding severity, prevalence, and test accuracy
- women’s thoughts and perspectives on ECS
- whether the marketing materials disseminated by commercial providers of ECS are accurate and balanced.
Genetic diseases identified by expanded carrier screening
Haque IS, Lazarin GA, Kang HP, Evans EA, Goldberg JD, Wapner RJ. Modeled fetal risk of genetic diseases identified by expanded carrier screening. JAMA. 2016;316(7):734-742.
Screening during pregnancy to determine if one or both parents are carriers of genetic disorders historically has involved testing for a limited number of conditions, such as cystic fibrosis, hemoglobinopathies, and Tay-Sachs disease. Patients usually are offered testing for 1 or 2 disorders, with test choices primarily based on patient race and ethnicity. Unfortunately, ancestry-based screening may result in inequitable distribution of genetic testing and resources, as it has significant limitations in our increasingly multicultural society, which includes many people of uncertain or mixed race and ethnicity.
Advantages of expanded carrier screening
Several commercial laboratories now offer ECS. Haque and colleagues used data from one of these laboratories and modeled the predicted number of potentially affected fetuses that would be identified with traditional, ethnicity-based screening as compared with ECS. In one of their hypothetical cohorts, of Northern European couples, traditional screening would identify 55 affected fetuses per 100,000 (1 in 1,800), and ECS would identify 159 per 100,000 (almost 3 times more). The numbers identified with ECS varied with race or ethnicity and ranged from 94 per 100,000 (about 1 in 1,000) for Hispanic couples to 392 per 100,000 (about 1 in 250) for Ashkenazi Jewish couples.
In Australia, Archibald and colleagues conducted a similar study, of panethnic screening of 12,000 women for cystic fibrosis, fragile X syndrome, and spinal muscular atrophy.1 The number of affected fetuses identified was about 1 per 1,000 screened couples--not much different from the ECS number, though comparison is difficult given the likely very different racial and ethnic backgrounds of the 2 cohorts.
Although these data suggest ECS increases detection of genetic disorders, and it seems almost self-evident that more screening is better, there are concerns about ECS.2 Traditional carrier screening methods focus on conditions that significantly affect quality of life--owing to cognitive or physical disabilities or required lifelong medical therapies--and that have a fetal, neonatal, or early-childhood onset and well-defined phenotype. In ECS panels, additional conditions may vary significantly in severity or age of onset. Although some genetic variants on ECS panels have a consistent phenotype, the natural history of others is less well understood. Panels often include conditions for which carrier screening of the general population is not recommended by current guidelines--for example, hemochromatosis and factor V Leiden. Moreover, almost by definition, ECS panels include rare conditions for which the natural history may not be well understood, and the carrier frequency as well as the proportion of condition-causing variants that can be detected may be unclear, leaving the residual risk unknown.
This study provides additional information on the number and type of conditions that can be detected with ECS in different populations. Although ever larger panels can detect more conditions, the veracity of the results and the types of conditions detected are important considerations as providers and patients weigh the risks and benefits of this screening.
Read about the ideal expanded carrier screening panel.
The ideal expanded carrier screening panel
Stevens B, Krstic N, Jones M, Murphy L, Hoskovec J. Finding middle ground in constructing a clinically useful expanded carrier screening panel. Obstet Gynecol. 2017;130(2):279-284.
Both the American College of Obstetricians and Gynecologists (ACOG) and the American College of Medical Genetics and Genomics (ACMG) have proposed criteria for including specific disorders on ECS panels.3,4 These criteria consider disorder characteristics, such as carrier prevalence, which should be at least 1 in 100; severity; early-childhood onset; and complete penetrance. In addition, they consider test characteristics, such as sensitivity, which should be at least 70%.
Details of the study
Stevens and colleagues evaluated the ECS panels offered by 6 commercial laboratories in the United States. They found that only 27% of included conditions met the recommended criteria, and concluded that these panels are putting patients at risk for undue anxiety, and that time and money are being spent on follow-up testing for rare and mild conditions for which the benefits of testing are unclear or unlikely. The potential benefits of the extra screening should be weighed against the significant resulting harms.
Across the 6 ECS panels, 96 conditions met the criteria. As some laboratories allow providers to customize their panels, members of my practice, after reviewing this thought-provoking article, agreed we should create a custom panel that includes only these 96 conditions. Unfortunately, no commercial laboratory includes all 96 conditions, so it is not feasible to create an "ideal" panel at this time.
Arguments favoring ECS include its low cost and the efficiency of screening with multigene panels. In a 2013 study, however, 24% of patients were identified as carriers, and in most cases this finding led to screening for the reproductive partner as well.5 If the rate of detection of the disorder is low, the utility of screening with the same panel may be limited, and couples may require more extensive testing, such as gene sequencing, which is far more expensive. These findings and the additional testing also will increase the need for genetic counseling, and may lead to invasive prenatal diagnostic testing with further increases in costs. If counseling and prenatal testing yield improved outcomes--increased detection of important findings--the benefit will justify the higher costs. However, if the increased costs are largely generated chasing down and explaining findings that are not important to patients or providers, the costs may be incurred without benefit.
For practices that want to offer ECS, it is important to consider the type of conditions on a given laboratory's panel. Panels that include more conditions will detect at least one condition in more patients. As each positive test requires follow-up (typically partner testing), careful consideration should be given up-front to which test is used.
Read about the pregnant women’s perspectives on ECS.
Pregnant women's perspectives on expanded carrier screening
Propst L, Connor G, Hinton M, Poorvu T, Dungan J. Pregnant women's perspectives on expanded carrier screening [published online February 23, 2018]. J Genet Couns. doi:10.1007/s10897-018-0232-x.
Although several authors have discussed ECS detection rates, less has been reported on how women perceive ECS or how they elect or decline screening. Studies have found that the decision to undergo screening for cystic fibrosis is influenced by factors that include age, sex, ethnicity, socioeconomic status, lack of family history, cost, fear of a blood test, lack of knowledge about the condition, already having children, wanting to avoid having a disabled child, abortion preferences, and feeling pressured by health care providers.6,7 Propst and colleagues asked women for their perspectives on ECS, on electing or declining screening, and on any anxiety associated with their decision.
Details of the study
Women who declined ECS said they did so because they:
- had no family history
- knew there was a very small chance their partner carried the same condition
- would not change the course of their pregnancy on the basis of the test results.
Women who elected ECS said they did so because they wanted to:
- know their risk of having a child with a genetic condition
- have all available information about their genetic risks
- be able to make decisions about continuing or terminating their pregnancy.
Women also were asked what they would do if they discovered their fetus had a genetic disorder. About 42% said they were unsure what they would do, 34% said they would continue their pregnancy and prepare for the birth of an affected child, and 24% said they likely would terminate their pregnancy.
The most common reason women gave for declining ECS was that they had no family history. However, ECS is not a good option for women with a positive family history, as they need genetic counseling and specific consideration of their own risks and what testing should be done. The majority of couples who have a child with a genetic disease have no other family history of the disorder. In a study of reproductive carrier screening in Australia, 88% of carriers had no family history.1 Careful pretest counseling is needed to explain the distinction between, on one hand, genetic counseling and testing for those with a family history of genetic disease and, on the other hand, population screening performed to identify unsuspecting individuals who are healthy carriers of genetic disorders.
Another crucial point about carrier screening is the need to consider how its results will be used, and what options the carrier couple will have. For women who are pregnant when a risk is identified, options include expectant management, with diagnosis after birth, or prenatal diagnosis with termination of an affected fetus, out-adoption of an affected fetus, or expectant management with preparation for caring for an affected child. For women who are not pregnant when they have ECS, additional options include use of a gamete (ovum or sperm) donor to achieve pregnancy, or preimplantation genetic diagnosis with implantation of only unaffected embryos.
Different pregnant women may have very different preferences regarding genetic testing. Although many are unsure how they would proceed following the diagnosis of a fetal genetic disorder, it is important to carefully explain their options before any testing is done.
Read about the marketing of ECS.
Marketing of expanded carrier screening
Chokoshvili D, Borry P, Vears DF. A systematic analysis of online marketing materials used by providers of expanded carrier screening [published online December 14, 2017]. Genet Med. doi:10.1038/gim.2017.222.
Prenatal carrier screening can be helpful to women and their families, but it is also a high-volume, lucrative business, with many commercial laboratories competing for the growing ECS market. Professional medical societies recommend making all screening candidates aware of the purpose, characteristics, and limitations of the tests, and of the potential significance of their results. As becoming familiar and comfortable with the tests and explaining them to each patient can be time-consuming, and daunting, many busy clinicians have started relying on marketing materials and other information from the commercial laboratories. Therefore analysis of the accuracy of such materials is in order.
Details of the study
Chokoshvili and colleagues performed a systematic analysis of the quality and accuracy of online marketing materials for ECS. They identified 18 providers: 16 commercial laboratories and 2 medical services providers. All described ECS as a useful tool for family planning, and some were very directive in stating that this testing is "one of the most important steps in preparing for parenthood." In their materials, most of the companies cover some limitations, such as residual risk, but none of the commercial laboratories indicate that ECS can overestimate risk (many variants have incomplete penetrance, meaning that some individuals with a positive test result may in fact be asymptomatic throughout their lifetime).
In addition, whereas a large amount of the marketing materials implies the test was developed in line with professional recommendations, none in fact complies with ACOG and ACMG guidance. Finally, though some of the online information provided by laboratories can be helpful, it is important for clinicians to remember that reproductive genetic counseling should be nondirective and balanced. Carrier testing should be based on patient (not provider) values regarding reproductive autonomy.
Determining that a woman carries a genetic disorder in the preconception period allows more time to evaluate her reproductive partner. If both partners in the couple carry the same genetic disorder, there are more options available to avoid an affected pregnancy. These options include the use of an ovum or sperm donor, or use of preimplantation genetic diagnosis on embryos conceived through in vitro fertilization. While obstetric providers commonly offer carrier screening, and most women are only screened during pregnancy, such genetic testing should be part of pregnancy planning. When gyn providers see patients who are considering a pregnancy, he or she should discuss the options of expanded carrier screening, or ethnicity-based screening.
Summary
ECS increasingly is being adopted into clinical practice. According to ACOG, traditional ethnicity-based screening, panethnic screening (the same limited panel of tests for all patients), and ECS are all acceptable alternatives for prenatal carrier screening.3 For providers who offer ECS, it is important to have a good understanding of each selected test and its limitations. Providers should have a plan for following up patients who have positive test results; this plan may include having genetic counseling and prenatal genetic diagnostic testing in place. Although treatment is available for a few genetic conditions, for the large majority, prenatal screening has not been proved to lead to improved therapeutic options. Providers should try to make sure that patients do not have unrealistic expectations of the outcomes of carrier screening.
Laboratories' educational materials can be useful, but clinicians must carefully assess them before recommending them to patients. Some commercial laboratory information is helpful and balanced; other information is directive or even coercive. Nonbiased information on prenatal genetic testing, for both patients and clinicians, is available in the Genetic Education Modules offered by the Perinatal Quality Foundation (https://www.perinatalquality.org).
Share your thoughts! Send your Letter to the Editor to rbarbieri@mdedge.com. Please include your name and the city and state in which you practice.
- Archibald AD, Smith MJ, Burgess T, et al. Reproductive genetic carrier screening for cystic fibrosis, fragile X syndrome, and spinal muscular atrophy in Australia: outcomes of 12,000 tests [published online October 26, 2017; published correction appears in Genet Med. 2018. doi:10.1038/gim.2017.266]. Genet Med. doi:10.1038/gim.2017.134.
- Edwards JG, Feldman G, Goldberg J, et al. Expanded carrier screening in reproductive medicine—points to consider: a joint statement of the American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, National Society of Genetic Counselors, Perinatal Quality Foundation, and Society for Maternal-Fetal Medicine. Obstet Gynecol. 2015;125(3):653–662.
- Committee on Genetics. Committee opinion no. 690: carrier screening in the age of genomic medicine. Obstet Gynecol. 2017;129(3):e35–e40.
- Grody WW, Thompson BH, Gregg AR, et al. ACMG position statement on prenatal/preconception expanded carrier screening. Genet Med. 2013;15(6):482–483.
- Lazarin GA, Haque IS, Nazareth S, et al. An empirical estimate of carrier frequencies for 400+ causal Mendelian variants: results from an ethnically diverse clinical sample of 23,453 individuals. Genet Med. 2013;15(3):178–186.
- Chen LS, Goodson P. Factors affecting decisions to accept or decline cystic fibrosis carrier testing/screening: a theory-guided systematic review. Genet Med. 2007;9(7):442–450.
- Ioannou L, McClaren BJ, Massie J, et al. Population-based carrier screening for cystic fibrosis: a systematic review of 23 years of research. Genet Med. 2014;16(3):207-216.

Dr. Norton is Professor of Obstetrics, Gynecology, and Reproductive Sciences at the University of California, San Francisco.
The author reports receiving grant or research support from Natera and being a consultant to Invitae.
Dr. Norton is Professor of Obstetrics, Gynecology, and Reproductive Sciences at the University of California, San Francisco.
The author reports receiving grant or research support from Natera and being a consultant to Invitae.
Dr. Norton is Professor of Obstetrics, Gynecology, and Reproductive Sciences at the University of California, San Francisco.
The author reports receiving grant or research support from Natera and being a consultant to Invitae.
Prenatal care has long included carrier screening for genetic diseases, such as cystic fibrosis and Tay-Sachs disease. Recently, advances in genetics technologies led to the development of multiplex panels that can be used to test for hundreds of genetic disorders simultaneously, and can be used to assess carrier status for expectant couples or those planning a pregnancy. Although such screening covers many more conditions than those recommended in traditional guidelines, the benefit of expanded carrier screening (ECS) over standard gene-by-gene testing is not clear.
In this Update, I review recent ECS research that can be helpful to those who practice reproductive endocrinology and infertility medicine, maternal–fetal medicine, and general ObGyn. This research considered some of the many complexities of ECS:
- number and type of severe autosomal recessive conditions identified by an ECS panel, or by panethnic screening for 3 common conditions (cystic fibrosis, fragile X syndrome, spinal muscular atrophy)
- whether the disorders covered by ECS panels meet recommended criteria regarding severity, prevalence, and test accuracy
- women’s thoughts and perspectives on ECS
- whether the marketing materials disseminated by commercial providers of ECS are accurate and balanced.
Genetic diseases identified by expanded carrier screening
Haque IS, Lazarin GA, Kang HP, Evans EA, Goldberg JD, Wapner RJ. Modeled fetal risk of genetic diseases identified by expanded carrier screening. JAMA. 2016;316(7):734-742.
Screening during pregnancy to determine if one or both parents are carriers of genetic disorders historically has involved testing for a limited number of conditions, such as cystic fibrosis, hemoglobinopathies, and Tay-Sachs disease. Patients usually are offered testing for 1 or 2 disorders, with test choices primarily based on patient race and ethnicity. Unfortunately, ancestry-based screening may result in inequitable distribution of genetic testing and resources, as it has significant limitations in our increasingly multicultural society, which includes many people of uncertain or mixed race and ethnicity.
Advantages of expanded carrier screening
Several commercial laboratories now offer ECS. Haque and colleagues used data from one of these laboratories and modeled the predicted number of potentially affected fetuses that would be identified with traditional, ethnicity-based screening as compared with ECS. In one of their hypothetical cohorts, of Northern European couples, traditional screening would identify 55 affected fetuses per 100,000 (1 in 1,800), and ECS would identify 159 per 100,000 (almost 3 times more). The numbers identified with ECS varied with race or ethnicity and ranged from 94 per 100,000 (about 1 in 1,000) for Hispanic couples to 392 per 100,000 (about 1 in 250) for Ashkenazi Jewish couples.
In Australia, Archibald and colleagues conducted a similar study, of panethnic screening of 12,000 women for cystic fibrosis, fragile X syndrome, and spinal muscular atrophy.1 The number of affected fetuses identified was about 1 per 1,000 screened couples--not much different from the ECS number, though comparison is difficult given the likely very different racial and ethnic backgrounds of the 2 cohorts.
Although these data suggest ECS increases detection of genetic disorders, and it seems almost self-evident that more screening is better, there are concerns about ECS.2 Traditional carrier screening methods focus on conditions that significantly affect quality of life--owing to cognitive or physical disabilities or required lifelong medical therapies--and that have a fetal, neonatal, or early-childhood onset and well-defined phenotype. In ECS panels, additional conditions may vary significantly in severity or age of onset. Although some genetic variants on ECS panels have a consistent phenotype, the natural history of others is less well understood. Panels often include conditions for which carrier screening of the general population is not recommended by current guidelines--for example, hemochromatosis and factor V Leiden. Moreover, almost by definition, ECS panels include rare conditions for which the natural history may not be well understood, and the carrier frequency as well as the proportion of condition-causing variants that can be detected may be unclear, leaving the residual risk unknown.
This study provides additional information on the number and type of conditions that can be detected with ECS in different populations. Although ever larger panels can detect more conditions, the veracity of the results and the types of conditions detected are important considerations as providers and patients weigh the risks and benefits of this screening.
Read about the ideal expanded carrier screening panel.
The ideal expanded carrier screening panel
Stevens B, Krstic N, Jones M, Murphy L, Hoskovec J. Finding middle ground in constructing a clinically useful expanded carrier screening panel. Obstet Gynecol. 2017;130(2):279-284.
Both the American College of Obstetricians and Gynecologists (ACOG) and the American College of Medical Genetics and Genomics (ACMG) have proposed criteria for including specific disorders on ECS panels.3,4 These criteria consider disorder characteristics, such as carrier prevalence, which should be at least 1 in 100; severity; early-childhood onset; and complete penetrance. In addition, they consider test characteristics, such as sensitivity, which should be at least 70%.
Details of the study
Stevens and colleagues evaluated the ECS panels offered by 6 commercial laboratories in the United States. They found that only 27% of included conditions met the recommended criteria, and concluded that these panels are putting patients at risk for undue anxiety, and that time and money are being spent on follow-up testing for rare and mild conditions for which the benefits of testing are unclear or unlikely. The potential benefits of the extra screening should be weighed against the significant resulting harms.
Across the 6 ECS panels, 96 conditions met the criteria. As some laboratories allow providers to customize their panels, members of my practice, after reviewing this thought-provoking article, agreed we should create a custom panel that includes only these 96 conditions. Unfortunately, no commercial laboratory includes all 96 conditions, so it is not feasible to create an "ideal" panel at this time.
Arguments favoring ECS include its low cost and the efficiency of screening with multigene panels. In a 2013 study, however, 24% of patients were identified as carriers, and in most cases this finding led to screening for the reproductive partner as well.5 If the rate of detection of the disorder is low, the utility of screening with the same panel may be limited, and couples may require more extensive testing, such as gene sequencing, which is far more expensive. These findings and the additional testing also will increase the need for genetic counseling, and may lead to invasive prenatal diagnostic testing with further increases in costs. If counseling and prenatal testing yield improved outcomes--increased detection of important findings--the benefit will justify the higher costs. However, if the increased costs are largely generated chasing down and explaining findings that are not important to patients or providers, the costs may be incurred without benefit.
For practices that want to offer ECS, it is important to consider the type of conditions on a given laboratory's panel. Panels that include more conditions will detect at least one condition in more patients. As each positive test requires follow-up (typically partner testing), careful consideration should be given up-front to which test is used.
Read about the pregnant women’s perspectives on ECS.
Pregnant women's perspectives on expanded carrier screening
Propst L, Connor G, Hinton M, Poorvu T, Dungan J. Pregnant women's perspectives on expanded carrier screening [published online February 23, 2018]. J Genet Couns. doi:10.1007/s10897-018-0232-x.
Although several authors have discussed ECS detection rates, less has been reported on how women perceive ECS or how they elect or decline screening. Studies have found that the decision to undergo screening for cystic fibrosis is influenced by factors that include age, sex, ethnicity, socioeconomic status, lack of family history, cost, fear of a blood test, lack of knowledge about the condition, already having children, wanting to avoid having a disabled child, abortion preferences, and feeling pressured by health care providers.6,7 Propst and colleagues asked women for their perspectives on ECS, on electing or declining screening, and on any anxiety associated with their decision.
Details of the study
Women who declined ECS said they did so because they:
- had no family history
- knew there was a very small chance their partner carried the same condition
- would not change the course of their pregnancy on the basis of the test results.
Women who elected ECS said they did so because they wanted to:
- know their risk of having a child with a genetic condition
- have all available information about their genetic risks
- be able to make decisions about continuing or terminating their pregnancy.
Women also were asked what they would do if they discovered their fetus had a genetic disorder. About 42% said they were unsure what they would do, 34% said they would continue their pregnancy and prepare for the birth of an affected child, and 24% said they likely would terminate their pregnancy.
The most common reason women gave for declining ECS was that they had no family history. However, ECS is not a good option for women with a positive family history, as they need genetic counseling and specific consideration of their own risks and what testing should be done. The majority of couples who have a child with a genetic disease have no other family history of the disorder. In a study of reproductive carrier screening in Australia, 88% of carriers had no family history.1 Careful pretest counseling is needed to explain the distinction between, on one hand, genetic counseling and testing for those with a family history of genetic disease and, on the other hand, population screening performed to identify unsuspecting individuals who are healthy carriers of genetic disorders.
Another crucial point about carrier screening is the need to consider how its results will be used, and what options the carrier couple will have. For women who are pregnant when a risk is identified, options include expectant management, with diagnosis after birth, or prenatal diagnosis with termination of an affected fetus, out-adoption of an affected fetus, or expectant management with preparation for caring for an affected child. For women who are not pregnant when they have ECS, additional options include use of a gamete (ovum or sperm) donor to achieve pregnancy, or preimplantation genetic diagnosis with implantation of only unaffected embryos.
Different pregnant women may have very different preferences regarding genetic testing. Although many are unsure how they would proceed following the diagnosis of a fetal genetic disorder, it is important to carefully explain their options before any testing is done.
Read about the marketing of ECS.
Marketing of expanded carrier screening
Chokoshvili D, Borry P, Vears DF. A systematic analysis of online marketing materials used by providers of expanded carrier screening [published online December 14, 2017]. Genet Med. doi:10.1038/gim.2017.222.
Prenatal carrier screening can be helpful to women and their families, but it is also a high-volume, lucrative business, with many commercial laboratories competing for the growing ECS market. Professional medical societies recommend making all screening candidates aware of the purpose, characteristics, and limitations of the tests, and of the potential significance of their results. As becoming familiar and comfortable with the tests and explaining them to each patient can be time-consuming, and daunting, many busy clinicians have started relying on marketing materials and other information from the commercial laboratories. Therefore analysis of the accuracy of such materials is in order.
Details of the study
Chokoshvili and colleagues performed a systematic analysis of the quality and accuracy of online marketing materials for ECS. They identified 18 providers: 16 commercial laboratories and 2 medical services providers. All described ECS as a useful tool for family planning, and some were very directive in stating that this testing is "one of the most important steps in preparing for parenthood." In their materials, most of the companies cover some limitations, such as residual risk, but none of the commercial laboratories indicate that ECS can overestimate risk (many variants have incomplete penetrance, meaning that some individuals with a positive test result may in fact be asymptomatic throughout their lifetime).
In addition, whereas a large amount of the marketing materials implies the test was developed in line with professional recommendations, none in fact complies with ACOG and ACMG guidance. Finally, though some of the online information provided by laboratories can be helpful, it is important for clinicians to remember that reproductive genetic counseling should be nondirective and balanced. Carrier testing should be based on patient (not provider) values regarding reproductive autonomy.
Determining that a woman carries a genetic disorder in the preconception period allows more time to evaluate her reproductive partner. If both partners in the couple carry the same genetic disorder, there are more options available to avoid an affected pregnancy. These options include the use of an ovum or sperm donor, or use of preimplantation genetic diagnosis on embryos conceived through in vitro fertilization. While obstetric providers commonly offer carrier screening, and most women are only screened during pregnancy, such genetic testing should be part of pregnancy planning. When gyn providers see patients who are considering a pregnancy, he or she should discuss the options of expanded carrier screening, or ethnicity-based screening.
Summary
ECS increasingly is being adopted into clinical practice. According to ACOG, traditional ethnicity-based screening, panethnic screening (the same limited panel of tests for all patients), and ECS are all acceptable alternatives for prenatal carrier screening.3 For providers who offer ECS, it is important to have a good understanding of each selected test and its limitations. Providers should have a plan for following up patients who have positive test results; this plan may include having genetic counseling and prenatal genetic diagnostic testing in place. Although treatment is available for a few genetic conditions, for the large majority, prenatal screening has not been proved to lead to improved therapeutic options. Providers should try to make sure that patients do not have unrealistic expectations of the outcomes of carrier screening.
Laboratories' educational materials can be useful, but clinicians must carefully assess them before recommending them to patients. Some commercial laboratory information is helpful and balanced; other information is directive or even coercive. Nonbiased information on prenatal genetic testing, for both patients and clinicians, is available in the Genetic Education Modules offered by the Perinatal Quality Foundation (https://www.perinatalquality.org).
Share your thoughts! Send your Letter to the Editor to rbarbieri@mdedge.com. Please include your name and the city and state in which you practice.
Prenatal care has long included carrier screening for genetic diseases, such as cystic fibrosis and Tay-Sachs disease. Recently, advances in genetics technologies led to the development of multiplex panels that can be used to test for hundreds of genetic disorders simultaneously, and can be used to assess carrier status for expectant couples or those planning a pregnancy. Although such screening covers many more conditions than those recommended in traditional guidelines, the benefit of expanded carrier screening (ECS) over standard gene-by-gene testing is not clear.
In this Update, I review recent ECS research that can be helpful to those who practice reproductive endocrinology and infertility medicine, maternal–fetal medicine, and general ObGyn. This research considered some of the many complexities of ECS:
- number and type of severe autosomal recessive conditions identified by an ECS panel, or by panethnic screening for 3 common conditions (cystic fibrosis, fragile X syndrome, spinal muscular atrophy)
- whether the disorders covered by ECS panels meet recommended criteria regarding severity, prevalence, and test accuracy
- women’s thoughts and perspectives on ECS
- whether the marketing materials disseminated by commercial providers of ECS are accurate and balanced.
Genetic diseases identified by expanded carrier screening
Haque IS, Lazarin GA, Kang HP, Evans EA, Goldberg JD, Wapner RJ. Modeled fetal risk of genetic diseases identified by expanded carrier screening. JAMA. 2016;316(7):734-742.
Screening during pregnancy to determine if one or both parents are carriers of genetic disorders historically has involved testing for a limited number of conditions, such as cystic fibrosis, hemoglobinopathies, and Tay-Sachs disease. Patients usually are offered testing for 1 or 2 disorders, with test choices primarily based on patient race and ethnicity. Unfortunately, ancestry-based screening may result in inequitable distribution of genetic testing and resources, as it has significant limitations in our increasingly multicultural society, which includes many people of uncertain or mixed race and ethnicity.
Advantages of expanded carrier screening
Several commercial laboratories now offer ECS. Haque and colleagues used data from one of these laboratories and modeled the predicted number of potentially affected fetuses that would be identified with traditional, ethnicity-based screening as compared with ECS. In one of their hypothetical cohorts, of Northern European couples, traditional screening would identify 55 affected fetuses per 100,000 (1 in 1,800), and ECS would identify 159 per 100,000 (almost 3 times more). The numbers identified with ECS varied with race or ethnicity and ranged from 94 per 100,000 (about 1 in 1,000) for Hispanic couples to 392 per 100,000 (about 1 in 250) for Ashkenazi Jewish couples.
In Australia, Archibald and colleagues conducted a similar study, of panethnic screening of 12,000 women for cystic fibrosis, fragile X syndrome, and spinal muscular atrophy.1 The number of affected fetuses identified was about 1 per 1,000 screened couples--not much different from the ECS number, though comparison is difficult given the likely very different racial and ethnic backgrounds of the 2 cohorts.
Although these data suggest ECS increases detection of genetic disorders, and it seems almost self-evident that more screening is better, there are concerns about ECS.2 Traditional carrier screening methods focus on conditions that significantly affect quality of life--owing to cognitive or physical disabilities or required lifelong medical therapies--and that have a fetal, neonatal, or early-childhood onset and well-defined phenotype. In ECS panels, additional conditions may vary significantly in severity or age of onset. Although some genetic variants on ECS panels have a consistent phenotype, the natural history of others is less well understood. Panels often include conditions for which carrier screening of the general population is not recommended by current guidelines--for example, hemochromatosis and factor V Leiden. Moreover, almost by definition, ECS panels include rare conditions for which the natural history may not be well understood, and the carrier frequency as well as the proportion of condition-causing variants that can be detected may be unclear, leaving the residual risk unknown.
This study provides additional information on the number and type of conditions that can be detected with ECS in different populations. Although ever larger panels can detect more conditions, the veracity of the results and the types of conditions detected are important considerations as providers and patients weigh the risks and benefits of this screening.
Read about the ideal expanded carrier screening panel.
The ideal expanded carrier screening panel
Stevens B, Krstic N, Jones M, Murphy L, Hoskovec J. Finding middle ground in constructing a clinically useful expanded carrier screening panel. Obstet Gynecol. 2017;130(2):279-284.
Both the American College of Obstetricians and Gynecologists (ACOG) and the American College of Medical Genetics and Genomics (ACMG) have proposed criteria for including specific disorders on ECS panels.3,4 These criteria consider disorder characteristics, such as carrier prevalence, which should be at least 1 in 100; severity; early-childhood onset; and complete penetrance. In addition, they consider test characteristics, such as sensitivity, which should be at least 70%.
Details of the study
Stevens and colleagues evaluated the ECS panels offered by 6 commercial laboratories in the United States. They found that only 27% of included conditions met the recommended criteria, and concluded that these panels are putting patients at risk for undue anxiety, and that time and money are being spent on follow-up testing for rare and mild conditions for which the benefits of testing are unclear or unlikely. The potential benefits of the extra screening should be weighed against the significant resulting harms.
Across the 6 ECS panels, 96 conditions met the criteria. As some laboratories allow providers to customize their panels, members of my practice, after reviewing this thought-provoking article, agreed we should create a custom panel that includes only these 96 conditions. Unfortunately, no commercial laboratory includes all 96 conditions, so it is not feasible to create an "ideal" panel at this time.
Arguments favoring ECS include its low cost and the efficiency of screening with multigene panels. In a 2013 study, however, 24% of patients were identified as carriers, and in most cases this finding led to screening for the reproductive partner as well.5 If the rate of detection of the disorder is low, the utility of screening with the same panel may be limited, and couples may require more extensive testing, such as gene sequencing, which is far more expensive. These findings and the additional testing also will increase the need for genetic counseling, and may lead to invasive prenatal diagnostic testing with further increases in costs. If counseling and prenatal testing yield improved outcomes--increased detection of important findings--the benefit will justify the higher costs. However, if the increased costs are largely generated chasing down and explaining findings that are not important to patients or providers, the costs may be incurred without benefit.
For practices that want to offer ECS, it is important to consider the type of conditions on a given laboratory's panel. Panels that include more conditions will detect at least one condition in more patients. As each positive test requires follow-up (typically partner testing), careful consideration should be given up-front to which test is used.
Read about the pregnant women’s perspectives on ECS.
Pregnant women's perspectives on expanded carrier screening
Propst L, Connor G, Hinton M, Poorvu T, Dungan J. Pregnant women's perspectives on expanded carrier screening [published online February 23, 2018]. J Genet Couns. doi:10.1007/s10897-018-0232-x.
Although several authors have discussed ECS detection rates, less has been reported on how women perceive ECS or how they elect or decline screening. Studies have found that the decision to undergo screening for cystic fibrosis is influenced by factors that include age, sex, ethnicity, socioeconomic status, lack of family history, cost, fear of a blood test, lack of knowledge about the condition, already having children, wanting to avoid having a disabled child, abortion preferences, and feeling pressured by health care providers.6,7 Propst and colleagues asked women for their perspectives on ECS, on electing or declining screening, and on any anxiety associated with their decision.
Details of the study
Women who declined ECS said they did so because they:
- had no family history
- knew there was a very small chance their partner carried the same condition
- would not change the course of their pregnancy on the basis of the test results.
Women who elected ECS said they did so because they wanted to:
- know their risk of having a child with a genetic condition
- have all available information about their genetic risks
- be able to make decisions about continuing or terminating their pregnancy.
Women also were asked what they would do if they discovered their fetus had a genetic disorder. About 42% said they were unsure what they would do, 34% said they would continue their pregnancy and prepare for the birth of an affected child, and 24% said they likely would terminate their pregnancy.
The most common reason women gave for declining ECS was that they had no family history. However, ECS is not a good option for women with a positive family history, as they need genetic counseling and specific consideration of their own risks and what testing should be done. The majority of couples who have a child with a genetic disease have no other family history of the disorder. In a study of reproductive carrier screening in Australia, 88% of carriers had no family history.1 Careful pretest counseling is needed to explain the distinction between, on one hand, genetic counseling and testing for those with a family history of genetic disease and, on the other hand, population screening performed to identify unsuspecting individuals who are healthy carriers of genetic disorders.
Another crucial point about carrier screening is the need to consider how its results will be used, and what options the carrier couple will have. For women who are pregnant when a risk is identified, options include expectant management, with diagnosis after birth, or prenatal diagnosis with termination of an affected fetus, out-adoption of an affected fetus, or expectant management with preparation for caring for an affected child. For women who are not pregnant when they have ECS, additional options include use of a gamete (ovum or sperm) donor to achieve pregnancy, or preimplantation genetic diagnosis with implantation of only unaffected embryos.
Different pregnant women may have very different preferences regarding genetic testing. Although many are unsure how they would proceed following the diagnosis of a fetal genetic disorder, it is important to carefully explain their options before any testing is done.
Read about the marketing of ECS.
Marketing of expanded carrier screening
Chokoshvili D, Borry P, Vears DF. A systematic analysis of online marketing materials used by providers of expanded carrier screening [published online December 14, 2017]. Genet Med. doi:10.1038/gim.2017.222.
Prenatal carrier screening can be helpful to women and their families, but it is also a high-volume, lucrative business, with many commercial laboratories competing for the growing ECS market. Professional medical societies recommend making all screening candidates aware of the purpose, characteristics, and limitations of the tests, and of the potential significance of their results. As becoming familiar and comfortable with the tests and explaining them to each patient can be time-consuming, and daunting, many busy clinicians have started relying on marketing materials and other information from the commercial laboratories. Therefore analysis of the accuracy of such materials is in order.
Details of the study
Chokoshvili and colleagues performed a systematic analysis of the quality and accuracy of online marketing materials for ECS. They identified 18 providers: 16 commercial laboratories and 2 medical services providers. All described ECS as a useful tool for family planning, and some were very directive in stating that this testing is "one of the most important steps in preparing for parenthood." In their materials, most of the companies cover some limitations, such as residual risk, but none of the commercial laboratories indicate that ECS can overestimate risk (many variants have incomplete penetrance, meaning that some individuals with a positive test result may in fact be asymptomatic throughout their lifetime).
In addition, whereas a large amount of the marketing materials implies the test was developed in line with professional recommendations, none in fact complies with ACOG and ACMG guidance. Finally, though some of the online information provided by laboratories can be helpful, it is important for clinicians to remember that reproductive genetic counseling should be nondirective and balanced. Carrier testing should be based on patient (not provider) values regarding reproductive autonomy.
Determining that a woman carries a genetic disorder in the preconception period allows more time to evaluate her reproductive partner. If both partners in the couple carry the same genetic disorder, there are more options available to avoid an affected pregnancy. These options include the use of an ovum or sperm donor, or use of preimplantation genetic diagnosis on embryos conceived through in vitro fertilization. While obstetric providers commonly offer carrier screening, and most women are only screened during pregnancy, such genetic testing should be part of pregnancy planning. When gyn providers see patients who are considering a pregnancy, he or she should discuss the options of expanded carrier screening, or ethnicity-based screening.
Summary
ECS increasingly is being adopted into clinical practice. According to ACOG, traditional ethnicity-based screening, panethnic screening (the same limited panel of tests for all patients), and ECS are all acceptable alternatives for prenatal carrier screening.3 For providers who offer ECS, it is important to have a good understanding of each selected test and its limitations. Providers should have a plan for following up patients who have positive test results; this plan may include having genetic counseling and prenatal genetic diagnostic testing in place. Although treatment is available for a few genetic conditions, for the large majority, prenatal screening has not been proved to lead to improved therapeutic options. Providers should try to make sure that patients do not have unrealistic expectations of the outcomes of carrier screening.
Laboratories' educational materials can be useful, but clinicians must carefully assess them before recommending them to patients. Some commercial laboratory information is helpful and balanced; other information is directive or even coercive. Nonbiased information on prenatal genetic testing, for both patients and clinicians, is available in the Genetic Education Modules offered by the Perinatal Quality Foundation (https://www.perinatalquality.org).
Share your thoughts! Send your Letter to the Editor to rbarbieri@mdedge.com. Please include your name and the city and state in which you practice.
- Archibald AD, Smith MJ, Burgess T, et al. Reproductive genetic carrier screening for cystic fibrosis, fragile X syndrome, and spinal muscular atrophy in Australia: outcomes of 12,000 tests [published online October 26, 2017; published correction appears in Genet Med. 2018. doi:10.1038/gim.2017.266]. Genet Med. doi:10.1038/gim.2017.134.
- Edwards JG, Feldman G, Goldberg J, et al. Expanded carrier screening in reproductive medicine—points to consider: a joint statement of the American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, National Society of Genetic Counselors, Perinatal Quality Foundation, and Society for Maternal-Fetal Medicine. Obstet Gynecol. 2015;125(3):653–662.
- Committee on Genetics. Committee opinion no. 690: carrier screening in the age of genomic medicine. Obstet Gynecol. 2017;129(3):e35–e40.
- Grody WW, Thompson BH, Gregg AR, et al. ACMG position statement on prenatal/preconception expanded carrier screening. Genet Med. 2013;15(6):482–483.
- Lazarin GA, Haque IS, Nazareth S, et al. An empirical estimate of carrier frequencies for 400+ causal Mendelian variants: results from an ethnically diverse clinical sample of 23,453 individuals. Genet Med. 2013;15(3):178–186.
- Chen LS, Goodson P. Factors affecting decisions to accept or decline cystic fibrosis carrier testing/screening: a theory-guided systematic review. Genet Med. 2007;9(7):442–450.
- Ioannou L, McClaren BJ, Massie J, et al. Population-based carrier screening for cystic fibrosis: a systematic review of 23 years of research. Genet Med. 2014;16(3):207-216.
- Archibald AD, Smith MJ, Burgess T, et al. Reproductive genetic carrier screening for cystic fibrosis, fragile X syndrome, and spinal muscular atrophy in Australia: outcomes of 12,000 tests [published online October 26, 2017; published correction appears in Genet Med. 2018. doi:10.1038/gim.2017.266]. Genet Med. doi:10.1038/gim.2017.134.
- Edwards JG, Feldman G, Goldberg J, et al. Expanded carrier screening in reproductive medicine—points to consider: a joint statement of the American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, National Society of Genetic Counselors, Perinatal Quality Foundation, and Society for Maternal-Fetal Medicine. Obstet Gynecol. 2015;125(3):653–662.
- Committee on Genetics. Committee opinion no. 690: carrier screening in the age of genomic medicine. Obstet Gynecol. 2017;129(3):e35–e40.
- Grody WW, Thompson BH, Gregg AR, et al. ACMG position statement on prenatal/preconception expanded carrier screening. Genet Med. 2013;15(6):482–483.
- Lazarin GA, Haque IS, Nazareth S, et al. An empirical estimate of carrier frequencies for 400+ causal Mendelian variants: results from an ethnically diverse clinical sample of 23,453 individuals. Genet Med. 2013;15(3):178–186.
- Chen LS, Goodson P. Factors affecting decisions to accept or decline cystic fibrosis carrier testing/screening: a theory-guided systematic review. Genet Med. 2007;9(7):442–450.
- Ioannou L, McClaren BJ, Massie J, et al. Population-based carrier screening for cystic fibrosis: a systematic review of 23 years of research. Genet Med. 2014;16(3):207-216.

Cell-free DNA screening for women at low risk for fetal aneuploidy
CASE: Low-risk patient requests cell-free DNA screening
Ms. Smith is a 25-year-old woman (G1P0) presenting at 10 weeks’ gestation for her first prenatal visit. She requests cell-free DNA (cfDNA) screening to test for fetal aneuploidy. You explain that the current recommendations are for traditional screening, and inform her that her insurance may not cover the cost of cfDNA screening. She is anxious to learn the sex of her fetus as early as possible, and indicates that she would like to pursue cfDNA. After further discussion of the pros and cons, you order the test.
Prenatal screening is currently recommended in pregnancy for a number of genetic disorders, chromosomal aneuploidy, and structural birth defects in the fetus, regardless of maternal age or family history. There is a broad range of sonographic and maternal serum-based options available for carrying out aneuploidy risk assessment in the first and/or second trimester.
In addition, cfDNA screening for fetal aneuploidy has been clinically available since 2011 and has seen tremendous uptake, particularly in the high-risk population. Recent data indicate that cfDNA screening likewise has very high sensitivity and specificity for trisomy 21 in the low-risk population.1,2
Many low-risk patients are asking providers about the pros and cons of cfDNA screening, and the appropriateness of this test as a primary screen, including in low-risk patients, is the focus of this article.
What is cfDNA?
cfDNA consists of small (<200 base pairs) fragments of DNA that are present in the maternal serum. After 10 weeks of gestation, about 10% of the total circulating cfDNA in the maternal serum is derived from the placenta and can therefore be used to test for fetal disorders (FIGURE).3
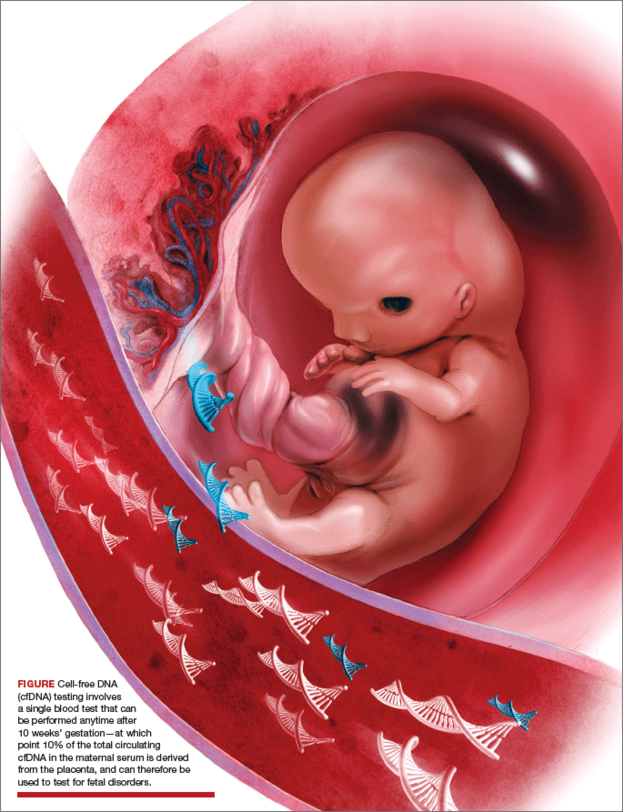
Although cfDNA screening has been reported to be possible for many different types of genetic conditions, such as RhD type and single-gene disorders such as achondroplasia,4 most clinical testing is done for fetal chromosomal disorders, including trisomies 13, 18, and 21 and the sex chromosomes. In addition, some laboratories provide testing for other trisomies (16 and 22), as well as some of the microdeletion syndromes (22q11.2, 1p36, Prader Willi syndrome, and others).5
Analysis of cfDNA to assess the risk for aneuploidy is done using a number of different approaches; these generally all include next-generation sequencing with advanced bioinformatics analyses.3,6–9 Although the laboratories use somewhat different techniques, all of them share very high sensitivity and specificity for detection of trisomy 21 (TABLE 1).10
Sensitivities for trisomy 13 and sex chromosomal abnormalities are somewhat lower, but the specificity is greater than 99% for each condition, meaning that false-positive rates are very low.
The accuracy of cfDNA in identifying chromosomal aneuploidy depends on several factors, including the relative amount of fetal to maternal DNA, the chance that a chromosome abnormality is present (that is, the risk based on maternal age or results of other screening), and other factors such as the presence of twins or a nonviable second fetus, or the presence of placental mosaicism.
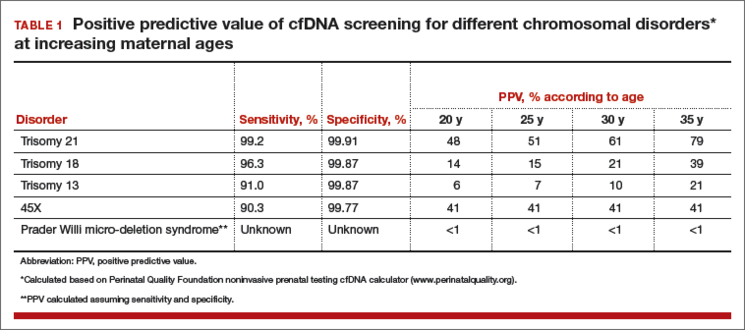
Because of these variables, both false-positive and false-negative results can occur, and the test is not diagnostic but rather is considered a screening test. A positive result does not mean that the fetus is definitely affected with aneuploidy.
What are the advantages of cfDNA screening for low-risk patients?
There are several benefits of cfDNA screening versus traditional screening or diagnostic testing, which are the other options available (TABLE 2). For Down syndrome, the detection rate is higher and the false-positive rate is lower than that seen with traditional aneuploidy screening using serum analytes and nuchal translucency ultrasonography.1,2
TABLE 2 Pros and cons of cfDNA screening in low-risk patients
Pros
- High detection rate and very low false-positive rate
- Can be performed any time after 10 weeks’ gestation
- Requires a single blood test at any gestational age
- Results presented in simple “Yes” or “No” format
- As with other screening tests, cfDNA provides a noninvasive determination of risk
Cons
- Tests for a limited range of conditions, which are rare in low-risk patients
- Is not as comprehensive or definitive as diagnostic testing with amniocentesis or chorionic villus sampling
- Results do not adjust for patient’s prior risk
- Positive results require calculation and interpretation of positive predictive value by provider
- Low fetal DNA and other factors can lead to test failure in some cases
- Cannot be used with vanishing twin
- Can reveal unsuspected maternal conditions of uncertain significance
The test can be done any time after 10 weeks’ gestation without the narrow gestational-age windows required or the need for accurate gestational age determination using traditional screening to accurately interpret results. cfDNA screening involves a single blood test that does not require integration with multiple serum markers or ultrasound findings. Finally, results are generally presented in a simple “Yes” or “No” format that is easy for providers and patients to understand.
CASE Continued
Your patient’s results are positive for trisomy 13. Her understanding is that the test is more than 99% accurate, and she interprets this to mean that the chance of trisomy 13 in her fetus is more than 99%. She is distraught and asks about pregnancy termination.
What are the limitations of cfDNA screening?
Similar to other noninvasive screening tests, cfDNA screening does not carry direct risk to the pregnancy. However, there are limitations to this testing. As a result, the American College of Obstetricians and Gynecologists (ACOG) and the Society for Maternal-Fetal Medicine (SMFM) recently have stated that traditional screening is the most appropriate option for most women.11,12
One reason that cfDNA screening may not be the best choice for low-risk women is that Down syndrome is quite uncommon in this group, so cfDNA screening is a very precise test for a rare condition. Traditional multiple marker screening, on the other hand, is more effective at signaling risk for the broad range of adverse perinatal outcomes that can affect a pregnancy, including other structural birth defects, as well as such obstetric complications as preterm birth, preeclampsia, and fetal growth restriction.13,14
Many women who undergo cfDNA screening are under the impression that they have had a definitive test for all birth defects when, in fact, the coverage of cfDNA for all possible birth defects in a low-risk woman is very limited; her residual risk for a birth defect is little changed by a normal cfDNA result.
The ease of obtaining a blood sample for cfDNA screening is an advantage of the test. However, because it is simple to perform, it often is done with inadequate pretest counseling or consideration. Just because the test is easily obtained does not negate the need for adequate discussion to assure that each woman understands what the test can and cannot measure, and the possible outcomes of testing.
Another perceived benefit of cfDNA screening is the simple presentation of results. While reports vary, they generally provide very dichotomized results. Aneuploidy risk is reported as “Positive” or “Negative,” or as “Detected” or “Not detected.”
Some laboratories report the chance of aneuploidy; this is almost always stated to be more than 99% in patients at increased risk, and less than 1 in 10,000 in patients at low risk.
All of these results suggest a near diagnostic certainty. However, this reporting is oversimplified and misleading, as it does not account for each patient’s prior or background risk. The chance that a positive result is a true positive is very different in a 20-year-old versus a 35-year-old woman, yet the reports do not reflect this difference in positive predictive value (PPV). See TABLE 1.
Accurate interpretation of risk for the individual patient, therefore, requires calculation by the provider; this can be done through an online calculator available through the Perinatal Quality Foundation (www.perina talquality.org).
CASE Continued
You explain to your patient that the chance her fetus has trisomy 13 is far lower than 99%, based in part on the very low prior risk given her age. You calculate the PPV using an online calculator, which estimates that there is only a 7% chance that this is a true positive result.
As mentioned earlier, there has been a tremendously rapid uptake of cfDNA screening. Given wider use by practitioners not as familiar with the complexities of genetic testing and statistical analysis, misunderstanding of the test characteristics carries risks if inappropriate recommendations or decisions are made or actions taken.
Most low-risk patients do not request or desire diagnostic testing. It is important during pretest counseling to explain that cfDNA cannot detect all significant chromosomal aneuploidies. Some serious abnormalities will be undetected; therefore, some women may prefer more comprehensive prenatal testing (TABLE 3).
TABLE 3 Checklist for pretest counseling for cfDNA28
- cfDNA screening is the most accurate screening test for trisomy 21
- cfDNA is a screening test, and false-positive and false-negative results can and do occur
- Diagnostic confirmation with chorionic villus sampling or amniocentesis is recommended for women with abnormal cfDNA results
- A negative cfDNA result decreases risk but does not rule out trisomy 21 and other chromosomal conditions
- cfDNA does not test for all chromosomal conditions
- Women who desire definitive information about chromosome conditions in the pregnancy should consider diagnostic testing with chorionic villus sampling or amniocentesis
- All genetic testing is optional. Whether a woman chooses to have a screening test, a diagnostic test, or no testing is a personal decision; any are reasonable options for any pregnant woman.
ACOG recommends that diagnostic testing should be available to all pregnant women, regardless of age.15 In prenatal series, trisomies 13, 18, and 21 make up approximately two-thirds of all clinically significant aneuploidies.16,17 Given that cfDNA detects only these aneuploidies, the other third will not be identified prenatally in patients who choose cfDNA. Traditional aneuploidy screening has been demonstrated to detect a broader range of these less common but clinically important chromosomal abnormalities.18
In one study of women found to be at increased risk based on traditional multiple marker screening, if cfDNA were chosen instead of diagnostic testing, 17% of the aneuploidies present in this group would not have been detected.18 Of all high-risk women in this study, 2%, or 1 in 50, had a chromosomal abnormality detectable by amniocentesis but not with cfDNA.
Successful tests require adequate placental DNA
Accurate interpretation of cfDNA screening also requires that an adequate quantity of placental DNA be present; this is often referred to as the “fetal fraction.” In some cases, the placental DNA volume is too low for accurate analysis, particularly in obese patients and women with specific chromosomal abnormalities.
Some laboratories measure this and do not report a result if the fetal fraction is below a specific cut-off, typically about 4%. Other laboratories do not measure or exclude cases with too little fetal DNA, raising concern that this could result in missing cases of aneuploidy. It has been noted that a placental DNA fraction of less than 8% is associated with less accurate test results, even if results are returned.8
Low fetal fraction also has been associated with maternal obesity, and in one study cfDNA failed to provide a result in 20% of women weighing more than 250 lb and 50% of women weighing more than 350 lb.19 Therefore, cfDNA is not the best option for obese women (TABLE 4).
TABLE 4 Appropriateness of cfDNA screeningin specific clinical circumstances
Optimal candidates for cfDNA screening
- High risk for trisomy based on maternal age (≥35 years)
- Ultrasound findings suggesting trisomy 13, 18, or 21
- History of prior pregnancy with trisomy 13, 18, or 21
- Positive traditional screening test
- Parental balanced Robertsonian translocation associated with risk for trisomy 13 or 21
Less optimal candidates
- Low risk for trisomy based on age and/or low risk traditional screening
- Ultrasound structural anomalies other than those specifically suggesting trisomy 13, 18, or 21
- High risk for nonchromosomal genetic disorder
- Comprehensive genetic diagnosis desired
- Maternal malignancy
- Maternal organ transplant
- Maternal sex chromosomal mosaicism or other chromosomal abnormality
- Maternal obesity
- Gestational age <10 weeks
While the free fraction is relatively constant from 10 to 22 weeks’ gestation, it is lower earlier than 10 weeks’ gestation and less likely to provide a result. For this reason, the test should not be attempted before 10 weeks’ gestation.
Recent evidence indicates that low fetal DNA fraction is associated with some chromosome abnormalities. Given this association, women with failed cfDNA results should be counseled and offered appropriate follow-up. As the association appears to be greater for trisomies 13 and 18, and triploidy, a careful ultrasound is likely to detect abnormalities in many such cases. However, it also is appropriate to offer the option of diagnostic testing, given the very high risk.
A repeat cfDNA test will be successful in some cases. Whether the patient chooses to attempt cfDNA again may depend in part on maternal body mass index (BMI), as well as gestational age—a patient at a more advanced gestation may not wish to delay obtaining definitive information given the high risk.
cfDNA screening has a low false-positive rate
One of the greatest benefits of cfDNA screening is a lower false-positive rate than is reported with traditional screening. However, when “no results” cases are also considered, the percentage of patients who require follow-up after cfDNA is close to that of traditional screening.
The chance of test failure is reported to be 0.9% to 8.1%,7,9,10 and varies in part by whether the laboratory measures fetal fraction and requires a minimum concentration.
A recent meta-analysis estimated the overall test failure rate at 3%.10 When comparing cfDNA to traditional screening, if “no results” cases are included with the “screen positive” group, the benefits of cfDNA over traditional screening are much less clear, particularly in a low-risk population.
ACOG: Offer traditional multiple-marker screening first
While multiple marker and cfDNA screening have differing performance characteristics, there are no data to support doing both tests concurrently. In fact, in a recent survey of nearly 200 women presented with different testing scenarios, women found it preferable and more reassuring to have a positive traditional screen followed by normal cfDNA results, rather that discrepant results of the 2 tests done concurrently.20
For many reasons, the approach recommended by ACOG and SMFM is to offer traditional multiple-marker screening first, and cfDNA screening or diagnostic testing as a follow-up for patients that screen positive. In that scenario, the benefits and limitations of diagnostic testing versus follow-up with cfDNA screening should be explained carefully.
In all patients who have a positive cfDNA result, diagnostic testing for confirmation should be offered and strongly recommended prior to pregnancy termination if that is considered. Even if a structural abnormality is present and a true positive result is highly likely, karyotyping is important to determine if there may be an inherited translocation putting subsequent pregnancies at higher risk.
Components of pretest counseling
A woman of any age can have a fetus with trisomy or another chromosomal abnormality, and some women prefer diagnostic testing or no testing regardless of age. It is therefore appropriate to offer diagnostic testing, screening, or the option of no testing to all women.
Recent studies have demonstrated that providing well-informed access to all prenatal tests results in more informed choices and no increase in uptake of invasive testing.22 However, the offer of prenatal testing requires discussion of the pros and cons of all test options, including the detection rates of all significant abnormalities, the screen positive rates, and recommended follow-up if an abnormal result is obtained. See TABLE 4.
Cost-effectiveness
Although the detection rate of cfDNA for trisomy 21 is higher than that of traditional screening, the detection rate of traditional screening is also quite high at lower cost. For low-risk women, therefore, traditional screening provides a less expensive alternative to cfDNA. Because aneuploidy is rare in low-risk patients, the residual chance of aneuploidy after a normal traditional screen is very low, and the cost per additional case of Down syndrome detected by cfDNA is very high.
In one study, this was estimated at $3.6 million.23 These authors suggested that, at present, cfDNA is optimally used as a secondary screen for high-risk women. Other cost analyses also have demonstrated that the most cost-effective strategy is a model in which cfDNA is used as a follow-up test in patients found to be screen positive by traditional screening.15,24 A recent cost utility analysis compared outcomes of 6 approaches to prenatal screening, including sequential screening, cfDNA screening, nuchal translucency only, and diagnostic testing with microarray (alone, in combination, or in sequence).
The clinical outcomes included fetal abnormalities detected, taking into account all chromosomal abnormalities, as well as failed cfDNA tests. For younger women (<40 yr), traditional sequential screening provided the highest detection of all abnormalities and was the optimal testing strategy, while cfDNA was preferable for women aged 40 or older, given the higher prevalence of trisomy 21.20
Incidental findings
Given that the cfDNA in maternal serum is a mixture of maternal and placental DNA, a number of biologic phenomena can cause a false-positive cfDNA result. In many cases, these false-positives reveal unanticipated or unexpected maternal conditions and information that the woman may have preferred not to know. A few cases of maternal malignancies with chromosomal abnormalities within the tumor have been reported in patients with false-positive cfDNA results.26
These case reports have raised the question about the need for further evaluation for maternal malignancy in women with false-positive results. Maternal genetic disorders also can cause false-positive results, and may lead to unanticipated detection of adult-onset conditions. In some cases, positive results for sex chromosomal aneuploidy can occur in pregnant women who themselves have a sex chromosomal abnormality, often in mosaic form and previously undiagnosed.27
Again, this has led to discussion of the possible health benefit of karyotyping women who have a false-positive cfDNA result to rule out a mosaic chromosomal abnormality in the mother.
At this time, the clinical utility of such investigations is unknown and there are no recommendations regarding appropriate follow-up for such cases.
CASE Resolved
Given the results of her cfDNA screening, your patient opts to undergo diagnostic testing. In that testing, trisomy 13 is ruled out and she goes on to have a healthy daughter.
Share your thoughts on this article! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
- Bianchi DW, Parker RL, Wentworth J, et al. DNA sequencing versus standard prenatal aneuploidy screening. N Engl J Med. 2014;370(9):799–808.
- Norton ME, Jacobsson B, Swamy GK, et al. Cell-free DNA analysis for noninvasive examination of trisomy. N Engl J Med. 2015;372(17):1589–1597.
- Norton ME, Brar H, Weiss J, et al. Non-Invasive Chromosomal Evaluation (NICE) study: results of a multicenter prospective cohort study for detection of fetal trisomy 21 and trisomy 18. Am J Obstet Gynecol. 2012;207(2):137.e1–e8.
- Chitty LS, Mason S, Barrett AN, et al. Non-invasive prenatal diagnosis of achondroplasia and thanatophoric dysplasia: next-generation sequencing allows for a safer, more accurate, and comprehensive approach. Prenat Diagn. 2015;35(7):656–662.
- Wapner RJ, Babiarz JE, Levy B, et al. Expanding the scope of noninvasive prenatal testing: detection of fetal microdeletion syndromes. Am J Obstet Gynecol. 2015;212(3):332.e1–e9.
- Bianchi DW, Platt LD, Goldberg JD, Abuhamad AZ, Sehnert AJ, Rava RP. Genome-wide fetal aneuploidy detection by maternal plasma DNA sequencing. Obstet Gynecol. 2012;119(5):890–901.
- Palomaki GE, Kloza EM, Lambert-Messerlian GM, et al. DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation study. Genet Med. 2011;13(11):913–920.
- Sparks AB, Wang ET, Struble CA, et al. DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation study [abstract]. Proceedings of the ISPD 16th International Conference on Prenatal Diagnosis and Therapy; Miami, Florida; June 3–6, 2012. Prenat Diagn. 2012;32(suppl 1):s3–s9.
- Zimmermann B, Hill M, Gemelos G, et al. Noninvasive prenatal aneuploidy testing of chromosomes 13, 18, 21, X, and Y, using targeted sequencing of polymorphic loci. Prenat Diagn. 2012;32(13):1233–1241.
- Gil MM, Quezada MS, Revello R, Akolekar R, Niclaides KH. Analysis of cell-free DNA in maternal blood in screening for fetal aneuploidies: updated meta-analysis. Ultrasound Obstet Gynecol. 2015;45(3):249–266.
- American College of Obstetricians and Gynecologists. Committee Opinion No. 640: cell-free DNA screening for fetal aneuploidy. Obstet Gynecol. 2015;126(3):e31–e37.
- Society for Maternal-Fetal Medicine (SMFM) Publications Committee. Prenatal aneuploidy screening using cell-free DNA. Am J Obstet Gynecol. 2015;212(6):711–716.
- Baer RJ, Currier RJ, Norton ME, et al. Obstetric, perinatal, and fetal outcomes in pregnancies with false-positive integrated screening results. Obstet Gynecol. 2014;123(3):603–609.
- Dugoff L; Society for Maternal-Fetal Medicine. First- and second-trimester maternal serum markers for aneuploidy and adverse obstetric outcomes. Obstet Gynecol. 2010;115(5):1052–1061.
- American College of Obstetricians and Gynecologists. Practice bulletin No. 88: invasive prenatal testing for aneuploidy. Obstet Gynecol. 2007;110(6):1459–1467.
- Alamillo CM, Krantz D, Evans M, Fiddler M, Pergament E. Nearly a third of abnormalities found after first-trimester screening are different than expected: 10-year experience from a single center. Prenat Diagn. 2013;33(3):251–256.
- Wellesley D, Dolk H, Boyd PA, et al. Rare chromosome abnormalities, prevalence and prenatal diagnosis rates from population-based congenital anomaly registers in Europe. Eur J Hum Genet. 2012;20(5):521–526.
- Norton ME, Jelliffe-Pawlowski LL, Currier RJ. Chromosome abnormalities detected by current prenatal screening and noninvasive prenatal testing. Obstet Gynecol. 2014;124(5):979–986.
- Ashoor G, Syngelaki A, Poon LC, Rezende JC, Nicolaides KH. Fetal fraction in maternal plasma cell-free DNA at 11-13 weeks’ gestation: relation to maternal and fetal characteristics. Ultrasound Obstet Gynecol. 2013;41(1):26–32.
- Kaimal AJ, Norton ME, Kuppermann M. Prenatal testing in the genomic age: clinical outcomes, quality of life, and costs. Obstet Gynecol. 2015;126(4):737–746.
- Wapner RJ, Martin CL, Levy B, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med. 2012;367(23):2175–2184.
- Kuppermann M, Pena S, Bishop JT, et al. Effect of enhanced information, values clarification, and removal of financial barriers on use of prenatal genetic testing: a randomized clinical trial. JAMA. 2014;312(12):1210–1217.
- Cuckle H, Benn P, Pergament E. Maternal cfDNA screening for Down syndrome—a cost sensitivity analysis. Prenat Diagn. 2013;33(7):636–642.
- Beulen L, Grutters JPC, Faas BH, Feenstra I, van Vugt JMG, Bekker MN. The consequences of implementing non-invasive prenatal testing in Dutch national health care: a cost-effectiveness analysis. Eur J Obstet Gynecol Reprod Biol. 2014;182:53–61.
- Okun N, Teitelbaum M, Huang T, Dewa CS, Hoch JS. The price of performance: a cost and performance analysis of the implementation of cell-free fetal DNA testing for Down syndrome in Ontario, Canada: Cost and performance analysis of cfDNA testing for Down syndrome in Ontario. Prenat Diagn. 2014;34(4):350–356.
- Bianchi DW, Chudova D, Sehnert AJ, et al. Noninvasive prenatal testing and incidental detection of occult maternal malignancies. JAMA. 2015;314(2):162–169.
- Wang Y, Chen Y, Tian F, et al. Maternal mosaicism is a significant contributor to discordant sex chromosomal aneuploidies associated with noninvasive prenatal testing. Clin Chem. 2014;60(1):251–259.
- Norton ME, Jelliffe-Pawlowski LL, Currier RJ. Chromosome abnormalities detected by current prenatal screening and noninvasive prenatal testing. Obstet Gynecol. 2014;124(5):979–986Society for Maternal-Fetal Medicine (SMFM) Publications Committee. Prenatal aneuploidy screening using cell-free DNA. Am J Obstet Gynecol. 2015;212(6):711–716.
CASE: Low-risk patient requests cell-free DNA screening
Ms. Smith is a 25-year-old woman (G1P0) presenting at 10 weeks’ gestation for her first prenatal visit. She requests cell-free DNA (cfDNA) screening to test for fetal aneuploidy. You explain that the current recommendations are for traditional screening, and inform her that her insurance may not cover the cost of cfDNA screening. She is anxious to learn the sex of her fetus as early as possible, and indicates that she would like to pursue cfDNA. After further discussion of the pros and cons, you order the test.
Prenatal screening is currently recommended in pregnancy for a number of genetic disorders, chromosomal aneuploidy, and structural birth defects in the fetus, regardless of maternal age or family history. There is a broad range of sonographic and maternal serum-based options available for carrying out aneuploidy risk assessment in the first and/or second trimester.
In addition, cfDNA screening for fetal aneuploidy has been clinically available since 2011 and has seen tremendous uptake, particularly in the high-risk population. Recent data indicate that cfDNA screening likewise has very high sensitivity and specificity for trisomy 21 in the low-risk population.1,2
Many low-risk patients are asking providers about the pros and cons of cfDNA screening, and the appropriateness of this test as a primary screen, including in low-risk patients, is the focus of this article.
What is cfDNA?
cfDNA consists of small (<200 base pairs) fragments of DNA that are present in the maternal serum. After 10 weeks of gestation, about 10% of the total circulating cfDNA in the maternal serum is derived from the placenta and can therefore be used to test for fetal disorders (FIGURE).3
Although cfDNA screening has been reported to be possible for many different types of genetic conditions, such as RhD type and single-gene disorders such as achondroplasia,4 most clinical testing is done for fetal chromosomal disorders, including trisomies 13, 18, and 21 and the sex chromosomes. In addition, some laboratories provide testing for other trisomies (16 and 22), as well as some of the microdeletion syndromes (22q11.2, 1p36, Prader Willi syndrome, and others).5
Analysis of cfDNA to assess the risk for aneuploidy is done using a number of different approaches; these generally all include next-generation sequencing with advanced bioinformatics analyses.3,6–9 Although the laboratories use somewhat different techniques, all of them share very high sensitivity and specificity for detection of trisomy 21 (TABLE 1).10
Sensitivities for trisomy 13 and sex chromosomal abnormalities are somewhat lower, but the specificity is greater than 99% for each condition, meaning that false-positive rates are very low.
The accuracy of cfDNA in identifying chromosomal aneuploidy depends on several factors, including the relative amount of fetal to maternal DNA, the chance that a chromosome abnormality is present (that is, the risk based on maternal age or results of other screening), and other factors such as the presence of twins or a nonviable second fetus, or the presence of placental mosaicism.
Because of these variables, both false-positive and false-negative results can occur, and the test is not diagnostic but rather is considered a screening test. A positive result does not mean that the fetus is definitely affected with aneuploidy.
What are the advantages of cfDNA screening for low-risk patients?
There are several benefits of cfDNA screening versus traditional screening or diagnostic testing, which are the other options available (TABLE 2). For Down syndrome, the detection rate is higher and the false-positive rate is lower than that seen with traditional aneuploidy screening using serum analytes and nuchal translucency ultrasonography.1,2
TABLE 2 Pros and cons of cfDNA screening in low-risk patients
Pros
- High detection rate and very low false-positive rate
- Can be performed any time after 10 weeks’ gestation
- Requires a single blood test at any gestational age
- Results presented in simple “Yes” or “No” format
- As with other screening tests, cfDNA provides a noninvasive determination of risk
Cons
- Tests for a limited range of conditions, which are rare in low-risk patients
- Is not as comprehensive or definitive as diagnostic testing with amniocentesis or chorionic villus sampling
- Results do not adjust for patient’s prior risk
- Positive results require calculation and interpretation of positive predictive value by provider
- Low fetal DNA and other factors can lead to test failure in some cases
- Cannot be used with vanishing twin
- Can reveal unsuspected maternal conditions of uncertain significance
The test can be done any time after 10 weeks’ gestation without the narrow gestational-age windows required or the need for accurate gestational age determination using traditional screening to accurately interpret results. cfDNA screening involves a single blood test that does not require integration with multiple serum markers or ultrasound findings. Finally, results are generally presented in a simple “Yes” or “No” format that is easy for providers and patients to understand.
CASE Continued
Your patient’s results are positive for trisomy 13. Her understanding is that the test is more than 99% accurate, and she interprets this to mean that the chance of trisomy 13 in her fetus is more than 99%. She is distraught and asks about pregnancy termination.
What are the limitations of cfDNA screening?
Similar to other noninvasive screening tests, cfDNA screening does not carry direct risk to the pregnancy. However, there are limitations to this testing. As a result, the American College of Obstetricians and Gynecologists (ACOG) and the Society for Maternal-Fetal Medicine (SMFM) recently have stated that traditional screening is the most appropriate option for most women.11,12
One reason that cfDNA screening may not be the best choice for low-risk women is that Down syndrome is quite uncommon in this group, so cfDNA screening is a very precise test for a rare condition. Traditional multiple marker screening, on the other hand, is more effective at signaling risk for the broad range of adverse perinatal outcomes that can affect a pregnancy, including other structural birth defects, as well as such obstetric complications as preterm birth, preeclampsia, and fetal growth restriction.13,14
Many women who undergo cfDNA screening are under the impression that they have had a definitive test for all birth defects when, in fact, the coverage of cfDNA for all possible birth defects in a low-risk woman is very limited; her residual risk for a birth defect is little changed by a normal cfDNA result.
The ease of obtaining a blood sample for cfDNA screening is an advantage of the test. However, because it is simple to perform, it often is done with inadequate pretest counseling or consideration. Just because the test is easily obtained does not negate the need for adequate discussion to assure that each woman understands what the test can and cannot measure, and the possible outcomes of testing.
Another perceived benefit of cfDNA screening is the simple presentation of results. While reports vary, they generally provide very dichotomized results. Aneuploidy risk is reported as “Positive” or “Negative,” or as “Detected” or “Not detected.”
Some laboratories report the chance of aneuploidy; this is almost always stated to be more than 99% in patients at increased risk, and less than 1 in 10,000 in patients at low risk.
All of these results suggest a near diagnostic certainty. However, this reporting is oversimplified and misleading, as it does not account for each patient’s prior or background risk. The chance that a positive result is a true positive is very different in a 20-year-old versus a 35-year-old woman, yet the reports do not reflect this difference in positive predictive value (PPV). See TABLE 1.
Accurate interpretation of risk for the individual patient, therefore, requires calculation by the provider; this can be done through an online calculator available through the Perinatal Quality Foundation (www.perina talquality.org).
CASE Continued
You explain to your patient that the chance her fetus has trisomy 13 is far lower than 99%, based in part on the very low prior risk given her age. You calculate the PPV using an online calculator, which estimates that there is only a 7% chance that this is a true positive result.
As mentioned earlier, there has been a tremendously rapid uptake of cfDNA screening. Given wider use by practitioners not as familiar with the complexities of genetic testing and statistical analysis, misunderstanding of the test characteristics carries risks if inappropriate recommendations or decisions are made or actions taken.
Most low-risk patients do not request or desire diagnostic testing. It is important during pretest counseling to explain that cfDNA cannot detect all significant chromosomal aneuploidies. Some serious abnormalities will be undetected; therefore, some women may prefer more comprehensive prenatal testing (TABLE 3).
TABLE 3 Checklist for pretest counseling for cfDNA28
- cfDNA screening is the most accurate screening test for trisomy 21
- cfDNA is a screening test, and false-positive and false-negative results can and do occur
- Diagnostic confirmation with chorionic villus sampling or amniocentesis is recommended for women with abnormal cfDNA results
- A negative cfDNA result decreases risk but does not rule out trisomy 21 and other chromosomal conditions
- cfDNA does not test for all chromosomal conditions
- Women who desire definitive information about chromosome conditions in the pregnancy should consider diagnostic testing with chorionic villus sampling or amniocentesis
- All genetic testing is optional. Whether a woman chooses to have a screening test, a diagnostic test, or no testing is a personal decision; any are reasonable options for any pregnant woman.
ACOG recommends that diagnostic testing should be available to all pregnant women, regardless of age.15 In prenatal series, trisomies 13, 18, and 21 make up approximately two-thirds of all clinically significant aneuploidies.16,17 Given that cfDNA detects only these aneuploidies, the other third will not be identified prenatally in patients who choose cfDNA. Traditional aneuploidy screening has been demonstrated to detect a broader range of these less common but clinically important chromosomal abnormalities.18
In one study of women found to be at increased risk based on traditional multiple marker screening, if cfDNA were chosen instead of diagnostic testing, 17% of the aneuploidies present in this group would not have been detected.18 Of all high-risk women in this study, 2%, or 1 in 50, had a chromosomal abnormality detectable by amniocentesis but not with cfDNA.
Successful tests require adequate placental DNA
Accurate interpretation of cfDNA screening also requires that an adequate quantity of placental DNA be present; this is often referred to as the “fetal fraction.” In some cases, the placental DNA volume is too low for accurate analysis, particularly in obese patients and women with specific chromosomal abnormalities.
Some laboratories measure this and do not report a result if the fetal fraction is below a specific cut-off, typically about 4%. Other laboratories do not measure or exclude cases with too little fetal DNA, raising concern that this could result in missing cases of aneuploidy. It has been noted that a placental DNA fraction of less than 8% is associated with less accurate test results, even if results are returned.8
Low fetal fraction also has been associated with maternal obesity, and in one study cfDNA failed to provide a result in 20% of women weighing more than 250 lb and 50% of women weighing more than 350 lb.19 Therefore, cfDNA is not the best option for obese women (TABLE 4).
TABLE 4 Appropriateness of cfDNA screeningin specific clinical circumstances
Optimal candidates for cfDNA screening
- High risk for trisomy based on maternal age (≥35 years)
- Ultrasound findings suggesting trisomy 13, 18, or 21
- History of prior pregnancy with trisomy 13, 18, or 21
- Positive traditional screening test
- Parental balanced Robertsonian translocation associated with risk for trisomy 13 or 21
Less optimal candidates
- Low risk for trisomy based on age and/or low risk traditional screening
- Ultrasound structural anomalies other than those specifically suggesting trisomy 13, 18, or 21
- High risk for nonchromosomal genetic disorder
- Comprehensive genetic diagnosis desired
- Maternal malignancy
- Maternal organ transplant
- Maternal sex chromosomal mosaicism or other chromosomal abnormality
- Maternal obesity
- Gestational age <10 weeks
While the free fraction is relatively constant from 10 to 22 weeks’ gestation, it is lower earlier than 10 weeks’ gestation and less likely to provide a result. For this reason, the test should not be attempted before 10 weeks’ gestation.
Recent evidence indicates that low fetal DNA fraction is associated with some chromosome abnormalities. Given this association, women with failed cfDNA results should be counseled and offered appropriate follow-up. As the association appears to be greater for trisomies 13 and 18, and triploidy, a careful ultrasound is likely to detect abnormalities in many such cases. However, it also is appropriate to offer the option of diagnostic testing, given the very high risk.
A repeat cfDNA test will be successful in some cases. Whether the patient chooses to attempt cfDNA again may depend in part on maternal body mass index (BMI), as well as gestational age—a patient at a more advanced gestation may not wish to delay obtaining definitive information given the high risk.
cfDNA screening has a low false-positive rate
One of the greatest benefits of cfDNA screening is a lower false-positive rate than is reported with traditional screening. However, when “no results” cases are also considered, the percentage of patients who require follow-up after cfDNA is close to that of traditional screening.
The chance of test failure is reported to be 0.9% to 8.1%,7,9,10 and varies in part by whether the laboratory measures fetal fraction and requires a minimum concentration.
A recent meta-analysis estimated the overall test failure rate at 3%.10 When comparing cfDNA to traditional screening, if “no results” cases are included with the “screen positive” group, the benefits of cfDNA over traditional screening are much less clear, particularly in a low-risk population.
ACOG: Offer traditional multiple-marker screening first
While multiple marker and cfDNA screening have differing performance characteristics, there are no data to support doing both tests concurrently. In fact, in a recent survey of nearly 200 women presented with different testing scenarios, women found it preferable and more reassuring to have a positive traditional screen followed by normal cfDNA results, rather that discrepant results of the 2 tests done concurrently.20
For many reasons, the approach recommended by ACOG and SMFM is to offer traditional multiple-marker screening first, and cfDNA screening or diagnostic testing as a follow-up for patients that screen positive. In that scenario, the benefits and limitations of diagnostic testing versus follow-up with cfDNA screening should be explained carefully.
In all patients who have a positive cfDNA result, diagnostic testing for confirmation should be offered and strongly recommended prior to pregnancy termination if that is considered. Even if a structural abnormality is present and a true positive result is highly likely, karyotyping is important to determine if there may be an inherited translocation putting subsequent pregnancies at higher risk.
Components of pretest counseling
A woman of any age can have a fetus with trisomy or another chromosomal abnormality, and some women prefer diagnostic testing or no testing regardless of age. It is therefore appropriate to offer diagnostic testing, screening, or the option of no testing to all women.
Recent studies have demonstrated that providing well-informed access to all prenatal tests results in more informed choices and no increase in uptake of invasive testing.22 However, the offer of prenatal testing requires discussion of the pros and cons of all test options, including the detection rates of all significant abnormalities, the screen positive rates, and recommended follow-up if an abnormal result is obtained. See TABLE 4.
Cost-effectiveness
Although the detection rate of cfDNA for trisomy 21 is higher than that of traditional screening, the detection rate of traditional screening is also quite high at lower cost. For low-risk women, therefore, traditional screening provides a less expensive alternative to cfDNA. Because aneuploidy is rare in low-risk patients, the residual chance of aneuploidy after a normal traditional screen is very low, and the cost per additional case of Down syndrome detected by cfDNA is very high.
In one study, this was estimated at $3.6 million.23 These authors suggested that, at present, cfDNA is optimally used as a secondary screen for high-risk women. Other cost analyses also have demonstrated that the most cost-effective strategy is a model in which cfDNA is used as a follow-up test in patients found to be screen positive by traditional screening.15,24 A recent cost utility analysis compared outcomes of 6 approaches to prenatal screening, including sequential screening, cfDNA screening, nuchal translucency only, and diagnostic testing with microarray (alone, in combination, or in sequence).
The clinical outcomes included fetal abnormalities detected, taking into account all chromosomal abnormalities, as well as failed cfDNA tests. For younger women (<40 yr), traditional sequential screening provided the highest detection of all abnormalities and was the optimal testing strategy, while cfDNA was preferable for women aged 40 or older, given the higher prevalence of trisomy 21.20
Incidental findings
Given that the cfDNA in maternal serum is a mixture of maternal and placental DNA, a number of biologic phenomena can cause a false-positive cfDNA result. In many cases, these false-positives reveal unanticipated or unexpected maternal conditions and information that the woman may have preferred not to know. A few cases of maternal malignancies with chromosomal abnormalities within the tumor have been reported in patients with false-positive cfDNA results.26
These case reports have raised the question about the need for further evaluation for maternal malignancy in women with false-positive results. Maternal genetic disorders also can cause false-positive results, and may lead to unanticipated detection of adult-onset conditions. In some cases, positive results for sex chromosomal aneuploidy can occur in pregnant women who themselves have a sex chromosomal abnormality, often in mosaic form and previously undiagnosed.27
Again, this has led to discussion of the possible health benefit of karyotyping women who have a false-positive cfDNA result to rule out a mosaic chromosomal abnormality in the mother.
At this time, the clinical utility of such investigations is unknown and there are no recommendations regarding appropriate follow-up for such cases.
CASE Resolved
Given the results of her cfDNA screening, your patient opts to undergo diagnostic testing. In that testing, trisomy 13 is ruled out and she goes on to have a healthy daughter.
Share your thoughts on this article! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
CASE: Low-risk patient requests cell-free DNA screening
Ms. Smith is a 25-year-old woman (G1P0) presenting at 10 weeks’ gestation for her first prenatal visit. She requests cell-free DNA (cfDNA) screening to test for fetal aneuploidy. You explain that the current recommendations are for traditional screening, and inform her that her insurance may not cover the cost of cfDNA screening. She is anxious to learn the sex of her fetus as early as possible, and indicates that she would like to pursue cfDNA. After further discussion of the pros and cons, you order the test.
Prenatal screening is currently recommended in pregnancy for a number of genetic disorders, chromosomal aneuploidy, and structural birth defects in the fetus, regardless of maternal age or family history. There is a broad range of sonographic and maternal serum-based options available for carrying out aneuploidy risk assessment in the first and/or second trimester.
In addition, cfDNA screening for fetal aneuploidy has been clinically available since 2011 and has seen tremendous uptake, particularly in the high-risk population. Recent data indicate that cfDNA screening likewise has very high sensitivity and specificity for trisomy 21 in the low-risk population.1,2
Many low-risk patients are asking providers about the pros and cons of cfDNA screening, and the appropriateness of this test as a primary screen, including in low-risk patients, is the focus of this article.
What is cfDNA?
cfDNA consists of small (<200 base pairs) fragments of DNA that are present in the maternal serum. After 10 weeks of gestation, about 10% of the total circulating cfDNA in the maternal serum is derived from the placenta and can therefore be used to test for fetal disorders (FIGURE).3
Although cfDNA screening has been reported to be possible for many different types of genetic conditions, such as RhD type and single-gene disorders such as achondroplasia,4 most clinical testing is done for fetal chromosomal disorders, including trisomies 13, 18, and 21 and the sex chromosomes. In addition, some laboratories provide testing for other trisomies (16 and 22), as well as some of the microdeletion syndromes (22q11.2, 1p36, Prader Willi syndrome, and others).5
Analysis of cfDNA to assess the risk for aneuploidy is done using a number of different approaches; these generally all include next-generation sequencing with advanced bioinformatics analyses.3,6–9 Although the laboratories use somewhat different techniques, all of them share very high sensitivity and specificity for detection of trisomy 21 (TABLE 1).10
Sensitivities for trisomy 13 and sex chromosomal abnormalities are somewhat lower, but the specificity is greater than 99% for each condition, meaning that false-positive rates are very low.
The accuracy of cfDNA in identifying chromosomal aneuploidy depends on several factors, including the relative amount of fetal to maternal DNA, the chance that a chromosome abnormality is present (that is, the risk based on maternal age or results of other screening), and other factors such as the presence of twins or a nonviable second fetus, or the presence of placental mosaicism.
Because of these variables, both false-positive and false-negative results can occur, and the test is not diagnostic but rather is considered a screening test. A positive result does not mean that the fetus is definitely affected with aneuploidy.
What are the advantages of cfDNA screening for low-risk patients?
There are several benefits of cfDNA screening versus traditional screening or diagnostic testing, which are the other options available (TABLE 2). For Down syndrome, the detection rate is higher and the false-positive rate is lower than that seen with traditional aneuploidy screening using serum analytes and nuchal translucency ultrasonography.1,2
TABLE 2 Pros and cons of cfDNA screening in low-risk patients
Pros
- High detection rate and very low false-positive rate
- Can be performed any time after 10 weeks’ gestation
- Requires a single blood test at any gestational age
- Results presented in simple “Yes” or “No” format
- As with other screening tests, cfDNA provides a noninvasive determination of risk
Cons
- Tests for a limited range of conditions, which are rare in low-risk patients
- Is not as comprehensive or definitive as diagnostic testing with amniocentesis or chorionic villus sampling
- Results do not adjust for patient’s prior risk
- Positive results require calculation and interpretation of positive predictive value by provider
- Low fetal DNA and other factors can lead to test failure in some cases
- Cannot be used with vanishing twin
- Can reveal unsuspected maternal conditions of uncertain significance
The test can be done any time after 10 weeks’ gestation without the narrow gestational-age windows required or the need for accurate gestational age determination using traditional screening to accurately interpret results. cfDNA screening involves a single blood test that does not require integration with multiple serum markers or ultrasound findings. Finally, results are generally presented in a simple “Yes” or “No” format that is easy for providers and patients to understand.
CASE Continued
Your patient’s results are positive for trisomy 13. Her understanding is that the test is more than 99% accurate, and she interprets this to mean that the chance of trisomy 13 in her fetus is more than 99%. She is distraught and asks about pregnancy termination.
What are the limitations of cfDNA screening?
Similar to other noninvasive screening tests, cfDNA screening does not carry direct risk to the pregnancy. However, there are limitations to this testing. As a result, the American College of Obstetricians and Gynecologists (ACOG) and the Society for Maternal-Fetal Medicine (SMFM) recently have stated that traditional screening is the most appropriate option for most women.11,12
One reason that cfDNA screening may not be the best choice for low-risk women is that Down syndrome is quite uncommon in this group, so cfDNA screening is a very precise test for a rare condition. Traditional multiple marker screening, on the other hand, is more effective at signaling risk for the broad range of adverse perinatal outcomes that can affect a pregnancy, including other structural birth defects, as well as such obstetric complications as preterm birth, preeclampsia, and fetal growth restriction.13,14
Many women who undergo cfDNA screening are under the impression that they have had a definitive test for all birth defects when, in fact, the coverage of cfDNA for all possible birth defects in a low-risk woman is very limited; her residual risk for a birth defect is little changed by a normal cfDNA result.
The ease of obtaining a blood sample for cfDNA screening is an advantage of the test. However, because it is simple to perform, it often is done with inadequate pretest counseling or consideration. Just because the test is easily obtained does not negate the need for adequate discussion to assure that each woman understands what the test can and cannot measure, and the possible outcomes of testing.
Another perceived benefit of cfDNA screening is the simple presentation of results. While reports vary, they generally provide very dichotomized results. Aneuploidy risk is reported as “Positive” or “Negative,” or as “Detected” or “Not detected.”
Some laboratories report the chance of aneuploidy; this is almost always stated to be more than 99% in patients at increased risk, and less than 1 in 10,000 in patients at low risk.
All of these results suggest a near diagnostic certainty. However, this reporting is oversimplified and misleading, as it does not account for each patient’s prior or background risk. The chance that a positive result is a true positive is very different in a 20-year-old versus a 35-year-old woman, yet the reports do not reflect this difference in positive predictive value (PPV). See TABLE 1.
Accurate interpretation of risk for the individual patient, therefore, requires calculation by the provider; this can be done through an online calculator available through the Perinatal Quality Foundation (www.perina talquality.org).
CASE Continued
You explain to your patient that the chance her fetus has trisomy 13 is far lower than 99%, based in part on the very low prior risk given her age. You calculate the PPV using an online calculator, which estimates that there is only a 7% chance that this is a true positive result.
As mentioned earlier, there has been a tremendously rapid uptake of cfDNA screening. Given wider use by practitioners not as familiar with the complexities of genetic testing and statistical analysis, misunderstanding of the test characteristics carries risks if inappropriate recommendations or decisions are made or actions taken.
Most low-risk patients do not request or desire diagnostic testing. It is important during pretest counseling to explain that cfDNA cannot detect all significant chromosomal aneuploidies. Some serious abnormalities will be undetected; therefore, some women may prefer more comprehensive prenatal testing (TABLE 3).
TABLE 3 Checklist for pretest counseling for cfDNA28
- cfDNA screening is the most accurate screening test for trisomy 21
- cfDNA is a screening test, and false-positive and false-negative results can and do occur
- Diagnostic confirmation with chorionic villus sampling or amniocentesis is recommended for women with abnormal cfDNA results
- A negative cfDNA result decreases risk but does not rule out trisomy 21 and other chromosomal conditions
- cfDNA does not test for all chromosomal conditions
- Women who desire definitive information about chromosome conditions in the pregnancy should consider diagnostic testing with chorionic villus sampling or amniocentesis
- All genetic testing is optional. Whether a woman chooses to have a screening test, a diagnostic test, or no testing is a personal decision; any are reasonable options for any pregnant woman.
ACOG recommends that diagnostic testing should be available to all pregnant women, regardless of age.15 In prenatal series, trisomies 13, 18, and 21 make up approximately two-thirds of all clinically significant aneuploidies.16,17 Given that cfDNA detects only these aneuploidies, the other third will not be identified prenatally in patients who choose cfDNA. Traditional aneuploidy screening has been demonstrated to detect a broader range of these less common but clinically important chromosomal abnormalities.18
In one study of women found to be at increased risk based on traditional multiple marker screening, if cfDNA were chosen instead of diagnostic testing, 17% of the aneuploidies present in this group would not have been detected.18 Of all high-risk women in this study, 2%, or 1 in 50, had a chromosomal abnormality detectable by amniocentesis but not with cfDNA.
Successful tests require adequate placental DNA
Accurate interpretation of cfDNA screening also requires that an adequate quantity of placental DNA be present; this is often referred to as the “fetal fraction.” In some cases, the placental DNA volume is too low for accurate analysis, particularly in obese patients and women with specific chromosomal abnormalities.
Some laboratories measure this and do not report a result if the fetal fraction is below a specific cut-off, typically about 4%. Other laboratories do not measure or exclude cases with too little fetal DNA, raising concern that this could result in missing cases of aneuploidy. It has been noted that a placental DNA fraction of less than 8% is associated with less accurate test results, even if results are returned.8
Low fetal fraction also has been associated with maternal obesity, and in one study cfDNA failed to provide a result in 20% of women weighing more than 250 lb and 50% of women weighing more than 350 lb.19 Therefore, cfDNA is not the best option for obese women (TABLE 4).
TABLE 4 Appropriateness of cfDNA screeningin specific clinical circumstances
Optimal candidates for cfDNA screening
- High risk for trisomy based on maternal age (≥35 years)
- Ultrasound findings suggesting trisomy 13, 18, or 21
- History of prior pregnancy with trisomy 13, 18, or 21
- Positive traditional screening test
- Parental balanced Robertsonian translocation associated with risk for trisomy 13 or 21
Less optimal candidates
- Low risk for trisomy based on age and/or low risk traditional screening
- Ultrasound structural anomalies other than those specifically suggesting trisomy 13, 18, or 21
- High risk for nonchromosomal genetic disorder
- Comprehensive genetic diagnosis desired
- Maternal malignancy
- Maternal organ transplant
- Maternal sex chromosomal mosaicism or other chromosomal abnormality
- Maternal obesity
- Gestational age <10 weeks
While the free fraction is relatively constant from 10 to 22 weeks’ gestation, it is lower earlier than 10 weeks’ gestation and less likely to provide a result. For this reason, the test should not be attempted before 10 weeks’ gestation.
Recent evidence indicates that low fetal DNA fraction is associated with some chromosome abnormalities. Given this association, women with failed cfDNA results should be counseled and offered appropriate follow-up. As the association appears to be greater for trisomies 13 and 18, and triploidy, a careful ultrasound is likely to detect abnormalities in many such cases. However, it also is appropriate to offer the option of diagnostic testing, given the very high risk.
A repeat cfDNA test will be successful in some cases. Whether the patient chooses to attempt cfDNA again may depend in part on maternal body mass index (BMI), as well as gestational age—a patient at a more advanced gestation may not wish to delay obtaining definitive information given the high risk.
cfDNA screening has a low false-positive rate
One of the greatest benefits of cfDNA screening is a lower false-positive rate than is reported with traditional screening. However, when “no results” cases are also considered, the percentage of patients who require follow-up after cfDNA is close to that of traditional screening.
The chance of test failure is reported to be 0.9% to 8.1%,7,9,10 and varies in part by whether the laboratory measures fetal fraction and requires a minimum concentration.
A recent meta-analysis estimated the overall test failure rate at 3%.10 When comparing cfDNA to traditional screening, if “no results” cases are included with the “screen positive” group, the benefits of cfDNA over traditional screening are much less clear, particularly in a low-risk population.
ACOG: Offer traditional multiple-marker screening first
While multiple marker and cfDNA screening have differing performance characteristics, there are no data to support doing both tests concurrently. In fact, in a recent survey of nearly 200 women presented with different testing scenarios, women found it preferable and more reassuring to have a positive traditional screen followed by normal cfDNA results, rather that discrepant results of the 2 tests done concurrently.20
For many reasons, the approach recommended by ACOG and SMFM is to offer traditional multiple-marker screening first, and cfDNA screening or diagnostic testing as a follow-up for patients that screen positive. In that scenario, the benefits and limitations of diagnostic testing versus follow-up with cfDNA screening should be explained carefully.
In all patients who have a positive cfDNA result, diagnostic testing for confirmation should be offered and strongly recommended prior to pregnancy termination if that is considered. Even if a structural abnormality is present and a true positive result is highly likely, karyotyping is important to determine if there may be an inherited translocation putting subsequent pregnancies at higher risk.
Components of pretest counseling
A woman of any age can have a fetus with trisomy or another chromosomal abnormality, and some women prefer diagnostic testing or no testing regardless of age. It is therefore appropriate to offer diagnostic testing, screening, or the option of no testing to all women.
Recent studies have demonstrated that providing well-informed access to all prenatal tests results in more informed choices and no increase in uptake of invasive testing.22 However, the offer of prenatal testing requires discussion of the pros and cons of all test options, including the detection rates of all significant abnormalities, the screen positive rates, and recommended follow-up if an abnormal result is obtained. See TABLE 4.
Cost-effectiveness
Although the detection rate of cfDNA for trisomy 21 is higher than that of traditional screening, the detection rate of traditional screening is also quite high at lower cost. For low-risk women, therefore, traditional screening provides a less expensive alternative to cfDNA. Because aneuploidy is rare in low-risk patients, the residual chance of aneuploidy after a normal traditional screen is very low, and the cost per additional case of Down syndrome detected by cfDNA is very high.
In one study, this was estimated at $3.6 million.23 These authors suggested that, at present, cfDNA is optimally used as a secondary screen for high-risk women. Other cost analyses also have demonstrated that the most cost-effective strategy is a model in which cfDNA is used as a follow-up test in patients found to be screen positive by traditional screening.15,24 A recent cost utility analysis compared outcomes of 6 approaches to prenatal screening, including sequential screening, cfDNA screening, nuchal translucency only, and diagnostic testing with microarray (alone, in combination, or in sequence).
The clinical outcomes included fetal abnormalities detected, taking into account all chromosomal abnormalities, as well as failed cfDNA tests. For younger women (<40 yr), traditional sequential screening provided the highest detection of all abnormalities and was the optimal testing strategy, while cfDNA was preferable for women aged 40 or older, given the higher prevalence of trisomy 21.20
Incidental findings
Given that the cfDNA in maternal serum is a mixture of maternal and placental DNA, a number of biologic phenomena can cause a false-positive cfDNA result. In many cases, these false-positives reveal unanticipated or unexpected maternal conditions and information that the woman may have preferred not to know. A few cases of maternal malignancies with chromosomal abnormalities within the tumor have been reported in patients with false-positive cfDNA results.26
These case reports have raised the question about the need for further evaluation for maternal malignancy in women with false-positive results. Maternal genetic disorders also can cause false-positive results, and may lead to unanticipated detection of adult-onset conditions. In some cases, positive results for sex chromosomal aneuploidy can occur in pregnant women who themselves have a sex chromosomal abnormality, often in mosaic form and previously undiagnosed.27
Again, this has led to discussion of the possible health benefit of karyotyping women who have a false-positive cfDNA result to rule out a mosaic chromosomal abnormality in the mother.
At this time, the clinical utility of such investigations is unknown and there are no recommendations regarding appropriate follow-up for such cases.
CASE Resolved
Given the results of her cfDNA screening, your patient opts to undergo diagnostic testing. In that testing, trisomy 13 is ruled out and she goes on to have a healthy daughter.
Share your thoughts on this article! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
- Bianchi DW, Parker RL, Wentworth J, et al. DNA sequencing versus standard prenatal aneuploidy screening. N Engl J Med. 2014;370(9):799–808.
- Norton ME, Jacobsson B, Swamy GK, et al. Cell-free DNA analysis for noninvasive examination of trisomy. N Engl J Med. 2015;372(17):1589–1597.
- Norton ME, Brar H, Weiss J, et al. Non-Invasive Chromosomal Evaluation (NICE) study: results of a multicenter prospective cohort study for detection of fetal trisomy 21 and trisomy 18. Am J Obstet Gynecol. 2012;207(2):137.e1–e8.
- Chitty LS, Mason S, Barrett AN, et al. Non-invasive prenatal diagnosis of achondroplasia and thanatophoric dysplasia: next-generation sequencing allows for a safer, more accurate, and comprehensive approach. Prenat Diagn. 2015;35(7):656–662.
- Wapner RJ, Babiarz JE, Levy B, et al. Expanding the scope of noninvasive prenatal testing: detection of fetal microdeletion syndromes. Am J Obstet Gynecol. 2015;212(3):332.e1–e9.
- Bianchi DW, Platt LD, Goldberg JD, Abuhamad AZ, Sehnert AJ, Rava RP. Genome-wide fetal aneuploidy detection by maternal plasma DNA sequencing. Obstet Gynecol. 2012;119(5):890–901.
- Palomaki GE, Kloza EM, Lambert-Messerlian GM, et al. DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation study. Genet Med. 2011;13(11):913–920.
- Sparks AB, Wang ET, Struble CA, et al. DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation study [abstract]. Proceedings of the ISPD 16th International Conference on Prenatal Diagnosis and Therapy; Miami, Florida; June 3–6, 2012. Prenat Diagn. 2012;32(suppl 1):s3–s9.
- Zimmermann B, Hill M, Gemelos G, et al. Noninvasive prenatal aneuploidy testing of chromosomes 13, 18, 21, X, and Y, using targeted sequencing of polymorphic loci. Prenat Diagn. 2012;32(13):1233–1241.
- Gil MM, Quezada MS, Revello R, Akolekar R, Niclaides KH. Analysis of cell-free DNA in maternal blood in screening for fetal aneuploidies: updated meta-analysis. Ultrasound Obstet Gynecol. 2015;45(3):249–266.
- American College of Obstetricians and Gynecologists. Committee Opinion No. 640: cell-free DNA screening for fetal aneuploidy. Obstet Gynecol. 2015;126(3):e31–e37.
- Society for Maternal-Fetal Medicine (SMFM) Publications Committee. Prenatal aneuploidy screening using cell-free DNA. Am J Obstet Gynecol. 2015;212(6):711–716.
- Baer RJ, Currier RJ, Norton ME, et al. Obstetric, perinatal, and fetal outcomes in pregnancies with false-positive integrated screening results. Obstet Gynecol. 2014;123(3):603–609.
- Dugoff L; Society for Maternal-Fetal Medicine. First- and second-trimester maternal serum markers for aneuploidy and adverse obstetric outcomes. Obstet Gynecol. 2010;115(5):1052–1061.
- American College of Obstetricians and Gynecologists. Practice bulletin No. 88: invasive prenatal testing for aneuploidy. Obstet Gynecol. 2007;110(6):1459–1467.
- Alamillo CM, Krantz D, Evans M, Fiddler M, Pergament E. Nearly a third of abnormalities found after first-trimester screening are different than expected: 10-year experience from a single center. Prenat Diagn. 2013;33(3):251–256.
- Wellesley D, Dolk H, Boyd PA, et al. Rare chromosome abnormalities, prevalence and prenatal diagnosis rates from population-based congenital anomaly registers in Europe. Eur J Hum Genet. 2012;20(5):521–526.
- Norton ME, Jelliffe-Pawlowski LL, Currier RJ. Chromosome abnormalities detected by current prenatal screening and noninvasive prenatal testing. Obstet Gynecol. 2014;124(5):979–986.
- Ashoor G, Syngelaki A, Poon LC, Rezende JC, Nicolaides KH. Fetal fraction in maternal plasma cell-free DNA at 11-13 weeks’ gestation: relation to maternal and fetal characteristics. Ultrasound Obstet Gynecol. 2013;41(1):26–32.
- Kaimal AJ, Norton ME, Kuppermann M. Prenatal testing in the genomic age: clinical outcomes, quality of life, and costs. Obstet Gynecol. 2015;126(4):737–746.
- Wapner RJ, Martin CL, Levy B, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med. 2012;367(23):2175–2184.
- Kuppermann M, Pena S, Bishop JT, et al. Effect of enhanced information, values clarification, and removal of financial barriers on use of prenatal genetic testing: a randomized clinical trial. JAMA. 2014;312(12):1210–1217.
- Cuckle H, Benn P, Pergament E. Maternal cfDNA screening for Down syndrome—a cost sensitivity analysis. Prenat Diagn. 2013;33(7):636–642.
- Beulen L, Grutters JPC, Faas BH, Feenstra I, van Vugt JMG, Bekker MN. The consequences of implementing non-invasive prenatal testing in Dutch national health care: a cost-effectiveness analysis. Eur J Obstet Gynecol Reprod Biol. 2014;182:53–61.
- Okun N, Teitelbaum M, Huang T, Dewa CS, Hoch JS. The price of performance: a cost and performance analysis of the implementation of cell-free fetal DNA testing for Down syndrome in Ontario, Canada: Cost and performance analysis of cfDNA testing for Down syndrome in Ontario. Prenat Diagn. 2014;34(4):350–356.
- Bianchi DW, Chudova D, Sehnert AJ, et al. Noninvasive prenatal testing and incidental detection of occult maternal malignancies. JAMA. 2015;314(2):162–169.
- Wang Y, Chen Y, Tian F, et al. Maternal mosaicism is a significant contributor to discordant sex chromosomal aneuploidies associated with noninvasive prenatal testing. Clin Chem. 2014;60(1):251–259.
- Norton ME, Jelliffe-Pawlowski LL, Currier RJ. Chromosome abnormalities detected by current prenatal screening and noninvasive prenatal testing. Obstet Gynecol. 2014;124(5):979–986Society for Maternal-Fetal Medicine (SMFM) Publications Committee. Prenatal aneuploidy screening using cell-free DNA. Am J Obstet Gynecol. 2015;212(6):711–716.
- Bianchi DW, Parker RL, Wentworth J, et al. DNA sequencing versus standard prenatal aneuploidy screening. N Engl J Med. 2014;370(9):799–808.
- Norton ME, Jacobsson B, Swamy GK, et al. Cell-free DNA analysis for noninvasive examination of trisomy. N Engl J Med. 2015;372(17):1589–1597.
- Norton ME, Brar H, Weiss J, et al. Non-Invasive Chromosomal Evaluation (NICE) study: results of a multicenter prospective cohort study for detection of fetal trisomy 21 and trisomy 18. Am J Obstet Gynecol. 2012;207(2):137.e1–e8.
- Chitty LS, Mason S, Barrett AN, et al. Non-invasive prenatal diagnosis of achondroplasia and thanatophoric dysplasia: next-generation sequencing allows for a safer, more accurate, and comprehensive approach. Prenat Diagn. 2015;35(7):656–662.
- Wapner RJ, Babiarz JE, Levy B, et al. Expanding the scope of noninvasive prenatal testing: detection of fetal microdeletion syndromes. Am J Obstet Gynecol. 2015;212(3):332.e1–e9.
- Bianchi DW, Platt LD, Goldberg JD, Abuhamad AZ, Sehnert AJ, Rava RP. Genome-wide fetal aneuploidy detection by maternal plasma DNA sequencing. Obstet Gynecol. 2012;119(5):890–901.
- Palomaki GE, Kloza EM, Lambert-Messerlian GM, et al. DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation study. Genet Med. 2011;13(11):913–920.
- Sparks AB, Wang ET, Struble CA, et al. DNA sequencing of maternal plasma to detect Down syndrome: an international clinical validation study [abstract]. Proceedings of the ISPD 16th International Conference on Prenatal Diagnosis and Therapy; Miami, Florida; June 3–6, 2012. Prenat Diagn. 2012;32(suppl 1):s3–s9.
- Zimmermann B, Hill M, Gemelos G, et al. Noninvasive prenatal aneuploidy testing of chromosomes 13, 18, 21, X, and Y, using targeted sequencing of polymorphic loci. Prenat Diagn. 2012;32(13):1233–1241.
- Gil MM, Quezada MS, Revello R, Akolekar R, Niclaides KH. Analysis of cell-free DNA in maternal blood in screening for fetal aneuploidies: updated meta-analysis. Ultrasound Obstet Gynecol. 2015;45(3):249–266.
- American College of Obstetricians and Gynecologists. Committee Opinion No. 640: cell-free DNA screening for fetal aneuploidy. Obstet Gynecol. 2015;126(3):e31–e37.
- Society for Maternal-Fetal Medicine (SMFM) Publications Committee. Prenatal aneuploidy screening using cell-free DNA. Am J Obstet Gynecol. 2015;212(6):711–716.
- Baer RJ, Currier RJ, Norton ME, et al. Obstetric, perinatal, and fetal outcomes in pregnancies with false-positive integrated screening results. Obstet Gynecol. 2014;123(3):603–609.
- Dugoff L; Society for Maternal-Fetal Medicine. First- and second-trimester maternal serum markers for aneuploidy and adverse obstetric outcomes. Obstet Gynecol. 2010;115(5):1052–1061.
- American College of Obstetricians and Gynecologists. Practice bulletin No. 88: invasive prenatal testing for aneuploidy. Obstet Gynecol. 2007;110(6):1459–1467.
- Alamillo CM, Krantz D, Evans M, Fiddler M, Pergament E. Nearly a third of abnormalities found after first-trimester screening are different than expected: 10-year experience from a single center. Prenat Diagn. 2013;33(3):251–256.
- Wellesley D, Dolk H, Boyd PA, et al. Rare chromosome abnormalities, prevalence and prenatal diagnosis rates from population-based congenital anomaly registers in Europe. Eur J Hum Genet. 2012;20(5):521–526.
- Norton ME, Jelliffe-Pawlowski LL, Currier RJ. Chromosome abnormalities detected by current prenatal screening and noninvasive prenatal testing. Obstet Gynecol. 2014;124(5):979–986.
- Ashoor G, Syngelaki A, Poon LC, Rezende JC, Nicolaides KH. Fetal fraction in maternal plasma cell-free DNA at 11-13 weeks’ gestation: relation to maternal and fetal characteristics. Ultrasound Obstet Gynecol. 2013;41(1):26–32.
- Kaimal AJ, Norton ME, Kuppermann M. Prenatal testing in the genomic age: clinical outcomes, quality of life, and costs. Obstet Gynecol. 2015;126(4):737–746.
- Wapner RJ, Martin CL, Levy B, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med. 2012;367(23):2175–2184.
- Kuppermann M, Pena S, Bishop JT, et al. Effect of enhanced information, values clarification, and removal of financial barriers on use of prenatal genetic testing: a randomized clinical trial. JAMA. 2014;312(12):1210–1217.
- Cuckle H, Benn P, Pergament E. Maternal cfDNA screening for Down syndrome—a cost sensitivity analysis. Prenat Diagn. 2013;33(7):636–642.
- Beulen L, Grutters JPC, Faas BH, Feenstra I, van Vugt JMG, Bekker MN. The consequences of implementing non-invasive prenatal testing in Dutch national health care: a cost-effectiveness analysis. Eur J Obstet Gynecol Reprod Biol. 2014;182:53–61.
- Okun N, Teitelbaum M, Huang T, Dewa CS, Hoch JS. The price of performance: a cost and performance analysis of the implementation of cell-free fetal DNA testing for Down syndrome in Ontario, Canada: Cost and performance analysis of cfDNA testing for Down syndrome in Ontario. Prenat Diagn. 2014;34(4):350–356.
- Bianchi DW, Chudova D, Sehnert AJ, et al. Noninvasive prenatal testing and incidental detection of occult maternal malignancies. JAMA. 2015;314(2):162–169.
- Wang Y, Chen Y, Tian F, et al. Maternal mosaicism is a significant contributor to discordant sex chromosomal aneuploidies associated with noninvasive prenatal testing. Clin Chem. 2014;60(1):251–259.
- Norton ME, Jelliffe-Pawlowski LL, Currier RJ. Chromosome abnormalities detected by current prenatal screening and noninvasive prenatal testing. Obstet Gynecol. 2014;124(5):979–986Society for Maternal-Fetal Medicine (SMFM) Publications Committee. Prenatal aneuploidy screening using cell-free DNA. Am J Obstet Gynecol. 2015;212(6):711–716.
In this Article
- Pros and cons of cfDNA in low-risk patients
- Optimal and less optimal candidates for cfDNA screening
- Checklist for pretest counseling for cfDNA
Does this new evidence for noninvasive prenatal testing to detect fetal aneuploidy move NIPT closer to universal use in pregnancy?
The introduction of cell-free fetal DNA (cfDNA) testing has had a tremendous impact on prenatal care. Numerous series reporting near-diagnostic accuracy for trisomy 21 detection have been reported,1 and several commercial laboratories offer clinical testing. Many patients now take advantage of these tests, and the very low false-positive rates have resulted in a marked decrease in the rate of invasive diagnostic testing.2 At present, most professional societies suggest that these tests be reserved for women at high risk for fetal aneuploidy.3
Details of the study
In this recent article by Porreco and colleagues, the researchers reported on a large cohort study of patients who had made the decision to undergo invasive diagnostic testing with chorionic villus sampling or amniocentesis prior to undergoing noninvasive testing, in order to validate the clinical performance of massively parallel genomic sequencing of cfDNA. As in several prior reports, the study authors found that the detection rate of cfDNA for trisomy 21 was 100%, and somewhat less for trisomy 18 (92%) and trisomy 13 (87%). The false-positive rate was very low, with only three false-positive results (all for trisomy 21) in 3,430 patients. Testing for fetal sex chromosomes reported 7 out of 3,322 errors in fetal sex and 100% detection of sex chromosomal aneuploidies, with 16 out of 3,200 false-positive results.
Study limitations
As in prior reports, limitations to the test were not clearly presented. Patients with “complex chromosomal abnormalities” not detectable by cfDNA were excluded from the reported cohort. Considering these cases, fewer of the total chromosomal abnormalities in the cohort were detected.
Also, adequate fetal DNA is necessary for accurate results, and patients with less than 4% fetal DNA were excluded. Low fetal DNA is associated with an increased risk of trisomy.4,5 Therefore, excluding such cases will bias results toward a higher detection rate.
The outcomes for cases of low fetal DNA were not included in this study, but in another recent paper 22% of cases of low fetal DNA had aneuploidy, and 16% of common aneuploidies were not detected because of failed testing.4
What this evidence means for practice
Cell-free fetal DNA is an exciting technology, and this study adds to the existing literature in the field. However, use of the test requires careful patient counseling regarding the limitations in detecting abnormalities other than trisomy 21, which comprises just 50% of all aneuploidies. Women who desire a comprehensive prenatal genetic assessment may prefer invasive diagnostic testing and should be counseled appropriately. Patients in whom the test fails should be informed that they are at high risk for a chromosomal abnormality.
Considering these outcomes, the benefits of prenatal screening with cfDNA over current testing alternatives, with serum analytes and/or invasive diagnostic testing, become less clear and the options more complex. Of primary importance is that patients understand the risks and benefits of alternative tests.
—Mary E. Norton, MD
Share your thoughts on this article! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
1. Gil MM, Akolekar R, Quezada MS, Bregant B, Nicolaides KH. Analysis of cell-free DNA in maternal blood in screening for aneuploidies: meta-analysis. Fetal Diagn Ther. 2014;35(3):156−173.
2. Wax JR, Cartin A, Chard R, Lucas FL, Pinette MG. Noninvasive prenatal testing: Impact on genetic counseling, invasive prenatal diagnosis, and trisomy 21 detection [published online ahead of print October 9, 2014]. J Clin Ultrasound. doi:`10.1002/jcu.22243. [Epub ahead of print]
3. American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 545: Noninvasive prenatal testing for fetal aneuploidy. Obstet Gynecol. 2012;120(6):1532−1534.
4. Pergament E, Cuckle H, Zimmermann B, et al. Single-nucleotide polymorphism-based noninvasive prenatal screening in a high-risk and low-risk cohort. Obstet Gynecol. 2014;124(2 pt 1):210−218.
5. Rava RP, Srinivasan A, Sehnert AJ, Bianchi DW. Circulating fetal cell-free DNA fractions differ in autosomal aneuploidies and monosomy X. Clin Chem. 2014;60(1):243−250.
The introduction of cell-free fetal DNA (cfDNA) testing has had a tremendous impact on prenatal care. Numerous series reporting near-diagnostic accuracy for trisomy 21 detection have been reported,1 and several commercial laboratories offer clinical testing. Many patients now take advantage of these tests, and the very low false-positive rates have resulted in a marked decrease in the rate of invasive diagnostic testing.2 At present, most professional societies suggest that these tests be reserved for women at high risk for fetal aneuploidy.3
Details of the study
In this recent article by Porreco and colleagues, the researchers reported on a large cohort study of patients who had made the decision to undergo invasive diagnostic testing with chorionic villus sampling or amniocentesis prior to undergoing noninvasive testing, in order to validate the clinical performance of massively parallel genomic sequencing of cfDNA. As in several prior reports, the study authors found that the detection rate of cfDNA for trisomy 21 was 100%, and somewhat less for trisomy 18 (92%) and trisomy 13 (87%). The false-positive rate was very low, with only three false-positive results (all for trisomy 21) in 3,430 patients. Testing for fetal sex chromosomes reported 7 out of 3,322 errors in fetal sex and 100% detection of sex chromosomal aneuploidies, with 16 out of 3,200 false-positive results.
Study limitations
As in prior reports, limitations to the test were not clearly presented. Patients with “complex chromosomal abnormalities” not detectable by cfDNA were excluded from the reported cohort. Considering these cases, fewer of the total chromosomal abnormalities in the cohort were detected.
Also, adequate fetal DNA is necessary for accurate results, and patients with less than 4% fetal DNA were excluded. Low fetal DNA is associated with an increased risk of trisomy.4,5 Therefore, excluding such cases will bias results toward a higher detection rate.
The outcomes for cases of low fetal DNA were not included in this study, but in another recent paper 22% of cases of low fetal DNA had aneuploidy, and 16% of common aneuploidies were not detected because of failed testing.4
What this evidence means for practice
Cell-free fetal DNA is an exciting technology, and this study adds to the existing literature in the field. However, use of the test requires careful patient counseling regarding the limitations in detecting abnormalities other than trisomy 21, which comprises just 50% of all aneuploidies. Women who desire a comprehensive prenatal genetic assessment may prefer invasive diagnostic testing and should be counseled appropriately. Patients in whom the test fails should be informed that they are at high risk for a chromosomal abnormality.
Considering these outcomes, the benefits of prenatal screening with cfDNA over current testing alternatives, with serum analytes and/or invasive diagnostic testing, become less clear and the options more complex. Of primary importance is that patients understand the risks and benefits of alternative tests.
—Mary E. Norton, MD
Share your thoughts on this article! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
The introduction of cell-free fetal DNA (cfDNA) testing has had a tremendous impact on prenatal care. Numerous series reporting near-diagnostic accuracy for trisomy 21 detection have been reported,1 and several commercial laboratories offer clinical testing. Many patients now take advantage of these tests, and the very low false-positive rates have resulted in a marked decrease in the rate of invasive diagnostic testing.2 At present, most professional societies suggest that these tests be reserved for women at high risk for fetal aneuploidy.3
Details of the study
In this recent article by Porreco and colleagues, the researchers reported on a large cohort study of patients who had made the decision to undergo invasive diagnostic testing with chorionic villus sampling or amniocentesis prior to undergoing noninvasive testing, in order to validate the clinical performance of massively parallel genomic sequencing of cfDNA. As in several prior reports, the study authors found that the detection rate of cfDNA for trisomy 21 was 100%, and somewhat less for trisomy 18 (92%) and trisomy 13 (87%). The false-positive rate was very low, with only three false-positive results (all for trisomy 21) in 3,430 patients. Testing for fetal sex chromosomes reported 7 out of 3,322 errors in fetal sex and 100% detection of sex chromosomal aneuploidies, with 16 out of 3,200 false-positive results.
Study limitations
As in prior reports, limitations to the test were not clearly presented. Patients with “complex chromosomal abnormalities” not detectable by cfDNA were excluded from the reported cohort. Considering these cases, fewer of the total chromosomal abnormalities in the cohort were detected.
Also, adequate fetal DNA is necessary for accurate results, and patients with less than 4% fetal DNA were excluded. Low fetal DNA is associated with an increased risk of trisomy.4,5 Therefore, excluding such cases will bias results toward a higher detection rate.
The outcomes for cases of low fetal DNA were not included in this study, but in another recent paper 22% of cases of low fetal DNA had aneuploidy, and 16% of common aneuploidies were not detected because of failed testing.4
What this evidence means for practice
Cell-free fetal DNA is an exciting technology, and this study adds to the existing literature in the field. However, use of the test requires careful patient counseling regarding the limitations in detecting abnormalities other than trisomy 21, which comprises just 50% of all aneuploidies. Women who desire a comprehensive prenatal genetic assessment may prefer invasive diagnostic testing and should be counseled appropriately. Patients in whom the test fails should be informed that they are at high risk for a chromosomal abnormality.
Considering these outcomes, the benefits of prenatal screening with cfDNA over current testing alternatives, with serum analytes and/or invasive diagnostic testing, become less clear and the options more complex. Of primary importance is that patients understand the risks and benefits of alternative tests.
—Mary E. Norton, MD
Share your thoughts on this article! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
1. Gil MM, Akolekar R, Quezada MS, Bregant B, Nicolaides KH. Analysis of cell-free DNA in maternal blood in screening for aneuploidies: meta-analysis. Fetal Diagn Ther. 2014;35(3):156−173.
2. Wax JR, Cartin A, Chard R, Lucas FL, Pinette MG. Noninvasive prenatal testing: Impact on genetic counseling, invasive prenatal diagnosis, and trisomy 21 detection [published online ahead of print October 9, 2014]. J Clin Ultrasound. doi:`10.1002/jcu.22243. [Epub ahead of print]
3. American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 545: Noninvasive prenatal testing for fetal aneuploidy. Obstet Gynecol. 2012;120(6):1532−1534.
4. Pergament E, Cuckle H, Zimmermann B, et al. Single-nucleotide polymorphism-based noninvasive prenatal screening in a high-risk and low-risk cohort. Obstet Gynecol. 2014;124(2 pt 1):210−218.
5. Rava RP, Srinivasan A, Sehnert AJ, Bianchi DW. Circulating fetal cell-free DNA fractions differ in autosomal aneuploidies and monosomy X. Clin Chem. 2014;60(1):243−250.
1. Gil MM, Akolekar R, Quezada MS, Bregant B, Nicolaides KH. Analysis of cell-free DNA in maternal blood in screening for aneuploidies: meta-analysis. Fetal Diagn Ther. 2014;35(3):156−173.
2. Wax JR, Cartin A, Chard R, Lucas FL, Pinette MG. Noninvasive prenatal testing: Impact on genetic counseling, invasive prenatal diagnosis, and trisomy 21 detection [published online ahead of print October 9, 2014]. J Clin Ultrasound. doi:`10.1002/jcu.22243. [Epub ahead of print]
3. American College of Obstetricians and Gynecologists Committee on Genetics. Committee Opinion No. 545: Noninvasive prenatal testing for fetal aneuploidy. Obstet Gynecol. 2012;120(6):1532−1534.
4. Pergament E, Cuckle H, Zimmermann B, et al. Single-nucleotide polymorphism-based noninvasive prenatal screening in a high-risk and low-risk cohort. Obstet Gynecol. 2014;124(2 pt 1):210−218.
5. Rava RP, Srinivasan A, Sehnert AJ, Bianchi DW. Circulating fetal cell-free DNA fractions differ in autosomal aneuploidies and monosomy X. Clin Chem. 2014;60(1):243−250.