User login
Pitted Depressions on the Hands and Elbows
The Diagnosis: Bazex‐Dupré‐Christol Syndrome
Bazex‐Dupré‐Christol syndrome (BDCS) is a rare X-linked dominant genodermatosis characterized by a triad of hypotrichosis, follicular atrophoderma, and multiple basal cell carcinomas (BCCs). Since first being described in 1964,1 there have been fewer than 200 reported cases of BDCS.2 Although a causative gene has not yet been identified, the mutation has been mapped to an 11.4-Mb interval in the Xq25-27.1 region of the X chromosome.3
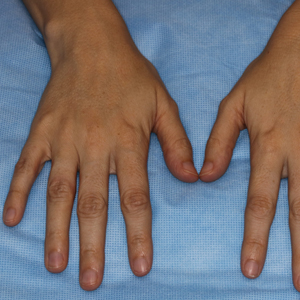
Classically, congenital hypotrichosis is the first observed symptom and can present shortly after birth.4 It typically is widespread, though sometimes it may be confined to the eyebrows, eyelashes, and scalp. Follicular atrophoderma, which occurs due to a laxation and deepening of the follicular ostia, is seen in 80% of cases and typically presents in early childhood as depressions lacking hair.2 It commonly is found on the face, extensor surfaces of the elbows and knees, and dorsal aspects of the hands and feet. Physical examination of our patient revealed follicular atrophoderma on both the dorsal surfaces of the hands and the extensor surfaces of the elbows. Hair shaft anomalies including pili torti, pili bifurcati, and trichorrhexis nodosa are infrequently observed symptoms of BDCS.2
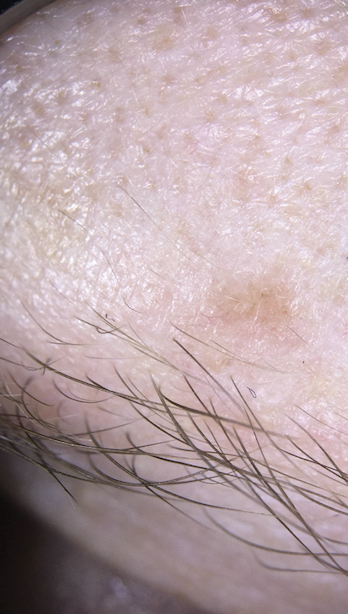
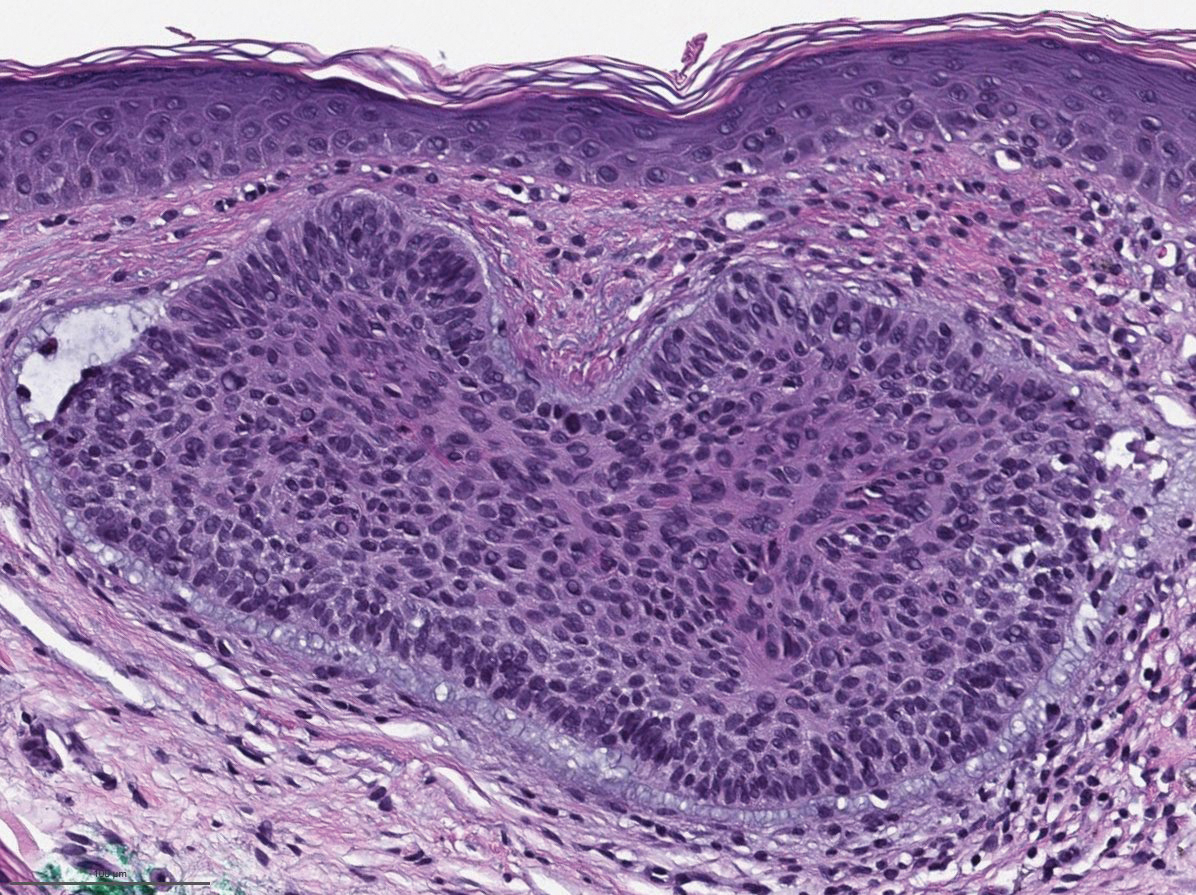
Basal cell carcinoma often manifests in the second or third decades of life, though there are reports of BCC developing in BDCS patients as young as 3 years. Basal cell carcinoma typically arises on sun-exposed areas, especially the face, neck, and chest. These lesions can be pigmented or nonpigmented and range from 2 to 20 mm in diameter.4 Our patient presented with a BCC on the forehead (Figure 1). Histopathologic evaluation showed a proliferation of basaloid cells with peripheral palisading (Figure 2), confirming the diagnosis of BCC.
Milia, which are not considered part of the classic BDCS triad, are seen in 70% of cases.2 They commonly are found on the face and often diminish with age. Milia may precede the formation of follicular atrophoderma and BCC. Hypohidrosis most commonly occurs on the forehead but can be widespread.2 Other less commonly observed features include epidermal cysts, hyperpigmentation of the face, and trichoepitheliomas.4 The management of BDCS involves frequent clinical examinations, BCC treatment, genetic counseling, and photoprotection.2,4
Nevoid BCC syndrome (NBCCS), also known as Gorlin-Goltz syndrome, is an autosomal-dominant disease characterized by multiple nevoid BCCs, macrocephaly with a large forehead, cleft lip or palate, jaw keratocysts, palmar and plantar pits, and calcification of the falx cerebri.5 Nevoid BCC syndrome is caused by a mutation in the PTCH1 gene in the hedgehog signaling pathway.6 The absence of common symptoms of NBCCS including macrocephaly, palmar or plantar pits, and cleft lip or palate, as well as negative genetic testing, suggested that our patient did not have NBCCS.
Rombo syndrome shares features with BDCS. Similar to BDCS, symptoms of Rombo syndrome include follicular atrophy, milialike papules, and BCC. Patients with Rombo syndrome typically present with atrophoderma vermiculatum on the cheeks and forehead in childhood.7 This atrophoderma presents with a pitted atrophic appearance in a reticular pattern on sun-exposed areas. Other distinguishing features from BDCS include cyanotic redness of sun-exposed skin and telangiectatic vessels.8
Multiple hereditary infundibulocystic BCC is another rare genodermatosis that is characterized by the presence of multiple infundibulocystic BCCs on the face and genitals. Infundibulocystic BCC is a well-differentiated subtype of BCC characterized by buds and cords of basaloid cells with scant stroma. Multiple hereditary infundibulocystic BCC is inherited in an autosomal-dominant fashion and has been linked to SUFU mutation in the sonic hedgehog pathway.9
Rothmund-Thomson syndrome is an autosomalrecessive disorder characterized by sparse hair, skeletal and dental abnormalities, and a high risk for developing keratinocyte carcinomas. It is differentiated from BDCS clinically by the presence of erythema, edema, and blistering, resulting in poikiloderma, plantar hyperkeratotic lesions, and bone defects.10
- Bazex A. Génodermatose complexe de type indéterminé associant une hypotrichose, un état atrophodermique généralisé et des dégénérescences cutanées multiples (épitheliomas baso-cellulaires). Bull Soc Fr Derm Syphiligr. 1964;71:206.
- Al Sabbagh MM, Baqi MA. Bazex-Dupre-Christol syndrome: review of clinical and molecular aspects. Int J Dermatol. 2018;57:1102-1106.
- Parren LJ, Abuzahra F, Wagenvoort T, et al. Linkage refinement of Bazex-Dupre-Christol syndrome to an 11.4-Mb interval on chromosome Xq25-27.1. Br J Dermatol. 2011;165:201-203.
- Abuzahra F, Parren LJ, Frank J. Multiple familial and pigmented basal cell carcinomas in early childhood--Bazex-Dupre-Christol syndrome. J Eur Acad Dermatol Venereol. 2012;26:117-121.
- Shevchenko A, Durkin JR, Moon AT. Generalized basaloid follicular hamartoma syndrome versus Gorlin syndrome: a diagnostic challenge. Pediatr Dermatol. 2018;35:E396-E397.
- Fujii K, Miyashita T. Gorlin syndrome (nevoid basal cell carcinoma syndrome): update and literature review. Pediatr Int. 2014;56:667-674.
- van Steensel MA, Jaspers NG, Steijlen PM. A case of Rombo syndrome. Br J Dermatol. 2001;144:1215-1218.
- Lee YC, Son SJ, Han TY, et al. A case of atrophoderma vermiculatum showing a good response to topical tretinoin. Ann Dermatol. 2018;30:116-118.
- Schulman JM, Oh DH, Sanborn JZ, et al. Multiple hereditary infundibulocystic basal cell carcinoma syndrome associated with a germline SUFU mutation. JAMA Dermatol. 2016;152:323-327.
- Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet J Rare Dis. 2010;5:2.
The Diagnosis: Bazex‐Dupré‐Christol Syndrome
Bazex‐Dupré‐Christol syndrome (BDCS) is a rare X-linked dominant genodermatosis characterized by a triad of hypotrichosis, follicular atrophoderma, and multiple basal cell carcinomas (BCCs). Since first being described in 1964,1 there have been fewer than 200 reported cases of BDCS.2 Although a causative gene has not yet been identified, the mutation has been mapped to an 11.4-Mb interval in the Xq25-27.1 region of the X chromosome.3
Classically, congenital hypotrichosis is the first observed symptom and can present shortly after birth.4 It typically is widespread, though sometimes it may be confined to the eyebrows, eyelashes, and scalp. Follicular atrophoderma, which occurs due to a laxation and deepening of the follicular ostia, is seen in 80% of cases and typically presents in early childhood as depressions lacking hair.2 It commonly is found on the face, extensor surfaces of the elbows and knees, and dorsal aspects of the hands and feet. Physical examination of our patient revealed follicular atrophoderma on both the dorsal surfaces of the hands and the extensor surfaces of the elbows. Hair shaft anomalies including pili torti, pili bifurcati, and trichorrhexis nodosa are infrequently observed symptoms of BDCS.2
Basal cell carcinoma often manifests in the second or third decades of life, though there are reports of BCC developing in BDCS patients as young as 3 years. Basal cell carcinoma typically arises on sun-exposed areas, especially the face, neck, and chest. These lesions can be pigmented or nonpigmented and range from 2 to 20 mm in diameter.4 Our patient presented with a BCC on the forehead (Figure 1). Histopathologic evaluation showed a proliferation of basaloid cells with peripheral palisading (Figure 2), confirming the diagnosis of BCC.
Milia, which are not considered part of the classic BDCS triad, are seen in 70% of cases.2 They commonly are found on the face and often diminish with age. Milia may precede the formation of follicular atrophoderma and BCC. Hypohidrosis most commonly occurs on the forehead but can be widespread.2 Other less commonly observed features include epidermal cysts, hyperpigmentation of the face, and trichoepitheliomas.4 The management of BDCS involves frequent clinical examinations, BCC treatment, genetic counseling, and photoprotection.2,4
Nevoid BCC syndrome (NBCCS), also known as Gorlin-Goltz syndrome, is an autosomal-dominant disease characterized by multiple nevoid BCCs, macrocephaly with a large forehead, cleft lip or palate, jaw keratocysts, palmar and plantar pits, and calcification of the falx cerebri.5 Nevoid BCC syndrome is caused by a mutation in the PTCH1 gene in the hedgehog signaling pathway.6 The absence of common symptoms of NBCCS including macrocephaly, palmar or plantar pits, and cleft lip or palate, as well as negative genetic testing, suggested that our patient did not have NBCCS.
Rombo syndrome shares features with BDCS. Similar to BDCS, symptoms of Rombo syndrome include follicular atrophy, milialike papules, and BCC. Patients with Rombo syndrome typically present with atrophoderma vermiculatum on the cheeks and forehead in childhood.7 This atrophoderma presents with a pitted atrophic appearance in a reticular pattern on sun-exposed areas. Other distinguishing features from BDCS include cyanotic redness of sun-exposed skin and telangiectatic vessels.8
Multiple hereditary infundibulocystic BCC is another rare genodermatosis that is characterized by the presence of multiple infundibulocystic BCCs on the face and genitals. Infundibulocystic BCC is a well-differentiated subtype of BCC characterized by buds and cords of basaloid cells with scant stroma. Multiple hereditary infundibulocystic BCC is inherited in an autosomal-dominant fashion and has been linked to SUFU mutation in the sonic hedgehog pathway.9
Rothmund-Thomson syndrome is an autosomalrecessive disorder characterized by sparse hair, skeletal and dental abnormalities, and a high risk for developing keratinocyte carcinomas. It is differentiated from BDCS clinically by the presence of erythema, edema, and blistering, resulting in poikiloderma, plantar hyperkeratotic lesions, and bone defects.10
The Diagnosis: Bazex‐Dupré‐Christol Syndrome
Bazex‐Dupré‐Christol syndrome (BDCS) is a rare X-linked dominant genodermatosis characterized by a triad of hypotrichosis, follicular atrophoderma, and multiple basal cell carcinomas (BCCs). Since first being described in 1964,1 there have been fewer than 200 reported cases of BDCS.2 Although a causative gene has not yet been identified, the mutation has been mapped to an 11.4-Mb interval in the Xq25-27.1 region of the X chromosome.3
Classically, congenital hypotrichosis is the first observed symptom and can present shortly after birth.4 It typically is widespread, though sometimes it may be confined to the eyebrows, eyelashes, and scalp. Follicular atrophoderma, which occurs due to a laxation and deepening of the follicular ostia, is seen in 80% of cases and typically presents in early childhood as depressions lacking hair.2 It commonly is found on the face, extensor surfaces of the elbows and knees, and dorsal aspects of the hands and feet. Physical examination of our patient revealed follicular atrophoderma on both the dorsal surfaces of the hands and the extensor surfaces of the elbows. Hair shaft anomalies including pili torti, pili bifurcati, and trichorrhexis nodosa are infrequently observed symptoms of BDCS.2
Basal cell carcinoma often manifests in the second or third decades of life, though there are reports of BCC developing in BDCS patients as young as 3 years. Basal cell carcinoma typically arises on sun-exposed areas, especially the face, neck, and chest. These lesions can be pigmented or nonpigmented and range from 2 to 20 mm in diameter.4 Our patient presented with a BCC on the forehead (Figure 1). Histopathologic evaluation showed a proliferation of basaloid cells with peripheral palisading (Figure 2), confirming the diagnosis of BCC.
Milia, which are not considered part of the classic BDCS triad, are seen in 70% of cases.2 They commonly are found on the face and often diminish with age. Milia may precede the formation of follicular atrophoderma and BCC. Hypohidrosis most commonly occurs on the forehead but can be widespread.2 Other less commonly observed features include epidermal cysts, hyperpigmentation of the face, and trichoepitheliomas.4 The management of BDCS involves frequent clinical examinations, BCC treatment, genetic counseling, and photoprotection.2,4
Nevoid BCC syndrome (NBCCS), also known as Gorlin-Goltz syndrome, is an autosomal-dominant disease characterized by multiple nevoid BCCs, macrocephaly with a large forehead, cleft lip or palate, jaw keratocysts, palmar and plantar pits, and calcification of the falx cerebri.5 Nevoid BCC syndrome is caused by a mutation in the PTCH1 gene in the hedgehog signaling pathway.6 The absence of common symptoms of NBCCS including macrocephaly, palmar or plantar pits, and cleft lip or palate, as well as negative genetic testing, suggested that our patient did not have NBCCS.
Rombo syndrome shares features with BDCS. Similar to BDCS, symptoms of Rombo syndrome include follicular atrophy, milialike papules, and BCC. Patients with Rombo syndrome typically present with atrophoderma vermiculatum on the cheeks and forehead in childhood.7 This atrophoderma presents with a pitted atrophic appearance in a reticular pattern on sun-exposed areas. Other distinguishing features from BDCS include cyanotic redness of sun-exposed skin and telangiectatic vessels.8
Multiple hereditary infundibulocystic BCC is another rare genodermatosis that is characterized by the presence of multiple infundibulocystic BCCs on the face and genitals. Infundibulocystic BCC is a well-differentiated subtype of BCC characterized by buds and cords of basaloid cells with scant stroma. Multiple hereditary infundibulocystic BCC is inherited in an autosomal-dominant fashion and has been linked to SUFU mutation in the sonic hedgehog pathway.9
Rothmund-Thomson syndrome is an autosomalrecessive disorder characterized by sparse hair, skeletal and dental abnormalities, and a high risk for developing keratinocyte carcinomas. It is differentiated from BDCS clinically by the presence of erythema, edema, and blistering, resulting in poikiloderma, plantar hyperkeratotic lesions, and bone defects.10
- Bazex A. Génodermatose complexe de type indéterminé associant une hypotrichose, un état atrophodermique généralisé et des dégénérescences cutanées multiples (épitheliomas baso-cellulaires). Bull Soc Fr Derm Syphiligr. 1964;71:206.
- Al Sabbagh MM, Baqi MA. Bazex-Dupre-Christol syndrome: review of clinical and molecular aspects. Int J Dermatol. 2018;57:1102-1106.
- Parren LJ, Abuzahra F, Wagenvoort T, et al. Linkage refinement of Bazex-Dupre-Christol syndrome to an 11.4-Mb interval on chromosome Xq25-27.1. Br J Dermatol. 2011;165:201-203.
- Abuzahra F, Parren LJ, Frank J. Multiple familial and pigmented basal cell carcinomas in early childhood--Bazex-Dupre-Christol syndrome. J Eur Acad Dermatol Venereol. 2012;26:117-121.
- Shevchenko A, Durkin JR, Moon AT. Generalized basaloid follicular hamartoma syndrome versus Gorlin syndrome: a diagnostic challenge. Pediatr Dermatol. 2018;35:E396-E397.
- Fujii K, Miyashita T. Gorlin syndrome (nevoid basal cell carcinoma syndrome): update and literature review. Pediatr Int. 2014;56:667-674.
- van Steensel MA, Jaspers NG, Steijlen PM. A case of Rombo syndrome. Br J Dermatol. 2001;144:1215-1218.
- Lee YC, Son SJ, Han TY, et al. A case of atrophoderma vermiculatum showing a good response to topical tretinoin. Ann Dermatol. 2018;30:116-118.
- Schulman JM, Oh DH, Sanborn JZ, et al. Multiple hereditary infundibulocystic basal cell carcinoma syndrome associated with a germline SUFU mutation. JAMA Dermatol. 2016;152:323-327.
- Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet J Rare Dis. 2010;5:2.
- Bazex A. Génodermatose complexe de type indéterminé associant une hypotrichose, un état atrophodermique généralisé et des dégénérescences cutanées multiples (épitheliomas baso-cellulaires). Bull Soc Fr Derm Syphiligr. 1964;71:206.
- Al Sabbagh MM, Baqi MA. Bazex-Dupre-Christol syndrome: review of clinical and molecular aspects. Int J Dermatol. 2018;57:1102-1106.
- Parren LJ, Abuzahra F, Wagenvoort T, et al. Linkage refinement of Bazex-Dupre-Christol syndrome to an 11.4-Mb interval on chromosome Xq25-27.1. Br J Dermatol. 2011;165:201-203.
- Abuzahra F, Parren LJ, Frank J. Multiple familial and pigmented basal cell carcinomas in early childhood--Bazex-Dupre-Christol syndrome. J Eur Acad Dermatol Venereol. 2012;26:117-121.
- Shevchenko A, Durkin JR, Moon AT. Generalized basaloid follicular hamartoma syndrome versus Gorlin syndrome: a diagnostic challenge. Pediatr Dermatol. 2018;35:E396-E397.
- Fujii K, Miyashita T. Gorlin syndrome (nevoid basal cell carcinoma syndrome): update and literature review. Pediatr Int. 2014;56:667-674.
- van Steensel MA, Jaspers NG, Steijlen PM. A case of Rombo syndrome. Br J Dermatol. 2001;144:1215-1218.
- Lee YC, Son SJ, Han TY, et al. A case of atrophoderma vermiculatum showing a good response to topical tretinoin. Ann Dermatol. 2018;30:116-118.
- Schulman JM, Oh DH, Sanborn JZ, et al. Multiple hereditary infundibulocystic basal cell carcinoma syndrome associated with a germline SUFU mutation. JAMA Dermatol. 2016;152:323-327.
- Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet J Rare Dis. 2010;5:2.
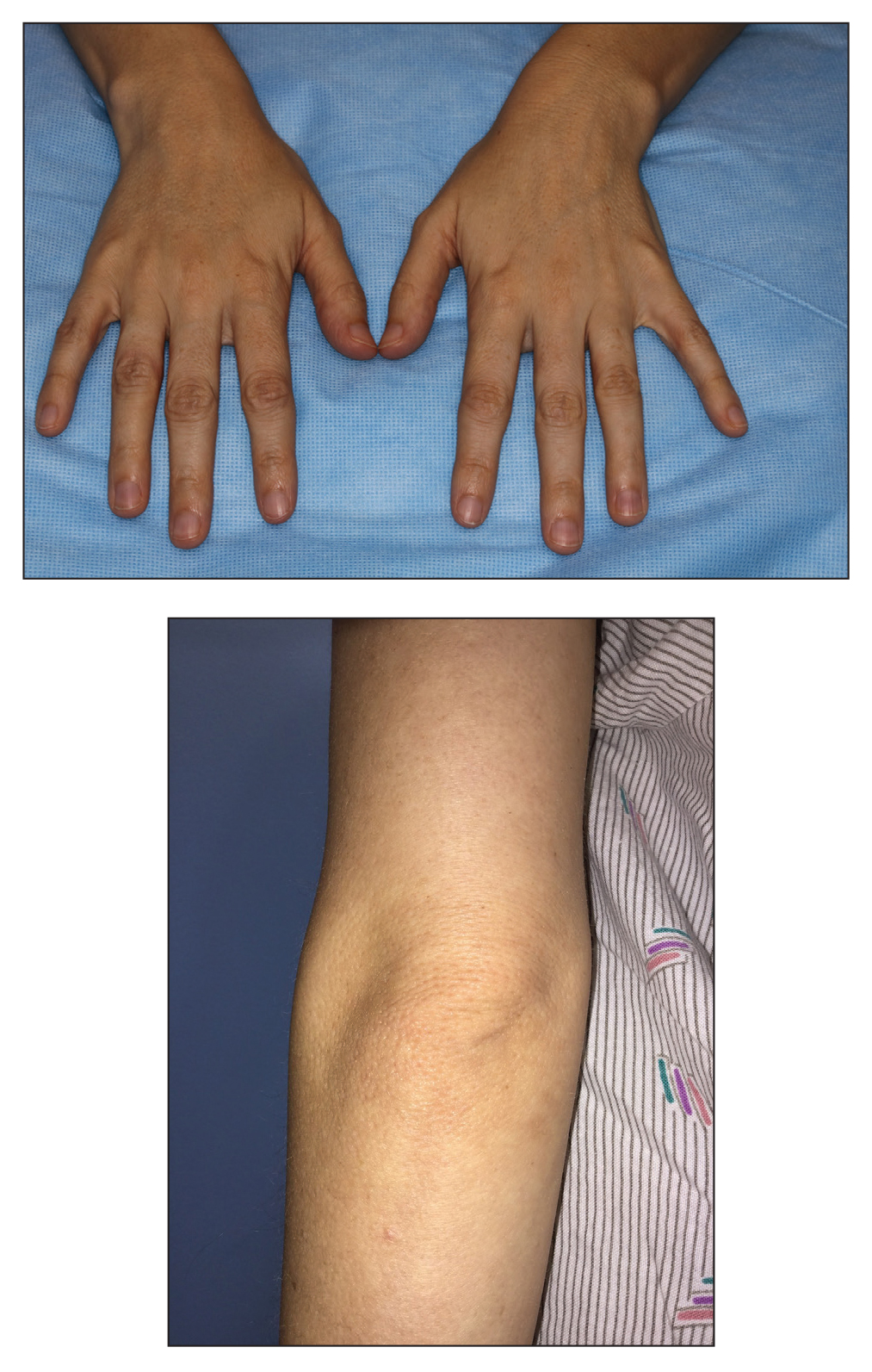
A 28-year-old woman presented for evaluation of a pearly papule on the forehead of several months’ duration that was concerning for basal cell carcinoma (BCC). She had a history of numerous BCCs starting at the age of 17 years. She denied radiation or other carcinogenic exposures and had no other notable medical history. The patient’s mother and grandmother also had numerous BCCs. Physical examination revealed hypotrichosis; numerous 3- to 5-mm white cystic papules on the face, chest, and upper arms; and 1- to 5-mm pitted depressions on the dorsal aspects of the hands (top) and extensor surfaces of the elbows (bottom). A proliferation of basaloid cells with peripheral palisading was seen on histopathologic evaluation. Genetic testing revealed no protein patched homolog 1, PTCH1, or suppressor of fused homolog, SUFU, gene mutations.