User login
Fragility fractures in chronic kidney disease: A clarification of views
To the Editor: I was pleased to see my article on fragility fractures in patients with chronic kidney disease (CKD) in the Cleveland Clinic Journal of Medicine1 and your preamble Letter from the Editor.2
However, Dr. Coco’s accompanying editorial3 misquoted a particular point I cautiously and consistently make—not only in the CCJM article, but in other invited papers on the topic of fractures in CKD. I specifically state that bisphosphonates should only be considered in stage 4–5 CKD in fracturing patients, not just those with “low bone mineral density,” who have clear-cut osteoporosis by exclusion of other causes of fractures in this population. Hence, Dr. Coco’s statement that “… the author advocates the use of bisphosphonate therapy in patients with chronic kidney disease who have low bone mineral density” is inaccurate.
If one carefully reads the last four paragraphs of my paper on page 721, one will see that I emphasize this caution repeatedly and even specifically state: “Treating only on the basis of low bone mineral density and other risk factors seems to be associated with greater risk than benefit.”
Thank you for your consideration.
1. Miller PD. Fragility fractures in chronic kidney disease: an opinion-based approach. Cleve Clin J Med 2009; 76:715–723.
2. Mandell BF. Low bone density is not always bisphosphonate deficiency (From the Editor). Cleve Clin J Med 2009; 76:683.
3. Coco M. Treating the renal patient who has a fracture: opinion vs evidence. Cleve Clin J Med 2009; 76:684–688.
To the Editor: I was pleased to see my article on fragility fractures in patients with chronic kidney disease (CKD) in the Cleveland Clinic Journal of Medicine1 and your preamble Letter from the Editor.2
However, Dr. Coco’s accompanying editorial3 misquoted a particular point I cautiously and consistently make—not only in the CCJM article, but in other invited papers on the topic of fractures in CKD. I specifically state that bisphosphonates should only be considered in stage 4–5 CKD in fracturing patients, not just those with “low bone mineral density,” who have clear-cut osteoporosis by exclusion of other causes of fractures in this population. Hence, Dr. Coco’s statement that “… the author advocates the use of bisphosphonate therapy in patients with chronic kidney disease who have low bone mineral density” is inaccurate.
If one carefully reads the last four paragraphs of my paper on page 721, one will see that I emphasize this caution repeatedly and even specifically state: “Treating only on the basis of low bone mineral density and other risk factors seems to be associated with greater risk than benefit.”
Thank you for your consideration.
To the Editor: I was pleased to see my article on fragility fractures in patients with chronic kidney disease (CKD) in the Cleveland Clinic Journal of Medicine1 and your preamble Letter from the Editor.2
However, Dr. Coco’s accompanying editorial3 misquoted a particular point I cautiously and consistently make—not only in the CCJM article, but in other invited papers on the topic of fractures in CKD. I specifically state that bisphosphonates should only be considered in stage 4–5 CKD in fracturing patients, not just those with “low bone mineral density,” who have clear-cut osteoporosis by exclusion of other causes of fractures in this population. Hence, Dr. Coco’s statement that “… the author advocates the use of bisphosphonate therapy in patients with chronic kidney disease who have low bone mineral density” is inaccurate.
If one carefully reads the last four paragraphs of my paper on page 721, one will see that I emphasize this caution repeatedly and even specifically state: “Treating only on the basis of low bone mineral density and other risk factors seems to be associated with greater risk than benefit.”
Thank you for your consideration.
1. Miller PD. Fragility fractures in chronic kidney disease: an opinion-based approach. Cleve Clin J Med 2009; 76:715–723.
2. Mandell BF. Low bone density is not always bisphosphonate deficiency (From the Editor). Cleve Clin J Med 2009; 76:683.
3. Coco M. Treating the renal patient who has a fracture: opinion vs evidence. Cleve Clin J Med 2009; 76:684–688.
1. Miller PD. Fragility fractures in chronic kidney disease: an opinion-based approach. Cleve Clin J Med 2009; 76:715–723.
2. Mandell BF. Low bone density is not always bisphosphonate deficiency (From the Editor). Cleve Clin J Med 2009; 76:683.
3. Coco M. Treating the renal patient who has a fracture: opinion vs evidence. Cleve Clin J Med 2009; 76:684–688.
Fragility fractures in chronic kidney disease: An opinion-based approach

But even in chronic kidney disease, many fractures are due to postmenopausal or agerelated osteoporosis, and estrogen-deficiency osteoporosis is the most common cause of fragility fractures overall.1–3 Osteoporosis can be diagnosed only after other causes of skeletal fragility have been ruled out. And how to diagnose and treat osteoporosis in the most severe stage of kidney disease is a matter of opinion, as we have almost no data to guide us.
Nevertheless, in the pages that follow, I will outline my admittedly opinion-based approach to diagnosing and managing the causes of fragility fractures in patients with chronic kidney disease.
T SCORES DO NOT DISTINGUISH THE CAUSES OF FRAGILITY
The most common cause of fragility fractures is osteoporosis due to estrogen deficiency.1–3 But since many other medical conditions can lead to osteoporosis, simple diagnostic criteria have been difficult to find.
Before 1994, the diagnosis of osteoporosis was made on the basis of low-trauma fractures.4 Now, we use the World Health Organization criteria,5 based on bone mineral density T scores:
- Normal—a T score of −1.0 standard deviations or higher
- Osteopenia—a T score of less than −1.0 but higher than −2.5
- Osteoporosis—a T score of −2.5 or less
- Severe osteoporosis—a T score of −2.5 or less with a fragility fracture.
However, fractures can also be due to metabolic bone diseases that are not osteoporosis, including renal bone diseases.6–7 While a low T score or a fracture provides a working diagnosis of osteoporosis, it does not help distinguish the different types of osteoporosis and nonosteoporotic metabolic bone diseases. For example, osteomalacia and osteogenesis imperfecta can also cause fragility fractures and can be associated with low bone density. Using these criteria to define osteoporosis is even more problematic in patients with chronic kidney disease.
FIVE STAGES OF CHRONIC KIDNEY DISEASE
The National Kidney Foundation8 classifies the severity of chronic kidney disease on the basis of the glomerular filtration rate (GFR), as measured by 24-hour urine for creatinine clearance, or as estimated by the Cockcroft-Gault equation or, preferably, the Modification of Diet in Renal Disease (MDRD) equation (calculators are available at www.kidney.org/professionals/kdoqi/gfr_calculator.cfm):
- Stage 1—GFR 90 mL/minute/1.73 m2 or higher
- Stage 2—GFR 60 to 89
- Stage 3—GFR 30 to 59
- Stage 4—GFR 15 to 29
- Stage 5—GFR lower than 15, or if the patient is on dialysis. (Another stage, called 5D, was added to the list to denote patients on dialysis, since the metabolic derangements in bone and systemic biology may differ between patients on dialysis vs those not on dialysis.)
This staging system is relevant to the discussion of bone fragility that follows.
CHRONIC KIDNEY DISEASE IS COMMON IN THE ELDERLY
The third National Health and Nutrition Examination Survey9 found that, at least as estimated by the Cockcroft-Gault equation, the GFR declines with age, so that by the age of 70 at least 20% of the US population has stage 4 or 5 chronic kidney disease.
Although the Cockcroft-Gault and MDRD equations may yield lower GFR values in the general population than one would get by measuring creatinine, inulin, or iothalamate clearance,10,11 the point is that both osteoporosis and chronic kidney disease are common.
THE GAMUT OF RENAL OSTEODYSTROPHY
In kidney failure (stage 5 chronic kidney disease), all forms of renal osteodystrophy may be associated with fragility fractures. Renal osteodystrophy can be defined by quantitative bone histomorphometry.12,13 The systemic conditions that may be associated with the bone disease and systemic vascular disease (chronic kidney disease–mineral and bone disorder) are characterized by one or more of the following14:
- Abnormalities of calcium, phosphorus, parathyroid hormone, or vitamin D metabolism
- Abnormalities in bone turnover, mineralization, volume, linear growth, or strength
- Vascular or other soft-tissue calcification.
The National Kidney Foundation14 classifies renal osteodystrophy on the basis of:
- Turnover—high, normal, or low
- Mineralization—normal or abnormal
- Volume—high, normal, or low.
Although this system helps us understand these diseases better, it does not provide a working diagnosis of osteoporosis.14
WHAT IS OSTEOPOROSIS?
In an attempt to define osteoporosis by a pathophysiologic mechanism, the National Institutes of Health15 have held two consensus conferences and have stated that “osteoporosis is a skeletal disorder characterized by impairment in bone strength predisposing a person to an increased risk of fracture. Bone strength primarily reflects the integration of bone density and bone quality.”15 However, the consensus statement also does not provide a working diagnosis of osteoporosis—one that clinicians can apply to management decisions, and one that is also accepted by the US International Classification of Disease codes for reimbursement purposes.
The 1994 World Health Organization criteria offer the most pragmatic operational definition of osteoporosis, and they can be applied in both men and women, as well as in younger patients with medical conditions associated with increased risk of low-trauma fracture.5,16 Although the main purpose of these criteria was to advise international health authorities of the potential future economic impact of osteoporosis, the T score also became the pragmatic diagnostic threshold for defining normal, osteopenia, and osteoporosis in clinical practice.
The T score also calls attention to an important observation: of people who have fractures and subsequently undergo bone densitometry, more are found to have osteopenia than osteoporosis. The reasons are that there are more people with osteopenia than osteoporosis,17,18 and many other factors independent of low bone mineral density contribute to bone strength.19,20
How is osteoporosis diagnosed in stage 1–3 chronic kidney disease?
In patients with chronic kidney disease who develop fragility fractures, the reasonable question is: Is the cause of the fracture osteoporosis or some other metabolic bone disease associated with chronic kidney disease?
The National Kidney Foundation guidelines14 say that the diagnosis of osteoporosis can be established in patients with stage 1, 2, or 3 chronic kidney disease on the basis of either of the World Health Organization criteria, ie, a T score of −2.5 or lower or fragility fractures, as in the postmenopausal population, as long as there are no biochemical abnormalities that suggest chronic kidney disease–mineral and bone disorder.
How is osteoporosis diagnosed in stage 4 or 5 chronic kidney disease?
The answer is neither straightforward nor clearly defined in severe (stage 4 or 5) chronic kidney disease.
In stage 5 and especially in patients on dialysis, the derangements in bone and mineral metabolism become serious enough to impair bone strength and increase the risk of lowtrauma fractures. The risk of hip fracture in stage 5 may be four times higher than in agematched controls.21–24
Adynamic, severe hyperparathyroid bone disease as well as osteomalacia can be associated with a higher risk of fragility fractures than in aged-matched controls in population studies of postmenopausal women or elderly men. These are bone fragility conditions that are not osteoporosis but that can mimic osteoporosis by the World Health Organization criteria.
Thus, when a patient in stage 5 has severe fragility fractures that by themselves may be life-threatening, it is reasonable to ask if the drugs that reduce the risk of fractures in many other osteoporotic conditions (postmenopausal, steroid-induced, elderly male osteoporosis, after solid organ transplantation) can also be used in patients with advanced chronic kidney disease.
The diagnosis of osteoporosis in these patients has no universally accepted criteria. The diagnosis is best suggested by excluding other forms of renal osteodystrophy by quantitative histomorphometry or by attempting to classify the form of renal osteodystrophy by noninvasive means of assessing bone turnover, mineralization, and volume. However, we lack clinical tools to make these distinctions in individual patients.
While many promising radiologic techniques that examine bone microarchitecture offer hope of being able to define turnover, mineralization, and volume noninvasively in severe chronic kidney disease, they are investigational and unproven at this time in discriminating between renal osteodystrophy and osteoporosis.6,25–27 As we increase our understanding of the relationships between turnover, mineralization, volume, and bone strength, these noninvasive imaging technologies may become the means to correlate turnover, mineralization, and volume to bone strength and open up an entirely new way to classify skeletal strength.
In the meantime, the clinician is left with quantitative bone histomorphometry (which requires biopsy) and biochemical markers of bone turnover to characterize the bone disease that may be responsible for low-trauma fractures in stage 5 chronic kidney disease. The clinician should first use biochemical markers before bone biopsy to distinguish the form of renal osteodystrophy, as this distinction may be able to prevent unnecessary biopsy.
Biochemical markers of bone metabolism
In chronic kidney disease, the bone biochemical tests that nephrologists usually assess during the course of declining renal function are the serum levels of:
- Phosphorus
- Parathyroid hormone
- Calcium
- Other electrolytes
- Total alkaline phosphatase or bone-specific alkaline phosphatase
- 1,25 dihydroxyvitamin D.
In postmenopausal osteoporosis, the biochemical markers of bone turnover that are measured to reflect baseline levels of bone turnover or change in bone turnover in response to drug therapy are:
- The serum or urine collagen cross-links N-telopeptide (NTx) and C-telopeptide (CTx), markers of bone resorption
- Bone-specific alkaline phosphatase (an osteoblast activity marker)
- Serum osteocalcin, a bone formation marker
- Propeptide type 1 collagen (P1NP), a marker of osteoblast activity, highly correlated with bone formation
- 25-hydroxyvitamin D levels.
Biochemical markers of bone turnover cannot be used to diagnose osteoporosis. They can, however, provide clinical guidance as to whether a patient has high or low bone turnover and whether therapy is affecting bone turnover.28–36 Although these markers have value in making these distinctions in groups of patients, they are less sensitive and specific for classifying an individual patient’s bone turnover status.
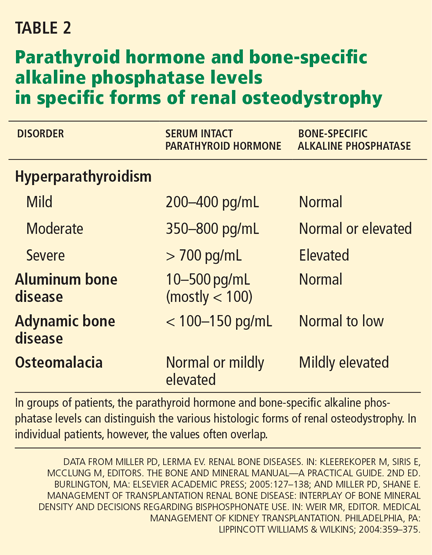
Bone-specific alkaline phosphatase, parathyroid hormone, and adynamic bone disease
If a patient’s bone-specific alkaline phosphatase level is elevated, adynamic bone disease is highly unlikely. Assuming that other causes of this elevated level (eg, Paget disease of bone, metastatic cancer) have already been excluded, the elevated level could represent either osteomalacia or hyperparathyroid bone disease.
However, a “normal” bone-specific alkaline phosphatase level does not exclude adynamic bone disease, whereas a low level is more often associated with low bone turnover.
An elevated parathyroid hormone level does not exclude adynamic renal bone disease, but a low level (< 150 pg/mL) suggests a lowbone-turnover state. A level six times or more greater than the upper limit of normal is far more likely to be associated with high bone turnover.
Thus, in clinical practice, patients with stage 4 or 5 chronic kidney disease who have elevated bone-specific alkaline phosphatase or very high parathyroid hormone values do not have adynamic bone disease. Furthermore, once other causes of these aberrant biochemical abnormalities have been defined, then “high-bone-turnover osteoporosis” may be a consideration. Certainly, in my opinion, if bone turnover markers suggest low bone turnover, bone biopsy is necessary before starting an antiresorptive agent.35
Quantitative bone histomorphometry
Double tetracycline-labeled quantitative histomorphometry is still the only accepted way to measure turnover, mineralization, and volume in clinical practice.43–45 A committee of the American Society for Bone and Mineral Research has developed histomorphometric criteria for distinguishing among the different types of metabolic bone diseases (osteomalacia, adynamic bone disease, hyperparathyroid bone disease).12 These criteria can be used to distinguish among the various metabolic bone diseases that accompany stage 5 chronic kidney disease, including adynamic bone disease.43,46–48
For patients in stage 5 who have had a fragility fracture, adynamic bone disease should be excluded before the off-label use of an osteoporosis drug that reduces bone turnover, such as a bisphosphonate, calcitonin, estrogen, a selective estrogen receptor modulator, or denosumab (anti-RANK ligand antibody). While there is no evidence, for example, that starting a bisphosphonate in a patient who already has adynamic bone disease is detrimental to either bone strength or systemic vascular calcification (which may be linked to low bone turnover),49 it seems unreasonable to do so until solid prospective data clarify the harm or benefit.50 Preliminary experimental and clinical data suggest that bisphosphonates may even reduce progression of extraosseous calcification and inhibit the development of atherosclerosis.50
Hence, quantitative bone histomorphometry can discriminate among the various forms of renal osteodystrophy. If a distinct form of renal osteodystrophy is not present in a patient with stage 4 or 5 chronic kidney disease who has had a fracture and who, on biopsy, has a low trabecular bone volume, the patient probably has osteoporosis by exclusion.
TREATING OSTEOPOROSIS IN STAGE 1–3 CHRONIC KIDNEY DISEASE
As previously mentioned, patients who have fragility fractures in stage 1, 2, or 3 chronic kidney disease are more likely to have osteoporosis than renal osteodystrophy as the cause of their impaired bone strength. Although several articles have described a higher risk of fragility fractures in patients with age-related reduction in renal function than in agematched patients with normal renal function, the specific metabolic bone disease other than osteoporosis accounting for this bone fragility has not been defined.6
Hence, patients with osteoporosis who are in stage 1, 2, or 3 chronic kidney disease and do not have a known biochemical abnormality that might suggest some form of renal osteodystrophy can and should be considered for treatment with approved drugs that reduce the risk of fractures in postmenopausal, male, or glucocorticoid-induced osteoporosis.51–53 In clinical trials, these agents were shown to be effective in patients with serum creatinine concentrations as high as 2.0 mg/dL or a GFR as low as 30 mL/min, as estimated by the Cockcroft-Gault equation.
While all of the approved agents show evidence of reducing the risk of vertebral fractures, patients at higher risk of fractures or those who have already suffered a nonvertebral fracture are more often considered candidates for treatment with a bisphosphonate or teriparatide (Forteo), both of which have shown evidence of reducing the risk of all fractures.
Bisphosphonates in stage 1–3 chronic kidney disease
There is prospective evidence that patients with an age-related reduction in GFR down to 30 mL/min benefit from oral and intravenous bisphosphonates, since all of the clinical trials that led to the approval of bisphosphonates included patients with GFRs as low as this.54–57 Bisphosphonates seem to have an excellent safety profile as measured by renal adverse events in patients with a GFR of 30 mL/min or greater.52–59
From the intravenous bisphosphonate studies, it appears that ibandronate (Boniva) at the approved dose of 3 mg intravenously every 3 months and zoledronic acid (Reclast) 5 mg once a year given over 15 minutes are safe in patients with a GFR greater than 30 or 35 mL/min.
However, the safety of these drugs might not be the same in patients with preexisting renal parenchymal disease (eg, in diabetes) or in patients using other agents that could affect renal function (eg, nonsteroidal antiinflammatory drugs). Therefore, caution is still needed when deciding to use intravenous bisphosphonates in specific higher-risk renal subpopulations.
In the clinical trials of zoledronic acid, a substantial proportion of patients had diabetes, and no difference was seen in adverse renal effects between diabetic and nondiabetic patients. Also, GFRs declined equally between the treated and placebo groups over time and were no different at the end of 3 years.55 However, in patients in whom serum creatinine was measured 9 to 11 days after the infusion of zoledronic acid, there was a small but statistically significant transient increase in serum creatinine concentration (0.5–2.0 mg/dL above baseline) after the second annual infusion. 58 The serum creatinine concentrations returned to their baseline values in all of these patients before the next annual infusion.
It is important that infusions of zoledronic acid be given no faster than over 15 minutes. More rapid infusion has been associated with acute renal failure, suggesting that the tubular damage that mimics acute tubular necrosis is related to the maximal concentration and not to the area under the curve. I infuse zoledronic acid over 30 minutes in patients with normal renal function or in those with stage 1, 2, or 3 chronic kidney disease.
Teriparatide
Teriparatide’s approval trial did not require baseline measurements of GFR, but patients were enrolled only if their baseline serum creatinine concentrations were less than 2.0 mg/dL.60 In a post hoc analysis, a small subset of patients had GFRs as low as 30 mL/min as estimated by the Cockcroft-Gault equation. In these patients, teriparatide 20 or 40 μg/day had an anabolic effect as measured by increases in osteoblast activity markers and bone mineral density, similar to that seen in patients with higher estimated GFRs and without any adverse renal effects.61
There are no data on using teriparatide in stage 4 or 5 chronic kidney disease, and I emphasize that in all of the clinical trials of teriparatide, all patients, even those with estimated GFRs as low as 30 mL/min, had normal baseline serum intact parathyroid hormone levels. It is possible that the bone biologic response could differ between patients with chronic kidney disease who have an elevated as compared with a normal serum parathyroid hormone level. This issue should be investigated.
TREATING OSTEOPOROSIS IN STAGE 4 OR 5 CHRONIC KIDNEY DISEASE
Treatment decisions are more difficult in patients with stage 4 and especially stage 5 chronic kidney disease who have had fragility fractures. This is even the case when the clinician has determined to the best of his or her ability that the patient has osteoporosis rather than renal osteodystrophy.
There are no prospective data showing any of the approved drugs to be effective in treating osteoporosis in patients whose GFRs are lower than 30 mL/min. However, a post hoc analysis of pooled data from nine clinical trials62 found that risedronate (Actonel) 5 mg/day reduced the incidence of new vertebral fractures. Another post hoc analysis, from the Fracture Intervention Trial,63 found that alendronate (Fosamax) 5 mg/day for the first 2 years and 10 mg/day for the 3rd year reduced the incidence of all clinical fractures. In neither of these post hoc analyses did the drug affect the serum creatinine concentration. The patients—postmenopausal women—had GFRs as low as 15 mL/min as estimated by the Cockcroft-Gault equation. Similar post hoc data have been published on raloxifene (Evista).64
There are no data on the efficacy (reduction in fracture risk) or safety of any bisphosphonate in patients with GFRs lower than 15 mL/min (stage 5 chronic kidney disease). Nevertheless, the question often arises when fragility fractures occur in this population. Here, only opinion and controversy exist, and we fervently await good science and randomized prospective data.
How to manage renal bone disease after transplantation is a distinctly separate issue in which bisphosphonate use may be even more controversial than in end-stage renal disease.65,66
In my opinion, patients without fractures with stage 5 chronic kidney disease should not be given bisphosphonates or teriparatide offlabel. Treating only on the basis of low bone mineral density and other risk factors seems to be associated with greater risk than benefit.
In stage 5 patients suffering fragility fractures, a bisphosphonate may be considered, but only after renal osteodystrophy has been thoroughly ruled out, which most often requires a bone biopsy.43,67,68 In skilled hands, transiliac bone biopsy is a safe procedure with little morbidity.
If osteoporosis appears to be the cause of the fracture, and if one chooses to use a bisphosphonate and the patient gives his or her informed consent, then I would give half the usual dose and restrict the therapy to no more than 3 years. The reason for halving the dose is based on the known pharmacokinetics of bisphosphonates in people with normal renal function: 50% of a given dose goes to bone and 50% is excreted by the kidney. Furthermore, the dialyzability of bisphosphonates has not been well studied. Limiting the treatment to 3 years is based on the unknown but probably greater bone retention of bisphosphonates when excretion is impaired.
I must emphasize that these approaches are not based on any evidence of efficacy, but rather are considered in extreme cases of often-recurrent fragility fractures in which the fractures per se pose a great risk of morbidity and death. These approaches should be clearly discussed with the patient, undertaken by specialists knowledgeable in complex metabolic bone disease management, and initiated only after the skeletal fragility disorder is well characterized.
SUMMING UP
No consensus exists on the criteria for diagnosing osteoporosis in stage 4 or 5 chronic kidney disease.
In higher-risk patients in stage 1, 2, or 3 chronic kidney disease who have osteoporosis, it appears that any drug approved for osteoporosis can be used, eg, a bisphosphonate, teriparatide, or both.
Considerations for management are far more complex in stage 4 or 5 because of the increased prevalence of other metabolic bone diseases and renal osteodystrophy, and because the World Health Organization criteria cannot be used to diagnose osteoporosis. In stage 5, the differential diagnosis requires careful analysis of a broad range of biochemical markers of bone turnover and, at times, quantitative bone histomorphometry, especially if one is considering using a bisphosphonate. It is unknown if bisphosphonates, by reducing bone turnover in a preexisting low-bone-turnover state, would help or harm bone or would lead to less or more cardiovascular disease. These questions must be addressed by better science and prospective data.
In the future, newer noninvasive radiologic tools to measure microstructure and mineralization of bone promise to help us better understand osteoporosis and renal osteodystrophy in a noninvasive manner.
In clinical practice, at the current time and with current limited knowledge, treatment of osteoporosis in stage 4 or 5 chronic kidney disease is opinion-based. Nevertheless, in very specific clinical cases of severe fragility fractures that, by themselves, may cause disability and death, bisphosphonates should be considered by experts in bone metabolism and, as with any off-label application, after careful informed discussions with the patient.
- Melton LJ. Epidemiology worldwide. Endocrinol Metab Clin North Am 2003; 32:1–13.
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J Bone Miner Res 2007; 22:465–475.
- Barrett-Connor E, Siris ES, Wehren LE, et al. Osteoporosis and fracture risk in women of different ethnic groups. J Bone Miner Res 2005; 20:185–194.
- Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. Report of a WHO Study Group. World Health Organ Tech Rep Ser 1994; 843:1–129.
- Miller PD, Bonnick SL. Clinical application of bone densitometry. In:Favus MJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 4th ed. American Society for Bone and Mineral Research. Philadelphia, PA: Lippincott Williams & Wilkins; 1999.
- Nickolas TL, Leonard MB, Shane E. Chronic kidney disease and bone fracture: a growing concern. Kidney Int 2008; 74:721–731.
- Gal-Moscovici A, Sprague SM. Osteoporosis and chronic kidney disease. Semin Dial 2007; 20:423–430.
- National Kidney Foundation. Clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(suppl 3):S1–S201.
- Coresh J, Astor BC, Greene T, Eknoyan G, Levey AS. Prevalence of chronic kidney disease and decreased kidney function in the adult US population: Third National Health and Nutrition Examination Survey. Am J Kidney Dis 2003; 41:1–12.
- Bennett WM. Reporting eGFR. Clin J Am Soc Nephrol 2008; 3:1561–1562.
- Glassock RJ, Winearls C. Screening for CKD with eGFR: doubts and dangers. Clin J Am Soc Nephrol 2008; 3:1563–1568.
- Parfitt AM, Drezner M, Glorieux F, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 1987; 2:595–610.
- Andress DL, Sherrard DJ. The osteodystrophy of chronic renal failure. In:Schrier RW, editor: Diseases of the Kidney and Urinary Tract. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:2431–2453.
- Moe S, Drueke T, Cunningham J, et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2006; 69:1945–1953.
- NIH Consensus Development Panel Osteoporosis Prevention Diagnosis and Therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA 2001; 285:785–795.
- Baim S, Binkley N, Bilezikian JP, et al. Official positions of the International Society for Clinical Densitometry and executive summary of the 2007 ISCD Position Development Conference. J Clin Densitom 2008; 11:75–91.
- Siris E, Miller P, Barrett-Connor E, et al. Identification and fracture outcomes of undiagnosed low bone mineral density in postmenopausal women: results from the National Osteoporosis Risk Assessment (NORA). JAMA 2001; 286:2815–2822.
- Schuit SCE, Oei HH, Witteman JC, et al. Fracture incidence and association with bone mineral density in elderly men and women: the Rotterdam Study. Bone 2004; 34:195–202.
- Bouxsein ML. Non-invasive measurements of bone strength: promise and peril. J Musculoskelet Neuronal Interact 2004; 4:404–405.
- Seeman E. Bone quality: the material and structural basis of bone strength. J Bone Miner Metab 2008; 26:1–8.
- Alem AM, Sherrard DJ, Gillen DL, et al. Increased risk of hip fracture among patients with end-stage renal disease. Kidney Int 2000; 58:396–399.
- Ball AM, Gillen DL, Sherrard D, et al. Risk of hip fracture among dialysis and renal transplant recipients. JAMA 2002; 288:3014–3018.
- Jadoul M, Albert JM, Akiba T, et al. Incidence and risk factors for hip or other bone fractures among hemodialysis patients in the Dialysis Outcomes and Practice Patterns Study. Kidney Int 2006; 70:1358–1366.
- Stehman-Breen CO, Sherrard DJ, Alem AM, et al. Risk factors for hip fracture among patients with end-stage renal disease. Kidney Int 2000; 58:2200–2205.
- Wehrli FW, Leonard MB, Saha PK, Gomberg BR. Quantitative highresolution magnetic resonance imaging reveals structural implications of renal osteodystrophy on trabecular and cortical bone. J Magn Reson Imaging 2004; 20:83–89.
- Genant HK, Lang TF, Engelke K, et al. Advances in the noninvasive assessment of bone density, quality, and structure. Calcif Tissue Int 1996; 59(suppl 1):S10–S15.
- Roschger P, Paschalis EP, Fratzl P, Klaushofer K. Bone mineralization density distribution in health and disease. Bone 2008; 42:456–466.
- Miller PD. Bone density and markers of bone turnover in predicting fracture risk and how changes in these measures predict fracture risk reduction. Curr Osteoporos Rep 2005; 3:103–110.
- Miller PD, Hochberg MC, Wehren LE, Ross PD, Wasnich RD. How useful are measures of BMD and bone turnover? Curr Med Res Opin 2005; 21:545–554.
- Chavassieux PM, Delmas PD. Bone remodeling: biochemical markers or bone biopsy? J Bone Miner Res 2006; 21:178–179.
- Garnero P. Biomarkers for osteoporosis management: utility in diagnosis, fracture risk prediction and therapy monitoring. Mol Diagn Ther 2008; 12:157–170.
- Hochberg M, Greenspan S, Wasnich R, Miller P, Thompson D, Ross P. Changes in bone density and turnover explain the reductions in incidence of nonvertebral fractures that occur during treatment with antiresorptive agents. J Clin Endocrinol Metab 2002; 87:1586–1592.
- Bouxsein ML, Delmas PD. Considerations for development of surrogate endpoints for antifracture efficacy of new treatments in osteoporosis: a perspective. J Bone Miner Res 2008; 23:1155–1167.
- Chen P, Satterwhite JH, Licatta AA, et al. Early changes in biochemical markers of bone formation predict BMD response to teriparatide in postmenopausal women with osteoporosis. J Bone Miner Res 2005; 20:962–970.
- Miller PD, Delmas PD, Lindsay R, et al. Early responsiveness of women with osteoporosis to teriparatide after therapy with alendronate or risedronate. J Clin Endocrinol Metab 2008; 93:3785–3793.
- Baim S, Miller PD. Assessing the clinical utility of serum CTX in postmenopausal osteoporosis and its use in predicting risk of osteonecrosis of the jaw. J Bone Miner Res 2009; 24:561–574.
- Miller PD, Lerma EV. Renal bone diseases. In:Kleerekoper M, Siris E, McClung M, editors. The Bone and Mineral Manual—A Practical Guide. 2nd ed. Burlington, MA: Elsevier Academic Press; 2005:127–138.
- Miller PD, Shane E. Management of transplantation renal bone disease: Interplay of bone mineral density and decisions regarding bisphosphonate use. In:Weir MR, editor. Medical Management of Kidney Transplantation. Philadelphia, PA: Lippincott Williams & Wilkins; 2004:359–375.
- Levin A, Bakris GL, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney int 2007; 71:31–38.
- Vassalotti JA, Uribarri J, Chen SC, et al. Trends in mineral metabolism: Kidney Early Evaluation Program (KEEP) and the National Health and Nutrition Examination Survey (NHANES) 1999–2004. Am J Kidney Dis 2008; 51(suppl 2):S56–S68.
- Meier C, Seibel MJ, Kraenzlin ME. Use of bone turnover markers in the real world: are we there yet? J Bone Miner Res 2009; 24:386–388.
- Lehmann G, Ott U, Kaemmerer D, Schuetze J, Wolf G. Bone histomorphometry and biochemical markers of bone turnover in patients with chronic kidney disease stages 3–5. Clin Nephrol 2008; 70:296–305.
- Miller PD. The role of bone biopsy in patients with chronic renal failure. Clin J Am Soc Nephrol 2008; 3(suppl 3):S140–S150.
- Frost HM. Tetracycline-based histological analysis of bone remodeling. Calcif Tissue Res 1969; 3:211–237.
- Hitt O, Jaworski ZF, Shimizu AG, Frost HM. Tissue-level bone formation rates in chronic renal failure, measured by means of tetracycline bone labeling. Can J Physiol Pharmacol 1970; 48:824–828.
- Coen G. Adynamic bone disease: an update and overview. J Nephrol 2005; 18:117–122.
- Parfitt AM. Renal bone disease: a new conceptual framework for the interpretation of bone histomorphometry. Curr Opin Nephrol Hypertens 2003; 12:387–403.
- Brandenburg VM, Floege J. Adynamic bone diseaseùbone and beyond. NDT Plus 2008; 3:135–147. doi:10.1093/ndtplus/sfn040.
- Hruska KA, Saab G, Mathew S, Lund R. Renal osteodystrophy, phosphate homeostasis, and vascular calcification. Semin Dial 2007; 20:309–315.
- Toussaint ND, Elder GJ, Kerr PG. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc Nephrol 2009; 4:221–233.
- Miller PD. Is there a role for bisphosphonates in chronic kidney disease? Semin Dial 2007; 20:186–190.
- Miller PD. Bisphosphonates: pharmacology and use in the treatment of osteoporosis. In:Marcus R, Feldman D, Nelson DA, Rosen CJ, editors. Osteoporosis. 3rd ed. Boston, MA: Elsevier Academic Press; 2008:1725–1736.
- Russell RG, Watts NB, Ebetino FH, Rogers MJ. Mechanisms of action of bisphosphonates: similarities and differences and their potential influence on clinical efficacy. Osteoporosis Int 2008; 19:733–759.
- Eisman JA, Civitelli R, Adami S, et al. Efficacy and tolerability of intravenous ibandronate injections in postmenopausal osteoporosis: 2-year results from the DIVA study. J Rheumatol 2008; 35:488–497.
- Black DM, Delmas PD, Eastell RR, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med 2007; 356:1809–1822.
- Lewiecki EM, Miller PD. Renal safety of intravenous bisphosphonates in the treatment of osteoporosis. Expert Opin Drug Saf 2007; 6:663–672.
- Miller PD. Anti-resorptives in the management of osteoporosis. Best Pract Res Clin Endocrinol Metab 2008; 22:849–868.
- Perazella MA, Markowitz GS. Bisphosphonate nephrotoxicity. Kidney Int 2008; 74:1385–1393.
- Boonen S, Sellmeyer DE, Lippuner K, et al. Renal safety of annual zoledronic acid infusions in osteoporotic postmenopausal women. Kidney Int 2008; 74:641–648.
- Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 2001; 344:1434–1441.
- Miller PD, Schwartz EN, Chen P, Misurski DA, Krege JH. Teriparatide in postmenopausal women with osteoporosis and mild or moderate renal impairment. Osteoporosis Int 2007; 18:59–68.
- Miller PD, Roux C, Boonen S, Barton I, Dunlap L, Burgio D. Safety and efficacy of risedronate in patients with age-related reduced renal function as estimated by the Cockcroft and Gault method: a pooled analysis of nine clinical trials. J Bone Miner Res 2005; 20:2105–2115.
- Jamal SA, Bauer DC, Ensrud KE, et al. Alendronate treatment in women with normal to severely impaired renal function: an analysis of the fracture intervention trial. J Bone Miner Res 2007; 22:503–508.
- Ishani A, Blackwell T, Jamal SA, Cummings SR, Ensrud KE; MORE Investigators. The effect of raloxifene treatment in postmenopausal women with CKD. J Am Soc Nephrol 2008; 19:1430–1438.
- Coco M, Glicklich D, Faugere MC, et al. Prevention of bone loss in renal transplant recipients: a prospective, randomized trial of intravenous pamidronate. J Am Soc Nephrol 2003; 14:2669–2676.
- Palmer SC, McGregor DO, Strippoli GF. Interventions for preventing bone disease in kidney transplant recipients. Cochrane Database Syst Rev 2007;CD005015.
- Ferreira MA. Diagnosis of renal osteodystrophy: when and how to use biochemical markers and non-invasive methods; when bone biopsy is needed. Nephrol Dial Transplant 2000; 15(suppl 5):8–14.
- Trueba D, Sawaya BP, Mawad H, Malluche HH. Bone biopsy: indications, techniques, and complications. Semin Dial 2003; 16:341–345.
But even in chronic kidney disease, many fractures are due to postmenopausal or agerelated osteoporosis, and estrogen-deficiency osteoporosis is the most common cause of fragility fractures overall.1–3 Osteoporosis can be diagnosed only after other causes of skeletal fragility have been ruled out. And how to diagnose and treat osteoporosis in the most severe stage of kidney disease is a matter of opinion, as we have almost no data to guide us.
Nevertheless, in the pages that follow, I will outline my admittedly opinion-based approach to diagnosing and managing the causes of fragility fractures in patients with chronic kidney disease.
T SCORES DO NOT DISTINGUISH THE CAUSES OF FRAGILITY
The most common cause of fragility fractures is osteoporosis due to estrogen deficiency.1–3 But since many other medical conditions can lead to osteoporosis, simple diagnostic criteria have been difficult to find.
Before 1994, the diagnosis of osteoporosis was made on the basis of low-trauma fractures.4 Now, we use the World Health Organization criteria,5 based on bone mineral density T scores:
- Normal—a T score of −1.0 standard deviations or higher
- Osteopenia—a T score of less than −1.0 but higher than −2.5
- Osteoporosis—a T score of −2.5 or less
- Severe osteoporosis—a T score of −2.5 or less with a fragility fracture.
However, fractures can also be due to metabolic bone diseases that are not osteoporosis, including renal bone diseases.6–7 While a low T score or a fracture provides a working diagnosis of osteoporosis, it does not help distinguish the different types of osteoporosis and nonosteoporotic metabolic bone diseases. For example, osteomalacia and osteogenesis imperfecta can also cause fragility fractures and can be associated with low bone density. Using these criteria to define osteoporosis is even more problematic in patients with chronic kidney disease.
FIVE STAGES OF CHRONIC KIDNEY DISEASE
The National Kidney Foundation8 classifies the severity of chronic kidney disease on the basis of the glomerular filtration rate (GFR), as measured by 24-hour urine for creatinine clearance, or as estimated by the Cockcroft-Gault equation or, preferably, the Modification of Diet in Renal Disease (MDRD) equation (calculators are available at www.kidney.org/professionals/kdoqi/gfr_calculator.cfm):
- Stage 1—GFR 90 mL/minute/1.73 m2 or higher
- Stage 2—GFR 60 to 89
- Stage 3—GFR 30 to 59
- Stage 4—GFR 15 to 29
- Stage 5—GFR lower than 15, or if the patient is on dialysis. (Another stage, called 5D, was added to the list to denote patients on dialysis, since the metabolic derangements in bone and systemic biology may differ between patients on dialysis vs those not on dialysis.)
This staging system is relevant to the discussion of bone fragility that follows.
CHRONIC KIDNEY DISEASE IS COMMON IN THE ELDERLY
The third National Health and Nutrition Examination Survey9 found that, at least as estimated by the Cockcroft-Gault equation, the GFR declines with age, so that by the age of 70 at least 20% of the US population has stage 4 or 5 chronic kidney disease.
Although the Cockcroft-Gault and MDRD equations may yield lower GFR values in the general population than one would get by measuring creatinine, inulin, or iothalamate clearance,10,11 the point is that both osteoporosis and chronic kidney disease are common.
THE GAMUT OF RENAL OSTEODYSTROPHY
In kidney failure (stage 5 chronic kidney disease), all forms of renal osteodystrophy may be associated with fragility fractures. Renal osteodystrophy can be defined by quantitative bone histomorphometry.12,13 The systemic conditions that may be associated with the bone disease and systemic vascular disease (chronic kidney disease–mineral and bone disorder) are characterized by one or more of the following14:
- Abnormalities of calcium, phosphorus, parathyroid hormone, or vitamin D metabolism
- Abnormalities in bone turnover, mineralization, volume, linear growth, or strength
- Vascular or other soft-tissue calcification.
The National Kidney Foundation14 classifies renal osteodystrophy on the basis of:
- Turnover—high, normal, or low
- Mineralization—normal or abnormal
- Volume—high, normal, or low.
Although this system helps us understand these diseases better, it does not provide a working diagnosis of osteoporosis.14
WHAT IS OSTEOPOROSIS?
In an attempt to define osteoporosis by a pathophysiologic mechanism, the National Institutes of Health15 have held two consensus conferences and have stated that “osteoporosis is a skeletal disorder characterized by impairment in bone strength predisposing a person to an increased risk of fracture. Bone strength primarily reflects the integration of bone density and bone quality.”15 However, the consensus statement also does not provide a working diagnosis of osteoporosis—one that clinicians can apply to management decisions, and one that is also accepted by the US International Classification of Disease codes for reimbursement purposes.
The 1994 World Health Organization criteria offer the most pragmatic operational definition of osteoporosis, and they can be applied in both men and women, as well as in younger patients with medical conditions associated with increased risk of low-trauma fracture.5,16 Although the main purpose of these criteria was to advise international health authorities of the potential future economic impact of osteoporosis, the T score also became the pragmatic diagnostic threshold for defining normal, osteopenia, and osteoporosis in clinical practice.
The T score also calls attention to an important observation: of people who have fractures and subsequently undergo bone densitometry, more are found to have osteopenia than osteoporosis. The reasons are that there are more people with osteopenia than osteoporosis,17,18 and many other factors independent of low bone mineral density contribute to bone strength.19,20
How is osteoporosis diagnosed in stage 1–3 chronic kidney disease?
In patients with chronic kidney disease who develop fragility fractures, the reasonable question is: Is the cause of the fracture osteoporosis or some other metabolic bone disease associated with chronic kidney disease?
The National Kidney Foundation guidelines14 say that the diagnosis of osteoporosis can be established in patients with stage 1, 2, or 3 chronic kidney disease on the basis of either of the World Health Organization criteria, ie, a T score of −2.5 or lower or fragility fractures, as in the postmenopausal population, as long as there are no biochemical abnormalities that suggest chronic kidney disease–mineral and bone disorder.
How is osteoporosis diagnosed in stage 4 or 5 chronic kidney disease?
The answer is neither straightforward nor clearly defined in severe (stage 4 or 5) chronic kidney disease.
In stage 5 and especially in patients on dialysis, the derangements in bone and mineral metabolism become serious enough to impair bone strength and increase the risk of lowtrauma fractures. The risk of hip fracture in stage 5 may be four times higher than in agematched controls.21–24
Adynamic, severe hyperparathyroid bone disease as well as osteomalacia can be associated with a higher risk of fragility fractures than in aged-matched controls in population studies of postmenopausal women or elderly men. These are bone fragility conditions that are not osteoporosis but that can mimic osteoporosis by the World Health Organization criteria.
Thus, when a patient in stage 5 has severe fragility fractures that by themselves may be life-threatening, it is reasonable to ask if the drugs that reduce the risk of fractures in many other osteoporotic conditions (postmenopausal, steroid-induced, elderly male osteoporosis, after solid organ transplantation) can also be used in patients with advanced chronic kidney disease.
The diagnosis of osteoporosis in these patients has no universally accepted criteria. The diagnosis is best suggested by excluding other forms of renal osteodystrophy by quantitative histomorphometry or by attempting to classify the form of renal osteodystrophy by noninvasive means of assessing bone turnover, mineralization, and volume. However, we lack clinical tools to make these distinctions in individual patients.
While many promising radiologic techniques that examine bone microarchitecture offer hope of being able to define turnover, mineralization, and volume noninvasively in severe chronic kidney disease, they are investigational and unproven at this time in discriminating between renal osteodystrophy and osteoporosis.6,25–27 As we increase our understanding of the relationships between turnover, mineralization, volume, and bone strength, these noninvasive imaging technologies may become the means to correlate turnover, mineralization, and volume to bone strength and open up an entirely new way to classify skeletal strength.
In the meantime, the clinician is left with quantitative bone histomorphometry (which requires biopsy) and biochemical markers of bone turnover to characterize the bone disease that may be responsible for low-trauma fractures in stage 5 chronic kidney disease. The clinician should first use biochemical markers before bone biopsy to distinguish the form of renal osteodystrophy, as this distinction may be able to prevent unnecessary biopsy.
Biochemical markers of bone metabolism
In chronic kidney disease, the bone biochemical tests that nephrologists usually assess during the course of declining renal function are the serum levels of:
- Phosphorus
- Parathyroid hormone
- Calcium
- Other electrolytes
- Total alkaline phosphatase or bone-specific alkaline phosphatase
- 1,25 dihydroxyvitamin D.
In postmenopausal osteoporosis, the biochemical markers of bone turnover that are measured to reflect baseline levels of bone turnover or change in bone turnover in response to drug therapy are:
- The serum or urine collagen cross-links N-telopeptide (NTx) and C-telopeptide (CTx), markers of bone resorption
- Bone-specific alkaline phosphatase (an osteoblast activity marker)
- Serum osteocalcin, a bone formation marker
- Propeptide type 1 collagen (P1NP), a marker of osteoblast activity, highly correlated with bone formation
- 25-hydroxyvitamin D levels.
Biochemical markers of bone turnover cannot be used to diagnose osteoporosis. They can, however, provide clinical guidance as to whether a patient has high or low bone turnover and whether therapy is affecting bone turnover.28–36 Although these markers have value in making these distinctions in groups of patients, they are less sensitive and specific for classifying an individual patient’s bone turnover status.
Bone-specific alkaline phosphatase, parathyroid hormone, and adynamic bone disease
If a patient’s bone-specific alkaline phosphatase level is elevated, adynamic bone disease is highly unlikely. Assuming that other causes of this elevated level (eg, Paget disease of bone, metastatic cancer) have already been excluded, the elevated level could represent either osteomalacia or hyperparathyroid bone disease.
However, a “normal” bone-specific alkaline phosphatase level does not exclude adynamic bone disease, whereas a low level is more often associated with low bone turnover.
An elevated parathyroid hormone level does not exclude adynamic renal bone disease, but a low level (< 150 pg/mL) suggests a lowbone-turnover state. A level six times or more greater than the upper limit of normal is far more likely to be associated with high bone turnover.
Thus, in clinical practice, patients with stage 4 or 5 chronic kidney disease who have elevated bone-specific alkaline phosphatase or very high parathyroid hormone values do not have adynamic bone disease. Furthermore, once other causes of these aberrant biochemical abnormalities have been defined, then “high-bone-turnover osteoporosis” may be a consideration. Certainly, in my opinion, if bone turnover markers suggest low bone turnover, bone biopsy is necessary before starting an antiresorptive agent.35
Quantitative bone histomorphometry
Double tetracycline-labeled quantitative histomorphometry is still the only accepted way to measure turnover, mineralization, and volume in clinical practice.43–45 A committee of the American Society for Bone and Mineral Research has developed histomorphometric criteria for distinguishing among the different types of metabolic bone diseases (osteomalacia, adynamic bone disease, hyperparathyroid bone disease).12 These criteria can be used to distinguish among the various metabolic bone diseases that accompany stage 5 chronic kidney disease, including adynamic bone disease.43,46–48
For patients in stage 5 who have had a fragility fracture, adynamic bone disease should be excluded before the off-label use of an osteoporosis drug that reduces bone turnover, such as a bisphosphonate, calcitonin, estrogen, a selective estrogen receptor modulator, or denosumab (anti-RANK ligand antibody). While there is no evidence, for example, that starting a bisphosphonate in a patient who already has adynamic bone disease is detrimental to either bone strength or systemic vascular calcification (which may be linked to low bone turnover),49 it seems unreasonable to do so until solid prospective data clarify the harm or benefit.50 Preliminary experimental and clinical data suggest that bisphosphonates may even reduce progression of extraosseous calcification and inhibit the development of atherosclerosis.50
Hence, quantitative bone histomorphometry can discriminate among the various forms of renal osteodystrophy. If a distinct form of renal osteodystrophy is not present in a patient with stage 4 or 5 chronic kidney disease who has had a fracture and who, on biopsy, has a low trabecular bone volume, the patient probably has osteoporosis by exclusion.
TREATING OSTEOPOROSIS IN STAGE 1–3 CHRONIC KIDNEY DISEASE
As previously mentioned, patients who have fragility fractures in stage 1, 2, or 3 chronic kidney disease are more likely to have osteoporosis than renal osteodystrophy as the cause of their impaired bone strength. Although several articles have described a higher risk of fragility fractures in patients with age-related reduction in renal function than in agematched patients with normal renal function, the specific metabolic bone disease other than osteoporosis accounting for this bone fragility has not been defined.6
Hence, patients with osteoporosis who are in stage 1, 2, or 3 chronic kidney disease and do not have a known biochemical abnormality that might suggest some form of renal osteodystrophy can and should be considered for treatment with approved drugs that reduce the risk of fractures in postmenopausal, male, or glucocorticoid-induced osteoporosis.51–53 In clinical trials, these agents were shown to be effective in patients with serum creatinine concentrations as high as 2.0 mg/dL or a GFR as low as 30 mL/min, as estimated by the Cockcroft-Gault equation.
While all of the approved agents show evidence of reducing the risk of vertebral fractures, patients at higher risk of fractures or those who have already suffered a nonvertebral fracture are more often considered candidates for treatment with a bisphosphonate or teriparatide (Forteo), both of which have shown evidence of reducing the risk of all fractures.
Bisphosphonates in stage 1–3 chronic kidney disease
There is prospective evidence that patients with an age-related reduction in GFR down to 30 mL/min benefit from oral and intravenous bisphosphonates, since all of the clinical trials that led to the approval of bisphosphonates included patients with GFRs as low as this.54–57 Bisphosphonates seem to have an excellent safety profile as measured by renal adverse events in patients with a GFR of 30 mL/min or greater.52–59
From the intravenous bisphosphonate studies, it appears that ibandronate (Boniva) at the approved dose of 3 mg intravenously every 3 months and zoledronic acid (Reclast) 5 mg once a year given over 15 minutes are safe in patients with a GFR greater than 30 or 35 mL/min.
However, the safety of these drugs might not be the same in patients with preexisting renal parenchymal disease (eg, in diabetes) or in patients using other agents that could affect renal function (eg, nonsteroidal antiinflammatory drugs). Therefore, caution is still needed when deciding to use intravenous bisphosphonates in specific higher-risk renal subpopulations.
In the clinical trials of zoledronic acid, a substantial proportion of patients had diabetes, and no difference was seen in adverse renal effects between diabetic and nondiabetic patients. Also, GFRs declined equally between the treated and placebo groups over time and were no different at the end of 3 years.55 However, in patients in whom serum creatinine was measured 9 to 11 days after the infusion of zoledronic acid, there was a small but statistically significant transient increase in serum creatinine concentration (0.5–2.0 mg/dL above baseline) after the second annual infusion. 58 The serum creatinine concentrations returned to their baseline values in all of these patients before the next annual infusion.
It is important that infusions of zoledronic acid be given no faster than over 15 minutes. More rapid infusion has been associated with acute renal failure, suggesting that the tubular damage that mimics acute tubular necrosis is related to the maximal concentration and not to the area under the curve. I infuse zoledronic acid over 30 minutes in patients with normal renal function or in those with stage 1, 2, or 3 chronic kidney disease.
Teriparatide
Teriparatide’s approval trial did not require baseline measurements of GFR, but patients were enrolled only if their baseline serum creatinine concentrations were less than 2.0 mg/dL.60 In a post hoc analysis, a small subset of patients had GFRs as low as 30 mL/min as estimated by the Cockcroft-Gault equation. In these patients, teriparatide 20 or 40 μg/day had an anabolic effect as measured by increases in osteoblast activity markers and bone mineral density, similar to that seen in patients with higher estimated GFRs and without any adverse renal effects.61
There are no data on using teriparatide in stage 4 or 5 chronic kidney disease, and I emphasize that in all of the clinical trials of teriparatide, all patients, even those with estimated GFRs as low as 30 mL/min, had normal baseline serum intact parathyroid hormone levels. It is possible that the bone biologic response could differ between patients with chronic kidney disease who have an elevated as compared with a normal serum parathyroid hormone level. This issue should be investigated.
TREATING OSTEOPOROSIS IN STAGE 4 OR 5 CHRONIC KIDNEY DISEASE
Treatment decisions are more difficult in patients with stage 4 and especially stage 5 chronic kidney disease who have had fragility fractures. This is even the case when the clinician has determined to the best of his or her ability that the patient has osteoporosis rather than renal osteodystrophy.
There are no prospective data showing any of the approved drugs to be effective in treating osteoporosis in patients whose GFRs are lower than 30 mL/min. However, a post hoc analysis of pooled data from nine clinical trials62 found that risedronate (Actonel) 5 mg/day reduced the incidence of new vertebral fractures. Another post hoc analysis, from the Fracture Intervention Trial,63 found that alendronate (Fosamax) 5 mg/day for the first 2 years and 10 mg/day for the 3rd year reduced the incidence of all clinical fractures. In neither of these post hoc analyses did the drug affect the serum creatinine concentration. The patients—postmenopausal women—had GFRs as low as 15 mL/min as estimated by the Cockcroft-Gault equation. Similar post hoc data have been published on raloxifene (Evista).64
There are no data on the efficacy (reduction in fracture risk) or safety of any bisphosphonate in patients with GFRs lower than 15 mL/min (stage 5 chronic kidney disease). Nevertheless, the question often arises when fragility fractures occur in this population. Here, only opinion and controversy exist, and we fervently await good science and randomized prospective data.
How to manage renal bone disease after transplantation is a distinctly separate issue in which bisphosphonate use may be even more controversial than in end-stage renal disease.65,66
In my opinion, patients without fractures with stage 5 chronic kidney disease should not be given bisphosphonates or teriparatide offlabel. Treating only on the basis of low bone mineral density and other risk factors seems to be associated with greater risk than benefit.
In stage 5 patients suffering fragility fractures, a bisphosphonate may be considered, but only after renal osteodystrophy has been thoroughly ruled out, which most often requires a bone biopsy.43,67,68 In skilled hands, transiliac bone biopsy is a safe procedure with little morbidity.
If osteoporosis appears to be the cause of the fracture, and if one chooses to use a bisphosphonate and the patient gives his or her informed consent, then I would give half the usual dose and restrict the therapy to no more than 3 years. The reason for halving the dose is based on the known pharmacokinetics of bisphosphonates in people with normal renal function: 50% of a given dose goes to bone and 50% is excreted by the kidney. Furthermore, the dialyzability of bisphosphonates has not been well studied. Limiting the treatment to 3 years is based on the unknown but probably greater bone retention of bisphosphonates when excretion is impaired.
I must emphasize that these approaches are not based on any evidence of efficacy, but rather are considered in extreme cases of often-recurrent fragility fractures in which the fractures per se pose a great risk of morbidity and death. These approaches should be clearly discussed with the patient, undertaken by specialists knowledgeable in complex metabolic bone disease management, and initiated only after the skeletal fragility disorder is well characterized.
SUMMING UP
No consensus exists on the criteria for diagnosing osteoporosis in stage 4 or 5 chronic kidney disease.
In higher-risk patients in stage 1, 2, or 3 chronic kidney disease who have osteoporosis, it appears that any drug approved for osteoporosis can be used, eg, a bisphosphonate, teriparatide, or both.
Considerations for management are far more complex in stage 4 or 5 because of the increased prevalence of other metabolic bone diseases and renal osteodystrophy, and because the World Health Organization criteria cannot be used to diagnose osteoporosis. In stage 5, the differential diagnosis requires careful analysis of a broad range of biochemical markers of bone turnover and, at times, quantitative bone histomorphometry, especially if one is considering using a bisphosphonate. It is unknown if bisphosphonates, by reducing bone turnover in a preexisting low-bone-turnover state, would help or harm bone or would lead to less or more cardiovascular disease. These questions must be addressed by better science and prospective data.
In the future, newer noninvasive radiologic tools to measure microstructure and mineralization of bone promise to help us better understand osteoporosis and renal osteodystrophy in a noninvasive manner.
In clinical practice, at the current time and with current limited knowledge, treatment of osteoporosis in stage 4 or 5 chronic kidney disease is opinion-based. Nevertheless, in very specific clinical cases of severe fragility fractures that, by themselves, may cause disability and death, bisphosphonates should be considered by experts in bone metabolism and, as with any off-label application, after careful informed discussions with the patient.
But even in chronic kidney disease, many fractures are due to postmenopausal or agerelated osteoporosis, and estrogen-deficiency osteoporosis is the most common cause of fragility fractures overall.1–3 Osteoporosis can be diagnosed only after other causes of skeletal fragility have been ruled out. And how to diagnose and treat osteoporosis in the most severe stage of kidney disease is a matter of opinion, as we have almost no data to guide us.
Nevertheless, in the pages that follow, I will outline my admittedly opinion-based approach to diagnosing and managing the causes of fragility fractures in patients with chronic kidney disease.
T SCORES DO NOT DISTINGUISH THE CAUSES OF FRAGILITY
The most common cause of fragility fractures is osteoporosis due to estrogen deficiency.1–3 But since many other medical conditions can lead to osteoporosis, simple diagnostic criteria have been difficult to find.
Before 1994, the diagnosis of osteoporosis was made on the basis of low-trauma fractures.4 Now, we use the World Health Organization criteria,5 based on bone mineral density T scores:
- Normal—a T score of −1.0 standard deviations or higher
- Osteopenia—a T score of less than −1.0 but higher than −2.5
- Osteoporosis—a T score of −2.5 or less
- Severe osteoporosis—a T score of −2.5 or less with a fragility fracture.
However, fractures can also be due to metabolic bone diseases that are not osteoporosis, including renal bone diseases.6–7 While a low T score or a fracture provides a working diagnosis of osteoporosis, it does not help distinguish the different types of osteoporosis and nonosteoporotic metabolic bone diseases. For example, osteomalacia and osteogenesis imperfecta can also cause fragility fractures and can be associated with low bone density. Using these criteria to define osteoporosis is even more problematic in patients with chronic kidney disease.
FIVE STAGES OF CHRONIC KIDNEY DISEASE
The National Kidney Foundation8 classifies the severity of chronic kidney disease on the basis of the glomerular filtration rate (GFR), as measured by 24-hour urine for creatinine clearance, or as estimated by the Cockcroft-Gault equation or, preferably, the Modification of Diet in Renal Disease (MDRD) equation (calculators are available at www.kidney.org/professionals/kdoqi/gfr_calculator.cfm):
- Stage 1—GFR 90 mL/minute/1.73 m2 or higher
- Stage 2—GFR 60 to 89
- Stage 3—GFR 30 to 59
- Stage 4—GFR 15 to 29
- Stage 5—GFR lower than 15, or if the patient is on dialysis. (Another stage, called 5D, was added to the list to denote patients on dialysis, since the metabolic derangements in bone and systemic biology may differ between patients on dialysis vs those not on dialysis.)
This staging system is relevant to the discussion of bone fragility that follows.
CHRONIC KIDNEY DISEASE IS COMMON IN THE ELDERLY
The third National Health and Nutrition Examination Survey9 found that, at least as estimated by the Cockcroft-Gault equation, the GFR declines with age, so that by the age of 70 at least 20% of the US population has stage 4 or 5 chronic kidney disease.
Although the Cockcroft-Gault and MDRD equations may yield lower GFR values in the general population than one would get by measuring creatinine, inulin, or iothalamate clearance,10,11 the point is that both osteoporosis and chronic kidney disease are common.
THE GAMUT OF RENAL OSTEODYSTROPHY
In kidney failure (stage 5 chronic kidney disease), all forms of renal osteodystrophy may be associated with fragility fractures. Renal osteodystrophy can be defined by quantitative bone histomorphometry.12,13 The systemic conditions that may be associated with the bone disease and systemic vascular disease (chronic kidney disease–mineral and bone disorder) are characterized by one or more of the following14:
- Abnormalities of calcium, phosphorus, parathyroid hormone, or vitamin D metabolism
- Abnormalities in bone turnover, mineralization, volume, linear growth, or strength
- Vascular or other soft-tissue calcification.
The National Kidney Foundation14 classifies renal osteodystrophy on the basis of:
- Turnover—high, normal, or low
- Mineralization—normal or abnormal
- Volume—high, normal, or low.
Although this system helps us understand these diseases better, it does not provide a working diagnosis of osteoporosis.14
WHAT IS OSTEOPOROSIS?
In an attempt to define osteoporosis by a pathophysiologic mechanism, the National Institutes of Health15 have held two consensus conferences and have stated that “osteoporosis is a skeletal disorder characterized by impairment in bone strength predisposing a person to an increased risk of fracture. Bone strength primarily reflects the integration of bone density and bone quality.”15 However, the consensus statement also does not provide a working diagnosis of osteoporosis—one that clinicians can apply to management decisions, and one that is also accepted by the US International Classification of Disease codes for reimbursement purposes.
The 1994 World Health Organization criteria offer the most pragmatic operational definition of osteoporosis, and they can be applied in both men and women, as well as in younger patients with medical conditions associated with increased risk of low-trauma fracture.5,16 Although the main purpose of these criteria was to advise international health authorities of the potential future economic impact of osteoporosis, the T score also became the pragmatic diagnostic threshold for defining normal, osteopenia, and osteoporosis in clinical practice.
The T score also calls attention to an important observation: of people who have fractures and subsequently undergo bone densitometry, more are found to have osteopenia than osteoporosis. The reasons are that there are more people with osteopenia than osteoporosis,17,18 and many other factors independent of low bone mineral density contribute to bone strength.19,20
How is osteoporosis diagnosed in stage 1–3 chronic kidney disease?
In patients with chronic kidney disease who develop fragility fractures, the reasonable question is: Is the cause of the fracture osteoporosis or some other metabolic bone disease associated with chronic kidney disease?
The National Kidney Foundation guidelines14 say that the diagnosis of osteoporosis can be established in patients with stage 1, 2, or 3 chronic kidney disease on the basis of either of the World Health Organization criteria, ie, a T score of −2.5 or lower or fragility fractures, as in the postmenopausal population, as long as there are no biochemical abnormalities that suggest chronic kidney disease–mineral and bone disorder.
How is osteoporosis diagnosed in stage 4 or 5 chronic kidney disease?
The answer is neither straightforward nor clearly defined in severe (stage 4 or 5) chronic kidney disease.
In stage 5 and especially in patients on dialysis, the derangements in bone and mineral metabolism become serious enough to impair bone strength and increase the risk of lowtrauma fractures. The risk of hip fracture in stage 5 may be four times higher than in agematched controls.21–24
Adynamic, severe hyperparathyroid bone disease as well as osteomalacia can be associated with a higher risk of fragility fractures than in aged-matched controls in population studies of postmenopausal women or elderly men. These are bone fragility conditions that are not osteoporosis but that can mimic osteoporosis by the World Health Organization criteria.
Thus, when a patient in stage 5 has severe fragility fractures that by themselves may be life-threatening, it is reasonable to ask if the drugs that reduce the risk of fractures in many other osteoporotic conditions (postmenopausal, steroid-induced, elderly male osteoporosis, after solid organ transplantation) can also be used in patients with advanced chronic kidney disease.
The diagnosis of osteoporosis in these patients has no universally accepted criteria. The diagnosis is best suggested by excluding other forms of renal osteodystrophy by quantitative histomorphometry or by attempting to classify the form of renal osteodystrophy by noninvasive means of assessing bone turnover, mineralization, and volume. However, we lack clinical tools to make these distinctions in individual patients.
While many promising radiologic techniques that examine bone microarchitecture offer hope of being able to define turnover, mineralization, and volume noninvasively in severe chronic kidney disease, they are investigational and unproven at this time in discriminating between renal osteodystrophy and osteoporosis.6,25–27 As we increase our understanding of the relationships between turnover, mineralization, volume, and bone strength, these noninvasive imaging technologies may become the means to correlate turnover, mineralization, and volume to bone strength and open up an entirely new way to classify skeletal strength.
In the meantime, the clinician is left with quantitative bone histomorphometry (which requires biopsy) and biochemical markers of bone turnover to characterize the bone disease that may be responsible for low-trauma fractures in stage 5 chronic kidney disease. The clinician should first use biochemical markers before bone biopsy to distinguish the form of renal osteodystrophy, as this distinction may be able to prevent unnecessary biopsy.
Biochemical markers of bone metabolism
In chronic kidney disease, the bone biochemical tests that nephrologists usually assess during the course of declining renal function are the serum levels of:
- Phosphorus
- Parathyroid hormone
- Calcium
- Other electrolytes
- Total alkaline phosphatase or bone-specific alkaline phosphatase
- 1,25 dihydroxyvitamin D.
In postmenopausal osteoporosis, the biochemical markers of bone turnover that are measured to reflect baseline levels of bone turnover or change in bone turnover in response to drug therapy are:
- The serum or urine collagen cross-links N-telopeptide (NTx) and C-telopeptide (CTx), markers of bone resorption
- Bone-specific alkaline phosphatase (an osteoblast activity marker)
- Serum osteocalcin, a bone formation marker
- Propeptide type 1 collagen (P1NP), a marker of osteoblast activity, highly correlated with bone formation
- 25-hydroxyvitamin D levels.
Biochemical markers of bone turnover cannot be used to diagnose osteoporosis. They can, however, provide clinical guidance as to whether a patient has high or low bone turnover and whether therapy is affecting bone turnover.28–36 Although these markers have value in making these distinctions in groups of patients, they are less sensitive and specific for classifying an individual patient’s bone turnover status.
Bone-specific alkaline phosphatase, parathyroid hormone, and adynamic bone disease
If a patient’s bone-specific alkaline phosphatase level is elevated, adynamic bone disease is highly unlikely. Assuming that other causes of this elevated level (eg, Paget disease of bone, metastatic cancer) have already been excluded, the elevated level could represent either osteomalacia or hyperparathyroid bone disease.
However, a “normal” bone-specific alkaline phosphatase level does not exclude adynamic bone disease, whereas a low level is more often associated with low bone turnover.
An elevated parathyroid hormone level does not exclude adynamic renal bone disease, but a low level (< 150 pg/mL) suggests a lowbone-turnover state. A level six times or more greater than the upper limit of normal is far more likely to be associated with high bone turnover.
Thus, in clinical practice, patients with stage 4 or 5 chronic kidney disease who have elevated bone-specific alkaline phosphatase or very high parathyroid hormone values do not have adynamic bone disease. Furthermore, once other causes of these aberrant biochemical abnormalities have been defined, then “high-bone-turnover osteoporosis” may be a consideration. Certainly, in my opinion, if bone turnover markers suggest low bone turnover, bone biopsy is necessary before starting an antiresorptive agent.35
Quantitative bone histomorphometry
Double tetracycline-labeled quantitative histomorphometry is still the only accepted way to measure turnover, mineralization, and volume in clinical practice.43–45 A committee of the American Society for Bone and Mineral Research has developed histomorphometric criteria for distinguishing among the different types of metabolic bone diseases (osteomalacia, adynamic bone disease, hyperparathyroid bone disease).12 These criteria can be used to distinguish among the various metabolic bone diseases that accompany stage 5 chronic kidney disease, including adynamic bone disease.43,46–48
For patients in stage 5 who have had a fragility fracture, adynamic bone disease should be excluded before the off-label use of an osteoporosis drug that reduces bone turnover, such as a bisphosphonate, calcitonin, estrogen, a selective estrogen receptor modulator, or denosumab (anti-RANK ligand antibody). While there is no evidence, for example, that starting a bisphosphonate in a patient who already has adynamic bone disease is detrimental to either bone strength or systemic vascular calcification (which may be linked to low bone turnover),49 it seems unreasonable to do so until solid prospective data clarify the harm or benefit.50 Preliminary experimental and clinical data suggest that bisphosphonates may even reduce progression of extraosseous calcification and inhibit the development of atherosclerosis.50
Hence, quantitative bone histomorphometry can discriminate among the various forms of renal osteodystrophy. If a distinct form of renal osteodystrophy is not present in a patient with stage 4 or 5 chronic kidney disease who has had a fracture and who, on biopsy, has a low trabecular bone volume, the patient probably has osteoporosis by exclusion.
TREATING OSTEOPOROSIS IN STAGE 1–3 CHRONIC KIDNEY DISEASE
As previously mentioned, patients who have fragility fractures in stage 1, 2, or 3 chronic kidney disease are more likely to have osteoporosis than renal osteodystrophy as the cause of their impaired bone strength. Although several articles have described a higher risk of fragility fractures in patients with age-related reduction in renal function than in agematched patients with normal renal function, the specific metabolic bone disease other than osteoporosis accounting for this bone fragility has not been defined.6
Hence, patients with osteoporosis who are in stage 1, 2, or 3 chronic kidney disease and do not have a known biochemical abnormality that might suggest some form of renal osteodystrophy can and should be considered for treatment with approved drugs that reduce the risk of fractures in postmenopausal, male, or glucocorticoid-induced osteoporosis.51–53 In clinical trials, these agents were shown to be effective in patients with serum creatinine concentrations as high as 2.0 mg/dL or a GFR as low as 30 mL/min, as estimated by the Cockcroft-Gault equation.
While all of the approved agents show evidence of reducing the risk of vertebral fractures, patients at higher risk of fractures or those who have already suffered a nonvertebral fracture are more often considered candidates for treatment with a bisphosphonate or teriparatide (Forteo), both of which have shown evidence of reducing the risk of all fractures.
Bisphosphonates in stage 1–3 chronic kidney disease
There is prospective evidence that patients with an age-related reduction in GFR down to 30 mL/min benefit from oral and intravenous bisphosphonates, since all of the clinical trials that led to the approval of bisphosphonates included patients with GFRs as low as this.54–57 Bisphosphonates seem to have an excellent safety profile as measured by renal adverse events in patients with a GFR of 30 mL/min or greater.52–59
From the intravenous bisphosphonate studies, it appears that ibandronate (Boniva) at the approved dose of 3 mg intravenously every 3 months and zoledronic acid (Reclast) 5 mg once a year given over 15 minutes are safe in patients with a GFR greater than 30 or 35 mL/min.
However, the safety of these drugs might not be the same in patients with preexisting renal parenchymal disease (eg, in diabetes) or in patients using other agents that could affect renal function (eg, nonsteroidal antiinflammatory drugs). Therefore, caution is still needed when deciding to use intravenous bisphosphonates in specific higher-risk renal subpopulations.
In the clinical trials of zoledronic acid, a substantial proportion of patients had diabetes, and no difference was seen in adverse renal effects between diabetic and nondiabetic patients. Also, GFRs declined equally between the treated and placebo groups over time and were no different at the end of 3 years.55 However, in patients in whom serum creatinine was measured 9 to 11 days after the infusion of zoledronic acid, there was a small but statistically significant transient increase in serum creatinine concentration (0.5–2.0 mg/dL above baseline) after the second annual infusion. 58 The serum creatinine concentrations returned to their baseline values in all of these patients before the next annual infusion.
It is important that infusions of zoledronic acid be given no faster than over 15 minutes. More rapid infusion has been associated with acute renal failure, suggesting that the tubular damage that mimics acute tubular necrosis is related to the maximal concentration and not to the area under the curve. I infuse zoledronic acid over 30 minutes in patients with normal renal function or in those with stage 1, 2, or 3 chronic kidney disease.
Teriparatide
Teriparatide’s approval trial did not require baseline measurements of GFR, but patients were enrolled only if their baseline serum creatinine concentrations were less than 2.0 mg/dL.60 In a post hoc analysis, a small subset of patients had GFRs as low as 30 mL/min as estimated by the Cockcroft-Gault equation. In these patients, teriparatide 20 or 40 μg/day had an anabolic effect as measured by increases in osteoblast activity markers and bone mineral density, similar to that seen in patients with higher estimated GFRs and without any adverse renal effects.61
There are no data on using teriparatide in stage 4 or 5 chronic kidney disease, and I emphasize that in all of the clinical trials of teriparatide, all patients, even those with estimated GFRs as low as 30 mL/min, had normal baseline serum intact parathyroid hormone levels. It is possible that the bone biologic response could differ between patients with chronic kidney disease who have an elevated as compared with a normal serum parathyroid hormone level. This issue should be investigated.
TREATING OSTEOPOROSIS IN STAGE 4 OR 5 CHRONIC KIDNEY DISEASE
Treatment decisions are more difficult in patients with stage 4 and especially stage 5 chronic kidney disease who have had fragility fractures. This is even the case when the clinician has determined to the best of his or her ability that the patient has osteoporosis rather than renal osteodystrophy.
There are no prospective data showing any of the approved drugs to be effective in treating osteoporosis in patients whose GFRs are lower than 30 mL/min. However, a post hoc analysis of pooled data from nine clinical trials62 found that risedronate (Actonel) 5 mg/day reduced the incidence of new vertebral fractures. Another post hoc analysis, from the Fracture Intervention Trial,63 found that alendronate (Fosamax) 5 mg/day for the first 2 years and 10 mg/day for the 3rd year reduced the incidence of all clinical fractures. In neither of these post hoc analyses did the drug affect the serum creatinine concentration. The patients—postmenopausal women—had GFRs as low as 15 mL/min as estimated by the Cockcroft-Gault equation. Similar post hoc data have been published on raloxifene (Evista).64
There are no data on the efficacy (reduction in fracture risk) or safety of any bisphosphonate in patients with GFRs lower than 15 mL/min (stage 5 chronic kidney disease). Nevertheless, the question often arises when fragility fractures occur in this population. Here, only opinion and controversy exist, and we fervently await good science and randomized prospective data.
How to manage renal bone disease after transplantation is a distinctly separate issue in which bisphosphonate use may be even more controversial than in end-stage renal disease.65,66
In my opinion, patients without fractures with stage 5 chronic kidney disease should not be given bisphosphonates or teriparatide offlabel. Treating only on the basis of low bone mineral density and other risk factors seems to be associated with greater risk than benefit.
In stage 5 patients suffering fragility fractures, a bisphosphonate may be considered, but only after renal osteodystrophy has been thoroughly ruled out, which most often requires a bone biopsy.43,67,68 In skilled hands, transiliac bone biopsy is a safe procedure with little morbidity.
If osteoporosis appears to be the cause of the fracture, and if one chooses to use a bisphosphonate and the patient gives his or her informed consent, then I would give half the usual dose and restrict the therapy to no more than 3 years. The reason for halving the dose is based on the known pharmacokinetics of bisphosphonates in people with normal renal function: 50% of a given dose goes to bone and 50% is excreted by the kidney. Furthermore, the dialyzability of bisphosphonates has not been well studied. Limiting the treatment to 3 years is based on the unknown but probably greater bone retention of bisphosphonates when excretion is impaired.
I must emphasize that these approaches are not based on any evidence of efficacy, but rather are considered in extreme cases of often-recurrent fragility fractures in which the fractures per se pose a great risk of morbidity and death. These approaches should be clearly discussed with the patient, undertaken by specialists knowledgeable in complex metabolic bone disease management, and initiated only after the skeletal fragility disorder is well characterized.
SUMMING UP
No consensus exists on the criteria for diagnosing osteoporosis in stage 4 or 5 chronic kidney disease.
In higher-risk patients in stage 1, 2, or 3 chronic kidney disease who have osteoporosis, it appears that any drug approved for osteoporosis can be used, eg, a bisphosphonate, teriparatide, or both.
Considerations for management are far more complex in stage 4 or 5 because of the increased prevalence of other metabolic bone diseases and renal osteodystrophy, and because the World Health Organization criteria cannot be used to diagnose osteoporosis. In stage 5, the differential diagnosis requires careful analysis of a broad range of biochemical markers of bone turnover and, at times, quantitative bone histomorphometry, especially if one is considering using a bisphosphonate. It is unknown if bisphosphonates, by reducing bone turnover in a preexisting low-bone-turnover state, would help or harm bone or would lead to less or more cardiovascular disease. These questions must be addressed by better science and prospective data.
In the future, newer noninvasive radiologic tools to measure microstructure and mineralization of bone promise to help us better understand osteoporosis and renal osteodystrophy in a noninvasive manner.
In clinical practice, at the current time and with current limited knowledge, treatment of osteoporosis in stage 4 or 5 chronic kidney disease is opinion-based. Nevertheless, in very specific clinical cases of severe fragility fractures that, by themselves, may cause disability and death, bisphosphonates should be considered by experts in bone metabolism and, as with any off-label application, after careful informed discussions with the patient.
- Melton LJ. Epidemiology worldwide. Endocrinol Metab Clin North Am 2003; 32:1–13.
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J Bone Miner Res 2007; 22:465–475.
- Barrett-Connor E, Siris ES, Wehren LE, et al. Osteoporosis and fracture risk in women of different ethnic groups. J Bone Miner Res 2005; 20:185–194.
- Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. Report of a WHO Study Group. World Health Organ Tech Rep Ser 1994; 843:1–129.
- Miller PD, Bonnick SL. Clinical application of bone densitometry. In:Favus MJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 4th ed. American Society for Bone and Mineral Research. Philadelphia, PA: Lippincott Williams & Wilkins; 1999.
- Nickolas TL, Leonard MB, Shane E. Chronic kidney disease and bone fracture: a growing concern. Kidney Int 2008; 74:721–731.
- Gal-Moscovici A, Sprague SM. Osteoporosis and chronic kidney disease. Semin Dial 2007; 20:423–430.
- National Kidney Foundation. Clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(suppl 3):S1–S201.
- Coresh J, Astor BC, Greene T, Eknoyan G, Levey AS. Prevalence of chronic kidney disease and decreased kidney function in the adult US population: Third National Health and Nutrition Examination Survey. Am J Kidney Dis 2003; 41:1–12.
- Bennett WM. Reporting eGFR. Clin J Am Soc Nephrol 2008; 3:1561–1562.
- Glassock RJ, Winearls C. Screening for CKD with eGFR: doubts and dangers. Clin J Am Soc Nephrol 2008; 3:1563–1568.
- Parfitt AM, Drezner M, Glorieux F, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 1987; 2:595–610.
- Andress DL, Sherrard DJ. The osteodystrophy of chronic renal failure. In:Schrier RW, editor: Diseases of the Kidney and Urinary Tract. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:2431–2453.
- Moe S, Drueke T, Cunningham J, et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2006; 69:1945–1953.
- NIH Consensus Development Panel Osteoporosis Prevention Diagnosis and Therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA 2001; 285:785–795.
- Baim S, Binkley N, Bilezikian JP, et al. Official positions of the International Society for Clinical Densitometry and executive summary of the 2007 ISCD Position Development Conference. J Clin Densitom 2008; 11:75–91.
- Siris E, Miller P, Barrett-Connor E, et al. Identification and fracture outcomes of undiagnosed low bone mineral density in postmenopausal women: results from the National Osteoporosis Risk Assessment (NORA). JAMA 2001; 286:2815–2822.
- Schuit SCE, Oei HH, Witteman JC, et al. Fracture incidence and association with bone mineral density in elderly men and women: the Rotterdam Study. Bone 2004; 34:195–202.
- Bouxsein ML. Non-invasive measurements of bone strength: promise and peril. J Musculoskelet Neuronal Interact 2004; 4:404–405.
- Seeman E. Bone quality: the material and structural basis of bone strength. J Bone Miner Metab 2008; 26:1–8.
- Alem AM, Sherrard DJ, Gillen DL, et al. Increased risk of hip fracture among patients with end-stage renal disease. Kidney Int 2000; 58:396–399.
- Ball AM, Gillen DL, Sherrard D, et al. Risk of hip fracture among dialysis and renal transplant recipients. JAMA 2002; 288:3014–3018.
- Jadoul M, Albert JM, Akiba T, et al. Incidence and risk factors for hip or other bone fractures among hemodialysis patients in the Dialysis Outcomes and Practice Patterns Study. Kidney Int 2006; 70:1358–1366.
- Stehman-Breen CO, Sherrard DJ, Alem AM, et al. Risk factors for hip fracture among patients with end-stage renal disease. Kidney Int 2000; 58:2200–2205.
- Wehrli FW, Leonard MB, Saha PK, Gomberg BR. Quantitative highresolution magnetic resonance imaging reveals structural implications of renal osteodystrophy on trabecular and cortical bone. J Magn Reson Imaging 2004; 20:83–89.
- Genant HK, Lang TF, Engelke K, et al. Advances in the noninvasive assessment of bone density, quality, and structure. Calcif Tissue Int 1996; 59(suppl 1):S10–S15.
- Roschger P, Paschalis EP, Fratzl P, Klaushofer K. Bone mineralization density distribution in health and disease. Bone 2008; 42:456–466.
- Miller PD. Bone density and markers of bone turnover in predicting fracture risk and how changes in these measures predict fracture risk reduction. Curr Osteoporos Rep 2005; 3:103–110.
- Miller PD, Hochberg MC, Wehren LE, Ross PD, Wasnich RD. How useful are measures of BMD and bone turnover? Curr Med Res Opin 2005; 21:545–554.
- Chavassieux PM, Delmas PD. Bone remodeling: biochemical markers or bone biopsy? J Bone Miner Res 2006; 21:178–179.
- Garnero P. Biomarkers for osteoporosis management: utility in diagnosis, fracture risk prediction and therapy monitoring. Mol Diagn Ther 2008; 12:157–170.
- Hochberg M, Greenspan S, Wasnich R, Miller P, Thompson D, Ross P. Changes in bone density and turnover explain the reductions in incidence of nonvertebral fractures that occur during treatment with antiresorptive agents. J Clin Endocrinol Metab 2002; 87:1586–1592.
- Bouxsein ML, Delmas PD. Considerations for development of surrogate endpoints for antifracture efficacy of new treatments in osteoporosis: a perspective. J Bone Miner Res 2008; 23:1155–1167.
- Chen P, Satterwhite JH, Licatta AA, et al. Early changes in biochemical markers of bone formation predict BMD response to teriparatide in postmenopausal women with osteoporosis. J Bone Miner Res 2005; 20:962–970.
- Miller PD, Delmas PD, Lindsay R, et al. Early responsiveness of women with osteoporosis to teriparatide after therapy with alendronate or risedronate. J Clin Endocrinol Metab 2008; 93:3785–3793.
- Baim S, Miller PD. Assessing the clinical utility of serum CTX in postmenopausal osteoporosis and its use in predicting risk of osteonecrosis of the jaw. J Bone Miner Res 2009; 24:561–574.
- Miller PD, Lerma EV. Renal bone diseases. In:Kleerekoper M, Siris E, McClung M, editors. The Bone and Mineral Manual—A Practical Guide. 2nd ed. Burlington, MA: Elsevier Academic Press; 2005:127–138.
- Miller PD, Shane E. Management of transplantation renal bone disease: Interplay of bone mineral density and decisions regarding bisphosphonate use. In:Weir MR, editor. Medical Management of Kidney Transplantation. Philadelphia, PA: Lippincott Williams & Wilkins; 2004:359–375.
- Levin A, Bakris GL, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney int 2007; 71:31–38.
- Vassalotti JA, Uribarri J, Chen SC, et al. Trends in mineral metabolism: Kidney Early Evaluation Program (KEEP) and the National Health and Nutrition Examination Survey (NHANES) 1999–2004. Am J Kidney Dis 2008; 51(suppl 2):S56–S68.
- Meier C, Seibel MJ, Kraenzlin ME. Use of bone turnover markers in the real world: are we there yet? J Bone Miner Res 2009; 24:386–388.
- Lehmann G, Ott U, Kaemmerer D, Schuetze J, Wolf G. Bone histomorphometry and biochemical markers of bone turnover in patients with chronic kidney disease stages 3–5. Clin Nephrol 2008; 70:296–305.
- Miller PD. The role of bone biopsy in patients with chronic renal failure. Clin J Am Soc Nephrol 2008; 3(suppl 3):S140–S150.
- Frost HM. Tetracycline-based histological analysis of bone remodeling. Calcif Tissue Res 1969; 3:211–237.
- Hitt O, Jaworski ZF, Shimizu AG, Frost HM. Tissue-level bone formation rates in chronic renal failure, measured by means of tetracycline bone labeling. Can J Physiol Pharmacol 1970; 48:824–828.
- Coen G. Adynamic bone disease: an update and overview. J Nephrol 2005; 18:117–122.
- Parfitt AM. Renal bone disease: a new conceptual framework for the interpretation of bone histomorphometry. Curr Opin Nephrol Hypertens 2003; 12:387–403.
- Brandenburg VM, Floege J. Adynamic bone diseaseùbone and beyond. NDT Plus 2008; 3:135–147. doi:10.1093/ndtplus/sfn040.
- Hruska KA, Saab G, Mathew S, Lund R. Renal osteodystrophy, phosphate homeostasis, and vascular calcification. Semin Dial 2007; 20:309–315.
- Toussaint ND, Elder GJ, Kerr PG. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc Nephrol 2009; 4:221–233.
- Miller PD. Is there a role for bisphosphonates in chronic kidney disease? Semin Dial 2007; 20:186–190.
- Miller PD. Bisphosphonates: pharmacology and use in the treatment of osteoporosis. In:Marcus R, Feldman D, Nelson DA, Rosen CJ, editors. Osteoporosis. 3rd ed. Boston, MA: Elsevier Academic Press; 2008:1725–1736.
- Russell RG, Watts NB, Ebetino FH, Rogers MJ. Mechanisms of action of bisphosphonates: similarities and differences and their potential influence on clinical efficacy. Osteoporosis Int 2008; 19:733–759.
- Eisman JA, Civitelli R, Adami S, et al. Efficacy and tolerability of intravenous ibandronate injections in postmenopausal osteoporosis: 2-year results from the DIVA study. J Rheumatol 2008; 35:488–497.
- Black DM, Delmas PD, Eastell RR, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med 2007; 356:1809–1822.
- Lewiecki EM, Miller PD. Renal safety of intravenous bisphosphonates in the treatment of osteoporosis. Expert Opin Drug Saf 2007; 6:663–672.
- Miller PD. Anti-resorptives in the management of osteoporosis. Best Pract Res Clin Endocrinol Metab 2008; 22:849–868.
- Perazella MA, Markowitz GS. Bisphosphonate nephrotoxicity. Kidney Int 2008; 74:1385–1393.
- Boonen S, Sellmeyer DE, Lippuner K, et al. Renal safety of annual zoledronic acid infusions in osteoporotic postmenopausal women. Kidney Int 2008; 74:641–648.
- Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 2001; 344:1434–1441.
- Miller PD, Schwartz EN, Chen P, Misurski DA, Krege JH. Teriparatide in postmenopausal women with osteoporosis and mild or moderate renal impairment. Osteoporosis Int 2007; 18:59–68.
- Miller PD, Roux C, Boonen S, Barton I, Dunlap L, Burgio D. Safety and efficacy of risedronate in patients with age-related reduced renal function as estimated by the Cockcroft and Gault method: a pooled analysis of nine clinical trials. J Bone Miner Res 2005; 20:2105–2115.
- Jamal SA, Bauer DC, Ensrud KE, et al. Alendronate treatment in women with normal to severely impaired renal function: an analysis of the fracture intervention trial. J Bone Miner Res 2007; 22:503–508.
- Ishani A, Blackwell T, Jamal SA, Cummings SR, Ensrud KE; MORE Investigators. The effect of raloxifene treatment in postmenopausal women with CKD. J Am Soc Nephrol 2008; 19:1430–1438.
- Coco M, Glicklich D, Faugere MC, et al. Prevention of bone loss in renal transplant recipients: a prospective, randomized trial of intravenous pamidronate. J Am Soc Nephrol 2003; 14:2669–2676.
- Palmer SC, McGregor DO, Strippoli GF. Interventions for preventing bone disease in kidney transplant recipients. Cochrane Database Syst Rev 2007;CD005015.
- Ferreira MA. Diagnosis of renal osteodystrophy: when and how to use biochemical markers and non-invasive methods; when bone biopsy is needed. Nephrol Dial Transplant 2000; 15(suppl 5):8–14.
- Trueba D, Sawaya BP, Mawad H, Malluche HH. Bone biopsy: indications, techniques, and complications. Semin Dial 2003; 16:341–345.
- Melton LJ. Epidemiology worldwide. Endocrinol Metab Clin North Am 2003; 32:1–13.
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J Bone Miner Res 2007; 22:465–475.
- Barrett-Connor E, Siris ES, Wehren LE, et al. Osteoporosis and fracture risk in women of different ethnic groups. J Bone Miner Res 2005; 20:185–194.
- Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. Report of a WHO Study Group. World Health Organ Tech Rep Ser 1994; 843:1–129.
- Miller PD, Bonnick SL. Clinical application of bone densitometry. In:Favus MJ, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 4th ed. American Society for Bone and Mineral Research. Philadelphia, PA: Lippincott Williams & Wilkins; 1999.
- Nickolas TL, Leonard MB, Shane E. Chronic kidney disease and bone fracture: a growing concern. Kidney Int 2008; 74:721–731.
- Gal-Moscovici A, Sprague SM. Osteoporosis and chronic kidney disease. Semin Dial 2007; 20:423–430.
- National Kidney Foundation. Clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(suppl 3):S1–S201.
- Coresh J, Astor BC, Greene T, Eknoyan G, Levey AS. Prevalence of chronic kidney disease and decreased kidney function in the adult US population: Third National Health and Nutrition Examination Survey. Am J Kidney Dis 2003; 41:1–12.
- Bennett WM. Reporting eGFR. Clin J Am Soc Nephrol 2008; 3:1561–1562.
- Glassock RJ, Winearls C. Screening for CKD with eGFR: doubts and dangers. Clin J Am Soc Nephrol 2008; 3:1563–1568.
- Parfitt AM, Drezner M, Glorieux F, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 1987; 2:595–610.
- Andress DL, Sherrard DJ. The osteodystrophy of chronic renal failure. In:Schrier RW, editor: Diseases of the Kidney and Urinary Tract. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:2431–2453.
- Moe S, Drueke T, Cunningham J, et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2006; 69:1945–1953.
- NIH Consensus Development Panel Osteoporosis Prevention Diagnosis and Therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA 2001; 285:785–795.
- Baim S, Binkley N, Bilezikian JP, et al. Official positions of the International Society for Clinical Densitometry and executive summary of the 2007 ISCD Position Development Conference. J Clin Densitom 2008; 11:75–91.
- Siris E, Miller P, Barrett-Connor E, et al. Identification and fracture outcomes of undiagnosed low bone mineral density in postmenopausal women: results from the National Osteoporosis Risk Assessment (NORA). JAMA 2001; 286:2815–2822.
- Schuit SCE, Oei HH, Witteman JC, et al. Fracture incidence and association with bone mineral density in elderly men and women: the Rotterdam Study. Bone 2004; 34:195–202.
- Bouxsein ML. Non-invasive measurements of bone strength: promise and peril. J Musculoskelet Neuronal Interact 2004; 4:404–405.
- Seeman E. Bone quality: the material and structural basis of bone strength. J Bone Miner Metab 2008; 26:1–8.
- Alem AM, Sherrard DJ, Gillen DL, et al. Increased risk of hip fracture among patients with end-stage renal disease. Kidney Int 2000; 58:396–399.
- Ball AM, Gillen DL, Sherrard D, et al. Risk of hip fracture among dialysis and renal transplant recipients. JAMA 2002; 288:3014–3018.
- Jadoul M, Albert JM, Akiba T, et al. Incidence and risk factors for hip or other bone fractures among hemodialysis patients in the Dialysis Outcomes and Practice Patterns Study. Kidney Int 2006; 70:1358–1366.
- Stehman-Breen CO, Sherrard DJ, Alem AM, et al. Risk factors for hip fracture among patients with end-stage renal disease. Kidney Int 2000; 58:2200–2205.
- Wehrli FW, Leonard MB, Saha PK, Gomberg BR. Quantitative highresolution magnetic resonance imaging reveals structural implications of renal osteodystrophy on trabecular and cortical bone. J Magn Reson Imaging 2004; 20:83–89.
- Genant HK, Lang TF, Engelke K, et al. Advances in the noninvasive assessment of bone density, quality, and structure. Calcif Tissue Int 1996; 59(suppl 1):S10–S15.
- Roschger P, Paschalis EP, Fratzl P, Klaushofer K. Bone mineralization density distribution in health and disease. Bone 2008; 42:456–466.
- Miller PD. Bone density and markers of bone turnover in predicting fracture risk and how changes in these measures predict fracture risk reduction. Curr Osteoporos Rep 2005; 3:103–110.
- Miller PD, Hochberg MC, Wehren LE, Ross PD, Wasnich RD. How useful are measures of BMD and bone turnover? Curr Med Res Opin 2005; 21:545–554.
- Chavassieux PM, Delmas PD. Bone remodeling: biochemical markers or bone biopsy? J Bone Miner Res 2006; 21:178–179.
- Garnero P. Biomarkers for osteoporosis management: utility in diagnosis, fracture risk prediction and therapy monitoring. Mol Diagn Ther 2008; 12:157–170.
- Hochberg M, Greenspan S, Wasnich R, Miller P, Thompson D, Ross P. Changes in bone density and turnover explain the reductions in incidence of nonvertebral fractures that occur during treatment with antiresorptive agents. J Clin Endocrinol Metab 2002; 87:1586–1592.
- Bouxsein ML, Delmas PD. Considerations for development of surrogate endpoints for antifracture efficacy of new treatments in osteoporosis: a perspective. J Bone Miner Res 2008; 23:1155–1167.
- Chen P, Satterwhite JH, Licatta AA, et al. Early changes in biochemical markers of bone formation predict BMD response to teriparatide in postmenopausal women with osteoporosis. J Bone Miner Res 2005; 20:962–970.
- Miller PD, Delmas PD, Lindsay R, et al. Early responsiveness of women with osteoporosis to teriparatide after therapy with alendronate or risedronate. J Clin Endocrinol Metab 2008; 93:3785–3793.
- Baim S, Miller PD. Assessing the clinical utility of serum CTX in postmenopausal osteoporosis and its use in predicting risk of osteonecrosis of the jaw. J Bone Miner Res 2009; 24:561–574.
- Miller PD, Lerma EV. Renal bone diseases. In:Kleerekoper M, Siris E, McClung M, editors. The Bone and Mineral Manual—A Practical Guide. 2nd ed. Burlington, MA: Elsevier Academic Press; 2005:127–138.
- Miller PD, Shane E. Management of transplantation renal bone disease: Interplay of bone mineral density and decisions regarding bisphosphonate use. In:Weir MR, editor. Medical Management of Kidney Transplantation. Philadelphia, PA: Lippincott Williams & Wilkins; 2004:359–375.
- Levin A, Bakris GL, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney int 2007; 71:31–38.
- Vassalotti JA, Uribarri J, Chen SC, et al. Trends in mineral metabolism: Kidney Early Evaluation Program (KEEP) and the National Health and Nutrition Examination Survey (NHANES) 1999–2004. Am J Kidney Dis 2008; 51(suppl 2):S56–S68.
- Meier C, Seibel MJ, Kraenzlin ME. Use of bone turnover markers in the real world: are we there yet? J Bone Miner Res 2009; 24:386–388.
- Lehmann G, Ott U, Kaemmerer D, Schuetze J, Wolf G. Bone histomorphometry and biochemical markers of bone turnover in patients with chronic kidney disease stages 3–5. Clin Nephrol 2008; 70:296–305.
- Miller PD. The role of bone biopsy in patients with chronic renal failure. Clin J Am Soc Nephrol 2008; 3(suppl 3):S140–S150.
- Frost HM. Tetracycline-based histological analysis of bone remodeling. Calcif Tissue Res 1969; 3:211–237.
- Hitt O, Jaworski ZF, Shimizu AG, Frost HM. Tissue-level bone formation rates in chronic renal failure, measured by means of tetracycline bone labeling. Can J Physiol Pharmacol 1970; 48:824–828.
- Coen G. Adynamic bone disease: an update and overview. J Nephrol 2005; 18:117–122.
- Parfitt AM. Renal bone disease: a new conceptual framework for the interpretation of bone histomorphometry. Curr Opin Nephrol Hypertens 2003; 12:387–403.
- Brandenburg VM, Floege J. Adynamic bone diseaseùbone and beyond. NDT Plus 2008; 3:135–147. doi:10.1093/ndtplus/sfn040.
- Hruska KA, Saab G, Mathew S, Lund R. Renal osteodystrophy, phosphate homeostasis, and vascular calcification. Semin Dial 2007; 20:309–315.
- Toussaint ND, Elder GJ, Kerr PG. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc Nephrol 2009; 4:221–233.
- Miller PD. Is there a role for bisphosphonates in chronic kidney disease? Semin Dial 2007; 20:186–190.
- Miller PD. Bisphosphonates: pharmacology and use in the treatment of osteoporosis. In:Marcus R, Feldman D, Nelson DA, Rosen CJ, editors. Osteoporosis. 3rd ed. Boston, MA: Elsevier Academic Press; 2008:1725–1736.
- Russell RG, Watts NB, Ebetino FH, Rogers MJ. Mechanisms of action of bisphosphonates: similarities and differences and their potential influence on clinical efficacy. Osteoporosis Int 2008; 19:733–759.
- Eisman JA, Civitelli R, Adami S, et al. Efficacy and tolerability of intravenous ibandronate injections in postmenopausal osteoporosis: 2-year results from the DIVA study. J Rheumatol 2008; 35:488–497.
- Black DM, Delmas PD, Eastell RR, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med 2007; 356:1809–1822.
- Lewiecki EM, Miller PD. Renal safety of intravenous bisphosphonates in the treatment of osteoporosis. Expert Opin Drug Saf 2007; 6:663–672.
- Miller PD. Anti-resorptives in the management of osteoporosis. Best Pract Res Clin Endocrinol Metab 2008; 22:849–868.
- Perazella MA, Markowitz GS. Bisphosphonate nephrotoxicity. Kidney Int 2008; 74:1385–1393.
- Boonen S, Sellmeyer DE, Lippuner K, et al. Renal safety of annual zoledronic acid infusions in osteoporotic postmenopausal women. Kidney Int 2008; 74:641–648.
- Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 2001; 344:1434–1441.
- Miller PD, Schwartz EN, Chen P, Misurski DA, Krege JH. Teriparatide in postmenopausal women with osteoporosis and mild or moderate renal impairment. Osteoporosis Int 2007; 18:59–68.
- Miller PD, Roux C, Boonen S, Barton I, Dunlap L, Burgio D. Safety and efficacy of risedronate in patients with age-related reduced renal function as estimated by the Cockcroft and Gault method: a pooled analysis of nine clinical trials. J Bone Miner Res 2005; 20:2105–2115.
- Jamal SA, Bauer DC, Ensrud KE, et al. Alendronate treatment in women with normal to severely impaired renal function: an analysis of the fracture intervention trial. J Bone Miner Res 2007; 22:503–508.
- Ishani A, Blackwell T, Jamal SA, Cummings SR, Ensrud KE; MORE Investigators. The effect of raloxifene treatment in postmenopausal women with CKD. J Am Soc Nephrol 2008; 19:1430–1438.
- Coco M, Glicklich D, Faugere MC, et al. Prevention of bone loss in renal transplant recipients: a prospective, randomized trial of intravenous pamidronate. J Am Soc Nephrol 2003; 14:2669–2676.
- Palmer SC, McGregor DO, Strippoli GF. Interventions for preventing bone disease in kidney transplant recipients. Cochrane Database Syst Rev 2007;CD005015.
- Ferreira MA. Diagnosis of renal osteodystrophy: when and how to use biochemical markers and non-invasive methods; when bone biopsy is needed. Nephrol Dial Transplant 2000; 15(suppl 5):8–14.
- Trueba D, Sawaya BP, Mawad H, Malluche HH. Bone biopsy: indications, techniques, and complications. Semin Dial 2003; 16:341–345.
KEY POINTS
- If the patient’s glomerular filtration rate (GFR) is at least 30 mL/min/1.73 m2 and if no biochemical test results suggest renal osteodystrophy, osteoporosis can be diagnosed if the T score is less than −2.5 or if the patient has had a fragility fracture. These criteria can also probably be applied, though with less certainty, if the patient’s GFR is as low as 15.
- If the patient’s GFR is less than 15 or if he or she is on dialysis, biochemical profiling often cannot distinguish among the heterogeneous forms of renal bone disease. In some cases of severe chronic kidney disease with fractures, bone biopsy is needed to rule out renal osteodystrophy and to diagnose osteoporosis by exclusion.
- In the author’s opinion, in patients with severe chronic kidney disease and fractures who have “osteoporosis” by exclusion, off-label use of bisphosphonates is an option, but only after very careful consideration.