User login
Systemic Therapy in Metastatic Melanoma
Melanoma is the most aggressive form of skin cancer, contributing to about 76,000 new cases and more than 9,000 deaths in 2014.1 Depending on the stage of the disease, 5-year melanoma survival can range from 15% to 97%. Patients with local and distant metastases have a 5-year survival of about 60% and 15%, respectively.2
The incidence of melanoma is rising, partly because of the increasing number of skin biopsies being performed.3 If melanoma is diagnosed early, surgical excision is the treatment of choice. In patients with oligometastatic disease (cancer that has spread, but only to 1 or a small number of sites), complete surgical excision of the metastases may provide prolonged overall survival (OS) and delay the need to use systemic therapy.4
Recently, many new drug therapies have shown promising results in clinical trials, which may improve the prognosis of metastatic disease. This article reviews currently available systemic treatment options for the management of metastatic melanoma, the role of cytotoxic chemotherapy and interleukin-2 (IL-2), and the latest therapies available, including immune checkpoint inhibitors.
Cytotoxic Chemotherapy and Interleukin-2
Cytotoxic chemotherapy does not have an established role in the initial treatment of metastatic melanoma. Currently, cytotoxic chemotherapy is used in patients who have not responded to immunotherapy or molecular targeted therapy. The most commonly used drugs include dacarbazine and its prodrug, temozolomide. Several studies have failed to demonstrate a survival benefit using a single-agent chemotherapy with either dacarbazine or temozolomide.5,6
Other agents used in metastatic melanoma include nitrosoureas (fotemustine), platinum compounds (cisplatin, carboplatin), vinca alkaloids (vincristine),
and taxanes (paclitaxel). None of these agents provide a survival benefit, but an objective response may be seen in a minority of cases. Combination chemotherapy regimens have not shown an advantage over singleagent dacarbazine or temozolomide.7,8
High-dose IL-2 has been used in cases of metastatic melanoma with good performance status (PS) and organ function. Studies have shown a complete response rate of 3% to 7% and a prolonged disease-free survival in a minority of patients.9-11 The use of highdose IL-2, however, is limited by the high incidence of adverse effects (AEs), which include bacterial sepsis, pulmonary edema, arrhythmias, fever, and on some occasions, death due to complications.10 The use of IL-2 requires admission of the patient to a specialized unit for AE monitoring and management. Because of its ability to “cure” a minority of patients, a role still exists for IL-2 therapy in the treatment of younger, healthy patients with no evidence of organ dysfunction at baseline.
Immune Checkpoint Inhibitors
Checkpoint inhibitors are a class of drugs that unmask the immune system to fight against cancer cells. This class of drugs has shown significant activity and survival advantage in recent phase 2 and 3 trials. The class includes the anticytotoxic T-lymphocyte antigen 4 (CTLA-4) antibody ipilimumab and monoclonal antibodies targeting the programmed death 1 protein (PD-1) or its ligand (PD-L1).
Anti-CTLA-4 Antibodies: Ipilimumab
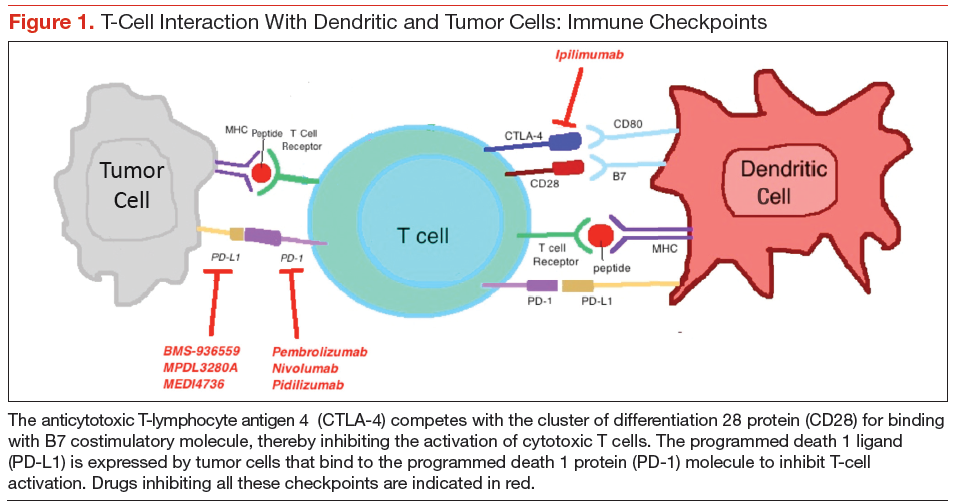
Cytotoxic T-lymphocyte antigen 4 is the antigen responsible for inhibition of cytotoxic T-cell-mediated immunity against foreign antigens presented by the antigen presenting cells (APCs). The APCs cause activation of the T cells when peptide fragments of intracellular proteins are presented in combination with mixed histocompatibility complex molecules. This step requires interaction of a costimulatory molecule (B7) on the APCs with a cluster of differentiation 28 protein (CD28) receptor located on T cells. CTLA-4 competes with CD28 to bind with the B7 molecule, thereby inhibiting the activation of the cytotoxic T cells (Figure 1). This pathway is thought to help with development of tolerance to host tissue antigens. Ipilimumab is a human monoclonal antibody that inhibits this CTLA-4 molecule and facilitates T-cell mediated antitumor activity.12 By blocking the CTLA-4 molecule, ipilimumab also mediates its autoimmune AEs on the host tissues.
Hodi and colleagues conducted a phase 3 trial of ipilimumab, including 676 patients who progressed after prior treatment for stage III or IV melanoma, and found that median OS was significantly better in the ipilimumab groups: 10 months in the ipilimumab plus gp100 peptide vaccine group vs 6.4 months in the gp100 vaccine alone group; 10.1 months in the ipilimumab alone group vs 6.4 months in the gp100 vaccine alone group.13 In another phase 3 trial comparing ipilimumab plus dacarbazine to dacarbazine alone, the ipilimumab group had a significantly improved OS (11.2 months vs 9.1 months).1 Survival rates with ipilimumab were prolonged for up to 3 years compared with the dacarbazine plus placebo group. However, the combination was associated with increased incidence of hepatotoxicity, thereby limiting its use.
A long-term survival analysis of 10 prospective and 2 retrospective studies of ipilimumab showed a median OS of 11.4 months and a long-term survival that began at 3 years with a plateau at 10 years of 21%, which was independent of prior therapy or ipilimumab dose.14 The immune-related AEs of ipilimumab are secondary to its activity against the host antigens and include dermatitis, enterocolitis, hepatitis, and endocrinopathies.15
A recent phase 2 trial studied the combination of ipilimumab with granulocyte-macrophage colonystimulating factor in 245 patients with stage III and IV melanoma. Median OS after 13 months was significantly higher with the combination compared with ipilimumab alone. The 1-year survival rate was 69% with
the combination and 53% with ipilimumab alone. There was no difference in the overall response rate (ORR) or progression-free survival (PFS) between the 2 groups. However, the AEs were significantly reduced with the combination (45% vs 58%).16 The dose of ipilimumab used in the trial was higher than the approved dose, making it difficult to apply the results in practice without further studies on the combination.
Anti-PD-1 Antibodies
Programmed death 1 ligands (PD-L1 and PD-L2) are expressed by tumor or stromal cells to inhibit the T-cell mediated antitumor activity. These ligands bind to the PD-1 protein on the surface of activated T cells to mediate their immunosuppressive effects. Interruption of this interaction by either anti-PD-1 antibodies or anti-PD-L1 antibodies facilitates tumor cell killing by activated T cells.17
Pembrozilumab and nivolumab are the 2 anti-PD-1 monoclonal antibodies that have been approved for treatment of metastatic melanoma. In a phase 1 trial
of pembrolizumab, 411 patients with advanced melanoma (consisting of both ipilimumab-naïve [IPI-N] and ipilimumab-treated [IPI-T] patients), ORR was 40% in IPI-N and 28% in IPI-T patients with a 1-year OS of 71% in all patients. Median PFS was 24 weeks in IPI-N and 23 weeks in IPI-T pts.18 There was no difference in outcomes and safety profiles across the various dosing regimens.18,19 Of note, pembrolizumab had antitumor activity irrespective of the PS, lactate dehydrogenase levels, BRAF (B-Raf proto-oncogene, serine/threonine kinase) gene mutation, metastatic stage, and number and type of prior therapy. In a subgroup analysis, 173 patients who had progression after treatment with ipilimumab were randomly assigned to pembrolizumab 2 mg/kg every 3 weeks (q3w) or 10 mg/kg q3w dosing regimens. Both groups had no significant difference in the ORR (26% in both) and safety profiles.20
In the 2012 KEYNOTE-002 clinical trial, a randomized phase 2 trial involving 540 patients with ipilimumab-refractory advanced melanoma, patients were randomized 1:1:1 to pembrolizumab 2 mg/kg or 10 mg/kg q3w or investigator-choice chemotherapy (control arm consisting of carboplatin plus paclitaxel, carboplatin, paclitaxel, dacarbazine, or temozolomide). The 6-month PFS was significantly improved with pembrolizumab (34% and 38% for pembrolizumab 2 mg/kg and 10 mg/kg, respectively) compared with 16% with chemotherapy. The ORR was significantly better with pembrolizumab (21% at 2 mg/kg, 25% at 10 mg/kg) compared with the control arm (4%).21 These findings led to the approval of pembrolizumab by the FDA for treatment of patients with advanced melanoma who have progressed on ipilimumab. Pembrolizumab is generally well tolerated. The most common AEs include fatigue, pruritus, and rash.
Nivolumab was studied in a recent phase 1 trial in which 107 patients with previously treated advanced melanoma were treated with escalated doses every
2 weeks.22 The 2-year and 3-year OS rates were 48% and 41%, respectively. Objective responses were seen in 32% of the patients. The median response duration was 23 months.23
The first phase 3 trial was conducted in 418 patients with previously untreated metastatic melanoma BRAF mutation. Patients were randomized to receive either nivolumab or dacarbazine. The PFS and OS were significantly better with nivolumab compared with dacarbazine (PFS 5.1 months vs 2.2 months; OS 73% vs 42% at 1 year).24 The AE profile of nivolumab is similar to pembrolizumab and includes lung, skin, endocrine, renal, and gastrointestinal tract toxicities.
Preliminary results of another phase 3 trial were presented at the European Society of Medical Oncology 2014 meeting. Patients with previously treated metastatic melanoma (ipilimumab or BRAF inhibitor) were randomized in a 2:1 ratio to receive either nivolumab or investigators’ choice chemotherapy (dacarbazine or carboplatin plus paclitaxel). The ORR was significantly better with nivolumab (32% vs 11%), and 95% of patients were still responding after 6 months. The nivolumab group showed a complete remission in 3% of the patients with 34% of the responses lasting ≥ 6 months.25 This led to the recent approval of nivolumab for patients with metastatic melanoma with a BRAF mutation who have advanced on ipilimumab. In the phase 3 NCT01844505 trial patients are being randomized to receive ipilimumab, nivolumab, or both.
A newer PD-1 inhibitor, pidilizumab, was studied in a phase 2 trial that included 103 patients with metastatic melanoma, 51% of whom had received therapy with ipilimumab. The ORR in the study group was relatively lower (6%), but the OS at 1 year was 64.5%.26 Further studies are underway to evaluate the role of this drug in metastatic melanoma.
The response with both nivolumab and pembrolizumab is durable as well as sustained, even after discontinuation of therapy. None of the deaths in the aforementioned studies were atributed to drug-related toxicities. As evidenced by current data, these 2 drugs hold a great promise for the management of patients who progress after therapy with anti-CTLA-4 antibodies.
Anti-PD-L1 Antibodies
The anti-PD-L1 monoclonal antibodies work in a similar way to the PD-1 inhibitors and block the interaction between the PD-1 and its ligand, PD-L1. This causes sustained activation of cytotoxic T cells and facilitates their antitumor activity. Two of PD-L1 inhibitors have shown clinical activity against metastatic melanoma.
BMS-936559, the first PD-L1 antibody, is being studied in a phase 1 trial that includes 55 patients with advanced melanoma along with 152 patients with other solid malignancies. Three patients achieved a complete response, and 5 patients had an objective response lasting 1 year. The ORR for melanoma was 17%, with disease stabilization of ≥ 24 weeks in 27% of the patients.27 Common AEs included infusion reactions, diarrhea, fatigue, rash, hypothyroidism, and hepatitis.
The second PD-L1 antibody, MPDL3280A, was studied in a phase 1 trial of 45 patients with metastatic melanoma. An ORR of 29% was observed, along with a 24-week PFS of 43%.28 Commonly noted AEs included hyperglycemia and elevated liver aminotransferases.
A newer PD-L1 inhibitor, MEDI4736, is being studied for advanced malignancies in 8 patients with melanoma. In preliminary analysis, MEDI4736 demonstrated a partial response in 1 out of 8 melanoma patients with a disease control rate of 46%.29 Although the PD-L1 inhibitors seem promising, more information will help discern their role in the management of metastatic melanoma.
Combined Anti-CTLA-4 Plus Anti-PD-1 Antibody
The combination of ipilimumab and the PD-1 inhibitor nivolumab was tested in a phase 1 trial in which both drugs were used concurrently as well as sequentially in metastatic melanoma.30 The 1- and 2-year OS in patients who were treated concurrently was 82% and 75%, respectively. Complete remission was seen in 17% of the patients, and the responses were seen irrespective of the BRAF mutation status. The responses were durable, and about 64% of the objective responses remain in remission at last follow-up.31 Grade 3 to grade 4 AEs were noted in 53% of the patients, with 11 patients requiring discontinuation of the medications. More studies are required to ascertain the optimum dosage of the combination prior to its approval for use in metastatic melanoma.
Molecular Targeted Therapy
The RAS-RAF–mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) signaling pathway is activated in almost 90% of patients
with melanoma.32 This pathway is normally required for the growth and survival of nonmalignant cells. In malignant transformation, mutations and/or overexpression is seen at various levels including KIT, NRAS, BRAF, and the MEK protein. This leads to activation of serine and threonine protein kinases, which lead to uncontrolled cell proliferation and survival.33
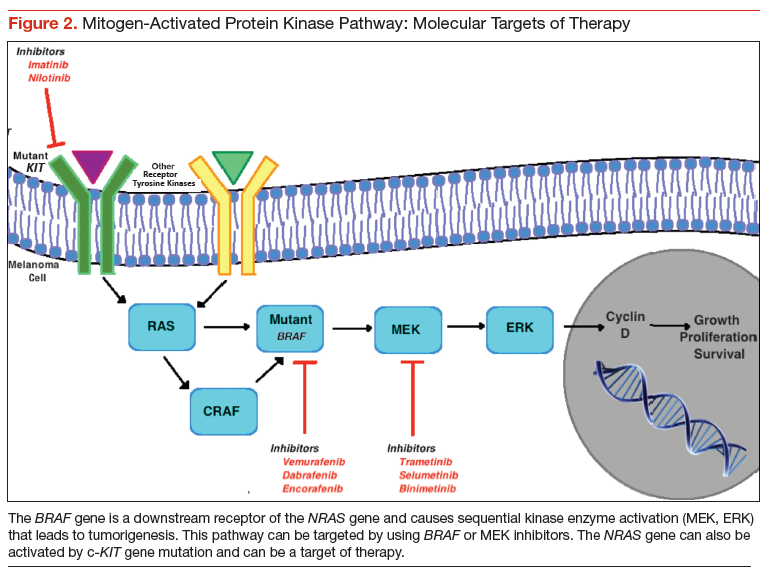
Novel therapeutic approaches have tried inhibiting one or more of these pathways for melanoma treatment. The most important mediator of tumorigenesis is BRAF, which is a downstream receptor of NRAS, and is mutated in almost 50% of melanoma cases.34 NRAS mutations are seen in 15% to 20% of cutaneous melanomas.35,36 After its activation, the RAF enzyme—coded by the BRAF gene—causes phosphorylation of the MEK protein, which activates ERK. This ERK activation leads to growth signaling and is the final pathway in several malignancies (Figure 2).37,38
BRAF Inhibitors
BRAF is the first mediator whose inhibition led to clinically significant outcomes in patients with melanoma. The most common BRAF mutation consists of the
substitution of glutamic acid for valine at amino acid 600 (V600E mutation) with majority of the remainder consisting of an alternate substitution (V600V or V600K).34 Vemurafenib and dabrafenib are the 2 BRAF inhibitors that have been shown to improve tumor regression, PFS, and OS considerably, especially in combination with a MEK protein inhibitor. In the phase 3 BRIM-3 trial, the vemurafenib group had a significantly prolonged PFS and OS compared with dacarbazine (13.6 months vs 9.7 months; 6.9 months vs 1.6 months, respectively). It was the first study to show improved survival with vemurafenib in both the V600E and V600K BRAF mutant melanomas.39
Another BRAF inhibitor, dabrafenib, was approved by the FDA for treatment of advanced melanoma with BRAF V600E mutation. It was tested in a phase 3 trial in which it was compared with dacarbazine in patients with advanced melanoma. Median OS in the dabrafenib arm was > 18 months and in dacarbazine arm > 15 months.40 Fifty-seven percent of the patients in dacarbazine arm were crossed over to the dabrafenib arm, thereby confounding the survival data for the former group. Another multicenter, phase 2 trial showed dabrafenib to have activity in melanoma patients with brain metastases, irrespective of previous therapy for the brain metastases.41 The long-term analysis of the BREAK-2 trial, which included 92 patients with metastatic melanoma treated with dabrafenib, showed a median OS of 12.9 months in BRAF V600K group and 13.1 months in BRAF V600E group.42
Adverse effects associated with BRAF inhibition include fatigue, rash, arthralgia, and photosensitivity reactions.43 Dermatologic complications may also include squamous cell carcinoma (SCC) (19%-26%), with keratoacanthoma being the most common subtype.44 These are believed to be likely secondary to the paradoxical activation of the MAPK signaling, since most of these lesions are found to have mutations in the RAS molecule.45 Other specific AEs of dabrafenib include hyperkeratosis (33%) and pyrexia (29%).42
Most patients treated with a BRAF inhibitor eventually have disease progression, likely secondary to reactivation of the MAPK pathway.46,47 This result has led to a heightened interest in combination therapies in an effort to improve outcomes. Combination therapy with ipilimumab and vemurafenib was studied and resulted in a higher incidence of hepatotoxicity (50%).48 However, no hepatotoxicity was seen in a phase 1 trial of combined dabrafenib and ipilimumab.49
Some studies have also suggested that extended BRAF inhibition after progression on a BRAF inhibitor may prolong survival.50,51 The phase 2 trial NCT01983124 is being conducted to evaluate the survival benefit with a combination of vemurafenib and a nitrosourea alkylating agent, fotemustine, in patients who have progressed on vemurafenib alone.
MEK Inhibitors
The inhibition of MEK can halt cell proliferation and induce apoptosis. The phase 3 METRIC trial, which compared the oral MEK inhibitor (trametinib) with chemotherapy, was conducted in 322 patients who had metastatic melanoma with a V600E or V600K BRAF mutation. The PFS and 6-month OS were significantly better with trametinib (4.8 months vs 1.5 months, 81% vs 66%) despite the crossover between the 2 groups.52 The AEs associated with trametinib included rash, diarrhea, and peripheral edema. Another phase 2 trial of trametinib including patients pretreated with a BRAF inhibitor showed no confirmed objective responses, 28% patients with stable disease, and minimal improvement in PFS (2 months). Among patients treated with prior chemotherapy and/or immunotherapy, trametinib showed significant improvement in complete responses, partial responses, stable disease, and the median PFS (2%, 23%, 51%, 4 months, respectively).53
The second MEK inhibitor, binimetinib, was studied in a phase 2 trial of advanced melanoma cases harboring a BRAF V600E or NRAS. Bimetinib demonstrated a PR in 20% cases of both the BRAF and NRAS mutant melanomas. Durable disease control was seen in 43% of the NRAS group and 32% of the BRAF group.54 The AE profile was similar to that seen with trametinib. Bimetinib is being studied in phase 1 and 2 trials with the CDK4/6 inhibitor as well as in the phase 3 trial NCT01763164 compared with dacarbazine in NRAS mutation positive melanomas.55
Selumetinib is a MEK inhibitor that has been compared with dacarbazine and temozolomide with no significant OS advantage. A novel highly specific inhibitor of MEK, cobimetinib, is currently being studied in combination with BRAF inhibitors.
Combined BRAF and MEK Inhibition
A randomized, double-blind, phase 3 study comparing the combination of dabrafenib and trametinib with dabrafenib and placebo in patients with advanced melanoma with a BRAF V600E mutation was presented at the 2014 American Society of Clinical Oncology meeting. Researchers found that after a median follow-up period of 9 months, there was a significant improvement with the combination in the PFS (9.3 months vs 8.8 months) and the ORR (67% vs 51%), with a similar incidence of AEs.56 The combination therapy group had fewer incidences of SCC of the skin but more incidence of pyrexia.
The combination of dabrafenib and trametinib was compared with vemurafenib monotherapy in a recent randomized phase 3 trial among 704 metastatic melanoma patients with a BRAF V600 mutation. Median PFS and ORR were significantly better with combination therapy compared with vemurafenib alone (11.4 months vs 7.3 months, 64% vs 51%, respectively). Overall survival rate at 1 year was significantly improved in the combination group as well (72% vs 65%).57 The incidence of SCC and keratoacanthoma was less in the combination (1%) compared with vemurafenib alone (18%). Another study investigating the coadministration and sequential administration of vemurafenib and trametinib is underway.58
The vemurafenib and cobimetinib combination was studied in a phase 3 trial of previously untreated unresectable locally advanced or metastatic BRAF V600
mutation-positive melanoma. The median PFS was 9.9 months in the combination group and 6.2 months in the control group. The interim analysis showed a 9-month survival rate of 81% in the combination group and 73% in the control group, with no significantly higher incidence of AEs in either arm.59 A longer follow-up will be needed to assess the OS benefit with the combination.
Encorafenib, a selective BRAF inhibitor, has been studied in a phase 1 trial in combination with binimetinib.60 This trial has paved the way to the initiation of a currently ongoing phase 3 trial (NCT01909453) comparing the combination with vemurafenib or encorafenib alone.
C-KIT Inhibitors
Mutations of c-KIT are seen more commonly in chronic sun damage-induced cutaneous melanomas, along with acral and mucosal melanomas.61,62 Earlier trials involving patients without selection for c-KIT mutation positivity failed to show benefit with imatinib. A single-arm, phase 2 trial of imatinib mesylate in patients with metastatic melanoma harboring the c-KIT mutation, an ORR of 23% was achieved, with a median PFS of 3.5 months.63 Imatinib showed an ORR of 29% in a phase 2 trial of mucosal, acral, and in chronic sun damage-induced melanoma patients with c-KIT amplifications and/or mutations. It was demonstrated that c-KIT amplification alone is not as responsive to imatinib compared with c-KIT mutation, suggesting that all patients with these specific melanomas should be tested for KIT mutation status.64
A second-generation c-KIT inhibitor, nilotinib, has shown some promising results with a favorable AE profile in small phase 2 trials.65,66 However, more clinical research will be needed before definite recommendations on its use in cutaneous melanomas can be made. Currently, its role seems to be limited to the management of acral, mucosal, and chronic sun damage-related melanomas with c-KIT mutations.
Future Directions
Angiogenesis promoters, such as vascular endothelial growth factor (VEGF), platelet-derived growth factor, fibroblast growth factor, and interleukin-8, are overexpressed in melanoma. Bevacizumab, an anti-VEGF antibody, has been shown to have some benefit in combination with carboplatin and paclitaxel as a triple therapy.67 However, grade 3 AEs were seen in a portion of patients.
The phosphatidylinositol-3 kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway has also been studied as a target for melanoma therapy. Everolimus, an mTOR inhibitor, was studied in a phase 2 trial in combination with bevacizumab for treatment of metastatic melanoma. The combination showed improved median PFS and OS with the combination (4 months and 8.6 months, respectively), with 43% of patients alive after 12 months of follow-up.68 This study points to the direction of possible benefits with the combination of anti-VEGF and immunotherapy. A recent study failed to show survival advantage with combination of bevacizumab and temozolomide.69
Buparlisib (BKM120), a PI3K inhibitor, has been shown to have activity in vivo and in vitro against melanoma brain metastases.70 More studies need to be done to assess the possible combination with other established therapies.
Oblimersen is an antisense oligonucleotide that suppresses B-cell lymphoma-2, thereby suppressing its anti-apoptotic effect. The triple combination of oblimersen with temozolomide and albumin-bound paclitaxel has shown to be safe and efficacious in a phase 1 trial, thereby creating a need for further clinical trials.71
Treatment Approach
Systemic therapy for metastatic melanoma depends on several factors, including BRAF mutation status, functional status of the patient, disease burden, and severity of symptoms. Assessing the BRAF mutation status has become an important component in the management of patients with metastatic melanoma. It can help recognize patients who will benefit from molecular targeted therapy. In case of a BRAF-positive melanoma, treatment can be initiated with either immunotherapy or BRAF inhibitors. There are no randomized studies comparing immunotherapy to molecular targeted therapy.
Patients who have good PS and lymph node metastases can be treated initially with IL-2, which has the advantage of inducing cure in a minority of patients but should only be considered in patients with well-preserved organ function who can be monitored in an intensive care setting. On the other hand, patients who have bulky, symptomatic disease and poor PS should be treated initially with BRAF inhibitors. Combination of BRAF and MEK inhibitors can also be used and has an improved PFS and OS with potential to cause early tumor regression. There are studies to suggest suboptimal outcomes in patients who are treated with ipilimumab after progression on a BRAF inhibitor compared with initial treatment with ipilimumab followed by a BRAF inhibitor.72-74 However, all these studies are retrospective and there is no prospective data to suggest the above. BRAF mutation-positive patients who progress on a BRAF inhibitor
can be treated with PD-1 inhibitors.
Patients who do not have a BRAF mutation are unlikely to benefit from a BRAF inhibitor and primarily receive immunotherapy with ipilimumab or IL-2. Whenever possible, such patients should be enrolled in a clinical trial, as they have a poor prognosis. Patients who progress on ipilimumab can be treated with one of the PD-1 inhibitors (pembrolizumab, nivolumab). These PD-L1 inhibitors are still being investigated for use in such situations.
The role of chemotherapy in the management of metastatic melanoma has been limited by numerous studies showing significantly better survival with immunotherapy and molecular targeted therapy. Dacarbazine is the only FDA-approved drug for the treatment of melanoma. Its use is reserved mainly for patients who are not candidates for any of the other therapies available, including enrollment in a clinical trial.
Conclusion
Therapies for metastatic melanoma are in a state of flux. In the past decade, several new therapeutic agents have been introduced for the management of this potentially lethal disease. The treatment of metastatic melanoma has gradually shifted from cytotoxic chemotherapy toward a more individualized treatment that has a definite survival advantage over traditional counterparts. The advent of novel therapies has led to initiation of further studies to determine their role in the treatment of advanced melanoma, singly or in combination with other agents. In addition to evaluating new agents, more studies are needed to compare existing treatment modalities so that definitive treatment protocols can be formulated.
Acknowledgement
The authors would like to thank Felicia Ratnaraj, MD, for her assistance in creating the figures.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin.2014;64(1):9-29.
2. Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27(36):6199-6206.
3. Welch HG, Woloshin S, Schwartz LM. Skin biopsy rates and incidence of melanoma:
population based ecological study. BMJ. 2005;331(7515):481.
4. Sosman JA, Moon J, Tuthill RJ, et al. A phase 2 trial of complete resection for stage IV melanoma: results of Southwest Oncology Group Clinical Trial S9430. Cancer. 2011;117(20):4740-4746.
5. Atkins MB. The role of cytotoxic chemotherapeutic agents either alone or in combination with biological response modifiers. In: Kirkwood JK, ed. Molecular Diagnosis, Prevention, & Therapy of Melanoma. New York, NY: Marcel Dekker;1997:219-225.
6. Patel PM, Suciu S, Mortier L, et al. Extended schedule, escalated dose temozolomide versus dacarbazine in stage IV melanoma: final results of a randomised phase III study (EORTC 18032). Eur J Cancer. 2011;47(10):1476-1483.
7. Chapman PB, Einhorn LH, Meyers ML, et al. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol. 1999;17(9):2745-2751.
8. Flaherty KT, Lee SJ, Zhao F, et al. Phase III trial of carboplatin and paclitaxel with
or without sorafenib in metastatic melanoma. J Clin Oncol. 2013;31(3):373-379.
9. Rosenberg SA, Yang JC, Topalian SL, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271(12):907-913.
10. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105-2116.
11. Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J Sci Am. 2000;6(suppl 1):S11-S14.
12. Hoos A, Ibrahim R, Korman A, et al. Development of ipilimumab: contribution to a new paradigm for cancer immunotherapy. Semin Oncol. 2010;37(5):533-546.
13. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711-723.
14. Schadendorf D, Hodi FS, Robert C, et. al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma [published online ahead of print February 9, 2015]. J Clin Oncol. pii:JCO.2014.56.2736.
15. Weber JS, Kähler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691-2697.
16. Hodi FS, Lee S, McDermott DF, et al. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: a randomized clinical trial. JAMA. 2014;312(17):1744-1753.
17. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443-2454.
18. Ribas A, Hodi FS, Kefford R, et al. Efficacy and safety of the anti-PD-1 monoclonal antibody pembrolizumab (MK-3475) in 411 patients (pts) with melanoma (MEL) (Abstract LBA9000). Paper presented at: 2014 American Society of Clinical Oncology (ASCO) meeting; May-June 2014; Chicago, IL.
19. Hamid O, Robert C, Ribas A, et al. Randomized comparison of two doses of the anti-PD-1 monoclonal antibody MK-3475 for ipilimumab-refractory (IPI-R) and IPI-naive (IPI-N) melanoma (MEL) (abstract 3000). Paper presented at: 2014 American Society of Clinical Oncology (ASCO) meeting; May-June 2014; Chicago, IL.
20. Robert C, Ribas A, Wolchok JD, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014; 384(9948):1109-1117.
21. Dummer R, Daud A, Puzanov I, et. al. A randomized controlled comparison of pembrolizumab and chemotherapy in patients with ipilimumab-refractory melanoma. J Transl Med. 2015;13(suppl 1):O5.
22. Topalian SL, Sznol M, McDermott DF, et. al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32(10):1020-1030.
23. Hodi FS, Sznol M, Kluger HM, et al. Long-term survival of ipilimumab-naive patients with advanced melanoma (MEL) treated with nivolumab (anti-PD-1, BMS-936558, ONO-4538) in a phase I trial (abstract 9002). Paper presented at: 2014 American Society of Clinical Oncology (ASCO) meeting; May-June 2014; Chicago, IL.
24. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320-330.
25. Weber J, D’Angelo S, Gutzmer R, et al. A phase 3 randomized, open-label study of nivolumab versus investigator’s choice of chemotherapy in patients with advanced melanoma after prior anti-CTLA4 therapy (abstract LBA3). Paper presented at: European Society of Medical Oncology 2014 meeting; September 2014; Madrid, Spain.
26. Atkins MB, Kudchadkar RR, Sznol M, et al. Phase 2, multicenter, safety and efficacy study of pidilizumab in patients with metastatic melanoma (abstract 9001). Paper presented at: 2014 American Society of Clinical Oncology (ASCO) meeting; May-June 2014; Chicago, IL.
27. Brahmer JR, Tykodi SS, Chow LQM, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455-2465.
28. Hamid O, Sosman JA, Lawrence DP, et. al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic melanoma (mM). J Clin Oncol. 2013;31(15)(suppl): Abstract 9010.
29. Lutzky J, Antonia SJ, Blake-Haskins A, et. al. A phase 1 study of MEDI4736, an anti–PD-L1 antibody, in patients with advanced solid tumors. J Clin Oncol. 2014;32(15)(suppl): Abstract 3001.
30. Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced
melanoma. N Engl J Med. 2013;369(2):122-133.
31. Sznol M, Kluger HM, Callahan MK, et al. Survival, response duration, and activity by BRAF mutation (MT) status of nivolumab (NIVO, anti-PD-1, BMS-936558, ONO-4538) and ipilimumab (IPI) concurrent therapy in advanced melanoma (MEL) (abstract LBA9003). Paper presented at: 2014 American Society of Clinical Oncology (ASCO) meeting; May-June 2014; Chicago, IL.
32. Omholt K, Platz A, Kanter L, Ringborg U, Hansson J. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9(17):6483-6488.
33. Wellbrock C, Hurlstone A. BRAF as therapeutic target in melanoma. Biochem Pharmacol. 2010;80(5):561-567.
34. Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29(10):1239-1246.
35. Ball NJ, Yohn JJ, Morelli JG, et al. Ras mutations in human melanoma: a marker of malignant progression. J Invest Dermatol. 1994;102(3):285-290.
36. Platz A, Ringborg U, Brahme EM, Lagerlöf B. Melanoma metastases from patients with hereditary cutaneous malignant melanoma contain a high frequency of N-ras activating mutations. Melanoma Res. 1994;4(3):169-177.
37. Beeram M, Patnaik A, Rowinsky EK. Raf: a strategic target for therapeutic development against cancer. J Clin Oncol. 2005;23(27):6771-6790.
38. Terai K, Matsuda M. The amino-terminal B-Raf-specific region mediates calcium-dependent homo- and hetero-dimerization of Raf. EMBO J. 2006;25(15):3556-3564.
39. McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15(3):323-332.
40. Hauschild A, Grob JJ, Demidov LV, et al. An update on BREAK-3, a phase III, randomized trial: dabrafenib versus dacarbazine in patients with BRAF V600E-positive mutation metastatic melanoma (Abstract 9013). Paper presented at: American Society of Clinical Oncology 2013 meeting; May-June 2013; Chicago, IL.
41. Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087-1095.
42. Ascierto PA, Minor DR, Ribas A, et. al., Long-term safety and overall survival update for BREAK-2, a phase 2, single-arm, open-label study of dabrafenib in previously treated metastatic melanoma (NCT01153763). J Clin Oncol. 2014;32(15)(suppl): Abstract 9034.
43. Larkin J, Del Vecchio M, Ascierto PA, et al. Vemurafenib in patients with
BRAF(V600) mutated metastatic melanoma: an open-label, multicentre, safety
study. Lancet Oncol. 2014;15(4):436-444.
44. Lacouture ME, Duvic M, Hauschild A, et al. Analysis of dermatologic events in vemurafenib-treated patients with melanoma. Oncologist. 2013;18(3):314-322.
45. Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366(3):207-215.
46. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507-2516.
47. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358-365.
48. Ribas A, Hodi FS, Callahan M, et. al. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2014;368(14):1365-1366.
49. Linette GP, Puzanov I, Callahan MK, et al. Phase 1 study of the BRAF inhibitor dabrafenib (D) with or without the MEK inhibitor trametinib (T) in combination with ipilimumab (Ipi) for V600E/K mutation–positive unresectable or metastatic melanoma (MM). J Clin Oncol. 2014;32(15)(suppl): Abstract 2511.
50. Chan MMK, Haydu LE, Menzies AM, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer. 2014;120(20):3142-3153.
51. Carlino MS, Gowrishankar K, Saunders CAB, et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther. 2013;12(7):1332-1342.
52. Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107-114.
53. Kim KB, Kefford R, Pavlick AC, et. al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. 2013;31(1):482-489.
54. Ascierto PA, Schadendorf D, Berking C, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14(3):249-256.
55. Sosman JA, Kittaneh M, Lolkema MP, et al. A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS-mutant melanoma: early encouraging clinical activity (abstract 9009). Paper presented at: 2014 American Society of Clinical Oncology meeting ; May-June 2014; Chicago, IL.
56. Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371(20):1877-1888.
57. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30-39.
58. Gogas H, Schadendorf D, Dummer R. Vemurafenib treatment in patients with BRAF-mutated melanoma failing MEK inhibition with trametinib. J Clin Oncol. 2014;32(15)(suppl): Abstract 9061.
59. Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867-1876.
60. Kefford R, Miller WH, Tan DS, et al. Preliminary results from a phase Ib/II, openlabel, dose-escalation study of the oral BRAF inhibitor LGX818 in combination with the oral MEK1/2 inhibitor MEK162 in BRAF V600-dependent advanced solid tumors (abstract 9019). Paper presented at: 2013 American Society of Clinical Oncology meeting; May-June 2014; Chicago, IL.
61. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct
subtypes of melanoma. J Clin Oncol. 2006;24(26):4340-4346.
62. Jin SA, Chun SM, Choi YD, et al. BRAF mutations and KIT aberrations and their clinicopathological correlation in 202 Korean melanomas. J Invest Dermatol. 2013;133(2):579-582.
63. Guo J, Si L, Kong Y et. al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29(21):2904-2909.
64. Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol. 2013;31(26):3182-3190.
65. Cho JH, Kim KM, Kwon M, Kim JH, Lee J. Nilotinib in patients with metastatic melanoma harboring KIT gene aberration. Invest New Drugs. 2012;30(5): 2008-2014.
66. Lebbe C, Chevret S, Jouary T, et. al. Phase II multicentric uncontrolled national trial assessing the efficacy of nilotinib in the treatment of advanced melanomas with c-KIT mutation or amplification. J Clin Oncol. 2014;32(15)(suppl): Abstract 9032.
67. Perez DG, Suman VJ, Fitch TR, et al. Phase 2 trial of carboplatin, weekly paclitaxel, and biweekly bevacizumab in patients with unresectable stage IV melanoma: a North Central Cancer Treatment Group study, N047A. Cancer. 2009;115(1):119-127.
68. Hainsworth JD, Infante JR, Spigel DR, et al. Bevacizumab and everolimus in the treatment of patients with metastatic melanoma. Cancer. 2010;116(17): 4122-4129.
69. Dronca RS, Allred JB, Perez DG, et. al. Phase II study of temozolomide (TMZ) and everolimus (RAD001) therapy for metastatic melanoma: a North Central Cancer Treatment Group study, N0675. Am J Clin Oncol. 2014;37(4):369-376.
70. Meier FE, Niessner H, Schmitz J, et al. The PI3K inhibitor BKM120 has potent antitumor activity in melanoma brain metastases in vitro and in vivo. J Clin Oncol. 2013;31(15)(suppl): Abstract e20050.
71. Ott PA, Chang J, Madden K, et al. Oblimersen in combination with temozolomide and albumin-bound paclitaxel in patients with advanced melanoma: a phase I trial. Cancer Chemother Pharmacol. 2013;71(1);183-191.
72. Ackerman A, Klein O, McDermott DF, et al. Outcomes of patients with metastatic
melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. 2014;120(11):1695-1701.
73. Ascierto PA, Margolin K. Ipilimumab before BRAF inhibitor treatment may be
more beneficial than vice versa for the majority of patients with advanced melanoma.
Cancer. 2014;120(11):1617-1619.
74. Ascierto PA, Simeone E, Sileni VC, et al. Sequential treatment with ipilimumab and BRAF inhibitors in patients with metastatic melanoma: data from the Italian cohort of the ipilimumab expanded access program. Cancer Invest. 2014;32(4):144-149.
Melanoma is the most aggressive form of skin cancer, contributing to about 76,000 new cases and more than 9,000 deaths in 2014.1 Depending on the stage of the disease, 5-year melanoma survival can range from 15% to 97%. Patients with local and distant metastases have a 5-year survival of about 60% and 15%, respectively.2
The incidence of melanoma is rising, partly because of the increasing number of skin biopsies being performed.3 If melanoma is diagnosed early, surgical excision is the treatment of choice. In patients with oligometastatic disease (cancer that has spread, but only to 1 or a small number of sites), complete surgical excision of the metastases may provide prolonged overall survival (OS) and delay the need to use systemic therapy.4
Recently, many new drug therapies have shown promising results in clinical trials, which may improve the prognosis of metastatic disease. This article reviews currently available systemic treatment options for the management of metastatic melanoma, the role of cytotoxic chemotherapy and interleukin-2 (IL-2), and the latest therapies available, including immune checkpoint inhibitors.
Cytotoxic Chemotherapy and Interleukin-2
Cytotoxic chemotherapy does not have an established role in the initial treatment of metastatic melanoma. Currently, cytotoxic chemotherapy is used in patients who have not responded to immunotherapy or molecular targeted therapy. The most commonly used drugs include dacarbazine and its prodrug, temozolomide. Several studies have failed to demonstrate a survival benefit using a single-agent chemotherapy with either dacarbazine or temozolomide.5,6
Other agents used in metastatic melanoma include nitrosoureas (fotemustine), platinum compounds (cisplatin, carboplatin), vinca alkaloids (vincristine),
and taxanes (paclitaxel). None of these agents provide a survival benefit, but an objective response may be seen in a minority of cases. Combination chemotherapy regimens have not shown an advantage over singleagent dacarbazine or temozolomide.7,8
High-dose IL-2 has been used in cases of metastatic melanoma with good performance status (PS) and organ function. Studies have shown a complete response rate of 3% to 7% and a prolonged disease-free survival in a minority of patients.9-11 The use of highdose IL-2, however, is limited by the high incidence of adverse effects (AEs), which include bacterial sepsis, pulmonary edema, arrhythmias, fever, and on some occasions, death due to complications.10 The use of IL-2 requires admission of the patient to a specialized unit for AE monitoring and management. Because of its ability to “cure” a minority of patients, a role still exists for IL-2 therapy in the treatment of younger, healthy patients with no evidence of organ dysfunction at baseline.
Immune Checkpoint Inhibitors
Checkpoint inhibitors are a class of drugs that unmask the immune system to fight against cancer cells. This class of drugs has shown significant activity and survival advantage in recent phase 2 and 3 trials. The class includes the anticytotoxic T-lymphocyte antigen 4 (CTLA-4) antibody ipilimumab and monoclonal antibodies targeting the programmed death 1 protein (PD-1) or its ligand (PD-L1).
Anti-CTLA-4 Antibodies: Ipilimumab
Cytotoxic T-lymphocyte antigen 4 is the antigen responsible for inhibition of cytotoxic T-cell-mediated immunity against foreign antigens presented by the antigen presenting cells (APCs). The APCs cause activation of the T cells when peptide fragments of intracellular proteins are presented in combination with mixed histocompatibility complex molecules. This step requires interaction of a costimulatory molecule (B7) on the APCs with a cluster of differentiation 28 protein (CD28) receptor located on T cells. CTLA-4 competes with CD28 to bind with the B7 molecule, thereby inhibiting the activation of the cytotoxic T cells (Figure 1). This pathway is thought to help with development of tolerance to host tissue antigens. Ipilimumab is a human monoclonal antibody that inhibits this CTLA-4 molecule and facilitates T-cell mediated antitumor activity.12 By blocking the CTLA-4 molecule, ipilimumab also mediates its autoimmune AEs on the host tissues.
Hodi and colleagues conducted a phase 3 trial of ipilimumab, including 676 patients who progressed after prior treatment for stage III or IV melanoma, and found that median OS was significantly better in the ipilimumab groups: 10 months in the ipilimumab plus gp100 peptide vaccine group vs 6.4 months in the gp100 vaccine alone group; 10.1 months in the ipilimumab alone group vs 6.4 months in the gp100 vaccine alone group.13 In another phase 3 trial comparing ipilimumab plus dacarbazine to dacarbazine alone, the ipilimumab group had a significantly improved OS (11.2 months vs 9.1 months).1 Survival rates with ipilimumab were prolonged for up to 3 years compared with the dacarbazine plus placebo group. However, the combination was associated with increased incidence of hepatotoxicity, thereby limiting its use.
A long-term survival analysis of 10 prospective and 2 retrospective studies of ipilimumab showed a median OS of 11.4 months and a long-term survival that began at 3 years with a plateau at 10 years of 21%, which was independent of prior therapy or ipilimumab dose.14 The immune-related AEs of ipilimumab are secondary to its activity against the host antigens and include dermatitis, enterocolitis, hepatitis, and endocrinopathies.15
A recent phase 2 trial studied the combination of ipilimumab with granulocyte-macrophage colonystimulating factor in 245 patients with stage III and IV melanoma. Median OS after 13 months was significantly higher with the combination compared with ipilimumab alone. The 1-year survival rate was 69% with
the combination and 53% with ipilimumab alone. There was no difference in the overall response rate (ORR) or progression-free survival (PFS) between the 2 groups. However, the AEs were significantly reduced with the combination (45% vs 58%).16 The dose of ipilimumab used in the trial was higher than the approved dose, making it difficult to apply the results in practice without further studies on the combination.
Anti-PD-1 Antibodies
Programmed death 1 ligands (PD-L1 and PD-L2) are expressed by tumor or stromal cells to inhibit the T-cell mediated antitumor activity. These ligands bind to the PD-1 protein on the surface of activated T cells to mediate their immunosuppressive effects. Interruption of this interaction by either anti-PD-1 antibodies or anti-PD-L1 antibodies facilitates tumor cell killing by activated T cells.17
Pembrozilumab and nivolumab are the 2 anti-PD-1 monoclonal antibodies that have been approved for treatment of metastatic melanoma. In a phase 1 trial
of pembrolizumab, 411 patients with advanced melanoma (consisting of both ipilimumab-naïve [IPI-N] and ipilimumab-treated [IPI-T] patients), ORR was 40% in IPI-N and 28% in IPI-T patients with a 1-year OS of 71% in all patients. Median PFS was 24 weeks in IPI-N and 23 weeks in IPI-T pts.18 There was no difference in outcomes and safety profiles across the various dosing regimens.18,19 Of note, pembrolizumab had antitumor activity irrespective of the PS, lactate dehydrogenase levels, BRAF (B-Raf proto-oncogene, serine/threonine kinase) gene mutation, metastatic stage, and number and type of prior therapy. In a subgroup analysis, 173 patients who had progression after treatment with ipilimumab were randomly assigned to pembrolizumab 2 mg/kg every 3 weeks (q3w) or 10 mg/kg q3w dosing regimens. Both groups had no significant difference in the ORR (26% in both) and safety profiles.20
In the 2012 KEYNOTE-002 clinical trial, a randomized phase 2 trial involving 540 patients with ipilimumab-refractory advanced melanoma, patients were randomized 1:1:1 to pembrolizumab 2 mg/kg or 10 mg/kg q3w or investigator-choice chemotherapy (control arm consisting of carboplatin plus paclitaxel, carboplatin, paclitaxel, dacarbazine, or temozolomide). The 6-month PFS was significantly improved with pembrolizumab (34% and 38% for pembrolizumab 2 mg/kg and 10 mg/kg, respectively) compared with 16% with chemotherapy. The ORR was significantly better with pembrolizumab (21% at 2 mg/kg, 25% at 10 mg/kg) compared with the control arm (4%).21 These findings led to the approval of pembrolizumab by the FDA for treatment of patients with advanced melanoma who have progressed on ipilimumab. Pembrolizumab is generally well tolerated. The most common AEs include fatigue, pruritus, and rash.
Nivolumab was studied in a recent phase 1 trial in which 107 patients with previously treated advanced melanoma were treated with escalated doses every
2 weeks.22 The 2-year and 3-year OS rates were 48% and 41%, respectively. Objective responses were seen in 32% of the patients. The median response duration was 23 months.23
The first phase 3 trial was conducted in 418 patients with previously untreated metastatic melanoma BRAF mutation. Patients were randomized to receive either nivolumab or dacarbazine. The PFS and OS were significantly better with nivolumab compared with dacarbazine (PFS 5.1 months vs 2.2 months; OS 73% vs 42% at 1 year).24 The AE profile of nivolumab is similar to pembrolizumab and includes lung, skin, endocrine, renal, and gastrointestinal tract toxicities.
Preliminary results of another phase 3 trial were presented at the European Society of Medical Oncology 2014 meeting. Patients with previously treated metastatic melanoma (ipilimumab or BRAF inhibitor) were randomized in a 2:1 ratio to receive either nivolumab or investigators’ choice chemotherapy (dacarbazine or carboplatin plus paclitaxel). The ORR was significantly better with nivolumab (32% vs 11%), and 95% of patients were still responding after 6 months. The nivolumab group showed a complete remission in 3% of the patients with 34% of the responses lasting ≥ 6 months.25 This led to the recent approval of nivolumab for patients with metastatic melanoma with a BRAF mutation who have advanced on ipilimumab. In the phase 3 NCT01844505 trial patients are being randomized to receive ipilimumab, nivolumab, or both.
A newer PD-1 inhibitor, pidilizumab, was studied in a phase 2 trial that included 103 patients with metastatic melanoma, 51% of whom had received therapy with ipilimumab. The ORR in the study group was relatively lower (6%), but the OS at 1 year was 64.5%.26 Further studies are underway to evaluate the role of this drug in metastatic melanoma.
The response with both nivolumab and pembrolizumab is durable as well as sustained, even after discontinuation of therapy. None of the deaths in the aforementioned studies were atributed to drug-related toxicities. As evidenced by current data, these 2 drugs hold a great promise for the management of patients who progress after therapy with anti-CTLA-4 antibodies.
Anti-PD-L1 Antibodies
The anti-PD-L1 monoclonal antibodies work in a similar way to the PD-1 inhibitors and block the interaction between the PD-1 and its ligand, PD-L1. This causes sustained activation of cytotoxic T cells and facilitates their antitumor activity. Two of PD-L1 inhibitors have shown clinical activity against metastatic melanoma.
BMS-936559, the first PD-L1 antibody, is being studied in a phase 1 trial that includes 55 patients with advanced melanoma along with 152 patients with other solid malignancies. Three patients achieved a complete response, and 5 patients had an objective response lasting 1 year. The ORR for melanoma was 17%, with disease stabilization of ≥ 24 weeks in 27% of the patients.27 Common AEs included infusion reactions, diarrhea, fatigue, rash, hypothyroidism, and hepatitis.
The second PD-L1 antibody, MPDL3280A, was studied in a phase 1 trial of 45 patients with metastatic melanoma. An ORR of 29% was observed, along with a 24-week PFS of 43%.28 Commonly noted AEs included hyperglycemia and elevated liver aminotransferases.
A newer PD-L1 inhibitor, MEDI4736, is being studied for advanced malignancies in 8 patients with melanoma. In preliminary analysis, MEDI4736 demonstrated a partial response in 1 out of 8 melanoma patients with a disease control rate of 46%.29 Although the PD-L1 inhibitors seem promising, more information will help discern their role in the management of metastatic melanoma.
Combined Anti-CTLA-4 Plus Anti-PD-1 Antibody
The combination of ipilimumab and the PD-1 inhibitor nivolumab was tested in a phase 1 trial in which both drugs were used concurrently as well as sequentially in metastatic melanoma.30 The 1- and 2-year OS in patients who were treated concurrently was 82% and 75%, respectively. Complete remission was seen in 17% of the patients, and the responses were seen irrespective of the BRAF mutation status. The responses were durable, and about 64% of the objective responses remain in remission at last follow-up.31 Grade 3 to grade 4 AEs were noted in 53% of the patients, with 11 patients requiring discontinuation of the medications. More studies are required to ascertain the optimum dosage of the combination prior to its approval for use in metastatic melanoma.
Molecular Targeted Therapy
The RAS-RAF–mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) signaling pathway is activated in almost 90% of patients
with melanoma.32 This pathway is normally required for the growth and survival of nonmalignant cells. In malignant transformation, mutations and/or overexpression is seen at various levels including KIT, NRAS, BRAF, and the MEK protein. This leads to activation of serine and threonine protein kinases, which lead to uncontrolled cell proliferation and survival.33
Novel therapeutic approaches have tried inhibiting one or more of these pathways for melanoma treatment. The most important mediator of tumorigenesis is BRAF, which is a downstream receptor of NRAS, and is mutated in almost 50% of melanoma cases.34 NRAS mutations are seen in 15% to 20% of cutaneous melanomas.35,36 After its activation, the RAF enzyme—coded by the BRAF gene—causes phosphorylation of the MEK protein, which activates ERK. This ERK activation leads to growth signaling and is the final pathway in several malignancies (Figure 2).37,38
BRAF Inhibitors
BRAF is the first mediator whose inhibition led to clinically significant outcomes in patients with melanoma. The most common BRAF mutation consists of the
substitution of glutamic acid for valine at amino acid 600 (V600E mutation) with majority of the remainder consisting of an alternate substitution (V600V or V600K).34 Vemurafenib and dabrafenib are the 2 BRAF inhibitors that have been shown to improve tumor regression, PFS, and OS considerably, especially in combination with a MEK protein inhibitor. In the phase 3 BRIM-3 trial, the vemurafenib group had a significantly prolonged PFS and OS compared with dacarbazine (13.6 months vs 9.7 months; 6.9 months vs 1.6 months, respectively). It was the first study to show improved survival with vemurafenib in both the V600E and V600K BRAF mutant melanomas.39
Another BRAF inhibitor, dabrafenib, was approved by the FDA for treatment of advanced melanoma with BRAF V600E mutation. It was tested in a phase 3 trial in which it was compared with dacarbazine in patients with advanced melanoma. Median OS in the dabrafenib arm was > 18 months and in dacarbazine arm > 15 months.40 Fifty-seven percent of the patients in dacarbazine arm were crossed over to the dabrafenib arm, thereby confounding the survival data for the former group. Another multicenter, phase 2 trial showed dabrafenib to have activity in melanoma patients with brain metastases, irrespective of previous therapy for the brain metastases.41 The long-term analysis of the BREAK-2 trial, which included 92 patients with metastatic melanoma treated with dabrafenib, showed a median OS of 12.9 months in BRAF V600K group and 13.1 months in BRAF V600E group.42
Adverse effects associated with BRAF inhibition include fatigue, rash, arthralgia, and photosensitivity reactions.43 Dermatologic complications may also include squamous cell carcinoma (SCC) (19%-26%), with keratoacanthoma being the most common subtype.44 These are believed to be likely secondary to the paradoxical activation of the MAPK signaling, since most of these lesions are found to have mutations in the RAS molecule.45 Other specific AEs of dabrafenib include hyperkeratosis (33%) and pyrexia (29%).42
Most patients treated with a BRAF inhibitor eventually have disease progression, likely secondary to reactivation of the MAPK pathway.46,47 This result has led to a heightened interest in combination therapies in an effort to improve outcomes. Combination therapy with ipilimumab and vemurafenib was studied and resulted in a higher incidence of hepatotoxicity (50%).48 However, no hepatotoxicity was seen in a phase 1 trial of combined dabrafenib and ipilimumab.49
Some studies have also suggested that extended BRAF inhibition after progression on a BRAF inhibitor may prolong survival.50,51 The phase 2 trial NCT01983124 is being conducted to evaluate the survival benefit with a combination of vemurafenib and a nitrosourea alkylating agent, fotemustine, in patients who have progressed on vemurafenib alone.
MEK Inhibitors
The inhibition of MEK can halt cell proliferation and induce apoptosis. The phase 3 METRIC trial, which compared the oral MEK inhibitor (trametinib) with chemotherapy, was conducted in 322 patients who had metastatic melanoma with a V600E or V600K BRAF mutation. The PFS and 6-month OS were significantly better with trametinib (4.8 months vs 1.5 months, 81% vs 66%) despite the crossover between the 2 groups.52 The AEs associated with trametinib included rash, diarrhea, and peripheral edema. Another phase 2 trial of trametinib including patients pretreated with a BRAF inhibitor showed no confirmed objective responses, 28% patients with stable disease, and minimal improvement in PFS (2 months). Among patients treated with prior chemotherapy and/or immunotherapy, trametinib showed significant improvement in complete responses, partial responses, stable disease, and the median PFS (2%, 23%, 51%, 4 months, respectively).53
The second MEK inhibitor, binimetinib, was studied in a phase 2 trial of advanced melanoma cases harboring a BRAF V600E or NRAS. Bimetinib demonstrated a PR in 20% cases of both the BRAF and NRAS mutant melanomas. Durable disease control was seen in 43% of the NRAS group and 32% of the BRAF group.54 The AE profile was similar to that seen with trametinib. Bimetinib is being studied in phase 1 and 2 trials with the CDK4/6 inhibitor as well as in the phase 3 trial NCT01763164 compared with dacarbazine in NRAS mutation positive melanomas.55
Selumetinib is a MEK inhibitor that has been compared with dacarbazine and temozolomide with no significant OS advantage. A novel highly specific inhibitor of MEK, cobimetinib, is currently being studied in combination with BRAF inhibitors.
Combined BRAF and MEK Inhibition
A randomized, double-blind, phase 3 study comparing the combination of dabrafenib and trametinib with dabrafenib and placebo in patients with advanced melanoma with a BRAF V600E mutation was presented at the 2014 American Society of Clinical Oncology meeting. Researchers found that after a median follow-up period of 9 months, there was a significant improvement with the combination in the PFS (9.3 months vs 8.8 months) and the ORR (67% vs 51%), with a similar incidence of AEs.56 The combination therapy group had fewer incidences of SCC of the skin but more incidence of pyrexia.
The combination of dabrafenib and trametinib was compared with vemurafenib monotherapy in a recent randomized phase 3 trial among 704 metastatic melanoma patients with a BRAF V600 mutation. Median PFS and ORR were significantly better with combination therapy compared with vemurafenib alone (11.4 months vs 7.3 months, 64% vs 51%, respectively). Overall survival rate at 1 year was significantly improved in the combination group as well (72% vs 65%).57 The incidence of SCC and keratoacanthoma was less in the combination (1%) compared with vemurafenib alone (18%). Another study investigating the coadministration and sequential administration of vemurafenib and trametinib is underway.58
The vemurafenib and cobimetinib combination was studied in a phase 3 trial of previously untreated unresectable locally advanced or metastatic BRAF V600
mutation-positive melanoma. The median PFS was 9.9 months in the combination group and 6.2 months in the control group. The interim analysis showed a 9-month survival rate of 81% in the combination group and 73% in the control group, with no significantly higher incidence of AEs in either arm.59 A longer follow-up will be needed to assess the OS benefit with the combination.
Encorafenib, a selective BRAF inhibitor, has been studied in a phase 1 trial in combination with binimetinib.60 This trial has paved the way to the initiation of a currently ongoing phase 3 trial (NCT01909453) comparing the combination with vemurafenib or encorafenib alone.
C-KIT Inhibitors
Mutations of c-KIT are seen more commonly in chronic sun damage-induced cutaneous melanomas, along with acral and mucosal melanomas.61,62 Earlier trials involving patients without selection for c-KIT mutation positivity failed to show benefit with imatinib. A single-arm, phase 2 trial of imatinib mesylate in patients with metastatic melanoma harboring the c-KIT mutation, an ORR of 23% was achieved, with a median PFS of 3.5 months.63 Imatinib showed an ORR of 29% in a phase 2 trial of mucosal, acral, and in chronic sun damage-induced melanoma patients with c-KIT amplifications and/or mutations. It was demonstrated that c-KIT amplification alone is not as responsive to imatinib compared with c-KIT mutation, suggesting that all patients with these specific melanomas should be tested for KIT mutation status.64
A second-generation c-KIT inhibitor, nilotinib, has shown some promising results with a favorable AE profile in small phase 2 trials.65,66 However, more clinical research will be needed before definite recommendations on its use in cutaneous melanomas can be made. Currently, its role seems to be limited to the management of acral, mucosal, and chronic sun damage-related melanomas with c-KIT mutations.
Future Directions
Angiogenesis promoters, such as vascular endothelial growth factor (VEGF), platelet-derived growth factor, fibroblast growth factor, and interleukin-8, are overexpressed in melanoma. Bevacizumab, an anti-VEGF antibody, has been shown to have some benefit in combination with carboplatin and paclitaxel as a triple therapy.67 However, grade 3 AEs were seen in a portion of patients.
The phosphatidylinositol-3 kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway has also been studied as a target for melanoma therapy. Everolimus, an mTOR inhibitor, was studied in a phase 2 trial in combination with bevacizumab for treatment of metastatic melanoma. The combination showed improved median PFS and OS with the combination (4 months and 8.6 months, respectively), with 43% of patients alive after 12 months of follow-up.68 This study points to the direction of possible benefits with the combination of anti-VEGF and immunotherapy. A recent study failed to show survival advantage with combination of bevacizumab and temozolomide.69
Buparlisib (BKM120), a PI3K inhibitor, has been shown to have activity in vivo and in vitro against melanoma brain metastases.70 More studies need to be done to assess the possible combination with other established therapies.
Oblimersen is an antisense oligonucleotide that suppresses B-cell lymphoma-2, thereby suppressing its anti-apoptotic effect. The triple combination of oblimersen with temozolomide and albumin-bound paclitaxel has shown to be safe and efficacious in a phase 1 trial, thereby creating a need for further clinical trials.71
Treatment Approach
Systemic therapy for metastatic melanoma depends on several factors, including BRAF mutation status, functional status of the patient, disease burden, and severity of symptoms. Assessing the BRAF mutation status has become an important component in the management of patients with metastatic melanoma. It can help recognize patients who will benefit from molecular targeted therapy. In case of a BRAF-positive melanoma, treatment can be initiated with either immunotherapy or BRAF inhibitors. There are no randomized studies comparing immunotherapy to molecular targeted therapy.
Patients who have good PS and lymph node metastases can be treated initially with IL-2, which has the advantage of inducing cure in a minority of patients but should only be considered in patients with well-preserved organ function who can be monitored in an intensive care setting. On the other hand, patients who have bulky, symptomatic disease and poor PS should be treated initially with BRAF inhibitors. Combination of BRAF and MEK inhibitors can also be used and has an improved PFS and OS with potential to cause early tumor regression. There are studies to suggest suboptimal outcomes in patients who are treated with ipilimumab after progression on a BRAF inhibitor compared with initial treatment with ipilimumab followed by a BRAF inhibitor.72-74 However, all these studies are retrospective and there is no prospective data to suggest the above. BRAF mutation-positive patients who progress on a BRAF inhibitor
can be treated with PD-1 inhibitors.
Patients who do not have a BRAF mutation are unlikely to benefit from a BRAF inhibitor and primarily receive immunotherapy with ipilimumab or IL-2. Whenever possible, such patients should be enrolled in a clinical trial, as they have a poor prognosis. Patients who progress on ipilimumab can be treated with one of the PD-1 inhibitors (pembrolizumab, nivolumab). These PD-L1 inhibitors are still being investigated for use in such situations.
The role of chemotherapy in the management of metastatic melanoma has been limited by numerous studies showing significantly better survival with immunotherapy and molecular targeted therapy. Dacarbazine is the only FDA-approved drug for the treatment of melanoma. Its use is reserved mainly for patients who are not candidates for any of the other therapies available, including enrollment in a clinical trial.
Conclusion
Therapies for metastatic melanoma are in a state of flux. In the past decade, several new therapeutic agents have been introduced for the management of this potentially lethal disease. The treatment of metastatic melanoma has gradually shifted from cytotoxic chemotherapy toward a more individualized treatment that has a definite survival advantage over traditional counterparts. The advent of novel therapies has led to initiation of further studies to determine their role in the treatment of advanced melanoma, singly or in combination with other agents. In addition to evaluating new agents, more studies are needed to compare existing treatment modalities so that definitive treatment protocols can be formulated.
Acknowledgement
The authors would like to thank Felicia Ratnaraj, MD, for her assistance in creating the figures.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
Melanoma is the most aggressive form of skin cancer, contributing to about 76,000 new cases and more than 9,000 deaths in 2014.1 Depending on the stage of the disease, 5-year melanoma survival can range from 15% to 97%. Patients with local and distant metastases have a 5-year survival of about 60% and 15%, respectively.2
The incidence of melanoma is rising, partly because of the increasing number of skin biopsies being performed.3 If melanoma is diagnosed early, surgical excision is the treatment of choice. In patients with oligometastatic disease (cancer that has spread, but only to 1 or a small number of sites), complete surgical excision of the metastases may provide prolonged overall survival (OS) and delay the need to use systemic therapy.4
Recently, many new drug therapies have shown promising results in clinical trials, which may improve the prognosis of metastatic disease. This article reviews currently available systemic treatment options for the management of metastatic melanoma, the role of cytotoxic chemotherapy and interleukin-2 (IL-2), and the latest therapies available, including immune checkpoint inhibitors.
Cytotoxic Chemotherapy and Interleukin-2
Cytotoxic chemotherapy does not have an established role in the initial treatment of metastatic melanoma. Currently, cytotoxic chemotherapy is used in patients who have not responded to immunotherapy or molecular targeted therapy. The most commonly used drugs include dacarbazine and its prodrug, temozolomide. Several studies have failed to demonstrate a survival benefit using a single-agent chemotherapy with either dacarbazine or temozolomide.5,6
Other agents used in metastatic melanoma include nitrosoureas (fotemustine), platinum compounds (cisplatin, carboplatin), vinca alkaloids (vincristine),
and taxanes (paclitaxel). None of these agents provide a survival benefit, but an objective response may be seen in a minority of cases. Combination chemotherapy regimens have not shown an advantage over singleagent dacarbazine or temozolomide.7,8
High-dose IL-2 has been used in cases of metastatic melanoma with good performance status (PS) and organ function. Studies have shown a complete response rate of 3% to 7% and a prolonged disease-free survival in a minority of patients.9-11 The use of highdose IL-2, however, is limited by the high incidence of adverse effects (AEs), which include bacterial sepsis, pulmonary edema, arrhythmias, fever, and on some occasions, death due to complications.10 The use of IL-2 requires admission of the patient to a specialized unit for AE monitoring and management. Because of its ability to “cure” a minority of patients, a role still exists for IL-2 therapy in the treatment of younger, healthy patients with no evidence of organ dysfunction at baseline.
Immune Checkpoint Inhibitors
Checkpoint inhibitors are a class of drugs that unmask the immune system to fight against cancer cells. This class of drugs has shown significant activity and survival advantage in recent phase 2 and 3 trials. The class includes the anticytotoxic T-lymphocyte antigen 4 (CTLA-4) antibody ipilimumab and monoclonal antibodies targeting the programmed death 1 protein (PD-1) or its ligand (PD-L1).
Anti-CTLA-4 Antibodies: Ipilimumab
Cytotoxic T-lymphocyte antigen 4 is the antigen responsible for inhibition of cytotoxic T-cell-mediated immunity against foreign antigens presented by the antigen presenting cells (APCs). The APCs cause activation of the T cells when peptide fragments of intracellular proteins are presented in combination with mixed histocompatibility complex molecules. This step requires interaction of a costimulatory molecule (B7) on the APCs with a cluster of differentiation 28 protein (CD28) receptor located on T cells. CTLA-4 competes with CD28 to bind with the B7 molecule, thereby inhibiting the activation of the cytotoxic T cells (Figure 1). This pathway is thought to help with development of tolerance to host tissue antigens. Ipilimumab is a human monoclonal antibody that inhibits this CTLA-4 molecule and facilitates T-cell mediated antitumor activity.12 By blocking the CTLA-4 molecule, ipilimumab also mediates its autoimmune AEs on the host tissues.
Hodi and colleagues conducted a phase 3 trial of ipilimumab, including 676 patients who progressed after prior treatment for stage III or IV melanoma, and found that median OS was significantly better in the ipilimumab groups: 10 months in the ipilimumab plus gp100 peptide vaccine group vs 6.4 months in the gp100 vaccine alone group; 10.1 months in the ipilimumab alone group vs 6.4 months in the gp100 vaccine alone group.13 In another phase 3 trial comparing ipilimumab plus dacarbazine to dacarbazine alone, the ipilimumab group had a significantly improved OS (11.2 months vs 9.1 months).1 Survival rates with ipilimumab were prolonged for up to 3 years compared with the dacarbazine plus placebo group. However, the combination was associated with increased incidence of hepatotoxicity, thereby limiting its use.
A long-term survival analysis of 10 prospective and 2 retrospective studies of ipilimumab showed a median OS of 11.4 months and a long-term survival that began at 3 years with a plateau at 10 years of 21%, which was independent of prior therapy or ipilimumab dose.14 The immune-related AEs of ipilimumab are secondary to its activity against the host antigens and include dermatitis, enterocolitis, hepatitis, and endocrinopathies.15
A recent phase 2 trial studied the combination of ipilimumab with granulocyte-macrophage colonystimulating factor in 245 patients with stage III and IV melanoma. Median OS after 13 months was significantly higher with the combination compared with ipilimumab alone. The 1-year survival rate was 69% with
the combination and 53% with ipilimumab alone. There was no difference in the overall response rate (ORR) or progression-free survival (PFS) between the 2 groups. However, the AEs were significantly reduced with the combination (45% vs 58%).16 The dose of ipilimumab used in the trial was higher than the approved dose, making it difficult to apply the results in practice without further studies on the combination.
Anti-PD-1 Antibodies
Programmed death 1 ligands (PD-L1 and PD-L2) are expressed by tumor or stromal cells to inhibit the T-cell mediated antitumor activity. These ligands bind to the PD-1 protein on the surface of activated T cells to mediate their immunosuppressive effects. Interruption of this interaction by either anti-PD-1 antibodies or anti-PD-L1 antibodies facilitates tumor cell killing by activated T cells.17
Pembrozilumab and nivolumab are the 2 anti-PD-1 monoclonal antibodies that have been approved for treatment of metastatic melanoma. In a phase 1 trial
of pembrolizumab, 411 patients with advanced melanoma (consisting of both ipilimumab-naïve [IPI-N] and ipilimumab-treated [IPI-T] patients), ORR was 40% in IPI-N and 28% in IPI-T patients with a 1-year OS of 71% in all patients. Median PFS was 24 weeks in IPI-N and 23 weeks in IPI-T pts.18 There was no difference in outcomes and safety profiles across the various dosing regimens.18,19 Of note, pembrolizumab had antitumor activity irrespective of the PS, lactate dehydrogenase levels, BRAF (B-Raf proto-oncogene, serine/threonine kinase) gene mutation, metastatic stage, and number and type of prior therapy. In a subgroup analysis, 173 patients who had progression after treatment with ipilimumab were randomly assigned to pembrolizumab 2 mg/kg every 3 weeks (q3w) or 10 mg/kg q3w dosing regimens. Both groups had no significant difference in the ORR (26% in both) and safety profiles.20
In the 2012 KEYNOTE-002 clinical trial, a randomized phase 2 trial involving 540 patients with ipilimumab-refractory advanced melanoma, patients were randomized 1:1:1 to pembrolizumab 2 mg/kg or 10 mg/kg q3w or investigator-choice chemotherapy (control arm consisting of carboplatin plus paclitaxel, carboplatin, paclitaxel, dacarbazine, or temozolomide). The 6-month PFS was significantly improved with pembrolizumab (34% and 38% for pembrolizumab 2 mg/kg and 10 mg/kg, respectively) compared with 16% with chemotherapy. The ORR was significantly better with pembrolizumab (21% at 2 mg/kg, 25% at 10 mg/kg) compared with the control arm (4%).21 These findings led to the approval of pembrolizumab by the FDA for treatment of patients with advanced melanoma who have progressed on ipilimumab. Pembrolizumab is generally well tolerated. The most common AEs include fatigue, pruritus, and rash.
Nivolumab was studied in a recent phase 1 trial in which 107 patients with previously treated advanced melanoma were treated with escalated doses every
2 weeks.22 The 2-year and 3-year OS rates were 48% and 41%, respectively. Objective responses were seen in 32% of the patients. The median response duration was 23 months.23
The first phase 3 trial was conducted in 418 patients with previously untreated metastatic melanoma BRAF mutation. Patients were randomized to receive either nivolumab or dacarbazine. The PFS and OS were significantly better with nivolumab compared with dacarbazine (PFS 5.1 months vs 2.2 months; OS 73% vs 42% at 1 year).24 The AE profile of nivolumab is similar to pembrolizumab and includes lung, skin, endocrine, renal, and gastrointestinal tract toxicities.
Preliminary results of another phase 3 trial were presented at the European Society of Medical Oncology 2014 meeting. Patients with previously treated metastatic melanoma (ipilimumab or BRAF inhibitor) were randomized in a 2:1 ratio to receive either nivolumab or investigators’ choice chemotherapy (dacarbazine or carboplatin plus paclitaxel). The ORR was significantly better with nivolumab (32% vs 11%), and 95% of patients were still responding after 6 months. The nivolumab group showed a complete remission in 3% of the patients with 34% of the responses lasting ≥ 6 months.25 This led to the recent approval of nivolumab for patients with metastatic melanoma with a BRAF mutation who have advanced on ipilimumab. In the phase 3 NCT01844505 trial patients are being randomized to receive ipilimumab, nivolumab, or both.
A newer PD-1 inhibitor, pidilizumab, was studied in a phase 2 trial that included 103 patients with metastatic melanoma, 51% of whom had received therapy with ipilimumab. The ORR in the study group was relatively lower (6%), but the OS at 1 year was 64.5%.26 Further studies are underway to evaluate the role of this drug in metastatic melanoma.
The response with both nivolumab and pembrolizumab is durable as well as sustained, even after discontinuation of therapy. None of the deaths in the aforementioned studies were atributed to drug-related toxicities. As evidenced by current data, these 2 drugs hold a great promise for the management of patients who progress after therapy with anti-CTLA-4 antibodies.
Anti-PD-L1 Antibodies
The anti-PD-L1 monoclonal antibodies work in a similar way to the PD-1 inhibitors and block the interaction between the PD-1 and its ligand, PD-L1. This causes sustained activation of cytotoxic T cells and facilitates their antitumor activity. Two of PD-L1 inhibitors have shown clinical activity against metastatic melanoma.
BMS-936559, the first PD-L1 antibody, is being studied in a phase 1 trial that includes 55 patients with advanced melanoma along with 152 patients with other solid malignancies. Three patients achieved a complete response, and 5 patients had an objective response lasting 1 year. The ORR for melanoma was 17%, with disease stabilization of ≥ 24 weeks in 27% of the patients.27 Common AEs included infusion reactions, diarrhea, fatigue, rash, hypothyroidism, and hepatitis.
The second PD-L1 antibody, MPDL3280A, was studied in a phase 1 trial of 45 patients with metastatic melanoma. An ORR of 29% was observed, along with a 24-week PFS of 43%.28 Commonly noted AEs included hyperglycemia and elevated liver aminotransferases.
A newer PD-L1 inhibitor, MEDI4736, is being studied for advanced malignancies in 8 patients with melanoma. In preliminary analysis, MEDI4736 demonstrated a partial response in 1 out of 8 melanoma patients with a disease control rate of 46%.29 Although the PD-L1 inhibitors seem promising, more information will help discern their role in the management of metastatic melanoma.
Combined Anti-CTLA-4 Plus Anti-PD-1 Antibody
The combination of ipilimumab and the PD-1 inhibitor nivolumab was tested in a phase 1 trial in which both drugs were used concurrently as well as sequentially in metastatic melanoma.30 The 1- and 2-year OS in patients who were treated concurrently was 82% and 75%, respectively. Complete remission was seen in 17% of the patients, and the responses were seen irrespective of the BRAF mutation status. The responses were durable, and about 64% of the objective responses remain in remission at last follow-up.31 Grade 3 to grade 4 AEs were noted in 53% of the patients, with 11 patients requiring discontinuation of the medications. More studies are required to ascertain the optimum dosage of the combination prior to its approval for use in metastatic melanoma.
Molecular Targeted Therapy
The RAS-RAF–mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) signaling pathway is activated in almost 90% of patients
with melanoma.32 This pathway is normally required for the growth and survival of nonmalignant cells. In malignant transformation, mutations and/or overexpression is seen at various levels including KIT, NRAS, BRAF, and the MEK protein. This leads to activation of serine and threonine protein kinases, which lead to uncontrolled cell proliferation and survival.33
Novel therapeutic approaches have tried inhibiting one or more of these pathways for melanoma treatment. The most important mediator of tumorigenesis is BRAF, which is a downstream receptor of NRAS, and is mutated in almost 50% of melanoma cases.34 NRAS mutations are seen in 15% to 20% of cutaneous melanomas.35,36 After its activation, the RAF enzyme—coded by the BRAF gene—causes phosphorylation of the MEK protein, which activates ERK. This ERK activation leads to growth signaling and is the final pathway in several malignancies (Figure 2).37,38
BRAF Inhibitors
BRAF is the first mediator whose inhibition led to clinically significant outcomes in patients with melanoma. The most common BRAF mutation consists of the
substitution of glutamic acid for valine at amino acid 600 (V600E mutation) with majority of the remainder consisting of an alternate substitution (V600V or V600K).34 Vemurafenib and dabrafenib are the 2 BRAF inhibitors that have been shown to improve tumor regression, PFS, and OS considerably, especially in combination with a MEK protein inhibitor. In the phase 3 BRIM-3 trial, the vemurafenib group had a significantly prolonged PFS and OS compared with dacarbazine (13.6 months vs 9.7 months; 6.9 months vs 1.6 months, respectively). It was the first study to show improved survival with vemurafenib in both the V600E and V600K BRAF mutant melanomas.39
Another BRAF inhibitor, dabrafenib, was approved by the FDA for treatment of advanced melanoma with BRAF V600E mutation. It was tested in a phase 3 trial in which it was compared with dacarbazine in patients with advanced melanoma. Median OS in the dabrafenib arm was > 18 months and in dacarbazine arm > 15 months.40 Fifty-seven percent of the patients in dacarbazine arm were crossed over to the dabrafenib arm, thereby confounding the survival data for the former group. Another multicenter, phase 2 trial showed dabrafenib to have activity in melanoma patients with brain metastases, irrespective of previous therapy for the brain metastases.41 The long-term analysis of the BREAK-2 trial, which included 92 patients with metastatic melanoma treated with dabrafenib, showed a median OS of 12.9 months in BRAF V600K group and 13.1 months in BRAF V600E group.42
Adverse effects associated with BRAF inhibition include fatigue, rash, arthralgia, and photosensitivity reactions.43 Dermatologic complications may also include squamous cell carcinoma (SCC) (19%-26%), with keratoacanthoma being the most common subtype.44 These are believed to be likely secondary to the paradoxical activation of the MAPK signaling, since most of these lesions are found to have mutations in the RAS molecule.45 Other specific AEs of dabrafenib include hyperkeratosis (33%) and pyrexia (29%).42
Most patients treated with a BRAF inhibitor eventually have disease progression, likely secondary to reactivation of the MAPK pathway.46,47 This result has led to a heightened interest in combination therapies in an effort to improve outcomes. Combination therapy with ipilimumab and vemurafenib was studied and resulted in a higher incidence of hepatotoxicity (50%).48 However, no hepatotoxicity was seen in a phase 1 trial of combined dabrafenib and ipilimumab.49
Some studies have also suggested that extended BRAF inhibition after progression on a BRAF inhibitor may prolong survival.50,51 The phase 2 trial NCT01983124 is being conducted to evaluate the survival benefit with a combination of vemurafenib and a nitrosourea alkylating agent, fotemustine, in patients who have progressed on vemurafenib alone.
MEK Inhibitors
The inhibition of MEK can halt cell proliferation and induce apoptosis. The phase 3 METRIC trial, which compared the oral MEK inhibitor (trametinib) with chemotherapy, was conducted in 322 patients who had metastatic melanoma with a V600E or V600K BRAF mutation. The PFS and 6-month OS were significantly better with trametinib (4.8 months vs 1.5 months, 81% vs 66%) despite the crossover between the 2 groups.52 The AEs associated with trametinib included rash, diarrhea, and peripheral edema. Another phase 2 trial of trametinib including patients pretreated with a BRAF inhibitor showed no confirmed objective responses, 28% patients with stable disease, and minimal improvement in PFS (2 months). Among patients treated with prior chemotherapy and/or immunotherapy, trametinib showed significant improvement in complete responses, partial responses, stable disease, and the median PFS (2%, 23%, 51%, 4 months, respectively).53
The second MEK inhibitor, binimetinib, was studied in a phase 2 trial of advanced melanoma cases harboring a BRAF V600E or NRAS. Bimetinib demonstrated a PR in 20% cases of both the BRAF and NRAS mutant melanomas. Durable disease control was seen in 43% of the NRAS group and 32% of the BRAF group.54 The AE profile was similar to that seen with trametinib. Bimetinib is being studied in phase 1 and 2 trials with the CDK4/6 inhibitor as well as in the phase 3 trial NCT01763164 compared with dacarbazine in NRAS mutation positive melanomas.55
Selumetinib is a MEK inhibitor that has been compared with dacarbazine and temozolomide with no significant OS advantage. A novel highly specific inhibitor of MEK, cobimetinib, is currently being studied in combination with BRAF inhibitors.
Combined BRAF and MEK Inhibition
A randomized, double-blind, phase 3 study comparing the combination of dabrafenib and trametinib with dabrafenib and placebo in patients with advanced melanoma with a BRAF V600E mutation was presented at the 2014 American Society of Clinical Oncology meeting. Researchers found that after a median follow-up period of 9 months, there was a significant improvement with the combination in the PFS (9.3 months vs 8.8 months) and the ORR (67% vs 51%), with a similar incidence of AEs.56 The combination therapy group had fewer incidences of SCC of the skin but more incidence of pyrexia.
The combination of dabrafenib and trametinib was compared with vemurafenib monotherapy in a recent randomized phase 3 trial among 704 metastatic melanoma patients with a BRAF V600 mutation. Median PFS and ORR were significantly better with combination therapy compared with vemurafenib alone (11.4 months vs 7.3 months, 64% vs 51%, respectively). Overall survival rate at 1 year was significantly improved in the combination group as well (72% vs 65%).57 The incidence of SCC and keratoacanthoma was less in the combination (1%) compared with vemurafenib alone (18%). Another study investigating the coadministration and sequential administration of vemurafenib and trametinib is underway.58
The vemurafenib and cobimetinib combination was studied in a phase 3 trial of previously untreated unresectable locally advanced or metastatic BRAF V600
mutation-positive melanoma. The median PFS was 9.9 months in the combination group and 6.2 months in the control group. The interim analysis showed a 9-month survival rate of 81% in the combination group and 73% in the control group, with no significantly higher incidence of AEs in either arm.59 A longer follow-up will be needed to assess the OS benefit with the combination.
Encorafenib, a selective BRAF inhibitor, has been studied in a phase 1 trial in combination with binimetinib.60 This trial has paved the way to the initiation of a currently ongoing phase 3 trial (NCT01909453) comparing the combination with vemurafenib or encorafenib alone.
C-KIT Inhibitors
Mutations of c-KIT are seen more commonly in chronic sun damage-induced cutaneous melanomas, along with acral and mucosal melanomas.61,62 Earlier trials involving patients without selection for c-KIT mutation positivity failed to show benefit with imatinib. A single-arm, phase 2 trial of imatinib mesylate in patients with metastatic melanoma harboring the c-KIT mutation, an ORR of 23% was achieved, with a median PFS of 3.5 months.63 Imatinib showed an ORR of 29% in a phase 2 trial of mucosal, acral, and in chronic sun damage-induced melanoma patients with c-KIT amplifications and/or mutations. It was demonstrated that c-KIT amplification alone is not as responsive to imatinib compared with c-KIT mutation, suggesting that all patients with these specific melanomas should be tested for KIT mutation status.64
A second-generation c-KIT inhibitor, nilotinib, has shown some promising results with a favorable AE profile in small phase 2 trials.65,66 However, more clinical research will be needed before definite recommendations on its use in cutaneous melanomas can be made. Currently, its role seems to be limited to the management of acral, mucosal, and chronic sun damage-related melanomas with c-KIT mutations.
Future Directions
Angiogenesis promoters, such as vascular endothelial growth factor (VEGF), platelet-derived growth factor, fibroblast growth factor, and interleukin-8, are overexpressed in melanoma. Bevacizumab, an anti-VEGF antibody, has been shown to have some benefit in combination with carboplatin and paclitaxel as a triple therapy.67 However, grade 3 AEs were seen in a portion of patients.
The phosphatidylinositol-3 kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway has also been studied as a target for melanoma therapy. Everolimus, an mTOR inhibitor, was studied in a phase 2 trial in combination with bevacizumab for treatment of metastatic melanoma. The combination showed improved median PFS and OS with the combination (4 months and 8.6 months, respectively), with 43% of patients alive after 12 months of follow-up.68 This study points to the direction of possible benefits with the combination of anti-VEGF and immunotherapy. A recent study failed to show survival advantage with combination of bevacizumab and temozolomide.69
Buparlisib (BKM120), a PI3K inhibitor, has been shown to have activity in vivo and in vitro against melanoma brain metastases.70 More studies need to be done to assess the possible combination with other established therapies.
Oblimersen is an antisense oligonucleotide that suppresses B-cell lymphoma-2, thereby suppressing its anti-apoptotic effect. The triple combination of oblimersen with temozolomide and albumin-bound paclitaxel has shown to be safe and efficacious in a phase 1 trial, thereby creating a need for further clinical trials.71
Treatment Approach
Systemic therapy for metastatic melanoma depends on several factors, including BRAF mutation status, functional status of the patient, disease burden, and severity of symptoms. Assessing the BRAF mutation status has become an important component in the management of patients with metastatic melanoma. It can help recognize patients who will benefit from molecular targeted therapy. In case of a BRAF-positive melanoma, treatment can be initiated with either immunotherapy or BRAF inhibitors. There are no randomized studies comparing immunotherapy to molecular targeted therapy.
Patients who have good PS and lymph node metastases can be treated initially with IL-2, which has the advantage of inducing cure in a minority of patients but should only be considered in patients with well-preserved organ function who can be monitored in an intensive care setting. On the other hand, patients who have bulky, symptomatic disease and poor PS should be treated initially with BRAF inhibitors. Combination of BRAF and MEK inhibitors can also be used and has an improved PFS and OS with potential to cause early tumor regression. There are studies to suggest suboptimal outcomes in patients who are treated with ipilimumab after progression on a BRAF inhibitor compared with initial treatment with ipilimumab followed by a BRAF inhibitor.72-74 However, all these studies are retrospective and there is no prospective data to suggest the above. BRAF mutation-positive patients who progress on a BRAF inhibitor
can be treated with PD-1 inhibitors.
Patients who do not have a BRAF mutation are unlikely to benefit from a BRAF inhibitor and primarily receive immunotherapy with ipilimumab or IL-2. Whenever possible, such patients should be enrolled in a clinical trial, as they have a poor prognosis. Patients who progress on ipilimumab can be treated with one of the PD-1 inhibitors (pembrolizumab, nivolumab). These PD-L1 inhibitors are still being investigated for use in such situations.
The role of chemotherapy in the management of metastatic melanoma has been limited by numerous studies showing significantly better survival with immunotherapy and molecular targeted therapy. Dacarbazine is the only FDA-approved drug for the treatment of melanoma. Its use is reserved mainly for patients who are not candidates for any of the other therapies available, including enrollment in a clinical trial.
Conclusion
Therapies for metastatic melanoma are in a state of flux. In the past decade, several new therapeutic agents have been introduced for the management of this potentially lethal disease. The treatment of metastatic melanoma has gradually shifted from cytotoxic chemotherapy toward a more individualized treatment that has a definite survival advantage over traditional counterparts. The advent of novel therapies has led to initiation of further studies to determine their role in the treatment of advanced melanoma, singly or in combination with other agents. In addition to evaluating new agents, more studies are needed to compare existing treatment modalities so that definitive treatment protocols can be formulated.
Acknowledgement
The authors would like to thank Felicia Ratnaraj, MD, for her assistance in creating the figures.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review the complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin.2014;64(1):9-29.
2. Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27(36):6199-6206.
3. Welch HG, Woloshin S, Schwartz LM. Skin biopsy rates and incidence of melanoma:
population based ecological study. BMJ. 2005;331(7515):481.
4. Sosman JA, Moon J, Tuthill RJ, et al. A phase 2 trial of complete resection for stage IV melanoma: results of Southwest Oncology Group Clinical Trial S9430. Cancer. 2011;117(20):4740-4746.
5. Atkins MB. The role of cytotoxic chemotherapeutic agents either alone or in combination with biological response modifiers. In: Kirkwood JK, ed. Molecular Diagnosis, Prevention, & Therapy of Melanoma. New York, NY: Marcel Dekker;1997:219-225.
6. Patel PM, Suciu S, Mortier L, et al. Extended schedule, escalated dose temozolomide versus dacarbazine in stage IV melanoma: final results of a randomised phase III study (EORTC 18032). Eur J Cancer. 2011;47(10):1476-1483.
7. Chapman PB, Einhorn LH, Meyers ML, et al. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol. 1999;17(9):2745-2751.
8. Flaherty KT, Lee SJ, Zhao F, et al. Phase III trial of carboplatin and paclitaxel with
or without sorafenib in metastatic melanoma. J Clin Oncol. 2013;31(3):373-379.
9. Rosenberg SA, Yang JC, Topalian SL, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271(12):907-913.
10. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105-2116.
11. Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J Sci Am. 2000;6(suppl 1):S11-S14.
12. Hoos A, Ibrahim R, Korman A, et al. Development of ipilimumab: contribution to a new paradigm for cancer immunotherapy. Semin Oncol. 2010;37(5):533-546.
13. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711-723.
14. Schadendorf D, Hodi FS, Robert C, et. al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma [published online ahead of print February 9, 2015]. J Clin Oncol. pii:JCO.2014.56.2736.
15. Weber JS, Kähler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691-2697.
16. Hodi FS, Lee S, McDermott DF, et al. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: a randomized clinical trial. JAMA. 2014;312(17):1744-1753.
17. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443-2454.
18. Ribas A, Hodi FS, Kefford R, et al. Efficacy and safety of the anti-PD-1 monoclonal antibody pembrolizumab (MK-3475) in 411 patients (pts) with melanoma (MEL) (Abstract LBA9000). Paper presented at: 2014 American Society of Clinical Oncology (ASCO) meeting; May-June 2014; Chicago, IL.
19. Hamid O, Robert C, Ribas A, et al. Randomized comparison of two doses of the anti-PD-1 monoclonal antibody MK-3475 for ipilimumab-refractory (IPI-R) and IPI-naive (IPI-N) melanoma (MEL) (abstract 3000). Paper presented at: 2014 American Society of Clinical Oncology (ASCO) meeting; May-June 2014; Chicago, IL.
20. Robert C, Ribas A, Wolchok JD, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014; 384(9948):1109-1117.
21. Dummer R, Daud A, Puzanov I, et. al. A randomized controlled comparison of pembrolizumab and chemotherapy in patients with ipilimumab-refractory melanoma. J Transl Med. 2015;13(suppl 1):O5.
22. Topalian SL, Sznol M, McDermott DF, et. al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32(10):1020-1030.
23. Hodi FS, Sznol M, Kluger HM, et al. Long-term survival of ipilimumab-naive patients with advanced melanoma (MEL) treated with nivolumab (anti-PD-1, BMS-936558, ONO-4538) in a phase I trial (abstract 9002). Paper presented at: 2014 American Society of Clinical Oncology (ASCO) meeting; May-June 2014; Chicago, IL.
24. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320-330.
25. Weber J, D’Angelo S, Gutzmer R, et al. A phase 3 randomized, open-label study of nivolumab versus investigator’s choice of chemotherapy in patients with advanced melanoma after prior anti-CTLA4 therapy (abstract LBA3). Paper presented at: European Society of Medical Oncology 2014 meeting; September 2014; Madrid, Spain.
26. Atkins MB, Kudchadkar RR, Sznol M, et al. Phase 2, multicenter, safety and efficacy study of pidilizumab in patients with metastatic melanoma (abstract 9001). Paper presented at: 2014 American Society of Clinical Oncology (ASCO) meeting; May-June 2014; Chicago, IL.
27. Brahmer JR, Tykodi SS, Chow LQM, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455-2465.
28. Hamid O, Sosman JA, Lawrence DP, et. al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic melanoma (mM). J Clin Oncol. 2013;31(15)(suppl): Abstract 9010.
29. Lutzky J, Antonia SJ, Blake-Haskins A, et. al. A phase 1 study of MEDI4736, an anti–PD-L1 antibody, in patients with advanced solid tumors. J Clin Oncol. 2014;32(15)(suppl): Abstract 3001.
30. Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced
melanoma. N Engl J Med. 2013;369(2):122-133.
31. Sznol M, Kluger HM, Callahan MK, et al. Survival, response duration, and activity by BRAF mutation (MT) status of nivolumab (NIVO, anti-PD-1, BMS-936558, ONO-4538) and ipilimumab (IPI) concurrent therapy in advanced melanoma (MEL) (abstract LBA9003). Paper presented at: 2014 American Society of Clinical Oncology (ASCO) meeting; May-June 2014; Chicago, IL.
32. Omholt K, Platz A, Kanter L, Ringborg U, Hansson J. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9(17):6483-6488.
33. Wellbrock C, Hurlstone A. BRAF as therapeutic target in melanoma. Biochem Pharmacol. 2010;80(5):561-567.
34. Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29(10):1239-1246.
35. Ball NJ, Yohn JJ, Morelli JG, et al. Ras mutations in human melanoma: a marker of malignant progression. J Invest Dermatol. 1994;102(3):285-290.
36. Platz A, Ringborg U, Brahme EM, Lagerlöf B. Melanoma metastases from patients with hereditary cutaneous malignant melanoma contain a high frequency of N-ras activating mutations. Melanoma Res. 1994;4(3):169-177.
37. Beeram M, Patnaik A, Rowinsky EK. Raf: a strategic target for therapeutic development against cancer. J Clin Oncol. 2005;23(27):6771-6790.
38. Terai K, Matsuda M. The amino-terminal B-Raf-specific region mediates calcium-dependent homo- and hetero-dimerization of Raf. EMBO J. 2006;25(15):3556-3564.
39. McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15(3):323-332.
40. Hauschild A, Grob JJ, Demidov LV, et al. An update on BREAK-3, a phase III, randomized trial: dabrafenib versus dacarbazine in patients with BRAF V600E-positive mutation metastatic melanoma (Abstract 9013). Paper presented at: American Society of Clinical Oncology 2013 meeting; May-June 2013; Chicago, IL.
41. Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087-1095.
42. Ascierto PA, Minor DR, Ribas A, et. al., Long-term safety and overall survival update for BREAK-2, a phase 2, single-arm, open-label study of dabrafenib in previously treated metastatic melanoma (NCT01153763). J Clin Oncol. 2014;32(15)(suppl): Abstract 9034.
43. Larkin J, Del Vecchio M, Ascierto PA, et al. Vemurafenib in patients with
BRAF(V600) mutated metastatic melanoma: an open-label, multicentre, safety
study. Lancet Oncol. 2014;15(4):436-444.
44. Lacouture ME, Duvic M, Hauschild A, et al. Analysis of dermatologic events in vemurafenib-treated patients with melanoma. Oncologist. 2013;18(3):314-322.
45. Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366(3):207-215.
46. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507-2516.
47. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358-365.
48. Ribas A, Hodi FS, Callahan M, et. al. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2014;368(14):1365-1366.
49. Linette GP, Puzanov I, Callahan MK, et al. Phase 1 study of the BRAF inhibitor dabrafenib (D) with or without the MEK inhibitor trametinib (T) in combination with ipilimumab (Ipi) for V600E/K mutation–positive unresectable or metastatic melanoma (MM). J Clin Oncol. 2014;32(15)(suppl): Abstract 2511.
50. Chan MMK, Haydu LE, Menzies AM, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer. 2014;120(20):3142-3153.
51. Carlino MS, Gowrishankar K, Saunders CAB, et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther. 2013;12(7):1332-1342.
52. Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107-114.
53. Kim KB, Kefford R, Pavlick AC, et. al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. 2013;31(1):482-489.
54. Ascierto PA, Schadendorf D, Berking C, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14(3):249-256.
55. Sosman JA, Kittaneh M, Lolkema MP, et al. A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS-mutant melanoma: early encouraging clinical activity (abstract 9009). Paper presented at: 2014 American Society of Clinical Oncology meeting ; May-June 2014; Chicago, IL.
56. Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371(20):1877-1888.
57. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30-39.
58. Gogas H, Schadendorf D, Dummer R. Vemurafenib treatment in patients with BRAF-mutated melanoma failing MEK inhibition with trametinib. J Clin Oncol. 2014;32(15)(suppl): Abstract 9061.
59. Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867-1876.
60. Kefford R, Miller WH, Tan DS, et al. Preliminary results from a phase Ib/II, openlabel, dose-escalation study of the oral BRAF inhibitor LGX818 in combination with the oral MEK1/2 inhibitor MEK162 in BRAF V600-dependent advanced solid tumors (abstract 9019). Paper presented at: 2013 American Society of Clinical Oncology meeting; May-June 2014; Chicago, IL.
61. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct
subtypes of melanoma. J Clin Oncol. 2006;24(26):4340-4346.
62. Jin SA, Chun SM, Choi YD, et al. BRAF mutations and KIT aberrations and their clinicopathological correlation in 202 Korean melanomas. J Invest Dermatol. 2013;133(2):579-582.
63. Guo J, Si L, Kong Y et. al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29(21):2904-2909.
64. Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol. 2013;31(26):3182-3190.
65. Cho JH, Kim KM, Kwon M, Kim JH, Lee J. Nilotinib in patients with metastatic melanoma harboring KIT gene aberration. Invest New Drugs. 2012;30(5): 2008-2014.
66. Lebbe C, Chevret S, Jouary T, et. al. Phase II multicentric uncontrolled national trial assessing the efficacy of nilotinib in the treatment of advanced melanomas with c-KIT mutation or amplification. J Clin Oncol. 2014;32(15)(suppl): Abstract 9032.
67. Perez DG, Suman VJ, Fitch TR, et al. Phase 2 trial of carboplatin, weekly paclitaxel, and biweekly bevacizumab in patients with unresectable stage IV melanoma: a North Central Cancer Treatment Group study, N047A. Cancer. 2009;115(1):119-127.
68. Hainsworth JD, Infante JR, Spigel DR, et al. Bevacizumab and everolimus in the treatment of patients with metastatic melanoma. Cancer. 2010;116(17): 4122-4129.
69. Dronca RS, Allred JB, Perez DG, et. al. Phase II study of temozolomide (TMZ) and everolimus (RAD001) therapy for metastatic melanoma: a North Central Cancer Treatment Group study, N0675. Am J Clin Oncol. 2014;37(4):369-376.
70. Meier FE, Niessner H, Schmitz J, et al. The PI3K inhibitor BKM120 has potent antitumor activity in melanoma brain metastases in vitro and in vivo. J Clin Oncol. 2013;31(15)(suppl): Abstract e20050.
71. Ott PA, Chang J, Madden K, et al. Oblimersen in combination with temozolomide and albumin-bound paclitaxel in patients with advanced melanoma: a phase I trial. Cancer Chemother Pharmacol. 2013;71(1);183-191.
72. Ackerman A, Klein O, McDermott DF, et al. Outcomes of patients with metastatic
melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. 2014;120(11):1695-1701.
73. Ascierto PA, Margolin K. Ipilimumab before BRAF inhibitor treatment may be
more beneficial than vice versa for the majority of patients with advanced melanoma.
Cancer. 2014;120(11):1617-1619.
74. Ascierto PA, Simeone E, Sileni VC, et al. Sequential treatment with ipilimumab and BRAF inhibitors in patients with metastatic melanoma: data from the Italian cohort of the ipilimumab expanded access program. Cancer Invest. 2014;32(4):144-149.
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin.2014;64(1):9-29.
2. Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27(36):6199-6206.
3. Welch HG, Woloshin S, Schwartz LM. Skin biopsy rates and incidence of melanoma:
population based ecological study. BMJ. 2005;331(7515):481.
4. Sosman JA, Moon J, Tuthill RJ, et al. A phase 2 trial of complete resection for stage IV melanoma: results of Southwest Oncology Group Clinical Trial S9430. Cancer. 2011;117(20):4740-4746.
5. Atkins MB. The role of cytotoxic chemotherapeutic agents either alone or in combination with biological response modifiers. In: Kirkwood JK, ed. Molecular Diagnosis, Prevention, & Therapy of Melanoma. New York, NY: Marcel Dekker;1997:219-225.
6. Patel PM, Suciu S, Mortier L, et al. Extended schedule, escalated dose temozolomide versus dacarbazine in stage IV melanoma: final results of a randomised phase III study (EORTC 18032). Eur J Cancer. 2011;47(10):1476-1483.
7. Chapman PB, Einhorn LH, Meyers ML, et al. Phase III multicenter randomized trial of the Dartmouth regimen versus dacarbazine in patients with metastatic melanoma. J Clin Oncol. 1999;17(9):2745-2751.
8. Flaherty KT, Lee SJ, Zhao F, et al. Phase III trial of carboplatin and paclitaxel with
or without sorafenib in metastatic melanoma. J Clin Oncol. 2013;31(3):373-379.
9. Rosenberg SA, Yang JC, Topalian SL, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271(12):907-913.
10. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105-2116.
11. Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J Sci Am. 2000;6(suppl 1):S11-S14.
12. Hoos A, Ibrahim R, Korman A, et al. Development of ipilimumab: contribution to a new paradigm for cancer immunotherapy. Semin Oncol. 2010;37(5):533-546.
13. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711-723.
14. Schadendorf D, Hodi FS, Robert C, et. al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma [published online ahead of print February 9, 2015]. J Clin Oncol. pii:JCO.2014.56.2736.
15. Weber JS, Kähler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691-2697.
16. Hodi FS, Lee S, McDermott DF, et al. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: a randomized clinical trial. JAMA. 2014;312(17):1744-1753.
17. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443-2454.
18. Ribas A, Hodi FS, Kefford R, et al. Efficacy and safety of the anti-PD-1 monoclonal antibody pembrolizumab (MK-3475) in 411 patients (pts) with melanoma (MEL) (Abstract LBA9000). Paper presented at: 2014 American Society of Clinical Oncology (ASCO) meeting; May-June 2014; Chicago, IL.
19. Hamid O, Robert C, Ribas A, et al. Randomized comparison of two doses of the anti-PD-1 monoclonal antibody MK-3475 for ipilimumab-refractory (IPI-R) and IPI-naive (IPI-N) melanoma (MEL) (abstract 3000). Paper presented at: 2014 American Society of Clinical Oncology (ASCO) meeting; May-June 2014; Chicago, IL.
20. Robert C, Ribas A, Wolchok JD, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014; 384(9948):1109-1117.
21. Dummer R, Daud A, Puzanov I, et. al. A randomized controlled comparison of pembrolizumab and chemotherapy in patients with ipilimumab-refractory melanoma. J Transl Med. 2015;13(suppl 1):O5.
22. Topalian SL, Sznol M, McDermott DF, et. al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32(10):1020-1030.
23. Hodi FS, Sznol M, Kluger HM, et al. Long-term survival of ipilimumab-naive patients with advanced melanoma (MEL) treated with nivolumab (anti-PD-1, BMS-936558, ONO-4538) in a phase I trial (abstract 9002). Paper presented at: 2014 American Society of Clinical Oncology (ASCO) meeting; May-June 2014; Chicago, IL.
24. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320-330.
25. Weber J, D’Angelo S, Gutzmer R, et al. A phase 3 randomized, open-label study of nivolumab versus investigator’s choice of chemotherapy in patients with advanced melanoma after prior anti-CTLA4 therapy (abstract LBA3). Paper presented at: European Society of Medical Oncology 2014 meeting; September 2014; Madrid, Spain.
26. Atkins MB, Kudchadkar RR, Sznol M, et al. Phase 2, multicenter, safety and efficacy study of pidilizumab in patients with metastatic melanoma (abstract 9001). Paper presented at: 2014 American Society of Clinical Oncology (ASCO) meeting; May-June 2014; Chicago, IL.
27. Brahmer JR, Tykodi SS, Chow LQM, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455-2465.
28. Hamid O, Sosman JA, Lawrence DP, et. al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic melanoma (mM). J Clin Oncol. 2013;31(15)(suppl): Abstract 9010.
29. Lutzky J, Antonia SJ, Blake-Haskins A, et. al. A phase 1 study of MEDI4736, an anti–PD-L1 antibody, in patients with advanced solid tumors. J Clin Oncol. 2014;32(15)(suppl): Abstract 3001.
30. Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced
melanoma. N Engl J Med. 2013;369(2):122-133.
31. Sznol M, Kluger HM, Callahan MK, et al. Survival, response duration, and activity by BRAF mutation (MT) status of nivolumab (NIVO, anti-PD-1, BMS-936558, ONO-4538) and ipilimumab (IPI) concurrent therapy in advanced melanoma (MEL) (abstract LBA9003). Paper presented at: 2014 American Society of Clinical Oncology (ASCO) meeting; May-June 2014; Chicago, IL.
32. Omholt K, Platz A, Kanter L, Ringborg U, Hansson J. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9(17):6483-6488.
33. Wellbrock C, Hurlstone A. BRAF as therapeutic target in melanoma. Biochem Pharmacol. 2010;80(5):561-567.
34. Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29(10):1239-1246.
35. Ball NJ, Yohn JJ, Morelli JG, et al. Ras mutations in human melanoma: a marker of malignant progression. J Invest Dermatol. 1994;102(3):285-290.
36. Platz A, Ringborg U, Brahme EM, Lagerlöf B. Melanoma metastases from patients with hereditary cutaneous malignant melanoma contain a high frequency of N-ras activating mutations. Melanoma Res. 1994;4(3):169-177.
37. Beeram M, Patnaik A, Rowinsky EK. Raf: a strategic target for therapeutic development against cancer. J Clin Oncol. 2005;23(27):6771-6790.
38. Terai K, Matsuda M. The amino-terminal B-Raf-specific region mediates calcium-dependent homo- and hetero-dimerization of Raf. EMBO J. 2006;25(15):3556-3564.
39. McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15(3):323-332.
40. Hauschild A, Grob JJ, Demidov LV, et al. An update on BREAK-3, a phase III, randomized trial: dabrafenib versus dacarbazine in patients with BRAF V600E-positive mutation metastatic melanoma (Abstract 9013). Paper presented at: American Society of Clinical Oncology 2013 meeting; May-June 2013; Chicago, IL.
41. Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087-1095.
42. Ascierto PA, Minor DR, Ribas A, et. al., Long-term safety and overall survival update for BREAK-2, a phase 2, single-arm, open-label study of dabrafenib in previously treated metastatic melanoma (NCT01153763). J Clin Oncol. 2014;32(15)(suppl): Abstract 9034.
43. Larkin J, Del Vecchio M, Ascierto PA, et al. Vemurafenib in patients with
BRAF(V600) mutated metastatic melanoma: an open-label, multicentre, safety
study. Lancet Oncol. 2014;15(4):436-444.
44. Lacouture ME, Duvic M, Hauschild A, et al. Analysis of dermatologic events in vemurafenib-treated patients with melanoma. Oncologist. 2013;18(3):314-322.
45. Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366(3):207-215.
46. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507-2516.
47. Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358-365.
48. Ribas A, Hodi FS, Callahan M, et. al. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2014;368(14):1365-1366.
49. Linette GP, Puzanov I, Callahan MK, et al. Phase 1 study of the BRAF inhibitor dabrafenib (D) with or without the MEK inhibitor trametinib (T) in combination with ipilimumab (Ipi) for V600E/K mutation–positive unresectable or metastatic melanoma (MM). J Clin Oncol. 2014;32(15)(suppl): Abstract 2511.
50. Chan MMK, Haydu LE, Menzies AM, et al. The nature and management of metastatic melanoma after progression on BRAF inhibitors: effects of extended BRAF inhibition. Cancer. 2014;120(20):3142-3153.
51. Carlino MS, Gowrishankar K, Saunders CAB, et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol Cancer Ther. 2013;12(7):1332-1342.
52. Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107-114.
53. Kim KB, Kefford R, Pavlick AC, et. al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. 2013;31(1):482-489.
54. Ascierto PA, Schadendorf D, Berking C, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14(3):249-256.
55. Sosman JA, Kittaneh M, Lolkema MP, et al. A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS-mutant melanoma: early encouraging clinical activity (abstract 9009). Paper presented at: 2014 American Society of Clinical Oncology meeting ; May-June 2014; Chicago, IL.
56. Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371(20):1877-1888.
57. Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30-39.
58. Gogas H, Schadendorf D, Dummer R. Vemurafenib treatment in patients with BRAF-mutated melanoma failing MEK inhibition with trametinib. J Clin Oncol. 2014;32(15)(suppl): Abstract 9061.
59. Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867-1876.
60. Kefford R, Miller WH, Tan DS, et al. Preliminary results from a phase Ib/II, openlabel, dose-escalation study of the oral BRAF inhibitor LGX818 in combination with the oral MEK1/2 inhibitor MEK162 in BRAF V600-dependent advanced solid tumors (abstract 9019). Paper presented at: 2013 American Society of Clinical Oncology meeting; May-June 2014; Chicago, IL.
61. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct
subtypes of melanoma. J Clin Oncol. 2006;24(26):4340-4346.
62. Jin SA, Chun SM, Choi YD, et al. BRAF mutations and KIT aberrations and their clinicopathological correlation in 202 Korean melanomas. J Invest Dermatol. 2013;133(2):579-582.
63. Guo J, Si L, Kong Y et. al. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol. 2011;29(21):2904-2909.
64. Hodi FS, Corless CL, Giobbie-Hurder A, et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol. 2013;31(26):3182-3190.
65. Cho JH, Kim KM, Kwon M, Kim JH, Lee J. Nilotinib in patients with metastatic melanoma harboring KIT gene aberration. Invest New Drugs. 2012;30(5): 2008-2014.
66. Lebbe C, Chevret S, Jouary T, et. al. Phase II multicentric uncontrolled national trial assessing the efficacy of nilotinib in the treatment of advanced melanomas with c-KIT mutation or amplification. J Clin Oncol. 2014;32(15)(suppl): Abstract 9032.
67. Perez DG, Suman VJ, Fitch TR, et al. Phase 2 trial of carboplatin, weekly paclitaxel, and biweekly bevacizumab in patients with unresectable stage IV melanoma: a North Central Cancer Treatment Group study, N047A. Cancer. 2009;115(1):119-127.
68. Hainsworth JD, Infante JR, Spigel DR, et al. Bevacizumab and everolimus in the treatment of patients with metastatic melanoma. Cancer. 2010;116(17): 4122-4129.
69. Dronca RS, Allred JB, Perez DG, et. al. Phase II study of temozolomide (TMZ) and everolimus (RAD001) therapy for metastatic melanoma: a North Central Cancer Treatment Group study, N0675. Am J Clin Oncol. 2014;37(4):369-376.
70. Meier FE, Niessner H, Schmitz J, et al. The PI3K inhibitor BKM120 has potent antitumor activity in melanoma brain metastases in vitro and in vivo. J Clin Oncol. 2013;31(15)(suppl): Abstract e20050.
71. Ott PA, Chang J, Madden K, et al. Oblimersen in combination with temozolomide and albumin-bound paclitaxel in patients with advanced melanoma: a phase I trial. Cancer Chemother Pharmacol. 2013;71(1);183-191.
72. Ackerman A, Klein O, McDermott DF, et al. Outcomes of patients with metastatic
melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. 2014;120(11):1695-1701.
73. Ascierto PA, Margolin K. Ipilimumab before BRAF inhibitor treatment may be
more beneficial than vice versa for the majority of patients with advanced melanoma.
Cancer. 2014;120(11):1617-1619.
74. Ascierto PA, Simeone E, Sileni VC, et al. Sequential treatment with ipilimumab and BRAF inhibitors in patients with metastatic melanoma: data from the Italian cohort of the ipilimumab expanded access program. Cancer Invest. 2014;32(4):144-149.
Colorectal Carcinoma and Emerging Targeted Therapies
Colorectal cancer (CRC) is the third most common cancer and the third leading cause of cancer death in the U.S.1-3 Only 40% of cases are diagnosed in a localized stage with an estimated 5-year survival of 90%, whereas 20% of cases present with metastatic disease with a 5-year survival of about 12.5%.1 With recent advances in cancer genetics and immunology as well as approval of targeted agents by the FDA, different treatment options are now available, even for progressive disease.
This article presents a brief review of CRC with a special focus on targeted therapies in metastatic CRC (mCRC). Colorectal cancers exhibit certain mutations, which affect the tumor responsiveness to various treatment options. This article describes the role of targeted therapies in various well-established mutations.
Epidemiology
The most common tumor location is the proximal colon (42%), followed by the rectum (28%). More than 90% of patients with CRCs are aged > 50 years at diagnosis.
Women have a higher percentage of proximal tumors compared with men (46% vs 38%) and a lower percentage of rectal tumors (24% vs 31%).Among both sexes, incidence and mortality are highest in African Americans and lowest in Asian/Pacific Islanders. The estimated 5-year survival is slightly higher for rectal cancer (66.5%) than for colon cancer (64.2%), although the stage-specific survival is similar. The difference in 5-year overall survival (OS) is attributed to the higher percentage of rectal tumors diagnosed at a localized stage (44% vs 38%). Patients aged < 65 years have higher 5-year survival rates than do those aged 65 years (68.9% vs 62.0%).1
Risk Factors
Like most human cancers, multiple genetic and environmental factors are believed to play a role in the development of colorectal carcinomas, with environmental factors playing the dominant role.4
Environmental risk factors include a low-fiber diet,5 red and processed meat intake,6 a high-fat diet,7 smoking,8 heavy alcohol consumption,9 obesity,10 physical inactivity,11 alteration in intestinal flora,12 and chronic inflammation.13 Aspirin (at doses > 300 mg/d), nonsteroidal anti-inflammatory drugs, and folic acid are believed to protect against colon cancer.14
Some of the genetic factors involved in colorectal cancers include (1) the loss of tumor suppressor genes, such as APC (most common tumor suppressor mutation), p53, SMAD4 pathways, or TGF-ß pathways; (2) DNA mismatch repair defects: mutations in MLH1, MSH2 in hereditary nonpolyposis colon cancer or methylation of MLH1 in sporadic cases; and (3) CpG island methylation (CIMP pathway), methylation of MLH1, MINT1, MINT2, MINT3; and (4) activation of oncogenes such as RAS and BRAF.15
Pathogenesis
The colonic mucosa consists of epithelial cells arranged in cylindrical structures called crypts. The human colon contains about 10 millioncrypts. The cellular proliferation and migration in each of the crypts is believed to be tightly regulated, with the majority of cells arising from a small number of stem cells (around 4-6) at the bottom of the crypt, which migrate upward after division and are eventually shed into the lumen.16 Each crypt is renewed in 2 to 8 days, which makes colonic mucosa one of the organs with the most cell proliferation in the human body and, hence, a target for various genetic and epigenetic alterations as well as environmental mutagenesis.7
Traditionally, the majority of colorectal carcinomas were believed to evolve from adenomatous polyps, which transform into an advanced adenoma with high-grade
dysplasia and then progress to an invasive cancer often referred to as the adenoma-to-carcinoma sequence.15 However, 2 other pathways, alternative and serrated,
have been described, and CRCs are now regarded as complex malignancies with a wide array of genetic and epigenetic mutations.17
About 70% to 85% of CRCs generally develop from chromosomal instability resulting from inactivation of tumor suppressor genes (APC gene, p53, etc). About 15% of cases are attributed to the failure of the DNA mismatch repair system either by germline/somatic mutations or by epigenetic silencing of gene transcription by CpG island methylation.18 Mutation in the BRAF or K-ras oncogenes are also believed to promote carcinogenesis.15 All these changes are believed to give rise to a precursor microscopic mucosal lesion that precedes the development of macroscopic adenomas.17
Clinical Features
The clinical features of CRCs can be widely variable, from incidental findings during screening colonoscopy to intestinal obstruction. The most common clinical presentation is rectal bleeding, followed by weight loss, abdominal pain, constipation, or diarrhea.19 The likelihood of CRC is higher with the combination of rectal bleeding and weight loss as well as rectal bleeding and change in bowel habits. Other clinical features may include bloating, abdominal pain, or anemia.20 (See Table 1.)
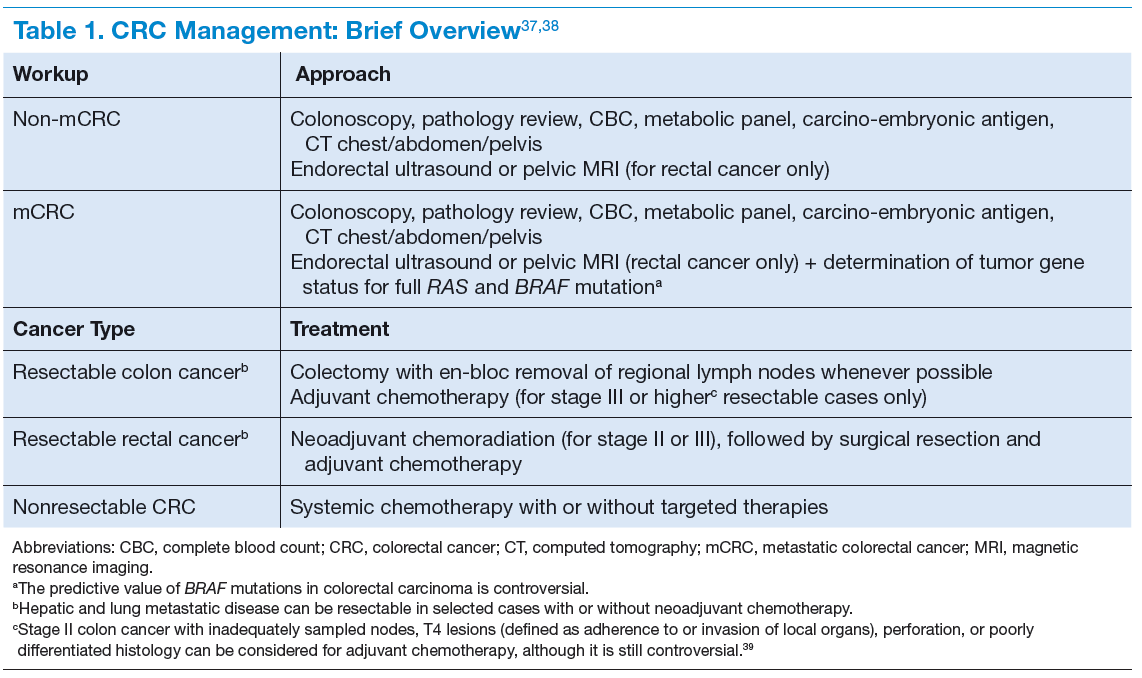
Targeted Therapy in CRC
The targeted therapies in CRC include (1) the antivascular endothelial growth factor-A (anti–VEGF-A) antibody bevacizumab; (2) the VEGF-A, VEGF-B, and placental growth factor inhibitor aflibercept; (3) the multikinase inhibitor regorafenib; and (4) the anti-epidermal growth factor receptor (anti-EGFR) antibodies cetuximab and panitumumab.
Bevacizumab is an anti-VEGF monoclonal antibody. Vascular endothelial growth factor promotes angiogenesisnecessary for tumor growth. Bevacizumab was approved by the FDA in February 2004 as a first-line treatment in combination with IFL (irinotecan plus 5-fluorouracil [5-FU]/leucovorin) regimen and in June 2006 as a second-line treatment in combination with 5-FU–based chemotherapy for patients with mCRC. In January 2013, bevacizumab was approved for use in combination with fluoropyrimidine–irinotecan- or fluoropyrimidine–oxaliplatin-based chemotherapy for the treatment of patients with mCRC whose disease has progressed while on first-line treatment with a bevacizumab-containing regimen.21 (See Table 2.)
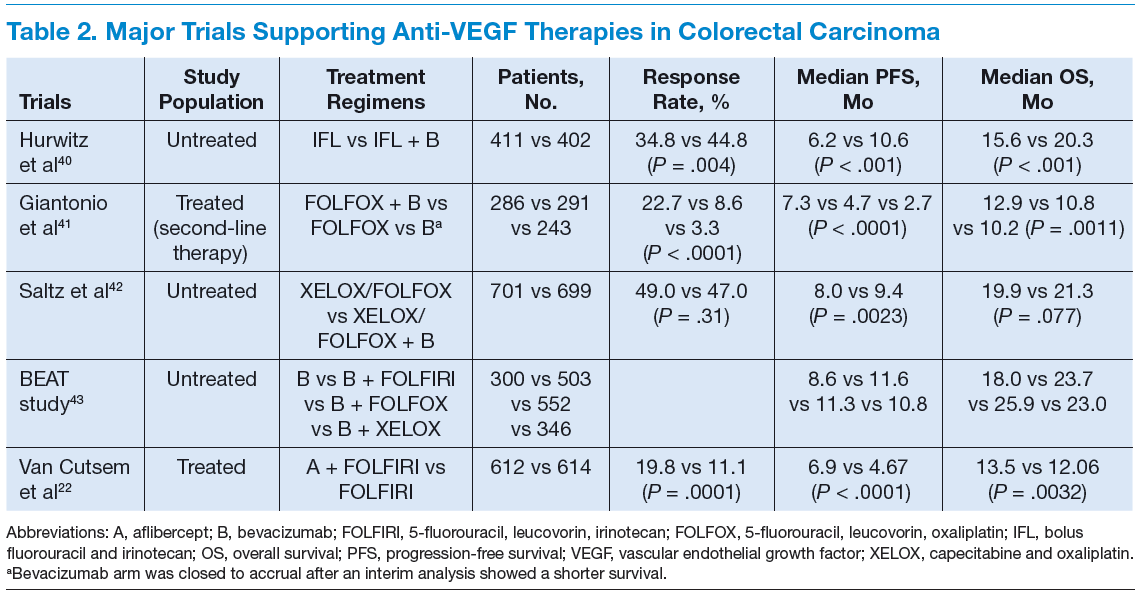
Aflibercept is a recombinant fusion protein, containing VEGF-binding portions from the extracellular domains of human VEGF receptors 1 and 2, fused to the Fc portion of human immunoglobulin IgG1 that blocks the activity of VEGF-A, VEGF-B, and placental growth factor by acting as a high-affinity ligand trap to prevent these ligands from binding to their endogenous receptors.22 It was approved by the FDA in August 2012 for use in combination with FOLFIRI (5-FU, leucovorin, irinotecan) for the treatment of patients with mCRC that is resistant to or has progressed following an oxaliplatin‑containing regimen.23
Cetuximab (a chimeric IgG1 anti-EGFR monoclonal antibody) and panitumumab (a human IgG2 anti-EGFR monoclonal antibody) have been shown to improve OS
and progression-free survival (PFS) in up to 20% of cases, either alone or in combination with chemotherapy.24,25 The FDA approved cetuximab in July 2012 for use in
combination with FOLFIRI for first-line treatment of patients with K-ras mutation-negative (wild-type), EGFRexpressing mCRC.26 The FDA approved panitumumab in
September 2006 for the treatment of patients with EGFRexpressing metastatic colorectal carcinoma with disease progression on or following FOLFOX (5-FU, leucovorin, oxaliplatin)/FOLFIRI.27 (See Table 3.)
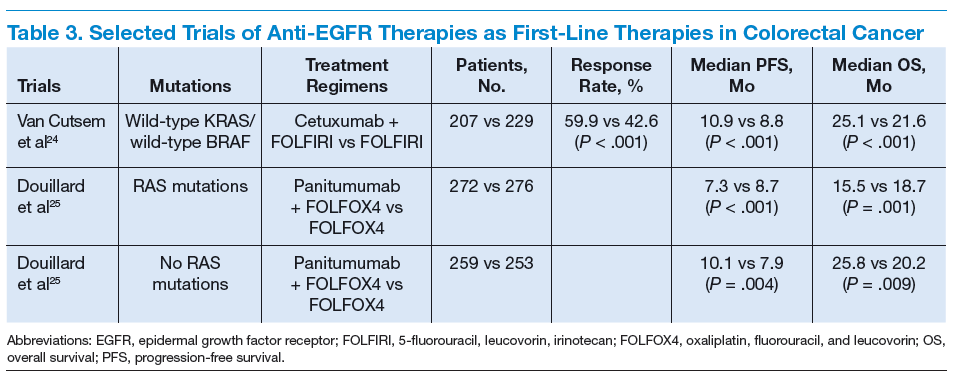
KRAS, a member of the rat sarcoma virus (ras) gene family of oncogenes, encodes for a small G protein downstream of EGFR. KRAS is mutated in CRC in up to 37% cases, resulting in activation of the different downstream signaling pathways.15,28 Therefore, KRAS mutations predict resistance to anti-EGFR therapy.28,29 Testing for KRAS mutation prior to treatment with cetuximab or panitumumab leads to targeted us of the very costly monoclonal antibodies and hence is considered a cost-effective practice.30
Even in cases with wild-type KRAS mutation, response to anti-EGFR therapies is seen in only less than half of patients.31 Up to 17% of tumors with wild-type for KRAS exon 2 at codons 12 and 13 can have a mutation in another of the ras pathway genes (eg, KRAS exon 3, 4 and NRAS exon 2, 3, 4).25 Mutations in the BRAF oncogene have been described in up to 13% of colorectal carcinoma cases.15 The data to suggest a lack of antitumor activity from anti-EGFR therapies in the presence of BRAF V600E mutation are still limited, but BRAF mutation is considered a poor prognostic factor.
A recent trial involving 1,137 patients with KRAS exon 2 wild-type mCRC randomly assigned to cetuximab or bevacizumab with standard chemotherapy (FOLFOX or FOLFIRI) found OS of 29.9 vs 29.0 months and median PFS 10.4 vs 10.8 months for cetuximab and bevacizumab, respectively.32 The OS for 5-FU–based therapies was about 11 months.33
Regorafenib is an oral multikinase inhibitor of angiogenic, stromal, and oncogenic receptor protein kinases, including those involved in the regulation of tumor angiogenesis (eg, VEGFR1-3 and TIE2 [tyrosine kinase with immunoglobulin and epidermal growth factor homology domain 2]), tumor microenvironment (plateletderived growth factor receptor-β and fibroblast growth factor receptor 1), as well as tumor oncogenesis (KIT, RET, RAF1, BRAF, BRAF V600E).34 A randomized phase 3 study involving 760 patients with documented progressive mCRC found a higher median OS with regorafenib vs placebo (6.4 mo vs 5.0 mo, P = .0052) and a
higher PFS (2.0 mo vs 1.7 mo, P < .0001).35 The FDA approved regorafenib in September 2012 for the treatment of patients with mCRC who have been previously treated with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy, with an anti-VEGF therapy and, if KRAS wild-type, with an anti-EGFR therapy.36
Conclusion
Targeted therapies in conjunction with newer chemotherapies have improved outcomes in metastatic colorectal carcinoma compared with those of conventional therapy (29 mo vs 11 mo).
Author disclosures
Peter T. Silberstein, MD, reports receiving payment for lectures from Bristol Myers and Celgene in the past. Drs. Khanal and Upadhyay report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
1. Siegel R, DeSantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. 2014;64(2):104-117.
2. American Cancer Society. Cancer Facts & Figures 2015. Atlanta, GA: American Cancer Society; 2015.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
4. Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343(2):78-85.
5. Bingham SA, Day NE, Luben R, et al; European Prospective Investigation into Cancer and Nutrition. Dietary fibre in food and protection against colorectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC): an observational study. Lancet. 2003;361(9368):1496-1501.
6. Bastide NM, Pierre FH, Corpet DE. Heme iron from meat and risk of colorectal cancer: a meta-analysis and a review of the mechanisms involved. Cancer Prev Res (Phila). 2011;4(2):177-184.
7. Raskov H, Pommergaard HC, Burcharth J, Rosenberg J. Colorectal carcinogenesis—update and perspectives. World J Gastroenterol. 2014;20(48):18151-18164.
8. Botteri E, Iodice S, Bagnardi V, Raimondi S, Lowenfels AB, Maisonneuve P. Smoking and colorectal cancer: a meta-analysis. JAMA. 2008;300(23):2765-2778.
9. Fedirko V, Tramacere I, Bagnardi V, et al. Alcohol drinking and colorectal cancer risk: an overall and dose-response meta-analysis of published studies. Ann Oncol. 2011;22(9):1958-1972.
10. Ma Y, Yang Y, Wang F, et al. Obesity and risk of colorectal cancer: a systematic review of prospective studies. PLoS One. 2013;8(1):e53916.
11. Samad AK, Taylor RS, Marshall T, Chapman MA. A meta-analysis of the association of physical activity with reduced risk of colorectal cancer. Colorectal Dis. 2005;7(3):204-213.
12. Candela M, Turroni S, Biagi E, et al. Inflammation and colorectal cancer, when
microbiota-host mutualism breaks. World J Gastroenterol. 2014;20(4):908-922.
13. Kraus S, Arber N. Inflammation and colorectal cancer. Curr Opin Pharmacol. 2009;9(4):405-410.
14. Tárraga López PJ, Albero JS, Rodríguez-Montes JA. Primary and secondary prevention of colorectal cancer. Clin Med Insights Gastroenterol. 2014;7:33-46.
15. Markowitz SD, Bertagnolli MM. Molecular origins of cancer: molecular basis of
colorectal cancer. N Engl J Med. 2009;361(25):2449-2460.
16. Zhao R, Michor F. Patterns of proliferative activity in the colonic crypt determine
crypt stability and rates of somatic evolution. PLoS Comput Biol. 2013;9(6):e1003082.
17. Pancione M, Remo A, Colantuoni V. Genetic and epigenetic events generate multiple
pathways in colorectal cancer progression. Pathol Res Int. 2012;2012:509348.
18. Worthley DL, Whitehall VL, Spring KJ, Leggett BA. Colorectal carcinogenesis: road maps to cancer. World J Gastroenterol. 2007;13(28):3784-3791.
19. Hamilton W, Round A, Sharp D, Peters TJ. Clinical features of colorectal cancer before diagnosis: a population-based case-control study. Br J Cancer. 2005;93(4):399-405.
20. Astin M, Griffin T, Neal RD, Rose P, Hamilton W. The diagnostic value of symptoms for colorectal cancer in primary care: a systematic review. Br J Gen Pract. 2011;61(586):e231-e243.
21. National Cancer Institute. FDA approval for bevacizumab: first-line treatment of metastatic colorectal cancer. National Cancer Institute Website. http://www.cancer.gov/cancertopics/druginfo/fda-bevacizumab#Anchor-Approva-23287. Updated December 4, 2014. Accessed July 6, 2015.
22. Van Cutsem E, Tabernero J, Lakomy R, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatinbased regimen. J Clin Oncol. 2012;30(28):3499-3506.
23. U.S. Food and Drug Administration. Ziv-Aflibercept. U.S. Food and Drug Administration Website. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm314438.htm. Updated August 3, 2012. Accessed July 6, 2015.
24. Van Cutsem E, Köhne CH, Láng I, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29(15):2011-2019.
25. Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369(11):1023-1034.
26. U.S. Food and Drug Administration. Cetuximab in combination with folfiri/therascreen. U.S. Food and Drug Administration Website. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm310933.htm. Updated July 9, 2012. Accessed July 6, 2015.
27. Giusti RM, Shastri KA, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary:
panitumumab (Vectibix). Oncologist. 2007;12(5):577-583.
28. Dahabreh IJ, Terasawa T, Castaldi PJ, Trikalinos TA. Systematic review: antiepidermal
growth factor receptor treatment effect modification by KRAS mutations in advanced colorectal cancer. Ann Intern Med. 2011;154(1):37-49.
29. Lièvre A, Bachet JB, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26(3):374-379.
30. Lange A, Prenzler A, Frank M, Kirstein M, Vogel A, von der Schulenburg JM. A systematic review of cost-effectiveness of monoclonal antibodies for metastatic colorectal cancer. Eur J Cancer. 2014;50(1):40-49.
31. Allegra CJ, Jessup JM, Somerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncology. 2009;27(12):2091-2096.
32. Venook AP, Niedzwiecki D, Lenz H-J, et al; Cancer and Leukemia Group B (Alliance); SWOG; ECOG. CALGB/SWOG 80405: phase III trial of irinotecan/5-FU/leucovorin (FOLFIRI) or oxaliplatin/5-FU/leucovorin (mFOLFOX6) with bevacizumab (BV) or cetuximab (CET) for patients (pts) with KRAS wild-type (wt) untreated metastatic adenocarcinoma of the colon or rectum (MCRC). J Clin Oncol. 2014;32(15)(suppl):Abstract LBA3.
33. Poon MA, O’Connell MJ, Moertel CG, et al. Biochemical modulation of fluorouracil: evidence of significant improvement of survival and quality of life in patients with advanced colorectal carcinoma. J Clin Oncol. 1989;7(10):1407-1418.
34. Wilhelm SM, Dumas J, Adnane L, et al. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011;129(1):245-255.
35. Grothey A, Van Cutsem E, Sobrero A, et al; CORRECT Study Group. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):303-312.
36. National Cancer Institute. FDA approval for regorafenib: previously treated metastatic colorectal cancer. National Cancer Institute Website. http://www.cancer.gov/cancertopics/druginfo/fda-regorafenib#Anchor-MCRC. Updated July 3, 2013. Accessed July 6, 2015.
37. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: colon cancer. National Comprehensive Cancer Network Website. http://www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Updated October 3, 2014. Accessed January 25, 2015.
38. National Comprehensive Cancer Network. Clinical practice guidelines in oncology: rectal cancer. National Comprehensive Cancer Network Website. http://www.nccn.org/professionals/physician_gls/pdf/rectal.pdf. Updated December 9, 2014. Accessed January 25, 2015.
39. Benson AB 3rd, Schrag D, Somerfield MR, et al. American Society of Clinical Oncology recommendations on adjuvant chemotherapy for stage II colon cancer. J Clin Oncol. 2004;22(16):3408-3419.
40. Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335-2342.
41. Giantonio BJ, Catalano PJ, Meropol NJ, et al; Eastern Cooperative Oncology Group Study E3200. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol. 2007;25(12):1539-1544.
42. Saltz LB, Clarke S, Díaz-Rubio E, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008;26(12):2013-2019.
43. Van Cutsem E, Rivera F, Berry S, et al; First BEAT investigators. Safety and efficacy of
first-line bevacizumab with FOLFOX, XELOX, FOLFIRI and fluoropyrimidines in
metastatic colorectal cancer: the BEAT study. Ann Oncol. 2009;20(11):1842-1847.
Colorectal cancer (CRC) is the third most common cancer and the third leading cause of cancer death in the U.S.1-3 Only 40% of cases are diagnosed in a localized stage with an estimated 5-year survival of 90%, whereas 20% of cases present with metastatic disease with a 5-year survival of about 12.5%.1 With recent advances in cancer genetics and immunology as well as approval of targeted agents by the FDA, different treatment options are now available, even for progressive disease.
This article presents a brief review of CRC with a special focus on targeted therapies in metastatic CRC (mCRC). Colorectal cancers exhibit certain mutations, which affect the tumor responsiveness to various treatment options. This article describes the role of targeted therapies in various well-established mutations.
Epidemiology
The most common tumor location is the proximal colon (42%), followed by the rectum (28%). More than 90% of patients with CRCs are aged > 50 years at diagnosis.
Women have a higher percentage of proximal tumors compared with men (46% vs 38%) and a lower percentage of rectal tumors (24% vs 31%).Among both sexes, incidence and mortality are highest in African Americans and lowest in Asian/Pacific Islanders. The estimated 5-year survival is slightly higher for rectal cancer (66.5%) than for colon cancer (64.2%), although the stage-specific survival is similar. The difference in 5-year overall survival (OS) is attributed to the higher percentage of rectal tumors diagnosed at a localized stage (44% vs 38%). Patients aged < 65 years have higher 5-year survival rates than do those aged 65 years (68.9% vs 62.0%).1
Risk Factors
Like most human cancers, multiple genetic and environmental factors are believed to play a role in the development of colorectal carcinomas, with environmental factors playing the dominant role.4
Environmental risk factors include a low-fiber diet,5 red and processed meat intake,6 a high-fat diet,7 smoking,8 heavy alcohol consumption,9 obesity,10 physical inactivity,11 alteration in intestinal flora,12 and chronic inflammation.13 Aspirin (at doses > 300 mg/d), nonsteroidal anti-inflammatory drugs, and folic acid are believed to protect against colon cancer.14
Some of the genetic factors involved in colorectal cancers include (1) the loss of tumor suppressor genes, such as APC (most common tumor suppressor mutation), p53, SMAD4 pathways, or TGF-ß pathways; (2) DNA mismatch repair defects: mutations in MLH1, MSH2 in hereditary nonpolyposis colon cancer or methylation of MLH1 in sporadic cases; and (3) CpG island methylation (CIMP pathway), methylation of MLH1, MINT1, MINT2, MINT3; and (4) activation of oncogenes such as RAS and BRAF.15
Pathogenesis
The colonic mucosa consists of epithelial cells arranged in cylindrical structures called crypts. The human colon contains about 10 millioncrypts. The cellular proliferation and migration in each of the crypts is believed to be tightly regulated, with the majority of cells arising from a small number of stem cells (around 4-6) at the bottom of the crypt, which migrate upward after division and are eventually shed into the lumen.16 Each crypt is renewed in 2 to 8 days, which makes colonic mucosa one of the organs with the most cell proliferation in the human body and, hence, a target for various genetic and epigenetic alterations as well as environmental mutagenesis.7
Traditionally, the majority of colorectal carcinomas were believed to evolve from adenomatous polyps, which transform into an advanced adenoma with high-grade
dysplasia and then progress to an invasive cancer often referred to as the adenoma-to-carcinoma sequence.15 However, 2 other pathways, alternative and serrated,
have been described, and CRCs are now regarded as complex malignancies with a wide array of genetic and epigenetic mutations.17
About 70% to 85% of CRCs generally develop from chromosomal instability resulting from inactivation of tumor suppressor genes (APC gene, p53, etc). About 15% of cases are attributed to the failure of the DNA mismatch repair system either by germline/somatic mutations or by epigenetic silencing of gene transcription by CpG island methylation.18 Mutation in the BRAF or K-ras oncogenes are also believed to promote carcinogenesis.15 All these changes are believed to give rise to a precursor microscopic mucosal lesion that precedes the development of macroscopic adenomas.17
Clinical Features
The clinical features of CRCs can be widely variable, from incidental findings during screening colonoscopy to intestinal obstruction. The most common clinical presentation is rectal bleeding, followed by weight loss, abdominal pain, constipation, or diarrhea.19 The likelihood of CRC is higher with the combination of rectal bleeding and weight loss as well as rectal bleeding and change in bowel habits. Other clinical features may include bloating, abdominal pain, or anemia.20 (See Table 1.)
Targeted Therapy in CRC
The targeted therapies in CRC include (1) the antivascular endothelial growth factor-A (anti–VEGF-A) antibody bevacizumab; (2) the VEGF-A, VEGF-B, and placental growth factor inhibitor aflibercept; (3) the multikinase inhibitor regorafenib; and (4) the anti-epidermal growth factor receptor (anti-EGFR) antibodies cetuximab and panitumumab.
Bevacizumab is an anti-VEGF monoclonal antibody. Vascular endothelial growth factor promotes angiogenesisnecessary for tumor growth. Bevacizumab was approved by the FDA in February 2004 as a first-line treatment in combination with IFL (irinotecan plus 5-fluorouracil [5-FU]/leucovorin) regimen and in June 2006 as a second-line treatment in combination with 5-FU–based chemotherapy for patients with mCRC. In January 2013, bevacizumab was approved for use in combination with fluoropyrimidine–irinotecan- or fluoropyrimidine–oxaliplatin-based chemotherapy for the treatment of patients with mCRC whose disease has progressed while on first-line treatment with a bevacizumab-containing regimen.21 (See Table 2.)
Aflibercept is a recombinant fusion protein, containing VEGF-binding portions from the extracellular domains of human VEGF receptors 1 and 2, fused to the Fc portion of human immunoglobulin IgG1 that blocks the activity of VEGF-A, VEGF-B, and placental growth factor by acting as a high-affinity ligand trap to prevent these ligands from binding to their endogenous receptors.22 It was approved by the FDA in August 2012 for use in combination with FOLFIRI (5-FU, leucovorin, irinotecan) for the treatment of patients with mCRC that is resistant to or has progressed following an oxaliplatin‑containing regimen.23
Cetuximab (a chimeric IgG1 anti-EGFR monoclonal antibody) and panitumumab (a human IgG2 anti-EGFR monoclonal antibody) have been shown to improve OS
and progression-free survival (PFS) in up to 20% of cases, either alone or in combination with chemotherapy.24,25 The FDA approved cetuximab in July 2012 for use in
combination with FOLFIRI for first-line treatment of patients with K-ras mutation-negative (wild-type), EGFRexpressing mCRC.26 The FDA approved panitumumab in
September 2006 for the treatment of patients with EGFRexpressing metastatic colorectal carcinoma with disease progression on or following FOLFOX (5-FU, leucovorin, oxaliplatin)/FOLFIRI.27 (See Table 3.)
KRAS, a member of the rat sarcoma virus (ras) gene family of oncogenes, encodes for a small G protein downstream of EGFR. KRAS is mutated in CRC in up to 37% cases, resulting in activation of the different downstream signaling pathways.15,28 Therefore, KRAS mutations predict resistance to anti-EGFR therapy.28,29 Testing for KRAS mutation prior to treatment with cetuximab or panitumumab leads to targeted us of the very costly monoclonal antibodies and hence is considered a cost-effective practice.30
Even in cases with wild-type KRAS mutation, response to anti-EGFR therapies is seen in only less than half of patients.31 Up to 17% of tumors with wild-type for KRAS exon 2 at codons 12 and 13 can have a mutation in another of the ras pathway genes (eg, KRAS exon 3, 4 and NRAS exon 2, 3, 4).25 Mutations in the BRAF oncogene have been described in up to 13% of colorectal carcinoma cases.15 The data to suggest a lack of antitumor activity from anti-EGFR therapies in the presence of BRAF V600E mutation are still limited, but BRAF mutation is considered a poor prognostic factor.
A recent trial involving 1,137 patients with KRAS exon 2 wild-type mCRC randomly assigned to cetuximab or bevacizumab with standard chemotherapy (FOLFOX or FOLFIRI) found OS of 29.9 vs 29.0 months and median PFS 10.4 vs 10.8 months for cetuximab and bevacizumab, respectively.32 The OS for 5-FU–based therapies was about 11 months.33
Regorafenib is an oral multikinase inhibitor of angiogenic, stromal, and oncogenic receptor protein kinases, including those involved in the regulation of tumor angiogenesis (eg, VEGFR1-3 and TIE2 [tyrosine kinase with immunoglobulin and epidermal growth factor homology domain 2]), tumor microenvironment (plateletderived growth factor receptor-β and fibroblast growth factor receptor 1), as well as tumor oncogenesis (KIT, RET, RAF1, BRAF, BRAF V600E).34 A randomized phase 3 study involving 760 patients with documented progressive mCRC found a higher median OS with regorafenib vs placebo (6.4 mo vs 5.0 mo, P = .0052) and a
higher PFS (2.0 mo vs 1.7 mo, P < .0001).35 The FDA approved regorafenib in September 2012 for the treatment of patients with mCRC who have been previously treated with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy, with an anti-VEGF therapy and, if KRAS wild-type, with an anti-EGFR therapy.36
Conclusion
Targeted therapies in conjunction with newer chemotherapies have improved outcomes in metastatic colorectal carcinoma compared with those of conventional therapy (29 mo vs 11 mo).
Author disclosures
Peter T. Silberstein, MD, reports receiving payment for lectures from Bristol Myers and Celgene in the past. Drs. Khanal and Upadhyay report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
Colorectal cancer (CRC) is the third most common cancer and the third leading cause of cancer death in the U.S.1-3 Only 40% of cases are diagnosed in a localized stage with an estimated 5-year survival of 90%, whereas 20% of cases present with metastatic disease with a 5-year survival of about 12.5%.1 With recent advances in cancer genetics and immunology as well as approval of targeted agents by the FDA, different treatment options are now available, even for progressive disease.
This article presents a brief review of CRC with a special focus on targeted therapies in metastatic CRC (mCRC). Colorectal cancers exhibit certain mutations, which affect the tumor responsiveness to various treatment options. This article describes the role of targeted therapies in various well-established mutations.
Epidemiology
The most common tumor location is the proximal colon (42%), followed by the rectum (28%). More than 90% of patients with CRCs are aged > 50 years at diagnosis.
Women have a higher percentage of proximal tumors compared with men (46% vs 38%) and a lower percentage of rectal tumors (24% vs 31%).Among both sexes, incidence and mortality are highest in African Americans and lowest in Asian/Pacific Islanders. The estimated 5-year survival is slightly higher for rectal cancer (66.5%) than for colon cancer (64.2%), although the stage-specific survival is similar. The difference in 5-year overall survival (OS) is attributed to the higher percentage of rectal tumors diagnosed at a localized stage (44% vs 38%). Patients aged < 65 years have higher 5-year survival rates than do those aged 65 years (68.9% vs 62.0%).1
Risk Factors
Like most human cancers, multiple genetic and environmental factors are believed to play a role in the development of colorectal carcinomas, with environmental factors playing the dominant role.4
Environmental risk factors include a low-fiber diet,5 red and processed meat intake,6 a high-fat diet,7 smoking,8 heavy alcohol consumption,9 obesity,10 physical inactivity,11 alteration in intestinal flora,12 and chronic inflammation.13 Aspirin (at doses > 300 mg/d), nonsteroidal anti-inflammatory drugs, and folic acid are believed to protect against colon cancer.14
Some of the genetic factors involved in colorectal cancers include (1) the loss of tumor suppressor genes, such as APC (most common tumor suppressor mutation), p53, SMAD4 pathways, or TGF-ß pathways; (2) DNA mismatch repair defects: mutations in MLH1, MSH2 in hereditary nonpolyposis colon cancer or methylation of MLH1 in sporadic cases; and (3) CpG island methylation (CIMP pathway), methylation of MLH1, MINT1, MINT2, MINT3; and (4) activation of oncogenes such as RAS and BRAF.15
Pathogenesis
The colonic mucosa consists of epithelial cells arranged in cylindrical structures called crypts. The human colon contains about 10 millioncrypts. The cellular proliferation and migration in each of the crypts is believed to be tightly regulated, with the majority of cells arising from a small number of stem cells (around 4-6) at the bottom of the crypt, which migrate upward after division and are eventually shed into the lumen.16 Each crypt is renewed in 2 to 8 days, which makes colonic mucosa one of the organs with the most cell proliferation in the human body and, hence, a target for various genetic and epigenetic alterations as well as environmental mutagenesis.7
Traditionally, the majority of colorectal carcinomas were believed to evolve from adenomatous polyps, which transform into an advanced adenoma with high-grade
dysplasia and then progress to an invasive cancer often referred to as the adenoma-to-carcinoma sequence.15 However, 2 other pathways, alternative and serrated,
have been described, and CRCs are now regarded as complex malignancies with a wide array of genetic and epigenetic mutations.17
About 70% to 85% of CRCs generally develop from chromosomal instability resulting from inactivation of tumor suppressor genes (APC gene, p53, etc). About 15% of cases are attributed to the failure of the DNA mismatch repair system either by germline/somatic mutations or by epigenetic silencing of gene transcription by CpG island methylation.18 Mutation in the BRAF or K-ras oncogenes are also believed to promote carcinogenesis.15 All these changes are believed to give rise to a precursor microscopic mucosal lesion that precedes the development of macroscopic adenomas.17
Clinical Features
The clinical features of CRCs can be widely variable, from incidental findings during screening colonoscopy to intestinal obstruction. The most common clinical presentation is rectal bleeding, followed by weight loss, abdominal pain, constipation, or diarrhea.19 The likelihood of CRC is higher with the combination of rectal bleeding and weight loss as well as rectal bleeding and change in bowel habits. Other clinical features may include bloating, abdominal pain, or anemia.20 (See Table 1.)
Targeted Therapy in CRC
The targeted therapies in CRC include (1) the antivascular endothelial growth factor-A (anti–VEGF-A) antibody bevacizumab; (2) the VEGF-A, VEGF-B, and placental growth factor inhibitor aflibercept; (3) the multikinase inhibitor regorafenib; and (4) the anti-epidermal growth factor receptor (anti-EGFR) antibodies cetuximab and panitumumab.
Bevacizumab is an anti-VEGF monoclonal antibody. Vascular endothelial growth factor promotes angiogenesisnecessary for tumor growth. Bevacizumab was approved by the FDA in February 2004 as a first-line treatment in combination with IFL (irinotecan plus 5-fluorouracil [5-FU]/leucovorin) regimen and in June 2006 as a second-line treatment in combination with 5-FU–based chemotherapy for patients with mCRC. In January 2013, bevacizumab was approved for use in combination with fluoropyrimidine–irinotecan- or fluoropyrimidine–oxaliplatin-based chemotherapy for the treatment of patients with mCRC whose disease has progressed while on first-line treatment with a bevacizumab-containing regimen.21 (See Table 2.)
Aflibercept is a recombinant fusion protein, containing VEGF-binding portions from the extracellular domains of human VEGF receptors 1 and 2, fused to the Fc portion of human immunoglobulin IgG1 that blocks the activity of VEGF-A, VEGF-B, and placental growth factor by acting as a high-affinity ligand trap to prevent these ligands from binding to their endogenous receptors.22 It was approved by the FDA in August 2012 for use in combination with FOLFIRI (5-FU, leucovorin, irinotecan) for the treatment of patients with mCRC that is resistant to or has progressed following an oxaliplatin‑containing regimen.23
Cetuximab (a chimeric IgG1 anti-EGFR monoclonal antibody) and panitumumab (a human IgG2 anti-EGFR monoclonal antibody) have been shown to improve OS
and progression-free survival (PFS) in up to 20% of cases, either alone or in combination with chemotherapy.24,25 The FDA approved cetuximab in July 2012 for use in
combination with FOLFIRI for first-line treatment of patients with K-ras mutation-negative (wild-type), EGFRexpressing mCRC.26 The FDA approved panitumumab in
September 2006 for the treatment of patients with EGFRexpressing metastatic colorectal carcinoma with disease progression on or following FOLFOX (5-FU, leucovorin, oxaliplatin)/FOLFIRI.27 (See Table 3.)
KRAS, a member of the rat sarcoma virus (ras) gene family of oncogenes, encodes for a small G protein downstream of EGFR. KRAS is mutated in CRC in up to 37% cases, resulting in activation of the different downstream signaling pathways.15,28 Therefore, KRAS mutations predict resistance to anti-EGFR therapy.28,29 Testing for KRAS mutation prior to treatment with cetuximab or panitumumab leads to targeted us of the very costly monoclonal antibodies and hence is considered a cost-effective practice.30
Even in cases with wild-type KRAS mutation, response to anti-EGFR therapies is seen in only less than half of patients.31 Up to 17% of tumors with wild-type for KRAS exon 2 at codons 12 and 13 can have a mutation in another of the ras pathway genes (eg, KRAS exon 3, 4 and NRAS exon 2, 3, 4).25 Mutations in the BRAF oncogene have been described in up to 13% of colorectal carcinoma cases.15 The data to suggest a lack of antitumor activity from anti-EGFR therapies in the presence of BRAF V600E mutation are still limited, but BRAF mutation is considered a poor prognostic factor.
A recent trial involving 1,137 patients with KRAS exon 2 wild-type mCRC randomly assigned to cetuximab or bevacizumab with standard chemotherapy (FOLFOX or FOLFIRI) found OS of 29.9 vs 29.0 months and median PFS 10.4 vs 10.8 months for cetuximab and bevacizumab, respectively.32 The OS for 5-FU–based therapies was about 11 months.33
Regorafenib is an oral multikinase inhibitor of angiogenic, stromal, and oncogenic receptor protein kinases, including those involved in the regulation of tumor angiogenesis (eg, VEGFR1-3 and TIE2 [tyrosine kinase with immunoglobulin and epidermal growth factor homology domain 2]), tumor microenvironment (plateletderived growth factor receptor-β and fibroblast growth factor receptor 1), as well as tumor oncogenesis (KIT, RET, RAF1, BRAF, BRAF V600E).34 A randomized phase 3 study involving 760 patients with documented progressive mCRC found a higher median OS with regorafenib vs placebo (6.4 mo vs 5.0 mo, P = .0052) and a
higher PFS (2.0 mo vs 1.7 mo, P < .0001).35 The FDA approved regorafenib in September 2012 for the treatment of patients with mCRC who have been previously treated with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy, with an anti-VEGF therapy and, if KRAS wild-type, with an anti-EGFR therapy.36
Conclusion
Targeted therapies in conjunction with newer chemotherapies have improved outcomes in metastatic colorectal carcinoma compared with those of conventional therapy (29 mo vs 11 mo).
Author disclosures
Peter T. Silberstein, MD, reports receiving payment for lectures from Bristol Myers and Celgene in the past. Drs. Khanal and Upadhyay report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
1. Siegel R, DeSantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. 2014;64(2):104-117.
2. American Cancer Society. Cancer Facts & Figures 2015. Atlanta, GA: American Cancer Society; 2015.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
4. Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343(2):78-85.
5. Bingham SA, Day NE, Luben R, et al; European Prospective Investigation into Cancer and Nutrition. Dietary fibre in food and protection against colorectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC): an observational study. Lancet. 2003;361(9368):1496-1501.
6. Bastide NM, Pierre FH, Corpet DE. Heme iron from meat and risk of colorectal cancer: a meta-analysis and a review of the mechanisms involved. Cancer Prev Res (Phila). 2011;4(2):177-184.
7. Raskov H, Pommergaard HC, Burcharth J, Rosenberg J. Colorectal carcinogenesis—update and perspectives. World J Gastroenterol. 2014;20(48):18151-18164.
8. Botteri E, Iodice S, Bagnardi V, Raimondi S, Lowenfels AB, Maisonneuve P. Smoking and colorectal cancer: a meta-analysis. JAMA. 2008;300(23):2765-2778.
9. Fedirko V, Tramacere I, Bagnardi V, et al. Alcohol drinking and colorectal cancer risk: an overall and dose-response meta-analysis of published studies. Ann Oncol. 2011;22(9):1958-1972.
10. Ma Y, Yang Y, Wang F, et al. Obesity and risk of colorectal cancer: a systematic review of prospective studies. PLoS One. 2013;8(1):e53916.
11. Samad AK, Taylor RS, Marshall T, Chapman MA. A meta-analysis of the association of physical activity with reduced risk of colorectal cancer. Colorectal Dis. 2005;7(3):204-213.
12. Candela M, Turroni S, Biagi E, et al. Inflammation and colorectal cancer, when
microbiota-host mutualism breaks. World J Gastroenterol. 2014;20(4):908-922.
13. Kraus S, Arber N. Inflammation and colorectal cancer. Curr Opin Pharmacol. 2009;9(4):405-410.
14. Tárraga López PJ, Albero JS, Rodríguez-Montes JA. Primary and secondary prevention of colorectal cancer. Clin Med Insights Gastroenterol. 2014;7:33-46.
15. Markowitz SD, Bertagnolli MM. Molecular origins of cancer: molecular basis of
colorectal cancer. N Engl J Med. 2009;361(25):2449-2460.
16. Zhao R, Michor F. Patterns of proliferative activity in the colonic crypt determine
crypt stability and rates of somatic evolution. PLoS Comput Biol. 2013;9(6):e1003082.
17. Pancione M, Remo A, Colantuoni V. Genetic and epigenetic events generate multiple
pathways in colorectal cancer progression. Pathol Res Int. 2012;2012:509348.
18. Worthley DL, Whitehall VL, Spring KJ, Leggett BA. Colorectal carcinogenesis: road maps to cancer. World J Gastroenterol. 2007;13(28):3784-3791.
19. Hamilton W, Round A, Sharp D, Peters TJ. Clinical features of colorectal cancer before diagnosis: a population-based case-control study. Br J Cancer. 2005;93(4):399-405.
20. Astin M, Griffin T, Neal RD, Rose P, Hamilton W. The diagnostic value of symptoms for colorectal cancer in primary care: a systematic review. Br J Gen Pract. 2011;61(586):e231-e243.
21. National Cancer Institute. FDA approval for bevacizumab: first-line treatment of metastatic colorectal cancer. National Cancer Institute Website. http://www.cancer.gov/cancertopics/druginfo/fda-bevacizumab#Anchor-Approva-23287. Updated December 4, 2014. Accessed July 6, 2015.
22. Van Cutsem E, Tabernero J, Lakomy R, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatinbased regimen. J Clin Oncol. 2012;30(28):3499-3506.
23. U.S. Food and Drug Administration. Ziv-Aflibercept. U.S. Food and Drug Administration Website. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm314438.htm. Updated August 3, 2012. Accessed July 6, 2015.
24. Van Cutsem E, Köhne CH, Láng I, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29(15):2011-2019.
25. Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369(11):1023-1034.
26. U.S. Food and Drug Administration. Cetuximab in combination with folfiri/therascreen. U.S. Food and Drug Administration Website. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm310933.htm. Updated July 9, 2012. Accessed July 6, 2015.
27. Giusti RM, Shastri KA, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary:
panitumumab (Vectibix). Oncologist. 2007;12(5):577-583.
28. Dahabreh IJ, Terasawa T, Castaldi PJ, Trikalinos TA. Systematic review: antiepidermal
growth factor receptor treatment effect modification by KRAS mutations in advanced colorectal cancer. Ann Intern Med. 2011;154(1):37-49.
29. Lièvre A, Bachet JB, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26(3):374-379.
30. Lange A, Prenzler A, Frank M, Kirstein M, Vogel A, von der Schulenburg JM. A systematic review of cost-effectiveness of monoclonal antibodies for metastatic colorectal cancer. Eur J Cancer. 2014;50(1):40-49.
31. Allegra CJ, Jessup JM, Somerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncology. 2009;27(12):2091-2096.
32. Venook AP, Niedzwiecki D, Lenz H-J, et al; Cancer and Leukemia Group B (Alliance); SWOG; ECOG. CALGB/SWOG 80405: phase III trial of irinotecan/5-FU/leucovorin (FOLFIRI) or oxaliplatin/5-FU/leucovorin (mFOLFOX6) with bevacizumab (BV) or cetuximab (CET) for patients (pts) with KRAS wild-type (wt) untreated metastatic adenocarcinoma of the colon or rectum (MCRC). J Clin Oncol. 2014;32(15)(suppl):Abstract LBA3.
33. Poon MA, O’Connell MJ, Moertel CG, et al. Biochemical modulation of fluorouracil: evidence of significant improvement of survival and quality of life in patients with advanced colorectal carcinoma. J Clin Oncol. 1989;7(10):1407-1418.
34. Wilhelm SM, Dumas J, Adnane L, et al. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011;129(1):245-255.
35. Grothey A, Van Cutsem E, Sobrero A, et al; CORRECT Study Group. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):303-312.
36. National Cancer Institute. FDA approval for regorafenib: previously treated metastatic colorectal cancer. National Cancer Institute Website. http://www.cancer.gov/cancertopics/druginfo/fda-regorafenib#Anchor-MCRC. Updated July 3, 2013. Accessed July 6, 2015.
37. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: colon cancer. National Comprehensive Cancer Network Website. http://www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Updated October 3, 2014. Accessed January 25, 2015.
38. National Comprehensive Cancer Network. Clinical practice guidelines in oncology: rectal cancer. National Comprehensive Cancer Network Website. http://www.nccn.org/professionals/physician_gls/pdf/rectal.pdf. Updated December 9, 2014. Accessed January 25, 2015.
39. Benson AB 3rd, Schrag D, Somerfield MR, et al. American Society of Clinical Oncology recommendations on adjuvant chemotherapy for stage II colon cancer. J Clin Oncol. 2004;22(16):3408-3419.
40. Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335-2342.
41. Giantonio BJ, Catalano PJ, Meropol NJ, et al; Eastern Cooperative Oncology Group Study E3200. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol. 2007;25(12):1539-1544.
42. Saltz LB, Clarke S, Díaz-Rubio E, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008;26(12):2013-2019.
43. Van Cutsem E, Rivera F, Berry S, et al; First BEAT investigators. Safety and efficacy of
first-line bevacizumab with FOLFOX, XELOX, FOLFIRI and fluoropyrimidines in
metastatic colorectal cancer: the BEAT study. Ann Oncol. 2009;20(11):1842-1847.
1. Siegel R, DeSantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. 2014;64(2):104-117.
2. American Cancer Society. Cancer Facts & Figures 2015. Atlanta, GA: American Cancer Society; 2015.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
4. Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343(2):78-85.
5. Bingham SA, Day NE, Luben R, et al; European Prospective Investigation into Cancer and Nutrition. Dietary fibre in food and protection against colorectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC): an observational study. Lancet. 2003;361(9368):1496-1501.
6. Bastide NM, Pierre FH, Corpet DE. Heme iron from meat and risk of colorectal cancer: a meta-analysis and a review of the mechanisms involved. Cancer Prev Res (Phila). 2011;4(2):177-184.
7. Raskov H, Pommergaard HC, Burcharth J, Rosenberg J. Colorectal carcinogenesis—update and perspectives. World J Gastroenterol. 2014;20(48):18151-18164.
8. Botteri E, Iodice S, Bagnardi V, Raimondi S, Lowenfels AB, Maisonneuve P. Smoking and colorectal cancer: a meta-analysis. JAMA. 2008;300(23):2765-2778.
9. Fedirko V, Tramacere I, Bagnardi V, et al. Alcohol drinking and colorectal cancer risk: an overall and dose-response meta-analysis of published studies. Ann Oncol. 2011;22(9):1958-1972.
10. Ma Y, Yang Y, Wang F, et al. Obesity and risk of colorectal cancer: a systematic review of prospective studies. PLoS One. 2013;8(1):e53916.
11. Samad AK, Taylor RS, Marshall T, Chapman MA. A meta-analysis of the association of physical activity with reduced risk of colorectal cancer. Colorectal Dis. 2005;7(3):204-213.
12. Candela M, Turroni S, Biagi E, et al. Inflammation and colorectal cancer, when
microbiota-host mutualism breaks. World J Gastroenterol. 2014;20(4):908-922.
13. Kraus S, Arber N. Inflammation and colorectal cancer. Curr Opin Pharmacol. 2009;9(4):405-410.
14. Tárraga López PJ, Albero JS, Rodríguez-Montes JA. Primary and secondary prevention of colorectal cancer. Clin Med Insights Gastroenterol. 2014;7:33-46.
15. Markowitz SD, Bertagnolli MM. Molecular origins of cancer: molecular basis of
colorectal cancer. N Engl J Med. 2009;361(25):2449-2460.
16. Zhao R, Michor F. Patterns of proliferative activity in the colonic crypt determine
crypt stability and rates of somatic evolution. PLoS Comput Biol. 2013;9(6):e1003082.
17. Pancione M, Remo A, Colantuoni V. Genetic and epigenetic events generate multiple
pathways in colorectal cancer progression. Pathol Res Int. 2012;2012:509348.
18. Worthley DL, Whitehall VL, Spring KJ, Leggett BA. Colorectal carcinogenesis: road maps to cancer. World J Gastroenterol. 2007;13(28):3784-3791.
19. Hamilton W, Round A, Sharp D, Peters TJ. Clinical features of colorectal cancer before diagnosis: a population-based case-control study. Br J Cancer. 2005;93(4):399-405.
20. Astin M, Griffin T, Neal RD, Rose P, Hamilton W. The diagnostic value of symptoms for colorectal cancer in primary care: a systematic review. Br J Gen Pract. 2011;61(586):e231-e243.
21. National Cancer Institute. FDA approval for bevacizumab: first-line treatment of metastatic colorectal cancer. National Cancer Institute Website. http://www.cancer.gov/cancertopics/druginfo/fda-bevacizumab#Anchor-Approva-23287. Updated December 4, 2014. Accessed July 6, 2015.
22. Van Cutsem E, Tabernero J, Lakomy R, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatinbased regimen. J Clin Oncol. 2012;30(28):3499-3506.
23. U.S. Food and Drug Administration. Ziv-Aflibercept. U.S. Food and Drug Administration Website. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm314438.htm. Updated August 3, 2012. Accessed July 6, 2015.
24. Van Cutsem E, Köhne CH, Láng I, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29(15):2011-2019.
25. Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369(11):1023-1034.
26. U.S. Food and Drug Administration. Cetuximab in combination with folfiri/therascreen. U.S. Food and Drug Administration Website. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm310933.htm. Updated July 9, 2012. Accessed July 6, 2015.
27. Giusti RM, Shastri KA, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary:
panitumumab (Vectibix). Oncologist. 2007;12(5):577-583.
28. Dahabreh IJ, Terasawa T, Castaldi PJ, Trikalinos TA. Systematic review: antiepidermal
growth factor receptor treatment effect modification by KRAS mutations in advanced colorectal cancer. Ann Intern Med. 2011;154(1):37-49.
29. Lièvre A, Bachet JB, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26(3):374-379.
30. Lange A, Prenzler A, Frank M, Kirstein M, Vogel A, von der Schulenburg JM. A systematic review of cost-effectiveness of monoclonal antibodies for metastatic colorectal cancer. Eur J Cancer. 2014;50(1):40-49.
31. Allegra CJ, Jessup JM, Somerfield MR, et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncology. 2009;27(12):2091-2096.
32. Venook AP, Niedzwiecki D, Lenz H-J, et al; Cancer and Leukemia Group B (Alliance); SWOG; ECOG. CALGB/SWOG 80405: phase III trial of irinotecan/5-FU/leucovorin (FOLFIRI) or oxaliplatin/5-FU/leucovorin (mFOLFOX6) with bevacizumab (BV) or cetuximab (CET) for patients (pts) with KRAS wild-type (wt) untreated metastatic adenocarcinoma of the colon or rectum (MCRC). J Clin Oncol. 2014;32(15)(suppl):Abstract LBA3.
33. Poon MA, O’Connell MJ, Moertel CG, et al. Biochemical modulation of fluorouracil: evidence of significant improvement of survival and quality of life in patients with advanced colorectal carcinoma. J Clin Oncol. 1989;7(10):1407-1418.
34. Wilhelm SM, Dumas J, Adnane L, et al. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011;129(1):245-255.
35. Grothey A, Van Cutsem E, Sobrero A, et al; CORRECT Study Group. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):303-312.
36. National Cancer Institute. FDA approval for regorafenib: previously treated metastatic colorectal cancer. National Cancer Institute Website. http://www.cancer.gov/cancertopics/druginfo/fda-regorafenib#Anchor-MCRC. Updated July 3, 2013. Accessed July 6, 2015.
37. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: colon cancer. National Comprehensive Cancer Network Website. http://www.nccn.org/professionals/physician_gls/pdf/colon.pdf. Updated October 3, 2014. Accessed January 25, 2015.
38. National Comprehensive Cancer Network. Clinical practice guidelines in oncology: rectal cancer. National Comprehensive Cancer Network Website. http://www.nccn.org/professionals/physician_gls/pdf/rectal.pdf. Updated December 9, 2014. Accessed January 25, 2015.
39. Benson AB 3rd, Schrag D, Somerfield MR, et al. American Society of Clinical Oncology recommendations on adjuvant chemotherapy for stage II colon cancer. J Clin Oncol. 2004;22(16):3408-3419.
40. Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335-2342.
41. Giantonio BJ, Catalano PJ, Meropol NJ, et al; Eastern Cooperative Oncology Group Study E3200. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol. 2007;25(12):1539-1544.
42. Saltz LB, Clarke S, Díaz-Rubio E, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008;26(12):2013-2019.
43. Van Cutsem E, Rivera F, Berry S, et al; First BEAT investigators. Safety and efficacy of
first-line bevacizumab with FOLFOX, XELOX, FOLFIRI and fluoropyrimidines in
metastatic colorectal cancer: the BEAT study. Ann Oncol. 2009;20(11):1842-1847.
New Treatment Options for Metastatic Thyroid Cancer
Thyroid cancer is the ninth most common malignancy in the U.S. At the time of diagnosis, thyroid cancer is mostly confined to the thyroid gland and regional lymph nodes. However, around 4% of patients with thyroid cancer present with metastatic disease. When compared with localized and regional thyroid cancer, 5-year survival rates for metastatic thyroid cancer are significantly worse (99.9%, 97.6%, and 54.7%, respectively).1 Treatment options for metastatic thyroid cancer are limited and largely depend on the pathology and the type of thyroid cancer.
Thyroid cancer can be divided into differentiated, medullary, and anaplastic subtypes based on pathology. The treatment for metastatic differentiated thyroid cancer (DTC) consists of radioactive iodine therapy, thyroid-stimulating hormone (TSH) suppression (thyroxine hormone) therapy, and external beam radiotherapy. Systemic therapy is considered in patients with metastatic DTC who progress despite the above treatment modalities. In the case of metastatic medullary thyroid cancer (MTC), patients who are not candidates for surgery or radiation are considered for systemic therapy, because MTC does not respond to radioactive iodine or TSH suppressive therapy. On the other hand, metastatic anaplastic thyroid cancer is a very aggressive subtype with no effective therapy available to date. Palliation of symptoms is the main goal for these patients, which can be achieved by loco-regional resection and palliative irradiation.2,3
This review focuses on the newer treatment options for metastatic DTC and MTC that are based on inhibition of cellular kinases.
Differentiated Thyroid Cancer
Differentiated thyroid cancer is the most common histologic type of thyroid cancer, accounting for 95% of all thyroid cancers and consists of papillary, follicular, and poorly differentiated thyroid cancer.2,3 Surgery is the treatment of choice for DTC. Based on tumor size and its local extension in the neck, treatment options include unilateral lobectomy and isthmectomy, total thyroidectomy, central neck dissection, and more extensive resection.2,3 After surgery, radioactive iodine is recommended in patients with known metastatic disease; locally invasive tumor, regardless of size; or primary tumor > 4 cm, in the absence of other high-risk features.2 This should be followed by TSH suppressive hormone therapy.2
About 7% to 23% of patients with DTC develop distant metastases.4 Two-thirds of these patients become refractory to radioactive iodine.5 Prognosis remains poor in these patients, with a 10-year survival rate from the time detection of metastasis of only 10%.5-7 Treatment options are limited. However, recently the understanding of cell biology in terms of key signaling pathways called kinases has been elucidated. The kinases that can stabilize progressive metastatic disease seem to be attractive therapeutic targets in treating patients whose disease no longer responds to radioiodine and TSH suppressive hormone therapy.
Papillary thyroid cancers frequently carry gene mutations and rearrangements that lead to activation of the mitogen-activated protein kinase (MAPK), which promotes cell division. The sequential components leading to activation of MAPK include rearrangements of RET and NTRK1 tyrosine kinases, activating mutations of BRAF, and activating mutations of RAS.8,9 Similarly, overexpression of normal c-myc and c-fos genes, as well as mutations of HRAS, NRAS, and KRAS genes, is found in follicular adenomas, follicular cancers, and occasionally papillary cancers.10-14 Increased expression of vascular endothelial growth factor (VEGF) and its receptors (VEGFRs) might have a role in thyroid carcinoma as well.15
These kinases (the serine kinase BRAF and tyrosine kinases RET and RAS, and the contributory roles of tyrosine kinases in growth factor receptors such as the VEGFR) stimulate tumor proliferation, angiogenesis, invasion, metastasis, and inhibit tumor cell apoptosis. Kinase inhibitors target these signaling kinases, affecting tumor cell biology and its microenvironment.16,17
A wide variety of multitargeted kinase inhibitors (MKIs) have entered clinical trials for patients with advanced or progressive metastatic thyroid cancers. Two such agents, sorafenib and lenvatinib, are approved by the FDA for use in selected patients with refractory metastatic DTC, whereas many other drugs remain investigational for this disease. In phase 2 and 3 trials, most of the treatment responses for MKIs were partial. Complete responses were rare, and no study has reported a complete analysis of overall survival (OS) outcomes. Results from some new randomized trials indicate an improvement in progression-free survival (PFS) compared with placebo, and additional trials are underway.
Sorafenib
Sorafenib was approved by the FDA in 2013 for the treatment of locally recurrent or metastatic, progressive DTC that no longer responds to radioactive iodine treatment.18 Sorafenib is an oral, small molecule MKI. It works on VEGFRs 1, 2, and 3; platelet-derived growth factor receptor (PDGFR); common RET/PTC subtypes; KIT; and less potently, BRAF.19 The recommended dose is 400 mg orally twice a day.
In April 2014, Brose and colleagues published the phase 3 DECISION study on sorafenib.20 It was a multicenter, randomized, double-blinded, placebo-controlled trial of 417 patients with radioactive iodine-refractory locally advanced or metastatic DTC that had progressed within the previous 14 months.20 The results of the trial were promising. The median PFS was 5 months longer in the sorafenib group (10.8 mo) than in the placebo group (5.8 mo; hazard ratio [HR], 0.59; 95% conidence interval [CI], 0.45-0.76; P < .0001). The primary endpoint of the trial was PFS, and crossover from placebo to sorafenib was permitted upon progression. Overall survival did not differ significantly between the treatment groups (placebo vs sorafenib) at the time of the primary analysis data cutoff. However, OS results may have been confounded by postprogression crossover from placebo to open-label sorafenib by the majority of placebo patients.
In subgroup analysis, patients with BRAF and RAS mutations and wild-type BRAF and RAS subgroups had a significant increase in median PFS in the sorafenib treatment group compared with the placebo group (Table 1).20
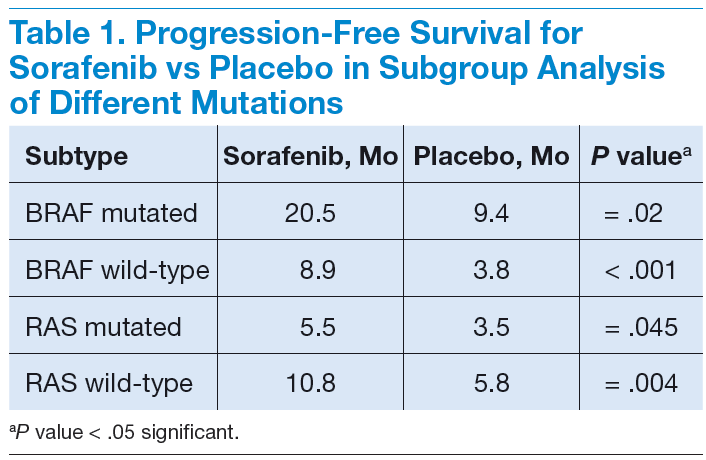
Adverse events (AEs) occurred in 98.6% of patients receiving sorafenib during the double-blind period and in 87.6% of patients receiving placebo. Most AEs were grade 1 or 2. The most common AEs were hand-foot-skin reactions (76.3%), diarrhea (68.6%), alopecia (67.1%), and rash or desquamation (50.2%). Toxicities led to dose modification in 78% of patients and permanent discontinuation of therapy in 19%.20 Like other BRAF inhibitors, sorafenib has been associated with an increased incidence of cutaneous squamous cell carcinomas (5%), keratoacanthomas, and other premalignant actinic lesions.21
Lenvatinib
In February 2015, lenvatinib was approved for the treatment of locally recurrent or metastatic, progressive DTC that no longer responds to radioactive iodine treatment.22 Lenvatinib is a MKI of VEGFRs 1, 2, and 3; fibroblast growth factor receptors 1 through 4; PDGFR-α; RET, and KIT.23,24 The recommended dose is 24 mg orally once daily.
Schlumberger and colleagues published results from the SELECT trial, a randomized, double-blinded, multicenter phase 3 study involving 392 patients with progressive thyroid cancer that was refractory to iodine-131.25 A total of 261 patients received lenvatinib, and 131 patients received a placebo. Upon disease progression, patients in the placebo group were allowed to receive open-label lenvatinib. The study’s primary endpoint was PFS. Secondary endpoints were the response rate (RR), OS, and safety. The median PFS was 18.3 months in the lenvatinib group and 3.6 months in the placebo group (HR, 0.21; 99% CI, 0.14-0.31; P < .001). The RR was 64.8% in the lenvatinib group (4 complete and 165 partial responses) and 1.5% in the placebo group (P < .001). There was no significant difference in OS between the 2 groups (HR for death, 0.73; 95% CI, 0.50-1.07; P = .10). This difference became larger when a potential crossover bias was considered (rank-preserving structural failure time model; HR, 0.62; 95% CI, 0.40-1.00; P = .05).25
In a subgroup analysis, median PFS was about 14 months in the absence of prior anti-VEGFR therapy and 11 months of prior therapy. The treatmentrelated AEs were 97.3% in the lenvatinib group, and 75.9% were grade 3 or higher. Common treatmentrelated AEs of any grade in the lenvatinib group included hypertension (67.8%), diarrhea (59.4%), fatigue or asthenia (59.0%), decreased appetite (50.2%), decreased weight (46.4%), and nausea (41.0%). The study drug had to be discontinued because of AEs in 14% of patients who received lenvatinib and 2% of patients who received placebo. In the lenvatinib group, 2.3% patients had treatment-related fatal events (6 patients).25
Summary
Patients with DTC who progress after radioactive iodine therapy, TSH suppressive therapy, and external beam radiotherapy should be considered for systemic therapy. Systemic therapy consists of MKIs, which can stabilize progressive metastatic disease. These newer drugs have significant toxicities. Therefore, it is important to limit the use of systemic treatments to patients at significant risk for morbidity or mortality due to progressive metastatic disease. Patients treated with systemic agents should have a good baseline performance status, such as being ambulatory at least 50% (Eastern Cooperative Oncology Group performance score of 2) of the day to tolerate these treatments.
Patients who have disease progression or are unable to tolerate sorafenib and lenvatinib can choose to participate in clinical trials with investigational multitarget inhibitors. Other alternatives include vandetinib, pazopanib, and sunitinib, which finished phase 2 trials and showed some partial responses.26-30 If the patients are unable to tolerate MKIs, they can try doxorubicin-based conventional chemotherapy regimens.31
Medullary Thyroid Cancer
Medullary thyroid cancer is a neuroendocrine tumor arising from the thyroid parafollicular cells, accounting for about 4% of thyroid carcinomas, most of which are sporadic. However, some are familial as part of the multiple endocrine neoplasia type 2 (MEN 2) syndromes, which are transmitted in an autosomal dominant fashion.32,33 Similar to DTC, the primary treatment option is surgery. Medullary thyroid cancer can be cured only by complete resection of the thyroid tumor and any local and regional metastases. Compared with DTC, metastatic MTC is unresponsive to radioiodine or TSH
suppressive treatment, because this cancer neither concentrates iodine nor is TSH dependent.34,35
The 10-year OS rate in MTC is ≤ 40% in patients with locally advanced or metastatic disease.32,36,37 In hereditary MTC, germline mutations in the c-ret proto-oncogene occur in virtually all patients. In sporadic MTC, 85% of patients have the M918T mutation, and somatic c-ret mutations are seen in about 50% of patients.38-42
Similar to DTC, due to the presence of mutations involving RET receptor tyrosine kinase, molecular targeted therapeutics with activity against RET demonstrate a potential therapeutic target in MTC.43-45 Other signaling pathways likely to contribute to the growth and invasiveness of MTC include VEGFR-dependent tumor angiogenesis and epidermal growth factor receptor (EGFR)-dependent tumor cell proliferation.46
In 2011 and 2012, the FDA approved tyrosine kinase inhibitors (TKIs) vandetanib and cabozantinib for metastatic MTC. Similar to treatment for DTC, systemic therapy is mainly based on targeted therapies. Patients with progressive or symptomatic metastatic disease who are not candidates for surgery or radiotherapy should be considered for TKI therapy.
Vandetanib
Vandetanib is approved for unresectable, locally advanced or metastatic sporadic or hereditary MTC.47 The daily recommended dose is 300 mg/d. It is an oral MKI that targets VEGFR, RET/PTC, and the EGFR.48
The ZETA trial was an international randomized phase 3 trial involving patients with unresectable locally advanced or metastatic sporadic or hereditary MTC.48 In a ZETA trial study by Wells Jr and colleagues, patients with advanced MTC were randomly assigned in a 2:1 ratio to receive vandetanib 300 mg/d or placebo. After objective disease progression, patients could elect to receive openlabel vandetanib. The primary endpoint was PFS, determined by independent central Response Evaluation Criteria in Solid Tumors assessments.
A total of 331 patients were randomly assigned to receive vandetanib (231 patients) or placebo (100 patients). At data cutoff, with median follow-up of 24 months, PFS was significantly prolonged in patients randomly assigned to vandetanib vs placebo (30.5 mo vs 19.3 mo; HR, 0.46; 95% CI, 0.31-0.69). The objective RR was significantly higher in the vandetanib group (45% vs 13%). The presence of a somatic RET M918T mutation predicted an improved PFS.
Common AEs (any grade) noted with vandetanib vs placebo include diarrhea (56% vs 26%), rash (45% vs 11%), nausea (33% vs 16%), hypertension (32% vs 5%), and headache (26% vs 9%). Torsades de pointes and sudden death were reported in patients receiving vandetanib. Data on OS were immature at data cutoff (HR, 0.89; 95% CI, 0.48-1.65). A final survival analysis will take place when 50% of the patients have died.48
Vandetanib is currently approved with a Risk Evaluation and Mitigation Strategy to inform health care professionals about serious heart-related risks. Electrocardiograms and serum potassium, calcium, magnesium, and TSH should be taken at 2 to 4 weeks and 8 to 12 weeks after starting treatment and every 3 months after that. Patients with diarrhea may require more frequent monitoring.
Cabozantinib
In 2012, the FDA approved cabozantinib for the treatment of progressive, metastatic MTC.49 It is an oral, small molecule TKI that targets VEGFRs 1 and 2, MET, and RET. The inhibitory activity against MET, the cognate receptor for the hepatocyte growth factor, may provide additional synergistic benefit in MTC.50 The daily recommended dose is 140 mg/d. A phase 3 randomized EXAM trial in patients with progressive, metastatic, or unresectable locally advanced MTC.51 Three hundred thirty patients were randomly assigned to receive either cabozantinib 140 mg or placebo once daily. Progressionfree survival was improved with cabozantinib compared with that of placebo (11.2 vs 4.0 mo; HR, 0.28; 95% CI, 0.19-0.40). Partial responses were observed in 27% vs 0% in placebo. A planned interim analysis of OS was conducted, including 96 (44%) of the 217 patient deaths required for the final analysis, with no statistically significant difference observed between the treatment arms (HR, 0.98; 95% CI, 0.63-1.52). Survival follow-up is planned to continue until at least 217 deaths have been observed.
There was markedly improved PFS in the subset of patients treated with cabozantinib compared with placebo whose tumors contained RET M918T mutations (61 vs 17 wk; HR, 0.15; 95% CI, 0.08-0.28) or RAS mutations (47 vs 8 wk; HR, 0.15; 95% CI, 0.02-1.10).51
The most common AEs, occurring in ≥ 25% of patients, were diarrhea, stomatitis, hand and foot syndrome, hypertension, and abdominal pain. Although uncommon, clinically significant AEs also included fistula formation and osteonecrosis of the jaw.
Summary
Patients with progressive or symptomatic metastatic disease who are not candidates for surgery or radiotherapy should be considered for TKI therapy. Though not curative, TKIs can only stabilize disease progression. Initiation of TKIs should be considered in rapidly progressive disease, because these drugs are associated with considerable AEs affecting the quality of life (QOL).
Patients who progressed or were unable to tolerate vandetanib or cabozantinib can choose to participate in clinical trials with investigational multitarget inhibitors. Other alternatives include pazopanib, sunitinib, and sorafenib, which finished phase 2 trials and showed some partial responses.29,52-57 If patients are unable to tolerate MKIs, they can try conventional chemotherapy consisting of dacarbazine with other agents or doxorubin.58-60
Conclusions
Molecular targeted therapy is an emerging treatment option for patients with metastatic thyroid cancer (Table 2). The authors suggest that such patients participate in clinical trials in the hope of developing more effective and tolerable drugs and recommend oral TKIs for patients with rapidly progressive disease who cannot participate in a clinical trial. For patients who cannot tolerate or fail one TKI, the authors recommend trying other TKIs before initiating cytotoxic chemotherapy.
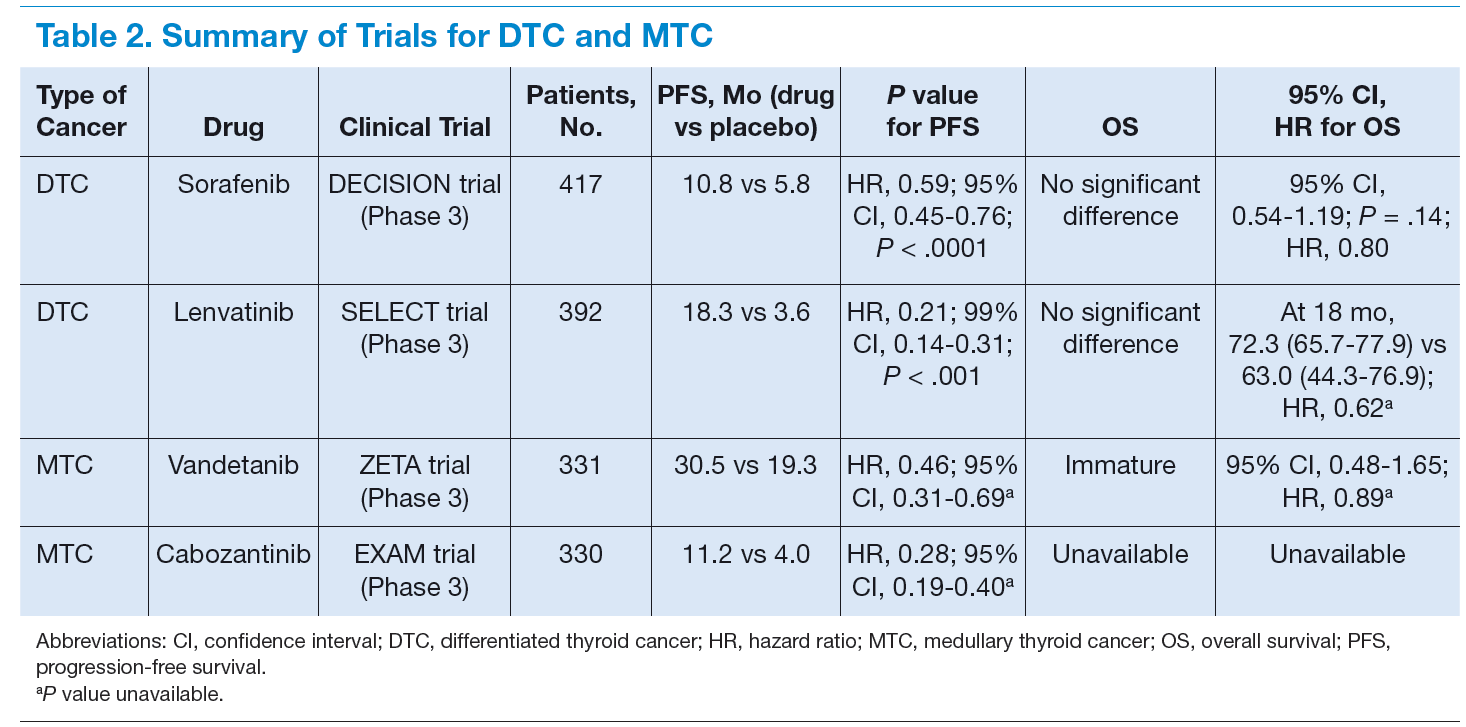
Before initiation of treatment for metastatic disease, an important factor to consider is the pace of disease progression. Patients who are asymptomatic and have the very indolent disease may postpone kinase inhibitor therapy until they become rapidly progressive or symptomatic, because the AEs of treatment will adversely affect the patient’s QOL. In patients with symptomatic and rapidly progressive disease, initiation of treatment with kinase inhibitor therapy can lead to stabilization of disease, although at the cost of some AEs. More structured clinical trials are needed, along with an evaluation of newer molecular targets for the management of this progressive metastatic disease with a dismal prognosis.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
1. Howlader N, Noone AM, Krapcho M, et al, eds. SEER Cancer Statistics Review, 1975-2012. Bethesda, MD: National Cancer Institute; 2015.
2. Cooper DS, Doherty GM, Haugen BR, et al; American Thyroid Association (ATA) Guidelines Taskforce on Thyroid Nodules and Differentiated Thyroid Cancer. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2009;19(11):1167-1214.
3. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: thyroid carcinoma. National Comprehensive Cancer Network Website. http://www.nccn.org/professionals/physician_gls/pdf/thyroid.pdf. Updated May 11, 2015. Accessed July 10, 2015.
4. Shoup M, Stojadinovic A, Nissan A, et al. Prognostic indicators of outcomes in patients with distant metastases from differentiated thyroid carcinoma. J Am Coll Surg. 2003;197(2):191-197.
5. Durante C, Haddy N, Baudin E, et al. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: benefits and limits of radioiodine therapy. J Clin Endocrinol Metab. 2006;91(8):2892-2899.
6. Busaidy NL, Cabanillas ME. Differentiated thyroid cancer: management of patients with radioiodine nonresponsive disease. J Thyroid Res. 2012;2012:618985.
7. Schlumberger M, Brose M, Elisei R, et al. Definition and management of radioactive iodine-refractory differentiated thyroid cancer. Lancet Diabetes Endocrinol. 2014;2(5):356-358.
8. Melillo RM, Castellone MD, Guarino V, et al. The RET/PTC-RAS-BRAF linear signaling cascade mediates the motile and mitogenic phenotype of thyroid cancer cells. J Clin Invest. 2005;115(4):1068-1081.
9. Ciampi R, Nikiforov YE. RET/PTC rearrangements and BRAF mutations in thyroid tumorigenesis. Endocrinology. 2007;148(3):936-941.
10. Lemoine NR, Mayall ES, Wyllie FS, et al. High frequency of ras oncogene activation in all stages of human thyroid tumorigenesis. Oncogene. 1989;4(2):159-164.
11. Namba H, Rubin SA, Fagin JA. Point mutations of ras oncogenes are an early event in thyroid tumorigenesis. Mol Endocrinol. 1990;4(10):1474-1479.
12. Suarez HG, du Villard JA, Severino M, et al. Presence of mutations in all three ras genes in human thyroid tumors. Oncogene. 1990;5(4):565-570.
13. Karga H, Lee JK, Vickery AL Jr, Thor A, Gaz RD, Jameson JL. Ras oncogene mutations in benign and malignant thyroid neoplasms. J Clin Endocrinol Metab. 1991;73(4):832-836.
14. Terrier P, Sheng ZM, Schlumberger M, et al. Structure and expression of c-myc and c-fos proto-oncogenes in thyroid carcinomas. Br J Cancer. 1988;57(1):43-47.
15. Klein M, Vignaud JM, Hennequin V, et al. Increased expression of the vascular endothelial growth factor is a pejorative prognosis marker in papillary thyroid carcinoma. J Clin Endocrinol Metab. 2001;86(2):656-658.
16. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9(1):28-39.
17. Haugen BR, Sherman SI. Evolving approaches to patients with advanced differentiated thyroid cancer. Endocr Rev. 2013;34(3):439-455.
18. U.S. Food and Drug Administration. FDA approves Nexavar to treat type of thyroid cancer [press release]. Silver Spring, MD: U.S. Food and Drug Administration; November 22, 2013.
19. Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099-7109.
20. Brose MS, Nutting CM, Jarzab B, et al; DECISION investigators. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet. 2014;384(9940):319-328.
21. Dubauskas Z, Kunishige J, Prieto VG, Jonasch E, Hwu P, Tannir NM. Cutaneous squamous cell carcinoma and inflammation of actinic keratoses associated with sorafenib. Clin Genitourin Cancer. 2009;7(1):20-23.
22. U.S. Food and Drug Administration. FDA approves Lenvima for a type of thyroid cancer [press release]. Silver Spring, MD: U.S. Food and Drug Administration; February 13, 2015.
23. Matsui J, Yamamoto Y, Funahashi Y, et al. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int J Cancer. 2008;122(3):664-671.
24. Matsui J, Funahashi Y, Uenaka T, Watanabe T, Tsuruoka A, Asada M. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factorreceptor (VEGF-R) 2 and VEGF-R3 kinase. Clin Cancer Res. 2008;14(17):5459-5465.
25. Schlumberger M, Tahara M, Wirth LJ, et al. Lenvatinib versus placebo in radioiodine- refractory thyroid cancer. N Engl J Med. 2015;372(7):621-630.
26. Leboulleux S, Bastholt L, Krause T, et al. Vandetanib in locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 2 trial. Lancet Oncol. 2012;13(9):897-905.
27. A randomised, double-blind, placebo-controlled, multi-centre phase III study to assess the efficacy and safety of vandetanib (CAPRELSA) 300 mg in patients with differentiated thyroid cancer that is either locally advanced or metastatic who are refractory or unsuitable for radioiodine (RAI) therapy. Trial number NCT01876784. ClinicalTrials.gov Website. https://clinicaltrials.gov/show/NCT01876784. Updated June 26, 2015. Accessed July 22, 2015.
28. Bible KC, Suman VJ, Molina JR, et al; Endocrine Malignancies Disease Oriented Group; Mayo Clinic Cancer Center; Mayo Phase 2 Consortium. Efficacy of pazopanib in progressive, radioiodine-refractory, metastatic differentiated thyroid cancers: results of a phase 2 consortium study. Lancet Oncol. 2010;11(10):962-972.
29. Kim DW, Jo YS, Jung HS, et al. An orally administered multitarget tyrosine kinase inhibitor, SU11248, is a novel potent inhibitor of thyroid oncogenic RET/papillary thyroid cancer kinases. J Clin Endocrinol Metab. 2006;91(10):4070-4076.
30. Dawson SJ, Conus NM, Toner GC, et al. Sustained clinical responses to tyrosine kinase inhibitor sunitinib in thyroid carcinoma. Anticancer Drugs. 2008;19(5):547-552.
31. Carter SK, Blum RH. New chemotherapeutic agents—bleomycin and adriamycin. CA Cancer J Clin. 1974;24(6):322-331.
32. Hundahl SA, Fleming ID, Fremgen AM, Menck HR. A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985-1995 [see comments]. Cancer. 1998;83(12):2638-2648.
33. Lakhani VT, You YN, Wells SA. The multiple endocrine neoplasia syndromes. Annu Rev Med. 2007;58:253-265.
34. Martins RG, Rajendran JG, Capell P, Byrd DR, Mankoff DA. Medullary thyroid cancer: options for systemic therapy of metastatic disease? J Clin Oncol. 2006;24(11):1653-1655.
35. American Thyroid Association Guidelines Task Force; Kloos RT, Eng C, Evans DB, et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid. 2009;19(6):565-612.
36. Roman S, Lin R, Sosa JA. Prognosis of medullary thyroid carcinoma: demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer. 2006;107(9):2134-2142.
37. Modigliani E, Cohen R, Campos JM, et al. Prognostic factors for survival and for biochemical cure in medullary thyroid carcinoma: results in 899 patients. The GETC Study Group. Groupe d’étude des tumeurs à calcitonine. Clin Endocrinol (Oxf). 1998;48(3):265-273.
38. Donis-Keller H, Dou S, Chi D, et al. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet. 1993;2(7):851-856.
39. Mulligan LM, Kwok JB, Healey CS, et al. Germ-line mutations of the RET protooncogene in multiple endocrine neoplasia type 2A. Nature. 363(6428):458-460.
40. Carlson KM, Dou S, Chi D, et al. Single missense mutation in the tyrosine kinase catalytic domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B. Proc Natl Acad Sci USA. 1994;91(4):1579-1583.
41. Marsh DJ, Learoyd DL, Andrew SD, et al. Somatic mutations in the RET protooncogene in sporadic medullary thyroid carcinoma. Clin Endocrinol (Oxf). 1996;44(3):249-257.
42. Elisei R, Cosci B, Romei C, et al. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. J Clin Endocrinol Metab. 2008;93(3):682-687.
43. Carlomagno F, Vitagliano D, Guida T, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002;62(24):7284-7290.
44. Carlomagno F, Anaganti S, Guida T, et al. BAY 43-9006 inhibition of oncogenic RET mutants. J Natl Cancer Inst. 2006;98(5):326-334.
45. Santoro M, Carlomagno F. Drug insight: small molecule inhibitors of protein kinases in the treatment of thyroid cancer. Nat Clin Pract Endocrinol Metab. 2006;2(1):42-52.
46. Rodríguez-Antona C, Pallares J, Montero-Conde C, et al. Overexpression and activation of EGFR and VEGFR2 in medullary thyroid carcinomas is related to metastasis. Endocr Relat Cancer. 2010;17(1):7-16.
47. U.S. Food and Drug Administration. FDA approves new treatment for rare form of thyroid cancer [press release]. Silver Spring, MD: U.S. Food and Drug Administration; April 6, 2011.
48. Wells SA Jr, Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30(2):134-141.
49. U.S. Food and Drug Administration. FDA approves Cometriq to treat rare type of thyroid cancer [press release]. Silver Spring, MD: U.S. Food and Drug Administration; November 29, 2012.
50. Cui JJ. Inhibitors targeting hepatocyte growth factor receptor and their potential therapeutic applications. Expert Opin Ther Pat. 2007;17(9):1035-1045.
51. Schoffski P, Elisei R, Müller S, et al. An international, double-blind, randomized, placebo-controlled phase III trial (EXAM) of cabozantinib (XL184) in medullary thyroid carcinoma (MTC) patients (pts) with documented RECIST progression at baseline. J Clin Oncol. 2012;30(suppl):5508.
52. Kober F, Hermann M, Handler A, Krotla G. Effect of sorafenib in symptomatic metastatic medullary thyroid cancer. J Clin Oncol. 2007;25(18S):14065.
53. Lam ET, Ringel MD, Kloos RT, et al. Phase II clinical trial of sorafenib in metastatic medullary thyroid cancer. J Clin Oncol. 2010;28(14):2323-2330.
54. Hong DS, Sebti SM, Newman RA, et al. Phase I trial of a combination of the multikinase inhibitor sorafenib and the farnesyltransferase inhibitor tipifarnib in advanced malignancies. Clin Cancer Res. 2009;15(22):7061-7068.
55. Kelleher FC, McDermott R. Response to sunitinib in medullary thyroid cancer. Ann Intern Med. 2008;148(7):567.
56. Carr LL, Mankoff DA, Goulart BH, et al. Phase II study of daily sunitinib in FDGPET- positive, iodine-refractory differentiated thyroid cancer and metastatic medullary carcinoma of the thyroid with functional imaging correlation. Clin Cancer Res. 2010;16(21):5260-5268.
57. Bible KC, Suman VJ, Molina JR, et al; Endocrine Malignancies Disease Oriented Group; Mayo Clinic Cancer Center; Mayo Phase 2 Consortium. A multicenter phase 2 trial of pazopanib in metastatic and progressive medullary thyroid carcinoma: MC057H. J Clin Endocrinol Metab. 2014;99(5):1687-1693.
58. Ball DW. Medullary thyroid cancer: monitoring and therapy. Endocrinol Metab Clin North Am. 2007;36(3):823-837, viii.
59. Nocera M, Baudin E, Pellegriti G, Cailleux AF, Mechelany-Corone C, Schlumberger M. Treatment of advanced medullary thyroid cancer with an alternating combination of doxorubicin-streptozocin and 5 FU-dacarbazine. Groupe d’Etude des Tumeurs à Calcitonine (GETC). Br J Cancer. 2000;83(6):715-718.
60. Shimaoka K, Schoenfeld DA, DeWys WD, Creech RH, DeConti R. A randomized trial of doxorubicin versus doxorubicin plus cisplatin in patients with advanced thyroid carcinoma. Cancer. 1985;56(9):2155-2160.
Thyroid cancer is the ninth most common malignancy in the U.S. At the time of diagnosis, thyroid cancer is mostly confined to the thyroid gland and regional lymph nodes. However, around 4% of patients with thyroid cancer present with metastatic disease. When compared with localized and regional thyroid cancer, 5-year survival rates for metastatic thyroid cancer are significantly worse (99.9%, 97.6%, and 54.7%, respectively).1 Treatment options for metastatic thyroid cancer are limited and largely depend on the pathology and the type of thyroid cancer.
Thyroid cancer can be divided into differentiated, medullary, and anaplastic subtypes based on pathology. The treatment for metastatic differentiated thyroid cancer (DTC) consists of radioactive iodine therapy, thyroid-stimulating hormone (TSH) suppression (thyroxine hormone) therapy, and external beam radiotherapy. Systemic therapy is considered in patients with metastatic DTC who progress despite the above treatment modalities. In the case of metastatic medullary thyroid cancer (MTC), patients who are not candidates for surgery or radiation are considered for systemic therapy, because MTC does not respond to radioactive iodine or TSH suppressive therapy. On the other hand, metastatic anaplastic thyroid cancer is a very aggressive subtype with no effective therapy available to date. Palliation of symptoms is the main goal for these patients, which can be achieved by loco-regional resection and palliative irradiation.2,3
This review focuses on the newer treatment options for metastatic DTC and MTC that are based on inhibition of cellular kinases.
Differentiated Thyroid Cancer
Differentiated thyroid cancer is the most common histologic type of thyroid cancer, accounting for 95% of all thyroid cancers and consists of papillary, follicular, and poorly differentiated thyroid cancer.2,3 Surgery is the treatment of choice for DTC. Based on tumor size and its local extension in the neck, treatment options include unilateral lobectomy and isthmectomy, total thyroidectomy, central neck dissection, and more extensive resection.2,3 After surgery, radioactive iodine is recommended in patients with known metastatic disease; locally invasive tumor, regardless of size; or primary tumor > 4 cm, in the absence of other high-risk features.2 This should be followed by TSH suppressive hormone therapy.2
About 7% to 23% of patients with DTC develop distant metastases.4 Two-thirds of these patients become refractory to radioactive iodine.5 Prognosis remains poor in these patients, with a 10-year survival rate from the time detection of metastasis of only 10%.5-7 Treatment options are limited. However, recently the understanding of cell biology in terms of key signaling pathways called kinases has been elucidated. The kinases that can stabilize progressive metastatic disease seem to be attractive therapeutic targets in treating patients whose disease no longer responds to radioiodine and TSH suppressive hormone therapy.
Papillary thyroid cancers frequently carry gene mutations and rearrangements that lead to activation of the mitogen-activated protein kinase (MAPK), which promotes cell division. The sequential components leading to activation of MAPK include rearrangements of RET and NTRK1 tyrosine kinases, activating mutations of BRAF, and activating mutations of RAS.8,9 Similarly, overexpression of normal c-myc and c-fos genes, as well as mutations of HRAS, NRAS, and KRAS genes, is found in follicular adenomas, follicular cancers, and occasionally papillary cancers.10-14 Increased expression of vascular endothelial growth factor (VEGF) and its receptors (VEGFRs) might have a role in thyroid carcinoma as well.15
These kinases (the serine kinase BRAF and tyrosine kinases RET and RAS, and the contributory roles of tyrosine kinases in growth factor receptors such as the VEGFR) stimulate tumor proliferation, angiogenesis, invasion, metastasis, and inhibit tumor cell apoptosis. Kinase inhibitors target these signaling kinases, affecting tumor cell biology and its microenvironment.16,17
A wide variety of multitargeted kinase inhibitors (MKIs) have entered clinical trials for patients with advanced or progressive metastatic thyroid cancers. Two such agents, sorafenib and lenvatinib, are approved by the FDA for use in selected patients with refractory metastatic DTC, whereas many other drugs remain investigational for this disease. In phase 2 and 3 trials, most of the treatment responses for MKIs were partial. Complete responses were rare, and no study has reported a complete analysis of overall survival (OS) outcomes. Results from some new randomized trials indicate an improvement in progression-free survival (PFS) compared with placebo, and additional trials are underway.
Sorafenib
Sorafenib was approved by the FDA in 2013 for the treatment of locally recurrent or metastatic, progressive DTC that no longer responds to radioactive iodine treatment.18 Sorafenib is an oral, small molecule MKI. It works on VEGFRs 1, 2, and 3; platelet-derived growth factor receptor (PDGFR); common RET/PTC subtypes; KIT; and less potently, BRAF.19 The recommended dose is 400 mg orally twice a day.
In April 2014, Brose and colleagues published the phase 3 DECISION study on sorafenib.20 It was a multicenter, randomized, double-blinded, placebo-controlled trial of 417 patients with radioactive iodine-refractory locally advanced or metastatic DTC that had progressed within the previous 14 months.20 The results of the trial were promising. The median PFS was 5 months longer in the sorafenib group (10.8 mo) than in the placebo group (5.8 mo; hazard ratio [HR], 0.59; 95% conidence interval [CI], 0.45-0.76; P < .0001). The primary endpoint of the trial was PFS, and crossover from placebo to sorafenib was permitted upon progression. Overall survival did not differ significantly between the treatment groups (placebo vs sorafenib) at the time of the primary analysis data cutoff. However, OS results may have been confounded by postprogression crossover from placebo to open-label sorafenib by the majority of placebo patients.
In subgroup analysis, patients with BRAF and RAS mutations and wild-type BRAF and RAS subgroups had a significant increase in median PFS in the sorafenib treatment group compared with the placebo group (Table 1).20
Adverse events (AEs) occurred in 98.6% of patients receiving sorafenib during the double-blind period and in 87.6% of patients receiving placebo. Most AEs were grade 1 or 2. The most common AEs were hand-foot-skin reactions (76.3%), diarrhea (68.6%), alopecia (67.1%), and rash or desquamation (50.2%). Toxicities led to dose modification in 78% of patients and permanent discontinuation of therapy in 19%.20 Like other BRAF inhibitors, sorafenib has been associated with an increased incidence of cutaneous squamous cell carcinomas (5%), keratoacanthomas, and other premalignant actinic lesions.21
Lenvatinib
In February 2015, lenvatinib was approved for the treatment of locally recurrent or metastatic, progressive DTC that no longer responds to radioactive iodine treatment.22 Lenvatinib is a MKI of VEGFRs 1, 2, and 3; fibroblast growth factor receptors 1 through 4; PDGFR-α; RET, and KIT.23,24 The recommended dose is 24 mg orally once daily.
Schlumberger and colleagues published results from the SELECT trial, a randomized, double-blinded, multicenter phase 3 study involving 392 patients with progressive thyroid cancer that was refractory to iodine-131.25 A total of 261 patients received lenvatinib, and 131 patients received a placebo. Upon disease progression, patients in the placebo group were allowed to receive open-label lenvatinib. The study’s primary endpoint was PFS. Secondary endpoints were the response rate (RR), OS, and safety. The median PFS was 18.3 months in the lenvatinib group and 3.6 months in the placebo group (HR, 0.21; 99% CI, 0.14-0.31; P < .001). The RR was 64.8% in the lenvatinib group (4 complete and 165 partial responses) and 1.5% in the placebo group (P < .001). There was no significant difference in OS between the 2 groups (HR for death, 0.73; 95% CI, 0.50-1.07; P = .10). This difference became larger when a potential crossover bias was considered (rank-preserving structural failure time model; HR, 0.62; 95% CI, 0.40-1.00; P = .05).25
In a subgroup analysis, median PFS was about 14 months in the absence of prior anti-VEGFR therapy and 11 months of prior therapy. The treatmentrelated AEs were 97.3% in the lenvatinib group, and 75.9% were grade 3 or higher. Common treatmentrelated AEs of any grade in the lenvatinib group included hypertension (67.8%), diarrhea (59.4%), fatigue or asthenia (59.0%), decreased appetite (50.2%), decreased weight (46.4%), and nausea (41.0%). The study drug had to be discontinued because of AEs in 14% of patients who received lenvatinib and 2% of patients who received placebo. In the lenvatinib group, 2.3% patients had treatment-related fatal events (6 patients).25
Summary
Patients with DTC who progress after radioactive iodine therapy, TSH suppressive therapy, and external beam radiotherapy should be considered for systemic therapy. Systemic therapy consists of MKIs, which can stabilize progressive metastatic disease. These newer drugs have significant toxicities. Therefore, it is important to limit the use of systemic treatments to patients at significant risk for morbidity or mortality due to progressive metastatic disease. Patients treated with systemic agents should have a good baseline performance status, such as being ambulatory at least 50% (Eastern Cooperative Oncology Group performance score of 2) of the day to tolerate these treatments.
Patients who have disease progression or are unable to tolerate sorafenib and lenvatinib can choose to participate in clinical trials with investigational multitarget inhibitors. Other alternatives include vandetinib, pazopanib, and sunitinib, which finished phase 2 trials and showed some partial responses.26-30 If the patients are unable to tolerate MKIs, they can try doxorubicin-based conventional chemotherapy regimens.31
Medullary Thyroid Cancer
Medullary thyroid cancer is a neuroendocrine tumor arising from the thyroid parafollicular cells, accounting for about 4% of thyroid carcinomas, most of which are sporadic. However, some are familial as part of the multiple endocrine neoplasia type 2 (MEN 2) syndromes, which are transmitted in an autosomal dominant fashion.32,33 Similar to DTC, the primary treatment option is surgery. Medullary thyroid cancer can be cured only by complete resection of the thyroid tumor and any local and regional metastases. Compared with DTC, metastatic MTC is unresponsive to radioiodine or TSH
suppressive treatment, because this cancer neither concentrates iodine nor is TSH dependent.34,35
The 10-year OS rate in MTC is ≤ 40% in patients with locally advanced or metastatic disease.32,36,37 In hereditary MTC, germline mutations in the c-ret proto-oncogene occur in virtually all patients. In sporadic MTC, 85% of patients have the M918T mutation, and somatic c-ret mutations are seen in about 50% of patients.38-42
Similar to DTC, due to the presence of mutations involving RET receptor tyrosine kinase, molecular targeted therapeutics with activity against RET demonstrate a potential therapeutic target in MTC.43-45 Other signaling pathways likely to contribute to the growth and invasiveness of MTC include VEGFR-dependent tumor angiogenesis and epidermal growth factor receptor (EGFR)-dependent tumor cell proliferation.46
In 2011 and 2012, the FDA approved tyrosine kinase inhibitors (TKIs) vandetanib and cabozantinib for metastatic MTC. Similar to treatment for DTC, systemic therapy is mainly based on targeted therapies. Patients with progressive or symptomatic metastatic disease who are not candidates for surgery or radiotherapy should be considered for TKI therapy.
Vandetanib
Vandetanib is approved for unresectable, locally advanced or metastatic sporadic or hereditary MTC.47 The daily recommended dose is 300 mg/d. It is an oral MKI that targets VEGFR, RET/PTC, and the EGFR.48
The ZETA trial was an international randomized phase 3 trial involving patients with unresectable locally advanced or metastatic sporadic or hereditary MTC.48 In a ZETA trial study by Wells Jr and colleagues, patients with advanced MTC were randomly assigned in a 2:1 ratio to receive vandetanib 300 mg/d or placebo. After objective disease progression, patients could elect to receive openlabel vandetanib. The primary endpoint was PFS, determined by independent central Response Evaluation Criteria in Solid Tumors assessments.
A total of 331 patients were randomly assigned to receive vandetanib (231 patients) or placebo (100 patients). At data cutoff, with median follow-up of 24 months, PFS was significantly prolonged in patients randomly assigned to vandetanib vs placebo (30.5 mo vs 19.3 mo; HR, 0.46; 95% CI, 0.31-0.69). The objective RR was significantly higher in the vandetanib group (45% vs 13%). The presence of a somatic RET M918T mutation predicted an improved PFS.
Common AEs (any grade) noted with vandetanib vs placebo include diarrhea (56% vs 26%), rash (45% vs 11%), nausea (33% vs 16%), hypertension (32% vs 5%), and headache (26% vs 9%). Torsades de pointes and sudden death were reported in patients receiving vandetanib. Data on OS were immature at data cutoff (HR, 0.89; 95% CI, 0.48-1.65). A final survival analysis will take place when 50% of the patients have died.48
Vandetanib is currently approved with a Risk Evaluation and Mitigation Strategy to inform health care professionals about serious heart-related risks. Electrocardiograms and serum potassium, calcium, magnesium, and TSH should be taken at 2 to 4 weeks and 8 to 12 weeks after starting treatment and every 3 months after that. Patients with diarrhea may require more frequent monitoring.
Cabozantinib
In 2012, the FDA approved cabozantinib for the treatment of progressive, metastatic MTC.49 It is an oral, small molecule TKI that targets VEGFRs 1 and 2, MET, and RET. The inhibitory activity against MET, the cognate receptor for the hepatocyte growth factor, may provide additional synergistic benefit in MTC.50 The daily recommended dose is 140 mg/d. A phase 3 randomized EXAM trial in patients with progressive, metastatic, or unresectable locally advanced MTC.51 Three hundred thirty patients were randomly assigned to receive either cabozantinib 140 mg or placebo once daily. Progressionfree survival was improved with cabozantinib compared with that of placebo (11.2 vs 4.0 mo; HR, 0.28; 95% CI, 0.19-0.40). Partial responses were observed in 27% vs 0% in placebo. A planned interim analysis of OS was conducted, including 96 (44%) of the 217 patient deaths required for the final analysis, with no statistically significant difference observed between the treatment arms (HR, 0.98; 95% CI, 0.63-1.52). Survival follow-up is planned to continue until at least 217 deaths have been observed.
There was markedly improved PFS in the subset of patients treated with cabozantinib compared with placebo whose tumors contained RET M918T mutations (61 vs 17 wk; HR, 0.15; 95% CI, 0.08-0.28) or RAS mutations (47 vs 8 wk; HR, 0.15; 95% CI, 0.02-1.10).51
The most common AEs, occurring in ≥ 25% of patients, were diarrhea, stomatitis, hand and foot syndrome, hypertension, and abdominal pain. Although uncommon, clinically significant AEs also included fistula formation and osteonecrosis of the jaw.
Summary
Patients with progressive or symptomatic metastatic disease who are not candidates for surgery or radiotherapy should be considered for TKI therapy. Though not curative, TKIs can only stabilize disease progression. Initiation of TKIs should be considered in rapidly progressive disease, because these drugs are associated with considerable AEs affecting the quality of life (QOL).
Patients who progressed or were unable to tolerate vandetanib or cabozantinib can choose to participate in clinical trials with investigational multitarget inhibitors. Other alternatives include pazopanib, sunitinib, and sorafenib, which finished phase 2 trials and showed some partial responses.29,52-57 If patients are unable to tolerate MKIs, they can try conventional chemotherapy consisting of dacarbazine with other agents or doxorubin.58-60
Conclusions
Molecular targeted therapy is an emerging treatment option for patients with metastatic thyroid cancer (Table 2). The authors suggest that such patients participate in clinical trials in the hope of developing more effective and tolerable drugs and recommend oral TKIs for patients with rapidly progressive disease who cannot participate in a clinical trial. For patients who cannot tolerate or fail one TKI, the authors recommend trying other TKIs before initiating cytotoxic chemotherapy.
Before initiation of treatment for metastatic disease, an important factor to consider is the pace of disease progression. Patients who are asymptomatic and have the very indolent disease may postpone kinase inhibitor therapy until they become rapidly progressive or symptomatic, because the AEs of treatment will adversely affect the patient’s QOL. In patients with symptomatic and rapidly progressive disease, initiation of treatment with kinase inhibitor therapy can lead to stabilization of disease, although at the cost of some AEs. More structured clinical trials are needed, along with an evaluation of newer molecular targets for the management of this progressive metastatic disease with a dismal prognosis.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
Thyroid cancer is the ninth most common malignancy in the U.S. At the time of diagnosis, thyroid cancer is mostly confined to the thyroid gland and regional lymph nodes. However, around 4% of patients with thyroid cancer present with metastatic disease. When compared with localized and regional thyroid cancer, 5-year survival rates for metastatic thyroid cancer are significantly worse (99.9%, 97.6%, and 54.7%, respectively).1 Treatment options for metastatic thyroid cancer are limited and largely depend on the pathology and the type of thyroid cancer.
Thyroid cancer can be divided into differentiated, medullary, and anaplastic subtypes based on pathology. The treatment for metastatic differentiated thyroid cancer (DTC) consists of radioactive iodine therapy, thyroid-stimulating hormone (TSH) suppression (thyroxine hormone) therapy, and external beam radiotherapy. Systemic therapy is considered in patients with metastatic DTC who progress despite the above treatment modalities. In the case of metastatic medullary thyroid cancer (MTC), patients who are not candidates for surgery or radiation are considered for systemic therapy, because MTC does not respond to radioactive iodine or TSH suppressive therapy. On the other hand, metastatic anaplastic thyroid cancer is a very aggressive subtype with no effective therapy available to date. Palliation of symptoms is the main goal for these patients, which can be achieved by loco-regional resection and palliative irradiation.2,3
This review focuses on the newer treatment options for metastatic DTC and MTC that are based on inhibition of cellular kinases.
Differentiated Thyroid Cancer
Differentiated thyroid cancer is the most common histologic type of thyroid cancer, accounting for 95% of all thyroid cancers and consists of papillary, follicular, and poorly differentiated thyroid cancer.2,3 Surgery is the treatment of choice for DTC. Based on tumor size and its local extension in the neck, treatment options include unilateral lobectomy and isthmectomy, total thyroidectomy, central neck dissection, and more extensive resection.2,3 After surgery, radioactive iodine is recommended in patients with known metastatic disease; locally invasive tumor, regardless of size; or primary tumor > 4 cm, in the absence of other high-risk features.2 This should be followed by TSH suppressive hormone therapy.2
About 7% to 23% of patients with DTC develop distant metastases.4 Two-thirds of these patients become refractory to radioactive iodine.5 Prognosis remains poor in these patients, with a 10-year survival rate from the time detection of metastasis of only 10%.5-7 Treatment options are limited. However, recently the understanding of cell biology in terms of key signaling pathways called kinases has been elucidated. The kinases that can stabilize progressive metastatic disease seem to be attractive therapeutic targets in treating patients whose disease no longer responds to radioiodine and TSH suppressive hormone therapy.
Papillary thyroid cancers frequently carry gene mutations and rearrangements that lead to activation of the mitogen-activated protein kinase (MAPK), which promotes cell division. The sequential components leading to activation of MAPK include rearrangements of RET and NTRK1 tyrosine kinases, activating mutations of BRAF, and activating mutations of RAS.8,9 Similarly, overexpression of normal c-myc and c-fos genes, as well as mutations of HRAS, NRAS, and KRAS genes, is found in follicular adenomas, follicular cancers, and occasionally papillary cancers.10-14 Increased expression of vascular endothelial growth factor (VEGF) and its receptors (VEGFRs) might have a role in thyroid carcinoma as well.15
These kinases (the serine kinase BRAF and tyrosine kinases RET and RAS, and the contributory roles of tyrosine kinases in growth factor receptors such as the VEGFR) stimulate tumor proliferation, angiogenesis, invasion, metastasis, and inhibit tumor cell apoptosis. Kinase inhibitors target these signaling kinases, affecting tumor cell biology and its microenvironment.16,17
A wide variety of multitargeted kinase inhibitors (MKIs) have entered clinical trials for patients with advanced or progressive metastatic thyroid cancers. Two such agents, sorafenib and lenvatinib, are approved by the FDA for use in selected patients with refractory metastatic DTC, whereas many other drugs remain investigational for this disease. In phase 2 and 3 trials, most of the treatment responses for MKIs were partial. Complete responses were rare, and no study has reported a complete analysis of overall survival (OS) outcomes. Results from some new randomized trials indicate an improvement in progression-free survival (PFS) compared with placebo, and additional trials are underway.
Sorafenib
Sorafenib was approved by the FDA in 2013 for the treatment of locally recurrent or metastatic, progressive DTC that no longer responds to radioactive iodine treatment.18 Sorafenib is an oral, small molecule MKI. It works on VEGFRs 1, 2, and 3; platelet-derived growth factor receptor (PDGFR); common RET/PTC subtypes; KIT; and less potently, BRAF.19 The recommended dose is 400 mg orally twice a day.
In April 2014, Brose and colleagues published the phase 3 DECISION study on sorafenib.20 It was a multicenter, randomized, double-blinded, placebo-controlled trial of 417 patients with radioactive iodine-refractory locally advanced or metastatic DTC that had progressed within the previous 14 months.20 The results of the trial were promising. The median PFS was 5 months longer in the sorafenib group (10.8 mo) than in the placebo group (5.8 mo; hazard ratio [HR], 0.59; 95% conidence interval [CI], 0.45-0.76; P < .0001). The primary endpoint of the trial was PFS, and crossover from placebo to sorafenib was permitted upon progression. Overall survival did not differ significantly between the treatment groups (placebo vs sorafenib) at the time of the primary analysis data cutoff. However, OS results may have been confounded by postprogression crossover from placebo to open-label sorafenib by the majority of placebo patients.
In subgroup analysis, patients with BRAF and RAS mutations and wild-type BRAF and RAS subgroups had a significant increase in median PFS in the sorafenib treatment group compared with the placebo group (Table 1).20
Adverse events (AEs) occurred in 98.6% of patients receiving sorafenib during the double-blind period and in 87.6% of patients receiving placebo. Most AEs were grade 1 or 2. The most common AEs were hand-foot-skin reactions (76.3%), diarrhea (68.6%), alopecia (67.1%), and rash or desquamation (50.2%). Toxicities led to dose modification in 78% of patients and permanent discontinuation of therapy in 19%.20 Like other BRAF inhibitors, sorafenib has been associated with an increased incidence of cutaneous squamous cell carcinomas (5%), keratoacanthomas, and other premalignant actinic lesions.21
Lenvatinib
In February 2015, lenvatinib was approved for the treatment of locally recurrent or metastatic, progressive DTC that no longer responds to radioactive iodine treatment.22 Lenvatinib is a MKI of VEGFRs 1, 2, and 3; fibroblast growth factor receptors 1 through 4; PDGFR-α; RET, and KIT.23,24 The recommended dose is 24 mg orally once daily.
Schlumberger and colleagues published results from the SELECT trial, a randomized, double-blinded, multicenter phase 3 study involving 392 patients with progressive thyroid cancer that was refractory to iodine-131.25 A total of 261 patients received lenvatinib, and 131 patients received a placebo. Upon disease progression, patients in the placebo group were allowed to receive open-label lenvatinib. The study’s primary endpoint was PFS. Secondary endpoints were the response rate (RR), OS, and safety. The median PFS was 18.3 months in the lenvatinib group and 3.6 months in the placebo group (HR, 0.21; 99% CI, 0.14-0.31; P < .001). The RR was 64.8% in the lenvatinib group (4 complete and 165 partial responses) and 1.5% in the placebo group (P < .001). There was no significant difference in OS between the 2 groups (HR for death, 0.73; 95% CI, 0.50-1.07; P = .10). This difference became larger when a potential crossover bias was considered (rank-preserving structural failure time model; HR, 0.62; 95% CI, 0.40-1.00; P = .05).25
In a subgroup analysis, median PFS was about 14 months in the absence of prior anti-VEGFR therapy and 11 months of prior therapy. The treatmentrelated AEs were 97.3% in the lenvatinib group, and 75.9% were grade 3 or higher. Common treatmentrelated AEs of any grade in the lenvatinib group included hypertension (67.8%), diarrhea (59.4%), fatigue or asthenia (59.0%), decreased appetite (50.2%), decreased weight (46.4%), and nausea (41.0%). The study drug had to be discontinued because of AEs in 14% of patients who received lenvatinib and 2% of patients who received placebo. In the lenvatinib group, 2.3% patients had treatment-related fatal events (6 patients).25
Summary
Patients with DTC who progress after radioactive iodine therapy, TSH suppressive therapy, and external beam radiotherapy should be considered for systemic therapy. Systemic therapy consists of MKIs, which can stabilize progressive metastatic disease. These newer drugs have significant toxicities. Therefore, it is important to limit the use of systemic treatments to patients at significant risk for morbidity or mortality due to progressive metastatic disease. Patients treated with systemic agents should have a good baseline performance status, such as being ambulatory at least 50% (Eastern Cooperative Oncology Group performance score of 2) of the day to tolerate these treatments.
Patients who have disease progression or are unable to tolerate sorafenib and lenvatinib can choose to participate in clinical trials with investigational multitarget inhibitors. Other alternatives include vandetinib, pazopanib, and sunitinib, which finished phase 2 trials and showed some partial responses.26-30 If the patients are unable to tolerate MKIs, they can try doxorubicin-based conventional chemotherapy regimens.31
Medullary Thyroid Cancer
Medullary thyroid cancer is a neuroendocrine tumor arising from the thyroid parafollicular cells, accounting for about 4% of thyroid carcinomas, most of which are sporadic. However, some are familial as part of the multiple endocrine neoplasia type 2 (MEN 2) syndromes, which are transmitted in an autosomal dominant fashion.32,33 Similar to DTC, the primary treatment option is surgery. Medullary thyroid cancer can be cured only by complete resection of the thyroid tumor and any local and regional metastases. Compared with DTC, metastatic MTC is unresponsive to radioiodine or TSH
suppressive treatment, because this cancer neither concentrates iodine nor is TSH dependent.34,35
The 10-year OS rate in MTC is ≤ 40% in patients with locally advanced or metastatic disease.32,36,37 In hereditary MTC, germline mutations in the c-ret proto-oncogene occur in virtually all patients. In sporadic MTC, 85% of patients have the M918T mutation, and somatic c-ret mutations are seen in about 50% of patients.38-42
Similar to DTC, due to the presence of mutations involving RET receptor tyrosine kinase, molecular targeted therapeutics with activity against RET demonstrate a potential therapeutic target in MTC.43-45 Other signaling pathways likely to contribute to the growth and invasiveness of MTC include VEGFR-dependent tumor angiogenesis and epidermal growth factor receptor (EGFR)-dependent tumor cell proliferation.46
In 2011 and 2012, the FDA approved tyrosine kinase inhibitors (TKIs) vandetanib and cabozantinib for metastatic MTC. Similar to treatment for DTC, systemic therapy is mainly based on targeted therapies. Patients with progressive or symptomatic metastatic disease who are not candidates for surgery or radiotherapy should be considered for TKI therapy.
Vandetanib
Vandetanib is approved for unresectable, locally advanced or metastatic sporadic or hereditary MTC.47 The daily recommended dose is 300 mg/d. It is an oral MKI that targets VEGFR, RET/PTC, and the EGFR.48
The ZETA trial was an international randomized phase 3 trial involving patients with unresectable locally advanced or metastatic sporadic or hereditary MTC.48 In a ZETA trial study by Wells Jr and colleagues, patients with advanced MTC were randomly assigned in a 2:1 ratio to receive vandetanib 300 mg/d or placebo. After objective disease progression, patients could elect to receive openlabel vandetanib. The primary endpoint was PFS, determined by independent central Response Evaluation Criteria in Solid Tumors assessments.
A total of 331 patients were randomly assigned to receive vandetanib (231 patients) or placebo (100 patients). At data cutoff, with median follow-up of 24 months, PFS was significantly prolonged in patients randomly assigned to vandetanib vs placebo (30.5 mo vs 19.3 mo; HR, 0.46; 95% CI, 0.31-0.69). The objective RR was significantly higher in the vandetanib group (45% vs 13%). The presence of a somatic RET M918T mutation predicted an improved PFS.
Common AEs (any grade) noted with vandetanib vs placebo include diarrhea (56% vs 26%), rash (45% vs 11%), nausea (33% vs 16%), hypertension (32% vs 5%), and headache (26% vs 9%). Torsades de pointes and sudden death were reported in patients receiving vandetanib. Data on OS were immature at data cutoff (HR, 0.89; 95% CI, 0.48-1.65). A final survival analysis will take place when 50% of the patients have died.48
Vandetanib is currently approved with a Risk Evaluation and Mitigation Strategy to inform health care professionals about serious heart-related risks. Electrocardiograms and serum potassium, calcium, magnesium, and TSH should be taken at 2 to 4 weeks and 8 to 12 weeks after starting treatment and every 3 months after that. Patients with diarrhea may require more frequent monitoring.
Cabozantinib
In 2012, the FDA approved cabozantinib for the treatment of progressive, metastatic MTC.49 It is an oral, small molecule TKI that targets VEGFRs 1 and 2, MET, and RET. The inhibitory activity against MET, the cognate receptor for the hepatocyte growth factor, may provide additional synergistic benefit in MTC.50 The daily recommended dose is 140 mg/d. A phase 3 randomized EXAM trial in patients with progressive, metastatic, or unresectable locally advanced MTC.51 Three hundred thirty patients were randomly assigned to receive either cabozantinib 140 mg or placebo once daily. Progressionfree survival was improved with cabozantinib compared with that of placebo (11.2 vs 4.0 mo; HR, 0.28; 95% CI, 0.19-0.40). Partial responses were observed in 27% vs 0% in placebo. A planned interim analysis of OS was conducted, including 96 (44%) of the 217 patient deaths required for the final analysis, with no statistically significant difference observed between the treatment arms (HR, 0.98; 95% CI, 0.63-1.52). Survival follow-up is planned to continue until at least 217 deaths have been observed.
There was markedly improved PFS in the subset of patients treated with cabozantinib compared with placebo whose tumors contained RET M918T mutations (61 vs 17 wk; HR, 0.15; 95% CI, 0.08-0.28) or RAS mutations (47 vs 8 wk; HR, 0.15; 95% CI, 0.02-1.10).51
The most common AEs, occurring in ≥ 25% of patients, were diarrhea, stomatitis, hand and foot syndrome, hypertension, and abdominal pain. Although uncommon, clinically significant AEs also included fistula formation and osteonecrosis of the jaw.
Summary
Patients with progressive or symptomatic metastatic disease who are not candidates for surgery or radiotherapy should be considered for TKI therapy. Though not curative, TKIs can only stabilize disease progression. Initiation of TKIs should be considered in rapidly progressive disease, because these drugs are associated with considerable AEs affecting the quality of life (QOL).
Patients who progressed or were unable to tolerate vandetanib or cabozantinib can choose to participate in clinical trials with investigational multitarget inhibitors. Other alternatives include pazopanib, sunitinib, and sorafenib, which finished phase 2 trials and showed some partial responses.29,52-57 If patients are unable to tolerate MKIs, they can try conventional chemotherapy consisting of dacarbazine with other agents or doxorubin.58-60
Conclusions
Molecular targeted therapy is an emerging treatment option for patients with metastatic thyroid cancer (Table 2). The authors suggest that such patients participate in clinical trials in the hope of developing more effective and tolerable drugs and recommend oral TKIs for patients with rapidly progressive disease who cannot participate in a clinical trial. For patients who cannot tolerate or fail one TKI, the authors recommend trying other TKIs before initiating cytotoxic chemotherapy.
Before initiation of treatment for metastatic disease, an important factor to consider is the pace of disease progression. Patients who are asymptomatic and have the very indolent disease may postpone kinase inhibitor therapy until they become rapidly progressive or symptomatic, because the AEs of treatment will adversely affect the patient’s QOL. In patients with symptomatic and rapidly progressive disease, initiation of treatment with kinase inhibitor therapy can lead to stabilization of disease, although at the cost of some AEs. More structured clinical trials are needed, along with an evaluation of newer molecular targets for the management of this progressive metastatic disease with a dismal prognosis.
Author disclosures
The authors report no actual or potential conflicts of interest with regard to this article.
Disclaimer
The opinions expressed herein are those of the authors and do not necessarily reflect those of Federal Practitioner, Frontline Medical Communications Inc., the U.S. Government, or any of its agencies. This article may discuss unlabeled or investigational use of certain drugs. Please review complete prescribing information for specific drugs or drug combinations—including indications, contraindications, warnings, and adverse effects—before administering pharmacologic therapy to patients.
Click here to read the digital edition.
1. Howlader N, Noone AM, Krapcho M, et al, eds. SEER Cancer Statistics Review, 1975-2012. Bethesda, MD: National Cancer Institute; 2015.
2. Cooper DS, Doherty GM, Haugen BR, et al; American Thyroid Association (ATA) Guidelines Taskforce on Thyroid Nodules and Differentiated Thyroid Cancer. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2009;19(11):1167-1214.
3. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: thyroid carcinoma. National Comprehensive Cancer Network Website. http://www.nccn.org/professionals/physician_gls/pdf/thyroid.pdf. Updated May 11, 2015. Accessed July 10, 2015.
4. Shoup M, Stojadinovic A, Nissan A, et al. Prognostic indicators of outcomes in patients with distant metastases from differentiated thyroid carcinoma. J Am Coll Surg. 2003;197(2):191-197.
5. Durante C, Haddy N, Baudin E, et al. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: benefits and limits of radioiodine therapy. J Clin Endocrinol Metab. 2006;91(8):2892-2899.
6. Busaidy NL, Cabanillas ME. Differentiated thyroid cancer: management of patients with radioiodine nonresponsive disease. J Thyroid Res. 2012;2012:618985.
7. Schlumberger M, Brose M, Elisei R, et al. Definition and management of radioactive iodine-refractory differentiated thyroid cancer. Lancet Diabetes Endocrinol. 2014;2(5):356-358.
8. Melillo RM, Castellone MD, Guarino V, et al. The RET/PTC-RAS-BRAF linear signaling cascade mediates the motile and mitogenic phenotype of thyroid cancer cells. J Clin Invest. 2005;115(4):1068-1081.
9. Ciampi R, Nikiforov YE. RET/PTC rearrangements and BRAF mutations in thyroid tumorigenesis. Endocrinology. 2007;148(3):936-941.
10. Lemoine NR, Mayall ES, Wyllie FS, et al. High frequency of ras oncogene activation in all stages of human thyroid tumorigenesis. Oncogene. 1989;4(2):159-164.
11. Namba H, Rubin SA, Fagin JA. Point mutations of ras oncogenes are an early event in thyroid tumorigenesis. Mol Endocrinol. 1990;4(10):1474-1479.
12. Suarez HG, du Villard JA, Severino M, et al. Presence of mutations in all three ras genes in human thyroid tumors. Oncogene. 1990;5(4):565-570.
13. Karga H, Lee JK, Vickery AL Jr, Thor A, Gaz RD, Jameson JL. Ras oncogene mutations in benign and malignant thyroid neoplasms. J Clin Endocrinol Metab. 1991;73(4):832-836.
14. Terrier P, Sheng ZM, Schlumberger M, et al. Structure and expression of c-myc and c-fos proto-oncogenes in thyroid carcinomas. Br J Cancer. 1988;57(1):43-47.
15. Klein M, Vignaud JM, Hennequin V, et al. Increased expression of the vascular endothelial growth factor is a pejorative prognosis marker in papillary thyroid carcinoma. J Clin Endocrinol Metab. 2001;86(2):656-658.
16. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9(1):28-39.
17. Haugen BR, Sherman SI. Evolving approaches to patients with advanced differentiated thyroid cancer. Endocr Rev. 2013;34(3):439-455.
18. U.S. Food and Drug Administration. FDA approves Nexavar to treat type of thyroid cancer [press release]. Silver Spring, MD: U.S. Food and Drug Administration; November 22, 2013.
19. Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099-7109.
20. Brose MS, Nutting CM, Jarzab B, et al; DECISION investigators. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet. 2014;384(9940):319-328.
21. Dubauskas Z, Kunishige J, Prieto VG, Jonasch E, Hwu P, Tannir NM. Cutaneous squamous cell carcinoma and inflammation of actinic keratoses associated with sorafenib. Clin Genitourin Cancer. 2009;7(1):20-23.
22. U.S. Food and Drug Administration. FDA approves Lenvima for a type of thyroid cancer [press release]. Silver Spring, MD: U.S. Food and Drug Administration; February 13, 2015.
23. Matsui J, Yamamoto Y, Funahashi Y, et al. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int J Cancer. 2008;122(3):664-671.
24. Matsui J, Funahashi Y, Uenaka T, Watanabe T, Tsuruoka A, Asada M. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factorreceptor (VEGF-R) 2 and VEGF-R3 kinase. Clin Cancer Res. 2008;14(17):5459-5465.
25. Schlumberger M, Tahara M, Wirth LJ, et al. Lenvatinib versus placebo in radioiodine- refractory thyroid cancer. N Engl J Med. 2015;372(7):621-630.
26. Leboulleux S, Bastholt L, Krause T, et al. Vandetanib in locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 2 trial. Lancet Oncol. 2012;13(9):897-905.
27. A randomised, double-blind, placebo-controlled, multi-centre phase III study to assess the efficacy and safety of vandetanib (CAPRELSA) 300 mg in patients with differentiated thyroid cancer that is either locally advanced or metastatic who are refractory or unsuitable for radioiodine (RAI) therapy. Trial number NCT01876784. ClinicalTrials.gov Website. https://clinicaltrials.gov/show/NCT01876784. Updated June 26, 2015. Accessed July 22, 2015.
28. Bible KC, Suman VJ, Molina JR, et al; Endocrine Malignancies Disease Oriented Group; Mayo Clinic Cancer Center; Mayo Phase 2 Consortium. Efficacy of pazopanib in progressive, radioiodine-refractory, metastatic differentiated thyroid cancers: results of a phase 2 consortium study. Lancet Oncol. 2010;11(10):962-972.
29. Kim DW, Jo YS, Jung HS, et al. An orally administered multitarget tyrosine kinase inhibitor, SU11248, is a novel potent inhibitor of thyroid oncogenic RET/papillary thyroid cancer kinases. J Clin Endocrinol Metab. 2006;91(10):4070-4076.
30. Dawson SJ, Conus NM, Toner GC, et al. Sustained clinical responses to tyrosine kinase inhibitor sunitinib in thyroid carcinoma. Anticancer Drugs. 2008;19(5):547-552.
31. Carter SK, Blum RH. New chemotherapeutic agents—bleomycin and adriamycin. CA Cancer J Clin. 1974;24(6):322-331.
32. Hundahl SA, Fleming ID, Fremgen AM, Menck HR. A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985-1995 [see comments]. Cancer. 1998;83(12):2638-2648.
33. Lakhani VT, You YN, Wells SA. The multiple endocrine neoplasia syndromes. Annu Rev Med. 2007;58:253-265.
34. Martins RG, Rajendran JG, Capell P, Byrd DR, Mankoff DA. Medullary thyroid cancer: options for systemic therapy of metastatic disease? J Clin Oncol. 2006;24(11):1653-1655.
35. American Thyroid Association Guidelines Task Force; Kloos RT, Eng C, Evans DB, et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid. 2009;19(6):565-612.
36. Roman S, Lin R, Sosa JA. Prognosis of medullary thyroid carcinoma: demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer. 2006;107(9):2134-2142.
37. Modigliani E, Cohen R, Campos JM, et al. Prognostic factors for survival and for biochemical cure in medullary thyroid carcinoma: results in 899 patients. The GETC Study Group. Groupe d’étude des tumeurs à calcitonine. Clin Endocrinol (Oxf). 1998;48(3):265-273.
38. Donis-Keller H, Dou S, Chi D, et al. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet. 1993;2(7):851-856.
39. Mulligan LM, Kwok JB, Healey CS, et al. Germ-line mutations of the RET protooncogene in multiple endocrine neoplasia type 2A. Nature. 363(6428):458-460.
40. Carlson KM, Dou S, Chi D, et al. Single missense mutation in the tyrosine kinase catalytic domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B. Proc Natl Acad Sci USA. 1994;91(4):1579-1583.
41. Marsh DJ, Learoyd DL, Andrew SD, et al. Somatic mutations in the RET protooncogene in sporadic medullary thyroid carcinoma. Clin Endocrinol (Oxf). 1996;44(3):249-257.
42. Elisei R, Cosci B, Romei C, et al. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. J Clin Endocrinol Metab. 2008;93(3):682-687.
43. Carlomagno F, Vitagliano D, Guida T, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002;62(24):7284-7290.
44. Carlomagno F, Anaganti S, Guida T, et al. BAY 43-9006 inhibition of oncogenic RET mutants. J Natl Cancer Inst. 2006;98(5):326-334.
45. Santoro M, Carlomagno F. Drug insight: small molecule inhibitors of protein kinases in the treatment of thyroid cancer. Nat Clin Pract Endocrinol Metab. 2006;2(1):42-52.
46. Rodríguez-Antona C, Pallares J, Montero-Conde C, et al. Overexpression and activation of EGFR and VEGFR2 in medullary thyroid carcinomas is related to metastasis. Endocr Relat Cancer. 2010;17(1):7-16.
47. U.S. Food and Drug Administration. FDA approves new treatment for rare form of thyroid cancer [press release]. Silver Spring, MD: U.S. Food and Drug Administration; April 6, 2011.
48. Wells SA Jr, Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30(2):134-141.
49. U.S. Food and Drug Administration. FDA approves Cometriq to treat rare type of thyroid cancer [press release]. Silver Spring, MD: U.S. Food and Drug Administration; November 29, 2012.
50. Cui JJ. Inhibitors targeting hepatocyte growth factor receptor and their potential therapeutic applications. Expert Opin Ther Pat. 2007;17(9):1035-1045.
51. Schoffski P, Elisei R, Müller S, et al. An international, double-blind, randomized, placebo-controlled phase III trial (EXAM) of cabozantinib (XL184) in medullary thyroid carcinoma (MTC) patients (pts) with documented RECIST progression at baseline. J Clin Oncol. 2012;30(suppl):5508.
52. Kober F, Hermann M, Handler A, Krotla G. Effect of sorafenib in symptomatic metastatic medullary thyroid cancer. J Clin Oncol. 2007;25(18S):14065.
53. Lam ET, Ringel MD, Kloos RT, et al. Phase II clinical trial of sorafenib in metastatic medullary thyroid cancer. J Clin Oncol. 2010;28(14):2323-2330.
54. Hong DS, Sebti SM, Newman RA, et al. Phase I trial of a combination of the multikinase inhibitor sorafenib and the farnesyltransferase inhibitor tipifarnib in advanced malignancies. Clin Cancer Res. 2009;15(22):7061-7068.
55. Kelleher FC, McDermott R. Response to sunitinib in medullary thyroid cancer. Ann Intern Med. 2008;148(7):567.
56. Carr LL, Mankoff DA, Goulart BH, et al. Phase II study of daily sunitinib in FDGPET- positive, iodine-refractory differentiated thyroid cancer and metastatic medullary carcinoma of the thyroid with functional imaging correlation. Clin Cancer Res. 2010;16(21):5260-5268.
57. Bible KC, Suman VJ, Molina JR, et al; Endocrine Malignancies Disease Oriented Group; Mayo Clinic Cancer Center; Mayo Phase 2 Consortium. A multicenter phase 2 trial of pazopanib in metastatic and progressive medullary thyroid carcinoma: MC057H. J Clin Endocrinol Metab. 2014;99(5):1687-1693.
58. Ball DW. Medullary thyroid cancer: monitoring and therapy. Endocrinol Metab Clin North Am. 2007;36(3):823-837, viii.
59. Nocera M, Baudin E, Pellegriti G, Cailleux AF, Mechelany-Corone C, Schlumberger M. Treatment of advanced medullary thyroid cancer with an alternating combination of doxorubicin-streptozocin and 5 FU-dacarbazine. Groupe d’Etude des Tumeurs à Calcitonine (GETC). Br J Cancer. 2000;83(6):715-718.
60. Shimaoka K, Schoenfeld DA, DeWys WD, Creech RH, DeConti R. A randomized trial of doxorubicin versus doxorubicin plus cisplatin in patients with advanced thyroid carcinoma. Cancer. 1985;56(9):2155-2160.
1. Howlader N, Noone AM, Krapcho M, et al, eds. SEER Cancer Statistics Review, 1975-2012. Bethesda, MD: National Cancer Institute; 2015.
2. Cooper DS, Doherty GM, Haugen BR, et al; American Thyroid Association (ATA) Guidelines Taskforce on Thyroid Nodules and Differentiated Thyroid Cancer. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2009;19(11):1167-1214.
3. National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: thyroid carcinoma. National Comprehensive Cancer Network Website. http://www.nccn.org/professionals/physician_gls/pdf/thyroid.pdf. Updated May 11, 2015. Accessed July 10, 2015.
4. Shoup M, Stojadinovic A, Nissan A, et al. Prognostic indicators of outcomes in patients with distant metastases from differentiated thyroid carcinoma. J Am Coll Surg. 2003;197(2):191-197.
5. Durante C, Haddy N, Baudin E, et al. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: benefits and limits of radioiodine therapy. J Clin Endocrinol Metab. 2006;91(8):2892-2899.
6. Busaidy NL, Cabanillas ME. Differentiated thyroid cancer: management of patients with radioiodine nonresponsive disease. J Thyroid Res. 2012;2012:618985.
7. Schlumberger M, Brose M, Elisei R, et al. Definition and management of radioactive iodine-refractory differentiated thyroid cancer. Lancet Diabetes Endocrinol. 2014;2(5):356-358.
8. Melillo RM, Castellone MD, Guarino V, et al. The RET/PTC-RAS-BRAF linear signaling cascade mediates the motile and mitogenic phenotype of thyroid cancer cells. J Clin Invest. 2005;115(4):1068-1081.
9. Ciampi R, Nikiforov YE. RET/PTC rearrangements and BRAF mutations in thyroid tumorigenesis. Endocrinology. 2007;148(3):936-941.
10. Lemoine NR, Mayall ES, Wyllie FS, et al. High frequency of ras oncogene activation in all stages of human thyroid tumorigenesis. Oncogene. 1989;4(2):159-164.
11. Namba H, Rubin SA, Fagin JA. Point mutations of ras oncogenes are an early event in thyroid tumorigenesis. Mol Endocrinol. 1990;4(10):1474-1479.
12. Suarez HG, du Villard JA, Severino M, et al. Presence of mutations in all three ras genes in human thyroid tumors. Oncogene. 1990;5(4):565-570.
13. Karga H, Lee JK, Vickery AL Jr, Thor A, Gaz RD, Jameson JL. Ras oncogene mutations in benign and malignant thyroid neoplasms. J Clin Endocrinol Metab. 1991;73(4):832-836.
14. Terrier P, Sheng ZM, Schlumberger M, et al. Structure and expression of c-myc and c-fos proto-oncogenes in thyroid carcinomas. Br J Cancer. 1988;57(1):43-47.
15. Klein M, Vignaud JM, Hennequin V, et al. Increased expression of the vascular endothelial growth factor is a pejorative prognosis marker in papillary thyroid carcinoma. J Clin Endocrinol Metab. 2001;86(2):656-658.
16. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9(1):28-39.
17. Haugen BR, Sherman SI. Evolving approaches to patients with advanced differentiated thyroid cancer. Endocr Rev. 2013;34(3):439-455.
18. U.S. Food and Drug Administration. FDA approves Nexavar to treat type of thyroid cancer [press release]. Silver Spring, MD: U.S. Food and Drug Administration; November 22, 2013.
19. Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099-7109.
20. Brose MS, Nutting CM, Jarzab B, et al; DECISION investigators. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet. 2014;384(9940):319-328.
21. Dubauskas Z, Kunishige J, Prieto VG, Jonasch E, Hwu P, Tannir NM. Cutaneous squamous cell carcinoma and inflammation of actinic keratoses associated with sorafenib. Clin Genitourin Cancer. 2009;7(1):20-23.
22. U.S. Food and Drug Administration. FDA approves Lenvima for a type of thyroid cancer [press release]. Silver Spring, MD: U.S. Food and Drug Administration; February 13, 2015.
23. Matsui J, Yamamoto Y, Funahashi Y, et al. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int J Cancer. 2008;122(3):664-671.
24. Matsui J, Funahashi Y, Uenaka T, Watanabe T, Tsuruoka A, Asada M. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factorreceptor (VEGF-R) 2 and VEGF-R3 kinase. Clin Cancer Res. 2008;14(17):5459-5465.
25. Schlumberger M, Tahara M, Wirth LJ, et al. Lenvatinib versus placebo in radioiodine- refractory thyroid cancer. N Engl J Med. 2015;372(7):621-630.
26. Leboulleux S, Bastholt L, Krause T, et al. Vandetanib in locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 2 trial. Lancet Oncol. 2012;13(9):897-905.
27. A randomised, double-blind, placebo-controlled, multi-centre phase III study to assess the efficacy and safety of vandetanib (CAPRELSA) 300 mg in patients with differentiated thyroid cancer that is either locally advanced or metastatic who are refractory or unsuitable for radioiodine (RAI) therapy. Trial number NCT01876784. ClinicalTrials.gov Website. https://clinicaltrials.gov/show/NCT01876784. Updated June 26, 2015. Accessed July 22, 2015.
28. Bible KC, Suman VJ, Molina JR, et al; Endocrine Malignancies Disease Oriented Group; Mayo Clinic Cancer Center; Mayo Phase 2 Consortium. Efficacy of pazopanib in progressive, radioiodine-refractory, metastatic differentiated thyroid cancers: results of a phase 2 consortium study. Lancet Oncol. 2010;11(10):962-972.
29. Kim DW, Jo YS, Jung HS, et al. An orally administered multitarget tyrosine kinase inhibitor, SU11248, is a novel potent inhibitor of thyroid oncogenic RET/papillary thyroid cancer kinases. J Clin Endocrinol Metab. 2006;91(10):4070-4076.
30. Dawson SJ, Conus NM, Toner GC, et al. Sustained clinical responses to tyrosine kinase inhibitor sunitinib in thyroid carcinoma. Anticancer Drugs. 2008;19(5):547-552.
31. Carter SK, Blum RH. New chemotherapeutic agents—bleomycin and adriamycin. CA Cancer J Clin. 1974;24(6):322-331.
32. Hundahl SA, Fleming ID, Fremgen AM, Menck HR. A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985-1995 [see comments]. Cancer. 1998;83(12):2638-2648.
33. Lakhani VT, You YN, Wells SA. The multiple endocrine neoplasia syndromes. Annu Rev Med. 2007;58:253-265.
34. Martins RG, Rajendran JG, Capell P, Byrd DR, Mankoff DA. Medullary thyroid cancer: options for systemic therapy of metastatic disease? J Clin Oncol. 2006;24(11):1653-1655.
35. American Thyroid Association Guidelines Task Force; Kloos RT, Eng C, Evans DB, et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid. 2009;19(6):565-612.
36. Roman S, Lin R, Sosa JA. Prognosis of medullary thyroid carcinoma: demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer. 2006;107(9):2134-2142.
37. Modigliani E, Cohen R, Campos JM, et al. Prognostic factors for survival and for biochemical cure in medullary thyroid carcinoma: results in 899 patients. The GETC Study Group. Groupe d’étude des tumeurs à calcitonine. Clin Endocrinol (Oxf). 1998;48(3):265-273.
38. Donis-Keller H, Dou S, Chi D, et al. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet. 1993;2(7):851-856.
39. Mulligan LM, Kwok JB, Healey CS, et al. Germ-line mutations of the RET protooncogene in multiple endocrine neoplasia type 2A. Nature. 363(6428):458-460.
40. Carlson KM, Dou S, Chi D, et al. Single missense mutation in the tyrosine kinase catalytic domain of the RET protooncogene is associated with multiple endocrine neoplasia type 2B. Proc Natl Acad Sci USA. 1994;91(4):1579-1583.
41. Marsh DJ, Learoyd DL, Andrew SD, et al. Somatic mutations in the RET protooncogene in sporadic medullary thyroid carcinoma. Clin Endocrinol (Oxf). 1996;44(3):249-257.
42. Elisei R, Cosci B, Romei C, et al. Prognostic significance of somatic RET oncogene mutations in sporadic medullary thyroid cancer: a 10-year follow-up study. J Clin Endocrinol Metab. 2008;93(3):682-687.
43. Carlomagno F, Vitagliano D, Guida T, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002;62(24):7284-7290.
44. Carlomagno F, Anaganti S, Guida T, et al. BAY 43-9006 inhibition of oncogenic RET mutants. J Natl Cancer Inst. 2006;98(5):326-334.
45. Santoro M, Carlomagno F. Drug insight: small molecule inhibitors of protein kinases in the treatment of thyroid cancer. Nat Clin Pract Endocrinol Metab. 2006;2(1):42-52.
46. Rodríguez-Antona C, Pallares J, Montero-Conde C, et al. Overexpression and activation of EGFR and VEGFR2 in medullary thyroid carcinomas is related to metastasis. Endocr Relat Cancer. 2010;17(1):7-16.
47. U.S. Food and Drug Administration. FDA approves new treatment for rare form of thyroid cancer [press release]. Silver Spring, MD: U.S. Food and Drug Administration; April 6, 2011.
48. Wells SA Jr, Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30(2):134-141.
49. U.S. Food and Drug Administration. FDA approves Cometriq to treat rare type of thyroid cancer [press release]. Silver Spring, MD: U.S. Food and Drug Administration; November 29, 2012.
50. Cui JJ. Inhibitors targeting hepatocyte growth factor receptor and their potential therapeutic applications. Expert Opin Ther Pat. 2007;17(9):1035-1045.
51. Schoffski P, Elisei R, Müller S, et al. An international, double-blind, randomized, placebo-controlled phase III trial (EXAM) of cabozantinib (XL184) in medullary thyroid carcinoma (MTC) patients (pts) with documented RECIST progression at baseline. J Clin Oncol. 2012;30(suppl):5508.
52. Kober F, Hermann M, Handler A, Krotla G. Effect of sorafenib in symptomatic metastatic medullary thyroid cancer. J Clin Oncol. 2007;25(18S):14065.
53. Lam ET, Ringel MD, Kloos RT, et al. Phase II clinical trial of sorafenib in metastatic medullary thyroid cancer. J Clin Oncol. 2010;28(14):2323-2330.
54. Hong DS, Sebti SM, Newman RA, et al. Phase I trial of a combination of the multikinase inhibitor sorafenib and the farnesyltransferase inhibitor tipifarnib in advanced malignancies. Clin Cancer Res. 2009;15(22):7061-7068.
55. Kelleher FC, McDermott R. Response to sunitinib in medullary thyroid cancer. Ann Intern Med. 2008;148(7):567.
56. Carr LL, Mankoff DA, Goulart BH, et al. Phase II study of daily sunitinib in FDGPET- positive, iodine-refractory differentiated thyroid cancer and metastatic medullary carcinoma of the thyroid with functional imaging correlation. Clin Cancer Res. 2010;16(21):5260-5268.
57. Bible KC, Suman VJ, Molina JR, et al; Endocrine Malignancies Disease Oriented Group; Mayo Clinic Cancer Center; Mayo Phase 2 Consortium. A multicenter phase 2 trial of pazopanib in metastatic and progressive medullary thyroid carcinoma: MC057H. J Clin Endocrinol Metab. 2014;99(5):1687-1693.
58. Ball DW. Medullary thyroid cancer: monitoring and therapy. Endocrinol Metab Clin North Am. 2007;36(3):823-837, viii.
59. Nocera M, Baudin E, Pellegriti G, Cailleux AF, Mechelany-Corone C, Schlumberger M. Treatment of advanced medullary thyroid cancer with an alternating combination of doxorubicin-streptozocin and 5 FU-dacarbazine. Groupe d’Etude des Tumeurs à Calcitonine (GETC). Br J Cancer. 2000;83(6):715-718.
60. Shimaoka K, Schoenfeld DA, DeWys WD, Creech RH, DeConti R. A randomized trial of doxorubicin versus doxorubicin plus cisplatin in patients with advanced thyroid carcinoma. Cancer. 1985;56(9):2155-2160.