User login
A 61-year-old man with fluctuating hypertension
A 61-year-old man with type 2 diabetes mellitus on glimepiride therapy presented with somnolence and slurred speech. His capillary glucose level was 17 mg/dL and his serum glucose level was 28 mg/dL. He was treated with intravenous dextrose, and his glucose level promptly returned to normal.
He had been adherent to his medication regimen and denied overmedicating or accidental overdosing. Over the past 7 months, he had noted redness on his palms, a rash on his legs, intermittent moderate to severe headaches, weight loss, and decreased appetite. In addition, his blood pressure had been labile, which his physicians had attributed to autonomic instability. He had continued on the same dose of glimepiride despite losing weight.
His history included multivessel coronary artery disease treated with angioplasty and placement of multiple coronary stents; ischemic cardiomyopathy with a left ventricular ejection fraction of 28%; implantation of a cardioverter-defibrillator for secondary prevention of ventricular arrhythmia; an ischemic stroke; and multiple sclerosis complicated by bilateral blindness, with optic nerve involvement and autonomic instability, present for over a year and manifested by labile blood pressure. He was a long-time tobacco user. His daily medications included ticagrelor 90 mg, aspirin 81 mg, metoprolol 50 mg, ramipril 10 mg, simvastatin 20 mg, glimepiride 2 mg, and esomeprazole 40 mg. He needed help taking his medications.
At the time of hospital admission, his heart rate was 69 beats per minute with a regular rhythm, blood pressure 115/73 mm Hg, respiratory rate 11 breaths per minute with an oxygen saturation of 99% on room air, and oral temperature 34.7°C (94.5°F). He appeared to be in no distress.
Cardiovascular examination revealed no murmurs or gallops; there was mild nonpitting edema of the lower extremities. Pulmonary, abdominal, and neurologic examinations were unrevealing except for bilateral blindness. Vascular examination revealed no bruits. Results of a complete blood cell count and metabolic panel were normal except for a hemoglobin level of 9.9 g/dL (reference range 13.5–17.5) and a platelet count of 477 × 109/L (150–450).
Although he continued to receive the same medications he had been taking at home, his blood pressure fluctuated. On the second hospital day, it reached 186/135 mm Hg, at which time he also had palpitations, dyspnea, and crackles in the lower lobes of both lungs. Volume resuscitation on admission was suspected to have played a role, and he received furosemide, which improved his symptoms. But several hours later, his blood pressure rose again, and he became diaphoretic. Despite aggressive treatment with different antihypertensive agents, his blood pressure remained high and his symptoms persisted. Chest radiography showed no evidence of pulmonary edema. Because of his progressive dyspnea, the diagnosis of pulmonary embolism was entertained.
CAUSES OF RESISTANT HYPERTENSION
1. What could explain this patient’s high blood pressure?
- A drug effect
- Renovascular disease
- Excess circulating catecholamines
- Obstructive sleep apnea
- Primary aldosteronism
Sympathomimetic drugs such as epinephrine, norepinephrine, dopamine, and vasopressin, which are used when hemodynamic support is required, can raise both systolic and diastolic blood pressure. Nonsteroidal anti-inflammatory drugs and nasal decongestants are common culprits in the community. However, our patient was using none of these drugs.
Renovascular disease is one of many causes of resistant hypertension, accounting for 8% of all cases.1,2 Despite fluctuations, the blood pressure often remains chronically elevated, its changes are less paroxysmal than in our patient, and a precipitating factor such as a dietary indiscretion is sometimes identified.1
Excess circulating catecholamines can be a result of stress, exogenous administration, or endogenous oversecretion. Our patient’s clinical presentation is highly suspicious for a high-catecholamine state, and this should be further evaluated.
Obstructive sleep apnea is common in patients with resistant hypertension, with an estimated prevalence as high as 60% in this group.3,4
Primary aldosteronism has an estimated prevalence of about 20% in patients evaluated for resistant hypertension.5
AN ADRENAL MASS IS INCIDENTALLY DISCOVERED
Computed tomographic angiography of the chest revealed no evidence of pulmonary emboli. There was mild dilation of the central pulmonary arteries and an incidental, incompletely imaged 4.7-by-3.4-cm mass of mixed attenuation in the right adrenal gland, with macroscopic fat within the lesion.
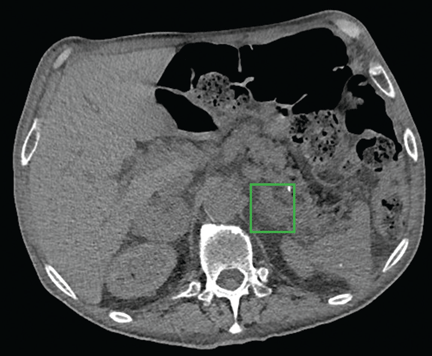
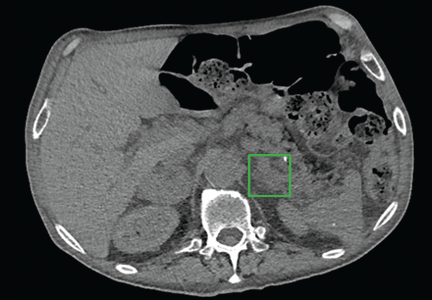
Computed tomography (CT) of the abdomen with dedicated cuts through the adrenal glands revealed a 4.7-cm heterogeneous right adrenal mass with a density of 34 Hounsfield units (HU). The left adrenal gland appeared diffusely enlarged without a discretely seen mass, consistent with hyperplasticity (Figure 1).
2. Based on the patient’s clinical presentation and findings on CT, what would be the most likely diagnosis for this incidentally found adrenal mass?
- Adrenocortical adenoma
- Adrenocortical carcinoma
- Metastatic mass
- Pheochromocytoma
Adrenocortical adenoma can present as a small homogeneous mass of variable size, with smooth margins, and rarely containing hemorrhagic tissue or calcifications. The typical density on nonenhanced CT is less than 10 HU. On enhanced CT, it is nonvascular. T2-weighted magnetic resonance imaging (MRI) shows a lesion of the same intensity as liver tissue.6
Adrenocortical adenoma is not classically associated with autologous activity and thus is less likely to explain our patient’s symptoms.
Adrenocortical carcinoma can present as a large heterogeneous mass, usually greater than 4 cm in diameter, with irregular margins and areas of necrosis, hemorrhage, or calcification. The typical density on nonenhanced CT is greater than 10 HU. On enhanced CT, the mass is usually vascular, and T2-weighted MRI will show a lesion more intense than liver tissue.6
Adrenocortical carcinoma is also not classically associated with autologous activity, and so is not likely to explain our patient’s symptoms.6
Metastatic disease can present with masses of variable size, often bilaterally, and occasionally with cysts or areas of hemorrhage. The typical density of metastatic lesions on nonenhanced CT is greater than 10 HU. On enhanced CT, they are usually vascular, and on T2-weighted MRI they are hyperintense.6 The characteristics of the mass and the absence of a primary malignancy on CT of the chest and abdomen do not support the diagnosis of metastatic disease.
Pheochromocytoma is a neuroendocrine tumor of the adrenal medulla that can present as a large heterogeneous mass, greater than 3 cm in diameter, with clear margins and cysts or areas of hemorrhage. Extra-adrenal neuroendocrine tumors are typically called paragangliomas and have features similar to those of pheochromocytoma. The typical density of pheochromocytoma on nonenhanced CT is greater than 10 HU. On enhanced CT, it is usually vascular, and T2-weighted MRI shows a hyperintense lesion. Pheochromocytoma can be biochemically active and thus can cause signs and symptoms that will lead to the diagnosis.6
Other imaging tests may play a role in the evaluation of adrenal masses but are not required for the diagnosis of pheochromocytoma. Functional positron emission tomography using metaiodobenzylguanidine labeled with iodine 123 or-iodine 131 or using the glucose analogue F-18 fluorodeoxyglucose has been used in the initial assessment of pheochromocytoma, with good sensitivity and specificity.7,8
Our patient’s pacemaker-defibrillator precluded him from undergoing MRI.
DIAGNOSIS: PHEOCHROMOCYTOMA
Pheochromocytoma was highly suspected on the basis of the patient’s clinical presentation, and metoprolol was immediately discontinued. He was started on the calcium channel blocker verapamil and the alpha-blocker phenoxybenzamine.
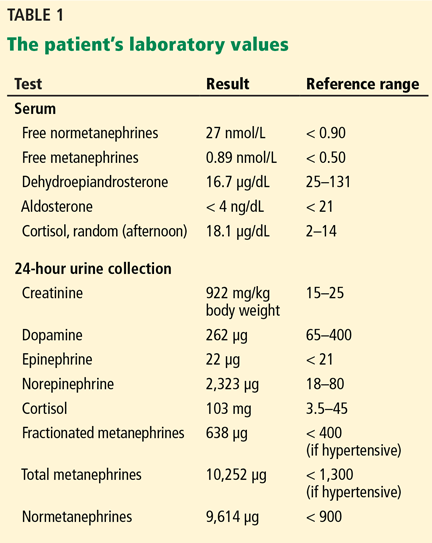
Serum samples were obtained to measure metanephrines, dehydroepiandrosterone, aldosterone, and cortisol, and a 24-hour urine collection was obtained to measure creatinine, dopamine, epinephrine, norepinephrine, cortisol, and metanephrines. Based on the results (Table 1) and on the findings on imaging, the patient was diagnosed with pheochromocytoma. A surgical consultation was obtained, and surgery was recommended.
WHEN DOES PHEOCHROMOCYTOMA CALL FOR SURGERY?
3. Which criterion is most important when determining the need for surgery for pheochromocytoma?
- Findings on fine-needle aspiration biopsy
- Biochemical activity
- Size of the mass
- Bilateral masses
Fine-needle aspiration biopsy can be done when a mass is found incidentally and no evidence of biochemical activity is detected, although it is not an essential part of the diagnostic workup.9 In most cases, the sampling from fine-needle aspiration is not sufficient to achieve a diagnosis.
Biochemical activity is the most important factor when determining the need for prompt surgical intervention. The excess circulating catecholamines have been associated with increased risk of cardiovascular morbidity and death independent of the morbidity associated with hypertension alone.10 Biochemical activity can be independent of the size of the mass, but larger masses typically present with symptoms.
Bilateral masses have been associated with metastatic disease.11 In retrospect, the patient’s history of hypertension and cerebrovascular accident could be associated with the development of a catecholamine-releasing tumor.
A GOOD OUTCOME FROM SURGERY
The patient was continued on phenoxybenzamine for 7 days and responded well to this therapy.
After this preoperative preparation, he underwent laparoscopic right adrenalectomy with excision of a retroperitoneal adrenal mass. His postoperative course was complicated by transient hypotension requiring low-dose vasopressin support for less than 24 hours. He was then restarted on his previous dosage of metoprolol and was discharged home on postoperative day 5 with stable blood pressure. Follow-up 24-hour urine collection 1 month after he was discharged showed normalization of metanephrine, normetanephrine, epinephrine, and norepinephrine levels.
Despite low suspicion for an underlying genetic syndrome, he was referred for genetic testing and was scheduled to have a repeat 24-hour urine collection and imaging in 6 months to follow his enlarged left adrenal gland, which did not appear to be metabolically hyperactive.
4. What is the most common perioperative complication of pheochromocytoma excision?
- Hypoglycemia
- Hypotension
- Hypocortisolism
- Hypertension
- Tachycardia
Hypoglycemia has been observed after removal of pheochromocytoma, as levels of catecholamines (which normally inhibit pancreatic beta cells) decrease and insulin secretion consequently increases.12 Our patient developed hypoglycemia before surgery, not after, and it was likely due to the combination of his antidiabetic therapy, weight loss, and decreased oral intake.
Hypotension is the most common complication in the perioperative period. It is associated with excessive loss of catecholamine secretion. It is usually short-lived but may require aggressive administration of intravenous fluids and use of sympathomimetic agents.
Hypocortisolism is unlikely in patients with pheochromocytoma, but it is likely after removal of adrenocortical adenoma.
Hypertension and tachycardia affect up to 40% of pheochromocytoma patients in some case series.12
PHEOCHROMOCYTOMA: A CATECHOLAMINE-SECRETING TUMOR
The pathophysiology of pheochromocytoma is complex. It is characterized by accelerated growth of cells producing catecholamines, which may produce symptoms when secreted into the bloodstream. The classic triad of symptoms is headache, hypertension, and hyperglycemia, although our patient had very low blood sugar levels. Other common symptoms are nausea, orthostasis, and tremor, although not all symptoms are invariably seen.
Genetic testing recommended
Genetic associations have been described and are thought to be responsible for 20% to 30% of cases of pheochromocytoma. All associated germline mutations are autosomal dominant, some with variable penetrance. These include:
- Succinate dehydrogenase subunit B, C, and D mutations
- von Hippel-Lindau syndrome
- Multiple endocrine neoplasia type 1 and type 2 syndromes
- Neurofibromatosis type 1.13,14
The succinate dehydrogenase subunit mutations have been associated with, but not limited to, extra-adrenal adenomas (paragangliomas) and carry a worse prognosis.
Some experts recommend genetic testing in all cases of pheochromocytoma, sporadic or familial, and this testing should be followed by counseling if a mutation is found.15 Others recommend genetic testing based on the patient’s age (under age 50), history, imaging, and biochemical features of the tumor (metanephrines predominate in multiple endocrine neoplasia syndromes, and normetanephrines in von Hippel-Lindau syndrome).13
Serious consequences
A thorough evaluation is recommended, since pheochromocytoma has been associated with increased cardiovascular morbidity. In a retrospective series, Stolk et al10 reported that patients with pheochromocytoma had a higher incidence of myocardial infarction, angina, and stroke in the years preceding the diagnosis than did patients with essential hypertension (13.8% vs 1.1%, P < .001).10
Catecholamine cardiomyopathy has been described and shares clinical features with Takotsubo or stress cardiomyopathy, with global left ventricular systolic and diastolic dysfunction that improve or resolve after the adrenergic insult is removed.16
Conditions that warrant further evaluation or that may suggest pheochromocytoma are malignant hypertension, hypertensive encephalopathy, ischemic stroke, subarachnoid hemorrhage, acute pulmonary edema, angina pectoris, myocardial infarction, aortic dissection, and kidney injury.
When to suspect pheochromocytoma
Pheochromocytoma should be suspected in a patient with resistant hypertension, family history, or imaging findings that suggest an adrenal mass with a heterogeneous appearance. The diagnostic algorithm follows the same pathway as for the evaluation of an incidentally found adrenal mass, with determination of its dimension and characteristics by CT or MRI, and with biochemical testing of urine catecholamines, plasma free metanephrines, renin, aldosterone, and cortisol.
The diagnosis of pheochromocytoma is established by obtaining fractionated metanephrines and catecholamines in a 24-hour urine collection (sensitivity 90%, specificity 98%). Analysis of plasma metanephrines has a higher sensitivity (97%) but lower specificity (85%).17 The combination of typical signs, symptoms, and laboratory findings makes the diagnosis likely, especially in combination with a unilateral adrenal mass.
Laparoscopic surgery after medical preparation for active tumors
If the mass appears benign and not biochemically hyperactive, then follow-up at 1 year is recommended, with repeat testing. Surgical evaluation and intervention is recommended for lesions that appear malignant or that are biochemically active and clinically symptomatic.9
Preoperative hemodynamic control is essential in the management of pheochromocytoma to prevent or minimize hemodynamic changes that can be driven by increased catecholamines. Control is typically achieved with initial alpha-blockade and then beta-blockade to avoid worsening hypertension and to prevent an acute hypertensive crisis during surgical intervention. Phenoxybenzamine, the mainstay of therapy, is a nonselective alpha-blocker with a long duration of action that requires titration over several days up to 3 weeks.
A selective alpha-1-blocker such as doxazosin can be used to control postoperative hypotension, as it has a shorter half-life than phenoxybenzamine. Alternative strategies include calcium channel blockers, centrally acting sympathetic blockers, and magnesium.18
Laparoscopic adrenalectomy by an experienced surgeon after excellent medical preparation is often considered the treatment of choice, but for larger or malignant masses, an open procedure is recommended. The risk of perioperative morbidity and death can be reduced by adequate medical management. With successful surgical resection, the long-term prognosis is favorable.
- Calhoun DA, Jones D, Textor S, et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension 2008; 51:1403–1419.
- Kumar N, Calhoun DA, Dudenbostel T. Management of patients with resistant hypertension: current treatment options. Integr Blood Press Control 2013; 6:139–151.
- Pedrosa RP, Drager LF, Gonzaga CC, et al. Obstructive sleep apnea: the most common secondary cause of hypertension associated with resistant hypertension. Hypertension 2011; 58:811–817.
- Marcus JA, Pothineni A, Marcus CZ, Bisognano JD. The role of obesity and obstructive sleep apnea in the pathogenesis and treatment of resistant hypertension. Curr Hypertens Rep 2014; 16:411.
- Calhoun DA, Nishizaka MK, Zaman MA, Thakkar RB, Weissmann P. Hyperaldosteronism among black and white subjects with resistant hypertension. Hypertension 2002; 40:892–896.
- Young WF Clinical practice. The incidentally discovered adrenal mass. N Engl J Med 2007; 356:601–610.
- Lin M, Wong V, Yap J, Jin R, Leong P, Campbell P. FDG PET in the evaluation of phaeochromocytoma: a correlative study with MIBG scintigraphy and Ki-67 proliferative index. Clin Imaging 2013; 37:1084–1088.
- Raja A, Leung K, Stamm M, Girgis S, Low G. Multimodality imaging findings of pheochromocytoma with associated clinical and biochemical features in 53 patients with histologically confirmed tumors. AJR Am J Roentgenol 2013; 201:825–833.
- Nieman LK. Approach to the patient with an adrenal incidentaloma. J Clin Endocrinol Metab 2010; 95:4106–4113.
- Stolk RF, Bakx C, Mulder J, Timmers HJ, Lenders JW. Is the excess cardiovascular morbidity in pheochromocytoma related to blood pressure or to catecholamines? J Clin Endocrinol Metab 2013; 98:1100–1106.
- Grumbach MM, Biller BM, Braunstein GD, et al. Management of the clinically inapparent adrenal mass (‘incidentaloma’). Ann Intern Med 2003; 138:424–429.
- Lentschener C, Gaujoux S, Tesniere A, Dousset B. Point of controversy: perioperative care of patients undergoing pheochromocytoma removal—time for a reappraisal? Eur J Endocrinol 2011; 165:365–373.
- Bryant J, Farmer J, Kessler LJ, Townsend RR, Nathanson KL. Pheochromocytoma: the expanding genetic differential diagnosis. J Natl Cancer Inst 2003; 95:1196–1204.
- Lee P, Leonard J. Textbook on endocrinology. BMJ 1994; 308:1512.
- Fishbein L, Merrill S, Fraker DL, Cohen DL, Nathanson KL. Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol 2013; 20:1444–1450.
- Agarwal G, Sadacharan D, Kapoor A, et al. Cardiovascular dysfunction and catecholamine cardiomyopathy in pheochromocytoma patients and their reversal following surgical cure: results of a prospective case-control study. Surgery 2011; 150:1202–1211.
- Sawka AM, Jaeschke R, Singh RJ, Young WF A comparison of biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab 2003; 88:553–558.
- Domi R, Laho H. Management of pheochromocytoma: old ideas and new drugs. Niger J Clin Pract 2012; 15:253–257.
A 61-year-old man with type 2 diabetes mellitus on glimepiride therapy presented with somnolence and slurred speech. His capillary glucose level was 17 mg/dL and his serum glucose level was 28 mg/dL. He was treated with intravenous dextrose, and his glucose level promptly returned to normal.
He had been adherent to his medication regimen and denied overmedicating or accidental overdosing. Over the past 7 months, he had noted redness on his palms, a rash on his legs, intermittent moderate to severe headaches, weight loss, and decreased appetite. In addition, his blood pressure had been labile, which his physicians had attributed to autonomic instability. He had continued on the same dose of glimepiride despite losing weight.
His history included multivessel coronary artery disease treated with angioplasty and placement of multiple coronary stents; ischemic cardiomyopathy with a left ventricular ejection fraction of 28%; implantation of a cardioverter-defibrillator for secondary prevention of ventricular arrhythmia; an ischemic stroke; and multiple sclerosis complicated by bilateral blindness, with optic nerve involvement and autonomic instability, present for over a year and manifested by labile blood pressure. He was a long-time tobacco user. His daily medications included ticagrelor 90 mg, aspirin 81 mg, metoprolol 50 mg, ramipril 10 mg, simvastatin 20 mg, glimepiride 2 mg, and esomeprazole 40 mg. He needed help taking his medications.
At the time of hospital admission, his heart rate was 69 beats per minute with a regular rhythm, blood pressure 115/73 mm Hg, respiratory rate 11 breaths per minute with an oxygen saturation of 99% on room air, and oral temperature 34.7°C (94.5°F). He appeared to be in no distress.
Cardiovascular examination revealed no murmurs or gallops; there was mild nonpitting edema of the lower extremities. Pulmonary, abdominal, and neurologic examinations were unrevealing except for bilateral blindness. Vascular examination revealed no bruits. Results of a complete blood cell count and metabolic panel were normal except for a hemoglobin level of 9.9 g/dL (reference range 13.5–17.5) and a platelet count of 477 × 109/L (150–450).
Although he continued to receive the same medications he had been taking at home, his blood pressure fluctuated. On the second hospital day, it reached 186/135 mm Hg, at which time he also had palpitations, dyspnea, and crackles in the lower lobes of both lungs. Volume resuscitation on admission was suspected to have played a role, and he received furosemide, which improved his symptoms. But several hours later, his blood pressure rose again, and he became diaphoretic. Despite aggressive treatment with different antihypertensive agents, his blood pressure remained high and his symptoms persisted. Chest radiography showed no evidence of pulmonary edema. Because of his progressive dyspnea, the diagnosis of pulmonary embolism was entertained.
CAUSES OF RESISTANT HYPERTENSION
1. What could explain this patient’s high blood pressure?
- A drug effect
- Renovascular disease
- Excess circulating catecholamines
- Obstructive sleep apnea
- Primary aldosteronism
Sympathomimetic drugs such as epinephrine, norepinephrine, dopamine, and vasopressin, which are used when hemodynamic support is required, can raise both systolic and diastolic blood pressure. Nonsteroidal anti-inflammatory drugs and nasal decongestants are common culprits in the community. However, our patient was using none of these drugs.
Renovascular disease is one of many causes of resistant hypertension, accounting for 8% of all cases.1,2 Despite fluctuations, the blood pressure often remains chronically elevated, its changes are less paroxysmal than in our patient, and a precipitating factor such as a dietary indiscretion is sometimes identified.1
Excess circulating catecholamines can be a result of stress, exogenous administration, or endogenous oversecretion. Our patient’s clinical presentation is highly suspicious for a high-catecholamine state, and this should be further evaluated.
Obstructive sleep apnea is common in patients with resistant hypertension, with an estimated prevalence as high as 60% in this group.3,4
Primary aldosteronism has an estimated prevalence of about 20% in patients evaluated for resistant hypertension.5
AN ADRENAL MASS IS INCIDENTALLY DISCOVERED
Computed tomographic angiography of the chest revealed no evidence of pulmonary emboli. There was mild dilation of the central pulmonary arteries and an incidental, incompletely imaged 4.7-by-3.4-cm mass of mixed attenuation in the right adrenal gland, with macroscopic fat within the lesion.
Computed tomography (CT) of the abdomen with dedicated cuts through the adrenal glands revealed a 4.7-cm heterogeneous right adrenal mass with a density of 34 Hounsfield units (HU). The left adrenal gland appeared diffusely enlarged without a discretely seen mass, consistent with hyperplasticity (Figure 1).
2. Based on the patient’s clinical presentation and findings on CT, what would be the most likely diagnosis for this incidentally found adrenal mass?
- Adrenocortical adenoma
- Adrenocortical carcinoma
- Metastatic mass
- Pheochromocytoma
Adrenocortical adenoma can present as a small homogeneous mass of variable size, with smooth margins, and rarely containing hemorrhagic tissue or calcifications. The typical density on nonenhanced CT is less than 10 HU. On enhanced CT, it is nonvascular. T2-weighted magnetic resonance imaging (MRI) shows a lesion of the same intensity as liver tissue.6
Adrenocortical adenoma is not classically associated with autologous activity and thus is less likely to explain our patient’s symptoms.
Adrenocortical carcinoma can present as a large heterogeneous mass, usually greater than 4 cm in diameter, with irregular margins and areas of necrosis, hemorrhage, or calcification. The typical density on nonenhanced CT is greater than 10 HU. On enhanced CT, the mass is usually vascular, and T2-weighted MRI will show a lesion more intense than liver tissue.6
Adrenocortical carcinoma is also not classically associated with autologous activity, and so is not likely to explain our patient’s symptoms.6
Metastatic disease can present with masses of variable size, often bilaterally, and occasionally with cysts or areas of hemorrhage. The typical density of metastatic lesions on nonenhanced CT is greater than 10 HU. On enhanced CT, they are usually vascular, and on T2-weighted MRI they are hyperintense.6 The characteristics of the mass and the absence of a primary malignancy on CT of the chest and abdomen do not support the diagnosis of metastatic disease.
Pheochromocytoma is a neuroendocrine tumor of the adrenal medulla that can present as a large heterogeneous mass, greater than 3 cm in diameter, with clear margins and cysts or areas of hemorrhage. Extra-adrenal neuroendocrine tumors are typically called paragangliomas and have features similar to those of pheochromocytoma. The typical density of pheochromocytoma on nonenhanced CT is greater than 10 HU. On enhanced CT, it is usually vascular, and T2-weighted MRI shows a hyperintense lesion. Pheochromocytoma can be biochemically active and thus can cause signs and symptoms that will lead to the diagnosis.6
Other imaging tests may play a role in the evaluation of adrenal masses but are not required for the diagnosis of pheochromocytoma. Functional positron emission tomography using metaiodobenzylguanidine labeled with iodine 123 or-iodine 131 or using the glucose analogue F-18 fluorodeoxyglucose has been used in the initial assessment of pheochromocytoma, with good sensitivity and specificity.7,8
Our patient’s pacemaker-defibrillator precluded him from undergoing MRI.
DIAGNOSIS: PHEOCHROMOCYTOMA
Pheochromocytoma was highly suspected on the basis of the patient’s clinical presentation, and metoprolol was immediately discontinued. He was started on the calcium channel blocker verapamil and the alpha-blocker phenoxybenzamine.
Serum samples were obtained to measure metanephrines, dehydroepiandrosterone, aldosterone, and cortisol, and a 24-hour urine collection was obtained to measure creatinine, dopamine, epinephrine, norepinephrine, cortisol, and metanephrines. Based on the results (Table 1) and on the findings on imaging, the patient was diagnosed with pheochromocytoma. A surgical consultation was obtained, and surgery was recommended.
WHEN DOES PHEOCHROMOCYTOMA CALL FOR SURGERY?
3. Which criterion is most important when determining the need for surgery for pheochromocytoma?
- Findings on fine-needle aspiration biopsy
- Biochemical activity
- Size of the mass
- Bilateral masses
Fine-needle aspiration biopsy can be done when a mass is found incidentally and no evidence of biochemical activity is detected, although it is not an essential part of the diagnostic workup.9 In most cases, the sampling from fine-needle aspiration is not sufficient to achieve a diagnosis.
Biochemical activity is the most important factor when determining the need for prompt surgical intervention. The excess circulating catecholamines have been associated with increased risk of cardiovascular morbidity and death independent of the morbidity associated with hypertension alone.10 Biochemical activity can be independent of the size of the mass, but larger masses typically present with symptoms.
Bilateral masses have been associated with metastatic disease.11 In retrospect, the patient’s history of hypertension and cerebrovascular accident could be associated with the development of a catecholamine-releasing tumor.
A GOOD OUTCOME FROM SURGERY
The patient was continued on phenoxybenzamine for 7 days and responded well to this therapy.
After this preoperative preparation, he underwent laparoscopic right adrenalectomy with excision of a retroperitoneal adrenal mass. His postoperative course was complicated by transient hypotension requiring low-dose vasopressin support for less than 24 hours. He was then restarted on his previous dosage of metoprolol and was discharged home on postoperative day 5 with stable blood pressure. Follow-up 24-hour urine collection 1 month after he was discharged showed normalization of metanephrine, normetanephrine, epinephrine, and norepinephrine levels.
Despite low suspicion for an underlying genetic syndrome, he was referred for genetic testing and was scheduled to have a repeat 24-hour urine collection and imaging in 6 months to follow his enlarged left adrenal gland, which did not appear to be metabolically hyperactive.
4. What is the most common perioperative complication of pheochromocytoma excision?
- Hypoglycemia
- Hypotension
- Hypocortisolism
- Hypertension
- Tachycardia
Hypoglycemia has been observed after removal of pheochromocytoma, as levels of catecholamines (which normally inhibit pancreatic beta cells) decrease and insulin secretion consequently increases.12 Our patient developed hypoglycemia before surgery, not after, and it was likely due to the combination of his antidiabetic therapy, weight loss, and decreased oral intake.
Hypotension is the most common complication in the perioperative period. It is associated with excessive loss of catecholamine secretion. It is usually short-lived but may require aggressive administration of intravenous fluids and use of sympathomimetic agents.
Hypocortisolism is unlikely in patients with pheochromocytoma, but it is likely after removal of adrenocortical adenoma.
Hypertension and tachycardia affect up to 40% of pheochromocytoma patients in some case series.12
PHEOCHROMOCYTOMA: A CATECHOLAMINE-SECRETING TUMOR
The pathophysiology of pheochromocytoma is complex. It is characterized by accelerated growth of cells producing catecholamines, which may produce symptoms when secreted into the bloodstream. The classic triad of symptoms is headache, hypertension, and hyperglycemia, although our patient had very low blood sugar levels. Other common symptoms are nausea, orthostasis, and tremor, although not all symptoms are invariably seen.
Genetic testing recommended
Genetic associations have been described and are thought to be responsible for 20% to 30% of cases of pheochromocytoma. All associated germline mutations are autosomal dominant, some with variable penetrance. These include:
- Succinate dehydrogenase subunit B, C, and D mutations
- von Hippel-Lindau syndrome
- Multiple endocrine neoplasia type 1 and type 2 syndromes
- Neurofibromatosis type 1.13,14
The succinate dehydrogenase subunit mutations have been associated with, but not limited to, extra-adrenal adenomas (paragangliomas) and carry a worse prognosis.
Some experts recommend genetic testing in all cases of pheochromocytoma, sporadic or familial, and this testing should be followed by counseling if a mutation is found.15 Others recommend genetic testing based on the patient’s age (under age 50), history, imaging, and biochemical features of the tumor (metanephrines predominate in multiple endocrine neoplasia syndromes, and normetanephrines in von Hippel-Lindau syndrome).13
Serious consequences
A thorough evaluation is recommended, since pheochromocytoma has been associated with increased cardiovascular morbidity. In a retrospective series, Stolk et al10 reported that patients with pheochromocytoma had a higher incidence of myocardial infarction, angina, and stroke in the years preceding the diagnosis than did patients with essential hypertension (13.8% vs 1.1%, P < .001).10
Catecholamine cardiomyopathy has been described and shares clinical features with Takotsubo or stress cardiomyopathy, with global left ventricular systolic and diastolic dysfunction that improve or resolve after the adrenergic insult is removed.16
Conditions that warrant further evaluation or that may suggest pheochromocytoma are malignant hypertension, hypertensive encephalopathy, ischemic stroke, subarachnoid hemorrhage, acute pulmonary edema, angina pectoris, myocardial infarction, aortic dissection, and kidney injury.
When to suspect pheochromocytoma
Pheochromocytoma should be suspected in a patient with resistant hypertension, family history, or imaging findings that suggest an adrenal mass with a heterogeneous appearance. The diagnostic algorithm follows the same pathway as for the evaluation of an incidentally found adrenal mass, with determination of its dimension and characteristics by CT or MRI, and with biochemical testing of urine catecholamines, plasma free metanephrines, renin, aldosterone, and cortisol.
The diagnosis of pheochromocytoma is established by obtaining fractionated metanephrines and catecholamines in a 24-hour urine collection (sensitivity 90%, specificity 98%). Analysis of plasma metanephrines has a higher sensitivity (97%) but lower specificity (85%).17 The combination of typical signs, symptoms, and laboratory findings makes the diagnosis likely, especially in combination with a unilateral adrenal mass.
Laparoscopic surgery after medical preparation for active tumors
If the mass appears benign and not biochemically hyperactive, then follow-up at 1 year is recommended, with repeat testing. Surgical evaluation and intervention is recommended for lesions that appear malignant or that are biochemically active and clinically symptomatic.9
Preoperative hemodynamic control is essential in the management of pheochromocytoma to prevent or minimize hemodynamic changes that can be driven by increased catecholamines. Control is typically achieved with initial alpha-blockade and then beta-blockade to avoid worsening hypertension and to prevent an acute hypertensive crisis during surgical intervention. Phenoxybenzamine, the mainstay of therapy, is a nonselective alpha-blocker with a long duration of action that requires titration over several days up to 3 weeks.
A selective alpha-1-blocker such as doxazosin can be used to control postoperative hypotension, as it has a shorter half-life than phenoxybenzamine. Alternative strategies include calcium channel blockers, centrally acting sympathetic blockers, and magnesium.18
Laparoscopic adrenalectomy by an experienced surgeon after excellent medical preparation is often considered the treatment of choice, but for larger or malignant masses, an open procedure is recommended. The risk of perioperative morbidity and death can be reduced by adequate medical management. With successful surgical resection, the long-term prognosis is favorable.
A 61-year-old man with type 2 diabetes mellitus on glimepiride therapy presented with somnolence and slurred speech. His capillary glucose level was 17 mg/dL and his serum glucose level was 28 mg/dL. He was treated with intravenous dextrose, and his glucose level promptly returned to normal.
He had been adherent to his medication regimen and denied overmedicating or accidental overdosing. Over the past 7 months, he had noted redness on his palms, a rash on his legs, intermittent moderate to severe headaches, weight loss, and decreased appetite. In addition, his blood pressure had been labile, which his physicians had attributed to autonomic instability. He had continued on the same dose of glimepiride despite losing weight.
His history included multivessel coronary artery disease treated with angioplasty and placement of multiple coronary stents; ischemic cardiomyopathy with a left ventricular ejection fraction of 28%; implantation of a cardioverter-defibrillator for secondary prevention of ventricular arrhythmia; an ischemic stroke; and multiple sclerosis complicated by bilateral blindness, with optic nerve involvement and autonomic instability, present for over a year and manifested by labile blood pressure. He was a long-time tobacco user. His daily medications included ticagrelor 90 mg, aspirin 81 mg, metoprolol 50 mg, ramipril 10 mg, simvastatin 20 mg, glimepiride 2 mg, and esomeprazole 40 mg. He needed help taking his medications.
At the time of hospital admission, his heart rate was 69 beats per minute with a regular rhythm, blood pressure 115/73 mm Hg, respiratory rate 11 breaths per minute with an oxygen saturation of 99% on room air, and oral temperature 34.7°C (94.5°F). He appeared to be in no distress.
Cardiovascular examination revealed no murmurs or gallops; there was mild nonpitting edema of the lower extremities. Pulmonary, abdominal, and neurologic examinations were unrevealing except for bilateral blindness. Vascular examination revealed no bruits. Results of a complete blood cell count and metabolic panel were normal except for a hemoglobin level of 9.9 g/dL (reference range 13.5–17.5) and a platelet count of 477 × 109/L (150–450).
Although he continued to receive the same medications he had been taking at home, his blood pressure fluctuated. On the second hospital day, it reached 186/135 mm Hg, at which time he also had palpitations, dyspnea, and crackles in the lower lobes of both lungs. Volume resuscitation on admission was suspected to have played a role, and he received furosemide, which improved his symptoms. But several hours later, his blood pressure rose again, and he became diaphoretic. Despite aggressive treatment with different antihypertensive agents, his blood pressure remained high and his symptoms persisted. Chest radiography showed no evidence of pulmonary edema. Because of his progressive dyspnea, the diagnosis of pulmonary embolism was entertained.
CAUSES OF RESISTANT HYPERTENSION
1. What could explain this patient’s high blood pressure?
- A drug effect
- Renovascular disease
- Excess circulating catecholamines
- Obstructive sleep apnea
- Primary aldosteronism
Sympathomimetic drugs such as epinephrine, norepinephrine, dopamine, and vasopressin, which are used when hemodynamic support is required, can raise both systolic and diastolic blood pressure. Nonsteroidal anti-inflammatory drugs and nasal decongestants are common culprits in the community. However, our patient was using none of these drugs.
Renovascular disease is one of many causes of resistant hypertension, accounting for 8% of all cases.1,2 Despite fluctuations, the blood pressure often remains chronically elevated, its changes are less paroxysmal than in our patient, and a precipitating factor such as a dietary indiscretion is sometimes identified.1
Excess circulating catecholamines can be a result of stress, exogenous administration, or endogenous oversecretion. Our patient’s clinical presentation is highly suspicious for a high-catecholamine state, and this should be further evaluated.
Obstructive sleep apnea is common in patients with resistant hypertension, with an estimated prevalence as high as 60% in this group.3,4
Primary aldosteronism has an estimated prevalence of about 20% in patients evaluated for resistant hypertension.5
AN ADRENAL MASS IS INCIDENTALLY DISCOVERED
Computed tomographic angiography of the chest revealed no evidence of pulmonary emboli. There was mild dilation of the central pulmonary arteries and an incidental, incompletely imaged 4.7-by-3.4-cm mass of mixed attenuation in the right adrenal gland, with macroscopic fat within the lesion.
Computed tomography (CT) of the abdomen with dedicated cuts through the adrenal glands revealed a 4.7-cm heterogeneous right adrenal mass with a density of 34 Hounsfield units (HU). The left adrenal gland appeared diffusely enlarged without a discretely seen mass, consistent with hyperplasticity (Figure 1).
2. Based on the patient’s clinical presentation and findings on CT, what would be the most likely diagnosis for this incidentally found adrenal mass?
- Adrenocortical adenoma
- Adrenocortical carcinoma
- Metastatic mass
- Pheochromocytoma
Adrenocortical adenoma can present as a small homogeneous mass of variable size, with smooth margins, and rarely containing hemorrhagic tissue or calcifications. The typical density on nonenhanced CT is less than 10 HU. On enhanced CT, it is nonvascular. T2-weighted magnetic resonance imaging (MRI) shows a lesion of the same intensity as liver tissue.6
Adrenocortical adenoma is not classically associated with autologous activity and thus is less likely to explain our patient’s symptoms.
Adrenocortical carcinoma can present as a large heterogeneous mass, usually greater than 4 cm in diameter, with irregular margins and areas of necrosis, hemorrhage, or calcification. The typical density on nonenhanced CT is greater than 10 HU. On enhanced CT, the mass is usually vascular, and T2-weighted MRI will show a lesion more intense than liver tissue.6
Adrenocortical carcinoma is also not classically associated with autologous activity, and so is not likely to explain our patient’s symptoms.6
Metastatic disease can present with masses of variable size, often bilaterally, and occasionally with cysts or areas of hemorrhage. The typical density of metastatic lesions on nonenhanced CT is greater than 10 HU. On enhanced CT, they are usually vascular, and on T2-weighted MRI they are hyperintense.6 The characteristics of the mass and the absence of a primary malignancy on CT of the chest and abdomen do not support the diagnosis of metastatic disease.
Pheochromocytoma is a neuroendocrine tumor of the adrenal medulla that can present as a large heterogeneous mass, greater than 3 cm in diameter, with clear margins and cysts or areas of hemorrhage. Extra-adrenal neuroendocrine tumors are typically called paragangliomas and have features similar to those of pheochromocytoma. The typical density of pheochromocytoma on nonenhanced CT is greater than 10 HU. On enhanced CT, it is usually vascular, and T2-weighted MRI shows a hyperintense lesion. Pheochromocytoma can be biochemically active and thus can cause signs and symptoms that will lead to the diagnosis.6
Other imaging tests may play a role in the evaluation of adrenal masses but are not required for the diagnosis of pheochromocytoma. Functional positron emission tomography using metaiodobenzylguanidine labeled with iodine 123 or-iodine 131 or using the glucose analogue F-18 fluorodeoxyglucose has been used in the initial assessment of pheochromocytoma, with good sensitivity and specificity.7,8
Our patient’s pacemaker-defibrillator precluded him from undergoing MRI.
DIAGNOSIS: PHEOCHROMOCYTOMA
Pheochromocytoma was highly suspected on the basis of the patient’s clinical presentation, and metoprolol was immediately discontinued. He was started on the calcium channel blocker verapamil and the alpha-blocker phenoxybenzamine.
Serum samples were obtained to measure metanephrines, dehydroepiandrosterone, aldosterone, and cortisol, and a 24-hour urine collection was obtained to measure creatinine, dopamine, epinephrine, norepinephrine, cortisol, and metanephrines. Based on the results (Table 1) and on the findings on imaging, the patient was diagnosed with pheochromocytoma. A surgical consultation was obtained, and surgery was recommended.
WHEN DOES PHEOCHROMOCYTOMA CALL FOR SURGERY?
3. Which criterion is most important when determining the need for surgery for pheochromocytoma?
- Findings on fine-needle aspiration biopsy
- Biochemical activity
- Size of the mass
- Bilateral masses
Fine-needle aspiration biopsy can be done when a mass is found incidentally and no evidence of biochemical activity is detected, although it is not an essential part of the diagnostic workup.9 In most cases, the sampling from fine-needle aspiration is not sufficient to achieve a diagnosis.
Biochemical activity is the most important factor when determining the need for prompt surgical intervention. The excess circulating catecholamines have been associated with increased risk of cardiovascular morbidity and death independent of the morbidity associated with hypertension alone.10 Biochemical activity can be independent of the size of the mass, but larger masses typically present with symptoms.
Bilateral masses have been associated with metastatic disease.11 In retrospect, the patient’s history of hypertension and cerebrovascular accident could be associated with the development of a catecholamine-releasing tumor.
A GOOD OUTCOME FROM SURGERY
The patient was continued on phenoxybenzamine for 7 days and responded well to this therapy.
After this preoperative preparation, he underwent laparoscopic right adrenalectomy with excision of a retroperitoneal adrenal mass. His postoperative course was complicated by transient hypotension requiring low-dose vasopressin support for less than 24 hours. He was then restarted on his previous dosage of metoprolol and was discharged home on postoperative day 5 with stable blood pressure. Follow-up 24-hour urine collection 1 month after he was discharged showed normalization of metanephrine, normetanephrine, epinephrine, and norepinephrine levels.
Despite low suspicion for an underlying genetic syndrome, he was referred for genetic testing and was scheduled to have a repeat 24-hour urine collection and imaging in 6 months to follow his enlarged left adrenal gland, which did not appear to be metabolically hyperactive.
4. What is the most common perioperative complication of pheochromocytoma excision?
- Hypoglycemia
- Hypotension
- Hypocortisolism
- Hypertension
- Tachycardia
Hypoglycemia has been observed after removal of pheochromocytoma, as levels of catecholamines (which normally inhibit pancreatic beta cells) decrease and insulin secretion consequently increases.12 Our patient developed hypoglycemia before surgery, not after, and it was likely due to the combination of his antidiabetic therapy, weight loss, and decreased oral intake.
Hypotension is the most common complication in the perioperative period. It is associated with excessive loss of catecholamine secretion. It is usually short-lived but may require aggressive administration of intravenous fluids and use of sympathomimetic agents.
Hypocortisolism is unlikely in patients with pheochromocytoma, but it is likely after removal of adrenocortical adenoma.
Hypertension and tachycardia affect up to 40% of pheochromocytoma patients in some case series.12
PHEOCHROMOCYTOMA: A CATECHOLAMINE-SECRETING TUMOR
The pathophysiology of pheochromocytoma is complex. It is characterized by accelerated growth of cells producing catecholamines, which may produce symptoms when secreted into the bloodstream. The classic triad of symptoms is headache, hypertension, and hyperglycemia, although our patient had very low blood sugar levels. Other common symptoms are nausea, orthostasis, and tremor, although not all symptoms are invariably seen.
Genetic testing recommended
Genetic associations have been described and are thought to be responsible for 20% to 30% of cases of pheochromocytoma. All associated germline mutations are autosomal dominant, some with variable penetrance. These include:
- Succinate dehydrogenase subunit B, C, and D mutations
- von Hippel-Lindau syndrome
- Multiple endocrine neoplasia type 1 and type 2 syndromes
- Neurofibromatosis type 1.13,14
The succinate dehydrogenase subunit mutations have been associated with, but not limited to, extra-adrenal adenomas (paragangliomas) and carry a worse prognosis.
Some experts recommend genetic testing in all cases of pheochromocytoma, sporadic or familial, and this testing should be followed by counseling if a mutation is found.15 Others recommend genetic testing based on the patient’s age (under age 50), history, imaging, and biochemical features of the tumor (metanephrines predominate in multiple endocrine neoplasia syndromes, and normetanephrines in von Hippel-Lindau syndrome).13
Serious consequences
A thorough evaluation is recommended, since pheochromocytoma has been associated with increased cardiovascular morbidity. In a retrospective series, Stolk et al10 reported that patients with pheochromocytoma had a higher incidence of myocardial infarction, angina, and stroke in the years preceding the diagnosis than did patients with essential hypertension (13.8% vs 1.1%, P < .001).10
Catecholamine cardiomyopathy has been described and shares clinical features with Takotsubo or stress cardiomyopathy, with global left ventricular systolic and diastolic dysfunction that improve or resolve after the adrenergic insult is removed.16
Conditions that warrant further evaluation or that may suggest pheochromocytoma are malignant hypertension, hypertensive encephalopathy, ischemic stroke, subarachnoid hemorrhage, acute pulmonary edema, angina pectoris, myocardial infarction, aortic dissection, and kidney injury.
When to suspect pheochromocytoma
Pheochromocytoma should be suspected in a patient with resistant hypertension, family history, or imaging findings that suggest an adrenal mass with a heterogeneous appearance. The diagnostic algorithm follows the same pathway as for the evaluation of an incidentally found adrenal mass, with determination of its dimension and characteristics by CT or MRI, and with biochemical testing of urine catecholamines, plasma free metanephrines, renin, aldosterone, and cortisol.
The diagnosis of pheochromocytoma is established by obtaining fractionated metanephrines and catecholamines in a 24-hour urine collection (sensitivity 90%, specificity 98%). Analysis of plasma metanephrines has a higher sensitivity (97%) but lower specificity (85%).17 The combination of typical signs, symptoms, and laboratory findings makes the diagnosis likely, especially in combination with a unilateral adrenal mass.
Laparoscopic surgery after medical preparation for active tumors
If the mass appears benign and not biochemically hyperactive, then follow-up at 1 year is recommended, with repeat testing. Surgical evaluation and intervention is recommended for lesions that appear malignant or that are biochemically active and clinically symptomatic.9
Preoperative hemodynamic control is essential in the management of pheochromocytoma to prevent or minimize hemodynamic changes that can be driven by increased catecholamines. Control is typically achieved with initial alpha-blockade and then beta-blockade to avoid worsening hypertension and to prevent an acute hypertensive crisis during surgical intervention. Phenoxybenzamine, the mainstay of therapy, is a nonselective alpha-blocker with a long duration of action that requires titration over several days up to 3 weeks.
A selective alpha-1-blocker such as doxazosin can be used to control postoperative hypotension, as it has a shorter half-life than phenoxybenzamine. Alternative strategies include calcium channel blockers, centrally acting sympathetic blockers, and magnesium.18
Laparoscopic adrenalectomy by an experienced surgeon after excellent medical preparation is often considered the treatment of choice, but for larger or malignant masses, an open procedure is recommended. The risk of perioperative morbidity and death can be reduced by adequate medical management. With successful surgical resection, the long-term prognosis is favorable.
- Calhoun DA, Jones D, Textor S, et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension 2008; 51:1403–1419.
- Kumar N, Calhoun DA, Dudenbostel T. Management of patients with resistant hypertension: current treatment options. Integr Blood Press Control 2013; 6:139–151.
- Pedrosa RP, Drager LF, Gonzaga CC, et al. Obstructive sleep apnea: the most common secondary cause of hypertension associated with resistant hypertension. Hypertension 2011; 58:811–817.
- Marcus JA, Pothineni A, Marcus CZ, Bisognano JD. The role of obesity and obstructive sleep apnea in the pathogenesis and treatment of resistant hypertension. Curr Hypertens Rep 2014; 16:411.
- Calhoun DA, Nishizaka MK, Zaman MA, Thakkar RB, Weissmann P. Hyperaldosteronism among black and white subjects with resistant hypertension. Hypertension 2002; 40:892–896.
- Young WF Clinical practice. The incidentally discovered adrenal mass. N Engl J Med 2007; 356:601–610.
- Lin M, Wong V, Yap J, Jin R, Leong P, Campbell P. FDG PET in the evaluation of phaeochromocytoma: a correlative study with MIBG scintigraphy and Ki-67 proliferative index. Clin Imaging 2013; 37:1084–1088.
- Raja A, Leung K, Stamm M, Girgis S, Low G. Multimodality imaging findings of pheochromocytoma with associated clinical and biochemical features in 53 patients with histologically confirmed tumors. AJR Am J Roentgenol 2013; 201:825–833.
- Nieman LK. Approach to the patient with an adrenal incidentaloma. J Clin Endocrinol Metab 2010; 95:4106–4113.
- Stolk RF, Bakx C, Mulder J, Timmers HJ, Lenders JW. Is the excess cardiovascular morbidity in pheochromocytoma related to blood pressure or to catecholamines? J Clin Endocrinol Metab 2013; 98:1100–1106.
- Grumbach MM, Biller BM, Braunstein GD, et al. Management of the clinically inapparent adrenal mass (‘incidentaloma’). Ann Intern Med 2003; 138:424–429.
- Lentschener C, Gaujoux S, Tesniere A, Dousset B. Point of controversy: perioperative care of patients undergoing pheochromocytoma removal—time for a reappraisal? Eur J Endocrinol 2011; 165:365–373.
- Bryant J, Farmer J, Kessler LJ, Townsend RR, Nathanson KL. Pheochromocytoma: the expanding genetic differential diagnosis. J Natl Cancer Inst 2003; 95:1196–1204.
- Lee P, Leonard J. Textbook on endocrinology. BMJ 1994; 308:1512.
- Fishbein L, Merrill S, Fraker DL, Cohen DL, Nathanson KL. Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol 2013; 20:1444–1450.
- Agarwal G, Sadacharan D, Kapoor A, et al. Cardiovascular dysfunction and catecholamine cardiomyopathy in pheochromocytoma patients and their reversal following surgical cure: results of a prospective case-control study. Surgery 2011; 150:1202–1211.
- Sawka AM, Jaeschke R, Singh RJ, Young WF A comparison of biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab 2003; 88:553–558.
- Domi R, Laho H. Management of pheochromocytoma: old ideas and new drugs. Niger J Clin Pract 2012; 15:253–257.
- Calhoun DA, Jones D, Textor S, et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension 2008; 51:1403–1419.
- Kumar N, Calhoun DA, Dudenbostel T. Management of patients with resistant hypertension: current treatment options. Integr Blood Press Control 2013; 6:139–151.
- Pedrosa RP, Drager LF, Gonzaga CC, et al. Obstructive sleep apnea: the most common secondary cause of hypertension associated with resistant hypertension. Hypertension 2011; 58:811–817.
- Marcus JA, Pothineni A, Marcus CZ, Bisognano JD. The role of obesity and obstructive sleep apnea in the pathogenesis and treatment of resistant hypertension. Curr Hypertens Rep 2014; 16:411.
- Calhoun DA, Nishizaka MK, Zaman MA, Thakkar RB, Weissmann P. Hyperaldosteronism among black and white subjects with resistant hypertension. Hypertension 2002; 40:892–896.
- Young WF Clinical practice. The incidentally discovered adrenal mass. N Engl J Med 2007; 356:601–610.
- Lin M, Wong V, Yap J, Jin R, Leong P, Campbell P. FDG PET in the evaluation of phaeochromocytoma: a correlative study with MIBG scintigraphy and Ki-67 proliferative index. Clin Imaging 2013; 37:1084–1088.
- Raja A, Leung K, Stamm M, Girgis S, Low G. Multimodality imaging findings of pheochromocytoma with associated clinical and biochemical features in 53 patients with histologically confirmed tumors. AJR Am J Roentgenol 2013; 201:825–833.
- Nieman LK. Approach to the patient with an adrenal incidentaloma. J Clin Endocrinol Metab 2010; 95:4106–4113.
- Stolk RF, Bakx C, Mulder J, Timmers HJ, Lenders JW. Is the excess cardiovascular morbidity in pheochromocytoma related to blood pressure or to catecholamines? J Clin Endocrinol Metab 2013; 98:1100–1106.
- Grumbach MM, Biller BM, Braunstein GD, et al. Management of the clinically inapparent adrenal mass (‘incidentaloma’). Ann Intern Med 2003; 138:424–429.
- Lentschener C, Gaujoux S, Tesniere A, Dousset B. Point of controversy: perioperative care of patients undergoing pheochromocytoma removal—time for a reappraisal? Eur J Endocrinol 2011; 165:365–373.
- Bryant J, Farmer J, Kessler LJ, Townsend RR, Nathanson KL. Pheochromocytoma: the expanding genetic differential diagnosis. J Natl Cancer Inst 2003; 95:1196–1204.
- Lee P, Leonard J. Textbook on endocrinology. BMJ 1994; 308:1512.
- Fishbein L, Merrill S, Fraker DL, Cohen DL, Nathanson KL. Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol 2013; 20:1444–1450.
- Agarwal G, Sadacharan D, Kapoor A, et al. Cardiovascular dysfunction and catecholamine cardiomyopathy in pheochromocytoma patients and their reversal following surgical cure: results of a prospective case-control study. Surgery 2011; 150:1202–1211.
- Sawka AM, Jaeschke R, Singh RJ, Young WF A comparison of biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab 2003; 88:553–558.
- Domi R, Laho H. Management of pheochromocytoma: old ideas and new drugs. Niger J Clin Pract 2012; 15:253–257.