User login
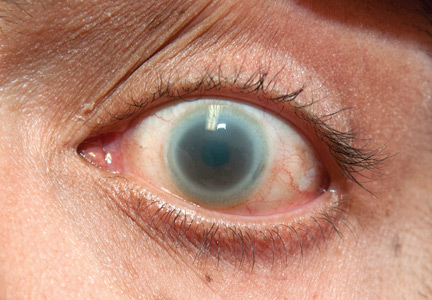
Corneal opacities in a man with chronic kidney disease
A 40-year-old man with end-stage renal disease on intermittent hemodialysis presented to the emergency department with a 1-week history of pain affecting his left lower back, left flank, and left lower abdomen, diagnosed as zoster prodrome.
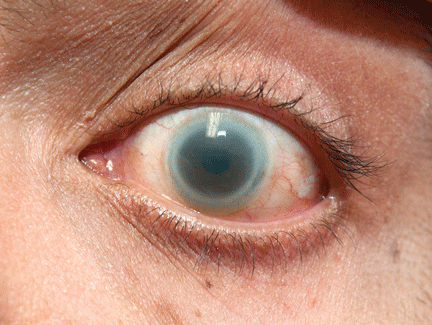
Of note, both corneas were cloudy, most severely in the limbus (Figure 1). His visual acuity and findings on funduscopic examination were normal.
CORNEAL OPACITY
The finding of corneal opacity should prompt an immediate ophthalmologic evaluation by the internist as well as an ophthalmologist. The initial examination should include visual acuity testing; gross examination with the naked eye; penlight examination of the pupil, conjunctiva, and anterior chamber; funduscopic examination to at least confirm a red reflex; and fluorescein examination of the cornea. Fluorescein testing is done last, as the dye may interfere with the other initial tests.1
A number of causes of opacity
A number of conditions can cause corneal opacity. Several genetic conditions can cause developmental anomalies of the cornea, leading to corneal defects present at birth.2 Causes of secondary corneal opacity in early infancy include infections such as herpes, iatrogenic injury during amniocentesis or forceps delivery, and infantile congenital glaucoma.2
Later in life, causes of corneal opacity include cataract, glaucoma, chemical exposure, foreign body injury, irradiation, infection (eg, syphilis, herpes, chlamydia), endophthalmitis, and metabolic genetic disorders such as Fabry disease, trisomy 18 syndrome, and lecithin-cholesterol acyltransferase (LCAT) deficiency.3
LCAT DEFICIENCY
LCAT is a key protein in reverse transport of cholesterol from the systemic circulation to the liver for excretion into the bile. Its deficiency results in low serum concentrations of high-density lipoprotein cholesterol (HDL-C).4 About 80 different mutations in the LCAT gene have been linked to LCAT deficiency.5
LCAT deficiency varies in severity. Patients with complete deficiency can have nearly undetectable levels of HDL-C, eruptive xanthoma, hepatosplenomegaly, and premature coronary artery disease (ie, by age 40).5,6 Features of coronary atherosclerosis can be lacking in patients with partial deficiency.
Regardless of the degree of LCAT deficiency, most patients have corneal opacification that is most severe near the limbus (thus, the term “fish eye syndrome”) and anemia.7 Although corneal opacification presents early in life and persists, it does not seem to affect vision.5 The anemia is associated with enhanced fractional clearance of red blood cells secondary to hypersplenism.8
LCAT deficiency and the kidneys
LCAT deficiency has its most devastating effect on the kidney. Renal disease begins early in life with mild proteinuria and microscopic hematuria. With increasing age, renal function deteriorates and proteinuria and hematuria worsen.9
Renal biopsy study may reveal foam cells in the glomerular tufts, arterioles with thickened intima and narrowed lumens, and subendothelial deposits of lipids in the renal arteries and arterioles.10 Some studies have suggested that kidney disease is most likely initiated by lipid deposition or cellular uptake of lipoproteins in the glomerular basement membrane, mesangium, and capillary subendothelium.
Treatment
There are few treatment options for patients with LCAT deficiency. Control of hypertension, if present, may halt or slow renal deterioration.5 Many patients eventually require dialysis, and some undergo renal transplantation, but the renal disease can recur.
OUR PATIENT
Our patient had a known diagnosis of LCAT deficiency. Five years before this presentation at our emergency department, he developed malignant hypertension, followed shortly by renal disease. Over the next 4 years, his kidney function deteriorated, culminating in the need for dialysis; his corneal opacities manifested and gradually worsened; and after extensive studies including kidney biopsies, he was finally diagnosed with LCAT deficiency.
He also exhibited a chronically low level of HDL-C (2 to 5 mg/dL) and significant coronary artery disease. Although unrelated, his zoster pain was treated with renally dosed acyclovir and gabapentin. He never demonstrated the characteristic rash, and his pain improved significantly within 5 days of treatment.
- Knox KA, McIntee J. Nurse management of corneal abrasion. Br J Nurs 1995; 4:440–460.
- Nischal KK. Congenital corneal opacities—a surgical approach to nomenclature and classification. Eye (Lond) 2007; 21:1326–1337.
- Chiapella AP, Rosenthal AR. One year in an eye casualty clinic. Br J Ophthalmol 1985; 69:865–870.
- Rosenson RS, Brewer HB Jr, Davidson WS, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation 2012; 125:1905–1919.
- Roshan B, Ganda OP, Desilva R, et al. Homozygous lecithin:cholesterol acyltransferase (LCAT) deficiency due to a new loss of function mutation and review of the literature. J Clin Lipidol 2011; 5:493–499.
- Kuivenhoven JA, van Voorst tot Voorst EJ, Wiebusch H, et al. A unique genetic and biochemical presentation of fish-eye disease. J Clin Invest 1995; 96:2783–2791.
- Palmiero PM, Sbeity Z, Liebmann J, Ritch R. In vivo imaging of the cornea in a patient with lecithin-cholesterol acyltransferase deficiency. Cornea 2009; 28:1061–1064.
- Norum KR, Gjone E. Familial serum-cholesterol esterification failure. A new inborn error of metabolism. Biochim Biophys Acta 1967; 144:698–700.
- Gjone E, Norum KR. Familial serum cholesterol ester deficiency. Clinical study of a patient with a new syndrome. Acta Med Scand 1968; 183:107–112.
- Lager DJ, Rosenberg BF, Shapiro H, Bernstein J. Lecithin cholesterol acyltransferase deficiency: ultrastructural examination of sequential renal biopsies. Mod Pathol 1991; 4:331–335.
A 40-year-old man with end-stage renal disease on intermittent hemodialysis presented to the emergency department with a 1-week history of pain affecting his left lower back, left flank, and left lower abdomen, diagnosed as zoster prodrome.
Of note, both corneas were cloudy, most severely in the limbus (Figure 1). His visual acuity and findings on funduscopic examination were normal.
CORNEAL OPACITY
The finding of corneal opacity should prompt an immediate ophthalmologic evaluation by the internist as well as an ophthalmologist. The initial examination should include visual acuity testing; gross examination with the naked eye; penlight examination of the pupil, conjunctiva, and anterior chamber; funduscopic examination to at least confirm a red reflex; and fluorescein examination of the cornea. Fluorescein testing is done last, as the dye may interfere with the other initial tests.1
A number of causes of opacity
A number of conditions can cause corneal opacity. Several genetic conditions can cause developmental anomalies of the cornea, leading to corneal defects present at birth.2 Causes of secondary corneal opacity in early infancy include infections such as herpes, iatrogenic injury during amniocentesis or forceps delivery, and infantile congenital glaucoma.2
Later in life, causes of corneal opacity include cataract, glaucoma, chemical exposure, foreign body injury, irradiation, infection (eg, syphilis, herpes, chlamydia), endophthalmitis, and metabolic genetic disorders such as Fabry disease, trisomy 18 syndrome, and lecithin-cholesterol acyltransferase (LCAT) deficiency.3
LCAT DEFICIENCY
LCAT is a key protein in reverse transport of cholesterol from the systemic circulation to the liver for excretion into the bile. Its deficiency results in low serum concentrations of high-density lipoprotein cholesterol (HDL-C).4 About 80 different mutations in the LCAT gene have been linked to LCAT deficiency.5
LCAT deficiency varies in severity. Patients with complete deficiency can have nearly undetectable levels of HDL-C, eruptive xanthoma, hepatosplenomegaly, and premature coronary artery disease (ie, by age 40).5,6 Features of coronary atherosclerosis can be lacking in patients with partial deficiency.
Regardless of the degree of LCAT deficiency, most patients have corneal opacification that is most severe near the limbus (thus, the term “fish eye syndrome”) and anemia.7 Although corneal opacification presents early in life and persists, it does not seem to affect vision.5 The anemia is associated with enhanced fractional clearance of red blood cells secondary to hypersplenism.8
LCAT deficiency and the kidneys
LCAT deficiency has its most devastating effect on the kidney. Renal disease begins early in life with mild proteinuria and microscopic hematuria. With increasing age, renal function deteriorates and proteinuria and hematuria worsen.9
Renal biopsy study may reveal foam cells in the glomerular tufts, arterioles with thickened intima and narrowed lumens, and subendothelial deposits of lipids in the renal arteries and arterioles.10 Some studies have suggested that kidney disease is most likely initiated by lipid deposition or cellular uptake of lipoproteins in the glomerular basement membrane, mesangium, and capillary subendothelium.
Treatment
There are few treatment options for patients with LCAT deficiency. Control of hypertension, if present, may halt or slow renal deterioration.5 Many patients eventually require dialysis, and some undergo renal transplantation, but the renal disease can recur.
OUR PATIENT
Our patient had a known diagnosis of LCAT deficiency. Five years before this presentation at our emergency department, he developed malignant hypertension, followed shortly by renal disease. Over the next 4 years, his kidney function deteriorated, culminating in the need for dialysis; his corneal opacities manifested and gradually worsened; and after extensive studies including kidney biopsies, he was finally diagnosed with LCAT deficiency.
He also exhibited a chronically low level of HDL-C (2 to 5 mg/dL) and significant coronary artery disease. Although unrelated, his zoster pain was treated with renally dosed acyclovir and gabapentin. He never demonstrated the characteristic rash, and his pain improved significantly within 5 days of treatment.
A 40-year-old man with end-stage renal disease on intermittent hemodialysis presented to the emergency department with a 1-week history of pain affecting his left lower back, left flank, and left lower abdomen, diagnosed as zoster prodrome.
Of note, both corneas were cloudy, most severely in the limbus (Figure 1). His visual acuity and findings on funduscopic examination were normal.
CORNEAL OPACITY
The finding of corneal opacity should prompt an immediate ophthalmologic evaluation by the internist as well as an ophthalmologist. The initial examination should include visual acuity testing; gross examination with the naked eye; penlight examination of the pupil, conjunctiva, and anterior chamber; funduscopic examination to at least confirm a red reflex; and fluorescein examination of the cornea. Fluorescein testing is done last, as the dye may interfere with the other initial tests.1
A number of causes of opacity
A number of conditions can cause corneal opacity. Several genetic conditions can cause developmental anomalies of the cornea, leading to corneal defects present at birth.2 Causes of secondary corneal opacity in early infancy include infections such as herpes, iatrogenic injury during amniocentesis or forceps delivery, and infantile congenital glaucoma.2
Later in life, causes of corneal opacity include cataract, glaucoma, chemical exposure, foreign body injury, irradiation, infection (eg, syphilis, herpes, chlamydia), endophthalmitis, and metabolic genetic disorders such as Fabry disease, trisomy 18 syndrome, and lecithin-cholesterol acyltransferase (LCAT) deficiency.3
LCAT DEFICIENCY
LCAT is a key protein in reverse transport of cholesterol from the systemic circulation to the liver for excretion into the bile. Its deficiency results in low serum concentrations of high-density lipoprotein cholesterol (HDL-C).4 About 80 different mutations in the LCAT gene have been linked to LCAT deficiency.5
LCAT deficiency varies in severity. Patients with complete deficiency can have nearly undetectable levels of HDL-C, eruptive xanthoma, hepatosplenomegaly, and premature coronary artery disease (ie, by age 40).5,6 Features of coronary atherosclerosis can be lacking in patients with partial deficiency.
Regardless of the degree of LCAT deficiency, most patients have corneal opacification that is most severe near the limbus (thus, the term “fish eye syndrome”) and anemia.7 Although corneal opacification presents early in life and persists, it does not seem to affect vision.5 The anemia is associated with enhanced fractional clearance of red blood cells secondary to hypersplenism.8
LCAT deficiency and the kidneys
LCAT deficiency has its most devastating effect on the kidney. Renal disease begins early in life with mild proteinuria and microscopic hematuria. With increasing age, renal function deteriorates and proteinuria and hematuria worsen.9
Renal biopsy study may reveal foam cells in the glomerular tufts, arterioles with thickened intima and narrowed lumens, and subendothelial deposits of lipids in the renal arteries and arterioles.10 Some studies have suggested that kidney disease is most likely initiated by lipid deposition or cellular uptake of lipoproteins in the glomerular basement membrane, mesangium, and capillary subendothelium.
Treatment
There are few treatment options for patients with LCAT deficiency. Control of hypertension, if present, may halt or slow renal deterioration.5 Many patients eventually require dialysis, and some undergo renal transplantation, but the renal disease can recur.
OUR PATIENT
Our patient had a known diagnosis of LCAT deficiency. Five years before this presentation at our emergency department, he developed malignant hypertension, followed shortly by renal disease. Over the next 4 years, his kidney function deteriorated, culminating in the need for dialysis; his corneal opacities manifested and gradually worsened; and after extensive studies including kidney biopsies, he was finally diagnosed with LCAT deficiency.
He also exhibited a chronically low level of HDL-C (2 to 5 mg/dL) and significant coronary artery disease. Although unrelated, his zoster pain was treated with renally dosed acyclovir and gabapentin. He never demonstrated the characteristic rash, and his pain improved significantly within 5 days of treatment.
- Knox KA, McIntee J. Nurse management of corneal abrasion. Br J Nurs 1995; 4:440–460.
- Nischal KK. Congenital corneal opacities—a surgical approach to nomenclature and classification. Eye (Lond) 2007; 21:1326–1337.
- Chiapella AP, Rosenthal AR. One year in an eye casualty clinic. Br J Ophthalmol 1985; 69:865–870.
- Rosenson RS, Brewer HB Jr, Davidson WS, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation 2012; 125:1905–1919.
- Roshan B, Ganda OP, Desilva R, et al. Homozygous lecithin:cholesterol acyltransferase (LCAT) deficiency due to a new loss of function mutation and review of the literature. J Clin Lipidol 2011; 5:493–499.
- Kuivenhoven JA, van Voorst tot Voorst EJ, Wiebusch H, et al. A unique genetic and biochemical presentation of fish-eye disease. J Clin Invest 1995; 96:2783–2791.
- Palmiero PM, Sbeity Z, Liebmann J, Ritch R. In vivo imaging of the cornea in a patient with lecithin-cholesterol acyltransferase deficiency. Cornea 2009; 28:1061–1064.
- Norum KR, Gjone E. Familial serum-cholesterol esterification failure. A new inborn error of metabolism. Biochim Biophys Acta 1967; 144:698–700.
- Gjone E, Norum KR. Familial serum cholesterol ester deficiency. Clinical study of a patient with a new syndrome. Acta Med Scand 1968; 183:107–112.
- Lager DJ, Rosenberg BF, Shapiro H, Bernstein J. Lecithin cholesterol acyltransferase deficiency: ultrastructural examination of sequential renal biopsies. Mod Pathol 1991; 4:331–335.
- Knox KA, McIntee J. Nurse management of corneal abrasion. Br J Nurs 1995; 4:440–460.
- Nischal KK. Congenital corneal opacities—a surgical approach to nomenclature and classification. Eye (Lond) 2007; 21:1326–1337.
- Chiapella AP, Rosenthal AR. One year in an eye casualty clinic. Br J Ophthalmol 1985; 69:865–870.
- Rosenson RS, Brewer HB Jr, Davidson WS, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation 2012; 125:1905–1919.
- Roshan B, Ganda OP, Desilva R, et al. Homozygous lecithin:cholesterol acyltransferase (LCAT) deficiency due to a new loss of function mutation and review of the literature. J Clin Lipidol 2011; 5:493–499.
- Kuivenhoven JA, van Voorst tot Voorst EJ, Wiebusch H, et al. A unique genetic and biochemical presentation of fish-eye disease. J Clin Invest 1995; 96:2783–2791.
- Palmiero PM, Sbeity Z, Liebmann J, Ritch R. In vivo imaging of the cornea in a patient with lecithin-cholesterol acyltransferase deficiency. Cornea 2009; 28:1061–1064.
- Norum KR, Gjone E. Familial serum-cholesterol esterification failure. A new inborn error of metabolism. Biochim Biophys Acta 1967; 144:698–700.
- Gjone E, Norum KR. Familial serum cholesterol ester deficiency. Clinical study of a patient with a new syndrome. Acta Med Scand 1968; 183:107–112.
- Lager DJ, Rosenberg BF, Shapiro H, Bernstein J. Lecithin cholesterol acyltransferase deficiency: ultrastructural examination of sequential renal biopsies. Mod Pathol 1991; 4:331–335.
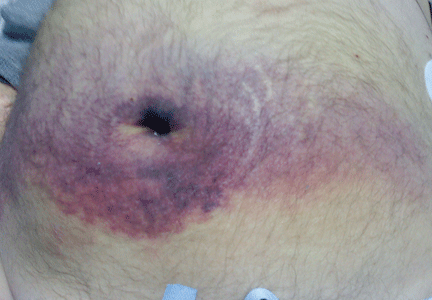
A 60-year-old man with abdominal bruising
A 60-year-old man with hepatocellular carcinoma was admitted to the hospital with pulmonary emboli secondary to inferior vena caval thrombosis that extended to the right atrium.
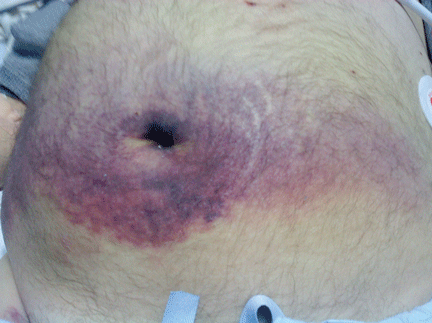
He became hypotensive on the second day, with a heart rate of 124 per minute, respiratory rate 44 per minute, pulse oxygen saturation 79% on room air, and systolic blood pressure 70 mm Hg. Physical examination revealed abdominal ecchymoses resembling the Cullen sign and flank ecchymoses resembling the Grey Turner sign (Figures 1 and 2).
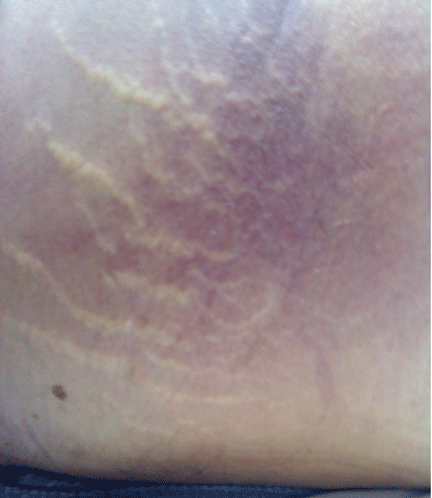
He was given a bolus of normal saline followed by infusion of fresh frozen plasma and packed red blood cells. His lactate level was 17.52 mg/dL (reference range 0.1–2.2). His hemoglobin and hematocrit decreased precipitously—the hemoglobin from 10.5 g/dL to 5.9 (reference range 14.0–17.5), and the hematocrit from 30.3% to 17.8% (reference range 41–50). He was transferred to the intensive care unit. A do-not-resuscitate order was instituted, and he died 12 hours later.
CONDITIONS RESULTING IN THE CULLEN AND GREY TURNER SIGNS
The Cullen sign, a bluish discoloration of the periumbilical skin, was originally described in 1918 by the gynecologist Thomas Cullen, MD, in a patient with a ruptured ectopic pregnancy.1 The Grey Turner sign, an ecchymotic discoloration of the lateral abdominal wall or flank, was first reported in 1920 by a surgeon, Dr. George Grey Turner, in a patient with acute pancreatitis.2
The signs occur in about 1% of patients with acute pancreatitis and predict a poor prognosis, with a reported death rate of 37%.3
The appearance of ecchymoses in the periumbilical area or flank has been taught as a hallmark of acute pancreatitis.4 However, the original patient described with the Cullen sign did not have pancreatitis,4,5 and it has been reported with many other conditions, including ruptured aortic aneurysm, splenic rupture, and rectus sheath hematoma, as a complication of anticoagulation or perforated duodenal ulcer, and, as in our patient, as a manifestation of liver disease.
How they occur
The common pathway leading to the occurrence of these subcutaneous ecchymoses is retroperitoneal bleeding followed by tracking of blood from the retroperitoneum through a defect in the transversalis fascia to the abdominal wall musculature and then to the periumbilical subcutaneous tissue. In the Cullen sign, blood diffuses from the retroperitoneum along the gastrohepatic and falciform ligaments to the umbilicus. In the Grey Turner sign, blood diffuses from the posterior pararenal space to the lateral edge of the quadratus lumborum muscle.6–8 Blood from a retroperitoneal hemorrhage may also diffuse and pool at the inguinal ligament (Fox sign) or at the scrotum (Bryant sign).5
The time to the appearance of the Cullen or the Grey Turner sign is thought to be at least 24 hours after the onset of retroperitoneal bleeding and averages about 3 days after the onset of pancreatitis.3
- Cullen TS. A new sign in ruptured extrauterine pregnancy. Am J Obstet Gynecol 1918; 78:457.
- Grey Turner G. Local discoloration of the abdominal wall as a sign of acute pancreatitis. Br J Surg 1910; 7:394–395.
- Dickson AP, Imrie CW. The incidence and prognosis of body wall ecchymosis in acute pancreatitis. Surg Gynecol Obstet 1984; 159:343–347.
- Harris S, Naina HV. Cullen’s sign revisited. Am J Med 2008; 121:682–683.
- Bosmann M, Schreiner O, Galle PR. Coexistence of Cullen’s and Grey Turner’s signs in acute pancreatitis. Am J Med 2009; 122:333–334.
- Bem J, Bradley EL. Subcutaneous manifestations of severe acute pancreatitis. Pancreas 1998; 16:551–555.
- Meyers MA, Feldberg MA, Oliphant M. Grey Turner’s sign and Cullen’s sign in acute pancreatitis. Gastrointest Radiol 1989; 14:31–37.
- Mabin TA, Gelfand M. Cullen’s sign, a feature in liver disease. Br Med J 1974; 1:493–494.
A 60-year-old man with hepatocellular carcinoma was admitted to the hospital with pulmonary emboli secondary to inferior vena caval thrombosis that extended to the right atrium.
He became hypotensive on the second day, with a heart rate of 124 per minute, respiratory rate 44 per minute, pulse oxygen saturation 79% on room air, and systolic blood pressure 70 mm Hg. Physical examination revealed abdominal ecchymoses resembling the Cullen sign and flank ecchymoses resembling the Grey Turner sign (Figures 1 and 2).
He was given a bolus of normal saline followed by infusion of fresh frozen plasma and packed red blood cells. His lactate level was 17.52 mg/dL (reference range 0.1–2.2). His hemoglobin and hematocrit decreased precipitously—the hemoglobin from 10.5 g/dL to 5.9 (reference range 14.0–17.5), and the hematocrit from 30.3% to 17.8% (reference range 41–50). He was transferred to the intensive care unit. A do-not-resuscitate order was instituted, and he died 12 hours later.
CONDITIONS RESULTING IN THE CULLEN AND GREY TURNER SIGNS
The Cullen sign, a bluish discoloration of the periumbilical skin, was originally described in 1918 by the gynecologist Thomas Cullen, MD, in a patient with a ruptured ectopic pregnancy.1 The Grey Turner sign, an ecchymotic discoloration of the lateral abdominal wall or flank, was first reported in 1920 by a surgeon, Dr. George Grey Turner, in a patient with acute pancreatitis.2
The signs occur in about 1% of patients with acute pancreatitis and predict a poor prognosis, with a reported death rate of 37%.3
The appearance of ecchymoses in the periumbilical area or flank has been taught as a hallmark of acute pancreatitis.4 However, the original patient described with the Cullen sign did not have pancreatitis,4,5 and it has been reported with many other conditions, including ruptured aortic aneurysm, splenic rupture, and rectus sheath hematoma, as a complication of anticoagulation or perforated duodenal ulcer, and, as in our patient, as a manifestation of liver disease.
How they occur
The common pathway leading to the occurrence of these subcutaneous ecchymoses is retroperitoneal bleeding followed by tracking of blood from the retroperitoneum through a defect in the transversalis fascia to the abdominal wall musculature and then to the periumbilical subcutaneous tissue. In the Cullen sign, blood diffuses from the retroperitoneum along the gastrohepatic and falciform ligaments to the umbilicus. In the Grey Turner sign, blood diffuses from the posterior pararenal space to the lateral edge of the quadratus lumborum muscle.6–8 Blood from a retroperitoneal hemorrhage may also diffuse and pool at the inguinal ligament (Fox sign) or at the scrotum (Bryant sign).5
The time to the appearance of the Cullen or the Grey Turner sign is thought to be at least 24 hours after the onset of retroperitoneal bleeding and averages about 3 days after the onset of pancreatitis.3
A 60-year-old man with hepatocellular carcinoma was admitted to the hospital with pulmonary emboli secondary to inferior vena caval thrombosis that extended to the right atrium.
He became hypotensive on the second day, with a heart rate of 124 per minute, respiratory rate 44 per minute, pulse oxygen saturation 79% on room air, and systolic blood pressure 70 mm Hg. Physical examination revealed abdominal ecchymoses resembling the Cullen sign and flank ecchymoses resembling the Grey Turner sign (Figures 1 and 2).
He was given a bolus of normal saline followed by infusion of fresh frozen plasma and packed red blood cells. His lactate level was 17.52 mg/dL (reference range 0.1–2.2). His hemoglobin and hematocrit decreased precipitously—the hemoglobin from 10.5 g/dL to 5.9 (reference range 14.0–17.5), and the hematocrit from 30.3% to 17.8% (reference range 41–50). He was transferred to the intensive care unit. A do-not-resuscitate order was instituted, and he died 12 hours later.
CONDITIONS RESULTING IN THE CULLEN AND GREY TURNER SIGNS
The Cullen sign, a bluish discoloration of the periumbilical skin, was originally described in 1918 by the gynecologist Thomas Cullen, MD, in a patient with a ruptured ectopic pregnancy.1 The Grey Turner sign, an ecchymotic discoloration of the lateral abdominal wall or flank, was first reported in 1920 by a surgeon, Dr. George Grey Turner, in a patient with acute pancreatitis.2
The signs occur in about 1% of patients with acute pancreatitis and predict a poor prognosis, with a reported death rate of 37%.3
The appearance of ecchymoses in the periumbilical area or flank has been taught as a hallmark of acute pancreatitis.4 However, the original patient described with the Cullen sign did not have pancreatitis,4,5 and it has been reported with many other conditions, including ruptured aortic aneurysm, splenic rupture, and rectus sheath hematoma, as a complication of anticoagulation or perforated duodenal ulcer, and, as in our patient, as a manifestation of liver disease.
How they occur
The common pathway leading to the occurrence of these subcutaneous ecchymoses is retroperitoneal bleeding followed by tracking of blood from the retroperitoneum through a defect in the transversalis fascia to the abdominal wall musculature and then to the periumbilical subcutaneous tissue. In the Cullen sign, blood diffuses from the retroperitoneum along the gastrohepatic and falciform ligaments to the umbilicus. In the Grey Turner sign, blood diffuses from the posterior pararenal space to the lateral edge of the quadratus lumborum muscle.6–8 Blood from a retroperitoneal hemorrhage may also diffuse and pool at the inguinal ligament (Fox sign) or at the scrotum (Bryant sign).5
The time to the appearance of the Cullen or the Grey Turner sign is thought to be at least 24 hours after the onset of retroperitoneal bleeding and averages about 3 days after the onset of pancreatitis.3
- Cullen TS. A new sign in ruptured extrauterine pregnancy. Am J Obstet Gynecol 1918; 78:457.
- Grey Turner G. Local discoloration of the abdominal wall as a sign of acute pancreatitis. Br J Surg 1910; 7:394–395.
- Dickson AP, Imrie CW. The incidence and prognosis of body wall ecchymosis in acute pancreatitis. Surg Gynecol Obstet 1984; 159:343–347.
- Harris S, Naina HV. Cullen’s sign revisited. Am J Med 2008; 121:682–683.
- Bosmann M, Schreiner O, Galle PR. Coexistence of Cullen’s and Grey Turner’s signs in acute pancreatitis. Am J Med 2009; 122:333–334.
- Bem J, Bradley EL. Subcutaneous manifestations of severe acute pancreatitis. Pancreas 1998; 16:551–555.
- Meyers MA, Feldberg MA, Oliphant M. Grey Turner’s sign and Cullen’s sign in acute pancreatitis. Gastrointest Radiol 1989; 14:31–37.
- Mabin TA, Gelfand M. Cullen’s sign, a feature in liver disease. Br Med J 1974; 1:493–494.
- Cullen TS. A new sign in ruptured extrauterine pregnancy. Am J Obstet Gynecol 1918; 78:457.
- Grey Turner G. Local discoloration of the abdominal wall as a sign of acute pancreatitis. Br J Surg 1910; 7:394–395.
- Dickson AP, Imrie CW. The incidence and prognosis of body wall ecchymosis in acute pancreatitis. Surg Gynecol Obstet 1984; 159:343–347.
- Harris S, Naina HV. Cullen’s sign revisited. Am J Med 2008; 121:682–683.
- Bosmann M, Schreiner O, Galle PR. Coexistence of Cullen’s and Grey Turner’s signs in acute pancreatitis. Am J Med 2009; 122:333–334.
- Bem J, Bradley EL. Subcutaneous manifestations of severe acute pancreatitis. Pancreas 1998; 16:551–555.
- Meyers MA, Feldberg MA, Oliphant M. Grey Turner’s sign and Cullen’s sign in acute pancreatitis. Gastrointest Radiol 1989; 14:31–37.
- Mabin TA, Gelfand M. Cullen’s sign, a feature in liver disease. Br Med J 1974; 1:493–494.