User login
Warfarin (coumadin) differs from most other drugs in that the dosage required to achieve a desired therapeutic effect varies greatly among individuals. This variability can lead to therapeutic failure, potentially resulting in new thrombosis, or, at the other extreme, to life-threatening bleeding.
Further, there is no reliable means to identify patients who require unusually high doses of warfarin, although genetic testing may become available in the future.
See related patient information
Warfarin, a coumarin derivative first synthesized in 1948, is still the only oral anticoagulant available for long-term use in the United States. Indications for its use include the treatment and, to a lesser extent, the prevention of arterial and venous thromboembolism. It is also used for long-term anticoagulation in patients with atrial arrhythmias (atrial fibrillation and atrial flutter) and mechanical heart valves.
In the paragraphs that follow, we review the causes of warfarin resistance and how to recognize and manage it.
WHAT IS WARFARIN RESISTANCE?
Resistance to warfarin has been described as the inability to prolong the prothrombin time or raise the international normalized ratio (INR) into the therapeutic range when the drug is given at normally prescribed doses.1
However, a higher warfarin requirement does not itself establish the diagnosis of warfarin resistance. The prevalence of warfarin resistance varies by patient population and is difficult to determine. The difficulty lies largely in accounting for dietary factors and in defining normal metabolic variations among individuals.
The range of normally recommended daily or weekly warfarin doses to maintain a therapeutic prothrombin time or INR depends on the study population. Nevertheless, patients who need more than 105 mg per week (15 mg/day) should be considered warfarin-resistant. These patients are likely to be in the top 5% for warfarin doses within an anticoagulated cohort.
Warfarin resistance is different than warfarin failure, which is defined as a new thrombotic event despite a therapeutic prothrombin time and INR. This situation is commonly seen in patients with malignant diseases.
An important characteristic of warfarin resistance is that patients need much smaller doses of vitamin K to reverse the effect of warfarin.2 Thijssen3 showed that, in warfarin-resistant rats, warfarin did not irreversibly inhibit vitamin K1 2,3-epoxide reductase (VKORC1) activity. This is consistent with the vitamin K hypersensitivity observed in warfarin-resistant people.2,3

WHAT CAUSES WARFARIN RESISTANCE?
Warfarin resistance can be classified in practical terms as acquired vs hereditary, or in mechanistic terms as pharmacokinetic vs pharmacodynamic.
Acquired vs hereditary resistance
Hulse4 categorizes warfarin resistance as either acquired or hereditary.
Acquired resistance to warfarin may result from:
- Poor patient compliance (the most common cause)
- High consumption of vitamin K

- Increased clearance (see Warfarin is metabolized by P450 enzymes5–11)
- Drug interactions (Table 1).12,13

Hereditary resistance has been postulated to be caused by genetic factors that result either in faster metabolism of the drug (a form of pharmacokinetic resistance) or in lower activity of the drug (pharmacodynamic resistance). Polymorphisms may play a role, as some VKORC1 and CYP2C9 variant alleles are known to be associated with increased sensitivity to warfarin.14
However, the genetic mechanisms of warfarin resistance are not clearly understood, despite several case reports of hereditary resistance confirmed by similar patterns of resistance in immediate family members.15–19 More than one mechanism is likely. There is ample room for further insight into genetic polymorphisms underlying hereditary warfarin resistance. More on this topic is included in the sections below.
Pharmacokinetic resistance
Pharmacokinetic resistance can result from diminished absorption or increased elimination of the drug. Causes of diminished absorption include emesis, diarrhea, and malabsorption syndrome.
The mechanism of increased warfarin clearance has not been delineated, although the following have been implicated.
Genetic factors. Duplication or multiplication of cytochrome P450 enzyme genes has been described as contributing to a phenotype of ultrarapid metabolism. Some people may carry multiple copies of the CYP2C9 gene, as has already been reported for cytochrome P450 CYP2D6 and CYP2A6.7,8 It is also plausible that rare allelic variants of CYP2C9 exist that are associated with higher-than-normal activity, given that there are alleles known to predispose to warfarin sensitivity.
Hypoalbuminemia may increase the free fraction of warfarin, leading to enhanced rates of clearance and a shorter plasma half-life.15
Hyperalbuminemia may paradoxically also contribute to warfarin resistance via drug binding.
Hyperlipidemia. Several observers have found that lowering serum lipids, primarily triglycerides, increases the sensitivity to warfarin irrespective of the means used to achieve this decrease.20 This most likely results in a decreased pool of vitamin K, some of which is bound to triglycerides.21 Conversely, patients receiving intravenous lipids with total parenteral nutrition have also been diagnosed clinically with warfarin resistance,22 and rat models have shown an association between a lipidrich diet and increased vitamin K-dependent factor activity.23
Diuretics may decrease the response to warfarin by reducing the plasma volume, with a subsequent increase in clotting factor activity.24
Pharmacodynamic resistance
Potential mechanisms of pharmacodynamic warfarin resistance described in rats and in people include:
- Increased affinity of vitamin K1, 2,3-epoxide reductase complex (VKOR) for vitamin K25,26 (see How warfarin works2,10,11,27–30)
- Prolongation of normal clotting factor activity16
- Production of clotting factors that is not dependent on vitamin K16
- Decreased VKOR sensitivity to warfarin.26
In rats, these mechanisms are manifested by relatively high doses of warfarin being required to achieve poisoning. In humans, they result in high doses being needed to achieve a therapeutic effect in the setting of normal warfarin pharmacokinetics, normal warfarin concentration, and normal half-lives of blood clotting proteins.
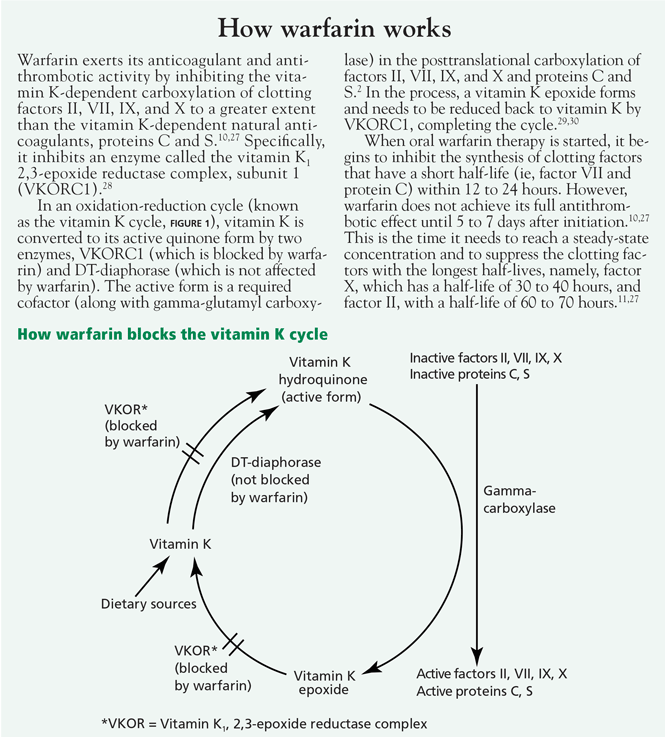
Genetics of pharmacodynamic resistance. Pharmacodynamic warfarin resistance has also been described with inheritance of a monogenetic dominant trait. An early study by O’Reilly24 traced anticoagulation resistance to a genetically linked abnormality of interaction between warfarin and a putative vitamin K receptor.
In one patient with hereditary resistance and high warfarin requirements, a heterozygous point mutation in the VKORC1 gene was identified.31 This results in a substitution that lies in a conserved (normally constant or unchanging DNA sequence in a genome) region of VKORC1 that contains three of four previously identified amino acid substitutions associated with warfarin resistance (Val29Leu, Val45Ala, and Arg58Gly). Further investigation is required to fully characterize the structure-function relationship for VKORC1 and to determine the relationship between the VKORC1 genotype and other pharmacogenetic determinants of warfarin dose-response.
Separately, Loebstein et al32 reported a new mutation, Asp36Tyr, which was common in Jewish ethnic groups of Ethiopian descent (in whom the prevalence is 5%) and Ashkenazi descent (prevalence 4%). In that study, Asp36Tyr carriers needed doses of more than 70 mg per week, placing them towards the high end of the usual warfarin dosing range.
Daly and Aithal7 discovered that warfarinresistant rats overexpressed a protein known as calumenin. This protein is situated in the endoplasmic reticulum and appears to interact with VKOR, decreasing the binding of warfarin. In mice, the calumenin gene is located on chromosome 7, where the gene for VKORC1 is also located.
DIAGNOSIS BY HISTORY AND LABORATORY STUDIES
A full drug and diet history is invaluable in diagnosing potential causes of warfarin resistance (Table 1).
Plasma warfarin levels that are subtherapeutic should raise suspicion of intestinal malabsorption or poor compliance. Poor compliance might be more appropriately seen as a mimic of warfarin resistance. Studies in humans suggest that a therapeutic total plasma warfarin level lies between 0.5 μg/mL and 3.0 μg/mL,10 though the range may vary among laboratories and patient populations.
Warfarin absorption and clearance can be evaluated by analyzing plasma levels at specific intervals after administration, eg, every 60 to 180 minutes. The drug’s half-life can be determined on the basis of its concentrations in different time samples. Normally, the S-enantiomer of warfarin is cleared at twice the rate of the R-enantiomer (5.2 vs 2.5 mL/min/70 kg).8 A normal clearance rate confirms that resistance to warfarin is not due to enhanced elimination.
Clotting assays of factors II, VII, IX, and X may be a more precise way to assess the pharmacodynamics of warfarin,10 although there is no strong evidence to support routine use of such assays. Some studies suggest targeting factor II and factor X activity levels of 10% to 30% of normal biologic activity for a therapeutic warfarin effect in patients with an unreliable or prolonged baseline prothrombin time and INR, such as those with lupus anticoagulant.

TREAT THE CAUSE
Once the type of warfarin resistance has been determined, treatment should be oriented toward the cause.
Educate the patient
The importance of compliance should be reinforced. Educating the patient about diet and other medications that may interact with warfarin is also important. (See an example of patient education material.)
Increase the warfarin dose
If the patient truly has hereditary resistance, there are two approaches to treatment.
The first is to increase the warfarin dose until the prothrombin time and INR are in the therapeutic ranges. When indicated, the warfarin dose can be safely titrated upward to more than 100 mg per day in patients who are monitored regularly—as all patients on chronic warfarin therapy should be—and whose other medications are otherwise stable. One such example is reported in a warfarinresistant patient who needed 145 mg/day to maintain a therapeutic prothrombin time.22
Try other anticoagulants?
The second approach is to change to another type of anticoagulant. However, there is no strong evidence in favor of this approach over prescribing larger dosages of warfarin.
Other anticoagulant drugs currently available in the United States include subcutaneous heparins (unfractionated and low-molecular-weight heparins) and the subcutaneous factor Xa inhibitor fondaparinux (Arixtra).
Agents not available in the United States include the following.
Dabigatran, an oral direct thrombin inhibitor, is undergoing phase 3 studies of its use for long-term anticoagulation.
Rivaroxaban (a direct factor Xa inhibitor) and dabigatran have been approved in Canada and the European Union to prevent venous thromboembolism after knee and hip arthroplasty, based on prospective comparisons with enoxaparin (Lovenox).34–37
Vitamin K antagonists other than warfarin that are not available in the United States include bishydroxycoumarin (which has limitations including slow absorption and high frequency of gastrointestinal side effects), phenprocoumon, and acenocoumarol. Another is phenindione, which has been associated with serious hypersensitivity reactions, some of which proved fatal and occurred within a few weeks of initiating therapy.
- Lefrere JJ, Horellou MH, Conard J, Samama M. Proposed classification of resistance to oral anticoagulant therapy. J Clin Pathol 1987; 40:242.
- Linder MW. Genetic mechanisms for hypersensitivity and resistance to the anticoagulant warfarin. Clin Chim Acta 2001; 308:9–15.
- Thijssen HH. Warfarin resistance. Vitamin K epoxide reductase of Scottish resistance gene is not irreversibly blocked by warfarin. Biochem Pharmacol 1987; 36:2753–2757.
- Hulse ML. Warfarin resistance: diagnosis and therapeutic alternative. Pharmacotherapy 1996; 16:1009–1017.
- Hirsh J, Dalen JE, Deykin D, Poller L, Bussey H. Oral anticoagulants. Mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 1995; 108( suppl 4):231S–234S.
- Daly AK, King BP. Pharmacogenetics of oral anticoagulants. Pharmacogenetics 2003; 13:247–252.
- Daly AK, Aithal GP. Genetic regulation of warfarin metabolism and response. Semin Vasc Med 2003; 3:231–238.
- Takahashi H, Echizen H. Pharmacogenetics of warfarin elimination and its clinical implications. Clin Pharmacokinet 2001; 40:587–603.
- Retti AE, Wienkers LC, Gonzalez FJ, Trager WF, Korezekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allele variant of CYP2C9. Pharmacogenetics 1994; 4:39–42.
- Porter RS, Sawyer WR. Warfarin. In:Evans WE, Shentag JJ, Jusko WJ, editors. Applied Pharmacokinetics. Principles of Therapeutics Drug Monitoring, 3rd ed. Washington, DC: Applied Therapeutics, 1992: 31.1–31.46.
- Warrell DA, Cox TM, Firth JD. Oxford Textbook of Medicine, 4th ed. Oxford University Press, 2003:734.
- Holbrook AM, Pereira JA, Labiris R, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med 2005; 165:1095–1106.
- Medical Economics Staff. Physicians’ Desk Reference, 55th Ed. Medical Economics, 2001:1139–1140.
- Schwarz UI, Ritchie MD, Bradford Y, et al. Genetic determinants of response to warfarin during initial anticoagulation. N Engl J Med 2008; 358:999–1008.
- Diab F, Feffer S. Hereditary warfarin resistance. South Med J 1994; 87:407–409.
- O’Reilly RA. The second reported kindred with hereditary resistance to oral anticoagulant drugs. N Engl J Med 1970; 282:1448–1451.
- O’Reilly RA, Aggeler PM, Hoag MS, Leong LS, Kropatkin ML. Hereditary transmission of exceptional resistance to coumarin anticoagulant drugs. The first reported kindred. N Engl J Med 1964; 271:809–815.
- Alving BM, Strickler MP, Knight RD, Barr CF, Berenberg JL, Peek CC. Hereditary warfarin resistance. Investigation of rare phenomenon. Arch Intern Med 1985; 145:499–501.
- Warrier L, Brennan CA, Lusher JM. Familial warfarin resistance in a black child. Am J Pediatr Hematol Oncol 1986; 8:346–347.
- Nikkila EA, Pelkonen R. Serum lipid-reducing agents and anticoagulant requirement. Lancet 1963; 1:332.
- Robinson A, Liau FO, Routledge PA, Backhouse G, Spragg BP, Bentley DP. Lipids and warfarin requirements. Thromb Haemost 1990; 63:148–149.
- MacLaren R, Wachsman BA, Swift DK, Kuhl DA. Warfarin resistance associated with intravenous lipid administration: discussion of propofol and review of the literature. Pharmacotherapy 1997; 17:1331–1337.
- DeCurtis A, D’Adamo MC, Amore C, et al. Experimental arterial thrombosis in genetically or diet induced hyperlipidemia in rats—role of vitamin K-dependent clotting factors and prevention by low-intensity oral anticoagulation. Thromb Haemost 2001; 86:1440–1448.
- O’Reilly RA. Drug interaction involving oral anticoagulation. In:Melmon KL, editor. Cardiovascular Drug Therapy, Philadelphia; FA Davis, 1975:23–41.
- O’ Reilly RA, Pool JG, Aggeler PM. Hereditary resistance to coumarin anticoagulation drugs in man and rat. Ann N Y Acad Sci 1968; 151:913–931.
- Cain D, Hutson SM, Wallin R. Warfarin resistance is associated with a protein component of the vitamin K 2,3-epoxide reductase enzyme complex in rat liver. Thromb Haemost 1998; 80:128–133.
- Rodvold KA, Quandt CM, Friedenberg WR. Thromboembolic disorders. In:DiPiro JT, Talbert RL, editors. Pharmacotherapy. A Pathophysiologic Approach, 2nd ed. New York: Elsevier, 1992:312–335.
- Park BK. Warfarin: metabolism and mode of action. Biochem Pharmacol 1988; 37:19–27.
- Cain D, Hutson SM, Wallin R. Assembly of the warfarin-sensitive vitamin K 2,3-epoxide reductase enzyme complex in the endoplasmic reticulum membrane. J Biol Chem 1997; 272:29068–29075.
- Gallop PM, Lian JB, Hauschka PV. Carboxylated calcium binding proteins and vitamin K. N Engl J Med 1980; 302:1460–1466.
- Rost S, Fregin A, Ivaskevicius V, et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 2004; 427:537–541.
- Loebstein R, Dovskin I, Halkin H, et al. A coding VKORC1 Asp36-Tyr polymorphism predisposes to warfarin resistance. Blood 2007; 109:2477–2480.
- Bentley DP, Backhouse G, Hutchings A, Haddon RL, Spragg B, Routledge PA. Investigation of patients with abnormal response to warfarin. Br J Clin Pharmacol 1986; 22:37–41.
- Eriksson BI, Borris LC, Friedman RJ, et al. RECORD1 Study Group. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med 2008; 358:2765–2775.
- Kakkar AK, Brenner B, Dahl OE, et al; RECORD2 Investigators. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial. Lancet 2008; 372:31–39.
- Lassen MR, Ageno W, Borris LC, et al; RECORD3 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med 2008; 358:2776–2786.
- Wolowacz SE, Roskell NS, Plumb JM, Caprini JA, Eriksson BI. Efficacy and safety of dabigatran etexilate for the prevention of venous thromboembolism following total hip or knee arthroplasty. A meta-analysis. Thromb Haemost 2009; 101:77–85.
Warfarin (coumadin) differs from most other drugs in that the dosage required to achieve a desired therapeutic effect varies greatly among individuals. This variability can lead to therapeutic failure, potentially resulting in new thrombosis, or, at the other extreme, to life-threatening bleeding.
Further, there is no reliable means to identify patients who require unusually high doses of warfarin, although genetic testing may become available in the future.
See related patient information
Warfarin, a coumarin derivative first synthesized in 1948, is still the only oral anticoagulant available for long-term use in the United States. Indications for its use include the treatment and, to a lesser extent, the prevention of arterial and venous thromboembolism. It is also used for long-term anticoagulation in patients with atrial arrhythmias (atrial fibrillation and atrial flutter) and mechanical heart valves.
In the paragraphs that follow, we review the causes of warfarin resistance and how to recognize and manage it.
WHAT IS WARFARIN RESISTANCE?
Resistance to warfarin has been described as the inability to prolong the prothrombin time or raise the international normalized ratio (INR) into the therapeutic range when the drug is given at normally prescribed doses.1
However, a higher warfarin requirement does not itself establish the diagnosis of warfarin resistance. The prevalence of warfarin resistance varies by patient population and is difficult to determine. The difficulty lies largely in accounting for dietary factors and in defining normal metabolic variations among individuals.
The range of normally recommended daily or weekly warfarin doses to maintain a therapeutic prothrombin time or INR depends on the study population. Nevertheless, patients who need more than 105 mg per week (15 mg/day) should be considered warfarin-resistant. These patients are likely to be in the top 5% for warfarin doses within an anticoagulated cohort.
Warfarin resistance is different than warfarin failure, which is defined as a new thrombotic event despite a therapeutic prothrombin time and INR. This situation is commonly seen in patients with malignant diseases.
An important characteristic of warfarin resistance is that patients need much smaller doses of vitamin K to reverse the effect of warfarin.2 Thijssen3 showed that, in warfarin-resistant rats, warfarin did not irreversibly inhibit vitamin K1 2,3-epoxide reductase (VKORC1) activity. This is consistent with the vitamin K hypersensitivity observed in warfarin-resistant people.2,3
WHAT CAUSES WARFARIN RESISTANCE?
Warfarin resistance can be classified in practical terms as acquired vs hereditary, or in mechanistic terms as pharmacokinetic vs pharmacodynamic.
Acquired vs hereditary resistance
Hulse4 categorizes warfarin resistance as either acquired or hereditary.
Acquired resistance to warfarin may result from:
- Poor patient compliance (the most common cause)
- High consumption of vitamin K
- Increased clearance (see Warfarin is metabolized by P450 enzymes5–11)
- Drug interactions (Table 1).12,13
Hereditary resistance has been postulated to be caused by genetic factors that result either in faster metabolism of the drug (a form of pharmacokinetic resistance) or in lower activity of the drug (pharmacodynamic resistance). Polymorphisms may play a role, as some VKORC1 and CYP2C9 variant alleles are known to be associated with increased sensitivity to warfarin.14
However, the genetic mechanisms of warfarin resistance are not clearly understood, despite several case reports of hereditary resistance confirmed by similar patterns of resistance in immediate family members.15–19 More than one mechanism is likely. There is ample room for further insight into genetic polymorphisms underlying hereditary warfarin resistance. More on this topic is included in the sections below.
Pharmacokinetic resistance
Pharmacokinetic resistance can result from diminished absorption or increased elimination of the drug. Causes of diminished absorption include emesis, diarrhea, and malabsorption syndrome.
The mechanism of increased warfarin clearance has not been delineated, although the following have been implicated.
Genetic factors. Duplication or multiplication of cytochrome P450 enzyme genes has been described as contributing to a phenotype of ultrarapid metabolism. Some people may carry multiple copies of the CYP2C9 gene, as has already been reported for cytochrome P450 CYP2D6 and CYP2A6.7,8 It is also plausible that rare allelic variants of CYP2C9 exist that are associated with higher-than-normal activity, given that there are alleles known to predispose to warfarin sensitivity.
Hypoalbuminemia may increase the free fraction of warfarin, leading to enhanced rates of clearance and a shorter plasma half-life.15
Hyperalbuminemia may paradoxically also contribute to warfarin resistance via drug binding.
Hyperlipidemia. Several observers have found that lowering serum lipids, primarily triglycerides, increases the sensitivity to warfarin irrespective of the means used to achieve this decrease.20 This most likely results in a decreased pool of vitamin K, some of which is bound to triglycerides.21 Conversely, patients receiving intravenous lipids with total parenteral nutrition have also been diagnosed clinically with warfarin resistance,22 and rat models have shown an association between a lipidrich diet and increased vitamin K-dependent factor activity.23
Diuretics may decrease the response to warfarin by reducing the plasma volume, with a subsequent increase in clotting factor activity.24
Pharmacodynamic resistance
Potential mechanisms of pharmacodynamic warfarin resistance described in rats and in people include:
- Increased affinity of vitamin K1, 2,3-epoxide reductase complex (VKOR) for vitamin K25,26 (see How warfarin works2,10,11,27–30)
- Prolongation of normal clotting factor activity16
- Production of clotting factors that is not dependent on vitamin K16
- Decreased VKOR sensitivity to warfarin.26
In rats, these mechanisms are manifested by relatively high doses of warfarin being required to achieve poisoning. In humans, they result in high doses being needed to achieve a therapeutic effect in the setting of normal warfarin pharmacokinetics, normal warfarin concentration, and normal half-lives of blood clotting proteins.
Genetics of pharmacodynamic resistance. Pharmacodynamic warfarin resistance has also been described with inheritance of a monogenetic dominant trait. An early study by O’Reilly24 traced anticoagulation resistance to a genetically linked abnormality of interaction between warfarin and a putative vitamin K receptor.
In one patient with hereditary resistance and high warfarin requirements, a heterozygous point mutation in the VKORC1 gene was identified.31 This results in a substitution that lies in a conserved (normally constant or unchanging DNA sequence in a genome) region of VKORC1 that contains three of four previously identified amino acid substitutions associated with warfarin resistance (Val29Leu, Val45Ala, and Arg58Gly). Further investigation is required to fully characterize the structure-function relationship for VKORC1 and to determine the relationship between the VKORC1 genotype and other pharmacogenetic determinants of warfarin dose-response.
Separately, Loebstein et al32 reported a new mutation, Asp36Tyr, which was common in Jewish ethnic groups of Ethiopian descent (in whom the prevalence is 5%) and Ashkenazi descent (prevalence 4%). In that study, Asp36Tyr carriers needed doses of more than 70 mg per week, placing them towards the high end of the usual warfarin dosing range.
Daly and Aithal7 discovered that warfarinresistant rats overexpressed a protein known as calumenin. This protein is situated in the endoplasmic reticulum and appears to interact with VKOR, decreasing the binding of warfarin. In mice, the calumenin gene is located on chromosome 7, where the gene for VKORC1 is also located.
DIAGNOSIS BY HISTORY AND LABORATORY STUDIES
A full drug and diet history is invaluable in diagnosing potential causes of warfarin resistance (Table 1).
Plasma warfarin levels that are subtherapeutic should raise suspicion of intestinal malabsorption or poor compliance. Poor compliance might be more appropriately seen as a mimic of warfarin resistance. Studies in humans suggest that a therapeutic total plasma warfarin level lies between 0.5 μg/mL and 3.0 μg/mL,10 though the range may vary among laboratories and patient populations.
Warfarin absorption and clearance can be evaluated by analyzing plasma levels at specific intervals after administration, eg, every 60 to 180 minutes. The drug’s half-life can be determined on the basis of its concentrations in different time samples. Normally, the S-enantiomer of warfarin is cleared at twice the rate of the R-enantiomer (5.2 vs 2.5 mL/min/70 kg).8 A normal clearance rate confirms that resistance to warfarin is not due to enhanced elimination.
Clotting assays of factors II, VII, IX, and X may be a more precise way to assess the pharmacodynamics of warfarin,10 although there is no strong evidence to support routine use of such assays. Some studies suggest targeting factor II and factor X activity levels of 10% to 30% of normal biologic activity for a therapeutic warfarin effect in patients with an unreliable or prolonged baseline prothrombin time and INR, such as those with lupus anticoagulant.
TREAT THE CAUSE
Once the type of warfarin resistance has been determined, treatment should be oriented toward the cause.
Educate the patient
The importance of compliance should be reinforced. Educating the patient about diet and other medications that may interact with warfarin is also important. (See an example of patient education material.)
Increase the warfarin dose
If the patient truly has hereditary resistance, there are two approaches to treatment.
The first is to increase the warfarin dose until the prothrombin time and INR are in the therapeutic ranges. When indicated, the warfarin dose can be safely titrated upward to more than 100 mg per day in patients who are monitored regularly—as all patients on chronic warfarin therapy should be—and whose other medications are otherwise stable. One such example is reported in a warfarinresistant patient who needed 145 mg/day to maintain a therapeutic prothrombin time.22
Try other anticoagulants?
The second approach is to change to another type of anticoagulant. However, there is no strong evidence in favor of this approach over prescribing larger dosages of warfarin.
Other anticoagulant drugs currently available in the United States include subcutaneous heparins (unfractionated and low-molecular-weight heparins) and the subcutaneous factor Xa inhibitor fondaparinux (Arixtra).
Agents not available in the United States include the following.
Dabigatran, an oral direct thrombin inhibitor, is undergoing phase 3 studies of its use for long-term anticoagulation.
Rivaroxaban (a direct factor Xa inhibitor) and dabigatran have been approved in Canada and the European Union to prevent venous thromboembolism after knee and hip arthroplasty, based on prospective comparisons with enoxaparin (Lovenox).34–37
Vitamin K antagonists other than warfarin that are not available in the United States include bishydroxycoumarin (which has limitations including slow absorption and high frequency of gastrointestinal side effects), phenprocoumon, and acenocoumarol. Another is phenindione, which has been associated with serious hypersensitivity reactions, some of which proved fatal and occurred within a few weeks of initiating therapy.
Warfarin (coumadin) differs from most other drugs in that the dosage required to achieve a desired therapeutic effect varies greatly among individuals. This variability can lead to therapeutic failure, potentially resulting in new thrombosis, or, at the other extreme, to life-threatening bleeding.
Further, there is no reliable means to identify patients who require unusually high doses of warfarin, although genetic testing may become available in the future.
See related patient information
Warfarin, a coumarin derivative first synthesized in 1948, is still the only oral anticoagulant available for long-term use in the United States. Indications for its use include the treatment and, to a lesser extent, the prevention of arterial and venous thromboembolism. It is also used for long-term anticoagulation in patients with atrial arrhythmias (atrial fibrillation and atrial flutter) and mechanical heart valves.
In the paragraphs that follow, we review the causes of warfarin resistance and how to recognize and manage it.
WHAT IS WARFARIN RESISTANCE?
Resistance to warfarin has been described as the inability to prolong the prothrombin time or raise the international normalized ratio (INR) into the therapeutic range when the drug is given at normally prescribed doses.1
However, a higher warfarin requirement does not itself establish the diagnosis of warfarin resistance. The prevalence of warfarin resistance varies by patient population and is difficult to determine. The difficulty lies largely in accounting for dietary factors and in defining normal metabolic variations among individuals.
The range of normally recommended daily or weekly warfarin doses to maintain a therapeutic prothrombin time or INR depends on the study population. Nevertheless, patients who need more than 105 mg per week (15 mg/day) should be considered warfarin-resistant. These patients are likely to be in the top 5% for warfarin doses within an anticoagulated cohort.
Warfarin resistance is different than warfarin failure, which is defined as a new thrombotic event despite a therapeutic prothrombin time and INR. This situation is commonly seen in patients with malignant diseases.
An important characteristic of warfarin resistance is that patients need much smaller doses of vitamin K to reverse the effect of warfarin.2 Thijssen3 showed that, in warfarin-resistant rats, warfarin did not irreversibly inhibit vitamin K1 2,3-epoxide reductase (VKORC1) activity. This is consistent with the vitamin K hypersensitivity observed in warfarin-resistant people.2,3
WHAT CAUSES WARFARIN RESISTANCE?
Warfarin resistance can be classified in practical terms as acquired vs hereditary, or in mechanistic terms as pharmacokinetic vs pharmacodynamic.
Acquired vs hereditary resistance
Hulse4 categorizes warfarin resistance as either acquired or hereditary.
Acquired resistance to warfarin may result from:
- Poor patient compliance (the most common cause)
- High consumption of vitamin K
- Increased clearance (see Warfarin is metabolized by P450 enzymes5–11)
- Drug interactions (Table 1).12,13
Hereditary resistance has been postulated to be caused by genetic factors that result either in faster metabolism of the drug (a form of pharmacokinetic resistance) or in lower activity of the drug (pharmacodynamic resistance). Polymorphisms may play a role, as some VKORC1 and CYP2C9 variant alleles are known to be associated with increased sensitivity to warfarin.14
However, the genetic mechanisms of warfarin resistance are not clearly understood, despite several case reports of hereditary resistance confirmed by similar patterns of resistance in immediate family members.15–19 More than one mechanism is likely. There is ample room for further insight into genetic polymorphisms underlying hereditary warfarin resistance. More on this topic is included in the sections below.
Pharmacokinetic resistance
Pharmacokinetic resistance can result from diminished absorption or increased elimination of the drug. Causes of diminished absorption include emesis, diarrhea, and malabsorption syndrome.
The mechanism of increased warfarin clearance has not been delineated, although the following have been implicated.
Genetic factors. Duplication or multiplication of cytochrome P450 enzyme genes has been described as contributing to a phenotype of ultrarapid metabolism. Some people may carry multiple copies of the CYP2C9 gene, as has already been reported for cytochrome P450 CYP2D6 and CYP2A6.7,8 It is also plausible that rare allelic variants of CYP2C9 exist that are associated with higher-than-normal activity, given that there are alleles known to predispose to warfarin sensitivity.
Hypoalbuminemia may increase the free fraction of warfarin, leading to enhanced rates of clearance and a shorter plasma half-life.15
Hyperalbuminemia may paradoxically also contribute to warfarin resistance via drug binding.
Hyperlipidemia. Several observers have found that lowering serum lipids, primarily triglycerides, increases the sensitivity to warfarin irrespective of the means used to achieve this decrease.20 This most likely results in a decreased pool of vitamin K, some of which is bound to triglycerides.21 Conversely, patients receiving intravenous lipids with total parenteral nutrition have also been diagnosed clinically with warfarin resistance,22 and rat models have shown an association between a lipidrich diet and increased vitamin K-dependent factor activity.23
Diuretics may decrease the response to warfarin by reducing the plasma volume, with a subsequent increase in clotting factor activity.24
Pharmacodynamic resistance
Potential mechanisms of pharmacodynamic warfarin resistance described in rats and in people include:
- Increased affinity of vitamin K1, 2,3-epoxide reductase complex (VKOR) for vitamin K25,26 (see How warfarin works2,10,11,27–30)
- Prolongation of normal clotting factor activity16
- Production of clotting factors that is not dependent on vitamin K16
- Decreased VKOR sensitivity to warfarin.26
In rats, these mechanisms are manifested by relatively high doses of warfarin being required to achieve poisoning. In humans, they result in high doses being needed to achieve a therapeutic effect in the setting of normal warfarin pharmacokinetics, normal warfarin concentration, and normal half-lives of blood clotting proteins.
Genetics of pharmacodynamic resistance. Pharmacodynamic warfarin resistance has also been described with inheritance of a monogenetic dominant trait. An early study by O’Reilly24 traced anticoagulation resistance to a genetically linked abnormality of interaction between warfarin and a putative vitamin K receptor.
In one patient with hereditary resistance and high warfarin requirements, a heterozygous point mutation in the VKORC1 gene was identified.31 This results in a substitution that lies in a conserved (normally constant or unchanging DNA sequence in a genome) region of VKORC1 that contains three of four previously identified amino acid substitutions associated with warfarin resistance (Val29Leu, Val45Ala, and Arg58Gly). Further investigation is required to fully characterize the structure-function relationship for VKORC1 and to determine the relationship between the VKORC1 genotype and other pharmacogenetic determinants of warfarin dose-response.
Separately, Loebstein et al32 reported a new mutation, Asp36Tyr, which was common in Jewish ethnic groups of Ethiopian descent (in whom the prevalence is 5%) and Ashkenazi descent (prevalence 4%). In that study, Asp36Tyr carriers needed doses of more than 70 mg per week, placing them towards the high end of the usual warfarin dosing range.
Daly and Aithal7 discovered that warfarinresistant rats overexpressed a protein known as calumenin. This protein is situated in the endoplasmic reticulum and appears to interact with VKOR, decreasing the binding of warfarin. In mice, the calumenin gene is located on chromosome 7, where the gene for VKORC1 is also located.
DIAGNOSIS BY HISTORY AND LABORATORY STUDIES
A full drug and diet history is invaluable in diagnosing potential causes of warfarin resistance (Table 1).
Plasma warfarin levels that are subtherapeutic should raise suspicion of intestinal malabsorption or poor compliance. Poor compliance might be more appropriately seen as a mimic of warfarin resistance. Studies in humans suggest that a therapeutic total plasma warfarin level lies between 0.5 μg/mL and 3.0 μg/mL,10 though the range may vary among laboratories and patient populations.
Warfarin absorption and clearance can be evaluated by analyzing plasma levels at specific intervals after administration, eg, every 60 to 180 minutes. The drug’s half-life can be determined on the basis of its concentrations in different time samples. Normally, the S-enantiomer of warfarin is cleared at twice the rate of the R-enantiomer (5.2 vs 2.5 mL/min/70 kg).8 A normal clearance rate confirms that resistance to warfarin is not due to enhanced elimination.
Clotting assays of factors II, VII, IX, and X may be a more precise way to assess the pharmacodynamics of warfarin,10 although there is no strong evidence to support routine use of such assays. Some studies suggest targeting factor II and factor X activity levels of 10% to 30% of normal biologic activity for a therapeutic warfarin effect in patients with an unreliable or prolonged baseline prothrombin time and INR, such as those with lupus anticoagulant.
TREAT THE CAUSE
Once the type of warfarin resistance has been determined, treatment should be oriented toward the cause.
Educate the patient
The importance of compliance should be reinforced. Educating the patient about diet and other medications that may interact with warfarin is also important. (See an example of patient education material.)
Increase the warfarin dose
If the patient truly has hereditary resistance, there are two approaches to treatment.
The first is to increase the warfarin dose until the prothrombin time and INR are in the therapeutic ranges. When indicated, the warfarin dose can be safely titrated upward to more than 100 mg per day in patients who are monitored regularly—as all patients on chronic warfarin therapy should be—and whose other medications are otherwise stable. One such example is reported in a warfarinresistant patient who needed 145 mg/day to maintain a therapeutic prothrombin time.22
Try other anticoagulants?
The second approach is to change to another type of anticoagulant. However, there is no strong evidence in favor of this approach over prescribing larger dosages of warfarin.
Other anticoagulant drugs currently available in the United States include subcutaneous heparins (unfractionated and low-molecular-weight heparins) and the subcutaneous factor Xa inhibitor fondaparinux (Arixtra).
Agents not available in the United States include the following.
Dabigatran, an oral direct thrombin inhibitor, is undergoing phase 3 studies of its use for long-term anticoagulation.
Rivaroxaban (a direct factor Xa inhibitor) and dabigatran have been approved in Canada and the European Union to prevent venous thromboembolism after knee and hip arthroplasty, based on prospective comparisons with enoxaparin (Lovenox).34–37
Vitamin K antagonists other than warfarin that are not available in the United States include bishydroxycoumarin (which has limitations including slow absorption and high frequency of gastrointestinal side effects), phenprocoumon, and acenocoumarol. Another is phenindione, which has been associated with serious hypersensitivity reactions, some of which proved fatal and occurred within a few weeks of initiating therapy.
- Lefrere JJ, Horellou MH, Conard J, Samama M. Proposed classification of resistance to oral anticoagulant therapy. J Clin Pathol 1987; 40:242.
- Linder MW. Genetic mechanisms for hypersensitivity and resistance to the anticoagulant warfarin. Clin Chim Acta 2001; 308:9–15.
- Thijssen HH. Warfarin resistance. Vitamin K epoxide reductase of Scottish resistance gene is not irreversibly blocked by warfarin. Biochem Pharmacol 1987; 36:2753–2757.
- Hulse ML. Warfarin resistance: diagnosis and therapeutic alternative. Pharmacotherapy 1996; 16:1009–1017.
- Hirsh J, Dalen JE, Deykin D, Poller L, Bussey H. Oral anticoagulants. Mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 1995; 108( suppl 4):231S–234S.
- Daly AK, King BP. Pharmacogenetics of oral anticoagulants. Pharmacogenetics 2003; 13:247–252.
- Daly AK, Aithal GP. Genetic regulation of warfarin metabolism and response. Semin Vasc Med 2003; 3:231–238.
- Takahashi H, Echizen H. Pharmacogenetics of warfarin elimination and its clinical implications. Clin Pharmacokinet 2001; 40:587–603.
- Retti AE, Wienkers LC, Gonzalez FJ, Trager WF, Korezekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allele variant of CYP2C9. Pharmacogenetics 1994; 4:39–42.
- Porter RS, Sawyer WR. Warfarin. In:Evans WE, Shentag JJ, Jusko WJ, editors. Applied Pharmacokinetics. Principles of Therapeutics Drug Monitoring, 3rd ed. Washington, DC: Applied Therapeutics, 1992: 31.1–31.46.
- Warrell DA, Cox TM, Firth JD. Oxford Textbook of Medicine, 4th ed. Oxford University Press, 2003:734.
- Holbrook AM, Pereira JA, Labiris R, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med 2005; 165:1095–1106.
- Medical Economics Staff. Physicians’ Desk Reference, 55th Ed. Medical Economics, 2001:1139–1140.
- Schwarz UI, Ritchie MD, Bradford Y, et al. Genetic determinants of response to warfarin during initial anticoagulation. N Engl J Med 2008; 358:999–1008.
- Diab F, Feffer S. Hereditary warfarin resistance. South Med J 1994; 87:407–409.
- O’Reilly RA. The second reported kindred with hereditary resistance to oral anticoagulant drugs. N Engl J Med 1970; 282:1448–1451.
- O’Reilly RA, Aggeler PM, Hoag MS, Leong LS, Kropatkin ML. Hereditary transmission of exceptional resistance to coumarin anticoagulant drugs. The first reported kindred. N Engl J Med 1964; 271:809–815.
- Alving BM, Strickler MP, Knight RD, Barr CF, Berenberg JL, Peek CC. Hereditary warfarin resistance. Investigation of rare phenomenon. Arch Intern Med 1985; 145:499–501.
- Warrier L, Brennan CA, Lusher JM. Familial warfarin resistance in a black child. Am J Pediatr Hematol Oncol 1986; 8:346–347.
- Nikkila EA, Pelkonen R. Serum lipid-reducing agents and anticoagulant requirement. Lancet 1963; 1:332.
- Robinson A, Liau FO, Routledge PA, Backhouse G, Spragg BP, Bentley DP. Lipids and warfarin requirements. Thromb Haemost 1990; 63:148–149.
- MacLaren R, Wachsman BA, Swift DK, Kuhl DA. Warfarin resistance associated with intravenous lipid administration: discussion of propofol and review of the literature. Pharmacotherapy 1997; 17:1331–1337.
- DeCurtis A, D’Adamo MC, Amore C, et al. Experimental arterial thrombosis in genetically or diet induced hyperlipidemia in rats—role of vitamin K-dependent clotting factors and prevention by low-intensity oral anticoagulation. Thromb Haemost 2001; 86:1440–1448.
- O’Reilly RA. Drug interaction involving oral anticoagulation. In:Melmon KL, editor. Cardiovascular Drug Therapy, Philadelphia; FA Davis, 1975:23–41.
- O’ Reilly RA, Pool JG, Aggeler PM. Hereditary resistance to coumarin anticoagulation drugs in man and rat. Ann N Y Acad Sci 1968; 151:913–931.
- Cain D, Hutson SM, Wallin R. Warfarin resistance is associated with a protein component of the vitamin K 2,3-epoxide reductase enzyme complex in rat liver. Thromb Haemost 1998; 80:128–133.
- Rodvold KA, Quandt CM, Friedenberg WR. Thromboembolic disorders. In:DiPiro JT, Talbert RL, editors. Pharmacotherapy. A Pathophysiologic Approach, 2nd ed. New York: Elsevier, 1992:312–335.
- Park BK. Warfarin: metabolism and mode of action. Biochem Pharmacol 1988; 37:19–27.
- Cain D, Hutson SM, Wallin R. Assembly of the warfarin-sensitive vitamin K 2,3-epoxide reductase enzyme complex in the endoplasmic reticulum membrane. J Biol Chem 1997; 272:29068–29075.
- Gallop PM, Lian JB, Hauschka PV. Carboxylated calcium binding proteins and vitamin K. N Engl J Med 1980; 302:1460–1466.
- Rost S, Fregin A, Ivaskevicius V, et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 2004; 427:537–541.
- Loebstein R, Dovskin I, Halkin H, et al. A coding VKORC1 Asp36-Tyr polymorphism predisposes to warfarin resistance. Blood 2007; 109:2477–2480.
- Bentley DP, Backhouse G, Hutchings A, Haddon RL, Spragg B, Routledge PA. Investigation of patients with abnormal response to warfarin. Br J Clin Pharmacol 1986; 22:37–41.
- Eriksson BI, Borris LC, Friedman RJ, et al. RECORD1 Study Group. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med 2008; 358:2765–2775.
- Kakkar AK, Brenner B, Dahl OE, et al; RECORD2 Investigators. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial. Lancet 2008; 372:31–39.
- Lassen MR, Ageno W, Borris LC, et al; RECORD3 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med 2008; 358:2776–2786.
- Wolowacz SE, Roskell NS, Plumb JM, Caprini JA, Eriksson BI. Efficacy and safety of dabigatran etexilate for the prevention of venous thromboembolism following total hip or knee arthroplasty. A meta-analysis. Thromb Haemost 2009; 101:77–85.
- Lefrere JJ, Horellou MH, Conard J, Samama M. Proposed classification of resistance to oral anticoagulant therapy. J Clin Pathol 1987; 40:242.
- Linder MW. Genetic mechanisms for hypersensitivity and resistance to the anticoagulant warfarin. Clin Chim Acta 2001; 308:9–15.
- Thijssen HH. Warfarin resistance. Vitamin K epoxide reductase of Scottish resistance gene is not irreversibly blocked by warfarin. Biochem Pharmacol 1987; 36:2753–2757.
- Hulse ML. Warfarin resistance: diagnosis and therapeutic alternative. Pharmacotherapy 1996; 16:1009–1017.
- Hirsh J, Dalen JE, Deykin D, Poller L, Bussey H. Oral anticoagulants. Mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 1995; 108( suppl 4):231S–234S.
- Daly AK, King BP. Pharmacogenetics of oral anticoagulants. Pharmacogenetics 2003; 13:247–252.
- Daly AK, Aithal GP. Genetic regulation of warfarin metabolism and response. Semin Vasc Med 2003; 3:231–238.
- Takahashi H, Echizen H. Pharmacogenetics of warfarin elimination and its clinical implications. Clin Pharmacokinet 2001; 40:587–603.
- Retti AE, Wienkers LC, Gonzalez FJ, Trager WF, Korezekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allele variant of CYP2C9. Pharmacogenetics 1994; 4:39–42.
- Porter RS, Sawyer WR. Warfarin. In:Evans WE, Shentag JJ, Jusko WJ, editors. Applied Pharmacokinetics. Principles of Therapeutics Drug Monitoring, 3rd ed. Washington, DC: Applied Therapeutics, 1992: 31.1–31.46.
- Warrell DA, Cox TM, Firth JD. Oxford Textbook of Medicine, 4th ed. Oxford University Press, 2003:734.
- Holbrook AM, Pereira JA, Labiris R, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med 2005; 165:1095–1106.
- Medical Economics Staff. Physicians’ Desk Reference, 55th Ed. Medical Economics, 2001:1139–1140.
- Schwarz UI, Ritchie MD, Bradford Y, et al. Genetic determinants of response to warfarin during initial anticoagulation. N Engl J Med 2008; 358:999–1008.
- Diab F, Feffer S. Hereditary warfarin resistance. South Med J 1994; 87:407–409.
- O’Reilly RA. The second reported kindred with hereditary resistance to oral anticoagulant drugs. N Engl J Med 1970; 282:1448–1451.
- O’Reilly RA, Aggeler PM, Hoag MS, Leong LS, Kropatkin ML. Hereditary transmission of exceptional resistance to coumarin anticoagulant drugs. The first reported kindred. N Engl J Med 1964; 271:809–815.
- Alving BM, Strickler MP, Knight RD, Barr CF, Berenberg JL, Peek CC. Hereditary warfarin resistance. Investigation of rare phenomenon. Arch Intern Med 1985; 145:499–501.
- Warrier L, Brennan CA, Lusher JM. Familial warfarin resistance in a black child. Am J Pediatr Hematol Oncol 1986; 8:346–347.
- Nikkila EA, Pelkonen R. Serum lipid-reducing agents and anticoagulant requirement. Lancet 1963; 1:332.
- Robinson A, Liau FO, Routledge PA, Backhouse G, Spragg BP, Bentley DP. Lipids and warfarin requirements. Thromb Haemost 1990; 63:148–149.
- MacLaren R, Wachsman BA, Swift DK, Kuhl DA. Warfarin resistance associated with intravenous lipid administration: discussion of propofol and review of the literature. Pharmacotherapy 1997; 17:1331–1337.
- DeCurtis A, D’Adamo MC, Amore C, et al. Experimental arterial thrombosis in genetically or diet induced hyperlipidemia in rats—role of vitamin K-dependent clotting factors and prevention by low-intensity oral anticoagulation. Thromb Haemost 2001; 86:1440–1448.
- O’Reilly RA. Drug interaction involving oral anticoagulation. In:Melmon KL, editor. Cardiovascular Drug Therapy, Philadelphia; FA Davis, 1975:23–41.
- O’ Reilly RA, Pool JG, Aggeler PM. Hereditary resistance to coumarin anticoagulation drugs in man and rat. Ann N Y Acad Sci 1968; 151:913–931.
- Cain D, Hutson SM, Wallin R. Warfarin resistance is associated with a protein component of the vitamin K 2,3-epoxide reductase enzyme complex in rat liver. Thromb Haemost 1998; 80:128–133.
- Rodvold KA, Quandt CM, Friedenberg WR. Thromboembolic disorders. In:DiPiro JT, Talbert RL, editors. Pharmacotherapy. A Pathophysiologic Approach, 2nd ed. New York: Elsevier, 1992:312–335.
- Park BK. Warfarin: metabolism and mode of action. Biochem Pharmacol 1988; 37:19–27.
- Cain D, Hutson SM, Wallin R. Assembly of the warfarin-sensitive vitamin K 2,3-epoxide reductase enzyme complex in the endoplasmic reticulum membrane. J Biol Chem 1997; 272:29068–29075.
- Gallop PM, Lian JB, Hauschka PV. Carboxylated calcium binding proteins and vitamin K. N Engl J Med 1980; 302:1460–1466.
- Rost S, Fregin A, Ivaskevicius V, et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 2004; 427:537–541.
- Loebstein R, Dovskin I, Halkin H, et al. A coding VKORC1 Asp36-Tyr polymorphism predisposes to warfarin resistance. Blood 2007; 109:2477–2480.
- Bentley DP, Backhouse G, Hutchings A, Haddon RL, Spragg B, Routledge PA. Investigation of patients with abnormal response to warfarin. Br J Clin Pharmacol 1986; 22:37–41.
- Eriksson BI, Borris LC, Friedman RJ, et al. RECORD1 Study Group. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med 2008; 358:2765–2775.
- Kakkar AK, Brenner B, Dahl OE, et al; RECORD2 Investigators. Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial. Lancet 2008; 372:31–39.
- Lassen MR, Ageno W, Borris LC, et al; RECORD3 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med 2008; 358:2776–2786.
- Wolowacz SE, Roskell NS, Plumb JM, Caprini JA, Eriksson BI. Efficacy and safety of dabigatran etexilate for the prevention of venous thromboembolism following total hip or knee arthroplasty. A meta-analysis. Thromb Haemost 2009; 101:77–85.
KEY POINTS
- The most common cause of warfarin resistance is noncompliance. Others include poor absorption, high vitamin K intake, hypersensitivity to vitamin K, and rapid drug deactivation.
- Patient education is necessary to improve compliance and to mitigate adverse effects of warfarin therapy, regardless of the dose.
- In time, it may be possible to individualize anticoagulant dosing on the basis of genetic testing for patients with warfarin resistance, although currently such tests are not routinely advocated and are usually done only in specialized laboratories.
- In true hereditary warfarin resistance, there are two approaches to treatment: increase the warfarin dosage (perhaps to as high as 100 mg/day or more), or switch to another anticoagulant.